
Farmacologia Generale
Prof.ssa Mariangela Serra Ricevimento studenti: tutti i giorni previo appuntamento [email protected] 070/6754136

l corso si propone di fornire agli studenti nozioni di base di
Farmacologia Generale, partendo dai principi di farmacocinetica
e farmacodinamica, per illustrare quindi gli effetti delle
principali classi di farmaci, sia a livello cellulare che sugli organi
e apparati. Verrà inoltre sottolineata l’importanza dei farmaci
quali strumenti di indagine per la comprensione dei meccanismi
cellulari e molecolari alla base dei processi fisiologici. Gli
obiettivi sopradescritti verranno perseguiti attraverso lo
svolgimento di lezioni frontali e lezioni in laboratorio.
Obiettivi del corso
Lo studente deve possedere conoscenze sulla biochimica e fisiologia della cellula e dei meccanismi fisiologici dei diversi apparati.

FARMACOLOGIA GENERALE Lezioni teoriche (7 CFU) Basi della farmacologia: Farmacocinetica, farmacodinamica, metabolismo dei farmaci Principi di farmacologia del sistema nervoso autonomo periferico: Sistema nervoso parasimpatico Agonisti e antagonisti del recettore muscarinico Farmaci anticolinesterasici Farmaci attivi sulla placca neuromuscolare e sui gangli autonomi Sistema nervoso simpatico Agonisti e antagonisti adrenergici Farmaci attivi sul sistema cardiovascolare: Inibitori del sistema renina-angiotensina Calcio-antagonisti Farmacologia del nitrossido

Farmacologia degli eicosanoidi: Metabolismo dell’acido arachidonico Farmaci antinfiammatori non steroidei (FANS) Farmacologia dell’istamina Agenti per il controllo dell’acidità gastrica: bloccanti della pompa protonica Analgesici oppiacei e loro antagonisti Farmacologia del sistema degli endocannabinoidi Principi di farmacologia endocrina Farmacologia della corteccia surrenale Estrogeni e progestinici Tossicodipendenza Effetti dell’esposizione ripetuta alle sostanze Sostanze d’Abuso Etanolo, Cannabis, Cocaina, Eroina, Anfetamine, Psichedelici Laboratorio

MODALITA’ D’ESAME PROVA INTERMEDIA ESAME FINALE Scritto I Sessione di giugno Orale Altre Sessioni
LEZIONI FRONTALI Inizio ore 9:00 LUNEDI’ MERCOLEDI’ VENERDI’ LABORATORIO POMERIGGIO MERCOLEDI’ calendario sarà pubblicato

Avviso
Le lezioni saranno disponibili sul sito docente: Didattica materiale didattico Password: farmaco2018


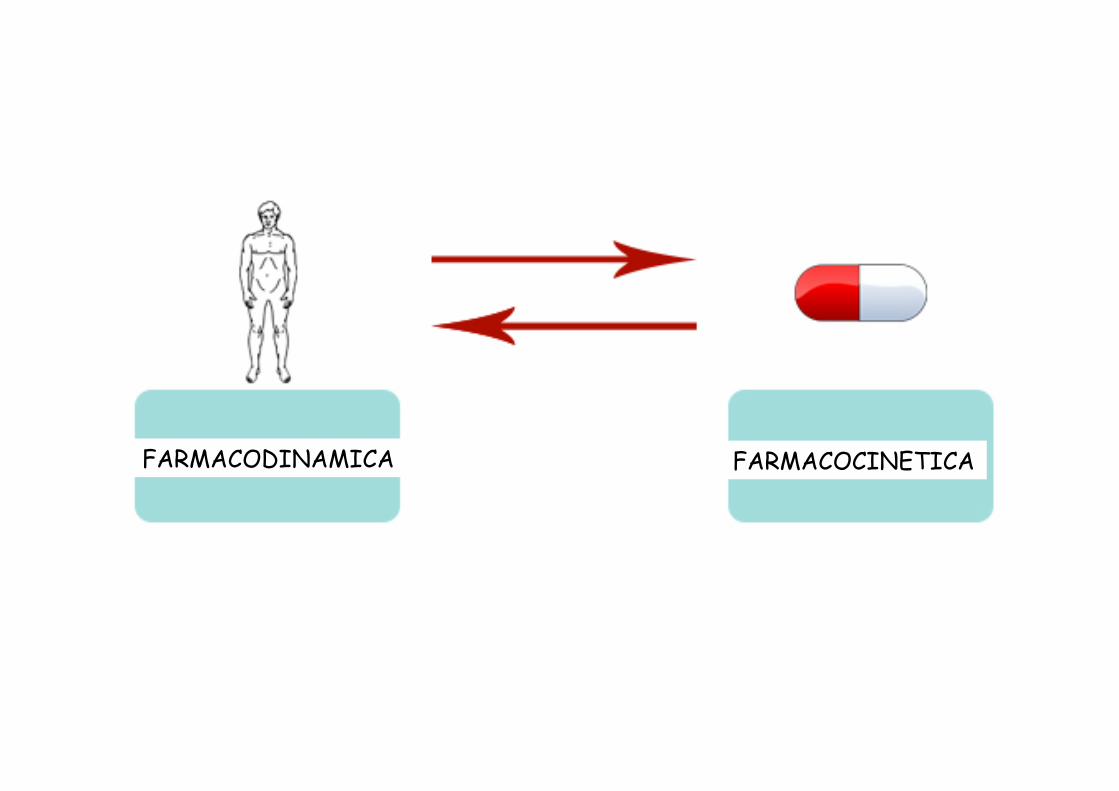
FARMACODINAMICA FARMACOCINETICA
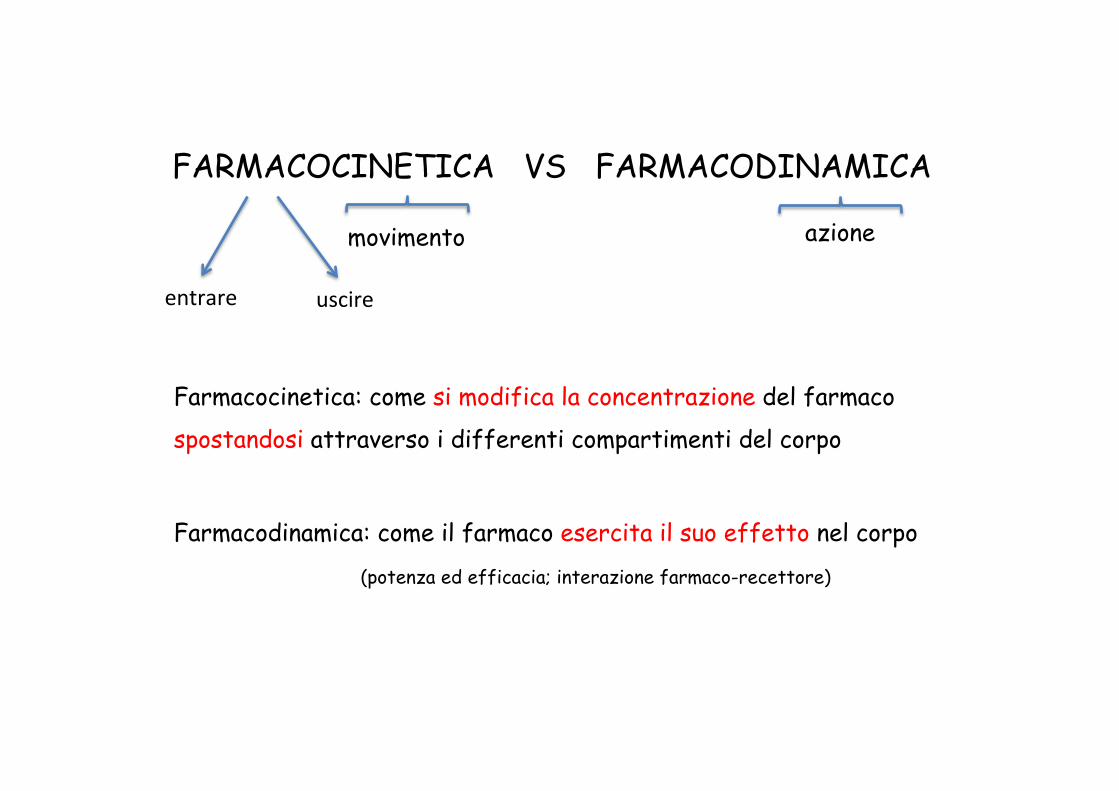
FARMACOCINETICA VS FARMACODINAMICA
movimento azione
Farmacocinetica: come si modifica la concentrazione del farmaco
spostandosi attraverso i differenti compartimenti del corpo
Farmacodinamica: come il farmaco esercita il suo effetto nel corpo
(potenza ed efficacia; interazione farmaco-recettore)
entrare uscire

Componenti della farmacocinetica (In e Out) A D
Processo attraverso il quale il farmaco entra nel circolo sanguigno
Processi che avvengono prima dell’ingresso del farmaco nella circolazione sistemica
Processi che disperdono il farmaco nei fluidi e nei tessuti dell’organismo
Assorbimento
Distribuzione
Metabolismo Biotrasformazioni
Eliminazione
M E
Trasformazione irreversibile di un farmaco in un metabolita
Eliminazione del farmaco dall’organismo
/Eliminazione (fegato)
Escrezione (fegato e rene)

PER OS Orale Farmaco inghio6to Sublinguale Farmaco so8o la lingua Buccale Farmaco all’interno della guancia INIEZIONE EV Endovenosa IM Intramuscolare SC So8ocutanea IA Intrarteriosa Intratecale Nello spazio subaracnoideo POLMONARE RETTALE TOPICA TRANSDERMICA Transre8ale transcutanea
Buccale
(Polmonare)
ASSORBIMENTO = PROCESSO ATTRAVERSO IL QUALE IL FARMACO ENTRA NELLA CIRCOLAZIONE SISTEMICA
ASSORBIMENTO DIPENDE DALLA VIA DI SOMMINISTRAZIONE

CONCETTI FONDAMENTALI
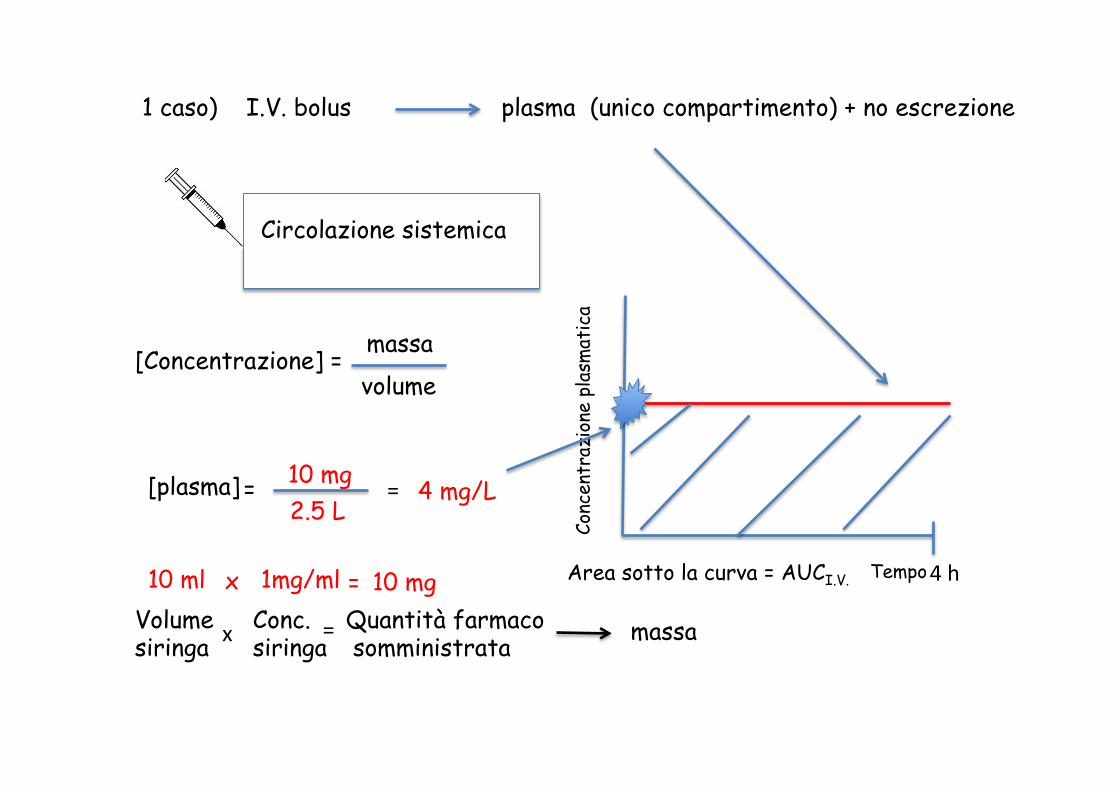
Massa = la quantità di farmaco (unità di misura = mg) Volume = unità di misura L, mL, cc (cc = 1 centimetro cubico = 1 mL) Volume del sangue = 5 L Volume del plasma = sangue – cellule = 2.5 L
Quale è la concentrazione plasmatica di 10 mg di farmaco X assorbito al 100% ?
[plasma] = 10 mg 2.5 L
= 4 mg/L
[Concentrazione] = massa volume
[plasma] = Volume sangue - ematocrito
Circolazione sistemica
Tempo Co
ncen
traz
ione
pla
smat
ica
[Concentrazione] = massa volume
Volume siringa x
Conc. siringa
= Quantità farmaco somministrata
10 ml 1mg/ml x = 10 mg
10 mg
[plasma] = 2.5 L
= 4 mg/L
massa
4 h Area sotto la curva = AUCI.V.
1 caso) I.V. bolus plasma (unico compartimento) + no escrezione
Circolazione sistemica
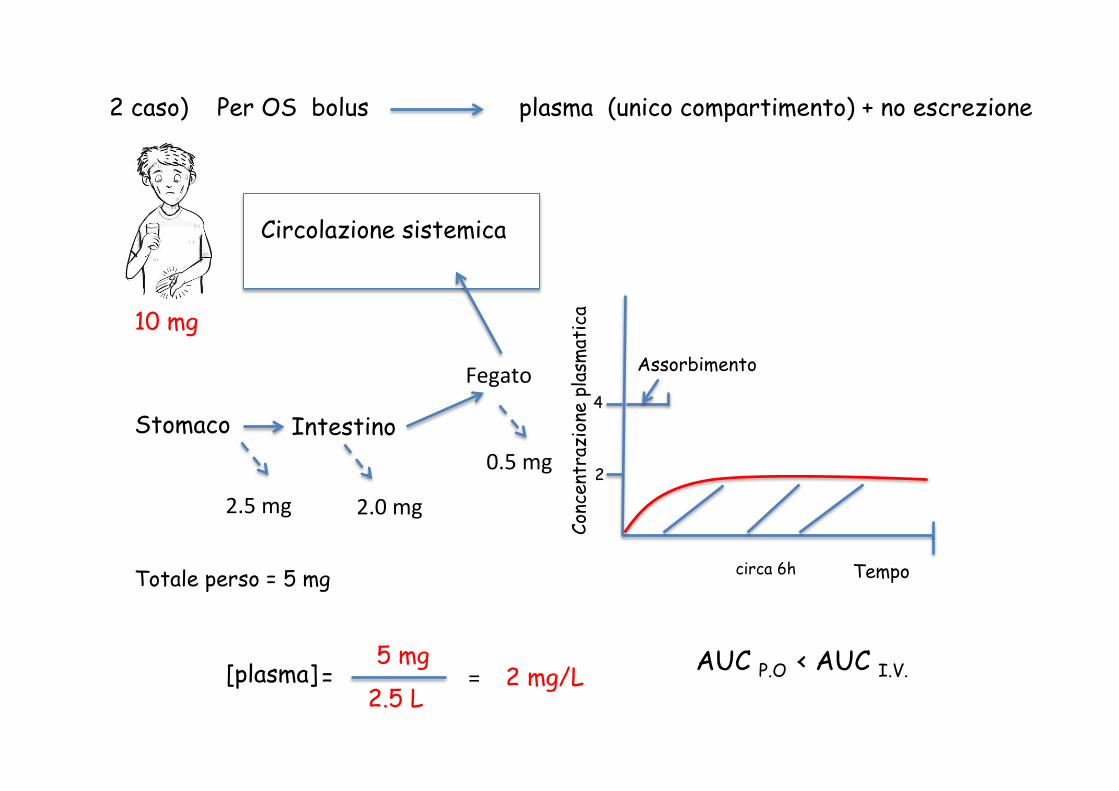
2 caso) Per OS bolus plasma (unico compartimento) + no escrezione
10 mg
Stomaco Intestino
2.5 mg
Fegato
0.5 mg
2.0 mg
Totale perso = 5 mg
[plasma] = 2.5 L
5 mg = 2 mg/L
Tempo Co
ncen
traz
ione
pla
smat
ica
4
2
AUC P.O < AUC I.V.
Assorbimento
circa 6h

