Embed Size (px)
Citation preview

1
Inżynieria genetyczna Ćwiczenie 3
Metoda PCR została opracowana w
1987 r. przez grupę Naukowców z
Cetus Corporation w USA. Firma
Roche odkupiła od Cetus Corp. prawa
do PCR za 300 mln $
Autor K. Mullis otrzymał w 1993r.
nagrodę Nobla w dziedzinie chemii.

2
Początkowo stosowano fragment Klenowa polimerazy DNA I z E. coli, który
wykazuje aktywność 5’→3’ polimerazy oraz 3’ →5’ egzonukleazy, brakuje
natomiast aktywności 5’ →3’ egzonukleazy, dzięki czemu nie występuje
niebezpieczeństwo degradacji primerów podczas reakcji. Polimeraza ta
była niestabilna w wyższych temperaturach, dlatego konieczne było
dodawanie jej kolejnej porcji w każdym kolejnym cyklu.
Niekorzystne cechy tej polimerazy
zostały wyeliminowane przez użycie
polimerazy Taq z termofilnych bakterii
Thermus aquaticus – temperatura
denaturacji matrycy nie pozbawia
enzymu aktywności.
1. Matryca DNA (RNA) 2. Startery 3. dNTP 4. MgCl2
5. Polimeraza 6. Bufor

3
najlepiej dodać najpierw wodę, a następnie kolejno pozostałe
składniki
mieszaninę reakcyjną przetrzymujemy w lodzie podczas pracy
gdy matrycą jest powszechnie używane DNA w laboratorium
lub gdy są jej śladowe ilości, to praca powinna odbywać się w
laminarnym przepływie sterylnego powietrza, a tipsy i pipety
powinny być zabezpieczone korkami
podczas pracy z małymi objętościami komponentów (1-2 μl)
trzeba uważać, aby do wszystkich prób dodana została
jednakowa ilość (szczególnie ważne przy ilościowym PCR)
Niekorzystna zbyt duża/zbyt niska liczba matryc
- Ilość DNA 1ng-1μg
- Wyjściowa liczba matryc:
DNA człowieka 3*10-5
DNA drożdży 1μg
DNA E. Coli 1ng
Odpowiedni stopień czystości (UV, elektroforeza ) -
Zanieczyszczenia matrycy: SDS, EDTA, DMSO,
heparyna, fenol, sole
Metoda izolacji - usunięcie inhibitorów reakcji

4
Efekt zbyt dużej
liczby matryc
Degradacja
DNA
Polimeraza Taq:
•Wyizolowana z bakterii Thermus aquaticus
•Aktywność polimerazy 5’-3’
•Aktywność egzonukleazy 5’-3’
•Brak aktywności egzonukleazy 3’-5’
•Aktywność polimerazy Taq – około 2000 nt/min (60 nt/s) w
optym. temperaturze 72-78°C
•Po reakcji na końcach 3' kilka nukleotydów adeninowych,
właściwość wykorzystywana jest przy klonowaniu produktów
PCR.
•Do swej aktywności wymaga jonów Mg2+
•Procesywność, wierność, szybkość

5
Cechy: - odpowiednia długość (18- 28 nt) - zawartość GC 50- 60 %
Tm [2n1(A+T)+4n2(G+C)]- 5 55-72˚C Ta 40- 55 ˚C
-wyznaczana doświadczalnie
-wpływ na specyficzność
Dobór stężenia roboczego 10 pmol/ μl
-zbyt duże stężenie- niespecyficzność produktów
Dimery starterów - komplementarność końców 3’
metoda oparta o %GC: Tm[oC] = 81,5 + 16,6 x log[Na] + 0,41 x (%GC) -
675/długość - 0,65 x (%formamidu) - (%niekomplementarnych)
metoda uproszczona: Tm = [2o x (A + T) + 4o x (G + C)]
metoda w oparciu o zasady sąsiadujące

