Embed Size (px)
Citation preview

Arzneimittelstudien beim Menschen:
Wissenswertes zur Prüfmedikation:
Inhalt der Investigator’s Brochure
16. Dezember 2009
Prof. Dr. Ingeborg Walter-Sack
Abt. Klinische Pharmakologie und Pharmakoepidemiologie
Medizinische KlinikUniversitätsklinikum Heidelberg
IWS 16.12.2009
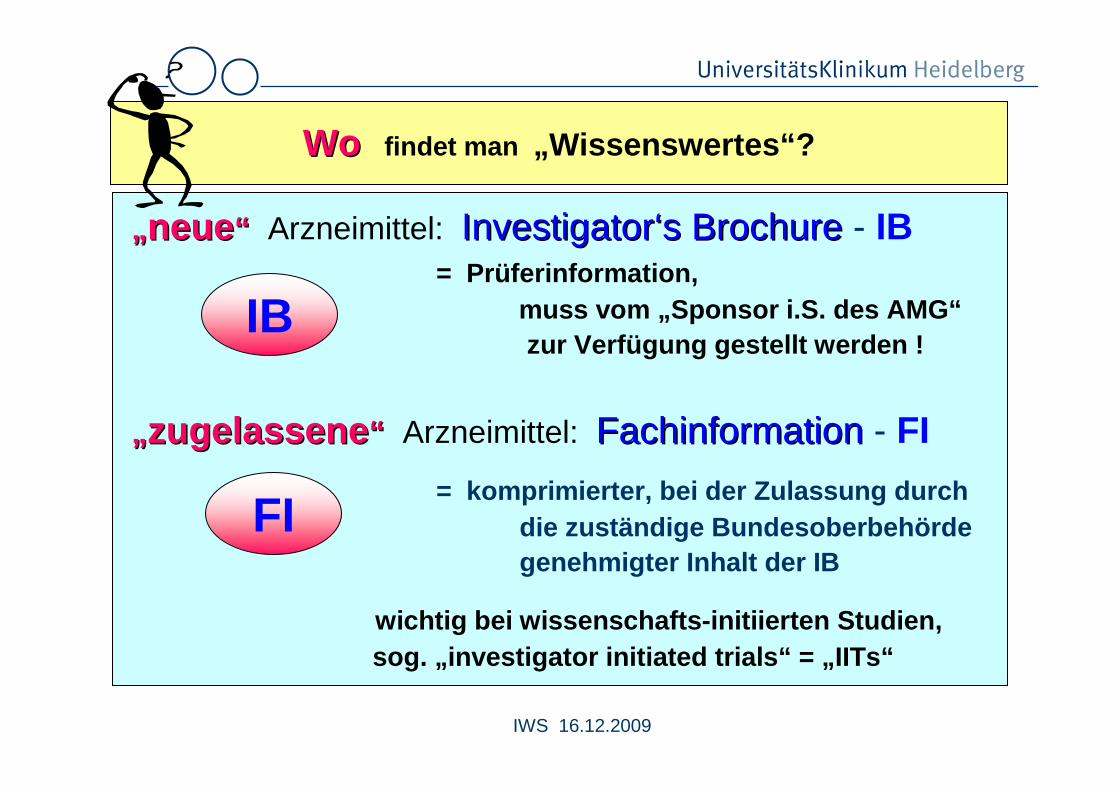
„„ neueneue ““ Arzneimittel: InvestigatorInvestigator‘‘ss BrochureBrochure - IB= Prüferinformation,
muss vom „Sponsor i.S. des AMG“zur Verfügung gestellt werden !
„„ zugelassenezugelassene ““ Arzneimittel: FachinformationFachinformation - FI
= komprimierter, bei der Zulassung durchdie zuständige Bundesoberbehördegenehmigter Inhalt der IB
wichtig bei wissenschafts-initiierten Studien,sog. „investigator initiated trials“ = „IITs“
WoWo findet man „Wissenswertes“?
IB
FI

