
H. ZULETA
1
Apuntes de Cinética y Diseño
de Reactores
Ing. Héctor Zuleta C.
Marzo - 2005

H. ZULETA
2
Estos apuntes, corresponden a una breve recopilación de la gran cantidad de
literatura existente para la enseñanza de pregrado en Reactores Químicos. Su
realización ha sido posible gracias al proyecto de apoyo a la docencia
“Confección de apuntes para la asignatura de Reactores químicos” dirigido
por la Sra. Jefe de la carrera de Ingeniería de ejecución en procesos químicos,
Loreto Otarola.
Se ha tomado como texto base, el libro “Ingeniería de las reacciones
químicas” del autor O. Levenspiel, principalmente en las unidades II, III, y V.
También se apoya en el texto de Missen, Mins y Saville, “Introducción a la
cinética e Ingeniería de las reacciones químicas”
En la primera parte se presentan los conceptos de cinética química
fundamentales para el diseño de reactores. Han sido elaborados considerando
apuntes propios del autor y de acuerdo al programa de la asignatura.
Estos apuntes no consideran el tema correspondiente a diseño en reacciones
múltiples, el cual se debe consultar en el texto de Levenspiel, sin embargo
incorpora la unidad IV acerca del diseño y configuración de sistemas de
reactores del autor Lee.
Se espera contribuir a la formación de los futuros ingenieros de procesos en
los conceptos de cinética química y diseño de reactores, siendo estos
fundamentos amplia y exclusivamente del dominio del ingeniero químico.
Como siempre es posible mejorar, se agradecerá cualquier comentario y/o
sugerencia acerca de estos apuntes.

H. ZULETA
3
CONTENIDO
I CINETICA DE REACCIONES 1
1.1 INTRODUCCION 1
1.2 REACCION QUIMICA 1
1.2.1 Reactores químicos 4
1.2.2 Principales funciones de los reactores químicos 6
1.3 CINETICA QUIMICA 6
1.3.1 Clasificación de las reacciones químicas 7
1.4 CINETICA E INGENIERIA DE LAS REACCIONES QUIMICAS 9
1.5 VELOCIDAD DE REACCION 10
1.5.1 Definición de velocidad de reacción 14
1.5.2 Factores que modifican la velocidad de reacción 15
1.5.3 Ecuación de velocidad y orden de reacción 17
1.5.4 Mecanismo de reacción 20
1.6 DEPENDENCIA DE LA VELOCIDAD DE REACCION CON
LA TEMPERATURA 23
1.6.1 Teoría de Arrhenius 25
1.6.2 Comparación de teorías con la ley de Arrhenius 26
1.6.3 Influencia de la energía de activación 27
1.7 MEDICIONES EXPERIMENTALES EN CINETICA DE
REACCION 28
1.7.1 Métodos experimentales para seguir la extensión de una
Reacción 28

H. ZULETA
4
1.8 REACTOR DISCONTINUO DE VOLUMEN CONSTANTE 30
1.8.1 Análisis de datos de presión total en un sistema de
Volumen constante 31
1.9 GRADO DE CONVERSIÓN 33
1.10 METODOS DE ANALISIS DE DATOS 34
1.11 METODO DE INTEGRAL 34
1.12 METODO INTEGRAL PARA MODELOS SENCILLOS DE
VELOCIDAD 35
1.12.1 Reacciones irreversibles de orden cero 35
1.12.2 Reacciones irreversibles de orden uno 38
1.12.3 Reacciones irreversibles de segundo orden 40
1.12.4 Ecuaciones empíricas de orden n 45
1.12.5 Método de Vida fraccional tf 50
1.13 REACCIONES EN SERIE O CONSECUTIVAS 51
1.14 REACCIONES IRREVERSIBLES EN PARALELO 55
1.15 REACCIONES REVERSIBLES O DE EQUILIBRIO 57
1.16 REACCIONES HOMOGENEAS CATALIZADAS 59
1.17 REACCIONES AUTOCATALITICAS 60
1.18 METODO DIFERENCIAL DE TRATAMIENTO DE DATOS 62
1.19 METODO DE LAS VELOCIDADES INICIALES 63
1.20 REACTOR DISCONTINUO DE VOLUMEN VARIABLE 64
1.20.1 Método integral de análisis 68
II INTRODUCCION AL DISEÑO DE REACTORES IDEALES

H. ZULETA
5
2.1 INTRODUCCION 72
2.2 CLASIFICACION DE REACTORES QUIMICOS 74
2.2.1 Reactores Batch ( discontinuos ) 76
2.2.2 Reactor de flujo Tanque agitado continuo 77
2.2.3 Reactor de flujo Tubular 78
III DISEÑO DE REACTORES IDEALES
3.1 ECUACION DE DISEÑO DEL REACTOR BATCH 79
3.1.1 Balance de materiales del Reactor batch 79
3.1.2 Balance de energía del Reactor batch 82
3.1.3 Operación no isotérmica 84
3.1.4 Operación adiabática 85
3.1.5 Operación no adiabática 86
3.2 REACTOR CONTINUO PERFECTAMENTE
MEZCLADO ( CSTR ) 86
3.3 ECUACIÓN DE DISEÑO DEL CSTR 88
3.3.1 Balance de materiales del CSTR o RTAC 89
3.3.2 Ecuación de diseño del CSTR para una reacción de
Primer orden 93
3.3.3 Ecuación de diseño del CSTR para una reacción de
Segundo orden 93
3.3.4 Balance de energía en el Reactor CSTR 94
3.3.5 Operación en estado no estacionario 96
3.4 REACTOR DE FLUJO TUBULAR ( RFP ) 97
3.5 ECUACIÓN DE DISEÑO DEL RFP 99

H. ZULETA
6
3.5.1 Balance de materiales del RFP 100
3.5.2 Balance de Energía del RFP 101
3.5.3 Balance de momento, operaciones no isobáricas 104
3.5.4 Sistemas con densidad constante 105
3.6 OPERACIÓN DEL RFP CON RECICLO 107
IV CASOS ESPECIALES DE DISEÑO DE REACTORES
4.1 COMPORTAMIENTO TRANSIENTE DE UN CSTR 114
4.2 COMPARACIÓN DE UN RFP Y UN CSTR 116
4.3 CONFIGURACION DE REACTORES 119
V FLUJO NO IDEAL
5.1 INTRODUCCIÓN 126
5.2 DISTRIBUCIÓN DEL TIEMPO DE RESIDENCIA 126
5.3 ESTADO DE AGREGACIÓN DE LAS CORRIENTES 128
5.4 RAPIDEZ DEL MEZCLADO 129
5.5 DISTRIBUCIÓN DE EDADES DEL FLUIDO E ( RTD ) 130
5.6 MÉTODOS EXPERIMENTALES FISICOS PARA
DETERMINAR E 132
5.6.1 Experimento Pulso 132
5.6.2 Experimento Escalón 135
5.6.3 Relación entre las curvas F y E 136

H. ZULETA
7
VI SISTEMAS REACTIVOS GAS – SÓLIDO
6.1 EJEMPLOS DE SISTEMAS 139
6.2 PARTÍCULAS DE TAMAÑO CONSTANTE 140
6.2.1 Modelo general 142
6.2.2 Modelo de Núcleo encogido 144
6.2.3 Deducción del Modelo 146
6.2.4 Parámetros de los procesos de velocidad y estimación de KAG 151
6.3 PARTICULA DE TAMAÑO DECRECIENTE 153
6.3.1 Modelo de partícula de tamaño decreciente 153
BIBLIOGRAFIA 157

H. ZULETA
8
CAPITULO I. CINÉTICA DE REACCIONES
1.1 INTRODUCCIÓN
Una gran variedad de productos de consumo directo o que sirven como
materia prima para la obtención de otros productos requieren para su uso de
alguna transformación o reacción química, Este proceso puede definirse como
una operación unitaria (operación unitaria química) que tiene por objeto
distribuir de formas distintas los átomos de moléculas (compuestos
reaccionantes o reactantes) para formar otras nuevas (compuestos producidos
o productos).
1.2 REACCION QUÍMICA
El estudio de una reacción química puede hacerse desde el punto de vista
termodinámico, para determinar el calor involucrado en la reacción o la
extensión máxima posible de la misma.
Es sabido que las reacciones cuya constante de equilibrio es grande, tienden a
realizarse totalmente. Si se considera el siguiente ejemplo:
CO(g) + NO(g) CO2(g) + ½ N2(g) (1.1)
A 25°C, la constante Kc de esta reacción es un valor muy grande, del orden de
1060
. Según esto, podría parecer que la reacción (1.1) es un buen método para
eliminar los dos gases más tóxicos (CO y NO) que expulsan los motores de
automóvil.
Tal como se planteo anteriormente el estudio termodinámico permite conocer
la posición en la cual la reacción alcanzará el equilibrio. Cuantitativamente la
posición de equilibrio viene definida por la constante de equilibrio, que
representa el cociente de las actividades de productos y reactantes:
A + B C + D K = aC aD / aA aB (1.2)
El valor de la constante es una indicación de la extensión en la que se
producirá la reacción. Sin embargo, no da ninguna información relacionada
con la duración del proceso. Los criterios termodinámicos no incluyen la
variable tiempo, puesto que sólo consideran la diferencia de propiedades del
sistema entre los estados inicial y final y, por lo tanto, no se ocupan de la

H. ZULETA
9
velocidad a la que tiene lugar la reacción ni los estados intermedios por los
que transcurre.
Para el ejemplo representado por la ecuación (1.1) se debería esperar casi una
eternidad para conseguir que el CO y el NO se conviertan completamente en
CO2 y N2.
Esta situación es bastante común. Muchas reacciones que se producirían, en
principio, hasta una conversión total, reaccionan muy lentamente.
Frecuentemente esto es un gran beneficio. Por ejemplo, en el caso de los
combustibles fósiles –carbón, petróleo y gas natural-. Desde el punto de vista
del equilibrio, estos combustibles se deberían convertir en CO2 y H2O al
exponerlos al aire.
Se concluye que no hay relación entre la velocidad de reacción y su constante
de equilibrio. Para predecir a qué velocidad se va a producir una reacción, se
debe profundizar en el conocimiento de la cinética química.
Por lo tanto para el logro de un mayor entendimiento acerca de cómo se
comporta la materia en una transformación química, será necesario tener
profundos conocimientos de Termodinámica y Cinética Química.
A partir de estas ramas de la ciencia es posible reproducir una transformación
química en un medio altamente controlado, en este caso el medio será el
recipiente conocido como Reactor Químico.
El reactor es considerado el corazón de un proceso químico. Es un elemento o
equipo en el cual ocurre la conversión de la materia prima en un o varios
productos deseados. El funcionamiento adecuado de este elemento es vital
para la buena operación de las unidades industriales.
Materias Productos
Primas
Recirculación
Fig. 1-1 Esquema general de un proceso Químico
Operaciones físicas de acondicionamiento
Reactor Químico
Operaciones físicas de
Separación

H. ZULETA
10
Los procesos químicos en general (Fig. 1-1) y cada operación unitaria en
particular tienen como objetivo el modificar las condiciones de una
determinada cantidad de materia en forma útil con propósitos previamente
definidos. Este cambio puede realizarse por tres caminos:
- Modificando su masa o composición (separación de fases, mezcla,
reacción química)
- Modificando el nivel o calidad de la energía que posee (enfriamiento,
vaporización, aumento de presión, etc.)
- Modificando sus condiciones de movimiento (aumentando o
disminuyendo su velocidad o dirección)
Los tres cambios mencionados anteriormente son los únicos cambios posibles
que un cuerpo puede experimentar. Un cuerpo estará absolutamente definido
si se especifican:
- La cantidad de materia y composición
- La energía total (interna, eléctrica, magnética, potencial, cinética)
- Los componentes de velocidad
Este hecho experimental tiene su expresión matemática en las tres leyes de
conservación:
- Ley de conservación de la materia
- Ley de conservación de la energía
- Ley de conservación de la cantidad de movimiento
Las operaciones unitarias se clasifican de acuerdo con las propiedades más
relevantes que se transfiera en la operación (materia, energía, cantidad de
movimiento)
Los aspectos de cinética de reacción y el diseño del reactor químico
comprenden lo fundamental del presente texto.

H. ZULETA
11
1.2.1 Reactores químicos
Consideremos que se debe llevar a cabo la reacción química
A + B C
Esta operación se desarrollará en un reactor químico, cuya selección ha sido
definida previamente, de acuerdo a los siguientes factores:
- Las condiciones de temperatura y presión deseadas
- En que grado se produce la reacción (Termodinámica)
- La velocidad de reacción (Cinética)
- Las fases presentes en la reacción
Generalmente existen muchas combinaciones de condiciones de operación y
tamaños de reactor que cumplen con los requerimientos de las leyes de la
termodinámica, química y cinética de reacción. El Ingeniero Químico debe
mantener un equilibrio entre el razonamiento analítico y su juicio o criterio
ingenieril para responder a una gran variedad de cuestionamientos, de los
cuales se puede mencionar los siguientes:
1. ¿Cuál es la composición de la alimentación? ¿Los reactivos estarán
en razón estequiometrica? ¿Será necesario un proceso de
purificación?
2. ¿Cuál es la capacidad requerida?
3. ¿Es necesario o deseable un catalizador?
4. ¿Será necesario añadir inertes para favorecer el rendimiento?
5. ¿Será un proceso intermitente, continuo o semicontinuo?
6. ¿Cuáles son los requerimientos térmicos? ¿Isotérmico o adiabático?
7. ¿Será conveniente la recirculación? ¿Un solo paso?

H. ZULETA
12
8. ¿Cuál será la conversión o composición de salida?
9. ¿Existen requerimientos especiales de materiales? ¿Corrosión?
¿Explosividad? ¿Toxicidad?
10. ¿Se presentan riesgos ambientales o ecológicos?
Las interrogantes anteriores con frecuencia, se condensan en tres tipos de
problemas y constituyen parte importante de las funciones del Ingeniero
Químico.
1. Cómo seleccionar el mejor tipo de reactor químico para una reacción
particular
2. Cómo estimar el tamaño necesario del reactor
3. Cómo determinar las mejores condiciones de operación del reactor
Las decisiones sólo pueden tomarse si se cuenta, previamente, con la
información correspondiente de la capacidad o escala de operación y de la
cinética de la reacción química involucrada.
Además, se puede requerir para un reactor que ya está en operación, los
siguientes tópicos:
1. Adaptar o realizar una nueva reacción con un requerimiento
determinado de producción en el equipo
2. Mejorar la calidad del producto
3. Determinar como se puede exigir mayor producción del reactor sin
gastos excesivos de capital, sin afectar el producto o producir
colapsos o inundaciones
En cuanto a la selección del reactor, el criterio a seguir debe conjugar diversos
aspectos que forman parte de las condiciones normales de trabajo; a saber: 1)
Costos del equipo y costos de operación. 2) Facilidad de manejo y seguridad

H. ZULETA
13
en la operación. 3) Efectos en el medio ambiente. 4) Disponibilidad y costo de
las materias primas.
Por supuesto, no existen reglas precisas en cuanto a la combinación apropiada
de esos factores, y cada caso se configurará en una decisión particular.
1.2.2 Principales funciones de los reactores químicos
Los reactores químicos tienen como principales funciones:
- Asegurar el tipo de contacto o modo de fluir de los reactantes en el
interior del reactor, para conseguir la mezcla deseada de las fases
presentes
- Proporcionar el tiempo suficiente de contacto entre las sustancias y el
catalizador (en el caso de reacciones heterogéneas), para conseguir la
extensión deseada de la reacción
- Permitir condiciones de presión, temperatura y composición de modo
que la reacción tenga lugar en el grado y velocidad deseados,
atendiendo a los aspectos termodinámicos y cinéticos de la reacción.
1.3 CINÉTICA QUÍMICA
El objeto de la cinética química es el estudio de las velocidades y los
mecanismos por los cuales unas sustancias se transforman en otras. Además
estudia los factores de que dependen dichas velocidades, explicando la causa
de la magnitud de esa velocidad de reacción. Entre las numerosas razones que
hacen importante su estudio, se pueden citar:
1. Para los fisicoquímicos, es la herramienta que les permite
profundizar en la naturaleza de los sistemas reaccionantes,
comprender como se forman y se rompen los enlaces químicos, y
estimar sus energías y estabilidades
2. Para los químico-orgánicos, el valor de la cinética es aún mayor
porque el modo en que reaccionan los compuestos les sirve de guía
sobre su estructura

H. ZULETA
14
3. Es la base de importantes teorías sobre combustión y disolución,
suministra un método para el estudio del transporte de calor y
materia, y sugiere métodos para tratar fenómenos de velocidad en
otros campos
4. El Ingeniero Químico ha de conocer la cinética de la reacción para
realizar un diseño satisfactorio del equipo en el que ésta ha de
efectuarse a escala técnica. Si la reacción es suficientemente rápida
para que el sistema esté prácticamente en equilibrio, el diseño es
muy sencillo ya que no es necesaria la información cinética y resulta
suficiente la información termodinámica.
1.3.1 Clasificación de las reacciones químicas
Una clasificación de los tipos de reacciones se puede hacer cuando menos
desde tres puntos de vista diferentes, en función del número de fases
presentes, en función de su complejidad y por la presencia de un
catalizador. También puede efectuarse en función de la manera como
representan resultados experimentales.
Cuando en el sistema reaccionante existe sólo una fase la reacción será
homogénea, y si están presentes dos o más será heterogénea. En ambos casos
puede o no haber la presencia de un catalizador. Los catalizadores actúan, en
cierto modo, como mediadores acelerando o retardando la reacción. Estos
materiales no necesitan estar presentes en grandes cantidades.
En la Tabla 1-1 se puede ver una clasificación general de reacciones químicas
distinguiendo entre sistemas homogéneos y heterogéneos, junto con la
posibilidad de que las reacciones sean catalíticas.
Tabla 1-1. Clasificación de reacciones químicas para el diseño de reactores
No catalizadas Catalizadas
Homogéneas La mayor parte de las
reacciones en fase gaseosa
La mayor parte de las
reacciones en fase líquida
Heterogéneas
Combustión de carbón Síntesis de amoniaco
Tostación de minerales Oxidación de amoniaco para
dar ácido nítrico
Ataque de sólidos por ácidos Cracking del petróleo
Oxidación gas-líquido con
reacción
Oxidación de SO2 a SO3

H. ZULETA
15
Además de la anterior se pueden establecer otro tipo de clasificaciones. En la
Fig.1-2 se expone una clasificación de las reacciones químicas atendiendo a
diferentes criterios
TIPOS DE REACCIONES
De acuerdo a la forma de su ecuación cinética:
Elementales (concentraciones de los reactantes con exponentes iguales a los
coeficientes estequiometricos)
No elementales (concentraciones de los reactantes con exponentes diferentes
a los coeficientes estequiometricos).
En función del número de fases:
Homogéneas (una sola fase)
Heterogéneas (más de una fase).
En función de su complejidad:
Simples (una sola ecuación estequiometrica, A + B R ).
Múltiples (o complejas, mas de una ecuación estequiometrica). Pueden ser en:
Serie ( A R S )
Paralelo ( A R; A S )
Serie-Paralelo ( A + B R; B + R S ).
En relación al equilibrio:
Irreversibles (conversión total).
Reversibles ( se llega al equilibrio antes de lograr el 100% de conversión).
Fig. 1-2 Clasificación de Reacciones químicas

H. ZULETA
16
1.4 INGENIERIA DE LAS REACCIONES QUÍMICAS Y CINETICA
En cinética química, el reactor químico es una herramienta utilizada para
determinaciones y estudios de sistemas reactivos. De estos últimos, los más
importantes son la velocidad de reacción y los efectos de la concentración de
especies y temperatura en dicha velocidad. En Ingeniería de reacciones
químicas, la información obtenida de la cinética química es un medio para
determinaciones acerca del reactor: el tamaño, configuraciones de flujo y
térmicas, distribución de productos, etc. La cinética, sin embargo, no provee
toda la información requerida para estos propósitos, y se debe considerar otros
procesos de velocidad que dificultan el diseño: la mecánica de fluidos, el
mezclado, la transferencia de calor, la difusión y la transferencia de masa.
Estas son restricciones de los Balances de materia (estequiometria) y energía,
y de equilibrio químico en algunos casos.
Se puede considerar tres niveles de tamaños de sistemas para apoyar la
comparación de la cinética y la ingeniería de reacciones químicas. En orden
creciente, estos niveles son:
(1) Microscópico o molecular- Un conjunto de moléculas
reactivas lo suficientemente grande para constituir un punto en
el espacio, caracterizado, en cualquier instante, por un valor
único de concentración Ci, temperatura T, presión P, y
densidad . Para fluidos se usa el término “elemento de
fluido” para describir este conjunto.
(2) Macroscópico local- Por ejemplo, una partícula de sólido en
reacción con un fluido, en la cual pueden existir gradientes de
concentración, temperatura, etc. al interior de la partícula; y
(3) Macroscópico global- Por ejemplo, un conjunto o un lecho de
partículas sólidas reaccionando con un fluido, en el cual,
además de gradientes locales en cada partícula, puede existir
gradientes globales a través del recipiente contenedor, entre
partículas y entre regiones de fluidos.

H. ZULETA
17
Estos niveles se ilustran en la Fig. 1-3. Los niveles (1) y (2) pertenecen al
dominio de la cinética, puesto que el interés se centra en la reacción
(velocidad, mecanismos, etc.). Posiblemente en conjunción con otros procesos
de velocidad, sujeto a restricciones de equilibrio y estequiometria. En el otro
extremo, el nivel (3) es del dominio de la Ingeniería de las reacciones
químicas, porque en general, es en este nivel que se requiere la información
suficiente para tomar decisiones acerca del reactor y su producción comercial.
No obstante estos comentarios, es posible bajo ciertas condiciones ideales del
nivel (3) lograr la información de las condiciones requeridas basándose sólo
en el nivel (1) o en los niveles (1) y (2) combinados.
1.5 VELOCIDAD DE REACCIÓN
Para una determinación eficaz de la velocidad de reacción hay que definir
previamente y con precisión lo que este término significa. La velocidad de
reacción es una magnitud positiva que expresa el cambio de la
concentración de un reactivo ( A ) o un producto ( R ) con el tiempo. La
mayoría de las veces, la velocidad de reacción se expresa en términos de las
concentraciones de los reactivos. En la Fig. 1-4 se muestra esquemáticamente
cómo puede variar con el tiempo esta concentración.
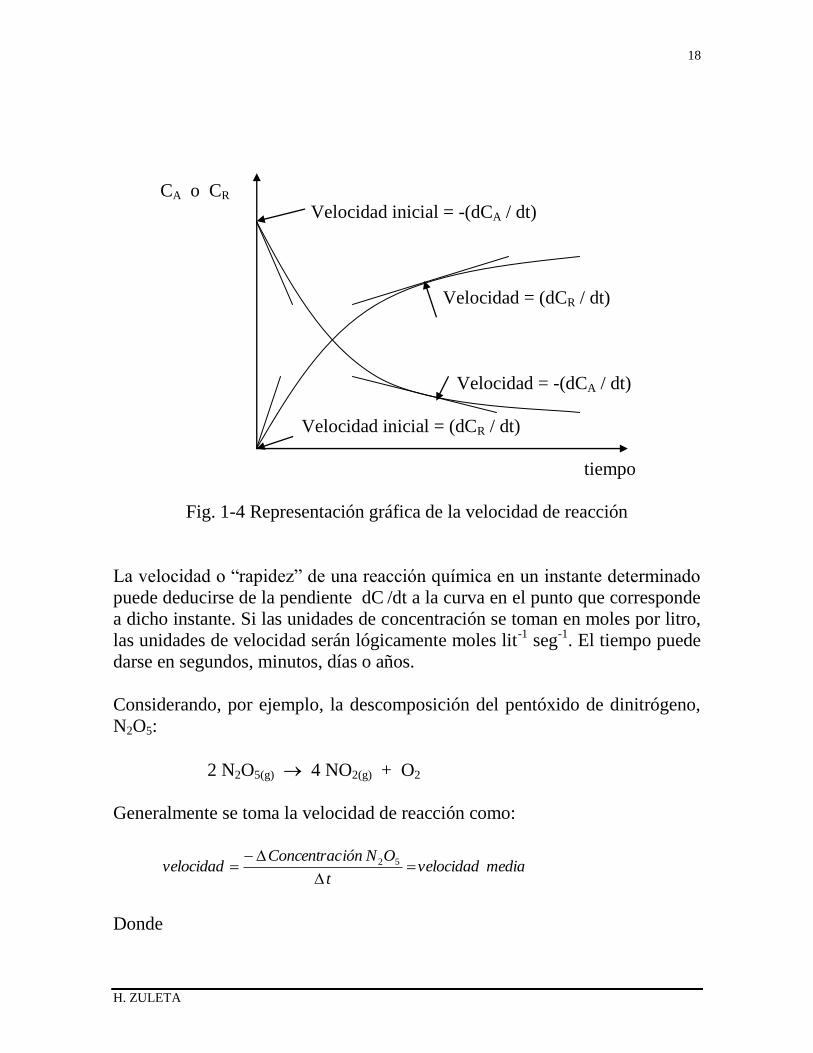
H. ZULETA
18
CA o CR
Velocidad inicial = -(dCA / dt)
Velocidad = (dCR / dt)
Velocidad = -(dCA / dt)
Velocidad inicial = (dCR / dt)
tiempo
Fig. 1-4 Representación gráfica de la velocidad de reacción
La velocidad o “rapidez” de una reacción química en un instante determinado
puede deducirse de la pendiente dC /dt a la curva en el punto que corresponde
a dicho instante. Si las unidades de concentración se toman en moles por litro,
las unidades de velocidad serán lógicamente moles lit-1
seg-1
. El tiempo puede
darse en segundos, minutos, días o años.
Considerando, por ejemplo, la descomposición del pentóxido de dinitrógeno,
N2O5:
2 N2O5(g) 4 NO2(g) + O2
Generalmente se toma la velocidad de reacción como:
mediavelocidadt
ONiónConcentracvelocidad
52
Donde

H. ZULETA
19
Concentración = Concentración final - Concentración inicial
t = tiempo final - tiempo inicial
El signo menos es necesario en la ecuación de velocidad para hacer que el
cambio de concentraciones sea positivo, ya que la concentración de N2O5
decrece con el tiempo.
Medida de la velocidad de reacción
Para medir la velocidad de reacción se debe determinar el cambio de las
concentraciones con el tiempo. En la reacción anterior, la velocidad se puede
medir de la siguiente forma: Se empieza poniendo 0,16 moles de N2O5 en un
recipiente de un litro a 67°C. A intervalos de 1 minuto se toman pequeñas
muestras de la mezcla en reacción y se analizan, para conocer la concentración
de N2O5. Así se obtienen los datos de concentración-tiempo que se dan en la
Tabla 1-2 (primera y segunda columna).
Tabla 1-2 Velocidad de descomposición de N2O5 a 67°C
Tiempo
(min)
[N2O5]
(moles / litro)
Velocidad
(moles / lit min)
0 0,160 0,054
1 0.113 0,040
2 0,080 0,028
3 0,056 0,019
4 0,040 0,014
Usando los datos de la tabla se puede calcular la velocidad media de la
reacción en un intervalo de tiempo. Debe aclararse que es un valor medio,
porque la velocidad cambia durante ese intervalo de tiempo.
Normalmente, lo que interesa no es la velocidad media, sino la velocidad
instantánea a un tiempo (o concentración) determinado. Para calcular esta
velocidad se puede recurrir a los siguientes métodos:

H. ZULETA
20
1. Métodos gráfico
2. Método del ajuste de polinomios
3. Método numérico de diferencias finitas.
1. Métodos gráfico. Se requiere construir la gráfica de concentración en
función del tiempo, se traza la tangente a la curva en un punto correspondiente
a un tiempo t determinado. La velocidad instantánea será la pendiente de la
recta tangente en el tiempo considerado. Se debe considerar que el valor de la
pendiente puede ser negativa o positiva según si se trate de reactivos o
productos respectivamente.
También es ampliamente conocido como método gráfico si se grafica C / t
en función del tiempo y luego se usa la diferenciación por áreas iguales para
obtener dC/dt.
2. Método del ajuste de polinomios. La técnica consiste en ajustar los datos de
concentración y tiempo a un polinomio de orden n:
C = ao + a1t + a2t2 +…+ ant
n (1.3)
Existe gran variedad de software para el calculo de los valores óptimos de las
constantes ai. Sólo se introducen los datos de concentración - tiempo y se
escoge el orden del polinomio. Una vez determinadas las constantes, se deriva
la ecuación polinómica (1.3).
dC / dt = a1 + 2 a2t + 3 a3t2 +… +n ant
n-1 (1.4)
Por lo tanto en cualquier instante t se puede conocer la concentración y la
velocidad instantánea de la reacción.
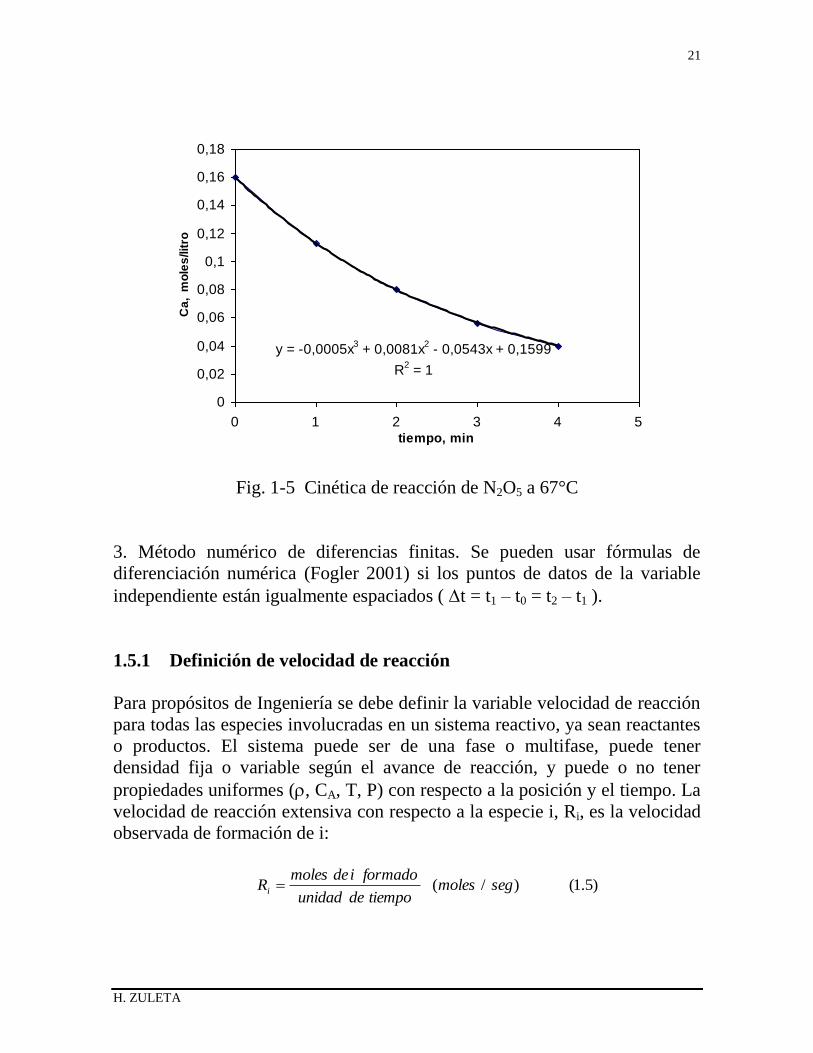
En la figura 1-5 se muestra la gráfica de concentración y tiempo para los datos
del ejemplo planteado además del polinomio de ajuste . Finalmente los valores
de velocidad obtenidos por este método se encuentran en la tercera columna
de la Tabla 1-2.
H. ZULETA
21
y = -0,0005x3 + 0,0081x
2 - 0,0543x + 0,1599
R2 = 1
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0 1 2 3 4 5tiempo, min
Ca
, m
ole
s/lit
ro
Fig. 1-5 Cinética de reacción de N2O5 a 67°C
3. Método numérico de diferencias finitas. Se pueden usar fórmulas de
diferenciación numérica (Fogler 2001) si los puntos de datos de la variable
independiente están igualmente espaciados ( t = t1 – t0 = t2 – t1 ).
1.5.1 Definición de velocidad de reacción
Para propósitos de Ingeniería se debe definir la variable velocidad de reacción
para todas las especies involucradas en un sistema reactivo, ya sean reactantes
o productos. El sistema puede ser de una fase o multifase, puede tener
densidad fija o variable según el avance de reacción, y puede o no tener
propiedades uniformes (, CA, T, P) con respecto a la posición y el tiempo. La
velocidad de reacción extensiva con respecto a la especie i, Ri, es la velocidad
observada de formación de i:
)5.1()/( segmolestiempodeunidad
formadoidemolesRi

