
DESACTIVACIÓN CATALÍTICA
AUTORES:
Chávez Cortavitarte, Taylor
Corcuera Valderrama, Carlos
Leyva Vallejos, Alejandro
Mego Monsalve, Alex

RESUMEN 3
INTRODUCCIÓN 7
CAPÍTULO 1: DESACTIVACIÓN CATALÍTICA 8
1.1.Envenenamiento 10
1.2.Ensuciamiento 13
1.3.Degradación térmica 22
1.4.Reacciones de vapor-sólido y/o sólido-sólido 27
1.5.Desgaste/trituración 32
CONCLUSIONES 40
REFERENCIAS BIBLIOGRÁFICAS 41
2013 DESACTIVACIÓN CATALÍTICA
Página 1Universidad Nacional de Trujillo

RESUMEN
Los mecanismos de desactivación del catalizador son muchas, sin embargo, se pueden
agrupar en seis mecanismos intrínsecos de deterioro del catalizador:
(i) intoxicación
(ii) ensuciamiento
(iii) degradación térmica
(iv) reacciones vapor - sólido y / o sólido-sólido
(v) desgaste / trituración
El envenenamiento es la fuerte quimisorción de los reactivos, productos o impurezas en
los sitios disponibles de otro modo para la catálisis. Dependiendo de la concentración de
veneno, la intoxicación puede ser rápida o lenta, dependiendo de la fuerza de adsorción
veneno, el envenenamiento puede ser reversible o irreversible. Mecánicamente, el
envenenamiento es un proceso complejo que involucra:
(a) bloqueo físico de uno o más sitios catalíticos por fuertemente adsorción.
(b) la modificación electrónica de los átomos vecinos más cercanos.
(c) la reestructuración de la superficie adsorbente.
(d) impedimento de la difusión superficial de reactantes, impidiendo de este modo la
reacción.
Dado que un número de los venenos comunes tales como los compuestos de coque,
azufre y arsénico son adsorbidos fuertemente y de forma irreversible , la intoxicación se
puede prevenir a través de la purificación de la corriente reactiva por medio de
depuradores o lechos de protección, en lugar de tratar de eliminar el veneno del
catalizador después de los hechos .
El ensuciamiento es la deposición física de las especies de carbón de fluido sobre la
superficie del catalizador, lo que resulta en la pérdida de actividad debida al bloque de
sitios y/o poros activos.
2013 DESACTIVACIÓN CATALÍTICA
Página 2Universidad Nacional de Trujillo

En sus etapas avanzadas puede resultar en la desintegración de partículas de catalizador
y el taponamiento de los huecos de los reactores. La deposición de carbono y la
formación de coque se producen a tasas relativamente bajas en condiciones de reacción
favorables, sin embargo, en condiciones desfavorables , las altas tasas pueden conducir a
una falla catastrófica del catalizador y el taponamiento de los huecos de los reactores que
llevan a la parada en cuestión de horas .
Mecánicamente, la deposición de carbono en metales soportados y la formación de coque
en zeolitas son muy diferentes. El primero consiste en la disociación de CO o
hidrocarburos en la superficie de metal en forma de carbono que puede polimerizar a
formas de carbono no deseado, tales como el grafito o de filamentos. Esta última (la
formación de coque sobre las zeolitas) se produce por una serie de reacciones de
carbocatión de radicales libres en los sitios del ácido, incluyendo la deshidrogenación,
ciclación, aromatización y la formación de compuestos aromáticos polinucleares.
Algunas claves para la prevención de la deposición de carbono y la formación de coque
incluyen : (a) servicio bajo condiciones que minimizan la formación, por ejemplo, en
proporciones suficientemente altas de H2/CO en la síntesis de FT para que los
precursores de la formación de carbono inactivo se mantengan a una baja cobertura por
gasificación con hidrógeno, (b) la optimización del diseño de catalizador, por ejemplo, en
el caso de las zeolitas, la optimización de la acidez a la formación de coque minimizar , y
(c) purificar la alimentación eliminando los precursores que aceleran la formación de
carbono o coque, por ejemplo, eliminación de compuestos aromáticos polinucleares de la
alimentación de un proceso de hidrocraqueo o de hidrotratamiento que de otro modo
reacciona rápidamente en sitios ácidos para formar coque.
Cuando la formación de coque no se evita fácilmente (por ejemplo, craqueo catalítico) la
desactivación es relativamente reversible por regeneración a través de combustión a baja
temperatura cuidadosamente controlada de coque depositado en el aire.
2013 DESACTIVACIÓN CATALÍTICA
Página 3Universidad Nacional de Trujillo

La desactivación térmicamente de catalizadores es resultado de (i) la pérdida de
superficie catalítica debido al crecimiento de cristal de la fase catalítica, (ii) la pérdida de
área de apoyo debido al colapso de apoyo y de la superficie catalítica debido al colapso
de los poros en cristalitos de la fase activa, y/o (iii) transformaciones químicas de fases
catalíticas a fases no catalíticos. Los dos primeros procesos se denominan típicamente
como "sinterización”. Los procesos de sinterización generalmente tienen lugar a altas
temperaturas de reacción (por ejemplo,> 500 ◦ C) y se aceleran generalmente por la
presencia de vapor de agua.
Tres mecanismos principales de crecimiento de los cristalitos de metal han sido
estudiadas: (1) la migración cristal, (2) la migración atómica, y (3) a temperaturas muy
altas de transporte de vapor. Redispersión, a la inversa del crecimiento de los cristalitos
en la presencia de O2 y/o Cl2, puede involucrar (a) la formación de óxido de metal o metal
volátil complejos de cloruro que se adhieren al soporte y, posteriormente, se
descomponen a pequeños cristalitos en la reducción y/o (b) la formación de partículas de
óxido o películas que rompen en pequeños cristalitos durante la posterior reducción. El
crecimiento de los cristalitos de metal sobre un soporte podría incluir, en principio, una
combinación de los tres mecanismos de sinterización que operan simultáneamente, a
pesar de las tasas relativas dependerán de las condiciones de reacción. Temperatura
ambiente, tipo de metal, la dispersión de metal, promotores / impurezas y soporte de área
de superficie, la textura y la porosidad, son los parámetros principales que afectan a las
tasas de sinterización y redispersión. Las tasas de sinterización aumentan de forma
exponencial con la temperatura; energías de activación son del orden de 15-100 kJ / mol.
Metales sinterizan relativamente rápidamente en oxígeno o vapor de agua y relativamente
lentamente en hidrógeno. Los promotores tales como MgO o BaO tasas de sinterización
inferiores al disminuir la movilidad átomo de metal, mientras que los defectos de la
superficie de apoyo o poros impedían la migración superficial de las partículas de metal.
En general, los procesos de sinterización son lentos a temperaturas de reacción
moderadas y sea irreversible o difícilmente reversible. Por lo tanto, la sinterización es más
fácil de prevenir que curar, la clave es maximizar catalítica actividad suficiente para
permitir el funcionamiento a temperaturas suficientemente bajas que las tasas de
sinterización son insignificantes.
2013 DESACTIVACIÓN CATALÍTICA
Página 4Universidad Nacional de Trujillo

Además de la intoxicación, hay otras rutas químicas que conducen a la desactivación del
catalizador:
(1) Las reacciones de la fase de vapor con la superficie del catalizador a las fases: (a) a
granel y de la superficie inactiva producir (en lugar de especies fuertemente
adsorbidos) y (b) compuestos volátiles que salen del catalizador y el reactor en la fase
de vapor.
(2) Soporte sólido o - catalíticos reacciones sólido – promotor.
(3) Transformaciones catalíticas de las fases catalíticas durante de reacción. Ejemplos de
estos cuatro fenómenos incluyen: la oxidación de metales Co soportado sobre sílice
por el agua producto a Co silicatos de superficie durante FT síntesis a alta conversión.
(4) La pérdida de Pt por formación de PtO2 volátil durante la oxidación del amoníaco
sobre catalizadores de gasa de Pt – Rh.
(5) La formación durante la síntesis de amoníaco en el catalizador de superficie
Fe/K/Al2O3 de KAlO2.
(6) La transformación reductiva de Mo18O52 a Mo4O11 durante la oxidación parcial de
propeno a acroleína. Estas formas de producto químico de desactivación se pueden
prevenir o moderados en gran parte a través del control cuidadoso de las condiciones
de reacción y el diseño apropiado del catalizador.
Se observó fallo mecánico de catalizadores en varias formas diferentes, incluyendo la
trituración de granulado, tabletas o formas catalizador monolítico debido a una carga, de
desgaste, la reducción de tamaño y/o ruptura de gránulos o pellets de catalizador para
producir multas, especialmente en el líquido o camas de lechada, y la erosión de
partículas de catalizador o revestimientos monolito a altas velocidades de fluido. Dos
mecanismos principales participan en el desgaste de los aglomerados de catalizador:
Fractura de aglomerados en aglomerados más pequeños de aproximadamente 0.2d0 -
0.8d0 y la erosión (o abrasión) de los agregados de partículas primarias que tienen
diámetros que van de 0,1 a 10 m de la superficie del aglomerado. Mientras que la erosión
es causada por tensiones mecánicas, la fractura puede ser debido a los esfuerzos
mecánicos, térmicos y/o químicos.
2013 DESACTIVACIÓN CATALÍTICA
Página 5Universidad Nacional de Trujillo