Con il termine biodisponibilità si fa riferimento alla
quantità percentuale della dose di farmaco somministrata
che raggiunge in forma attiva il circolo ed è quindi
disponibile per le azioni sistemiche
AUC PO / AUC I.V = Biodisponibilità.

AUC PO / AUC I.V = Biodisponibilità.
AUC I.V
quantità di farmaco che arriva al plasma, è il 100% di quella somministrata = DOSE
AUC P.O
reale dose del farmaco che arriva al plasma
Prima dell’assorbimento
Dopo l’assorbimento
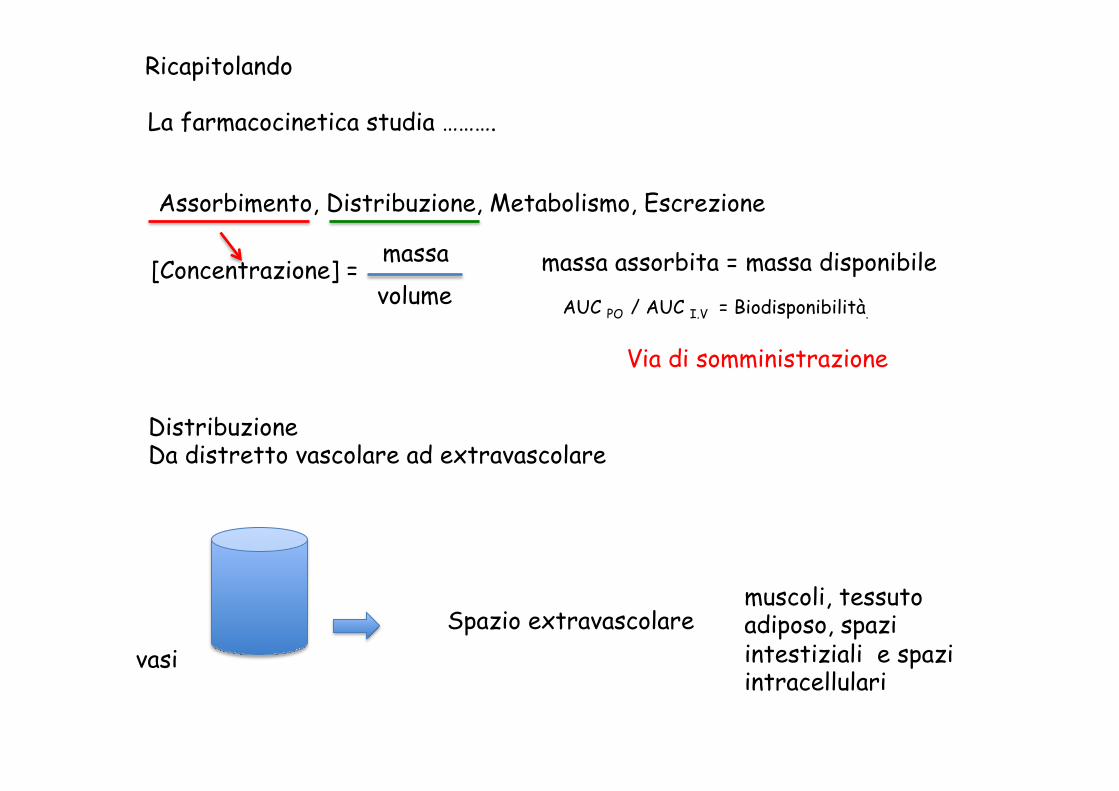
Ricapitolando
La farmacocinetica studia ……….
Assorbimento, Distribuzione, Metabolismo, Escrezione
[Concentrazione] = massa volume AUC PO / AUC I.V = Biodisponibilità.
massa assorbita = massa disponibile
Via di somministrazione
Distribuzione Da distretto vascolare ad extravascolare
vasi Spazio extravascolare
muscoli, tessuto adiposo, spazi intestiziali e spazi intracellulari
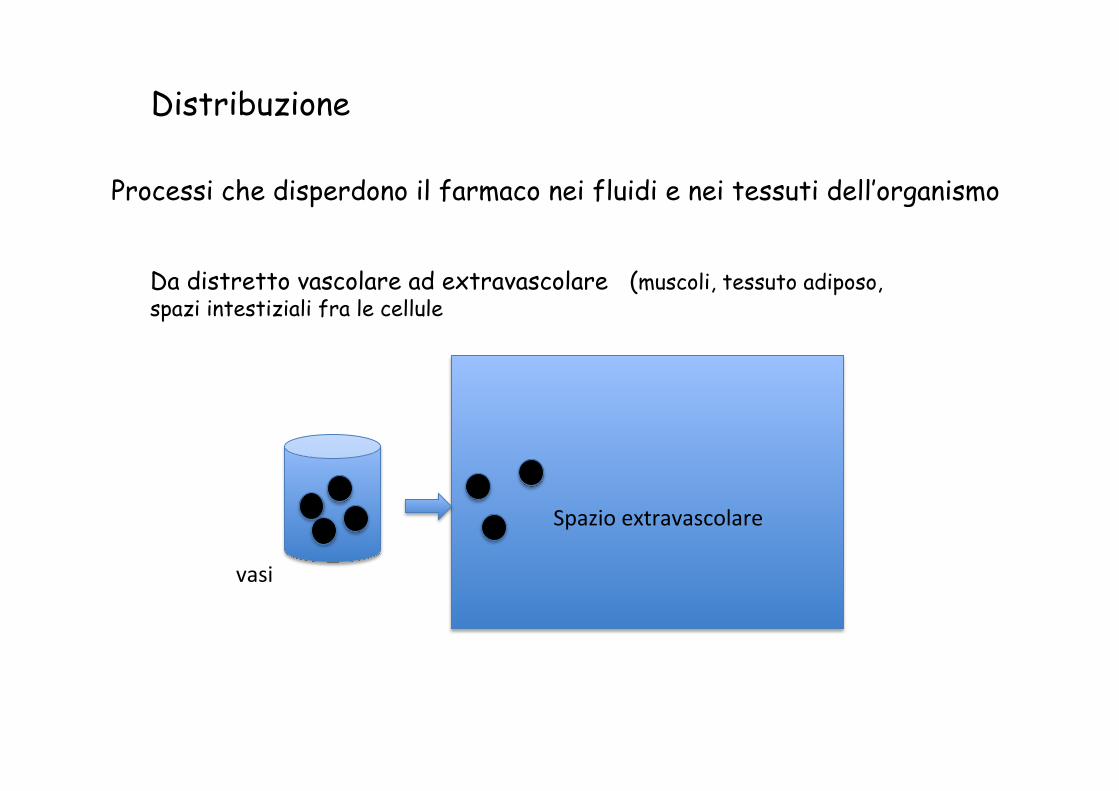
Da distretto vascolare ad extravascolare (muscoli, tessuto adiposo, spazi intestiziali fra le cellule
vasi
Spazio extravascolare
Distribuzione
Processi che disperdono il farmaco nei fluidi e nei tessuti dell’organismo
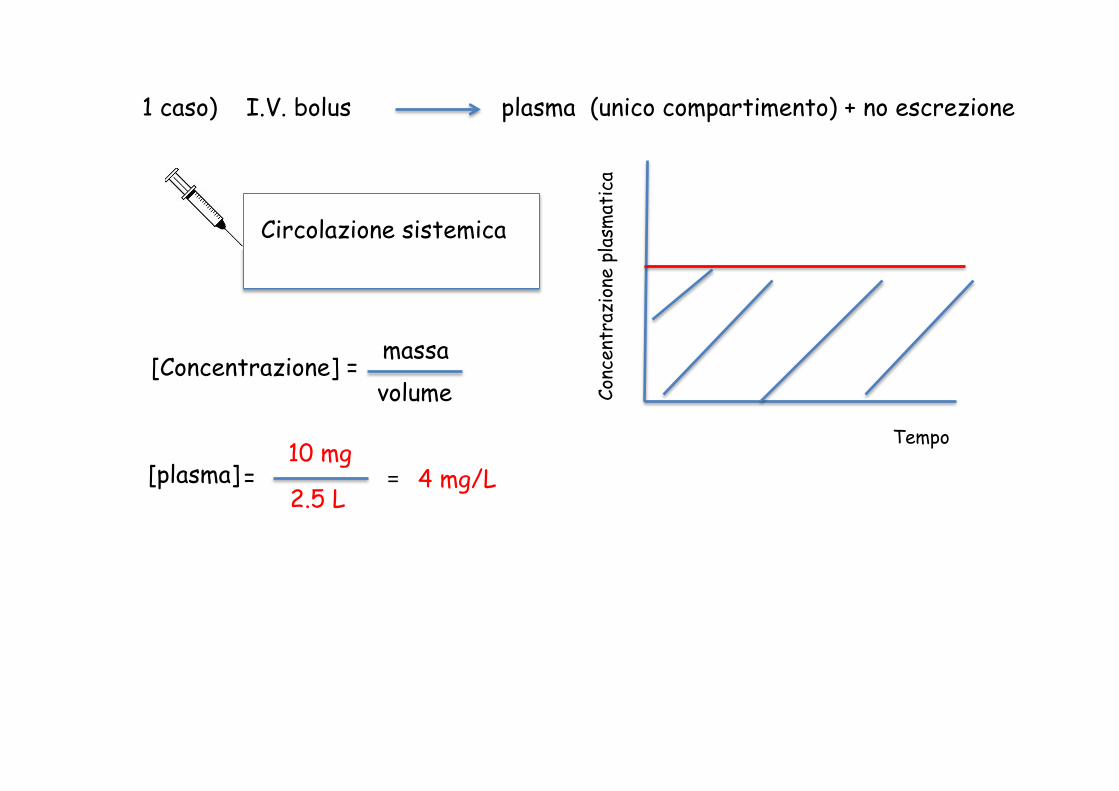
Circolazione sistemica
1 caso) I.V. bolus plasma (unico compartimento) + no escrezione
Tempo
Conc
entr
azio
ne p
lasm
atic
a
10 mg [plasma] = 2.5 L
= 4 mg/L
[Concentrazione] = massa volume
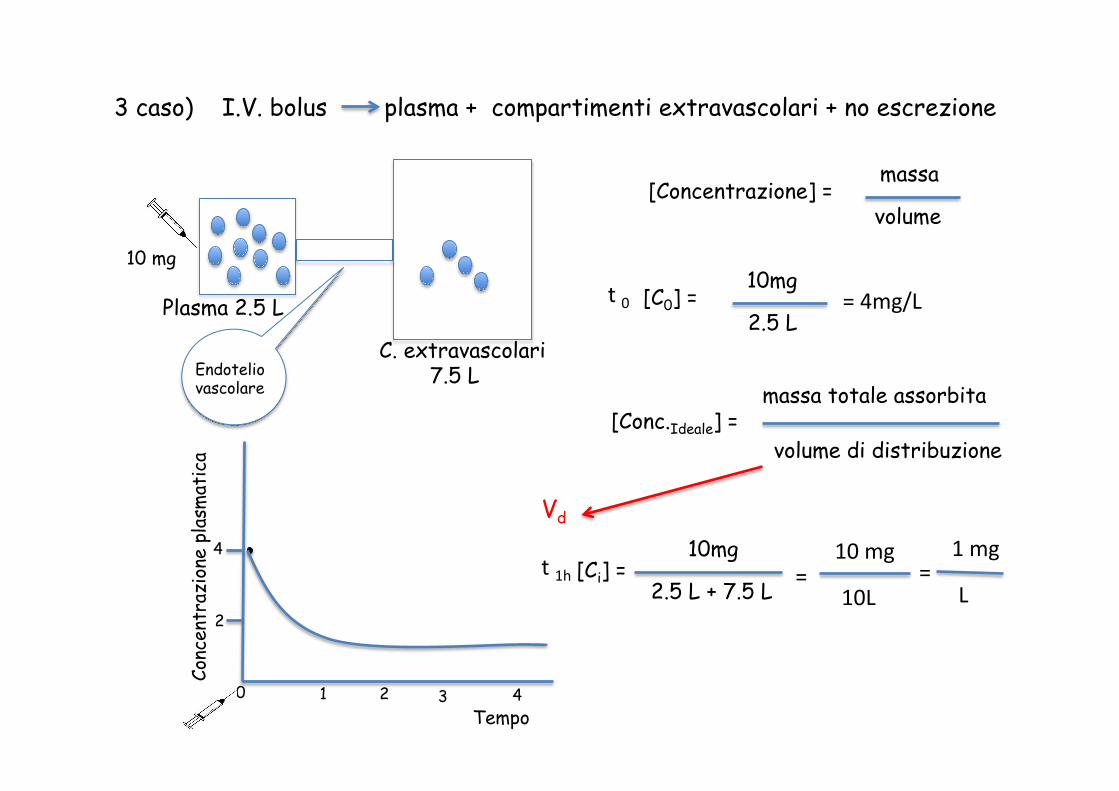
3 caso) I.V. bolus plasma + compartimenti extravascolari + no escrezione
Plasma 2.5 L
C. extravascolari 7.5 L Endotelio
vascolare
[Concentrazione] = massa volume
10 mg
t 0 [C0] = 10mg 2.5 L
= 4mg/L
Tempo
Conc
entr
azio
ne p
lasm
atic
a
4
2
0
[Conc.Ideale] = massa totale assorbita volume di distribuzione
t 1h [Ci] = 10mg 2.5 L + 7.5 L =
10 mg
10L =
1 mg
L
1 2 4 3
Vd
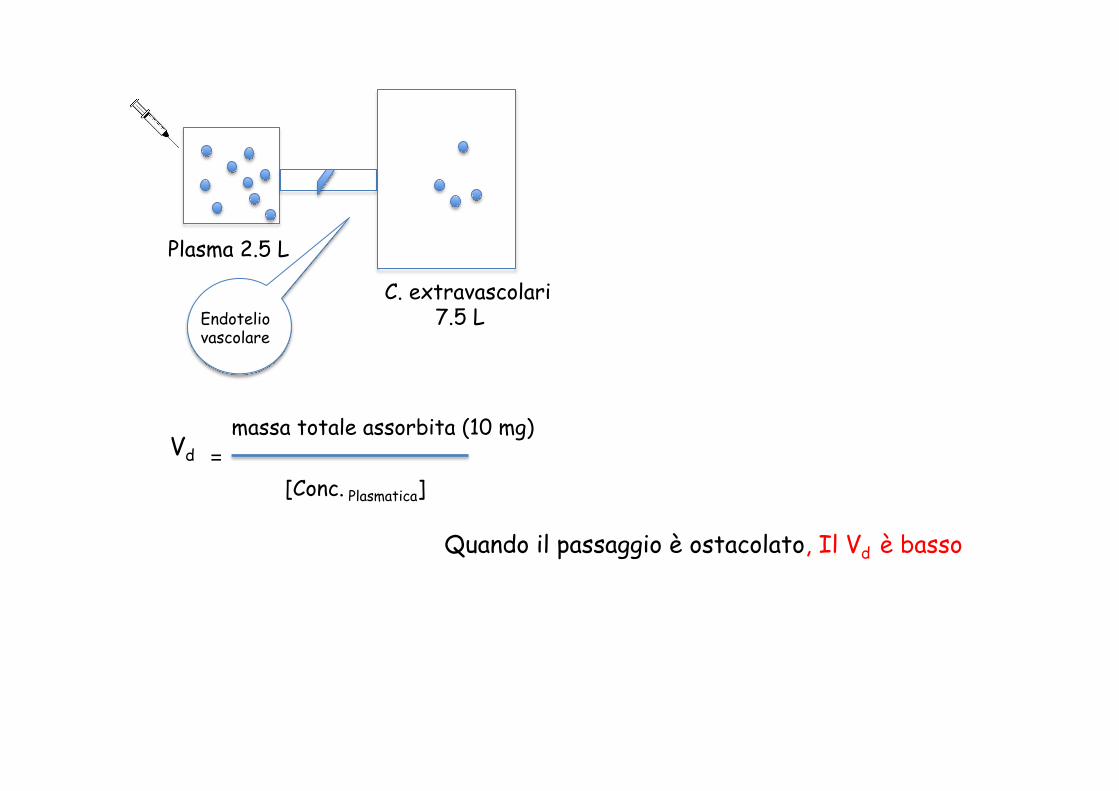
Vd massa totale assorbita (10 mg)
[Conc. Plasmatica] =
Quando il passaggio è ostacolato, Il Vd è basso
Plasma 2.5 L
C. extravascolari 7.5 L Endotelio
vascolare
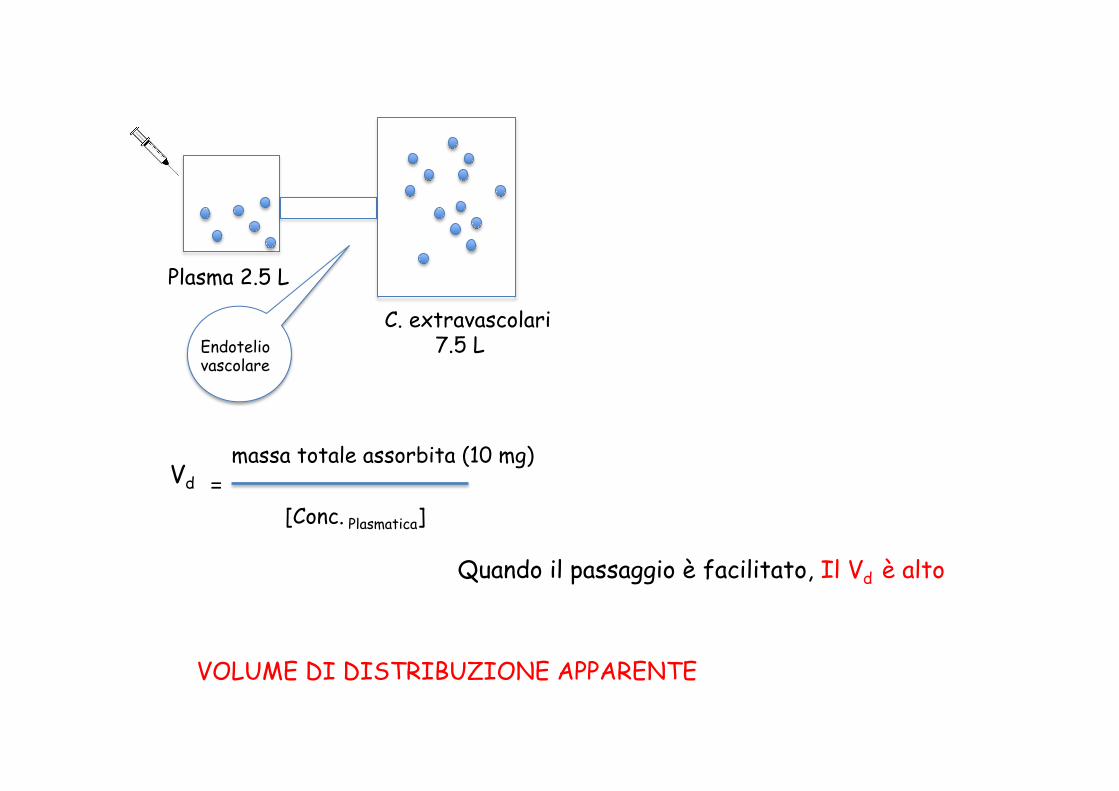
Vd massa totale assorbita (10 mg)
[Conc. Plasmatica] =
Plasma 2.5 L
C. extravascolari 7.5 L Endotelio
vascolare
Quando il passaggio è facilitato, Il Vd è alto
VOLUME DI DISTRIBUZIONE APPARENTE

