
VETAGRO SUP CAMPUS VETERINAIRE DE LYON
Année 2017 - Thèse n° 045
CONTRIBUTION À L’ÉTUDE DE LA PHARMACOCINÉTIQUE ET DE LA TOLÉRANCE DE LA DOXORUBICINE CHEZ LE
CHIEN
THESE
Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I (Médecine - Pharmacie)
et soutenue publiquement le 13 octobre 2017 pour obtenir le grade de Docteur Vétérinaire
par
CHAVALLE Thomas Né le 12 mai 1992
à Marcq-en-Barœul (59)


VETAGRO SUP
CAMPUS VETERINAIRE DE LYON
Année 2017 - Thèse n° 045
CONTRIBUTION À L’ÉTUDE DE LA PHARMACOCINÉTIQUE ET DE LA TOLÉRANCE DE LA DOXORUBICINE CHEZ LE
CHIEN
THESE
Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I (Médecine - Pharmacie)
et soutenue publiquement le 13 octobre 2017 pour obtenir le grade de Docteur Vétérinaire
par
CHAVALLE Thomas Né le 12 mai 1992
à Marcq-en-Barœul (59)



3
LISTE DES ENSEIGNANTS DU CAMPUS VÉTÉRINAIRE DE LYON Mise à jour le 13 avril 2017

4

5
RemerciementsRemerciementsRemerciementsRemerciements
Au jury de thèse,
A Monsieur le Professeur Serge LEBECQUE,
De la Faculté de Médecine de Lyon Sud, Pour m’avoir fait l’honneur d’accepter la présidence de mon jury de thèse, Pour l’intérêt porté à mon travail, Hommages respectueux. A Monsieur le Professeur Philippe BERNY,
Du campus Vétérinaire de Lyon, VetAgro Sup, Pour son importante contribution à ce travail de thèse mais aussi du master 2, Pour avoir su être toujours à mon écoute, et ce à tout moment, Un immense merci. A Madame le Professeur Frédérique PONCE,
Du campus Vétérinaire de Lyon, VetAgro Sup, Pour avoir participé à la conception et au développement de cette thèse, Pour votre gentillesse et vos conseils dans mes choix d’orientations Très sincères remerciements.

6

7
Aux personnes ayant contribué à la réalisation de ce travail,
A l’équipe du service de toxicologie-pharmacologie de VetAgro Sup, sous la responsabilité de Monsieur le Professeur Philippe BERNY, et tout particulièrement à Danièle VEY et Bernadette ESPAÑA, Pour leur patience et leur aide dans la réalisation de cette étude, Pour le financement de la phase analytique de notre étude. A l’équipe du service de cancérologie clinique de VetAgro Sup, sous la responsabilité de Madame le Professeur Frédérique PONCE, et notamment Gabriel CHAMEL et Pauline DENOEUX, Pour leur aide précieuse dans la réalisation des prélèvements et pour tout ce qu’ils m’ont transmis. A toutes les personnes m’ayant assistées dans la réalisation des prélèvements, dont Sandrine FRANCHEQUIN, Eglantine NOBLET, Léa PU, Romain MALLET, Boris DEPRÉ, Pour avoir toujours répondu présents malgré la nuit, la fatigue et mes blagues. A Rita, Alba, Alma, Cléo, Sam, Cooki, Ibie, Baya et à leurs propriétaires, Pour leurs participations toujours enthousiastes à notre étude.

8

9
Table des matièresTable des matièresTable des matièresTable des matières
TABLE DES ANNEXES................................................................................................. 13
TABLE DES FIGURES ..................................................................................... 15
TABLE DES TABLEAUX ................................................................................. 17
LISTE DES ABRÉVIATIONS ........................................................................... 19
INTRODUCTION ........................................................................................... 21
PARTIE I : ETUDE BIBLIOGRAPHIQUE DE LA DOXORUBICINE EN MEDECINE VETERINAIRE ................................................................................................. 23
I. LA DOXORUBICINE, PHARMACIE CHIMIQUE .......................................................................... 25
A. Historique, formule chimique et synthèse.............................................................. 25
1. Historique .................................................................................................................... 25
2. Structure chimique de la doxorubicine .......................................................................... 26
3. Synthèse de la doxorubicine ......................................................................................... 27
B. Principales propriétés physico-chimiques et stabilité de la doxorubicine ................ 30
1. Principales caractéristiques physico-chimiques de la doxorubicine ................................. 30
2. Stabilité de la doxorubicine en solution ......................................................................... 31
C. Principales méthodes d’analyse de la doxorubicine ............................................... 31
1. La spectrophotométrie infrarouge ................................................................................. 31
2. La spectrophotométrie ultraviolet et visible .................................................................. 32
3. La spectrofluorimétrie .................................................................................................. 33
4. La spectrométrie de masse ........................................................................................... 33
II. PHARMACOCINETIQUE DE LA DOXORUBICINE ................................................................ 34
A. Voie d’administration et absorption ...................................................................... 34
B. Distribution .......................................................................................................... 34
C. Métabolisme ........................................................................................................ 35
1. Le doxorubicinol ........................................................................................................... 36

10
2. Le dérivé semi-quinone ................................................................................................. 36 3. Les dérivés aglycones .................................................................................................... 36
D. Elimination ........................................................................................................... 37
III. PHARMACODYNAMIE DE LA DOXORUBICINE ................................................................. 42
A. Interactions directes de la doxorubicine avec l’ADN et sa dynamique .................... 42
1. Transport nucléaire de la doxorubicine ......................................................................... 42
2. Inhibition de l’ADN topoisomérase de type II ................................................................ 43 a. La structure topologique de la molécule d’ADN chez les eucaryotes ............................ 43
b. Les ADN topoisomérases ........................................................................................... 45
c. Action de la doxorubicine sur les ADN topoisomérases de type II ................................ 49
3. Intercalation de la doxorubicine au sein de la molécule d’ADN ...................................... 50
a. Mécanisme d’intercalation de la doxorubicine ........................................................... 51
b. Conséquences de l’intercalation de la doxorubicine au sein de la molécule d’ADN ...... 51
B. Réactivité chimique de la doxorubicine et interaction avec les composés
cellulaires ................................................................................................................. 53
1. Doxorubicine et réactions d’oxydo-réduction ................................................................ 53 a. Formation de ROS par réduction à un électron de la doxorubicine .............................. 53
b. Formation d‘espèces alkylantes par réduction de la doxorubicine .............................. 54
c. Formation de complexes doxorubicine-métal ............................................................. 55
2. Le stress oxydatif, une conséquence de la réduction de la doxorubicine ......................... 58
a. Le concept de stress oxydatif ..................................................................................... 58
b. L’inhibition de la thiorédoxine réductase par la doxorubicine ..................................... 59
c. Perturbation de la chaîne mitochondriale de transport d’électrons par la doxorubicine60
d. Les conséquences du stress oxydatif sur les composés cellulaires ............................... 61
e. Importance du stress oxydatif dans la cytotoxicité tumorale de la doxorubicine ......... 64
3. La formation d’adduits, conséquence de la réactivité de la doxorubicine ....................... 65
a. Adduit à l’ADN par chélation directe de la doxorubicine ............................................. 66
b. Adduits à l’ADN par formation d’agents alkylants ...................................................... 68
C. Effets de la doxorubicine sur les membranes plasmiques ....................................... 71
1. Rappel sur la structure des membranes plasmiques ....................................................... 71
2. Effets de la doxorubicine sur les membranes plasmiques ............................................... 72 a. Perturbation de la fluidité et de la perméabilité membranaires par la doxorubicine .... 72
b. Modifications de la voie de signalisation des céramides ............................................. 73
D. Les différents mécanismes de mort cellulaire doxorubicine-induits ........................ 75
1. L’apoptose cellulaire ..................................................................................................... 75

11
2. L'autophagie cellulaire .................................................................................................. 77 a. Rôle de l’inhibition de mTOR dans la régulation de l’autophagie cellulaire .................. 77
b. Rôle de l’activation de la c-Jun N-terminal kinase (JNK) dans l’autophagie cellulaire ... 78
3. La sénescence cellulaire ................................................................................................ 79
4. La nécrose cellulaire ..................................................................................................... 80
E. Les mécanismes de résistance des cellules tumorales à la doxorubicine ................. 82
1. La résistance à la doxorubicine par augmentation de son efflux cellulaire ...................... 84
2. La résistance à la doxorubicine associée aux ADN topoisomérases de type II .................. 85 a. Résistance par altération de la concentration en ADN topoisomérase de type II .......... 86
b. Résistance altération de la sensibilité de l’ADN topoisomérase de type II à la
doxorubicine ................................................................................................................. 86
3. La résistance associée à une modulation des processus enzymatiques ........................... 87
a. Altération du métabolisme de la doxorubicine ........................................................... 87
b. Résistance par stimulation des mécanismes de détoxification cellulaire ..................... 87
4. La résistance par réparation des dommages à l’ADN ...................................................... 88
5. Résistance par inhibition de l’apoptose ......................................................................... 89
IV. LA DOXORUBICINE EN MEDECINE VETERINAIRE ............................................................. 91
A. Principales indications de la doxorubicine en médecine vétérinaire ....................... 91
B. Les principales interactions médicamenteuses avec la doxorubicine ...................... 92
C. Toxicité et effets indésirables de la doxorubicine ................................................... 93
1. Le choc histaminique .................................................................................................... 94
2. La toxicité gastro-intestinale ......................................................................................... 94
3. Toxicité hématologique ................................................................................................ 96
4. Toxicité cutanée lors d’extravasation ............................................................................ 97
5. L’alopécie cutanée ........................................................................................................ 98
6. La toxicité cardiaque ..................................................................................................... 98 a. Epidémiologie et symptomatologie de la cardiotoxicité doxorubicine-induite ............................................. 99 b. Mécanismes à l’origine de la cardiotoxicité doxorubicine-induite .............................................................. 100 c. Diagnostic de la cardiotoxicité doxorubicine-induite .................................................................................. 108 d. Traitement et prévention de la cardiotoxicité doxorubicine-induite .......................................................... 109
7. Principales contre-indications de la doxorubicine en médecine vétérinaire ................... 109
8. Mutation et toxicité ..................................................................................................... 110

12
PARTIE II : ETUDE EXPERIMENTALE DE LA PHARMACOCINETIQUE ET DE LA TOXICITE DE LA DOXORUBICINE EN MEDECINE VETERINAIRE ...................... 115
I. MISE AU POINT D’UNE METHODE ANALYTIQUE DE DOSAGE DE LA DOXORUBICINE PLASMATIQUE
PAR HPLC-FLUORESCENCE ..........................................................................................117
A. Mise au point de la méthode analytique .............................................................. 117
1. Description des conditions d’analyse par chromatographie ........................................... 117
2. Préparation des solutions ............................................................................................ 117
3. Préparation des échantillons de plasma ....................................................................... 118
4. Vérification de la méthode de dosage .......................................................................... 119
B. Discussion ............................................................................................................ 126
II. APPROCHE NON COMPARTIMENTALE DE LA PHARMACOCINETIQUE ET DE LA TOLERANCE DE LA
DOXORUBICINE CHEZ LE CHIEN......................................................................................128
A. Matériels et méthodes ......................................................................................... 128
1. Recrutement des cas .................................................................................................... 128
2. Echantillonnage ........................................................................................................... 128
3. Analyse des échantillons .............................................................................................. 129
B. Résultats ............................................................................................................. 129
1. Population d’étude et suivi clinique et biologique des animaux .................................... 129
2. Pharmacocinétique ...................................................................................................... 132
3. Discussion ................................................................................................................... 133 a. Pharmacocinétique et ses implications ..................................................................... 133
b. Limitations du protocole d’étude .............................................................................. 135
c. Perspectives de l’étude ............................................................................................. 136
CONCLUSION ............................................................................................... 141
BIBLIOGRAPHIE ........................................................................................... 141
ANNEXES ..................................................................................................... 165

13
TABLE DES TABLE DES TABLE DES TABLE DES ANNEXESANNEXESANNEXESANNEXES
Annexe 1 : Classification VCOG-CTCAE (Veterinary cooperative oncology group –
common terminology criteria for adverse events) version 1.1 ............................................. 167
Annexe 2 : Accord par le Comité d’éthique en expérimentation animale de VetAgro Sup
(n°1648) .................................................................................................................................. 175
Annexe 3 : Questionnaire présenté au propriétaire permettant d’évaluer les effets
secondaires de l’administration de doxorubicine. ................................................................. 177
Annexe 4 : Numération-formule sanguine et biochimie sanguine lors de l’admission
des cas dans l’étude et au cours des suivis à une semaine et trois semaines. ...................... 179

14

15
TABLE DES FIGURESTABLE DES FIGURESTABLE DES FIGURESTABLE DES FIGURES
Figure 1 : Formule semi-développée de la doxorubicine ......................................................... 27
Figure 2 : Isolement du chlorhydrate de doxorubicine à partir de la souche Streptomyces
peucetius var. caesisus ............................................................................................................. 27 Figure 3 : Mécanismes biochimiques de formation de la ε -rhodomycinone ......................... 28 Figure 4 : Mécanismes biochimiques de formation de la (TDP)-ι-daunosamine ..................... 28 Figure 5 : Mécanismes biochimiques de formation de la doxorubicine .................................. 29 Figure 6 : Schématisation de la formation de la doxorubicine à partir de la daunorubicine .. 29 Figure 7 : Spectre d'absorption infrarouge de la doxorubicine dans une solution de bromure de potassium 0,6% .................................................................................................... 32 Figure 8 : Spectre d'absorption ultraviolet et visible de la doxorubicine dans du méthanol .. 32 Figure 9 : Spectre d’émission (en rouge) et d’excitation (en noir) de fluorescence de la doxorubicine ............................................................................................................................. 33 Figure 10 : Spectre de masse de la doxorubicine..................................................................... 33 Figure 11 : Schéma des voies métaboliques de la doxorubicine ............................................. 39 Figure 12 : Schématisation des modèles de pharmacocinétique bi-compartimental et tri-compartimental de la doxorubicine .................................................................................... 40 Figure 13 : Schéma ADME de la doxorubicine (Absorption, Distribution, Métabolisme, Elimination), détaillant les transporteurs cellulaires ............................................................... 41 Figure 14 : Modalités d’action du complexe protéasome-doxorubicine ................................. 43 Figure 15 : Caténanes au cours de la réplication (à gauche) et nœuds d’ADN (à droite)........ 44 Figure 16 : La réaction de transestérification caractérisant l’activité des topoisomérases .... 45 Figure 17 : Mécanisme de la relaxation du surenroulement par les ADN topoisomérases de type I .................................................................................................................................... 46 Figure 18 : Cycle de topo-isomérisation par une ADN topoisomérase de type II .................... 48 Figure 19 : Modèles cinétiques d’intercalation de la doxorubicine au sein de l’ADN. (A) Modèle à 3 étapes. (B) Modèle à cinq étapes.......................................................................... 51 Figure 20 : Formation de ROS par réduction à un électron de la doxorubicine ...................... 54 Figure 21 : Formation de radicaux alkylants par réduction de la doxorubicine ...................... 55 Figure 22 : Formation de ROS par formation du complexe doxorubicine-fer(III) .................... 56 Figure 23 : Libération de fer à partir de l’aconitase cytoplasmique et formation de ROS ...... 57 Figure 24 : Régulation de l’homéostasie ferrique et implication du doxorubicinol ................ 58 Figure 25 : Les systèmes antioxydants enzymatiques ............................................................. 59 Figure 26 : Mécanisme de peroxydation des lipides ................................................................ 62 Figure 27 : Oxydation des protéines porteuses d’un groupement thiol .................................. 64 Figure 28 : Structure du complexe doxorubicine-ADN par liaison covalente (en rouge) ........ 66 Figure 29 : Formation du composé intermédiaire DOXform, hydrolyse en un métabolite actif, et mécanismes de formation d’adduits doxorubicine-ADN .................................................... 67 Figure 30 : Adduits à l’ADN formé par alkylation monofonctionnelle à partir du radical méthide quinone ...................................................................................................................... 68 Figure 31 : Rôle de la réactivité chimique de la doxorubicine à l’origine de sa cytotoxicité cellulaire ................................................................................................................................... 69 Figure 32 : Organisation de la membrane plasmatique et notion de radeaux lipidiques ....... 71 Figure 33 : Métabolisme des céramides et effets de la doxorubicine ..................................... 74 Figure 34 : Rôle de la doxorubicine dans l’activation des voies extrinsèque et intrinsèque de l’apoptose cellulaire ............................................................................................................ 76 Figure 35 : Rôle de la doxorubicine dans l’induction de l’autophagie cellulaire ..................... 78

16
Figure 36 : Rôle de la doxorubicine dans l’induction de la sénescence prématurée .............. 80 Figure 37 : Rôle de la doxorubicine dans la nécrose cellulaire ................................................ 81 Figure 38 : Les différents mécanismes de réparation de l’ADN selon le mécanisme de cytotoxicité doxorubicine-induit .............................................................................................. 88 Figure 39 : Les différents mécanismes de résistance à la doxorubicine .................................. 90 Figure 40 : Nécrose cutanée due à l’extravasation de doxorubicine chez une chienne de 12 ans, deux semaines après l’injection de doxorubicine ....................................................... 97 Figure 41 : Signalisation β-adrénergique cardiaque et doxorubicine .................................... 105 Figure 42: Principaux mécanismes de cardiotoxicité de la doxorubicine .............................. 113 Figure 43 : (A) Chromatogramme de plasma blanc. (B) Chromatogramme de plasma supplémenté en doxorubicine (DOX) et daunorubicine (DNR) à 0,1 µg/mL. ...................... 119 Figure 44 : Courbes d’étalonnage moyenne évaluant la linéarité de la méthode analytique ............................................................................................................................... 121 Figure 45 : Chromatogramme de plasma supplémenté en doxorubicinol, doxorubicine et daunorubicine à 1 µg/mL. .................................................................................................. 125 Figure 46 : Evolution du comptage des polynucléaires neutrophiles au cours des suivis chez quatre chiens ................................................................................................................. 130 Figure 47 : Courbes de pharmacocinétique ........................................................................... 133

17
TABLE DES TABLEAUXTABLE DES TABLEAUXTABLE DES TABLEAUXTABLE DES TABLEAUX
Tableau I : Principales propriétés physico-chimiques de la doxorubicine ............................... 30
Tableau II : Principales bande d'absorption infrarouge de la doxorubicine dans une
solution de KBr 0,6% ................................................................................................................ 32
Tableau III : Principales indications de la chimiothérapie à base de doxorubicine ................. 91
Tableau IV : Principales interactions médicamenteuses de la doxorubicine ........................... 93
Tableau V : Effets indésirables de la doxorubicine à court-terme ........................................... 93
Tableau VI : Fréquence de l’allèle mdr1 mutée et des génotypes ........................................ 110
Tableau VII : Gradient d’élution utilisée lors de la chromatographie par HPLC .................... 117
Tableau VIII : Méthode de préparation de la série d’étalonnage .......................................... 118
Tableau IX : Validation des critères de linéarité de la méthode analytique .......................... 121
Tableau X : Résultats de l’étude de précision et d’exactitude de la méthode analytique .... 123
Tableau XI : Résultats de l’étude de rendement d’extraction de la méthode analytique ..... 124
Tableau XII : Résultats de l’étude de stabilité de la doxorubicine dans du plasma ............... 125
Tableau XIII : Méthodes d’analyses par HPLC de détermination de la concentration en
doxorubicine dans des liquides biologiques et des tissus. ..................................................... 126
Tableau XIV : Description des cas inclus dans notre étude .................................................... 131
Tableau XV : Effets secondaires observés suite à la séance de chimiothérapie et
thérapeutiques mises en place .............................................................................................. 132
Tableau XVI : Paramètres pharmacocinétiques calculés pour chaque individu de l’étude ... 132
Tableau XVII : Comparatif des paramètres pharmacocinétiques des différentes études
portant sur des animaux non sains ........................................................................................ 134

18

19
LISTE DES ABRÉVIATIONSLISTE DES ABRÉVIATIONSLISTE DES ABRÉVIATIONSLISTE DES ABRÉVIATIONS
°C : Degrés Celsius
ADN : Acide désoxyribonucléique
ARN : Acide ribonucléique
ATP : Adénosine-Triphosphate
AUC : Aire sous la courbe (Area under the curve)
BCRP : Breast cancer resistance protein
CV : Coefficient de variation
DNR : Daunorubicine
DOX : Doxorubicine
DOXol : Doxorubicinol
EMA : Agence européenne du médicament (European Medecine Agency)
IV : Intra-veineuse
IL-2 : Interleukine-2
IRE : Iron responsive elements
Kapp : Coefficient d’affinité
MDR : Multidrug resistance
MRP : Multidrug resistance related-protein
NADPH : Nicotinamide adenine dinucleotide phosphate
P-gp : Glycoprotéine-P
PO : Per os ou voie orale
ROS : Dérivés réactifs de l’oxygène (Reactive oxygen species)
SD : Ecart-type
TfR : Récepteur à la transferrine
Trr : Thiorédoxine réductase
VCOG-CTCAE : Veterinary cooperative oncology group – common terminology criteria for
adverse events

20

21
INTRODUCTIONINTRODUCTIONINTRODUCTIONINTRODUCTION
La doxorubicine est un antibiotique anticancéreux apparu dans les années 1960 et qui
s’est illustré comme outil thérapeutique essentiel dans le traitement de nombreuses
tumeurs chez l’Homme, notamment dans le cadre de la thérapeutique des carcinomes
mammaires, ovariens ou vésicaux, des ostéosarcomes et des sarcomes des tissus mous, des
lymphomes hodgkiniens ou non, des tumeurs solides chez l’enfant, des cancers du poumon
et des leucémies aiguës et chroniques.
Son utilisation en médecine vétérinaire est actée depuis de nombreuses années pour le
traitement des lymphomes et de diverses tumeurs solides, mais malgré cela, il n’existe que
peu d’études sur sa pharmacocinétique et sa pharmacodynamie chez le chien sain comme
chez le chien non sain et la posologie proposée (en mg/kg ou mg/m² selon la taille du chien)
ne repose que sur des données anciennes et non validées. Les protocoles analytiques utilisés
lors de ces études sont basés sur des protocoles développés chez l’Homme ou dans l’espèce
murine. Une méthode de dosage de la doxorubicine validée chez le chien serait donc
intéressante à mettre en place, afin d’être appliquée à des études de pharmacocinétique de
la doxorubicine dans cette espèce.
De plus, il existe de nombreuses études sur l’efficacité ainsi que sur la toxicité à plus ou
moins long terme de la doxorubicine chez le chien, sans qu’aucun lien ne soit fait avec le
schéma posologique proposé ni la pharmacocinétique au sein de cette espèce. L’approche
de la relation pharmacocinétique/pharmacodynamie et pharmacocinétique/toxicité pourrait
permettre la mise en place d’une thérapeutique personnalisée et non pas d’une
thérapeutique de groupe.
Dans une première partie, nous ferons un état des lieux des connaissances sur la
doxorubicine, en nous attachant à présenter particulièrement sa pharmacocinétique, ses
mécanismes d’action et son utilisation thérapeutique en médecine vétérinaire. La mise en
place de notre protocole expérimental et son application à une étude de pharmacocinétique
seront développées dans une seconde partie, pour aboutir sur les différentes perspectives
que notre étude permet d’envisager.

22

23
Partie IPartie IPartie IPartie I : Etude bibliographique de la : Etude bibliographique de la : Etude bibliographique de la : Etude bibliographique de la
doxorubicine en médecine vétérinairedoxorubicine en médecine vétérinairedoxorubicine en médecine vétérinairedoxorubicine en médecine vétérinaire

24

25
I. La doxorubicine, pharmacie chimiqueI. La doxorubicine, pharmacie chimiqueI. La doxorubicine, pharmacie chimiqueI. La doxorubicine, pharmacie chimique
La doxorubicine, ou adriamycine, fait partie de la famille des anthracyclines, médicaments
anti-cancéreux d’origine naturelle et possédant des propriétés antibactériennes. Les
anthracyclines ont été isolées à partir de bactéries Gram positive du genre Streptomyces sp
(Lown, 1993).
A. Historique, formule chimique et synthèseA. Historique, formule chimique et synthèseA. Historique, formule chimique et synthèseA. Historique, formule chimique et synthèse
1. Historique1. Historique1. Historique1. Historique
Le premier antibiotique à activité antitumorale, l’actinomycine A, produite par Actinomyces
antibioticus, fut découvert par Waksman et Woodruff en 1940. Suite à cette découverte, une
entreprise de recherche italienne, Farmitalia Research Laboratories, commence à investir
dans la recherche de composés anti-cancéreux issus de bactéries à partir du début des
années 50. La première anthracycline, la β-rhodomycine II ou rhodomycine A, est isolée en
1950 par Brockmann et Bauer à partir d’une souche de l’espèce Streptomyces purpurascens
(Morrison, 2010). Par la suite, à la fin des années 50, un pigment glycosidique rouge, la
rhodomycine B, est isolé à partir de Streptomyces sp au niveau d’un échantillon de sol indien
par Frederico Arcamone. Malgré l’activité antibactérienne importante de cette molécule, sa
forte toxicité, notamment une paralysie de la motilité intestinale découverte lors d’études
sur des souris, empêche son utilisation en tant qu’agent antitumoral. Cette première
découverte entraîne des études poussées sur les pigments glycosidiques issus de la bactérie
du genre Streptomyces sp (Lown, 1993).
En 1963, une équipe de recherche italienne du laboratoire Farmitalia découvre une nouvelle
molécule issue d’une souche de Streptomyces peucetius qu’elle nomme daunomycine, du
nom des Dauni, une tribu préromaine qui occupait la région où cette souche a été mise en
évidence. A la même période, une équipe française du laboratoire Rhône-Poulenc découvre
une molécule issue d’une souche de Streptomyces caeruleorubidus qu’elle nomme
rubidomycine, en référence à la couleur du composé. Ces deux molécules se révèleront être
une seule et unique molécule, alors renommée daunorubicine en 1968 (Morrison, 2010). Des
essais cliniques sur des souris ont lieu dans le cadre d’un traitement de carcinomes d’Ehrlich
et de sarcomes 180 et montrent une inhibition de la croissance tumorale et une
augmentation de la durée de survie des animaux. Les premiers essais thérapeutiques en
médecine humaine se déroulent en 1964 et montrent une efficacité en tant que traitement
anti-cancéreux, notamment contre les leucémies aigues. Malgré cela, des effets indésirables
majeurs tels qu’une aplasie médullaire sévère, une immunosuppression et une cardiotoxicité
à dose cumulative, empêchent son utilisation en tant que traitement anti-cancéreux en
phase d’entretien. Après comparaison des propriétés physicochimiques et biologiques de la
rhodomycine B et de la daunorubicine, il semblerait qu’une faible modification de la
structure des anthracyclines soit à l’origine d’une amélioration remarquable des propriétés
pharmacologiques. Ces découvertes orientent alors la recherche vers de nouveaux
analogues biosynthétiques, dérivés de Streptomyces peucetius (Di Marco et al., 1981).

26
En 1967, la doxorubicine (ou adriamycine), un analogue 14-hydroxy de la daunorubicine, est isolée à partir de la culture d’une des variétés dérivées de la souche originale de Streptomyces peucetius ATCC29050, obtenue par mutation après utilisation du N-nitroso-N-méthyl uréthane. La nouvelle souche est nommée Streptomyces peucetius var. caesius
ATCC27952. Le professeur A.DiMarco est le premier à mettre en évidence les propriétés anti-cancéreuses de la doxorubicine sur différentes tumeurs expérimentales chez la souris et le rat, qu’il présente lors du symposium sur l’adriamycine à Milan en 1971 (Carter et al., 1972). La doxorubicine est à l’origine d’une inhibition de la croissance tumorale et d’une augmentation de la durée de survie avec une meilleure activité antitumorale que la daunorubicine contre les tumeurs murines, notamment les tumeurs solides, et un index thérapeutique plus élevé (1,21 contre 0,67 pour la daunorubicine). Cependant, de nombreux effets indésirables dont la toxicité cardiaque dose-cumulative sont toujours présents (Arcamone et al., 1969).
La première utilisation de la doxorubicine chez le chien remonte à 1976 lors d’une étude sur
les effets systémiques, mettant en évidence les effets indésirables de celle-ci sur l’organisme
(Smith, Kirk, 1976).
Depuis les années 70, plus de 2000 analogues de la famille des anthracyclines ont été
développés au cours d’études in vitro et in vivo. Parmi les analogues testés dans des études
pré-cliniques, beaucoup se sont avérés décevants en phase I et II d’essais cliniques avec une
toxicité élevée et une activité antitumorale inférieure ou égale à celle de la doxorubicine
(Muggia, Green, 1991). Parmi ceux ayant montré un intérêt médical dans le domaine
vétérinaire, on peut citer :
� L’épirubicine, un dérivé semi-synthétique de la doxorubicine avec un index
thérapeutique plus élevé, une dose-cumulative plus importante permise par une
clairance plus rapide et un temps de demi-vie plus court et ce, pour une activité
antitumorale identique et une cardiotoxicité moindre. Son spectre d’activité et
similaire à celui de la doxorubicine. Cependant, peu d’études sont disponibles
chez le chien et l’épirubicine semble présenter une toxicité gastro-intestinale plus
importante.
� L’idarubicine, un analogue de la daunorubicine avec une efficacité thérapeutique
supérieure ou égale à celle-ci mais avec une cardiotoxicité encore mal
caractérisée. De plus, l’idarubicine présente une meilleure biodisponibilité
gastrique, permettant son usage per os. Elle présente cependant un spectre
d’activité plus réduit, limité au lymphome, et peu d’études sont disponibles sur
son intérêt dans le domaine vétérinaire.
2. Structure chimique de la doxorubicine2. Structure chimique de la doxorubicine2. Structure chimique de la doxorubicine2. Structure chimique de la doxorubicine La doxorubicine ou (8S,10S)-10-{[(2R,4S,5S,6S)-4-Amino-5-hydroxy-6-methyltetrahydro-2H-
pyran-2-yl]oxy}-8-glycoloyl-6,8,11-trihydroxy-1-methoxy-7,8,9,10-tetrahydro-5,12 tetracène-
dione (C27H29O11N), est un membre de la famille des anthracyclines.

27
Figure 1 : Formule semi-développée de la doxorubicine
Elle possède une structure poly-aromatique plane (Figure 1). Elle est composée d’une
fraction sucre, la daunosamine ou 3-amino-2.3.6-trideoxy-L-fucosyl, liée par une liaison
glycosique à une fraction chromophore aglycone, l’adriamycinone (ou 14-
hydroxydaunomycine), tétracycle avec des groupements adjacents quinone-hydroquinone,
un substituant méthoxy et une chaîne courte avec un groupement carbonyl se terminant par
un alcool primaire (Bakker et al., 1995).
3. Synthèse de la doxorubicine3. Synthèse de la doxorubicine3. Synthèse de la doxorubicine3. Synthèse de la doxorubicine
On distingue 2 méthodes empiriques de synthèse de la doxorubicine :
� La synthèse microbiologique
A partir de la fermentation de Streptomyces peucetius var. caesius ATCC2795, seule souche
capable de produire de la doxorubicine, suivi de son extraction et sa purification par
chromatographie à partition (Figure 2)(Malla et al., 2010).
Figure 2 : Isolement du chlorhydrate de doxorubicine à partir de la souche Streptomyces
peucetius var. caesisus Traduit et adapté de l’anglais d’après Arcamone et al. (1981).

28
D’un point de vue biochimique, on distingue 3 étapes à la formation de la doxorubicine au
sein de la souche Streptomyces peucetius var. caesius, toutes dépendantes de gènes
spécifiques (Arcamone et al., 1969).
1/ Formation de la ε-rhodomycinone (partie aglycone) à partir d’un propionyl-coenzyme A et
de 9 précurseurs malonyl-coenzyme A (Figure 3).
Figure 3 : Mécanismes biochimiques de formation de la ε -rhodomycinone
Traduit et adapté de l’anglais d’après Malla et al. (2009) AAME : acide aclanonique méthyl ester ; dnrC : acide-O-méthyltransférase ; dnrD : AAME cyclase ;
dnrE : aclavicétone réductase ; dnrF : aclavinone-11-hydroxylase ; dnrG : C-12 oxygénase ; dpsA : transférase ;
dpsB and dpsC : cétosynthases ; dpsD : acyltransférase ; dpsE : 9-cétoréductase ; dpsF : aromatase ; dpsG :
protéine porteuse d’acyl ; dpsY : cyclase ; SAM : S-adénosyl méthionine.
2/ Formation de la thymidine diphosphate (TDP)-ι-daunosamine (partie glycosamine) à partir
du glucose-1-phosphate (Figure 4).
Figure 4 : Mécanismes biochimiques de formation de la (TDP)-ι-daunosamine
Traduit et adapté de l’anglais d’après Malla et al. (2009). dnmJ : 3-aminotransférase ; dnmL : glucose-1-phosphate thymidylyltransférase ; dnmM : TDP-D-glucose 4,6-
déshydratase ; dnmT : 2,3-déshydratase ; dnmU : 3,5-épimérase ; dnmV : 4-cétoréductase.

29
3/ Glycosylation et modifications par méthylation, décarboxylation et hydroxylation de la
partie aglycone, aboutissant à la formation de doxorubicine (Figure 5).
Figure 5 : Mécanismes biochimiques de formation de la doxorubicine
Traduit et adapté de l’anglais d’après Malla et al. (2009). dnrK : S-adénosyl-L-méthionine-dépendante 4-O-méthyltransférase ; dnrP : rhodomycine D méthylestérase ;
dnrQ et dnrS : glycosyltransférases ; doxA : cytochrome P450 hydroxylase.
� La synthèse chimique
La doxorubicine est formée par semi-synthèse à partir de la daunorubicine, obtenue par
fermentation de différentes souches de Streptomyces sp. Une première réaction entre le
chlorhydrate de daunorubicine dans un solvant équimolaire méthanol/dioxane et une
solution chloroformique de dibrome est à l’origine de la formation du 14-bromo-
daunorubicine. S’en suit une hydrolyse par une solution aqueuse de formiate de sodium. Le
mélange est acidifié jusqu’à pH 3 avec extraction par du dichlorométhane. La phase aqueuse
est amenée à un pH 8,6 et extraite avec du dichlorométhane. La phase organique est alors
récupérée et séchée avec du sulfate de sodium anhydre puis traitée avec de l’acide
chlorhydrique et enfin diluée dans de l’éther. Le précipité formé correspond alors au
chlorhydrate de doxorubicine, purifié par cristallisation dans un mélange équimolaire de
méthanol et de 1-propanol. Cette méthode de synthèse présente un rendement de 36%
(Horton et al., 1988).
Figure 6 : Schématisation de la formation de la doxorubicine à partir de la daunorubicine
Traduit et adapté de l’anglais, d’après Horton et al. (1988).

30
Depuis, avec le développement de la génomique, de nouvelles méthodes de synthèse ont
été proposées, notamment à partir d’un transfert du gène doxA. Le gène doxA est cloné sur
un plasmide qui est ensuite introduit au sein du micro-organisme hôte. Ce gène permet
l’acquisition de la capacité de convertir la daunorubicine en doxorubicine. En effet, le gène
doxA code une enzyme cytochrome P450 hydroxylase qui catalyse l’hydroxylation de la
daunorubicine au niveau du carbone 14. On réalise ensuite l’extraction de la doxorubicine
des cultures de micro-organismes hôtes. Cette méthode de synthèse présente un rendement
d’environ 80%, selon les souches hôtes utilisées (Dickens, Strohl, 1996).
B. Principales propriétés physicoB. Principales propriétés physicoB. Principales propriétés physicoB. Principales propriétés physico----chimiques et stabilité de la doxorubicinechimiques et stabilité de la doxorubicinechimiques et stabilité de la doxorubicinechimiques et stabilité de la doxorubicine
1. Principales caractéristiques physico1. Principales caractéristiques physico1. Principales caractéristiques physico1. Principales caractéristiques physico----chimchimchimchimiques de la doxorubicineiques de la doxorubicineiques de la doxorubicineiques de la doxorubicine
Les principales caractéristiques physico-chimiques sont décrites dans le Tableau I.
Tableau I : Principales propriétés physico-chimiques de la doxorubicine
Caractéristiques
Aspect macroscopique Poudre cristallisée rouge
Poids moléculaire 543,52 Dalton
Constantes d’acidité logarithmiques pKa 1 (amine) = 8,15 pKa 2 (phénol en C11) = 10,16 pKa 3 (phénol en C6) = 13,2
Solubilité 10mg/mL dans l’eau 10 mg/mL dans DMSO Soluble dans les solutions alcooliques Modérément soluble dans le méthanol Non soluble dans solvants organiques non polaires
Température de fusion 230°C
Pression de vaporisation 8,99.10-25 mmHg
Pouvoir rotatoire spécifique + 248° à 20°C (0,1% dans le méthanol)
Coefficient de partage octanol/eau Log Ko/w = 1,27
Surface polaire topologique (TPSA) 206,07 Ų
Avec une température de fusion de 230°C, la doxorubicine se présente sous forme de
poudre cristallisée rouge à l’état pur. Celle-ci est soluble dans l’eau (10 mg/mL) et la plupart
des solutions alcooliques, modérément soluble dans le méthanol et non soluble dans les
solutions organiques polaires.
Cette base faible, de 543,52 Daltons, possède trois pKa, pKa1 (amine) = 8,15, pKa2 (phénol
en C11) = 10,16 et pKa3 (phénol en C6) = 13,2 et est donc chargée positivement à pH sanguin
(Bouma et al., 1986). Etant donné les différentes constantes d’acidité, la doxorubicine est
instable en milieu acide, expliquant son absence d’administration par voie orale. De plus, elle
présente des propriétés caustiques, contre-indiquant son administration par voie intra-
musculaire ou sous-cutanée (Gallois et al., 1996).

31
Etant donné son coefficient de partage octanol/eau légèrement supérieur à un, la
doxorubicine présente un caractère peu lipophile, à l’origine d’une faible absorption par voie
orale et de l’absence de passage de la barrière hémato-méningée. De plus, sa surface polaire
topologique supérieure à 140 Ų montre sa capacité naturelle faible à traverser les
membranes (Fernandes, Gattass, 2009).
2. Stabilité de la doxorubicine en solution2. Stabilité de la doxorubicine en solution2. Stabilité de la doxorubicine en solution2. Stabilité de la doxorubicine en solution
En ce qui concerne la stabilité de la doxorubicine, plusieurs caractéristiques sont à prendre en compte :
� La doxorubicine est une molécule photo-labile. Sa photo-dégradation est
supérieure à 20% après exposition à une source lumineuse pendant 3 jours pour
des concentrations inférieures à 100 µg/mL (Tavoloni et al., 1980). Sa dégradation
à la lumière est inversement proportionnelle à la concentration de la solution et
est accélérée lors d’une augmentation du pH. Cependant, les concentrations
utilisées en chimiothérapie étant élevées (au minimum 500 µg/mL), il n’y pas de
précautions particulières à prendre (Wood et al., 1990).
� La doxorubicine est également une molécule thermosensible. En solution saline à
pH 6,1, celle-ci se dégrade de 10% en quatorze jours à 37°C (Bosanquet, 1986).
Cependant, en solution à 2 mg/mL, aucune dégradation significative n’est
observée à 4°C pendant six mois ou à -20°C pendant un mois. Elle peut également
subir jusqu’à 7 cycles de congélation/décongélation à -20°C sans dégradation
significative, à condition que la décongélation soit réalisée à température
ambiante (Hoffman et al., 1979).
� Pour la stabilité pH-dépendante, la doxorubicine est considérée comme stable
dans des solutions entre pH 3 et 6,5, avec une dégradation proportionnelle à
l’augmentation du pH. La stabilité maximale en solution est observée pour un pH
égal à 4, avec absence de dégradation de la doxorubicine après 60 jours. Lors du
passage à une solution basique et donc à une stabilité moindre, on observe un
changement de couleur, du rouge-orangé pour le bleu-violet (Janssen et al.,
1985).
C. Principales méthodes d’analyse de la doxorubicine C. Principales méthodes d’analyse de la doxorubicine C. Principales méthodes d’analyse de la doxorubicine C. Principales méthodes d’analyse de la doxorubicine
1. La spectrophotométrie infrarouge1. La spectrophotométrie infrarouge1. La spectrophotométrie infrarouge1. La spectrophotométrie infrarouge
Cette méthode est basée sur la différence d’excitation des molécules sous la lumière
infrarouge, permettant de mettre en évidence les différentes liaisons chimiques présentes
dans la molécule. Les principales bandes d’absorption infrarouge de la doxorubicine sont
décrites ci-dessous (Figure 7 et Tableau II)(Bouma et al., 1986).

32
Figure 7 : Spectre d'absorption infrarouge de la doxorubicine dans une solution de bromure
de potassium 0,6%
Tableau II : Principales bande d'absorption infrarouge de la doxorubicine dans une solution de KBr 0,6%
Traduit et adapté de l’anglais d’après Bouma et al. (1986). Fréquences des bandes d’absorption infrarouge (en
cm-1) Interprétation
1008 Liaison alcool primaire C-OH 1071 Liaison alcool secondaire C-OH 1115 Liaison alcool tertiaire C-OH 1282 Liaison éther C-O-C
1580 et 1613 Liaison quinone C = O 1724 Liaison cétone C = O
2300 à 3160 Liaison amine primaire N-H 3160 à 3560 Liaison O-H (alcool lié)
2. La spectrophotométrie ultraviolet et visible2. La spectrophotométrie ultraviolet et visible2. La spectrophotométrie ultraviolet et visible2. La spectrophotométrie ultraviolet et visible
Cette méthode est basée sur la capacité d’une molécule à subir une ou plusieurs transitions
électroniques suite à un rayonnement électromagnétique dont le spectre d’ondes recouvre
les domaines de l’ultraviolet (100 à 400 nm) et du visible (400 à 750 nm).
Les principaux pics d’absorption de la doxorubicine en solution dans du méthanol sont 233,
252, 288, 479, 496 et 529 nm (Figure 8)(Abraham et al., 2005).
Figure 8 : Spectre d'absorption ultraviolet et visible de la doxorubicine dans du méthanol
D’après Abraham et al. (2005).
33
3. 3. 3. 3. La spectrofluorimétrieLa spectrofluorimétrieLa spectrofluorimétrieLa spectrofluorimétrie
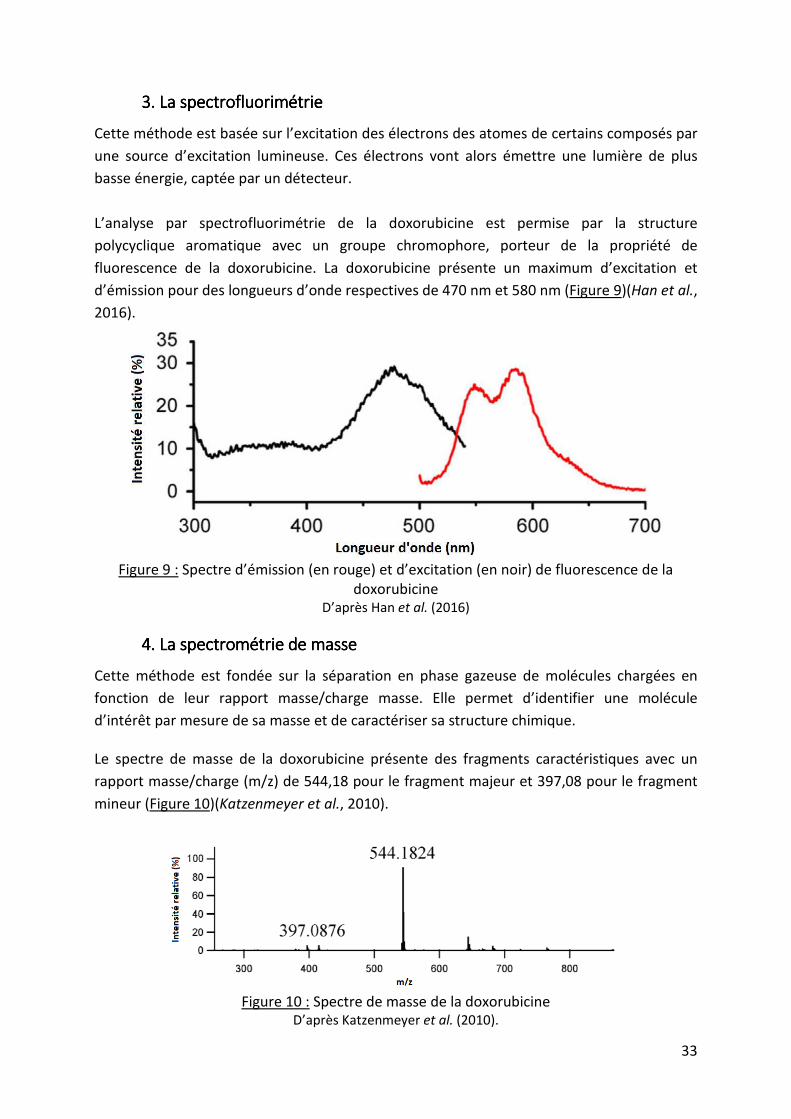
Cette méthode est basée sur l’excitation des électrons des atomes de certains composés par
une source d’excitation lumineuse. Ces électrons vont alors émettre une lumière de plus
basse énergie, captée par un détecteur.
L’analyse par spectrofluorimétrie de la doxorubicine est permise par la structure
polycyclique aromatique avec un groupe chromophore, porteur de la propriété de
fluorescence de la doxorubicine. La doxorubicine présente un maximum d’excitation et
d’émission pour des longueurs d’onde respectives de 470 nm et 580 nm (Figure 9)(Han et al.,
2016).
Figure 9 : Spectre d’émission (en rouge) et d’excitation (en noir) de fluorescence de la
doxorubicine D’après Han et al. (2016)
4. La spectrométrie4. La spectrométrie4. La spectrométrie4. La spectrométrie de massede massede massede masse
Cette méthode est fondée sur la séparation en phase gazeuse de molécules chargées en
fonction de leur rapport masse/charge masse. Elle permet d’identifier une molécule
d’intérêt par mesure de sa masse et de caractériser sa structure chimique.
Le spectre de masse de la doxorubicine présente des fragments caractéristiques avec un
rapport masse/charge (m/z) de 544,18 pour le fragment majeur et 397,08 pour le fragment
mineur (Figure 10)(Katzenmeyer et al., 2010).
Figure 10 : Spectre de masse de la doxorubicine
D’après Katzenmeyer et al. (2010).

34
II. Pharmacocinétique de la doxorubicineII. Pharmacocinétique de la doxorubicineII. Pharmacocinétique de la doxorubicineII. Pharmacocinétique de la doxorubicine
La pharmacocinétique permet d’étudier le comportement de la molécule d’intérêt et son
devenir dans l’organisme, afin de déterminer son schéma posologique optimal et
éventuellement la fenêtre thérapeutique. Pour cela, on étudie quatre paramètres :
l’absorption, la distribution dans l’organisme, le métabolisme et l’élimination.
Dans le cas de la doxorubicine chez le chien, il existe peu d’études de pharmacocinétique.
A. Voie d’administration et absorption A. Voie d’administration et absorption A. Voie d’administration et absorption A. Voie d’administration et absorption
La structure de la doxorubicine, une base faible, ionisée à pH acide et neutre, lui confère une
liposolubilité très limitée. De plus, du fait de son instabilité en milieu acide, la
biodisponibilité de la doxorubicine par voie orale est inférieure à 5 % (Kim et al., 2013). La
doxorubicine n’est donc pas utilisée per os chez le chien. La doxorubicine présente
également des propriétés caustiques, interdisant son administration par voie sous-cutanée
ou intra-musculaire. L’administration de la doxorubicine se réalise donc par voie
intraveineuse stricte, l’injection péri-veineuse étant à l’origine d’une nécrose tissulaire
importante (Venable et al., 2012).
B. DistributionB. DistributionB. DistributionB. Distribution
La phase de distribution correspond à la répartition plasmatique et à l’incorporation
tissulaire et cellulaire de la doxorubicine.
Dans le sang, 50 à 85% de la doxorubicine est lié aux protéines plasmatiques,
particulièrement à l’albumine et à l’α1-glycoprotéine (Speth et al., 1988). De nombreuses
études montrent qu’après administration de la doxorubicine, celle-ci est rapidement et
largement distribuée à la plupart des tissus de l’organisme, hormis le cerveau.
La doxorubicine a, chez le chien, un volume de distribution variant de 17,8 à 76 L/kg selon
les études et la concentration utilisée, et un temps de demi-vie de distribution de l’ordre de
quelques minutes, correspondant à un profil de distribution intracellulaire très rapide de la
molécule, malgré ce que laissent supposer ses caractéristiques physico-chimiques
(Oosterbaan et al., 1984)(Gustafson, Thamm, 2010). Cela peut être expliqué par la méthode
de calcul du coefficient de partage octanol-eau.
La répartition de la doxorubicine au sein des tissus semble être en corrélation avec la
concentration en ADN des tissus (Terasaki et al., 1984) et avec la concentration en lipides
anioniques, et plus particulièrement en cardiolipides (Nicolay et al., 1984). Cela est expliqué
par la forte affinité de la doxorubicine pour le cardiolipide (Kapp = 1,6.106) et l’ADN (Kapp =
2,2.106). Le cardiolipide est un glycérophospholipide double représentant environ 20 % des
molécules constituant la membrane interne de la mitochondrie et qui est impliqué dans le
métabolisme énergétique de la mitochondrie (Schlame et al., 1993).