Tensiones mecánicas que conducen a la fractura o la erosión en lechos fluidos o
suspensión puede ser el resultado de la colisión de partículas entre sí o con las paredes
reactor o las fuerzas de cizallamiento creadas por remolinos turbulentos o burbujas
colapsan (cavitación) a altas velocidades de fluido
El grado en que un mecanismo, es decir la fractura o la erosión, participa en la reducción
de tamaño de aglomerado depende de varios factores: la magnitud de una tensión, la
fuerza y la tenacidad a la fractura del aglomerado, el tamaño de aglomeración y el área
superficial y grieta o tamaño de poro y el radio. Parece que existe un potencial
considerable para el fortalecimiento de los aglomerados de catalizador, ya que sus puntos
fuertes son generalmente factores de 3-50 inferiores para cerámicas convencionales, con
tratamiento térmico de porosidad similar.
Los cambios sutiles en la preparación, el tratamiento previo, y la fabricación pueden
mejorar en gran medida la fuerza de aglomerado catalizador. Algunas alternativas
prometedoras para aumentar catalizador desgaste resistencia y la fuerza incluyen el
aumento de la fuerza agregado/aglomerado por medio de métodos de preparación
avanzadas , por ejemplo, granulación de sol-gel , secado por pulverización y métodos de
precipitación cuidadosamente controlados, la adición de aglutinantes para mejorar la
fuerza y tenacidad de revestimiento agregados con un material poroso pero muy fuerte,
tal como ZrO2 , y química o templado térmico de aglomerados de introducir tensiones de
compresión que aumentan la fuerza y la resistencia a la atrición .
2013 DESACTIVACIÓN CATALÍTICA
Página 6Universidad Nacional de Trujillo

INTRODUCCIÓN
La desactivación del catalizador, la pérdida en el tiempo de la actividad catalítica y/o
selectividad, es un problema de gran preocupación en la práctica de los procesos
catalíticos industriales. Los costos para la sustitución de catalizadores implican miles de
millones de dólares en total por año para las industrias. Las escalas de tiempo para la
desactivación del catalizador varían considerablemente, por ejemplo, en el caso de
catalizadores de craqueo, la mortalidad del catalizador puede estar en el orden de
segundos, mientras que en la síntesis de amoniaco, el catalizador de hierro puede durar
5-10 años. Pero es inevitable que todos los catalizadores decaigan.
Típicamente, la pérdida de actividad en un proceso bien controlado se produce
lentamente. Sin embargo, los trastornos del proceso pueden provocar una falla
catastrófica. Por ejemplo, en la reforma con vapor de metano o de nafta se debe tomar
gran cuidado para evitar la operación del reactor a temperaturas excesivamente. De
hecho, estas condiciones pueden causar la formación de grandes cantidades de
filamentos de carbono que se conectan a los poros del catalizador y espacios vacíos,
llevando a un proceso de apagado dentro de unas pocas horas.
Mientras que la desactivación del catalizador es inevitable para la mayoría de los
procesos, algunas de sus inmediatas y drásticas consecuencias pueden evitarse,
aplazarse o incluso invertirse. Por lo tanto, los problemas de desactivación (es decir, nivel,
frecuencia y reactivación) tendrán gran influencia en la investigación, desarrollo, diseño y
el funcionamiento de los procesos comerciales. En consecuencia, hay una considerable
motivación para entender y tratar este tema. De hecho, en las últimas tres décadas, la
ciencia de la desactivación del catalizador se ha venido desarrollando de forma constante.
Una fracción importante de esta literatura se ocupa de los mecanismos de desactivación.
Esta área de investigación proporciona una comprensión crítica que es la base para los
procesos de desactivación.
2013 DESACTIVACIÓN CATALÍTICA
Página 7Universidad Nacional de Trujillo

CAPÍTULO 1
2013 DESACTIVACIÓN CATALÍTICA
Página 8Universidad Nacional de Trujillo

DESACTIVACIÓN CATALÍTICA
La prevención de la degradación del catalizador plantea retos sustanciales en el diseño y
el funcionamiento de un proceso catalítico a gran escala. Hay muchos caminos para el
decaimiento del catalizador. Por ejemplo, un catalizador puede ser envenenado por uno o
varios contaminantes presentes en la alimentación; su superficie, los poros y huecos
pueden ser ensuciados por carbono o coque producido por craqueo y/o reacciones de
condensación de los reactivos de hidrocarburos, productos intermedios y/o productos. En
el tratamiento de un gas de combustión en una planta de energía, el catalizador puede ser
espolvoreado o erosionado por cenizas volantes. Los convertidores catalíticos utilizados
para reducir las emisiones de los motores de gasolina o diésel pueden ser envenenados.
Si la reacción catalítica se lleva a cabo a altas temperaturas, la degradación térmica se
puede producir en la forma de crecimiento de los cristales. Además, la presencia de
oxígeno o de cloro en el gas de alimentación puede conducir a la formación de óxidos o
cloruros de la fase activa seguida por el transporte en fase gaseosa del reactor volátil. Del
mismo modo, los cambios en el estado de oxidación de la fase catalítica activa pueden
ser inducidos por la presencia de gases reactivos en la alimentación.
Por lo tanto, los mecanismos de desactivación del catalizador son muchas, sin embargo,
se pueden agrupar en seis mecanismos intrínsecos de deterioro del catalizador:
i. Envenenamiento
ii. Ensuciamiento
iii. Degradación térmica
iv. Reacciones de vapor-sólido y/o sólido-sólido
v. Desgaste/trituración
Los mecanismos (i), (iv), y (v) son de naturaleza química, mientras que (ii) y (v) son
mecánicos; las causas de desactivación son básicamente de tres tipos: química,
mecánica y térmica.
2013 DESACTIVACIÓN CATALÍTICA
Página 9Universidad Nacional de Trujillo

Cada uno de los seis mecanismos básicos se define brevemente en la Tabla 1 y se tratan
con cierto detalle en las subsecciones que siguen, con énfasis en los tres primeros. Los
mecanismos (iv) y (v) se tratan conjuntamente, ya que (iv) es un subconjunto de (v).
Tabla 1. Mecanismos de desactivación del catalizador
Tipo de mecanismoBreve
definiciónDescripción
Envenenamiento QuímicoFuerte quimisorción de las especies en los sitios catalíticos, bloqueando los sitios de reacción catalítica
Ensuciamiento MecánicoDeposición física de las especies de fase fluida en el catalizador superficie y en los poros del catalizador
Degradación térmica TérmicoPérdida térmica de la superficie catalítica, área de apoyo, yreacciones de fase de asistencia activos
Formación de vapor QuímicoLa reacción de fase de gas con el catalizador para producir el compuesto volátil
Reacciones vapor-sólidos y sólidos-sólidos
QuímicoLa reacción del fluido, el apoyo o promotor con la fase catalítica paraproducir fase inactiva
Desgaste / trituración Mecánico
La pérdida de material catalítico debido a la abrasión.La pérdida de área de superficie interna debido a la trituración mecánica inducidade la partícula de catalizador
1. ENVENENAMIENTO
Es la fuerte quimisorción de reactivos, productos o impurezas en los sitios catalíticos,
bloqueando los sitios de reacción catalítica. Por lo tanto, el envenenamiento tiene
significado operativo, es decir, si una especie actúa como un veneno depende de su
fuerza de adsorción en relación con las otras especies que compiten por los sitios
catalíticos.
2013 DESACTIVACIÓN CATALÍTICA
Página 10Universidad Nacional de Trujillo

Por ejemplo, el oxígeno puede ser un reactivo en la oxidación parcial de etileno a óxido de
etileno sobre un catalizador de plata y un veneno en la hidrogenación de etileno en el
níquel. Además para el bloqueo físico de los sitios de adsorción, el veneno adsorbido
puede inducir cambios en la estructura electrónica o geométrica de la superficie.
Los mecanismos por los que un veneno puede afectar a la actividad catalítica son
múltiples, como se ilustra para un modelo de envenenamiento por azufre en la
hidrogenación de etileno sobre una superficie de metal en la figura 1. Para empezar, un
átomo de azufre fuertemente adsorbido físicamente forma bloques de al menos una de
tres o cuatro veces el sitio de adsorción / reacción. En segundo lugar, en virtud de su
fuerte enlace químico, modifica electrónicamente sus átomos más cercanos de metal
vecino y, posiblemente, sus próximos átomos vecinos más cercanos, modificando de este
modo su capacidad para adsorber y/o disociar moléculas de reactivo (en este caso H2 y
etileno moléculas. Un tercer efecto puede ser la reestructuración de la superficie por la
fuerte adsorción del veneno, lo que puede causar cambios dramáticos en las propiedades
catalíticas, especialmente para reacciones sensibles a la estructura de la superficie.
Además, el veneno adsorbido bloquea el acceso del reactivo (cuarto efecto) y, finalmente,
impide o retarda la difusión superficial del reactivo adsorbido (quinto efecto).
Figura 1. Modelo conceptual de la intoxicación por átomos de azufre de una superficie de metal
durante la hidrogenación de etileno.
Los venenos de catalizador pueden ser clasificados de acuerdo con su composición
química, la selectividad para los sitios activos y los tipos de reacciones envenenadas. La
tabla 2 enumera los cuatro grupos de venenos de catalizador clasificados según su origen
químico y su tipo de interacción con metales.
2013 DESACTIVACIÓN CATALÍTICA
Página 11Universidad Nacional de Trujillo