VOLUME DI DISTRIBUZIONE
Non è una variabile
Per i farmaci che “preferiscono” il plasma = Vd basso
Per i farmaci che “preferiscono” il compartimento extravascolare= Vd alto
Molecole ad alto peso molecolare Molecole ad
basso peso molecolare
Altri fattori Legame alle proteine plasmatiche Lipofilicità Patologie
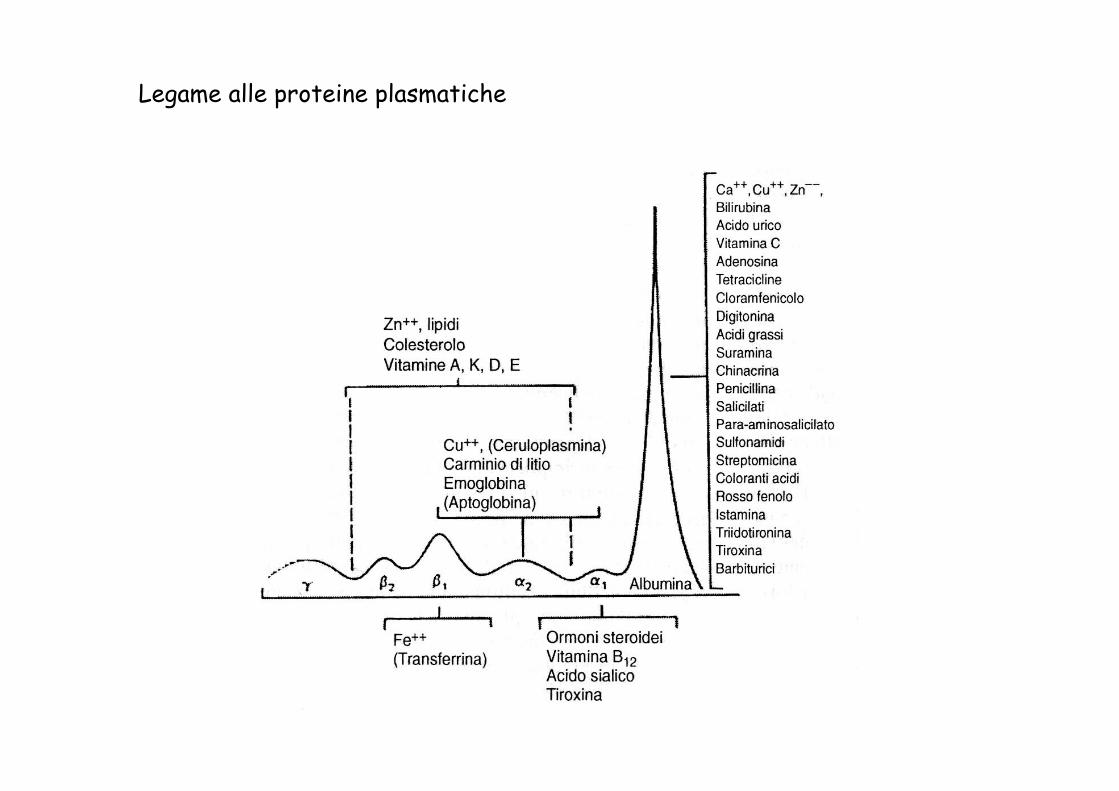
Legame alle proteine plasmatiche
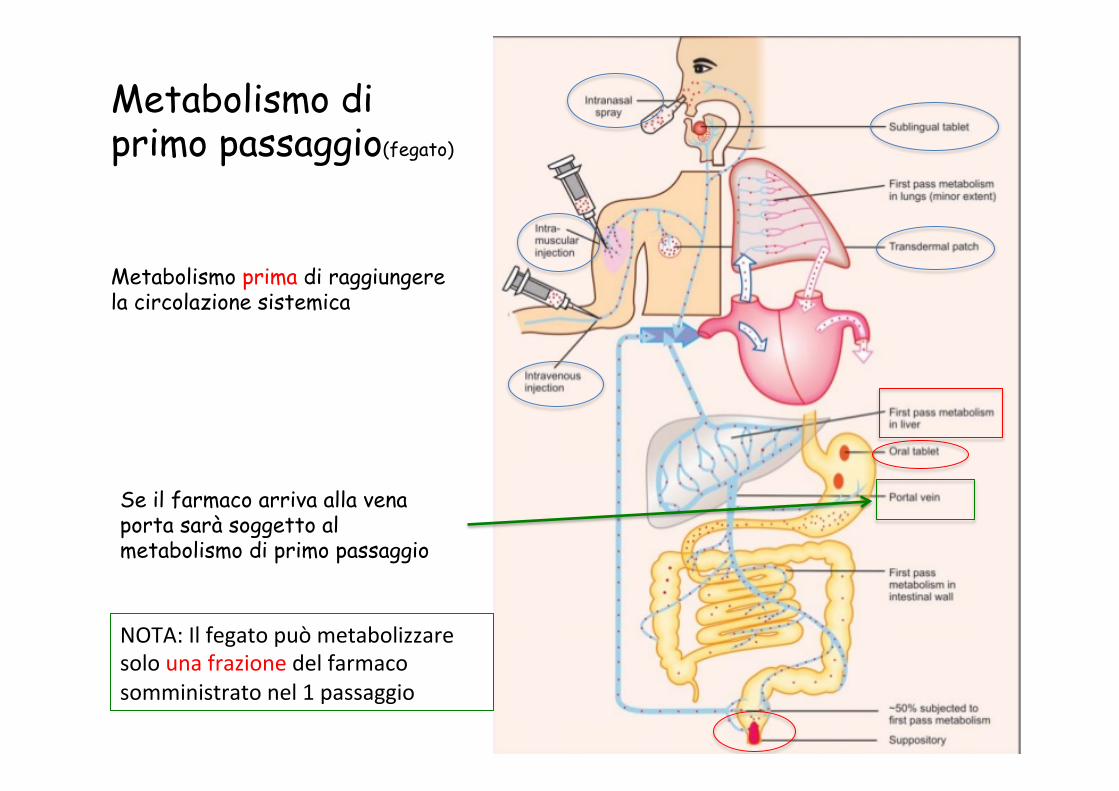
Metabolismo di primo passaggio(fegato)
Metabolismo prima di raggiungere la circolazione sistemica
Se il farmaco arriva alla vena porta sarà soggetto al metabolismo di primo passaggio
NOTA: Il fegato può metabolizzare solo una frazione del farmaco somministrato nel 1 passaggio