6
startery - najczęściej to ich sekwencja i stężenie decyduje o powodzeniu reakcji:
nie powinny tworzyć wewnętrznych struktur 2-rzędowych
Powinny mieć wyrównaną zawartość zasad G/C i A/T
nie mogą komplementować między sobą na 3’ końcu
nie mogą zbyt silnie wiązać na 3’ końcu
do 5’ można dodawać: miejsca cięcia dla enzymów restrykcyjnych, tzw.
lepkie końce
stężenie 0,1-0,5 M (za dużo - pomyłki, za mało - mniej produktu)
startery zdegenerowane,
modyfikacje: biotynowanie, zabezpieczanie przed 3’ degradacją - wiązania
fosfotiolowe, fosforylacja
Źle zaprojektowane startery
http://www.bioinformatics.org/sms2/pcr_primer_stats.html

7
Wpływ temperatury wiązania starterów
- termocykler z gradientem temp.
- wyznaczanie optymalnej Tm
dNTP-trifosforany deoksyrybonukleotydów
niezrównoważony skład obniża dokładność polimerazy,
stężenie wyjściowe: 5-10 mM stężenie reakcji: 20-200 µM
jednakowe stężenie wszystkich dNTP
pH 7,

8
MgCl2 - 0,5-5,0 mM - wpływa na
aktywność enzymu, zwiększa Tm dsDNA,
tworzy kompleksy z dNTP dając substrat
dla polimerazy,
Stężenie jonów Mg > stężenie dNTP
Wrażliwość na degradację
Rozkład- przyczyna braku produktu
dATP:dGTP: dCTP: dTTP
Stosowanie jednakowych stężeń
dNTP
Stężenie optymalne 20- 200µM
Wzrost dokładności polimerazy
Stężenie MgCl2 nieco wyższe od steżenia dNTP
Dokładność reakcji
Proporcjonalna do stężenia wolnych MgCl2
MgCl2
Wiązanie przez: matrycę, startery, polimerazę Taq, dNTP

9
Należy zwrócić uwagę na:
- Stężenie 0,5- 5U na 100µl zbyt duże- niespecyficzne produkty zbyt małe- brak produktów - Przechowywanie -20˚C
- Okres półtrwania 92,5 ˚C 2h 95,0 ˚C 40 min 97,5 ˚C 5 min (!)

10
DMSO Żelatyna BSA Tween 20 PEG
Fenol SDS Proteinaza K Porfiryna EDTA Etanol

11
Zapewnia odpowiednie warunki dla działania polimerazy
pH 8,3-8,8
Tris-HCl w zakresie 10-50 mM
Czynniki ujemnie wpływające na PCR: 1. Jakość i zanieczyszczenia matrycy 2. Startery - powstawanie dimerów starterów - niska specyficzność starterów 3. Niewłaściwy dobór stężeń dNTP i MgCl2 4. Błędy polimerazy 5. Zanieczyszczenia odczynników 6. Ilość cykli

12
ETAP CZAS °C
początkowa denaturacja 2-5 min 94
denaturacja 1-60 s 94
przyłączanie starterów 30-60 s 54
wydłużanie starterów 30-90 s 72
ostatnie wydłużanie 5-10 min 72
schładzanie b/o 4
x 20-35
Aktywność polimerazy Taq – około 2000 nt/min (60 nt/s) w
optym. temperaturze 72-78°C
Denaturacja – nie powinna być dłuższa niż 60 s, ponieważ
wpływałoby to na aktywność enzymu
Początkowo czas wydłużania ustalamy 1kb -1 min, 2kb – 2
min itd…., przy kolejnych reakcjach można skracać
Przyłączanie starterów – temp. zależy od temp. topnienia obu
oligonukleotydów
Liczba cykli – 30 , więcej cykli nie wpływa znacząco na ilość
produktu
OPTYMALIZACJA – usunięcie niespecyficznych produktów

13
Specyficzność – eliminowanie niespecyficznych produktów
podwyższyć temperaturę annealingu (przyłączania starterów), obniżyć stężenie jonów Mg2+ zmienić skład nukleotydowy primera obniżyć ilość cykli zmienić polimerazę
Długość produktu – amplifikacja długich fragmentów
wydłużyć czas syntezy dobrać odpowiednie stężenie jonów Mg2+ zmienić polimerazę skrócić czas denaturacji podwyższyć stężenie matrycy zmienić pH buforu do PCR
Wierność – redukcja błędów syntezy
obniżyć liczbę cykli podwyższyć stężenie matrycy obniżyć stężenie dNTP zmienić polimerazę
Ilość produktu – zwiększenie ilości amplifikowanych
fragmentów zwiększyć liczbę cykli podwyższyć stężenie matrycy zmienić polimerazę obniżyć temperaturę annilingu wydłużyć czas syntezy