Debe hacerse hincapié en que las interacciones de los elementos del grupo VA - VIIIA con
fases de metal catalíticos depende en el estado de oxidación, es decir, cuántos pares de
electrones están disponibles para la unión y el grado de protección de los iones de azufre
por ligandos. Por lo tanto, el orden de disminución de la toxicidad para el envenenamiento
de un metal dado por diferentes especies de azufre es de H2S, SO2, SO4-2, es decir, en el
orden de aumento de blindaje por el oxígeno. La toxicidad aumenta con el aumento de
tamaño y la electronegatividad atómica o molecular, pero disminuye si el veneno puede
ser gasificado por O2, H2O o H2 presente en la corriente de reaccionantes.
Tabla 2. Cuatro grupos de venenos de catalizador
Tipo de química Ejemplo Tipo de interacción con metales
Grupos VA y VIAN, P, As, Sb, O, S, Se, Te
A través de s-y p-orbitales; estructuras apantallados son menos tóxicos
Grupo VIIA F, Cl, Br, IA través de s-y p-orbitales; formación de haluros volátiles
Metales y iones pesados tóxicos
As, Pb, Hg, Bi, Sn, Zn, Cd, Cu, Fe
Ocupar orbitales d; puede formar aleaciones
Las moléculas que se adsorben con
CO, NO, HCN, benceno, acetileno,
Quimisorción través de enlaces múltiples y la vinculación de nuevo
Enlaces múltiplesOtros hidrocarburos insaturados
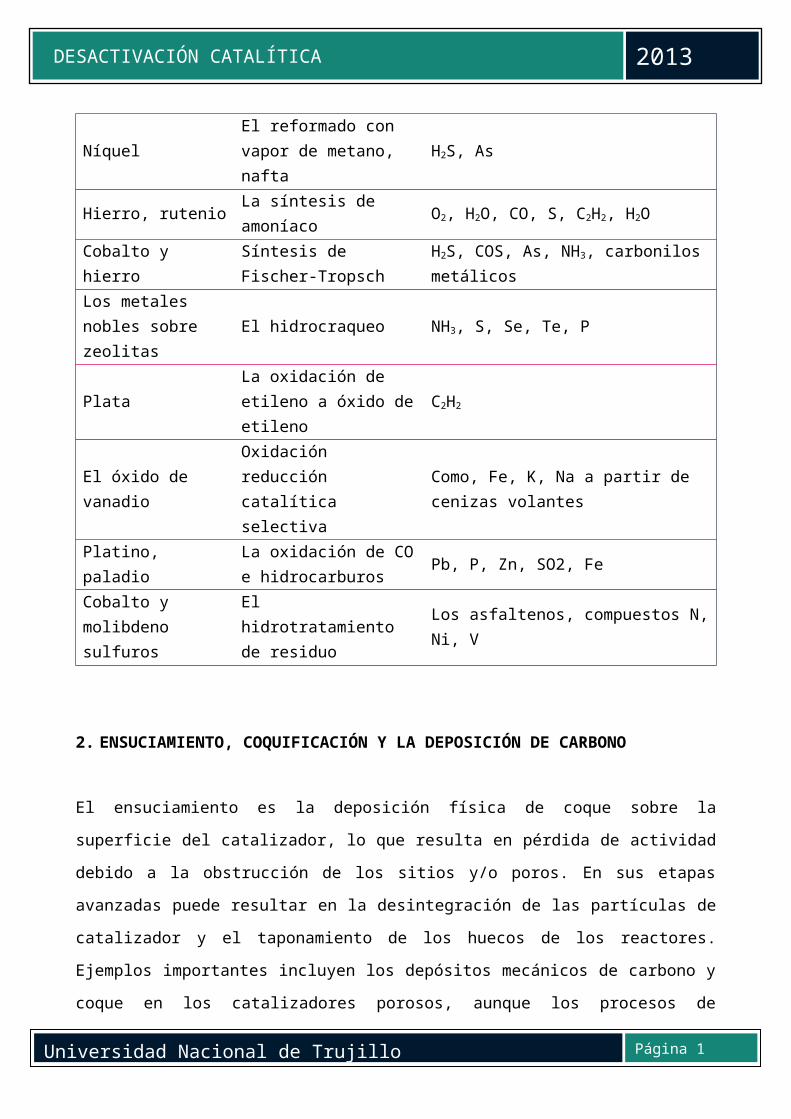
La tabla 3 enumera una serie de venenos comunes para catalizadores seleccionados. Es
evidente que las bases orgánicas (por ejemplo aminas y amoniaco) son venenos
comunes para sólidos ácidos tales como sílice - alúmina y zeolitas en agrietamiento,
mientras azufre y compuestos que contienen arsénico son venenos típicos para metales
en hidrogenación, deshidrogenación y reacciones de reformado con vapor.
Compuestos de metal (por ejemplo, Ni, Pb, V y Zn) son venenos en el control de
emisiones de los automóviles, el craqueo catalítico e hidrotratamiento. El acetileno es un
veneno para la oxidación de etileno, mientras que los asfaltenos son venenos en el
hidrotratamiento de residuales de petróleo.
2013 DESACTIVACIÓN CATALÍTICA
Página 12Universidad Nacional de Trujillo

Tabla 3. Venenos para los catalizadores seleccionados en importantes reacciones representativas
Catalizador Reacción VenenosLa sílice-alúmina, zeolitas
AgrietamientoBases orgánicas, hidrocarburos metales pesados
Níquel, platino, paladio
La hidrogenación deshidrogenación
Los compuestos de S, P, As, Zn, Hg, halogenuros, Pb, NH3, C2H2
NíquelEl reformado con vapor de metano, nafta
H2S, As
Hierro, rutenio La síntesis de amoníaco O2, H2O, CO, S, C2H2, H2O
Cobalto y hierroSíntesis de Fischer-Tropsch
H2S, COS, As, NH3, carbonilos metálicos
Los metales nobles sobre zeolitas
El hidrocraqueo NH3, S, Se, Te, P
PlataLa oxidación de etileno a óxido de etileno
C2H2
El óxido de vanadioOxidación reducción catalítica selectiva
Como, Fe, K, Na a partir de cenizas volantes
Platino, paladioLa oxidación de CO e hidrocarburos
Pb, P, Zn, SO2, Fe
Cobalto y molibdeno sulfuros
El hidrotratamiento de residuo
Los asfaltenos, compuestos N, Ni, V
2. ENSUCIAMIENTO, COQUIFICACIÓN Y LA DEPOSICIÓN DE CARBONO
El ensuciamiento es la deposición física de coque sobre la superficie del catalizador, lo
que resulta en pérdida de actividad debido a la obstrucción de los sitios y/o poros. En sus
etapas avanzadas puede resultar en la desintegración de las partículas de catalizador y el
taponamiento de los huecos de los reactores. Ejemplos importantes incluyen los depósitos
mecánicos de carbono y coque en los catalizadores porosos, aunque los procesos de
formación de coque de carbono también implican quimisorción de diferentes tipos de
átomos de carbono o hidrocarburos condensados que pueden actuar como venenos de
catalizador. El carbono es típicamente un producto de CO mientras que el coque se
produce por la descomposición o la condensación de hidrocarburos en superficies
catalíticas y por lo general consiste de hidrocarburos pesados polimerizados. Sin
embargo, las formas de coque pueden variar de hidrocarburos de alto peso molecular a
principalmente átomos de carbono, como el grafito, dependiendo de la condiciones bajo
las que se formó el coque.
2013 DESACTIVACIÓN CATALÍTICA
Página 13Universidad Nacional de Trujillo

Las estructuras químicas de coques o átomos de carbono formadas en los procesos
catalíticos varían con el tipo de reacción, tipo de catalizador, y las condiciones de
reacción. Menon menciona que las reacciones catalíticas acompañadas de la formación
de carbón o coque pueden ser ampliamente clasificadas como coque sensible o coque
insensible. Ejemplos de reacciones de coque sensibles incluyen craqueo catalítico e
hidrogenólisis, por el otro lado, la síntesis de Fischer-Tropsch, de reformado catalítico y de
síntesis de metanol son ejemplos de reacciones de coque insensible. De acuerdo con la
clasificación de Menon, también se observó que en general no sólo la estructura y la
ubicación de coque varían, sino también su mecanismo de formación varía con el tipo de
catalizador. Debido a estas diferencias significativas en el mecanismo, la formación de
carbono y el coque es discontinua.
a) Formación de carbón y coque en los catalizadores metálicos soportados
Uno de los posibles efectos de ensuciamiento en el funcionamiento de un catalizador
metálico soportado se ilustra en la figura 2. El carbono puede unirse fuertemente como
una capa físicamente o adsorberse en multicapas, y en cualquier caso bloquear el acceso
de los reactivos a los sitios de la superficie de metal, encapsular totalmente una partícula
de metal y por lo tanto completamente desactivar esa partícula y el enchufe micro y meso-
poros, de tal manera que el acceso de los reactivos se niega a muchos cristalitos dentro
de estos poros.
Figura 2. Modelo conceptual de la incrustación, la encapsulación de los cristalitos y obstrucción
del poro de un catalizador metálico soportado debido a la deposición de carbono.
2013 DESACTIVACIÓN CATALÍTICA
Página 14Universidad Nacional de Trujillo