H. ZULETA
22
La velocidad de reacción intensiva, ri, es la velocidad referida a una cantidad
especifica de normalización (CN), o base de velocidad, tal como el volumen
del sistema de reacción o masa de catalizador.
)6.1()/()()(
3msegmolesCNdeunidadtiempodeunidad
formadoidemolesri
Las velocidades de reacción, Ri o ri serán negativas si i se consume , y positiva
si aparece como producto. Se pueden definir velocidades de reacción de
especies independientes para reacciones simples o etapas de un mecanismo,
pero esto requiere consideraciones de estequiometria.
La velocidad ri es independiente del tamaño y de las circunstancias físicas del
sistema reactivo, mientras que Ri no lo es. Por lo tanto, ri, que es una cantidad
más ampliamente usada, se puede considerar una velocidad “puntual” o
“intrínseca” en un nivel molecular. Las dos velocidades se pueden relacionar,
considerando el volumen como la CN, de la siguiente forma:
Para un sistema uniforme, como un estanque bien mezclado,
)7.1(VrR ii
1.5.2 Factores que modifican la velocidad de reacción
La velocidad de reacción depende de varios parámetros, siendo los más
importantes:
(1) La naturaleza de las especies involucradas en la reacción.
(2) La concentración de las especies
(3) La temperatura
(4) La actividad catalítica
(5) La naturaleza de contacto de reactantes; y
(6) La longitud de onda de la radiación incidente.
(1) Existen muchos ejemplos de reacciones muy rápidas relacionadas con
iones en solución, tales como la neutralización de ácidos y bases fuertes y
explosiones. En este caso la velocidad de reacción depende de la velocidad
con que los reactantes se ponen en contacto íntimo. En el otro extremo,
reacciones muy lentas pueden ser de tipo heterogéneo, tal como la oxidación
del carbono a temperatura ambiente. Otros ejemplos de reacciones muy

H. ZULETA
23
rápidas son la producción de polietileno, uno de los plásticos más importantes,
o la producción de gasolina a partir del crudo de petróleo. Las reacciones se
completan en menos de un segundo, mientras que las reacciones en el
tratamiento de aguas residuales, pueden tomar días o semanas.
En la Figura 1-6 se indican velocidades relativas de reacciones. Se puede
observar de estos valores que las velocidades en las plantas de tratamiento de
desechos, respecto de motores de cohetes, son equivalentes a una relación de 3
años a un segundo.
10-4
10-3
10-2
0,1 1 10
trabajo exigente
reacciones celulares en Plantas de Gases en partículas porosas de
tratamiento de Ser humano en reposo catalizador Hornos de combustión
Aguas
103 10
4 10
9 10
10
Motores de aviones Motores de cohetes reacciones bimoleculares, en las Cuales cada colisión ocurre a 1 atm
Y 400°C
Fig. 1- 6 Velocidades relativas de reacción
-rA’’’
= moles de A consumidos / m3 seg.
(2) La velocidad de reacción generalmente aumenta al incrementarse las
concentraciones de reactantes ( y algunas veces de los productos).
Muchas reacciones de combustión ocurren más rápidamente con
oxígeno puro que con aire a la misma presión total.
(3) La velocidad de reacción usualmente aumenta de forma exponencial
con el incremento de la temperatura. Una excepción importante es la
oxidación de óxido nítrico, el cual se usa en la fabricación de ácido
nítrico; en este caso, la velocidad disminuye con la temperatura.
(4) Muchas reacciones proceden más rápidamente en presencia de una
substancia que no se consume ni genera en la reacción. Este es el
fenómeno de la catálisis y muchos procesos biológicos e industriales

H. ZULETA
24
dependen de ellos. Por ejemplo, la oxidación de SO2 a SO3 se acelera
enormemente en presencia de V2O5 como catalizador, por lo tanto el
proceso de producción de ácido sulfúrico depende de este hecho.
(5) La naturaleza o facilidad de contacto de reactantes puede afectar
grandemente la velocidad de reacción, es decir, el carbono finamente
dividido se quema mucho más rápido que trozos de carbón. La
titulación de un ácido con una base ocurre mucho más rápido si se
agita la mezcla, en vez del escurrimiento natural de la base en la
solución ácida.
(6) Algunas reacciones ocurren mucho más rápido si el sistema de
reacción se expone a radiación incidente de una frecuencia apropiada.
Por ejemplo, una mezcla de cloro e hidrógeno puede mantenerse en la
sombra, y la reacción para formar HCl puede ser muy lenta; sin
embargo, si la mezcla se expone a la luz ordinaria, la reacción ocurre
con una velocidad explosiva. Tales reacciones se conocen
generalmente como reacciones fotoquímicas.
La manera en la cual la velocidad de reacción depende de estos parámetros se
expresa matemáticamente en la forma de una ley de velocidad; esto es, para
especies A en una determinada reacción, la ley de velocidad toma la forma
general
rA = rA (concentración, temperatura, actividad catalítica, etc)
La forma de esta ley debe ser determinada experimentalmente, y su expresión
completa puede ser compleja, y en varios casos, muy difícil, sino imposible de
formular explícitamente.
1.5.3 Ecuación de velocidad y velocidad de reacción
La extensión en la cual ha progresado una reacción se conoce como grado de
avance de la reacción. Para reacciones a volumen constante puede definirse
como:
)8.1()( 0
i
ii CtC

H. ZULETA
25
En esta expresión Ci(t) es la concentración del componente i que hay en el
tiempo t, Cio la inicial y i el coeficiente estequiometrico con signo, positivo
para productos y negativo para reactantes.
El grado de avance es independiente de la especie que se mida, por ello la
velocidad de reacción también se puede definir como la variación del grado de
avance con el tiempo:
)9.1()(1
dt
tdC
dt
d i
i
Por ejemplo, en la reacción trimolecular (tres moléculas reactantes):
A + 2 B 3 D (1.10)
La velocidad se puede expresar convenientemente como:
)11.1(32
.dt
dC
dt
dC
dt
dCvel DBA
La división por los coeficientes estequiométricos es necesaria para unificar el
concepto de velocidad de reacción, haciéndolo independiente de la especie
química considerada. Además asegura que la velocidad de reacción sea
siempre positiva, tanto si se considera un reactivo como un producto.
La expresión que relaciona la velocidad de reacción con las concentraciones
de los compuestos que toman parte en ella se denomina ley de velocidad o
ecuación de velocidad r (ley de potencia de la velocidad).
)12.1(in
iCKr
En donde
K = coeficiente o constante de velocidad
[conc.]1 – m
[tiempo]-1
ni = orden parcial con respecto al reactante i
puede ser positivo o negativo, números
enteros o fraccionarios

H. ZULETA
26
m =
N
i
in1
= orden global (orden de la reacción)
En el caso de modelos empíricos: ni no siempre puede ser interpretado
físicamente. Solamente para las reacciones elementales el orden ni
corresponde al coeficiente estequiométrico i. Las concentraciones de
productos también pueden aparecer en la ecuación cinética.
nproducto > 0 reacciones autocatalíticas
nproducto < 0 inhibición por productos
Ejemplo:
La ley de velocidad para la reacción C2H4Br2 + 3 KI C2H4 + 2 KBr + KI3
en un solvente inerte, la cual puede escribirse como A + 3 B Productos, se
ha determinado igual a (-rA) = KACACB, con KA = 1,34 L mol-1
h-1
a 74,9°C
(a) Para la velocidad de desaparición de KI (-rB), ¿Cuál es el valor
de la constante KB?
(b) ¿A que velocidad se está consumiendo el KI cuando las
concentraciones son CA = 0,022 y CB = 0,22 moles / litro?
Respuestas:
(a) Según las velocidades de reacción para A y B
(-rA) = dCA / dt (-rB) = dCB / dt
aplicando las ecuaciones (1.9), (1.11) y la estequiometria de la reacción
)(3
1)(
31BA
BA rrdt
dC
dt
dC
Por lo tanto (-rB) = 3 (-rA) = 3 KA CA CB = KB CA CB
KB = 3 KA = 3(1,34) = 4,02 lt mol-1
h-1

H. ZULETA
27
(b) (-rB) = KB CA CB = 4,02*0,022*0,22 = 0,0195 mol/(L min).
1.5.4 Mecanismos de reacción
Un mecanismo de reacción es una descripción del camino o secuencia de
pasos por los que transcurre la reacción. Las diversas etapas de una reacción
químicas son frecuentemente muy complicadas y no pueden predecirse a partir
de la ecuación completa que normalmente se escribe para la reacción. Por
ejemplo, la ecuación que se escribe para la reacción del hidrógeno con el
oxígeno para formar agua, parece indicar que dos moléculas de hidrógeno y
una de oxígeno se ponen en contacto simultáneamente y que de este encuentro
se producen inmediatamente dos moléculas de agua. Mediante numerosos
experimentos se ha comprobado que este caso no se da en absoluto. Existen
partículas muy reactivas como H, HO2, OH y O que aparecen durante un breve
instante antes de que choquen y reaccionen con otras.
Otro ejemplo puede ser la reacción del CO con NO2
CO(g) + NO2(g) NO(g) + CO2(g)
La experiencia indica que, a bajas temperaturas, por debajo de 500°K, la
reacción ocurre en dos etapas:
Etapa 1: NO2(g) + NO2(g) NO3(g) + NO(g)
Etapa 2: NO3(g) + CO(g) NO2(g) + CO2(g)
______________________________
CO(g) + NO2(g) NO(g) + CO2(g)
La reacción total, sumando las etapas 1 y 2, es la misma que la reacción en
una sola etapa anteriormente planteada. Sin embargo, las dos expresiones de la
velocidad de reacción son bastante diferentes.
Expresión de velocidad a partir del mecanismo

H. ZULETA
28
Las expresiones de velocidad de las reacciones deben ser determinadas
experimentalmente. Después de esto, es posible deducir un posible mecanismo
que explique la expresión de velocidad encontrada. Es un proceso complicado
y no se tratará en estos apuntes. En su lugar, se analizará el proceso inverso,
que es mucho más sencillo. Dado el mecanismo de la reacción del ejemplo
anterior, se deducirá la expresión de la velocidad de la reacción. Para ello se
seguirán dos reglas:
1. Para cualquier etapa se supone reacciones elementales y que
ocurren vía colisiones
Etapa 1: velocidad = K1 CNO2 CNO2 = K1 (CNO2)2
Etapa 2: velocidad = K2 CNO3 CCO
Donde K1 y K2 son las constantes de velocidad de las etapas individuales.
2. Frecuentemente, uno de los pasos del mecanismo es más lento que los
demás. En tales casos, la etapa más lenta es la que controla la velocidad.
Esto es, la velocidad de todo el proceso está determinada por la etapa más
lenta. La situación es análoga a la de una carrera de relevos en la que hay un
corredor de relevos más lento y dos más rápidos. El tiempo que el equipo
necesita para alcanzar la meta depende fundamentalmente del corredor más
lento.
En el proceso de dos etapas anteriormente mencionado, la primera etapa es
mucho más lenta que la segunda, de donde se deduce que la velocidad del
proceso total es la de la etapa 1:
Velocidad = K1 (CNO2)2
Experimentalmente se ha encontrado que la reacción entre el CO y el NO2 a
baja temperatura es, en efecto, de segundo orden en NO2 y de orden cero en
CO, como este análisis predice. Esta situación es diferente a la que ocurre a
alta temperatura. Por encima de 600°C, la reacción tiene lugar en una sola
etapa; es decir, a través de colisiones producidas entre una molécula de CO y
una molécula de NO2. La reacción a alta temperatura es, como se ha
planteado, de primer orden con respecto al NO2, y de primer orden con
respecto al CO.

H. ZULETA
29
Los tipos de productos intermedios que se supone están relacionados con la
naturaleza química de las sustancias, pueden clasificarse en los siguientes
grupos:
Radicales libres. Son átomos libres o fragmentos estables de moléculas (CH3,
C2H5, I, CCl3 )
Iones y sustancias polares. Son átomos, moléculas o fragmentos de
moléculas cargados eléctricamente (N3-, Na
+, OH
-, H3O
+)
Complejos de transición.
Los esquemas de las reacciones supuestas, con los tres tipos de productos
intermedios que se han considerado, pueden ser de dos clases:
Reacciones sin mecanismo en cadena
Reactantes (productos intermedios)*
(productos intermedios)* Productos
Reacciones con mecanismo en cadena. En las reacciones con mecanismo en
cadena el producto intermedio se forma en la primera reacción, llamada
eslabón de iniciación; después reacciona con el reactante dando el producto y
más producto intermedio en el eslabón de propagación ( a veces el producto
intermedio se consume en el eslabón final)
Reactante ( Prod. Inter..)* Iniciación
( Prod. Interm.)* + Reactante ( Prod. Interm.)
* + Producto Propagación
( Prod. Interm.)* Producto Terminación
La etapa de propagación es la característica esencial de la reacción en cadena.
En esta etapa el producto intermedio no se consume sino que actúa
simplemente como un catalizador para la conversión de la sustancia; de este
modo, cada molécula del producto intermedio puede catalizar una larga
cadena de reacciones antes de que se destruya.

H. ZULETA
30
1.6 DEPENDENCIA DE LA VELOCIDAD DE REACCION CON LA
TEMPERATURA
En las expresiones cinéticas estudiadas se ha observado la variación de la
velocidad de reacción como dependiente de las concentraciones de reactantes
y productos a temperatura constante. Para obtener la ecuación cinética
completa se necesita conocer también el efecto de la temperatura sobre la
velocidad de reacción.
La velocidad de la mayor parte de las reacciones aumenta con la temperatura.
Cualquier persona con apuros para cocinar aplica este principio cuando usa
una olla a presión, que aumenta la temperatura de cocción de los alimentos.
De la misma forma cuando se guardan los alimentos en el frigorífico, se
pretende que los procesos químicos de alteración o descomposición se
produzcan más lentamente.
Como regla general aproximada se puede decir que un aumento de la
temperatura de 10ºC duplica la velocidad de reacción. Según esta regla, las
comidas deben cocinarse en la mitad de tiempo en una olla a presión a 110ºC
que en una olla abierta, y deben deteriorarse cuatro veces más rápido a la
temperatura ambiente (25ºC) que en el frigorífico a 5ºC.
El efecto de la temperatura sobre la velocidad de reacción se puede explicar en
términos de la teoría cinética de los gases. Un aumento de temperatura
aumenta la fracción de moléculas con valores altos de energía cinética. Esto
significa que a temperatura superior, una fracción mayor de moléculas tendrá
energía suficiente para reaccionar cuando colisionen. Por lo tanto, la reacción
será más rápida. En otras palabras la constante de velocidad K, se hará más
grande cuando la temperatura aumente.
Ej. CO(g) + NO2 (g) CO2 (g) + NO (g)
T / (ºK) 600 650 700 750 800
K / (lt/mol s) 0,028 0,22 1,3 6,0 23
H. ZULETA
31
La constante aumenta en un factor de casi 1000 cuando la temperatura
aumenta de 600 a 800°K.
Presentan comportamiento excepcional las reacciones complejas o aquellas
limitadas por factores físicos tales como la difusión, la adsorción o la acción
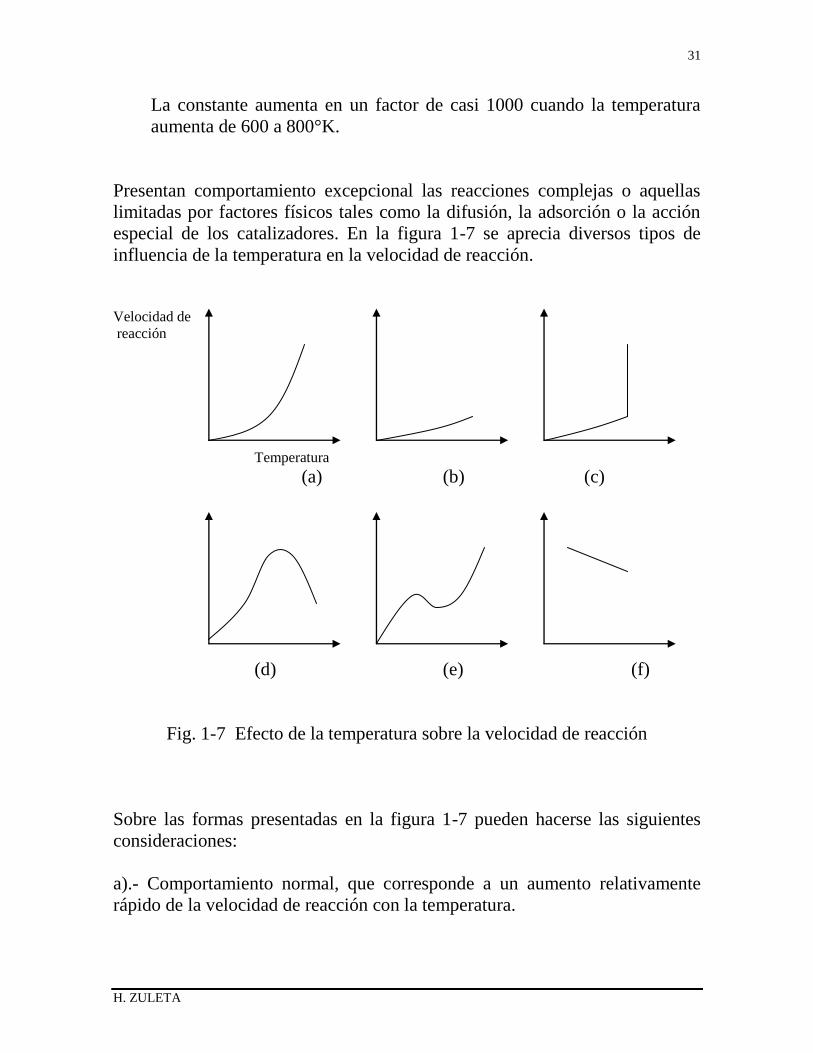
especial de los catalizadores. En la figura 1-7 se aprecia diversos tipos de
influencia de la temperatura en la velocidad de reacción.
Velocidad de
reacción
Temperatura
(a) (b) (c)
(d) (e) (f)
Fig. 1-7 Efecto de la temperatura sobre la velocidad de reacción
Sobre las formas presentadas en la figura 1-7 pueden hacerse las siguientes
consideraciones:
a).- Comportamiento normal, que corresponde a un aumento relativamente
rápido de la velocidad de reacción con la temperatura.

H. ZULETA
32
b).- Algunas reacciones heterogéneas regidas por la resistencia de difusión
entre fases presentan un aumento pequeño de la velocidad con la temperatura.
c).- En las reacciones explosivas típicas, la velocidad aumenta rápidamente a
la temperatura de ignición.
d).- Comportamiento típico de las reacciones catalíticas controladas por la
velocidad de adsorción (en las que la cantidad de sustancia adsorbida
disminuye a temperaturas altas) y de las reacciones enzimáticas (en las que se
destruye la enzima a temperaturas altas).
e).- Curva típica de algunas reacciones, por ejemplo, la oxidación del carbono,
complicadas por reacciones secundarias que se hacen más significativas a
medida que se eleva la temperatura.
f).- La velocidad disminuye al elevarse la temperatura, por ejemplo, en la
reacción entre el oxígeno y el óxido nítrico, por favorecerse el equilibrio de
conversión a temperaturas bajas y depender la velocidad del alejamiento del
equilibrio.
1.6.1 Teoría de Arrhenius
En muchas reacciones, y en particular las reacciones elementales, la expresión
de velocidad puede escribirse como producto de un factor dependiente de la
temperatura y otro dependiente de la composición.
Velocidad = 1(temperatura) 2(composición)
Velocidad = K 2 (composición)
Para tales reacciones el término dependiente de la temperatura, la constante de
velocidad de reacción, se ajusta a la ecuación conocida como Ley de
Arrhenius
)13.1(exp0
RT
EKK A

H. ZULETA
33
donde Ko se conoce como factor de frecuencia o factor pre-exponencial y EA
es la energía de activación [ J/mol]. Las unidades de Ko son las mismas de K y
R, la constante de los gases ideales puede adoptar los valores de 8,314 J/mol
°K o 1,987 cal/mol °K.
La ecuación de Arrhenius se ajusta muy bien a resultados experimentales en
un amplio rango de temperaturas y se considera como una primera
aproximación para el estudio del efecto de la temperatura sobre la ecuación
cinética.
De la ecuación se puede ver que a temperatura constante cuanto mayor es la
Energía de activación, más pequeña será la constante de velocidad y por lo
tanto más lenta será la velocidad de reacción. Por el contrario velocidades de
reacción rápida tendrán una EA pequeña.
Para una misma concentración, pero a dos temperaturas diferentes, la
aplicación de la ley de Arrhenius indica:
)14.1(11
lnln211
2
1
2
TTR
E
K
K
r
r A
1.6.2 Comparación de teorías con la Ley de Arrhenius
La ecuación
RT
E
mA
eTKK
'
0 0 m 1 (1.15)
Complementa los planteamientos más simples de la teoría de colisiones y la
teoría del estado de transición, sobre la variación del coeficiente cinético con
la temperatura. Como el término exponencial es mucho más sensible a la
temperatura que el término Tm, la variación de K debido a este último término
esta enmascarada y en consecuencia resulta que
K e –E/RT
K = Ko e –E/RT

H. ZULETA
34
1.6.3 Influencia de la energía de activación
El efecto de la temperatura sobre la velocidad viene dado por la energía de
activación y por el nivel de temperaturas. De la figura 1-8 que relaciona ln K y
1/T, de acuerdo con la ecuación 1.14, se pueden plantear las siguientes
conclusiones:
Ln K
E alta
-E/R
E baja
1/T
Fig. 1-8 Dependencia de la constante cinética con la temperatura
1. Si se cumple la ecuación de Arrhenius, representando ln K frente a
1/T se obtiene una recta de pendiente elevada si E es grande, y
pendiente pequeña si E es pequeña.
2. Las reacciones con energía de activación grande son muy sensibles a
la temperatura; las reacciones con energía de activación pequeñas
son muy poco sensibles a la temperatura.
3. El efecto de la temperatura sobre una reacción es mucho mayor a
temperatura baja que a temperatura alta.
4. Se deduce de la ecuación de Arrhenius, que el factor de frecuencia,
Ko no afecta a la influencia de la temperatura sobre la reacción. En

H. ZULETA
35
casos reales puede haber una pequeña influencia de la temperatura
sobre este factor, como predice la ecuación (1.15), sin embargo es
muy pequeña y puede despreciarse.
1.7 MEDICIONES EXPERIMENTALES EN CINÉTICA DE
REACCIÓN
Establecer la ley de velocidad de una reacción en forma experimental
involucra determinar los valores de los parámetros cinéticos, tales como el
orden de reacción, la constante cinética, la energía de activación y el factor de
frecuencia. Un enfoque general para sistemas simples requiere resolver o
seleccionar los siguientes factores:
(1) Seleccionar las especies (reactantes o productos) a las cuales seguir
la extensión de la reacción o especificaciones de velocidad; si la
estequiometria no es conocida, será necesario su determinación
experimental, además de la comprobación de si se trata de reacciones
simples.
(2) Elegir el tipo de reactor a ser utilizado y sus características de
operación. Esto permite la interpretación numérica de la velocidad a
través de las ecuaciones de balances de materiales apropiadas.
(3) Elegir el método para seguir la extensión de la reacción con respecto
al tiempo o una cantidad relacionada con el tiempo(por ejemplo,
análisis químico)
(4) Elegir una estrategia experimental que considere el desarrollo de los
puntos (1) al (3), es decir, cómo efectuar los experimentos y el
número y las características de ellos.
(5) Elegir el método para la determinación numérica de los valores de
los parámetros, y como se relacionan en la forma real de la ley de
velocidad.
1.7.1 Métodos experimentales para medir la extensión de una reacción
Para sistemas simples, sólo es necesario seguir la extensión (el progreso) de
una reacción mediante un tipo de medida. Esta puede ser la concentración de
una especie u otra propiedad dependiente de la concentración. Las formas más
antiguas consideran un método químico de análisis con muestreo intermitente,
y los más actuales son método físicos con instrumentos que pueden
monitorear en forma continua la característica elegida del sistema.

H. ZULETA
36
Una gran variedad de técnicas utilizan métodos físicos y químicos. Las
técnicas clasificadas ex situ requieren la remoción y análisis de mezcla
reaccionante. Los métodos in situ consisten en medidas instantáneas del
estado del sistema reaccionante sin perturbaciones de este.
METODOS QUÍMICOS
La titulación de un ácido con una base, o viceversa, y la precipitación de un
ion en un compuesto insoluble son ejemplos de métodos químicos de análisis
usados para determinar la concentración de especies en muestras de líquido
removidas del reactor. Dichos métodos son usados en reacciones
relativamente lentas. Esto es debido al tiempo que se ocupa en el análisis; la
simple colección de la muestra no detiene el progreso de la reacción, por lo
tanto se puede requerir una forma de detención de la reacción. Para reactores
batch la dificultad de establecer el tiempo real de medición de la
concentración es más notoria que en el caso de reactores de flujo.
METODOS FISICOS
A medida que una reacción progresa, las propiedades físicas del sistema
varian debido a cambios en la composición química. Si una propiedad cambia
de una forma mensurable esta se puede relacionar con la composición, por lo
tanto la velocidad de cambio de la propiedad es una medida de la velocidad de
reacción. La relación entre la propiedad física y la composición puede ser
conocida de antemano por un modelo simple y aproximado, o puede
establecerse por un procedimiento de calibración. Una ventaja del método
físico es la posibilidad de monitoreo continuo de la propiedad del sistema
usando instrumentos de medición sin perturbar el sistema con toma de
muestras.
Los siguientes son ejemplos de cambios de propiedades físicas que pueden
utilizarse en mediciones cinéticas.
(1) Cambios de presión en una reacción en fase gaseosa que involucra
cambios en los moles totales de gas en un reactor de volumen
constante; en este caso, se mide la presión total y se relaciona con la
concentración de especies. Los instrumentos usados son manómetros
de variados tipos.

H. ZULETA
37
(2) Cambios de volumen en reacciones en fase líquida; la densidad de
sistemas reaccionantes pueden cambiar muy levemente, y el efecto
se puede trasladar a cambios considerables de volumen mediante un
tubo capilar montado en el reactor (el cambio en el volumen es un
porcentaje pequeño del volumen total del reactor). Este tipo de
reactor se conoce como dilatómetro. El cambio en el volumen se
relaciona con un cambio en el volumen de líquido en el capilar, el
cual se sigue mediante un microscopio corredizo.
(3) Cambios en la rotación óptica en un sistema reactivo con isomeros
óptimamente activos (ejemplo, la inversión de sucrosa); el
instrumento usado es un polarímetro para medir el ángulo de
rotación de la luz polarizada que pasa a través del sistema.
(4) Cambios de la conductividad eléctrica en sistemas con especies
iónicas (ejemplo, la hidrólisis del acetato de etilo); la reacción se
efectúa en una celda de conductividad provista de un circuito para
mediciones de resistencia.
(5) Cambios en el índice de refracción medido en un refractómetro (para
sistemas líquidos) o en un interferómetro (para sistemas gaseosos).
(6) Cambios de color, usando celda de un espectrofotómetro.
(7) Electrodos de ión único para mediciones de concentración de
especies individuales.
(8) Medidas continuas de masa para reactantes sólidos, o absorbentes de
productos.
1.8 REACTOR DISCONTINUO DE VOLUMEN CONSTANTE
Es un recipiente que se llena con reactantes en un tiempo inicial (t = 0) y se
mezclan intensamente, de tal manera, que la concentración de especies sea
homogénea, es decir, el mismo valor en cualquier punto del reactor. Por lo
tanto la concentración depende únicamente del tiempo medido del proceso.

H. ZULETA
38
La característica de volumen constante, se refiere a la mezcla reaccionante. Se
dice que es un sistema reaccionante a volumen constante o de densidad
constante. En este reactor ocurren principalmente reacciones en fase líquida y
reacciones en fase gaseosa que se efectúan en bombas de volumen constante.
Para este tipo de sistemas, la medida de la velocidad de reacción de un
componente i será:
dt
dC
dt
V
Nd
dt
dN
Vr i
i
i
i
1 (1.16)
Considerando comportamiento de gas ideal
dt
dP
RTr
CV
n
RT
PRTnVP
i
i
i
ii
ii
1
Para medir la velocidad de reacción, que viene expresada por la velocidad de
cambio de la concentración o de su presión, no importa la magnitud que se
escoja para seguir el progreso de la reacción. Lo que se debe procurar es
relacionar estas medidas con la concentración o la presión parcial.
1.8.1 Análisis de datos de presión total en un sistema de volumen
constante
Para reacciones isotérmicas en fase gaseosa en las que el número de moles
varía durante la reacción. Las variables en este caso que relacionan
concentración y tiempo son ahora la presión total o presión parcial.