IWS 16.12.2009
Im Zusammenhang mit Arzneimittelstudien gilt in der Regel:
„neueneue“ Arzneimittel sind (i.d.R.) neue WirkstoffeWirkstoffe ,die zu Handelspräparaten entwickelt werden
Europa: „IInvestigational MMedicinal PProduct“ - „IMPIMP“
nicht zu verwechseln mit Medizinprodukt = „medical device“ !!
USA: „I nvestigational New Drug“ – „ IND“
Was sind „ neue Arzneimittelneue Arzneimittel ““ ??
Mehrfach-Bedeutung des Begriffes „Arzneimittel“ beachten:„Substanz“, „Zubereitung“ [ inkl. Placebo‘s ! ], „Fertigarzneimittel“
IB

IWS 16.12.2009
1. Prüfarzneimittel:Prüf-Substanz: physikalische + chemische EigenschaftenDarreichungsformen: pharmazeutische Charakteristika
2. Prä-klinische Studien: ausschließlich tierexperimentelleund in vitro-Daten: Pharmakologie, Pharmakokinetik,
Toxikologie
3. Klinische Daten = Daten beim Menschen ( ! )3.1 Klinische Prüfung (Interventionsstudien)
3.1.1 bei gesunden Probanden3.1.2 bei Patienten
3.2 Verordnete Therapie: Post-marketing -Daten
WasWas ist ist wissenswert: IB IB -- InhaltInhaltIB

IWS 16.12.2009
► EMEAEMEA Europäische Arzneimittelbehörde
http://www.emea.europa.eu ► Human Medicines ►
Scientific Guidelines ► ICH ► Efficacy ►
(R1), Topic E6 Step 5 (July 2002) ►
Note for Guidance on Good Clinical Practice, GCPDokumenten – Code [Doc.Ref. ]: CPMP/ICH/135/95
http://www.emea.europa.eu/pdfs/human/ich/013595en.p df
IB
Kapitel 7: IB
IB:IB: Inhalt Inhalt gemäß GCPGCP--EmpfehlungenEmpfehlungen

IWS 16.12.2009
„„ SynthetischerSynthetischer ““ Stoff, „small molecule“pH-Wert, Aussehen u.a.; oft aufschlussreich: Löslichkeit
hydrophil, gut wasserlöslich: oft Substanzen, die in erheblichem
Umfang unverändert über die Niere ausgeschieden werden
lipophil, fettlöslich: Substanzen, die in erheblichem Umfang
(v.a. in der Leber) metabolisiert werden
► PharmakokinetikPharmakokinetik ► Arzneimittelinteraktionen !Arzneimittelinteraktionen !
„„ BiologikaBiologika ““ , , Proteine u.a. ► PharmakokinetikPharmakokinetik ????Spaltung z.B. von Peptidbindungen fast ubiquitär mög lich !!
Physikalische und chemische Eigenschaften der
Prüfsubstanz
IB
IWS 16.12.2009
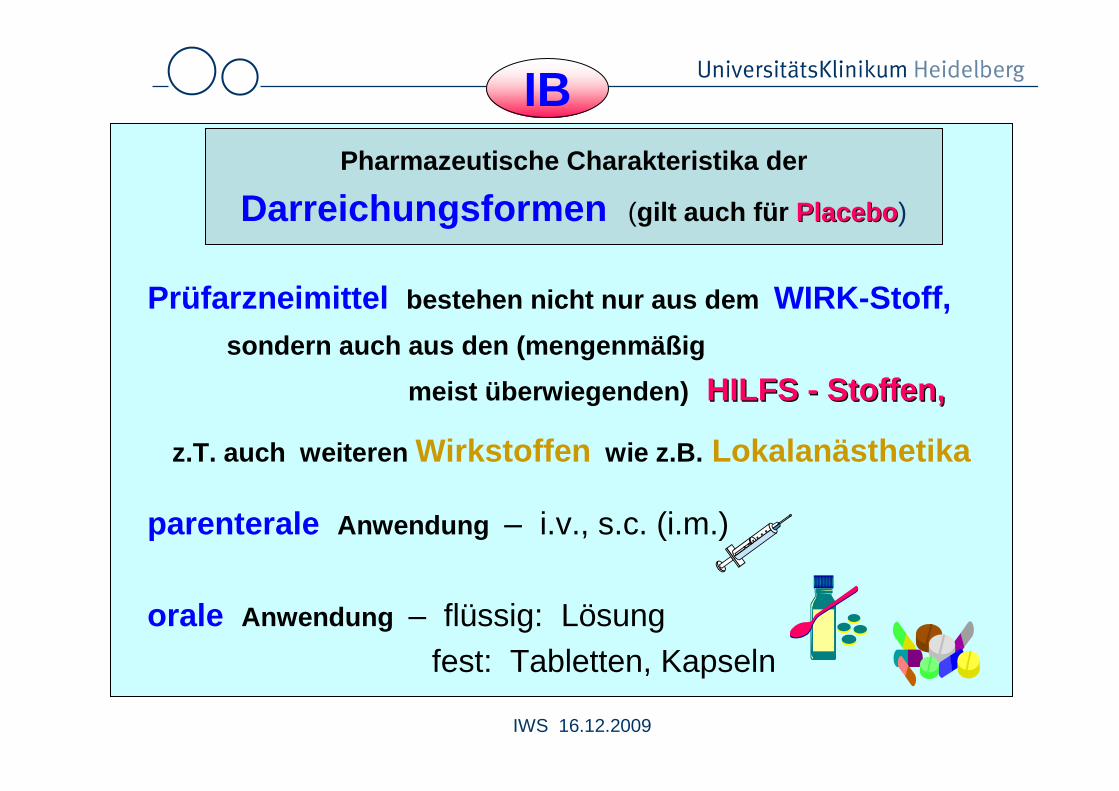
Prüfarzneimittel bestehen nicht nur aus dem WIRK-Stoff,sondern auch aus den (mengenmäßig
meist überwiegenden) HILFS HILFS -- Stoffen,Stoffen,
z.T. auch weiteren Wirkstoffen wie z.B. Lokalanästhetika
parenterale Anwendung – i.v., s.c. (i.m.)
orale Anwendung – flüssig: Lösung fest: Tabletten, Kapseln
IBPharmazeutische Charakteristika der
Darreichungsformen (gilt auch für PlaceboPlacebo )