14
Hot start - unikanie niespecyficznej amplifikacji podczas
pierwszego cyklu (RT 94oC):
wkładać probówki do gorącego bloku
dodawać polimerazę lub startery do gorącej mieszaniny
odseparować startery od reszty reakcji parafiną
korzystać z polimerazy zabezpieczonej przeciwciałem
inne zabezpieczenia dysocjujące w wyższej temperaturze
Touch-down PCR – pierwsze kilka cykli wykonane przy dużo
wyższej temp. przył. startera – zwiększa specyficzność
Kłopoty z subklonowaniem produktu PCR
po Taq polimerazie zostaje ok. 70% końców tępych, a reszta ma 1
zasadę wystającą na końcu 3’ (zwykle A) - można ligować na lepko (!)
większość starterów zdefosforylowanych na końcu 5’ (PNK).

15
Cechy PCR
PCR w zastosowaniu analitycznym (diagnostycznym)
PCR jako metoda syntezy (zastosowanie laboratoryjne)
Wykrywanie sekwencji o
niewielu kopiach
Analizy genetyczne
Sądownic-two
Synteza sond
Synteza matryc do sekwencjono-wania
Synteza insertów przeznaczonych do klonowania
Czułość +++ + +++ - - -
Specyficz-ność
+++ +++ +++ ++ ++ +++
Wierność - - - - - +++
Ilość produktu
- (+) - +++ +++ _
+++, niezwykle istotna; ++, bardzo istotna; +, istotna; (+), może być istotna; -, nieistotna.
Jedna zasada, wiele modyfikacji

16
nested - „zagnieżdżony” PCR,
multiplex PCR - jednoczesna
amplifikacja kilku fragmentów
(dłuższe startery),
long (expanded, extended)
PCR - dla fragmentów > 5kb -
mieszanki polimeraz, octany,
dwie temp. (np.99/68),
inverted PCR,
mutageneza kierowana -
wprowadzanie zmian,
asymetryczny PCR (zmiana
proporcji starterów po 15-25
cyklach),
LCR - ligase chain reaction,
RT-PCR,
in situ PCR (genomowy i RT) –
w tkance,
Real time PCR
AMV-RT - avian myeloblastosis virus reverse transcriptase, zachowuje aktywność do 55°C, eliminuje problemy związane strukturą II-rzędową
MMLV-RT - moloney murine leukaemia virus reverse transcriptase, ma mniejszą aktywność RNAzy H niż AMV-RT i jest lepsza do syntezy pełnej długości cząsteczek cDNA
synteza cDNA:
hybrydyzacja ze specyficznym starterem
hybrydyzacja z oligo (dT)
hybrydyzacja z heksamerami
AAAA
AAAA
TTTT
AAAA

17
dwie lub więcej docelowych sekwencji jest namnażanych w jednej probówce
jedna z tych sekwencji to najczęściej wewnętrzna kontrola poprawności warunków reakcji a druga jest sekwencją docelową
zastosowanie diagnostyczne – polimorfizmy analizy mikrobiologiczne
wykorzystywany w celu
wykrywania punktowych
mutacji w genomowym DNA
w war.natywnych DNA
przyjmuje określoną
konformację zależną od
sekwencji nukleotydowych
szybkość zależna od
wielkości i konformacji
>95% wykrywalność dla DNA
o wielkości 100-300 pz
Do detekcji reakcja
srebrzenia, radioizotopy,
rzadziej bromek etydyny
SSCP- polimorfizm konformacji jednoniciowych fragmentów DNA (ang. single strand conformation polymorphism)

18
1. Do produktów PCR powyżej 400 pz
2. Układ heterozygotyczny( 2 allele zmutowany i prawidłowy)
3. Amplifikacjainkubacja w celu utworzenia heterodupleksów
4. Heterodupleks- hybrydyzacja nie w pełni komplementarna
5. Substytucja-obustronne wybrzuszenie
6. Insercja lub delecja- jednostronne wybrzuszenie lub ugięcie nici
PCR-HD
Wykrywanie mutacji metodą heterodupleksów
Polimorfizmy RFLP to dziedziczne warianty sekwencji DNA, które objawiają się utworzeniem bądź zanikiem miejsca rozpoznawalnego przez endonukleazy restrykcyjne lub zmianą liczby nukleotydów między takimi miejscami . Amplifikacja fragmentu DNA, w obrębie którego występuje marker RFLP a potem jego hydroliza enzymem restrykcyjnym.