H. ZULETA
39
Si se considera una ecuación estequiométrica general del tipo:
aA + bB +..... rR + sS +....
t= 0 NA0 NB0 NR0 NS0 inerte
t= t NA0 – ax NB0 – bx NR0 + rx NS0 + sx inerte
Inicialmente el número total de moles N0 es
N0 = NA0 + NB0 + ... + NR0 + NS0 + .... + inertes
En el instante t es : N = No + x ( r + s + ...+..-a –b..) = No + x ( n )
Con n = r + s ... –a –b ...
Suponiendo comportamiento de gas ideal para el reactante A ( por
ejemplo)
V
xaN
V
N
RT
PC AAA
A
0 es decir
V
xa
V
NC A
A 0 Reemplazando n
NNx
0(
RTn
NN
V
a
V
NC A
A /*)( 00
RTn
NN
V
a
V
NPRTC A
AA
)( 00
)( 00
n
aPP AA (1.17)
= presión total en el instante t
o = presión total inicial del sistema

H. ZULETA
40
Análogamente, para cualquier producto R
)( 00
n
rPRTCP RRR (1.18)
Obs.: Debe conocerse la estequiometria de la reacción.
El tratamiento anterior ha estado limitado a sistemas sin flujo a
temperatura constante y volumen constante, por lo que la velocidad
puede representarse por medio de la velocidad de cambio de
concentración de un reactante o producto. Con estas restricciones, la
presión total de una reacción gaseosa varía muy poco, a menos que
cambie el número de moles totales. Sin embargo, si se presenta un
aumento del número de moles durante el transcurso de la reacción, se
producirá un aumento de presión. Este aumento esta relacionado
exclusivamente con el grado de reacción. Por lo tanto, la medición de la
presión total en función del tiempo es un método adecuado para estudiar
la cinética de este tipo de sistemas.
1.9 GRADO DE CONVERSION
Una variable ampliamente usada en ingeniería de reacciones es la conversión
fraccional, definida como la fracción de reactante que se ha convertido ( Xi ).
Al suponer que NA0 es la cantidad inicial de reactante A en el reactor en el
tiempo t=0, y que NA es la cantidad actual de A en el tiempo t. La conversión
de A en el sistema de volumen constante esta dada por:
)19.1(1/
/1
000
0
A
A
A
A
A
AAA
C
C
VN
VN
N
NNX
Por lo tanto )20.1(0A
AA
C
dCdX
En los métodos que se desarrollan en el siguiente capitulo se usarán como
variables la concentración y la conversión. Para el caso de sistemas con
volumen variable se usará la relación entre CA y XA.

H. ZULETA
41
1.10 METODOS DE ANALISIS DE DATOS
Una vez obtenida la curva cinética Ci = f (t), a través de experimentos
realizados a temperatura constante, y en el caso que no existan puntos de
inflexión, se considera que la ecuación de velocidad de reacción sigue la
forma
n
i
i
i KCdt
dCr (1.21)
en donde Ci = concentración del compuesto i
t = tiempo
K = constante de velocidad
n = orden de la reacción
Para comprobar la validez de esta ecuación, se necesita conocer los valores de
K y n. Para ello se plantean los siguientes métodos:
1. Método Integral
2. Método diferencial
3. Método de las velocidades iniciales
4. Método de la vida fraccional
1.11 METODO INTEGRAL
a) Se elige una forma de la expresión de velocidad ( se supone “n”
=0,1,2,3,1/2 etc.) y se reemplaza en la ecuación básica anterior.
b) Se integra la ecuación y se comparan los datos “modelo” de C en
función de t, con los datos experimentales respectivos.
c) Si el ajuste no es satisfactorio (constancia de K), será necesario
probar con otra cinética (otro orden n)

H. ZULETA
42
PROCEDIMIENTO
i) Para sistemas isotérmicos a volumen constante, la expresión cinética general
para la desaparición de un reactante A es
)(),( AAA
A CfkCkfdt
dCr
ii) Separando variables
dtkCf
dC
A
A )(
iii) Integrando:
dt
Cf
dC
A
A
)(
Límites de integración t = 0 CA = CA0 , t = t CA = CA
La ecuación integrada tiene la forma de una recta que pasa por el origen con
pendiente K.
1.12 METODO INTEGRAL PARA MODELOS SENCILLOS DE
VELOCIDAD
1.12.1 Reacción irreversible de orden cero. En esta situación la velocidad es
independiente de la concentración. Puede presentarse en dos situaciones:
Cuando la velocidad es intrínsicamente independiente de la concentración y
cuando la especie es tan abundante que su concentración es prácticamente
constante durante la reacción y prevalece un orden cero aparente. Algunas de
estas reacciones se presentan en catálisis heterogénea, cuando la reacción se
lleva a cabo sobre una superficie saturada de reactivos o bien en reacciones
catalíticas con suficiente exceso de substratos para saturar el catalizador.

H. ZULETA
43
Ejemplo A Productos
dtkdCCk
dt
dCr AA
AA
0
Aplicando límites de integración
tkCC AA )( 0
Obteniéndose la ecuación integrada de la ley de velocidad para reacción de
orden cero )22.1(0 tkCC AA
CA
“La concentración del reactante disminuye
linealmente con el tiempo”
“m” = -K
t = CA0 / K
t
Fig. 1-9 Representación de la ecuación integrada de orden cero
También la representación de la velocidad de reacción en función del tiempo
toma la forma siguiente:
-rA
t
Fig. 1-10 Velocidad de reacción constante – orden cero

H. ZULETA
44
Ecuación integrada en función de la conversión
De acuerdo con la definición de grado de conversión ( ecuación 1.19 ).
)1(0 AAA XCC
La ecuación integrada quedará
tkCXC AAA 00 )1(
)23.1(0 tkXC AA
Los datos de conversión y tiempo deberán ajustar la siguiente gráfica
XA
1
“m” = K / CA0
CA0/K t
Fig. 1-11 Cinética de conversión para la reacción de orden cero
Existe un tiempo característico en reacciones químicas de cero, primer y
segundo orden; el tiempo de vida medio ( t1/2 ), el cual se define como el
tiempo requerido para que la concentración de un reactante alcance la mitad
del valor inicial CA0.
Para la reacción de orden cero t1/2 = CA0 / 2K (1.24)
H. ZULETA
45
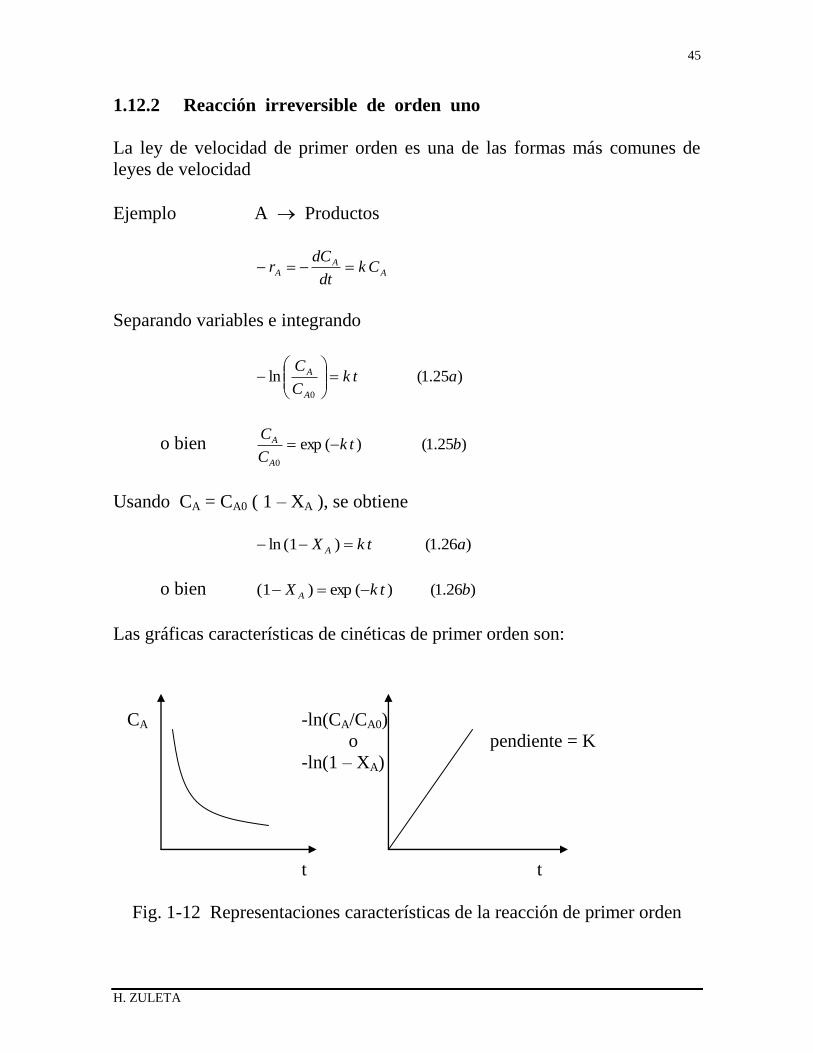
1.12.2 Reacción irreversible de orden uno
La ley de velocidad de primer orden es una de las formas más comunes de
leyes de velocidad
Ejemplo A Productos
AA
A Ckdt
dCr
Separando variables e integrando
)25.1(ln0
atkC
C
A
A
o bien )25.1()(exp0
btkC
C
A
A
Usando CA = CA0 ( 1 – XA ), se obtiene
)26.1()1(ln atkX A
o bien )26.1()(exp)1( btkX A
Las gráficas características de cinéticas de primer orden son:
CA -ln(CA/CA0)
o pendiente = K
-ln(1 – XA)
t t
Fig. 1-12 Representaciones características de la reacción de primer orden

H. ZULETA
46
el tiempo de vida medio es t1/2 = ln 2 / K = 0,693 / k (1.27)
Se puede apreciar que el tiempo de vida medio es independiente de la
concentración inicial. Esto significa que el tiempo de vida medio es el mismo
a cualquier tiempo durante la reacción. Para reacciones de orden distinto de
uno, el tiempo de vida medio no es constante, y cambia según la extensión en
la cual ocurre. Por esta razón, el tiempo de vida medio, generalmente se usa
para describir solamente reacciones de primer orden.
Los decaimientos nucleares corresponden a reacciones de primer orden. Una
muestra de cobalto-60 que se utiliza para eliminar células cancerosas de un
tumor tiene un tiempo de vida medio de 5,7 años.
Ejemplo de velocidad de Decaimiento
El mercurio-206 es un isótopo radiactivo, con vida media de 7,7 minutos.
¿Qué fracción de los núcleos de mercurio-206 ha decaído después de 30
minutos?
Solución: Cada 7,7 minutos decae la mitad del mercurio-206, por lo tanto se
puede construir una tabla que indique la cantidad que queda después de cada
7,7 minutos.
Tiempo t/min 0 7,7 15,4 23,1 30,8
Fracción remanente 1 0,5 0,25 0,125 0,0625
Estos datos nos permiten estimar que a los 30 minutos quedará un poco más
del 6,25%. Por lo tanto ha decaído cerca del 94% del mercurio-206 inicial (el
resultado exacto es 93,3%)
t1/2 = 0,693/ K K = 0,693 / 7,7 = 0,09 min-1
Por lo tanto -ln(1 – XA) = K t
XA = 1 – exp ( -K t ) = 1 – exp ( -0,09*30) = 0,933
La desintegración de un isótopo radiactivo puede servir también para medir la
antigüedad de muchos objetos. La “edad” del objeto por determinar, decide el

H. ZULETA
47
isótopo que se debe usar. En particular es común el uso de carbono 14, que
sufre una desintegración .
eNC 0
1
14
7
14
6
Con un tiempo de vida medio de 5770 años. La radiación cósmica en la
atmósfera superior sintetiza 14
C que equilibra la pérdida por desintegración
radiactiva. La materia viva mantiene un nivel de 14
C que produce 15,3
desintegraciones por minuto por cada gramo de carbono. Los organismos
muertos no intercambian carbono por CO2 en la atmósfera, así que la cantidad
de 14
C en la materia sin vida disminuye con el tiempo debido a la
desintegración.
Tiempo de caducidad de un fármaco
El principio activo de los medicamentos se descompone siguiendo cinéticas de
primer orden. Con este mismo procedimiento se estima el tiempo de
caducidad de los alimentos, como lácteos, galletas, cereales, etc.
Persistencia ambiental relativa de residuos peligrosos
En las concentraciones relativamente bajas encontradas en los RSU (residuos
sólidos urbanos) la descomposición de un constituyente individual de residuo
peligroso, se puede describir adecuadamente como una función de primer
orden.
1.12.3 Reacciones irreversible de segundo orden
Las leyes de velocidad de segundo orden típicamente pueden considerar:
(a.1) Un solo Reactante A Productos
(a.2) Reactantes idénticos A + A Productos
( 2 A Productos )
(b) Reactantes diferentes AA + BB Productos

H. ZULETA
48
Para los casos (a.1) y (a.2)
)28.1(2
AA
A Ckdt
dCr
Ecuaciones integradas
)29.1()1(
11
00
tkXC
X
CC AA
A
AA
1/ CA XA/(1-XA)
“m”= K “m”= CA0 K
1/CA0
t t
Fig. 1-13 Representaciones características de la reacción de segundo orden
Para el caso (b)
Reactantes diferentes: alimentación no estequiometrica
AA + BB Productos
En este caso, la ley de velocidad es de primer orden con respecto de A, de
primer orden con respecto de B y de orden global igual a dos. Por lo tanto la
ecuación de velocidad es:
BAA CCk
dt
dC o bien )30.1(tk
CC
dC
BA
A
Considerando el grado de avance o extensión de la reacción, según la
estequiometria.

H. ZULETA
49
)31.1(00
B
BB
A
AA CCCC
Esta última ecuación permite eliminar CA y CB en la ecuación (1.30),
obteniéndose
dtk
CC
d
ABBAA
)()( 00
La integración por el método de fracciones parciales, considerando
evaluación de la constante de integración y además de convertir en CA y CB,
resulta en
)32.1()(lnln 00
0
0 tCCk
C
C
C
CBAAB
AB
A
B
A
Se puede apreciar que ln(CA/CB) es una función lineal de t, pudiendo
determinarse la constante K a partir de la pendiente de la recta. Todas las otras
cantidades son conocidas.
Ecuaciones equivalentes a la ecuación (1.32) se pueden obtener usando las
siguientes relaciones:
Razón molar inicial = M = CB0 / CA0
Concentración de B en un tiempo t = CB = CB0 ( 1 – XB )
Concentración de A en un tiempo t = CA = CA0 ( 1 – XA )
Cantidades en reacción A
B
AA
BB
XC
XC
0
0
A
B
AB
AB
A
AAB
A
B
CM
C
CC
CC
XM
XM
X
Xlnln
)1(
)/(ln
1
1ln
0
0
(1.33)
tMCk
tCCk
BA
A
A
ABBA
A
)()( 0
00

H. ZULETA
50
Se puede observar que si la alimentación esta de acuerdo con la estequiometria
(alimentación estequiometrica), las ecuaciones anteriores no pueden aplicarse,
es decir,
M B / A
Las figuras (1-14 a) y (1-14b) son características de este tipo de cinéticas.
ln (CB/CA)
“m” = (K/A) (CB0 A – CA0 B)
ln (CB0 / CA0) = ln M
t (a)
ln (CBCA0/CACB0)
“m” = (K/A) (CB0A – CA0B)
t
(b)
Fig. 1-14 Representación de cinéticas de segundo orden, reactantes diferentes

H. ZULETA
51
Reactantes diferentes: alimentación estequiometrica
AA + BB Productos
CA0 / CB0 = A / B
La expresión de velocidad es:
BAA
A CCkdt
dCr
Puede integrarse la ecuación si se expresa CB en función de CA. Para ello se
recurre a la estequiometria, en donde también se cumple:
CA / CB = A / B
Por lo tanto 2
A
A
BA
A
BA
AA CkCCk
dt
dCr
Separando variables e integrando
)34.1(11
0
tkCC A
B
AA
Debe observarse que si los coeficientes estequiometricos son iguales, la
ecuación integrada corresponde exactamente a reacciones de segundo
orden del tipo A Productos.
Observación 1: La forma de la expresión cinética integrada depende tanto
de la estequiometria como de la cinética

H. ZULETA
52
Observación 2: Para el caso de reacciones con concentraciones iniciales
desiguales, cuya expresión integrada es:
tCCk
CC
CCABBA
AAB
AB )(ln 00
0
0
Si CB0 >>>> (B / A) CA0 CB permanece aproximadamente constante
tCCk
X
XbienotktkC
C
CABBA
AA
BB
A
A )()1(
)1(ln'ln 000
0
tkXXSi AB ')1(ln0
La reacción de Segundo orden se convierte en una reacción de pseudo
primer orden.
1.12.4 Ecuaciones empíricas de orden n
Cuando se tiene un solo reactante y no se conoce el mecanismo de reacción,
los datos cinéticos obtenidos en un reactor isotérmico de volumen constante,
se pueden ajustar con la ley cinética de orden n.
)35.1(n
AA
A Ckdt
dCr
La integración de esta ecuación entre los límites de CA0 a t = 0, y CA a t
permite obtener:
)36.1()1(1
0
1 tknCC n
A
n
A
Esta ecuación aparece representada en la figura (1.15)

H. ZULETA
53
CA1 – n
Pendiente = ( n – 1 ) K
CA01 - n
t
Fig.1-15 Representación de cinéticas de orden n
Inicialmente n es desconocido, por lo tanto, se debe suponer un valor y
comprobar el ajuste (se debe verificar con el calculo del intercepto y verificar
con los datos del gráfico).
También se puede proceder sin el ajuste gráfico. Se supone un valor de n, y se
calcula la constante cinética K para cada uno de los puntos. El valor de n que
implique la menor variación de K, será el valor correcto.
Observaciones:
Reacciones con n > 1, no se completan en un tiempo finito
Reacciones con n < 1, se completan en un tiempo finito ( CA = 0 )
En este caso al reemplazar CA = 0 en la ecuación (1.36) se obtiene
El tiempo de término )37.1()1(
1
0
)0(kn
Ct
n
A
CA
La ecuación (1.36) no debe usarse para tiempos mayores al tiempo de termino,
debido a que se obtendrían valores de CA negativos.

H. ZULETA
54
Ejemplo:
Se ha encontrado experimentalmente que en 10 minutos se convierte en
producto el 75% de un líquido reactante con un orden de reacción igual a ½.
Calcular la cantidad convertida en media hora.
Solución:
Para una reacción de orden n n
AA Ckr n 1
Si se considera que )1(0 AAA XCC y se reemplaza en la ecuación integrada
1)1()1( 11
0 n
A
n
A XtkCn
Entonces, reemplazando para los tiempos considerados
)38.1(1)1(
1)1(
)1(
)1(1
1,
1
2,
1
2
1
1
0
2
1
0
n
A
n
A
n
A
n
A
X
X
t
t
tkCn
tkCn
si t1 = 10 min, t2 = 30 min y XA,1 = 0,75, se obtiene
1)75,01(
1)1(3
10
302/1
2/1
2,
AX
Resolviendo
( 1 – XA,2 )1/2
= - 0,5 ¡imposible!
El tiempo de término no se puede conocer de su ecuación de definición, pues
no se conocen K y CA0. Sin embargo se puede usar la misma ecuación (1.38)
para calcular un tiempo equivalente, es decir el tiempo de conversión
completa o t(X =1)
2/1
1,
1)1()1(1
1
A
XX
ttA
Reemplazando los valores conocidos
.min20)75,01(1
110
2/1)1(
AXt

H. ZULETA
55
Por lo tanto, a los 20 minutos la reacción ya ha terminado, en consecuencia al
cabo de media hora se ha convertido toda la cantidad inicial ( XA = 1 ).
Orden global de reacciones irreversibles usando el tiempo de vida medio
Algunas reacciones pueden tener la forma
oductosCBA Pr....
La ley de velocidad se puede plantear
)39.1(....c
C
b
B
a
AA
A CCCkdt
dCr
Si los reactantes se encuentran presentes en proporción estequiometrica, la
razón permanece constante durante toda la reacción. En consecuencia, para los
reactantes A, B y C en cualquier tiempo se cumple que
entoncesC
C
C
C
A
C
A
B ,,
)40.1(........ .....cba
A
cbc
A
b
A
a
AA
A CkCCCkdt
dCr
La ecuación anterior se puede simplificar, definiendo ...
cb
kK
y
considerando que el orden global es ...cban
)41.1(n
A
A CKdt
dC
Integrando para n 1 se obtiene
)42.1()1(1
0
1 tnKCC n
A
n
A
Aplicando la definición de tiempo de vida media t1/2 ( CA = CA0 / 2 )

H. ZULETA
56
)43.1()1()1(
12 1
0
1
2/1
nCnK
t n
A
n
Recordando que para n = 1
K
t2ln
2/1
Se puede concluir de las ecuaciones anteriores
t1/2 CA0n – 1
= constante (para cualquier valor de n)
Lo anterior permite probar o establecer el valor de n, mediante tanteo, a partir
de una serie de experimentos efectuados para medir t1/2 a diferentes valores de
CA0. El valor de K se puede calcular con los valores de n obtenidos. Otra
alternativa consiste en aplicar logaritmos a la ecuación (1.43) y efectuar la
representación gráfica de ln t ½ en función de ln CA0.
ln t1/2
n < 1
pendiente = 1 - n
n > 1
n = 1
ln CA0
Fig. 1- 16 Representación de experimentos de periodo medio y orden global
)44.1(ln)1()1(
12ln 0
1
2/1 A
n
CnnK
t
Si se efectúan experimentos, cada uno de concentración iniciales diferente.
Para n > 1, la conversión fraccional en un tiempo dado se eleva al aumentar la
Concentración inicial.

H. ZULETA
57
Para n < 1, la conversión disminuye al aumentar la concentración inicial
Para n = 1, la conversión es independiente de la concentración inicial.
Una variación al procedimiento, se puede presentar si todos los componentes
están en exceso, excepto uno, por ejemplo el componente A. Será posible
calcular el orden con respecto de A.
)('' 0
b
B
a
AA CkKCK
dt
dC CB CB0
1.12.5 Método de vida fraccional tf
El método de tiempo de vida medio se puede ampliar a cualquier fracción de
vida tal que la concentración de reactante disminuye una fracción F = CA/CA0
en un tiempo tf. La relación en este caso será:
)45.1()1(
1 1
0
1n
A
n
f Cnk
Ft
A través de un gráfico de ln tf y ln CA0 es posible determinar el orden de
reacción.

H. ZULETA
58
1.13 REACCIONES EN SERIE O CONSECUTIVAS
Un sistema de reacciones químicas de mucha importancia industrial, lo
constituyen las reacciones en serie o consecutivas. En la práctica las
reacciones no ocurren según una sola ecuación estequiometrica, sino a través
de combinaciones de ellas. En las reacciones en serie irreversibles las
reacciones son etapas secuenciales caracterizadas cada una por sus respectivas
constantes cinéticas.
Un buen ejemplo lo constituyen todas las reacciones de oxidación con
moléculas orgánicas.
Hidrocarburos Producto deseado Producto final no deseado
( propileno ) (acroleína) ( CO2 , CO )
Considerando un esquema sencillo de reacción consistente en dos etapas de
primer orden.
K1 K2
A R S ( 1.46 )
Las ecuaciones de velocidad para cada una de las especies son:
)47.1(1 AA
A Ckdt
dCr
)48.1(21 RAR
R CkCkdt
dCr
)49.1(2 R
S
S Ckdt
dCr
Para obtener la distribución de cada componente con el tiempo, es necesario
realizar la integración de las ecuaciones anteriores. Comenzando con la
expresión para A, se obtiene el siguiente resultado:
)50.1(exp 1
0
tk
AA CC

H. ZULETA
59
Si esta ecuación se reemplaza en la ecuación (1.48)
R
tk
A
R CkCkdt
dC201
1exp
Ordenando
)51.1(exp 1
012
tk
ARR CkCk
dt
dC
Esta última expresión es una ecuación diferencial de primer orden en R. Su
resolución puede lograrse fácilmente usando cualquier texto básico de Calculo
diferencial o bien Manuales de formulas de integración. En cualquier caso
debe considerarse:
21 kk ; CR(t=0) = CR0 = 0
La solución es:
)52.1(expexp
2112
0
21
kkkkCC
tktk
AR
Considerando el Balance de materiales para el sistema a volumen constante
CA0 = CA + CR + CS al despejar CR
CR = CA0 - CA - CS (1.53)
Reemplazando las expresiones de CA y CR en la ecuación (1.53) y despejar CS
)54.1(expexp1 21
12
1
21
20
tktk
ASkk
k
kk
kCC

H. ZULETA
60
Observaciones :
Si K2 >>> K1, la ecuación (1.54) se convierte en CS = CA0 ( 1 – exp (-K1t) ),
es decir la velocidad está controlada por K1 o la primera de las reacciones.
Si K1 >>> K2, la ecuación ( 1.54 ) se convierte en CS = CA0 ( 1 – exp (-K2t) ),
es decir la velocidad está controlada por K2 o la segunda de las dos reacciones.
Se puede concluir que para cualquier número de reacciones en serie, la etapa
más lenta es la que tiene mayor influencia en la velocidad global de reacción.
Se puede representar el cambio de CA, CR y CS con el tiempo, según las
ecuaciones (1.50), (1.52), y (1.54). El resultado obtenido se puede observar en
la Fig. 1-17. La concentración de A disminuye en forma exponencial con el
tiempo. La concentración de R comienza en cero, pasa por un máximo y
nuevamente vuelve a cero, ya que al final de la reacción sólo queda producto
S. Por último la velocidad de formación de S depende en todo momento de R.
Al principio es cero y alcanza su máximo valor (punto de inflexión de la
curva) cuando la concentración de R es máxima. Al principio la formación de
S es lenta. Se puede decir que existe inicialmente un periodo de inducción. La
duración de este periodo de inducción se suele tomar como el tiempo
necesario para alcanzar el punto de inflexión de la curva concentración-
tiempo, que es igual al tiempo necesario para que la concentración de R sea
máxima.
Las concentraciones máximas y sus respectivos tiempos, el tiempo de
inducción, es decir la forma y tendencia de las curvas, dependen de la razón de
K2 / K1.