IWS 16.12.2009
Für jedes Prüfarzneimittel muss die IB u.a. enthalte n:
→ Lagerungsvorschriften (z.B. Kühlschrank)
→ Haltbarkeitsdauer→ Auflistung aller (!) Inhaltsstoffe
► auf Begleitstoff Begleitstoff -- UnvertrUnvertr ääglichkeitenglichkeiten achten !z.B. Konservierungsstoffe, Sulfite, Lokalanästhetika u.a.
[Sojabohnenöl in Generika: NEJM 2009;361:1317-8.]
Tabletten: oft Laktose-haltigLaktosegehalt bis zu 70-80 % des Tablettengewichtes ,oft keine genauen Angaben aus patentrechtlichen Gründen!
Pharmazeutische Charakteristika der
Darreichungsformen
IB

IWS 16.12.2009
Patienten mit Laktose-Intoleranz (Laktasemangel):
Bedine und Bayless, Gastroenterology 1973;65:735-43:
bei ED von 3 g Laktose: nur 2/20 Pat. symptomatisch
Vesa et al., Am J Clin Nutr 1996;64:197-201:
bis zu ED von 7g Laktose: 1/3 der Pat. symptomfrei; 2/3: keine Korrelation der Symptome zur Laktosedosis
Mengenvergleich: 1 Stück Würfelzucker = 3 g
► Laktose-Intoleranz (bei Laktasemangel):
Tabletten Tabletten klein klein ►► wenig Laktose wenig Laktose ►► in der Regelin der Regel irrelevantirrelevant
[ nicht zu verwechseln mit der hereditären Galaktose-In toleranz ]
Prüfarzneimittel - Zusammensetzung
IB

IWS 16.12.2009
→ Pharmakologie
Therapeutisches Potential
Sicherheitspharmakologie
(im Dosis- bzw. Konzentrationsbereichpotentiell therapeutischer Effekte)
→ Pharmakokinetik
→ Toxikologie
Fehlende Daten ≠ fehlende Wirkungen !
IBPrä-klinische Daten - ÜÜbersichtbersicht

