
Bioinformática estructuralPredicción de estructuras de proteínas y ARN
Dr. Eduardo A. RODRÍGUEZ TELLO
CINVESTAV-Tamaulipas
25 de julio del 2013
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 1 / 134

1 Conceptos básicos de bioinformática estructural
2 Predicción de la estructura secundaria de proteínas
3 Predicción de la estructura terciaria de proteínas
4 Predicción de la estructura secundaria de ARN
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 2 / 134

Conceptos básicos de bioinformática estructural Introducción
Introducción
Las proteínas realizan la mayoría de las funciones biológicas yquímicas esenciales en una célula
Juegan un papel importante en las funciones estructurales,enzimáticas, de transporte y regulación
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 3 / 134

Conceptos básicos de bioinformática estructural Introducción
Introducción
Estructura 3D→ Funcionalidad
La estructura está codificada en lasecuencia de aminoácidos[Anfinsen, 1973]
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 4 / 134
Conceptos básicos de bioinformática estructural Ángulos diedrales
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 5 / 134

Conceptos básicos de bioinformática estructural Ángulos diedrales
Ángulos diedrales
Los átomos asociados al enlace peptídico se encuentran en elmismo plano
Por esta razón el enlace peptídico no puede girar libremente
El ángulo de rotación de un enlace se conoce como diedral o detorsión
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 6 / 134
Conceptos básicos de bioinformática estructural Ángulos diedrales
Ángulos diedrales
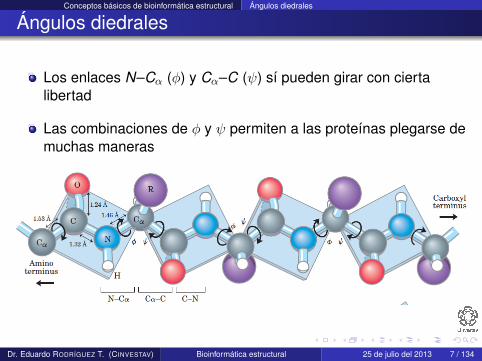
Los enlaces N–Cα (φ) y Cα–C (ψ) sí pueden girar con ciertalibertad
Las combinaciones de φ y ψ permiten a las proteínas plegarse demuchas maneras
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 7 / 134
Conceptos básicos de bioinformática estructural Ángulos diedrales
Gráfica de Ramachandran
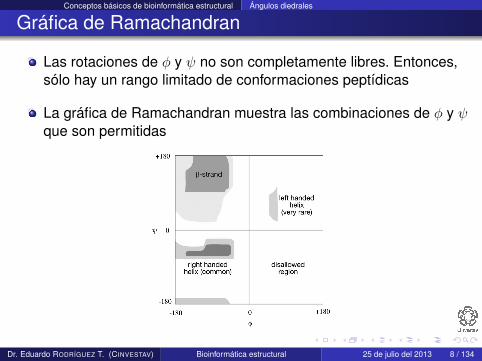
Las rotaciones de φ y ψ no son completamente libres. Entonces,sólo hay un rango limitado de conformaciones peptídicas
La gráfica de Ramachandran muestra las combinaciones de φ y ψque son permitidas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 8 / 134

Conceptos básicos de bioinformática estructural Jerarquía
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 9 / 134

Conceptos básicos de bioinformática estructural Jerarquía
Estructura de las proteínas
La estructura primaria es la secuencia de aminoácidos unidos porenlaces peptídicos
El polipéptido resultante se puede plegar en unidades deestructura secundaria como las hélices alfa
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 10 / 134
Conceptos básicos de bioinformática estructural Jerarquía
Estructura de las proteínas
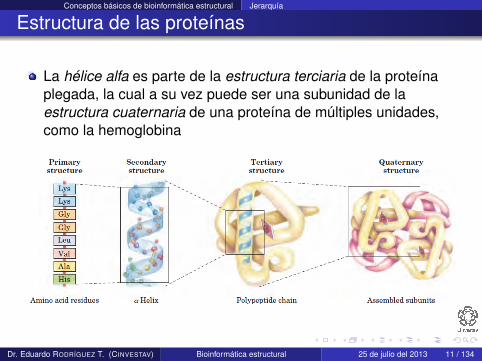
La hélice alfa es parte de la estructura terciaria de la proteínaplegada, la cual a su vez puede ser una subunidad de laestructura cuaternaria de una proteína de múltiples unidades,como la hemoglobina
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 11 / 134

Conceptos básicos de bioinformática estructural Jerarquía
Fuerzas de estabilización
La estructura de las proteínas se mantiene por fuerzas deestabilización como las interacciones electrostáticas, las fuerzasde Van der Waals y los enlaces de hidrógeno
Las interacciones electrostáticas ocurren cuando el exceso decarga negativa en una región es neutralizado por cargas positivasen otra región formando puentes salinos entre residuos de cargaopuesta
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 12 / 134

Conceptos básicos de bioinformática estructural Jerarquía
Fuerzas de estabilización
Los enlaces de hidrógeno son un tipo de interaccioneselectrostáticas que involucran a un átomo de hidrógeno de unresiduo y a un átomo de oxígeno de otro residuo
El hidrógeno con carga positiva se une parcialmente al oxígenocon carga negativa
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 13 / 134

Conceptos básicos de bioinformática estructural Jerarquía
Fuerzas de estabilización
Las fuerzas de Van der Waals son las fuerzas de atracción orepulsión entre moléculas o entre partes de una misma molécula
Los electrones de un átomo crean un dipolo eléctrico que atrae aotro dipolo de un átomo cercano
Pero cuando están muy cerca los átomos, se comienzan a repeler
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 14 / 134

Conceptos básicos de bioinformática estructural Jerarquía
Fuerzas de estabilización
El radio de Van der Waals es la distancia a la que un átomopuede estar cerca de otro
Los puentes disulfuro también intervienen en la estabilización dela estructura de una proteína
Estos puentes se forman entre los átomos de azufre de la cisteína
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 15 / 134
Conceptos básicos de bioinformática estructural Estructura secundaria
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 16 / 134

Conceptos básicos de bioinformática estructural Estructura secundaria
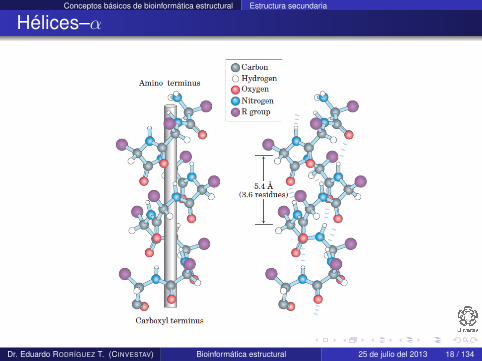
Hélices–α
Una hélice–α tiene una conformación de la cadena principalparecida a un sacacorchos
En esta hélice existen 3.6 residuos en cada giro
La estructura se estabiliza mediante enlaces de hidrógeno entreátomos de la cadena principal i e i + 4, que son casi paralelos aleje de la hélice
φ y ψ son de 60o y 45o
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 17 / 134
Conceptos básicos de bioinformática estructural Estructura secundaria
Hélices–α
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 18 / 134
Conceptos básicos de bioinformática estructural Estructura secundaria
Hojas–β
Una hoja–β es una configuración completamente extendida quese construye de varias regiones espacialmente adyacentes de unpolipéptido
Cada región que la forma se conoce como hebra–β
Esta estructura se estabiliza por medio de enlaces de hidrógenoque se forman entre residuos de hebras adyacentes
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 19 / 134
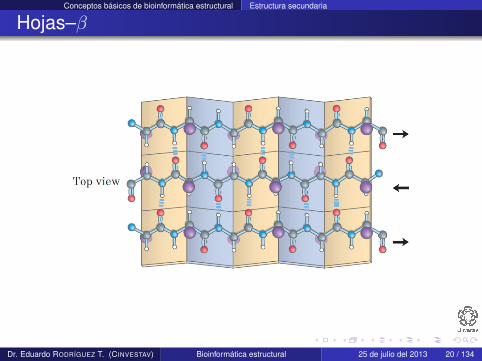
Conceptos básicos de bioinformática estructural Estructura secundaria
Hojas–β
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 20 / 134