H. ZULETA
61
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 0,5 1 1,5 2 2,5 3 3,5 4
tiempo / min.
C /
Ca
o
Reactante A
Producto R
Producto S
Fig. 1-17 Distribución de Productos A, R y S. K1=2, K2= 1 ( min
-1 )
Localización del máximo de R
Diferenciando la ecuación para R e igualando a cero ( dCR / dt = 0 ), se
determina el tiempo en donde se obtiene el máximo valor de concentración de
R
)55.1(
ln
12
1
2
kk
k
k
tmáx
El correspondiente valor de concentración será
)56.1(12
2
2
10,
kk
k
AmáxRk
kCC

H. ZULETA
62
Estos dos parámetros son frecuentemente importantes para la ejecución de
procesos técnicos, si se pretende:
- La operación en el máximo
- La obtención de resultados conocidos.
1.14 REACCIONES IRREVERSIBLES EN PARALELO
En este tipo de reacciones una sustancia puede reaccionar por varios caminos
en forma simultánea. La descomposición del etanol es un buen ejemplo de
este sistema de reacciones.
K1 C2H4 + H2O
C2H5OH
K2 CH3CHO + H2
Considerando el caso más simple de reacciones elementales en paralelo
B
K1
A
K2
C
Las ecuaciones de velocidad para cada uno de los compuestos son:
)57.1()( 21 AA
A Ckkdt
dCr
)58.1(1 AB
B Ckdt
dCr
)59.1(2 A
C
C Ckdt
dCr
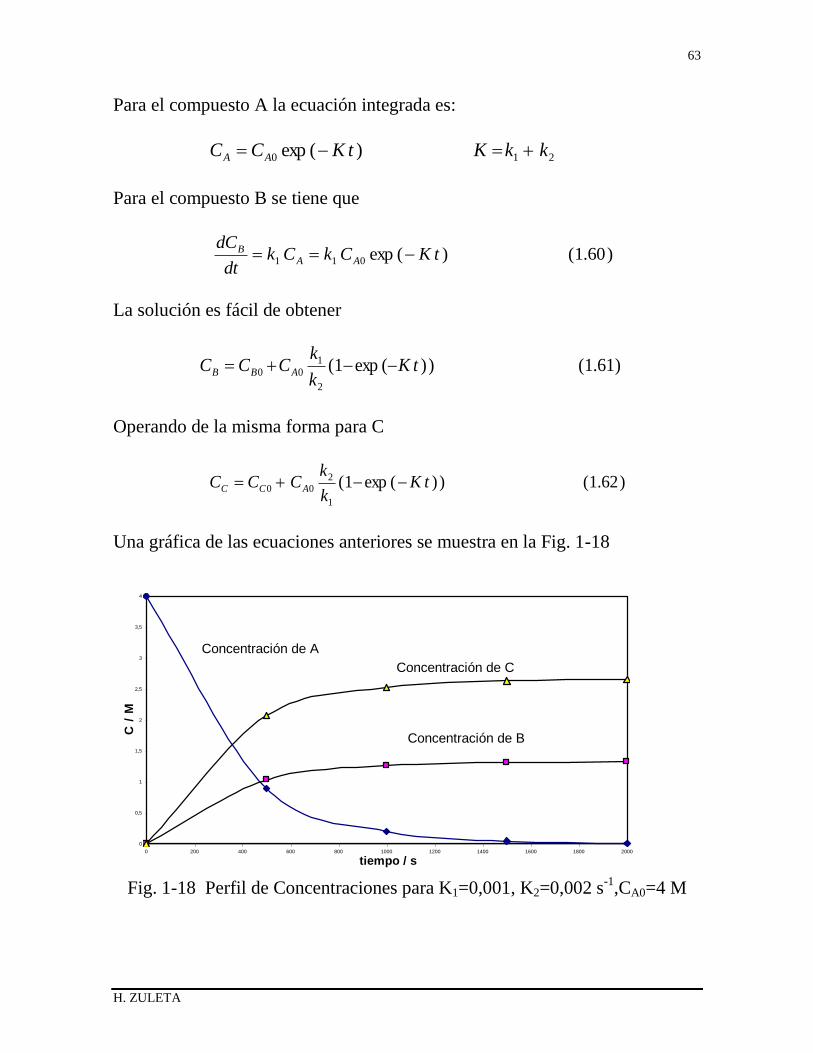
H. ZULETA
63
Para el compuesto A la ecuación integrada es:
210 )(exp kkKtKCC AA
Para el compuesto B se tiene que
)60.1()(exp011 tKCkCkdt
dCAA
B
La solución es fácil de obtener
)61.1())(exp1(2
100 tK
k
kCCC ABB
Operando de la misma forma para C
)62.1())(exp1(1
200 tK
k
kCCC ACC
Una gráfica de las ecuaciones anteriores se muestra en la Fig. 1-18
0
0,5
1
1,5
2
2,5
3
3,5
4
0 200 400 600 800 1000 1200 1400 1600 1800 2000
tiempo / s
C /
M
Concentración de A
Concentración de B
Concentración de C
Fig. 1-18 Perfil de Concentraciones para K1=0,001, K2=0,002 s
-1,CA0=4 M

H. ZULETA
64
Al dividir la diferencia de concentraciones de las ecuaciones 1.61 y 1.62 se
obtiene:
)63.1(2
1
0
0 adSelectividSk
k
CC
CC
CC
BB
En este caso, la selectividad depende solo de la razón de constantes. La mayor
proporción que exista de B o de C depende de la diferencia entre las
constantes.
1.15 REACCIONES REVERSIBLES O DE EQUILIBRIO
Cualquier reacción es básicamente reversible (una reacción nunca transcurre
hasta conversión completa). En la práctica, será reversible solo si las
reacciones directa e inversa pueden ser medidas o detectadas.
Considerando una reacción reversible monomolecular elemental
equilibriodeteconsKKRA C
KC tan,
)(tan
)(tan
2
1
2
1
ARinversateconsk
RAdirectateconsk
k
k
Iniciando la reacción con una razón M = CR0 / CA0, la ecuación cinética será
)()(
)64.1(
002001
210
AAAAAA
RAAAAR
XCMCkXCCk
CkCkdt
dXC
dt
dC
dt
dC
Se sabe que en el equilibrio dCA/dt = 0
)65.1(1 2
1
00
00
k
k
X
XM
XCC
XCMC
C
CK
eqA
eqA
eqAAA
eqAAA
eqA
eqR

H. ZULETA
65
Combinando las dos ecuaciones anteriores, se obtiene la ecuación cinética en
función de la conversión de equilibrio
)66.1()()1(1
AeqA
eqA
A XXXM
Mk
dt
dX
A partir de la integración de la ecuación 1.66 se obtiene:
)67.1(1
ln1ln 1
0
tkXM
M
CC
CC
X
X
AeqeqAA
eqAA
eqA
A
El cambio de conversiones de equilibrio a concentraciones de equilibrio se
realiza usando
0
1A
eqA
eqAC
CX
Los datos cinéticos de reacciones reversibles del tipo anteriormente planteado
deben ajustar la ecuación 1.67, según la Figura 1-19
- ln (1 – XA / XA eq)
o
- ln ((CA-CA eq)/(CA0-CA eq))
Pendiente = K1(M + 1)/(M + XA eq)
t
Fig. 1-19 Ajuste para reacciones reversibles de tipo monomolecular
Las reacciones reversibles se pueden considerar como irreversibles si se mide
la concentración en (CA – CA eq), es decir concentración en exceso sobre la de
equilibrio. La conversión se mide como una fracción del valor máximo

H. ZULETA
66
alcanzable. En consecuencia la reacción irreversible es un caso especial de la
reversible en la que CA eq = 0 o XA eq = 1 o bien K =.
1.16 REACCIONES HOMOGENEAS CATALIZADAS
Si se considera que la velocidad de reacción para un sistema homogéneo
catalizado es igual a la suma de las reacciones no catalizada y catalizada.
K1
A R (Rx 1) no catalizada
K2
A + C R + C (Rx 2) catalizada
Las correspondientes ecuaciones de velocidad son
CAA
AA CCk
dt
dCCk
dt
dC2
2
1
1
es decir, la velocidad de reacción se expresa con y sin catalizador
ACCAAA CCkkCCkCk
dt
dC)( 2121
Integrando y si la concentración del catalizador permanece constante
tKtCkkXC
CobservadoCA
A
A
)()1(lnln 21
0
Las constantes K1 y K2 se calculan realizando una serie de experiencias con
diferentes concentraciones de catalizador y representando el valor de Kobservado
frente a CC ( Fig. 1-20 )

H. ZULETA
67
Kobservado
pendiente = K2
K1
CC
Fig. 1-20 Ajuste para la determinación de coeficientes cinéticos para reacción
homogénea catalizada
1.17 REACCIONES AUTOCATALITICAS
Se conocen como reacciones autocataliticas aquellas en la que uno de los
productos actúa como catalizador. La reacción autocatalitica más sencilla es
A + R R + R
Para esta reacción la ecuación cinética es
RAA
A CCkdt
dCr
A partir del balance de materiales
C0 = CA + CR = CA0 + CR0 = constante ( 1.68 )
Reemplazando el valor de CR de la ecuación anterior en la ecuación cinética,
se obtiene
)69.1()( 0 AAA
A CCCkdt
dCr

H. ZULETA
68
Arreglando y expresando en fracciones parciales
dtkCC
dC
C
dC
CCCC
dC
A
A
A
A
AA
A
000
1
)(
La integración permite obtener la siguiente expresión:
)70.1()(/
/ln
)(
)(ln 000
0
0
00
00 tkCCtkCCC
CC
CCC
CCCRA
AA
RR
AA
AA
En función de la relación inicial de reactante, es decir M = CR0 / CA0, y de la
conversión, la expresión anterior se puede escribir:
)71.1()()1()1(
ln 000 tkCCtkMCXM
XMRAA
A
A
Una de las características de las reacciones autocatalíticas es que la velocidad
se incrementa inicialmente debido a que la concentración del producto de la
catálisis aumenta, pero eventualmente una vez que alcanza un máximo,
disminuye debido al agotamiento de reactante. El comportamiento inicial
puede ser definido como cinética “anormal”, y tiene consecuencias
importantes en la selección del tipo de reactor.
Obteniendo la derivada de la expresión de velocidad, es decir 0)(
A
A
dC
rd
junto con la ecuación (1.68), se obtiene el máximo de velocidad y la
concentración óptima para el máximo ( Fig 1-21 )
-rA cinética anormal
CA,op CA0 CA
Fig. 1-21 Comportamiento óptimo de velocidad de reacciones autocataliticas

H. ZULETA
69
1.18 METODO DIFERENCIAL DE TRATAMIENTO DE DATOS
Al igual que en el método integral, se realiza experimentación en reactores de
volumen y temperatura constantes, para la obtención de datos cinéticos. Se
considera que la ley de velocidad es de la forma
CBAA CCCkr
Donde A, B y C reactantes. Por simplicidad, se puede considerar un solo
reactante, y ahora la ley se expresa
n
AA CKr
Al aplicar logaritmos a la ecuación anterior
)72.1(lnln)(ln AA CnKr
Ya se ha planteado que para sistemas con densidad constante
dt
dCr A
A
El método diferencial consiste en que a partir de una diferenciación gráfica o
numérica, se pueden obtener valores de (-rA) en función de CA, lo cual permite
determinar n y K a través de la regresión lineal de ln(-rA) y ln CA
ln (-rA)
Pendiente = n
ln CA
También se puede usar papel log-log y representar directamente –rA y CA, la
pendiente de la recta será el orden de reacción.
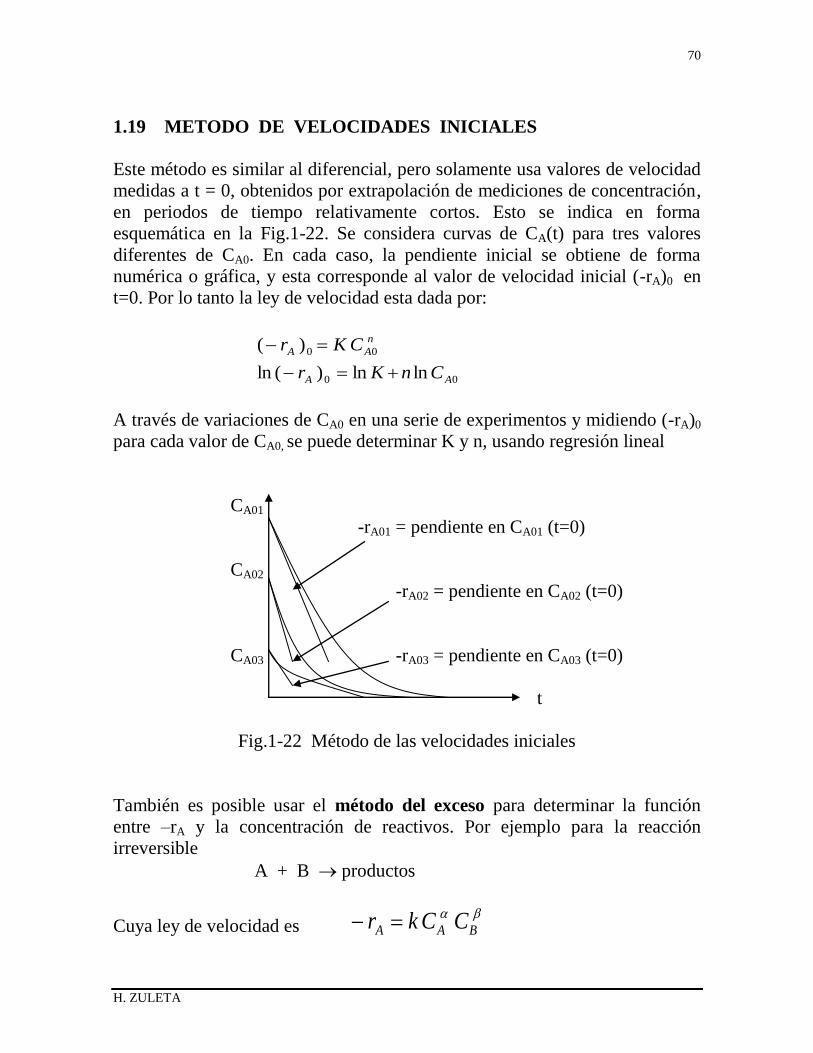
H. ZULETA
70
1.19 METODO DE VELOCIDADES INICIALES
Este método es similar al diferencial, pero solamente usa valores de velocidad
medidas a t = 0, obtenidos por extrapolación de mediciones de concentración,
en periodos de tiempo relativamente cortos. Esto se indica en forma
esquemática en la Fig.1-22. Se considera curvas de CA(t) para tres valores
diferentes de CA0. En cada caso, la pendiente inicial se obtiene de forma
numérica o gráfica, y esta corresponde al valor de velocidad inicial (-rA)0 en
t=0. Por lo tanto la ley de velocidad esta dada por:
00
00
lnln)(ln
)(
AA
n
AA
CnKr
CKr
A través de variaciones de CA0 en una serie de experimentos y midiendo (-rA)0
para cada valor de CA0, se puede determinar K y n, usando regresión lineal
CA01
-rA01 = pendiente en CA01 (t=0)
CA02
-rA02 = pendiente en CA02 (t=0)
CA03 -rA03 = pendiente en CA03 (t=0)
t
Fig.1-22 Método de las velocidades iniciales
También es posible usar el método del exceso para determinar la función
entre –rA y la concentración de reactivos. Por ejemplo para la reacción
irreversible
A + B productos
Cuya ley de velocidad es
BAA CCkr

H. ZULETA
71
La reacción se puede efectuar primero con gran exceso de B, para que CB
prácticamente no cambie durante el curso de la reacción, es decir
AA Ckr '
En donde
0' BB CkCkk
Se debe disponer de datos de –rA y CA o bien otra información indirecta que
permita determinar
Una vez calculado la reacción se efectúa con A en exceso, y en ese caso la
ley de velocidad se aproxima con
BA Ckr ''
Procediendo de la misma forma anterior se puede calcular . La constante k se
puede calcular a partir de –rA a concentraciones conocidas de A y B
)(
BA
A
CC
rk
1.20 REACTOR DISCONTINUO DE VOLUMEN VARIABLE
Hasta el momento se ha considerado sistemas isotérmicos de volumen
constante, es decir, a medida que la reacción progresa el volumen de reacción
no cambia. La mayoría de los recipientes por lote, en fase líquida y algunos en
fase gaseosa, pertenecen a esta categoría. En las reacciones en fase gaseosa
con cambio en el número de moles, existe cambio en el volumen (sólo
números iguales de moles ocupan volúmenes iguales en fase gaseosa a igual
temperatura y presión). Otro caso de volumen variable es el de reactores por
lote en donde cambia el volumen con el tiempo (Cámaras de combustión de
motores).
Un ejemplo de reactor de volumen variable lo constituye un microreactor, el
cual esta formado por un pequeño tubo capilar provisto de una esfera que se
desplaza a medida que la reacción progresa (Fig. 1-23).

H. ZULETA
72
Reactor de volumen variable
Fig. 1-23 Reactor batch de volumen variable
Definiendo V0 = volumen inicial del reactor
V = volumen en un instante t
Considerando que este tipo de reactor se puede usar en reacciones de
estequiometria simple a temperatura y presión constante. El volumen se puede
relacionar en forma lineal con la conversión, es decir
)73.1(
)73.1()1(
0
0
0
bV
VVXbieno
aXVV
A
A
AA
De esta última ecuación
)74.1(0 A
AV
dVdX
En donde A es el cambio fraccional de volumen del sistema o coeficiente de
variación relativa del sistema entre la no conversión y la conversión completa
de reactivo, es decir
)75.1(0
01
A
AA
X
XX
AV
VV
También A puede calcularse usando la siguiente relación
A = yA0 ( 1.76 )

H. ZULETA
73
con = n / a
n = diferencia de coeficientes estequiometricos de productos y reactivos
a = coeficiente estequiometrico de reactante A
yA0 = fracción molar inicial de A
Ejemplo: Considerar la reacción en fase gaseosa isotérmica
A ½ C
Determinar A:
a) considerando que inicialmente se tiene A puro.
b) Si la alimentación tiene un 30% de un componente inerte
Solución:
a) Según la estequiometria inicialmente se tienen V0 volúmenes de A. Esto
equivale a decir que cuando aún no ha comenzado la reacción (XA= 0), el
volumen del sistema es V0.
Cuando la reacción ha terminado (XA=1) el volumen total será ½ V0. Por lo
tanto
2/12/1
0
00
V
VVA
Usando la otra formula alternativa n = ½ - 1 = -1/2, a = 1 y como se trata de
un componente puro, yA0 = 1
A = -1/2 * 1 = -1/2
El signo negativo indica una compresión (disminución en el número de
moles), se puede plantear entonces que si A es positivo, se trata de una
expansión o aumento en el volumen o número de moles.
b) Al considerar para el mismo ejemplo que la alimentación tiene un 30% de
un componente inerte, el volumen total inicial será V0, pero ahora
V0 = volumen inicial de A + volumen inicial de inerte

H. ZULETA
74
V0 = 0,7 V0 + 0,3 V0 ¡ Es el volumen de no conversión!
Una vez producida la conversión completa el volumen del sistema será
V (XA=1) = volumen de C producido + volumen de inerte
=. ½ (0,7 V0) + 0,3 V0 = 0,65 V0
por lo tanto: A = (0,65 V0 – V0) / V0 = - 0,35
Se concluye claramente que en la determinación de A, se debe tomar en
cuenta la estequiometria y la presencia de inertes.
Por otra parte si: )77.1()1(0 AAAA
A XNNyV
NC
Considerando el volumen variable
AA
AA
AA
AA
AX
XC
XV
XNC
1
1
)1(
)1(0
0
0
Por lo tanto )78.1(1
/1
1
1 0
0 AA
AA
A
AA
A
A
A
X
CCXbieno
X
X
C
C
Estas expresiones caracterizan la relación entre la concentración y conversión
para sistemas de volumen(o densidad) variable.
La velocidad de reacción (desaparición del componente A), es, en general
dt
dN
Vr A
A
1
Reemplazando en esta ecuación la definición de volumen variable V (ecuación
1.73a) y la derivada de la ecuación 1.77, se obtiene

H. ZULETA
75
)79.1(1
0
dt
dX
X
Cr A
AA
A
A
o en términos del volumen según la ecuaciones 1.74 y 1.73a
)80.1()ln(00
dt
VdC
dtV
dVCr
A
A
A
A
A
1.20.1 Método integral de análisis
Desgraciadamente, sólo algunas de las formas integradas más simples pueden
generar expresiones de V en función de t. Las principales son:
Reacciones de orden cero. Para reacciones homogéneas de orden cero, la
velocidad de cambio de cualquier reactante A es independiente de la
concentración de las sustancias, es decir:
kdt
VdCr
A
A
A )ln(0
Integrando se obtiene
)81.1(ln0
0 tkV
VC
A
A
Como se observa en la Fig. 1-24, el logaritmo del cambio fraccional de
Volumen frente el tiempo es una línea recta de pendiente KA / CA0

H. ZULETA
76
pendiente = K A / CA0
ln (V/V0)
A > 0
t
pendiente = K A / CA0
A < 0
Fig. 1-24 Ajuste de una reacción de orden cero a presión constante y volumen
variable
Reacciones de primer orden. Para reacciones unimoleculares de primer
orden la ecuación cinética para el reactante A, de acuerdo a la ecuación
1.79es:
AA
AAA
A
AA
AA
X
XCkCk
dt
dX
X
Cr
1
1
10
0
Separando variables e integrando se obtiene
0
0
)82.1(1ln)1(ln
VVVcon
tkV
VX
A
A
Se puede concluir, de acuerdo a la última ecuación, que la conversión
fraccional es la misma independientemente de si el volumen es constante o
variable.
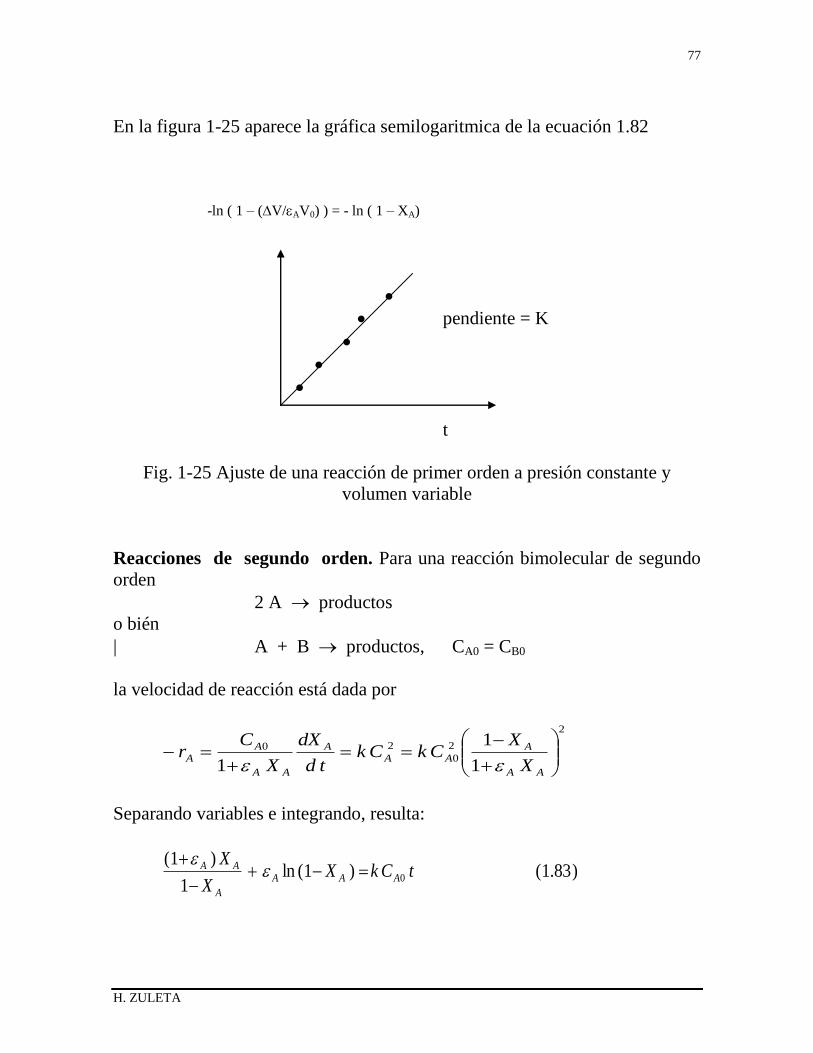
H. ZULETA
77
En la figura 1-25 aparece la gráfica semilogaritmica de la ecuación 1.82
-ln ( 1 – (V/AV0) ) = - ln ( 1 – XA)
pendiente = K
t
Fig. 1-25 Ajuste de una reacción de primer orden a presión constante y
volumen variable
Reacciones de segundo orden. Para una reacción bimolecular de segundo
orden
2 A productos
o bién
| A + B productos, CA0 = CB0
la velocidad de reacción está dada por
2
2
0
20
1
1
1
AA
AAA
A
AA
A
AX
XCkCk
td
dX
X
Cr
Separando variables e integrando, resulta:
)83.1()1(ln1
)1(0 tCkX
X
XAAA
A
AA
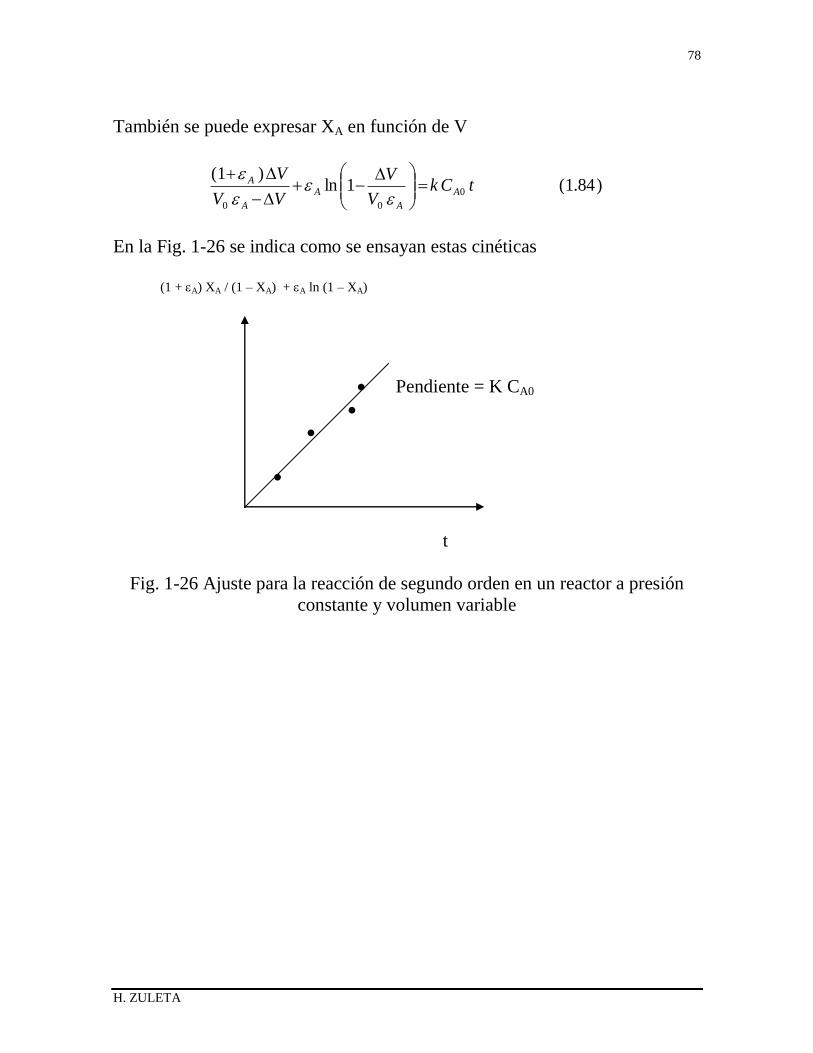
H. ZULETA
78
También se puede expresar XA en función de V
)84.1(1ln)1(
0
00
tCkV
V
VV
VA
A
A
A
A
En la Fig. 1-26 se indica como se ensayan estas cinéticas
(1 + A) XA / (1 – XA) + A ln (1 – XA)
Pendiente = K CA0
t
Fig. 1-26 Ajuste para la reacción de segundo orden en un reactor a presión
constante y volumen variable

H. ZULETA
79
CAPITULO II: INTRODUCCION AL DISEÑO DE REACTORES
IDEALES
2.1 INTRODUCCION
El diseño de reactores considera muchas facetas y disciplinas. En estos
apuntes el análisis esta centrado en el diseño de procesos en vez del diseño
mecánico de equipos, instrumentación y control. El diseño de reactores es un
término aplicado a una nueva instalación o para alguna modificación con
propósitos de optimización, en este caso se habla del análisis de operación de
un reactor existente.
Parámetros que afectan el funcionamiento de un reactor
El término “comportamiento de reactores” generalmente se refiere a la
condición de operación alcanzada por un reactor, particularmente con respecto
la cantidad de reactante convertida o distribución de productos en un
determinado tamaño de equipo y configuración; alternativamente, se puede
referir al tamaño y configuración para una determinada conversión o
distribución. En cualquiera de los casos depende de dos tipos de
comportamientos principales: (1) las velocidades de los procesos
involucrados, incluyendo reacción y procesos de transferencia de calor y
masa, algunas veces influenciado por limitaciones de equilibrio; y (2) el
movimiento relativo de elementos de fluido (situaciones de una fase o
multiples fases ) y particulas sólidas en sistemas con o sin flujo.
Ecuaciones de balance
Una de las herramientas más usadas para el diseño y analisis de
comportamiento son las ecuaciones de balance. En este tipo de relaciones se
expresan cantidades conservativas, tales como la masa y la energía en un
sistema especifico.
Los balances se realizan con respecto a un “volumen de control” que puede
ser finito (V) o diferencial (dV), como se indican en la Fig. 2-1. El volumen de
control está limitado por una “superficie de control”. En la Fig. 2-1, m, F y q
son los flujos másicos (kg), molares (moles) y volumetricos (m3)
respectivamente, las cuales atraviesan las partes especificadas del volumen de
control y Q es la velocidad de transferencia de calor hacia o desde el volumen

H. ZULETA
80
de control. En (a), el volumen de control puede ser el contenido de un
estanque, y en (b), puede ser una lámina fina de un tubo cilindrico.
Q Q
ment msal ment msal
Fent Fsal Fent Fsal
qent qsal qent dV qsal
(a) (b)
Fig. 2-1 (a) Volumen de control con tamaño finito (V) y (b) Volumen de
control con tamaño diferencial, con corrientes de entrada y salida de
materiales y transferencia de calor ( Q y Q ).
La ecuación de balance, expresada para masa y energía es de la forma:
)1.2()(
.
)(
.
)(
.
controldevolumenel
enenergíaomasade
nacumulaciódevel
controldevolumen
delenergíaomasade
salidadevel
controldevolumen
alenergíaomasade
entradadevel
La ecuación anterior, usada como un balance de masas, se aplica normalmente
a especies químicas. Para sistemas simples, sólo se requiere una ecuación para
un componente seleccionado por conveniencia. Para sistemas complejos, el
número máximo de ecuaciones de balance independientes es igual a N, el
número de ecuaciones químicas. En este caso también se eligen las especies
en forma arbitraria. Ya sea si el sistema es simple o complejo, usualmente hay
un solo balance de energía.
Los términos de entrada y salida de la ecuación pueden tener más de una
contribución. Las entradas de especies pueden ser mediante flujo convectivo,
por un tipo de difusión que cruce el punto de entrada, y por formación por
reacción química en el volumen de control. La salida de especies puede incluir
consumo por reacción. También existen términos correspondientes en el
balance de energía (generación o consumo de entalpía por reacción),
agregando además transferencia de calor, lo cual no incluye flujo de
materiales. El término de acumulación es el resultado neto de entradas y
salidas; y solo para operaciones en estado estacionario es cero.

H. ZULETA
81
2.2 CLASIFICACION DE REACTORES QUÍMICOS
Antes de presentar varios tipos de aparatos para reacciones químicas, es
necesaria una breve introducción de las características de los procesos
involucrados.
Importancia del conocimiento de los procesos
La importancia del conocimiento de los procesos esta en la posibilidad de
establecer analogías entre soluciones o procesos existentes con problemas
nuevos por resolver. Además debido a que existe una gran variedad de
reacciones químicas y a las complejas interrelaciones entre ellas, se dispone de
una gran cantidad de configuraciones diferentes de equipos para reacciones
químicas y para modos de operación específicos.
Clasificación de Reactores sobre la base del modo de operación
Operación discontinua (Reactor batch)
En la forma de operación batch, los reactantes y cualquier sustancia adicional
tales como catalizadores e inertes se cargan en el reactor, donde permanecen
por un tiempo de reacción definido. En el curso de este proceso, la
composición del contenido del reactor cambia continuamente, es decir, el
reactor opera en estado no estacionario.
Ventajas: alta flexibilidad, grandes conversiones puesto que el tiempo de
reacción puede ser arbitrariamente alto.
Desventajas: periodos de detención por carga, descarga, limpieza y
calentamiento. El control y regulación de los procesos no estacionarios
requiere esfuerzo e instrumentación considerable.
En la industria química y farmacéutica el reactor batch es ampliamente usado
cuando se obtienen pequeñas cantidades de productos (drogas, colorantes y
cosméticos) o si el reactor se usa en la producción de productos diferentes.

H. ZULETA
82
Operación continua
En este tipo de operación los reactantes se alimentan continuamente en el
reactor y los productos son extraídos en forma continua del reactor. Todos los
parámetros de reacción (presión, temperatura, concentración) son constantes
con respecto al tiempo, es decir, el reactor opera en estado estacionario.
Ventajas: facilidad en alcanzar la automatización, volumen reducido de
equipos en comparación al mismo nivel de producción del reactor batch,
debido a que se considera periodos reducidos de detención de planta. Las
condiciones constantes de operación implican calidad constante de productos.
Desventajas: baja flexibilidad, los parámetros pueden ser modificados
solamente en limitada extensión ( por Ej. : temperatura ). La calidad de la
materia de entrada debe permanecer constante. Alto nivel de inversión.
Operación semicontinua
En la operación semicontinua, algunos reactantes pueden ingresar en forma
discontinua mientras que otros se alimentan continuamente, de igual manera
los productos pueden ser descargados continua o discontinuamente. Este
método de operación es muy conveniente cuando se producen efectos térmicos
notables, ya que permite retrasar las reacciones tanto endotérmicas como
exotérmicas por limitación de la concentración de uno de los reactivos,
manteniéndose así la reacción dentro de los límites adecuados para la
transmisión del calor.
También es conveniente este tipo de operación cuando la formación de
productos en concentraciones elevadas puede originar productos secundarios
indeseables, o bien cuando uno de los reactivos es un gas de solubilidad
limitada, que solamente puede entrar como alimentación a la velocidad de
disolución.
Algunos ejemplos para formas de operación semicontinua:
1. Un reactante inicialmente presente, siendo adicionado un segundo en
forma continua.