Los mecanismos de la deposición de carbón y coque en los catalizadores de metales de
monóxido de carbono e hidrocarburos se ilustran en las figuras 3 y 4. Se aprecian
diferentes tipos de carbono y coque que varían en la morfología y la reactividad (ver
Tablas 6 y 7). Por ejemplo, el CO se disocia en los metales para formar C, un carbono
atómico adsorbido; C puede reaccionar en una película de carbono polimérico. Las formas
más reactivas son carbonos amorfos formados a temperaturas bajas, que pueden ser
convertidos en altas temperaturas durante un período de tiempo a formas menos
reactivas, como el grafito.
Figura 3. Formación, transformación y gasificación de carbón en níquel (a, g, s se refieren a
estados adsorbidos, gaseosos y sólidos, respectivamente)
Figura 4. Formación y transformación de coque en las superficies metálicas (a, g, s se refieren a
adsorbido, estados gaseosos y sólidos, respectivamente), reacciones en fase gaseosa no se
consideran.
2013 DESACTIVACIÓN CATALÍTICA
Página 15Universidad Nacional de Trujillo

Tabla 4. Formas y reactividad de las especies de carbono formado por la descomposición de CO
en el níquel.
Tipo estructural DesignaciónTemperatura de formación (°C)
Temperatura pico (°C) para reacción con H2
Adsorbido, atómica (carburo de superficie)
C 200 - 400 200
Películas o filamentos poliméricos amorfos
C 250 - 500 400
Vermicular filamentos, fibras y/o barbas
Cv 300 - 1000 400 - 600
Carburo de níquel(a granel)
C 150 - 250 275
Grafíticos (cristalinos) plaquetas o películas
cc 500 - 550 550 - 850
Tabla 5. Especies de carbono formado en el reformado con vapor de hidrocarburos en
catalizadores de níquel.
Encapsulaciónde la película
Barba similar Carbón pirolítico
Formación
Lenta polimerización de radicales CnHm en la superficie de Ni.
Difusión de C a través del cristal de Ni, la nucleación y crecimiento de la barba con el cristal de Ni en la parte superior
El craqueo térmico de hidrocarburo; deposición de precursores C sobre el catalizador
EfectosDesactivación progresiva
Sin desactivación de la superficie de Ni; ruptura de catalizador y el aumento de P
La encapsulación de las partículas de catalizador;la desactivación y el aumento de P
Temperatura (°C)
< 500 > 450 > 600
Parámetros críticos
Baja temperatura Temperatura alta Temperatura altaBajo H2O/CnHm Bajo H2O/CnHm Alta fracción de vacío
Bajo H2/CnHmNo hay mayor adsorción de H2O
Bajo H2O/CnHm
Alimentaciónaromática
Alimentaciónaromática
Catalizador ácido
b) Formación de coque en los catalizadores de óxido y sulfuro de metal
2013 DESACTIVACIÓN CATALÍTICA
Página 16Universidad Nacional de Trujillo

En las reacciones que implican hidrocarburos, el coque se puede formar en la fase
gaseosa y en superficies catalíticas y no catalíticas. Sin embargo, la formación de coque
sobre óxidos y sulfuros es principalmente un resultado de la formación de grietas por las
reacciones que implican precursores de coque (típicamente olefinas o compuestos
aromáticos). Reacciones de deshidrogenación y ciclación de carbocationes intermedios
formados en sitios ácidos conducen a los compuestos aromáticos que reaccionan con
más aromáticos de mayor peso molecular condensándose como coque (véase. Fig. 5).
2013 DESACTIVACIÓN CATALÍTICA
Página 17Universidad Nacional de Trujillo

Figura 5. Reacciones de formación de coque de alquenos y compuestos aromáticos sobre
catalizadores de óxido y sulfuro de: (a) polimerización de alquenos; (b) la ciclación a partir de
alquenos; (c) la formación de compuestos aromáticos polinucleares a partir de benceno (cortesía:
Kluwer Academic Publishers).
Las reacciones 1 - 3 en la figura 5 ilustran la polimerización de olefinas, las reacciones 4 -
8 ilustran ciclación a partir de olefinas y las reacciones 9 - 14 ilustran la formación de la
reacción en cadena de aromáticos polinucleares que se condensan como coque sobre la
superficie del catalizador. Debido a la alta estabilidad de los carbocationes polinucleares
(formados en reacciones de 10 a 13), puede seguir creciendo en la superficie durante un
tiempo relativamente largo antes de que ocurra una reacción final a través de la donación
de la parte de atrás de un protón.
2013 DESACTIVACIÓN CATALÍTICA
Página 18Universidad Nacional de Trujillo
A partir de este esquema (Fig. 5), está claro que las olefinas, el benceno y sus derivados
y compuestos aromáticos polinucleares son precursores de la formación de coque. Sin
embargo, el orden de reactividad de formación de coque es claramente dependiente de la
estructura, es decir:
Compuestos aromáticos polinucleares > aromáticos > olefinas ramificadas > alcanos >
alcanos normales.
Por ejemplo, la cantidad de coque formado sobre sílice/alúmina a 500 °C es 0,06, 3,8,
12,5, y 23 en %peso para el benceno, naftaleno, antraceno y antraceno, respectivamente.
Las reacciones de coque en los procesos que implican hidrocarburos pesados son muy
complejas; se pueden formar diferentes tipos de coque, estos pueden variar en
composición a partir de CH a C y tienen una amplia gama de reactividades con el oxígeno
e hidrógeno en función del tiempo de corrida y la temperatura a los que están expuestos.
Por ejemplo, los depósitos de coque que ocurren en la hidrodesulfuración de residuo se
han clasificado en tres tipos:
1. Tipo I Los depósitos son compuestos aromáticos normales reversiblemente adsorbidos
depositados durante la primera parte del ciclo a baja temperatura.
2. Depósitos de tipo II se adsorben reversiblemente asfaltenos depositados temprano en
el proceso de coquización.
3. Tipo III depósitos resultan de la condensación de los concentrados aromáticos en
grupos y luego los cristales que constituyen una "mesofase". Esta fase cristalina se forma
después de largos tiempos de reacción a alta temperatura. Este coque endurecido causa
la desactivación severa del catalizador.
En general, la velocidad y el grado de aumento de la formación de coque aumentan con la
fuerza del ácido y la concentración. El rendimiento de coque disminuye con la disminución
de tamaño de poro (para una intensidad de ácido y concentración fija); esto es
especialmente cierto en zeolitas donde la selectividad de forma juega un papel importante
en la formación de coque.
2013 DESACTIVACIÓN CATALÍTICA
Página 19Universidad Nacional de Trujillo

Por ejemplo, rendimiento de coque en el craqueo catalítico fluido es sólo 0,4 % de ZSM -
5 (diámetros de poro de 0,54 mm x 0,56 nm) en comparación con el 2,2 % para la
faujasita (diámetro de apertura de 0,72 nm). Sin embargo, en los poros de diámetro
molecular, una relativamente pequeña cantidad de coque puede causar pérdida sustancial
de actividad. Debe hacerse hincapié en que el rendimiento de coque puede variar
considerablemente en los poros interiores de una partícula de catalizador o a lo largo de
un lecho de catalizador dependiendo de la medida en que las principales reacciones de
desactivación se ven afectados por el transporte de masa de película y resistencia a la
difusión de poros.
Los mecanismos por los que el coque desactiva catalizadores de óxidos y sulfuros son,
como en el caso de metales soportados, tanto químicos como físicos. Sin embargo,
algunos aspectos de la química son muy diferentes. La pérdida química de actividad en
óxidos y sulfuros se debe a la fuerte adsorción de moléculas de coque sobre los sitios
ácidos.
Las pérdidas físicas de la actividad también se producen como acumulación de coque, en
última instancia, parcial o totalmente el bloqueo de poros del catalizador como en
catalizadores metálicos soportados.
Varios estudios recientes se han centrado en la formación de coque durante las
reacciones de hidrocarburos en zeolitas incluyendo (1) la química detallada de
precursores de coque y moléculas de coque formado en los poros de zeolita y las
intersecciones de poro (o súper-jaulas) y (2) la relación importancia de adsorción en sitios
ácidos frente a la obstrucción del poro. Las principales conclusiones de estos estudios se
pueden resumir de la siguiente manera: la formación de coque y la manera en que se
desactiva un catalizador de zeolita son procesos selectivos a la forma, la desactivación es
principalmente debida a la formación y la retención de grupos aromáticos pesados en los
poros y las intersecciones de los poros, y mientras que tanto la intoxicación con ácido
participan en la desactivación y las altas temperaturas, mientras que el último proceso
domina a altas velocidades de reacción , temperaturas bajas y altas coberturas de coque.
2013 DESACTIVACIÓN CATALÍTICA
Página 20Universidad Nacional de Trujillo