35
L’entrée de la doxorubicine dans les cellules résulte de la diffusion passive de celle-ci sous
forme non ionisée, sous contrôle du pH et du gradient de concentration en doxorubicine. Le
pH intracellulaire étant légèrement plus faible que le pH sanguin et extracellulaire, et la
doxorubicine étant une base faible, celle-ci est plus ionisée en intracellulaire, ce qui est à
l’origine d’une diffusion passive vers le milieu intracellulaire et d’un piégeage ionique
intracellulaire. De plus, la doxorubicine diffuse selon un gradient de concentration. En effet,
même si la doxorubicine s’accumule fortement en milieu intracellulaire, elle se lie à l’ADN et
aux membranes plasmiques. Elle est donc très peu présente sous forme libre au niveau
cytoplasmique, ce qui permet le maintien d’un gradient entre l’extérieur et l’intérieur de la
cellule (Gallois et al., 1996). L’entrée de la doxorubicine au sein de la cellule peut être
influencée par le potentiel de membrane, le gradient de pH, la composition de la membrane
lipidique et la fluidité membranaire (Frézard, Garnier-Suillerot, 1998).
Plus précisément, la doxorubicine traverserait la membrane cellulaire par un mouvement de
« flip-flop » passif. Il y aurait une liaison massive et rapide de la doxorubicine à la membrane
cellulaire par des interactions électrostatiques avec la partie « tête » hydrophile des
phospholipides. Cette liaison serait suivie d’un long temps de résidence à la surface
membranaire suivi d’un mouvement de « flip-flop » à travers la membrane et d’un équilibre
rapide avec le milieu intracellulaire (Regev, Eytan, 1997).
Un flux de la doxorubicine par un transport facilité a également été mis en évidence chez
l’homme au niveau de certaines lignées cellulaires tumorales et de la moelle osseuse. Ce
transport serait permis par le solute carrier family 22 member 16 ou SLC22A16, transporteur
de la carnitine, de la bétaïne mais aussi de certaines molécules organiques telles que la
bléomycine ou la doxorubicine (Okabe et al., 2005). Ce transporteur est également présent
dans certaines lignées cellulaires tumorales canines et au niveau de la moelle osseuse mais
aucune étude n’a montré son implication dans le flux cellulaire de la doxorubicine.
Après entrée dans la cellule, la doxorubicine est prise en charge par le complexe protéasome
et s’accumule dans le noyau cellulaire (cf partie III.A.1) (Kiyomiya et al., 2001). Finalement, la
concentration nucléaire en doxorubicine est environ cinquante fois plus importante que la
concentration cytoplasmique. La doxorubicine cytoplasmique est distribuée équitablement
entre les organites dont les membranes sont constituées de lipides anioniques : les
mitochondries, l’appareil de Golgi et les lysosomes (Tacar et al., 2013).
C. MétabolismeC. MétabolismeC. MétabolismeC. Métabolisme
La doxorubicine présente un métabolisme principalement hépatique, avec des réactions de
phase I et de phase II, et constitué de trois voies métaboliques, à l’origine de la production
de divers métabolites (Figure 11)(Thorn et al., 2011).

36
1. Le doxorubicinol1. Le doxorubicinol1. Le doxorubicinol1. Le doxorubicinol
La doxorubicine subit une réduction à deux électrons, stéréospécifique, de la fonction
cétone de la chaîne latérale, à l’origine de la synthèse de son principal métabolite, le
doxorubicinol. Cette réaction est catalysée par la NADPH-dépendante carbonyle réductase
et dans une moindre mesure par des aldo-céto réductases (Sturgill et al., 2000). Cette voie
métabolique correspond à la voie principale de métabolisation de la doxorubicine.
Le doxorubicinol est un métabolite hydrophile, se liant également aux protéines
plasmatiques et avec une clairance plasmatique (19,6 mL/min/kg) et un volume de
distribution (0,464 L/kg) moindres par rapport à la doxorubicine (Gilbert et al., 2006). Il
présente une activité antitumorale environ dix fois moindre par rapport au composé parent,
avec une action d’agent alkylant de l’ADN. C’est un des acteurs de la cardiotoxicité et de la
myélotoxicité de la doxorubicine, celui-ci pouvant lui aussi former un radical semi-quinone
(Schaupp et al., 2015).
2. Le dérivé semi2. Le dérivé semi2. Le dérivé semi2. Le dérivé semi----quinonequinonequinonequinone
La doxorubicine peut également subir une réduction à un électron, pour former un radical
doxorubicine semi-quinone. Cette réaction est catalysée par différentes enzymes
oxydoréductases, selon le compartiment concerné. Ces enzymes incluent les NADH-
déshydrogénases mitochondriales, cytoplasmique ou sarcoplasmique, la xanthine oxydase
ou l’oxyde nitrique synthase (Bates, Winterbourn, 1982).
En milieu aérobie, la ré-oxydation du radical semi-quinone en doxorubicine est à l’origine de
la production de dérivés réactifs de l’oxygène, plus communément appelés reactive oxygene
species (ROS). Ces ROS jouent un rôle dans la cytotoxicité tumorale de la doxorubicine, mais
également dans sa toxicité (Riddick et al., 2005). En milieu anaérobie, le radical semi-
quinone présente une stabilité plus importante, permettant son auto-oxydation en un
radical semi-quinone centré en C7 (Airley, 2009).
3. Les dérivés aglycones3. Les dérivés aglycones3. Les dérivés aglycones3. Les dérivés aglycones
Une dernière voie, minoritaire, a été décrite, basée sur la déglycosidation de la doxorubicine, ou de son principal métabolite, le doxorubicinol. Elle ne représente que 1 à 2% du métabolisme de la doxorubicine (Thorn et al., 2011). Deux formes de dérivés aglycones sont décrites. L’une, issue de la déglycosidation par
hydrolyse catalysée par la glycoside hydrolase, est à l’origine de composés 7-hydroxy-
aglycones. L’autre est issue de la déglycosidation réductrice par la NADPH-cytochrome c
réductase, menant à la formation de composés 7-déoxy-aglycones. Le dérivé aglycone 7-
déoxydoxorubicinol peut également subir une déméthylation par un cytochrome P450. Ces
dérivés aglycones ne sont pas décelables chez tous les patients mais pourraient être eux-
aussi à l’origine de la toxicité cardiaque de la doxorubicine (Gewirtz, Yanovich, 1987).

37
La doxorubicine et ses métabolites subissent ensuite des réactions de phase II de type
sulfatation ou glucurono-conjugaison afin d’être sous forme conjuguée pour être excrétés
(Sturgill et al., 2000).
D. EliminationD. EliminationD. EliminationD. Elimination
La concentration en doxorubicine diminue rapidement après son administration. Son
élimination est principalement biliaire (environ 90 % de la dose administrée) et, dans une
moindre mesure, rénale. Elle est excrétée sous forme inchangée par élimination biliaire à
hauteur de 40 à 50 % et par élimination rénale à hauteur d’environ 5%. La clairance
plasmatique totale moyenne de la doxorubicine varie de 28,5 à 57,8 mL/min/kg. Des temps
de demi-vie plasmatique moyens de 3,2 minutes, 24 minutes et 11,14 heures ont été décrits
en utilisant un modèle tri-compartimental (Gustafson, Thamm, 2010). La première phase de
demi-vie serait due à la distribution tissulaire de la molécule, la deuxième phase au
métabolisme hépatique de la molécule et la troisième phase à la libération du médicament à
partir des sites de liaison dans les tissus (Tannock, Hill, 1987).
Le doxorubicinol présente un comportement similaire à son composé parent avec une demi-
vie de 48 minutes avec 12% du doxorubicinol excrété sous forme inchangée par voie urinaire
en deux heures et une clairance rénale équivalent à environ 9 à 17% de la clairance
plasmatique totale (Gilbert et al., 2006).
En ce qui concerne les résidus, la doxorubicine est détectable dans le sérum jusqu’à 7 jours
post-injection pour une dose de 30 mg/m² administrée sur 30 minutes (Knobloch et al.,
2010a). Dans les urines, la doxorubicine reste détectable pendant 21 jours, avec une valeur
inférieure à 10 % à la valeur de base après une semaine et inférieure à 1 % à la valeur de
base après deux semaines. (Knobloch et al., 2010b).
L’efflux cellulaire de la doxorubicine, contrairement à l’influx, se fait contre son gradient de
concentration et nécessite donc un transport actif de la molécule contre celui-ci. Il existe
différentes classes de transporteurs, les principaux appartenant à la famille des
transporteurs ATP-binding cassette (ou transporteurs ABC). Ces transporteurs sont impliqués
dans la translocation contre un gradient de concentration d’un large panel de substrats tels
que des ions, des peptides, des hormones stéroïdiennes, des phospholipides et certains
médicaments, dont la doxorubicine. Cet efflux est un mécanisme ATP-dépendant (Li et al.,
2008).

38
Parmi ces transporteurs ABC, la doxorubicine et ses métabolites sont majoritairement pris
en charge par :
� La glycoprotéine-P (ou P-gp), aussi appelée protéine MDR1 ou ABCB1. Premier
transporteur décrit de la famille des transporteurs ABC, c’est une protéine d’efflux de
170 kDa codée par le gène mdr1. Ce transporteur présente une grande variété de
substrats, la plupart neutres ou cationiques à pH physiologique, liposolubles,
organiques avec parfois des cycles aromatiques, et pour un poids moléculaire de 200
à 1900 Da. La P-gp est hautement conservée entre les espèces, avec 93 %
d’homologie entre le chien et l’Homme (Steingold et al., 1998). Ce transporteur est
fortement exprimé au pôle canaliculaire des hépatocytes, dans l’épithélium du tubule
proximal des reins, au niveau des voies biliaires et pancréatiques et les cellules
endothéliales des vaisseaux de l’encéphale (Ginn, 1996). Il présente donc un rôle
dans l’absorption intestinale, la sécrétion biliaire, la sécrétion tubulaire rénale et
dans la prévention du passage de la barrière hémato-méningée. Il est responsable à
lui seul d’environ 90 % de l’élimination de la doxorubicine et de ses métabolites (van
Asperen et al., 2000).
� La multidrug resistance related-protein 1 (MRP1) ou ABCC1. C’est un transporteur de
190 kDa, présent au niveau de la membrane cellulaire mais également au sein de la
membrane de certains organites. Elle prend en charge des substrats anioniques
organiques dont certains anti-cancéreux, conjugués ou non, comme la doxorubicine
et ses métabolites. Elle présente un haut degré d’homologie entre les espèces mais
l’expression tissulaire peut fortement varier (Li et al., 2008). Chez le chien, elle est
fortement exprimée au niveau de la barrière hémato-encéphalique, des reins, du foie
et des testicules (Conrad et al., 2001). Elle présente un rôle dans la sécrétion biliaire
et rénale.
� La breast cancer resistance protein (BCRP) ou ABCG2. C’est un transporteur de 72 kDa
prenant en charge des xénobiotiques et des contaminants environnementaux. Chez
l’Homme, seuls les mutants de la protéine BCRP peuvent prendre en charge la
doxorubicine, par modification de spécificité de substrat (Honjo et al., 2001). Chez le
chien, il semblerait que la protéine BCRP non mutée joue un rôle dans le transport de
la doxorubicine (Honscha et al., 2009). BCRP est fortement exprimée au niveau des
cellules tubulaires du rein, au pôle caniculaire des hépatocytes et au pôle luminal des
cellules endothéliales des capillaires de l’encéphale (Li et al., 2008). Elle serait
responsable d’un blocage de l’absorption au niveau intestinal et cérébral et d’une
sécrétion rénale et hépatique.
Ces transporteurs sont également impliqués dans le phénomène de résistance à la
doxorubicine des cellules tumorales (cf partie III.E.1) (Withrow, Page, 2013).

Figure 11 : Schéma des voies métaboliques de la doxorubicine Par souci de lisibilité, les cofacteurs permettant les différentes réactions du métabolisme n’ont pas été précisés.
39

40
En plus de ces transporteurs ABC, d’autres transporteurs, accessoires, sont responsable de
l’efflux et de l’élimination de la doxorubicine et de ses métabolites chez l’Homme : la RalA-
binding protein 1 (RALBP1) ou la lung resistance protein (LRP). Même si leur présence est
attestée, il n’existe aucune donnée sur le rôle de ces transporteurs chez le chien (Zandvliet,
Teske, 2015).
Chez le chien, on utilise la plupart du temps des modèles bi-compartimental ou tri-
compartimental pour décrire la pharmacocinétique de la doxorubicine (Figure 12). Le
modèle tri-compartimental correspond à un compartiment central et deux compartiments
périphériques, selon la vitesse d’équilibration rapide (les organes splanchniques et le tissu
musculaire) ou lente (le tissu adipeux). Celui-ci est la plupart du temps simplifié en modèle
bi-compartimental, menant à une approximation des données mais d’une plus grande
simplicité d’interprétation.
Figure 12 : Schématisation des modèles de pharmacocinétique bi-compartimental et tri-
compartimental de la doxorubicine kélim : constante d’élimination, kxy : constante de transfert entre les compartiments x et y.

Figure 13 : Schéma ADME de la doxorubicine (Absorption, Distribution, Métabolisme, Elimination), détaillant les transporteurs cellulaires
ALB : albumine ; DOX : doxorubicine ; DOXol : doxorubicinol
41

42
III. Pharmacodynamie de la doxorubicineIII. Pharmacodynamie de la doxorubicineIII. Pharmacodynamie de la doxorubicineIII. Pharmacodynamie de la doxorubicine
Suite à sa diffusion passive au sein des cellules, la doxorubicine va pouvoir atteindre les différents sites potentiels de son action cytotoxique : le noyau, les mitochondries et les membranes cellulaires
A. Interactions directes de la doxorubicine avec l’ADN et sa dynamiqueA. Interactions directes de la doxorubicine avec l’ADN et sa dynamiqueA. Interactions directes de la doxorubicine avec l’ADN et sa dynamiqueA. Interactions directes de la doxorubicine avec l’ADN et sa dynamique
1. Transport nucléaire de la doxorubicine1. Transport nucléaire de la doxorubicine1. Transport nucléaire de la doxorubicine1. Transport nucléaire de la doxorubicine
Une fois le cytoplasme atteint, la majeure partie de la doxorubicine s’accumule dans le
noyau des cellules tumorales. En effet, par quantification par marquage radioactif sur
différentes lignées cellulaires tumorales, une distribution non équitable est mise en évidence
avec une forte accumulation au niveau nucléaire (Merlin et al., 1995).
Ce processus d’accumulation nucléaire est permis par le protéasome 28S. Les protéasomes
sont des complexes protéiques intracellulaires impliqués dans des processus non
lysosomiques de dégradation des protéines. Parmi les protéasomes, le protéasome 26S est
essentiel dans le turnover normal des protéines cytosoliques et nucléaires, et joue un rôle
dans le traitement et la dégradation des protéines régulatrices de la croissance cellulaire et
du métabolisme. Il est constitué de trois sous-unités, un cœur catalytique, le
protéasome 20S, et deux complexes régulateurs 19S, de chaque côté du protéasome 20S
(Adams, 2003).
Après diffusion passive de la doxorubicine dans le cytoplasme de la cellule, un complexe
protéasome-doxorubicine est formé, grâce à la forte affinité de la doxorubicine pour le
protéasome. Une fois le complexe formé, celui-ci subit une translocation du cytoplasme vers
le noyau, via les pores nucléaires, le protéasome présentant des signaux de localisation
nucléaire. La doxorubicine présentant une affinité pour l’ADN supérieure à celle pour le
protéasome, celle-ci se dissocie de ce dernier pour rentrer en interaction avec l’ADN
(Figure 14) (Kiyomiya et al., 2001).
De plus, la doxorubicine se lie à un site allostérique de l’activité protéase chymotrypsine-like
de la sous-unité catalytique 20S, agissant alors comme un inhibiteur réversible non
compétitif de l’activité protéase (Figueiredo-Pereira et al., 1996).
Le complexe protéasome-doxorubicine présente donc deux types d’avantages. Tout d’abord,
il permet une augmentation de l’accumulation de doxorubicine dans le noyau, à l’origine
d’un de ses principaux modes d’action. De plus, cela permet l’accumulation de protéines non
dégradées, à l’origine d’un signal de l’apoptose cellulaire (Minotti et al., 2004).

43
Figure 14 : Modalités d’action du complexe protéasome-doxorubicine
Une fois dans le noyau, la doxorubicine peut interagir avec la molécule d’ADN et les
différents acteurs intervenant dans la dynamique de l’ADN.
2. Inhibition de l’ADN topoisomérase de type II2. Inhibition de l’ADN topoisomérase de type II2. Inhibition de l’ADN topoisomérase de type II2. Inhibition de l’ADN topoisomérase de type II
a. La structure topologique de la molécule d’ADN chez les eucaryotesa. La structure topologique de la molécule d’ADN chez les eucaryotesa. La structure topologique de la molécule d’ADN chez les eucaryotesa. La structure topologique de la molécule d’ADN chez les eucaryotes
L’ADN est une macromolécule polynucléotide double brin linéaire dont les nucléotides sont
formés d’un phosphate, d’un sucre et d’une base purique ou pyrimidique. Dans un état
relaxé idéal de moindre énergie, l’ADN suit une conformation de type B, c’est-à-dire une
structure en double-hélice dextrogyre avec un diamètre de 20 Å, un pas de 3,4 Å et avec 10
paires de bases par tour d’hélice. Cependant, la molécule d’ADN étant très longue, celle-ci se
condense grâce à la formation d’une structure mixte d’ADN et de protéines, appelée
nucléosome. Ces nucléosomes sont à l’origine d’une modification de la structure
topologique (ou structure tertiaire) de l’ADN (Charvin, 2004).
La topologie de l’ADN, c’est-à-dire la configuration tridimensionnelle complexe de la
molécule d’ADN au sein de la cellule, est définie par ses principales figures topologiques : les
surenroulements, les nœuds et les caténanes (Watt, Hickson, 1994).
� Les surenroulements ou supertours
Les surenroulements (ou supertours) correspondent aux torsions ou aux contraintes
appliquées à l’ADN, qui modifient les rapports géométriques dans l’hélice et sont à l’origine
d’une surtension. Le surenroulement est défini par différents éléments :
• Le nombre d’enlacements Lk, c’est à dire le nombre de fois où les deux
brins d’ADN sont liés.

44
• Le nombre de vrilles Wr, c’est-à-dire le nombre de fois où l’axe central de
la double hélice tourne autour de lui-même.
• Le nombre de torsades Tw, c’est-à-dire le nombre de tours qu’effectue un
simple brin d’ADN autour de l’autre brin simple d’ADN.
L’équation de White ΔLk = ΔWr + ΔTw relie la variation du nombre d’enlacements à celles
du nombre de vrilles et de torsades (White, 1969). Cette formule permet de définir la
distribution des contraintes au sein de la molécule d’ADN. Dans une molécule d’ADN seule
qui se déforme continuellement, le nombre d’enlacement reste constant tandis que les
changements de Wr et de Tw sont couplés.
Dans une cellule, les extrémités de l’ADN linéaire étant fixées à la membrane nucléaire, le
nombre d’enlacements n’est pas stable et provoque un surenroulement de l’ADN. On pourra
différencier les surenroulements positifs (dans le sens de torsion de la double hélice d’ADN),
des surenroulements négatifs (dans le sens inverse de torsion de la double hélice d’ADN).
Le surenroulement de l’ADN intervient lors de la réplication et de la transcription en ARN. Il
résulte du déroulement de la double hélice par l’ADN ou l’ARN polymérase, déroulement qui
induit des supertours positifs en amont et négatifs en aval.
� Les nœuds et caténanes
Les nœuds correspondent à des extrémités de la molécule d’ADN liées l’une à l’autre (Figure
15). Les nœuds fragilisent l’ADN et bloquent certains processus cellulaires tels que la mitose
ou la méiose, en empêchant la séparation des deux brins d’ADN (Sumners, 1995).
Lors d’une concentration importante en ADN, il y a une forte probabilité d’obtenir des
molécules d’ADN liées entre elles, nommées caténanes (Figure 15). En pratique, les
caténanes apparaissent lors de la réplication de l’ADN, où les molécules d’ADN nouvellement
synthétisées s’enroulent l’une autour de l’autre. Les caténanes empêchent la séparation et
la ségrégation des chromosomes au cours des divisions cellulaires (Sumners, 1995).
Figure 15 : Caténanes au cours de la réplication (à gauche) et nœuds d’ADN (à droite)
D’après Sumners et al. (1995).

45
L’ADN présente donc plusieurs figures topologiques, les surenroulements, à l’origine d’une
topologie désirable permettant à l’ADN d’être compacté et qui jouent un rôle au cours de la
réplication et de la transcription, et les nœuds et caténanes, à l’origine d’une topologie
indésirable et qui doivent être supprimés pour permettre le déroulement des processus
biologiques.
Pour que les processus biologiques cellulaires de réplication, de transcription et de division
puissent avoir lieu, l’hélicité de l’ADN doit être maintenue et les contraintes de torsions
régulées, en fonction de la phase du cycle cellulaire et de l’activité transcriptionnelle. De
plus, dans tous les cas, les nœuds et caténanes doivent être supprimés. Cela nécessite un
clivage simple ou double brin de l’ADN, opérations réalisées par les ADN topoisomérases
(Champoux, 2001).
b. Les ADN topoisomérases b. Les ADN topoisomérases b. Les ADN topoisomérases b. Les ADN topoisomérases
En 1971, Wang et al mettent en évidence, chez une bactérie Escherichia Coli, une nouvelle
classe d’enzyme ayant la capacité de modifier la topologie de la molécule d’ADN : les ADN
topoisomérases (Wang, 1971). Ces enzymes ubiquitaires et fortement conservées dans
toutes les espèces, sont capables de générer des clivages transitoires de l’ADN et de
catalyser le passage de segments d’ADN au travers de ces zones de clivage avant de les
refermer (Lodish et al., 2000). Elles sont essentielles pour de nombreux processus de la vie
cellulaire, incluant la transcription, la réplication, la séparation des chromosomes lors de la
mitose et de la méiose, etc (Nitiss, 2009a).
� Mécanisme d’action général des ADN topoisomérases
Pour prévenir et corriger la topologie de la double hélice d’ADN, les ADN topoisomérases
réalisent une réaction de transestérification réversible catalysée pour le groupement
hydroxyle d’une tyrosine appartenant à son site actif (Figure 16). Ce groupement réagit avec
la liaison phosphodiester de l’ADN et provoque une coupure de brin d’ADN et la formation
d’un intermédiaire covalent entre la tyrosine de l’enzyme et une des extrémités du brin
d’ADN. La réaction inverse reforme la liaison phosphodiester et libère la tyrosine. Ainsi, la
réaction de coupure/religation ne nécessite pas d’apport d’énergie (Duguet, Riou, 1994).
Figure 16 : La réaction de transestérification caractérisant l’activité des topoisomérases
D’après Duguet et Riou (1994). ENZ : enzyme ; Tyr : tyrosine.

46
� Classification des ADN topoisomérases
En fonction du nombre de brins d’ADN concernés par le clivage transitoire, on distingue deux
classes de topoisomérases :
• Les ADN topoisomérases de type I, quasi-exclusivement monomériques.
Ces enzymes clivent un seul des deux brins de la double hélice d’ADN,
permettant soit le passage du brin d’ADN non clivé au niveau de la cassure
simple brin (enzyme de type IA), soit la libre rotation du brin clivé autour
de l’autre simple brin (enzyme de type IB)(Dekker et al., 2002). Le brin
clivé est ensuite réparé, formant une structure identique à celle avant
clivage, mais avec un ΔLk = ± 1 (Figure 17). La plupart de ces enzymes,
exceptée la réverse gyrase, ne nécessite pas la présence d’un cofacteur
énergétique ATP, l’énergie conduisant la réaction étant apportée par le
surenroulement de la molécule d’ADN (Charvin, 2004).
Figure 17 : Mécanisme de la relaxation du surenroulement par les ADN topoisomérases de
type I Traduit et adapté de l’anglais, d’après Dekker et al. (2002).
En haut : l’ADN topoisomérase de type IA s’insère dans la double hélice en la faisant « fondre » localement. Elle
opère ensuite une cassure simple-brin et fait passer l’autre brin à travers. En bas : l’ADN topoisomérase de type
IB se fixe sur le brin d’ADN et réalise une cassure simple brin, permettant la libre rotation de l’ADN autour du
squelette sucre-phosphate.
• Les ADN topoisomérases de type II eucaryotes sont des homodimères. Ces
enzymes présentent la capacité de cliver les deux brins de la double hélice
d’ADN afin de permettre le passage d’une autre portion de la double
hélice d’ADN au sein de la zone de cassure. Les brins clivés sont ensuite
réparés par un mécanisme nécessitant l’hydrolyse de deux molécules
d’ATP. Les ADN topoisomérases de type II seront à l’origine d’un ΔLk = ± 2.
Les ADN topoisomérases de type II sont aussi à l’origine de la décaténation
des chromosomes au cours de la réplication. On distingue les ADN
topoisomérases de type IIα, exprimée dans les cellules prolifératrices et

47
impliquée dans la réplication et la ségrégation chromosomique, des ADN
topoisomérases de type IIβ, impliquées dans la transcription et la
différenciation cellulaire (Drake et al., 1989). Les ADN topoisomérases de
type IIα sont dépendantes du cycle cellulaire. Leur expression et leur
activité catalytique augmente au cours de la phase S pour être maximales
lors de la phase G2 et décroître ensuite au cours de la phase G1.
L’augmentation de l’activité catalytique semble être contrôlée par la
phosphorylation de résidus sérine au cours du cycle cellulaire (Larsen et
al., 1996).
Dans la suite, nous nous intéresserons principalement aux topoisomérases eucaryotes de
type II, celles-ci étant inhibées par la doxorubicine.
� Structure des ADN topoisomérases eucaryotes de type II
Toutes les sous-classes d’ADN topoisomérases de type II présentent des domaines communs
au sein de leur structure (Corbett, Berger, 2004) :
• Un domaine 5Y-CAP (Catabolite Activator Protein), portant l’acide aminé
catalytique tyrosine qui se lie de façon covalente à l’extrémité 5’ du brin
d’ADN après attaque nucléophilique du pont phosphodiester. Un acide
aminé arginine conservé proche de la tyrosine permettrait de positionner
la molécule d’ADN et de stabiliser l’état de transition pentavalent au cours
de la réaction.
• Un domaine Toprim (Topoisomérase-primase), constitué d’une centaine
d’acide aminés et présentant deux motifs conservés : l’un centré autour
d’un acide aminé glutamate et présentant un rôle dans la religation du
brin d’ADN, l’autre centré autour de deux acides aminés aspartate et
permettant la liaison avec Mg2+, cofacteur requis pour la fonction
catalytique (Aravind et al., 1998).
• Un domaine GHKL ATPase qui se dimérise après fixation de l’ATP pour
former une « porte » à travers laquelle les duplex d’ADN passent.
• Un domaine de transduction, essentiel pour l’hydrolyse de l’ATP,
notamment grâce à un acide aminé lysine qui se projette sur le site actif
du domaine GHKL et stabilise l’état de transition de l’ATP au cours de son
hydrolyse.
� Mécanisme d’action de l’ADN topoisomérase de type II
Au cours d’un cycle de topo-isomérisation, chaque sous-unité de l’enzyme dimérique clive
un brin de l’ADN. Cette réaction nécessite la fixation et l’hydrolyse de l’ATP. Les
intermédiaires clivés, aussi nommés « complexes de clivage » ont normalement une durée
de vie très courte, le temps de passage de l’ADN à travers cette coupure.

48
Figure 18 : Cycle de topo-isomérisation par une ADN topoisomérase de type II
D’après Duguet et Riou (1994).
Les différentes étapes du cycle de l’ADN topoisomérase de type II sont les suivantes (Figure 18) (Duguet, Riou, 1994) :
(1) Etape de fixation : l’ADN topoisomérase de type II (en rouge), constituée d’un
homodimère, se fixe en 5’ du brin d’ADN et forme un complexe covalent au niveau d’un
croisement de deux doubles brins d’ADN, dont l’un (en gris) porte une séquence consensus
dégénérée.
(2) Etape de clivage : les deux brins de ce dernier sont clivés respectivement par
chacune des deux sous-unités de l’ADN topoisomérase de type II, grâce à des réactions de
transestérification, produisant l’intermédiaire clivé a. Cette étape est catalysée par un
cofacteur Mg2+, indispensable.
(3) Etape de modifications topologiques : le double brin d’ADN intact passe au travers
de la brèche, sans doute grâce à un changement conformationnel de l’enzyme qui a fixé
deux molécules d’ATP, formant l’intermédiaire clivé b.
(4) Etape de religation : l’intermédiaire clivé b reforme les liaisons phosphodiesters
mais l’ADN topoisomérase de type II reste fixée sur l’ADN sous forme d’un complexe non
covalent.
(5) Etape de régénération de l’enzyme : l’ATP est hydrolysée, régénérant la forme
active de l’enzyme qui se dissocie de l’ADN.

49
Chaque cycle catalytique modifie d’une valeur de deux le nombre de liens topologiques
d’une molécule d’ADN.
Ces enzymes sont des cibles pharmacologiquement importantes d’un certain nombre
d’agents anticancéreux. Parmi ceux-ci, la famille des anthracyclines, et plus particulièrement
la doxorubicine, sont responsables de l’inhibition de l’ADN topoisomérase de type II
(Chabner, Longo, 2011).
c. Action de la doxorubicine sur les ADN topoisomérases de type IIc. Action de la doxorubicine sur les ADN topoisomérases de type IIc. Action de la doxorubicine sur les ADN topoisomérases de type IIc. Action de la doxorubicine sur les ADN topoisomérases de type II
En 1981, Ross et al décrivent l’induction d’une rupture des brins d’ADN dans des cellules
murines leucémiques L1210 par la doxorubicine pour des concentrations allant de 0,4 à 5
µM (Ross, Bradley, 1981). Les cassures de brins d’ADN, simple ou doubles brins sont
lentement et incomplètement réparées après retrait de la doxorubicine des cellules. En
1984, Tewey et al. identifient l’ADN topoisomérase de type II comme cible de la
doxorubicine et démontrent que les sous-unités de l’enzyme restent bloquées sur
l’extrémité 5’ de la molécule d’ADN après avoir complété la réaction de clivage (Tewey et al.,
1984). La doxorubicine est donc un inhibiteur réversible de l’ADN topoisomérase de type II,
et plus exactement, elle stabilise l’intermédiaire clivé en se fixant à celui-ci, permettant la
formation d’un complexe ternaire de clivage doxorubicine/ADN/ADN topoisomérase de
type II (Wang, 1971).
La formation et la stabilité de ce complexe ternaire reposent sur des déterminants
structuraux définis et notamment sur la fraction sucre de la doxorubicine, déterminant
critique de l’action de la doxorubicine en tant qu’inhibiteur de l’ADN topoisomérase de type
(Minotti et al., 2004). La formation du complexe ternaire serait séquence-dépendante et
favorisée par la présence d’une base azotée adénine en -1 du site de cassure (Pommier et al.,
2010). Ce complexe ternaire de clivage bloque la transcription et la réplication cellulaire. De
plus, en stabilisant l’ADN topoisomérase de type II, la doxorubicine empêche la religation de
l’ADN et est à l’origine de lésions doubles brins de l’ADN. D’un point de vue mécanistique, on
peut imaginer un modèle d’interaction dans lequel la réaction de religation est inhibée par le
positionnement de l’agent au centre de la région où l’ADN topoisomérase de type II interagit
avec l’ADN et le clive. Le groupement hydroxyle de l’extrémité du brin clivé serait alors placé
dans une position trop éloignée du site actif pour que la réaction de religation puisse
s’effectuer.
Il est cependant difficile de relier le degré d’induction de cassures de brins d’ADN avec la
cytotoxicité de la doxorubicine, celle-ci ne produisant qu’une faible quantité de dommage à
l’ADN en comparaison avec d’autres inhibiteurs de l’ADN topoisomérase de type II (tels que
l’amsacrine ou les épipodophyllotoxines). On a donc un conflit entre le haut degré de
cytotoxicité de la doxorubicine et le faible taux de cassures de l’ADN. Une hypothèse serait
que le site de cassure est un facteur critique de l’action de la doxorubicine et que celle-ci
présente un site spécifique de clivage, différent des autres inhibiteurs de l’ADN
topoisomérase de type II (Gewirtz, 1999).

50
� Quelle est la part d’inhibition de l’ADN topoisomérase de type II dans la cytotoxicité de la
doxorubicine ?
Trois conditions inhibent l’ADN topoisomérase de type II : une température faible, des
concentrations en sels importantes et des concentrations en doxorubicine excédant une
valeur critique entre 0,5 et 2,5 µM. Pour ces trois conditions, il a été mis en évidence une
inhibition de la cytotoxicité de la doxorubicine, en faveur d’un rôle majeur de l’ADN
topoisomérase de type II dans cette cytotoxicité (Keizer et al., 1990). On observe qu’à
faible concentration, la doxorubicine est à l’origine d’une induction de la formation de
complexes de clivage mais qu’à forte concentration, elle permet l’inhibition de la fixation
de l’ADN topoisomérase de type II à l’ADN, sans former de complexe ternaire (Chabner,
Longo, 2011).
� Comment l’action de la doxorubicine concernant la topoisomérase de type II conduit-elle
à la cytotoxicité de la doxorubicine ?
La cytotoxicité est liée à l’accumulation de complexes de clivage stables dans lesquels l’ADN
est clivé : plus il y a d’ADN topoisomérase de type II au voisinage de l’ADN, plus ces
complexes de clivage se forment en grand nombre sur l’ADN et plus grande est la
cytotoxicité (Duguet, Riou, 1994). C’est également cela qui explique la relative sélectivité de
la doxorubicine vis-à-vis des cellules tumorales. En effet, celles-ci présentent une
concentration plus élevée en ADN topoisomérase de type II que les cellules non tumorales.
La réplication, la transcription et d’autres processus comme la recombinaison pourraient
transformer les complexes de clivage initiaux en lésions secondaires de l’ADN. Ces lésions
secondaires peuvent être la conséquence directe (par recombinaison de brins) ou indirecte
(par réparation anarchique) de la présence de complexes de clivage. A ce stade, deux
possibilités se présentent, soit l’ADN est réparé, soit la lésion persiste. La persistance de
cette lésion mène à un blocage du cycle cellulaire, préférentiellement entre la phase G2 et la
phase M, préférence due à la plus forte activité de l’ADN topoisomérase de type IIα à cette
phase du cycle cellulaire. La cellule rentre ensuite dans l’un des processus de mort cellulaire
(Swift et al., 2006).
3. Intercalation de la doxorubicine au sein de la molécule d’ADN3. Intercalation de la doxorubicine au sein de la molécule d’ADN3. Intercalation de la doxorubicine au sein de la molécule d’ADN3. Intercalation de la doxorubicine au sein de la molécule d’ADN
La doxorubicine est un agent mono-intercalant de l’ADN, c’est à dire une molécule
organique de petite taille dont une partie est plane, et qui s’intercale entre les paires de
bases de l’ADN. Au sein de la molécule de doxorubicine, les cycles B, C et D de la partie
chromophore aglycone constituent la partie intercalante de la molécule (Figure 1). Cette
intercalation de la doxorubicine au sein de la molécule d’ADN aurait lieu pour des
concentrations allant de 0,01 à 4 µM, selon les études (Minotti et al., 2004).

51
a. Mécanisa. Mécanisa. Mécanisa. Mécanisme d’intercalation de la doxome d’intercalation de la doxome d’intercalation de la doxome d’intercalation de la doxorubrubrubrubicineicineicineicine
L’intercalation de la doxorubicine au sein de la molécule d’ADN est un processus avec
plusieurs étapes, présentant des évènements non spécifiques montrant une forte affinité
(Kapp = 2,2.106) pour des séquences spécifiques riches en motifs guanine-cytosine et des
régions transcriptionnellement actives de l’ADN. Le complexe doxorubicine-ADN est stabilisé
par au moins trois forces de liaisons : des interactions hydrophobes, des liaisons hydrogènes
au squelette phosphaté de l’ADN avec la chaine latérale et le cycle d, et l’insertion du
groupement amino-sucre au sein du petit sillon de l’ADN. Cela donne lieu à un temps de
séjour long qui semble être un facteur critique pour différencier un agent intercalant avec
une activité antitumorale ou non (Cummings, Anderson, et al., 1991).
Le mécanisme d’intercalation, étudié par cristallographie, se déroule en trois étapes selon
Chaires et al : une interaction extérieure rapide type liaison faible suivi d’une intercalation
de la doxorubicine et enfin d’un ajustement conformationnel lent du complexe ADN-
doxorubicine (Chaires et al., 1985). D’autres auteurs suggèrent l’existence d’un modèle
cinétique à 5 étapes (Rizzo et al., 1989). Celui-ci inclurait une disposition en parallèle des
étapes 1 et 2 (étapes d’interaction type liaison faible), suivie par une étape d’intercalation de
la doxorubicine et d’une nouvelle disposition en parallèle des étapes 4 et 5, étapes de
réarrangement conformationnel du complexe doxorubicine-ADN et de redistribution des
liaisons à des sites privilégiés, sans dissociation. B5 correspond au complexe final
doxorubicine-ADN (Figure 19)(Lei et al., 2012).
Figure 19 : Modèles cinétiques d’intercalation de la doxorubicine au sein de l’ADN. (A)
Modèle à 3 étapes. (B) Modèle à cinq étapes
D’après Chaires et al. (1985) et Rizzo et al. (1989).
b. Conséquences de l’intercalation de la doxorubicine au sein de la molécule b. Conséquences de l’intercalation de la doxorubicine au sein de la molécule b. Conséquences de l’intercalation de la doxorubicine au sein de la molécule b. Conséquences de l’intercalation de la doxorubicine au sein de la molécule d’ADNd’ADNd’ADNd’ADN
L’intercalation de la doxorubicine au sein de la molécule d’ADN présente des effets directs
sur la chromatine, à l’origine de la mort des cellules cancéreuses. En effet, la doxorubicine va
venir s’intercaler préférentiellement entre les deux brins de l’ADN, dans des sites adjacents
aux paires de bases guanine-cytosine à cause de la formation de liaisons hydrogènes
spécifiques entre la doxorubicine et la guanine. On observe alors un écartement des paires
de bases supérieure et inférieure, l’espace entre deux paires de bases passant de 3,4 Å

52
à environ 6,8 Å (Yang et al., 2014). Cet écartement permet l’intercalation de la doxorubicine
et de ce fait, empêche la fixation de certaines enzymes impliquées dans des processus
cellulaires, dont les ADN polymérases et les hélicases (Chabner, Longo, 2011). Les hélicases
sont des enzymes permettant la séparation des deux brins de l’ADN par hydrolyse des
liaisons hydrogènes entre ceux-ci (Matson, Kaiser-Rogers, 1990). La séparation des brins
d’ADN est une étape indispensable aux processus de réplication, de transcription et de
réparation de l’ADN. La doxorubicine inhibe donc la fixation des hélicases sur l’ADN et la
séparation des deux brins d’ADN. De plus, tout comme avec les ADN topoisomérases de type
II, la doxorubicine formerait des complexes ternaires irréversibles doxorubicine/ADN/ADN
hélicases (Bachur et al., 1992).
De plus, dans une molécule d’ADN de conformation B, chaque paire de bases présente une
rotation d’environ -36° avec la paire de bases suivante. L’introduction d’une seule molécule
de doxorubicine est à l’origine d’une modification de la périodicité de la molécule d’ADN
avec une modification de la rotation de -27° entre les deux paires de bases adjacentes.
L’intercalation de la doxorubicine est aussi à l’origine d’une augmentation de la longueur de
l’ADN ainsi que de sa viscosité (Mukherjee, Sasikala, 2013). On observe alors une altération
de la structure topologique de l’ADN avec induction des torsions positives compensatrices,
pouvant être à l’origine de la modification de nombreux mécanismes cellulaires tels que la
réplication de l’ADN ou sa transcription.
L’inhibition des processus de réplication, transcription et réparation de l’ADN mène à
l’inhibition de la synthèse de macromolécules essentielles au fonctionnement cellulaire et
donc à la mort cellulaire (Cummings, Anderson, et al., 1991).
En raison de sa forte affinité pour l’ADN, la doxorubicine s’accumule dans le noyau et
réagit avec l’ADN. Ces interactions expliquent certains des mécanismes d’action de la
doxorubicine à l’origine de sa cytotoxicité :
� L’inhibition de l’ADN topoisomérase de type II, enzyme responsable de la
topologie de la molécule d’ADN au cours du cycle cellulaire. Cette inhibition est
soit directe, soit indirecte par la formation d’un complexe ternaire
doxorubicine/ADN/ADN topoisomérase de type II. L’inhibition résulte en la
formation de cassures double-brins de l’ADN, de cross-links et en l’inhibition des
processus cellulaires de réplication et de transcription.
� L’intercalation de la doxorubicine au sein de la molécule d’ADN. Ce mécanisme
est à l’origine de l’inhibition des processus cellulaires de réplication, transcription
et réparation de l’ADN par modification des caractéristiques de la molécule
d’ADN.

53
B. Réactivité chimique de la doxorubicine et interaction aveB. Réactivité chimique de la doxorubicine et interaction aveB. Réactivité chimique de la doxorubicine et interaction aveB. Réactivité chimique de la doxorubicine et interaction avec les composés c les composés c les composés c les composés cellulairescellulairescellulairescellulaires
Durant l’intercalation ou l’inhibition de l’ADN topoisomérase de type II, la doxorubicine agit
comme un composé inerte, son activité étant due à sa capacité à se lier à des
macromolécules clés et à modifier la structure tridimensionnelle de ses cibles. Cependant, la
doxorubicine est également capable d’agir en tant que réactif chimique. En effet, la
doxorubicine peut subir différentes réactions d’oxydoréduction, à l’origine d’effets variés sur
la cellule.
1. Doxorubicine et réact1. Doxorubicine et réact1. Doxorubicine et réact1. Doxorubicine et réactions d’oxydoions d’oxydoions d’oxydoions d’oxydo----réductionréductionréductionréduction
Le cycle d’oxydo-réduction de la doxorubicine est décrit pour la première fois lors de
l’observation d’une consommation de dioxygène en présence de NADPH, de la cytochrome
P450 réductase et de doxorubicine (Goodman, Hochstein, 1977).
La doxorubicine est capable de subir une réduction à un ou deux électrons, à l’origine de la
production de ROS et d’un radical semi-quinone. Ces composés réactifs peuvent causer de
larges dommages aux macromolécules intracellulaires et sont à l’origine de l’activité
antitumorale de la doxorubicine, mais aussi de sa cardiotoxicité (Doroshow, 1986) (Mizutani
et al., 2005). De plus, la doxorubicine peut également se complexer avec l’ion fer(III), et
mener à la formation de ROS. Au cours de son cycle d’oxydo-réduction, elle peut aussi
former des agents alkylants.
a. Formation de ROS par réduction à un électron de la doxorubicinea. Formation de ROS par réduction à un électron de la doxorubicinea. Formation de ROS par réduction à un électron de la doxorubicinea. Formation de ROS par réduction à un électron de la doxorubicine
La réduction à un électron de la doxorubicine peut se dérouler dans la plupart des
compartiments intracellulaires, incluant les mitochondries, le réticulum endoplasmique et le
cytoplasme. Cette réaction est catalysée par des déshydrogénases liées à la flavine ou des
réductases incluant la cytochrome P450 réductase, la NADH déshydrogénase (ou complexe I
de la chaine mitochondriale de transport d’électrons), la xanthine oxydase et la cytochrome
b5 réductase (Doroshow, 1986) (Vásquez-Vivar et al., 1997). De plus, plusieurs isoformes de
l’oxyde nitrique synthase sont capables de catalyser la réduction à un électron de la
doxorubicine avec la production subséquente de superoxyde et une diminution de l’oxyde
nitrique (Garner et al., 1999). Ces enzymes sont largement distribuées dans les tissus et la
formation de radicaux libres se déroule donc dans de nombreux organes et lignées
cellulaires tumorales.
L’addition d’un électron libre convertit le groupement quinone du cycle C de la doxorubicine
en un radical libre semi-quinone. Ce radical peut être à l’origine de dégâts oxydatifs directs,
résultant en un clivage ou à la dégradation de l’ADN ou des désoxyriboses (Bates,
Winterbourn, 1982). En conditions anoxiques, ce radical est relativement stable. En présence
de dioxygène, ce radical libre est rapidement oxydé et donne son électron au dioxygène
moléculaire pour former un anion superoxyde O2.-. Bien que peu toxique en lui-même, sa
dismutation spontanée est à l’origine de la formation de peroxyde d’hydrogène H2O2

54
(Wagner et al., 2005). L’anion superoxyde peut alors réagir avec le peroxyde d’hydrogène
selon la réaction de Haber-Weiss, formant ainsi le radical hydroxyle HO-. Cette réaction est
catalysée par l’ion fer(III). Ce radical hydroxyle est l’une des espèces chimiques les plus
réactives et peut être à l’origine de l’oxydation délétère de constituants cellulaires
(Kalyanaraman et al., 1991). L’anion superoxyde peut également réagir avec le monoxyde
d’azote pour produire l’anion peroxynitrite, agent oxydant très instable, menant à la
formation de radicaux hydroxyles (Figure 20) (Powis, 1987).
Figure 20 : Formation de ROS par réduction à un électron de la doxorubicine
b. Formation d‘espèces alkylantes par réductionb. Formation d‘espèces alkylantes par réductionb. Formation d‘espèces alkylantes par réductionb. Formation d‘espèces alkylantes par réduction de la doxorubicinede la doxorubicinede la doxorubicinede la doxorubicine
La doxorubicine peut également être à l’origine de la formation d’espèces alkylantes par
réduction à un ou deux électrons.
La doxorubicine peut subir une réaction de réduction à deux électrons, menant à la
formation d’une quinone méthide instable, de demi-vie très courte (23 secondes). Cette
réaction est catalysée par la NAD(P)H oxydoréductase en présence de réducteurs forts
(Powis, 1987). Ce dérivé quinone méthide subit ensuite une série de réaction menant à la
formation d’un métabolite 7-déoxy-aglycone (Figure 21)(Moore, 1977). Ce dérivé quinone
méthide instable présenterait un rôle d’agent alkylant monofonctionnel de l’ADN et des
protéines, mais avec une cytotoxicité moindre que le composé parent. Même si l’implication
de cette quinone méthide concernant la cytotoxicité de la doxorubicine n’est pas certaine,

55
cette réduction à deux électrons de la doxorubicine présente une application thérapeutique.
En effet, l’utilisation de réducteurs forts lors d’extravasation sanguine permettrait de
diminuer les dégâts tissulaires en formant des dérivés 7-déoxy-aglycones, moins toxiques
que le composé parent (Chabner, Longo, 2011).
La doxorubicine peut également subir une réduction à un électron, menant à la formation du
radical semi-quinone. En présence de dioxygène moléculaire, la semi-quinone réagit
rapidement avec ce dernier, à l’origine de la production de ROS (Figure 20). En milieu hypo-
aérobie ou anaérobie, le radical semi-quinone présente une stabilité bien plus importante,
donnant lieu à son auto-oxydation de la liaison entre le cycle A et la fraction daunosamine,
et résultant en la formation d’un radical libre semi-quinone centré en C7. Ce radical serait
ensuite transformé en métabolite 7-déoxy-aglycone (Figure 21) (Airley, 2009). Ce radical
présente lui aussi un rôle d’agent alkylant monofonctionnel de l’ADN.
Figure 21 : Formation de radicaux alkylants par réduction de la doxorubicine
Le radical libre semi-quinone centré en C7 et le radical quinone-méthide vont pouvoir alkyler
la molécule d’ADN afin de former des adduits à l’ADN.
c. Formation de complexes doxorubicinec. Formation de complexes doxorubicinec. Formation de complexes doxorubicinec. Formation de complexes doxorubicine----métalmétalmétalmétal
Certaines études ont montré que la peroxydation de lipides par la doxorubicine nécessitait la
présence de fer dans le milieu réactionnel (Sterrenberg et al., 1985). De plus, bien que les
ROS semblent être à l’origine de la cardiotoxicité de la doxorubicine, les anti-radicalaires se
sont révélés inefficaces comme cardio-protecteur. Ces résultats indiquent l’implication d’une
autre variable : l’interaction de la doxorubicine avec le fer.