H. ZULETA
83
A + B P Ejemplo: Nitración
2. Continuamente se remueve un producto como parte de un proceso para
producir un cambio en el equilibrio.
A B + X
Ejemplo: Remoción por destilación de agua durante el proceso de
esterificación.
3. Inicialmente se ingresa un reactante, adicionando enseguida otro en forma
continua. Los productos se extraen continuamente.
A + B P
Ejemplo: Reacciones gas-sólido (combustión de carbono en hornos
refractarios).
En la operación semicontinua el reactor opera en estado no estacionario.
Ejemplos de Reactores para el diseño de procesos específicos
Existen reactores de variados tipos con diferentes formas de operación y
distintos procesos o sistemas de reacción. A continuación se presentan algunos
de estos, en esquemas, en sentido genérico y algunos de procesos específicos.
2.2.1 Reactores Batch (discontinuos)
Una representación esquemática del reactor batch se indica en la Figura 2-2
observándose sus características principales. Un recipiente cilíndrico provisto
con boquillas para agregar reactantes y remover el contenido del reactor. Para
asegurar una mezcla adecuada, el recipiente tiene un agitador (turbina)
accionado por un motor externo, además de placas deflectoras verticales para
eliminar el vórtice alrededor del extremo del impulsor. El control de
temperatura se alcanza con un serpentín de calentamiento/enfriamiento interno
o con un intercambiador de calor externo o chaqueta. El equipo se puede usar
a presión, en tal caso se requiere aparatos para aliviar las sobrepresiones, o
como un reactor “atmosférico”, el cual debe tener venteo.

H. ZULETA
84
Fig. 2-2 Reactor Batch: (a) Esquema con características principales; (b)
Reactor de planta piloto para la producción de aluminosilicato de sodio
2.2.2 Reactores de flujo tanque agitado continuo
El reactor de tanque agitado continuo puede ser de simple o multietapa. Es un
reactor de mezcla ideal y se lo identifica como CSTR o RTAC (reactor tanque
agitado continuo).
En la Fig. 2-3 se aprecian reactores CSTR en tres etapas. Las características de
mezcla y transferencia de calor pueden ser similares a las del reactor batch,
pero además se pueden usar intercambiadores de calor externo. La mayor
diferencia entre un reactor batch y un CSTR es que, en este último se debe
tener en cuenta el flujo continuo de materiales (entrada y salida) por gravedad
o circulación forzada con bombas.

H. ZULETA
85
2.2.3 Reactores de flujo tubular
El término “reactor de flujo tubular” se usa genéricamente para referirse a un
reactor en el cual el flujo es esencialmente en una dirección (flujo axial en un
recipiente cilíndrico) sin promover retromezcla por agitación. Las formas
idealizadas son el Reactor en flujo Pistón (RFP) y el Reactor en flujo Laminar.
La configuración puede variar ampliamente desde una muy alta o una muy
baja relación longitud/diámetro (L/D), como se indica en el esquema de la
figura 2-4

H. ZULETA
86
CAPITULO III. DISEÑO DE REACTORES IDEALES
3.1 ECUACIÓN DE DISEÑO DEL REACTOR BATCH
Consideraciones generales
El diseño de procesos de un reactor batch implica determinar el tiempo (t)
requerido para lograr un grado de conversión determinado XA ( del reactivo
limitante A), o el volumen (V) de un sistema reaccionante requerido para
lograr una velocidad especifica de producción en una base continua.. El
término “base continua” significa una operación cíclica, es decir sucesivos
procesos batch. Esto incluye el tiempo (td) para carga, descarga y limpieza.
La operación puede ser con densidad () constante o variable, a temperatura
(T) constante o variable. En el primer caso se requiere una ecuación de estado
para determinar V y en el segundo se requiere del balance de energía para
determinar la temperatura.
Para sistemas de una sola fase, el volumen se refiere al volumen del sistema
de reacción, pero no es necesariamente el volumen del reactor. Por ejemplo
para reacciones en fase líquida en un reactor batch existe un margen de
tolerancia para un espacio vacío por encima del líquido, es decir, el volumen
real del reactor es más grande que el volumen del sistema. En cualquier caso,
la variable V en forma convencional se referirá al “volumen del reactor”,
teniendo en cuenta tal consideración. Cuando exista diferencia considerable
entre el volumen de fluido reaccionante (V), el volumen del reactor se indicara
como VR. Por ejemplo, en un reactor de relleno de un catalizador sólido de
fracción hueca , el volumen disponible para el fluido en reacción es V = VR.
3.1.1 Balance de materiales del reactor batch
Se considera una reacción representada por A + … C C + ... la cual se
efectúa en un reactor batch. El Balance de materias general expresado en la
ecuación 2.1 se puede plantear para el reactivo limitante A con referencia a un
volumen de control especifico (el volumen del fluido).
Para este tipo de sistema, los únicos términos posibles de entrada y salida son
por reacción química, ya que no existen flujos de entrada y salida. En este
caso para el reactante A, sólo existe “salida”. Al aplicar en la ecuación se
obtiene:

H. ZULETA
87
AdenacumulaciódevelocidadAdeóndesaparicidevelocidad
)1.3()(dt
dNVr A
A
Sabiendo que AAAAAA dXNdNXNN 00 )1(
Reemplazando en la Ec. 3.1
)2.3()( 0
dt
dXNVr AA
A
Integrando
)3.3()(
2
10
A
A
X
XA
AA
Vr
dXNt
Esta es una ecuación general que permite calcular el tiempo requerido para
aumentar la conversión desde XA1 a XA2 en un reactor batch, en un sistema
que puede ser de volumen y temperatura constantes o variables.
Para los casos en que la densidad permanece constante
)4.3()(
0
2
1
0
A
A
A
A
C
C A
A
X
X A
AA
r
dC
r
dXCt
Para sistemas con densidad variable, la ecuación (3.3) se expresa
)5.3()1()()1(
0
0
0 0
0
AA X
AAA
AA
X
AAA
AA
Xr
dXC
XVr
dXNt
La interpretación gráfica de la ecuación general del reactor batch se muestra
en la figura 3-1
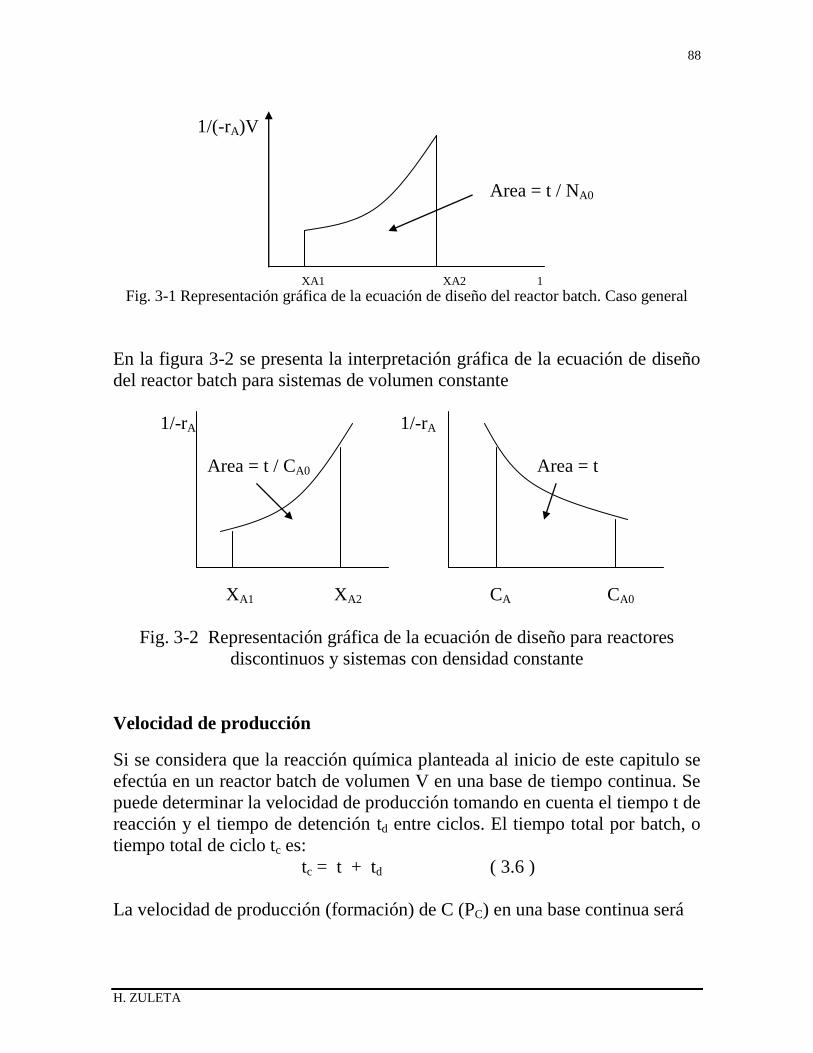
H. ZULETA
88
1/(-rA)V
Area = t / NA0
XA1 XA2 1
Fig. 3-1 Representación gráfica de la ecuación de diseño del reactor batch. Caso general
En la figura 3-2 se presenta la interpretación gráfica de la ecuación de diseño
del reactor batch para sistemas de volumen constante
1/-rA 1/-rA
Area = t / CA0 Area = t
XA1 XA2 CA CA0
Fig. 3-2 Representación gráfica de la ecuación de diseño para reactores
discontinuos y sistemas con densidad constante
Velocidad de producción
Si se considera que la reacción química planteada al inicio de este capitulo se
efectúa en un reactor batch de volumen V en una base de tiempo continua. Se
puede determinar la velocidad de producción tomando en cuenta el tiempo t de
reacción y el tiempo de detención td entre ciclos. El tiempo total por batch, o
tiempo total de ciclo tc es:
tc = t + td ( 3.6 )
La velocidad de producción (formación) de C (PC) en una base continua será

H. ZULETA
89
PC = (moles de C formado / batch) (batch / tiempo)
)7.3(12
d
AC
C
C
C
CC
Ctt
N
t
N
t
NNP
y en términos de la conversión fraccional de A
)8.3()( 120
d
AAAC
Ctt
XXNP
El volumen de reactor V está relacionado con NA0 a través de una ecuación de
estado (V = V (NA, T, P)). En la mayoría de los casos, XA1 = 0, y XA2 se
escribe simplemente XA. En la última ecuación, t se obtiene de la ecuación de
diseño. Las otras cantidades, NA0, XA y td, deben ser especificadas.
3.1.2 Balance de Energía del reactor batch
El balance de energía en general, de acuerdo a la ecuación 2.1, es:
)9.3(
energíade
nacumulaciódevelocidad
energíade
salidadevelocidad
energíade
entradadevelocidad
El volumen de control para el reactor batch es el volumen del sistema de
reacción. La entrada de energía puede ser por transferencia de calor mediante
serpentines o chaquetas y/o mediante generación por reacción. Similarmente,
las salidas de energía pueden ser por transferencia de calor (serpentines o
chaquetas), y/o mediante pérdidas por reacción. La acumulación es el
resultado neto de salidas o entradas, y puede significar un aumento o
disminución de la temperatura del sistema.
El flujo de calor transferido, Q, puede ser una entrada o salida de energía, y se
puede expresar como
Q = U AC ( TC – T )m ( 3.10 )
U = coeficiente global de transferencia de calor, J m-2
s-1
K-1
o W/m2 K
-1
Obtenido experimentalmente o de una correlación apropiada.
AC = área del medio de calentamiento o enfriamiento, m2

H. ZULETA
90
TC = temperatura del medio (serpentín o chaqueta), no necesariamente
Constante con el tiempo o la posición,°K
(TC – T)m = Una diferencia media de temperatura Tm apropiada para
Transferencia de calor.
Si Q > 0 (TC > T), la transferencia de calor es una entrada (al sistema
reaccionante), todo lo contrario se aplica si Q < 0.
Las transferencias pueden ser entradas o salidas significativas debido a lo
energético de la reacción. Una reacción exotérmica implica una generación
(una entrada) de energía. Una reacción endotérmica implica una pérdida o
consumo (una salida) de energía. La velocidad de generación o pérdida es el
producto de la energía de reacción y de la velocidad de reacción. La energía
de reacción se representa por la entalpía de reacción (HR) en procesos a
presión constante, y por la energía interna (UR) para procesos a volumen
constante.
Para la reacción química propuesta, con la energía y la velocidad de reacción
en términos del reactante A, se tiene:
)11.3()()()()()(.
VrUoVrHreacciónporenergíade
pérdidaogeneracióndeVelARAARA
Dependiendo del signo de HRA (o URA), este término será una entrada o
salida de energía.
HRA > 0 Reacción endotérmica, se trata de una salida
HRA < 0 Reacción exotérmica, se trata de una entrada
Puesto que el Balance de energía representa un balance de entalpías, la
velocidad de acumulación de energía es la velocidad de cambio de entalpía, H
del sistema reaccionante.

H. ZULETA
91
dt
dTcm
dt
dTCn
dt
dH
nacumulaciódeVelocidad
Pt
Pt (3.12)
en donde nt y mt representan los moles totales y la masa total del sistema
respectivamente, CP y cP corresponden a las capacidades caloricas molar y
especificas respectivamente.
Reemplazando las ecuaciones (3.10), (3.11) y (3.12) en la ecuación del
Balance de energía se obtiene
)13.3()()()(dt
dTCnVrHTTAU PtARAmCC
Esta ecuación es válida tanto si el calor se transfiere al sistema o si se retira de
este, o si la reacción es exotérmica o endotérmica. Además, en cualquier caso,
siempre se debe mantener la consistencia dimensional, usualmente J/s-1
3.1.3 Operación no-isotérmica
La condición de operación isotérmica puede no ser realista en el diseño de un
reactor batch, particularmente para reacciones muy exotérmicas o
endotérmicas. Aunque la temperatura puede cambiar considerablemente si se
descuida el control, y puede ser necesario evitar aumentos o disminuciones
apreciables. Esto implica temperaturas no necesariamente constantes. Sin
embargo, en muchos casos, puede ser ventajoso desde el punto de vista
cinético, si la temperatura aumenta en forma controlada.
Con el propósito de estimar los diseños del reactor y el intercambiador de
calor para el control de la temperatura, es necesario usar el balance de
materias y el balance de energía simultáneamente, además de la información
de la cinética de reacción y la transferencia de calor, debido a que existe
interacción entre conversión ( XA ) y temperatura.

H. ZULETA
92
3.1.4 Operación Adiabática
En la condición adiabática, no se debe calentar ni enfriar el contenido del
reactor (no existe intercambio de calor). Por lo tanto, la temperatura se eleva
en una reacción exotérmica y disminuye en una reacción endotérmica. Este
caso puede ser usado como limitante en el comportamiento no isotérmico,
para determinar si la temperatura cambia lo suficiente como para requerir
costos adicionales de intercambio de calor y el control respectivo.
Para un sistema adiabático con Q = 0, el Balance de energía queda:
)14.3()()(dt
dTCnVrH PtARA
Sustituyendo (-rA) V por el balance de materiales en términos de XA, se
obtiene:
)15.3()( 0dt
dTCn
dt
dXNH Pt
AARA
ya que la relación entre dXA/dt y dT/dt es implícita con respecto a t, la
ecuación anterior se puede escribir:
)16.3()( 0 dTCndXNH PtAARA
la integración de esta ecuación permite obtener T en función de XA
)17.3()(
00 A
Pt
RAA dX
Cn
HNTT
Donde T0 y NA0 son las condiciones iniciales. El resultado más simple para la
ecuación 3.17, considerando (-HRA ), CP y nt constantes es
)18.3()()(
00
0 AA
Pt
ARA XXCn
NHTT

H. ZULETA
93
El tiempo t requerido para lograr una conversión XA se obtiene por integración
del balance de materiales (ecuación de diseño), ecuación 3.3
Vr
dXNt
A
AA
)(0
En donde (-rA) se obtiene de la ley de velocidad y V de una ecuación de
estado. Se requiere una solución simultánea con la ecuación 3.17, ya que la
integral de la ecuación 3.3 depende de T y XA.
3.1.5 Operación no-adiabática
Si la operación del reactor batch es no-adiabática y no-isotérmica, el balance
de energía completo de la ecuación (3.13) se debe usar junto con el balance de
materiales de la ecuación (3.2). Esto constituye un set de dos ecuaciones
diferenciales ordinarias simultáneas, no lineales, de primer orden con T y XA
como variables dependientes y t como variable independiente. Las dos
condiciones límites son T = T0 y XA = XA0 (generalmente cero) en t = 0. Las
dos ecuaciones comúnmente se resuelven con procedimientos numéricos.
3.2 REACTOR CONTINUO PERFECTAMENTE MEZCLADO (CSTR)
El reactor continuo perfectamente mezclado (CSTR o RTAC) se usa
normalmente en reacciones en fase líquida al nivel de laboratorio o a gran
escala. Aunque también puede ser usado en reacciones en fase gaseosa en
laboratorios de investigación, particularmente cuando se tienen catalizadores
sólidos. Las características esenciales del RTAC son las siguientes:
(1) El flujo a través del recipiente, y en las corrientes de entrada y salida, es
continuo, pero no necesariamente constante.
(2) El fluido al interior del reactor está perfectamente mezclado, por lo tanto
sus propiedades son uniformes en cualquier tiempo.
(3) La densidad del sistema de flujo no es necesariamente constante, es decir,
la densidad de la corriente de salida puede diferir de la corriente de entrada.

H. ZULETA
94
(4) Se puede disponer, junto al reactor de un intercambiador de calor, para el
control de temperatura.
Existen varias consecuencias importantes que se pueden deducir de los puntos
anteriores, además del perfil de concentración planteado en la figura 3-3
CA0 CA0 CA1
CAf CA2
CA CA
CA0 CA0
CAf CA1
CA2
Fig. 3-3 Perfil de Concentraciones en un CSTR y en dos CSTR en serie
(1) Puesto que el fluido al interior del recipiente se encuentra uniformemente
mezclado (elementos de fluido uniformemente distribuidos), todos los
elementos de fluido tienen la misma probabilidad de abandonar el recipiente
por la corriente de salida en cualquier instante.
(2) Como consecuencia de (1), la corriente de salida tiene las mismas
propiedades que el fluido al interior del reactor.
(3) Como consecuencia de (2) se produce un cambio escalón en la entrada en
cualquier propiedad del sistema, la cual cambia inmediatamente desde la
entrada a la salida; esto se aprecia en la figura 3-3 para la concentración de A.
(4) Existe una distribución continua (amplitud) de tiempos de residencia (t) de
elementos de fluido; la extensión se puede apreciar intuitivamente
considerando dos casos extremos: (i) el fluido se mueve directamente de la

H. ZULETA
95
entrada a la salida (tiempo corto), y (ii) el fluido adopta un movimiento de
recirculación debido a la agitación (tiempo largo).
(5) El tiempo de residencia medio, t del fluido en estado estacionario al
interior del recipiente es
Vt (3.19)
en donde es el flujo (por ejemplo en m3/s) de fluido que abandona el reactor;
es una consecuencia de (2).
(6) El tiempo especial, en estado estacionario es
)20.3(0
V
En donde 0 es el flujo en estado estacionario de alimentación; se debe
observar que para sistemas a densidad constante, ty,0
Es importante entender las implicancias entre el punto (3) y el (5). El punto
(3) implica que hay una mezcla instantánea en el punto de entrada entre la
corriente de entrada y el contenido del recipiente; es decir la corriente de
entrada instantáneamente se mezcla con el contenido del recipiente. Esto no
significa que cualquier reacción que tiene lugar al interior del fluido ocurre
instantáneamente. El tiempo requerido para el cambio en la composición de la
entrada a la salida es t , punto (5), el cual puede ser pequeño o grande.
3.3 ECUACIÓN DE DISEÑO DEL CSTR
Consideraciones generales
El proceso del diseño de un CSTR primordialmente involucra la
determinación del volumen del recipiente necesario para obtener una
velocidad de producción requerida. Los parámetros a investigar incluyen el
número de etapas necesarias para una operación óptima, y la conversión
fraccional y temperatura entre cada etapa.

H. ZULETA
96
3.3.1 Balance de materiales del CSTR o RTAC
Para la operación continua de un CSTR como un recipiente “cerrado”, la
ecuación general del balance de materiales para el reactante A (según la
reacción A +… C C + … ), con un volumen de control definido por el
volumen de fluido en el reactor, se puede escribir
Entrada de A – Salida de A – Desaparición de A = Acumulación de A
)21.3()(0dt
dNVrFF A
AAA
Considerando que FA0 y FA corresponden a los flujos molares (moles/tiempo)
de entrada y salida del reactante A respectivamente. Además en la ecuación se
ha establecido que XA0 = 0.
Si se expresa la ecuación en término de los flujos volumétricos
)22.3()(000dt
dNVrCC A
AAA
Utilizando la conversión fraccional de A, XA = ( FA0 – FA ) / FA0
)23.3()(0dt
dNVrXF A
AAA
Puesto que el reactor opera en estado estacionario dNA / dt = 0, las ecuaciones
(3.22) y (3.23) quedarán:
)25.3()(
)24.3()(
)(
0
00
A
AA
A
AA
r
XFV
r
CCV
La interpretación de las diferentes cantidades con respecto a la ubicación en el
reactor se indica en la Fig. 3-4

H. ZULETA
97
FA0
CA0
0 NA0
V FA
CA
XA
Fig. 3-4 Cantidades en la ecuación del Balance de materiales para el CSTR
Arreglando la ecuación 3.25
)()( 000 A
A
AA r
X
C
V
F
V
Sabiendo además que 0
V
Por lo tanto
)()(00 A
A
A
A
AA r
X
r
X
CF
V
También
)26.3(0
0
0
0 A
AA
A
A
r
XC
F
CVV
Donde XA y –rA se evalúan en las condiciones de la corriente de salida, que
son iguales a las existentes al interior del reactor.
De modo general, si la alimentación sobre la que se basa la conversión ingresa
al reactor parcialmente convertida (que expresamos con el subíndice i) y sale
en las condiciones que se expresan con el subíndice f, se tiene:
)27.3()(
)(
)(0 fA
AiAf
fA
A
A r
XX
r
X
F
V

H. ZULETA
98
)28.3()(
)(0
0
0
fA
AiAfA
A
A
r
XXC
F
CV
Las ecuaciones anteriores y principalmente la ecuación (3.25), se pueden
interpretar gráficamente si se representan la velocidad de reacción reciproca,
(1/-rA) en función de la conversión XA. Esto se puede apreciar en la curva EB
de la figura 3-5. El punto B se conoce como “punto de operación” del reactor;
esto es, representa la condición en estado estacionario en el reactor ( y en la
corriente de salida). El área ABCD, el producto de 1/(-rA) y XA en las
condiciones de operación, representa la razón V/FA0 para el CSTR (según la
ecuación (3.25). La figura se aplica a cinéticas normales, y es válida ya sea en
sistemas con densidad constante o variable. Debe destacarse que para el CSTR
sólo tiene importancia el punto B.
1/-rA
Punto de operación
A B
Area = V / FA0
E
D C XA
Fig. 3-5 Interpretación gráfica de la ecuación de diseño del CSTR
Para el caso especial de sistemas de densidad constante (XA = 1- (CA/CA0)), la
ecuación de diseño para los reactores de mezcla completa puede escribirse
también en función de las concentraciones, es decir:

H. ZULETA
99
)29.3()(0
0
0 AA
AA
A
A
A rC
CC
r
X
F
V
)30.3(00
0 A
AA
A
AA
r
CC
r
XCV
Estas expresiones relacionan de manera sencilla los cuatro términos XA, -rA, V
y FA0; por lo tanto, conociendo tres de ellos se obtiene directamente el cuarto.
Por lo general se fijan las condiciones de entrada ( FA0 =CA0 0 ) y a través de
estudios cinéticos, es posible evaluar -rA. De esta manera es posible calcular el
volumen del reactor necesario para obtener un grado de conversión
determinado.
En la figura 3-4 se representa gráficamente la ecuación de diseño para
sistemas de densidad constante.
1/-rA
Punto de operación
Área = = V CA0 / FA0
CA
CA CA0
Fig. 3-4 Interpretación gráfica de la ecuación de diseño del CSTR para
sistemas con densidad constante

H. ZULETA
100
3.3.2 Ecuación de diseño del CSTR para una reacción de primer orden
Remplazando en la ecuación de diseño (ec.3.26) la ley cinética, -rA= k CA y
considerando la aplicación en sistemas de fase líquida
)31.3(1
1 0
0 A
AA
A
AA
A
A
C
CC
X
XkX
C
C
La ecuación (3.31) corresponde a la ecuación de diseño de un CSTR en donde
se verifica una reacción de primer orden
Para sistemas con variación de volumen, se utiliza la ecuación (1.78)
AA
A
A
A
X
X
C
C
1
1
0
La ecuación de diseño resultante es:
)32.3()1(
)1(
A
AAA
X
XXk
o su equivalente
)33.3()(
)(
0
00
AAAA
AAA
CCC
CCCk
3.3.3 Ecuación de diseño del CSTR para una reacción de segundo orden
Ecuación de diseño
)34.3()1( 2
0
2
0
AA
A
A
AA
XC
Xk
Ck
XC
)35.3(2
0
A
AA
C
CCk

H. ZULETA
101
Con variación de volumen
)36.3()(
)(
)1(
)1(
0
2
2
00
2
2
0
AAAA
AAA
A
AAAA
CCC
CCC
X
XXCk
3.3.4 Balance de energía en el reactor CSTR
Para reactores de flujo, como es el caso del CSTR, el balance de energía es un
balance de entalpías, si se desprecia cualquier cambio en energía cinética y
potencial de las corrientes, y cualquier trabajo de eje entre los puntos de salida
y entrada. Sin embargo, en comparación con el reactor batch, el balance debe
incluir las entradas y salidas de entalpía en las corrientes fluyentes, además de
la transferencia de calor de o hacia el volumen de control, y la generación o
pérdida de entalpía por reacción. Por lo tanto la ecuación de energía (entalpía)
quedará:
)37.3(
.
,
.
,
.
controldevolumen
elenentalpíade
nacumulaciódevel
químicareacciónocalor
deciatransferenflujopor
entalpíadesalidadevel
químicareacciónocalor
deciatransferenflujopor
entalpíadeentradadevel
Para expresar esta ecuación en términos operacionales, se debe usar la
velocidad de flujo másico total, m, en vez de la velocidad de flujo molar. Una
ventaja de este tratamiento es que en la operación en estado estacionario, m es
constante, mientras que la velocidad de flujo molar no lo es. Por lo tanto
usando Tref como la temperatura de referencia para evaluar la entalpía para las
corrientes de entrada y salida, se tiene:
)38.3()(
))(()(00
0
dt
TCmd
dt
dHVrHTTUAdTmCdTCm Pt
A
T
T
RAmCCPp
T
Tref
En esta ecuación, el subíndice “0” representa las condiciones de entrada, Cp es
el calor específico total del sistema, y mt es la masa total contenida en el
volumen de control en el tiempo t; la interpretación de estas cantidades se
muestra en la figura 3-5

H. ZULETA
102
Hint Hout
Cpo Cp
mo m
To UAc TC T
Fig. 3-5 Cantidades de la ecuación del balance de energía
La ecuación (anterior) puede ser simplificada en varios de sus términos. Para
la operación en estado estacionario, m = m0, y el lado derecho se anula;
además se considera CP y TC constantes y estableciendo Tref = T0, por lo tanto
la entalpía del flujo de entrada se hace cero, por lo tanto,
0))(()()( 0 VrHTTUATTmC ARACCP (3.39)
Además, si se reemplaza FA0 XA por (-rA)V,
0)()()( 00 AARACCP XFHTTUATTmC (3.40)
La ecuación (3.40) relaciona XA y T para la operación en estado estacionario;
resolviendo para XA, se tiene:
TFH
UAmC
FH
TUATmCX
ARA
CP
ARA
CCPA
00
0
)()( ( 3.41 )
El uso del balance de energía en el diseño de un CSTR dependerá de lo que se
especifique inicialmente. Si se fija la temperatura T, el balance de energía no
se acopla con el balance de materiales, y se usa en primer lugar para el diseño
del intercambiador de calor requerido para el control de temperatura; el
volumen está determinado por la ecuación de diseño. Si la temperatura T, no
esta especificada, como en la operación adiabática, los balances de materia y
energía se acoplan ( a través de T ), y deben ser resueltos simultáneamente.

H. ZULETA
103
3.3.5 Operación en estado no estacionario
Para la operación en estado no estacionario (transitorio) del RTAC, se debe
usar la forma completa de la ecuación del balance de materiales.
Ejemplo:
Considere la puesta en marcha de un CSTR para la reacción en fase líquida
A productos. El reactor esta inicialmente lleno con alimentación cuando
comienza la alimentación en forma continua. Determine el tiempo t
requerido para alcanzar el 99% de la conversión en estado estacionario.
Datos: V = 8000 lts. = 2 lts/s CA0 = 1,5 M
K = 1,5*10-4
s-1
Solución:
Aunque la alimentación es un flujo en estado estacionario, el reactor está en
estado no estacionario en un tiempo t, debido a que la concentración de salida
CA ( y también XA ) cambia continuamente ( CA disminuye a partir de CA0, y
XA aumenta a partir de cero ).
En primer lugar se determina el valor de XA en estado estacionario. Usando la
ecuación general del balance de materiales (3.22), con = 0
C C r VdN
dtA A A
A
0 0 0 ( )
Esta ecuación puede expresarse en términos de = V/0 , además de
CA = CA0 ( 1 – XA ) y en combinación con la ley de velocidad, el resultado es:
C C X KC X dC dtA A A A A A0 0 01 1 ( ) ( ) /
Para la operación en estado estacionario dCA/dt =0, por lo tanto

H. ZULETA
104
X K X
XK
K
A A
A
( )
. ( )( / )
. ( )( / ),
1 0
1
15 10 8000 2
1 15 10 8000 20 375
4
4
El tiempo requerido para alcanzar el 99% de este valor, se obtiene por
integración de la ecuación (A), expresada en una forma equivalente:
C X K C X CdX
dtA A A A A
A
0 0 01 ( )
K K XdX
dtA
A ( )1
o bien
dtdX
K K X
A
A
( )1
con la condición XA = 0 a t = 0 se integra la ecuación anterior, resultando
en:
tK
K
K K X A
1 1ln
( )
Se obtiene t = 11500 s. ( 3,2 horas ) para XA = 0,371. ( Esta no es
necesariamente la manera más eficiente para poner en marcha un CSTR. ¿De
que manera se podría reducir el tiempo requerido?)
3.4 REACTOR DE FLUJO TUBULAR ( RFP )
Características generales
El reactor en flujo pistón puede ser usado para reacciones en fases líquida o
gaseosa, a escala de laboratorio o investigación y a escala de producción
industrial. El reactor puede consistir de un tubo o recipiente vacío o relleno
con empaque o un lecho fijo de partículas (partículas de catalizador).