De hecho, la desactivación es típicamente más rápida en zeolitas que tienen pequeños
poros o aberturas y/o una estructura mono-dimensional. La figura. La figura 6 ilustra
cuatro posibles modos de desactivación de HZSM - 5 por depósitos carbonosos con
aumento de la gravedad de coquización de acuerdo.
Estas conclusiones (en el párrafo anterior) se confirman, por ejemplo, en el estudio
realizado por Cerqueira et al. de la desactivación ushy-zeolita durante transformación de
metilciclohexano a 450 °C que muestra lo siguiente:
1. El coque se forma probablemente por la rápida transformación de los iones carbenio
C7 etilénicos con menores contribuciones de reacciones de ciclopentadieno, olefinas
C3 - C6, y compuestos aromáticos.
2. El coque soluble consiste en grupos aromáticos polinucleares que contienen seis y
cincuenta y siete de cinco y seis miembros anillos que tienen una composición típica
de C30H40 a C40H44 y con unas dimensiones de 0,9 mm x 1,1 nm a 1,1 mm × 1,5 nm ,
es decir, los tamaños que hacen ser atrapado en los superjaulas de Y - zeolita .
3. En tiempo de contacto corto, la coquización es relativamente lenta y la desactivación
se debe principalmente a envenenamiento por ácido - sitio, mientras que en tiempos
de contacto largos, la coquización es mucho más rápido debido a las altas
concentraciones de precursores de coque; bajo estas últimas condiciones de coque
se deposita preferentemente en el exterior de las aberturas de los poros de cristalitos
de zeolita y la desactivación está dominada por la obstrucción de la boca de los
poros. El coque formado en tiempos de contacto grandes no sólo bloquea poros y/o
las intersecciones de poros dentro de la zeolita, sino también migra hacia el exterior
de los cristalitos de zeolita bloqueando las entradas. Sin embargo, la cantidad,
estructura y localización de coque en ZSM - 5 depende en gran medida del precursor
de coque, por ejemplo, coque formado a partir de mesitileno se deposita sobre la
superficie de la zeolita externa, mientras que coque de isobuteno conduce a depósitos
en gran medida parafínicos dentro de los poros; el coque a partir de tolueno, por otro
lado, es poliaromático y se deposita tanto en las superficies externas e internas de
zeolita.
2013 DESACTIVACIÓN CATALÍTICA
Página 21Universidad Nacional de Trujillo

Figura 6. Esquema de los cuatro posibles modos de desactivación por depósitos carbonosos en
HZSM-5: (1) de adsorción reversible de los sitios de ácido, (2) adsorción irreversible de los sitios
con bloqueo parcial de las intersecciones de poro, (3) de bloqueo estérico parcial de los poros, y
(4) extenso bloqueo estérico de los poros por los depósitos exteriores
3. LA DEGRADACIÓN TÉRMICA Y SINTERIZACIÓN
La desactivación térmicamente de catalizadores, resultados de (i) la pérdida de área de
superficie catalítica debido al crecimiento de los cristalitos de la fase catalítica, (ii) la
pérdida de área de soporte debido al colapso de apoyo y de área de superficie catalítica
debido al colapso de los poros en cristalitos de la fase activa, y/o (iii) las transformaciones
químicas de las fases catalíticas a fases no catalíticas. Los dos primeros procesos se
denominan típicamente como "sinterización". El tercero se discute en la siguiente sección
bajo las reacciones sólido-sólido. Procesos de sinterización tienen lugar generalmente a
temperaturas de reacción altas (por ejemplo > 500 °C) y se aceleran generalmente por la
presencia de vapor de agua.
Tres mecanismos principales de crecimiento de los cristalitos de metal han sido
estudiados: (1) la migración de los cristalitos, (2) la migración atómica, y (3) a
temperaturas muy altas de transporte de vapor. Los procesos de los cristalitos y la
migración atómica se ilustran en la figura. 7.
2013 DESACTIVACIÓN CATALÍTICA
Página 22Universidad Nacional de Trujillo

La migración de cristalito implica la migración de cristalitos completos por la superficie de
soporte seguido por colisión y coalescencia. La migración atómica implica
desprendimiento de átomos de metal de cristalitos, la migración de estos átomos sobre la
superficie de apoyo y en última instancia, la captura por cristalitos más grandes.
Figura 7. Dos modelos conceptuales para el crecimiento de los cristalitos debido a la sinterización
por (A) la migración atómica o (B) la migración de los cristalitos.
La redispersión, la inversa de crecimiento de los cristalitos en la presencia de O2 y/o Cl2,
puede implicar (1) la formación de óxido de metal volátil o complejos de cloruro de metal
que se adhieren al soporte y, posteriormente, se descomponen a los pequeños cristalitos
después de la reducción y/o (2) la formación de partículas de óxido o películas que
rompen en pequeños cristalitos durante la reducción posterior. Ha habido cierta
controversia en la literatura con respecto a qué mecanismo de sinterización (o
redispersión) funciona a una serie de condiciones dadas. Sin embargo, cada uno de los
tres mecanismos de sinterización (y dos mecanismos de dispersión) es una simplificación
que no tiene en cuenta la posibilidad de que se produzcan todos los mecanismos al
mismo tiempo y se puede acoplar entre sí a través de procesos físico-químicos complejos
que incluyen lo siguiente:
(1) la disociación y la emisión de metales átomos o moléculas que contienen metales de
cristalitos de metal,
(2) de adsorción y atrapamiento de átomos de metal o moléculas que contienen metales
sobre la superficie de apoyo,
(3) difusión de los átomos de metal, moléculas y/o cristalitos de metal a través de
superficies de soporte que contiene un metal,
2013 DESACTIVACIÓN CATALÍTICA
Página 23Universidad Nacional de Trujillo

(4) de partículas de óxido de metal o de metal difusión,
(5) humectación de la superficie de apoyo por metal o partículas de óxido de metal,
(6) la nucleación de partículas de metal,
(7), o coalescencia de puente entre, dos partículas de metal,
(8) de captura de átomos o moléculas por partículas de metal,
(9) la formación de líquido,
(10) la volatilización de metal a través de la formación de compuestos volátiles,
(11) de división de cristalitos en atmósfera de O2 debido a la formación de óxidos de un
volumen específico diferente, y
(12) de vaporización átomo de metal.
Dependiendo de las condiciones de reacción o de redispersión, algunos o todos estos
procesos pueden ser importantes, por lo tanto, la complejidad de los procesos de
sinterización/redispersión se enfatiza. En general, los procesos de sinterización son
cinéticamente lentos (a temperaturas de reacción moderadas) e irreversibles o difíciles de
revertir. Por lo tanto, la sinterización se evita más fácilmente que curar.
a) Los factores que afectan el crecimiento de partículas de metal y redispersión en
metales soportados.
Las tasas de sinterización aumentan de forma exponencial con la temperatura. Metales
sinterizan relativamente rápido en oxígeno y relativamente lentamente en hidrógeno,
aunque dependiendo de la ayuda, la redispersión de metal puede ser facilitada por la
exposición a alta temperatura (por ejemplo 500-550 °C durante Pt/Al2O3) al oxígeno y
cloro seguido de reducción. El vapor de agua también aumenta la velocidad de
sinterización de metales soportados.
La dispersión normalizada (porcentaje de la exposición en cualquier momento, dividido
por el porcentaje inicial de metal expuesto) de datos y al tiempo de la figura 4 muestran
que a temperaturas de 650 °C o más altas, tasas de superficie metálica con la pérdida de
área (medida por quimisorción de hidrógeno), debido a la sinterización de Ni/sílice en
atmósfera de hidrógeno son significativas, causando la pérdida de 70% del área de
superficie de metal original en el plazo 50 horas a 750 °C.
2013 DESACTIVACIÓN CATALÍTICA
Página 24Universidad Nacional de Trujillo

En atmósferas oxidantes, la estabilidad de los cristalitos depende de la volatilidad de los
óxidos metálicos y la fuerza de la interacción de óxido metálico de soporte.
Figura 8. Área normalizada de superficie de níquel (basado en la adsorción de H2) frente a los
datos de tiempo durante la sinterización de 13,5% Ni/SiO2 en H2 a 650, 700 y 750 °C
b) La sinterización de soportes de catalizadores
La sinterización de los transportistas ha sido revisado por Baker et al y Trimm. Soportes
de óxido de fase individual de sinterización por uno o más de los siguientes procesos:
(1) difusión superficial,
(2) difusión en estado sólido,
(3) evaporación/condensación de átomos o moléculas volátiles,
(4) la difusión del límite de grano, y
(5) de fase transformaciones.
El vapor acelera la sinterización mediante la formación de grupos hidroxilo superficial
móvil que se volatilizan posteriormente a temperaturas más altas. El cloro también
promueve la sinterización y grano crecimiento de magnesia y óxido de titanio durante
calcinación a alta temperatura. Esto se ilustra en la figura 9.
2013 DESACTIVACIÓN CATALÍTICA
Página 25Universidad Nacional de Trujillo