56
� Formation de complexe Fe(III)-doxorubicine et production de ROS
Comme vu précédemment, l’ion fer(III) peut catalyser la réaction de Haber-Weiss à l’origine
de la formation de radicaux hydroxyles (Figure 20). Alternativement, une interaction directe
entre l’ion fer(III) et la doxorubicine pourrait être impliquée, formant un complexe fort entre
l’ion et trois molécules de doxorubicine, de constante de stabilité d’environ 1030 (Fiallo et al.,
1999). La chélation de l’ion fer(III) rend la doxorubicine plus électrophile, permettant son
auto-réduction en un complexe radical doxorubicine semi-quinone-fer(II), par transfert
intramoléculaire d’électron (Gianni et al., 1985). Cette réaction peut également être
catalysée par le système cytochrome P450. Phénomène normalement lent, la réduction du
complexe avec le fer(III) est accélérée en présence de résidus thiols. En présence de
dioxygène moléculaire, le fer(II) est rapidement oxydé en fer(III), avec la formation de
radicaux superoxydes (Figure 22)(Powis, 1987).
Contrairement à la réduction de la doxorubicine menant à la formation de dérivés 7-déoxy-
aglycones, la réduction du complexe avec le fer(III) n’est pas associée à une perte de
doxorubicine ou à la formation de métabolites aglycones.
D’autres ions métalliques tels que le cuivre Cu(II) ou le magnésium Mg(II) peuvent se
substituer au fer(III) pour former un complexe avec la doxorubicine et mener à la formation
de ROS. De plus, des ions métalliques réduits tels que le fer (II) ou le cuivre (I) peuvent
également cliver le peroxyde d’hydrogène afin de former des radicaux hydroxyles (Powis,
1987).
Figure 22 : Formation de ROS par formation du complexe doxorubicine-fer(III)
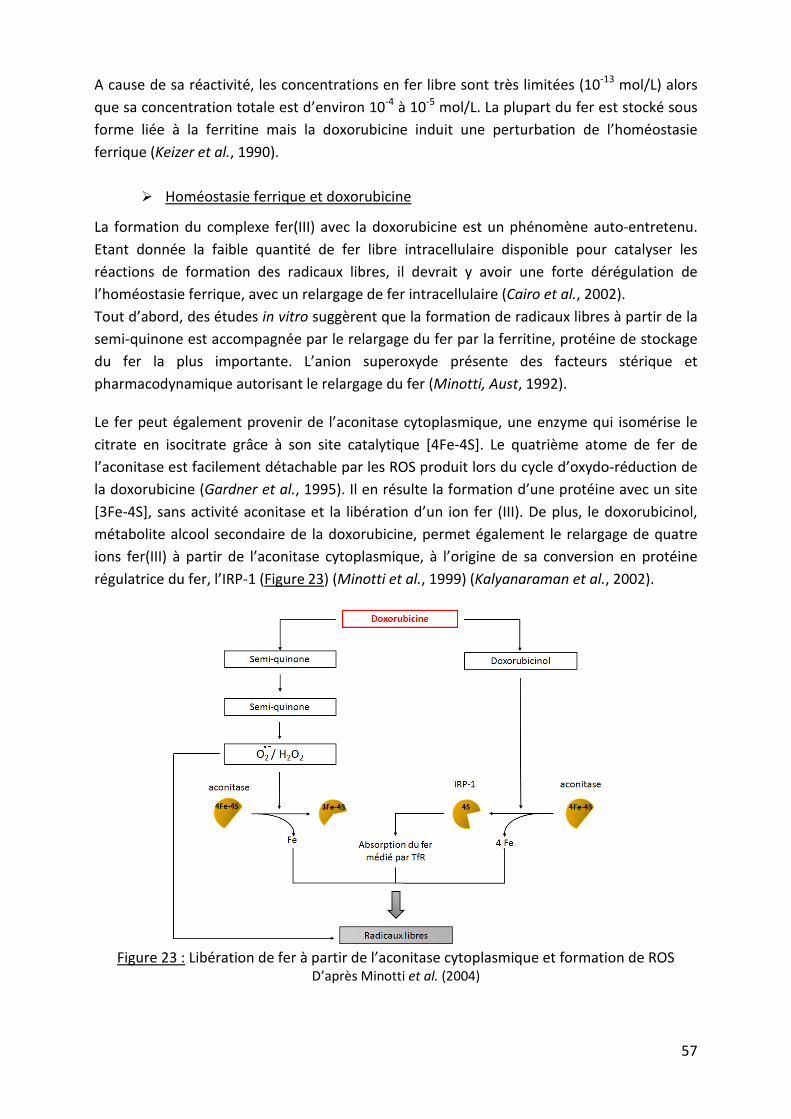
57
A cause de sa réactivité, les concentrations en fer libre sont très limitées (10-13 mol/L) alors
que sa concentration totale est d’environ 10-4 à 10-5 mol/L. La plupart du fer est stocké sous
forme liée à la ferritine mais la doxorubicine induit une perturbation de l’homéostasie
ferrique (Keizer et al., 1990).
� Homéostasie ferrique et doxorubicine
La formation du complexe fer(III) avec la doxorubicine est un phénomène auto-entretenu.
Etant donnée la faible quantité de fer libre intracellulaire disponible pour catalyser les
réactions de formation des radicaux libres, il devrait y avoir une forte dérégulation de
l’homéostasie ferrique, avec un relargage de fer intracellulaire (Cairo et al., 2002).
Tout d’abord, des études in vitro suggèrent que la formation de radicaux libres à partir de la
semi-quinone est accompagnée par le relargage du fer par la ferritine, protéine de stockage
du fer la plus importante. L’anion superoxyde présente des facteurs stérique et
pharmacodynamique autorisant le relargage du fer (Minotti, Aust, 1992).
Le fer peut également provenir de l’aconitase cytoplasmique, une enzyme qui isomérise le
citrate en isocitrate grâce à son site catalytique [4Fe-4S]. Le quatrième atome de fer de
l’aconitase est facilement détachable par les ROS produit lors du cycle d’oxydo-réduction de
la doxorubicine (Gardner et al., 1995). Il en résulte la formation d’une protéine avec un site
[3Fe-4S], sans activité aconitase et la libération d’un ion fer (III). De plus, le doxorubicinol,
métabolite alcool secondaire de la doxorubicine, permet également le relargage de quatre
ions fer(III) à partir de l’aconitase cytoplasmique, à l’origine de sa conversion en protéine
régulatrice du fer, l’IRP-1 (Figure 23) (Minotti et al., 1999) (Kalyanaraman et al., 2002).
Figure 23 : Libération de fer à partir de l’aconitase cytoplasmique et formation de ROS
D’après Minotti et al. (2004)

58
L’IRP-1 ainsi formée intervient dans la modulation de l’expression du récepteur à la
transferrine (TfR) et de la ferritine. En effet, celle-ci se lie avec une haute affinité à des iron-
conserved elements (IRE) dans des régions non transcrites de l’ARNm du TfR et de la
ferritine, augmentant la stabilité du premier et bloquant la transcription du second. Il en
résulte une augmentation de l’expression du récepteur à la transferrine et une accumulation
d’ions ferriques dans la cellule, sous forme non liée, pouvant de nouveau complexer trois
molécules de doxorubicine et produire des ROS. La conversion de l’aconitase facilite donc
l’absorption sur la séquestration du fer (Figure 24). Ce processus est spontané dans des
cellules en déplétion ferrique et sert de mécanisme adaptatif pour restaurer les fonctions
métaboliques nécessitant du fer (Cairo et al., 2002). Ce même processus est toxique dans
des cellules avec suffisamment de fer, comme lorsque la conversion de l’aconitase en IRP-1
est induite par la doxorubicine (Minotti et al., 2004).
Figure 24 : Régulation de l’homéostasie ferrique et implication du doxorubicinol
D’après Minotti et al. (2004).
2. Le stress oxydatif, une conséquence de la réduction de la 2. Le stress oxydatif, une conséquence de la réduction de la 2. Le stress oxydatif, une conséquence de la réduction de la 2. Le stress oxydatif, une conséquence de la réduction de la doxorubicinedoxorubicinedoxorubicinedoxorubicine
a. Le concept de stress oxydatif a. Le concept de stress oxydatif a. Le concept de stress oxydatif a. Le concept de stress oxydatif
L’utilisation de dioxygène moléculaire par un organisme mène inévitablement à la formation
de ROS qui, quand ils sont laissés actifs, sont à l’origine d’effets délétères oxydatifs sur les
principaux constituants cellulaires : les acides nucléiques, les protéines et les lipides
(Nordberg, Arnér, 2001). Etant donné que la formation de ces dérivés réactifs peut se
dérouler lors de processus métaboliques biologiques, incluant la respiration mitochondriale,
et qu’elle est un mécanisme d’action commun de plusieurs toxines naturelles, la plupart des
cellules ont élaboré des mécanismes de défense contre ces radicaux (Muggia, Green, 1991).
La superoxyde dismutase, la catalase et la glutathion peroxydase agissent de concert pour

59
réduire les ROS ainsi que les lipides hydro-peroxydés (Figure 25). Il existe également des
systèmes antioxydants non enzymatiques tels que les oligoéléments, cofacteurs des
enzymes antioxydantes, le glutathion réduit, les vitamines C et E, les ubiquinones, le
cytochrome c, … De plus, il existe des systèmes de réparation spécifique des dommages
oxydatifs à l’ADN (Breimer, 1991). Cependant, l’activité des enzymes capables d’activer la
doxorubicine ainsi que les défenses anti-oxydantes ne sont pas équitablement réparties dans
les différents tissus et types cellulaires, expliquant les différences de toxicité et d’action
antitumorale (Chabner, Longo, 2011).
Figure 25 : Les systèmes antioxydants enzymatiques
Le terme de stress oxydatif se réfère à une situation au cours de laquelle la formation de
ROS submerge la capacité de détoxification du système antioxydant, provoquant une
accumulation de ces dérivés réactifs oxygénés. Cela est à l’origine de conséquences
préjudiciables pour la cellule, allant du dysfonctionnement à la mort cellulaire lors d’un
stress oxydatif majeur (Li et al., 2016). Les conditions de stress oxydatif peuvent être causées
par une augmentation de la formation de dérivés réactifs oxygénés, une diminution de
l’activité anti-oxydante, voir une combinaison des deux.
Le stress oxydatif médié par la doxorubicine résulte de plusieurs mécanismes (Zhu et al.,
2016):
� La production de ROS par un cycle d’oxydoréduction, principal mécanisme de stress
oxydatif doxorubicine-induit (Figure 20).
� Une diminution des défenses cellulaires anti-oxydantes, principalement par inhibition
de la thiorédoxine réductase.
� La perturbation de la chaîne mitochondriale de transport d’électrons, à l’origine
d’une perte d’électrons captés pour augmenter la formation de ROS mitochondriaux.
Cette perturbation peut être directe, ou indirecte par modification du génome
mitochondrial.
b. L’inhibition de la thiorédoxine réductase par la doxorubicineb. L’inhibition de la thiorédoxine réductase par la doxorubicineb. L’inhibition de la thiorédoxine réductase par la doxorubicineb. L’inhibition de la thiorédoxine réductase par la doxorubicine La thiorédoxine réductase (Trr) est une flavo-enzyme présente dans la quasi-totalité des
cellules de l’organisme. Elle présente trois iso-enzymes, la forme cytosolique Trr1, la forme
mitochondriale Trr2, et la forme Trr3 localisée principalement dans les testicules.

60
Ces enzymes sont des sélénoprotéines avec un résidu sélénocystéine présent dans un motif
rédox C-terminal contenant la séquence d’acides aminés -Gly-Cys-Sec-Gly-COOH. Ce résidu
est essentiel pour l’activité catalytique de l’enzyme. Trr1 présente un rôle prédominant pour
l’homéostasie oxydo-réductrice, les défenses antioxydantes et la régulation rédox de
nombreux processus intracellulaires (Koedrith, Seo, 2011).
Cette enzyme est la seule à pouvoir prendre en charge la protéine thiorédoxine, gardée sous
forme réduite par l’enzyme, en présence du cofacteur NADPH. La thiorédoxine est une
protéine d’oxydo-réduction, composée de deux résidus cystine en son centre actif. Elle
existe sous forme dithiol-réduite ou sous forme disulfure oxydée. La forme oxydée est
réduite par l’enzyme Trr1, donnant la forme réduite qui participe à de nombreux processus
cellulaires. Il peut agit en tant que donneur d’électron pour de nombreuses enzymes, telles
que la ribonucléotide réductase, la vitamine K époxyde réductase, et la disulfure isomérase.
Il intervient également comme fixateur de radicaux libres et comme facteur endogène
activateur des récepteurs cytosoliques aux glucocorticoïdes (Mau, Powis, 1992a).
L’ensemble enzyme-substrat forme le système thiorédoxine, source importante
d’équivalents réducteurs, permettant le piégeage des radicaux libres (Mustacich, Powis,
2000).
La doxorubicine est un inhibiteur irréversible, temps-dépendant, de la thiorédoxine
réductase, avec une inhibition maximale à 24 heures. Elle mène à la diminution de la
production d’équivalents réducteurs et donc des défenses anti-oxydantes de la cellule. De
plus, étant donné que ce système thiorédoxine permet le fonctionnement de la
ribonucléotide réductase, seule enzyme catalysant la première étape de synthèse de l’ADN,
son inhibition conduit à l’arrêt de la croissance cellulaire (Mau, Powis, 1992b).
cccc. Perturbation de la chaîne mitochondriale de transport d’électrons par la . Perturbation de la chaîne mitochondriale de transport d’électrons par la . Perturbation de la chaîne mitochondriale de transport d’électrons par la . Perturbation de la chaîne mitochondriale de transport d’électrons par la doxorubicinedoxorubicinedoxorubicinedoxorubicine
La doxorubicine peut être à l’origine de la perturbation, directe ou indirecte, de la chaîne
mitochondriale de transport d’électrons.
� Effet direct
La doxorubicine présente une forte affinité pour les cardiolipides, molécules essentielles au
fonctionnement des complexes I, III et IV de la chaîne mitochondriale (Fry, Green, 1981).
L’inhibition des enzymes NADH-oxydase et succinate-oxydase pourrait donc être due à la
séquestration des cardiolipides par la doxorubicine, inhibant alors le transfert d’électrons au
sein de la chaîne respiratoire (Gilliam et al., 2013). De plus, la modification de l’activité
enzymatique de la chaîne mitochondriale pourrait être due à l’effet chaotropique de la
doxorubicine sur la membrane interne de la mitochondrie, à l’origine de l’inactivation des
enzymes succinate déshydrogénase et cytochrome c oxydase (Marcillat et al., 1989).

61
� Effet indirect
La doxorubicine peut également être à l’origine d’une perturbation indirecte de la chaîne
mitochondriale par modification du génome mitochondrial. En effet, l’intercalation au sein
de la double hélice d’ADN mitochondrial empêche la réplication et la transcription,
processus nécessaires à la synthèse des enzymes (Lebrecht et al., 2003).
Suite aux effets de la doxorubicine, la chaîne mitochondriale est non-fonctionnelle. Les
électrons sont alors détournés de leur but premier pour former des radicaux superoxydes à
partir du dioxygène moléculaire (Gilliam et al., 2013).
d. Les conséquences du stress oxydatif sur les composés cellulairesd. Les conséquences du stress oxydatif sur les composés cellulairesd. Les conséquences du stress oxydatif sur les composés cellulairesd. Les conséquences du stress oxydatif sur les composés cellulaires
La formation importante de ROS, dépassant les défenses anti-oxydantes de la cellule, est à
l’origine de dommages sur les différents constituants cellulaires. Cependant, ces ROS
n’auront qu’une action sur leur lieu de synthèse, ceux-ci étant très peu mobiles dans la
cellules (Halliwell, Gutteridge, 1984).
� La peroxydation lipidique
Au niveau membranaire, les ROS, notamment les plus toxiques, les radicaux hydroxyles, sont
à l’origine de la peroxydation des lipides. Les lipides peroxydés se forment selon un
mécanisme de réactions en chaîne (Benchekroun, Robert, 1992). Cette peroxydation
lipidique affecte le plus souvent les acides gras polyinsaturés, ceux-ci contenant plusieurs
doubles liaisons entre lesquels se trouvent des ponts méthylènes (-CH2-) présentant des
atomes d’hydrogènes fortement réactifs. La réaction consiste en trois étapes : l’initiation, la
propagation et la terminaison (Figure 26) (Ayala et al., 2014).
L’initiation est l’étape au cours de laquelle le radical acide gras est produit par réaction entre
un acide gras et des ROS.
Lors de la propagation, le radical acide gras est instable et réagit directement avec le
dioxygène moléculaire pour former un radical peroxyl-acide gras. Ce radical est également
instable et réagit avec un acide gras libre, produisant un radical acide gras différent et un
lipide hydroperoxyde, ou un peroxyde cyclique si le radical acide gras réagit avec lui-même.
Le nouveau radical acide gras réagit de même, ce qui est à l’origine d’une réaction en chaîne.
La phase de terminaison a lieu lorsque deux radicaux acides gras réagissent entre eux et
produisent une espèce non radical libre. Cela se produit lorsque la concentration en radicaux
est assez élevée pour qu’il y ait une forte probabilité de rencontre de deux radicaux. Les
antioxydants tels que la vitamine E peuvent également donner un atome d’hydrogène au
radical acide gras, formant un radical vitamine E qui va réagir avec un autre radical acide
gras, formant un produit non-radical.

62
Figure 26 : Mécanisme de peroxydation des lipides
1 : Phase d’initiation ; 2 : Phase de propagation ; 3 : Phase de terminaison par réaction de deux radicaux acide
gras ; 3’ : Phase de terminaison par réaction d’un radical acide gras et d’un radical peroxyl-acide gras ; 3’’ :
Phase de terminaison par réaction de deux radicaux peroxyl-acide gras.
Les principaux produits primaires de la peroxydation lipidique sont les lipides
hydroperoxydes. Ces lipides peuvent être la cible de réactions de réduction, à l’origine de
l’inhibition ou de l’induction de dégâts peroxydatifs, pouvant mener à la perte de l’intégrité
membranaire par altération de sa fluidité. De plus, contrairement aux ROS, les lipides
hydroperoxydes peuvent diffuser à travers les membranes et se lier aux lipides cellulaires du
cytoplasme et du noyau (Negre-Salvayre et al., 2008).
Parmi les nombreux dérivés carbonylés formés, les aldéhydes sont les produits secondaires
de la peroxydation lipidique les plus nombreux. Le malondialdéhyde (MDA) et le 4-
hydroxynonenal (4-HNE) ont été largement étudiés, le premier pour son effet mutagène et
le second pour sa forte toxicité cellulaire (Esterbauer, Cheeseman, 1990). Le MDA peut
former en l’absence de réparation, des adduits à l’ADN, à l’origine de mutations, de cassures
de brins, de l’arrêt du cycle cellulaire et de l’apoptose cellulaire (Marnett, 1999). Le 4-HNE
est quant à lui un messager cellulaire à l’origine de l’activation des différents processus de
mort cellulaire : apoptose, autophagie, nécrose et sénescence cellulaire.
En absence d’agents réducteurs, cette réaction de peroxydation peut aussi être médiée par
le complexe doxorubicine-fer(III). Le mécanisme est mal connu mais semble impliquer la
réduction du fer par la chaîne latérale de la doxorubicine. En effet, lors de l’utilisation de la
daunorubicine, analogue de la doxorubicine ne possédant pas de chaîne latérale oxydable, la
peroxydation lipidique est bien moindre que lors de l’utilisation de doxorubicine (Keizer et
al., 1990).

63
� Les dommages à l’ADN médiés par les ROS
Les agressions radicalaires créées au sein de la molécule d’ADN peuvent entraîner des
ruptures de brins, des modifications de bases azotées, de désoxyriboses et de groupements
phosphates ainsi qu’à la mise en place de cross-links ou d’adduits à l’ADN (Evans et al.,
2004). Ces effets entraînent alors une instabilité génétique et des mutations ponctuelles, qui
peuvent avoir de fortes conséquences sur la synthèse de protéines, la transmission de
l’intégrité du patrimoine génétique et la survie cellulaire (Vergely, Rochette, 2003). Par
exemple, la doxorubicine peut réduire l’expression des gènes gclc et gss codant pour le
glutathion, protéine impliquée dans la détoxification des ROS, et donc améliorer son action
cytotoxique (Leonel et al., 2014).
Cela nécessite la production de ROS au sein du noyau ou de la mitochondrie. Concernant la
mitochondrie, les dérivés réactifs sont produits à proximité de l’ADN. Pour le noyau, la
plupart des flavo-enzymes responsables de la réduction de la doxorubicine en semi-quinone
sont situées à l’extérieur de celui-ci et la semi-quinone n’est pas suffisamment stable pour
atteindre le noyau (Kalyanaraman, Baker, 1990). Les ROS ne diffusent pas non plus au sein
de la cellule, avec un rayon d’action effectif d’environ 30 Å pour les radicaux superoxydes et
hydroxyles. Il est donc nécessaire que les ROS soient formés à proximité de l’ADN. Il a été
suggéré que la formation de ROS par cycle d’oxydo-réduction de la doxorubicine puisse être
à l’origine de dommages sélectifs de l’ADN (Lown et al., 1982). Cependant, l’intercalation de
la doxorubicine au sein de la double-hélice d’ADN prévient sa réduction enzymatique et
diminue la production de ROS à proximité de l’ADN (Peters et al., 1986). Il se pourrait que la
doxorubicine intercalée puisse être réduite par une espèce réductrice diffusible de petite
taille telle que le peroxyde d’hydrogène. De même, il est également possible que la
production de ROS soit due à l’activation d’adduits à l’ADN de doxorubicine, dont la partie
chromophore est plus disponible que lorsque la doxorubicine s’intercale (Youngman, Elstner,
1984).
� Oxydation et inactivation des protéines
L’électron non apparié des ROS peut s’attaquer à la structure de certaines protéines, en
particulier les protéines porteuses d’un groupement thiol (-SH). C’est le cas de nombreuses
enzymes cellulaires et de protéines de transport qui vont ainsi être oxydées et pour
lesquelles on observe la formation de ponts disulfures intra- et intermoléculaire, ou une
dénaturation (Figure 27). Ces réactions peuvent altérer de façon marquée le métabolisme
cellulaire en modifiant la conformation et/ou l’activité biologique de ces protéines (Vergely,
Rochette, 2003).
Le stress oxydatif peut également être à l’origine de la carbonylation des protéines par
oxydation. Outre leur formation indirecte lors de la peroxydation lipidique, les dérivés
carbonylés peuvent être formés directement par oxydation des chaînes latérales des acides
aminés par des métaux ou le peroxyde d’hydrogène (Grimsrud et al., 2008). Les résidus
lysine, arginine et proline sont particulièrement concernés (Stadtman, Oliver, 1991).

64
Figure 27 : Oxydation des protéines porteuses d’un groupement thiol
L’oxydation des protéines est à l’origine de la modification de la structure et de la fonction
de la membrane, de la modification de récepteurs protéiques et des jonctions gap ainsi que
la stimulation ou de l’inhibition de l’activité enzymatique. Toutes ces modifications sont
susceptibles de déclencher les différents mécanismes de mort cellulaire (Suzuki et al., 2010).
e. Importance du stress oxydatif dans la cytotoxicité tumorale de la doxorubicinee. Importance du stress oxydatif dans la cytotoxicité tumorale de la doxorubicinee. Importance du stress oxydatif dans la cytotoxicité tumorale de la doxorubicinee. Importance du stress oxydatif dans la cytotoxicité tumorale de la doxorubicine Le stress oxydatif présente-t-il réellement un rôle dans la cytotoxicité tumorale de la
doxorubicine ? Son rôle exact est difficile à déterminer étant donné le nombre d’études
s’intéressant au sujet et présentant des résultats contradictoires.
Il subsiste des doutes sur la réalité du rôle des ROS dans la mort cellulaire des cellules
tumorales. Certaines études démontrent que la formation de radicaux hydroxyles résulte de
concentrations non pertinentes cliniquement en doxorubicine (Minotti et al., 2004). De plus,
la plupart des tumeurs solides contre lesquelles la doxorubicine présente une bonne
efficacité, sont clairement hypoxiques, remettant en cause la faisabilité de la chimie oxydo-
réductrice. Cependant, sous pression partielle faible en dioxygène, la peroxydation lipidique
médiée par le fer et les dommages à l’ADN par la semi-quinone sont effectivement
renforcés. Enfin, une supposition commune a été que les effets des ROS sur l’ADN sont peu
probables car les molécules intercalées ne pourraient pas être réduites. Cependant, la
doxorubicine liée de façon covalente aux oligonucléotides peut tout de même être
accessible aux dérivés réactifs (Peters et al., 1986).

65
Néanmoins, plusieurs faits viennent étayer le rôle du stress oxydatif. Premièrement,
certaines études montrent la présence de ROS intracellulaire lors du traitement avec de la
doxorubicine de cultures de cellules tumorales. De plus, la formation des ROS a été détectée
dans de nombreuses lignées tumorales, en intra- comme en extra-cellulaire (Ubezio, Civoli,
1994). Certaines lignées cellulaires résistantes à la doxorubicine montrent également une
augmentation des systèmes de défense contre le stress oxydatif, incluant une augmentation
du glutathion, molécule anti-oxydante, et cette résistance peut être diminuée par des agents
diminuant la concentration en glutathion (Dusre et al., 1989). Une augmentation d’un
rapport de quatre de la surexpression de la superoxyde dismutase dans certaines lignées
tumorales est à l’origine d’une augmentation de la résistance à la doxorubicine de 250 %,
tandis que l’inhibition de la superoxyde dismutase augmente sa cytotoxicité (Chabner,
Longo, 2011). Finalement, des bases oxydées de l’ADN par les produits du cycle
d’oxydoréduction sont présentes dans les urines et les cellules mononuclées du sang
périphérique chez des patients recevant de la doxorubicine (Doroshow et al., 2001).
Il y a donc bien un cycle redox dans les cellules après traitement par de la doxorubicine.
Cependant on ne sait pas si cela a lieu dans tous les types tumoraux et son importance réelle
dans les mécanismes d’action divers de la doxorubicine reste méconnue.
3333. La formation d’adduits, conséquence de la réactivité de la doxorubicine. La formation d’adduits, conséquence de la réactivité de la doxorubicine. La formation d’adduits, conséquence de la réactivité de la doxorubicine. La formation d’adduits, conséquence de la réactivité de la doxorubicine
En génétique moléculaire, les adduits à l’ADN résultent de la fixation d’une molécule à un
site nucléophile de l’ADN par liaison covalente. Ces adduits peuvent modifier l’expression de
gènes et participer à la cancérogénèse (Hemminki, 1993).
La doxorubicine peut être à l’origine de la formation d’adduits à l’ADN par différents
mécanismes :
� Par chélation directe de la molécule de doxorubicine avec l’ADN, médiée par la
formation de formaldéhyde.
� Par réduction à un ou deux électrons de la doxorubicine, à l’origine de la formation
d’agents alkylants.
L’apparition des adduits à l’ADN induit principalement des cross-links au sein de l’ADN à
l’origine de cassures double-brins et de l’inhibition des processus de transcription et de
réplication, à l’origine d’un arrêt du cycle cellulaire, principalement en phase S. Cela stimule
également les mécanismes de réparation de l’ADN et les voies de signalisation responsables
de l’induction de l’apoptose et de la nécrose cellulaire (Cutts et al., 2005). Cependant, cette
formation d’adduits ne semble pas être le mécanisme d’action majeur de la doxorubicine
étant donné que les doses cliniques résultent en la formation de seulement 4,4 +/- 1 adduits
pour 107 paires de bases (Yang et al., 2014).

66
a. Adduit à l’ADN par chélation directe de la doxorubicinea. Adduit à l’ADN par chélation directe de la doxorubicinea. Adduit à l’ADN par chélation directe de la doxorubicinea. Adduit à l’ADN par chélation directe de la doxorubicine
Il a longtemps été pensé que toutes les molécules de doxorubicine présentes dans le noyau
s’intercalaient au sein de l’ADN. Des études de microscopie par fluorescence sur des cellules
tumorales traitées par de la doxorubicine ont révélé la présence d’un intense noyau
fluorescent, interprété à l’époque comme une mesure de l’intercalation de la doxorubicine
au sein de l’ADN. Or, il a été démontré depuis que ce n’est pas l’augmentation de la
fluorescence mais sa diminution qui signe l’intercalation au sein de l’ADN. La fluorescence
serait probablement due à une liaison de la doxorubicine à des composants nucléaires par
un mécanisme non-intercalant. En réalité, la doxorubicine forme des adduits à l’ADN, sous
des conditions favorisant la chimie oxydo-réductrice fer-dépendante (Chabner, Longo, 2011).
Ce mécanisme aurait lieu pour des concentrations entre 50 µM et 1 mM (Cummings, et al.,
1991).
Lors de la formation d’adduits à l’ADN, la doxorubicine se lie par une liaison covalente à la
guanine d’un des brins d’ADN et par des liaisons hydrogènes à une guanine de l’autre brin de
la double hélice d’ADN (Figure 28) (Yang et al., 2014).
Figure 28 : Structure du complexe doxorubicine-ADN par liaison covalente (en rouge)
Plus précisément, une étude de footprinting à l’ADN in vitro en 1990 révèle l’existence de
sites spécifiques où ces adduits se forment sur l’ADN (Cullinane, Phillips, 1990). En présence
de séquences 5’-GCN-3’, le chromophore de la doxorubicine s’intercale entre les bases
azotées C et N de la séquence. La doxorubicine est liée à l’ADN par une liaison covalente
unique, se formant entre la fonction amine en 3’ de la partie daunosamine de la
doxorubicine et la fonction amine exo-cyclique en 2 de la base azotée guanine. Cette liaison
covalente nécessite la présence de formaldéhyde, lequel réagit avec la fonction amine de la
doxorubicine pour former un composé intermédiaire correspondant à deux molécules de
doxorubicine liées entre elles par trois groupements méthylènes, et appelé DOXform (Yang
et al., 2014).Cet intermédiaire est ensuite hydrolysé pour former une base de Schiff réactive,
métabolite actif se liant alors à l’ADN (Figure 29) (Minotti et al., 2004). L’adduit alors formé
est stabilisé par des liaisons hydrogènes avec le second brin de la double hélice d’ADN.

67
Ce composé intermédiaire DOXform présente une absorption accélérée par les cellules, avec
un temps de rétention nucléaire et une cytotoxicité plus importants. Ce composé est 200
fois plus cytotoxique que la doxorubicine seule et est particulièrement cytotoxique pour les
cellules cancéreuses résistantes à la doxorubicine (Swift et al., 2006).
Figure 29 : Formation du composé intermédiaire DOXform, hydrolyse en un métabolite actif,
et mécanismes de formation d’adduits doxorubicine-ADN D’après Minotti et al. (2004).

68
Les conditions optimales de formation in vitro et in vivo de ces adduits ne sont pas encore
connues en détail mais la formation optimale se déroulerait à pH 7 et nécessite la présence
d’ADN double-brins et de formaldéhyde. Le formaldéhyde provient de l’oxydo-réduction
médiée par le fer d’une source cellulaire de carbone (lipides, spermine), formant des
radicaux libres (Taatjes et al., 1996). Le formaldéhyde peut également provenir de
l’oxydation de la doxorubicine. D’importants niveaux en formaldéhyde ont été décelé dans
les cellules tumorales, en comparaison avec des cellules normales, suggérant la formation de
plus d’adduits au niveau des cellules tumorales, en addition de la sélectivité primaire offerte
par la vitesse de réplication des cellules tumorales (Cutts et al., 2005).
Ces adduits présentent une labilité à la chaleur et dans les milieux alcalins. La liaison
covalente est instable avec une demi-vie entre 5 et 40 heures à 37°C, dépendant de la
séquence d’ADN concernée, de la longueur du fragment et de la méthode d’analyse. Celle-ci
peut être maintenue à basse température (Cutts et al., 2003).
Chez l’homme, des pro-drogues à l’origine d’un relargage de formaldéhyde, tel que le
pivaloyloxymethyl butyrate, peuvent être utilisés pour améliorer la concentration
intracellulaire en formaldéhyde. Le formaldéhyde réagit avec la doxorubicine et induit la
formation d’adduits doxorubicine-ADN et une réponse cytotoxique synergique des cellules
tumorales (Swift et al., 2006).
b. Adduits à l’ADN par formation d’agents alkylantsb. Adduits à l’ADN par formation d’agents alkylantsb. Adduits à l’ADN par formation d’agents alkylantsb. Adduits à l’ADN par formation d’agents alkylants La réduction à un ou deux électrons de la doxorubicine est à l’origine de la formation de
composés radicaux libres, le radical quinone méthode et le radical libre semi-quinone en C7.
Ces deux espèces réactives peuvent agir en tant qu’agent alkylant monofonctionnel de l’ADN
et former des adduits à l’ADN en réagissant avec l’un des deux brins de la double hélice
d’ADN (Figure 30) (Powis, 1987).
Figure 30 : Adduits à l’ADN formé par alkylation monofonctionnelle à partir du radical
méthide quinone

Figure 31 : Rôle de la réactivité chimique de la doxorubicine à l’origine de sa cytotoxicité cellulaire
69

70
La doxorubicine est une espèce chimique réactive, subissant diverses réactions
(Figure 31) :
� Une réduction à un électron
• En milieu aérobie, à l’origine de la formation d’un radical semi-quinone
très réactif, menant à la formation de ROS, dont le radical hydroxyle,
composé cellulaire hautement réactif.
• En milieu anaérobie, à l’origine de la formation d’un radical libre en C7
mono-alkylant après auto-oxydation du radical semi-quinone.
� Une réduction à deux électrons, à l’origine de la formation d’une quinone
méthide instable, agent mono-alkylant.
� Une complexation avec le fer (III) intracellulaire, à l’origine de la formation de
ROS.
Cette réactivité de la doxorubicine présente plusieurs conséquences :
� Un stress oxydatif, dû aux ROS, à une diminution des défenses anti-oxydantes
de la cellule ou à une perturbation de la chaine mitochondriale. Ce stress
cellulaire présente de nombreux effets à l’origine de la cytotoxicité de la
doxorubicine et de la mort cellulaire :
→ La peroxydation lipidique, principalement due aux ROS. Elle est à
l’origine d’une perturbation des membranes cellulaires et à la formation
de seconds messagers déclenchants les différentes voies de mort
cellulaire.
→ Les dommages à l’ADN médiés par les ROS.
→ L’oxydation et l’inactivation des protéines cellulaires, principalement
thiols. Ce phénomène est à l’origine de la perturbation de nombreuses
réactions enzymatiques et voies de communications intracellulaires.
� La formation d’adduits à l’ADN, soit directement par la doxorubcine, soit par les
dérivés mono-alkylants formés lors de la réduction à un ou deux électrons de la
doxorubicine.

71
C. Effets de la doxorubicine sur les membranes plasmiquesC. Effets de la doxorubicine sur les membranes plasmiquesC. Effets de la doxorubicine sur les membranes plasmiquesC. Effets de la doxorubicine sur les membranes plasmiques
1. Rappel sur la structure des membranes plasmiques1. Rappel sur la structure des membranes plasmiques1. Rappel sur la structure des membranes plasmiques1. Rappel sur la structure des membranes plasmiques
La membrane plasmique correspond à une enveloppe limitant la cellule ou différents
organites cellulaires. Elle est constituée d’une double couche de phospholipides dans
laquelle se trouvent insérées de nombreuses molécules (protéines, glycoprotéines et glyco-
phospholipides). L’ensemble constitue une « mosaïque fluide » (Singer, Nicolson, 1972).
Dans les membranes, les lipides sont distribués de manière hétérogène au niveau de
microdomaines dynamiques. Les domaines enrichis en cholestérol, en sphingolipides et en
lipides saturés formeraient une phase dite « solide », et appelée radeaux lipidiques. Ces
radeaux lipidiques sont des structures d’environ 70 nm et représentant 10 à 30 % de la
surface membranaire. Ils sont insérés au sein d’une matrice membranaire moins ordonnée,
dite phase « fluide » (Figure 32) (Gerlier et al., 2004). La fonction de plusieurs protéines
membranaires serait fortement dépendante de leur association avec ces domaines, comme
par exemple la glycoprotéine P (Eckford, Sharom, 2008).
Figure 32 : Organisation de la membrane plasmatique et notion de radeaux lipidiques
D’après Gerlier et al. (2004).
La membrane plasmique présente de nombreux rôles. Elle sépare deux milieux tout en
maintenant la communication et les échanges entre ces deux milieux. Elle assure la
reconnaissance de signaux et de molécules provenant du milieu extérieur grâce à des
récepteurs moléculaires spécifiques. Elle régule également les flux membranaires d’ions et
de molécules. Ces différentes fonctions sont permises par la fluidité membranaire,
dépendante de facteurs externes tels que la température, et de facteurs internes :
composition en acides gras, proportion de cholestérol, proportion de protéines (Kraft, 2013).
Une altération de cette fluidité membranaire est à l’origine d’un dysfonctionnement de la
membrane et donc à une perte de ses principales fonctions.

72
2. Effets de la doxorubicine sur les membranes plasmiques2. Effets de la doxorubicine sur les membranes plasmiques2. Effets de la doxorubicine sur les membranes plasmiques2. Effets de la doxorubicine sur les membranes plasmiques
La membrane plasmique présente plusieurs avantages en tant que cible de l’action des
anticancéreux (Tritton, Hickman, 1985). En effet, celle-ci correspond à la première structure
cellulaire rencontrée par la molécule. De plus, la surface membranaire est intimement
impliquée dans la régulation des fonctions biologiques et dans les voies de signalisation
intracellulaires, la perturbation de celle-ci peut donc être cytotoxique. Enfin, les surfaces
membranaires de cellules tumorales incluent des substrats pour produits oncogènes,
absents au niveau des cellules non tumorales (Amini, Kaji, 1983). Cette différence peut être
exploitée par les molécules anti-cancéreuses.
L’action de la doxorubicine au niveau des membranes lipidiques est de plus renforcée par
une forte affinité pour les lipides membranaires chargés négativement, et notamment les
cardiolipides, constituant majeur de la membrane interne mitochondriale (Goormaghtigh et
al., 1980). Les principaux effets de la doxorubicine au niveau des membranes plasmiques
sont une perturbation de la fluidité et de la perméabilité membranaire ainsi qu’une
modification de certaines voies de signalisation intracellulaires.
a. Perturbation de la fluidité et de la perméabilité membranairea. Perturbation de la fluidité et de la perméabilité membranairea. Perturbation de la fluidité et de la perméabilité membranairea. Perturbation de la fluidité et de la perméabilité membranairessss par la par la par la par la doxorubicinedoxorubicinedoxorubicinedoxorubicine
La perturbation de la fluidité membranaire représenterait le mode d’action principal de la
doxorubicine au niveau de la membrane plasmique. Il est cependant difficile d’interpréter ce
phénomène étant donné que de nombreuses molécules non anticancéreuses influencent la
fluidité membranaire par des interactions non spécifiques (Jain et al., 1975). Cette
perturbation de la fluidité membranaire est liée à plusieurs mécanismes, dont les plus
importants sont la peroxydation lipidique et l’intercalation de la doxorubicine au sein de la
bicouche lipidique.
� La peroxydation lipidique
Une des actions de la doxorubicine sur la fluidité membranaire est la peroxydation lipidique
par production de ROS (cf partie III.B). Ces espèces réactives augmentent les teneurs en
acides gras présentant un groupement acyle, résultant en un réarrangement de l’état
physique de la membrane, diminuant alors la fluidité membranaire. D’autres modifications
par les ROS peuvent être à l’origine d’une variation de la fluidité membranaire : des
changements conformationnels des protéines membranaires, une agrégation des protéines
du cytosquelette ou bien des modifications entre les différents constituants de la membrane
(Peetla et al., 2010).
� L’intercalation de la doxorubicine au sein de la bicouche lipidique
La doxorubicine s’intercale dans la partie hydrocarbonée de la bicouche lipidique, avec une
meilleure pénétration dans la phase fluide que dans la phase solide. Cela augmenterait la
rigidité de la membrane, indépendamment de la concentration de doxorubicine (Speelmans
et al., 1997). Plus précisément, on observe deux comportements de la doxorubicine selon

73
les différentes régions membranaires. La doxorubicine augmente la fluidité membranaire
des chaînes acyl des phospholipides dans la phase dite « solide », et diminue la micro-
viscosité dans la phase « fluide » de la bicouche lipidique. En parallèle, la doxorubicine
diminue la micro-viscosité des têtes de phospholipides. L’ensemble de ces modifications
résultent en une diminution de la fluidité membranaire (Alves et al., 2017).
La doxorubicine est également à l’origine d’une modification de la perméabilité
membranaire par altération de la fluidité membranaire. Celle-ci modifie les interactions des
lipides entre eux et avec les autres constituants membranaires, à l’origine d’une
modification des caractéristiques physico-chimiques de la membrane, influençant alors de la
diffusion passive de certaines molécules (Oth et al., 1987).
b. Modifications de la voie de signalisation des céramidesb. Modifications de la voie de signalisation des céramidesb. Modifications de la voie de signalisation des céramidesb. Modifications de la voie de signalisation des céramides
La doxorubicine est à l’origine d’une modification de certaines voies de signalisation
intracellulaires par modification de structures membranaires, dont celle des céramides.
Un céramide est un sphingolipide, résultant de la combinaison d’une base à longue chaîne
(sphingosine ou dihydrosphingosine) et d’un acide gras lié par une liaison amide. Ces
molécules sont présentes dans les membranes cellulaires, où elles entrent dans la
constitution des sphingomyélines. En plus de jouer un rôle structurel dans les membranes
biologiques, elles peuvent intervenir dans la signalisation lipidique (Kitatani et al., 2008).
L’accumulation de céramides peut induire la différenciation, l’apoptose ou bien la
sénescence cellulaire. Dans tous les cas, la modification de la concentration intracellulaire en
céramides précède ces effets cellulaires lors d’un stress. Différents agents exogènes peuvent
être à l’origine de ce stress cellulaire : le TNF-α, l’interleukine-1, l’acide rétinoïque, les
prostaglandines, les radiations ionisantes et certains agents de chimiothérapie dont la
doxorubicine (Hannun, 1996).
Il existe différentes sources d’accumulation de céramides en réponse à ces stimuli, par
modulation des enzymes impliquées dans leur synthèse ou leur métabolisme (Figure 33) :
� L’induction de la sphingomyélinase neutre : la sphingomyéline, lipide membranaire,
est considérée comme le précurseur majeur de production des céramides. La
sphingomyélinase neutre est l’enzyme principale de production des céramides à
partir de la sphingomyéline.
� L’activation de la céramide synthase : située dans le réticulum endoplasmique, la
céramide synthase permet la production de céramides à partir de la sphinganine.
� La modification du métabolisme des céramides : la production de céramide peut
également être accrue par l’inhibition de la céramidase, ou par l’activation de la
céramide synthase.

74
Figure 33 : Métabolisme des céramides et effets de la doxorubicine
Deux théories s’affrontent concernant l’origine de l’augmentation de la concentration en
céramides intracellulaires lors de l’utilisation de doxorubicine. D’une part, les céramides
s’accumuleraient selon une cinétique d’apparition biphasique transitoire, dans les minutes
suivant l’exposition à la doxorubicine. Cela impliquerait une augmentation modeste de
l’accumulation de céramide, de l’ordre de 1,5 fois. Cette augmentation résulterait de
l’activation de la sphingomyélinase neutre (Jaffrézou et al., 1996). D’autre part, les
céramides s’accumuleraient selon une cinétique d’apparition progressive durant plusieurs
heures, pour une augmentation de trois à cinq fois la concentration initiale. Cette
augmentation résulterait principalement de l’activation de la céramide synthase, mais
n’exclut pas l’intervention de l’induction de sphingomyélinases dans une phase initiale (Lucci
et al., 1999) (Liu et al., 2001).
L’accumulation de céramides en intracellulaire est à l’origine du déclenchement de
l’apoptose cellulaire même si le mécanisme précis n’est pas connu. Il semblerait que les
céramides puissent activer l’apoptose en stimulant à la fois la voie intrinsèque et la voie
extrinsèque. Les céramides seraient à l’origine d’une régulation positive du ligand FasL,
stimulant alors les récepteurs de mort cellulaire et la voie extrinsèque. De plus, ils seraient à
l’origine du relargage du cytochrome c de la membrane mitochondriale, stimulant la voie
intrinsèque de l’apoptose (cf partie III.D.1) (Delpy et al., 1999).
La doxorubicine interagit également avec la membrane cellulaire, à l’origine d’une
perturbation de la fluidité et de la perméabilité membranaire, soit à cause de la
peroxydation lipidique, provoquant des modifications de la composition et des
interactions entre composants de la membrane, soit par intercalation directe au sein de
la bicouche lipidique.
La doxorubicine est également à l’origine d’une modification de voies de signalisation
intracellulaires, dont celle des céramides. Elle augmente la concentration intracellulaire
de céramides, stimulant ainsi l’apoptose.

75
D. Les différents mécanismes de mort cellulaire doxorubicineD. Les différents mécanismes de mort cellulaire doxorubicineD. Les différents mécanismes de mort cellulaire doxorubicineD. Les différents mécanismes de mort cellulaire doxorubicine----induitsinduitsinduitsinduits
Plusieurs études ont suggéré que les mécanismes d’action moléculaire décrits
précédemment tels que l’inhibition de la synthèse d’ADN par intercalation de la
doxorubicine au sein de la molécule d’ADN, l’inhibition des ADN topoisomérases de type II
ou la production de ROS, seraient à l’origine de la mort ou de l’arrêt de la croissance
cellulaire (Gewirtz, 1999). Outre l’apoptose, d’autres mécanismes cellulaires cellulaire ont
été décrits : l’autophagie cellulaire, la sénescence cellulaire et la nécrose cellulaire (Di et al.,
2009).
1. L’apoptose cellulaire1. L’apoptose cellulaire1. L’apoptose cellulaire1. L’apoptose cellulaire
Aussi appelée mort cellulaire programmée, l’apoptose cellulaire est un processus
physiologique d’autodestruction cellulaire sous l’impulsion d’un signal, permettant
l’élimination de cellules dites « inutiles » pour l’organisme. C’est un processus biologique
naturel impliqué dans la croissance normale et le développement de l’organisme. Elle est
habituellement considérée comme une forme de défense de l’organisme. Elle est activée en
réponse à des dommages ou des altérations de l’ADN, tuant la cellule afin de prévenir la
réplication et l’apparition d’autres mutants (Edinger, Thompson, 2004). La cellule
apoptotique est caractérisée par la condensation et la fragmentation nucléaires avec la
formation de corps apoptotiques, identifiés et éliminés par les phagocytes. Au cours du
processus apoptotique, les membranes sont conservées, évitant ainsi toute réaction
inflammatoire.
La plupart, si ce ne sont toutes les cellules, contiennent des facteurs nécessaires au
déclenchement de l’apoptose. Cependant, ces facteurs, tels que les caspases, sont à l’état
inactif dans la cellule. La mort cellulaire provient alors de l’activation de ces facteurs, initiée
par une large variété de conditions qui sont considérées comme imparfaites par la cellule.
L’apoptose cellulaire se déroule principalement via des voies caspases-dépendantes, celles-ci
pouvant être extrinsèque ou intrinsèque.
La voie extrinsèque est initiée par des ligands extra-cellulaires divers tels que le Fas ligand
(FasL), le Tumor Necrosis Factor-α (TNF-α) ou le TNF-Related Apoptosis-Inducing Ligand
(TRAIL). Ceux-ci se lient à des récepteurs de mort pro-apoptotiques situés au niveau de la
membrane cellulaire. Ces récepteurs de mort sont couplés à la procaspase-8 par une
protéine transmembranaire associée, nommée Fas-Associated Death Domain (FADD). La
liaison d’un ligand au niveau du récepteur induit la formation d’un complexe de haut poids
moléculaire appelé Death Inducing Signal Complex (DISC). Ce complexe est constitué de la
protéine FADD et de la procaspase-8, à l’origine de l’auto-clivage de celle-ci en caspase-8
initiatrice, activant à son tour les caspases effectrices -3, -6 et -7. Celles-ci sont à l’origine de
l’apoptose par activation du système de DNases activées par les caspases Figure 34(Tacar et
al., 2013). La doxorubicine joue le rôle d’inducteur de la voie extrinsèque, via la régulation
de Fas. Plusieurs hypothèses existent sur le rôle exact de la doxorubicine dans l’induction de
la voie extrinsèque. Son action pourrait se faire via la régulation négative de l’expression de
Fas soluble, un inhibiteur de FasL (Zhang et al., 2009). Une autre possibilité serait l’activation

76
par la doxorubicine de la voie calcium/calcineurine, à l’origine de l’activation du Nuclear
Factor Activated T-cell 4 (NFATc4), lui-même à l’origine d’une régulation positive de FasL
(Kalivendi et al., 2005). La doxorubicine est également à l’origine de l’activation du Nuclear
Factor-kappa B (NF-κB) via la production de ROS, menant à la régulation positive de
plusieurs gènes pro-apoptotiques tels que fas, myc ou p53 (Wang et al., 2002).
Figure 34 : Rôle de la doxorubicine dans l’activation des voies extrinsèque et intrinsèque de
l’apoptose cellulaire Traduit et adapté de l’anglais d’après Meredith et al (2016).
AMPK : AMP-activated protein kinase ; Apaf-1 : apoptotic protease-activating factor-1 ; Bax : Bcl-2-associated X
; Bcl-2 : B-cell leukaemia/lymphoma 2 ; FADD : Fas-associated death domain ; FasL : Fas ligand ; JNK : c-Jun N-
terminal kinase ; mTORC1 : mammalian target for rapamycin complex 1 ; TNF-a : tumour necrosis factor alpha ;
and TRAIL : TNF-related apoptosis-inducing ligand.
La voie intrinsèque est quant à elle médiée par la famille de protéines anti-apoptotiques de
la famille B-cell leukaemia/lymphoma 2 (Bcl-2), dont les membres sont présents à la surface
membranaire des mitochondries. La doxorubicine initie la mort cellulaire par apoptose en
induisant l’activation de l’AMP kinase (AMPK), activant à son tour p53 et la c-Jun N-terminal
kinase (JNK), et inactivant la mammalian target for rapamycin complex 1 (mTORC1).
L’augmentation d’expression du gène p53 induit une régulation négative de la protéine anti-
apoptotique Bcl-2 et une régulation positive de la protéine pro-apoptotique Bcl-2-associated
X (Bax). La doxorubicine est donc à l’origine d’une altération du ratio Bcl-2/Bax, facteur clé
du devenir de la cellule (Chen et al., 2011). Cette altération mène à la libération du
cytochrome c et d’autres facteurs mitochondriaux de la membrane interne mitochondriale
tels que l’apoptosis inducing factor (AIF). Le cytochrome c forme alors un complexe de
l’apoptosome avec l’apoptotic protease-activating factor-1 (Apaf-1) et la procaspase-9,
permettant l’activation de cette dernière en caspase-9, initiatrice de l’apoptose. Une fois
activée, celle-ci quitte le complexe de l’apoptosome et active les caspases effectrices -3, -6
et -7, à l’origine de l’apoptose cellulaire (Figure 34) (Meredith, Dass, 2016).