H. ZULETA
105
El RFP es similar al CSTR en cuanto a que es un reactor de flujo y esta misma
característica lo diferencia del reactor batch, pero es similar en el cambio
progresivo de las propiedades con la posición en vez del tiempo. Las
siguientes características definen a un RFP:
(1) Los flujos a través del reactor de las corrientes de entrada y salida son
continuos, pero no necesariamente a velocidad constante; el flujo en el
recipiente es tipo tapón.
(2) La masa del sistema al interior del reactor no es necesariamente fija.
(3) No hay mezclado axial al interior del reactor (es decir, en la dirección
del flujo).
(4) Hay un mezclado radial completo del fluido al interior del reactor (es
decir, en un plano perpendicular a la dirección del flujo); por lo tanto
las propiedades del fluido, incluyendo su velocidad, son uniformes en
este plano.
(5) La densidad del sistema puede variar en la dirección del flujo.
(6) El sistema puede operar en estado estacionario o en estado transciente.
(7) Puede existir intercambio de calor a través de las paredes del reactor
entre el sistema y los alrededores.
Algunas consecuencias del modelo descrito en los siete puntos anteriores, son
las siguientes:
[1] Cada elemento de fluido tiene el mismo tiempo de residencia t; es decir, no
existe difusión o dispersión de t.
[2] Las propiedades pueden cambiar continuamente en la dirección del flujo.
[3] En la dirección axial, cada porción de fluido, actúa como un sistema
cerrado en movimiento, no existiendo intercambio de materias con porciones
anteriores ni posteriores.
[4] El volumen de un elemento de fluido no necesariamente permanece
constante a través del reactor; puede cambiar por cambios de T, P o nt, el
número de moles totales.

H. ZULETA
106
En la siguiente figura se muestran los perfiles de concentración y conversión
con respecto a la posición en el reactor
F
C
X
A
A
A
F dF
C dC
X dX
A A
A A
A A
CA XA
longitud
Fig. 3-6 Perfil de concentración y conversión en un RFP
3.5 ECUACIONES DE DISEÑO DEL RFP
Consideraciones generales; Balances de materiales, energía y momento
El proceso de diseño de un RFP, típicamente involucra determinar el tamaño
de un recipiente requerido para lograr una velocidad específica de producción.
El tamaño se determina básicamente como un volumen, el cual se expresa en
términos de, por ejemplo, la longitud y diámetro de un reactor cilíndrico, la
longitud o número de tubos de un tamaño determinado. Aspectos adicionales a
considerar son los efectos de temperatura resultantes de la naturaleza
energética de la reacción, y los efectos de presión resultante por fricción. Si
los efectos de temperatura son importantes, ya sea en operaciones adiabáticas,

H. ZULETA
107
o no adiabáticas con una superficie de transferencia de calor apropiada, los
balances de materiales y energía se deben considerar simultáneamente.
Además, si los efectos de presión son considerables, el balance de momento
también se debe tomar en cuenta. Este puede ser el caso de reacciones en fase
gaseosa con un fluido circulando a alta velocidad en un recipiente de longitud
considerable, como en la deshidrogenación de etano a etileno.
3.5.1 Balance de materiales del RFP
El balance de materiales del RFP se desarrolla de manera similar al CSTR,
excepto que el volumen de control es un volumen diferencial dV, ya que sus
propiedades cambian continuamente en dirección axial.
Considerando una reacción representada por A + .... CC +... , que
transcurre en un reactor de volumen V y en estado estacionario, el balance
para A alrededor de dV es
Operación no estacionaria F F dF r dVdN
dtA A A A
A ( ) (3.42)
Operación estacionaria F F dF r dVA A A A ( ) 0 (3.43)
A partir de esta última ecuación y considerando que dF F dXA A A 0
Se obtiene:
dV
dX
F
rA
A
A
0
0( )
(3.44)
Para obtener V se integra para todo el volumen
V FdX
rA
A
A
0 ( )
(3.45) Ecuación de diseño
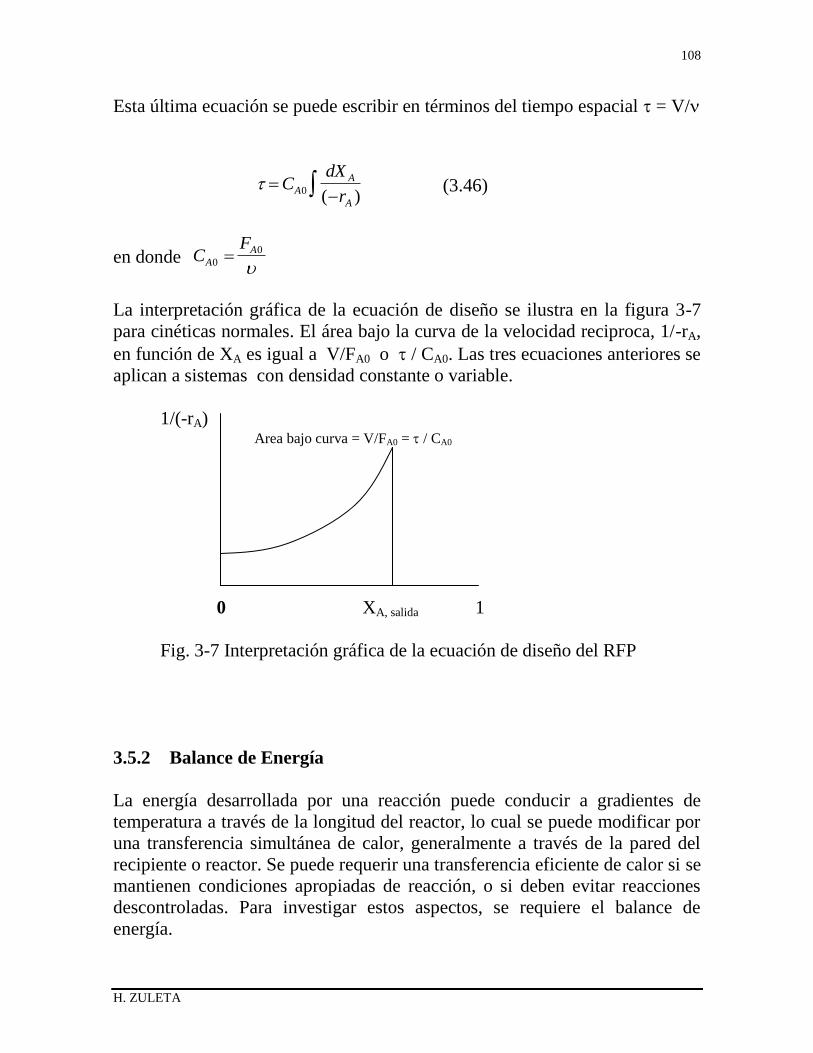
H. ZULETA
108
Esta última ecuación se puede escribir en términos del tiempo espacial = V/
CdX
rA
A
A
0 ( ) (3.46)
en donde CF
A
A
0
0
La interpretación gráfica de la ecuación de diseño se ilustra en la figura 3-7
para cinéticas normales. El área bajo la curva de la velocidad reciproca, 1/-rA,
en función de XA es igual a V/FA0 o / CA0. Las tres ecuaciones anteriores se
aplican a sistemas con densidad constante o variable.
1/(-rA) Area bajo curva = V/FA0 = / CA0
0 XA, salida 1
Fig. 3-7 Interpretación gráfica de la ecuación de diseño del RFP
3.5.2 Balance de Energía
La energía desarrollada por una reacción puede conducir a gradientes de
temperatura a través de la longitud del reactor, lo cual se puede modificar por
una transferencia simultánea de calor, generalmente a través de la pared del
recipiente o reactor. Se puede requerir una transferencia eficiente de calor si se
mantienen condiciones apropiadas de reacción, o si deben evitar reacciones
descontroladas. Para investigar estos aspectos, se requiere el balance de
energía.

H. ZULETA
109
El balance de energía de un RFP, es un balance de entalpías que puede ser
desarrollado de una manera similar al CSTR, excepto que el volumen de
control es un volumen diferencial. Esto se ilustra en la figura 3-8 junto con la
simbología usada. Para desarrollar el balance, se considera operación en
estado estacionario. Las velocidades de entrada y salida de entalpías por: (a)
flujo, (b) transferencia de calor y (c) por reacción, se pueden obtener sobre la
base de un volumen de control dV de la figura.
X TS dX
m T T dT m
H H dH
F F F dF FA A A A A sal
0
0 ,
D
dV
Fig. 3-8 Volumen de control y simbología para el balance de energía del RFP
dTCmdHdHHHflujoporentalpía
desalidadeVel
flujoporentalpía
deentradadeVela P
)(..
)(
En donde m es la velocidad de flujo másico en estado estacionario, CP es el
calor específico de las corrientes, y dT es el cambio en la temperatura, que
puede ser positivo o negativo;
(b) Velocidad de transferencia de calor al (desde) volumen de control
Q U T T dAS p( )
Donde U es el coeficiente de transferencia de calor global, TS es la
temperatura de los alrededores al reactor tubular en un punto determinado, y
dAp es el área diferencial de transferencia de calor circunferencial;

H. ZULETA
110
( c ) Velocidad de entrada / generación ( o salida / absorción) de entalpía por
reacción química
( )( )H r dVRA A
Aplicando estos términos en la ecuación del balance de energía en estado
estacionario, se obtiene:
mC dT U T T dA H r dVp S p RA A( ) ( )( ) 0 (3.47)
Esta ecuación puede ser convenientemente transformada para relacionar T y
XA. Puesto que
dA Ddxp (3.48)
y
dV D dx ( / ) 2 4 (3.49)
donde D es el diámetro del tubo o recipiente,
dA D dVp ( / )4 (3.50)
Usando esta última ecuación junto con la ecuación de diseño para eliminar
dV y dAp, de la ecuación del balance de energía, se obtiene, previos arreglos la
ecuación
mC dT HU T T
D rF dXP RA
S
A
A A
( )
( )
( )
40 (3.51)
Alternativamente, la ecuación del balance de energía (3.47), se puede
transformar para obtener el perfil de temperatura en términos de x, la distancia
a lo largo del reactor a partir de la entrada. Esto considera el uso de las
ecuaciones (3.48) y (3.49) para eliminar dAp y dV. Por lo tanto,
mC dT H r D U T T DdxP RA A S ( )( )( / ) ( ) 4 ( 3.52 )
Se debe enfatizar que todas las formas desarrolladas anteriormente del balance
de energía se refieren a arreglos en los cuales los flujos reaccionantes están al
interior de un simple tubo o recipientes, o en el interior de un conjunto de
tubos. Si las corrientes o flujos reaccionantes se encuentran por fuera de los

H. ZULETA
111
tubos en un arreglo particular, la geometría es diferente y conduce a diferentes
formas de la ecuación.
Para operaciones adiabáticas (Q = 0), las dos últimas ecuaciones se
simplifican eliminando el término que involucra el coeficiente de transferencia
de calor U.
3.5.3 Balance de momento; operaciones no isobáricas
En muchos casos, la longitud y el diámetro de un reactor cilíndrico se pueden
seleccionar para obtener condiciones aproximadamente isobáricas. Sin
embargo, a veces, la caída friccional de presión es considerable como para
afectar el comportamiento del reactor. Como una regla empírica, para fluidos
compresibles, si la diferencia de presión entre la entrada y salida es más
grande que 10 o 15%, el cambio de presión probablemente afectara la
conversión, y debería ser considerado en el diseño del reactor. En esta
situación, el gradiente de presión a lo largo del reactor debe ser determinado
simultáneamente con los gradientes de XA y T.
Para determinar el gradiente de presión a lo largo del reactor, se puede usar la
ecuación de Fanning o Darcy para flujo en tubos cilíndricos ( Knudsen y Katz,
1958,p.80):
dP
dx
u f
D
q f
D
2 322 2
2 5
Donde P es la presión, x es la posición axial en el reactor, es la densidad del
fluido, u es la velocidad lineal, es el factor de fricción de Fanning, D es el
diámetro del reactor, y q es el flujo volumétrico; , u y q puede variar con la
posición.
El factor de fricción de Fanning se puede determinar de cartas para tubos
rugosos y lisos o partir de correlaciones (Knudsen y Katz, 1958). Las
siguientes correlaciones se aplican para flujo turbulento en tubos lisos y para
Re entre 3000 y 3.000.000:
f 0 00140 0125 0 32, , (Re) ,
Se debe considerar que Re , y por lo tanto , pueden variar a lo largo del
reactor, si la velocidad y la densidad del fluido cambian.

H. ZULETA
112
Para evaluar el efecto de la caída de presión en el comportamiento del reactor,
las ecuaciones diferenciales de caída de presión, balance de materiales y
balance de energía deben ser integradas simultáneamente para resolver P, XA y
T como funciones de la posición axial, x.
3.5.4 Sistemas de densidad constante
Operación isotérmica
Para estos sistemas X
C C
Cpor lo tanto dX
CdCA
A A
A
A
A
A
( )0
0 0
1
Reemplazando en la ecuación de diseño (3.46) ))(
( 0
A
AA
r
dXC
Se obtiene
V dC
r
A
A( ) (3.53)
El signo (-) antes de la integral se puede eliminar invirtiendo los límites de
integración. Una representación gráfica de esta última ecuación se muestra en
la figura 3-9.
1/(-rA) Area bajo la curva = V/ =
0 CA salida CA0 CA
Fig. 3-9 Interpretación gráfica de la ecuación de diseño del RFP- Densidad
constante
Operación No isotérmica
Para caracterizar el comportamiento del RFP, sujeto a gradientes axiales de
temperatura, los balances de materia y energía deben ser resueltos
simultáneamente. Esto puede requerir integración gráfica usando algún
software determinado. En el siguiente ejemplo, se ilustra el desarrollo de

H. ZULETA
113
ecuaciones y los perfiles resultantes de conversión XA con respecto a la
posición (x), para una reacción con densidad constante.
Ejemplo
Una reacción en fase líquida A + B 2C se efectúa en un RFP no
isotérmico multitubular. Los tubos del reactor ( 7 m. de longitud, 2 cm.de
diámetro) están rodeados por un refrigerante que mantiene constante la
temperatura de la pared. La reacción es de pseudo primer orden con respecto
de A, con k = 4,03*105exp(-5624/T) s
-1. La velocidad de flujo másico es
constante a 0,06 Kg/s, la densidad es constante (1,025 g/cm3), y la temperatura
a la entrada del reactor (T0) es 350°C.
(a) Desarrollar una expresión para dXA/dx y dT/dx.
(b) Graficar el perfil XA(x) para las siguientes temperaturas de pared (TS):
350°K, 365°K, 400°K, y 425°K.
Datos : CA0 = 0,5 M; CP = 4,2 J / g °K;
HRA = -210 KJ/mol; U = 1,59 KW/m2 °K
Solución
(a) La ley de velocidad es ( ) ( ) r K C K C XA A A A A A0 1 ( 3.54 )
Donde KA puede ser expresada según la ley de Arrhenius. Si se asume que el
reactor es un cilindro de radio R. El volumen del reactor desde la entrada hasta
la posición x (longitud x) es V x R x o dV R dx( ) , 2 2 reemplazando este
valor en la ecuación de diseño fundamental (3.44) y arreglando, se obtiene:
dX
dx
R r
F
A A
A
2
0
0( )
( 3.55 )
Al despejar (-rA) de esta última ecuación se obtiene:
dx
dX
R
Fr AA
A 2
0)(
(3.56)

H. ZULETA
114
Igualando con la ecuación (3.54) y despejando (dXA / dx), se obtiene ( con
R=D/2 y FA0 / CA0 = )
dX
dx
D K XA A A
2 1
4
( ) (3.57)
La ecuación (3.57) corresponde a la expresión requerida.
Para desarrollar una expresión para dT/dx, se debe usar la ecuación del
balance de energía ( 3.51)
mC dT HU T T
D rF dXP RA
S
A
A A
( )
( )
( )
40 (3.51)
la cual se divide por dx y eliminando (-rA) de la ecuación (3.54), y dXA/dx por
la ecuación (3.57), se logra.
dT
dx
D K C X H
mC
UD
mCT T
A A A RA
P P
S
2
0 1
4
( )( )( )
(3.58)
Donde TS es la temperatura de la pared. Se debe observar que dT/dx está
implícitamente relacionada con dXA/dx.
(b) Se puede usar algún tipo de software para integrar numéricamente las
ecuaciones diferenciales (3.57) y (3.58) desde x = 0 hasta x = 7m, usando las
cuatro temperaturas de pared especificadas. Del perfil resultante XA (x) se
debería observar que como la temperatura TS se incrementa de 350 a 425, la
longitud del reactor requerido para lograr una conversión determinada se
reduce.
3.6 OPERACIÓN DEL RFP CON RECICLO
En muchos procesos químicos, el uso del reciclo, esto es, el regreso de una
porción de la corriente de salida a la entrada para unirse con alimentación
fresca, puede tener los siguientes propósitos: (1) recuperar alimentación de
reactante que no se ha convertido completamente,(2) recuperar
catalizadores,(3) diluir el flujo de un proceso, (4) controlar una variable de

H. ZULETA
115
proceso. El reciclo implica retromezcla de la corriente de producto con la
alimentación fresca, y este efecto produce un acercamiento del RFP al CSTR.
El grado de retromezcla, y la extensión del efecto de recirculación esta
controlado por la razón de reciclo R, definido por:
productodecorrientelaenAdemolarflujodevelocidad
reciclodecorrientelaenAdemolarflujodevelocidadR
)59.3(111
1
1
R
A
RA
A
AR
C
C
F
F
productosdecorrienteladeovolumetricflujodevelocidad
reciclodecorrienteladeovolumetricflujodevelocidad
El subíndice R se refiere al reciclo y el subíndice 1 a la salida del reactor. La
ecuación (3.59) es aplicable a sistemas con densidad constante y variable.
La razón R puede variar de 0 (sin reciclo) a un valor muy grande (virtualmente
reflujo total), es decir el comportamiento del reactor estará entre un RFP y un
CSTR (reflujo total), dependiendo del valor de R. El problema se reduce
entonces a determinar las condiciones en las cuales el RFP con reciclo es más
ventajoso. En esta condición se encuentran los casos con cinéticas anormales,
para reducir el volumen; las cinéticas complejas, para el control de la
selectividad; y reacciones fuertemente exotérmicas, para reducir los gradientes
de temperatura a lo largo del reactor y evitar posibles “puntos o sitios
calientes”.
Sistemas con densidad constante
Para sistemas con densidad constante, puesto que 1 = 0 , la ecuación 3.59 se
puede escribir como:
)tan(0
teconsdensidadR R

H. ZULETA
116
Un reactor con reciclo, operando en estado estacionario, de acuerdo a la
reacción A + .... productos, se muestra en el esquema de la figura 3-10,
junto con las corrientes asociadas y la terminología. En el punto de separación
S, la corriente de salida se divide en una corriente de reciclo (con velocidad de
flujo R0 ) y una corriente de reflujo (con velocidad de reflujo 0), ambas con
una concentración de salida CA1. En el punto de mezcla M, la corriente de
reciclo se une con alimentación fresca ( flujo 0 y concentración CA0) para
formar la corriente real que ingresa al reactor ( velocidad de flujo (1+R) 0, y
concentración C’A0). La concentración de entrada C’A0 puede ser relacionada
con CA0, CA1, y R por un balance de materiales para A alrededor de M:
)60.3()1( '
001000 AAA CRCRC
dV
0
0
0
A
A
C
F
M C’A0 CA CA+dCA S
CA1
0
1
1
A
A
C
F
(1+R)0
Fig.3-10 Diagrama de flujo para el reactor flujo pistón con reciclo
A partir de la última ecuación R
RCCC AA
A
1
10'
0 (3.61)
El volumen del RFP con reciclo se puede obtener mediante un balance de
materiales para A alrededor de un volumen diferencial de control dV
dVrdCCRCR AAAA )()()1()1( 00 (3.62)
en consecuencia
1
'0' )(
)1(0
A
A
C
CA
A
r
dCR
V
(3.63)

H. ZULETA
117
Una interpretación gráfica de la ecuación (3.63) se muestra en la figura 3-11,
la cual es una representación de la velocidad de reacción reciproca para
cinéticas normales (curva GE’) y la concentración. En la figura, el área
achurada bajo la curva GE’ desde C’A0 hasta CA1, BFGA, representa el valor
de la integral en la ecuación (la integral tiene un valor negativo). El tiempo
espacial , en la ecuación es igual a –(1+R) veces el valor de esta integral, la
cual está basada en C’A0 y CA1. Se puede determinar en términos de CA0 y
CA1, como se indica en los siguientes cálculos:
)64.3(1
tan
)()1(
61.3.)1(
10
'
00
010
0010
10
'
0
R
R
CC
CC
toloPor
CCRCR
RCRCRCC
ecladepartiraRCCCR
AA
AA
AAA
AAAA
AAA
G E’’
F’
D E
1/(-rA)
F
E’
A B C
0 CA1 C’A0 CA0 CA
1 R
1 + R
Fig. 3-11 Interpretación gráfica de la ecuación del RFP con reciclo

H. ZULETA
118
La construcción en la figura, usa las relaciones anteriores, gráficamente. Si la
distancia CA0 – CA1 se representa por (1+R), a partir de la ecuación 3.64, la
distancia CA0 – C’A0 está representada por R, y C’A0 – CA1 por 1 ( la unidad ).
CA0 – CA1 es por lo tanto (1+R) veces C’A0 – CA1. Por otra parte se puede
relacionar CA0 – CA1 y C’A0 – CA1 usando rectángulos de alturas iguales, como
se aprecia en la figura. Se puede elegir el punto D de tal manera que el
rectángulo DF’BA tenga la misma área que el área sombreada.
Consecuentemente, sobre la base de la misma ecuación, el área rectangular
DECA es (1+R) veces el área DF’BA, puesto que CA0 – CA1 es (R+1) veces
C’A0 – CA1.
Los dos casos límite para R ( R 0 y R ) pueden ser analizados
cualitativamente en la figura 3-11. Si R 0, el punto F E’, y el área bajo la
curva representa V/ 0 para un RFP sin reciclo. Si R , el punto F G, y
el área rectangular CE’’GA representa V/ 0 para el reactor con reciclo, y es
equivalente al CSTR operando en estado estacionario para las mismas
condiciones.
Una demostración analítica del primer caso límite se puede obtener
directamente de las ecuaciones (3.61) y (3.63). Es decir si R 0, C’A0 CA0,
y V/0 equivale a la ecuación del reactor sin reciclo. Una demostración
analítica del segundo caso límite no se puede obtener directamente de estas
dos ecuaciones. Esto es si R , C’A0 CA1 ( de la ecuación 3.61), y V/0
-()(0) ( de la ecuación 3.63), lo cual es una forma “indeterminada”. Esto
último puede ser evaluado con la ayuda de la regla de L’Hôpital.
El tratamiento de un RFP con reciclo hasta ahora, basado en cinéticas
“normales”, no revela alguna ventaja relativa al RFP simple. De hecho, esto es
una desventaja respecto del tamaño del reactor, el cual es mayor para
cualquier valor de reciclo, debido al efecto de retromezcla: el rectángulo
CEDA en la figura 3-11, para cualquier posición posible de la línea ED, es
mayor que el área CE’GA bajo la curva de velocidad reciproca.
Para cinéticas “anormales”, sin embargo, la situación se invierte, y existe la
posibilidad de un reactor más pequeño. Un ejemplo de cinéticas anormales

H. ZULETA
119
corresponde a las reacciones autocatalíticas ( Cap.I, Fig.1-21) . En la Fig.3-12,
se representa el reciproco de la velocidad de reacción, observándose un
mínimo que equivale a un máximo de la Fig.1-21. La porción de la curva GE’
desde H a E’ representa cinética “anormal”, y desde G hasta H, la cinética
“normal”. El resto de la Fig.3-12 corresponde a lo que se interpreta como el
efecto del reciclo en la Fig. 3-11 y la interpretación de la ecuación (3.63).
El área CEDA en la Fig.3-12 representa para un RFP con reciclo R. Esto
varia de una manera compleja desde un RFP sin reciclo ( representado por el
área bajo la curva GE’ ) hasta un CSTR (área rectangular CE’’GA). Debido a
la forma de la curva GE’, el problema sería determinar en que condición se
tiene un valor óptimo de R para el cual es mínimo, y como es este mínimo
comparado con para un RFP sin reciclo.
'E G ''E
Ar
1
D 'F E
H A B C 001 ' AAA CCC
1 R
1+ R
Fig.3-12 interpretación gráfica de la ecuación de diseño para un RFP con
reciclo con una reacción autocatalitica (densidad constante)

H. ZULETA
120
Ejemplo 1 a) Para una reacción autocatalitica en fase líquida A + ... B + ...
que se efectúa en un RFP con reciclo en estado estacionario, derivar una
expresión para el valor óptimo de la razón de reciclo, Ropt, que minimice el
volumen o tiempo espacial del reactor. La ley de velocidad es (-rA) = k CA CB
con CA >> CB0, CB = CA0 - CA
b) Expresar el volumen mínimo o tiempo espacial del reactor en términos de
Ropt.
Ejemplo 2 Una reacción autocatalitica en fase líquida irreversible, A B + ..,
se deberá efectuar en un reactor de flujo continuo. La conversión fraccional de
A es del 90%, el flujo es 0,5 lts/s, y la concentración de alimentación es 1,5 M
La ley de velocidad es (-rA) = kA CA CB, con kA= 0,002 lt mol-1
s-1
. Si no hay B
en la alimentación.
a) ¿Cuál es el volumen de CSTR requerido?
b) ¿Cuál es el volumen de RFP requerido?
c) ¿Cuál es el volumen mínimo requerido del RFP con
reciclo?.

H. ZULETA
121
CAPITULO IV. CASOS ESPECIALES DE DISEÑO DE REACTORES
4.1 COMPORTAMIENTO TRANSIENTE DE UN CSTR
Considerando una reacción de primer orden, en fase líquida e isotérmica
BA k2 r=kCA
La concentración de la alimentación de A, CAf = 2 M, el tiempo de residencia
es τ = 100 min, y la constante cinética k = 0.1 min-1
.
1. Determinar la concentración de A en el efluente en estado estacionario.
2. Graficar la concentración de A versus tiempo para una concentración de
Alimentación CAf = 2 M, si el reactor esta inicialmente lleno con un
Inerte y CA0 = 0 M .
3. Graficar la concentración de A versus tiempo para una concentración de
Alimentación CAf = 2 M, si el reactor esta inicialmente lleno y CA0 = 2 M
SOLUCION
Parte 1: Fase líquida: se asume constante la densidad del fluido.
Sustituyendo la velocidad de reacción -rA=kCA y resolviendo para CAS, la
concentración en estado estacionario.
k
CC
Af
As
1
Sustituyendo los valores numéricos dados:
*)( AAfA rCC

H. ZULETA
122
)/(182.0min)100(*)min1.0(1
)/(2
1ltmol
ltmolCAs
Parte 2 y 3: AAAfA CkCC
dt
dC*)(
1
(ecuación del balance de materiales)
0)0( AA CC
Esta ecuación es de variables separables y la solución analítica es
tkAftk
AA ek
CeCtC )/1()/1(
0 1*1
*)(
0
0.5
1
1.5
2
2.5
0 20 40 60 80 100
t (min)
Ca (
t) (
mo
l/lt
)
Ambas soluciones convergen al mismo estado estacionario aún cuando las
condiciones iniciales sean completamente diferentes.