IWS 16.12.2009
Wirkmechanismen: in vitro-Effekte
z.B. Rezeptorbindung agonistisch/ antagonistisch?reversibel/ irreversibel (Antidot möglich?) , Effekte selektiv/ nicht-selektiv, bei welchen Konzentrationen?
Wirkungen (Tiermodelle): in vivo- Effektez.B. ZNS-Effekte (Anxiolyse?) ,bei welcher Tierspezies (Nager und Nicht-Nager,
z.B. Hund, Affe)bei welchen Dosierungen bzw. Konzentrationen?
IBPrä-klinische Daten
Pharmakologie: Therapeutisches Potential

IWS 16.12.2009
Vitale Funktionen:Vitale Funktionen: in vitro-Zell- und Tiermodelle
Kardiale Effekte? z.B. Hemmung von hERG-Kanal-Strömen/EKG-Veränderungen beim Tier→→→→ ventrikuläre Rhythmusstörungen ?
Respiratorische Effekte? z.B. →→→→ Bronchospastik?
ZNS-Effekte? z.B. Senkung der Krampfschwelle→→→→ epileptische Anfälle?
unerwünschte Sedierung?
→→→→→→→→ Mögliche UE bzw. UAW nicht nur durch Toxikologie , sondern auch aus Pharmakologie erkennbar [ Unterschied: Dosisbereiche! ]
IBPrä-klinische Daten
Sicherheits - Pharmakologie

IWS 16.12.2009
in vitro-ModelleMetabolisierung über CYP450-System , v.a. CYP3A4
(für viele Arzneimittelinteraktionen relevant!)
Arzneimitteltransporter (Proteine, die AM aktiv aus der Zelle transportieren), bestimmend z.B. für Ausmaß der Aufnah meeiner Substanz aus dem GI-Trakt (z.B. Pgp; Digoxin)
Tiermodelle (verschiedenen Spezies)
unterschiedliches Metabolitenmuster?Einfluss der Nierenfunktion?
Passage der Blut-Hirn-Schranke? (Sedierung?)
Konzentrationen im Zielgewebe? z.B. bei Antibiotika
IBPrä-klinische Daten - Pharmakologie: Pharmakokinetik

IWS 16.12.2009
bei hohen Substanz-Dosen bzw. –Konzentrationen (!)und allen geplanten Darreichungsformen (oral, parenter al)
Einzeldosis -Studien →→→→→→→→ akute Effekteund „nicht-letale Einzeldosis“
Mehrfach -Exposition (über unterschiedlich lange Zeiten)
→→→→→→→→ Identifizierung von „Zielorganen“, sog. „Target-Organen“einer möglichen Schädigung
Dosis- und Zeit- Abhängigkeit bestimmter Effekte;Korrelation zu Blut- oder Gewebekonzentrationen ? Spezifisches Schädigungs-Muster ? Reversibilität ?
Spezies -spezifische Effekte ? (Nager oder Nicht-Nager)
Prä-klinische Daten: Toxikologie inkl. ToxikokinetikIB

IWS 16.12.2009
NOAELNOAEL = NNo OObservable AAdverse EEvent (Effect) LLevelDosierung/ Konzentration, bei der geradenoch keine Schädigung zu beobachten ist
(toxikologisch unterschiedlich sensitive Spezies! )
Achten auf Unterschiede zwischen dem NOAEL und
der Dosierung/ Konzentrationen, bei der die gewünschte, potentiell therapeutische Wirkung
zu beobachten ist
z.B. Thrombin-Inhibitor: Unterschied zwischenDosis/ Konzentration bei Thrombin-Hemmung
und bei Transaminasen-Anstieg
IBPrä-klinische Daten: Toxikologie + Pharmakologie

IWS 16.12.2009
SystematikSystematik der toxikologische Untersuchungen
The European Agency for the Evaluation of Medicinal P roducts,Committee for Proprietary Medicinal Products (CPMP).
Note for guidance on non-clinical safety studies for the conduct ofhuman clinical trials and marketing authorization for p harmaceuticals.ICH topic M3 (R2). Step 3.
Doc. Ref. CPMP/ICH/286/95 (revised version).Coming into operation: December 2009.
http://www.emea.europa.eu/pdfs/human/ich/028695en.p df
Prä-klinische Daten: Toxikologie
IB

