
UNIVERSITA’ DEGLI STUDI DI CATANIA
DEPARTIMENTO DI SCIENZE BIOLOGICHE, GEOLOGICHE E AMBIENTALI
PhD in BIOTECHNOLOGY
course XXV
Building a β-structured artificial pore-
forming protein by PA83 Bacillus
anthracis toxin
Dott. CLAUDIA A. FICHERA
Supervisor and Tutor prof. Vito De Pinto

Solo la ricerca dell'impossibile può condurre
a ciò che è realizzabile.

ABSTRACT
Le porine naturali sono delle proteine che si trovano principalmente nella
membrana esterna dei batteri Gram negativi, come Escherichia coli, ma che
sono state ritrovate anche nelle cellule vegetali e nelle membrane di organuli
cellulari eucariotici come mitocondri e cloroplasti. Le porine generano dei
canali acquosi poco o nulla selettivi, a volte voltaggio dipendenti, e alcune
rivestono molta importanza in determinati pathway cellulari. L‟utilizzo sempre
più frequente di peptidi strutturalmente definiti con funzioni chimiche
specifiche ha portato alla realizzazione di questo lavoro. Attraverso una
preliminare analisi bioinformatica è stato individuato un pattern proteico di
base con una struttura a forcina (β-strand/loop/ β-strand) partendo dalla
proteina PA83 di Bacillus anthracis. Mediante la realizzazione di un protocollo
sperimentale semplice e molto rapido è stato possibile costruire multimeri del
modulo di base, ovvero costrutti contenenti varie ripetizioni dello stesso. I dati
dell‟analisi bioinformatica suggerivano che il giusto numero di ripetizioni del
modulo di base necessario alla produzione di una proteina formante poro fosse
sette, che tra l‟altro è il numero di oligomeri che si assemblano nella proteina
naturale da cui siamo partiti. Il piano sperimentale messo a punto ci ha
permesso di ottenere in pochi giorni una proteina chimerica completamente
artificiale, la quale è stata clonata in un vettore di espressione batterico.
L‟espressione della proteina è stata ottenuta difficilmente e inoltre le colture
batteriche in cui veniva indotta l‟espressione della proteina chimerica
risultavano meno torbide, indice di una ridotta crescita cellulare. In accordo
con la recente letteratura siamo propensi a credere che la proteina agisca in
qualche modo da antibiotico, inserendosi massivamente in membrana e
determinando la lisi cellulare dell‟ospite batterico. Studi futuri saranno volti a
valutare le strutture secondaria e terziaria della porina artificiale prodotta e le
sue proprietà elettrofisiologiche.

CONTENTS
I
CONTENTS
1 INTRODUCTION .......................................................................................... 1
1.1 Porins ............................................................................................... 1
1.1.1 Natural porins: description ........................................................... 2
1.1.2 Natural porins: structures ............................................................. 5
1.1.3 Natural porins: proprieties ........................................................... 8
1.2 Building artificial pore-forming proteins ........................................... 9
1.2.1 Why build an artificial porin?........................................................ 10
1.2.2 New, artificial porins. .................................................................... 12
1.3 Anthrax toxin of Bacillus anthracis ....................................................... 13
1.3.1 Protective antigen (PA) – The binding and translocation moiety ... 14
1.3.2 Edema factor (EF) and Lethal factor (LF) ........................................ 15
1.3.3 Intoxication pathway of Anthrax toxin .......................................... 16
1.3.4 Bacillus anthracis - Pharmacology of treatments ........................... 17
2 AIM OF THE WORK................................................................................. 19
3 METHODS .............................................................................................. 20
3.1 Bioinformatic analysis ....................................................................... 20
3.2 PCR amplification of the basic module ............................................ 20
3.3 Multimerization and insertion in pET52b expression vector ............ 21
3.4 PCR-based colony screening of cloned multimers ........................... 23
3.5 Prokaryotic strain used ................................................................... 24
3.6 Site-directed mutagenesis of T293-I334(7x) clone........................... 24
3.7 Expression protein .......................................................................... 25
3.7.1 Standard protocol for the expression of recombinant protein ....... 26

CONTENTS
II
3.7.2 Lysis of the cellular pellet ............................................................. 26
4 RESULTS ................................................................................................. 28
4.1 Bioinformatics analysis ...................................................................... 28
4.2 PCR amplification of the basic module ................................................ 33
4.3 Multimerization and insertion in pET52b expression vector ................ 34
4.4 Site-directed mutagenesis of T293-I334(7x) clone........................... 37
4.5 Expression of T293-I334(7x) artificial porine ................................... 38
5 DISCUSSION ........................................................................................... 40
5.1 Conclusion ........................................................................................ 40
5.2 Future investigation ........................................................................... 41
REFERENCES ................................................................................................. 44

INTRODUCTION
1
1 INTRODUCTION
1.1 Porins
Gram-negative bacteria, such as Escherichia coli, are surrounded by two
membranes, the inner, or cytoplasmic, and the outer membrane. Both of them
contain transport systems that allow the passage of solutes across the
membranes. In the inner membrane mostly primary or secondary active
transporters are present. In the outer membrane, typically, the transport
proteins allow passive diffusion. These passive diffusion pathways across the
outer membranes are formed by the family of porins.
Outer and inner membrane are profoundly different both for structural
and functional properties. The internal membrane is formed by phospholipids,
mainly phosphatidylethanolamine (70%-80 %), but also phosphatidilglycerol
and cardiolipin (1; 2) all evenly distributed between the citoplasmatic side and
the periplasmatic side of the membrane. The outer membrane is, additionally,
highly asymmetrical: indeed, the main component of the external layer are not
phospholipids, but the lipopolysaccharide (LPS, also known as lipoglycans, are
large molecules consisting of a lipid and a polysaccharide joined by a covalent
bond), while the periplasmatic side has a phospholipidic composition
comparable with the internal membrane (Fig. 1.1.1).
Figure 1.1.1: Gram-negative bacteria envelope.

INTRODUCTION
2
Approximately 50% of the mass of outer membrane are proteins, both
integral membrane proteins and proteins anchored to lipideA of the
lipopolysaccharide (O-antigen). In E. coli were identified about a dozen of
different outer membrane proteins. Some of these proteins are constitutively
expressed at high levels; most, however, are inducible and produced in case of
necessity (PhoE and for LamB example). In Tab.1.1 there is a short list of
proteins expressed on the outer membrane (3).
Table 1.1: Summary of the most important proteic families. Each protein has a unique
PDB ID (4).
1.1.1 Natural porins: description
General porin form aqueous channels with an exclusion limit of typically
600 Da and extremes of 5000 Da. Due to their high copy number they form the
major integral protein component of the outer membrane in Gram-negative
bacteria and turn it into a molecular sieve (5).
Structural studies show that many or most of these proteins exist as beta-
barrels with the β-strands traversing the thickness of the outer membrane (6).
Owing to the strong hydrophilicity of their amino acid sequence and the nature
of their secondary structure (beta strands), conventional hydropathy methods
for predicting membrane topology are useless for this class of protein. The
large number of available porin amino acid sequences was exploited to

INTRODUCTION
3
improve the accuracy of the prediction in combination with tools detecting
amphipathicity of secondary structure.
The channels often have diameters in the range of 1 nm, and thus the
penetration rates of solutes through porin channels are likely to be affected
strongly by what appear to be minor differences in the size, shape,
hydrophobicity or charge of the solute molecule.
Porins can be divided into the following three classes:
(I) Non-specific or general porins. Porins produce water-filled channels across
the membrane. The gross physicochemical parameters of the solute obviously
influence its rate of penetration through these channels, but the channels do not
appear to contain specific ligand binding sites.
(II) Specific channels. These proteins also produce water-filled channels,
which contain stereospecific binding sites. The presence of specific binding
sites has important consequences. The diffusion of the solutes of a specific
class is accelerated when the solute concentration is low, but it is slowed down
when the concentration is high, producing saturation-type kinetics very similar
to Michaelis-Menten enzyme kinetics. This behavior is very different from that
of the non-specific porin channel, where the solute diffusion rate increases
proportionally to the solute concentration on one side of the membrane if the
concentration on the other side is zero (Fig. 1.1.2).
Figure 1.1.2: Solute diffusion through ponn channels and specific
channels. Initial rates of solute diffusion from a compartment filled with a solution of
concentration [c] into a compartment tilled with water are plotted. The penetration through
porin channels follows Fick's law of diffusion, and therefore its rate is expected to be
proportional to [c]. In contrast, that through the specific channels follows a saturation curve,
and thus measurement of flux at one arbitrarily chosen value of [c] can be quite misleading.

INTRODUCTION
4
(III) High-affinity, energy-dependent transport systems. The transport of iron-
chelator complexes and vitamin B12 is also carried out by specific systems in
Escherichia coli
However, they are quite different from the simple, specific channels
mentioned above, because the proteins bind the ligands with much higher
affinity, and because the systems apparently carry out uphill transport through
energy coupling via TonB protein (7).
All porins have high stability: they can tolerate treatments with strong
denaturing agents and high temperatures. General porins, also, have a certain
sensitivity to the membrane potential, which is indeed not present in substrate-
specific porins.
A more widespread property of porins and specific channels, and indeed
of most intrinsic outer membrane proteins, appears to be the predominance of
β-structure (6) (Fig. 1.1.3).
Figure 1.1.3: General prokaryotic porin: (A) Porin inserted into lipid bilayer; (B)
Top view of the channel formed; (C) Secondary structure of generic porin.
Porins are present in bacterial cell walls, as well as in plant, fungal,
mammalian and other vertebrate cell membranes and mitochondrial
membranes.

INTRODUCTION
5
1.1.1.1 Eukaryotic porins
A voltage-dependent anion channel protein (VDAC) in the mitochondrial
outer membrane (8) may have evolved from “general diffusion prokaryotic
porins” as suggested by Zalman et al. in the 1980. The voltage-dependent
anion channel (VDAC) is a small family of integral membrane proteins of the
mitochondrial outer membrane (MOM) whose role is to allow the flow of
hydrophilic metabolites such as substrates, ATP and ADP between the
mitochondrion and the cytosol (9)(10)(11). The functional features of VDAC1
are well characterized (9)(10)(11), and, furthermore, it has been implicated as
an important factor in cell processes including apoptosis (12) (13), calcium
homeostasis (14) and diseases such as cancer (15). Deficiency of VDAC1 has
been associated with a let halencephalomyopathy (16). The intensive research
culminated in the proposal of the 3D structure by different groups almost in
the same time (17). The topology of the protein in the membrane has been
determined (18).
Porins from chloroplast outer membranes (19) and mycobacteria (20)
have also been reported.
1.1.2 Natural porins: structures
Most of the 'classical' porins from enteric bacteria studied so far,
including the OmpF, OmpC, and PhoE porins of E.coli exist as tight, trimeric
complexes that are not dissociated even in sodium dodecyl sulphate (SDS),
unless the protein is denatured by heating (Fig. 1.1.4).
Figure1.1.4. Trimeric complexes in the Gram-negative outer membrane.