77
Bien que les voies extrinsèque et intrinsèque présentent un fonctionnement indépendant, le
chevauchement des deux voies à parfois lieu. En effet, il semblerait par exemple que la
protéine p53 puisse réguler positivement certains récepteurs pro-apoptotiques de la voie
extrinsèque ou que la caspase-8 puisse augmenter l’expression du gène codant pour la
protéine Bax, déclenchant alors la voie intrinsèque de l’apoptose (Ashkenazi, Herbst, 2008).
2. L'autophagie cellulaire2. L'autophagie cellulaire2. L'autophagie cellulaire2. L'autophagie cellulaire
L’autophagie cellulaire, ou autophagocytose, correspond à la dégradation d’une partie du
cytoplasme de la cellule par ses propres lysosomes (Lockshin, Zakeri, 2004). C’est un
processus biologique catabolique naturel impliqué dans le maintien de l’homéostasie
cellulaire, communément impliqué dans le turn-over des organites et des protéines longue
vie, ainsi que dans la dégradation des organites non fonctionnels et des agrégats protéiques
(Lee et al., 2012). Au cours de ce processus, les macromolécules et les organites sont
dégradés par les lysosomes et recyclés en énergie et nutriments. Selon les conditions de vie
cellulaire, l’autophagie peut être cytotoxique ou cytoprotectrice. Lors des stades précoces de
développement tumoral, l’autophagie permettrait l’élimination des protéines non
fonctionnelles qui pourrait concourir à la transformation d’une cellule saine en cellule
tumorale (Karantza-Wadsworth et al., 2007). Cependant, lors du développement tumoral,
l’autophagie permettrait la survie des cellules tumorales en les protégeant des ROS et en
supprimant les organites et protéines non fonctionnels (Hart et al., 2012).
La doxorubicine peut induire le processus d’autophagie via le stress oxydatif résultant de la
production de ROS au niveau de la mitochondrie. Ces dérivés provoquent un
dysfonctionnement de la chaîne respiratoire de la mitochondrie et une perturbation de la
production d’énergie, à l’origine d’un état de faible énergie cellulaire et de l’augmentation
du ratio AMP/ATP. On observe alors une activation de la kinase calmoduline-dépendante et
de l’AMPK, responsable de l’activation de la JNK et de l’inhibition de mTOR.
a. Rôle de l’inhibition de mTOR dans la régulation de l’autophagie cellulairea. Rôle de l’inhibition de mTOR dans la régulation de l’autophagie cellulairea. Rôle de l’inhibition de mTOR dans la régulation de l’autophagie cellulairea. Rôle de l’inhibition de mTOR dans la régulation de l’autophagie cellulaire
La régulation de l’autophagie est réalisée par une famille de gènes, appelés Autophagy-
related genes (Atg), via des interactions avec des complexes protéiques formés durant le
processus d’autophagie. L’activation ou l’inhibition de l’autophagie cellulaire est dépendante
de la protéine mTOR, normalement responsable de la phosphorylation des protéines Atg13
et Unc-like-51 kinase 1 (Ulk 1). Durant la privation en mTOR, la dissociation de celui-ci avec le
complexe Atg13-FIP200-Ulk1 résulte en la déphosphorylation d’Atg13 et d’Ulk1. Cela active
Ulk1 qui phosphoryle à son tour Atg13 et la focal adhesion kinase family interacting protein
of 200 kDa (FIP200), aboutissant à la formation d’une membrane pré-autophagosomale
(Figure 35).

78
b. Rôle de l’activation de la cb. Rôle de l’activation de la cb. Rôle de l’activation de la cb. Rôle de l’activation de la c----Jun NJun NJun NJun N----terminal kinase (JNK) dans l’autophagie terminal kinase (JNK) dans l’autophagie terminal kinase (JNK) dans l’autophagie terminal kinase (JNK) dans l’autophagie cellulairecellulairecellulairecellulaire
La suite du processus nécessite la maturation de l’autophagosome, sous contrôle de la
vacuolar-sorting protein 34 (Vps34), aussi connue sous le nom de phosphoinositide 3-kinase
de classe III (PI3K). La protéine Vps34 est activée en formant un complexe protéique avec
beclin-1, Vps15 et est renforcée par le Bax-interacting factor-1 (Bif-1) et le gène associé à la
résistance aux radiations UV (UVRAG). Vps34 interagit alors avec la protéine Atg1 pour
former l’autophagosome (Figure 35). La formation de ce complexe protéique est régulée par
la dissociation de Bcl-2 avec beclin-1 par JNK, stoppant l’inhibition de beclin-1 par Bcl-2 et
permettant sa complexation avec Vps34.
Figure 35 : Rôle de la doxorubicine dans l’induction de l’autophagie cellulaire
Traduit et adapté de l’anglais d’après Meredith et al (2016). AMP : adenosine monophosphate ; AMPK :AMP-activated protein kinase ; Atg : AuTophaGy-related ; ATP :
adenosine triphosphate ; Bcl-2 : B-cell leukaemia/lymphoma 2 ; Bif-1 : Bax interacting factor-1 ; FIP200 : focal
adhesion kinase family interacting protein of 200kDa ; JNK : c-Jun N-terminal kinase ; mTOR : mammalian target
for rapamycin ; PARP-1 : poly(ADP-ribose) polymerase-1 ; Ulk-1 : Unc-like-51 kinase 1 ; UVRAG : ultraviolet
radiation resistance-associated gene ; and Vps : vacuolar-sorting protein.

79
Une voie alternative de stimulation de l’autophagie cellulaire par la doxorubicine est
possible par l’activation de la poly(ADP-ribose) polymérase-1 (PARP-1), responsable de
l’inhibition de mTOR durant des périodes de stress cellulaire dues à une carence en
nutriments (Rodríguez-Vargas et al., 2012).
Il a été suggéré qu’un faible niveau d’autophagie promeut la survie cellulaire en prévenant
l’apoptose cellulaire, tandis qu’une augmentation significative de l’autophagie est à l’origine
de la mort cellulaire par dégradation excessive des organites et des macromolécules,
modifiant l’homéostasie cellulaire. La présence ou non de caspases semble également être
un facteur important dans le mécanisme cellulaire de cytotoxicité, l’absence ou l’inhibition
de ces protéines pouvant donner lieu au passage d’un processus d’apoptose à celui
d’autophagie (Edinger, Thompson, 2004).
3. La sénescence cellula3. La sénescence cellula3. La sénescence cellula3. La sénescence cellulaireireireire
La sénescence cellulaire est un état cellulaire dans lequel la cellule ne peut plus se diviser
mais reste métaboliquement active. En pratique, cela correspond à un blocage du cycle
cellulaire entre la phase G2 et la phase M et entre la phase G1 et la phase S. Deux types de
sénescence cellulaire sont classiquement décrites. La sénescence réplicative, ou
télomérique, impliquant un arrêt de la croissance due à un raccourcissement télomérique
après un nombre prédéterminé de divisions cellulaires dans des cellules non modifiées
(Gewirtz, 2014). La sénescence prématurée, aussi appelée sénescence accélérée ou induite
par le stress, pouvant se produire en présence d’oncogènes afin de tenter d’empêcher ou
bien de reporter la transformation cellulaire ou lors d’utilisation d’agents de chimiothérapie
(Gewirtz et al., 2008). Lors du développement tumoral, la sénescence prématurée présente
une dualité de fonction. Celle-ci permet un arrêt de croissance prolongée des cellules
tumorales mais elle peut aussi refléter une forme de dormance pour les cellules tumorales
qui pourrait être annulée par des stimuli exogènes (Di et al., 2009).
L’induction de la sénescence prématurée présente des similarités avec l’apoptose et d’autres
voies inhibant la croissance cellulaire. L’administration de doxorubicine induit la synthèse de
la protéine p53, laquelle régule positivement la protéine p21, à l’origine de l’arrêt du cycle
entre les phases G1 et S, et négativement la cyclin-dependant kinase-1 (cdk1), aussi appelée
cell-division cycle-2 (cdc2), à l’origine de l’arrêt du cycle entre les phases G2 et M (Figure 36).
Ce processus cellulaire a lieu en l’absence de la protéine p53, indiquant que p21 et cdk1 ont
un rôle vital dans l’induction de la sénescence alors que la protéine p53 ne semble pas jouer
un rôle critique.
La sénescence prématurée ne semble pas être le mécanisme d’action cellulaire principal de
la doxorubicine mais plutôt être un mécanisme alternatif utilisé dans les cellules où
l’apoptose est inhibée (Rebbaa et al., 2003).

80
Figure 36 : Rôle de la doxorubicine dans l’induction de la sénescence prématurée
Traduit et adapté de l’anglais d’après Meredith et al (2016). Cdc2 : cell-division cycle 2 ; and cdk1 : cyclin-dependent kinase-1
4. La nécrose cellul4. La nécrose cellul4. La nécrose cellul4. La nécrose cellulaireaireaireaire
La nécrose cellulaire est un processus passif résultant de la diminution de production d’ATP à
un niveau rendant la survie cellulaire impossible, menant à une catastrophe bio-énergétique
et à la mort cellulaire prématurée et non programmée. Cela pourrait être dû à des
dommages physiques ou toxiques, suggérant que les activités cytotoxiques de la
doxorubicine, incluant les dommages à l’ADN et le stress oxydatif, pourraient mener à une
nécrose cellulaire (Edinger, Thompson, 2004).
Lors d’un déficit en protéine p53 ou en protéines de la famille Bcl-2, notamment la protéine
Bax, le mécanisme d’apoptose est altéré. Le processus de nécrose cellulaire autorise à la
doxorubicine de rester active, indépendamment de p53 ou de Bax.
La doxorubicine induit la nécrose cellulaire par deux voies distinctes (Figure 37) :
� La doxorubicine est à l’origine de la synthèse de ROS et d’une perturbation de
l’homéostasie calcique, menant à l’ouverture cyclophiline D-dépendante des
pores de transition de perméabilité membranaire et à un œdème
mitochondrial. Cela perturbe la production d’énergie mitochondriale et
diminue le pool d’ATP cellulaire, déclenchant la nécrose cellulaire (Wallace,
2007).
� Par ses différents mécanismes d’action moléculaire, la doxorubicine
endommage l’ADN. Cela est à l’origine de l’activation de PARP-1 et du H2A
histone family member X (H2AX). PARP-1 est une protéine réparatrice de
l’ADN qui initie le processus de nécrose cellulaire (Feoktistova, Leverkus,
2015). Son activation excessive est à l’origine d’une déplétion cellulaire en
nicotinamide adénine dinucléotide (NAD+) et de l’inhibition de la glycolyse
(Ying, Padanilam, 2016). Cela perturbe la production d’énergie et diminue le
pool d’ATP cellulaire. Les cellules végétatives non-proliférantes seront plus
sujettes à la réparation de l’ADN qu’à la nécrose cellulaire, la production
d’énergie cellulaire étant principalement due à la phosphorylation oxydative

81
et au catabolisme des acides aminés et des lipides au niveau des
mitochondries. Au contraire, les cellules proliférantes, telles que les cellules
tumorales, sont dépendantes de la glycolyse pour la production d’ATP, ces
cellules utilisant les acides aminés et les lipides pour la synthèse de protéines
et de membranes.
Une autre voie de nécrose programmée est initiée par l’activation de TRAIL ou de TNF et
l’inhibition de la caspase-8. Cependant, le rôle de la doxorubicine au sein de cette voie n’est
pas clairement défini (Meredith, Dass, 2016).
Etant donné que de nombreuses cellules tumorales proviennent de mutations inhibant
l’apoptose et/ou autorisant les cellules à passer les check-points du cycle cellulaire, cette
voie alternative de nécrose cellulaire pourrait expliquer pourquoi les anticancéreux tels que
la doxorubicine induisent tout de même la mort cellulaire, malgré le blocage des autres voies
(Edinger, Thompson, 2004).
Figure 37 : Rôle de la doxorubicine dans la nécrose cellulaire
Traduit et adapté de l’anglais d’après Meredith et al (2016). ATP : adenosine triphosphate ; H2AX : H2A histone family member X ; MLKL : mixed-lineage kinase domain-like
protein ; MPT : mitochondrial permeability transition ; NAD : nicotinamide adenine dinucleotide ; PARP-1 : poly
(ADP-ribose) polymerase-1 ; RIPK : receptor-interacting serine/threonine protein kinase ; TNF : tumour necrosis
factor ; and TRAIL : TNF-related apoptosis-inducing ligand.
Etant donné la palette de mécanismes d’action de la doxorubicine, il est possible que la cible
cellulaire varie selon le type tumoral, et ainsi, l’importance de chaque mécanisme serait
individu-dépendant. Cependant, la concentration intracellulaire en doxorubicine semble être
le facteur déclenchant, avec une concentration importante nécessaire à la mort cellulaire
(Tacar et al., 2013). Cela nécessite cependant des études supplémentaires pour déterminer
la concentration la plus à même de tuer tout type de cellules cancéreuses afin d’obtenir un
traitement efficace mais présentant également une toxicité moindre (Jackson, 2003).

82
De plus, même si l’apoptose semble être le mécanisme cellulaire prédominant lors de
l’utilisation de la doxorubicine, cela reste soumis aux différents mécanismes de résistance
mis en place par les cellules tumorales.
E. Les mécanismesE. Les mécanismesE. Les mécanismesE. Les mécanismes de résistance des cellules tumorales à la doxorubicinede résistance des cellules tumorales à la doxorubicinede résistance des cellules tumorales à la doxorubicinede résistance des cellules tumorales à la doxorubicine
La résistance aux agents de chimiothérapie anticancéreuse est définie comme l’impossibilité
d’obtenir ou de maintenir le résultat thérapeutique escompté et constitue une des causes
principales des échecs de la chimiothérapie anticancéreuse.
On peut définir deux types de résistance :
� La résistance intrinsèque, due aux caractéristiques de la tumeur, à sa vascularisation
et à sa cinétique de croissance. La tumeur est d’emblée résistante ou peu sensible
aux médicaments, ou présente une capacité de réparation supérieure à celle des
tissus sains. Cette résistance est préalable à tout traitement.
� La résistance acquise, c’est-à-dire exprimée après le traitement, par échappement.
Les agents anticancéreux ne sont plus actifs par émergence de cellules tumorales
mutées résistantes.
La doxorubicine agit sur différents mécanismes de mort cellulaire dont :
� L’apoptose cellulaire :
• Par la voie intrinsèque en activant p53, menant à l’activation de la
caspase-9 initiatrice.
• Par la voie extrinsèque en activant les récepteurs de mort, menant à
l’activation de la caspase-8 initiatrice.
Ces deux voies aboutissent à l’activation des caspases-3, -6 et -7 effectrices,
permettant l’activation de DNases et l’apoptose cellulaire.
� L’autophagie cellulaire :
• Par le stress oxydatif, à l’origine de l’activation de l’AMPK, responsable
de la formation et de la maturation de l’autophagosome.
• Par les dommages à l’ADN, menant à l’activation de PARP-1 et donc à
l’inhibition de mTORC1 et à la formation de la membrane pré-
autophagosomale.
� La sénescence cellulaire, par régulation positive de p21 et régulation
négative de cdk1, à l’origine d’un arrêt de la croissance cellulaire.
� La nécrose cellulaire, par diminution de la production d’énergie ATP, due au
stress oxydatif ou aux dommages à l’ADN.

83
Bien que ces deux types de résistance représentent deux entités cliniques distinctes, les
mécanismes sous-jacents à ces situations sont probablement similaires (Zandvliet, Teske,
2015).
Certains mécanismes de résistance à la chimiothérapie ont été identifiés à différents
niveaux, anatomophysiologique, cellulaire et/ou moléculaire. La plupart de ces mécanismes
de résistance sont issues de recherches in vitro, par le développement de lignées cellulaires
présentant un phénotype de résistance, mais leurs rôles in vivo restent encore parfois
controversés.
Il est possible de distinguer deux types de résistance cellulaire (Housman et al., 2014) :
� La résistance cellulaire spécifique à une molécule ou une classe de molécules, qui
s’appuie sur une altération du transport spécifique, une modification du métabolisme
ou une altération des cibles intracellulaires.
� La résistance cellulaire pléiotrope, c’est-à-dire une résistance à plusieurs molécules
ou classes de molécules, souvent appelée multidrug resistance, ou MDR. Elle peut
résulter de l’induction d’un seul mécanisme de résistance capable d’influencer
plusieurs molécules mais peut aussi découler d’une seule molécule qui va déclencher
de multiples mécanismes de résistance. Elle prend en compte les modifications de
l’expression de gènes et de protéines, l’activation de mécanisme de réparation ou de
détoxification et l’altération de certaines protéines de transport.
Les informations disponibles sur la résistance aux anticancéreux chez le chien sont limitées.
Cependant, contrairement à la pharmacocinétique à l’origine de la résistance, qui varie entre
les espèces à cause des différences de métabolisme, les mécanismes cellulaires semblent
être similaires à ceux présent chez l’Homme, étant donnée la conservation inter-espèces des
voies impliquées.
Cinq mécanismes de résistance peuvent être identifiés concernant la doxorubicine (Thorn et
al., 2011) :
� La résistance due à une diminution de la concentration intracellulaire de la
doxorubicine par augmentation de l’efflux. C’est le mécanisme de résistance le plus
étudié dans l’espèce canine.
� La résistance due à l’altération quantitative et/ou qualitative d’une des protéines
cibles, l’ADN topoisomérase de type II.
� La résistance due à une modulation des processus enzymatiques impliqués dans le
métabolisme de la doxorubicine.
� La résistance due à l’activation des systèmes de réparation de l’ADN.
� La résistance due à l’inactivation de l’apoptose.

84
1. La 1. La 1. La 1. La résistance à la doxorubicine par augmentation de son efflux cellulairerésistance à la doxorubicine par augmentation de son efflux cellulairerésistance à la doxorubicine par augmentation de son efflux cellulairerésistance à la doxorubicine par augmentation de son efflux cellulaire
Le phénotype MDR implique une diminution de la concentration intracellulaire en
doxorubicine. Ce phénomène se traduit par un efflux actif de celle-ci hors de la cellule,
associé ou non à une altération de sa distribution subcellulaire par la séquestration
cytoplasmique.
L’efflux cellulaire est souvent considéré comme la phase III du métabolisme de la
doxorubicine. Celui-ci est permis par une grande variété de transporteurs, dont les plus
importants appartiennent à la superfamille des ATP-binding cassette (ABC). Ce sont des
transporteurs membranaires impliqués dans la translocation de la doxorubicine contre son
gradient, par un mécanisme ATP-dépendant. Parmi ces transporteurs, on peut citer la
glycoprotéine-P (P-gp) et les protéines MRP1 et BCRP (cf partie II.D).
Les cellules tumorales qui expriment initialement à un haut niveau les transporteurs à la
doxorubicine peuvent être intrinsèquement résistantes à celle-ci. Alternativement, la
sélection de la résistance en présence de la doxorubicine peut résulter en une élévation de
l’expression des transporteurs, le plus souvent par amplification du gène codant pour le
transporteur.
Dans la suite, nous nous attarderons à décrire le rôle potentiel des différents transporteurs
dans la résistance des cellules tumorales à la doxorubicine chez le chien.
� La glycoprotéine-P
La P-gp est exprimée dans certaines lignées de cellules tumorales canines, incluant les
leucémies lymphoïdes, les mastocytomes et les ostéosarcomes (Page et al., 2000), ainsi que
dans des tumeurs in vivo, incluant certains carcinomes (Petterino et al., 2006), les
mastocytomes et les lymphomes malins (Bergman et al., 1996). L’expression de la P-gp a été
associée à la résistance cellulaire à la doxorubicine pour des lignées cellulaires de lymphome
B canins (Uozurmi et al., 2005). Cependant, même si la concentration en ARNm est en
corrélation avec la sensibilité à la doxorubicine in vitro, cette corrélation n’est pas retrouvée
in vivo (Steingold et al., 1998).
Le mécanisme génétique derrière l’augmentation de l’expression du gène mdr1 dans les
lignées cellulaires résistantes in vitro est variable. Les cellules faiblement résistantes
présentent une activation importante de la transcription et de la traduction. Pour les cellules
hautement résistantes, on observe également une amplification génique, soit sous forme
libre (DM, double minute chromosome fragment), soit intégré de façon linéaire sur le
chromosome au locus originel ou à un autre locus (HSR, homogeneous staining region)
(Storlazzi et al., 2010).

85
� La protéine MRP1
Dans l’espèce canine, la protéine MRP1 est exprimée au niveau de certains lymphomes B
multicentriques (Tomiyasu et al., 2010), de mastocytomes cutanés (Miyoshi et al., 2002) et
de certains carcinomes (Lee et al., 2007) (Honscha et al., 2009).
Chez l’homme, le mécanisme d’apparition du phénotype MDR impliquant MRP1 n’est pas
encore totalement déterminé. Des amplifications géniques ont été relevées dans divers
modèles cellulaires mais il est possible que des mutations d’éléments cis-régulateur ou de
facteurs de trans-activation augmentent la transcription du gène mrp1. Dans l’espèce
canine, le gène mrp1 ne semble pas être associé à une résistance à la doxorubicine (Ma et
al., 2002).
� La protéine BCRP
Chez le chien, la protéine BCRP est exprimée au niveau de certaines tumeurs mammaires,
son niveau d’expression augmentant avec le grade de la tumeur (Honscha et al., 2009)
(Nowak et al., 2009). Celle-ci est aussi exprimée dans le cas de lymphomes B et T (Zandvliet
et al., 2015).
Chez l’homme, seule la protéine BCRP mutée transporte la doxorubicine. Dans l’espèce
canine, certaines études montrent qu’une résistance médiée par BCRP est possible par
augmentation de son expression, sans mutation de cette dernière (Honscha et al., 2009). Il
existe cependant peu d’études sur l’implication de BCRP dans la résistance des cellules
tumorales à la doxorubicine.
Bien que ces transporteurs ABC soient typiquement localisés au niveau de la membrane
cellulaire, ils peuvent aussi être présents au sein des membranes des différents organites
intracellulaires tels que le réticulum endoplasmique, les mitochondries et le peroxysome,
permettant alors une séquestration de la doxorubicine. Celle-ci est alors transportée du
cytoplasme au réticulum endoplasmique et n’exerce plus son action cytotoxique, menant
alors à la résistance de la cellule à la doxorubicine (Arancia et al., 1998). L’expression de la
transport associated with antigen processing (TAP), ou ABCB2, a été associée à une faible
résistance des cellules tumorales à la doxorubicine (Izquierdo et al., 1996) (Zandvliet et al.,
2015).
2. La résistance à la doxorubicine associée aux ADN 2. La résistance à la doxorubicine associée aux ADN 2. La résistance à la doxorubicine associée aux ADN 2. La résistance à la doxorubicine associée aux ADN topoisomérases detopoisomérases detopoisomérases detopoisomérases de type IItype IItype IItype II
Bien que la synthèse de l’ADN topoisomérase de type II soit dépendante du cycle cellulaire
(cf partie III.A.2), des altérations quantitatives et/ou qualitatives de cette enzyme nucléaire
ont pu être observées, pouvant expliquer la résistance de certaines cellules tumorales à la
doxorubicine. La résistance résulterait d’une diminution de l’expression de l’ADN
topoisomérase de type II ou du développement de mutations de cette dernière (Pommier et
al., 2010).

86
a. Résistance par altération de la concentration ea. Résistance par altération de la concentration ea. Résistance par altération de la concentration ea. Résistance par altération de la concentration en ADN topoisomérase de typen ADN topoisomérase de typen ADN topoisomérase de typen ADN topoisomérase de type IIIIIIII
Les niveaux d’expression des isoformes des ADN topoisomérases de type II présentent
d’importantes variations dans les cellules tumorales et non-tumorales. Des études sur
lignées cellulaires ont montré qu’un haut niveau d’expression de l’isoforme α conférerait
une sensibilité accrue à la doxorubicine, et serait un marqueur de prolifération (Booser,
Hortobagyi, 1994). L’un des mécanismes généraux de résistance à la doxorubicine
correspondrait donc à la présence en plus faible concentration d’ADN topoisomérase de
type II nucléaire, à l’origine d’une diminution de la cytotoxicité de la doxorubicine.
La concentration en ADN topoisomérase de type II peut être réduite par diminution de la
transcription. Alors que l’expression d’au moins une des deux isoformes est nécessaire pour
la viabilité cellulaire, la cellule peut réduire drastiquement l’expression de l’une de ces deux
isoformes, avec peu d’effets sur la croissance. Pour la doxorubicine, il semblerait que la
résistance soit inversement proportionnelle à l’expression de l’ADN topoisomérase de type
IIβ (Brown et al., 1995). Cependant, certaines études sont contradictoires et ne montrent
pas de corrélation entre la résistance à la doxorubicine et le niveau d’expression des
isoformes, cela pouvant être due à la multitude de mécanismes d’action de la doxorubicine
(Houlbrook et al., 1995). Plusieurs facteurs influencent le niveau d’expression de ces
isoenzymes. Une diminution de la transcription du gène top2α est associée à un niveau élevé
en Sp3, un facteur de transcription pouvant activer ou réprimer l’expression de certains
gènes (Harris, Hochhauser, 1992). Le mode d’action de l’ADN topoisomérase de type II
repose sur la formation d’un complexe ternaire avec la doxorubicine et l’ADN. Sa localisation
se doit donc d’être nucléaire. Certaines lignées cellulaires tumorales résistantes à la
doxorubicine présente une localisation cytoplasmique de l’ADN topoisomérase de type IIα
(Harker et al., 1995). La localisation nucléaire de l’ADN topoisomérase de type IIα nécessite
des signaux de localisation nucléaire, présents à son extrémité C-terminale. La mutation de
cette portion inhibe alors sa translocation nucléaire. Cependant, la cellule ne présente pas
d’anomalies de croissance en l’absence de l’ADN topoisomérase de type IIα à localisation
nucléaire (Walker, Nitiss, 2002).
b. Résistance altération de la sensibilité de l’ADN topoisomérase de type II à la b. Résistance altération de la sensibilité de l’ADN topoisomérase de type II à la b. Résistance altération de la sensibilité de l’ADN topoisomérase de type II à la b. Résistance altération de la sensibilité de l’ADN topoisomérase de type II à la doxorubicinedoxorubicinedoxorubicinedoxorubicine
Les modifications décrites précédemment induisent une diminution de la concentration en
ADN topoisomérase de type II mais pas d’altération de sa sensibilité vis-à-vis de la
doxorubicine. Pourtant, certaines mutations peuvent conduire à cette absence de sensibilité,
avec une concentration normale en ADN topoisomérase de type II (Bergman, 2003). Il existe
plusieurs mécanismes par lesquels les mutations de l’ADN topoisomérase de type II mènent
à la résistance des cellules tumorales à la doxorubicine. L’une d’entre elle correspond aux
modifications du site de liaison entre la doxorubicine et l’enzyme, prévenant l’inhibition de
cette dernière ou la formation du complexe ternaire. Une autre possibilité réside dans
l’altération du site actif de l’enzyme, à l’origine d’une réduction de l’activité enzymatique
(Nitiss, 2009b).

87
La mutation dans le gène top2 conférant la résistance à la doxorubicine est récessive au type
sauvage. Autrement dit, si une cellule est hétérozygote, la cellule sera toujours sensible à
l’anti-cancéreux. En effet, la réintroduction du gène sauvage dans une cellule résistante
restaure la sensibilité de cette dernière (Eder et al., 1993). Il est possible d’avoir une
résistance par dominance partielle. Etant donné que l’ADN topoisomérase de type II est un
dimère, il est possible que l’une des deux sous-unités soit résistante et que l’autre soit
sensible à la doxorubicine. Dans cette situation, le niveau total en enzymes sensibles sera
réduit (Nitiss et al., 1994).
Il existe de nombreuses études sur les mutations du gène top2α, l’analyse de celles-ci ne
révélant pas de locus particulier menant à la résistance des cellules tumorales à la
doxorubicine (Walker, Nitiss, 2002).
L’état de phosphorylation semble également être impliqué dans la résistance à la
doxorubicine. Puisque la phosphorylation de l’ADN topoisomérase de type II durant le cycle
cellulaire régule l’activité de l’enzyme, son rôle dans la résistance des cellules tumorales à la
doxorubicine a été étudié (Kohno et al., 1991). L’hyperphosphorylation de l’ADN
topoisomérase de type II semble être à l’origine de la résistance des cellules. Puisque la
phosphorylation est essentielle aux évènements catalytiques de décaténation,
l’hyperphosphorylation de l’enzyme est à l’origine d’une moindre sensibilité de celle-ci à la
doxorubicine (Ganapathi, Ganapathi, 2013).
3. La résistance associée à3. La résistance associée à3. La résistance associée à3. La résistance associée à une modulation des processus enzymatiques une modulation des processus enzymatiques une modulation des processus enzymatiques une modulation des processus enzymatiques
Les enzymes impliquées dans le métabolisme sont des déterminants importants de la
concentration intracellulaire et systémique de la doxorubicine. Les réactions d’oxydation, de
réduction et d’hydrolyse (réactions de phase I), ainsi que les réactions de conjugaison
(réactions de phase II), jouent un rôle crucial dans la protection des cellules contre les
toxines cellulaires. Ces mêmes réactions peuvent mener à la résistance cellulaire à la
doxorubicine dans les cellules tumorales, par diminution de son métabolisme ou par
détoxification de certains de ses métabolites (Zandvliet, Teske, 2015).
a. Altération du métabolisme de la doxorubicinea. Altération du métabolisme de la doxorubicinea. Altération du métabolisme de la doxorubicinea. Altération du métabolisme de la doxorubicine
Une diminution de l’activité enzymatique du système cytochrome P450 et de la flavine
réductase est à l’origine d’une diminution de la formation du radical semi-quinone dans les
cellules tumorales. Cette diminution est à l’origine d’une moindre production de ROS et
donc d’une cytotoxicité moindre (Walker, Nitiss, 2002). De même, certaines cellules
tumorales surexpriment l’enzyme de glucurono-conjugaison UDP-glucuronyl transférase qui
est associée à l’inactivation des métabolites de la doxorubicine (Zandvliet, Teske, 2015).
b. Résistance par stimulation des mécanismes de détoxification cellulaireb. Résistance par stimulation des mécanismes de détoxification cellulaireb. Résistance par stimulation des mécanismes de détoxification cellulaireb. Résistance par stimulation des mécanismes de détoxification cellulaire
L’acquisition de la résistance à la doxorubicine par les cellules tumorales peut également
provenir d’une augmentation de l’activité des enzymes impliquées dans la détoxification
cellulaire (Kumaraguruparan et al., 2006). Les principaux systèmes enzymatiques concernés

88
sont les système glutathion, ainsi que les enzymes d’élimination des radicaux libres : la
superoxyde dismutase, la catalase et la glutathion peroxydase.
Le système glutathion cellulaire est responsable de la métabolisation et de la détoxification
de certains agents nocifs pour la cellule tels que les ROS. Certaines cellules tumorales vont
présenter une expression plus importante des gènes codant pour ces enzymes, intrinsèque
ou acquise. Cette plus forte expression va être à l’origine d’une moindre sensibilité aux ROS
et donc d’une moindre cytotoxicité de la doxorubicine envers ces cellules (Leonel, Gelaleti,
2014).
Le système glutathion est composé d’une famille d’iso-enzymes, les glutathion-S-
transférases, et du glutathion. Le glutathion (GSH) est un tripeptide (γ-glutamyl-cystéine-
glycine), jouant le rôle de nucléophile dans de nombreuses réactions de détoxification. Le
maintien du taux intracellulaire constant s’effectue par une synthèse de novo et un système
de réparation. Certaines cellules tumorales résistantes à la doxorubicine présentent un taux
en glutathion plus élevé. Les glutathion-S-transférases (GSTs) sont quant à elle des
isoenzymes qui catalysent l’addition nucléophile du GSH réduit sur un composé électrophile.
Chaque isoenzyme est associée à un type de résistance. L’isoenzyme π, surexprimée dans
certaines cellules tumorales, est associée aux mécanismes de résistance à la doxorubicine
par conjugaison du radical semi-quinone (Tew, 1994).
4. La résistance par réparation des dommages à l’ADN4. La résistance par réparation des dommages à l’ADN4. La résistance par réparation des dommages à l’ADN4. La résistance par réparation des dommages à l’ADN
Il existe très peu d’études sur la résistance à la doxorubicine par activation des systèmes de
réparation au niveau de l’ADN cellulaire chez le chien. Cependant, il semblerait que ces
mécanismes soient similaires à ceux présents chez l’Homme.
Les cellules ont développé des mécanismes de réparation de l’ADN. L’augmentation de ces
mécanismes de réparation de l’ADN dans certaines lignées tumorales est liée à la résistance
à la doxorubicine. Les mécanismes de réparation incluent l’excision de nucléotide, l’excision
de base et la recombinaison homologue ou non homologue par jonction des extrémités
(Figure 38) (Slupphaug et al., 2003). En fonction du mécanisme de cytotoxicité impliqué, les
dommages à l’ADN et donc les mécanismes de réparation seront différents.
Figure 38 : Les différents mécanismes de réparation de l’ADN selon le mécanisme de
cytotoxicité doxorubicine-induit Traduit et adapté d’après Slupphaug et al. (2003).

89
L’inhibition de l’ADN topoisomérase de type II est à l’origine de cassures double-brins et de
cross-links. La réparation des dommages à l’ADN causés par ce mécanisme d’action doit
accomplir plusieurs tâches. Tout d’abord, le système de réparation doit reconnaître le
complexe ternaire formé comme un dommage à l’ADN et non pas comme une enzyme
active pouvant se dissocier de l’ADN. La détection du complexe covalent comme lésion de
l’ADN a sans doute lieu car ce complexe covalent correspond à un blocage de la transcription
et de la réplication. Cependant, ce complexe ternaire est réversible jusqu’à ce que certains
processus interviennent. Tant que la reconnaissance et la réparation ne sont pas initiées, la
formation du complexe covalent peut s’inverser sans effet délétère. Une fois ces processus
enclenchés, le complexe est irréversible et mène soit vers une réparation des lésions à
l’ADN, soit vers la mort cellulaire. La réparation nécessite ensuite le retrait de l’enzyme.
Celui-ci peut être réalisé soit par l’excision endonucléolytique des adduits protéiques, soit
par dégradation de l’ADN topoisomérase de type II par protéolyse. La cellule répare ensuite
les lésions à l’ADN (Nitiss, 2009b).
Les ROS synthétisés à partir de la doxorubcine sont quant à eux à l’origine de cassures
simple ou double-brins, de modifications de bases azotées, de désoxyriboses et de
groupements phosphates ainsi qu’à la mise en place de cross-links ou d’adduits à l’ADN
(Evans et al., 2004).
5. Résistance par inhibition de l’apoptose5. Résistance par inhibition de l’apoptose5. Résistance par inhibition de l’apoptose5. Résistance par inhibition de l’apoptose
De nombreux mécanismes cellulaires peuvent être à l’origine d’une résistance des cellules
tumorales par inhibition de l’apoptose et des autres processus de mort cellulaire. Nous ne
traiterons ici que des mécanismes les plus souvent rencontrés lors de résistance à la
doxorubicine.
Dans de nombreux types tumoraux, la famille de protéine Bcl-2, Akt et d’autres facteurs anti-
apoptotiques sont présents en forte concentration, ce qui est à l’origine d’une résistance à la
doxorubicine (Housman et al., 2014). Cette élévation de facteurs anti-apoptotiques peut être
due à des mutations, une amplification, une translocation ou une surexpression des gènes
codants pour ces facteurs (Holohan et al., 2013). De plus, ces gènes sont des cibles pour des
facteurs de transcription tels que NF-κB, activés par des mutations oncogéniques lors de la
tumorigénèse.
L’inhibition de l’apoptose peut également être due à une mutation du gène codant pour la
protéine p53, à l’origine d’une perte de ses capacités antiprolifératives et pro-apoptotiques
(Walker, Nitiss, 2002).

90
Figure 39 : Les différents mécanismes de résistance à la doxorubicine
Il existe de nombreux mécanismes de résistance à la doxorubicine chez l’animal, la
plupart des mécanismes ayant été extrapolé depuis l’Homme (Figure 39) :
1- L’augmentation de l’efflux de la doxorubicine par différents transporteurs
membranaires, à l’origine d’une diminution de sa concentration cellulaire. Ces
transporteurs sont également à l’origine d’une altération (1’) de la distribution
subcellulaire de doxorubicine.
2- L’altération des ADN topoisomérases de type II, cible de la doxorubicine. Cette
altération peut être quantitative et/ou qualitative, soit par diminution de son
expression ou de sa localisation nucléaire, soit par diminution de sa sensibilité.
3- La modulation des processus enzymatiques, impliqués dans le métabolisme de la
doxorubicine. Cela résulte en une moindre production de ROS ou en une
détoxification de ces derniers.
4- La réparation des dommages à l’ADN, dépendant des mécanismes cytotoxiques
impliqués.
5- L’inhibition de l’apoptose, mécanisme de résistance commun à de nombreux anti-
cancéreux. Cette inhibition est principalement due à l’augmentation des facteurs pro-
apoptotiques et aux mutations du gène p53.
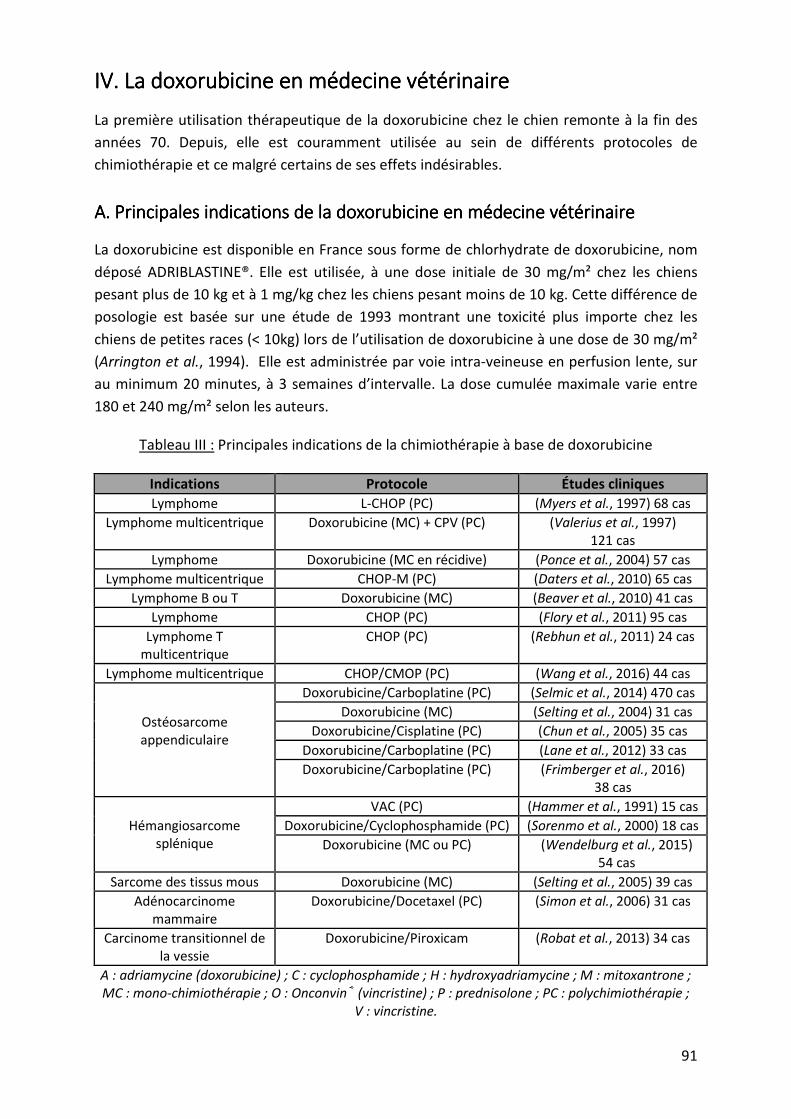
91
IV.IV.IV.IV. La doxorubicine en La doxorubicine en La doxorubicine en La doxorubicine en médecinemédecinemédecinemédecine vétérinairevétérinairevétérinairevétérinaire
La première utilisation thérapeutique de la doxorubicine chez le chien remonte à la fin des
années 70. Depuis, elle est couramment utilisée au sein de différents protocoles de
chimiothérapie et ce malgré certains de ses effets indésirables.
A. A. A. A. PrincipalesPrincipalesPrincipalesPrincipales iiiindicationndicationndicationndicationssss de la doxorubicine en médecine vétérinaire de la doxorubicine en médecine vétérinaire de la doxorubicine en médecine vétérinaire de la doxorubicine en médecine vétérinaire
La doxorubicine est disponible en France sous forme de chlorhydrate de doxorubicine, nom
déposé ADRIBLASTINE®. Elle est utilisée, à une dose initiale de 30 mg/m² chez les chiens
pesant plus de 10 kg et à 1 mg/kg chez les chiens pesant moins de 10 kg. Cette différence de
posologie est basée sur une étude de 1993 montrant une toxicité plus importe chez les
chiens de petites races (< 10kg) lors de l’utilisation de doxorubicine à une dose de 30 mg/m²
(Arrington et al., 1994). Elle est administrée par voie intra-veineuse en perfusion lente, sur
au minimum 20 minutes, à 3 semaines d’intervalle. La dose cumulée maximale varie entre
180 et 240 mg/m² selon les auteurs.
Tableau III : Principales indications de la chimiothérapie à base de doxorubicine
Indications Protocole Études cliniques
Lymphome L-CHOP (PC) (Myers et al., 1997) 68 cas
Lymphome multicentrique Doxorubicine (MC) + CPV (PC) (Valerius et al., 1997) 121 cas
Lymphome Doxorubicine (MC en récidive) (Ponce et al., 2004) 57 cas
Lymphome multicentrique CHOP-M (PC) (Daters et al., 2010) 65 cas
Lymphome B ou T Doxorubicine (MC) (Beaver et al., 2010) 41 cas
Lymphome CHOP (PC) (Flory et al., 2011) 95 cas
Lymphome T multicentrique
CHOP (PC) (Rebhun et al., 2011) 24 cas
Lymphome multicentrique CHOP/CMOP (PC) (Wang et al., 2016) 44 cas
Ostéosarcome appendiculaire
Doxorubicine/Carboplatine (PC) (Selmic et al., 2014) 470 cas
Doxorubicine (MC) (Selting et al., 2004) 31 cas
Doxorubicine/Cisplatine (PC) (Chun et al., 2005) 35 cas
Doxorubicine/Carboplatine (PC) (Lane et al., 2012) 33 cas
Doxorubicine/Carboplatine (PC) (Frimberger et al., 2016) 38 cas
Hémangiosarcome splénique
VAC (PC) (Hammer et al., 1991) 15 cas
Doxorubicine/Cyclophosphamide (PC) (Sorenmo et al., 2000) 18 cas
Doxorubicine (MC ou PC) (Wendelburg et al., 2015) 54 cas
Sarcome des tissus mous Doxorubicine (MC) (Selting et al., 2005) 39 cas
Adénocarcinome mammaire
Doxorubicine/Docetaxel (PC) (Simon et al., 2006) 31 cas
Carcinome transitionnel de la vessie
Doxorubicine/Piroxicam (Robat et al., 2013) 34 cas
A : adriamycine (doxorubicine) ; C : cyclophosphamide ; H : hydroxyadriamycine ; M : mitoxantrone ;
MC : mono-chimiothérapie ; O : Onconvin® (vincristine) ; P : prednisolone ; PC : polychimiothérapie ;
V : vincristine.

92
Les principales indications d’une chimiothérapie à base de doxorubicine sont présentées
dans le Tableau III, accompagnées de références d’études cliniques illustrant chacune des
indications.
La doxorubicine est principalement employée dans le cadre de protocoles de
polychimiothérapie pour le traitement de lymphomes, ou en alternance avec des dérivés de
platine (cisplatine ou carboplatine) dans la prise en charge des ostéosarcomes
appendiculaires. Elle est aussi utilisée en monochimiothérapie ou polychimiothérapie
adjuvante à la chirurgie lors d’adénocarcinomes mammaires avec emboles ou lors
d’envahissement métastatique des ganglions loco-régionaux. Elle peut également être
utilisée dans la cadre du traitement des hémangiosarcomes spléniques, des sarcomes des
tissus mous très indifférenciés, des carcinomes thyroïdiens avec emboles ou d’autres
carcinomes.
Cette liste n’est pas exhaustive et ne prend pas en compte les associations en cours
d’évaluation dans le cadre d’essais cliniques. Elle permet néanmoins de prendre conscience
de l’ampleur de l’utilisation de la doxorubicine en pratique médicale vétérinaire.
La doxorubicine existe également sous forme encapsulée dans des liposomes encapsulés et
pégylés (DOXIL®), permettant une réduction de la cardiotoxicité. En effet, l’encapsulation
permet une moindre distribution aux tissus tels que le cœur et le tractus gastro-intestinal et
un ciblage des cellules tumorales grâce à l’enhanced permeability and retention effect.
Cependant cette forme n’est pas disponible en France et ses indications thérapeutiques
restent à définir chez le chien. Il n’existe que peu d’études cliniques sur l’efficacité et
l’innocuité de la forme liposomale chez le chien, mettant en avant une cardiotoxicité plus
faible mais à l’origine d’une toxicité cutanée plus importante : l’érythrodysesthésie palmo-
plantaire (Sorenmo et al., 2007) (Teske et al., 2011).
BBBB.... Les principales interactions médicamenteuses avec la doxorubicineLes principales interactions médicamenteuses avec la doxorubicineLes principales interactions médicamenteuses avec la doxorubicineLes principales interactions médicamenteuses avec la doxorubicine
Les principales interactions médicamenteuses de la doxorubicine et leurs effets sont décrits
dans le Tableau IV.

93
Tableau IV : Principales interactions médicamenteuses de la doxorubicine
Molécules Effets Mécanismes suspectés
Barbituriques (phénobarbital)
Diminution modérée possible des effets
pharmacologiques de la doxorubicine
Augmentation du métabolisme de la doxorubicine par induction du
système cytochrome P450
Bevacizumab Augmentation possible de la cardiotoxicité
Inconnu
Inhibiteurs calciques (amlodipine, diltiazem,
vérapamil)
Augmentation possible de la cardiotoxicité
Toxicité additive
Cyclophosphamide Augmentation possible de la cardiotoxicité
Toxicité additive
Ciclosporine Augmentation des effets pharmacologiques de la
doxorubicine
Diminution du métabolisme de la doxorubicine par compétition pour le
système cytochrome P450 ou l’inhibition de la P-gp
Digoxine Diminution modérée possible des effets
pharmacologiques de la digoxine
Diminution de l’absorption par altération de la muqueuse intestinale
Paclitaxel Augmentation des effets pharmacologiques de la
doxorubicine
Diminution de la clairance de la doxorubicine par compétition pour le système cytochrome P450 ou la P-gp
Quinolones Diminution possible de l’action antimicrobienne
Diminution de l’absorption par altération de la muqueuse intestinale
Trastuzumab Augmentation possible de la cardiotoxicité
Inconnu
C. Toxicité et effets indésirables C. Toxicité et effets indésirables C. Toxicité et effets indésirables C. Toxicité et effets indésirables de la doxorubicinede la doxorubicinede la doxorubicinede la doxorubicine
Comme tous les agents de chimiothérapie, la doxorubicine ne présente pas une cytotoxicité
spécifique aux cellules tumorales, mais a également une certaine toxicité pour les cellules
non tumorales, à l’origine d’effets secondaires délétères. Ces effets secondaires peuvent
être non spécifiques et à mettre en lien avec la cytotoxicité sur les cellules des tissus à
renouvellement rapide, ou spécifiques, en lien avec une particularité métabolique ou une
concentration préférentielle dans certains tissus (Barranco, 1984).
Tableau V : Effets indésirables de la doxorubicine à court-terme D’après Ogilvie et al. (1989).
Effets indésirables Nombre de chiens
Vomissements 26/185 (14 %)
Diarrhée 19/185 (10,3 %)
Colite 11/185 (5,9 %)
Anorexie 7/185 (3,8 %)
Prurit 6/185 (3,2 %)
Alopécie 2/185 (1,1 %)
Sepsis secondaire à la myélosuppression
2/185 (1,1 %)

94
Il existe peu d’études de grande ampleur concernant les effets secondaires de la
doxorubicine chez le chien, la plus complète portant sur les effets à court-terme sur 185
chiens (Tableau V) (Ogilvie, Richardson, et al., 1989).
Ces effets secondaires devront être évalués au cours du suivi de l’animal, pour permettre
une adaptation du traitement et l’ajout d’agents thérapeutiques supplémentaires au besoin.
Les différents effets secondaires sont gradés selon la classification VCOG-CTCAE (Veterinary
cooperative oncology group – common terminology criteria for adverse events) (Annexe 1).
Les différents effets secondaires seront présentés par ordre chronologique d’apparition
après injection chez le chien.
1111. Le choc histaminique. Le choc histaminique. Le choc histaminique. Le choc histaminique
Toxicité spécifique mais rare de la doxorubicine et d’apparition suraiguë, le choc
histaminique est lié à une injection trop rapide de la doxorubicine. L’injection provoque une
réaction anaphylactoïde qui résulte de la libération immédiate d’histamine et d’autres
substances vaso-actives par les polynucléaires basophiles, mais qui, à la différence de la
réaction anaphylactique, n’implique pas de réaction immunologique (Moore, Frimberger,
2009).
Les symptômes sont l’hypersalivation, des tremblements, des nausées et vomissements, une
hyperhémie cutanée, la présence de plaques urticaires et dans les cas les plus graves, une
hypotension sévère.
� Traitement
Le traitement repose sur l’arrêt immédiat de l’injection de doxorubicine et l’utilisation de
méthylprednisolone ou de dexaméthasone (1 à 2 mg/kg IV), voire d’adrénaline lors d’un
choc hypotensif sévère (0,01 mg/kg IV). On pourra également utiliser des antihistaminiques
(diphenhydramine 0,5 à 1 mg/kg en IV lente ou 1 à 4 mg/kg IM) même si leur effet en phase
clinique n’est pas certain (Eschalier et al., 1988).
� Prévention
Il est possible de prémédiquer l’animal avec de la diphenhydramine à 2 mg/kg en sous-
cutané et/ou de la dexaméthasone à 0,2 mg/kg en intraveineux, 15 à 20 minutes avant
administration de la doxorubicine. Le meilleur moyen de prévention du choc histaminique
reste la réalisation d’une perfusion lente de doxorubicine sur 20 minutes au minimum.
2222. La toxicité gastro. La toxicité gastro. La toxicité gastro. La toxicité gastro----intestinaleintestinaleintestinaleintestinale
La toxicité gastro-intestinale est une toxicité non spécifique de la doxorubicine, et qui reste
modérée chez 80 à 90 % des animaux traités. Celle-ci peut être la conséquence de la
cytotoxicité directe de la doxorubicine sur les cellules des cryptes intestinales, ou bien de la
stimulation de la trigger zone par la sérotonine et la substance P. Les vomissements par
stimulation de la trigger zone sont des phénomènes aigus, apparaissant dans les 24 heures

95
suivant l’administration de la doxorubicine. L’anorexie, les nausées, les vomissements et la
diarrhée dus à la toxicité affectant les entérocytes immatures apparaissent quant à eux un à
cinq jours après administration de la doxorubicine (Tageja, Groninger, 2014).
La présence de diarrhée induite par la chimiothérapie est plus rare que les vomissements,
son apparition signe alors des lésions étendues de la muqueuse intestinale. Il existe deux
formes de diarrhées chimio-induites. Une diarrhée de l’intestin grêle, banale, ou une colite,
touchant 10 à 15 % des animaux traités. La colite se traduit par une diarrhée importante,
plus ou moins hémorragique avec une déshydratation rapide en absence de traitement.
La sévérité des effets secondaires gastro-intestinaux est gradée de 1 à 4 selon le système
VCOG-CTCAE (Annexe 1).
� Traitement
L’anorexie peut être compensée par l’effet orexigène des corticoïdes lors d’une
administration conjointe au sein du protocole de chimiothérapie. En cas de persistance de
l’anorexie, il est possible d’utiliser des stimulants de la prise alimentaire comme la
cyproheptadine (0,2 mg/kg PO, SID à BID) ou de la mirtazapine (0,5 mg/kg PO SID).
Pour des vomissements faibles, inférieurs à trois épisodes par 24 heures, une diète hydrique
d’une journée peut suffire. Pour des vomissements modérés, un traitement peut être mis en
place (Rau et al., 2010) :
� Le citrate de maropitant (1 mg/kg IV ou SC SID, ou 2 mg/kg PO SID) : c’est un
antiémétique central, par inhibition des récepteurs centraux à la neurokinine (NK1).
� Le métoclopramide (0,3-0,5 mg/kg IV ou PO, BID à TID) : c’est un antiémétique à
action centrale et périphérique. Il présente également un effet gastrokinétique.
� Les « setrons », antagonistes sélectifs de la sérotonine à effet antiémétique central.
Parmi eux, on utilise principalement l’ondansetron (0,1 à 0,2 mg/kg IV BID).
On pourra également ajouter des antiacides (oméprazole 0,5 à 1 mg/kg PO SID) qui, même
s’ils n’ont pas d’effet antiémétique propre, peuvent être bénéfiques. En cas de
vomissements sévères avec déshydratation, une hospitalisation de l’animal peut être
nécessaire.
En cas de diarrhée légère, une diète hydrique de 24 heures puis le passage à un aliment
hyper-digestible peut permettre une amélioration. En cas de diarrhée modérée, un
traitement à base de pansements digestifs et de lopéramide (0,1 mg/kg PO BID, si le chien
est porteur du gène mdr1 non muté) peut être mis en place. En cas de diarrhée importante
avec déshydratation, une hospitalisation de l’animal peut être nécessaire.
De plus, en cas de neutropénie concomitante, la mise en place d’une antibiothérapie peut se
justifier. On utilise le plus souvent du triméthoprime-sulfamide (12,5 mg/kg PO BID), associé
à du métronidazole (10 à 15 mg/kg PO BID) dans les cas les plus graves.