Metabolismo del farmaco prima di raggiungere la circolazione sistemica
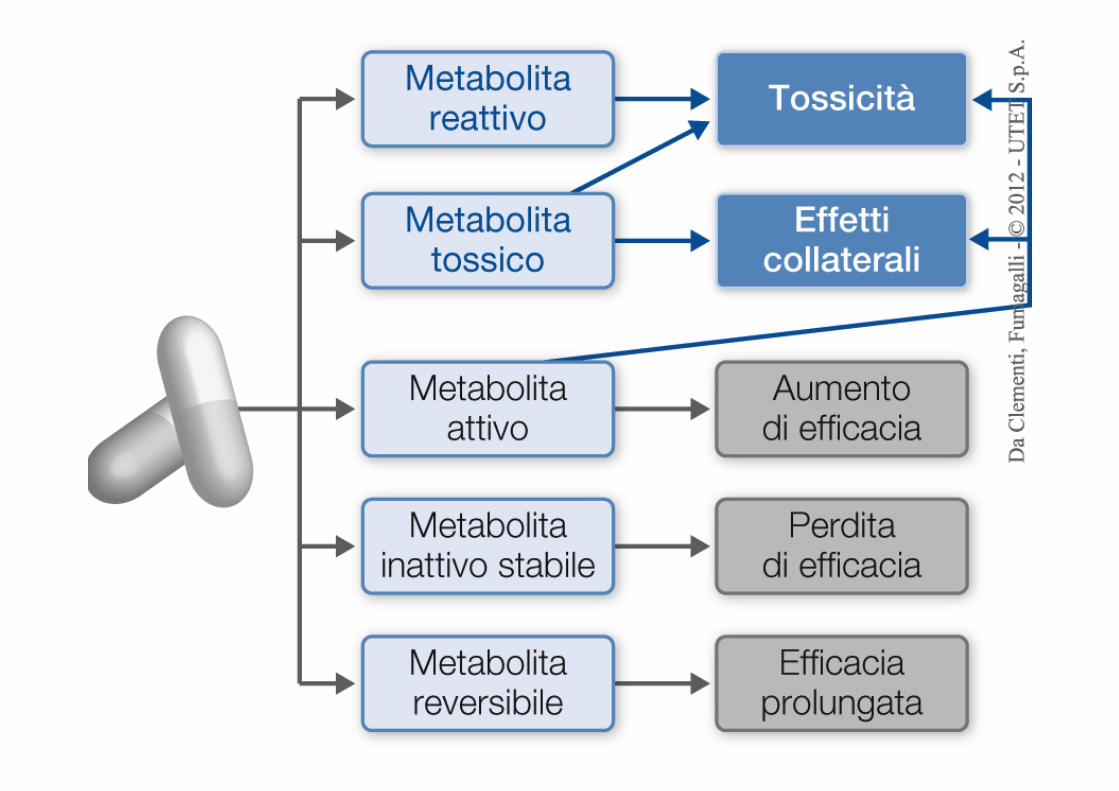
Farmaco Attivo Farmaco Inattivo
Farmaco Inattivo Farmaco Attivo
Perché e come metabolizziamo i farmaci ?
Farmaco A Farmaco B = Aumento polarità Enzimi
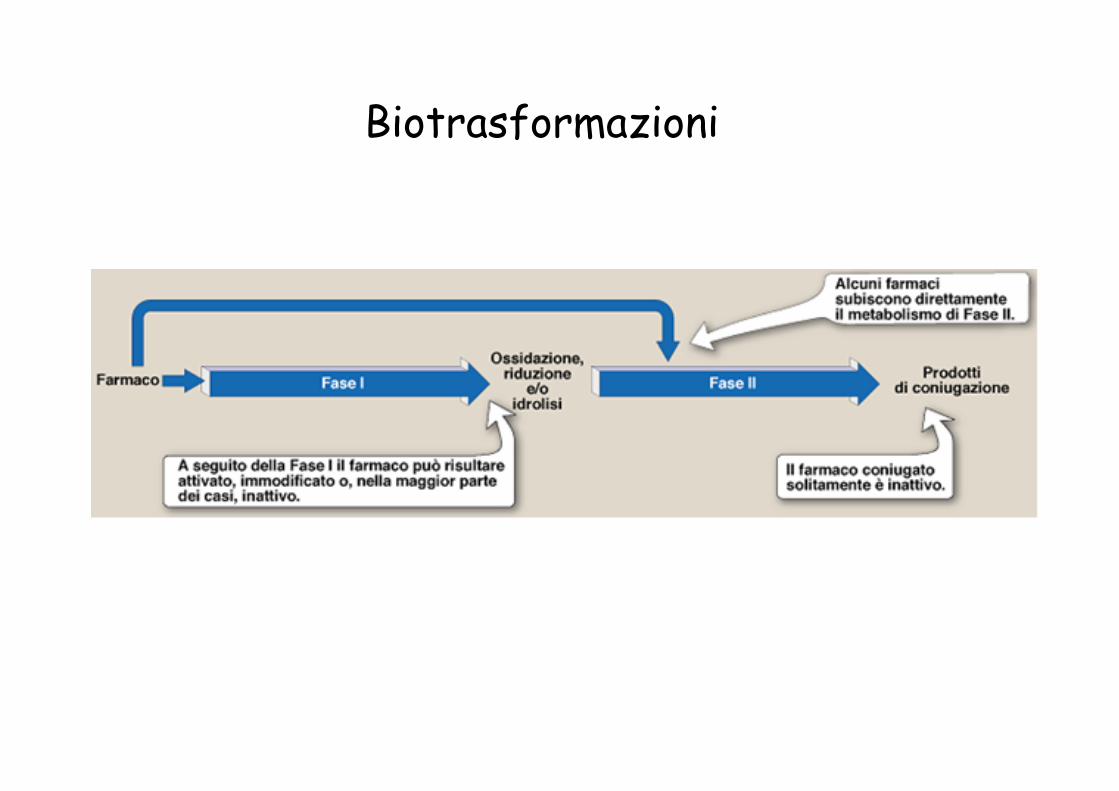
Biotrasformazioni