INTRODUCTION
6
At least one of the specific channels, the maltose channel protein of E
coli. LamB, also shares this property. However, there is little evidence for
trimeric structure for some other porins, including the F porin of Pseudomonas
aeruginosa.
Since the second half of the 70s Jurg Rosenbusch had proposed, for the
prokaryotic porin, a beta-barrel structure formed by amphipathic filaments,
from 16 (for OmpF, OmpC and PhoE) to 18 (LamB), characterized by the
presence of polar aminoacidic residues (protruding from the internal side of the
wall into the water channel), alternating with apolar residues (hydrophobic)
inserted into the double layer of lipid membrane. This assumption was correct,
as was confirmed when the structure of the protein was resolved by X-ray
diffraction of the purified protein. Using this technique, different porins
structures have been solved including LamB, OmpF and PhoE. This analysis
revealed that the structure of general bacterial porins is trimeric. Each subunit
is made up of a beta-barrel, usually consisting of 16 beta-strand. Two
consecutive antiparallel beta-strands are linked by very short aminoacidic
sequences in the periplasmatic side (turns), and through longer sequences in
the opposite side (loops) (Fig. 1.1.5).
The three channels of the trimer have axes almost parallel to each other
and perpendicular to the double-layer lipid plane. As has been seen in other
transmembrane proteins, also in porins there are two ‟belts‟ of aromatic
aminoacids which are designed to anchor the protein to the membrane.
Between the two belts the surface of the barrel is mainly composed of
hydrofobic aminoacids. The distance between the belts is 25°A, corresponding
to the thickness of the outer membrane. Each monomer has a molecular weight
of between 30 and 50 KDa.
Figure 1.1.5: β-barrels formed from different numbers of β-strands. (1) The
E. coli OmpA protein (8 β-strands), which serves as a receptor for a bacterial virus. (2) The E.
coli OMPLA protein (12 beta-strands), is a lipase that hydrolyses lipid molecules. The amino

INTRODUCTION
7
acids that catalyze the enzymatic reaction (shown in red) protrude from the outside surface of
the barrel. (3) A porin from the bacterium Rhodobacter capsulatus, which forms water-filled
pores across the outer membrane (16 β-strands). The diameter of the channel is restricted by
loops (shown in blue) that protrude into the channel. (4) The E. coli FepA protein (22 β-
strands), which transports iron ions. The inside of the barrel is completely filled by a globular
protein domain (shown in blue) that contains an iron-binding site. This domain is thought to
change its conformation to transport the bound iron, but the molecular details of the changes
are not known (21).
Electron diffraction studies of the two-dimensional crystalline arrays of PhoE
porin showed that the polypeptide chain traverses the membrane interior more
than a dozen times (22) and the Fourier transform infrared study of OmpF
porin showed β -strands somewhat tilted from the membrane normal (23).
The most exciting development in this area is the determination, at 0.18
nm resolution, of the structure of Rhodobacter capsulatus porin by the group
led by G. E. Schuiz (24). This porin exists also as a tightly assembled trimer,
and each of the subunits produces a water-filled channel. The polypeptide
chain of a subunit traverses the membrane 16 times as antiparallel β-strands.
The loops on one side, presumably the periplasmic side, are all very short: they
are usually much longer on the outer side. Interestingly, one of the external
loops folds back into the channel, and produces a significant narrowing of the
channel ('eyelet'). This eyelet region is only about 1 nm in thickness, and the
channel is much wider at other places. This construction seems ideally suited
for the physiological function of porins, i.e. to produce a narrow eyelet to
exclude toxic compounds, yet to maximize the influx of nutrients by
minimizing the friction between solute molecules and the walls of the pore.
The major problem in the prediction of the tertiary structure of porins has
been the lack of crystallographic structures for β-sheet-rich membrane proteins.
But the comparison of the X-ray structure with the primary structure (25) now
allows the evaluation of various prediction methods. It appears that prediction
of the locations of β-sheets is impossible by the methods of Chou and Fasman
or Eisenberg. Turns are predicted much more successfully, and the Chou-
Fasman procedure predicts 13 out of the total of 15 actual turns in this protein,
although portions of several membrane-spanning β-strands are incorrectly
predicted as turns.
In comparing the sequences of various related porin genes, it is
remarkable that short deletions (or insertions) always seemed to have occurred

INTRODUCTION
8
in the externally exposed parts of the molecule. Undoubtedly these sequences
exposed on the bacteria outer surface are undergoing an extremely rapid
evolutionary change, because they are the targets of recognition by components
of the host immune systems as well as phages and bacteriocins found in the
environment. It is not known how the narrowing of the channel is produced in
the enterobacterial porin channel (22). However, OmpF porin, which produces
a larger channel than the OmpC porin, lacks a 15-residue stretch present in
loop 4 of OmpC. In addition, several deletions occur in loop 3 of OmpF porin
in mutants selected for the production of still larger pores (26). These data
suggest that one, or perhaps both, of these loops may be involved in narrowing
of the E. coli porin channel.
The folding pattern of mitochondria porin was predicted from the
primary sequence as well as the alteration of ion selectivity caused by site-
directed mutagenesis (27). Many prediction efforts were dedicated to this
protein (17) but they were all disowned by the real structure that showed to be
a 19-strands β-barrel (28)(29)(30).
1.1.3 Natural porins: proprieties
General porins are passive channels in which the speed of transport of
solutes is proportional to the chemical gradient, but also depends on the eyelet
size and on the property of the solute. Eyelet size and distribution of charges on
it act as a filter for selecting ions (Fig. 1.1.6) (31).
Charged solutes are retained by the pore for the presence of opposite charges
on the eyelet surface. Schulz et al. showed that the presence of opposite
charges on the inner surface of the barrel produces a cross-growing electric

INTRODUCTION
9
field that can generate a force capable of expelling the neutral molecules (Fig.
1.1.7).
Figure 1.1.7: Cross-growing electric field through the pore eyelet. (31)
Substrate-specific porine, however, have a binding site for solutes and their
transport rate follows kinetic kinetic features similar to Michaelis-Menten
enzyme kinetics: at low concentrations diffusion rate is proportional to the
concentration of solute; at high concentrations there is a saturation effect and
the rate remains constant (32).
1.2 Building artificial pore-forming proteins
During the last twenty years, several studies were performed with beta-
barreled transmembrane protein channels from different issues: from the
genetic characterization to biofunctional assays (2)(3) and structural analysis
(33). Objects of study were not only prokaryotic porins, but eukaryotic
(mitochondrial) ones too(17)(18). These studies have provided a basic
understanding of the main biological mechanisms underlying solute diffusion
through porins across a biological membrane. A growing area of interest
concerns now the possible use of these protein channel in industrial
applications. In agreement with this new investigation field, the basic idea of
my PhD thesis, held in the molecular biology laboratory of Prof. Vito De Pinto
at the Department of Biological, Geological and Environmental Sciences
(DBGES - University of Catania, Italy) was to build completely artificial
chimeric porins (34). These porins should have been based upon natural
structure but endowed with constrains to form pores with pre-defined diameters

INTRODUCTION
10
in membranes. Another long-range aim was to understand whether there is a
folding rule governing the formation of the beta barrel in the membrane, thus
the basic mechanisms of its molecular evolution in nature (35). From the point
of view of applied biology chimeric porins may allow (us) to set the basis for
producing future advanced techniques for rapid diagnosis of diseases
(biosensors and large scale sequencing) (36)(37) or for developing new
aspecific antibiotics (38)(39)(40) or anticancer peptides.
1.2.1 Why build an artificial porin?
Channels and pores with altered functional properties and with built-in
triggers and switches have been created. Progress in applications has been
greatest in sensor technology, where sensor elements based on ligand
activation, channel selectivity and channel block have been made, but growing
interest is now directed to use these protein channels for a lot of other
biotechnological applications.
1.2.1.1 Analyte recognition
In nanopore analytics, individual molecules pass through a single
nanopore giving rise to detectable temporary blockades in ionic pore current.
Reflecting its simplicity, nanopore analytics has gained popularity and can be
conducted with natural protein as well as man-made polymeric and inorganic
pores. The spectrum of detectable analytes ranges from nucleic acids, peptides,
proteins, and biomolecular complexes to organic polymers and small
molecules. Apart from being an analytical tool, nanopores have developed into
a general platform technology to investigate the biophysics, physicochemistry,
and chemistry of individual molecules. Electrophorescing biopolymers across
nanopores modulates the ionic current through the pore, revealing the
polymer's diameter, length, and conformation. The rapidity of polymer
translocation ( 30000 bp/ms) in this geometry greatly limits the information
that can be obtained for each base (41).
1.2.1.2 DNA sequencing

INTRODUCTION
11
A nanopore-based device provides single-molecule detection and
analytical capabilities that are achieved by electrophoretically driving
molecules in solution through a nano-scale pore. The nanopore provides a
highly confined space within which single nucleic acid polymers can be
analyzed at high throughput by one of a variety of means, and the perfect
processivity that can be enforced in a narrow pore ensures that the native order
of the nucleobases in a polynucleotide is reflected in the sequence of signals
that is detected. Kilobase length polymers (single-stranded genomic DNA or
RNA) or small molecules (e.g., nucleosides) can be identified and
characterized without amplification or labeling, a unique analytical capability
that makes inexpensive, rapid DNA sequencing a possibility. Further research
and development to overcome current challenges to nanopore identification of
each successive nucleotide in a DNA strand offers the prospect of 'third
generation' instruments that will sequence a diploid mammalian genome for ~
1,000 dollar in ~24 h (42).
1.2.1.3. Biosensor production
Single nanopores have attracted interest for their use as biosensing
devices. In general, methods involve measuring ionic current blockades
associated with translocation of analytes through the nanopore, but the
detection of such short time lasting events requires complex equipment and
setup that are critical for convenient routine biosensing. A novel biosensing
concept based on a single nanopore in a silicon nitride membrane and two
anchor-linked DNA species that forms trans-pore hybrids, realizing a stable
blockade of ionic current through the pore has been presented. Molecular
recognition events affecting the DNA hybrids cause a pore opening and the
consequent establishment of an ionic current (43) (44).
1.2.1.4 Molecular filtration devices
Filtration of molecules by nanometer-sized structures is ubiquitous in our
everyday life, but our understanding of such molecular filtration processes is
far less than desired. Until recently, one of the main reasons was the lack of
experimental methods that can help provide detailed, microscopic pictures of

INTRODUCTION
12
molecule–nanostructure interactions. Several innovations in experimental
methods, such as nuclear track-etched membranes developed in the 70s, and
more recent development of nanofluidic molecular filters, played pivotal roles
in advancing our understanding. With the ability to make truly molecular-scale
filters and pores with well-defined sizes, shapes, and surface properties, now
we are well positioned to engineer better functionality in molecular sieving,
separation and other membrane applications (45).
1.2.1.4 Antimicrobial peptide
The increasing resistance of bacteria to conventional antibiotics makes
the development of new modes of treatment essential. Over the past few years,
antimicrobial peptides were presented as a potential solution: whereas classical
antibiotics act specifically on biosynthetic pathways, antimicrobial peptides
may directly destabilize the lipid membrane and constitute a promising
alternative strategy for fighting the actions of microorganisms (46).
1.2.2 New, artificial porins.
The abundance of porin sequences and 3D structures (porins are the
membrane proteins that were crystallized with the most success, thus are the
class of membrane proteins showing the largest number of high resolution 3D
structures, see PDB database) prompted us to design a new pore-forming
structure with variable but controlled functions starting from a natural example.
The first step was the choice of a candidate sequence to become the basic
module for the artificial porin(s). We used a conserved sequence of the B-
component of Anthrax toxin, called protective antigen (PA), due to its use as a
vaccine, produced by Bacillus anthracis. The selected sequence, forming a
couple of antiparallel beta-strands with the connecting loops, is into domain II,
as a flexible loop and inserts in the membrane.
The overall sequence is 42 amino acid long. This constituted the basic Lego
brick. The idea was to repeat this conserved motif or module as a building
block. The polymerization of this module was envisaged as a building
procedure to get various beta-barrels with an increasing number of beta
strands.