19
pozwala na amplifikację długich fragmentów DNA,
można w ten sposób uzyskiwać fragmenty nawet do 27 kb, rutynowo służy do otrzymywania fragmentów 10 – 20 kb
długie fragmenty uzyskuje się stosując mieszaninę termostabilnych polimeraz DNA, zwykle:
polimerazę Taq – zapewnia wysoką procesywność (aktywność 5’-3’ polimerazy)
polimerazę Pwo – zapewnia poprawność (aktywność 3’-5’ egzonukleazy)
dwa zestawy primerów: zewnetrzne i wewnętrzne, ale dwie rundy amplifikacji.
cel: zwiększenie czułości (pg) i specyficzności PCR
Nested PCR

20
pozwala zamplifikować
obszary wokół transgenu
transgen
enz enz
trawienie restrykcyjne a następnie ligacja
enz., który nie trawi transgenu (to nie jest plazmid !)
przecinamy w obszarze transgenu
projektujemy primery
amplifikacja,
klonowanie i
sekwencjonowanie
Koliste fragmenty DNA stają się matrycą
produkty są w mieszaninach, w których formy
koliste zawierają oba miejsca przyłączania starterów
21
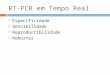
niskie tło – znakowanie po specyficznej hybrydyzacji, startery nie są znakowane szybkość – hybrydyzacja i elongacja w 30 min., cała procedura 2h wydłużanie nie jest limitowane długością startera
22
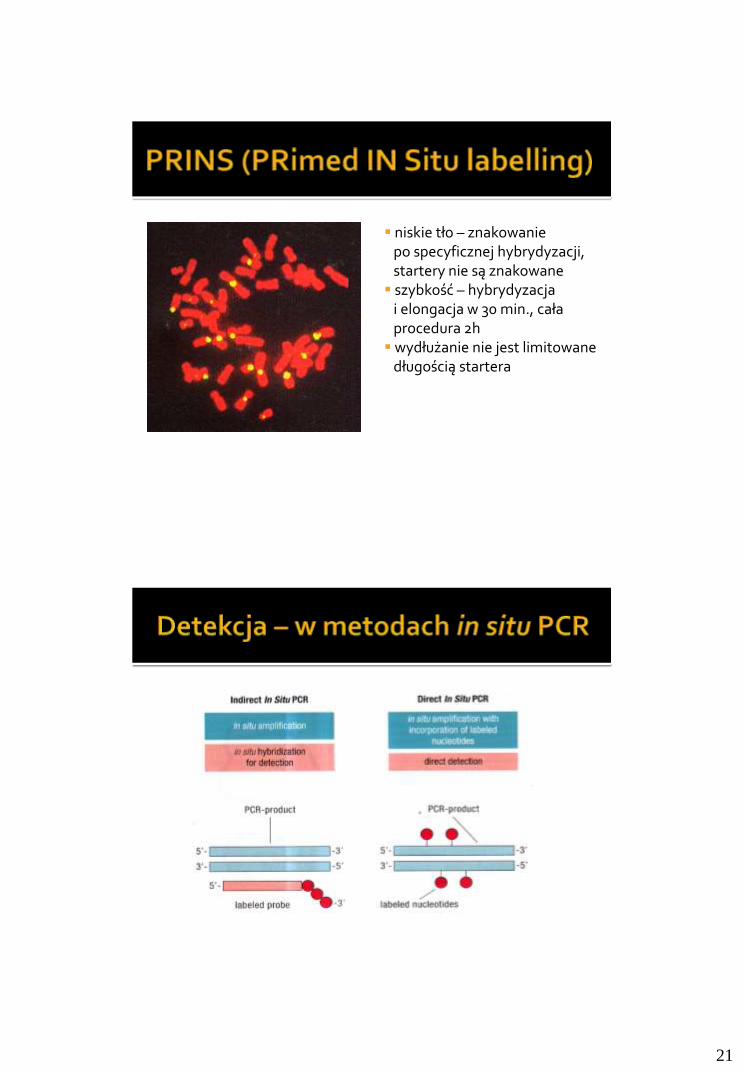
• Ocena ilościowa produktu
• Wysoka czułość i powtarzalność
• Szeroki zakres oznaczanych stężeń
Budowa:
- źródło światła wzbudzającego cząsteczkę reporterową - układ detekcji fluorescencji - termocyler