IWS 16.12.2009
Besonders beachten:beachten:
→ Genotoxizität („Mutagenität“, Schädigung des Erbgutes)
→ Reproduktions- und Entwicklungstoxikologie:Fertilität?Embryo- und Feto-Toxizität ? ► Fehlbildungen
= teratogene Effekte? typisches Muster?Prä- und post-natale Entwicklung?
→ Fototoxizität
→ Kanzerogenität - Exposition dauert 2 Jahre!daher erst spät im Rahmen der AM-Entwicklung verfügbar
Prä-klinische Daten: ToxikologieIB

IWS 16.12.2009
oft komplexe Proteine [oder -gemische ], z.B. monoklonale AK,
aber auch „körpereigene“ Stoffe wie Insulin, Epo u.a.
► Spektrum der Untersuchungen zur Pharmakologie, Toxikologie und Kinetik
meist nicht so vollständig wie bei konventionellen synthetischen
Arzneimitteln [ unter „small molecules“ zusammengefasst ]
Auswahl erfolgt jeweils individuell in Absprache mit der
zuständigen Bundesoberbehörde = Genehmigungsbehörde für
Klinische Prüfungen: Paul Ehrlich-Institut [ PEI ] in Langen
IBPrä-klinische Daten: „Biologika“
[ biotechnologisch hergestellte Arzneimittel ]

IWS 16.12.2009
► http://www.emea.europa.eu ► Human Medicines ►
Scientific Guidelines ► Biologicals ► diverse Themen
► The European Agency for the Evaluation of Medicinal P roducts,Committee for Proprietary Medicinal Products (CPMP).
Note for guidance on preclinical safety evaluation o f biotechnology-derived pharmaceuticals. ICH Topic S6. Step 5.
Doc. Ref. CPMP/ICH/302/95.
Date for coming into operation: March 1998.
http://www.emea.europa.eu/pdfs/human/ich/030295en.p df
Prä-klinische Daten: „Biologika“
IB

IWS 16.12.2009
Überblick über Systematik der klinischen Entwicklung
► Klinischer Teil der IB:The European Agency for the Evaluation of Medicinal P roducts,Committee for Proprietary Medicinal Products (CPMP).
Note for Guidance on general considerations for clinical tri als.
Doc. Ref. CPMP/ICH/291/95.Date for coming into operation: March1998.
http://www.emea.europa.eu/pdfs/human/ich/029195en.p df
IB
Entwicklung neuer Arzneimittel:
Klinische Studien der Phasen I - IV

IWS 16.12.2009
IB
EMEA Doc. Ref. : CPMP/ICH/291/95

IWS 16.12.2009
→ „Human-Pharmakologie“Phase I-Studien
(potentieller Dosisbereich beim Menschen)
→ Interventionsstudien bei PatientenPhase II- und Phase III-Studien zu
• Sicherheit und Verträglichkeit• therapeutische Wirksamkeit
• Phase IV- Studien (!)
→ Verordnete TherapiePost-marketing-Erfahrungen
Klinische Daten - ÜÜbersichtbersicht
IB

IWS 16.12.2009
Einzeldosis + Mehrfachgabe der Prüfsubstanzv.a. bei gesunden Probanden; falls nicht vertretbar,z.B. zytotoxische Substanzen: Patienten mit der
zu behandelnden Grundkrankheit
Sicherheit und Verträglichkeit: UE-Dokumentation
Pharmakodynamik (Wirkung auf RR, EKG, Blutgerinnung,Reaktionsvermögen u.a.)
Pharmakokinetik: Resorption, Metabolisierung [ EM, PM ] inkl.Plausibilitätsprüfung der in vitro-Daten, Arzneimitte l-Interaktionen
Subgruppen z.B. Patienten mit Niereninsuffizienz/ Leberzirrhose
IBKlinische Studien - Phase I
sog. Humanpharmakologieallgemein wichtige (nicht-therapeutische) Aspekte