INTRODUCTION
13
This should provide growing diameters. For example the repetition of two of
these bricks in principle should form a small barrel of four beta strands and the
fusion of three bricks a barrel of six. A bioinformatic simulation was
performed to predict the potential folding pattern of the multimers. These
simulations were used as an additional criteria to decide the sequence of the
module. Bioinformatics software able to perform fold recognition, secondary
structures predictions and alignment between 3D model structures were used.
1.3 Anthrax toxin of Bacillus anthracis
The rod-shaped Gram-positive bacterium Bacillus anthracis produces
Anthrax toxin as its main virulence factor. One possible Symptom of an
Anthrax infection is large black necrotic patches on the skin. Therefore, the
name Anthrax is derived from the Greek word for coal “ánthrax“. An infection
is caused by the uptake of Bacillus anthracis spores in skin bruises, the lung or
gastrointestinal. Depending on the site of infection different phenotypes of the
disease evolve, of which the one in the lungs is the most dangerous and leads to
death with nearly 100% probability if not treated. These spores are resistant to
environmental stresses and could last for more than 100 years, still able to start
bacterial growth and the cycle of infection (47)(48). The uptake of the spores is
followed by germination and proliferation of vegetative bacteria, which invade
the lymphatic system. There, they eliminate host immune cells and enter the
bloodstream. Finally, death occurs due to septicemia and toxemia. With the
restriction of nutrients a high amount of spores is produced afterwards.
During the assaults of September 2001 in the USA, letters containing
Anthrax spores have been sent to persons involved in the government and led
to cases of infection and death, conjuring up Anthrax toxin in the public again
(49).
This toxin is classified, as a binary AB7-type toxin comprised of three
components. Protective antigen (PA) is the binding and translocation unit,
which transports edema factor (EF) and lethal factor (LF) into target cell‟s
cytosol (50)(51)(48). Thereby, in contrast to other members of the AB-family
Anthrax contains two enzymatically active moieties. Additionally, another

INTRODUCTION
14
virulence factor, the poly-D-glutamyl capsule, inhibits the phagocytosis of B.
anthracis by host immune system.
1.3.1 Protective antigen (PA) – The binding and translocation
moiety
The B-component of Anthrax toxin is called protective antigen (PA) due to its
use as a vaccine. It is secreted as a 83 kDa monomeric protein (PA83) to the
external media and consists of four domains. Correlated to their function,
domain I is proteolytically cleaved by furin-like cell bound proteases during
activation, domain II, a flexible loop, inserts in the membrane, oligomerization
takes place in domain III and domain IV binds to the receptors
(52)(53)(54)(55). Activation of PA83 leads to a 20 kDa (PA20) and a 63 kDa
(PA63) fragment, of which the larger one represents the active PA. Lately, a
vital discussion in this field of work is going on about the number of
monomers, which form the water-soluble so-called prepore and later on the
channel. The structures of a homo-heptameric and a homo-octameric prepore
were published (52)(56). Concerning the electrophysiological results and the
possibility of crystallization artifacts, this work considers the heptameric
symmetry as the prominent form. For the membrane active PA-pore only a
model exists (57), based on the mushroom shaped resolved structure of α-
hemolysin (58). Accordingly, the 14-stranded β-barrel, forming the channel is
created by unfolding β-hairpins in a Greek-key motif (strands 2β1-2β4)
(59)(60). The important process of prepore to pore transition is triggered by
acid pH in the endosome (61)(62). Recent progress in electron microscopy
made it possible to take pictures of PA-channels in membrane disks. These
show a slightly different shaped protein, especially in the head region (Fig.
1.3.1) (63).

INTRODUCTION
15
Figure 1.3.1: Structures and hypothetical models of Anthrax’s protective
antigen: A: Top and side view of the heptameric PA prepore (52); B: Hypothetical model of
membrane-inserted PA-heptamer with membrane-spanning β-barrel (57); C: Three-
dimensional reconstruction of PA-pore, inserted in nano-discs (63).
1.3.2 Edema factor (EF) and Lethal factor (LF)
The 89 kDa protein edema factor (EF) is one of the enzymatically active A-
moieties of Anthrax toxin and represents a calmodulin- and Ca2+-dependent
adenylate-cyclase. It is named edema factor because it is believed to cause
edemas in cutaneous Anthrax. The toxic potential of EF reasons in the
transformation of ADP in cAMP. Accumulated cAMP interferes with water
homeostasis and many intracellular signaling pathways, causing the cell to die
(47)(48)(55)(64). Cell death is the final effect of the second A-component
lethal factor (LF), as well. LF represents a 90 kDa Zn2+-dependent
metalloprotease, that specifically cleaves mitogen-activated protein kinase
kinases (MAPKKs), thereby not only blocking the MAPK-pathway, but also
leading to a special form of apoptosis (64). Especially when attacking
macrophages, this death does not spread chemokines and cytokines, which
normally alert the host immune system (65)(66). Even specialized killers like
dendritic cells and T-cells are inhibited (55)(67)(68), which explains, why
antibiotics including penicillin and doxicyclin only show effect during early

INTRODUCTION
16
stages of an Anthrax infection in which no obvious symptoms could be
detected.
1.3.3 Intoxication pathway of Anthrax toxin
After the release of the enzymatic components and the PA83, this
precursor binds to cellular expressed surface receptors. Two receptors represent
prominent targets, ATR (Anthrax toxin receptor), an alternative splice product
of TEM8 (tumor endothelial marker 8) and CMG2 (capillary morphogenesis
gene transcript 2) (69)(70), which are also called ANTXR1 and ANTXR2.
Afterwards, LRP6 (low density lipoprotein receptor-related protein 6) interacts
with ATR or CMG2 , on the cell surface to initiate internalization of the whole
complex (71). As mentioned before, PA83 is proteolytically activated by furin-
like proteases on the cell surface, rendering it possible to form heptameric
PA63-prepores (52). The 20 kDa PA20 fragment is supposed to play a role in
uptake of the complex, as well (72). Up to three EF and LF molecules attach to
the prepore with their N-terminal end (73)(74)(75), which exhibits a significant
homology. In other studies the steric complexity of the complex leads to the
assumption that only one effector molecule is able to bind each prepore
(76)(72) . It is generally proven, that ion-ion interaction between the positively
charged N-terms of the A-components and the negative charges of the PA-
heptamer enhance the affinity of the binding process. This is further reasoned
in experiments, which use charged tags to increase this effect (77)(78)(79).
Subsequently, the whole (PA63)7-EF/LF-complex is endocytosed by receptor-
mediated and/or clatherin-coated pits (80). During the maturation of the
endosome it‟s acidification prompts changes in both, the prepore and the
enzymatic-units. While the first undergoes a transition to a β-barrel formed
pore, the later partially unfold and pass through the channel, driven by voltage
and pH-gradient (59) (60)(81). The transport through the pore occurs in a
molten-globular state, in which the N-terminal end of the effectors inserts in
the Lumen first. Further unfolding leads - together with Brownian molecular
movement and cytosolic uncharging of some amino acids – to the translocation
to the cytosol, where the toxic impact unravels. The whole intoxication
pathway is depicted in figure 1.3.2.
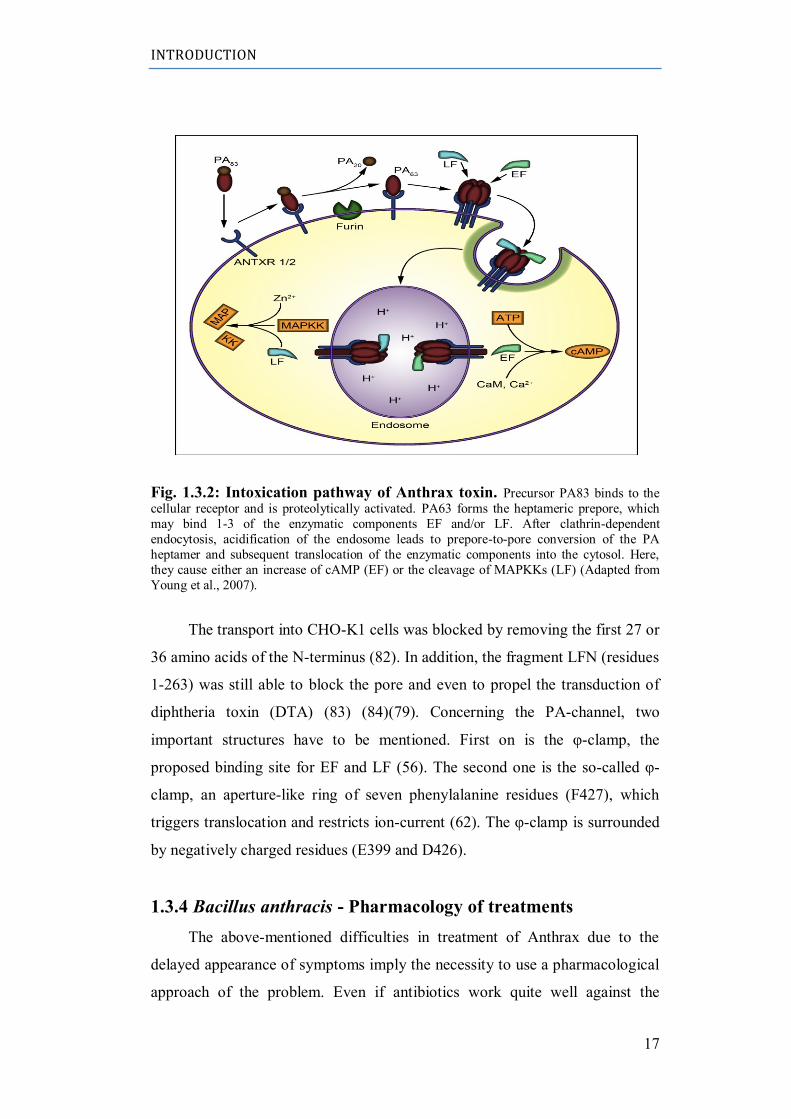
INTRODUCTION
17
Fig. 1.3.2: Intoxication pathway of Anthrax toxin. Precursor PA83 binds to the
cellular receptor and is proteolytically activated. PA63 forms the heptameric prepore, which
may bind 1-3 of the enzymatic components EF and/or LF. After clathrin-dependent
endocytosis, acidification of the endosome leads to prepore-to-pore conversion of the PA
heptamer and subsequent translocation of the enzymatic components into the cytosol. Here,
they cause either an increase of cAMP (EF) or the cleavage of MAPKKs (LF) (Adapted from
Young et al., 2007).
The transport into CHO-K1 cells was blocked by removing the first 27 or
36 amino acids of the N-terminus (82). In addition, the fragment LFN (residues
1-263) was still able to block the pore and even to propel the transduction of
diphtheria toxin (DTA) (83) (84)(79). Concerning the PA-channel, two
important structures have to be mentioned. First on is the φ-clamp, the
proposed binding site for EF and LF (56). The second one is the so-called φ-
clamp, an aperture-like ring of seven phenylalanine residues (F427), which
triggers translocation and restricts ion-current (62). The φ-clamp is surrounded
by negatively charged residues (E399 and D426).
1.3.4 Bacillus anthracis - Pharmacology of treatments
The above-mentioned difficulties in treatment of Anthrax due to the
delayed appearance of symptoms imply the necessity to use a pharmacological
approach of the problem. Even if antibiotics work quite well against the