Conceptos básicos de bioinformática estructural Estructura secundaria
Espirales y rizos
También hay estructuras locales que no pertenecen a estructurassecundarias regulares
Estas estructuras son las espirales y los rizos
Los rizos se caracterizan por ser giros bruscos
Las espirales se forman por regiones de conexión completamenteirregulares
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 21 / 134
Conceptos básicos de bioinformática estructural Estructura terciaria
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 22 / 134

Conceptos básicos de bioinformática estructural Estructura terciaria
Tipos de proteínas
El arreglo y empaque completo de estructuras secundarias formala estructura terciaria de la proteína
La estructura terciaria generalmente se clasifica en proteínasglobulares o de membrana
Las globulares existen en solventes a través de interaccioneshidrofílicas con moléculas solventes
Las de membrana existen en lípidos de membrana y seestabilizan por medio de interacciones hidrofóbicas con lasmoléculas de lípidos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 23 / 134

Conceptos básicos de bioinformática estructural Estructura terciaria
Proteínas globulares
Son solubles y están rodeadas por moléculas de agua
Tienen estructuras compactas de forma esférica con residuoshidrofílicos en la superficie e hidrofóbicos en el núcleo
Minimiza el contacto con el agua en el centro y maximiza lasinteracciones con agua en el exterior
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 24 / 134

Conceptos básicos de bioinformática estructural Estructura terciaria
Proteínas globulares
Algunos ejemplos: enzimas, mioglobinas y hormonas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 25 / 134

Conceptos básicos de bioinformática estructural Estructura terciaria
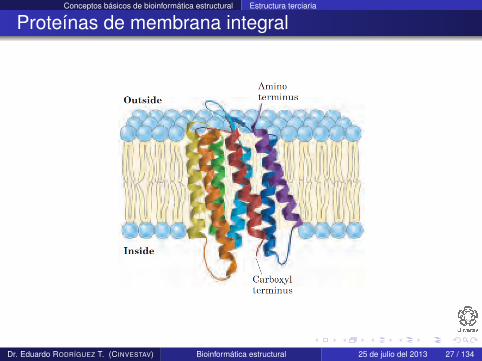
Proteínas de membrana integral
Existen en las bicapas de lípidos de la membrana de la célula
Como están rodeadas de lípidos, el exterior debe ser hidrofóbicopara ser estable
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 26 / 134
Conceptos básicos de bioinformática estructural Estructura terciaria
Proteínas de membrana integral
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 27 / 134
Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 28 / 134

Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
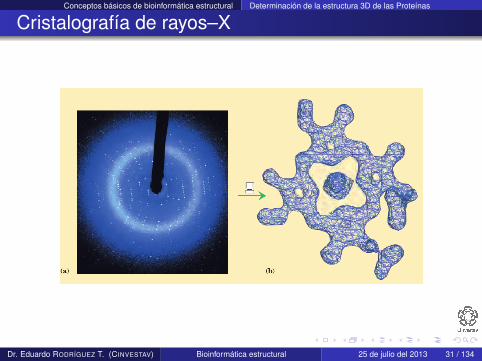
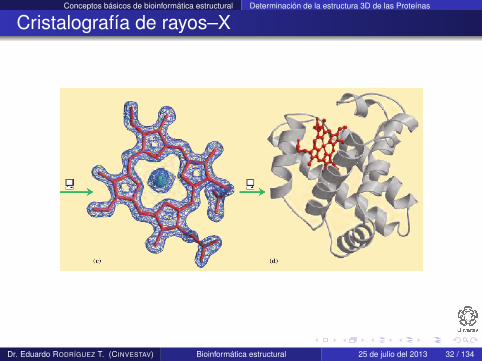
Cristalografía de rayos–X
Requiere que las proteínas formen cristales con posiciones fijasde una manera repetida y ordenada
Los cristales se iluminan con un haz intenso de rayos–X
Los electrones que rodean a los átomos desvían los rayos–Xproduciendo un patrón regular de difracción
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 29 / 134

Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
Cristalografía de rayos–X
El patrón está compuesto de miles de puntos grabados en unaplaca de rayos–X
El patrón se convierte a un mapa de densidad de electrones
La estructura se modela con los aminoácidos que mejor seajustan al mapa
Una limitante que existe es la necesidad de obtener cristales apartir de las proteínas, lo que no siempre es posible
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 30 / 134
Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
Cristalografía de rayos–X
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 31 / 134
Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
Cristalografía de rayos–X
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 32 / 134

Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
Espectroscopia NMR
La espectroscopia de resonancia magnética nuclear (NMR)detecta patrones de giro de núcleos atómicos en un campomagnético
Utiliza radiación para inducir transiciones entre estados de giro delos núcleos en un campo magnético
Las interacciones entre pares de isótopos producen señales deradio que están correlacionadas con la distancia entre ellos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 33 / 134

Conceptos básicos de bioinformática estructural Determinación de la estructura 3D de las Proteínas
Espectroscopia NMR
Interpretando estas señales se puede determinar la proximidadentre átomos y con esto se puede construir un modelo para laproteína
No tiene la limitación de generar cristales, pero solamente puededeterminar estructuras con menos de 200 residuos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 34 / 134
Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 35 / 134

Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
Banco de datos de proteínas
Las estructuras de proteínas que se obtienen por cristalografía yespectroscopia, entre otros métodos, se almacenan en el Bancode Datos de Proteínas (PDB)
Las estructuras definen la posición, en un espacio tridimensional,de cada átomo de la proteína
El sitio en Internet de PDB permite subir, buscar y bajar datos deproteínas
Aunque PDB tiene miles de estructuras almacenadas, lainformación es redundante, existen muchas entradas para unamisma proteína, ya que se reportan con diferentes resoluciones,con mutaciones en un residuo, etc
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 36 / 134

Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
Formato PDB
Cada descripción de una proteína tiene un código de 4 símbolosalfanuméricos
Las líneas tienen 80 caracteres de longitud
Consta de un encabezado y una sección de coordenadasatómicas
El encabezado puede incluir información de método deldeterminación, resolución, parámetros de cristalografía,referencias bibliográficas, etc
Las coordenadas incluyen el nombre del átomo, del residuo,número del residuo, coordenadas en x , y , z, factor detemperatura, entre otros datos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 37 / 134

Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
Formato PDB
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 38 / 134

Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
Formato mmCIF y MMDB
Las limitaciones del formato PDB han permitido el desarrollo denuevos formatos como mmCIF y MMDB que son más fáciles deanalizar por una computadora y permiten describir estructurasmás complejas
Cada línea describe un campo de la descripción de la estructura,primero se escribe el nombre del campo y luego el valor
Un archivo MMDB utiliza el formato ASN.1 para describir unaestructura
Incluye información de enlaces para cada molécula, llamadagráfica química, permitiendo que las estructuras se dibujen másrápido
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 39 / 134

Conceptos básicos de bioinformática estructural BD de estructuras de proteínas
Formato mmCIF y MMDB
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 40 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
1 Conceptos básicos de bioinformática estructuralIntroducciónÁngulos diedralesJerarquíaEstructura secundariaEstructura terciariaDeterminación de la estructura 3D de las ProteínasBD de estructuras de proteínasVisualización de estructuras proteínicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 41 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Visualización de estructuras proteínicas
La característica más básica de un software de visualización es lacapacidad de crear conectividad entre átomos para simular lavista de una molécula
El programa de visualización puede ofrecer diferentespresentaciones de visualización:
1 Tramas de alambres (wire-frame)2 Esferas y líneas (balls and sticks)3 Esferas (space-filling o CPK - Corey, Pauling, and Koltan)4 Listones (ribbons)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 42 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Tramas de alambres (wire-frame)
Es un diagrama de líneas que representa los enlaces entreátomos (representación más simple)
Es útil para localizar residuos específicos en una estructura deproteína
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 43 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Esferas y líneas (balls and sticks)
Representan átomos y sus enlaces respectivamente
Pueden representar la columna vertebral de una estructura
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 44 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Esferas (space-filling o CPK
Cada átomo se describe usando una esfera grande cuyo radiocorresponde a su radio de van der Waals
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 45 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Listones (ribbons)
Usa listones en forma de espiral para representar las hélices-α yflechas planas para representar las hebras-β
Permiten identificar fácilmente las estructuras secundarias
Ofrece una vista general de toda la topología de la estructura
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 46 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Software
RasMol. Lee formatos PDB y mmCIF. Puede desplegar unamolécula completa o partes específicas de ella. Es un programade línea de comandos y se encuentra disponible en plataformasUNIX, Windows y Mac.
RasTop. Es una nueva versión de RasMol disponible enplataforma Windows. Posee una mejor interface de usuario.
Swiss-PDBViewer. Es un visor de estructuras disponible paraMac y Windows. Posee mucha funcionalidad para ser unshareware. Capaz de visualizar múltiples estructuras, analizar ymodelar. Puede medir distancias, potencial electrostático, ploteode Ramachandran, etc.
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 47 / 134

Conceptos básicos de bioinformática estructural Visualización de estructuras proteínicas
Visualización de estructuras proteínicas
Molscript. Despliega estructuras tridimensionalmente y ofrecevarios formatos de salida. Disponible en plataformas UNIX.Ofrecer diferentes presentaciones de visualización. Sin embargo,es una aplicación de línea de comandos.
JMol. Es un applet para visualuzar estructuras químicas queemplea representación de esferas.
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 48 / 134
Predicción de la estructura secundaria de proteínas Introducción
2 Predicción de la estructura secundaria de proteínasIntroducciónMétodos ab initioMétodos basados en homologíaPredicción con redes neuronales
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 49 / 134

Predicción de la estructura secundaria de proteínas Introducción
Introducción
Las estructuras secundarias son conformaciones locales establesde una cadena polipeptídica
Son esenciales en la determinación de la estructuratridimensional de proteínas
Incluyen elementos estructurales regulares y altamente repetidoscomo las hélices-α y las hojas-β
Se estima que cerca del 50 % de los residuos de una proteína sepliegan en alguna de esas dos formas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 50 / 134

Predicción de la estructura secundaria de proteínas Introducción
Introducción
La predicción de estructuras secundarias de proteínas se refiere ala identificación del estado de conformación de cada residuo delos aminoácidos en la secuencia de una proteína
Dichos estados de conformación pueden ser de tres tipos: Hélices(H), Hebras (E) o Rizos (C).
La predicción está basada en el hecho de que las estructurassecundarias tienen un arreglo regular de los aminoácidos,estabilizado por los enlaces de hidrógeno
Esta regularidad sirve de base a los algoritmos de predicción
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 51 / 134

Predicción de la estructura secundaria de proteínas Introducción
Introducción
La predicción de estructuras secundarias de proteínas tieneaplicación en la clasificación de proteínas y en la separación dedominios de proteínas y de motivos funcionales
Además es un paso intermedio para determinar la estructuraterciaria de proteínas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 52 / 134

Predicción de la estructura secundaria de proteínas Introducción
Introducción
Los métodos para la predicción de la estructura secundaria deproteínas (globulares) son de dos tipos:
Basados en ab initio. Predicen la estructura secundaria empleandoinformación estadística calculada a partir de una sola secuencia
Basados en homología. No sólo toman en cuenta estadísticas delos residuos de una secuencia, además también consideranpatrones comunes conservados entre múltiples secuenciashomólogas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 53 / 134
Predicción de la estructura secundaria de proteínas Métodos ab initio
2 Predicción de la estructura secundaria de proteínasIntroducciónMétodos ab initioMétodos basados en homologíaPredicción con redes neuronales
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 54 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Métodos ab initio
Este tipo de métodos mide la tendencia relativa de cadaaminoácido de pertenecer a cierto tipo de elemento de estructurasecundaria
Las puntuaciones de propensión fueron derivadas de estructurasconocidas de cristales
Algunos ejemplos: Chou-Fasman y Ganier, Osguthorpe y Robson(GOR)
Estos pertenecen a la primera generación de métodos depredicción (1970s)
La información estructural de proteínas era limitada y lasestadísticas eran derivadas de conjuntos de datos restringidos(baja exactitud)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 55 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método Chou-Fasman
Determina la tendencia de cada residuo a encontrarse en unahélice, una hebra o un giro usando frecuencias observadas encristales de proteínas
El cálculo de la puntuación de propensión es simple.
Supongamos que hay n residuos en la estructura de la proteínade los cuales m son residuos en hélices
El número total de residuos de Alanina es y de los cuales x estánen hélices
La puntuación de propensión para la Alanina de estar en unahélice está dada por la siguiente relación:
(x/m)
(y/n)(1)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 56 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método Chou-Fasman
Si la puntuación de propensión para un residuo es igual a 1.0para hélices (P(hélice-α)) significa que el residuo tiene igualprobabilidad de ser encontrado en una hélice o en cualquier otraestructura
Si P(hélice-α) < 1,0 entonces el residuo tiene poca oportunidadde ser encontrado en una hélice
Si P(hélice-α) > 1,0 entonces es altamente probable que elresiduo se encuentre en una hélice
Usando este concepto Chou y Fasman crearon la siguiente tabla
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 57 / 134
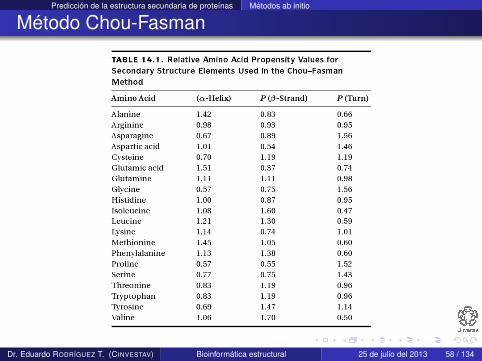
Predicción de la estructura secundaria de proteínas Métodos ab initio
Método Chou-Fasman
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 58 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método Chou-Fasman
El algoritmo Chou-Fasman toma la secuencia y la divide enventanas de tamaño fijo para determinar el número de residuospertenecientes a cada estructura usando la puntuación depropensión
Para hélices-α la ventana es de tamaño 6, si una región tiene 4residuos contiguos cada uno con P(hélice-α) > 1,0, se concluyeque el conjunto forma parte de una hélice
Esta región en hélice se extiende en ambas direcciones hasta queP(hélice-α) < 1,0
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 59 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método Chou-Fasman
Para las hebras-β utiliza una ventana de 5 residuos, si se tienenal menos 3 residuos cada uno con P(hebra-β) > 1,0, se concluyeque el conjunto forma parte de una hebra-β
Si ambos tipos de estructuras se traslapan en cierta región, setoma la siguiente decisión
Si∑
P(hélice-α) >∑
P(hebra-β) entonces se concluye unahélice-α
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 60 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método GOR
También se basa en la puntuación de propensión de cada residuode estar en cada una de las 4 siguientes estructuras: Hélices (H),Hebras (E), Giros (T) o Rizos (C).
Sin embargo, toma en cuenta para este cálculo las interaccionescon los residuos vecinos
Examina una ventana de 17 residuos y suma la propensión paralos residuos para las 4 posibles estructuras (4 sumatorias)
La puntuación más alta define el tipo de estructura al quepertenece el residuo al centro de la ventana (noveno residuo)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 61 / 134