Figura 9. Área de superficie BET del óxido de titanio como una función del tratamiento térmico y el
cloro contenido de muestras frescas (antes de pre tratamiento). Las muestras se trataron a la
temperatura indicada durante 2 h.
c) Efectos de sinterización sobre la actividad del catalizador
Baker et al. Han examinado los efectos de sinterización en la actividad catalítica. La
actividad específica (basada en la superficie catalítica) puede aumentar o disminuir con el
aumento de tamaño de los cristalitos de metal durante la sinterización si la reacción es
sensible a la estructura, o puede ser independiente de los cambios en tamaño de los
cristalitos de metal si la reacción es insensible estructura.
Por lo tanto, para una reacción de estructura sensible, el impacto de la sinterización
puede ser ya sea ampliada o moderado, mientras que para una reacción insensible
estructura, la sinterización tiene en principio ningún efecto sobre la actividad específica
(por unidad de superficie). En este último caso, la disminución en la actividad basada en
la masa es proporcional a la disminución de metal en el área de superficie. Hidrogenólisis
etano y vapor de etano reforma son ejemplos de reacciones sensibles a la estructura,
mientras que la hidrogenación de CO en el apoyo de cobalto, níquel, hierro y rutenio es la
estructura insensible.
2013 DESACTIVACIÓN CATALÍTICA
Página 26Universidad Nacional de Trujillo

4. REACCIONES DE VAPOR - SÓLIDO Y SÓLIDO - SÓLIDO
Además de la intoxicación, hay una serie de rutas químicas que conducen a la
desactivación del catalizador: (1) reacciones de la fase de vapor con la superficie del
catalizador para producir (a) mayor inactividad y fases de superficie (en lugar de especies
fuertemente adsorbidos) o (b) compuestos volátiles que salen del catalizador y el reactor
en la fase de vapor , (2) catálisis de soporte sólido o catálisis de reacciones sólido -
promotor, y (3) transformaciones de estado sólido de las fases catalíticas durante la
reacción . Cada una de estas rutas se discute en detalle a continuación.
a) Reacciones de vapor (gas) - sólido
Reacciones de gas/vapor con sólido para producir fases inactivas
Metales dispersos, óxidos metálicos, sulfuros metálicos y carburos metálicos son fases
catalíticas típicas, las superficies de los cuales son similares en composición a las fases a
granel. Para una reacción dada, uno de estos tipos de catalizadores es en general
sustancialmente más activo que los otros, por ejemplo, sólo los metales Fe y Ru son
activos para la síntesis de amoníaco, mientras que los óxidos, sulfuros y carburos están
inactivos. Si, por lo tanto, uno de estos catalizadores de metal se oxida, sulfura o carbura,
perderá esencialmente toda su actividad.
Si bien estas modificaciones químicas están estrechamente relacionadas con intoxicación,
la distinción aquí es que en lugar de perder actividad debido a la presencia de una
especie adsorbida, la pérdida de actividad es debido a la formación de una nueva fase en
conjunto.
Reacciones de gas/vapor con sólido para producir compuestos volátiles
La pérdida de metal a través de la vaporización directa es generalmente una ruta
insignificante para la desactivación del catalizador.
2013 DESACTIVACIÓN CATALÍTICA
Página 27Universidad Nacional de Trujillo

Por el contrario, la pérdida de metal a través de la formación de compuestos volátiles, por
ejemplo, carbonilos metálicos, óxidos, sulfuros y haluros en CO, O2, H2S, y entornos que
contienen halógenos, pueden ser importantes en una amplia gama de condiciones,
incluyendo condiciones relativamente suaves. Algunos ejemplos de compuestos volátiles
se enumeran en la Tabla 6. Los carbonilos se forman a temperatura relativamente baja,
pero a altas presiones de CO; los haluros pueden formarse a temperaturas relativamente
bajas y a baja concentración de los halógenos. Sin embargo, las condiciones bajo las
cuales se forman óxidos volátiles varían considerablemente con el metal. Por ejemplo,
RuO3 puede formarse a temperatura ambiente, mientras que PtO2 se forma a tasas
medibles sólo a temperaturas superiores a aproximadamente 500 °C.
Mientras que las propiedades químicas de los metales carbonilos volátiles, óxidos y
haluros son bien conocidas, hay sorprendentemente poca información disponible sobre
sus tasas de formación durante las reacciones catalíticas. No se han realizado
comentarios sobre este tema y los relativamente pocos estudios reportados para definir
los efectos de la pérdida de metal sobre la actividad catalítica, la mayoría de los trabajos
anteriores se ha centrado en la volatilización de Ru en automoción convertidores, la
formación de carbonilo de níquel en catalizadores de níquel durante metanización de CO
o durante la quimisorción de CO a 25 °C, la formación de carbonilos Ru durante la síntesis
de Fischer-Tropsch, y la volatilización de Pt durante la oxidación de amoniaco sobre Pt -
Rh catalizadores de gasa.
Tabla 6. Tipos y ejemplos de compuestos volátiles formados en reacciones catalíticas
Ambiente gaseoso Tipo de compuesto Ejemplo de compuesto
CO, NOCarbonilos ycarbonilos de nitrosilo
Ni(CO)4, Fe(CO)5 (0-300°C)
O2 ÓxidosRuO3 (25◦C), PbO (>850◦C),PtO2 (>700◦C)
H2S Sulfuros MoS2 (>550◦C)
Halógenos Haluros PdBr2, PtCl4, PtF6
2013 DESACTIVACIÓN CATALÍTICA
Página 28Universidad Nacional de Trujillo

Agnelli et al. Investigaron el modelado de la sinterización debido a la formación y la
migración de carbonilo en especies de níquel. Sobre la base de su trabajo, se proponen
dos soluciones para reducir la pérdida de níquel: (1) aumento de la temperatura de
reacción y disminución de la presión parcial de CO con el fin de reducir la tasa de
formación de carbonilo, y (2) cambiar la composición del catalizador, por ejemplo,
aleación de níquel con cobre o la adición de álcali para inhibir la migración de las especies
de carbonilo.
La pérdida de níquel metal durante la quimisorción de CO en catalizadores de níquel a
temperaturas superiores a 0 °C es también un serio problema, por otra parte, esta pérdida
es catalizada por envenenamiento por azufre. En vista de la toxicidad de tetracarbonilo de
níquel, la rápida pérdida de metal de níquel y las estequiometrías de adsorción mal
definidas, los investigadores aconsejan evitar el uso de quimisorción de CO para medir
áreas de superficie de níquel; en su lugar, usar quimisorción de hidrógeno, un método de
la norma ASTM aceptada. Se recomienda estequiometria de adsorción.
La figura 10 ilustra un mecanismo para la formación de Ni(CO)4 en un cristalito de níquel
en atmósfera de CO conteniendo cristalitos de metal de gran tamaño (3 nm) con respecto
a los que contienen cristalitos metálicos pequeños (1,3 nm).
Figura 10. La formación de carbonilo volátil de tetra-níquel en la superficie de los cristalitos de
níquel en atmósfera de CO.
2013 DESACTIVACIÓN CATALÍTICA
Página 29Universidad Nacional de Trujillo

Por otra parte, la pérdida de metal se inhibió en parte a temperaturas de reacción más
altas como resultado de la deposición de carbono.
Por lo tanto, si bien es evidente que la pérdida de rutenio podría ser un problema grave en
la síntesis de Fischer-Tropsch, hay medidas en términos de diseño del catalizador y la
elección de condiciones de reacción que se pueden tomar para minimizar la pérdida.
Uno de los ejemplos más dramáticos de pérdida de fase de vapor del catalizador se
produce durante la oxidación de NH3 en gasa Pt-Rh, una reacción importante en la
fabricación de óxido nítrico. A la temperatura de reacción alta (~ 900 ◦ C), la formación de
un óxido de platino volátil (PtO2) se produce a una velocidad muy significativa; de hecho,
la tasa de pérdida de 0,05-0,3 g de Pt/t de HNO3 es suficientemente alta para
proporcionar un incentivo económico considerable para la recuperación de Pt. El proceso
de recuperación más eficaz consiste en colocar una aleación de tejido rico en Pd
inmediatamente debajo de la gasa Pt-Rh para capturar el Pt mediante la formación de una
aleación Pd-Pt. La pérdida de Pt es también la más importante causa de la desactivación
del catalizador, lo que requiere el apagado y sustitución del catalizador cada 3-12 meses.
Otros ejemplos de la desactivación del catalizador debido a formación de compuestos
volátiles incluyen (1) la pérdida de promotor fósforo del catalizador VPO utilizado en la
producción de anhídrido maleico en lecho fluidizado con una consiguiente pérdida de
selectividad del catalizador, (2) pérdida de fase de vapor del promotor de potasio en
catalizadores de reformado con vapor de alta temperatura , entorno que contiene vapor, y
(3) la pérdida de Mo debido a la formación de una especie volátil de Mo durante
oxideshidrogenación de ácido isobutírico a ácido metacrílico.
Aunque relativamente pocos estudios definitivos de desactivación por la formación de
compuesto volátil se ha informado, el trabajo previo provee la base para enumerar
algunos principios generales. Un mecanismo generalizado de desactivación por formación
de compuestos metálicos volátiles se pueden postular (ver Fig. 11) Además, los papeles
de la cinética y termodinámica se puede afirmar en términos generales:
2013 DESACTIVACIÓN CATALÍTICA
Página 30Universidad Nacional de Trujillo