INTRODUCTION
18
infection, when applied on time, and vaccination offers some protection against
the intoxication, the existence of multi-resistant strains and the high probability
of death reason the research for so-called blocker-substrates. These form a plug
in the lumen of the pore (presumably on the φ-clamp), thereby hindering the
effectors from being translocated. Experiments performed with anti-bodies
(85)(86) and 4-aminoquinolines or cyclodextrins seem to be promising as a
complementary medication (87)(88)(89)(90)(91).

AIM OF THE WORK
19
2 AIM OF THE WORK
My PhD thesis, held in the molecular biology laboratory of Prof. Vito
De Pinto at the Department of Biological, Geological and Environmental
Sciences (DBGES - University of Catania), stemmed from the idea of building
a β-structure pore-forming protein completely artificial. A previous approach to
this matter resulted in the construction of multimers of a OmpF module (34).
Unfortunately, this structure was only poorly able to form pores in artificial
membranes, thus we decided to utilize a new template.
In agreement with the newest discovery about potentiality of β-structured
peptides (36) (43) (44) the goal of this project was find a perfect candidate
basic module as β-hairpin motif (β-strand/loop/β-strand) to perform an artificial
protein chemically, functionally a structurally defined.
The first step was the choice, by bioinformatics analysis, of a candidate
sequence to become the basic module for the artificial porin(s).
In a second phase was developed an experimental protocol, simple and
fast, aimed to obtaining the plasmid constructs containing the sequence
encoding our artificial protein.
At the end we performed the expression of the produced protein in order
to study the new artificial molecule, its behavior in membrane, its ability to
form pores and, if possible, to analyze its electrophysiological properties
(conductance, estimated pore diameter, voltage dependence, selectivity, etc.).

METHODS
20
3 METHODS
3.1 Bioinformatic analysis
The basic sequence module was chosen after a thorough screening of the
domain II of PA83 of Bacillus anthracis. Virtual multimers were simply
constructed repeating the single module 2 to 10 times and the structures
resulting from the fused sequences were modeled using the SP5Fold
Recognition server (http://sparks.informatics.iupui.edu/SP5/) (92).
On the basis of mentioned analysis we identified an ideal candidate to
become the “basic module”. Models were visualized using the open source
software PyMol (http://pymol.sourceforge.net/).
Resulting models were then submitted on the web-server PDBeFold, a
free software for fast protein structure alignment in three dimensions, and
predicted 3D model of our chimeric protein was compared with all resolved
structures present in the PDB database through iterative three-dimensional
alignment of protein backbone C atoms (http://www.ebi.ac.uk/msd-
srv/ssm/cgi-bin/ssmserver).
The sequence of the putative artificial protein was also analyzed by the
web-server PRED-TMBB (http://bioinformatics.biol.uoa.gr/PRED-TMBB/) to
check whether the construct can form transmembrane β-strands (93)(94).
3.2 PCR amplification of the basic module
The predicted single module identified above was 126 bp long,
corresponding to 42 amino acids. It was amplified using as a template the wild
type protein PA83 cloned in pET21(a), kindly provided by Prof. R. Benz of
Rudolf-Virchow- Zentrum für Biomedizin (Würzburg, Gerrmany) and the
following primers: forward: 5‟-AAA CGG ACC GGA ACT TCT ACA AGT
AGG ACA CAT-3‟; reverse: 5‟- AAA CGG TCC GAA TTG CGA CCG TAC
TTG AAT T-3‟. A CpoI restriction sites were added at the extremities. In the
forward primer was added a GA, between the recognition site for CpoI

METHODS
21
restriction enzyme and the starting basic module sequence, in order to maintain
the correct frame between multimers after the ligation.
The PCR was carried out with the following program: 1 cycle at 95°C x
3min., 25 cycles at: 95°C x 20 sec. - 58°C x 10 sec. - 72°C x 20 sec., 1 cycle at
72°C x 5 min.
3.3 Multimerization and insertion in pET52b
expression vector
The multimerization reaction was performed after digestion with CpoI
enzyme (Fermentas) of basic module amplified.
A ligation reaction among basic modules present in PCR mixture was
carried out for 2h at 22°C and with 300 rpm shaking. The ligation reaction was
checked in 2% TAE Agarose gel.
The vector used in heterologous expression experiments in this work was
the phagemide pET-52b (Novagen) (Fig. 3.1). This vector is 5227bp long and
it was chosen for the presence of the recognition site for SanDI restriction
enzyme (Fig. 3.2). This enzyme, same as CpoI, allow an unidirectional cloning,
furthermore both produce sticky ends compatible each other. Therefore when
the multimers, which have sticky ends CpoI-generated, were ligated with
expression vector pET52(b) linearized by SanDI (Fermentas), the ligation
reaction between multimers and vector form hybrid sites SanDI/CpoI not more
recognizable by either (Fig. 3.3).
Figure 3.1: The phagemide pET52(b) with the recognition site SanDI (in
the MCS) in evidence.

METHODS
22
Figure 3.2: Cloning/expression region: SanDI recognition site is localized near the
Strep-TAG region. In this work cloning experiment was made to express recombinant protein
with fused Strep-TAG region useful for recombinant protein tagging or purification.
Figure 3.3: Scheme of ligation reaction between pET52(b) vector and multimers: Ligation of multimers CpoI-sticky-ending and the vector linearized by SanDI restriction enzyme. Junction form hybrid sites not more recognizable by either.

METHODS
23
The ultimate goal of this experiment is cloning the multimer containing 7
repeats of basic module. Reaction of ligation produce different multimers with
several repeats of the basic module and it is very difficult purify the researched
multimer and insert it in the expression vector pET52b. The applied strategy
developed a novel protocol to cloning and purify putative porine T297-I334
derived by 7 repeats of basic module, as bioinformatics software predicted.
Ligation mixture of multimers was ligated into the expression vector pET52b
linearized by SanDI and dephopholirated as follow:
- 5µL multimers ligation mixture;
- 100 ng pET52b/SanDI-deP
- T4 DNA ligase
- Buffer T4 DNA ligase
- Water up to 10 µL
This ligation was used to transform competent cell XL10GOLD.
3.4 PCR-based colony screening of cloned multimers
This procedure uses PCR to determine the size of DNA cloned into a
plasmid from a single colony on a transformation plate, while reserving some
of the bacteria for further growth and plasmid preparation.
In its simplest form, transformed colonies are directly tooth-picked into a
small volume of PCR reaction mix that includes primers that flank the cloning
site. Primer used in this work are T7 promoter primer as forward and T7
terminator primer as reverse (Fig. 3.2). Following in vitro amplification,
aliquots of each reaction are analyzed by agarose gel electrophoresis, which
reveals both the presence and size of cloned inserts.
The PCR was carried out with the following program: 1 cycle at 95°C x
10 min., 25 cycles: 95°C x 30 sec. - 50°C x 30 sec. - 72°C x 30 sec., 1 cycle at
72°C x 10 min.
The researched clone should includes a multimer with 7 repetition (1186
bp) and the resulting one was confirmed by sequencing.

METHODS
24
3.5 Prokaryotic strain used
Bacterial cells used for the expression are cells of Escherichia coli, strain BL21
(ΔE3), with a copy of the chromosomal gene for T7 RNA polymerase. This
gene is under the control of promoter lac UV5 insensitive to inhibition by
glucose and inducible by lactose or similar compound like isopropyl-β-D-1-
thiogalactopyranoside (IPTG). The pET-52b vector allows protein expression
at high levels in bacterial cells by induction
with IPTG. The IPTG is used in research laboratories because, unlike
allolactose (natural inducer), is not hydrolysed by the cell and, thus, its
concentration does not change during the experiment. Genotype of this strain is
the following: F− ompT gal dcm lon hsdSB (r−B m−B ) "(ΔE3 [lacI lacUV5-
T7 gene 1 ind1 sam7 nin5]).
3.6 Site-directed mutagenesis of T293-I334(7x) clone
The artificial porin, definitely cloned in pET52B, must be mutated in two
position to allow the correct expression: the first one (I) should remove the
nucleotides GA, added in the forward primer after CpoI recognition site (see
par. 3.2) in order to have the sequence in frame with Strep-TAG vector (Fig.
3.2). The latter mutation (II) is required to add a stop codone (TAA) at the
ending of multimer T293-I334(7x).
In this work site-directed mutagenesis was performed using QuikChange
site-directed mutagenesis (Stratagene) with PfuTurbo™ DNA polymerase that
replicates both plasmid strands with high fidelity and without displacing the
mutant oligonucleotide primers. The basic procedure utilizes a supercoiled
double-stranded DNA (dsDNA) vector with an insert of interest and two
synthetic oligonucleotide primers containing the desired mutation (Fig. 3.4).
Incorporation of the oligonucleotide primers generates a mutated plasmid
containing staggered nicks. Following temperature cycling, the product is
treated with Dpn I. The Dpn I endonuclease is specific for methylated and
hemimethylated DNA and is used to digest the parental DNA template and to
select for mutation-containing synthesized DNA. DNA isolated from almost all

METHODS
25
E. coli strains is dam methylated and therefore susceptible to Dpn I digestion.
The nicked vector DNA incorporating the desired mutations is then
transformed into XL10GOLD competent cells.
(I) Primers used for the first site-directed mutagenenesis are:
Forward (Mut GA pET/TI Fw):
5’-CTTGAAGTCCTCTTTCAGGGACCG ACTTCTACAAGTAGGA-3’
Reverse (Mut GA pET/TI rev):
5’-TCCTACTTGTAGAAGTCGGTCCCTGAAAGAGGACTTCAAG-3’
(II) Primer used for stop codon adding:
Forward (T293I334 STOP Fw):
5’-AGTACGGTCGCAATTTAACGGACCCGGGTACCA-3’
Reverse (T293I334 STOP rev):
5’-TGGTACCCGGGTCCGTTAAATTGCGACCGTACT-3’
3.7 Expression protein
Bacterial protein expression systems are popular because bacteria are
easy to culture, grow fast and produce high yields of recombinant protein.
However, multi-domain eukaryotic proteins expressed in bacteria often are