Tipi di reazioni Enzima
Fase I Ossidazione Riduzione Idrolisi Fase II Coniugazione
Citocromo P450 Alcol deidrogenasi Aldeide deidrogenasi Monossigenasi flaviniche Monoaminossidasi Chinone reduttasi (DT diaforasi) Citocromo P450 reduttasi Epossido idrolasi UDP glucuronil transferasi Solfotransferasi N-acetil transferasi Metiltransferasi Coniugazione con amminoacidi Glutatione transferasi
Enzimi del metabolismo degli xenobiotici
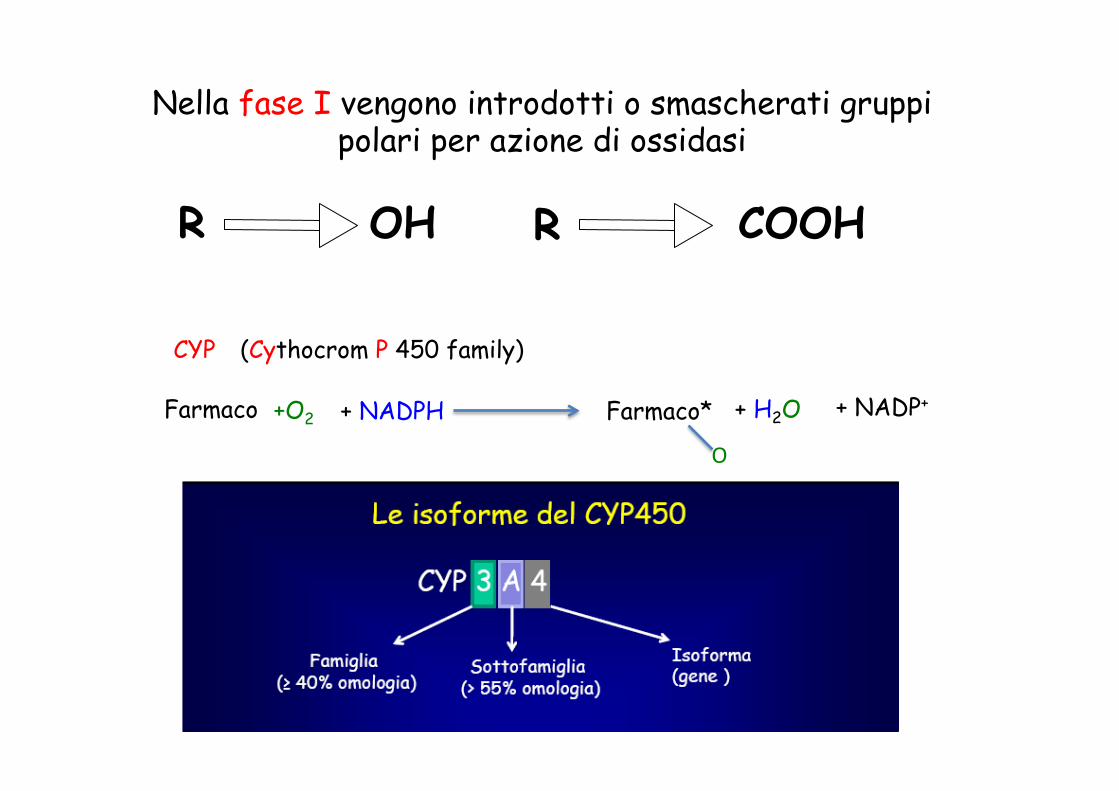
R COOH R OH
Nella fase I vengono introdotti o smascherati gruppi polari per azione di ossidasi
CYP (Cythocrom P 450 family)
+O2 + NADPH Farmaco Farmaco* + NADP+
O
+ H2O
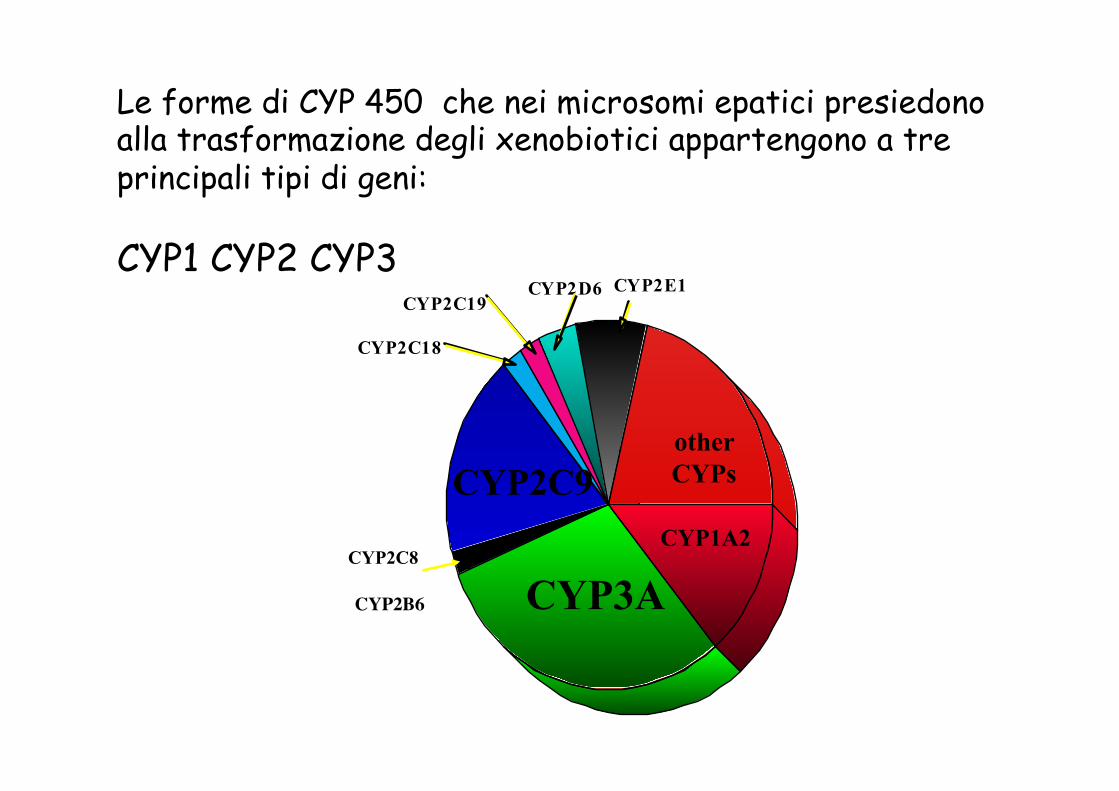
Le forme di CYP 450 che nei microsomi epatici presiedono alla trasformazione degli xenobiotici appartengono a tre principali tipi di geni: CYP1 CYP2 CYP3
C Y P 2 E 1 C Y P 2 D 6
C Y P 2 C 1 8
C Y P 2 C 1 9
CYP3A CYP1A2
other CYPs CYP2C9
CYP2C8
CYP2B6
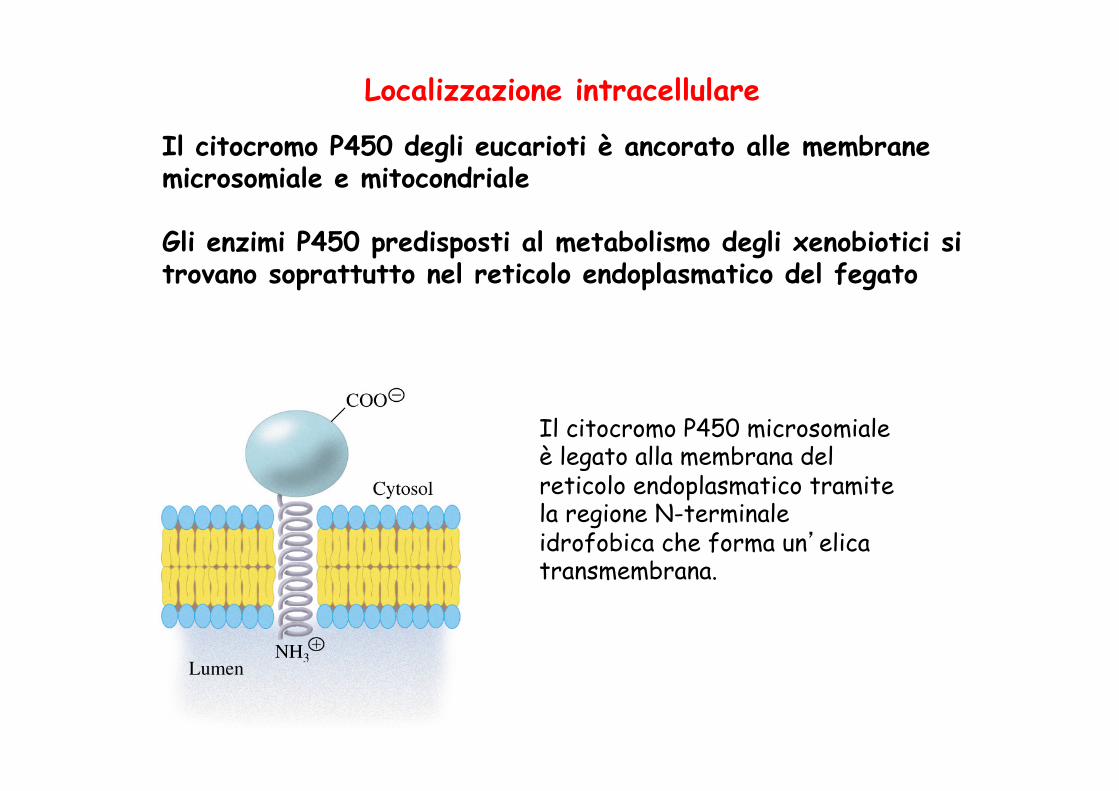
Localizzazione intracellulare
Il citocromo P450 degli eucarioti è ancorato alle membrane microsomiale e mitocondriale Gli enzimi P450 predisposti al metabolismo degli xenobiotici si trovano soprattutto nel reticolo endoplasmatico del fegato
Il citocromo P450 microsomiale è legato alla membrana del reticolo endoplasmatico tramite la regione N-terminale idrofobica che forma un’elica transmembrana.

Tipi di reazioni Enzima
Fase I Ossidazione Riduzione Idrolisi Fase II Coniugazione
Citocromo P450 Alcol deidrogenasi Aldeide deidrogenasi Monossigenasi flaviniche Monoaminossidasi Chinone reduttasi (DT diaforasi) Citocromo P450 reduttasi Epossido idrolasi UDP glucuronil transferasi Solfotransferasi N-acetil transferasi Metiltransferasi Coniugazione con amminoacidi Glutatione transferasi
Enzimi del metabolismo degli xenobiotici

D+ENDOX DX+ENDO
Nella fase II la molecola endogena (ENDOX) dona una sua porzione allo xenobiotico (D)
CONIUGAZIONE
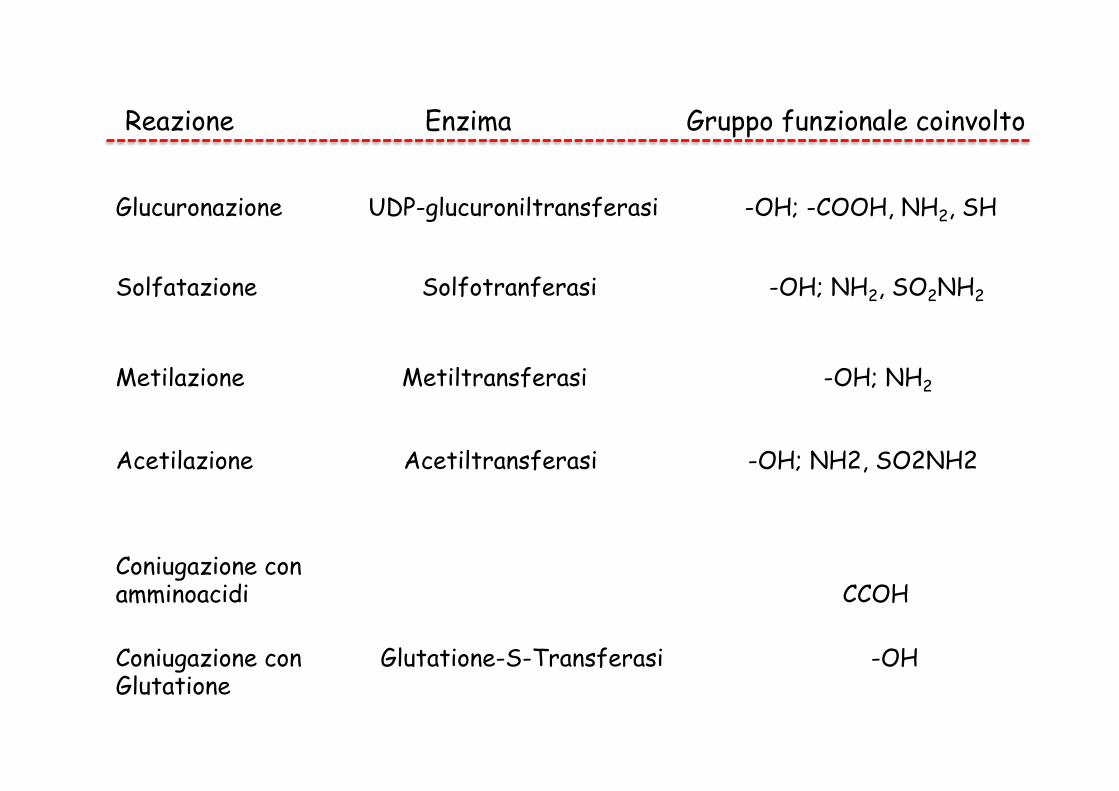
Reazione Enzima Gruppo funzionale coinvolto
Glucuronazione UDP-glucuroniltransferasi -OH; -COOH, NH2, SH
Solfatazione Solfotranferasi -OH; NH2, SO2NH2
Metilazione Metiltransferasi -OH; NH2
Acetilazione Acetiltransferasi -OH; NH2, SO2NH2
Coniugazione con amminoacidi CCOH
Coniugazione con Glutatione-S-Transferasi -OH Glutatione

eliminazione del farmaco dall’organismo Escrezione:
1) Escrezione renale
2) Escrezione biliare e fecale 3) Escrezione attraverso il sudore 4) Escrezione nel latte 5) Escrezione polmonare
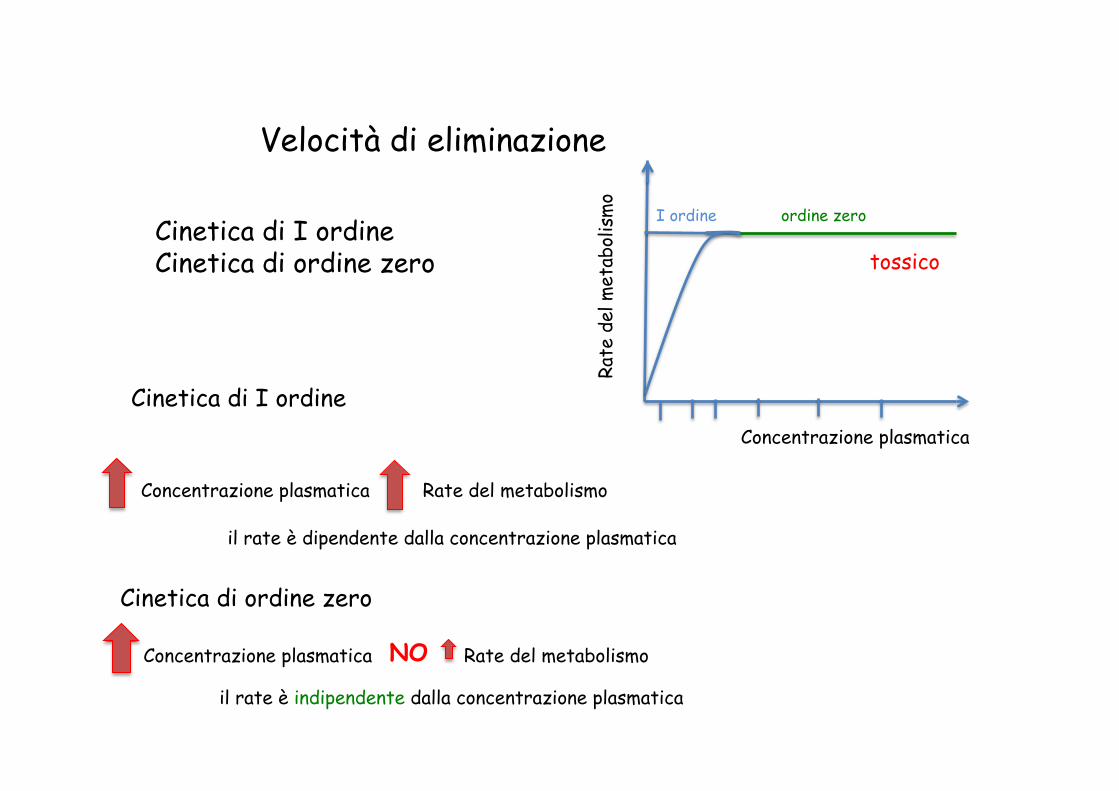
Velocità di eliminazione
Cinetica di I ordine Cinetica di ordine zero
Concentrazione plasmatica
Rate
del
met
abol
ism
o Cinetica di ordine zero
Concentrazione plasmatica Rate del metabolismo NO
Concentrazione plasmatica Rate del metabolismo
Cinetica di I ordine
I ordine
il rate è dipendente dalla concentrazione plasmatica
ordine zero
il rate è indipendente dalla concentrazione plasmatica
tossico