1. A bajas temperaturas y presiones parciales de los agentes de volatilización (VA), el
índice general del proceso está limitada por la tasa de compuesto volátil en formación.
2. A temperaturas intermedias y las presiones parciales de los VA, la velocidad de
formación del compuesto volátil excede la velocidad de descomposición. Por lo tanto,
la tasa de vaporización es alta, el vapor es estable y la pérdida de metal es alta.
3. A altas temperaturas y presiones parciales de los VA, la velocidad de formación es
igual a la velocidad de descomposición, es decir, se logra el equilibrio. Sin embargo, el
compuesto volátil puede ser demasiado inestable para formarse o puede
descomponerse antes de que haya una oportunidad para ser transportados desde el
sistema. Desde el anterior trabajo, también es evidente que, además de la temperatura
y composición de la fase de gas, las propiedades del catalizador (El tamaño y el apoyo
del cristalito) pueden jugar un papel importante en la determinación de la tasa de
pérdida de metal.
Figura 11. Mecanismos y la cinética generalizadas para la desactivación por pérdida de metal
2013 DESACTIVACIÓN CATALÍTICA
Página 31Universidad Nacional de Trujillo

b) Reacciones de estado sólido.
La desactivación del catalizador por difusión en estado sólido y la reacción parece ser un
mecanismo importante para la degradación de los catalizadores de varios componentes
complejos en la deshidrogenación, la síntesis, la oxidación parcial y reacciones de
oxidación total. Sin embargo, es difícil en la mayoría de estas reacciones conocer el
alcance a la que los procesos de estado sólido, tales como la difusión y la reacción de
estado sólido se ven afectados por las reacciones de superficie.
Por ejemplo, la velocidad de difusión de Al2O3 a la superficie para formar un aluminato se
puede mejorar por la presencia de oxígeno en fase gaseosa o agua o la nucleación de
una fase diferente puede ser inducida ya sea por reducción o por condiciones oxidantes.
5. FALLA MECÁNICA DE LOS CATALIZADORES
a) Las formas y mecanismos de fallo
Se observó el fallo mecánico de catalizadores en varias formas diferentes, incluyendo (1)
la trituración de granulado, tabletas o formas catalizador monolítico debido a una carga,
(2) de desgaste, la reducción de tamaño y/o ruptura de gránulos o pellets de catalizador
para producir multas, especialmente en el líquido o camas de lechada, y (3) la erosión de
partículas de catalizador o revestimientos monolito a altas velocidades de fluido.
El desgaste es evidente por una reducción en el tamaño de partícula o un redondeo o
suavizado de la partícula de catalizador fácilmente observado bajo un microscopio óptico
o electrónico.
La pérdida de recubrimiento por inmersión se observa mediante el escaneo de la pared
del canal de nido de abeja, ya sea con una óptica o microscopio electrónico. Grandes
aumentos en la caída de presión en un proceso catalítico son a menudo indicativos de
ensuciamiento, enmascaramiento o la fractura y la acumulación de desgaste del
catalizador en el lecho del reactor.
2013 DESACTIVACIÓN CATALÍTICA
Página 32Universidad Nacional de Trujillo

Los catalizadores comerciales son vulnerables a la falla mecánica, en gran parte debido a
la manera en la que se forman, es decir gránulos de catalizador, esferas, extruidos, y
gránulos con diámetros de 50 µm hasta varios centímetros se preparan en general por
aglomeración de 0,02-2 µm agregados de partículas primarias mucho más pequeñas que
tienen diámetros de 10-100 nm por medio de la formación de precipitación o gel seguido
por secado por pulverización, extrusión, o compactación.
Estos aglomerados tienen, en general, considerablemente fortalezas más bajas que las
partículas primarias y agregados de partículas a partir de los cuales se forman.
Dos mecanismos principales participan en la falla mecánica de aglomerados de
catalizador: (1) la fractura de aglomerados en aglomerados más pequeños de
aproximadamente 0.2d0 - 0.8d0 y (2) la erosión (o abrasión) de agregados de partículas
primarias que tienen diámetros que van desde 0,1 a 10 mm desde la superficie de la
aglomeración.
Figura 12. Representación esquemática de la reducción / oxidación cíclica de pares gemelos de
octaedros MoO6 entre la esquina y los acuerdos de reparto de borde (las cajas representan
octaedros de MoO6 con el intercambio de átomos de oxígeno en las esquinas de MoO3 o bordes
para MoO2).
2013 DESACTIVACIÓN CATALÍTICA
Página 33Universidad Nacional de Trujillo

Figura 13. Representación esquemática de las estructuras de MoO3, Mo18O52 y Mo4O11. Los planos
de corte en Mo18O52 y Mo4O11 representados por las flechas oblicuas (las cajas con una "X"
representan octaedros de MoO5).
b) Papel de las propiedades físicas y químicas de los aglomerados de cerámica en
la determinación de la fuerza y la resistencia de desgaste
2013 DESACTIVACIÓN CATALÍTICA
Página 34Universidad Nacional de Trujillo

Factores que afectan a la magnitud de la tensión necesaria para la rotura
del aglomerado y los mecanismos por los que se produce.
El grado en que una fractura o la erosión, participa en la reducción de tamaño de
aglomeración que depende de varios factores:
(1) La magnitud de una tensión.
(2) La fuerza y la tenacidad a la fractura del aglomerado.
(3) Aglomerarse tamaño y superficie.
(4) Tamaño de la grieta y el radio.
La erosión (abrasión) se produce cuando el estrés (por ejemplo, fuerza por área debido a
la colisión o la presión de cavitación) excede la fuerza de aglomerado, es decir, la
resistencia de la unión entre las partículas primarias. La tasa de erosión se informa,
proporcional al área de superficie externa del catalizador, por lo tanto, aumenta la
velocidad de erosión con la disminución del tamaño de aglomerado.
Tenacidad a la fractura de los aglomerados cerámicos.
La mayoría de los catalizadores heterogéneos son materiales complejos, de múltiples
fases que consisten en gran parte de los materiales cerámicos porosos, es decir, son
típicamente óxidos, sulfuros, o metales sobre un soporte de óxido o de apoyo. Cuando se
aplica un esfuerzo de tracción de una magnitud cerca del punto de fluencia, cerámicas
casi siempre se someten a la rotura frágil antes se puede producir deformación plástica.
La fractura frágil se produce a través de la formación y propagación de grietas a través de
la sección transversal de un material en una dirección perpendicular a la tensión aplicada.
Un aglomerado de fractura debido a un esfuerzo de tensión se produce por la
propagación de defectos internos y superficiales ; estos defectos creados por tensiones
externas o defectos inherentes son multiplicadores de tensión , es decir, la tensión se
multiplica por 2(a/r) 0.5 , donde a es la longitud de la grieta y r es el radio de curvatura de
la punta de la grieta; a/r puede variar de 2 a 1000 , la tensión efectiva en la punta de una
grieta puede ser 4-60 veces la tensión aplicada . Los multiplicadores de esfuerzo de
tracción pueden ser microfisuras, poros internos, y las esquinas de grano.
2013 DESACTIVACIÓN CATALÍTICA
Página 35Universidad Nacional de Trujillo

La capacidad de un material para resistir la fractura se denomina resistencia a la fractura.
La resistencia a la fractura cepa llanura KIc se define como:
KIc=Yσ (πa )0,5(¿)
Donde Y es un parámetro adimensional (a menudo cerca de 1,0-2,0) cuya magnitud
depende tanto de la muestra y el crack geometrías, σ la tensión aplicada, y a es la
longitud de una grieta superficial o la mitad de la longitud de una grieta interna. La
propagación de grietas y fracturas son probable si el lado derecho de la ecuación (*)
supera el valor experimental de la tensión normal fractura tenacidad (lado izquierdo de la
ecuación (*)).
Valores de tenacidad a la fractura de deformación plana para los materiales cerámicos
son significativamente menores que para los metales y por lo general por debajo de 10
MPa (m) 0,5; valores, alúmina no poroso cristalino (99,9 %), la sílice fundida, y zirconio (3
mol % Y2O3) reportado se 4-6 , 0,8 , y 7-12 MPa (m) 0,5, respectivamente; fortalezas
(análogos a límites elásticos para metales) de flexión para los mismos materiales son 280
a 550, 100, y 800-1500 MPa. Por lo tanto, basándose tanto en tenacidad a la fractura y
resistencia a la flexión, no poroso, óxido de circonio cristalino es mucho más fuerte a las
salas de fractura que la alúmina que a su vez es mucho más fuerte que la sílice fundida.
Resistencias a la compresión de los materiales cerámicos
Hasta ahora, la discusión se ha centrado principalmente en la resistencia a la tracción, en
la medida de que se reduce considerablemente por la presencia de grietas o poros. Sin
embargo, para los materiales cerámicos en la compresión, no hay amplificación de estrés
debido a defectos o poros; materiales de este modo cerámicos (incluidos los materiales
catalíticos) en la compresión son mucho más fuertes (aproximadamente un factor de 10)
que en la tensión. Además, la resistencia de los materiales cerámicos se puede mejorar
drásticamente mediante la imposición de una tensión de compresión residual en la
superficie a través de templado térmico o químico. Por otra parte, introducción de
aglutinantes tales como el grafito permite aglomerados de polvos cerámicos para
someterse a deformación plástica significativa antes de la fractura.
2013 DESACTIVACIÓN CATALÍTICA
Página 36Universidad Nacional de Trujillo