METHODS
26
non-functional because the cells are not equipped to accomplish the required
post-translational modifications or molecular folding. Also, many proteins
become insoluble as inclusion bodies that are very difficult to recover without
harsh denaturants and subsequent cumbersome protein-refolding procedures.
Since we suppose that the artificial porin cloned is a membrane protein,
we developed a protocol for bypassing all resulting problems. Membrane
proteins are especially difficult to produce in quantity and usually, 70%-80% of
proteins produced by recombinant techniques in E. coli form inclusion bodies
(i.e., protein aggregates).
3.7.1 Standard protocol for the expression of recombinant
protein
The first step (preculture) consisted in the inoculums of a single bacterial
colony in 10 ml of sterile LB broth and its incubation for 16 hours at 37°C at a
speed of 250 rpm. Following day, 3 ml of the preculture were added to a flask
containing 60 mL (1:20 dilution) of sterile LB broth in the presence of
antibiotics and this culture was incubated at 37°C at 250 rpm until the optical
density ot the broth did not reach the value of OD600 = 0.6. Just prior to
induction, the culture was splitted into 2 × 30 ml cultures. IPTG was added to
one of the 30 ml cultures and the other culture has been used as an uninduced
control. Both cultures were incubated over night at 18°C at 220 rpm shaking.
Following day, a 3 ml aliquot of the cultures, induced and not, were
centrifuged at 16.000g for 5 minutes and the pellet is lysed with a denaturant
solution (Par. 3.6.2).
3.7.2 Lysis of the cellular pellet
Lysis of cellular pellet allowed the solubilization of most cellular
proteins. In standard conditions it is used a solution containing high
concentration of denaturing agents. These experiment was performed with a
strong denaturant lysis buffer prepared with guanidine-HCl 6M as follow:
10 mM Tris-HCl (pH=8.0);
0.1 M Na-phosphate;

METHODS
27
6 M Guanidine-HCl;
10% (w/v) Sarkosyl (N-laurosylsarcosine);
Deionized H2O .
For each lysis was added an amount of denaturing buffer equal to 1/20 of
original volume of broth. Next step was the incubation at 4°C over night and
subsequently centrifugation for 30min at 9.300g. The collected samples were
checked on 12% SDS-PAGE, after washing cellular pellet lised by Guanidine-
HCl buffer with 10% TCA solution.

RESULTS
28
4 RESULTS
4.1 Bioinformatics analysis
The basic sequence module was chosen after a thorough screening of the
domain II of PA83 of Bacillus anthracis (PDB code: 1TZO). Accordingly, the
14-stranded β-barrel, forming the channel is created by unfolding β-hairpins in
a Greek-key motif (strands 2β1-2β4) (59)(60) (Fig. 4.1). The β-structured
hairpins is formed by 100 aminoacidic residues between Valine 263 and
Alanine 362, that, at low pH, insert itself in plasmatic membrane and form a
pore (Fig. 4.2).
Figure 4.1: PA83 domains: PA domain IV is top right, domain II is top left. The
elements in domain II predicted to undergo a large conformational change upon pore
conversion are colored red (95).

RESULTS
29
Figure 4.2: Model of acidification-induced pore formation by PA63. (A)
Unfolding of the Greek-key motif (strands 2β1-2β4 ) to form a β-hairpin. (B) Association of the
seven β-hairpins in the PA63 heptamer to form a 14-stranded, membrane-inserted β-barrel
(60).
Several trials was performed by analyzing a lot of combination among
the 100 residues forming the β-structure in B. anthracis. The consensus
sequence was finally found between Tyrosine 293 and Isoleucine 334 residues
(T293-I334) (Fig. 4.3):
NH2–RTGT293STSRTHTSEVHGNAEVHASFFDIGGSVSAGFSNSNSSTVAI334RT-COOH
where the highlighted aminoacids are sequences added to build the CpoI
restriction site in the PCR primers.
This sequence has been defined as the „basic module‟ throughout this
work. The module contains two β-strands and one connecting loop. Virtual
multimers were simply built repeating the single module from six to 10 times
and the structures resulting from the fused sequences were modeled using
theSP5 Fold Recognition server (http://sparks-
lab.org/yueyang/server/SPARKS-X/). This software was developed by Yang et
al. in 2011, after a series of single fold recognition methods (SPARKS, SP2,
SP3, SP
4 and SP
5) that are based on weighted matching of multiple profiles.
Number of repeats tested derive by assumption that membrane-inserted β-

RESULTS
30
barrel forming pore of PA63 originates by association of seven β-hairpins (Fig.
4.2).
Resulting models were visualized using the open source software
PyMOL, a Python-enhanced molecular graphics tool (http://www.pymol.org/)
(Fig. 4.4).

RESULTS
31
These models were saved as pdb files and submitted to free software
PDBeFold to evaluate their alignment with other structures, present in the
whole database, previously resolved by crystallography. The surprising results
was a very high alignment of T293-I334(7x) with the Fbw7-Skp1-Cyclin E
Complex (PDB code: 2ovr:B), that is an ubiquitin-mediated proteolysis of
cyclin E which plays a central role in cell-cycle progression, when cyclin E
accumulation is a common event in cancer (Fig. 4.5). This results suggest that
building artificial protein may help the understanding of potential role of key
proteins in particular cellular pathways.
Figure 4.5. Protein structure alignment in three dimensions obtained by
PDBeFold free software: (A) T293-I334(7x).pdb; (B) Fbw7-Skp1-Cyclin E Complex
(2ovr:B.pdb); (C) View superposed of both structures aligned; (D) Data of alignment sorted by
Q-score that represent the quality function of Cα-alignment, maximized by the SSM alignment
algorithm. Q-score reaches 1 only in the case of identical structures, and drops down with
increasing RMSD or decreasing alignment length.
In order to obtain more data about the virtual protein that we were creating, the
consensus sequence was submitted to topology predicting programs. The

RESULTS
32
topology of a protein structure is a highly simplified description of its fold
including only the sequence of secondary structure elements, and their relative
spatial positions and approximate orientations. This information can be
embodied in a two-dimensional diagram of protein topology, called a TOPS
cartoon. These cartoons are useful for the understanding of particular folds and
making comparisons between folds.
Sequence of putative artificial protein was also analyzed by the web-
server PRED-TMBB (http://bioinformatics.biol.uoa.gr/PRED-TMBB/). The
program is based on a Hidden Markov Model, capable of predicting the
transmembrane beta-strands of the gram-negative bacteria outer membrane
proteins and of discriminating such proteins from water-soluble ones when
screening large datasets. The model is trained in a discriminative manner,
aiming at maximizing the probability of the correct prediction rather than the
likelihood of the sequences. The training is performed on a non-redundant
database consisting of 16 outer membrane proteins (OMP's) with their
structures known at atomic resolution. This web-server can achieve predictions
at least as good comparing with other existing methods, using as input only the
amino-acid sequence, without the need of evolutionary information included in
multiple alignments. The method is also powerful when used for discrimination
purposes, as it can discriminate with a high accuracy the outer membrane
proteins from water soluble in large datasets, making it a quite reliable solution
for screening entire genomes. Prediction output file scored a value of 2.847,
which is lower than the threshold value of 2.965. The difference between the
value and the threshold indicates the possibility of the protein being an outer
membrane protein. Output comes with a graphical plot of the posterior
probabilities (Fig. 4.6)
The 'TransMembrane protein Re-Presentation in 2 Dimensions' tool,
automates the creation of uniform, two-dimensional, high analysis graphical
images/models of alpha-helical or beta-barrel transmembrane proteins. Protein
sequence data and structural information may be acquired from public protein
knowledge bases, emanate from prediction algorithms, or even be defined by

RESULTS
33
the user. Several important biological and physical sequence attributes can be
embedded in the graphical representation (Fig.4.7).
Figure 4.6. Posterior probability plot file elaborated by PRED-TMBB: 3
different labels to define the triple-state in which a residue can be: [green] for periplasmic
space, [red] for transmembrane strand and [blue] for extracellular space.
Figure 4.7. Graphical 2D model of putative T293-I334(7x) porin: putative
artificial porin is represented in a 2D image colored 'by Electrostatic Potential', according to the
electrical charge (assuming pH=7), by using blue for positive, red for negative and gray for
uncharged residues.
4.2 PCR amplification of the basic module
The basic module was amplified using specific primer designed on
chosen sequence. In the sequence of primers was added a recognition site for
CpoI restriction enzyme at the ends of each one and furthermore, in the
forward primer, a couple of nucleotides (GA) was attached after CpoI site and

RESULTS
34
before the PA83 sequence in order to maintain the correct reading frame
among basic modules following ligation reaction to create multimers (Fig. 4.8).
Figure 4.8. Scheme of primers designed on chosen sequence: Forward primer
has a couple of nucleotide added (GA) after CpoI site to maintain the correct reading frame
among modules following ligation reaction.
In the figure 4.9 is showed the band amplified by PCR reaction. The
molecular weight of this band derives from the sum of 126 bp of basic module
plus 16 bp from CpoI recognition sites and GA (in the forward primer).
4.3 Multimerization and insertion in pET52b
expression vector After PCR reaction, basic modules were first digested to create CpoI-
sticky-ends and then ligated to each other in order to form multimers with a
variable repetition numbers (Fig. 4.10).

RESULTS
35
Since the multimer of interest has 7 repetition, as predicted by
bioinformatics analysis, we load on 2% agarose gel the ligation mixture in
order to purify from the gel the correct multimer (Fig. 4.10). Unfortunately the
concentration of discrete bands was too low to allowing the purification from
agarose gel of T293-I334(7x) so as cloning straight in the expression vector.
For this reason the whole ligation reaction containing multimers with various
repeats was used to cloning.
Figure 4.10. Representation of ligation reaction among basic modules.