96
� Prévention
En prévention, la chimiothérapie est pratiquée à jeun. Il est également possible
d’administrer un antiémétique 30 minutes avant la séance de chimiothérapie ainsi qu’un
pansement gastrique à base de phosphate d’alumine de la veille à quatre jours post-
chimiothérapie. Lors d’effets indésirables gastro-intestinaux modérés à sévères, il est
également possible de diminuer la dose de chimiothérapie de 10 à 20 % ou de modifier le
protocole (Moore, Frimberger, 2009).
3333. Toxicité hématologique. Toxicité hématologique. Toxicité hématologique. Toxicité hématologique
La doxorubicine présente une myélotoxicité dite forte, par rapport à d’autres agents
anticancéreux. Celle-ci est due à sa cytotoxicité envers les cellules avec un index prolifératif
élevé. La toxicité hématologique apparait après un temps de latence, correspondant au
temps de demi-vie des différentes cellules sanguines. De plus, cette toxicité est dose-
dépendante. Ceci explique que la toxicité hématologique de la doxorubicine concerne
principalement la lignée granulocytaire, à l’origine d’une neutropénie et d’une
thrombocytopénie. Plus rarement, elle pourra donner lieu à une anémie régénérative,
même si celle-ci est rarement objectivée étant donné la durée de vie des érythrocytes et la
dose nécessaire pour inhiber la lignée érythroïde (Reagan et al., 1993). Le nadir de la
neutropénie et de la thrombopénie est de sept à dix jours, avec une récupération d’environ
trois semaines.
La sévérité des cytopénies est gradée de 1 à 4 selon le système VCOG-CTCAE (Annexe 1).
Une numération formule sanguine est réalisée avec chaque administration. On considère
généralement qu’une molécule potentiellement myélotoxique ne doit pas être administrée
si les neutrophiles sont inférieurs à 2000 cellules/mm3 ou si les plaquettes sont inférieures à
75000 cellules/mm3. Dans ce cas, la séance de chimiothérapie doit être reportée de deux à
sept jours, et une numération-formule sanguine doit de nouveau être réalisée.
� Traitement
La mise en place d’un traitement lors de neutropénie sans signe clinique n’est pas forcément
conseillée et dépend des auteurs. Une antibioprophylaxie par voie orale peut être mise en
place en utilisant un antibiotique large spectre tel que le triméthoprime-sulfamide (15 à 20
mg/kg PO BID), l’amoxicilline-acide clavulanique (20 mg/kg PO BID) ou la céfalexine (15 à 30
mg/kg PO BID). Lors d’une neutropénie accompagnée de signes cliniques (fièvre, léthargie,
anorexie), il est conseillé par certains auteurs de réaliser un traitement en hospitalisation
avec une antibiothérapie par voie intraveineuse et avec une fluidothérapie.
Lors de thrombocytopénie, aucun traitement n’est conseillé, les signes cliniques étant
rarement observés pour un comptage supérieur à 25000 plaquettes/mm3. En cas de
thrombocytopénie majeure, l’animal doit être placé sous surveillance accrue pour prévenir
les ecchymoses, les pétéchies et les saignements (saignements gastro-intestinaux, gingivaux,
épistaxis) (Sorenmo et al., 2010).

97
� Prévention
La prévention de la toxicité hématologique de la doxorubicine repose sur le respect des
posologies avec une réévaluation de la posologie avant chaque séance, un respect du
rythme d’administration et un contrôle de la numération-formule sanguine avant chaque
injection. Lors d’une neutropénie sévère (< 1000 cellules/mm3), il est également possible de
diminuer la dose de chimiothérapie de 10 à 20 % ou de modifier le protocole (Moore,
Frimberger, 2009).
4444. Toxicité cutanée lors d’extravasation. Toxicité cutanée lors d’extravasation. Toxicité cutanée lors d’extravasation. Toxicité cutanée lors d’extravasation
C’est une toxicité spécifique des agents vésicants tels que la doxorubicine. L’extravasation
est définie comme une fuite par inadvertance d’une molécule à partir de l’espace vasculaire,
se répandant alors dans les tissus environnants. Lors de cette extravasation, les dommages
locaux sont la plupart du temps minimes. Lors de l’utilisation d’agents anticancéreux
vésicants, tels que la doxorubicine, l’extravasation est à l’origine de dommages plus
importants apparaissant comme une irritation et évoluant vers la cellulite et la nécrose
cutanée sévère. Les signes cliniques apparaissent sous sept à dix jours, avec une extension
de la nécrose pendant plusieurs semaines, sans cicatrisation spontanée (Figure 40) (Venable
et al., 2012). Ces signes cliniques sont dus aux mécanismes de cytotoxicité de la
doxorubicine sur les cellules cutanées.
Figure 40 : Nécrose cutanée due à l’extravasation de doxorubicine chez une chienne de 12
ans, deux semaines après l’injection de doxorubicine D’après Venable et al. (2012).
Lors d’une extravasation constatée au cours de la chimiothérapie, le praticien doit arrêter
l’administration puis essayer d’aspirer le maximum de liquide extravasé par le cathéter avant
de retirer celui-ci. Une application de glace 30 minutes, quatre fois par jour pendant 72
heures est conseillée.
� Traitement
Lors de l’installation de lésions, le traitement repose principalement sur un parage
chirurgical en tissu sain accompagné si nécessaire d’une chirurgie reconstructrice.
Le dexrazoxane est le seul traitement médical recommandé en humaine lors de
l’extravasation de doxorubicine, mais son utilité reste contestée. Le dexrazoxane est un
chélateur fort d’ions métalliques qui protège contre le stress oxydatif.

98
C’est un inhibiteur réversible du cycle catalytique de l’ADN topoisomérase de type II,
protégeant les cellules des cassures double-brins létales de l’ADN. Il est également utilisé en
humaine comme cardioprotecteur lors de l’utilisation d’anthracyclines (Kane et al., 2008). Il
n’existe cependant que peu de données sur l’utilisation de dexrazoxane chez le chien, la
plupart issues de cas cliniques ou de recommandations pour l’utilisation en médecine
humaine. En médecine vétérinaire, les doses sont variables, de 300 (ou dix fois la dose de
doxorubicine) à 1000 mg/m² par voie intraveineuse. L’utilisation de dexrazoxane dans les six
heures suivant l’extravasation semble être un facteur pronostic positif (Kassner, 2000). En
médecine, il est recommandé de réitérer le traitement après 24 et 48 heures.
L’utilisation de diméthyl-sulfoxyde (DMSO) est communément suggérée lors de
l’extravasation de doxorubicine. Le DMSO est un réducteur de ROS et son application locale
pourrait diminuer les dommages locaux. Il est également vasodilatateur et présente des
propriétés anti-inflammatoires et analgésiques. Cependant, aucune étude chez le chien n’a
été menée et le DMSO semble réduire l’effet du dexrazoxane (Reeves, 2007).
L’utilisation de hyaluronidase en intra-lésionnel a également été décrite chez six chiens,
permettant le maintien de la fonction locomotrice du membre malgré le développement de
fibrose au site d’extravasation (Spugnini, 2002).
� Prévention
La prévention de l’extravasation repose sur une administration intraveineuse stricte, avec
l’utilisation systématique d’un cathéter et d’une perfusion, sur une veine non abimée dans la
mesure du possible, afin d’éviter le risque de fuite vasculaire en amont et en aval. Les prises
de sang autres devront être réalisées au niveau des veines jugulaires. Un contrôle du
cathéter sera réalisé juste avant l’injection, et la doxorubicine est injectée diluée dans la
tubulure de la perfusion sur 15 à 20 minutes. A la fin de la chimiothérapie, la tubulure et le
cathéter sont abondamment rincés avec du NaCl 0,9%, avant son retrait. Un pansement
légèrement compressif est mis en place après retrait du cathéter afin d’éviter une
extravasation modérée et une sclérose veineuse ultérieure (Villalobos, 2006).
5555. L’alopécie cutanée. L’alopécie cutanée. L’alopécie cutanée. L’alopécie cutanée
Toxicité non spécifique de la doxorubicine, elle est due à la mise au repos des follicules
pileux et est réversible à l’arrêt du traitement, avec repousse des poils en un à deux mois. La
doxorubicine peut entraîner une chute importante de poils après une seule injection.
Cependant, cet effet secondaire de la doxorubicine est beaucoup moins fréquent chez le
chien que chez l’homme (5% contre 80%). L’alopécie est plus fréquente chez les races à poils
longs ou laineux (Caniche, Bobtail, Bearded Collie, etc …) et plus rare chez les races à poils
courts ou durs (Boxer, Fox Terrier, Schnauzer, etc …) (Morrison, 2002).
6666. La toxicité cardiaque. La toxicité cardiaque. La toxicité cardiaque. La toxicité cardiaque
La toxicité cardiaque est une toxicité spécifique de la doxorubicine et des anthracyclines,
largement étudiée étant donné son implication dans le devenir du patient.

99
Cette spécificité est expliquée par plusieurs caractéristiques. Premièrement, les cellules
cardiaques sont caractérisées par un déficit relatif des défenses anti-oxydantes, avec une
activité enzymatique plus faible de la superoxyde dismutase et de la catalase ainsi qu’un
niveau constitutif plus faible en glutathion (Quiles et al., 2002). De plus, les cardiomyocytes
présentent une teneur constitutive plus importante en enzymes réductases et
déshydrogénases, responsables de la production de ROS (Green, Leeuwenburgh, 2002).
Cette formation de ROS est également augmentée par la forte concentration en fer. Enfin, le
tissu cardiaque est riche en mitochondries et la doxorubicine présente une forte affinité
pour les cardiolipides, constituants majeurs de la membrane interne des mitochondries.
Cette affinité provoque des lésions site-spécifiques de la mitochondrie (Nicolay et al., 1984).
La doxorubicine est à l’origine d’une cardiotoxicité de type I, c’est-à-dire irréversible par
mort de la cellule cardiaque par apoptose ou nécrose cellulaire. En médecine, elle est définie
soit par une diminution de plus de 20 % de la fraction d’éjection systolique gauche pour une
valeur initiale supérieure à 50 %, ou de plus de 10 % pour une valeur initiale inférieure à 50
%, soit par la présence de signes cliniques d’insuffisance cardiaque congestive : tachycardie,
intolérance à l’effort, toux, etc … (Rahman et al., 2007).
La sévérité des effets secondaires cardiaque est gradée de 1 à 4 selon le système VCOG-CTCAE (Annexe 1).
aaaa. Epidémiologie et symptomatologie de la cardiotoxicité doxorubicine. Epidémiologie et symptomatologie de la cardiotoxicité doxorubicine. Epidémiologie et symptomatologie de la cardiotoxicité doxorubicine. Epidémiologie et symptomatologie de la cardiotoxicité doxorubicine----induiteinduiteinduiteinduite
Chez le chien, on distingue la cardiotoxicité aiguë de la cardiotoxicité chronique (Ratterree et
al., 2012) :
� La cardiotoxicité aiguë, au cours de l’injection et jusqu’à une semaine post-
chimiothérapie. Cette forme est la plupart du temps asymptomatique et auto-
résolutive en quelques heures à quelques jours, avec un très bon pronostic vital. Elle
se manifeste par des anomalies non spécifiques de l’échocardiogramme telles qu’une
augmentation du segment ST, une diminution du voltage de l’onde QRS, un
allongement de l’intervalle QT, un bloc atrio-ventriculaire, une tachycardie sinusale
ou des extrasystoles supra-ventriculaires ou ventriculaires.
� La cardiotoxicité chronique dose-dépendante, caractérisée par cardiomyopathie
dilatée avec une diminution de l’épaisseur de la paroi libre du ventricule gauche, de
la masse myocardique et de la compliance ventriculaire. Celle-ci évolue vers une
insuffisance cardiaque congestive irréversible. Les animaux présentent un tonus
sympathique plus élevé et un dysfonctionnement diastolique précoce, suivi d’un
dysfonctionnement systolique plus tardif. Cliniquement, la cardiotoxicité chronique
est caractérisée par de la toux, une intolérance à l’effort, de la fatigue et une
tachycardie, accompagnée par une diminution du voltage de l’onde QRS à
l’électrocardiogramme. Aucune étude chez le chien n’a permis de déterminer les
facteurs de risque de la cardiotoxicité chronique, même si certains sont extrapolés
depuis des études réalisées en médecine humaine : un pic plasmatique important,

100
une dose cumulative supérieure à 240 mg/m² et l’utilisation de la doxorubicine lors
d’une polychimiothérapie (Gillings et al., 2009).
Trois études permettent d’établir l’incidence d’effets secondaires cardiaques à l’administration de doxorubicine chez le chien et révèlent des modifications de l’électrocardiogramme dans 7,4 à 17,7 % des cas, et l’apparition d’une insuffisance cardiaque congestive dans 2,1 à 4 % des cas (Mauldin et al., 1992) (Gillings et al., 2009) (Ratterree et
al.,2012).
bbbb. Mécanismes à l’origine de la cardiotoxicité doxorubicine. Mécanismes à l’origine de la cardiotoxicité doxorubicine. Mécanismes à l’origine de la cardiotoxicité doxorubicine. Mécanismes à l’origine de la cardiotoxicité doxorubicine----induiteinduiteinduiteinduite
La toxicité cardiaque de la doxorubicine repose sur un ensemble de mécanismes cellulaires,
parfois communs avec les mécanismes de cytotoxicité de la doxorubicine envers les cellules
tumorales (Figure 42).
b1b1b1b1. Le stress oxydatif . Le stress oxydatif . Le stress oxydatif . Le stress oxydatif
Bien que les mécanismes mis en jeu lors de la cardiotoxicité ne soient pas totalement
élucidés, il est maintenant admis qu’ils reposent essentiellement sur une majoration d’un
processus oxydatif mettant en jeu la production de ROS. Cette production s’opère via
différents mécanismes (cf partie III.B) :
� La réduction de la doxorubicine en radical semi-quinone, qui en présence de
dioxygène moléculaire est à l’origine de la formation de radicaux superoxydes. Ces
radicaux forment ensuite des radicaux hydroxyles, sous la dépendance d’un cofacteur
Fe (III).
� La complexation du fer (III) avec la doxorubicine et l’oxydation du complexe par auto-
oxydation ou par le système cytochrome P450 en radical semi-quinone et fer (II) qui,
au contact de dioxygène moléculaire, réagit pour former des ROS. Ce phénomène
s’auto-entretient par dérégulation de l’homéostasie ferrique.
Cette surproduction de ROS induit au niveau de toutes les structures cellulaires du
cardiomyocyte, une augmentation de la peroxydation lipidique, aboutissant à des altérations
membranaires. Ces anomalies de la perméabilité membranaire ont de multiples
conséquences sur l’homéostasie ionique. Les perturbations du flux calcique qui en découlent
altèrent la contractilité myocardique. De plus, les lésions mitochondriales induites
participent à l’initiation de processus apoptotiques par libération, à partir de la
mitochondrie, du cytochrome c qui active les caspases pro-apoptotiques. Par ailleurs, la
majoration du stress oxydatif participe à la modulation de différentes voies de signalisation
intracellulaires dont :
� L’activation des voies des mitogen-activated protein kinases (MAPK) et des stress-
activated protein kinases (SAPK), dont certaines, comme celle de JNK, à l’origine de
l’activation de facteurs pro-apoptotiques (Bax, Bad) et de l’inactivation de facteurs
anti-apoptotiques de la famille des Bcl-2 (Sordet et al., 2003).

101
� L’activation de la voie du facteur de transcription nuclear factor-kappa B (NF-κB), à
l’origine de la transcription de gènes de la réponse inflammatoire (cytokines,
chémokines), mais également des gènes codant pour de protéines modulant
l’apoptose telles que Bcl-XL et FasL (Wang et al., 2002).
Le processus oxydatif se poursuit bien après l’arrêt du traitement, expliquant l’apparition
retardée de la cardiomyopathie induite par la doxorubicine.
On observe également une augmentation de l’expression de l’enzyme monoxyde d’azote
synthase inductible (iNOS) ainsi que de son ARNm au niveau du myocarde (Delemasure et
al., 2006) (Li et al., 2006). Le monoxyde d’azote formé diffuse facilement au travers des
membranes et des différents compartiments cellulaires, ce qui permet son interaction avec
l’anion superoxyde, produisant en grande quantité du peroxynitrite, qui potentialise les
altérations initialement induites par les ROS : peroxydation lipidique, inactivation d’enzymes
et de protéines intervenant dans la contractilité cardiaque ou dans la chaîne respiratoire
mitochondriale.
Il se pourrait également que les métabolites aglycones de la doxorubicine interviennent dans
le stress oxydatif au sein des cardiomyocytes. En effet, comme ils ne possèdent plus de
groupe daunosamine, ils sont plus liposolubles et diffusent donc mieux au sein des
compartiments cellulaires que le composé parent. Il se peut que ceux-ci soient à l’origine de
la production de ROS, notamment au niveau des mitochondries. Ce mécanisme est un des
mécanismes suspectés expliquant la cardiotoxicité aiguë de la doxorubicine (Sokolove,
1994).
L’échec thérapeutique de l’utilisation de certains antioxydants laisse penser que la
production de ROS n’est pas le seul mécanisme à l’origine de la cardiotoxicité de la
doxorubicine. On observe alors l’émergence d’hypothèses alternatives et/ou
complémentaires.
b2b2b2b2. L’inhibition de la topoisomérase de type II. L’inhibition de la topoisomérase de type II. L’inhibition de la topoisomérase de type II. L’inhibition de la topoisomérase de type IIββββ
Contrairement aux cellules tumorales proliférantes, riches en ADN topoisomérase de type
IIα, les cellules cardiaques contiennent principalement l’isomère β (Vavrova et al., 2013).
L’inhibition de l’ADN topoisomérase de type IIβ par la doxorubicine conduit à la répression
des gènes de facteurs de transcription impliqués dans la survie cellulaire et dans la synthèse
de protéines du sarcomère telles que l’actine-α, les troponines ou la chaîne légère
régulatrice de la myosine. De plus, cette interaction avec l’isomère β pourrait être à l’origine
d’une diminution de l’expression génique de la pompe Ca2+-ATPase du réticulum
sarcoplasmique (SERCA), conduisant à des perturbations des flux calciques, à l’origine d’une
perturbation de la contractilité cardiaque (Appel et al., 2007).

102
b3b3b3b3. Le rôle du métabolite doxorubicinol dans la cardiotoxicité. Le rôle du métabolite doxorubicinol dans la cardiotoxicité. Le rôle du métabolite doxorubicinol dans la cardiotoxicité. Le rôle du métabolite doxorubicinol dans la cardiotoxicité
Le métabolite alcool secondaire de la doxorubicine, le doxorubicinol, semble avoir un rôle
prépondérant dans la cardiotoxicité de la doxorubicine. En effet, l’analyse post-mortem
montre que le doxorubicinol est le métabolite le plus abondant retenu au sein des
cardiomyocytes (Stewart et al., 1993). La formation prépondérante de ce métabolite au sein
des cellules cardiaques s’expliquerait par l’induction par le peroxyde d’hydrogène de
certaines aldo-céto réductases cardiaques, similaires à celles réduisant la chaîne latérale de
la doxorubicine. Cette conversion facilitée pourrait être accompagnée par une diminution de
la formation de ROS, expliquant la réduction de la concentration en ROS au sein des cellules
cardiaques et de la sensibilité aux antioxydants au cours de l’installation de la
cardiomyopathie (Gervasi et al., 1986) (Licata et al., 2000).
Des études sur des modèles murins montrent l’implication du doxorubicinol dans la
cardiomyopathie. En effet, une augmentation de la susceptibilité à la cardiomyopathie est
corrélée avec une conversion plus importante de la doxorubicine en son métabolite (Lanza
et al., 1989). De plus, les anthracyclines ne présentant pas de chaîne latérale carbonylée, ou
avec une affinité moindre pour les réductases induisent une cardiomyopathie moins sévère
et plus progressive (Cirillo et al., 2000).
Le doxorubicinol semble pouvoir induire une cardiotoxicité par des mécanismes pouvant
impliquer ou non le fer intracellulaire.
Concernant les mécanismes fer-indépendants, des études montrent que le doxorubicinol est
plus apte que la doxorubicine à inhiber la SERCA, l’échangeur sodium/calcium de la
membrane plasmique et le récepteur à la ryanodine 2 (RyR2), perturbant le cycle
excitation/contraction des myocytes. Le doxorubicinol est également à l’origine de la
suppression de certains gènes, anciennement attribuée à la doxorubicine (Olson, Mushlin,
1990).
Concernant les mécanismes fer-dépendants, le doxorubicinol convertit l’aconitase
cytoplasmique en IRP-1, permettant la libération de fer et la production de ROS (cf partie
III.B.1.c). Cependant, la conversion chronique de la doxorubicine en doxorubicinol perturbe
le mécanisme de conversion de l’aconitase en IRP-1, qu’il convertit en une protéine non
fonctionnelle, dénuée d’activité de liaison avec l’ARNm et incapable de retrouver l’activité
aconitase. Ces modifications sont attribuées aux modifications oxydatives des résidus
cystéines (Minotti et al., 1998) (Brazzolotto et al., 2003).
La protéine non fonctionnelle sera incapable de jouer le rôle de senseur du niveau de fer et
d’adapter les processus cellulaires de séquestration et/ou relargage du fer aux besoins de la
cellule. En effet, en plus de ne plus moduler l’expression de la ferritine et du TfR (cf partie
III.B.1.c), IRP-1 ne régule plus l’expression d’autres enzymes reliées à l’utilisation (erythroid
aminolevulinate synthase), l’absorption (DMT1/Nramp2) et le relargage (ferroportin
1/IREG1/MTP1) du fer.

103
De plus, l’inactivation permanente de l’aconitase cytoplasmique réduit l’apport en substrat
de la forme cytoplasmique de l’isocitrate déshydrogénase, diminue la formation de NADH
par cette enzyme et contribue à l’altération de la balance oxydo-réductrice et métabolique
de la cellule (Narahari et al., 2000).
b4b4b4b4. Altérations des mitochondries et du métabolisme énergétique. Altérations des mitochondries et du métabolisme énergétique. Altérations des mitochondries et du métabolisme énergétique. Altérations des mitochondries et du métabolisme énergétique
Les mitochondries, qui représentent entre 20 et 40 % du volume des cardiomyocytes, sont
une des cibles préférentielles de la doxorubicine, en raison d’une forte affinité pour le
cardiolipide, présent en quantité importante dans la membrane interne des mitochondries.
Elles sont aussi la source la plus importante de ROS cellulaires en conditions
physiopathologiques.
La doxorubicine est donc fortement concentrée dans la mitochondrie et plus
particulièrement au niveau des protéines associées au cardiolipide dont la NADH
déshydrogénase et le complexe I de la chaîne respiratoire mitochondriale (Goormaghtigh et
al., 1990). Les complexes III et IV de la chaîne respiratoire mitochondriale sont également
altérés par la doxorubicine, ce qui conduit à la réduction du gradient électrochimique de
protons et donc à une diminution de la synthèse d’ATP par l’ATP synthase (Tokarska-
Schlattner et al., 2006) (Berthiaume, Wallace, 2007).
L’altération du métabolisme énergétique conduit à une baisse des réserves énergétiques des
cellules cardiaques. En plus de la diminution de production d’ATP par la chaîne respiratoire,
la doxorubicine est à l’origine d’une diminution de l’utilisation des acides gras par inhibition
de la carnitine O-palmitoyltransférase. On observe également une diminution de
l’expression et de l’activité de l’AMP-activated protein kinase (AMPK), senseur du
métabolisme énergétique et du peroxisome proliferator-activated receptor gamma
coactivator 1-α (PGC1-α), régulateur de la biogénèse mitochondriale d’énergie. Cela aboutit
à l’arrêt des imports mitochondriaux de sources d’énergie et à l’inhibition de la β-oxydation
des acides gras (Tokarska-Schlattner et al., 2002). Enfin, la doxorubicine régule également de
manière négative la glycolyse par l’inhibition de la phosphofructokinase, diminuant la
capacité compensatrice de la cellule par le shift glucidique.
Les ROS conduisent à l’augmentation de la perméabilité de la membrane interne de la
mitochondrie par peroxydation lipidique, à l’origine du dysfonctionnement de la
mitochondrie et donc de la diminution de la production d’énergie cellulaire.
b5b5b5b5. Le dysfonctionnement de la voie β. Le dysfonctionnement de la voie β. Le dysfonctionnement de la voie β. Le dysfonctionnement de la voie β----adrénergiqueadrénergiqueadrénergiqueadrénergique
Le système nerveux sympathique joue un rôle déterminant dans le contrôle de la fonction
cardiaque, notamment par une réponse β-adrénergique, médiée par trois récepteurs β-
adrénergique (β-AR).

104
� Les récepteurs β1-adrénergiques (β1-ARs)
Les β1-ARs sont majoritaires au sein des cellules cardiaques. Ils sont couplés à une protéine
G stimulatrice (Gs) à l’origine de l’activation de l’adénylate cyclase (AC). Celle-ci est à
l’origine de la production du second messager AMPc, qui va activer la protéine kinase A
(PKA). Sa sous-unité catalytique phosphoryle de nombreux effecteurs, à l’origine d’un effet
inotrope et lusitrope positifs. La stimulation de la voie Gs/AMPc/PKA conduit également à
l’activation de la glycogène synthase kinase 3β (GSK3β), intervenant dans le métabolisme du
glycogène et des protéines, et pouvant activer la protéine Bax pro-apoptotique. Lors d’une
stimulation soutenue des β1-ARs, ceux-ci se découplent de Gs, au profit de l’activation de la
protéine kinase calcium/calmoduline-dépendante II (CaMKII), principal effecteur des effets
pro-apoptotiques récepteurs β1-dépendants (Bernstein et al., 2011).
La stimulation de β1-AR est donc à l’origine d’un effet inotrope positif mais est également
pro-apoptotique.
� Les récepteurs β2-adrénergiques (β2-ARs)
Les β2-ARs sont couplés à la protéine Gs, mais également à la protéine G inhibitrice (Gi). L’activation de Gi conduit à l’inhibition de la voie Gs/AC/AMPc mais également au recrutement d’autres voies de signalisation dont la voie anti-apoptotique PI3K/Akt, à l’origine de la phosphorylation de GSK3β, de l’inhibition de la caspase 9 et de l’activation de Bcl. L’activation de Gi induit également la voie des phospholipases A2 cytosoliques (PLA2c), présentant un rôle dans l’inflammation cellulaire. Les β2-ARs peuvent également activer la NADPH oxydase ce qui conduit à l'activation de la voie MAPK (Mitogen-activated protein
kinase) et à la production de ROS pouvant être responsables de l'induction des voies apoptotiques (Di Lisa et al., 2011). Les β2-ARs contribuent donc à la modulation des voies de l’apoptose, principalement par activation via la voie Gs/AMPc/PKA ou inhibition via la voie Gi/PI3K/Akt (Zhu et al., 2001).
� Les récepteurs β3-adrénergiques (β3-AR)
Les β3-ARs sont peu exprimés dans un cœur sain, leur impact sur la contraction cardiaque
est donc minoritaire. Ils sont couplés aux protéines Gi et induisent l’activation de la voie du
monoxyde d’azote (NO) qui conduit à la production de la guanylyl cyclase (GC) par
l’activation de l’oxyde nitrique synthase endothéliale (eNOS). La GC permet ensuite la
production de GMPc, activant la protéine kinase G (PKG) et à l’origine d’un effet inotrope
négatif (Gauthier et al., 1998) (Birenbaum et al., 2008).
La doxorubicine est à l’origine d’une dysfonction β-adrénergique en agissant de façon
chronique sur les différents récepteurs. On observe une augmentation de la noradrénaline
circulante avec une régulation négative de l’expression des β1-ARs et de la protéine Gs ainsi
qu’une régulation positive des β3-ARs et de la protéine Gi (Figure 41). On observe alors une
inversion du ratio d’expression entre β1-AR et β3-AR, qui limite l’effet inotrope positif. La
stimulation prolongée des β1-Ars provoque leur découplage de la protéine Gs. L’expression
des β2-ARs est peu modifiée, permettant le maintien d’une activité anti-apoptotique
(Fajardo et al., 2006).

105
Figure 41 : Signalisation β-adrénergique cardiaque et doxorubicine
AA : acide arachidonique ; Akt : protéine kinase B ; AC : adénylate cyclase ; AMPc : adénosine monophosphate
cyclique ; Bax : Bcl-2 associated X protein ; CaMKIIδC : protéine kinase calcium/calmoduline-dépendante II δC ;
eNOS : oxyde nitrique synthase endothéliale ; GC : guanylate cyclase ; Gi : protéine G inhibitrice ; GMPc :
guanosine monophosphate cyclique ; Gs : protéine G stimulatrice ; GSK3β : glycogène synthase kinase 3β ;
PI3K : phosphoinositide-3 kinase ; PKA : protéine kinase A ; PKG : protéine kinase G ; PLA2c : phospholipases A2
cytosoliques
b6b6b6b6. L’altération de l’homéostasie calcique. L’altération de l’homéostasie calcique. L’altération de l’homéostasie calcique. L’altération de l’homéostasie calcique
Au cours d’un cycle cardiaque, la dépolarisation de la membrane des cardiomyocytes
pendant le potentiel d’action permet l’ouverture des canaux calciques de type L. L’entrée de
calcium par ces canaux et donc l’élévation locale du calcium induit l’activation des
récepteurs RyR2, entraînant une sortie massive de calcium du réticulum sarcoplasmique. Le
calcium ainsi libéré se fixe à la troponine C, ce qui entraine l’interaction des myofilaments
d’actine et de myosine et le raccourcissement des sarcomères, responsable de la contraction
cardiaque (Bers, 2002).
La calséquestrine est une protéine de stockage du calcium, présente dans le réticulum
sarcoplasmique. Elle peut interagir directement ou indirectement avec le récepteur RyR2 et
joue un rôle dans la régulation de son activité (Gaburjakova et al., 2013).
Pour permettre la relaxation cardiaque, la concentration en calcium cytosolique doit
diminuer rapidement. Le calcium est libéré par la troponine C et capté majoritairement par
la SERCA dans le réticulum sarcoplasmique, ou extrudé de la cellule par l’échangeur
sodium/calcium présent au niveau de la membrane plasmique. L’ensemble de ce processus
est nommé couplage excitation-contraction.
La doxorubicine peut induire une altération de l’homéostasie calcique conduisant à un
dysfonctionnement cardiaque diastolique, puis systolique au cours de l’évolution de la
cardiomyopathie.

106
L’expression de tous les acteurs de l’homéostasie calcique et notamment de la SERCA, de
l’échangeur sodium/calcium et du RyR2 est diminuée, hormis celle du canal calcique de
type L. De plus, la doxorubicine et surtout son métabolite principal, le doxorubicinol, se
fixent au récepteur RyR2 et à la calséquestrine, induisant une désynchronisation des
ouvertures et fermetures du canal calcique et une libération plus importante de calcium en
intracytoplasmique. L’ouverture du canal récepteur RyR2 est due à une action directe par
fixation, ou indirecte par oxydation de ses résidus thiols par les ROS. La fonction de
recapture du calcium par la SERCA est également inhibée, par oxydation de ses groupements
thiols (Gambliel et al., 2002).
Elle induit une augmentation de la concentration cytoplasmique de calcium par diminution
du captage de celui-ci par le réticulum sarcoplasmique tout en maintenant sa sortie. On
observe alors une altération de l’homéostasie calcique et de la relaxation cardiaque, ou
dysfonction diastolique. Secondairement, le stock de calcium mobilisable lors de la
contraction se réduit, ce qui peut expliquer une réduction de la contractilité cardiaque et
une dysfonction systolique (Appel et al., 2007).
L’altération du couplage excitation-contraction cardiaque implique également la
phosphorylation de la protéine kinase calcium/calmoduline dépendante, protéine
intervenant dans le maintien de l’homéostasie calcique.
Cette perturbation de l’homéostasie calcique peut être à l’origine de la cardiotoxicité aigue
de la doxorubicine. En effet¸ la doxorubicine conduit à une diminution de l’amplitude des
transitoires calciques et ralentit ainsi la contraction et la relaxation, et cela avant même la
modification d’expression des différents acteurs de l’homéostasie calcique. De plus, le
doxorubicinol inhibe le courant potassique sortant, responsable de la phase de
repolarisation du potentiel d’action cardiaque, ce qui se traduit par un allongement de la
durée du potentiel d’action et donc par un allongement de l’intervalle QT sur l’ECG (Wang et
al., 2001).
Les altérations de l’homéostasie calcique par la doxorubicine concernent toutes les
protéines impliquées dans le couplage excitation contraction, ce qui a pour conséquence
une altération de la contractilité et de la relaxation cardiaque. Cependant, les mécanismes à
long terme et le rôle de la signalisation calcique dans le développement de la cardiotoxicité
chronique restent à être caractérisés.
b7b7b7b7. Remodelage de la matrice extracellulaire. Remodelage de la matrice extracellulaire. Remodelage de la matrice extracellulaire. Remodelage de la matrice extracellulaire
L’altération dans la structure et la composition de la matrice extracellulaire contribue au
développement de l’insuffisance cardiaque (Spinale, 2007).
La doxorubicine augmente la transcription et la traduction des métalloprotéinases 2 et 9
(MMP-2, MMP-9). Les métalloprotéinases sont des enzymes protéolytiques qui dégradent
les composants de la matrice extracellulaire ainsi que des substrats non matriciels comme
certaines protéines cardiaques spécifiques (Octavia et al., 2012).

107
L’induction du stress oxydatif, du dysfonctionnement mitochondrial, de l’inhibition d’un
certain nombre de gènes codants pour des protéines cardiaques, de l’altération de
l’homéostasie calcique et de la signalisation β-adrénergique, contribuent à l’activation des
différentes voies de mort cellulaire des cardiomyocytes.
b8b8b8b8. La modulation des voie. La modulation des voie. La modulation des voie. La modulation des voies de mort cellulaire par la doxorubicines de mort cellulaire par la doxorubicines de mort cellulaire par la doxorubicines de mort cellulaire par la doxorubicine
La doxorubicine va principalement être à l’origine d’une stimulation de l’apoptose et de la
nécrose cellulaire. Des biopsies endomyocardiques lors de cardiomyopathie doxorubicine-
induite révèlent des altérations caractéristiques de la nécrose cellulaire telles que la
déformation du réticulum sarcoplasmique, la formation de vacuoles cytoplasmiques, des
distorsions myofibrillaires ou la raréfaction des myofibrilles. De plus, environ 1% des cellules
cardiaques sont concernées par l’apoptose cellulaire (Mauldin et al., 1992).
� L’apoptose cellulaire
Il va y avoir à la fois une altération de la voie extrinsèque et de la voie intrinsèque de la
doxorubicine :
� Concernant la voie intrinsèque, l’induction d’un stress oxydatif et l’altération de
l’homéostasie calcique cytosolique ont pour conséquence l’augmentation de la
concentration calcique dans les mitochondries. Celle-ci conduit à l’ouverture des
pores de transition de la perméabilité membranaire, responsable de la perte de
potentiel mitochondrial, du gonflement de la mitochondrie et de la rupture de la
membrane externe. On observe alors le relargage du cytochrome c et de
l’apoptosis inducing facteur (AIF) dans le cytoplasme, à l’origine de l’apoptose
cellulaire (Childs et al., 2002).
� Concernant la voie extrinsèque, les cardiomyocytes sont généralement résistants
à l’induction de l’apoptose via le récepteur de mort Fas. Cependant, plusieurs
études ont montré que l’apoptose des cardiomyocytes induite par la
doxorubicine peut se produire via la voie Fas. En effet, la voie calcineurine et plus
particulièrement le Nuclear Factor Activated T-cells (NFAT-4) ainsi que le facteur
de transcription NF-κB activé par les ROS conduisent à une surexpression de Fas
et à l’activation des gènes pro-apoptotiques (Kalivendi et al., 2005).
A la suite de l’activation de la voie extrinsèque et intrinsèque, l’activation des caspases mène
au clivage de protéines impliqués dans la réparation de l’ADN, dont PARP-1 (Pacher et al.,
2002). D’autres molécules comme la JNK, la p38 kinase (Zhu et al., 1999) ou les facteurs de
transcriptions NF-κB (Wang et al., 2002) et GATA-4 (Kim et al., 2003) sont activés lors de
l’apoptose des cardiomyocytes. A l’inverse, des molécules anti-apoptotiques comme Bcl-xL,
PI3K et Akt sont inhibées (Negoro et al., 2001).

108
� La nécrose cellulaire
La nécrose serait provoquée par l’induction du stress oxydatif et l’accumulation de calcium
dans les mitochondries, à l’origine d’une déplétion en énergie (Lebrecht, Walker, 2007). De
plus, la doxorubicine active les calpaines, enzymes responsables de la protéolyse calcium-
dépendante, responsables de la dégradation de la connectine, protéine du myofilament
impliquée dans le contrôle de l’assemblage des protéines sarcomériques et qui régule
l’élasticité du sarcomère. Ceci mène à l’accélération de la dégradation des myofilaments et à
la nécrose induite par la doxorubicine (Lim et al., 2004).
� L’autophagie cellulaire
Les résultats concernant le rôle de l’autophagie dans la cardiotoxicité de la doxorubicine
sont contradictoires. Sur quatre études in vivo sur des modèles murins, seule la moitié ont
montré une activation du processus d’autophagie par la doxorubicine (Dirks-Naylor, 2013)
(Sishi et al., 2013). Les études montrant une stimulation de l’autophagie associent cette
activation à une diminution de l’expression de Bcl-2 et du facteur de transcription GATA-4
(Kobayashi et al., 2010).
cccc. Diagnostic de la cardiotoxicité doxorubicine. Diagnostic de la cardiotoxicité doxorubicine. Diagnostic de la cardiotoxicité doxorubicine. Diagnostic de la cardiotoxicité doxorubicine----induiteinduiteinduiteinduite
Chez le chien, étant donné le coût et la disponibilité des différentes techniques, il existe peu
de méthodes diagnostiques de la cardiomyopathie.
Le gold standard du diagnostic de cardiomyopathie induite par la doxorubicine repose sur
la biopsie endomyocardique du ventricule droit. Cette technique est rarement utilisée étant
donné son caractère invasif (Gharib, Burnett, 2002).
La technique la plus couramment employée est l’échocardiographie, permettant l’évaluation
de la fonction systolique et diastolique. La mesure la plus utilisée sera celle de la fraction de
raccourcissement du ventricule gauche, dépendant des diamètres télésystolique et
télédiastolique du ventricule. On considère que l’animal présente un dysfonctionnement
cardiaque si on observe une fraction de raccourcissement inférieure à 25%, même si des
variations intraspécifiques existent. Cette mesure présente cependant une sensitivité limitée
pour la détection précoce de la cardiomyopathie doxorubicine-induite (Lotrionte et al.,
2013). Il est également possible de mesurer le volume d’éjection systolique gauche. Pour la
mesure de la fonction diastolique, on peut mesurer le temps de relaxation isovolumique
(IVRT) et le ratio vélocité de l’onde E/ vélocité de l’onde A, une diminution de celui-ci avec
une augmentation de l’IVRT montrant un dysfonctionnement diastolique. Cette méthode
présente une meilleure sensibilité de diagnostic précoce mais son aspect prédictif dans
l’évaluation de la cardiomyopathie nécessite d’autres études (Tater et al., 2017).
L’utilisation de biomarqueurs est encore peu courante chez le chien pour détecter une
cardiomyopathie induite par les anthracyclines. Les troponines sont des protéines
régulatrices impliquées dans la contraction cardiaque, présentes en concentration
importante et rapidement libérées dans le sang après la survenue des lésions cardiaques

109
(Langhorn, Willesen, 2016). D’après une étude menée sur 44 chiens, l’augmentation de la
troponine I est le marqueur biologique et clinique le plus précoce de l’insuffisance cardiaque
induite par la doxorubicine le chien (Selting et al., 2004). Le facteur natriurétique cardiaque
de type B (BNP) est un peptide sécrété par les cellules cardiaques ventriculaires lors d’une
augmentation du travail cardiaque. La fraction terminale de ce facteur, appelée NT-proBNP
est un biomarqueur utilisé pour détecter certaines cardiomyopathies dilatées. Une étude
portant sur 13 chiens recevant de la doxorubicine pendant 3 mois n’a pas révélé
d’augmentation plasmatique du marqueur NT-proBNP (Gallay-Lepoutre et al., 2016).
dddd. Traitement. Traitement. Traitement. Traitement et prévention de la cardiotoxicité doxorubicineet prévention de la cardiotoxicité doxorubicineet prévention de la cardiotoxicité doxorubicineet prévention de la cardiotoxicité doxorubicine----induiteinduiteinduiteinduite
Il n’existe pas de traitement spécifique de la cardiomyopathie induite par la doxorubicine. La
plupart du temps, les patients sont réfractaires aux traitements conventionnels : inhibiteurs
des enzymes de conversion de l’angiotensine (IECA), furosémide et pimobendane (Atkins et
al., 2009).
La prévention repose sur le respect d’une dose-cumulative maximale de de 180 mg/m², soit
six séances avec une dose de 30 mg/m², afin de limiter la cardiotoxicité (Mauldin et al.,
1992). Il convient également d’injecter lentement la doxorubicine (sur 20 à 30 minutes) afin
de diminuer le pic plasmatique responsable de la formation de radicaux libres.
Il est également possible d’administrer 30 minutes avant la doxorubicine un chélateur du fer,
le dexrazoxane, à une concentration égale à dix fois la dose de doxorubicine injectée, afin de
diminuer le risque de cardiotoxicité (FitzPatrick et al., 2010). Cette molécule est cependant
très peu utilisée en médecine vétérinaire étant donné son coût et sa non disponibilité en
officine. On réalise également un suivi par échocardiographie avant les première et
quatrième séances de chimiothérapie à base de doxorubicine.
L’utilisation de doxorubicine encapsulée permettrait d’augmenter les doses cumulées sans
cardiotoxicité, mais cette présentation n’est pas disponible en médecine vétérinaire et
nécessite des études supplémentaires autre que des essais pré-cliniques (Vail et al., 1997).
7777. Principales contre. Principales contre. Principales contre. Principales contre----indications de la doxorubicine en médecine indications de la doxorubicine en médecine indications de la doxorubicine en médecine indications de la doxorubicine en médecine vétérinairevétérinairevétérinairevétérinaire
Il existe plusieurs contre-indications à l’utilisation de la doxorubicine chez l’animal, liées à la
toxicité et aux effets indésirables de celle-ci :
� Une hypersensibilité connue à la doxorubicine, à d’autres anthracyclines ou aux
anthracènediones, dont l’épirubicine et la mitoxantrone.
� Une hypoplasie médullaire passagère ou persistante.
� Une insuffisance hépatique marquée.

110
� Une atteinte cardiaque, pouvant être une insuffisance myocardique grave, des
antécédents de cardiopathie grave ou la présence d’arythmies non sinusales.
� Des antécédents thérapeutiques comportant l’administration de doxorubicine ou
d’autres anthracyclines et anthracènediones, jusqu’à concurrence de la dose
cumulée maximale.
8888. Mutation et toxicité. Mutation et toxicité. Mutation et toxicité. Mutation et toxicité
Dans certaines races de chien, on observe une sensibilité plus importante à certaines
molécules, à l’origine d’une toxicité plus importante, notamment à la doxorubicine. Cela est
dû à une modification de l’expression des transporteurs d’efflux P-gp, à l’origine d’une
accumulation plus importante de la doxorubicine au sein des cellules. Cette sensibilité est
due à une mutation du gène mdr1 correspondant à une délétion de quatre paires de base
dans l’exon 4 et appelée mutation ABCB1-1Δ ou MDR1-1Δ. Cette mutation engendre
l’apparition de multiples codons stop prématurés, à l’origine de la production d’une protéine
P-gp tronquée et non fonctionnelle, avec seulement 10 % de la séquence d’acide aminés en
commun avec la protéine non mutée (Mealey et al., 2001).
Cette mutation touche en premier lieu les chiens de race, particulièrement les races
apparentées aux Colleys. On peut citer parmi les races les plus touchées par cette mutation
le Colley, le Border Collie, le Berger Australien, le Berger Australien miniature, le Berger
Anglais, le Whippet à poil long, le MacNab, le Berger Anglais ancestral, le Berger de Shetland,
le Silken Windhound et le Berger Blanc Suisse (Tableau VI) (Neff et al., 2004). Dans certaines
de ces races, les chiens porteurs de cette mutation représentent une part prédominante de
la population de cette race. En France, sur 83 colleys testés, 80% portent au moins un allèle
muté ABCB1-1Δ, dont 32 % sont hétérozygotes et 48 % sont homozygotes mutés (Hugnet et
al., 2004).
Tableau VI : Fréquence de l’allèle mdr1 mutée et des génotypes D’après l’étude de Neff et al. (2004).
Races Nombre de chiens
Allèle mutée (%)
-/- (%) -/+ (%) +/+ (%)
Berger australien 178 16,6 1,7 29,8 68,5
Berger australien miniature 56 25,9 3 ,6 44,6 51,8
Colley 263 54,6 31,2 46,8 22,0
Berger anglais 91 7,1 0 14,3 85,7
Whippet à poils longs 89 41,6 15,7 51,7 32,6
McNab 35 17,1 2,8 28,6 68,6
Bobtail 151 3,6 0 7,3 92,7
Shetland 190 8,4 1,1 14,7 84,2
Silken Windhound 84 17,9 1,2 33,3 65,5

111
Chez le chien, la mutation du gène mdr1 est à l’origine d’une augmentation de la toxicité de
la doxorubicine chez les individus porteurs. Cela est expliqué par une augmentation
d’environ 25 % de l’AUC de la doxorubicine plasmatique chez des chiens homozygotes
mutés. De plus, par extrapolation, il faudrait utiliser la dose de 10,7 +/- 5,9 mg/m² chez les
chiens homozygotes mutés pour obtenir une toxicité gastro-intestinale équivalente à celle
des chiens homozygotes non mutés traités à 30 mg/m² (Gustafson, Thamm, 2010).
La cytotoxicité de la doxorubicine et de son principal métabolite, le doxorubicinol, est à l’origine
de l’activation de nombreux mécanismes menant à différents effets indésirables, spécifiques ou
non, à court-terme ou à plus long terme. Ces effets indésirables doivent être gradés selon la
classification VCOG-CTCAE et pris en compte dans la réévaluation possible du protocole de
chimiothérapie.
Parmi ces effets indésirables, on peut citer :
� Le choc histaminique, toxicité spécifique d’apparition suraiguë et de gravité variable. Il
est lié à la dégranulation des mastocytes dans l’organisme.
� La toxicité gastro-intestinale, toxicité non spécifique liée soit à la stimulation de la
trigger zone et responsable de vomissements dans les 24 heures suivant l’injection, soit
à la cytotoxicité de la doxorubicine directement sur les entérocytes immatures, à
l’origine de vomissements, d’anorexie et de diarrhée dans les deux à cinq jours suivant
l’injection.
� La toxicité hématologique, toxicité non spécifique de la doxorubicine liée à la forte
prolifération des cellules souches des différentes lignées cellulaires sanguines. Elle est
principalement à l’origine d’une neutropénie et d’une thrombocytopénie transitoires,
avec un nadir entre sept et dix jours pour la neutropénie.
� La toxicité cutanée par extravasation de la doxorubicine, toxicité spécifique à l’origine
d’un érythème pouvant évoluer vers une nécrose à extension locale en plusieurs
semaines. Le traitement le plus efficace reste le parage chirurgical des zones nécrosées
afin d’éviter l’extension de celles-ci.
� L’alopécie, effet indésirable spécifique de la doxorubicine mais rare chez le chien, elle
peut survenir près une seule injection de doxorubicine.