H. ZULETA
123
4.2 COMPARACION DE UN RFP y UN CSTR
Comparación en estado estacionario de la eficiencia de conversión de
reactantes a productos en los reactores continuos CSTR y RFP
Por simplicidad, se considera densidad constante, reacción irreversible en fase
líquida y de orden n.
BA k
n
AkCr
Para esta situación, el balance de material de un RFP en estado estacionario
esta dado por la ecuación de diseño.
Se reordena y se determina el tiempo espacial requerido, cambiando la
condición inicial CAf para algunas concentraciones de salida CA
,
, )(
1A
C
CA
dCCr
A
Af
El área bajo la curva 1/-r (CA) es el tiempo total requerido para alcanzar la
concentración final.
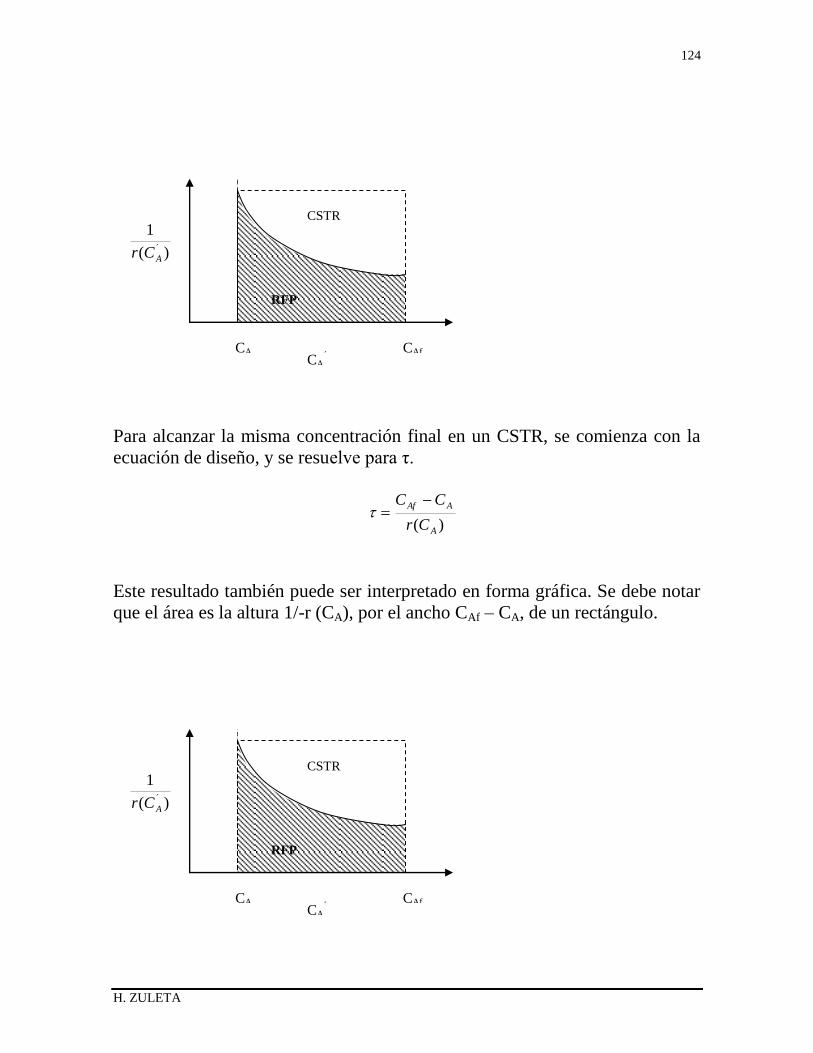
H. ZULETA
124
Para alcanzar la misma concentración final en un CSTR, se comienza con la
ecuación de diseño, y se resuelve para τ.
)( A
AAf
Cr
CC
Este resultado también puede ser interpretado en forma gráfica. Se debe notar
que el área es la altura 1/-r (CA), por el ancho CAf – CA, de un rectángulo.
CSTR
RFP
CA CAf CA
´
)(
1´
ACr
CSTR
RFP
CA CAf CA
´
)(
1´
ACr
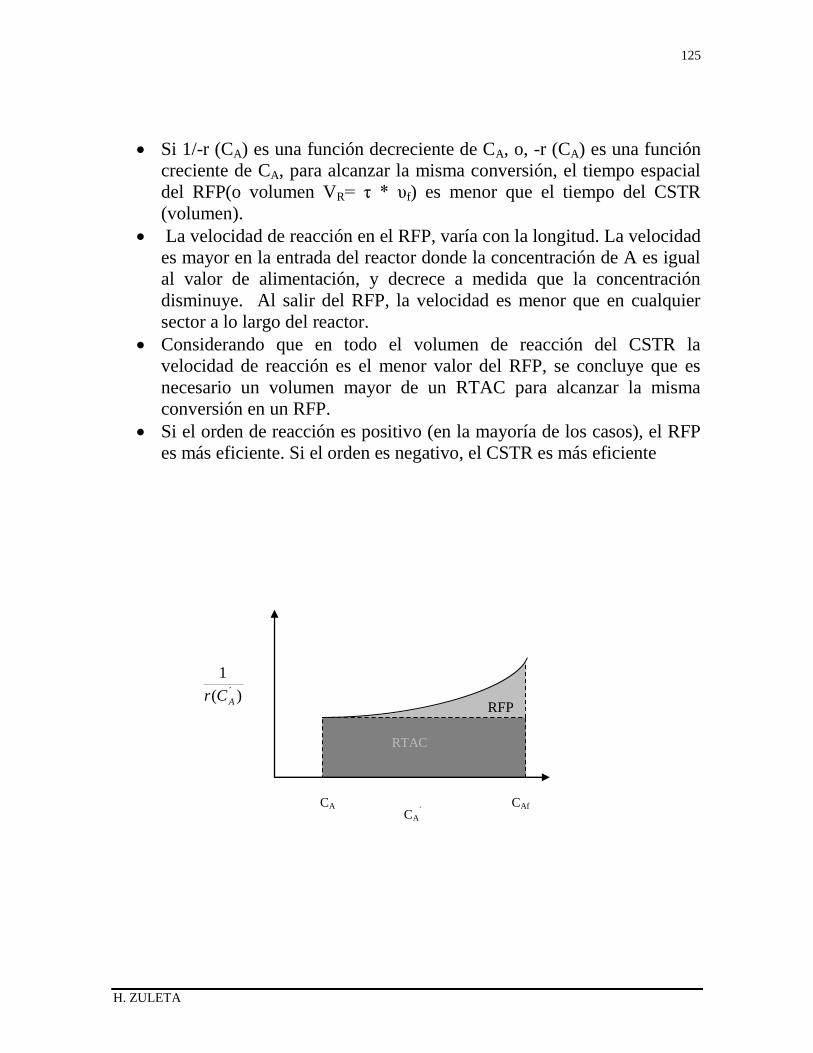
H. ZULETA
125
Si 1/-r (CA) es una función decreciente de CA, o, -r (CA) es una función
creciente de CA, para alcanzar la misma conversión, el tiempo espacial
del RFP(o volumen VR= τ * υf) es menor que el tiempo del CSTR
(volumen).
La velocidad de reacción en el RFP, varía con la longitud. La velocidad
es mayor en la entrada del reactor donde la concentración de A es igual
al valor de alimentación, y decrece a medida que la concentración
disminuye. Al salir del RFP, la velocidad es menor que en cualquier
sector a lo largo del reactor.
Considerando que en todo el volumen de reacción del CSTR la
velocidad de reacción es el menor valor del RFP, se concluye que es
necesario un volumen mayor de un RTAC para alcanzar la misma
conversión en un RFP.
Si el orden de reacción es positivo (en la mayoría de los casos), el RFP
es más eficiente. Si el orden es negativo, el CSTR es más eficiente
RTAC
RFP )(
1´
ACr
CA CAf
CA´

H. ZULETA
126
4.3 CONFIGURACION DE REACTORES
Secuencia de Reactores
A menudo los reactores son conectados en serie permitiendo que la
corriente de salida de un reactor sea la alimentación a otro. Es más
conveniente analíticamente, definir la conversión en términos de material
convertido en un punto aguas abajo en lugar de cualquier otro reactor.
En otras palabras, XA es el numero total de moles de A que reaccionan en un
punto determinado sobre los moles de A alimentados al primer reactor.
Esta definición se aplica solamente cuando no hay corrientes laterales y
la alimentación se introduce solamente en el primer reactor en serie.
Ejemplo: dos CSTR en serie
)1(* 101 AAA xFF
)1(* 202 AAA xFF La alimentación al segundo reactor está
Parcialmente convertida
El Balance de materiales en el segundo reactor esta dado por:
22
12021
*
)(*
Vr
xxFFF
A
AAAAA
FA0
XA1
FA0
XA=0
FA0
XA2
V1 V2

H. ZULETA
127
Por lo tanto 2
120
2
)(*
A
AAA
r
xxFV
O una forma general Af
AiAfA
r
xxFV
)(*0
La ecuación de diseño del RFP puede ser modificada para una conversión
parcial de alimentación:
A
A
X
X
Ar
dxFV
AF
AI
0
RFP en serie
xAi para el i-ésimo reactor en serie se define como:
)1(*0 AiAAi xFF
En este contexto, la ecuación de diseño para todos los reactores en serie
puede ser escrita de la siguiente manera:
FA0
xA=0
FA1 FA,n-1 FAn
xA1 xA,n-1 xAn
A
A
X
Ar
dxFV
A
1
0
01

H. ZULETA
128
A
A
X
X
Ar
dxFV
A
A
2
1
02
.
.
.
.
A
A
X
X
ANr
dxFV
AN
NA
1,
0
Sumando ambos lados
A
A
X
ANr
dxFVVV
AN
0
021 .....
De la misma manera la conversión puede ser descrita para un solo reactor de
volumen V, entonces
A
A
X
Ar
dxFV
AN
0
0
nota: N RFP(de distinto tamaño) en serie es equivalente a un solo RFP,
de un volumen igual a la suma de los volúmenes individuales.
NVVVV .....21

H. ZULETA
129
RFP en paralelo
Donde 1.....21 N
Balance de material en el punto A, esta dado como:
)1()1(.....)1()1( 00202101 AfAANANAAAA xFxFxFxF
Si todas las corrientes salen con la misma composición
Entonces AfANAA xxxx .....21
.
.
.
2FA0
1FA0
NFA0
FA0 xA1
xA2
xAN
xAf
V1
V2
VN
A
A
A
X
Ar
dxFV
Af
0
011
A
A
X
Ar
dxFV
Af
0
022
A
A
X
ANNr
dxFV
Af
0
0
AfANNAA xxxx .....2211

H. ZULETA
130
Sumando,
A
A
X
ANNr
dxFVVV
Af
0
02121 )....(.....
A
A
X
Ar
dxF
AN
0
0
Nuevamente se puede decir que el sistema de reactores RFP es equivalente a
un solo reactor de volumen V= V1+V2+…+VN
En este caso, todos los tiempos espaciales deben ser iguales, lo cual no es el
caso de reactores RFP en serie.
Sin embargo N .....21 ¿Por qué?
NVVV .....21 ¿Por qué?
CSTR en serie – análisis complementario
1110 VrxF AAA
22120 )( VrxxF AAAA
.
.
.
NANNAANA VrxxF )( 1,0
FA0
xA=0
FA1 FA,n-1 FAn
xA1 xA,n-1 xAn
V1 V2 VN

H. ZULETA
131
Sumando,
NANAAANA VrVrVrxF )(.....)()( 22110
Con un solo CSTR es posible obtener la conversión deseada teniendo un
volumen V, como sigue:
VrxF ANANA )(0
N
AN
A
AN
A VVr
rV
r
rV
.....
)(
)(
)(
)(2
21
1
Para n › 0 reacciones, donde -rAi ›-rAN
NVVVV .....21
Recíprocamente, para n ‹ 0 reacciones
NVVVV .....21
CSTR en paralelo
2FA0
1FA0
NFA0
FA0 xA1
xA2
xAN
xAf
A

H. ZULETA
132
1.....21 N
Nuevamente considerar el caso especial de xA1 = xA2 = ….. = xAN = xAf, lo cual
implica que todos los τ son iguales,
101 VrxF AfAfA
202 VrxF AfAfA
.
.
.
NAfAfAN VrxF 0
Sumando,
)......().....( 21021 NAfAfAN VVVrxF
)......( 210 NAfAfA VVVrxF
Usando un solo CSTR de volumen V
NAfAfA VVVVtoloPorVrxF ...tan 210

H. ZULETA
133
CAPITULO V. FLUJO NO IDEAL
5.1 INTRODUCCION
Hasta aquí se han analizados dos modelos de flujos, el flujo pistón y el flujo
en mezcla completa. Su comportamiento es muy diferente (tamaños de
reactores, grado de conversión, distribución del productos). Interesa
considerar estos modelos de flujo y en muchos casos se tratan de diseñar
equipos para aproximarse a ellos. Esto porque
Un sistema o el otro puede ser óptimo para una situación determinada
Estos dos modelos son simples de utilizar matemáticamente.
Sin embargo, un equipo real siempre se desvía del comportamiento ideal.
¿Cómo sucede esto?. Esta pregunta se analizara en el siguiente capitulo.
Hay tres factores relacionados que determinan las características de flujo y
contacto en sistemas continuos.
1. La RTD o distribución del tiempo de residencia del material, que fluye
a través del reactor.
2. El estado de agregación del material que fluye, es la tendencia a
aglomerarse y formar un grupo de moléculas que se mueven juntas.
3. El mezclado temprano (anticipado) o tardío del material en el
reactor.
La RTD describe CUANTO tiempo permanecieron las moléculas dentro del
sistema, el estado de agregación expresa COMO permanecieron las moléculas,
mientras que el mezclado temprano o tardío se refiere al momento en que
deben ponerse en contacto las moléculas.
5.2 DISTRIBUCION DEL TIEMPO DE RESIDENCIA, RTD
La desviación del los dos modelos ideales de flujo puede ser causado por
canalización de fluidos, por recirculación del fluido, o por la formación de una
región estancada en el reactor. En la figura 5-1 se muestran estos
comportamientos. En todos los tipos de equipamiento de procesos, tal como
intercambiadores de calor, columnas empacadas, y reactores, este tipo de flujo
debe ser evitado puesto que siempre disminuyen la eficiencia de la unidad.

H. ZULETA
134
Si se conoce con precisión lo que sucede dentro de un reactor, entonces se
tendría un completo esquema de la distribución de la velocidad del fluido en el
recipiente, por lo que se debería, en principio, ser capaz de predecir el
comportamiento de un recipiente como un reactor. Desafortunadamente, esta
aproximación es impracticable, incluso con los adelantos tecnológicos
actuales.
Estableciendo, por otro lado, eficientemente el conocimiento completo del
flujo, se debe ser menos ambicioso y observar que es lo que realmente se
necesita conocer. En muchos casos no es necesario conocer bastante,
simplemente cuanto tiempo tardan las moléculas en quedarse en el reactor, o
más precisamente, la distribución del tiempo de residencia del fluido. Esta
LECHO
EMPACADO
Canalización,
especialmente
en operaciones de dos
fases.
Corto
circuito
y bypass
cortocircuito
Región estancada
Figura 5-1 Modelos de flujo no ideal que existen en los equipos de procesos.

H. ZULETA
135
información puede determinarse de manera fácil y directa, por medio de un
método de investigación, llamado Estimulo-Respuesta.
Este capítulo trata, en gran parte de la aproximación de la distribución del
tiempo de residencia (o RTD) al flujo no ideal. Se muestra cuando puede ser
legítimamente utilizado, como se usa y cuando no es aplicable que alternativas
considerar. Al desarrollar el “lenguaje” para este capitulo de flujo no ideal
( Danckwerts, 1953), se consideró solamente flujo en estado estacionario, sin
reacción y densidad constante, de un solo fluido a través del reactor.
5.3 ESTADO DE AGREGACION DE LA CORRIENTE
La corriente de materiales se alimenta en un particular estado de agregación,
dependiendo de su naturaleza. Los casos extremos de esos estados pueden
llamarse microfluido y macro fluido, como se indica en la figura 5-2.
Sistema de una fase. Este se encuentra entre los extremos llamados micro
fluido y macro fluido.
Sistema de dos fases. Una corriente de sólidos siempre se comporta como
un macro fluido, pero para un gas reaccionando con un líquido, una de las dos
fases puede ser un macro o un microfluido dependiendo del tipo de contacto
usado. En la figura 5-3, se muestra completamente dicho comportamiento.
Microfluido Macrofluido
Gases y líquidos ordinarios no
muy viscosos
Partículas sólidas
Gotas sin coalescer y líquidos
muy viscosos
Las moléculas individuales se mueven
libremente en la mezcla.
Las moléculas se mantienen
unidas en agregados o paquetes de
composición uniforme.
Figura 5-2 Casos extremos de agregación del fluido

H. ZULETA
136
5.4 RAPIDEZ DEL MEZCLADO
Los elementos del fluido en una corriente pueden mezclarse rápida o
lentamente con cada uno de ellos a través del reactor. Por ejemplo, ver figura
5-4.
Generalmente este factor tiene un efecto menor en el comportamiento de una
corriente. Sin embargo, para un sistema con dos corriente de alimentación
puede ser muy importante, como se aprecia en la figura 5-5.
No hay mezcla
de las distintas
edades del fluido
(elementos jóvenes y viejos)
Igual grado de
mezcla a lo largo
del reactor
Perfil de
Velocidad
plano
Mezcla anticipada Mezcla uniforme Mezcla tardía
Se mezcla distintas
edades de fluido Región bien
mezclada
Figura 5-4 Ejemplos de mezclas anticipadas y tardías de fluidos.
Figura 5-3 Ejemplos de comportamientos de macro y microfluidos.
G
L
G
L
El gas es un macrofluido, mientras
que el líquido es un microfluido
Reactor de burbujeo
El gas es un microfluido, mientras
que el líquido es un macrofluido
L
Gotas de líquido
en el gas
L
G
G
Reactor de ducha

H. ZULETA
137
La función de la RTD, estado de agregación de la materia, y rapidez de
mezclado en la determinación del comportamiento del reactor.
En algunas situaciones uno de estos tres factores puede ser ignorado; en otros
puede llegar a ser crucial. Frecuentemente, muchos dependen del tiempo de
reacción, rxt , el tiempo de mezclado mixt y el tiempo de permanencia en el
reactor stayt . En muchos casos stayt tiene un significado algo parecido al mixt
pero esto es rigurosamente incorrecto.
5.5 E, DISTRIBUCION DE EDADES DEL FLUIDO, RTD
Es evidente que elementos de fluido que toman diferentes caminos a través
del reactor pueden tomar distintos tiempos en pasar a través de el. La
distribución de los tiempos de las corrientes del fluido es llamada distribución
de la edad a la salida E, distribución del tiempo de residencia RTD del fluido.
E tiene unidades de tiempo-1
.
Es conveniente representar la RTD de manera que el área bajo la curva sea la
unidad, o
0
1Edt [ adim ]
Este procedimiento se conoce como normalización de la distribución, ver
figura 5-6. Se puede apreciar una restricción en la curva E -el fluido entra y
sale solamente una vez del recipiente. Esto significa que no debe existir flujo o
difusión de remolinos a la entrada o a la salida del reactor. Esto se conoce
como recipiente de extremo cerrado. Cuando los elementos de fluido pueden
Atravesar el límite del reactor más de una vez, esto se denomina recipiente de
extremo abierto.
A
B
A
B
Buena mezcla a la entrada,
A y B tiene n bastante
tiempo para reaccionar
A y B fluyen en forma separada
De modo que no pueden
reaccionar La mezcla ocurre muy
tarde. A la salida del
reactor A y B no tienen
tiempo para reaccionar
Figura 5-5 Efectos de una mezcla anticipada y tardía en el comportamiento de un
reactor

H. ZULETA
138
Con esta representación la fracción de corriente de salida cuya edad esta
comprendida entre t y dtt es
Edt [ adim ]
La fracción con edad inferior a 1t es
1
0
t
Edt [ adim ] (5.1)
Mientras que la fracción de material con edad superior a 1t , representada por el
área sombreada de la figura anterior.
1
1 01
t
tEdtEdt [ adim ] (5.2 )
La curva E es la distribución necesaria que se debe tener en cuenta para el
flujo no ideal.
Área total =1
t1
Fracción de la corriente
De salida, con edad superior a t1
RTD, o curva E
0
E
Figura 5-6 La distribución de la edad (curva E ) del flujo través del reactor,
También se denomina distribución del tiempo de residencia, o RTD.

H. ZULETA
139
5.6 METODOS EXPERIMENTALES PARA DETERMINAR E
( FISICOS )
La manera más simple y directa de encontrar la curva E usa trazadores físicos
o no reactivos. Para propósitos especiales, no obstante, se puede usar un
trazador reactivo. En este capitulo se consideran los trazadores no reactivos, y
la clase de experimentos en donde se pueden utilizar. La figura 5-7
muestra algunas señales características. Debido a que las señales pulso y
escalón son fáciles de interpretar en comparación con una señal periódica y
una señal al azar, solamente se consideran las dos primeras.
En la próxima sección se discute como encontrar la curva E por medio de los
dos métodos experimentales. Se indica como determinar el comportamiento
del reactor, conociendo la curva E del reactor.
5.6.1 Experimento Pulso
Determinación de la curva E para un reactor de volumen V m3 a través del
cual fluye υ m3/s de un fluido. Instantáneamente se introduce M unidades de
trazador (kg o mol) en la corriente que entra al reactor, registrándose la
concentración del trazador que abandona el reactor. Esta es la curva Cpulso.
A través de un balance de materiales se encuentra.
Área bajo la curva Cpulso :
30 m
skgMtCCdtA
i
ii
(5.3)
Entrada
Pulso
Entrada
escalón
Entrada
periódica
Entrada
Al azar
Figura 5-7 Diferentes maneras de estudiar los modelos de flujo en el reactor.
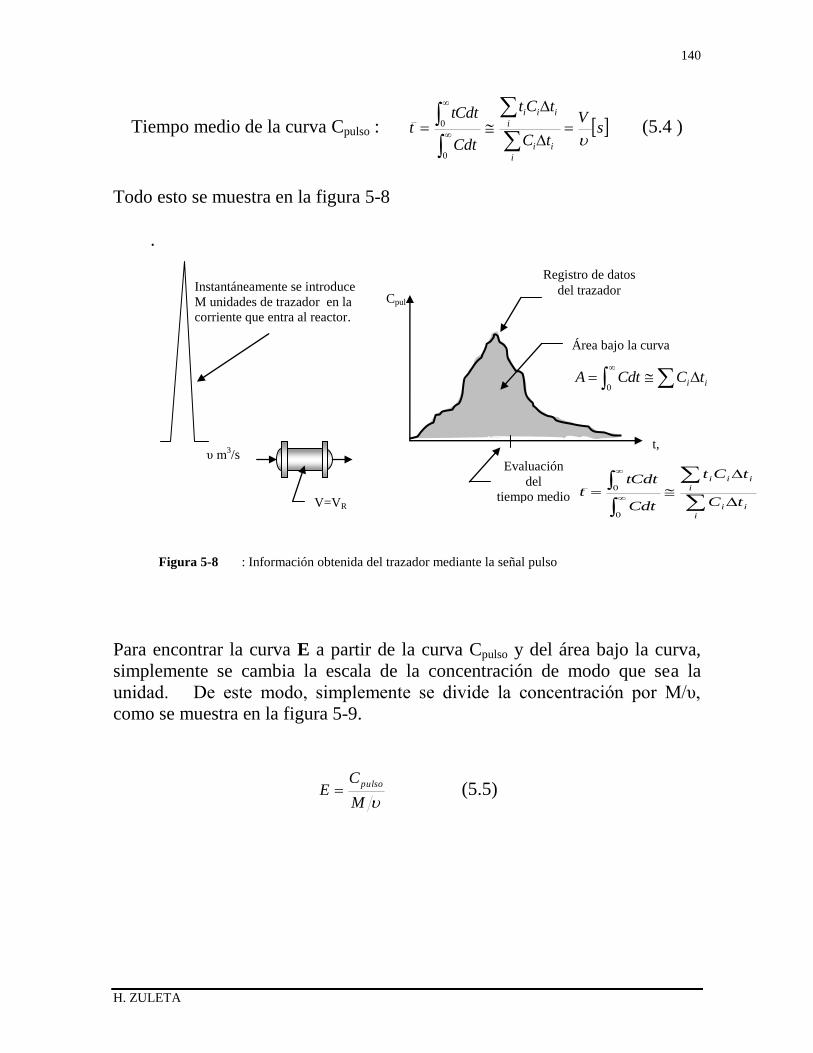
H. ZULETA
140
Tiempo medio de la curva Cpulso : sV
tC
tCt
Cdt
tCdtt
i
ii
i
iii
0
0 (5.4 )
Todo esto se muestra en la figura 5-8
.
Para encontrar la curva E a partir de la curva Cpulso y del área bajo la curva,
simplemente se cambia la escala de la concentración de modo que sea la
unidad. De este modo, simplemente se divide la concentración por M/υ,
como se muestra en la figura 5-9.
M
CE
pulso (5.5)
υ m3/s
V=VR
Registro de datos
del trazador
i
ii
i
iii
tC
tCt
Cdt
tCdtt
0
0
t,
s
Cpul
so
kg/
m3 Área bajo la curva
ii tCCdtA0
Evaluación
del
tiempo medio
Instantáneamente se introduce
M unidades de trazador en la
corriente que entra al reactor.
Figura 5-8 : Información obtenida del trazador mediante la señal pulso
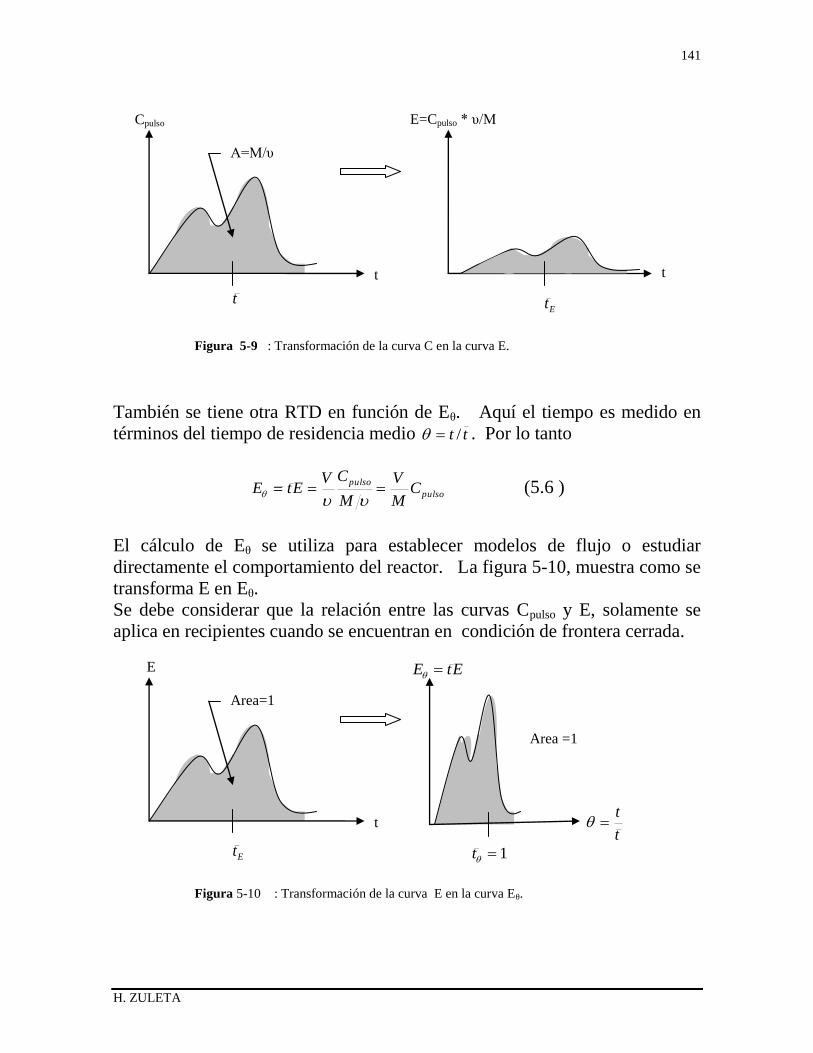
H. ZULETA
141
También se tiene otra RTD en función de Eθ. Aquí el tiempo es medido en
términos del tiempo de residencia medio tt / . Por lo tanto
pulso
pulsoC
M
V
M
CVEtE
(5.6 )
El cálculo de Eθ se utiliza para establecer modelos de flujo o estudiar
directamente el comportamiento del reactor. La figura 5-10, muestra como se
transforma E en Eθ.
Se debe considerar que la relación entre las curvas Cpulso y E, solamente se
aplica en recipientes cuando se encuentran en condición de frontera cerrada.
E
Area=1
Et
t
Figura 5-10 : Transformación de la curva E en la curva Eθ.
EtE
1t
t
t
Area =1
Cpulso
A=M/υ
t
t
E=Cpulso * υ/M
Et
t
Figura 5-9 : Transformación de la curva C en la curva E.