Predicción de la estructura secundaria de proteínas Métodos ab initio
Método GOR
Tanto este método como el de Chou-Fasman tienen la desventajade tener baja precisión de predicción (aprox. 50 %)
Sin embargo, han surgido algunas nuevas versiones como GORII, GOR III y GOR IV (1980s e inicio de 1990s)
Integran estadísticas más refinadas basadas en un número másgrande de proteínas conocidas e incorporan más interaccioneslocales entre residuos
Su precisión de predicción mejoró 10 %
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 62 / 134
Predicción de la estructura secundaria de proteínas Métodos basados en homología
2 Predicción de la estructura secundaria de proteínasIntroducciónMétodos ab initioMétodos basados en homologíaPredicción con redes neuronales
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 63 / 134

Predicción de la estructura secundaria de proteínas Métodos basados en homología
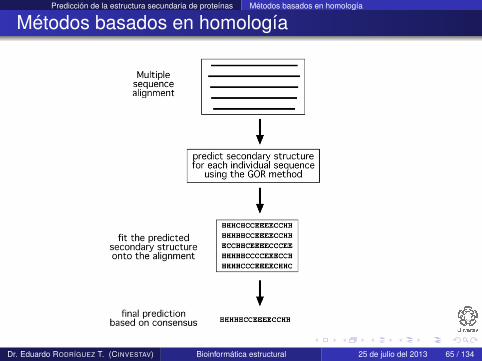
Métodos basados en homología
Son la tercera generación de métodos (finales de 1990s) yemplean información evolutiva
Combinan métodos ab initio para predicción de la estructurasecundaria de secuencias individuales e información dealineamiento múltiple de secuencias homologas (identidad> 35%)
La idea detrás de este enfoque es que proteínas homologasadoptan la misma estructura secundaria y terciaria
Este tipo de métodos han ayudado a mejorar la precisión depredicción en 10 % con respecto a los métodos de segundageneración
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 64 / 134
Predicción de la estructura secundaria de proteínas Métodos basados en homología
Métodos basados en homología
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 65 / 134

Predicción de la estructura secundaria de proteínas Predicción con redes neuronales
2 Predicción de la estructura secundaria de proteínasIntroducciónMétodos ab initioMétodos basados en homologíaPredicción con redes neuronales
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 66 / 134

Predicción de la estructura secundaria de proteínas Predicción con redes neuronales
Predicción con redes neuronales
La tercera generación de algoritmos de predicción también haceuso de redes neuronales para analizar patrones de substituciónen alineamientos de múltiples secuencias
Esto ha permitido aumentar la precisión de predicción a un 75 %
Algunos ejemplos de aplicaciones que utilizan redes neuronales:PHD, PSIPRED, SSpro, PROF, HMMMSTR
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 67 / 134
Predicción de la estructura terciaria de proteínas Introducción
3 Predicción de la estructura terciaria de proteínasIntroducciónMétodos basados en homologíaMétodos basados en plegado (threading)Modelos Ab InitioModelo HP (Hydrophobic-Polar)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 68 / 134