RESULTS
36
Cloning experiment worked well and we found a lot of possible
candidate to became the correct clone of putative porine T293-I334. Thanks to
colony PCR it was easy discriminate and find clone with the researched
multimer inserted (Fig 4.12).
Figure 4.12. PCR-based colony screening of cloned multimers: Lane1: 100bp;
Lane 1-8: positive clone amplified by primers designed on vector (see Par. 3.4). Amplified
band showed in Lane 3 is from the clone containing the insert with 7 repeats of T293-I334
basic module. Molecular weigh of this band (1500bp) derive by molecular weigh of multimer
(1186bp) plus a small portion of vector amplified flanking the insert.
Sequence of amplified band of putative porin was confirmed by sequencing
(Fig.4.13)
Figure 4.13. Sequence of putative porin T297-I334(7x): figures shows how
multimer is correctly inserted into expression vector. In the yellow circle the hybrid restriction
site SanDI/CpoI following ligation vector/insert is highlighted. In red CpoI restriction sites

RESULTS
37
resulting from the ligation among basic modules are showed. Sequences in green show 7
repeats of T293-I334 basic module.
4.4 Site-directed mutagenesis of T293-I334(7x) clone
in vitro mutagenesis is a very powerful tool for studying protein
structure-function relationships, altering protein activity, and for modifying
vector sequences to incorporate affinity tags and correct frame shift errors.
Site-directed mutagenesis is the method of choice for altering a gene or
vector sequence at a selected location. Point mutations, insertions, or deletions
are introduced by incorporating primers containing the desired modification(s)
with a DNA polymerase in an amplification reaction.
In order to express the protein correctly, a needful adjustment to the
sequence showed in figure 4.12 is required, that is the deletion of the GA bases
added to forward primer (see Par. 3.2 and Fig. 4.8) at the beginning of the
sequence. This step is necessary in order to get T297-I334(7x) sequence in
frame with the Strep-TAG of the vector for its subsequent purification.
The second mutagenesis added a STOP codon (TAA) at the end of
putative porine sequence.
The occurred mutagenesis were confirmed by sequencing and thanks to a
free software (http://web.expasy.org/translate/) it was possible translate DNA
sequence in a aminoacidic sequence and check if the goal of experiment is
reached, that is the putative porin sequence is in frame with the vector Strep-
TAG and if is present a STOP at the its end (Fig. 4.14).
Figure 4.14. Aminoacidic sequence of T293-I334(7x) artificial porine after
mutagenesis: In green vector sequence is highlighted; orange letters shows coding sequence

RESULTS
38
as 7 repeats of basic module linked by RTG (in red) aminoacids (see par 3.2 and Fig. 4.8). At
the end of sequence the STOP codonTAA was added by mutagenesis.
4.5 Expression of T293-I334(7x) artificial porine
Traditional strategies for recombinant protein expression involve
transforming cells with a DNA vector that contains the template and then
culturing the cells so that they transcribe and translate the desired protein.
Typically, the cells are then lysed to extract the expressed protein for
subsequent purification.
Since we suppose that the artificial T293-I334(7x) porin cloned is a
membrane protein, we developed a protocol for bypassing all resulting
problems. Membrane proteins are especially difficult to produce in quantity
and usually, 70%-80% of proteins produced by recombinant techniques in E.
coli localize in inclusion bodies (i.e., protein aggregates).
Many tests were carried out in order to find the best conditions for
expression of our recombinant protein. A time course experiment was
performed at different IPTG concentration and at various temperature (18°C-
37°C) and pellets were lysated with different denaturing buffer (Urea 8M and
Guanidne-HCl 6M). A special protocol for recovering of inclusion bodies was
assayed. A very small amount of recombinant protein was recovered after the
induction with 1mM IPTG when the cells were over-night cultured at 18°C,
with 220rpm shaking (Fig. 4.15).
Interestingly, after the over-night incubation, the culture of induced
sample was less cloudy then the control not induced and the pellets were,
consequently, less abundant. Accordingly with experiments of Jean-Francxois
F. et al. (46) and starting from the assumption of this work, this result might
suggest a potential role of chimera T293-I334(7x) protein as inductor of
cellular lysis thanks to its capability to insert itself in membrane with high
efficiency. Since in a expression system for recombinant protein the chimera is
over-expressed, if it really is a forming-pore protein it‟s possible that the
formation of numerous pores in the membrane may determine the cell break.

RESULTS
39

DISCUSSION
40
5 DISCUSSION
5.1 Conclusion
During the last twenty years, several studies were performed with β-
barreled transmembrane protein channels from different issues: from the
genetic characterization to bio-functional assays(3) and structural analysis (33).
Objects of studies were not only prokaryotic porins, but eukaryotic
(mitochondrial) ones too (96)(97). These studies have provided a basic
understanding of the main biological mechanisms underlying solute diffusion
through porins across a biological membrane. A growing area of interest
concerns now the possible use of these protein channel in industrial
applications.
In agreement with this new investigation field, the basic idea of my PhD
thesis was to build completely artificial chimeric porins able to fold in a β-
barrel like structures with defined characteristics. This genomic construct
derived from a β-hairpins motif belonging to a protein that is a naturally pore-
forming protein and should have been based upon natural structure but
endowed with constrains to form pores with pre-defined diameters in
membranes. Thanks to bioinformatics analysis was found the ideal candidate to
basic module from Domain II of PA83 B. anthracis toxin. The greek-key
motif was replicated in vitro in serial arrangement(s) to form artificial pore-
forming structure. A similar approach has been adopted by Arnold and
colleagues who duplicated an eight-stranded β-barreled protein, producing a
functional pore(1). In literature there are studies about synthetic nanopores (98)
or about semi-natural proteins produced from α-hemolysin (58)(99).
Sequence of basic module was definitely find between Tyrosine 293 and
Isoleucine 334:
NH2–RTGT293STSRTHTSEVHGNAEVHASFFDIGGSVSAGFSNSNSSTVAI334RT-COOH
A module of 7 repeats of basic module was constructed and inserted in a
pET system expression vector; the expression vector used in this work was

DISCUSSION
41
adopted because it allows the design of a fusion protein with attached, in the N-
terminus side of the protein, the so called ”Strep-tag” which is used in
purification of the expressed protein by affinity chromatography.
The artificial protein was extracted from the inclusion bodies using a
strong denaturing solution containing 6M Guanidine-HCl, but the low level of
expression resulting suggest that another method for the expression of this
protein should be needed. Furthermore was noticed that induced cell culture
was, after an over-night incubation, less cloudy then the un-induced control.
These results both suggest that the over-expression of recombinant protein
induce some cellular response leading degradation of protein or cell lysis.
Maybe its mRNA is unstable or recombinant protein is toxic. From assumption
of this work, I designed and produced a porine, so the most likely hypothesis is
that the over-expression of the T293-I334(7x) artificial protein in E.coli leads
a cellular lysis because it massively insert itself into outer membrane and acts
as a kind of antibiotic.
After protein purification in recombinant systems, the most important
aim of future work will be investigate whether these artificial proteins are
really able to form pores.
This construct should be analyzed in a Black Lipid Membrane system
(BLMs-lipidic bilayer) in order to analyze its electrophysiology propriety
(pore‟s dimension, voltage-dependance, selectivity).
From the point of view of applied biology chimeric porins may allow
(us) to set the basis for producing future advanced techniques for rapid
diagnosis of diseases (biosensors and large scale sequencing) or for developing
new a-specific antibiotics(100)(101) or anticancer peptides.
5.2 Future investigation
To understand finally, in an absolutely way, how is the behavior of these
channels is required, at least, another year of laboratory tests. In addition, to
deepening the electrophysiological characterization of these proteins (such as
by studying the ionic selectivity), it would be appropriate some experiments
aimed at understanding the fold of the chimera with studies by circular

DISCUSSION
42
dichroism (CD) and/or calorimetry. After discovering all the features of these
proteins, the last step is undoubtedly the crystallization and the resolution of
the structure by X-ray or NMR.
Let me therefore outline the guidelines for a future project. The following
studies could be carried out:
- Ionic selectivity and other functional aspects.
- Circular Dichroism experiments (secondary structure information).
- Calorimetry (folding information).
- X-ray crystallography (tertiary structure information).

43
AKNOWLEDGEMENTS
Questi anni in cui si è svolto il mio lavoro di dottorato sono stati
veramente speciali. Ho vissuto molte cose meravigliose nella mia vita privata e
molte persone splendide sono entrate nella mia vita. Il dono più grande che io
abbia mai ricevuto si sveglia ogni mattina accanto a me e con il suo faccino
morbido si accoccola accanto al mio viso e mi dice “mamma…io mi sono
sbegliato” e io ringrazio per quanto amore ho la fortuna di vivere.
Ringrazio dal profondo del cuore il Prof. De Pinto che mi ha permesso di
vivere fino in fondo ancora una volta la vita di laboratorio e mi ha dato la
possibilità da fare un percorso di ricerca profondamente formativo, esperienza
tra l‟altro per me molto importante, in quanto probabilmente è stata l‟ultima
della mia lunga carriera accademica.
Ringrazio tutto lo “staff” del laboratorio, la Prof. Messina e la Prof.
Guarino, sempre prodighe di consigli utili; non ho parole per dire quanto mi
sono affezionata ad Andrea, Agata e Simona, per me compagni di avventura e
di risate interminabili. Grazie a tutti, senza di voi non ce l‟avrei mai fatta!
Ringrazio infine mio marito Francesco per avermi sopportato e
supportato senza sosta e per avermi aiutato in tutti i modi possibili a
completare il mio percorso. Questo lavoro è per te amore mio.

REFERENCES
44
REFERENCES
1. Gene duplication of the eight-stranded b-barrel OmpX produces a functional
pore: a scenario for the evolution of transmembrane beta-barrels. Arnold, T.,
Poynor, M., Nussberger, S., Lupas, A.N., and Linke, D.: J. Mol. Biol. 366,
1174–1184, 2007.
2. Structure and function of porins from gram-negative bacteria. Benz, R.:
Annu. Rev. Microbiol. 42, 359–393, 1988.
3. Permeation of hydrophilic molecules through the outer membrane of Gram-
negative bacteria. Benz R., Bauer K.: Eur. J. Biochem. 176, 1–19, 1988.
4. Structure and function of bacterial outer membrane proteins: barrels in a
nutshell. Koebnik R., KP. Locher, Van Gelder P.: Molecular Microbiology,
37(2):239–253, 2000.
5. Porins and specific channels of bacterial outer membranes. Nikaido, H.:
Molecular Microbiology 6(4), 435-442, 1992.
6. Evolutionary relationship between the TonB-dependent outer membrane
transport pro- teins: nucleotide and amino acid sequences of the Escherichia
coli colicin I receptor gene. Nau, CD, and Konisky, J.: J. Bacteriol.
171:1041-1047, 1989.
7. TonB and the Gram-negative dilemma. Postle, K.: Mol Microbiol 4: 2019-
2025, 1990.
8. Mitochondrial outer membrane contains a protein producing nonspecific
diffusion channels. Zalman Ls, Nikaido H, Kagawa Y.: Biol Chem 255:1771-
1774, 1980.
9. Structure and mode of action of a voltagedependentanion-selectivechannel
(VDAC) located in the outermitochondrial membrane. Colombini, M.: Ann.
N.Y.Acad. Sci. 341, 552–563, 1980.
10. VDAC, a multi-functional lmitochondrial protein regulating cell life and
death. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan
N, et al.: Mol. AspectsMed. 31: 227-285, 2010.