IWS 16.12.2009
NOAELNOAEL bei der toxikologisch sensitivsten Test-(Tier-) Species;
daraus wird häufig dieHEDHED = HHuman EEquivalent DDose ermittelt
muss um einen bestimmtem Sicherheitsfaktor (ausunterschiedlichen Daten ermittelt) unterschritten werd en
► MRSD MRSD = = MMaximum RRecommended SStarting DDose
== maximale vertretbare Startdosis
für die Erstanwendung einer Substanz beim Menschen
IBKlinische Studien – Beginn der Phase I
ErsteErste Anwendung einer neuen Substanz beim Menschen

IWS 16.12.2009
USA - Arzneimittelbehörde
Empfehlungen Empfehlungen der der FDAFDA ffüürr DosiswahlDosiswahlbei der Erstanwendung einer Substanz beim Menschen( v.a. bei sog. „small molecules“ )
► U.S. Department of Health and Human Services. Food a nd DrugAdministration. Center for Drug Evaluation and Research (CDER).
Guidance for Industry: Estimating the maximum safe startin g dosein initial clinical trials for therapeutics in adult health y volunteers.
[ Version July 2005 ]
http://www.fda.gov/cder/guidance/5541fnl.pdf
IBKlinische Studien – Beginn der Phase I
ErsteErste Anwendung einer neuen Substanz beim Menschen

IWS 16.12.2009
BESSER:BESSER: EMEAEMEA neue Empfehlungen,neue Empfehlungen,insbesondere auch für sog. Biologika (!)
um unter Einbeziehung aller relevanten Datender Präklinik, auch mithilfe von „PK-PD-modelling“ den
MABELMABEL = MMinimum AAnticipated BBiological EEffect LLevelzu ermitteln
als die niedrigste Dosis, bei der voraussichtlich gerad e noch ein biologischer Effekt nachweisbar ist
vor allem wichtig bei Arzneimitteln,
deren „Toxizität“ durch Übersteigerung der Hauptwirkung entsteht
IBKlinische Studien – Beginn der Phase I
ErsteErste Anwendung einer neuen Substanz beim Menschen

IWS 16.12.2009
► The European Agency for the Evaluation of Medicinal P roducts, Committee for Proprietary Medicinal Products (CPMP).
Guideline on strategies to identify and mitigate risks for f irst-in-humanclinical trials with investigational medicinal products.
Doc. Ref. EMEA/CHMP/SWP/28367/07. Date for coming into effect: September 2007.
http://www.emea.europa.eu/pdfs/human/swp/2836707enfin .pdf
► Agoram BM.
Use of pharmacokinetic/ pharmacodynamic modelling for st artingdose selection in first-in-human trials of high-risk biol ogics.
Br J Clin Pharmacol 2009;67:153-60.
Klinische Studien – Beginn der Phase I
ErsteErste Anwendung einer neuen Substanz beim Menschen
IB

IWS 16.12.2009
Ziel:Ziel: Klärung der Eignung einer Substanz für bestimmte
Indikationen: Sicherheit + Verträglichkeit; Wirksamkeit (ist überhaupt eine therapeutische Wirkung erkennbar?)
Studien bei → kleineren, gut definierten Patientengruppen
(nach Möglichkeit ohne relevante Begleiterkrankung)
→ mit relativ kurzer Anwendungsdauer
→ mit klinischen oder Surrogat- (!)-Endpunkten(z.B. Senkung des HbA1c in 3 Mon.)
z.T. in Form von Dosisfindungs- Studien
AusmaAusma ßß + Dauer+ Dauer erwerw üünschter Wirkungen?nschter Wirkungen? UEUE--Profil?Profil?
Klinische Studien - Phase II
Therapeutisches Potential - explorativ
IB