112
� La toxicité cardiaque, toxicité spécifique de la doxorubicine. On distingue deux types de
toxicité cardiaque :
• Aiguë, elle survient au cours de l’injection ou dans les jours qui suivent. La
plupart du temps asymptomatique et auto-résolutive, elle est rarement
objectivée chez le chien. Son origine exacte reste indéterminée mais elle
pourrait être due à la formation de ROS via les métabolites aglycones de la
doxorubicine et/ou aux perturbations des flux calcique et potassique,
modifiant le couplage excitation-contraction des cardiomyocytes.
• Chronique et dose-cumulative, elle survient plusieurs mois après la fin
de la chimiothérapie. Elle est due à l’apparition d’une cardiomyopathie
dilatée par remodelage du cœur, via de nombreux mécanismes
cellulaires (Figure 42). Sa prévention repose principalement sur une
limitation de la dose cumulative à 180 mg/m².
Ces effets indésirables sont parfois exacerbés chez certains individus, notamment ceux
porteurs de la mutation du gène mdr1 codant pour la glycoprotéine G, à l’origine d’une
diminution de l’efflux de la doxorubicine en dehors des cellules.

Figure 42: Principaux mécanismes de cardiotoxicité de la doxorubicine
β-AR : récepteur β-adrénergique ; CalS : calséquestrine ; cTn : troponine cardiaque ; cyt c : cytochrome c ; DOX : doxorubicine ; DOX° : radical semi-quinone ;
DOXol : doxorubicinol ; MAPK : mitogen-activted protein kinase ; MLC-2 : chaîne légère régulatrice de la myosine ; NCX : échangeur calcium/sodium ; NF-κB :
nuclear factor-κB ; RyR2 : récepteur à la ryanodine 2 ; SERCA : pompe Ca2+
-ATPase du réticulum sarcoplasmique ; Top IIβ : ADN topoisomérase de type IIβ.
113

114

115
Partie IIPartie IIPartie IIPartie II : Etude expérimentale de la : Etude expérimentale de la : Etude expérimentale de la : Etude expérimentale de la
pharmacocinétique et de la toxicité de la pharmacocinétique et de la toxicité de la pharmacocinétique et de la toxicité de la pharmacocinétique et de la toxicité de la doxorubicine en médecine vétérinairedoxorubicine en médecine vétérinairedoxorubicine en médecine vétérinairedoxorubicine en médecine vétérinaire

116
La pharmacocinétique de la doxorubicine a été étudiée dans de nombreuses espèces
(Homme, chat, souris, rat, lapin, perroquet, etc.…) mais les études chez le chien restent
sporadiques. Malgré l’utilisation fréquente de la doxorubicine au sein de cette espèce, les
études de pharmacocinétique sur des chiens non sains sont rares (Gustafson et al., 2002)
(Selting et al., 2006). La plupart des données de pharmacocinétique sont principalement
issues d’études sur des chiens sains (Arrington et al., 1994) (Oosterbaan et al., 1984) (Timour
et al., 1988) ou se reposant sur des simulations de populations de chiens malades
(Gustafson, Thamm, 2010). La posologie (30 mg/m² ou 1 mg/kg) de la doxorubicine utilisée
chez le chien repose sur des études de 1989 et 1994, montrant une augmentation de la
toxicité chez les chiens de moins de dix kilogrammes lors de l’utilisation de la doxorubicine à
la dose standard de 30 mg/m² (Ogilvie, Reynolds, et al., 1989) (Arrington et al., 1994).
Depuis, aucune étude sur l’intérêt de cette distinction de posologie n’a été réalisée.
Plusieurs méthodes de quantification de la doxorubicine et de son métabolite principal, le
doxorubicinol, dans des liquides biologiques ou des tissus ont été mises au point. La plupart
du temps, ces méthodes reposent sur une technique de chromatographie en phase liquide
couplée à la spectrométrie de masse en tandem (LC/MS/MS), ou de chromatographie en
phase liquide à haute performance (HPLC) avec détection par ultraviolet ou fluorimétrie
(Tableau XIII). Malheureusement, certaines de ces études manquent de sensibilité et/ou de
spécificité et présentent des méthodes d’extraction complexes (Al-Abd et al., 2009). D’autres
méthodes sont plus rarement utilisées telles que le dosage radio-immunologique sur urine
ou plasma (Rahmani et al., 1983), la voltammétrie sur urine (Chaney, Baldwin, 1985), la
spectrofluorimétrie sur sérum ou l’électrophorèse capillaire (Gavenda et al., 2001).
Parmi les méthodes analytiques LC/MS/MS et HPLC, seules quelques-unes ont été testées
sur du plasma de chien et la plupart des méthodes utilisées dans les études de
pharmacocinétique chez le chien reposent sur des méthodes HPLC testées sur d’autres
espèces mais pas chez le chien.
Les différents objectifs de cette étude sont de :
� Valider une méthode d’analyse sensible en HPLC avec détection par fluorescence
pour déterminer la concentration de doxorubicine dans le plasma de chien.
� Appliquer cette méthode d’analyse à une étude de pharmacocinétique et de toxicité
de la doxorubicine chez une population de chiens non sains. Le choix de cette
population nous permettant de limiter le recours à l’expérimentation animale et
d’évaluer la pharmacocinétique dans des conditions réelles d’emploi de la
doxorubicine.

117
I. Mise au point d’une méthode analytique de dosage de la I. Mise au point d’une méthode analytique de dosage de la I. Mise au point d’une méthode analytique de dosage de la I. Mise au point d’une méthode analytique de dosage de la doxorubicine plasmatique par HPLCdoxorubicine plasmatique par HPLCdoxorubicine plasmatique par HPLCdoxorubicine plasmatique par HPLC----fluorescencefluorescencefluorescencefluorescence
A. Mise au point de la A. Mise au point de la A. Mise au point de la A. Mise au point de la méthode analytiqueméthode analytiqueméthode analytiqueméthode analytique
1. Description des conditions d’analyse par chromatographie1. Description des conditions d’analyse par chromatographie1. Description des conditions d’analyse par chromatographie1. Description des conditions d’analyse par chromatographie
Les analyses ont été réalisées par un système de chromatographie en phase liquide à haute
performance (HPLC, Elite Lachrom) équipé d’une colonne à chromatographie Merck
LiChrospher RP-18 Endcapped (4 mm x 250 mm, 5 µm) et avec une détection par
fluorescence. Les longueurs d’ondes d’excitation (470 nm) et d’émission (580 nm) ont été
déterminées à partir de l’analyse de spectrofluorimétrie de la doxorubicine (Figure 9).
Concernant la phase mobile, plusieurs solutions ont été envisagées. La plupart des méthodes
d’analyse basées sur la chromatographie en phase liquide utilise comme phase mobile un
mélange d’une solution tampon de phosphate de potassium KH2PO4 (0,06 mmol/L, pH 3)
avec de l’acétonitrile. Cependant, l’utilisation de cette phase mobile était à l’origine de
l’encrassement notre système d’HPLC par précipitation chimique de cristaux de potassium.
La phase mobile a alors été modifiée et il a été opté pour un mélange de tampon de
phosphate mono-ammonique (NH4)H2PO4 (0,05 mmol/L, pH 3) et d’acétonitrile (70 : 30, v/v),
délivrée à un débit constant de 1 mL/min et ce à température ambiante. Le volume injecté
était de 50 µL.
Une élution avec un mode de gradient isocratique avait d’abord été envisagée mais les
temps de rétention de la doxorubicine et de l’étalon interne étaient inférieurs à cinq minutes
avec une séparation des pics non satisfaisante. L’élution a donc été réalisée en mode
gradient sur 20 minutes selon le schéma décrit dans le Tableau VII, avec un mélange phase
mobile : acétonitrile.
Tableau VII : Gradient d’élution utilisée lors de la chromatographie par HPLC
Phase mobile (en %) Acétonitrile (en %)
0 minute 100 0
5 minutes 88,2 11,8
10 minutes 70,6 29,4
15 minutes 64,7 35,3
20 minutes 100 0
2. Préparation des solutions2. Préparation des solutions2. Préparation des solutions2. Préparation des solutions
La doxorubicine hydrochloride, la daunorubicine hydrochloride (Sigma Aldrich Chemie), le
trifluoroacétate de doxorubicinol (Alsachim) et les autres composés chimiques utilisés sont
de pureté analytique.

118
Des solutions-mères de doxorubicine à 100 µg/mL et de daunorubicine à 100 µg/mL ont été
préparées par dilution du composé d’intérêt dans de l’acétonitrile. Ces solutions ont été
stockées à l’abri de la lumière à -21°C.
De plus, des solutions de travail à base de doxorubicine ou de daunorubicine à 10 µg/mL ont
été préparées en diluant les solutions-mères dans la phase mobile. Une solution de
doxorubicinol à 10 µg/mL a également été préparée en diluant le trifluoroacétate de
doxorubicinol dans de l’acétonitrile. Toutes les solutions de travail ont été conservées à
l’abri de la lumière à 4°C.
La daunorubicine est utilisée comme étalon interne lors de notre étude.
On définit également trois concentrations témoins en doxorubicine, qui serviront de
contrôle au cours de la validation de la méthode analytique. On définit ainsi les solutions
basse concentration (LQC, 0,1 µg/mL), moyenne concentration (MQC, 0,5 µg/mL) et haute
concentration (HQC, 1,5 µg/mL).
3. Préparation des échantillons de plasma3. Préparation des échantillons de plasma3. Préparation des échantillons de plasma3. Préparation des échantillons de plasma
Les échantillons plasmatiques canins utilisés pour la validation de la méthode analytique ont
été récupérés à partir d’échantillons sanguins qui ont été centrifugés à 3000 tours/minute
pendant cinq minutes puis stockés à 4°C jusqu’à analyse.
Une série d’échantillons plasmatiques supplémentés en doxorubicine, de concentrations
finales allant de 0,01 µg/mL à 1,5 µg/mL, a été préparée afin de servir d’échantillons
d’étalonnage pour vérifier la linéarité de la méthode analytique. La préparation de ces
solutions est décrite dans le Tableau VIII.
Tableau VIII : Méthode de préparation de la série d’étalonnage
Volume
solution de
travail/mère
(µL)
Volume de
phase
mobile (µL)
Concentration
finale en
solution
(µg/mL)
Concentration
plasmatique
finale (µg/mL)
10 (10 µg/mL) 990 0,10 0,01
25 (10 µg/mL) 975 0,25 0,025
50 (10 µg/mL) 950 0,5 20 µL solution 0,05
100 (10 µg/mL) 900 1 0,1
250 (10 µg/mL) 750 2,5 + 180 µL plasma 0,25
250 (10 µg/mL) 250 5 0,5
500 (10 µg/mL) 0 10 1
150 (100 µg/mL) 850 15 1,5

119
L’extraction a ensuite été réalisée par précipitation des protéines. Pour cela, on a prélevé 190 µL du plasma d’intérêt auquel on a ajouté 10 µL de l’étalon interne à 10 µg/mL, 0,4 mL d’acétonitrile et 0,4 mL de la phase mobile. L’ensemble a été homogénéisé grâce à un agitateur puis le tube à essai a été centrifugé à 3000 tours par minute pendant 10 minutes à 4°C. Le surnageant a été filtré par un filtre de porosité 0,45 µm dans un vial pour analyseur HPLC.
4. Vérification de la méthode de dosage4. Vérification de la méthode de dosage4. Vérification de la méthode de dosage4. Vérification de la méthode de dosage
La validation de la méthode analytique a été réalisée selon les recommandations de l’Agence
Européenne du Médicament (EMA, 2011).
� Spécificité de la méthode analytique
Une méthode est dite spécifique lorsqu’elle permet de mesurer l’analyte avec la garantie
que le signal analytique ne provient que de l’analyte sans interférence avec des éléments de
la matrice ou des impuretés. Ce critère certifie que la procédure analytique donne de façon
précise la quantité d’analyte présente dans l’échantillon.
La spécificité de la méthode est évaluée par la recherche de l’effet matrice. En
chromatographie, elle est évaluée moyennant les temps de rétention.
Pour tester la spécificité de la méthode vis-à-vis de la doxorubicine et de l’étalon interne, six
échantillons plasmatiques ont été analysés, sans supplémentation en doxorubicine et en
étalon interne, puis avec supplémentation à 0,1 µg/mL.
Figure 43 : (A) Chromatogramme de plasma blanc. (B) Chromatogramme de plasma
supplémenté en doxorubicine (DOX) et daunorubicine (DNR) à 0,1 µg/mL.
La Figure 43 présente deux chromatogrammes, le premier correspondant à un échantillon
plasmatique non supplémenté et le second à un échantillon plasmatique supplémenté.
D’après les résultats obtenus, les pics des substances d’intérêts ne semblent pas en
interférence avec de possibles impuretés présentes dans le plasma. De plus, le temps de
rétention pour la doxorubicine est de 6,09 ± 0,16 minutes (CV = 1,64 %) et celui de la
daunorubicine de 9,04 ± 0,15 minutes (CV = 1,64 %). On peut donc conclure que notre
méthode est sélective.

120
� Sélectivité de la méthode analytique
La sélectivité est l’aptitude d’un système analytique à discerner un analyte donné dans un
mélange complexe. Ce critère permet de garantir que la réponse analytique n’est pas
affectée par d’autres substances. On cherche ici à vérifier l’écart entre le pic de doxorubicine
(DOX) et celui de l’étalon interne (DNR).
La sélectivité est évaluée par le degré de résolution (Rs), dépendant des temps de rétention
(tr) des deux pics chromatographiques et de la largeur des deux pics (W), selon la formule
suivante :
Pour considérer que deux pics ont un recouvrement inférieur ou égal à 2 %, le degré de
résolution doit être supérieur à 1. Et si le degré de résolution est supérieur à 1,5, on
considère que la séparation des pics est totale. Lors de notre étude, le degré de résolution
est évalué à 1,55. On peut donc conclure que notre méthode est sélective pour la
doxorubicine.
� Linéarité de la méthode analytique
La linéarité est la capacité d’une méthode analytique, à l’intérieur d’un certain intervalle, à
fournir une réponse instrumentale ou des résultats proportionnels à la quantité en analyte à
doser dans l’échantillon.
La linéarité de cette méthode de dosage de la doxorubicine a été évaluée pour des
concentrations allant de 0,01 à 1,5 µg/mL. Trois courbes d’étalonnage ont été réalisées sur
trois jours pour s’assurer de la linéarité de la méthode (Figure 44). Les étalons étaient
préparés chaque jour.
Afin de quantifier la doxorubicine dans les échantillons plasmatiques, on a utilisé le ratio de
l’aire du pic correspondant à la doxorubicine sur celle du pic de l’étalon interne, coefficient
de réponse relatif de la doxorubicine par rapport à l’étalon interne. Cette approche permet,
lorsque deux substances sont extraites de façon comparable, d’avoir un témoin de
l’extraction et de ne pas tenir compte du pourcentage d’extraction réel à chaque nouvelle
analyse. De plus, toute variation du système chromatographique entraîne une perturbation
du même ordre sur la réponse de la doxorubicine et de l’étalon interne, permettant une
meilleure estimation de la concentration en doxorubicine.
Pour valider la méthode, les concentrations calculées doivent être inférieures à plus ou
moins 15 % de la concentration nominale, exception pour la LOQ pour laquelle une valeur de
plus ou moins 20 % est acceptée. Au moins 75 % des étalons doivent remplir ces critères de
validation de la linéarité. De plus le coefficient de corrélation linéaire doit être supérieur
à 0,98.
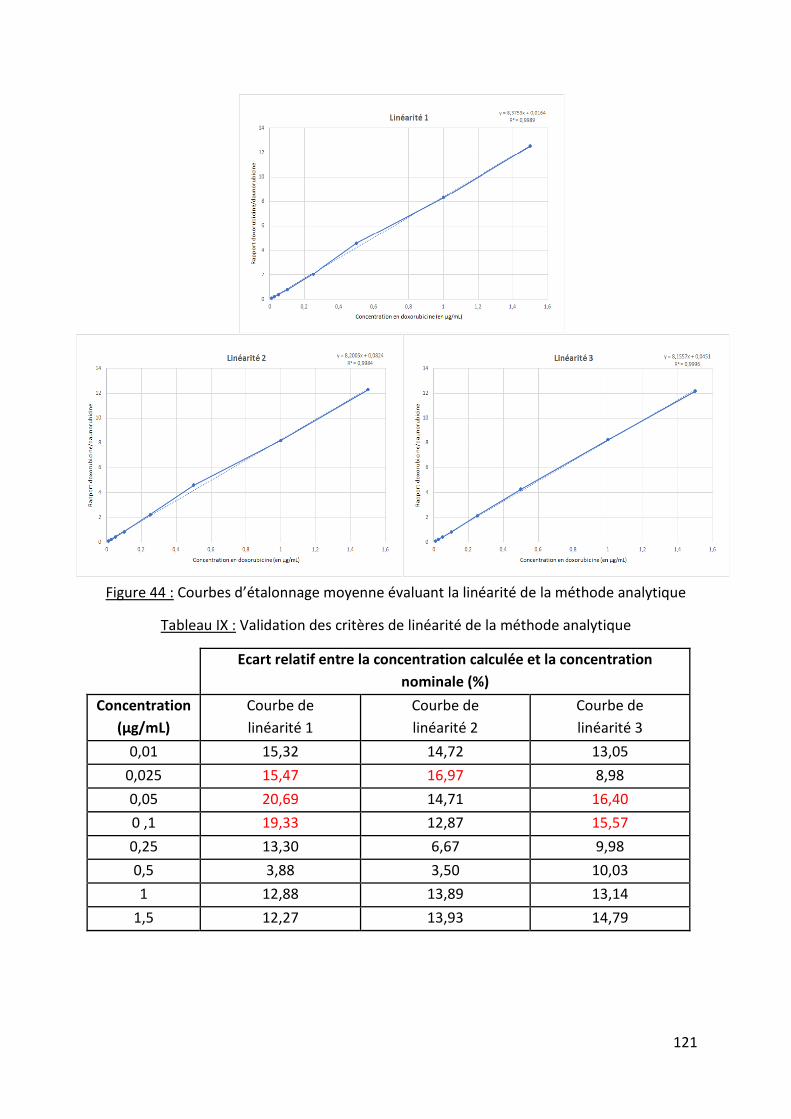
121
Figure 44 : Courbes d’étalonnage moyenne évaluant la linéarité de la méthode analytique
Tableau IX : Validation des critères de linéarité de la méthode analytique
Ecart relatif entre la concentration calculée et la concentration
nominale (%)
Concentration
(µg/mL)
Courbe de
linéarité 1
Courbe de
linéarité 2
Courbe de
linéarité 3
0,01 15,32 14,72 13,05
0,025 15,47 16,97 8,98
0,05 20,69 14,71 16,40
0 ,1 19,33 12,87 15,57
0,25 13,30 6,67 9,98
0,5 3,88 3,50 10,03
1 12,88 13,89 13,14
1,5 12,27 13,93 14,79

122
La courbe d’étalonnage moyenne obtenue à partir des trois courbes d’étalonnage était
linéaire pour des concentrations allant de 0,01 à 1,5 µg/mL. L’équation de régression de la
courbe est la suivante :
Avec x la concentration en doxorubicine et y le ratio de l’aire du pic de doxorubicine sur celle
du pic de l’étalon interne. Le coefficient de corrélation linéaire est supérieur à 0,998 pour
toutes les courbes d’étalonnage. De plus, les critères de validation de la linéarité sont bien
respectés (Tableau IX).
La méthode développée est donc bien linéaire sur l’intervalle de concentration 0,01 à 1,5
µg/mL.
� Limite de détection (LOD)
La limite de détection est définie comme la plus faible quantité de doxorubicine pouvant
être détectée dans la matrice, ici le plasma, avec certitude. Pour les méthodes analytiques
chromatographiques, la limite de détection est établie soit en utilisant la proportion 3 :1
(signal : bruit), soit par méthode mathématique à partir de l’équation de linéarité.
Dans notre étude, la limite de détection a été calculée à 0,006 µg/mL.
� Limite de quantification (LOQ)
La limite de quantification est définie comme la plus faible quantité d’analyte pouvant être
quantifiée dans la matrice avec certitude. C’est la plus basse concentration sur la courbe
d’étalonnage. Dans les méthodes chromatographiques, la détermination de cette limite est
faite soit par rapport au bruit de fond, en utilisant la proportion 10 : 1 (signal : bruit), soit par
méthode mathématique à partir de l’équation de linéarité.
Dans notre étude, la limite de quantification a été calculée à 0,01 µg/mL.
� Précision et exactitude de la méthode analytique
La précision est définie par l’étroitesse de l’accord entre des mesures effectuées sur des
prises multiples d’un échantillon homogène. Elle exprime le degré de dispersion entre une
série de dosage provenant de multiples prises d’essai de l’échantillon dans des conditions
expérimentales bien précises.
Ce critère est évalué par la mesure de la répétabilité et de la reproductibilité et est exprimé
comme le coefficient de variation (CV) d’une série de mesure.
La répétabilité, ou précision des essais internes, est la mesure de la précision lorsqu’elle est
réalisé dans les même conditions et dans un court laps de temps.

123
Celle-ci est réalisée sur une série de quatre concentrations de doxorubicine (LOQ, LQC, MQC
et HQC), chaque concentration est réalisée en cinq extractions. Elle est exprimée par le
coefficient de variation (CV).
La reproductibilité exprime la précision inter-laboratoires et est la mesure de la précision
lorsque n’importe quelle condition opératoire change.
Elle est évaluée sur une série de quatre concentrations de doxorubicine (LOQ, LQC, MQC et
HQC), et analysée pendant trois jours différents. Chaque concentration est réalisée en cinq
extractions. Elle est exprimée par le coefficient de variation (CV).
L’exactitude (accuracy) est définie par l’étroitesse d’accord entre la valeur trouvée et la
valeur de référence. Elle correspond à l’écart entre la valeur obtenue et la valeur considérée
comme exacte.
Celle-ci est réalisée sur une série de quatre concentrations de doxorubicine (LOQ, LQC, MQC,
HQC), analysée pendant trois jours différents et chaque concentration est réalisée en cinq
extractions. Elle est exprimée par l’erreur relative (ER).
Pour valider la méthode, les coefficients de variation de répétabilité et de reproductibilité,
ainsi que les erreurs relatives doivent être inférieurs à 15 %, exception faite pour les
échantillons de concentration LOQ pour lesquelles les valeurs doivent être inférieures à
20 %.
Tableau X : Résultats de l’étude de précision et d’exactitude de la méthode analytique
Concentration
théorique
(µg/mL)
Concentration moyenne en doxorubicine (µg/mL)
Répétabilité (n=5) Reproductibilité (n=3)
Concentration
trouvée ± SD
CV (%) ER (%) Concentration
trouvée ± SD
CV (%) ER (%)
0,01 (LOQ) 0,0084 ± 0,0005 5,76 16,13 0,0082 ± 0,0004 4,77 18,22
0,1 (LQC) 0,1057 ± 0,0012 1,13 5,72 0,1124 ± 0,0058 5,13 12,39
0,5 (MQC) 0,5542 ± 0,0025 0,45 10,85 0,5658 ± 0,0184 3,25 13,16
1,5 (HQC) 1,4580 ± 0,0339 2,33 2,80 1,3878 ± 0,0835 6,02 7,48
Le coefficient de variation de la répétabilité varie entre 0,45 et 5,76 %, avec une erreur
relative variant de 2,80 à 16,13 %. Le coefficient de variation de la reproductibilité varie
entre 3,25 et 6,02 %, avec une erreur relative variant de 7,48 à 18, 22 % (Tableau X).

124
Notre méthode analytique remplit donc les critères de précision et d’exactitude requis par
l’EMA pour être validée.
� Rendement d’extraction de la méthode
La détermination du rendement d’extraction permet de connaitre le pourcentage de
doxorubicine et de daunorubicine récupéré et d’évaluer la reproductibilité de la méthode.
En chromatographie, le rendement d’extraction (R) exprimé en pourcentage s’obtient en
appliquant la formule suivante :
Le rendement d’extraction de la doxorubicine et de l’étalon interne ont été évalués en
comparant l’aire moyenne des pics de la doxorubicine et de la daunorubicine des solutions
LQC, MQC et HQC avec l’aire moyenne des pics de six solutions de références contenant la
même concentration en doxorubicine et en daunorubicine.
Tableau XI : Résultats de l’étude de rendement d’extraction de la méthode analytique
Molécule Concentration
(µg/mL)
Rendement ± SD (%) CV (%)
Doxorubicine 0,1 103,2 ± 1,3 1,21
0,5 98,9 ± 7,3 7,43
1,5 104,4 ± 4,6 4,44
Daunorubicine 0,1 97,1 ± 5,0 2,85
La méthode d’extraction par précipitation des protéines par un mélange d’acétonitrile et de
phase mobile permet d’obtenir un rendement de 97,1 % pour la daunorubicine et variant de
98,9 à 104,4 % pour la doxorubicine (Tableau XI). Ces valeurs sont supérieures ou égales à
celle d’autres méthodes analytiques de la doxorubicine (Tableau XIII).
� Stabilité de la doxorubicine
La stabilité d’un analyte en solution correspond à sa capacité à résister à la dégradation
lorsque la solution est soumise à différentes conditions. On considère qu’une molécule est
stable dans la matrice si la variation observée entre le début des conditions et la fin de
celles-ci est inférieure à 15 %.
La stabilité de la doxorubicine dans le plasma de chien a été étudiée en réalisant des lots de
six échantillons, trois contenant de la doxorubicine à 0,1 µg/mL (LQC) et trois à 1,5 µg/mL
(HQC). Chaque lot a ensuite été soumis à une condition différente pour pouvoir évaluer la
stabilité de la doxorubicine selon la température, et donc d’optimiser sa conservation.

125
Les différentes conditions de température sont :
� Exposition à trois cycles de congélation puis décongélation à température ambiante.
� Exposition à la température ambiante (20°C en moyenne) pendant 1 heure, 5 heures
et 24 heures.
� Exposition à une température de 4°C pendant 24 heures, mimant les conditions de
conservation dans un réfrigérateur.
� Exposition à une température de -21°C pendant 1 mois, mimant les conditions de
congélation des échantillons avant analyse.
Tableau XII : Résultats de l’étude de stabilité de la doxorubicine dans du plasma
Conditions
Rapport de la concentration sur la concentration initiale ± SD (%)
Doxorubicine Daunorubicine
1,5 µg/mL 0,1 µg/mL 1,5 µg/mL 0,1 µg/mL
Température ambiante (1h) 99,95 ± 0,32 99,42 ± 0,76 100,54 ± 0,70 99,54 ± 1,13
Température ambiante (5h) 96,36 ± 1,37 99,87 ± 0,11 98,90 ± 2,43 100,42 ± 0,18
Température ambiante
(24h)
94,45 ± 1,62 95,30 ± 1,12 92,64 ± 1,43 97,28 ± 1,05
4°C pendant 24 heures 97,70 ± 0,16 99,74 ± 1,81 99,81 ± 0,35 100,46 ± 0,62
3 cycles de congélation 100,81 ± 0,41 99,88 ± 1,13 99,13 ± 0,47 99,22 ± 0,46
-21°C pendant 1 mois 97,65 ± 0,65 94,13 ± 0,45 95,93 ± 0,92 91,87 ± 0,68
Les variations de concentrations de la doxorubicine et de la daunorubicine dans du plasma
canin n’excèdent pas 9% (Tableau XII). On considère donc que sous ces conditions, ces
molécules sont stables dans le plasma.
� Détection du principal métabolite de la doxorubicine, le doxorubicinol
Des plasmas canins supplémentés en doxorubicine, doxorubicinol et daunorubicine à la
concentration 1 µg/mL ont été préparées afin d’évaluer la sélectivité de la méthode
analytique lors de la présence de doxorubicinol dans le plasma, après métabolisation par le
foie.
Figure 45 : Chromatogramme de plasma supplémenté en doxorubicinol, doxorubicine et
daunorubicine à 1 µg/mL.

126
B. DiscussionB. DiscussionB. DiscussionB. Discussion
Plusieurs méthodes analytiques pour déterminer la concentration en doxorubicine dans un
échantillon de plasma ont été décrites. La plupart des méthodes utilisées dans des études de
pharmacocinétique de la doxorubicine sont décrites dans le Tableau XIII.
Tableau XIII : Méthodes d’analyses par HPLC de détermination de la concentration en doxorubicine dans des liquides biologiques et des tissus
Auteurs Méthode
de
détection
Matrice de
calibration
Matrice de
validation
LOQ Rendement
d’extraction
(van Asperen et al.,
2000)
FL Plasma humain Plasma de souris
Tissus de souris
1,3 ng/mL
96 ng/mg
86-99 %
(de Bruijn et al., 1999) FL Plasma humain Plasma humain 100 ng/mL 112 ± 8,6 %
(Alvarez-Cedrón et al.,
1999)
FL Plasma humain Plasma de rat
Tissus de rat
5 ng/mL
100 ng/mg
89,33-109,5 %
(Kümmerle et al., 2003) FL Plasma humain Plasma de rat
Tissus de rat
2 ng/mL
100 ng/mg
/
(Ricciarello et al., 1998) ECD Plasma humain Plasma humain 1 ng/mL 89-93 %
(Pérez-Blanco et al.,
2014)
FL Plasma humain Plasma humain 8 ng/mL 94,82-104,38 %
(Gilbert et al., 2005) FL Plasma
perroquet
Plasma perroquet 20 ng/mL 71-87 %
(de Jong et al., 1991) FL Tissus de souris / 25ng/mg 74,9-79,1 %
(Arnold et al., 2004) MS/MS Plasma de rat
Tissus de rat
Plasma de rat
Tissus de rat
0,2 ng/mL
0,15-15ng/mg
84-112 %
(Ahmed et al., 2009) CLD Plasma de souris Plasma de rat / 90,7-104 %
(Urva et al., 2009) FL Plasma de souris
Tissus de souris
Plasma de souris
Tissus de souris
/ 94,7-101 %
(Zhou, Chowbay, 2002) FL Plasma de rat Plasma de rat 10 ng/mL 89,6-94 %
(Al-Abd et al., 2009) FL Plasma de souris
Tissus de souris
Plasma de souris
Tissus de souris
25 ng/mL 52,4-95,2 %
(Dharmalingam et al.,
2014)
UV Plasma de rat / 1000 ng/mL 86,86-91,76 %
(Cox et al., 1991) FL Plasma canin
Tissus canins
/ 25 ng/mL
10 ng/mg
80-90 %
(Reddy et al., 2005) FL Sang total canin Sang total canin 12,38 ng/mL 95,7-100,4 %
Notre méthode FL Plasma canin Plasma canin 10 ng/mL 98,9-104,4 %
CLD = détection par chimiluminescence, ECD = détection électrochimique, FL = fluorescence, MS/MS =
spectrométrie de masse, U-HPLC = ultra-HPLC, UV = ultra-violet.

127
Notre méthode analytique développée est une des seules avec celle de Cox et al. (1991) à
utiliser du plasma de chien comme matrice pour validation. La plupart du temps, les études
de pharmacocinétique chez le chien sont basées sur des méthodes analytiques n’utilisant
pas de plasma canin pour leur calibration.
Plusieurs des méthodes présentées dans le Tableau XIII sont basées sur des méthodes
d’extraction en phase solide ou d’extraction liquide/liquide, utilisant des solvants organiques
non polaires comme le dichlorométhane ou le chloroforme.
La doxorubicine est une base faible et pour une meilleure extraction, le pH doit être ajusté à
un pH alcalin mais la doxorubicine est instable sous des conditions alcalines. De plus, utiliser
ces méthodes d’extraction pourrait constituer un risque d’extraction incomplète de la
molécule étant donné le nombre d’étapes et la haute volatilité des solvants utilisés. Notre
méthode utilise une technique simple et rapide de précipitation des protéines avec un
mélange d’acétonitrile et de phase mobile (v/v). La précipitation de protéines est considérée
comme la méthode d’extraction de choix à partir d’une matrice biologique (Loadman,
Calabrese, 2001). Notre méthode présente également un rendement d’extraction élevé
(98,9 à 104,4 %), similaire voire supérieur aux autres méthodes d’analyse. Cela indique
l’efficacité de la méthode d’extraction utilisée en plus de sa simplicité de mise en place de sa
rapidité et de son moindre coût.
Les coefficients de corrélation supérieurs à 0,998 pour des concentrations allant de 0,01 à
1,5 µg/mL indiquent une très bonne linéarité des courbes expérimentales. La limite de
quantification (0,01 µg/mL) de la méthode testée est similaire aux valeurs retrouvées dans la
littérature. Cependant, une meilleure sensitivité est obtenue pour des techniques de
détection basées sur la spectrométrie de masse et la détection électrochimique.
Plusieurs conditions de stabilité de la doxorubicine dans le plasma canin ont été testées et
sont similaires avec la littérature (Reddy, 2005). Malgré l’apparente stabilité de la
doxorubicine dans le plasma, les échantillons prélevés devraient tout de même être congelés
aussi vite que possible afin d’éviter des pertes et il est recommandé de réaliser leur analyse
dans les semaines suivants sa congélation.
La méthode d’analyse d’échantillons plasmatiques développée présente donc plusieurs
avantages, incluant la précision et l’exactitude des mesures réalisées. De plus, le matériel et
les infrastructures nécessaires pour réaliser cette méthode sont présents dans la plupart des
laboratoires de biologie et de chimie analytique et l’ensemble de la méthode peut être
réalisée en interne, sans nécessiter d’une formation supplémentaire.
Cette méthode d’analyse de la doxorubicine dans des échantillons plasmatiques une fois
développée peut être appliquée dans une étude de pharmacocinétique.

128
II. Approche non compartimentale de la pharmacocinétique et II. Approche non compartimentale de la pharmacocinétique et II. Approche non compartimentale de la pharmacocinétique et II. Approche non compartimentale de la pharmacocinétique et de la tolérance de la doxorubicine chez le chiende la tolérance de la doxorubicine chez le chiende la tolérance de la doxorubicine chez le chiende la tolérance de la doxorubicine chez le chien
A. Matériels et méthodesA. Matériels et méthodesA. Matériels et méthodesA. Matériels et méthodes
1. Recrutement des cas1. Recrutement des cas1. Recrutement des cas1. Recrutement des cas
Les échantillons plasmatiques proviennent d’animaux traités lors d’une séance de
chimiothérapie à base de doxorubicine à l’école vétérinaire de Lyon (VetAgro Sup, France).
Pour chaque chien, un consentement éclairé des propriétaires été obtenu. Ce projet
d’expérimentation clinique de pharmacocinétique a été approuvé par le Comité d’éthique en
expérimentation animale de VetAgro Sup (n°1648) (Annexe 2).
Un animal est inclus dans l’étude si celui-ci est traité par de la doxorubicine au cours de son
protocole de chimiothérapie. Pour chaque animal engagé dans l’étude, les données
suivantes issues de son dossier médical sont consignées : âge, poids, sexe, race, diagnostic
histologique de la tumeur et nombre total de séances de chimiothérapie à base de
doxorubicine. Une numération-formule sanguine, une analyse biochimique de base
(glycémie, urée, créatinine, phosphatase alcaline (PAl), alanine amino-transférase (AlAT),
protéines totales, albumine et globuline) et une échocardiographie sont réalisées avant la
séance de chimiothérapie.
Les animaux sont exclus de l’étude en cas d’insuffisance cardiaque (fraction de
raccourcissement inférieure à 25 %), rénale (présence d’une azotémie avec la créatinine >
180 µmol/L) ou hépatique (paramètres hépatiques augmentés avec AlAT > 210 UI/L et PAl >
600 UI/L) sévères.
Un suivi est réalisé à une semaine et trois semaines après la séance de chimiothérapie avec
un contrôle de la numération-formule sanguine. Dans les deux cas, un questionnaire sur
l’état de santé de l’animal ainsi que les éventuels effets indésirables (Annexe 3) est rempli
par le propriétaire et le clinicien.
2. Echantillonnage2. Echantillonnage2. Echantillonnage2. Echantillonnage
Les animaux sont traités par une perfusion de doxorubicine pendant environ 25 minutes, à
une dose de 30 mg/m² pour les chiens de plus de 10 kg et à une dose de 1 mg/m² pour les
chiens de moins de 10 kg.
Les échantillons sanguins sont collectés à 0, 5, 10, 30 minutes, 1, 2, 4, 8, 12, 16 et 24 heures
après le début de l’injection de la doxorubicine. A chaque temps d’échantillonnage, une
ponction de la veine jugulaire ou saphène externe est effectuée (site préalablement tondu et
désinfecté). Entre 2 et 3 mL de sang sont prélevés, à l’aide d’une seringue en plastique de 5
mL et d’une aiguille de 21 Gauges, et transférés dans un tube sous vide contenant de
l’héparinate de lithium. Les prélèvements sanguins sont ensuite centrifugés à

129
3000 tours/minute pendant 5 minutes puis le plasma est séparé par pipetage et transféré
dans un tube sec. Ces tubes sont identifiés et conservés par congélation à -21°C jusqu’à la
phase analytique.
Lors du suivi à une semaine, les chiens étaient de nouveau prélevés afin de déterminer la
présence éventuelle de résidus de doxorubicine.
3. Analyse des échantillons3. Analyse des échantillons3. Analyse des échantillons3. Analyse des échantillons
Les échantillons sont décongelés à température ambiante et analysés par HPLC-fluorescence
selon le protocole développé dans la partie I.
A partir des résultats obtenus, différents paramètres de pharmacocinétique ont été
déterminés selon les recommandations de l’Agence Européenne du Médicament (EMA,
2010) pour une étude non-compartimentale :
� La concentration plasmatique maximale (Cmax) de la doxorubicine dans le plasma.
� L’aire sous la courbe (AUC), calculée par intégration approchée grâce à la méthode
linéaire des trapèzes, L’AUC représente la biodisponibilité de la doxorubicine pour
un individu et une voie d’administration donnés.
� Le temps de demi-vie de distribution (T1/2), qui représente le temps nécessaire
pour que la concentration de doxorubicine du compartiment vasculaire diminue
de moitié, par distribution vers les autres compartiments de l’organisme.
� La clairance plasmatique (CL), qui correspond au volume plasmatique totalement
épuré de la doxorubicine par unité de temps au cours de la phase de distribution.
� Le volume de distribution du compartiment central (Vc), qui représente la capacité
de la doxorubicine à diffuser dans les différents tissus cibles.
B. RésultatsB. RésultatsB. RésultatsB. Résultats
1. Population d’étude et suivi clinique et biologique des animaux1. Population d’étude et suivi clinique et biologique des animaux1. Population d’étude et suivi clinique et biologique des animaux1. Population d’étude et suivi clinique et biologique des animaux
Huit chiens ont reçu une séance de chimiothérapie à base de doxorubicine pendant la
période de notre étude. Aucun d’entre eux ne présentaient de critères d’exclusion et ils ont
donc été inclus dans notre étude. L’ensemble des cas est référencé dans le Tableau XIV.
Les résultats des analyses effectuées lors de leur insertion dans le protocole d’étude ainsi
que le suivi hématologique des chiens sont présentés en Annexe 4.
Au cours du suivi des animaux, l’un d’entre eux décède avant le suivi à une semaine et l’un
d’entre eux est retiré de l’étude sur décision des propriétaires. De plus, deux d’entre eux ne
sont pas présent pour un suivi à une semaine.

130
Le suivi hématologique chez les quatre animaux restants montre la présence d’une
leucopénie sévère avec une neutropénie sévère de grade 3 et d’une légère
thrombocytopénie de grade 1 selon la classification VCOG-CTCAE chez l’un d’entre eux à une
semaine après la séance de chimiothérapie. Cet individu présentait déjà une leucopénie lors
de son admission dans l’étude et celle-ci est persistante au cours du suivi à trois semaines,
mais s’aggrave lors du contrôle à une semaine. Afin de limiter le risque de surinfection
bactérienne, une antibiothérapie à base d’amoxicilline et d’acide clavulanique (20 mg/kg PO
BID) pendant deux semaines est mis en place. De plus, l’un d’entre eux présente une
thrombocytopénie sévère de grade 3, et un autre une légère anémie de grade 1 selon la
classification VCOG-CTCAE une semaine après le traitement. L’ensemble de ces anomalies
est résolu à trois semaines post-chimiothérapie.
Figure 46 : Evolution du comptage des polynucléaires neutrophiles au cours des suivis chez
quatre chiens
Concernant les réponses aux questionnaires, certains effets secondaires à court-terme de la
doxorubicine ont été observés et sont présentés dans le Tableau XV, accompagnés des
mesures thérapeutiques mises en place.

Tableau XIV : Description des cas inclus dans notre étude
Nom Sexe Race Âge
(années) Poids (kg)
Surface
corporelle (m²) Diagnostic
Dose reçue
(mg)
Numéro de
séance à base de
doxorubicine
Thérapeutique
complémentaire
Rita Femelle non
stérilisée
Yorkshire
Terrier 12,1 4 0,25 Mésothéliome péricardique 4 1 Firocoxib
Alba Femelle non
stérilisée
Yorkshire
Terrier 11,9 4,6 0,27
Lymphome B diffus à
grandes cellules, variant
centroblastique
polymorphe
4,6 5 Prednisolone
Cooki Mâle entier Berger blanc
suisse 9,8 48,9 1,32
Hémangiosarcome
splénique 39,6 1 /
Ibie Femelle
stérilisée
Labrador
Retriever 3,7 22,8 0,8
Ostéosarcome
télangiectasique métastasé
de l’humérus droit
24 4 Firocoxib
Alma Femelle
stérilisée
Croisé
Berger
Allemand
11,9 30,4 0,96
Récidive de lymphome B
diffus à grandes cellules,
variant immunoblastique, de
stade initial IVa
28,8 1 Prednisolone
Cléo Femelle
stérilisée
Labrador
Retriever 13 31,9 0,99
Mélanome achromique
malin de la cavité orale de
stade T2N1M0
29,7 1 /
Sam Mâle castré Braque
hongrois 10,8 26,6 0,88
Récidive de lymphome B
diffus à grandes cellules,
variant immunoblastique, de
stade initial Va
26,4 1 /
Baya Femelle
stérilisée
Bouvier
Bernois 11,2 37,3 1,09
Carcinome épidermoïde
digital de stade T4N0M0 et
sarcome dermique de stade
T1N0M0
32,7 1 /
131

132
Tableau XV : Effets secondaires observés suite à la séance de chimiothérapie et thérapeutiques mises en place
2. Pharmacocinétique2. Pharmacocinétique2. Pharmacocinétique2. Pharmacocinétique
Après traitement des données brutes individuelles, les courbes pharmacocinétiques pour
chaque chien sont tracées, sans aucune modélisation (Figure 47).
Les différents paramètres pharmacocinétiques individuels sont alors calculés et présentés
dans le Tableau XVI.
Tableau XVI : Paramètres pharmacocinétiques calculés pour chaque individu de l’étude
Animal Posologie AUC
(nmol.h.L-1)
Cmax
(nmol.L-1)
T1/2
(min)
CL
(mL.min-1.kg-1)
Vc
(L.kg-1)
Rita 1 mg/kg 109,56 559,53 6,92 279,92 1,26
Alba 1 mg/kg 184,97 684,56 2,69 165,80 1,71
Cooki 30 mg/m² 141,40 424,21 6,46 175,62 1,70
Ibie 30 mg/m² 164,76 680,62 4,45 195,91 1,86
Alma 30 mg/m² 258,46 1097,17 5,26 112,40 1,12
Cléo 30 mg/m² 101,80 372,49 10,52 280,44 2,61
Sam 30 mg/m² 195,19 389,47 8,63 155,92 3,04
Baya 30 mg/m² 206,45 615,31 6,16 130,21 2,46
Moyenne 170,32 602,92 6,39 187,03 1,97
SD 52,32 235,92 2,42 62,99 0,67
CV 30,72 39,13 37,96 33,68 34,22
SD correspond à l’écart-type à la moyenne et CV est le coefficient de variation exprimé en pourcentage.
Effets secondaires Grade selon la
classification
VCOG-CTCAE
Nombre
d’occurrences
Thérapeutiques mises en place
Anorexie Grade 2 3/6 /
Léthargie Grade 1 1/6 /
Prurit sur zone
d’injection
Grade 1 1/6 /
Diarrhée Grade 1 1/6 Silicilate de magnésium et
d’aluminium Grade 2 1/6
Colite Grade 2 1/6 Silicilate de magnésium et
d’aluminium
Métronidazole 15 mg/kg PO BID
Vomissement Grade 1 1/6 Citrate de maropitant
2 mg/kg PO SID

133
Lors du suivi à une semaine, les quatre chiens présents ne présentaient pas de résidus
plasmatiques détectables de doxorubicine.
Figure 47 : Courbes de pharmacocinétique
3. Discussion 3. Discussion 3. Discussion 3. Discussion
a. Pharmacocinétique et ses implicationsa. Pharmacocinétique et ses implicationsa. Pharmacocinétique et ses implicationsa. Pharmacocinétique et ses implications
Cette étude réalisée chez des individus malades est une des seules de son genre. Les
paramètres pharmacocinétiques étudiés présentent une forte variabilité inter-individuelle
(entre 30,72 et 39,13 % de variabilité selon les paramètres), tout comme dans les autres
études sur des individus malades. Nos paramètres pharmacocinétiques sont du même ordre
de grandeur que ceux présentés par Gustafson et al., (2002) et par Selting et al., (2006)

134
même si notre concentration maximale Cmax est plus faible (Tableau XVII). Cela pourrait
être expliqué notamment par un temps de perfusion plus important. Cependant, de grandes
disparités subsistent entre les études, cette disparité pouvant être expliquée par l’important
volume de distribution (variant de 17,8 à 76 L/kg), signe d’une distribution très rapide au
sein des différents tissus de l’organisme. De plus, les différentes études, sur animal sain ou
malade, présentent un design différent en termes de dose administrée et de durée de la
perfusion (de dix à trente minutes). Une variation de la durée de perfusion influence la
concentration maximale atteinte, mais également l’AUC, pouvant alors expliquer une partie
des variations entre les études. La comparaison des études nécessite donc des précautions.
Tableau XVII : Comparatif des paramètres pharmacocinétiques des différentes études portant sur des animaux non sains
Notre étude
(n=8)
Gustafson et al., 2002
(n=4), lymphome
Selting et al., 2006
(n=9), lymphome
Dose administrée 30 mg/kg ou 1 mg/m² 30 mg/kg 30 mg/kg
Durée de la perfusion 25 minutes 20 minutes 20 minutes
AUC (nmol.h.L-1) 170,32 ± 52,32 66,7 ± 66,1 1103,20 ± 256,66
T1/2 (min) 6,39 ± 2,42 2,58 ± 0,78 /
Cmax (nmol.L-1) 602,92 ± 235,92 / 2135,01 ± 672,85
CL (mL.min-1.kg-1) 187,03 ± 62,99 51,66 ± 31,67 27,85 ± 4,68
Vc (L.kg-1) 1,97 ± 0,67 / /
Dans notre étude, la doxorubicine n’est plus dosable après la première heure, contrairement
aux autres études où la doxorubicine reste dosable jusqu’à 240 minutes. Cette différence
pourrait être due à une concentration plasmatique en doxorubicine proche de la limite de
détection et/ou de quantification, malgré une limite de détection comparable à celle des
autres méthodes de dosage de la doxorubicine. Il se pourrait également que la
concentration en doxorubicine soit influencée par les conditions de stockage des
échantillons plasmatiques, bien que pour certains échantillons, la durée de stockage n’ait
pas excédé les deux semaines et il semblerait donc que la dégradation de la doxorubicine
soit minime (cf partie I.A.4 étude de stabilité).
La diminution très rapide de la concentration en doxorubicine plasmatique nous empêche de
pouvoir observer avec évidence la phase d’élimination, et donc de calculer les paramètres
pharmacocinétiques inhérents à cette phase. Cette phase de distribution rapide permet tout
de même de confirmer l’intérêt d’une phase d’échantillonnage plus courte en se limitant à
une pharmacocinétique sur une heure chez le chien. Le coût et le refus par le propriétaire de
participer à une étude à cause de la procédure de prélèvement font partie des facteurs
restrictifs limitant les études sur de grandes populations en oncologie vétérinaire. Une étude
avec une phase de prélèvement limitée pourrait permettre, avec peu d’échantillons, de
prédire l’exposition globale à la doxorubicine et d’initier des études à grande échelle sur la
relation entre la pharmacocinétique et la pharmacodynamique (Wittenburg et al., 2014).