c) Resistencias a la tracción y resistencia al desgaste de los soportes de
catalizadores y catalizadores
Datos de resistencia a la tracción de los aglomerados de soporte del
catalizador
Las fortalezas citadas anteriormente para materiales no porosos, cristalinas o
policristalinas recocido no se aplican necesariamente a catalizadores porosos
aglomerados incluso bajo compresión, sino que la fuerza aglomerada depende de las
fortalezas de los enlaces químicos y físicos, incluyendo la energía cohesiva entre las
partículas primarias. La fuerza aglomerada dependería en gran medida de la preparación
del compacto. Los datos representativos de catalizadores aglomerados (ver Tabla 8)
sugieren que son en general sustancialmente más débiles que los materiales
policristalinos cerámicos elaborados por sinterización a alta temperatura, tales como
alúmina, citada en la fig. 24. Por ejemplo, Pham encontraron que la resistencia a la rotura
de un aglomerado de alúmina vista durante la compactación uniaxial está en el intervalo
de 5-10 MPa
Sustancialmente son más bajos que los valores reportados para un tratamiento térmico de
alúmina policristalina de 280-550 MPa. Una gran parte de esta diferencia (85-95%
aproximadamente) puede atribuirse a la porosidad, sin embargo, el 5-15% restante debe
ser debido a diferencias en la unión entre las partículas primarias. En otras palabras, los
enlaces entre partículas primarias en aglomerados de catalizador (y algunos aglomerados
cerámicos preparados por métodos similares) son típicamente de naturaleza física (por
ejemplo, involucran las fuerzas de van der Waals) mientras que los aglomerados
cerámicos policristalinos sinterizados son principalmente química debido al sólido puente
de partículas primarias. Por lo tanto, parece que hay un gran potencial para el
fortalecimiento de los aglomerados del catalizador, ya que sus puntos fuertes son factores
típicamente de 3-50 inferior a la de las cerámicas convencionales, con tratamiento térmico
de porosidad similar.
2013 DESACTIVACIÓN CATALÍTICA
Página 37Universidad Nacional de Trujillo

Tabla 8. Resistencias mecánicas y las tasas de desgaste de soportes catalíticos en comparación
con los de aglomerados cerámicos sinterizados
Catalizador o soporte de cerámica
Preparación / pre-tratamiento / propiedades
Fuerza (MPa)
Índice desgaste (% p/h)
GRAN ÁREA SUPERFICIAL SOPORTES DE CATALIZADORES
Al2O3, esferas 01.02 a 04.25 mm
Granulación sol-gel / secó 10 horas a 40 ◦C, se calcinó 3 h
11,6 ± 1,9 0.033
Al2O3, esferas de 4,25 mm
A 450 ◦C/389 m2 / g,dpore = 3,5 nmAlcoa LD-350 0.7 0.177
Al2O3, 100 m VISTA-B-965-500C 6,2 ± 1,3
TiO2 (anatasa), 30 mHidrólisis térmica / seca 110 ◦C,se calcina 2 h
28a
500 ◦C/92 m2/g, <10 cristalitos primarios nm
15a
TiO2 (anatasa), 90 mPrecipitación Base / seca 110 ◦C, se calcina 2 h500 ◦C/81 m2/g, 10-14 cristalitos primarios nm
TiO2 (anatasa 75%, 25% de rutilo)
Degussa P25 ahumada / 4 mm extrudates/48 m2/g,
0.9
V de poros = 0,34 cm3 / g, dpore = 21 nm
0.9
TiO2 (anatasa)Rhône-Poulenc DT51, precipitar / 4 extruidos mm /92 m2/g, V poros = 0,40 cm3/g, dpore = 8,65 nm
CERÁMICAS DE BAJA SUPERFICIE
Al2O3Rocíe seca con un aglutinante orgánico, plástico
2.3
deformación observado
Al2O3Tratamiento térmico (sinterizada), el 99,9%
282-551
TiO2 (rutilo) parcialmente sinterizado 194
ZrO2 (aditivo itrio)Muestras comerciales de tres empresas,
0,035-,43
secada por pulverizaciónZrO2 (3% Y2O3) Tratada térmicamente (sinterizado) 800-1500
2013 DESACTIVACIÓN CATALÍTICA
Página 38Universidad Nacional de Trujillo

Implicaciones del conocimiento mecanicista de desgaste para el diseño de
catalizador
La comprensión de los mecanismos son importantes en el desgaste de los soportes de
catalizadores, la relación entre la fuerza y la tasa de desgaste de un material dado, y los
datos de prueba se puede utilizar con gran ventaja en el diseño de catalizadores
resistentes a desgaste. Varias alternativas se siguen para aumentar la resistencia al
desgaste:
(1) aumento de la fuerza agregado/aglomerado por medio de métodos de preparación
avanzadas, por ejemplo, granulación sol- gel, secado por aspersión, y precipitación
métodos cuidadosamente controlados.
(2) la adición de aglutinantes para mejorar la fuerza y resistencia, por ejemplo, la adición
de un aglutinante de polivinilpirrolidona a aglomerados de aumentos de arena de
cuarzo aglomerado fuerza de 0,1 a 3 MPa.
(3) el revestimiento de los áridos con un material poroso pero muy fuerte, tal como ZrO2,
por ejemplo, la incrustación de un catalizador de lecho fluidizado para la oxidación
parcial de n-butano a anhídrido maleico en una matriz sólida, amorfa de fosfato de
hidrógeno de zirconio mejora significativamente su resistencia de desgaste.
(4) templado térmico de aglomerados para introducir a la compresión tensiones que
aumentan la fuerza y la resistencia al desgaste, por ejemplo, de calefacción y de
refrigeración partículas rápidamente pasándolos a través de un tiempo de residencia
bajo, horno de alta temperatura para endurecer el exterior aglomerado, mientras que la
prevención de sinterización significativa de o cambios de fase en el interior porosa.
2013 DESACTIVACIÓN CATALÍTICA
Página 39Universidad Nacional de Trujillo

CONCLUSIONES
El desarrollo en las últimas dos décadas de espectroscopias de superficie más
sofisticadas y tecnologías informáticas poderosas brindan oportunidades para obtener
sustancialmente mejor comprensión de los mecanismos desactivación y la construcción
de este conocimiento en modelos matemáticos integrales que permitirá el diseño y la
optimización de los procesos que implican la desactivación más eficaz catalizadores.
El envenenamiento de catalizadores es un fenómeno relativamente bien estudiado,
razonablemente bien entendido. Intoxicaciones por venenos reversibles e irreversibles por
lo general se puede modelar con éxito moderado en el nivel de proceso.
Los mecanismos por los que diversas especies de carbono se forman en metales
soportados y por la que varios tipos de coque se forman sobre óxidos ácidos y sulfuros
son sólo moderadamente bien entendidos. De mayor importancia es la investigación el
establecimiento de vínculos específicos entre la cantidad de ciertas estructuras de
carbono o coque en la superficie o en multicapas y el grado de desactivación.
Mientras que las herramientas espectroscópicas sofisticados se han utilizado con eficacia
durante las últimas dos décadas para avance nuestra comprensión fundamental de
sinterización y redispersión, se necesitan nuevos datos sobre los procesos atómicos y
moleculares que ocurren durante la sinterización y redispersión para desarrollar modelos
más sofisticados y realistas.
Un fallo mecánico debido al desgaste o colapso de los aglomerados es un problema grave
que limita muchos procesos catalíticos comerciales. Sin embargo, nuestro presente
comprensión de estos problemas apenas supera lo que se enseña en las materias de
segundo año de ciencias. Hay necesidades críticas para el desarrollo de materiales
catalíticos más resistentes al desgaste más fuertes.
2013 DESACTIVACIÓN CATALÍTICA
Página 40Universidad Nacional de Trujillo

REFERENCIAS BIBLIOGRÁFICAS
- J.L. Figuerido (Ed.), Progress in Catalyst Deactivation, NATO Advanced Study Institute
Series E, Marunus Nijhoff, Boston, 1982.
- R. Hughes, Deactivation of Catalysts, Academic Press, London, 1984 (Chapter 8).
- J. Oudar, H. Wise, Deactivation and Poisoning of Catalysts, Marcel Dekker, New York,
1985, p. 1.
- J.B. Butt, E.E. Petersen, Activation, Deactivation, and Poisoning of Catalysts,
Academic Press, San Diego, 1988.
- P.J. Denny, M.V. Twigg, in: B. Delmon, G.F. Froment (Eds.), Catalyst Deactivation
1980, Stud. Surf. Sci. Catal., Vol. 6, Elsevier, Amsterdam, 1980, p. 577.
- C.H. Bartholomew, Chem. Eng. 91 (1984) 96.
- J.B. Butt, in: J.R. Anderson, M. Boudart (Eds.), Catalysis, Science and Technology,
Springer, New York, 1984, p. 1.
- R.J. Farrauto, C.H. Bartholomew, Fundamentals of Industrial Catalytic Processes,
Chapman & Hall, Kluwer Academic Publishers, London, 1997 (Chapter 5).
- B. Delmon, G.F. Froment, Catalyst Deactivation 1980, Stud. Surf. Sci. Catal., Vol. 6,
Elsevier, Amsterdam, 1980.
- B. Delmon, G.F. Froment, Catalyst Deactivation 1987, Stud. Surf. Sci. Catal., Vol. 34,
Elsevier, Amsterdam, 1987.
2013 DESACTIVACIÓN CATALÍTICA
Página 41Universidad Nacional de Trujillo
Recommended

