REFERENCES
45
11. VDAC channels mediate and gate the flow of ATP: implications for the
regulation of mitochondrial function. Rostovtseva, T. and Colombini, M.:
Biophys J. 72: 1954-62, 1997.
12. Mitochondrial Membrane Permeabilization in Cell Death. Kroemer G.,
Galluzzi L. and Brenner C.: Physiol. Rev. 87: 99-163, 2007.
13. Outer membrane VDAC1 controls permeability transition of the inner
mitochondrial membrane in cellulo durings stress-induced apoptosis.
Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina, S, et
al.: Cell. Res. 19: 1363-76, 2009.
14. VDAC1 selectively transfers apoptotic Ca(2+) signals to mitochondria. De
Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, and
Rizzuto R.: Cell Death Differ 19: 267-73, 2012.
15. VDAC1: from structure to cancer therapy. D., Shoshan-Barmatz V. and
Mizrachi.: Front.Oncol. 2: 164, 2012.
16. Deficiency of the Voltage-Dependent Anion Channel (VDAC): a novel
cause of mitochondrial myopathies. Huizing M, Ruitenbeek W,Thinnes F
and De Pinto V.: The Lancet 344: 762, 1994.
17. The structure of Voltage-Dependent Anion selective Channel: state of the
art. De Pinto V., Reina S., Guarino F., and Messina A.: J. Bioenerg.
Biomembr. 40, 139-147, 2008.
18. The Voltage-Dependent Anion selective Channel 1 (VDAC1) topography in
the mitochondrial outer membrane as detected in intact cell. Tomasello F.M.,
Guarino F., Messina A., Reina S., De Pinto V . (2013).: PlosOne, in press,
2013.
19. Pore-forming activity in the outer membrane of the chloroplast envelope.
FJ.OGGE UI, BENZ R.: FF.RS Lett 169:85-89, 1984.
20. Porins in the cell wall of mycobacteria. Trias J, Jarlier Y, Benz R.:
Science 258:1479-1481, 1992.
21. Alberts B., Johnson A., Lewis J. Raff M. Roberts K. Walter P.
Molecular Biology of the Cell.: Garland Science, 2002.

REFERENCES
46
22. Structural architecture of an outer membrane channel as determined by
electron crystallography. B. K. Jap, P. J. Walian & K. Gehring.: Nature 350:
167-169, 1991.
23. The orientation of beta-sheets in porin. A polarized Fourier transform
infrared spectroscopic investigation. E. Nabedryk, R.M. Garavito and J.
Breton.: Biophys J 53: 671-676, 1988.
24. Prediction of the general structure of porin from Rhodobacter capsulatus.
Orientation of porin in the membrane. Welte W, Weiss Ms, Nestel U,
Weckesser J, Schiltz E, Schulz Ge.: Biochim Biophys Acta 1080:271-274,
1991.
25. Primary structure of porin from Rhodobacter capsulatus. Schiltz E,
Kreusch A, Nestel U, Schu Lz Ge.: Eur J Biochem 199:587-594, 1991.
26. Mutations that alter the pore function of the OmpF porin of Escherichia
coli K12. Benson SA, Occi JL, Sampson BA.: J Mol Biol 203: 961-70, 1988.
27. Selectivity changes in site-directed mutants of the VDAC ion channel:
structural implications. E. Blachly-Dyson, S. Peng, M. Colombini, M.
Forte.: Science 247:1233-1236, 1990.
28. Solution structure of the integral human membrane protein VDAC-1 in
detergent micelles. Hiller S, Garces RG, Malia, TJ, Orekhov VY,
Colombini M and Wagner G.: Science 321: 1206-1210, 2008.
29. Structure of the human voltage-dependent anion channel. Bayrhuber M,
Meins T, Habeck M, Becker S, Giller K et al.: Proc. Natl. Acad. Sci. USA
105: 15370-15375, 2008.
30. The crystal structure of mouse VDAC1 at 2.3 angstrom resolution reveals
mechanisticin sights into metabolite gating. Ujwal R, Cascio D, Colletier JP,
Faham S, Zhang J et al.: Proc.Natl. Acad. Sci. USA 105: 17742-17747, 2008.
31. Bertil, Hille. Ionic Channels of Excitable Membranes.: Sinauer Associates
Inc., 1984.
32. Roland, Benz. Bacterial and Eukariotic Porins. Wiley-VCH, 2004.

REFERENCES
47
33. A 3D Model of the Voltage-dependent Anion Channel (VDAC). Casadio
R., Jacoboni I., Messina A. De Pinto V.: FEBS Lett., 520(1-3):1–7, 2002.
34. Generation of artificial channels by multimerization of beta-strands from
natural porin. Marco Lolicato, Simona Reina, Angela Messina, Francesca
Guarino, Mathias Winterhalter, Roland Benz and Vito De Pinto.: Biol.
Chem., Vol. 392, pp. 617–624, 2011.
35. A alpha/beta-barrel built by the combination of fragments from different
folds. Tanmay A. M. Bharat, S. Eisenbeis, K. Zeth B. Hoecker.: PNAS,
105(29):9942–9947, 2008.
36. Nanopores and nucleic acids: prospects for ultrarapid sequencing.
Deamer D, Akeson D.: Tibtech, 18(4):147–151, 2000.
37. Voltage-Driven DNA translocation through a Nanopore. Meller A, Nivon
L, Branton D.: Phys Rev Lett., 86(15):3435–8, 2001.
38. Electrically gated ionic channels in lipid bilayers. Ehrenstein G., Lecar
H.: Quarterly reviews of biophysics, 10(1):1–34, 1977.
39. Rational combinatorial design of pore-forming beta-sheet peptides.
Rausch JM, Marks JR, Wimley WC.: Proceedings of the National Academy
of Sciences, 102(30):10511–10515, 2005.
40. Nanopores: maltoporin channel as a sensor for maltodextrines and lamda-
phage. Berkane E., Orlik F., Danelon C. Fournier D. Benz R.Winterhalter
M.: Journal of Nanobiotechnology, 3(1):3, 2005.
41. Modulation of magainin 2-lipid bilayer interactions by peptide charge.
Matsuzaki K, Nakamura A , Murase O , Sugishita K , Fujii N , Miyajima
K .: Biochemistry: 36 (8), 1997.
42. The potential and challenges of nanopore sequencing. Daniel Branton,
David W Deamer, Andre Marziali, Hagan Bayley, Steven A Benner,
Thomas Butler, Massimiliano Di Ventra, Slaven Garaj, Andrew Hibbs,
Xiaohua Huang, Stevan B Jovanovich, Predrag S Krstic, Stuart Lindsay,
Xinsheng Sean Ling.: Nature Biotechnology 26, 1146 - 1153 , 2008.

REFERENCES
48
43. A biosensor that uses ion-channel switches. Cornell BA, Braach-
Maksvytis VL, King LG, Osman PD, Raguse B, Wieczorek L, Pace RJ.:
Nature: 387(6633):580-3., 1997.
44. Early Lung Cancer Diagnosis by Biosensors. Yuqian Zhang, Dongliang
Yang, Lixing Weng, Lianhui Wang.: Int J Mol Sci. 14(8): 15479–15509,
2013.
45. Designed to penetrate: time-resolved interaction of single antibiotic
molecules with bacterial pores. Nestorovich EM, Danelon C, Winterhalter
M, Bezrukov SM.: Proc Natl Acad Sci U S A.:99(15):9789-94, 2002.
46. Pore Formation Induced by an Antimicrobial Peptide: Electrostatic
Effects. Frantz Jean-Francxois, Juan Elezgaray, Pascal Berson,Pierre
Vacher,Erick J. Dufourc.: Biophysical Journal Volume 95:5748–5756, 2008.
47. Anthrax. Dixon, T.C., Meselson, M., Guillemin, J. and Hanna, P.C.: N.
Engl. J. Med., 1999.
48. Anthrax. Mock, M. and Fouet, A.: Annu. Rev. Microbiol. 55, 647-671,
2001.
49. Responding to the threat of bioterrorism: a microbial ecology perspective.
The case of anthrax. Atlas, R.M.: International microbiology: the official
journal of the Spanish Society for Microbiology 5, 161-167, 2002.
50. Anthrax toxin. Collier, R.J. and Young, J.A.: Annu. Rev. Cell Dev. Biol.
19, 45-70, 2003.
51. Macrophages are sensitive to anthrax lethal toxin through an acid
dependent process. Friedlander, A.M.: J. Biol. Chem. 261, 7123-7126, 1986.
52. Crystal structure of the anthrax toxin protective antigen. Petosa, C.,
Collier, R.J., Klimpel, K.R., Leppla, S.H. and Liddington, R.C.: Nature
385, 833-838, 1997.
53. Stoichiometry of anthrax toxin complexes. Mogridge, J., Cunningham, K.
and Collier, R.J.: Biochemistry (Mosc). 41, 1079-1082, 2002.
54. The lethal and edema factors of anthrax toxin bind only to oligomeric
forms of the protective antigen. Mogridge, J., Cunningham, K., Lacy, D.B.,

REFERENCES
49
Mourez, M. and Collier, R.J.: Proc. Natl. Acad. Sci. U. S. A. 99, 7045-7048,
2002.
55. Structure and function of anthrax toxin. Lacy, D.B. and Collier, R.J.:
Curr. Top. Microbiol. Immunol. 271, 61-85, 2002.
56. Structural basis for the unfolding of anthrax lethal factor by protective
antigen oligomers. Feld, G.K., Thoren, K.L., Kintzer, A.F., Sterling, H.J.,
Tang, Greenberg, S.G., et al.: Nature structural & molecular biology 17,
1383-1390, 2010.
57. Three-dimensional model of the pore form of anthrax protective antigen.
Structure and biological implications. Nguyen, T.L.: J. Biomol. Struct. Dyn.
22, 253-265, 2004.
58. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane
pore. Song, L., Hobaugh, M.R., Shustak, C., Cheley, S., Bayley, H. and
Gouaux, J.E.: Science: 274, 1859-1866, 1996.
59. Identification of residues lining the anthrax protective antigen channel.
Benson, E.L., Huynh, P.D., Finkelstein, A. and Collier, R.J.: Biochemistry
(Mosc). 37, 3941-3948, 1998.
60. PA63 channel of anthrax toxin: an extended beta-barrel. Nassi, S., Collier,
R.J. and Finkelstein, A.: Biochemistry (Mosc). 41, 1445-1450, 2002.
61. Acid-induced unfolding of the amino-terminal domains of the lethal and
edema factors of anthrax toxin. Krantz, B.A., Trivedi, A.D., Cunningham,
K., Christensen, K.A. and Collier, R.J.: J. Mol. Biol. 344, 739-756, 2004.
62. A phenylalanine clamp catalyzes protein translocation through the anthrax
toxin pore. Krantz, B.A., Melnyk, R.A., Zhang, S., Juris, S.J., Lacy, D.B.,
Wu, Z., et al.: Science: 309, 777-781, 2005.
63. Three-dimensional structure of the anthrax toxin pore inserted into lipid
nanodiscs and lipid vesicles. Katayama, H., Wang, J., Tama, F., Chollet, L.,
Gogol, E.P., Collier, R.J. and Fisher, M.T.: Proc. Natl. Acad. Sci. U. S. A.
107, 3453-3457, 2010.