Predicción de la estructura terciaria de proteínas Introducción
Introducción
Existen tres enfoques computacionales para el modelado ypredicción de estructuras tridimensionales de proteínas
Homología
Plegado (Threading)
Ab initio
Los dos primeros se basan en el conocimiento estructural de laproteína obtenido de las BD, mientras que el tercero no requierede ninguna información adicional
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 69 / 134
Predicción de la estructura terciaria de proteínas Métodos basados en homología
3 Predicción de la estructura terciaria de proteínasIntroducciónMétodos basados en homologíaMétodos basados en plegado (threading)Modelos Ab InitioModelo HP (Hydrophobic-Polar)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 70 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
Métodos basados en homología
Como su nombre lo indica, predice las estructuras de lasproteínas mediante la comparación con estructuras de proteínashomólogas conocidas
También es llamado Modelo Comparativo
Se basa en el principio de que si dos proteínas tienen un altogrado de similitud es muy probable que tengan estructurastridimensionales similares
El modelo de homología general consta de 6 pasos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 71 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
1. Selección de plantilla
Consiste en encontrar las estructuras principales y sirve comobase para el proceso de modelado
Este paso consiste en la búsqueda en el Banco de Datos deProteínas (PDB) para seleccionar aquellas proteínas homólogas
Esta búsqueda se pude llevar a cabo mediante cualquier métodode alineamiento de pares tales como BLAST o FASTA.
Por lo general, es posible encontrar varias estructuras con unporcentaje de similitud considerable, sin embargo se recomiendausar sólo aquella con el porcentaje más alto
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 72 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
2. Alineamiento de secuencias
Una vez identificada la secuencia con mayor similitud, se lleva acabo un reajuste, para ello se usa un algoritmo de alineamientopara obtener una adaptación óptima entre las secuencias
Se considera como el paso más critico, ya que un alineamientoincorrecto conducirá a una designación incorrecta de los residuos
Los algoritmos usados en este paso pueden ser T-Coffe o Praline
De ser necesario se puede llevar a cabo un perfeccionamientomanual del resultado arrojado por el algoritmo
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 73 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
3. Creación del esqueleto del modelo
Una vez teniendo el alineamiento óptimo, existen tresposibilidades para los residuos en las regiones alienadas:
1 Residuos similares. Las coordenadas de los residuos de la plantillapueden ser copiadas directamente a la proteína objetivo (query)
2 Residuos idénticos. Las coordenadas de los átomos de la cadenalateral se copian junto con los átomos de la cadena principal
3 Residuos diferentes. Sólo los átomos de la columna vertebral sepueden copiar
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 74 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
4. Modelado de bucles
Como sabemos, el resultado de un alineamiento de secuenciascausa la inserción de huecos, los cuales son el resultado por elalineamiento mismo
Estos huecos no pueden ser directamente modelados, por lo quese requiere de un modelo para “cerrar” estos huecos
Existen dos técnicas para abordar este problemaMétodo de búsqueda en BDMétodo ab initio
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 75 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
4. Modelado de bucles
El método de búsqueda en BD propone buscar “piezas derepuesto”, de estructuras conocidas de proteínas que se acoplenen el hueco
La secuencias de átomos que preceden y continúan a esta regiónse suelen llamar tallo.
El procedimiento inicia midiendo la orientación y distancia de lasregiones entre los tallos y buscar en PDB los segmentos de lamisma longitud que coincidan
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 76 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
4. Modelado de bucles
Suelen existir diferentes segmentos alternativos que se adapten aesta región
El mejor fragmento se copia en los puntos de anclaje de los tallos
El método ab initio genera muchos bucles y búsquedas al azar
Si los huecos son relativamente cortos (de 3 a 5 residuos) los dosmétodos producen modelos correctos
Si los huecos son muy largos, es muy difícil lograr un modelofiable
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 77 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
4. Modelado de bucles
FREADwww.cryst.bioc.cam.ac.uk/cgi-bin/coda/fread.cgi,usa el método de BD
PETRAwww.cryst.bioc.cam.ac.uk/cgi-bin/coda/pet.cgiemplea el método ab initio
CODA www.cryst.bioc.cam.ac.uk/~charlotte/Coda/search_coda.html utiliza consenso basado en los resultadosde los dos sitios anteriores
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 78 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
5. Perfeccionamiento de la cadena lateral
Una vez que la cadena principal de átomos está construida, lasposiciones de las cadenas laterales deben ser determinadas
La cadena lateral puede ser construida mediante la búsqueda decada ángulo de torsión, seleccionando aquellos que tengan lamenor interacción de energía con sus vecinos
Sin embargo, esto no se puede llevar a cabo en la mayoría de loscasos (computacionalmente prohibitivo)
Para ello ha surgido el concepto de rotamers, el cual usa losángulos de torsión extraídos de estructuras de proteínasconocidas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 79 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
5. Perfeccionamiento de la cadena lateral
Teniendo una librería de rotamers se reduce el tiempo decómputo debido a que sólo unos cuantos ángulos de torsión sonexaminados
Sin embargo, aún es necesario reducir más el tiempo de cómputo,mediante observaciones se ha visto que la columna vertebral estarelacionada con ciertas conformaciones de la cadena lateral
Haciendo uso de la existencia de esta correlación, es posibleeliminar aún más ángulos innecesarios
Uno de los paquetes que ha demostrado presentar un buendesempeño es SCWRLwww.fccc.edu/research/labs/dunbrack/scwrl/
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 80 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
6. Refinamiento mediante funciones de energía
Hasta este paso no se garantiza que la estructura este libre deirregularidades
Para tratar de solucionar esto, se hace uso de la minimización deenergía, esto tiene como objetivo reducir la energía al mínimopara aliviar tensiones y colisiones sin afectar significativamente laestructura
Este paso debe aplicarse cuidadosamente, ya que en ocasioneses posible que residuos se muevan a otras posiciones incorrectas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 81 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
6. Refinamiento mediante funciones de energía
Otro método hace uso del proceso de simulación de dinámicamolecular
Este hecho se basa en que la minimización de la energía seobtiene moviendo los átomos de un mínimo local sin necesidadde buscar todas las posibles combinaciones
Requiere de cálculos termodinámicos con los átomos
GROMOS www.igc.ethz.ch/gromos/ es un programa el cualusa simulación de dinámica molecular
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 82 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en homología
Evaluación del modelo
El modelo obtenido tiene que ser evaluado para asegurarse deque las características estructurales del modelo son coherentescon las normas físico-químicas
Para ello se detectan los errores haciendo uso de perfilesestadísticos, características espaciales e interacción de energía através de estructuras determinadas experimentalmente
Si se detectan irregularidades estructurales, la región seconsidera con errores y tiene que ser perfeccionada
Procheck www.biochem.ucl.ac.uk/~roman/procheck/procheck.html es un programa el cual es capaz de comprobarlos parámetros físico-químicos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 83 / 134
Predicción de la estructura terciaria de proteínas Métodos basados en homología
Evaluación del modelo
WHAT IF www.cmbi.kun.nl:1100/WIWWWI/ es un servidor deanálisis de proteínas que valida una proteína mediante correcciónquímica.
ANOLEA http://protein.bio.puc.cl/cardex/servers/anolea/index.html es un servidor web que utiliza el métodode evaluación estadística
Verify3D www.doe-mbi.ucla.edu/Services/Verify3D/ esotro servidor que utiliza el enfoque estadístico
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 84 / 134
Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
3 Predicción de la estructura terciaria de proteínasIntroducciónMétodos basados en homologíaMétodos basados en plegado (threading)Modelos Ab InitioModelo HP (Hydrophobic-Polar)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 85 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
Métodos basados en plegado (threading)
En ocasiones muchas proteínas pueden compartir la mismaestructura aunque no exista mucha similitud en las secuencias
Esta propiedad permitió desarrollar métodos computacionalespara poder predecir estructuras de las proteínas sin importar lasimilitud de las secuencias
Para determinar si una secuencia adopta una estructuratridimensional conocida se hacen uso de los métodos dereconocimiento de plegado (threading)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 86 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
Métodos basados en plegado (threading)
Dicha comparación hace hincapié en la congruencia de lasestructuras secundarias, ya que estas son las más conservadasevolutivamente
Gracias a este enfoque se pueden identificar proteínasestructuralmente similares, incluso sin detectarse similitud algunaen la secuencia
Estos algoritmos se pueden clasificar en dos grupos: basados enpares de energías y basados en perfiles
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 87 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
Métodos de pares de energía
Estos métodos buscan en una BD estructural la mejorcoincidencia, haciendo uso de un alineamiento con la secuenciade consulta
Este alineamiento se hace a nivel de perfil de las secuenciasusando programación dinámica. En ocasiones también se sueleusar un alineamiento local
El siguiente paso es construir un modelo el cual lleve a cabo unasustitución de residuos
Se calcula la energía, la cual consiste en la interacción de energíaentre los residuos
Finalmente se clasifican en base a la energía para encontrar lamenor de ellas (la estructura más compatible)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 88 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
Métodos de perfil
Se construye un perfil para un grupo de proteínas relacionadas,usando información estadística de cada residuo
Este perfil contiene la probabilidad de ocurrencia de cada uno delos veinte aminoácidos por cada posición
El puntaje de este perfil contiene información para tipos deestructuras secundarias
Para predecir el pliegue estructural, primero se predice suestructura secundaria y a partir de esta información se comparacon estructuras de perfiles conocidos
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 89 / 134