23

24
SYBR Green I – interkaluje do dsDNA podczas reakcji, związane z DNA
ma silną fluorescencję (1000x silniej niż nie związane – stąd (+) niskie tło)
technika FRET - (fluorescence resonanse energy transfer) – do detekcji
specyficznych sekwencji. Przygotowuje się 2 sondy hybrydyzujące blisko
na matrycy. Górna sonda jest wyznakowana fluoresceiną na końcu 3’,
która działa jako FRET. Koniec 5’ dolnej sondy jest wyznakowany
akceptorem. Jeżeli dojdzie do hybrydyzacji obu - jest fluorescencja
technika Molecular Beacons – podwójnie znakowany starter, gdy jest nie
związany jest wygaszanie1 (struktura spinki do włosów), gdy się przyłączy
jest fluorescencja
TaqMan – wykorzystanie aktywności egzonukleolitycznej 5’-3’. Starter jest
podwójnie wyznakowany: fluoresceiną i wygaszaczem. Następuje
wycięcie barwnika, co odblokowuje fluorescencję.
SYBR Green I (metoda niespecyficznej detekcji fluorescencji)

25
TaqMan (metoda specyficznej detekcji fluorescencji)

26

27
Geny:
miR482d
miR6023
U6
Matryce:
3dpi K
3dpi Inf

28
Geny:
β tubulina
Lsd1
Matryce:
Col0
Lsd1(Col0)

29
Gene Expression Std. Error 95% C.I. P(H1) Result
ACT2 1
APX1 1,584 0,909 0,663 - 2,870
0,065
CAT2 0,687 0,586 0,560 - 0,885
0,008 DOWN
EIN2 1,795 1,065 0,903 - 4,560
0,045 UP
ERF1 195,904 131,464 103,877 - 338,734
0 UP
GA3 0,474 0,4 0,357 - 0,629
0,007 DOWN
LOX2 3,35 2,099 0,993 - 9,826
0,014 UP
ORA47 1,01 0,477 0,423 - 2,022
0,963
OXI1 1,209 0,702 0,601 - 2,150
0,363
PDF1.2 400,04 198,096 48,870 -
2 351,125 0,001 UP
PR1 8,044 2,83 2,589 - 42,579
0,002 UP
RRTF 6,84 5,689 5,203 - 10,045
0,001 UP
SAG2 2,486 1,398 0,453 - 8,868
0,058
UBQ 1,261 0,54 0,212 - 6,047
0,661
WRKY33 4,335 2,269 1,760 - 10,719
0 UP
0,1
1
10
100
1000
Ge
ne
ex
pre
ssio
n le
vel
ROOT