Cinetica di I ordine
Il rate è dipendente dalla concentrazione plasmatica
Concentrazione plasmatica rate del metabolismo
Cinetica di ordine zero
Concentrazione plasmatica rate del metabolismo NO
Il rate è indipendente dalla concentrazione plasmatica
Il rate del metabolismo è costante Il rate del metabolismo è proporzionale alla concentrazione del farmaco
Emivita del farmaco è costante
tempo necessario per metabolizzare il 50% del farmaco
Proporzione costante tempo
= 50%
1 h
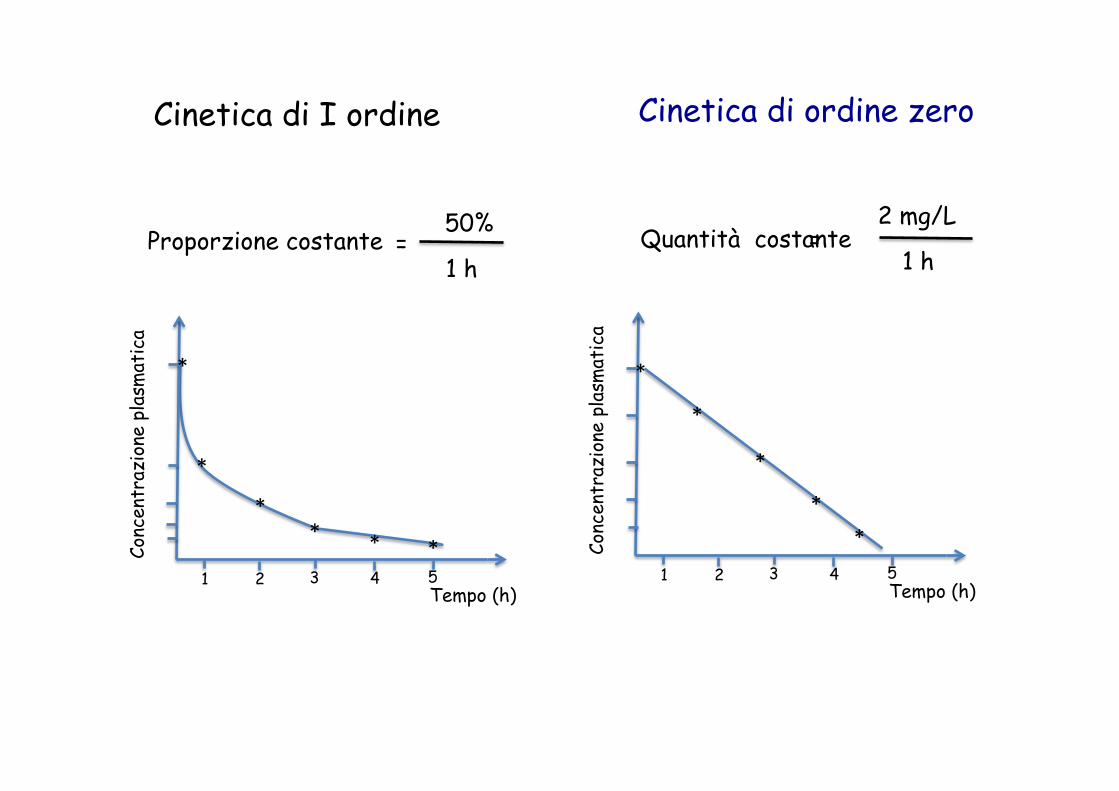
Proporzione costante = 50%
1 h Quantità costante =
2 mg/L
1 h
Tempo (h) Co
ncen
traz
ione
pla
smat
ica
1 2 3 4 5
*
*
* *
*
Tempo (h)
Conc
entr
azio
ne p
lasm
atic
a
1 2 3 4 5
*
* * * *
*
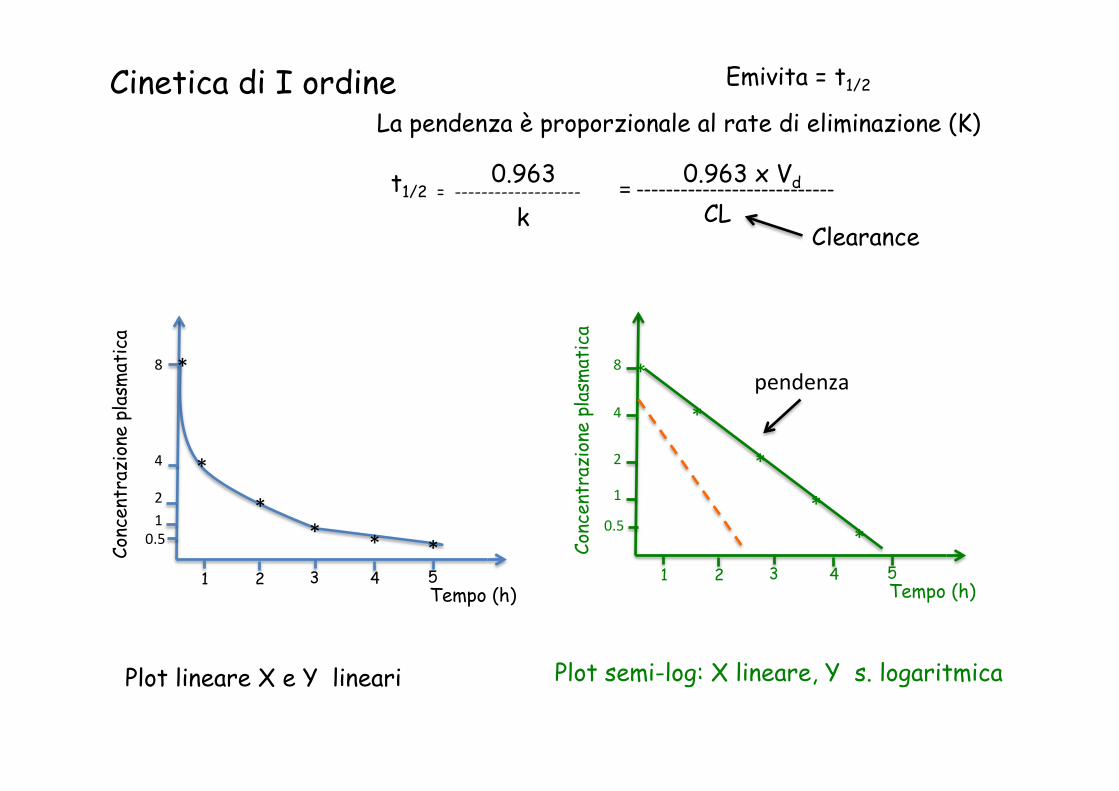
Cinetica di I ordine
Cinetica di ordine zero
Tempo (h) Co
ncen
traz
ione
pla
smat
ica
1 2 3 4 5
*
*
* *
*
8
4
2
1
0.5
Tempo (h)
Conc
entr
azio
ne p
lasm
atic
a
1 2 3 4 5
*
* * * *
* 8
4
2 1
0.5
Plot lineare X e Y lineari Plot semi-log: X lineare, Y s. logaritmica
Emivita = t1/2
pendenza
La pendenza è proporzionale al rate di eliminazione (K)
0.963 t1/2 = ------------------- k
= -‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐-‐ 0.963 x Vd CL
Clearance
Cinetica di I ordine

Clearance è una misura dell’eliminazione
Viene definita come = Velocità di eliminazione
Conc. plasmatica del farmaco
= mg/min
= mg/ml
= ml/min
È il volume del plasma da cui il farmaco è completamente rimosso nell’unità di tempo
Escrezione renale
Escrezione biliare e fecale Escrezione polmonare
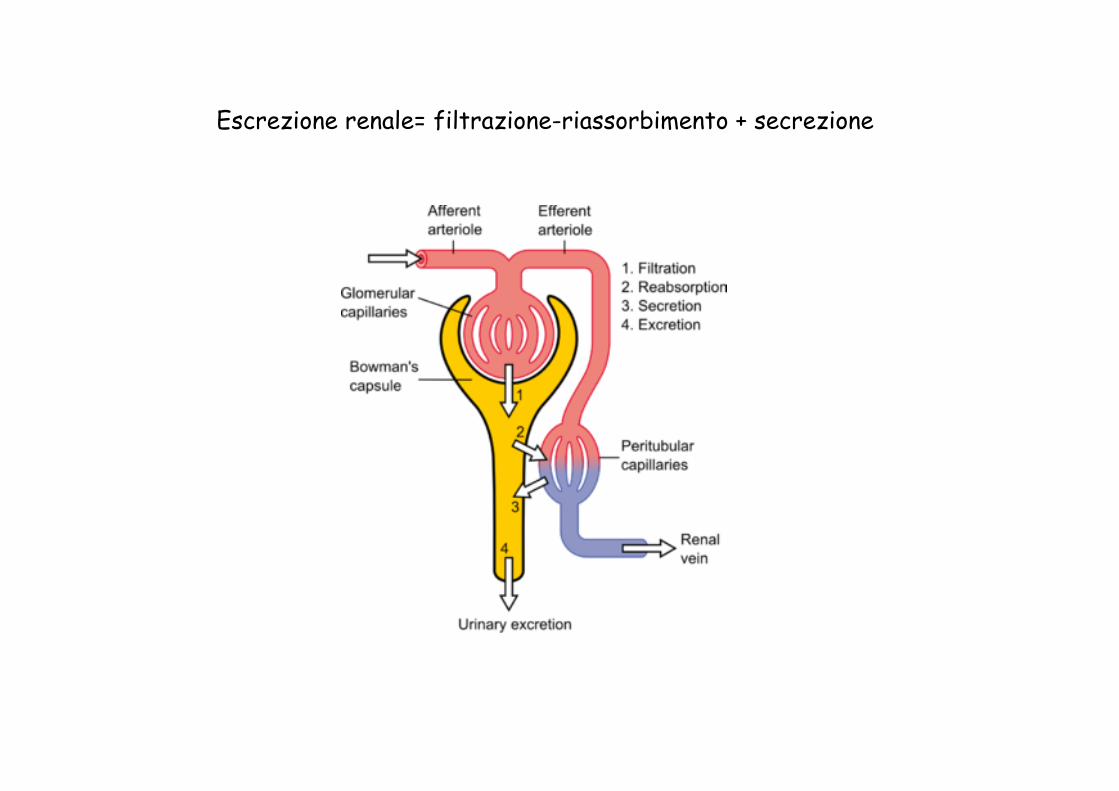
Escrezione renale= filtrazione-riassorbimento + secrezione