Predicción de la estructura terciaria de proteínas Métodos basados en plegado (threading)
Métodos basados en plegado (threading)
3D-PSSM www.bmm.icnet.uk/~3dpssm/ es un programabasado en perfiles para identificar estructuras.
GenThreaderhttp://bioinf.cs.ucl.ac.uk/psipred/index.html esun programa híbrido (perfiles y pares de energía)
Fugewww.cryst.bioc.cam.ac.uk/~fugue/prfsearch.html esun servidor el cual hace uso del método de perfiles
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 90 / 134
Predicción de la estructura terciaria de proteínas Modelos Ab Initio
3 Predicción de la estructura terciaria de proteínasIntroducciónMétodos basados en homologíaMétodos basados en plegado (threading)Modelos Ab InitioModelo HP (Hydrophobic-Polar)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 91 / 134

Predicción de la estructura terciaria de proteínas Modelos Ab Initio
Modelos Ab Initio
En los métodos vistos anteriormente se requiere de ladisponibilidad de plantillas en BD para poder lograr predicciones.Al no existir estructuras suficientes para ello, los métodos fallan
En estos caso se debe considerar otro tipo de información la cualpermita encontrar la estructura
El poco conocimiento de estas estructuras es la base del métodoab initio
Este trata de predecir todas las secuencias de átomos de laproteína sin la ayuda de estructuras de proteínas ya conocidas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 92 / 134

Predicción de la estructura terciaria de proteínas Modelos Ab Initio
Modelos Ab Initio
Una de las ventajas de este método es que la predicción no selimita a los pliegues ya conocidos
Sin embargo, las leyes fisicoquímicas que rigen estecomportamiento aún no son bien conocidas, lo cual sigue siendoun gran reto de la bioinformática
Estos métodos trabajan con algún tipo de heurística, siguiendo elprincipio de minimización de energía, para lo que se lleva a cabouna búsqueda de todos los sitios posibles para encontrar dicharegión
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 93 / 134

Predicción de la estructura terciaria de proteínas Modelos Ab Initio
Modelos Ab Initio
Esta búsqueda global no es factible computacionalmente, ya queaún usando una supercomputadora (1× 1012 operaciones porseg) está se tardaría en muestrear todas las posiblesconformaciones para una proteína de 20 residuos entre 10 y 20años
Es por esta razón que se requiere hacer uso de heurísticas quepermitan reducir el espacio de búsqueda
Algunos de estos métodos fragmentan dicho espacio y combinandiversos tipos de búsqueda para producir un modelo
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 94 / 134

Predicción de la estructura terciaria de proteínas Modelos Ab Initio
Modelos Ab Initio
Rosettawww.bioinfo.rpi.edu/~bystrc/hmmstr/server.php esun servidor el cual permite predecir estructuras tridimensionalesusando el método ab initio.
Para ello rompe la secuencia en segmentos cortos (3 a 9residuos) prediciendo la estructura de estos segmentos haciendouso de modelos ocultos de Markov.
Los resultados para cada uno de estos segmentos se juntan parallevar a cabo la configuración en tres dimensiones (todas lascombinaciones posibles)
La conformación con la menor energía global es la elegida
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 95 / 134
Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
3 Predicción de la estructura terciaria de proteínasIntroducciónMétodos basados en homologíaMétodos basados en plegado (threading)Modelos Ab InitioModelo HP (Hydrophobic-Polar)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 96 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)
Predicción de la estructura de proteínas (PSP)Es el problema de encontrar una conformación funcional para unaproteína dada únicamente su secuencia de aminoácidos.Formalmente:
Dado un modelo de energía E : C → R, encontrar laconformación c ∈ C que minimice E(c).
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 97 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Modelo HP (Hydrophobic-Polar) [Dill, 1985]
Las proteínas son cadenas lineales formadas por aminoácidos
Los aminoácidos se abstraen y clasifican en: Hidrófobos (H) yPolares (P)
Dada la secuencia HP de una proteína S ∈ {H,P}L, lasconformaciones son modeladas como caminatas no traslapadasen una malla:
1 cada nodo de la malla puede ser asignado a máximo unaminoácido
2 aminoácidos consecutivos en S deben ser adyacentes en la malla
Principalmente se enfoca en mallas 2D cuadradas y 3D cúbicas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 98 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Modelo HP (Hydrophobic-Polar) [Dill, 1985]
La meta es maximizar la interacción entre aminoácidos H en lamalla, i.e., minimizar:
E(c) =∑
si ,sj∈S
e(si , sj) , donde
e(si , sj) =
−1 si si y sj son ambos del tipo H
y forman un contacto topológico
0 de otro modo
Dos aminoácidos si , sj ∈ S forman un contacto topológico sison no consecutivos en S, pero adyacentes en la malla
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 99 / 134
Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
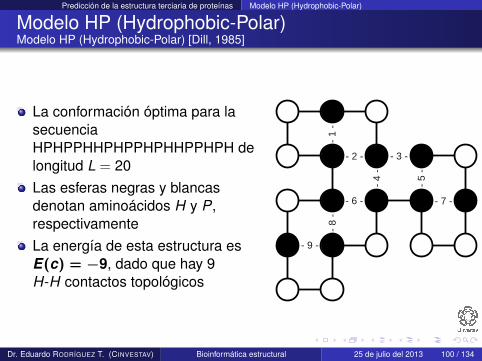
Modelo HP (Hydrophobic-Polar)Modelo HP (Hydrophobic-Polar) [Dill, 1985]
La conformación óptima para lasecuenciaHPHPPHHPHPPHPHHPPHPH delongitud L = 20Las esferas negras y blancasdenotan aminoácidos H y P,respectivamenteLa energía de esta estructura esE(c) = −9, dado que hay 9H-H contactos topológicos
- 7 -
- 3 -
- 6 -
- 2 -
- 1
-
- 9 -
- 4
-
- 8
-
- 5
-
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 100 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)
Estructura generada aleatoriamente
Estructura óptima
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 101 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
1 22
2
2
RLU
D
Codificación movimientos absolutos:las estructuras se codifican comosecuencias en {U,D,L,R}L−1
Por qué L− 1? la posición del primeraminoácido es fija
Por lo tanto, el tamaño del espacio de búsqueda es: 4L−1
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 102 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
Asumamos que tenemos una computadora capaz de explorar1,000 soluciones por segundo
L Soluciones (4L−1) Tiempo
5 256 0.256 sec.
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 103 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
Asumamos que tenemos una computadora capaz de explorar1,000 soluciones por segundo
L Soluciones (4L−1) Tiempo
5 256 0.256 sec.10 262,144 4.370 min.
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 103 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
Asumamos que tenemos una computadora capaz de explorar1,000 soluciones por segundo
L Soluciones (4L−1) Tiempo
5 256 0.256 sec.10 262,144 4.370 min.20 274,877,906,944 8.720 años
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 103 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
Asumamos que tenemos una computadora capaz de explorar1,000 soluciones por segundo
L Soluciones (4L−1) Tiempo
5 256 0.256 sec.10 262,144 4.370 min.20 274,877,906,944 8.720 años30 288,230,376,151,712,000 9,139,725 años
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 103 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)Espacio de búsqueda 2D
L Soluciones (4L−1) Tiempo
5 256 0.256 sec.10 262,144 4.370 min.20 274,877,906,944 8.720 años30 288,230,376,151,712,000 9,139,725 años50 316,912,650,057,057,000,000,000,000,000 -
Se trabaja comúnmente con secuencias de proteínas de longitudentre 18 y 136....
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 103 / 134