135
La dose de doxorubicine administrée ne semble pas influencer les paramètres
pharmacocinétiques de la doxorubicine. En effet, il n’y a pas de différence visible entre les
animaux recevant la dose de 1 mg/kg (AUCmoyen = 147,27 nmol.h.L-1, Cmax = 622,05 nmol.L-1)
et ceux traités à la dose de 30 mg/m² (soit une posologie entre 0,80 et 1 mg/m², AUCmoyen =
178,01 nmol.h.L-1, Cmax = 595,55 nmol.L-1). En médecine humaine, cette absence de
corrélation entre la dose de doxorubicine administrée et l’exposition (mesurée par l’AUC) est
également notée (Ackland et al., 1989).
Dans notre étude, les paramètres de pharmacocinétique calculés ne semblent pas être en
corrélation avec la toxicité observée. En effet, l’animal présentant la neutropénie au cours
du suivi présente une Cmax (389,47 nmol.L-1) plus faible que la moyenne des animaux de cette
étude et une exposition à la doxorubicine (AUC = 195,19 nmol.h.L-1) similaire ou inférieure à
celle d’autres animaux, ne présentant pas pour autant de signes de toxicité à court terme.
Cela va à l’encontre de certaines études en médecine humaine montrant une corrélation
entre l’exposition et la toxicité hématologique de la doxorubicine (Piscitelli et al., 1993).
Cependant, la relation entre exposition et toxicité à tendance à être remise en cause par des
études plus récentes (Joerger et al., 2007).
Le doxorubicinol, principal métabolite de la doxorubicine, n’est pas dosable dans notre
étude, malgré son extraction possible (cf partie I.A). Peu d’études discutent de la
pharmacocinétique du doxorubicinol et dans la plupart des cas, le doxorubicinol était
directement injecté à haute dose (30 mg/m²), donnant une concentration plasmatique bien
plus importante que lors de l’injection de doxorubicine et sa métabolisation (Gilbert et al.,
2005). Dans une étude plus ancienne, la concentration en doxorubicinol mesuré à cinq
minutes après injection de doxorubicine à 1,27 mg/kg (soit 28,4mg/m²) est de 10 ng/mL.
Cette valeur n’a pu être mesurée dans notre étude et n’est retrouvée que sporadiquement
chez le chien dans une étude (Cox et al., 1991).
b. Limitations du protob. Limitations du protob. Limitations du protob. Limitations du protocole d’étudecole d’étudecole d’étudecole d’étude
Tout protocole expérimental comporte des limites. Nous tenterons de souligner les points
qui nous semblent critiquables, permettant une meilleure interprétation de notre étude.
� Représentativité de l’échantillonnage
Notre étude repose sur une population de petite taille (huit individus), empêchant la
réalisation d’une analyse statistique et nous limitant à une analyse purement descriptive. De
plus, la population est très hétérogène avec un âge moyen de 10 ans et demi (allant de 3 ans
et 8 mois à 12 ans et 1 mois) pour un poids moyen de 25,8 kg (allant de 4 à 39,6 kg), de races
différentes et de types tumoraux différents. Cette très grande hétérogénéité est inhérente à
notre choix de travailler sur des animaux malades. Cela pourrait expliquer la grande
variabilité inter-individuelle.

136
L’hétérogénéité de type tumoral influence la distribution de la doxorubicine, dépendant de
la perfusion, de la vascularisation, de la concentration en protéines de liaison et en ADN
pourrait également expliquer partiellement la grande variabilité inter-individuelle
concernant les paramètres pharmacocinétiques.
� Design de l’étude
Notre protocole d’administration de la doxorubicine influence également les paramètres
pharmacocinétiques. Une durée d’administration plus longue résulte en une AUC plus
importante avec une Cmax plus faible (Gustafson et al., 2002). Dans notre étude, les chiens
étaient traités sur 25 minutes pour permettre la normalisation de l’échantillonnage. On
observe également une certaine variabilité pour les temps d’échantillonnage, dépendant des
différents patients et des cliniciens.
c. Perspectives de l’étudec. Perspectives de l’étudec. Perspectives de l’étudec. Perspectives de l’étude L’utilisation de la posologie en se basant sur la surface corporelle (mg/m²) remonte à des
études des années 50 montrant une normalisation de la dose maximale tolérée des agents
de chimiothérapie chez la souris, le rat, le chien et l’Homme (Pinkel, 1958). Cette posologie
calculée en utilisant la surface corporelle serait plus en corrélation avec les valeurs
physiologiques (vitesse de métabolisation, débit cardiaque, volume sanguin, clairance
plasmatique, etc…) que le poids de l’animal. En effet, le poids de l’animal représente un
facteur variant de 1 à 50 selon la race du chien, alors que sa surface corporelle passe d’un
facteur 1 à 15. Même si la surface corporelle présente une meilleure approche pour calculer
la dose, on constate chez le chien un manque de corrélation entre la posologie selon la
surface corporelle de la doxorubicine et l’efficacité ou la toxicité. Il est donc nécessaire
d’avoir une meilleure connaissance des causes de variabilité de la pharmacodynamique et la
relation avec la pharmacocinétique individu-dépendant. Cette approche utilisant la surface
corporelle est basée sur celle utilisée en médecine humaine pour le calcul de posologie.
Cependant, la surface corporelle est une mesure calculée, estimée et non mesurée et
dépendant de la formule employée. De plus, cette méthode ne tient pas compte des
particularités raciales et physiologiques, l’espèce canine présentant une large variation de
morphologie influençant la surface corporelle. Plusieurs facteurs contribuent à
l’augmentation de la concentration totale en doxorubicine, incluant la distribution aux
différents tissus, le métabolisme et l’excrétion. Tous ces processus devraient être corrélés à
la surface corporelle si l’on voulait que cette méthode de calcul de posologie soit normalisée
à la toxicité et à l’exposition à la doxorubicine dans tous les cas. Cependant, le métabolisme
contribue grandement à la variation de la toxicité de la doxorubicine lorsque la posologie se
base sur la surface corporelle. Par exemple, la doxorubicine est en partie métabolisée par
l’aldo-céto réductase pour former le doxorubicinol. Cette enzyme présente une affinité et un
taux d’activité maximal différents pour la doxorubicine selon l’espèce et les tissus (Loveless
et al., 1978). Le manque de corrélation entre le métabolisme de la doxorubicine et la surface
corporelle est illustré par le fait que l’affinité de la forme humaine est plus importante que

137
celle de la forme canine mais que le taux d’activité maximal est inférieur. Ainsi les fortes
disparités du métabolisme ne permettent pas d’envisager de prendre la surface corporelle
pour déterminer la posologie et il semble préférable de privilégier une posologie liée au
poids corporel. A cet égard, malgré la faible taille de l’échantillon, l’absence de différence
entre les petits et les grands chiens dans notre étude (qui reçoivent une posologie
comparable en mg/kg de poids corporel) confirme la validité de cette approche.
Dans ce cadre-là, le concept de dosage de la doxorubicine basé sur la pharmacocinétique
semble logique. Bien que cette approche ait démontré des améliorations par rapport à
l’usage de la surface corporelle chez l’homme dans certains cas, elle n’est que rarement
utilisée car la relation entre l’exposition du patient à la molécule et son effet n’est pas
élucidé pour toutes les molécules de chimiothérapie. De plus, cette relation est encore
moins développée en médecine vétérinaire. Bien que certaines études montrent une
corrélation entre la toxicité et le devenir du patient, notamment dans le cadre du lymphome
canin, aucune n’évalue l’exposition à la doxorubicine avec le devenir du patient (Sorenmo et
al., 2010). Ces études démontrent une survie prolongée chez les patients ayant développé
une neutropénie de grade III ou IV après la chimiothérapie, preuve d’une meilleure
exposition. Cependant, le critère utilisé en médecine vétérinaire pour le réajustement de la
dose est une réduction de la dose de 20 % lors d’une neutropénie de stade III ou plus, ces
réductions étant la plupart du temps instaurées en permanence pour éviter une future
toxicité (Vaughan et al., 2007). A l’inverse, il est rare d’effectuer une augmentation de la
dose pour des patients ne présentant pas d’effets secondaires indésirables à la dose
standard. Certaines études suggèrent l’importance de la posologie dans les lymphomes
canins, mais avec un ajustement thérapeutique individuel basé sur la tolérance maximale
propre à chaque individu. Cependant, certaines limites à cette méthode existent concernant
la doxorubicine, notamment l’importante variabilité intra-individuelle des paramètres
pharmacocinétiques pour la doxorubicine, celle-ci variant de 6 à 59 % selon les études en
médecine humaine (Jacquet et al., 1990), ce que tend à confirmer notre étude. D’autres
études sont donc nécessaires mais il pourrait être intéressant de réaliser une adaptation
thérapeutique personnalisée à chaque patient, limitant la toxicité de la doxorubicine tout en
maintenant une efficacité thérapeutique maximale. L’utilisation de cette méthode pour
déterminer la posologie idéale de la doxorubicine pour chaque individu serait d’autant plus
intéressante dans le cas des chiens présentant une mutation du gène mdr1, ceux-ci
présentant une très grande sensibilité à la doxorubicine. Une étude de 2010 a déterminé
que la concentration idéale pour traiter un chien homozygote muté serait de 10,7 ± 5,9
mg/m² pour éviter la toxicité hématologique et gastro-intestinale (Gustafson, Thamm, 2010).
Le développement de l’étude de la pharmacocinétique pourrait donc permettre d’identifier
des facteurs de risque ou d’individualiser l’approche thérapeutique en médecine vétérinaire.
Une gestion améliorée de la posologie des agents anti-cancéreux pourrait aider à réduire la
toxicité de ces derniers. Dans ce sens, notre étude décrit certains paramètres de la
pharmacocinétique de la doxorubicine administrée par voie intraveineuse chez les chiens

138
non-sains et peut être une base pour un travail futur sur l’amélioration des stratégies
thérapeutiques en cancérologie, incluant, par exemple, le développement des formes
liposomales et des nanoparticules en médecine vétérinaire.
Plusieurs articles évoquent le sujet et suggèrent une meilleure diffusion, avec une bonne
tolérance des particules. C’est notamment le cas des nano-particules de chitosane greffé
avec du glycol ou des folates (Alavi et al., 2017) (Motiei, Kashanian, 2017). Le
développement de cette technologie permet une réduction des doses de doxorubicine tout
en ayant une meilleure diffusion, et une moindre exposition du propriétaire et de
l’environnement. On observe une concentration en doxorubicine accrue dans les cellules,
voire un ciblage des cellules tumorales grâce à l’enhanced permeability and retention effect
ou au greffage de groupements de type folate (Wu et al., 2017). Il y a également une
amélioration de la persistance dans les cellules tumorales en comparaison avec les autres
types cellulaires, avec une modification de l’AUC (Ren et al., 2016). L’association avec des
complexes moléculaires au sein de ces particules peut également augmenter l’efficacité
combinée et donc permettre de réduire les doses et les effets toxiques, comme lors de la
complexation avec l’IL-2 (Wu et al., 2017). Il est également possible que l’utilisation de ces
liposomes pegylés permettent de dépasser certaines résistances tumorales, notamment
celle due à la glycoprotéine P.
L’étude de ces nouvelles technologies nécessite cependant une connaissance de la
pharmacocinétique de la doxorubicine chez le chien malade afin de comparer l’efficacité et
l’innocuité de ces nouvelles formes de délivrance avec la forme non encapsulée de la
doxorubicine. En ce sens, notre étude apporte des éléments sur la cinétique de la forme non
encapsulée et fournit ainsi une base de comparaison pour le développement de projets
ultérieurs.

139
ConclusionConclusionConclusionConclusion
La doxorubicine est un agent anticancéreux employé depuis des décennies en oncologie
humaine et vétérinaire dans le cadre du traitement de divers types tumoraux.
Le plus souvent classée comme inhibiteur des ADN topoisomérases de type II ou comme
agent intercalant, la doxorubicine présente également une réactivité chimique avec le
dioxygène cellulaire, à l’origine de la synthèse de dérivés réactifs de l’oxygène. Ces dérivés
provoquent un stress oxydatif cellulaire, causant la peroxydation des lipides constituant les
membranes cellulaires, des dommages à l’ADN ainsi l’oxydation de nombreuses protéines
cytoplasmiques. Tous ces mécanismes activent les différentes voies de mort cellulaire :
l’apoptose, l’autophagie, la sénescence et la nécrose cellulaire. L’orientation vers l’une ou
l’autre de ces voies semble dépendre de la concentration cellulaire en doxorubicine et donc
de sa pharmacocinétique.
Certaines cellules tumorales peuvent échapper aux différents mécanismes de cytotoxicité
de la doxorubicine, ce qui est à l’origine d’une résistance de la tumeur à la chimiothérapie.
Cette résistance passe parfois également par une modification de la pharmacocinétique de
la doxorubicine en augmentant son efflux cellulaire par surexpression de la glycoprotéine-P.
Hormis son implication dans la cytotoxicité des cellules tumorales, la pharmacocinétique
de la doxorubicine permet également d’expliquer une partie de sa toxicité concernant les
cellules non tumorales, et principalement sa toxicité cardiaque dose-dépendante par une
distribution importante de la doxorubicine dans le tissu cardiaque, expliquée par sa forte
affinité pour les cardiolipides.
Pour autant, il existe peu d’études sur la pharmacocinétique de la doxorubicine chez le
chien sain ou malade permettant d’adapter la posologie aux besoins thérapeutiques de
l’animal.
Notre étude permet de dessiner une vue d’ensemble de la pharmacocinétique de la
doxorubicine chez l’animal malade. Ceci a été réalisé grâce à la mise en place d’une méthode
analytique validée de dosage de la doxorubicine plasmatique chez le chien et à son
application sur une population de patients non sains. Tout comme chez l’homme, la
variabilité inter-individuelle est importante et la corrélation entre la pharmacocinétique et la
toxicité générale à court terme de la doxorubicine ne semble pas être établie.

140
La poursuite de cette étude en impliquant une population plus importante est nécessaire
pour déterminer d’éventuels paramètres pharmacocinétiques pouvant servir de facteur de
prédiction des effets indésirables de la doxorubicine chez les patients et d’ajuster au mieux
la posologie au patient. Thèse de M CHAVALLE Thomas Le Professeur responsable Le Directeur général VetAgro Sup campus vétérinaire VetAgro Sup Philippe BERNY
Le Président de la thèse
Professeur Serge Lebecque
Vu et permis d’imprimer Lyon, le Pour Le Président de l’Université, Le Président du Comité de Coordination des Etudes Médicales Professeur Pierre COCHAT

141
BiblioBiblioBiblioBibliographiegraphiegraphiegraphie
ABRAHAM S.A., WATERHOUSE D.N., MAYER L.D., CULLIS P.R., MADDEN T.D. et BALLY M.B. The liposomal formulation of doxorubicin. Methods in Enzymology. 2005. Vol. 391, pp. 71�97.
ACKLAND S.P., RATAIN M.J., VOGELZANG N.J., CHOI K.E., RUANE M. et SINKULE J.A. Pharmacokinetics and pharmacodynamics of long-term continuous-infusion doxorubicin. Clinical Pharmacology and
Therapeutics. 1989. Vol. 45, n° 4, pp. 340�347.
ADAMS J. The proteasome: structure, function, and role in the cell. Cancer Treatment Reviews. 2003. Vol. 29 Suppl 1, pp. 3�9.
AHMED S., KISHIKAWA N., OHYAMA K., WADA M., NAKASHIMA K. et KURODA N. Selective determination of doxorubicin and doxorubicinol in rat plasma by HPLC with photosensitization reaction followed by chemiluminescence detection. Talanta. 2009. Vol. 78, n° 1, pp. 94�100.
AIRLEY R., 2009. Chapter 8 - Classical anticancer agents. In : Cancer Chemotherapy : Basic Science to
the Clinic. John Wiley & Sons. pp. 67�116.
AL-ABD A.M., KIM N.H., SONG S.-C., LEE S.J. et KUH H.-J. A simple HPLC method for doxorubicin in plasma and tissues of nude mice. Archives of Pharmacal Research. 2009. Vol. 32, n° 4, pp. 605�611.
ALAVI M., KARIMI N. et SAFAEI M. Application of Various Types of Liposomes in Drug Delivery Systems. Advanced Pharmaceutical Bulletin. 2017. Vol. 7, n° 1, pp. 3�9.
ALVAREZ-CEDRÓN L., SAYALERO M.L. et LANAO J.M. High-performance liquid chromatographic validated assay of doxorubicin in rat plasma and tissues. Journal of Chromatography. B, Biomedical
Sciences and Applications. 1999. Vol. 721, n° 2, pp. 271�278.
ALVES A.C., MAGARKAR A., HORTA M., LIMA J.L.F.C., BUNKER A., NUNES C. et al. Influence of doxorubicin on model cell membrane properties: insights from in vitro and in silico studies. Scientific
Reports. 2017. Vol. 7.
AMINI S. et KAJI A. Association of pp36, a phosphorylated form of the presumed target protein for the src protein of Rous sarcoma virus, with the membrane of chicken cells transformed by Rous sarcoma virus. Proceedings of the National Academy of Sciences of the United States of America. 1983. Vol. 80, n° 4, pp. 960�964.
APPEL J.M., NIELSEN D., ZERAHN B., JENSEN B.V. et SKAGEN K. Anthracycline-induced chronic cardiotoxicity and heart failure. Acta Oncologica. 2007. Vol. 46, n° 5, pp. 576�580.
ARANCIA G., CALCABRINI A., MESCHINI S. et MOLINARI A. Intracellular distribution of anthracyclines in drug resistant cells. Cytotechnology. 1998. Vol. 27, n° 1�3, pp. 95�111.
ARAVIND L., LEIPE D.D. et KOONIN E.V. Toprim--a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids
Research. 1998. Vol. 26, n° 18, pp. 4205�4213.
ARCAMONE F., CASSINELLI G., FANTINI G., GREIN A., OREZZI P., POL C. et al. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnology and
Bioengineering. 1969. Vol. 11, n° 6, pp. 1101�1110.

142
ARNOLD R.D., SLACK J.E. et STRAUBINGER R.M. Quantification of Doxorubicin and metabolites in rat plasma and small volume tissue samples by liquid chromatography/electrospray tandem mass spectroscopy. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life
Sciences. 2004. Vol. 808, n° 2, pp. 141�152.
ARRINGTON K.A., LEGENDRE A.M., TABELING G.S. et FRAZIER D.L. Comparison of body surface area-based and weight-based dosage protocols for doxorubicin administration in dogs. American Journal
of Veterinary Research. 1994. Vol. 55, n° 11, pp. 1587�1592.
ASHKENAZI A. et HERBST R.S. To kill a tumor cell: the potential of proapoptotic receptor agonists. The
Journal of Clinical Investigation. 2008. Vol. 118, n° 6, pp. 1979�1990.
ATKINS C., BONAGURA J., ETTINGER S., FOX P., GORDON S., HAGGSTROM J. et al. Guidelines for the Diagnosis and Treatment of Canine Chronic Valvular Heart Disease. Journal of Veterinary Internal
Medicine. 2009. Vol. 23, n° 6, pp. 1142�1150.
AYALA A., MUÑOZ M.F. et ARGÜELLES S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Medicine and Cellular
Longevity. 2014. Vol. 2014, pp. 360438.
BACHUR N.R., YU F., JOHNSON R., HICKEY R., WU Y. et MALKAS L. Helicase inhibition by anthracycline anticancer agents. Molecular Pharmacology. 1992. Vol. 41, n° 6, pp. 993�998.
BAKKER M., GRAAF W.T.A.V.D., GROEN H.J.M., SMIT E.F. et VRIES E.G.E.D. Anthracyclines - Pharmacology and resistance, a review. ResearchGate. 1995. Vol. 1, n° 1, pp. 133�144.
BARRANCO S.C. Cellular and molecular effects of adriamycin on dividing and nondividing cells. Pharmacology & Therapeutics. 1984. Vol. 24, n° 3, pp. 303�319.
BATES D.A. et WINTERBOURN C.C. Deoxyribose breakdown by the adriamycin semiquinone and H2O2: evidence for hydroxyl radical participation. FEBS letters. 1982. Vol. 145, n° 1, pp. 137�142.
BEAVER L.M., STROTTNER G. et KLEIN M.K. Response rate after administration of a single dose of doxorubicin in dogs with B-cell or T-cell lymphoma: 41 cases (2006-2008). Journal of the American
Veterinary Medical Association. 2010. Vol. 237, n° 9, pp. 1052�1055.
BENCHEKROUN M.N. et ROBERT J. Measurement of doxorubicin-induced lipid peroxidation under the conditions that determine cytotoxicity in cultured tumor cells. Analytical Biochemistry. 1992. Vol. 201, n° 2, pp. 326�330.
BERGMAN P.J. Mechanisms of anticancer drug resistance. Veterinary Clinics: Small Animal Practice. 2003. Vol. 33, n° 3, pp. 651�667.
BERGMAN P.J., OGILVIE G.K. et POWERS B.E. Monoclonal antibody C219 immunohistochemistry against P-glycoprotein: sequential analysis and predictive ability in dogs with lymphoma. Journal of
Veterinary Internal Medicine. 1996. Vol. 10, n° 6, pp. 354�359.
BERNSTEIN D., FAJARDO G. et ZHAO M. The role of β-adrenergic receptors in heart failure : differential regulation of cardiotoxicity and cardioprotection. Progress in pediatric cardiology. 2011. Vol. 31, n° 1, pp. 35�38.
BERS D.M. Calcium and cardiac rhythms: physiological and pathophysiological. Circulation Research. 2002. Vol. 90, n° 1, pp. 14�17.

143
BERTHIAUME J.M. et WALLACE K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell
Biology and Toxicology. 2007. Vol. 23, n° 1, pp. 15�25.
BIRENBAUM A., TESSE A., LOYER X., MICHELET P., ANDRIANTSITOHAINA R., HEYMES C. et al. Involvement of beta 3-adrenoceptor in altered beta-adrenergic response in senescent heart: role of nitric oxide synthase 1-derived nitric oxide. Anesthesiology. 2008. Vol. 109, n° 6, pp. 1045�1053.
BOOSER D.J. et HORTOBAGYI G.N. Anthracycline antibiotics in cancer therapy. Focus on drug resistance. Drugs. 1994. Vol. 47, n° 2, pp. 223�258.
BOSANQUET A.G. Stability of solutions of antineoplastic agents during preparation and storage for in vitro assays. Cancer Chemotherapy and Pharmacology. 1986. Vol. 17, n° 1, pp. 1�10.
BOUMA J., BEIJNEN J.H., BULT A. et UNDERBERG W.J. Anthracycline antitumour agents. A review of physicochemical, analytical and stability properties. Pharmaceutisch Weekblad. Scientific Edition. 1986. Vol. 8, n° 2, pp. 109�133.
BRAZZOLOTTO X., ANDRIOLLO M., GUIRAUD P., FAVIER A. et MOULIS J.-M. Interactions between doxorubicin and the human iron regulatory system. Biochimica Et Biophysica Acta. 2003. Vol. 1593, n° 2�3, pp. 209�218.
BREIMER L.H. Repair of DNA damage induced by reactive oxygen species. Free Radical Research
Communications. 1991. Vol. 14, n° 3, pp. 159�171.
BROWN G.A., MCPHERSON J.P., GU L., HEDLEY D.W., TOSO R., DEUCHARS K.L. et al. Relationship of DNA Topoisomerase IIα and β Expression to Cytotoxicity of Antineoplastic Agents in Human Acute Lymphoblastic Leukemia Cell Lines. Cancer Research. 1995. Vol. 55, n° 1, pp. 78�82.
CAIRO G., RECALCATI S., PIETRANGELO A. et MINOTTI G. The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Radical Biology & Medicine. 2002. Vol. 32, n° 12, pp. 1237�1243.
CHABNER, BA et LONGO, DL. Cancer Chemotherapy and Biotherapy: Principles and Practice. Lippincott Williams & Wilkins. 2011.
CHAIRES J.B., DATTAGUPTA N. et CROTHERS D.M. Kinetics of the daunomycin-DNA interaction. Biochemistry. 1985. Vol. 24, n° 2, pp. 260�267.
CHAMPOUX J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annual Review of
Biochemistry. 2001. Vol. 70, n° 1, pp. 369�413.
CHANEY E.N. et BALDWIN R.P. Voltammetric determination of doxorubicin in urine by adsorptive preconcentration and flow injection analysis. Analytica Chimica Acta. 1985. Vol. 176, pp. 105�112.
CHARVIN G. Etudes des topoisomérases de type II par micromanipulation d’ADN. Université Paris-Diderot - Paris VII. 2004
CHEN M.-B., WU X.-Y., GU J.-H., GUO Q.-T., SHEN W.-X. et LU P.-H. Activation of AMP-Activated Protein Kinase Contributes to Doxorubicin-Induced Cell Death and Apoptosis in Cultured Myocardial H9c2 Cells. Cell Biochemistry and Biophysics. 2011. Vol. 60, n° 3, pp. 311�322.
CHILDS A.C., PHANEUF S.L., DIRKS A.J., PHILLIPS T. et LEEUWENBURGH C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Research. 2002. Vol. 62, n° 16, pp. 4592�4598.

144
CHUN R., GARRETT L.D., HENRY C., WALL M., SMITH A. et AZENE N.M. Toxicity and efficacy of cisplatin and doxorubicin combination chemotherapy for the treatment of canine osteosarcoma. Journal of the American Animal Hospital Association. 2005. Vol. 41, n° 6, pp. 382�387.
CIRILLO R., SACCO G., VENTURELLA S., BRIGHTWELL J., GIACHETTI A. et MANZINI S. Comparison of doxorubicin- and MEN 10755-induced long-term progressive cardiotoxicity in the rat. Journal of
Cardiovascular Pharmacology. 2000. Vol. 35, n° 1, pp. 100�108.
CONRAD S., VIERTELHAUS A., ORZECHOWSKI A., HOOGSTRAATE J., GJELLAN K., SCHRENK D. et al. Sequencing and tissue distribution of the canine MRP2 gene compared with MRP1 and MDR1. Toxicology. 2001. Vol. 156, n° 2�3, pp. 81�91.
CORBETT K.D. et BERGER J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annual Review of Biophysics and Biomolecular Structure. 2004. Vol. 33, n° 1, pp. 95�118.
COX S.K., WILKE A.V. et FRAZIER D. Determination of adriamycin in plasma and tissue biopsies. Journal of Chromatography. 1991. Vol. 564, n° 1, pp. 322�329.
CULLINANE C. et PHILLIPS D.R. Induction of stable transcriptional blockage sites by Adriamycin: GpC specificity of apparent Adriamycin-DNA adducts and dependence on iron(III) ions. Biochemistry. 1990. Vol. 29, n° 23, pp. 5638�5646.
CUMMINGS J., ANDERSON L., WILLMOTT N. et SMYTH J.F. The molecular pharmacology of doxorubicin in vivo. European Journal of Cancer. 1991. Vol. 27, n° 5, pp. 532�535.
CUMMINGS J., BARTOSZEK A. et SMYTH J.F. Determination of covalent binding to intact DNA, RNA, and oligonucleotides by intercalating anticancer drugs using high-performance liquid chromatography. Studies with doxorubicin and NADPH cytochrome P-450 reductase. Analytical
Biochemistry. 1991. Vol. 194, n° 1, pp. 146�155.
CUTTS S.M., NUDELMAN A., REPHAELI A. et PHILLIPS D.R. The power and potential of doxorubicin-DNA adducts. IUBMB life. 2005. Vol. 57, n° 2, pp. 73�81.
CUTTS S.M., SWIFT L.P., REPHAELI A., NUDELMAN A. et PHILLIPS D.R. Sequence Specificity of Adriamycin-DNA Adducts in Human Tumor Cells. Molecular Cancer Therapeutics. 2003. Vol. 2, n° 7, pp. 661�670.
DATERS A.T., MAULDIN G.E., MAULDIN G.N., BRODSKY E.M. et POST G.S. Evaluation of a multidrug chemotherapy protocol with mitoxantrone based maintenance (CHOP-MA) for the treatment of canine lymphoma. Veterinary and Comparative Oncology. 2010. Vol. 8, n° 1, pp. 11�22.
DE BRUIJN P., VERWEIJ J., LOOS W.J., KOLKER H.J., PLANTING A.S., NOOTER K. et al. Determination of doxorubicin and doxorubicinol in plasma of cancer patients by high-performance liquid chromatography. Analytical Biochemistry. 1999. Vol. 266, n° 2, pp. 216�221.
DE JONG J., GUÉRAND W.S., SCHOOFS P.R., BAST A. et VAN DER VIJGH W.J. Simple and sensitive quantification of anthracyclines in mouse atrial tissue using high-performance liquid chromatography and fluorescence detection. Journal of Chromatography. 1991. Vol. 570, n° 1, pp. 209�216.
DEKKER N.H., RYBENKOV V.V., DUGUET M., CRISONA N.J., COZZARELLI N.R., BENSIMON D. et al. The mechanism of type IA topoisomerases. Proceedings of the National Academy of Sciences of the
United States of America. 2002. Vol. 99, n° 19, pp. 126�131.

145
DELEMASURE S., VERGELY C., ZELLER M., COTTIN Y. et ROCHETTE L. Preventing the cardiotoxic effects of anthracyclins. From basic concepts to clinical data. Annales De Cardiologie Et D’angeiologie. 2006. Vol. 55, n° 2, pp. 104�112.
DELPY E., HATEM S.N., ANDRIEU N., DE VAUMAS C., HENAFF M., RÜCKER-MARTIN C. et al. Doxorubicin induces slow ceramide accumulation and late apoptosis in cultured adult rat ventricular myocytes. Cardiovascular Research. 1999. Vol. 43, n° 2, pp. 398�407.
DHARMALINGAM S.R., RAMAMURTHY S., CHIDAMBARAM K. et NADARAJU S. A Simple HPLC Bioanalytical Method for the Determination of Doxorubicin Hydrochloride in Rat Plasma: Application to Pharmacokinetic Studies. Tropical Journal of Pharmaceutical Research. 2014. Vol. 13, n° 3, pp. 409�415.
DI LISA F., KALUDERCIC N. et PAOLOCCI N. β₂-Adrenoceptors, NADPH oxidase, ROS and p38 MAPK: another « radical » road to heart failure? British Journal of Pharmacology. 2011. Vol. 162, n° 5, pp. 1009�1011.
DI MARCO A., CASSINELLI G. et ARCAMONE F. The discovery of daunorubicin. Cancer Treatment
Reports. 1981. Vol. 65 Suppl 4, pp. 3�8.
DI X., SHIU R.P., NEWSHAM I.F. et GEWIRTZ D.A. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to adriamycin. Biochemical
Pharmacology. 2009. Vol. 77, n° 7, pp. 1139�1150.
DICKENS M.L. et STROHL W.R. Isolation and characterization of a gene from Streptomyces sp. strain C5 that confers the ability to convert daunomycin to doxorubicin on Streptomyces lividans TK24. Journal of Bacteriology. 1996. Vol. 178, n° 11, pp. 3389�3395.
DIRKS-NAYLOR A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sciences. 2013.
DOROSHOW J.H. Role of hydrogen peroxide and hydroxyl radical formation in the killing of Ehrlich tumor cells by anticancer quinones. Proceedings of the National Academy of Sciences of the United
States of America. 1986. Vol. 83, n° 12, pp. 4514�4518.
DOROSHOW J.H., SYNOLD T.W., SOMLO G., AKMAN S.A. et GAJEWSKI E. Oxidative DNA base modifications in peripheral blood mononuclear cells of patients treated with high-dose infusional doxorubicin. Blood. 2001. Vol. 97, n° 9, pp. 2839�2845.
DRAKE F.H., HOFMANN G.A., BARTUS H.F., MATTERN M.R., CROOKE S.T. et MIRABELLI C.K. Biochemical and pharmacological properties of p170 and p180 forms of topoisomerase II. Biochemistry. 1989. Vol. 28, n° 20, pp. 8154�8160.
DUGUET M. et RIOU J.-F. De la topologie de l’ADN aux médicaments antibiotiques et anticancéreux.
Médecine sciences. 1994. Vol. 10, n° 10, pp. 962�972.
DUSRE L., MIMNAUGH E.G., MYERS C.E. et SINHA B.K. Potentiation of doxorubicin cytotoxicity by buthionine sulfoximine in multidrug-resistant human breast tumor cells. Cancer Research. 1989. Vol. 49, n° 3, pp. 511�515.
ECKFORD P.D.W. et SHAROM F.J. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: effects on ATPase activity, drug binding and transport. Biochemistry. 2008. Vol. 47, n° 51, pp. 13686�13698.

146
EDER J.P., CHAN V.T., NIEMIERKO E., TEICHER B.A. et SCHNIPPER L.E. Conditional expression of wild type topoisomerase II complements a mutant enzyme in mammalian cells. The Journal of Biological
Chemistry. 1993. Vol. 268, n° 19, pp. 13844�13849.
EDINGER A.L. et THOMPSON C.B. Death by design: apoptosis, necrosis and autophagy. Current
Opinion in Cell Biology. 2004. Vol. 16, n° 6, pp. 663�669.
ESCHALIER A., LAVARENNE J., BURTIN C., RENOUX M., CHAPUY E. et RODRIGUEZ M. Study of histamine release induced by acute administration of antitumor agents in dogs. Cancer
Chemotherapy and Pharmacology. 1988. Vol. 21, n° 3, pp. 246�250.
ESTERBAUER H. et CHEESEMAN K.H. Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods in Enzymology. 1990. Vol. 186, pp. 407�421.
EVANS M.D., DIZDAROGLU M. et COOKE M.S. Oxidative DNA damage and disease: induction, repair and significance. Mutation Research. 2004. Vol. 567, n° 1, pp. 1�61.
FAJARDO G., ZHAO M., POWERS J. et BERNSTEIN D. Differential cardiotoxic/cardioprotective effects of beta-adrenergic receptor subtypes in myocytes and fibroblasts in doxorubicin cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2006. Vol. 40, n° 3, pp. 375�383.
FEOKTISTOVA M. et LEVERKUS M. Programmed necrosis and necroptosis signalling. FEBS Journal. 2015. Vol. 282, n° 1, pp. 19�31.
FERNANDES J. et GATTASS C.R. Topological Polar Surface Area Defines Substrate Transport by Multidrug Resistance Associated Protein 1 (MRP1/ABCC1). Journal of Medicinal Chemistry. 2009. Vol. 52, n° 4, pp. 1214�1218.
FIALLO M.M., GARNIER-SUILLEROT A., MATZANKE B. et KOZLOWSKI H. How Fe3+ binds anthracycline antitumour compounds. The myth and the reality of a chemical sphinx. Journal of Inorganic
Biochemistry. 1999. Vol. 75, n° 2, pp. 105�115.
FIGUEIREDO-PEREIRA M.E., CHEN W.E., LI J. et JOHDO O. The antitumor drug aclacinomycin A, which inhibits the degradation of ubiquitinated proteins, shows selectivity for the chymotrypsin-like activity of the bovine pituitary 20 S proteasome. The Journal of Biological Chemistry. 1996. Vol. 271, n° 28, pp. 16455�16459.
FITZPATRICK W.M., DERVISIS N.G. et KITCHELL B.E. Safety of concurrent administration of dexrazoxane and doxorubicin in the canine cancer patient. Veterinary and Comparative Oncology. 2010. Vol. 8, n° 4, pp. 273�282.
FLORY A.B., RASSNICK K.M., ERB H.N., GARRETT L.D., NORTHRUP N.C., SELTING K.A. et al. Evaluation of factors associated with second remission in dogs with lymphoma undergoing retreatment with a cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy protocol: 95 cases (2000-2007). Journal of the American Veterinary Medical Association. 2011. Vol. 238, n° 4, pp. 501�506.
FRÉZARD F. et GARNIER-SUILLEROT A. Permeability of lipid bilayer to anthracycline derivatives. Role of the bilayer composition and of the temperature. Biochimica Et Biophysica Acta. 1998. Vol. 1389, n° 1, pp. 13�22.
FRIMBERGER A.E., CHAN C.M. et MOORE A.S. Canine Osteosarcoma Treated by Post-Amputation Sequential Accelerated Doxorubicin and Carboplatin Chemotherapy: 38 Cases. Journal of the
American Animal Hospital Association. 2016. Vol. 52, n° 3, pp. 149�156.

147
FRY M. et GREEN D.E. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. The Journal of Biological Chemistry. 1981. Vol. 256, n° 4, pp. 1874�1880.
GABURJAKOVA M., BAL N.C., GABURJAKOVA J. et PERIASAMY M. Functional interaction between calsequestrin and ryanodine receptor in the heart. Cellular and molecular life sciences: CMLS. 2013. Vol. 70, n° 16, pp. 2935�2945.
GALLAY-LEPOUTRE J., BÉLANGER M.C. et NADEAU M.E. Prospective evaluation of Doppler echocardiography, tissue Doppler imaging and biomarkers measurement for the detection of doxorubicin-induced cardiotoxicity in dogs: A pilot study. Research in Veterinary Science. 2016. Vol. 105, pp. 153�159.
GALLOIS L., FIALLO M., LAIGLE A., PRIEBE W. et GARNIER-SUILLEROT A. The Overall Partitioning of Anthracyclines into Phosphatidyl-Containing Model Membranes Depends Neither on the Drug Charge Nor the Presence of Anionic Phospholipids. European Journal of Biochemistry. 1996. Vol. 241, n° 3, pp. 879�887.
GAMBLIEL H.A., BURKE B.E., CUSACK B.J., WALSH G.M., ZHANG Y.L., MUSHLIN P.S. et al. Doxorubicin and C-13 deoxydoxorubicin effects on ryanodine receptor gene expression. Biochemical and
Biophysical Research Communications. 2002. Vol. 291, n° 3, pp. 433�438.
GANAPATHI R.N. et GANAPATHI M.K. Mechanisms regulating resistance to inhibitors of topoisomerase II. Frontiers in Pharmacology. 2013. Vol. 4, pp. 89.
GARDNER P.R., RAINERI I., EPSTEIN L.B. et WHITE C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. The Journal of Biological Chemistry. 1995. Vol. 270, n° 22, pp. 13399�13405.
GARNER A.P., PAINE M.J., RODRIGUEZ-CRESPO I., CHINJE E.C., ORTIZ DE MONTELLANO P., STRATFORD I.J. et al. Nitric oxide synthases catalyze the activation of redox cycling and bioreductive anticancer agents. Cancer Research. 1999. Vol. 59, n° 8, pp. 1929�1934.
GAUTHIER C., LEBLAIS V., KOBZIK L., TROCHU J.N., KHANDOUDI N., BRIL A. et al. The negative inotropic effect of β3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. The Journal of Clinical Investigation. 1998. Vol. 102, n° 7, pp. 1377�1384.
GAVENDA A., ŠEVČÍK J., PSOTOVÁ J., BEDNÁŘ P., BARTÁK P., ADAMOVSKÝ P. et al. Determination of anthracycline antibiotics doxorubicin and daunorubicin by capillary electrophoresis with UV absorption detection. ELECTROPHORESIS. 2001. Vol. 22, n° 13, pp. 2782�2785.
GERLIER D., ALAIS S. et CHAZAL N. Les radeaux membranaires : des plates-formes de choix pour l’entrée, l’assemblage ou le bourgeonnement de virus. Virologie. 2004. Vol. 8, n° 3, pp. 199�214.
GERVASI P.G., AGRILLO M.R., CITTI L., DANESI R. et DEL TACCA M. Superoxide anion production by adriamycinol from cardiac sarcosomes and by mitochondrial NADH dehydrogenase. Anticancer
Research. 1986. Vol. 6, n° 5, pp. 1231�1235.
GEWIRTZ D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochemical Pharmacology. 1999. Vol. 57, n° 7, pp. 727�741.
GEWIRTZ D.A. The four faces of autophagy: implications for cancer therapy. Cancer Research. 2014. Vol. 74, n° 3, pp. 647�651.

148
GEWIRTZ D.A., HOLT S.E. et ELMORE L.W. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochemical Pharmacology. 2008. Vol. 76, n° 8, pp. 947�957.
GEWIRTZ D.A. et YANOVICH S. Metabolism of adriamycin in hepatocytes isolated from the rat and the rabbit. Biochemical Pharmacology. 1987. Vol. 36, n° 11, pp. 1793�1798.
GHARIB M.I. et BURNETT A.K. Chemotherapy-induced cardiotoxicity: current practice and prospects of prophylaxis. European Journal of Heart Failure. 2002. Vol. 4, n° 3, pp. 235�242.
GIANNI L., ZWEIER J.L., LEVY A. et MYERS C.E. Characterization of the cycle of iron-mediated electron transfer from Adriamycin to molecular oxygen. The Journal of Biological Chemistry. 1985. Vol. 260, n° 11, pp. 6820�6826.
GILBERT C.M., FILIPPICH L.J., MCGEARY R.P. et CHARLES B.G. Pharmacokinetics of doxorubicinol in dogs. Journal of Veterinary Pharmacology and Therapeutics. 2006. Vol. 29, n° 5, pp. 433�435.
GILBERT C.M., MCGEARY R.P., FILIPPICH L.J., NORRIS R.L.G. et CHARLES B.G. Simultaneous liquid chromatographic determination of doxorubicin and its major metabolite doxorubicinol in parrot plasma. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 2005. Vol. 826, n° 1�2, pp. 273�276.
GILLIAM L.A.A., FISHER-WELLMAN K.H., LIN C.-T., MAPLES J.M., CATHEY B.L. et NEUFER P.D. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free radical biology & medicine. 2013. Vol. 65.
GILLINGS S., JOHNSON J., FULMER A. et HAUCK M. Effect of a 1-hour IV infusion of doxorubicin on the development of cardiotoxicity in dogs as evaluated by electrocardiography and echocardiography. Veterinary Therapeutics: Research in Applied Veterinary Medicine. 2009. Vol. 10, n° 1�2, pp. 46�58.
GINN P.E. Immunohistochemical detection of P-glycoprotein in formalin-fixed and paraffin-embedded normal and neoplastic canine tissues. Veterinary Pathology. 1996. Vol. 33, n° 5, pp. 533�541.
GOODMAN J. et HOCHSTEIN P. Generation of free radicals and lipid peroxidation by redox cycling of adriamycin and daunomycin. Biochemical and Biophysical Research Communications. 1977. Vol. 77, n° 2, pp. 797�803.
GOORMAGHTIGH E., CHATELAIN P., CASPERS J. et RUYSSCHAERT J.M. Evidence of a complex between adriamycin derivatives and cardiolipin: possible role in cardiotoxicity. Biochemical Pharmacology. 1980. Vol. 29, n° 21, pp. 3003�3010.
GOORMAGHTIGH E., HUART P., PRAET M., BRASSEUR R. et RUYSSCHAERT J.M. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophysical Chemistry. 1990. Vol. 35, n° 2�3, pp. 247�257.
GREEN P.S. et LEEUWENBURGH C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochimica Et Biophysica Acta. 2002. Vol. 1588, n° 1, pp. 94�101.
GRIMSRUD P.A., XIE H., GRIFFIN T.J. et BERNLOHR D.A. Oxidative Stress and Covalent Modification of Protein with Bioactive Aldehydes. The Journal of Biological Chemistry. 2008. Vol. 283, n° 32, pp. 21837�21841.

149
GUSTAFSON D.L., RASTATTER J.C., COLOMBO T. et LONG M.E. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. Journal of
Pharmaceutical Sciences. 2002. Vol. 91, n° 6, pp. 1488�1501.
GUSTAFSON D.L. et THAMM D.H. Pharmacokinetic modeling of doxorubicin pharmacokinetics in dogs deficient in ABCB1 drug transporters. Journal of Veterinary Internal Medicine. 2010. Vol. 24, n° 3, pp. 579�586.
HALLIWELL B. et GUTTERIDGE J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. The Biochemical Journal. 1984. Vol. 219, n° 1, pp. 1�14.
HAMMER A.S., COUTO C.G., FILPPI J., GETZY D. et SHANK K. Efficacy and toxicity of VAC chemotherapy (vincristine, doxorubicin, and cyclophosphamide) in dogs with hemangiosarcoma. Journal of Veterinary Internal Medicine. 1991. Vol. 5, n° 3, pp. 160�166.
HAN J., ZHANG J., ZHAO H., LI Y. et CHEN Z. Simultaneous determination of doxorubicin and its dipeptide prodrug in mice plasma by HPLC with fluorescence detection. Journal of Pharmaceutical
Analysis. 2016. Vol. 6, n° 3, pp. 199�202.
HANNUN Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 1996. Vol. 274, n° 5294, pp. 1855�1859.
HARKER W.G., SLADE D.L., PARR R.L., FELDHOFF P.W., SULLIVAN D.M. et HOLGUIN M.H. Alterations in the topoisomerase II alpha gene, messenger RNA, and subcellular protein distribution as well as reduced expression of the DNA topoisomerase II beta enzyme in a mitoxantrone-resistant HL-60 human leukemia cell line. Cancer Research. 1995. Vol. 55, n° 8, pp. 1707�1716.
HARRIS A.L. et HOCHHAUSER D. Mechanisms of multidrug resistance in cancer treatment. Acta
Oncologica. 1992. Vol. 31, n° 2, pp. 205�213.
HART L.S., CUNNINGHAM J.T., DATTA T., DEY S., TAMEIRE F., LEHMAN S.L. et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. The Journal of Clinical
Investigation. 2012. Vol. 122, n° 12, pp. 4621�4634.
HEMMINKI K. DNA adducts, mutations and cancer. Carcinogenesis. 1993. Vol. 14, n° 10, pp. 2007�2012.
HOFFMAN D.M., GROSSANO D.D., DAMIN L. et WOODCOCK T.M. Stability of refrigerated and frozen solutions of doxorubicin hydrochloride. American Journal of Hospital Pharmacy. 1979. Vol. 36, n° 11, pp. 1536�1538.
HOLOHAN C., VAN SCHAEYBROECK S., LONGLEY D.B. et JOHNSTON P.G. Cancer drug resistance: an evolving paradigm. Nature Reviews. Cancer. 2013. Vol. 13, n° 10, pp. 714�726.
HONJO Y., HRYCYNA C.A., YAN Q.W., MEDINA-PÉREZ W.Y., ROBEY R.W., VAN DE LAAR A. et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Research. 2001. Vol. 61, n° 18, pp. 6635�6639.
HONSCHA K.U., SCHIRMER A., REISCHAUER A., SCHOON H.-A., EINSPANIER A. et GÄBEL G. Expression of ABC-transport proteins in canine mammary cancer : consequences for chemotherapy. Reproduction in Domestic Animals = Zuchthygiene. 2009. Vol. 44 Suppl 2, pp. 218�223.
HORTON D., PRIEBE W. et SZNAIDMAN M. Preparative procedures for conversion of daunorubicin into doxorubicin (Adriamycin) and 14-O-acetyldoxorubicin by way of 14-bromodaunorubicin. Carbohydrate Research. 1988. Vol. 184, pp. 231�235.

150
HOULBROOK S., ADDISON C.M., DAVIES S.L., CARMICHAEL J., STRATFORD I.J., HARRIS A.L. et al. Relationship between expression of topoisomerase II isoforms and intrinsic sensitivity to topoisomerase II inhibitors in breast cancer cell lines. British Journal of Cancer. 1995. Vol. 72, n° 6, pp. 1454�1461.
HOUSMAN G., BYLER S., HEERBOTH S., LAPINSKA K., LONGACRE M., SNYDER N. et al. Drug Resistance in Cancer: An Overview. Cancers. 2014. Vol. 6, n° 3, pp. 1769�1792.
HUGNET C., BENTJEN S.A. et MEALEY K.L. Frequency of the mutant MDR1 allele associated with multidrug sensitivity in a sample of collies from France. Journal of Veterinary Pharmacology and
Therapeutics. 2004. Vol. 27, n° 4, pp. 227�229.
IZQUIERDO M.A., NEEFJES J.J., MATHARI A.E., FLENS M.J., SCHEFFER G.L. et SCHEPER R.J. Overexpression of the ABC transporter TAP in multidrug-resistant human cancer cell lines. British
Journal of Cancer. 1996. Vol. 74, n° 12, pp. 1961�1967.
JACKSON T.L. Intracellular Accumulation and Mechanism of Action of Doxorubicin in a Spatio-temporal Tumor Model. Journal of Theoretical Biology. 2003. Vol. 220, n° 2, pp. 201�213.
JACQUET J.-M., BRESSOLLE F., GALTIER M., BOURRIER M., DONADIO D., JOURDAN J. et al. Doxorubicin and doxorubicinol: intra-and inter-individual variations of pharmacokinetic parameters. Cancer Chemotherapy and Pharmacology. 1990. Vol. 27, n° 3, pp. 219�225.
JAFFRÉZOU J.P., LEVADE T., BETTAÏEB A., ANDRIEU N., BEZOMBES C., MAESTRE N. et al. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. The EMBO journal. 1996. Vol. 15, n° 10, pp. 2417�2424.
JAIN M.K., WU N.Y. et WRAY L.V. Drug-induced phase change in bilayer as possible mode of action of membrane expanding drugs. Nature. 1975. Vol. 255, n° 5508, pp. 494�496.
JANSSEN M.J.H., CROMMELIN D.J.A., STORM G. et HULSHOFF A. Doxorubicin decomposition on storage. Effect of pH, type of buffer and liposome encapsulation. International Journal of
Pharmaceutics. 1985. Vol. 23, n° 1, pp. 1�11.
JOERGER M., HUITEMA A.D.R., RICHEL D.J., DITTRICH C., PAVLIDIS N., BRIASOULIS E. et al. Population pharmacokinetics and pharmacodynamics of doxorubicin and cyclophosphamide in breast cancer patients: a study by the EORTC-PAMM-NDDG. Clinical Pharmacokinetics. 2007. Vol. 46, n° 12, pp. 1051�1068.
KALIVENDI S.V., KONOREV E.A., CUNNINGHAM S., VANAMALA S.K., KAJI E.H., JOSEPH J. et al. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. The Biochemical Journal. 2005. Vol. 389, n° Pt 2, pp. 527�539.
KALYANARAMAN B. et BAKER J.E. On the detection of paramagnetic species in the adriamycin-perfused rat heart: A reappraisal. Biochemical and Biophysical Research Communications. 1990. Vol. 169, n° 1, pp. 30�38.
KALYANARAMAN B., JOSEPH J., KALIVENDI S., WANG S., KONOREV E. et KOTAMRAJU S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Molecular and Cellular Biochemistry. 2002. Vol. 234�235, n° 1�2, pp. 119�124.
KALYANARAMAN B., MOREHOUSE K.M. et MASON R.P. An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers. Archives of Biochemistry and Biophysics. 1991. Vol. 286, n° 1, pp. 164�170.