ASSORBIMENTO L’entità e la velocità di assorbimento di un farmaco dipendono essenzialmente da: ü Via di somministrazione ü Forma farmaceutica ü Liposolubilità ü Per via orale dal pH dell’ambiente e dalla costante di
dissociazione (pKa) ü Flusso sanguigno nel sito di assorbimento ü Superficie totale disponibile per l’assorbimento ü Tempo di contatto con la superficie di assorbimento
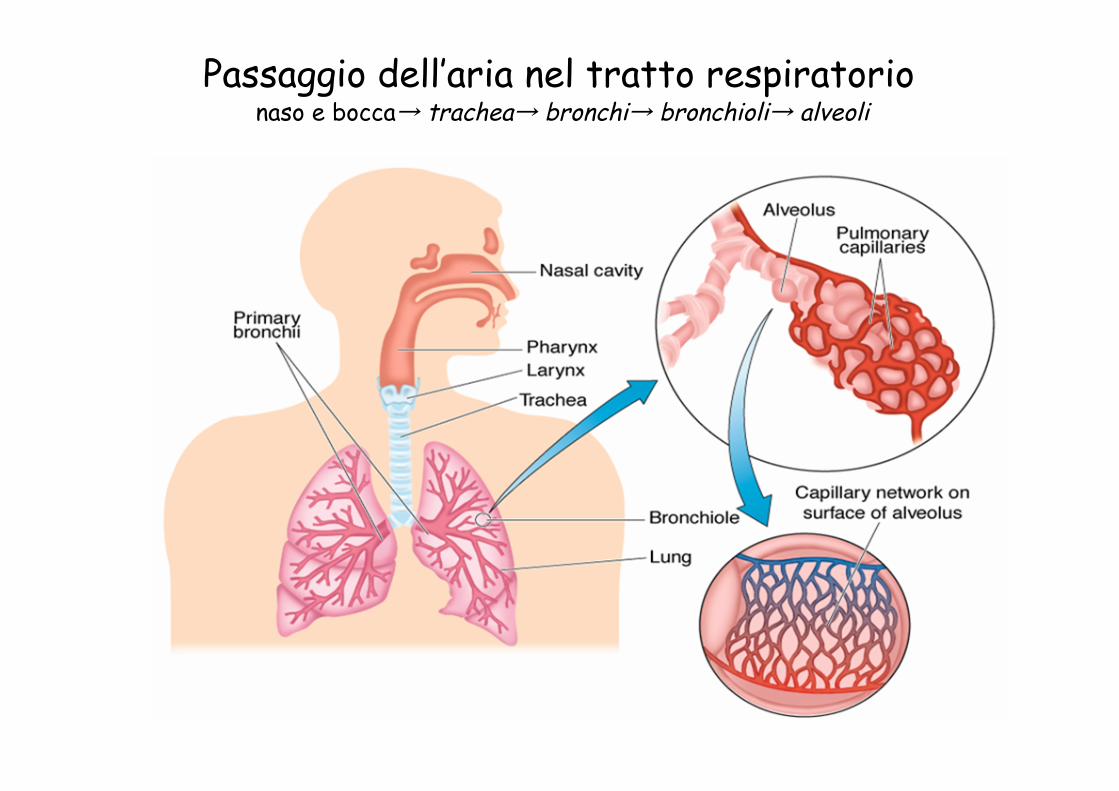
Passaggio dell’aria nel tratto respiratorio naso e bocca→ trachea→ bronchi→ bronchioli→ alveoli
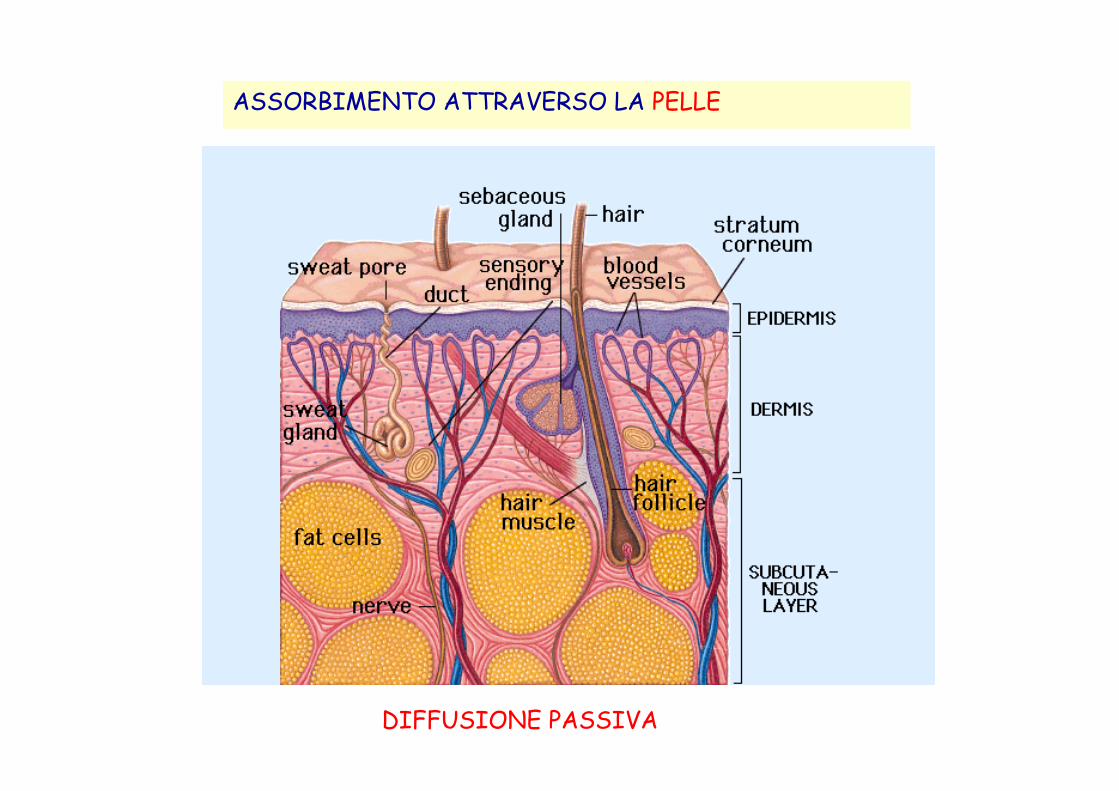
ASSORBIMENTO ATTRAVERSO LA PELLE
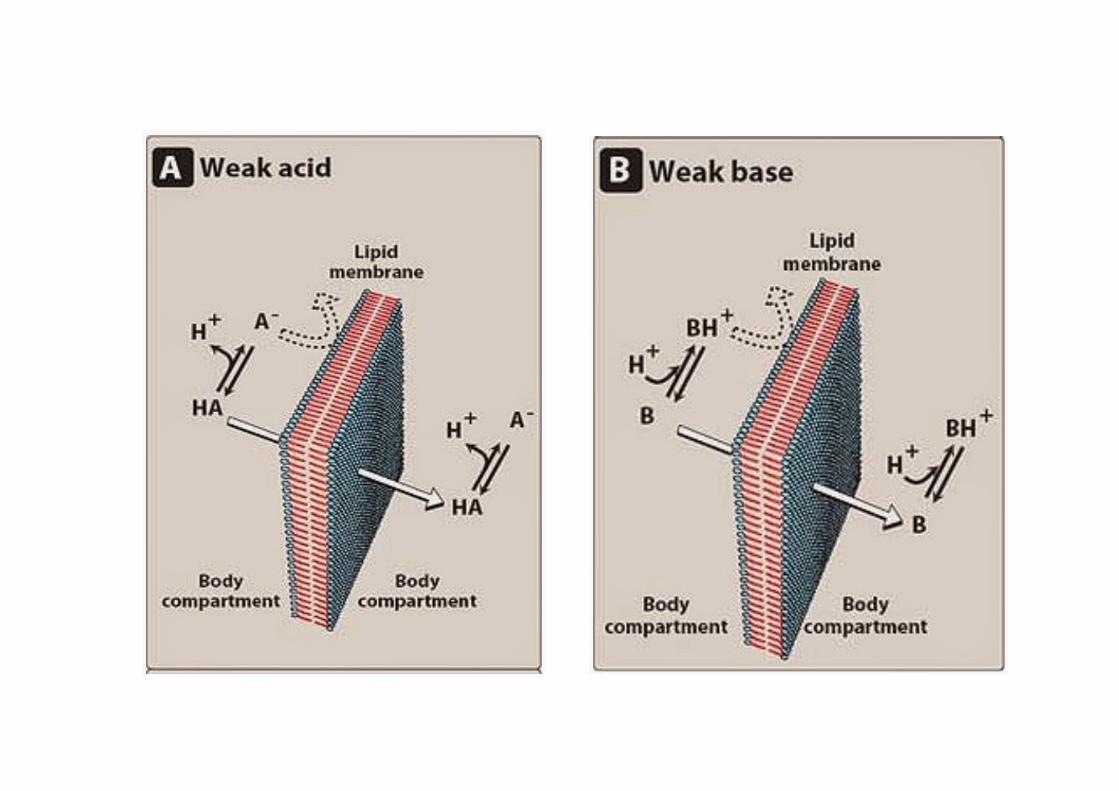
DIFFUSIONE PASSIVA
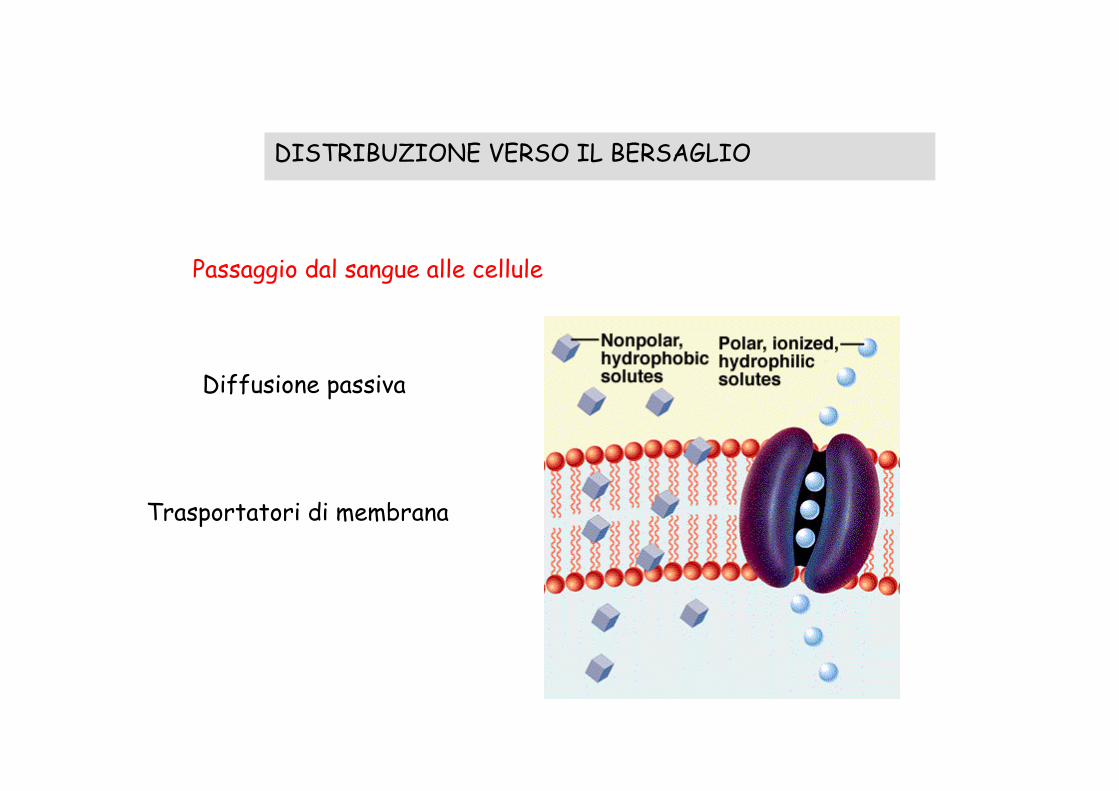
DISTRIBUZIONE VERSO IL BERSAGLIO
Passaggio dal sangue alle cellule
Diffusione passiva
Trasportatori di membrana

La velocità e l’entità di distribuzione dipendono da vari fattori: il flusso ematico liposolubilità affinità per i diversi compartimenti legame alle proteine plasmatiche
Le concentrazioni nel sangue e nei tessuti possono essere diverse tra loro
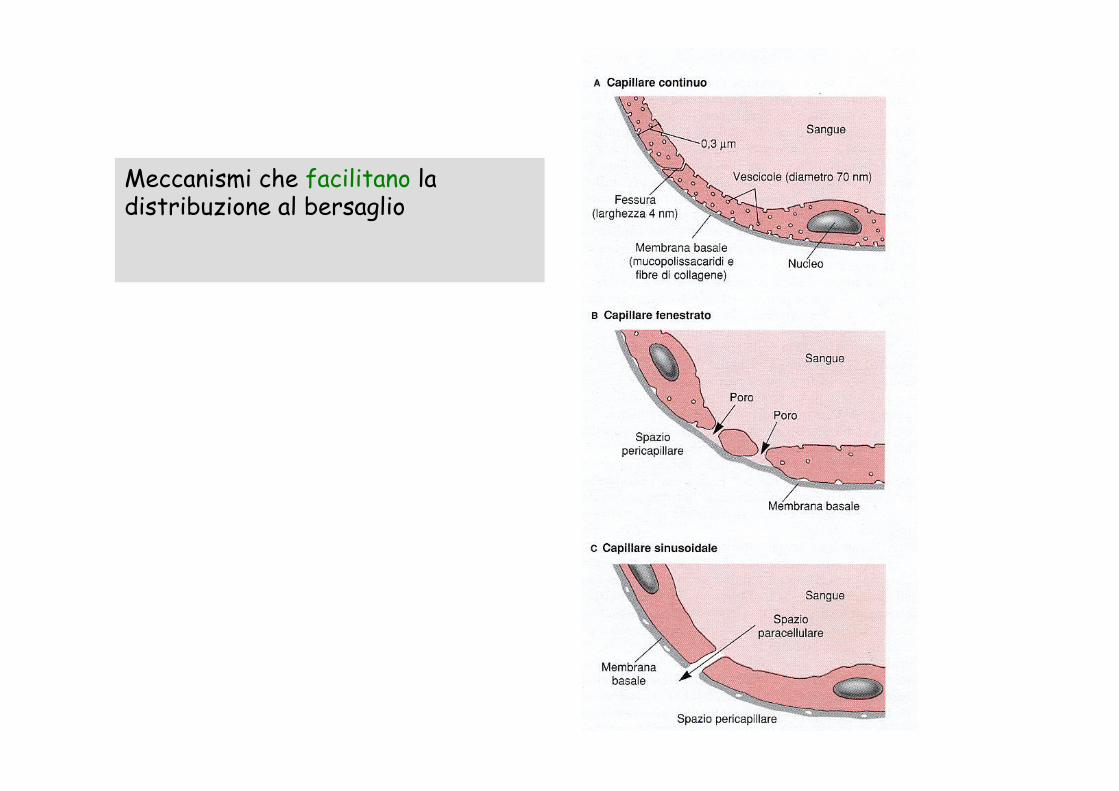
Meccanismi che facilitano la distribuzione al bersaglio