Predicción de la estructura terciaria de proteínas Modelo HP (Hydrophobic-Polar)
Modelo HP (Hydrophobic-Polar)
La alternativa es utilizar metaheurísticasAlgoritmos GenéticosBúsqueda TabuRecocido Simulado ...
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 104 / 134
Predicción de la estructura secundaria de ARN Introducción
4 Predicción de la estructura secundaria de ARNIntroducciónTipos de estructuras de ARNMétodos de predicción
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 105 / 134

Predicción de la estructura secundaria de ARN Introducción
Predicción de la estructura secundaria de ARN
El ARN es un portador de información genética y existe en tresformas: ARN mensajero (ARNm), ARN ribosomal (ARNr) y ARNde transferencia (ARNt)
A diferencia del ADN, el ARN se integra de una sola hebra,aunque una molécula de ARN puede auto-hibridarse en ciertasregiones para formar estructuras de doble hebra
El ARNm es más o menos lineal y no estructurado, mientras queel ARNr y el ARNt sólo pueden funcionar formando estructurassecundarias y terciarias particulares
Es por ello que el conocimiento de las estructuras de dichasmoléculas es particularmente importante
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 106 / 134

Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
4 Predicción de la estructura secundaria de ARNIntroducciónTipos de estructuras de ARNMétodos de predicción
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 107 / 134

Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
Tipos de estructuras de ARN
Las estructuras de ARN pueden ser descritas en tres niveles:primario, secundario y terciario
La estructura primaria es la secuencia lineal de ARN integrada porcuatro bases: adenina (A), citosina (C), guanina (G) y uracilo (U)
La estructura secundaria se refiere a la representación planar quecontiene regiones de bases apareadas entre regiones de una solahebra
La estructura terciaria es el arreglo tridimensional de bases deuna molécula de ARN
Dado a que la estructura terciaria de una molécula de ARN esdifícil de predecir, se ha prestado particular atención a lapredicción de la estructura secundaria
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 108 / 134

Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
Tipos de estructuras de ARN
Figura: Estructuras primaria, secundaria y terciaria de una molécula de ARNt
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 109 / 134

Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
Tipos de estructuras de ARN
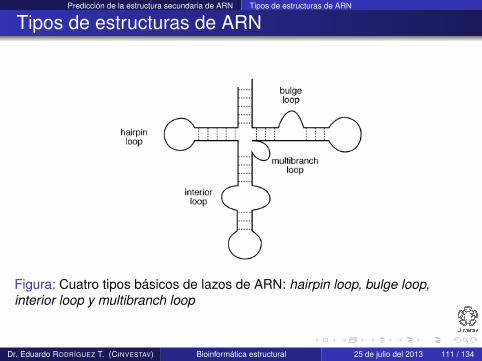
Se pueden identificar cuatro subtipos de estructura secundaria:hairpin loop, bulge loop, interior loop y multibranch loop
Adicionalmente, el apareamiento de bases entre lazos dediferentes elementos de la estructura secundaria puede resultaren estructuras de más alto nivel como pseudoknot loop, kissinghairpin y hairpin-bulge
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 110 / 134
Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
Tipos de estructuras de ARN
Figura: Cuatro tipos básicos de lazos de ARN: hairpin loop, bulge loop,interior loop y multibranch loop
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 111 / 134
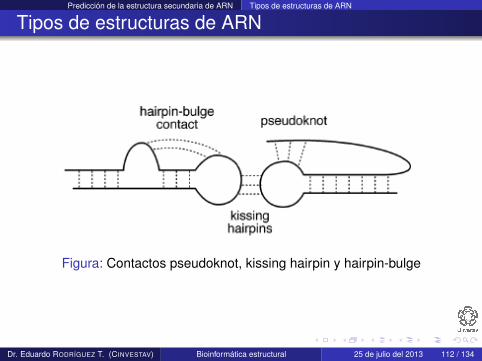
Predicción de la estructura secundaria de ARN Tipos de estructuras de ARN
Tipos de estructuras de ARN
Figura: Contactos pseudoknot, kissing hairpin y hairpin-bulge
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 112 / 134
Predicción de la estructura secundaria de ARN Métodos de predicción
4 Predicción de la estructura secundaria de ARNIntroducciónTipos de estructuras de ARNMétodos de predicción
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 113 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Predicción de la estructura secundaria de ARN
Esencialmente, existen dos enfoques de predicción de laestructura secundaria del ARN: el enfoque ab initio y el enfoquecomparativo
El enfoque ab initio se basa en el cálculo de la mínima energíaliberada de la estructura estable derivada de una secuencia deARN
El enfoque comparativo infiere estructuras en base a lacomparación evolutiva de múltiples secuencias de ARNrelacionadas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 114 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Este enfoque realiza predicciones estructurales basadas en unasola secuencia de ARN
Generalmente, cuando se efectúa un apareamiento entre bases,la energía de la molécula disminuye debido a las interacciones deatracción entre las dos hebras
La energía necesaria para formar pares de bases individuales esinfluenciada por los pares de bases adyacentes a través defuerzas de apilamiento (cooperatividad en la formación dehélices)
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 115 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Se han determinado parámetros para calcular la cooperatividaden la formación de pares de bases para la predicción de laestructura secundaria
Las interacciones de atracción conducen a un estado de aúnmenor energía
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 116 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Sin embargo, si el par de bases es adyacente a lazos (loops) osalientes (bulges), los lazos y salientes vecinas tienden adesestabilizar la formación del par de bases
La fuerza desestabilizadora en una estructura helicoidal tambiéndepende del tipo de lazos cercanos
Pueden utilizarse los parámetros para calcular las diferentesenergías desestabilizadoras como penalizaciones en el cálculo delas estructuras secundarias
Los esquemas de puntaje de las interacciones de estabilización ydesestabilización representan la base del enfoque de predicciónab initio
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 117 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
El método ab initio funciona de la siguiente manera:Primero busca todos los posibles patrones de apareamiento debases de una secuencia
Calcula la energía total de una estructura secundaria potencialtomando en cuenta las fuerzas estabilizadoras y desestabilizadoras
Si hay múltiples alternativas de estructuras secundarias, el métododetermina la conformación con la menor energía
Existen varias técnicas para encontrar todas las posibles regionesde bases apareadas a partir de una secuencia de ácidosnucleicos: la matriz de puntos y la programación dinámica
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 118 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Una matriz de puntos puede encontrar todos los posiblespatrones de apareamiento de bases comparando la secuenciaconsigo misma
Las diagonales perpendiculares a la diagonal principalrepresentan regiones que pueden auto-hibridarse para formarestructuras de doble hebra
Sin embargo, la detección de patrones es a menudo oscurecidapor altos niveles de ruido
Una manera de reducir el ruido es seleccionando una ventana detamaño apropiado
Si la matriz revela más de una estructura factible, se elige la demenor energía
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 119 / 134
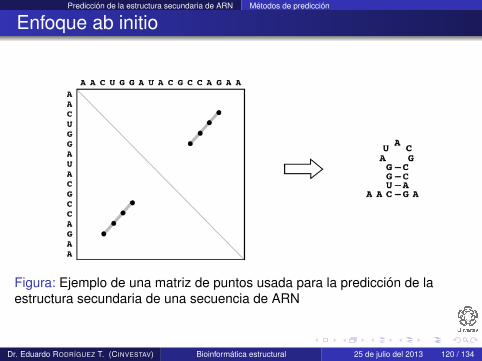
Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Figura: Ejemplo de una matriz de puntos usada para la predicción de laestructura secundaria de una secuencia de ARN
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 120 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Si una molécula grande contiene múltiples segmentos deestructura secundaria, elegir la combinación más estable puedeser una tarea abrumadora
Por ello puede utilizarse un enfoque cuantitativo como laprogramación dinámica
Al igual que en la matriz de puntos, la secuencia de ARN escomparada consigo misma
Se utiliza un esquema de puntaje para llenar la matriz conpuntajes de correspondencia
Después de tomar en cuenta toda la información de la secuencia,se determina el camino con el puntaje máximo dentro de la matrizde puntajes
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 121 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
El método de programación dinámica produce una estructura conun único mejor puntaje
Sin embargo, lo anterior representa una desventaja potencial yaque en realidad una molécula de RNA puede existir en múltiplesformas alternativas con energías cercanas a la mínima y nonecesariamente con el máximo número de pares de bases
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 122 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
La desventaja de la programación dinámica puede ser superadaadicionando una función de distribución de probabilidad, conocidacomo función de partición
La función de partición calcula la distribución matemática depares de bases probables en equilibrio termodinámico
Gracias a esta función es posible seleccionar un número deestructuras subóptimas dentro de un rango de energíadeterminado
Mfold y RNAfold son dos ejemplos populares de aplicaciones queutilizan el enfoque de predicción ab initio
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 123 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
Mfold(http://www.bioinfo.rpi.edu/applications/mfold/)es una aplicación web para la predicción de estructurassecundarias de ARN
Combina programación dinámica con cálculos termodinámicospara identificar la estructura secundaria más estable con la menorenergía
También produce matrices de puntos junto con términos deenergía
Este método es confiable para secuencias cortas, pero suprecisión decrece conforme crece la longitud de la secuencia
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 124 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque ab initio
RNAfold(http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi)es otra aplicación web y forma parte del paquete Vienna
RNAfold extiende el alineamiento de secuencia a la vecindad delas diagonales óptimas para calcular la estabilidad de estructurasalternativas
Incorpora una función de partición para seleccionar el número deestructuras secundarias estadísticamente más probables
En base a cálculos termodinámicos y a la función de partición, seprovee un conjunto de estructuras subóptimas
Debido al gran número de estructuras secundarias computadas,se utiliza una regla de energía simplificada
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 125 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
El enfoque comparativo utiliza múltiples secuenciasevolutivamente relacionadas para inferir una estructura consenso
Para distinguir la estructura secundaria conservada entre lassecuencias múltiples de RNA se utiliza el concepto de covariación
Para conservar la estructura secundaria cuando secuenciashomólogas evolucionan, una mutación en una posiciónresponsable de un apareamiento se compensa con la mutaciónen la posición de apareamiento correspondiente
Basados en esta regla, pueden escribirse algoritmos que busquenpatrones de covariación en un conjunto de secuencias homólogasapropiadamente alineadas
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 126 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Figura: Ejemplo de covariacion de residuos entre tres secuencias homólogasde ARN
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 127 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Otro aspecto del método comparativo es la selección de unaestructura común a través de un consenso
Al comparar todas las estructuras predichas de un grupo desecuencias de ARN alineadas es posible adoptar la estructuraconsenso
Los algoritmos que siguen el enfoque comparativo puedendividirse en dos categorías, dependiendo del tipo de entrada:aquellos que requieren un alineamiento predefinido y aquellosque no lo necesitan
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 128 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Los algoritmos que usan prealineamiento requieren que el usuarioprovea un alineamiento múltiple de secuencias como entrada
Estos programas computan los patrones de mutación como lacovariacion, y derivan una estructura consenso, común a todaslas secuencias
Este tipo de algoritmos son relativamente exitosos parasecuencias razonablemente conservadas
El requerimiento para usarlos es un conjunto apropiado desecuencias homólogas suficientemente similares y divergentes
También dependen de la calidad de la entrada
La selección de una única estructura consenso representa unadesventaja
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 129 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
RNAalifold(http://rna.tbi.univie.ac.at/cgi-bin/alifold.cgi)es un programa que forma parte del paquete Vienna
Utiliza un alineamiento múltiple de secuencias como entrada yanaliza los patrones de covariación en las secuencias
Luego crea una matriz de puntajes que es utilizada para aplicarprogramación dinámica con el objetivo de seleccionar laestructura con la mínima energía
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 130 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Los algoritmos que no utilizan prealineamiento alineansimultáneamente un conjunto de secuencias e infieren unaestructura consenso
El alineamiento es realizado utilizando programación dinámicacon un esquema de puntaje que incorpora la similaridad de lassecuencias así como términos de energía
Debido al costo computacional de la programación dinámica, losprogramas que se encuentran actualmente disponibles limitan laentrada a dos secuencias
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 131 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Foldalign(http://foldalign.kvl.dk/server/index.html) es unaaplicación web para el alineamiento y la predicción de estructurassecundarias
El usuario provee un par de secuencias no alineadas y utiliza unacombinación de Clustal y programación dinámica con esquemasde puntaje que incluyen información de covariación para construirel alineamiento
La estructura secundaria conservada en ambas secuencias esposteriormente calculada
Para reducir el costo computacional, el programa ignora losmultibranch loops
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 132 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Enfoque comparativo
Dynalign (http://rna.urmc.rochester.edu/) es unprograma UNIX libre
El programa calcula las posibles estructuras secundariasutilizando un método similar a Mfold
Comparando estructuras alternativas para cada secuencia, laestructura común a ambas secuencias con menor energía eselegida
No requiere que las secuencias sean similares por lo que puedemanejar secuencias altamente divergentes
Sin embargo, solo sirve para predecir secuencias pequeñas deARN con una precisión razonable, como secuencias de ARNt
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 133 / 134

Predicción de la estructura secundaria de ARN Métodos de predicción
Anfinsen, C. (1973).Principles that Govern the Folding of Protein Chains.Science, 181(4096):223–230.
Dill, K. (1985).Theory for the Folding and Stability of Globular Proteins.Biochemistry, 24(6):1501–9.
Dr. Eduardo RODRÍGUEZ T. (CINVESTAV) Bioinformática estructural 25 de julio del 2013 134 / 134
Recommended

