H. ZULETA
142
5.6.2 Experimento Escalón
Se considera υ m3/s de un fluido, el cual fluye a través de un reactor de
volumen V en m3. En un tiempo t=0, se alimenta un fluido ordinario con una
cierta concentración de trazador 3
max mmolokgC , y se mide la
concentración del trazador a la salida Eescalón en función del tiempo t. Esto se
muestra en la figura 5-11.
Un balance de materiales relaciona las diferentes cantidades medidas de la
curva de salida ante una señal de entrada escalón.
max
max
max
0max
0
0
3
2
2max
3max
1 C
escalonC
escalon
C
escalon
tdCCdC
tdCt
m
skgVmtC
figura
sombreada
area
m
skgmC
(5.7)
Donde skgm es la razón de flujo del trazador en el fluido de entrada.
Entrada escalón, t=0
t<0…no hay trazador
t>0… skgm de
trazador
υ
m3/s
V
m3
Señal de
salida
Cmax
Cescalon
0
t
t
Fluido
viejo
Fluido
Nuevo
Señal de
salida
Figura 5-11 Información obtenida experimentalmente a partir de una señal escalón

H. ZULETA
143
La forma adimensional de la curva Cescalon es llamada curva F. En esta
se observa un aumento en la razón de concentraciones F de cero a la unidad,
como se muestra en la figura 5-12
.
5.6.3 Relación Entre las Curvas F y E.
Para relacionar E con F, se considera un flujo constante de color blanco. En
el instante t=0, se introduce un fluido rojo y se registra el aumento de la
concentración del fluido en la corriente de salida, obteniendo la curva F.
Para cualquier instante t > 0 el fluido rojo, y solamente el fluido rojo, de la
corriente de salida tiene una edad inferior a t. En consecuencia, se tiene que:
Pero, el primer término es simplemente el valor de F, mientras que el segundo
viene dado por la ecuación (5.1). Por lo que se tiene, para cualquier instante t,
EdtFt
0 (5.8)
y en forma diferencial
Edt
dF (5.9)
En forma gráfica esta relación se muestra en la figura 5-13 .
escalonCm
F
Figura 5-12 Transformación experimental de la curva Cescalon a una curva F
0
t
t
Cmax
C
es
cal
on 0
t
t
3maxm
kgsCtArea
1
fracción del fluido rojo
en la corriente de salida
= fracción de la corriente de
salida con edad inferior a t
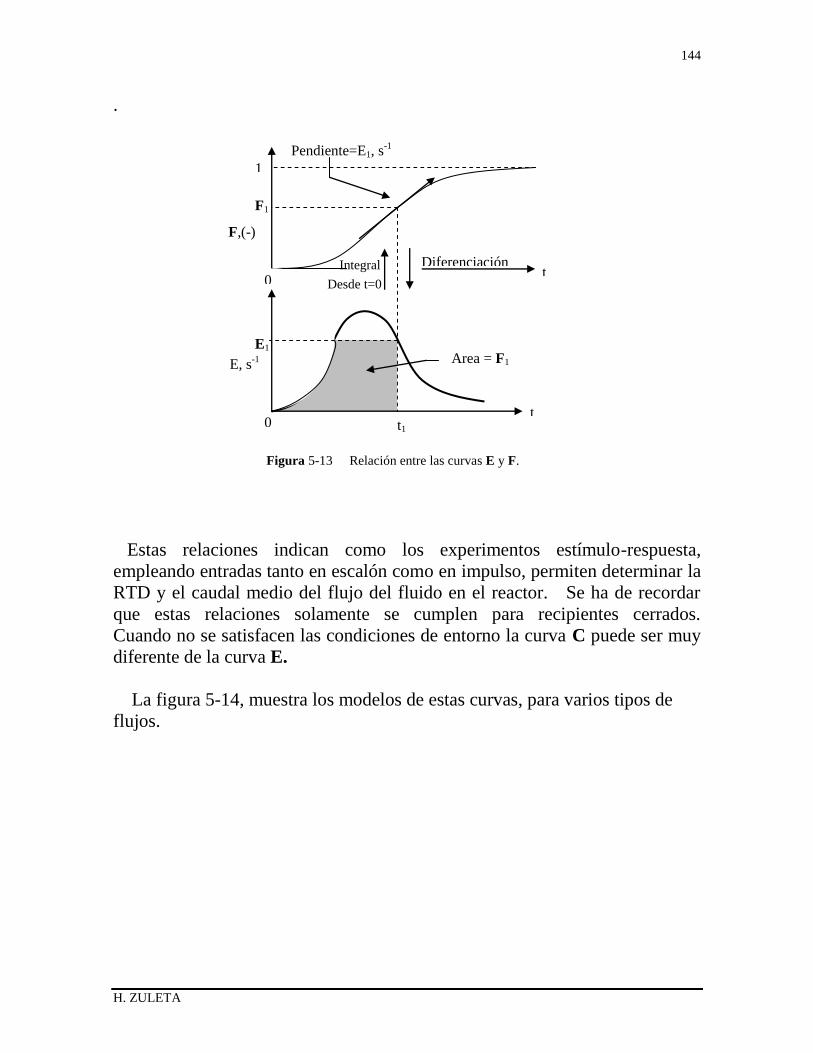
H. ZULETA
144
.
Estas relaciones indican como los experimentos estímulo-respuesta,
empleando entradas tanto en escalón como en impulso, permiten determinar la
RTD y el caudal medio del flujo del fluido en el reactor. Se ha de recordar
que estas relaciones solamente se cumplen para recipientes cerrados.
Cuando no se satisfacen las condiciones de entorno la curva C puede ser muy
diferente de la curva E.
La figura 5-14, muestra los modelos de estas curvas, para varios tipos de
flujos.
Pendiente=E1, s-1
Integral
Desde t=0 0
F,(-)
F1
1
t Diferenciación
E1
E, s-1
t
t1 0
Area = F1
Figura 5-13 Relación entre las curvas E y F.
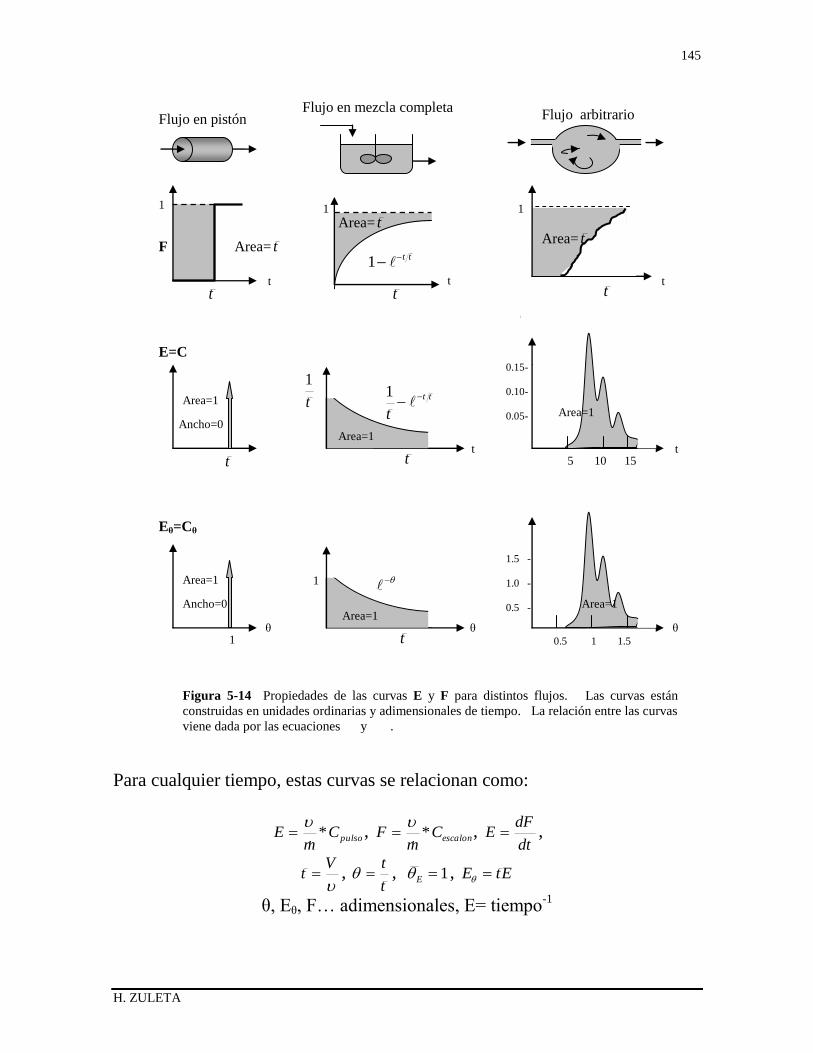
H. ZULETA
145
Para cualquier tiempo, estas curvas se relacionan como:
pulsoCm
E *
, escalonC
mF *
,
dt
dFE ,
Vt ,
t
t , 1E , EtE
θ, Eθ, F… adimensionales, E= tiempo-1
Flujo en pistón Flujo en mezcla completa
Flujo arbitrario
F
E=C
Eθ=Cθ
Area= t
Area= t
Area=1
Area=1
Area=1
Area=1
Area=1
Area=1
tt 1
tt
t
1
1 1 1
t
1
0.15-
0.10-
0.05-
Area= t
5 10 15
t t
t
t t
t t
t
t t
Ancho=0
Ancho=0
t
1
1 θ θ θ
1.5 -
1.0 -
0.5 -
Figura 5-14 Propiedades de las curvas E y F para distintos flujos. Las curvas están
construidas en unidades ordinarias y adimensionales de tiempo. La relación entre las curvas
viene dada por las ecuaciones y .
0.5 1 1.5

H. ZULETA
146
CAPITULO VI. SISTEMAS REACTIVOS GAS – SÓLIDO
6.1 EJEMPLOS DE SISTEMAS
Se pueden considerar dos tipos de sistemas gas-sólido. En el primer tipo, el
sólido se convierte en uno o más sólidos, en el otro, desaparece formando
productos gaseosos.
Ejemplos del primer tipo:
)(222
)(
)(434
)(2232
)(4)(2)(2)(
)()(2)(2)(2
)(2)()(2)(43
)(2)()(2)(
DCaSOOSOCaO
CCCaCNNCaC
BOHFeHOFe
ASOZnOOZnS
sggs
ssgs
gsgs
gsgs
Aunque no todos estos ejemplos cumplen el siguiente modelo de reacción, se
requiere escribirlos en una forma estándar
)1.6()(),()()( gsproductosbBA sg
En donde b es el coeficiente estequiometrico del reactante B. Algunos
ejemplos del segundo tipo, en los cuales los productos son todos gaseosos, y el
sólido se va reduciendo y puede eventualmente desaparecer son los siguientes:
)(
)(
)(2)()(2)(
)(2)(2)(
FHCOOHC
ECOOC
gggs
ggs
La forma estándar para este tipo de reacciones es
)2.6()()()( gproductosbBA sg

H. ZULETA
147
6.2 PARTICULAS DE TAMAÑO CONSTANTE
Consideraciones generales para el modelo cinético
Para desarrollar un modelo cinético (una ley de velocidad) para la reacción
dada por la ecuación (6.1), se debe considerar una sola partícula de la
substancia B, reaccionando con especies gaseosas A (en cantidad ilimitada).
Este es un nivel de tamaño local macroscópico, nivel 2, planteado en el
capitulo I y mostrado en la Fig.1-3. Se asume que el tamaño de partícula
permanece constante durante la reacción. Esto significa que la integridad de la
partícula se mantiene, y requiere que las densidades del reactante sólido B y el
producto sólido (que rodea a B) sean aproximadamente iguales. El tamaño de
partícula es un parámetro y no una variable. El supuesto de tamaño constante,
entre otras cosas, simplifica consideraciones de velocidad de reacción, la cual
puede ser normalizada con respecto a una unidad constante de área superficial
externa o por unidad de volumen de partícula.
La partícula simple actúa como un reactor batch en el cual cambian las
condiciones con respecto al tiempo. Este comportamiento inestable de
partículas reaccionantes difiere del comportamiento de estado estacionario de
partículas de catalizador en catálisis heterogénea. El propósito de este
tratamiento es el desarrollo de una ley de velocidad integrada, en la cual se
exprese la fracción de B convertida, XB, como una función del tiempo, o a la
inversa.
Los modelos de reacción o cinéticos toman en cuenta los diferentes procesos
individuales involucrados en un proceso global. Se puede considerar que la
reacción tiene lugar en alguna parte de la superficie del sólido B al interior de
la partícula, pero para llegar a esta superficie, el reactante A debe desplazarse
desde el seno de la fase gas al interior de la partícula. Esto sugiere la
posibilidad de resistencias en fase gaseosa similar a las que se presentan en
partículas de catalizadores: resistencias externas de transferencia de masa en la
vecindad de la superficie exterior de la partícula, y resistencia de difusión
interior a través de los poros de producto formado y reactante sin convertir. La
situación se ilustra en la figura 6-1 para una partícula isotérmica de radio R en
un instante determinado, para un caso general y dos casos extremos. Estos
últimos casos forman la base de modelos relativamente simples, con sus
correspondientes perfiles de concentración de A y B.

H. ZULETA
148
En la Fig.6-1 se muestra una película de gas que afecta la transferencia de A
en los tres casos. Una importancia adicional del tamaño constante de partícula
es que cualquier efecto de transferencia de masa externa es el mismo en todos
los casos, independiente de la situación al interior de la partícula.
En la Fig.6-1(b), se muestra el caso general en el cual los reactantes y
productos sólidos son relativamente porosos, y los perfiles de concentración
para A y B con respecto a la posición radial (r) cambian continuamente, es
decir, CB, como se muestra a la izquierda del eje central, aumenta, y CA, a la
derecha, disminuye desde la superficie exterior al centro de la partícula. La
concentración de B es el número de moles de B (no reaccionados) por unidad
de volumen de partícula
)4.6()(
)3.6(
puro
V
nC
B
p
BB
B
Donde B es la densidad molar de una partícula de B puro ( mol / m3 )

H. ZULETA
149
En la Fig.6-1(a), se muestra el caso extremo de un sólido B no poroso. En este
caso, el reactante A reacciona inicialmente con la superficie exterior de B, y
se va formando el producto sólido (se supone poroso). A debe difundir a
través de un espesor de producto que se incrementa progresivamente, para
alcanzar la superficie de B que progresivamente retrocede. Existe un límite
claro entre la capa externa de producto poroso y el rectante poroso B no
reaccionado o el corazón o núcleo reducido de reactante B. Los perfiles de
concentración reflejan esto: el valor de CB es cero (capa externa
completamente reaccionada) o B (núcleo sin reaccionar de B puro); CA
decrece continuamente porque aumenta la resistencia difusional a través de la
capa externa, pero es cero en el corazón sin reaccionar. Este caso es la base
para el modelo simplificado llamado modelo de núcleo o corazón reducido
(encogido) (SCM).
En la figura 6-1(c), se observa el caso extremo opuesto de B sólido muy
poroso. En esta situación no existe resistencia interna difusional, todas las
partes del interior de B son igualmente accesibles al reactante A, y la reacción
ocurre uniformemente (pero no instantáneamente) a través de la partícula. Los
perfiles de concentración son planos con respecto a la posición radial, pero CB
decrece con respecto al tiempo, como esta indicado por la flecha. Este modelo
puede ser llamado modelo de reacción uniforme (URM).
6.2.1 Modelo general
Partícula esférica isotérmica. Considerando una partícula isotérmica de
radio R en la fig. 6-1(b) en reacción (a la temperatura del seno del gas) de
acuerdo con la ecuación (6.1). Con el balance de materiales para el reactante
A(g) alrededor de una corteza (cubierta) delgada (volumen de control) de
radio interno r y espesor dr, tomando en cuenta la reacción y la difusión, se
obtiene la ecuación de continuidad para A:
controlde
volumenelenAde
nacumulaciódevel
cortezala
enóndesaparicipor
Adesalidadevel
radifusiónpor
Adesalidadevel
drrendifusiónpor
Adeentradadevel..
..
es decir,

H. ZULETA
150
)4(4)(4)4(4 22222 drrCtt
ndrrr
r
CDrdr
r
CDr
rr
CDr A
AA
Ae
Ae
Ae
En donde se usa la ley de Fick de la difusión, con una difusión efectiva de A a
través de la estructura del sólido De, y (-rA) es la velocidad de desaparición de
A; Esta velocidad se encuentra normalizada con respecto al volumen de
partícula y cada termino tiene unidades de moles de A / s. Si la estructura del
poro es uniforme a través de la partícula, De es constante; de otra manera
depende de la posición radial r. En consecuencia, la ecuación de continuidad
se puede simplificar:
t
Cr
r
C
rr
CD A
AAA
e
)(
22
2
( 6.5)
La ecuación de continuidad para B, escrita para toda la partícula, es
)6.6()(
t
CV
t
nR B
pB
B
o bien )7.6()(t
C
V
Rr B
p
BB
De acuerdo a la estequiometria de la ecuación (6.1), (-rA) y (-rB) están
relacionados por:
)8.6()()(
)8.6()()(
bRbR
o
arbr
AB
AB
En donde (-RA) es la velocidad de reacción extensiva para toda la partícula
correspondiente a (-RB).
Las ecuaciones ( 6.5 ) y ( 6.7 ) son dos ecuaciones diferenciales parciales
acopladas con las siguientes condiciones inicial y de frontera:

H. ZULETA
151
)11.6()(,
)10.6(
)9.6(,0 0
AsAgAg
Rr
Ae
AgA
BBB
CCkr
CDRren
CC
CCten
En la última condición se toma en cuenta la transferencia de masa externa de
película; kAg es el coeficiente de transferencia de masa; la condición de
frontera establece que la velocidad de difusión de A a través de la superficie
exterior de partícula es igual a la velocidad de transporte de A desde el seno
del gas hasta la superficie de sólido por transferencia de masa;
)12.6(0,0
0
r
A
r
Cren
Correspondiente a la transferencia nula de masa en el centro de la partícula,
por consideraciones de simetría.
En general no existe solución analítica para las ecuaciones diferenciales
anteriores, debe emprenderse la solución numérica. Sin embargo, se puede
obtener solución analítica para el caso simplificado del modelo de núcleo
encogido.
6.2.2 Modelo de núcleo encogido
Partícula esférica isotérmica. El modelo de núcleo encogido (SCM) para
una partícula isotérmica esférica se ilustra en la Fig.6-1(a) para un instante
determinado. Se muestra también en la Fig.6-2 para dos tiempos diferentes
para evidenciar el tamaño del corazón y los perfiles de concentración a medida
que avanza la reacción.
En la Fig.6-2(a) o (b) se observa la esencia del modelo SCM para una
partícula parcialmente reaccionada. Existe un límite definido (la superficie de
reacción) entre el corazón no poroso de sólido B sin reaccionar y la carcaza
externa porosa de producto sólido (frecuentemente conocida como capa de
“cenizas”, y también a veces constituye el producto deseado). Por fuera de la
partícula, existe una película gaseosa que implica resistencia a la transferencia
de masa de A desde el seno del gas hasta la superficie exterior de la partícula.
A medida que transcurre el tiempo, la superficie de reacción se mueve
progresivamente hacia el centro de la partícula; esto es, el corazón reducido

H. ZULETA
152
sin reaccionar de B. El SCM es un modelo idealizado ya que el límite entre
zona reaccionada y sin reaccionar puede no ser muy definido, lo cual puede
ser revelado dividiendo la partícula y examinando la sección de corte. El SCM
difiere del modelo general en un aspecto muy importante, como consecuencia
del límite claro y definido entre zonas. De acuerdo con este modelo, los
procesos de transferencia de masa de A, difusión de A, y reacción de A con B
en la superficie del núcleo, son tres procesos en serie. En el caso general, la
transferencia de masa se produce en serie con los otros dos procesos, pero
estos últimos no lo son entre ellos, ocurriendo simultáneamente a través de la
partícula de alguna manera. Esto, junto con supuestos adicionales acerca de
las velocidades relativas de difusión y movimiento de la superficie de
reacción, permite simplificaciones considerables para la solución de la
ecuación (6.5).
En la Fig.6-2, CAg es la concentración de A en el seno del gas que rodea la
partícula, CAs en la superficie exterior de la partícula, y CAc en la superficie del
corazón sin reaccionar de B en el interior de la partícula; R es el radio
(constante) de la partícula y rc es el radio (variable) del núcleo sin reaccionar.
Las concentraciones CAg y CAs son constantes, pero CAc al igual que rc,
decrecen a medida que el tiempo aumenta.

H. ZULETA
153
6.2.3 Deducción del modelo
Se obtendrá la relación que permita calcular el tiempo t requerido para
alcanzar una fracción de B convertido, XB, de acuerdo al SCM. Se considerara
partícula esférica de la especie B con radio R, en reacción con la especie
gaseosa A según la ecuación (6.1). También se asume reacción de superficie
de primer orden.
Para obtener el resultado deseado, )( BXtt , se puede proceder de dos
maneras. En la primera, debido a que los tres procesos de velocidad son en
serie, se pueden tratar cada uno por separado y sumar los resultados para
obtener el tiempo total. En la otra forma, se puede resolver la forma
simplificada de la ecuación (6.5) para los tres procesos y obtener un resultado,
con lo cual se puede demostrar la aditividad de los tres resultados del primer
método.
Se adoptará el segundo método y la base de este análisis se muestra en la
Fig.6-3. La película gaseosa, la capa de producto (cenizas) y el núcleo de B
sin reaccionar son las tres regiones distintas. Se deriva la ecuación de
continuidad para A mediante un balance de materiales a través de una capa
esférica en la zona de producto en una posición radial r y con un espesor dr. El
procedimiento es igual al empleado en la deducción de la ecuación (6.5),
excepto que no se incluye el término (-rA) ya que no existe reacción en esta
capa. El resultado es:

H. ZULETA
154
)13.6(2
2
2
t
C
rr
C
r
CD AAA
e
La ecuación (6.13) representa la difusión de A en la capa de ceniza en estado
no estacionario, es decir, ),( trCC AA . Para simplificar el tratamiento se asume
que la velocidad de movimiento de la interfase de reacción en rc es pequeña
con respecto a la velocidad de difusión de A a través de la capa de producto.
La difusión se considera en estado estacionario para un valor fijo de rc; es
decir, )(rCC AA
La ecuación diferencial parcial (6.13) se transforma en una ecuación
diferencial ordinaria, con 0/ tCA :
)14.6(02
2
2
rd
dC
rrd
Cd AA
El supuesto realizado se conoce como aproximación de estado cuasi
estacionario (QSSA) o simplemente estado seudoestable . Esto es válido
principalmente por la gran diferencia en las densidades de las especies
reaccionantes ( A gas y B sólido). Por lo general, esto se cumple.
La solución de la ecuación (6.13) se obtiene por un procedimiento de dos
etapas:
Etapa 1. Resolver la ecuación (6.14) en la cual las variables son CA y r ( t y rc
están fijos ). Esto resulta en una expresión para el flujo de A, NA, como una
función de rc.
Etapa 2. Usando el resultado de la etapa 1, junto con la ecuación (6.13) y la
ecuación (6.8) se obtiene t = t ( rc ), lo cual se puede transformar al resultado
deseado t = t ( XB ). En esta etapa las variables son t y rc.
En la etapa 1, la solución de la ecuación (6.14) requiere dos condiciones
de frontera, cada una de las cuales puede ser expresada de dos formas; cada
una de estas formas considera los otros dos procesos de velocidad, la ecuación

H. ZULETA
155
de velocidad de difusión de A igual a la velocidad de transporte de A en la
superficie de partícula (ecuación 6.11), y también la velocidad de difusión en
la superficie del núcleo igual a la velocidad de reacción en la superficie,
ecuación (6.16) respectivamente. Por lo tanto,
)17.6(
)16.6(,
)15.6(
)11.6()(,
ACA
ACAs
rr
AeAC
AsA
AsAgAg
Rr
AeA
CCo
Ckr
CDNrren
CCo
CCkr
CDNRren
C
En donde kAS es la constante de velocidad de primer orden de la reacción de
superficie, con la velocidad de reacción dada por:
)18.6()(4
)18.6(4)(
2
2
brr
aCkrR
AsC
ACAsCA
con
sm
molanormalizadr
s
mk
s
molR AsAsA 2
)()(,,)(
La solución de la ecuación (6.14) se puede lograr usando la sustitución
dr
dCy A
Se integra una vez y se obtiene drdCA / , con la segunda integración se obtiene
CA, usando las condiciones de frontera para evaluar las constantes de
integración, y eliminando CAs para obtener CAc.
La primera integración, junto con la ecuación (6.11) resulta en:

H. ZULETA
156
)(1
)(2
2
Ar
CCD
Rk
rd
dCAsAg
e
AgA
La integración de (A), junto con la ecuación (6.17)
)(11
)(
2
Brr
CCD
RkCC
c
AsAg
e
Ag
AcA
Aplicando la ecuación (A) en la superficie del núcleo (r = rc), junto con la
ecuación (6.16), se obtiene una expresión para CAs en términos de CAc:
)(2
2
CCRk
rkCC Ac
Ag
CAs
AgAs
Similarmente, a partir de la ecuación (B), junto con la ecuación (6.15), en la
superficie de la partícula, se puede obtener otra expresión:
)(11
)(
2
DRr
CCD
RkCC
c
AsAg
e
Ag
AcAs
Eliminando CAs de las ecuaciones ( C ) y ( D ) y reemplazando la expresión
resultante para CAc en la ecuación (6.18), se obtiene (previos arreglos):
)19.6(11
4)(
22
cAsce
c
Ag
Ag
A
rkrRD
rR
Rk
CR
Con esto termina la etapa 1, resultando en una expresión para (-RA) en
términos de rc (fijo).
En la etapa 2 de la solución de la ecuación (6.13), se alcanza la superficie del
núcleo, fijada en la etapa 1, para cambiar a B, e integrar su ecuación de
continuidad, se usa la primera parte de la ecuación (6.6). Para este propósito,
se remplazan las ecuaciones (6.19) y (6.6). Esta última se escribe en la forma,

H. ZULETA
157
)20.6(4)3
4()( 23
td
rdrr
dt
d
dt
dnR C
CBCBB
B
De acuerdo a la relación estequiometrica, ecuación (6.8b), se obtiene
)21.6(11
2
2
2
C
As
C
C
eAg
C
Ag
B drkR
rr
DRk
r
Cbdt
De la integración de la ecuación (6.21), de t = 0, rc = R a t, rc, se logra
)22.6(11
2316
13
1323
R
r
kR
r
R
r
D
R
R
r
kCb
Rt C
As
CC
e
C
AgAg
B
Se puede expresar rc de la ecuación anterior, en términos de XB, a partir de una
relación del volumen de una esfera.
)23.6(1
3
0
0
R
r
n
nnX C
B
BB
B
Con lo cual se obtiene
)24.6()1(11
)1(2)1(3163
3/13/2
B
As
BB
eAg
B
Ag
B Xk
XXD
R
k
X
Cb
Rt
Si se define como el tiempo requerido para conversión completa ( XB = 1),
entonces de la ecuación (6.24)
)25.6(1
63
1
AseAgAg
B
kD
R
kCb
R
es un parámetro cinético, propio de la reacción y depende de los parámetros
característicos de los procesos individuales, kAg, De, y kAs, y del tamaño de
partícula R.

H. ZULETA
158
Con las ecuaciones 6.24 y 6.25 se puede analizar casos especiales en donde un
término (es decir, un proceso de velocidad) o bien los otros dos, dominan. Por
ejemplo, si De es pequeño comparado con kAg o kAs, esto significa que la
difusión en la capa de productos es la velocidad o etapa controlante. Entonces,
el valor de t o , se determina completamente con el segundo término de cada
ecuación. Por lo tanto, ya que cada término refiere un solo proceso de
velocidad en cada ecuación, se puede escribir, globalmente, la relación
aditiva:
)26.6(sup
erficielaen
reacciónladecontrolt
cenizasdecapalaen
difusionladecontrolt
masadeciatransferen
laapeliculaladecontroltt
De manera similar se puede plantear para
6.2.4 Parámetros de los procesos de velocidad; estimación de kAg para
partículas esféricas
Los tres parámetros de los procesos de velocidad en la expresión para t ( XB )
(kAg, De, y kAs), requieren mediciones experimentales para situaciones
particulares. Sin embargo, se considera una correlación para estimar kAg de
partículas esféricas dada por Ranz y Marshall (1952).
Para una partícula esférica de radio R en caída libre, con una velocidad u
relativa al fluido de densidad y viscosidad , en la cual el coeficiente
molecular de difusión ( para las especies A) es DA, la correlación de Ranz
Marshall relaciona el número de Sherwood (Sh), el cual incorpora kAg, con el
número de Schmidt (Sc) y el número de Reynolds (Re).
)27.6(Re6.02 2/13/1ScSh
es decir
)28.6(2
6,022
2/13/1
uR
DD
kR
AA
Ag
Esta correlación puede ser usada para determinar kAg, conociendo la
información suficiente de las otras cantidades.

H. ZULETA
159
Para un fluido determinado y su velocidad relativa, se puede escribir la
ecuación (6.28), centrando la dependencia de kAg en R como un parámetro:
)29.6(
2/1
21
R
K
R
KkAg
En donde K1 y K2 son constantes. Existen dos casos límite en la ecuación
(6.29), en los cuales el primer o segundo término predomina (referidos como
los casos de partícula “pequeña” o “grande” respectivamente). Una
consecuencia de esto, y de la correlación en general, se origina de reexaminar
el tiempo de reacción t (XB), como sigue:
De las ecuaciones (6.24) y (6.25) para partículas esféricas, kAg es una
constante, independiente de R. Esto es válido para un valor particular de R.
Sin embargo, si R cambia de un tamaño de partícula a otro como un
parámetro, se puede comparar el efecto de este cambio en el tiempo t ( XB ).
Suponiendo, por simplicidad, que la película de gas es la etapa controlante.
Por lo tanto:
)32.6()(3
)31.6()(3
)30.6(3
2
2/3
1
2
grandepartículaKCb
XR
pequeñaparticulaKCb
XR
kCb
XRt
Ag
BB
Ag
BB
AgAg
BB
A partir de la ecuación (6.29). Por lo tanto, dependiendo de la región de
cambio de la partícula, la dependencia de t en R, la contribución de la película
gaseosa puede ser R2 o R
3/2.

H. ZULETA
160
6.3 PARTICULAS DE TAMAÑO DECRECIENTE
Consideraciones generales
Cuando una partícula sólida de especies B reacciona con especies gaseosas A
para formar sólo productos gaseosos, el sólido puede desaparecer por un
desarrollo interno de porosidad, mientras mantiene su forma macroscópica.
Un ejemplo es la reacción de carbón con vapor de agua para producir carbón
activado; la velocidad intrínseca depende solamente del desarrollo de sitios
para la reacción. Alternativamente, el sólido puede desaparecer solamente de
la superficie mientras la partícula progresivamente se va reduciendo y
reaccionando hasta desaparecer cuando la reacción se completa ( XB = 1 ). Un
ejemplo es la combustión de carbón en aire o bien oxígeno.
Una diferencia importante entre partícula decreciente en reacción para formar
solo producto(s) gaseosos y partícula en reacción con tamaño constante con
formación de capa de producto es que la inexistencia de esta implica que no
existe resistencia para la difusión de A por la capa de cenizas. Por lo tanto sólo
existen dos procesos de velocidad, transferencia de masa en la película de gas,
y reacción química de A y B.
Modelo de partícula de tamaño decreciente
Se puede desarrollar este modelo considerando los dos procesos de velocidad
en etapas en serie, con un procedimiento más simple que el usado para el
SCM, aunque algunos de lo supuestos son los mismos:
(1) La reacción en la partícula es isotérmica
(2) La partícula es no porosa, por lo tanto la reacción ocurre en la
superficie exterior
(3) La reacción de superficie entre el gas A y el sólido B es de primer
orden.

H. ZULETA
161
Ejemplo
Para la reacción representada por )()()( gproductosBbgA , derivar la
relación entre el tiempo (t) de reacción y la fracción de B convertida ( XB ), si
la partícula es esférica con un radio inicial R0, y es válida la correlación de
Ranz-Marshall para kAg(R), donde R es el radio al tiempo t.
Solución:
La velocidad de reacción de A, (-RA), se puede expresar en forma
independiente en términos de la velocidad de transporte de A por transferencia
de masa y la velocidad de reacción de superficie:
)33.6()(4)()( 2
AsAgAgA CCRRkR
)34.6(4)( 2
AsAsA CRkR
La velocidad de reacción de B es (ec.6.20):
)35.6()/(4)( 2 dtdRRR BB
y (-RA) y (-RB) están relacionados por
)()( AB RbR
Se puede eliminar (-RA), (-RB) y CAs (concentración en la superficie) de las
cuatro ecuaciones anteriores para obtener una ecuación diferencial para dR/dt,
la cual por integración genera la relación deseada. La ecuación resultante para
dR/dt es:
)36.6(1
)(
1
/
AsAg
BAg
kRk
Cb
dt
dR

H. ZULETA
162
la cual conduce a
dRkRkCb
t
R
R AsAgAg
B
0
1
)(
1
)37.6(10
1
2/1
21 dRkR
K
R
K
Cbt
R
R AsAg
B
Por simplicidad, se considera resultados para los dos casos límite de la
ecuación (6.29) (ignorando K2/R1/2
o K1/R ) como se describió anteriormente.
Para partículas pequeñas, de la integración de la ecuación (6.37) sin el término
K2/R1/2
se obtiene
)38.6(11
12 0
2
01
00
R
R
kR
R
K
R
Cb
Rt
AsAg
B
Puesto que para una partícula esférica
)39.6(1
3
0
R
RX B
)40.6()1(11
)1(12
3/13/2
1
00
B
As
B
Ag
B Xk
XK
R
Cb
Rt
El tiempo de conversión completa ( XB = 1 ) es
)41.6(1
2 1
00
AsAg
B
kK
R
Cb
R

H. ZULETA
163
Para partículas grandes, el resultado correspondiente (eliminando el término
K1/R en la ecuación (6.37) ) es
)42.6(11
13
2
0
2/3
02
2/1
00
R
R
kR
R
K
R
Cb
Rt
AsAg
B
)43.6()1(11
)1(13
2 3/12/1
2
2/1
00
B
As
B
Ag
B Xk
XK
R
Cb
Rt
)44.6(1
3
2
2
2/1
00
AsAg
B
kK
R
Cb
R
Ecuaciones correspondientes para los dos casos especiales de control (la
película gaseosa y la reacción química), se pueden obtener de estos resultados
(también se pueden derivar en forma individual). La solución para el último
caso (control por reacción) tiene la misma forma que en el SCM, en donde se
reemplaza R0 por R ( y R (variable) reemplaza rc en el desarrollo).
Recommended

