IWS 16.12.2009
BewertungBewertung von von UEsUEs: : Vergleich
► Verum - Placebo mit der Frage:welche UE unterscheiden sich in ihrer Häufigkeit von Placebo,
sind also potentiell Arzneimittel-bedingt ?
► verschiedener Dosisstufen von Verum mit der Frage:welche UE nehmen mit steigender Dosierung zu,
sind also potentiell dosisabhängig = = vermeidbar?[ Häufigkeit der UE? Intensität? ]
zutreffend v.a. für UE, die auf verstärkte Hauptwirkung(en) zurückgehen;nicht zutreffend z.B. für allergische Reaktionen !
Klinische Studien: Phase II
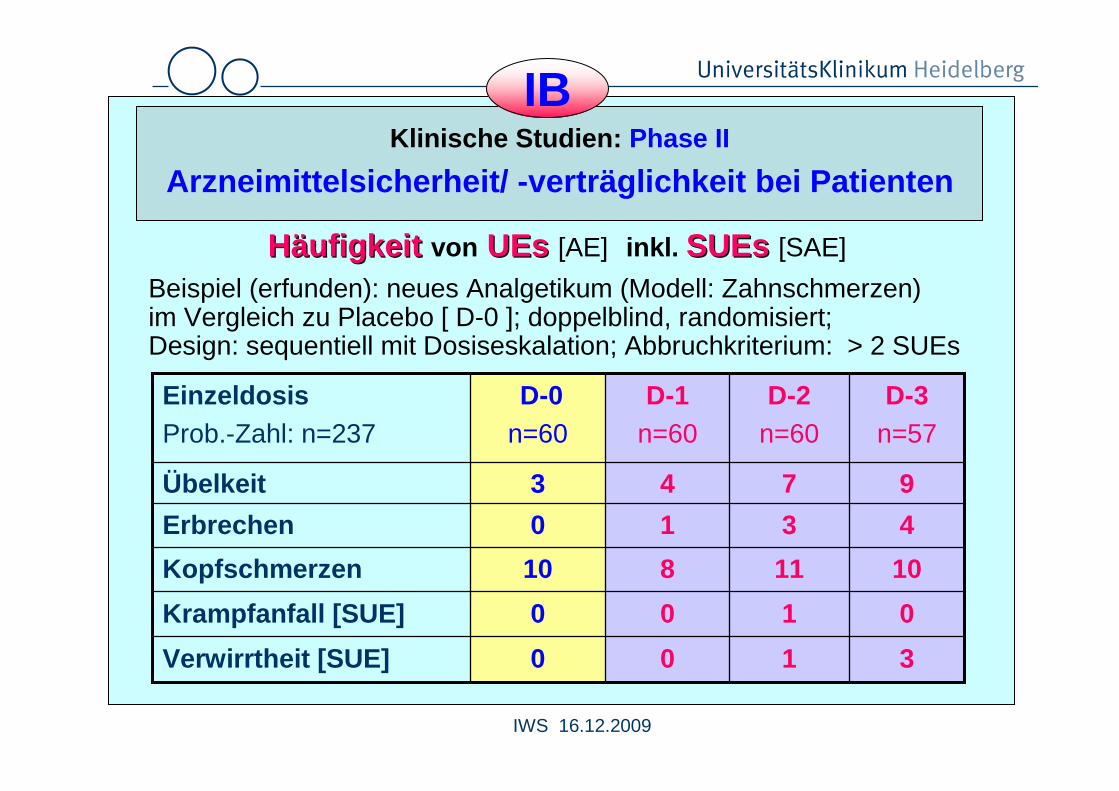
Arzneimittelsicherheit/ -verträglichkeit bei Patien ten
IB
IWS 16.12.2009
HHääufigkeit ufigkeit von UEsUEs [AE] inkl. SUEsSUEs [SAE]
Beispiel (erfunden): neues Analgetikum (Modell: Zahnschmerzen)im Vergleich zu Placebo [ D-0 ]; doppelblind, randomisiert; Design: sequentiell mit Dosiseskalation; Abbruchkriterium: > 2 SUEs
Klinische Studien: Phase II
Arzneimittelsicherheit/ -verträglichkeit bei Patien ten
3100Verwirrtheit [SUE]
0100Krampfanfall [SUE]
1011810Kopfschmerzen
4310Erbrechen
9743Übelkeit
D-3n=57
D-2n=60
D-1n=60
D-0n=60
EinzeldosisProb.-Zahl: n=237
IB