30
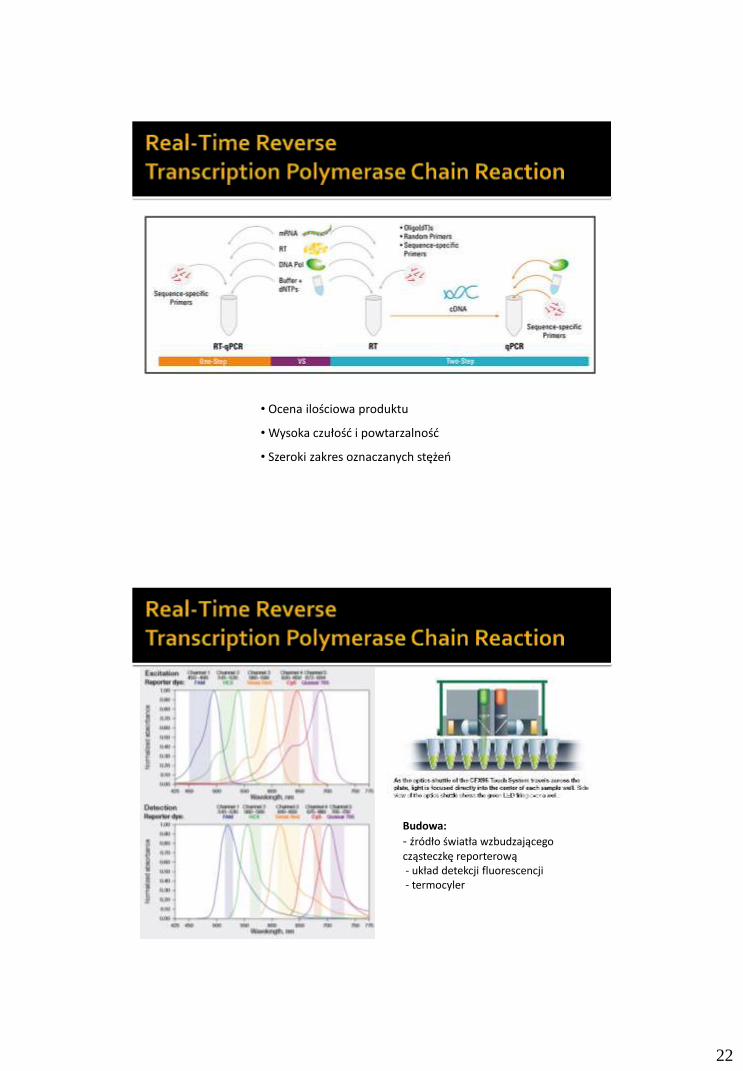
Elektorforeza i metody elucji

31
rodzaj agarozy
przezroczystość
rozdział krótkich prążków
cena
czas (dla prążków krótszych niż 600pz)
lepiej krócej tzn. 3-4 h dla żelu 15-20 cm,
przy dłuższym czasie 14-16h (niskie napięcie)
prążki nie są ostre, rozdział gorszy
rozdział wielu prążków (polimorficzne loci):
lepsze żele poliakrylamidowe, jeżeli prążki różnią się kilkoma pz (np.
markery mikrosatelitalne) 6-10%
niedenaturujące – dobry dla pojedynczych loci
denaturujące 6%/7M mocznik - żele do sekwencjonowania
multiplex PCR – 2 - 3% żel agarozowy
Agaroza Wielkość rozdzielanych fragmentów DNA
0,3% 5-60 kpz
0,5% 1-30 kpz
0,7% 0,8-12 kpz
1,0% 0,5-10 kpz
1,2% 0,4-7 kpz
1,5% 0,2-3 kpz
2,0% 0,05-2 kpz
Elektroelucja konieczne przeprowadzenie kolejne elektroforezy, w specjalnie przygotowanym aparacie, pracochłonna
Szkło kwarcowe – sole chaotropowe odpowiednia do oczyszczania dużych i małych fragmentów DNA, pracochłonna, zanieczyszczenia NaI, trudności przy agarozie opartej na TBE
Modyfikowana celuloza DEAE nadaje się do oczyszczania długich odcinków DNA, skomplikowana i pracochłonna, konieczność wykonania kolejnej elektroforezy, możliwość nieodwracalnego związania się DNA z celulozą po wyschnięciu
Zamrażanie, wyciskanie, wirowanie tania, styczność z BrEt podczas wyciskania
Trawienie -agarazą odpowiednia do oczyszczania dużych i małych fragmentów DNA, tylko do agarozy o niskiej temperaturze topnienia, trudności przy wykorzystaniu bufora TBE, konieczne utrzymywanie odpowiedniej temp. (42-45C) oraz pH 5-8
Metody z wykorzystaniem kolumienek wygodna, w kontrolowanych warunkach, powtarzalna, kosztowne, większe cząsteczki DNA mogą zahaczać się w kolumienkach
32
Dziękuję za uwagę !