151
KANE R.C., MCGUINN W.D., DAGHER R., JUSTICE R. et PAZDUR R. Dexrazoxane : FDA review and approval for the treatment of accidental extravasation following intravenous anthracycline chemotherapy. The Oncologist. 2008. Vol. 13, n° 4, pp. 445�450.
KARANTZA-WADSWORTH V., PATEL S., KRAVCHUK O., CHEN G., MATHEW R., JIN S. et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes & Development. 2007. Vol. 21, n° 13, pp. 1621�1635.
KASSNER E. Evaluation and treatment of chemotherapy extravasation injuries. Journal of Pediatric
Oncology Nursing: Official Journal of the Association of Pediatric Oncology Nurses. 2000. Vol. 17, n° 3, pp. 135�148.
KATZENMEYER J.B., EDDY C.V. et ARRIAGA E.A. Tandem Laser-Induced Fluorescence and Mass Spectrometry Detection for High-Performance Liquid Chromatography Analysis of the in Vitro Metabolism of Doxorubicin. Analytical Chemistry. 2010. Vol. 82, n° 19, pp. 8113�8120.
KEIZER H.G., PINEDO H.M., SCHUURHUIS G.J. et JOENJE H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacology & Therapeutics. 1990. Vol. 47, n° 2, pp. 219�231.
KIM J.-E., CHO H.-J., KIM J.S., SHIM C.-K., CHUNG S.-J., OAK M.-H. et al. The limited intestinal absorption via paracellular pathway is responsible for the low oral bioavailability of doxorubicin. Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 2013. Vol. 43, n° 7, pp. 579�591.
KIM Y., MA A.-G., KITTA K., FITCH S.N., IKEDA T., IHARA Y. et al. Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Molecular
Pharmacology. 2003. Vol. 63, n° 2, pp. 368�377.
KITATANI K., IDKOWIAK-BALDYS J. et HANNUN Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cellular signalling. 2008. Vol. 20, n° 6, pp. 1010�1018.
KIYOMIYA K., MATSUO S. et KUREBE M. Mechanism of Specific Nuclear Transport of Adriamycin. Cancer Research. 2001. Vol. 61, n° 6, pp. 2467�2471.
KNOBLOCH A., MOHRING S. a. i., EBERLE N., NOLTE I., HAMSCHER G. et SIMON D. Drug Residues in Serum of Dogs Receiving Anticancer Chemotherapy. Journal of Veterinary Internal Medicine. 2010a. Vol. 24, n° 2, pp. 379�383.
KNOBLOCH A., MOHRING S. a. i., EBERLE N., NOLTE I., HAMSCHER G. et SIMON D. Cytotoxic Drug Residues in Urine of Dogs Receiving Anticancer Chemotherapy. Journal of Veterinary Internal
Medicine. 2010b. Vol. 24, n° 2, pp. 384�390.
KOBAYASHI S., VOLDEN P., TIMM D., MAO K., XU X. et LIANG Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. The Journal of Biological Chemistry. 2010. Vol. 285, n° 1, pp. 793�804.
KOEDRITH P. et SEO Y.R. Induction of doxorubicin-mediated apoptosis via thioredoxin reductase 1 RNAi in human colon cancer cells. Molecular & Cellular Toxicology. 2011. Vol. 7, n° 2, pp. 112�119.
KOHNO K., TAKANO H. et KUWANO M. Mechanisms of resistance to DNA topoisomerase II inhibitors. Gan to Kagaku Ryoho. Cancer & Chemotherapy. 1991. Vol. 18, n° 10, pp. 1568�1573.
KRAFT M.L. Plasma membrane organization and function: moving past lipid rafts. Molecular Biology
of the Cell. 2013. Vol. 24, n° 18, pp. 2765�2768.

152
KUMARAGURUPARAN R., SUBAPRIYA R., BALACHANDRAN C., MANOHAR B.M., THANGADURAI A. et NAGINI S. Xenobiotic-metabolizing enzymes in canine mammary tumours. Veterinary Journal
(London, England: 1997). 2006. Vol. 172, n° 2, pp. 364�368.
KÜMMERLE A., KRUEGER T., DUSMET M., VALLET C., PAN Y., RIS H.B. et al. A validated assay for measuring doxorubicin in biological fluids and tissues in an isolated lung perfusion model: matrix effect and heparin interference strongly influence doxorubicin measurements. Journal of
Pharmaceutical and Biomedical Analysis. 2003. Vol. 33, n° 3, pp. 475�494.
LANE A., BLACK M. et WYATT K. Toxicity and efficacy of a novel doxorubicin and carboplatin chemotherapy protocol for the treatment of canine appendicular osteosarcoma following limb amputation. Australian Veterinary Journal. 2012. Vol. 90, n° 3, pp. 69�74.
LANGHORN R. et WILLESEN J.L. Cardiac Troponins in Dogs and Cats. Journal of Veterinary Internal
Medicine. 2016. Vol. 30, n° 1, pp. 36�50.
LANZA E., ROZZA A., FAVALLI L., MONTI E., POGGI P. et VILLANI F. The rat model in the comparative evaluation of anthracyclines cardiotoxicity. Tumori. 1989. Vol. 75, n° 6, pp. 533�536.
LARSEN A.K., SKLADANOWSKI A. et BOJANOWSKI K. The roles of DNA topoisomerase II during the cell cycle. Progress in Cell Cycle Research. 1996. Vol. 2, pp. 229�239.
LEBRECHT D., SETZER B., KETELSEN U.-P., HABERSTROH J. et WALKER U.A. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003. Vol. 108, n° 19, pp. 2423�2429.
LEBRECHT D. et WALKER U.A. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovascular
Toxicology. 2007. Vol. 7, n° 2, pp. 108�113.
LEE J., GIORDANO S. et ZHANG J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. The Biochemical Journal. 2012. Vol. 441, n° 2, pp. 523�540.
LEE J.-Y., TANABE S., SHIMOHIRA H., KOBAYASHI Y., OOMACHI T., AZUMA S. et al. Expression of cyclooxygenase-2, P-glycoprotein and multi-drug resistance-associated protein in canine transitional cell carcinoma. Research in Veterinary Science. 2007. Vol. 83, n° 2, pp. 210�216.
LEI H., WANG X. et WU C. Early stage intercalation of doxorubicin to DNA fragments observed in molecular dynamics binding simulations. Journal of Molecular Graphics and Modelling. 2012. Vol. 38, pp. 279�289.
LEONEL C., GELALETI G.B., JARDIM B.V., MOSCHETTA M.G., REGIANI V.R., OLIVEIRA J.G. et al. Expression of glutathione, glutathione peroxidase and glutathione S-transferase pi in canine mammary tumors. BMC Veterinary Research. 2014. Vol. 10, pp. 49.
LI D., QU Y., TAO L., LIU H., HU A., GAO F. et al. Inhibition of iNOS Protects the Aging Heart Against β-Adrenergic Receptor Stimulation-Induced Cardiac Dysfunction and Myocardial Ischemic Injury. Journal of Surgical Research. 2006. Vol. 131, n° 1, pp. 64�72.
LI M., YUAN H., LI N., SONG G., ZHENG Y., BARATTA M. et al. Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. European Journal of
Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2008. Vol. 35, n° 1�2, pp. 114�126.
LI Y.R., JIA Z. et TRUSH M.A. Defining ROS in Biology and Medicine. Reactive Oxygen Species. 2016. Vol. 1, n° 1, pp. 9�21.

153
LICATA S., SAPONIERO A., MORDENTE A. et MINOTTI G. Doxorubicin Metabolism and Toxicity in Human Myocardium: Role of Cytoplasmic Deglycosidation and Carbonyl Reduction. Chemical
Research in Toxicology. 2000. Vol. 13, n° 5, pp. 414�420.
LIM C.C., ZUPPINGER C., GUO X., KUSTER G.M., HELMES M., EPPENBERGER H.M. et al. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. The Journal of Biological
Chemistry. 2004. Vol. 279, n° 9, pp. 8290�8299.
LIU Y.Y., HAN T.Y., GIULIANO A.E. et CABOT M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB journal: official publication of the Federation of American Societies for
Experimental Biology. 2001. Vol. 15, n° 3, pp. 719�730.
LOADMAN P.M. et CALABRESE C.R. Separation methods for anthraquinone related anti-cancer drugs. Journal of Chromatography. B, Biomedical Sciences and Applications. 2001. Vol. 764, n° 1�2, pp. 193�206.
LOCKSHIN R.A. et ZAKERI Z. Apoptosis, autophagy, and more. The International Journal of
Biochemistry & Cell Biology. 2004. Vol. 36, n° 12, pp. 2405�2419.
LODISH H., BERK A., ZIPURSKY S.L., MATSUDAIRA P., BALTIMORE D. et DARNELL J. The Role of Topoisomerases in DNA Replication. [en ligne]. 2000. [Consulté le 8 juin 2017].
LOTRIONTE M., BIONDI-ZOCCAI G., ABBATE A., LANZETTA G., D’ASCENZO F., MALAVASI V. et al. Review and meta-analysis of incidence and clinical predictors of anthracycline cardiotoxicity. The
American Journal of Cardiology. 2013. Vol. 112, n° 12, pp. 1980�1984.
LOVELESS H., ARENA E., FELSTED R.L. et BACHUR N.R. Comparative mammalian metabolism of adriamycin and daunorubicin. Cancer Research. 1978. Vol. 38, n° 3, pp. 593�598.
LOWN J.W. Discovery and development of anthracycline antitumour antibiotics. . 1993. Vol. 22, n° 3, pp. 165�176.
LOWN J.W., CHEN H.-H., PLAMBECK J.A. et ACTON E.M. Further studies on the generation of reactive oxygen species from activated anthracyclines and the relationship to cytotoxic action and cardiotoxic effects. Biochemical Pharmacology. 1982. Vol. 31, n° 4, pp. 575�581.
LUCCI A., HAN T.Y., LIU Y.Y., GIULIANO A.E. et CABOT M.C. Modification of ceramide metabolism increases cancer cell sensitivity to cytotoxics. International Journal of Oncology. 1999. Vol. 15, n° 3, pp. 541�546.
MA L., PRATT S.E., CAO J., DANTZIG A.H., MOORE R.E. et SLAPAK C.A. Identification and characterization of the canine multidrug resistance-associated protein. Molecular Cancer
Therapeutics. 2002. Vol. 1, n° 14, pp. 1335�1342.
MALLA S., PRASAD NIRAULA N., SINGH B., LIOU K. et KYUNG SOHNG J. Limitations in doxorubicin production from Streptomyces peucetius. Microbiological Research. 2010. Vol. 165, n° 5, pp. 427�435.
MARCILLAT O., ZHANG Y. et DAVIES K.J. Oxidative and non-oxidative mechanisms in the inactivation of cardiac mitochondrial electron transport chain components by doxorubicin. Biochemical Journal. 1989. Vol. 259, n° 1, pp. 181�189.
MARNETT L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutation Research. 1999. Vol. 424, n° 1�2, pp. 83�95.

154
MATSON S. et KAISER-ROGERS and K.A. DNA Helicases. Annual Review of Biochemistry. 1990. Vol. 59, n° 1, pp. 289�329.
MAU B.L. et POWIS G. Mechanism-based inhibition of thioredoxin reductase by antitumor quinoid compounds. Biochemical Pharmacology. 1992a. Vol. 43, n° 7, pp. 1613�1620.
MAU B.L. et POWIS G. Inhibition of cellular thioredoxin reductase by diaziquone and doxorubicin. Relationship to the inhibition of cell proliferation and decreased ribonucleotide reductase activity. Biochemical Pharmacology. 1992b. Vol. 43, n° 7, pp. 1621�1627.
MAULDIN G.E., FOX P.R., PATNAIK A.K., BOND B.R., MOONEY S.C. et MATUS R.E. Doxorubicin-lnduced Cardiotoxicosis Clinical Features in 32 Dogs. Journal of Veterinary Internal Medicine. 1992. Vol. 6, n° 2, pp. 82�88.
MEALEY K.L., BENTJEN S.A., GAY J.M. et CANTOR G.H. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics. 2001. Vol. 11, n° 8, pp. 727�733.
MEREDITH A.-M. et DASS C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. The Journal of Pharmacy and Pharmacology. 2016. Vol. 68, n° 6, pp. 729�741.
MERLIN J.L., MARCHAL S., RAMACCI C., DIETERLEN A., SCHULTZ G., LUCAS C. et al. Influence of S9788, a new modulator of multidrug resistance, on the cellular accumulation and subcellular distribution of daunorubicin in P-glycoprotein-expressing MCF7 human breast adenocarcinoma cells. Cytometry. 1995. Vol. 20, n° 4, pp. 315�323.
MINOTTI G. et AUST S.D. Redox cycling of iron and lipid peroxidation. Lipids. 1992. Vol. 27, n° 3, pp. 219�226.
MINOTTI G., CAIRO G. et MONTI E. Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB journal: official publication of the Federation of American Societies for Experimental
Biology. 1999. Vol. 13, n° 2, pp. 199�212.
MINOTTI G., MENNA P., SALVATORELLI E., CAIRO G. et GIANNI L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacological Reviews. 2004. Vol. 56, n° 2, pp. 185�229.
MINOTTI G., RECALCATI S., MORDENTE A., LIBERI G., CALAFIORE A.M., MANCUSO C. et al. The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB journal: official publication of the
Federation of American Societies for Experimental Biology. 1998. Vol. 12, n° 7, pp. 541�552.
MIYOSHI N., TOJO E., OISHI A., FUJIKI M., MISUMI K., SAKAMOTO H. et al. Immunohistochemical detection of P-glycoprotein (PGP) and multidrug resistance-associated protein (MRP) in canine cutaneous mast cell tumors. The Journal of Veterinary Medical Science. 2002. Vol. 64, n° 6, pp. 531�533.
MIZUTANI H., TADA-OIKAWA S., HIRAKU Y., KOJIMA M. et KAWANISHI S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sciences. 2005. Vol. 76, n° 13, pp. 1439�1453.
MOORE, AS et FRIMBERGER, AE. Oncology for Veterinary Technicians and Nurses. John Wiley & Sons. 2009.
MOORE H.W. Bioactivation as a model for drug design bioreductive alkylation. Science. 1977. Vol. 197, n° 4303, pp. 527�532.

155
MORRISON W.B. Cancer chemotherapy: an annotated history. Journal of Veterinary Internal
Medicine. 2010. Vol. 24, n° 6, pp. 1249�1262.
MORRISON, WB. Cancer in Dogs and Cats: Medical and Surgical Management. 2002.
MOTIEI M. et KASHANIAN S. Novel amphiphilic chitosan nanocarriers for sustained oral delivery of hydrophobic drugs. European Journal of Pharmaceutical Sciences: Official Journal of the European
Federation for Pharmaceutical Sciences. 2017. Vol. 99, pp. 285�291.
MUGGIA F.M. et GREEN M.D. New anthracycline antitumor antibiotics. Critical Reviews in
Oncology/Hematology. 1991. Vol. 11, n° 1, pp. 43�64.
MUKHERJEE A. et SASIKALA W.D., 2013. Chapter One - Drug–DNA Intercalation: From Discovery to the Molecular Mechanism. In Advances in Protein Chemistry and Structural Biology. Academic Press. Dynamics of Proteins and Nucleic Acids. pp. 1�62.
MUSTACICH D. et POWIS G. Thioredoxin reductase. Biochemical Journal. 2000. Vol. 346, n° 1, pp. 1�8.
MYERS N.C., MOORE A.S., RAND W.M., GLIATTO J. et COTTER S.M. Evaluation of a multidrug chemotherapy protocol (ACOPA II) in dogs with lymphoma. Journal of Veterinary Internal Medicine. 1997. Vol. 11, n° 6, pp. 333�339.
NARAHARI J., MA R., WANG M. et WALDEN W.E. The aconitase function of iron regulatory protein 1. Genetic studies in yeast implicate its role in iron-mediated redox regulation. The Journal of Biological
Chemistry. 2000. Vol. 275, n° 21, pp. 16227�16234.
NEFF M.W., ROBERTSON K.R., WONG A.K., SAFRA N., BROMAN K.W., SLATKIN M. et al. Breed distribution and history of canine mdr1-1Delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proceedings of the National Academy of Sciences of the
United States of America. 2004. Vol. 101, n° 32, pp. 11725�11730.
NEGORO S., OH H., TONE E., KUNISADA K., FUJIO Y., WALSH K. et al. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation. 2001. Vol. 103, n° 4, pp. 555�561.
NEGRE-SALVAYRE A., COATRIEUX C., INGUENEAU C. et SALVAYRE R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. British Journal of Pharmacology. 2008. Vol. 153, n° 1, pp. 6�20.
NICOLAY K., TIMMERS R.J., SPOELSTRA E., VAN DER NEUT R., FOK J.J., HUIGEN Y.M. et al. The interaction of adriamycin with cardiolipin in model and rat liver mitochondrial membranes. Biochimica Et Biophysica Acta. 1984. Vol. 778, n° 2, pp. 359�371.
NITISS J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nature reviews.
Cancer. 2009a. Vol. 9, n° 5, pp. 327�337.
NITISS J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nature Reviews. Cancer. 2009b. Vol. 9, n° 5, pp. 338�350.
NITISS J.L., VILALTA P.M., WU H. et MCMAHON J. Mutations in the gyrB domain of eukaryotic topoisomerase II can lead to partially dominant resistance to etoposide and amsacrine. Molecular
Pharmacology. 1994. Vol. 46, n° 4, pp. 773�777.

156
NORDBERG J. et ARNÉR E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology & Medicine. 2001. Vol. 31, n° 11, pp. 1287�1312.
NOWAK M., MADEJ J.A. et DZIEGIEL P. Expression of Breast Cancer Resistance Protein (BCRP-1) in canine mammary adenocarcinomas and adenomas. In Vivo (Athens, Greece). 2009. Vol. 23, n° 5, pp. 705�709.
OCTAVIA Y., TOCCHETTI C.G., GABRIELSON K.L., JANSSENS S., CRIJNS H.J. et MOENS A.L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. Journal of Molecular
and Cellular Cardiology. 2012. Vol. 52, n° 6, pp. 1213�1225.
OGILVIE G.K., REYNOLDS H.A., RICHARDSON R.C., WITHROW S.J., NORRIS A.M., HENDERSON R.A. et al. Phase II evaluation of doxorubicin for treatment of various canine neoplasms. Journal of the
American Veterinary Medical Association. 1989a. Vol. 195, n° 11, pp. 1580�1583.
OGILVIE G.K., RICHARDSON R.C., CURTIS C.R., WITHROW S.J., REYNOLDS H.A., NORRIS A.M. et al. Acute and short-term toxicoses associated with the administration of doxorubicin to dogs with malignant tumors. Journal of the American Veterinary Medical Association. 1989b. Vol. 195, n° 11, pp. 1584�1587.
OKABE M., UNNO M., HARIGAE H., KAKU M., OKITSU Y., SASAKI T. et al. Characterization of the organic cation transporter SLC22A16: a doxorubicin importer. Biochemical and Biophysical Research
Communications. 2005. Vol. 333, n° 3, pp. 754�762.
OLSON R.D. et MUSHLIN P.S. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB
journal: official publication of the Federation of American Societies for Experimental Biology. 1990. Vol. 4, n° 13, pp. 3076�3086.
OOSTERBAAN M.J., DIRKS R.J., VREE T.B. et VAN DER KLEIJN E. Pharmacokinetics of anthracyclines in dogs: evidence for structure-related body distribution and reduction to their hydroxy metabolites. Pharmaceutical Research. 1984. Vol. 1, n° 1, pp. 33�38.
OTH D., BÉGIN M., BISCHOFF P., LEROUX J.Y., MERCIER G. et BRUNEAU C. Induction, by adriamycin and mitomycin C, of modifications in lipid composition, size distribution, membrane fluidity and permeability of cultured RDM4 lymphoma cells. Biochimica Et Biophysica Acta. 1987. Vol. 900, n° 2, pp. 198�208.
PACHER P., LIAUDET L., BAI P., VIRAG L., MABLEY J.G., HASKÓ G. et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. The Journal of
Pharmacology and Experimental Therapeutics. 2002. Vol. 300, n° 3, pp. 862�867.
PAGE R.L., HUGHES C.S., HUYAN S., SAGRIS J. et TROGDON M. Modulation of P-glycoprotein-mediated doxorubicin resistance in canine cell lines. Anticancer Research. 2000. Vol. 20, n° 5B, pp. 3533�3538.
PEETLA C., BHAVE R., VIJAYARAGHAVALU S., STINE A., KOOIJMAN E. et LABHASETWAR V. Drug Resistance in Breast Cancer Cells: Biophysical Characterization of and Doxorubicin Interactions with Membrane Lipids. Molecular pharmaceutics. 2010. Vol. 7, n° 6, pp. 2334�2348.
PÉREZ-BLANCO J.S., FERNÁNDEZ DE GATTA M. del M., HERNÁNDEZ-RIVAS J.M., GARCÍA SÁNCHEZ M.J., SAYALERO MARINERO M.L. et GONZÁLEZ LÓPEZ F. Validation and clinical evaluation of a UHPLC method with fluorescence detector for plasma quantification of doxorubicin and doxorubicinol in haematological patients. Journal of Chromatography. B, Analytical Technologies in the Biomedical
and Life Sciences. 2014. Vol. 955�956, pp. 93�97.

157
PETERS J.H., GORDON G.R., KASHIWASE D., LOWN J.W., YEN S.F. et PLAMBECK J.A. Redox activities of antitumor anthracyclines determined by microsomal oxygen consumption and assays for superoxide anion and hydroxyl radical generation. Biochemical Pharmacology. 1986. Vol. 35, n° 8, pp. 1309�1323.
PETTERINO C., ROSSETTI E., BERTONCELLO D., MARTINI M., ZAPPULLI V., BARGELLONI L. et al. Immunohistochemical detection of P-glycoprotein (clone C494) in canine mammary gland tumours. Journal of Veterinary Medicine. A, Physiology, Pathology, Clinical Medicine. 2006. Vol. 53, n° 4, pp. 174�178.
PINKEL D. The use of body surface area as a criterion of drug dosage in cancer chemotherapy. Cancer
Research. 1958. Vol. 18, n° 7, pp. 853�856.
PISCITELLI S.C., RODVOLD K.A., RUSHING D.A. et TEWKSBURY D.A. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clinical Pharmacology and
Therapeutics. 1993. Vol. 53, n° 5, pp. 555�561.
POMMIER Y., LEO E., ZHANG H. et MARCHAND C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chemistry & Biology. 2010. Vol. 17, n° 5, pp. 421�433.
PONCE F., MAGNOL J.-P., LEDIEU D., MARCHAL T., TURINELLI V., CHALVET-MONFRAY K. et al. Prognostic significance of morphological subtypes in canine malignant lymphomas during chemotherapy. Veterinary Journal (London, England: 1997). 2004. Vol. 167, n° 2, pp. 158�166.
POWIS G. Metabolism and reactions of quinoid anticancer agents. Pharmacology & Therapeutics. 1987. Vol. 35, n° 1�2, pp. 57�162.
QUILES J.L., HUERTAS J.R., BATTINO M., MATAIX J. et RAMÍREZ-TORTOSA M.C. Antioxidant nutrients and adriamycin toxicity. Toxicology. 2002. Vol. 180, n° 1, pp. 79�95.
RAHMAN A.M., YUSUF S.W. et EWER M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. International Journal of Nanomedicine. 2007. Vol. 2, n° 4, pp. 567�583.
RAHMANI R., GIL P., MARTIN M., DURAND A., BARBET J. et CANO J.P. Quantitation of adriamycin in plasma and urine: comparative study of radioimmunoassay and high-performance liquid chromatography methods. Journal of Pharmaceutical and Biomedical Analysis. 1983. Vol. 1, n° 3, pp. 301�309.
RATTERREE W., GIEGER T., PARIAUT R., SAELINGER C. et STRICKLAND K. Value of echocardiography and electrocardiography as screening tools prior to Doxorubicin administration. Journal of the
American Animal Hospital Association. 2012. Vol. 48, n° 2, pp. 89�96.
RAU S.E., BARBER L.G. et BURGESS K.E. Efficacy of maropitant in the prevention of delayed vomiting associated with administration of doxorubicin to dogs. Journal of Veterinary Internal Medicine. 2010. Vol. 24, n° 6, pp. 1452�1457.
REAGAN W.J., HANDY V., MCKAMEY A., BLOOM J.C., DELDAR A., STEVENS C. et al. Effects of doxorubicin on the canine erythroid and myeloid progenitor cells and bone marrow microenvironment. Comparative Haematology International. 1993. Vol. 3, n° 2, pp. 96�101.
REBBAA A., ZHENG X., CHOU P.M. et MIRKIN B.L. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene. 2003. Vol. 22, n° 18, pp. 2805�2811.

158

159
REBHUN R.B., KENT M.S., BORROFKA S. a. E.B., FRAZIER S., SKORUPSKI K. et RODRIGUEZ C.O. CHOP chemotherapy for the treatment of canine multicentric T-cell lymphoma. Veterinary and
Comparative Oncology. 2011. Vol. 9, n° 1, pp. 38�44.
REDDY L.H., MEDA N. et MURTHY R.R. Rapid and sensitive HPLC method for the estimation of doxorubicin in dog blood--the silver nitrate artifact. Acta Pharmaceutica. 2005. Vol. 55, n° 1, pp. 81�91.
REEVES D. Management of anthracycline extravasation injuries. The Annals of Pharmacotherapy. 2007. Vol. 41, n° 7, pp. 1238�1242.
REGEV R. et EYTAN G.D. Flip-Flop of Doxorubicin across Erythrocyte and Lipid Membranes. Biochemical Pharmacology. 1997. Vol. 54, n° 10, pp. 1151�1158.
REN S., DAI Y., LI C., QIU Z., WANG X., TIAN F. et al. Pharmacokinetics and pharmacodynamics evaluation of a thermosensitive chitosan based hydrogel containing liposomal doxorubicin. European
Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical
Sciences. 2016. Vol. 92, pp. 137�145.
RICCIARELLO R., PICHINI S., PACIFICI R., ALTIERI I., PELLEGRINI M., FATTOROSSI A. et al. Simultaneous determination of epirubicin, doxorubicin and their principal metabolites in human plasma by high-performance liquid chromatography and electrochemical detection. Journal of Chromatography. B,
Biomedical Sciences and Applications. 1998. Vol. 707, n° 1�2, pp. 219�225.
RIDDICK D.S., LEE C., RAMJI S., CHINJE E.C., COWEN R.L., WILLIAMS K.J. et al. Cancer Chemotherapy and Drug Metabolism. Drug Metabolism and Disposition. 2005. Vol. 33, n° 8, pp. 1083�1096.
RIZZO V., SACCHI N. et MENOZZI M. Kinetic studies of anthracycline-DNA interaction by fluorescence stopped flow confirm a complex association mechanism. Biochemistry. 1989. Vol. 28, n° 1, pp. 274�282.
ROBAT C., BURTON J., THAMM D. et VAIL D. Retrospective evaluation of doxorubicin-piroxicam combination for the treatment of transitional cell carcinoma in dogs. The Journal of Small Animal
Practice. 2013. Vol. 54, n° 2, pp. 67�74.
RODRÍGUEZ-VARGAS J.M., RUIZ-MAGAÑA M.J., RUIZ-RUIZ C., MAJUELOS-MELGUIZO J., PERALTA-LEAL A., RODRÍGUEZ M.I. et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Research. 2012. Vol. 22, n° 7, pp. 1181�1198.
ROSS W.E. et BRADLEY M.O. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochimica Et Biophysica Acta. 1981. Vol. 654, n° 1, pp. 129�134.
SCHAUPP C.M., WHITE C.C., MERRILL G.F. et KAVANAGH T.J. Metabolism of Doxorubicin to the Cardiotoxic Metabolite Doxorubicinol Is Increased in a Mouse Model of Chronic Glutathione Deficiency: A Potential Role for Carbonyl Reductase 3. Chemico-biological interactions. 2015. Vol. 234, pp. 154�161.
SCHLAME M., BRODY S. et HOSTETLER K.Y. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. European Journal of Biochemistry. 1993. Vol. 212, n° 3, pp. 727�735.
SELMIC L.E., BURTON J.H., THAMM D.H., WITHROW S.J. et LANA S.E. Comparison of carboplatin and doxorubicin-based chemotherapy protocols in 470 dogs after amputation for treatment of appendicular osteosarcoma. Journal of Veterinary Internal Medicine. 2014. Vol. 28, n° 2, pp. 554�563.

160
SELTING K.A., LANA S.E., OGILVIE G.K., OLMSTEAD A., MYKLES D.L., BRIGHT J. et al. Cardiac troponin I in canine patients with lymphoma and osteosarcoma receiving doxorubicin: comparison with clinical heart disease in a retrospective analysis. Veterinary and Comparative Oncology. 2004. Vol. 2, n° 3, pp. 142�156.
SELTING K.A., OGILVIE G.K., GUSTAFSON D.L., LONG M.E., LANA S.E., WALTON J.A. et al. Evaluation of the effects of dietary n-3 fatty acid supplementation on the pharmacokinetics of doxorubicin in dogs with lymphoma. American Journal of Veterinary Research. 2006. Vol. 67, n° 1, pp. 145�151.
SELTING K.A., POWERS B.E., THOMPSON L.J., MITTLEMAN E., TYLER J.W., LAFFERTY M.H. et al. Outcome of dogs with high-grade soft tissue sarcomas treated with and without adjuvant doxorubicin chemotherapy: 39 cases (1996-2004). Journal of the American Veterinary Medical
Association. 2005. Vol. 227, n° 9, pp. 1442�1448.
SIMON D., SCHOENROCK D., BAUMGÄRTNER W. et NOLTE I. Postoperative adjuvant treatment of invasive malignant mammary gland tumors in dogs with doxorubicin and docetaxel. Journal of
Veterinary Internal Medicine. 2006. Vol. 20, n° 5, pp. 1184�1190.
SINGER S.J. et NICOLSON G.L. The fluid mosaic model of the structure of cell membranes. Science
(New York, N.Y.). 1972. Vol. 175, n° 4023, pp. 720�731.
SISHI B.J.N., LOOS B., VAN ROOYEN J. et ENGELBRECHT A.-M. Doxorubicin induces protein ubiquitination and inhibits proteasome activity during cardiotoxicity. Toxicology. 2013. Vol. 309, pp. 23�29.
SLUPPHAUG G., KAVLI B. et KROKAN H.E. The interacting pathways for prevention and repair of oxidative DNA damage. Mutation Research. 2003. Vol. 531, n° 1�2, pp. 231�251.
SMITH D. et KIRK G. Some systemic effects of adriamycin on the dog. Journal of the American Animal
Hospital Association. 1976. Vol. 12, pp. 92�97.
SOKOLOVE P.M. Interactions of Adriamycin aglycones with mitochondria may mediate Adriamycin cardiotoxicity. International Journal of Biochemistry. 1994. Vol. 26, n° 12, pp. 1341�1350.
SORDET O., KHAN Q.A., KOHN K.W. et POMMIER Y. Apoptosis induced by topoisomerase inhibitors. Current Medicinal Chemistry. Anti-Cancer Agents. 2003. Vol. 3, n° 4, pp. 271�290.
SORENMO K., DUDA L., BARBER L., CRONIN K., SAMMARCO C., USBORNE A. et al. Canine hemangiosarcoma treated with standard chemotherapy and minocycline. Journal of Veterinary
Internal Medicine. 2000. Vol. 14, n° 4, pp. 395�398.
SORENMO K., OVERLEY B., KRICK E., FERRARA T., LABLANC A. et SHOFER F. Outcome and toxicity associated with a dose-intensified, maintenance-free CHOP-based chemotherapy protocol in canine lymphoma: 130 cases. Veterinary and Comparative Oncology. 2010. Vol. 8, n° 3, pp. 196�208.
SORENMO K., SAMLUK M., CLIFFORD C., BAEZ J., BARRETT J.S., POPPENGA R. et al. Clinical and pharmacokinetic characteristics of intracavitary administration of pegylated liposomal encapsulated doxorubicin in dogs with splenic hemangiosarcoma. Journal of Veterinary Internal Medicine. 2007. Vol. 21, n° 6, pp. 1347�1354.
SPEELMANS G., STAFFHORST R. et DE KRUIJFF B. The anionic phospholipid-mediated membrane interaction of the anti-cancer drug doxorubicin is enhanced by phosphatidylethanolamine compared to other zwitterionic phospholipids. Biochemistry. 1997. Vol. 36, n° 28, pp. 8657�8662.

161
SPETH P.A., VAN HOESEL Q.G. et HAANEN C. Clinical pharmacokinetics of doxorubicin. Clinical
Pharmacokinetics. 1988. Vol. 15, n° 1, pp. 15�31.
SPINALE F.G. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiological Reviews. 2007. Vol. 87, n° 4, pp. 1285�1342.
SPUGNINI E.P. Use of hyaluronidase for the treatment of extravasation of chemotherapeutic agents in six dogs. Journal of the American Veterinary Medical Association. 2002. Vol. 221, n° 10.
STADTMAN E.R. et OLIVER C.N. Metal-catalyzed oxidation of proteins. Physiological consequences. The Journal of Biological Chemistry. 1991. Vol. 266, n° 4, pp. 2005�2008.
STEINGOLD S.F., SHARP N.J., MCGAHAN M.C., HUGHES C.S., DUNN S.E. et PAGE R.L. Characterization of canine MDR1 mRNA: its abundance in drug resistant cell lines and in vivo. Anticancer Research. 1998. Vol. 18, n° 1A, pp. 393�400.
STERRENBERG L., JULICHER R.H., GOOSSENS P.A., BAST A. et NOORDHOEK J. Anthracycline-induced oxygen consumption and oxidative damage in rat liver microsomes are not necessarily coupled. A study with 8 structurally related anthracyclines. Free Radical Research Communications. 1985. Vol. 1, n° 1, pp. 41�54.
STEWART D.J., GREWAAL D., GREEN R.M., MIKHAEL N., GOEL R., MONTPETIT V.A. et al. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Research. 1993. Vol. 13, n° 6A, pp. 1945�1952.
STORLAZZI C.T., LONOCE A., GUASTADISEGNI M.C., TROMBETTA D., D’ADDABBO P., DANIELE G. et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Research. 2010. Vol. 20, n° 9, pp. 1198�1206.
STURGILL M.G., AUGUST D.A. et BRENNER D.E. Hepatic enzyme induction with phenobarbital and doxorubicin metabolism and myelotoxicity in the rabbit. Cancer Investigation. 2000. Vol. 18, n° 3, pp. 197�205.
SUMNERS D.W. Lifting the curtain: using topology to probe the hidden action of enzymes. Notices of
the AMS. 1995. Vol. 42, pp. 528–537.
SUZUKI Y.J., CARINI M. et BUTTERFIELD D.A. Protein Carbonylation. Antioxidants & Redox Signaling. 2010. Vol. 12, n° 3, pp. 323�325.
SWIFT L.P., REPHAELI A., NUDELMAN A., PHILLIPS D.R. et CUTTS S.M. Doxorubicin-DNA adducts induce a non-topoisomerase II-mediated form of cell death. Cancer Research. 2006. Vol. 66, n° 9, pp. 4863�4871.
TAATJES D.J., GAUDIANO G., RESING K. et KOCH T.H. Alkylation of DNA by the anthracycline, antitumor drugs adriamycin and daunomycin. Journal of Medicinal Chemistry. 1996. Vol. 39, n° 21, pp. 4135�4138.
TACAR O., SRIAMORNSAK P. et DASS C.R. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. The Journal of Pharmacy and Pharmacology. 2013. Vol. 65, n° 2, pp. 157�170.
TAGEJA N. et GRONINGER H. Chemotherapy-Induced Nausea and Vomiting. Journal of Palliative
Medicine. 2014. Vol. 17, n° 12, pp. 1400�1402.
TANNOCK, I et HILL, RP. The Basic Science of Oncology. Elsevier Science Limited. 1987.

162
TATER G., EBERLE N., HUNGERBUEHLER S., JOETZKE A., NOLTE I., WESS G. et al. Assessment of cardiac troponin I (cTnI) and tissue velocity imaging (TVI) in 14 dogs with malignant lymphoma undergoing chemotherapy treatment with doxorubicin. Veterinary and Comparative Oncology. 2017. Vol. 15, n° 1, pp. 55�64.
TAVOLONI N., GUARINO A.M. et BERK P.D. Photolytic degradation of adriamycin. Journal of
Pharmacy and Pharmacology. 1980. Vol. 32, n° 1, pp. 860�862.
TERASAKI T., IGA T., SUGIYAMA Y. et HANANO M. Pharmacokinetic study on the mechanism of tissue distribution of doxorubicin: interorgan and interspecies variation of tissue-to-plasma partition coefficients in rats, rabbits, and guinea pigs. Journal of Pharmaceutical Sciences. 1984. Vol. 73, n° 10, pp. 1359�1363.
TESKE E., RUTTEMAN G.R., KIRPENSTEIN J. et HIRSCHBERGER J. A randomized controlled study into the efficacy and toxicity of pegylated liposome encapsulated doxorubicin as an adjuvant therapy in dogs with splenic haemangiosarcoma. Veterinary and Comparative Oncology. 2011. Vol. 9, n° 4, pp. 283�289.
TEW K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Research. 1994. Vol. 54, n° 16, pp. 4313�4320.
TEWEY K.M., ROWE T.C., YANG L., HALLIGAN B.D. et LIU L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984. Vol. 226, n° 4673, pp. 466�468.
THORN C.F., OSHIRO C., MARSH S., HERNANDEZ-BOUSSARD T., MCLEOD H., KLEIN T.E. et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenetics and Genomics. 2011. Vol. 21, n° 7, pp. 440�446.
TIMOUR Q., NONY P., LANG J., LAKHAL M., TRILLET V. et FAUCON G. Doxorubicin concentrations in plasma and myocardium and their respective roles in cardiotoxicity. Cardiovascular Drugs and
Therapy. 1988. Vol. 1, n° 5, pp. 559�560.
TOKARSKA-SCHLATTNER M., WALLIMANN T. et SCHLATTNER U. Alterations in myocardial energy metabolism induced by the anti-cancer drug doxorubicin. Comptes Rendus Biologies. 2006. Vol. 329, n° 9, pp. 657�668.
TOKARSKA-SCHLATTNER M., WALLIMANN T. et SCHLATTNER U. Multiple interference of anthracyclines with mitochondrial creatine kinases: preferential damage of the cardiac isoenzyme and its implications for drug cardiotoxicity. Molecular Pharmacology. 2002. Vol. 61, n° 3, pp. 516�523.
TOMIYASU H., GOTO-KOSHINO Y., TAKAHASHI M., FUJINO Y., OHNO K. et TSUJIMOTO H. Quantitative analysis of mRNA for 10 different drug resistance factors in dogs with lymphoma. The Journal of
Veterinary Medical Science. 2010. Vol. 72, n° 9, pp. 1165�1172.
TRITTON T.R. et HICKMAN J.A., 1985. Cell Surface Membranes as a Chemotherapeutic Target. In : Experimental and Clinical Progress in Cancer Chemotherapy [en ligne]. Springer, Boston, MA. Cancer Treatment and Research. pp. 81�131.
UBEZIO P. et CIVOLI F. Flow cytometric detection of hydrogen peroxide production induced by doxorubicin in cancer cells. Free Radical Biology & Medicine. 1994. Vol. 16, n° 4, pp. 509�516.
UOZURMI K., NAKAICHI M., YAMAMOTO Y., UNE S. et TAURA Y. Development of multidrug resistance in a canine lymphoma cell line. Research in veterinary science, 2005.

163

164
URVA S.R., SHIN B.S., YANG V.C. et BALTHASAR J.P. Sensitive high performance liquid chromatographic assay for assessment of doxorubicin pharmacokinetics in mouse plasma and tissues. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 2009. Vol. 877, n° 8�9, pp. 837�841.
VAIL D.M., KRAVIS L.D., COOLEY A.J., CHUN R. et MACEWEN E.G. Preclinical trial of doxorubicin entrapped in sterically stabilized liposomes in dogs with spontaneously arising malignant tumors. Cancer Chemotherapy and Pharmacology. 1997. Vol. 39, n° 5, pp. 410�416.
VALERIUS K.D., OGILVIE G.K., MALLINCKRODT C.H. et GETZY D.M. Doxorubicin alone or in combination with asparaginase, followed by cyclophosphamide, vincristine, and prednisone for treatment of multicentric lymphoma in dogs: 121 cases (1987-1995). Journal of the American
Veterinary Medical Association. 1997. Vol. 210, n° 4, pp. 512�516.
VAN ASPEREN J., VAN TELLINGEN O. et BEIJNEN J.H. The role of mdr1a P-glycoprotein in the biliary and intestinal secretion of doxorubicin and vinblastine in mice. Drug Metabolism and Disposition: The
Biological Fate of Chemicals. 2000. Vol. 28, n° 3, pp. 264�267.
VÁSQUEZ-VIVAR J., MARTASEK P., HOGG N., MASTERS B.S., PRITCHARD K.A. et KALYANARAMAN B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry. 1997. Vol. 36, n° 38, pp. 11293�11297.
VAUGHAN A., JOHNSON J.L. et WILLIAMS L.E. Impact of chemotherapeutic dose intensity and hematologic toxicity on first remission duration in dogs with lymphoma treated with a chemoradiotherapy protocol. Journal of Veterinary Internal Medicine. 2007. Vol. 21, n° 6, pp. 1332�1339.
VAVROVA A., JANSOVA H., MACKOVA E., MACHACEK M., HASKOVA P., TICHOTOVA L. et al. Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PloS One. 2013. Vol. 8, n° 10, pp. e76676.
VENABLE R.O., SABA C.F., ENDICOTT M.M. et NORTHRUP N.C. Dexrazoxane treatment of doxorubicin extravasation injury in four dogs. Journal of the American Veterinary Medical Association. 2012. Vol. 240, n° 3, pp. 304�307.
VERGELY C. et ROCHETTE L. Stress oxydant dans le domaine cardiovasculaire. Médecine
thérapeutique Cardiologie. 2003. Vol. 1, n° 3, pp. 131�139.
VILLALOBOS A. Dealing with chemotherapy extravasations: a new technique. Journal of the American
Animal Hospital Association. 2006. Vol. 42, n° 4, pp. 321�325.
WAGNER B.A., EVIG C.B., RESZKA K.J., BUETTNER G.R. et BURNS C.P. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Archives of Biochemistry and Biophysics. 2005. Vol. 440, n° 2, pp. 181�190.
WALKER J.V. et NITISS J.L. DNA topoisomerase II as a target for cancer chemotherapy. Cancer
Investigation. 2002. Vol. 20, n° 4, pp. 570�589.
WALLACE K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovascular Toxicology. 2007. Vol. 7, n° 2, pp. 101�107.
WANG G.X., WANG Y.X., ZHOU X.B. et KORTH M. Effects of doxorubicinol on excitation--contraction coupling in guinea pig ventricular myocytes. European Journal of Pharmacology. 2001. Vol. 423, n° 2�3, pp. 99�107.

165
WANG J.C. Interaction between DNA and an Escherichia coli protein ω. Journal of Molecular Biology. 1971. Vol. 55, n° 3, pp. 523-IN16.
WANG S., KOTAMRAJU S., KONOREV E., KALIVENDI S., JOSEPH J. et KALYANARAMAN B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochemical Journal. 2002. Vol. 367, n° Pt 3, pp. 729�740.
WANG S.-L., LEE J.-J. et LIAO A.T. Comparison of efficacy and toxicity of doxorubicin and mitoxantrone in combination chemotherapy for canine lymphoma. The Canadian Veterinary Journal, 2016. Vol. 57, n° 3, pp. 271�276.
WATT P.M. et HICKSON I.D. Structure and function of type II DNA topoisomerases. Biochemical
Journal. 1994. Vol. 303, n° Pt 3, pp. 681�695.
WENDELBURG K.M., PRICE L.L., BURGESS K.E., LYONS J.A., LEW F.H. et BERG J. Survival time of dogs with splenic hemangiosarcoma treated by splenectomy with or without adjuvant chemotherapy: 208 cases (2001-2012). Journal of the American Veterinary Medical Association. 2015. Vol. 247, n° 4, pp. 393�403.
WHITE J.H. Self-Linking and the Gauss Integral in Higher Dimensions. American Journal of
Mathematics. 1969. Vol. 91, n° 3, pp. 693�728.
WITHROW, SJ et PAGE, RL. Withrow and MacEwen’s Small Animal Clinical Oncology. Elsevier Health Sciences. 2013.
WITTENBURG L.A., THAMM D.H. et GUSTAFSON D.L. Development of a limited-sampling model for prediction of doxorubicin exposure in dogs. Veterinary and Comparative Oncology. 2014. Vol. 12, n° 2, pp. 114�119.
WOOD M.J., IRWIN W.J. et SCOTT D.K. Photodegradation of doxorubicin, daunorubicin and epirubicin measured by high-performance liquid chromatography. Journal of Clinical Pharmacy and
Therapeutics. 1990. Vol. 15, n° 4, pp. 291�300.
WU J., TANG C. et YIN C. Co-delivery of doxorubicin and interleukin-2 via chitosan based nanoparticles for enhanced antitumor efficacy. Acta Biomaterialia. 2017. Vol. 47, pp. 81�90.
YANG F., TEVES S.S., KEMP C.J. et HENIKOFF S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochimica et Biophysica Acta - Reviews on Cancer. 2014. Vol. 1845, n° 1, pp. 84�89.
YING Y. et PADANILAM B.J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cellular and molecular life sciences: CMLS. 2016. Vol. 73, n° 11�12, pp. 2309�2324.
YOUNGMAN R.J. et ELSTNER E.F. On the interaction of adriamycin with DNA: investigation of spectral changes. Archives of Biochemistry and Biophysics. 1984. Vol. 231, n° 2, pp. 424�429.
ZANDVLIET M. et TESKE E. Mechanisms of Drug Resistance in Veterinary Oncology— A Review with an Emphasis on Canine Lymphoma. Veterinary Sciences. 2015. Vol. 2, n° 3, pp. 150�184.
ZANDVLIET M., TESKE E., SCHRICKX J.A. et MOL J.A. A longitudinal study of ABC transporter expression in canine multicentric lymphoma. Veterinary Journal. 2015. Vol. 205, n° 2, pp. 263�271.
ZHANG Y.-W., SHI J., LI Y.-J. et WEI L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Archivum immunologiae et therapiae experimentalis. 2009. Vol. 57, n° 6, pp. 435�445.

166
ZHOU Q. et CHOWBAY B. Determination of doxorubicin and its metabolites in rat serum and bile by LC: application to preclinical pharmacokinetic studies. Journal of Pharmaceutical and Biomedical
Analysis. 2002. Vol. 30, n° 4, pp. 1063�1074.
ZHU H., SARKAR S., SCOTT L., DANELISEN I., TRUSH M.A., JIA Z. et al. Doxorubicin Redox Biology: Redox Cycling, Topoisomerase Inhibition, and Oxidative Stress. Reactive Oxygen Species. 2016. Vol. 1, n° 3, pp. 189�198.
ZHU W., ZOU Y., AIKAWA R., HARADA K., KUDOH S., UOZUMI H. et al. MAPK superfamily plays an important role in daunomycin-induced apoptosis of cardiac myocytes. Circulation. 1999. Vol. 100, n° 20, pp. 2100�2107.
ZHU W.Z., ZHENG M., KOCH W.J., LEFKOWITZ R.J., KOBILKA B.K. et XIAO R.P. Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proceedings of
the National Academy of Sciences of the United States of America. 2001. Vol. 98, n° 4, pp. 1607�1612.
Committee for Veterinary Medicinal Products. Guidelines for the conduct of pharmacokinetic studies in target animal species, 2000 [Internet]. Disponible sur : http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004355.pdf
Committee for Medicinal Products for Human Use. Guideline on bioanalytical method validation, 2011 [Internet]. Disponible sur : http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf Veterinary cooperative oncology group– common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1, 2011 [Internet]. Disponible sur : http://onlinelibrary.wiley.com/doi/10.1111/j.1476-5810.2004.0053b.x/pdf

167
ANNEXESANNEXESANNEXESANNEXES

166

Annexe 1 : Classification VCOG-CTCAE (Veterinary cooperative oncology group – common terminology criteria for adverse events) version 1.1, des effets indésirables secondaires à la chimiothérapie (VCOG, 2001). Ne sont présentes que les parties concernant la toxicité liée à l’injection, la toxicité gastro-intestinale, la myélotoxicité, la toxicité cutanée et la toxicité cardiaque, correspondants aux effets indésirables de la doxorubicine.
167

168

169

170

171

172

173

176

177
Annexe 2 : Accord par le Comité d’éthique en expérimentation animale de VetAgro-Sup (n°1648).

176

179
Annexe 3 : Questionnaire présenté au propriétaire permettant d’évaluer les effets secondaires de l’administration de doxorubicine.

178

Annexe 4 : Numération-formule sanguine et biochimie sanguine lors de l’admission des cas dans l’étude et au cours des suivis à une semaine et trois semaines. Les valeurs en rouge correspondent aux valeurs significativement différentes de l’intervalle de référence.
179

180

183

CHAVALLE Thomas
CONTRIBUTION À L’ÉTUDE DE LA PHARMACOCINÉTIQUE ET D E LA TOLÉRANCE DE LA DOXORUBICINE CHEZ LE CHIEN Thèse d’Etat de Doctorat Vétérinaire : Lyon, le 13 octobre 2017
RESUME : La doxorubicine est un anti-cancéreux communément utilisé en médecine vétérinaire pour le traitement des lymphomes et de diverses tumeurs solides. Les mécanismes d’action sont présentés de façon détaillée, ainsi que les éléments de pharmacocinétique chez les rongeurs et l’Homme. Cependant, malgré son utilisation répandue, il n’existe que très peu d’études sur la pharmacocinétique de cet anticancéreux chez le chien non sain. Les protocoles analytiques utilisés lors de ces études sont basés sur des protocoles développés chez l’Homme et dans l’espèce murine. Le développement d’une méthode analytique chez le chien permet son application à des études de pharmacocinétique de la doxorubicine dans l’espèce cible. Cette étude expérimentale permet une première description de la pharmacocinétique de la doxorubicine chez le chien non sain et les éventuelles relations entre sa pharmacocinétique et sa toxicité. C’est une étude préliminaire pour un travail futur sur l’améliorati on des stratégies thérapeutiques en cancérologie.
MOTS CLES : - Doxorubicine - Toxicité - Chimiothérapie - Chien - Pharmacocinétique
JURY : Président : Monsieur le Professeur Serge LEBECQUE 1er Assesseur : Monsieur le Professeur Philippe BERNY 2ème Assesseur : Madame le Professeur Frédérique PONCE
DATE DE SOUTENANCE : 13 octobre 2017
ADRESSE DE L’AUTEUR : 245 chemin des Marronniers
69280 Marcy l’étoile
Recommended

