IWS 16.12.2009
Nachweis/ Bestätigung von therapeutischer Wirksamkeitsowie Sicherheit und Verträglichkeit
(inkl. Dosis-Wirksamkeits-Beziehung) bei bestimmten Indikationen
Große (!) kontrollierte randomisierte Studien mitParallelgruppen-Vergleich
→ für bestimmte Indikationen, z.B. Hypertonie-Pat.→ bei gut definierten Patientengruppen, z.B. ohne Diab etes→ mit längerer (!) Anwendungsdauer
Endpunktstudien (!)Endpunktstudien (!) zu Morbidität und Mortalität ?
► adäquate Wirksamkeit? UE-Profil? ►
Nutzen / Risiko Nutzen / Risiko --VerhVerh äältnisltnis ? ► Zulassung ?Zulassung ?
Klinische Studien - Phase III
Therapeutisches Potential - konfirmativ
IB

IWS 16.12.2009
WirkstoffWirkstoff1.1. zugelassen in geprüfter Darreichungsform ,
z.B. in wässriger Lösung ; in 0.9% NaCl ► nicht zugelassen
2.2. in zugelassener, weil geprüfter Dosierungz.B. 2 ng/kg/min ; überschrittene Dosis ► nicht zugelassen
3.3. für zugelassene, weil geprüfte Anwendungsweisez.B. iv-Infusion ; inhaliert ► nicht zugelassen
4.4. für zugelassene, weil geprüfte Indikationz.B. pAVK ; pulmonale Hypertonie ► nicht zugelassen
Was ist einein „„ zugelasseneszugelassenes ““ Arzneimittel ?Arzneimittel ?
[ oft verwechselt mit [ oft verwechselt mit „„ handelshandels üüblichblich ““ ]]
IB

IWS 16.12.2009
Daten aus der VerordnungVerordnungfür individualisierte Therapie
Relevant z.B. bei Entwicklung einer neuen Indikation oder beiArzneimitteln aus anderen Ländern (USA, Japan); Zulassung in BRD
angestrebt [ethnische Unterschiede beachten! ]
→ Einzelfallberichte zu unerwünschten Wirkungen
→ Anwendungsbeobachtungen zur Arzneimittelsicherheitin den ersten 5 Jahren nach Beginn der Vermarktung:Berichterstattung bei der Zulassungsbehörde verpflichten d
für den pharmazeutischen Unternehmer
Post-Marketing ErfahrungenIB

IWS 16.12.2009
bezieht sich auf alle Charakteristika eines Handelspräparates,die im Rahmen der Zulassungsstudien ermittelt wurden
Reihenfolge der Informationen anders als in IB !
Abschnitte 1-3: Bezeichnung (Handelsname), Zusammensetzung, Darreichungsformen
Abschnitt 4: Klinische Angaben für die Anwendung
Abschnitt 5: Pharmakologische EigenschaftenPharmakodynamik (z.B. Wirkmechanismen),
z.T. Ergebnisse klinischer Studien
Pharmakokinetik bei verschiedenen Populationen
Fachinformation Fachinformation [ gem. AMG § 11a ]
EU: „Summary of product characteristics“ [ „SPC“ ]
FI

IWS 16.12.2009
[ Forts. Abs. 5: Pharmakologische Eigenschaften ]
Prä-klinische Daten zur Sicherheit:Organ- bzw. Gewebeschädigung (KM)?Spermiogenese? Genotoxizität?Teratogenität?
Abschnitt 6: Pharmazeutische Angabeninkl. Hilfsstoffen und anderenWirkstoffen, z.B. Lokalanästhetika !
Abschnitt 9: Datum der Zulassungbzw. Verlängerung der Zulassung
Informationen über Handelspräparate: www.fachinfo.de ; AiD-Klinik
Ausgezeichnetes Beispiel: Rebetol® Hartkapseln (14 Seiten!)
Fachinformation [Forts.]FI

IWS 16.12.2009
Viel Erfolg und
danke für IhreAufmerksamkeit !





























![Vergleichende tierexperimentelle Studie zur ossären ... · [40, 95, 129] Diabetes mellitus stellt damit die häufigste endokrine Stoffwechselerkrankung in Deutschland dar. Die hohe](https://img.pdfslide.tips/doc/110x75/5ba0001609d3f267388c2627/vergleichende-tierexperimentelle-studie-zur-ossaeren-40-95-129-diabetes.jpg)