REFERENCES
50
64. Manipulation of host signalling pathways by anthrax toxins. Turk, B.E.:
Biochem. J. 402,405-417, 2007.
65. On the role of macrophages in anthrax. Hanna, P.C., Acosta, D. and
Collier, R.J.: Proc. Natl. Acad. Sci. U. S. A. 90, 10198-10201, 1993.
66. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the
LPS/IFNgammainduced release of NO and TNFalpha. Pellizzari, R., Guidi-
Rontani, C., Vitale, G., Mock, M. and Montecucco, C.: FEBS Lett. 462,
199-204, 1999.
67. Impairment of dendritic cells and adaptive immunity by anthrax lethal
toxin. Agrawal, A., Lingappa, J., Leppla, S.H., Agrawal, S., Jabbar, A.,
Quinn, C. and Pulendran, B.: Nature 424, 329-334, 2003.
68. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine
receptor signalling. Rossi Paccani, S., Tonello, F., Patrussi, L., Capitani, N.,
Simonato, M., Montecucco, C.: Cellular microbiology 9, 924-929, 2007.
69. Identification of the cellular receptor for anthrax toxin. Bradley, K.A.,
Mogridge, J., Mourez, M., Collier, R.J. and Young, J.A.: Nature 414, 225-
229, 2001.
70. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin
pore formation. Scobie, H.M., Marlett, J.M., Rainey, G.J., Lacy, D.B.,
Collier, R.J. and Young, J.A.: PloS one 2, e329, 2007.
71. The LDL receptor-related protein LRP6 mediates internalization and
lethality of anthrax toxin. Wei, W., Lu, Q., Chaudry, G.J., Leppla, S.H. and
Cohen, S.N.: Cell 124,1141-1154, 2006.
72. Lethal factor of anthrax toxin binds monomeric form of protective antigen.
Chvyrkova, I., Zhang, X.C. and Terzyan, S.: Biochem. Biophys. Res.
Commun. 360, 690-695, 2007.
73. Anthrax protective antigen interacts with a specific receptor on the surface
of CHO-K1 cell. Escuyer, V. and Collier, R.J.: Infect. Immun. 59, 3381-3386,
1991.

REFERENCES
51
74. A quantitative study of the interactions of Bacillus anthracis edema factor
and lethal factor with activated protective antigen. Elliott, J.L., Mogridge, J.
and Collier, R.J. 2000, Vols. Biochemistry (Mosc). 39, 6706-6713.
75. Mapping the lethal factor and edema factor binding sites on oligomeric
anthrax protective antigen. Cunningham, K., Lacy, D.B., Mogridge, J. and
Collier, R.J.: Proc. Natl. Acad. Sci. U. S. A. 99, 7049-7053, 2002.
76. Large-scale structural changes accompany binding of lethal factor to
anthrax protective antigen: a cryo-electron microscopic study. Ren, G.,
Quispe, J., Leppla, S.H. and Mitra, A.K.: Structure 12, 2059-2066, 2004.
77. Anthrax edema factor, voltage-dependent binding to the protective antigen
ion channel and comparison to LF binding. Neumeyer, T., Tonello, F., Dal
Molin, F., Schiffler, B. and Benz, R.: J. Biol. Chem. 281, 32335-32343, 2006.
78. Anthrax lethal factor (LF) mediated block of the anthrax protective antigen
(PA) ion channel: effect of ionic strength and voltage. Neumeyer, T., Tonello,
F., Dal Molin, F., Schiffler, B., Orlik, F. and Benz, R.: Biochemistry (Mosc).
45, 3060-3068, 2006.
79. Fused polycationic peptide mediates delivery of diphtheria toxin A chain to
the cytosol in the presence of anthrax protective antigen. Blanke, S.R., Milne,
J.C., Benson, E.L. and Collier, R.J.: Proc. Natl. Acad. Sci. U. S. A. 93, 8437-
8442, 1996.
80. Anthrax toxin: the long and winding road that leads to the kill. Abrami, L.,
Reig, N. and van der Goot, F.G.: Trends Microbiol. 13, 72-78, 2005.
81. Anthrax toxin: receptor binding, internalization, pore formation, and
translocation . Young, J.A. and Collier, R.J.: Annu. Rev. Biochem. 76, 243-
265, 2007.
82. Evidence that translocation of anthrax toxin's lethal factor is initiated by
entry of its N terminus into the protective antigen channel. Zhang, S.,
Finkelstein, A. and Collier, R.J.: Proc. Natl. Acad. Sci. U. S. A. 101, 16756-
16761, 2004.

REFERENCES
52
83. Anthrax protective antigen: efficiency of translocation is independent of the
number of ligands bound to the prepore. Zhang, S., Cunningham, K. and
Collier, R.J.: Biochemistry (Mosc). 43, 6339-6343, 2004.
84. Anthrax biosensor, protective antigen ion channel asymmetric blockade.
Halverson, K.M., Panchal, R.G., Nguyen, T.L., Gussio, R., Little, S.F.,
Misakian, M., et al.: J. Biol. Chem. 280, 34056-34062, 2005.
85. Passive protection by polyclonal antibodies against Bacillus anthracis
infection in guinea pigs. Little, S.F., Ivins, B.E., Fellows, P.F. and
Friedlander, A.M.: Infect. Immun. 65, 5171-5175, 1997.
86. Efficiency of protection of guinea pigs against infection with Bacillus
anthracis spores by passive immunization. Kobiler, D., Gozes, Y., Rosenberg,
H., Marcus, D., Reuveny, S. and Altboum, Z.: Infect. Immun. 70, 544-560,
2002.
87. Blocking anthrax lethal toxin at the protective antigen channel by using
structure-inspired drug design. Karginov, V.A., Nestorovich, E.M., Moayeri,
M., Leppla, S.H. and Bezrukov, S.M.: Proc. Natl. Acad. Sci. U. S. A. 102,
15075-15080, 2005.
88. Beta-cyclodextrin derivatives that inhibit anthrax lethal toxin . Karginov,
V.A., Yohannes, A., Robinson, T.M., Fahmi, N.E., Alibek, K. and Hecht,
S.M.: Bioorg. Med. Chem. 14, 33-40, 2006.
89. Anthrax toxin protective antigen: inhibition of channel function by
chloroquine and related compounds and study of binding kinetics using the
current noise analysis. Orlik, F., Schiffler, B. and Benz, R.: Biophys. J. 88,
1715-1724, 2005.
90. Chloroquine prevents T lymphocyte suppression induced by anthrax lethal
toxin. Hirsh, M.I. and Cohen, V. s.l.: J. Infect. Dis. 194, 1003-1007, 2006.
91. Blockage of anthrax PA63 pore by a multicharged high-affinity toxin
inhibitor. Nestorovich, E.M., Karginov, V.A., Berezhkovskii, A.M. and
Bezrukov, S.M.: Biophys. J. 99, 134-143, 2010.
92. Improving protein fold recognition and template-based modeling by
employing probabilistic-based matching between predicted one-dimensional

REFERENCES
53
structural properties of the query and corresponding native properties of
templates. Yuedong Yang, Eshel Faraggi, Huiying Zhao, Yaoqi Zhou.:
Bioinformatics 27:2076-82, 2011.
93. PRED-TMBB: a web server for predicting the topology of beta-barrel outer
membrane proteins. Bagos PG, Liakopoulos TD, Spyropoulos IC and
Hamodrakas SJ.: Nucleic Acids Res;32, 2004.
94. A Hidden Markov Model method, capable of predicting and discriminating
beta-barrel outer membrane proteins. Bagos PG, Liakopoulos TD,
Spyropoulos IC and Hamodrakas SJ.: BMC Bioinformatics, 5:29, 2004.
95. Crystal structure of a complex between anthrax toxin and its host cell
receptor. Santelli E, Bankston LA, Leppla SH, Liddington RC.: Nature;
430(7002):905-8, 2004.
96. Structure of the voltage dependent anion channel: state of the art. V De
Pinto, S Reina, F Guarino A Messina.: J Bioenerg Biomembr, 40(3):139–
147, 2008.
97. A genetic analysis of the porin gene encoding a voltage-dependent anion
channel protein in Drosophila melanogaster. Oliva M, De Pinto V, Barsanti
P Caggese C.: Mol Genet Genomics, 267(6):746–756, 2002.
98. Detecting single stranded DNA with a solid state nanopore. Fologea D,
Gershow M, Ledden B McNabb DS, Golovchenko JA Li J.: Nano Letters,
5(10):1905–1909, 2005.
99. Driven DNA Transport into an Asymmetric Nanometer-Scale Pore.
Henrickson S, Misakian M, Robertson B Kasianowicz J.: Phys Rev Lett.,
85(14):3057–3060, 2000.
100. Rational combinatorial design of pore-forming beta-sheet peptides.
Rausch JM, Marks JR, Wimley WC.: Proceedings of the National Academy
of Sciences, 102(30):10511–10515, 2005.
101. Interaction of zwitterionic penicillins with the OmpF channel facilitates
their translocation. Danelon C, Nestorovich EM, Winterhalter M Ceccarell
iM Bezrukov SM.: Biophysical Journaly, 90(5):1617–1627, 2006.

REFERENCES
54
102. Regulation of the mitochondrial outer membrane channel, VDAC. M.,
Colombini.: J Bioenerg Biomemb 19:309-320, 1987.
103. Electron microscopy and image analysis of the mitochondrial outer
membrane channel, VDAC. Ca, Mannella.: J Bioenerg Biomemb 19:329-340,
1987.
104. Determination of the number of polypeptide subunits in a functional
VDAC channel from Saccharomyces cerevisiae. Peno S, Blachly-Dyson E,
Colombini M, Forte M.: J Bionerget Biomemb 24:27-31, 1992.
105. Interaction of mitochondrial porin with cytosolic proteins. D, Brdiczka.:
Experentia 46:161-167, 1990.
106. Intracellular compartmentation, structure and function of cratine kinase
isozymes in tissues with high and fluctuating energy demands: The
'phophocreatine circuit' for cellular energy homeostasis. Wallimann T, Wyss
M, Brdlczka D, Nicolay K, Eppenberger Mh.: Biochem J 281:21-40, 1992.
107. Peripheral-type benzodiazepine receptors mediate translocation of
cholesterol from outer to inner mitochondrial membranes in adrenocortical
cells. Krueger Ke, Papadopoulos Y.: J Biol Chem 265: 15015-15022, 1990.
108. Characterization and partial purification of the VDAC-channel-
modulating protein from calf liver mitochondria. Llu My, Torgrimson A,
Colombini M.: Biochim Biophys Acta 1185:203- 212, 1994.
109. Cloning and in situ localization of a brain-derived porin that constitutes a
large-conductance anion channel in astrocytic plasma membranes. .
Dermietzel R, Hwang Tk, Bu Ettner R, Hofer A, Dotzler E, Kremer M,
Deutzmann R, Thinnes F, Fishman Gi, Spray Dc, Siemen D.: Proc Natl
Acad Sci USA 91:499-503, 1994.
Recommended

