Meccanismi che ostacolano il raggiungimento del bersaglio
distribuzione a siti di deposito
legame con proteine intracellulari
escrezione dalla cellula
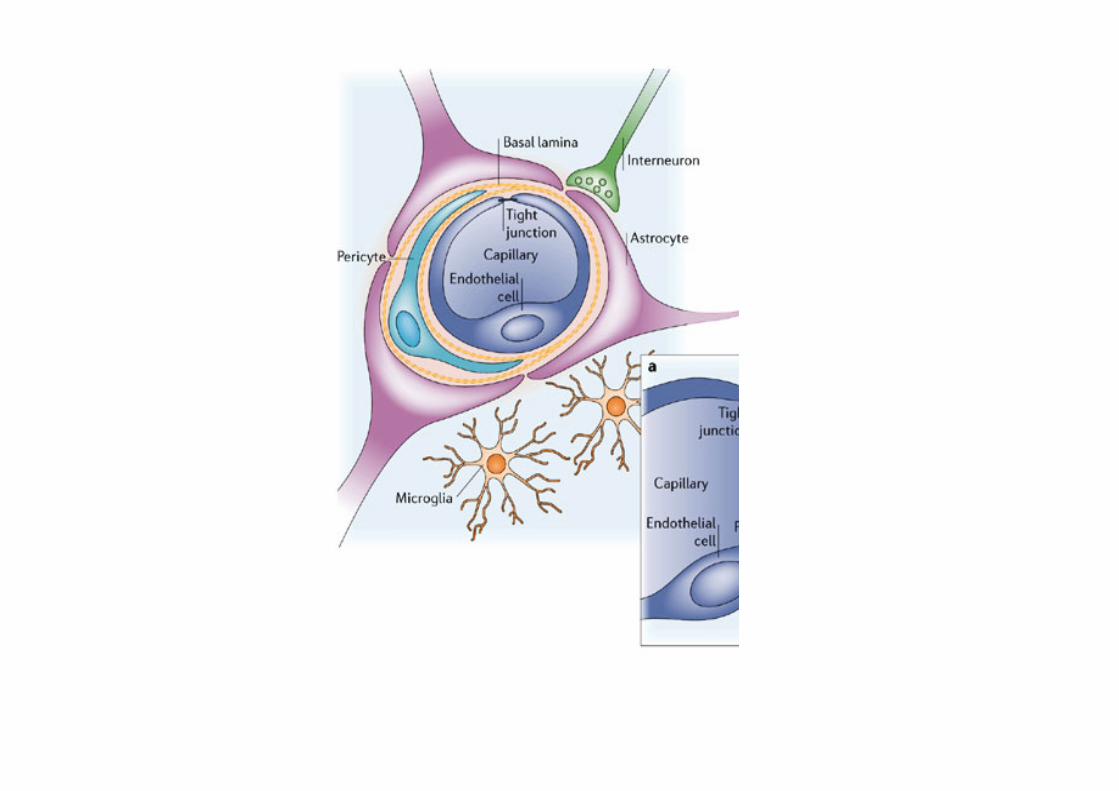
barriere specializzate
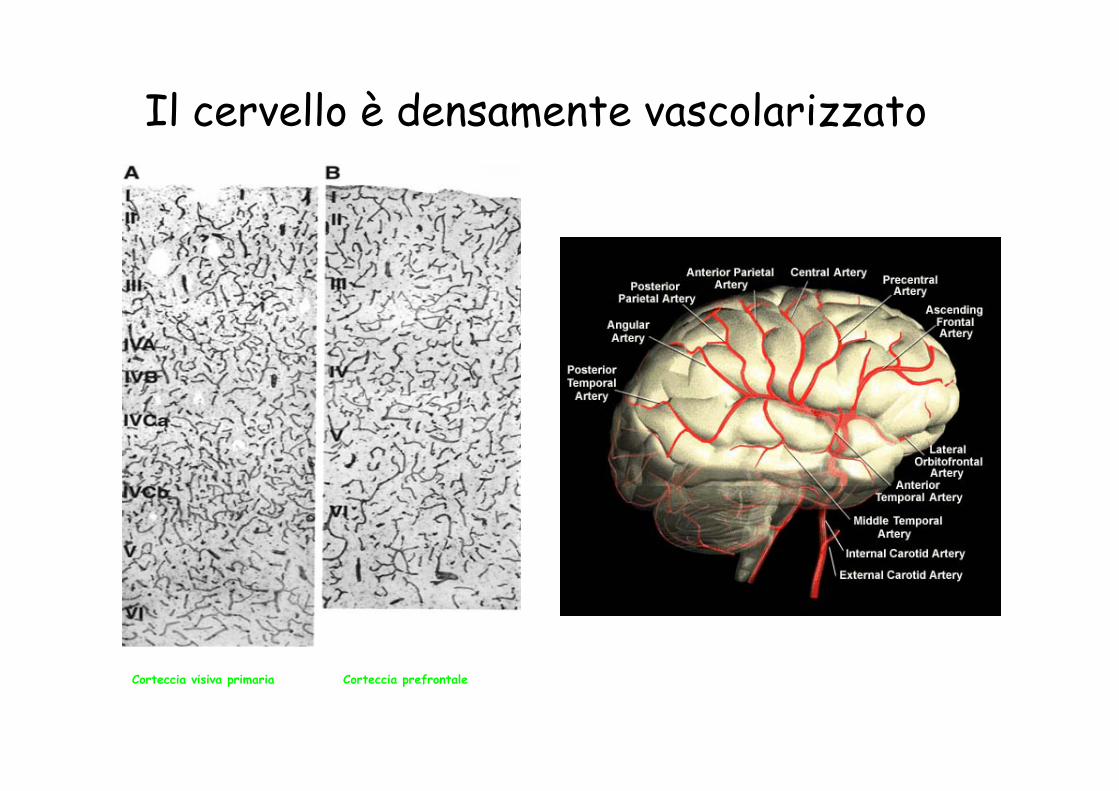
Il cervello è densamente vascolarizzato
Corteccia visiva primaria Corteccia prefrontale
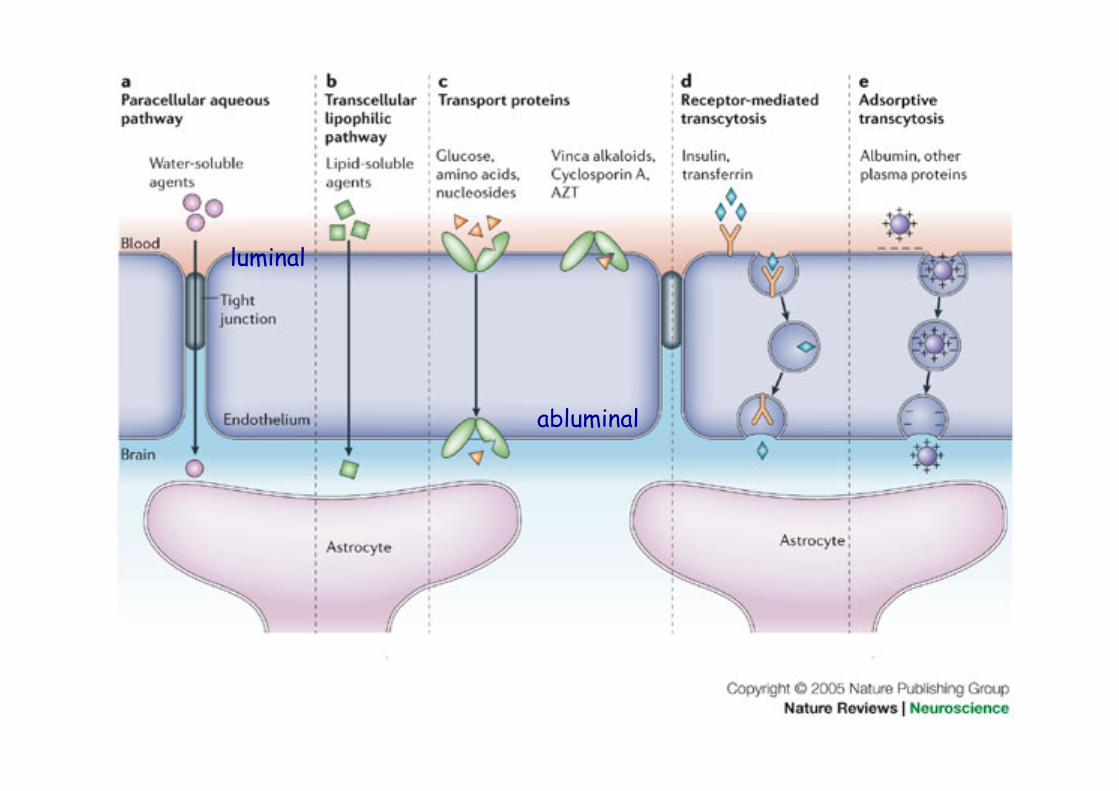
abluminal
luminal

Ø Induzione o inibizione da farmaci concomitanti o fattori ambientali
Ø Stati fisiologici (età, sesso) Ø Stati patologici Ø Polimorfismi genetici (variazioni a livello dei geni
presenti in >1% popolazione)
PRINCIPALI FATTORI RESPONSABILI DELLA VARIABILITÀ NEL METABOLISMO DEI
FARMACI

Inibizione del metabolismo
Induzione del metabolismo
Alterazione nell’assorbimento
Alterazione dell’escrezione
Interazioni farmacodinamiche
INTERAZIONI: cause

INTERAZIONI: effetto
Effetto additivo à 10 + 10 = 20
Effetto sinergico à 10 + 10 = 50
Potenziamento à 0 + 10 = 30
Antagonismo à 0-2 + 10 = <12
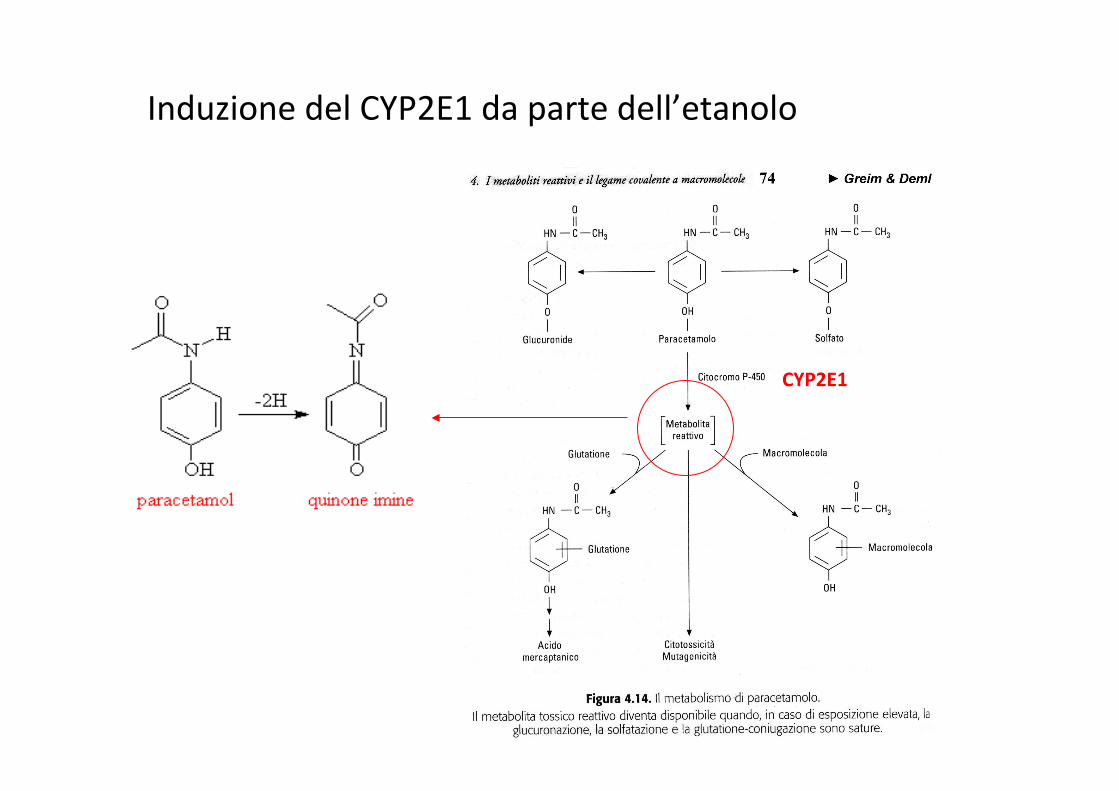
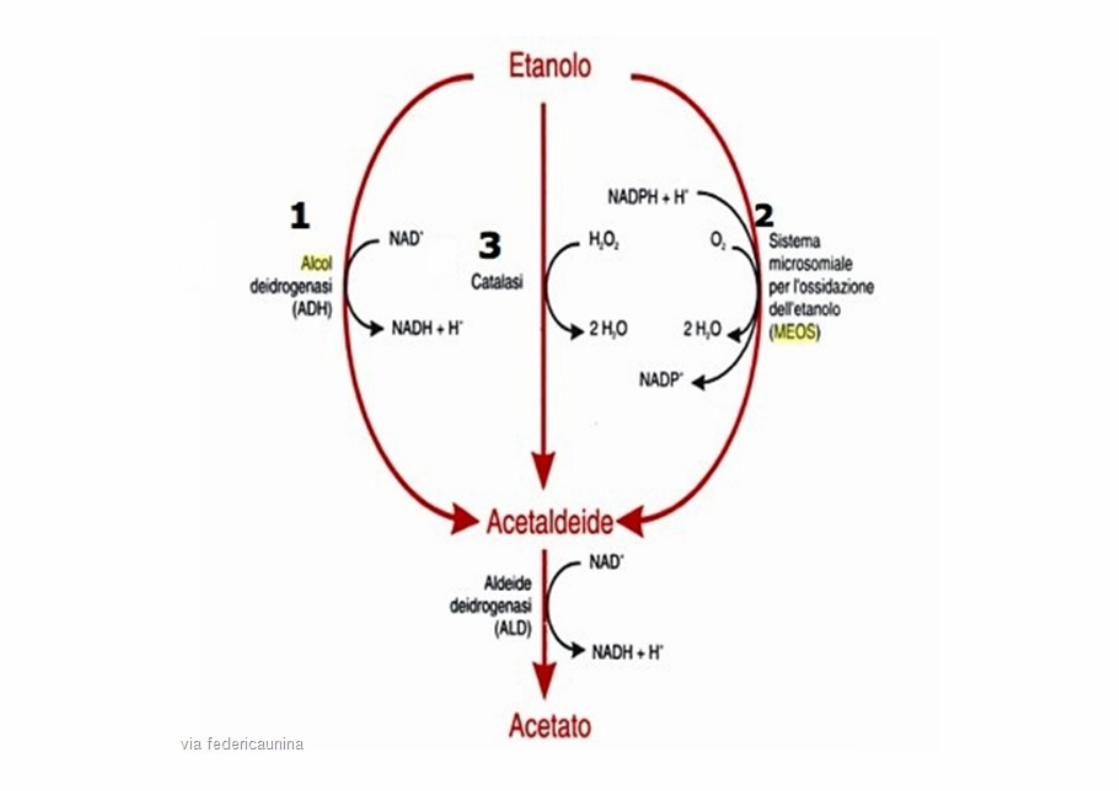
Induzione del CYP2E1 da parte dell’etanolo
CYP2E1
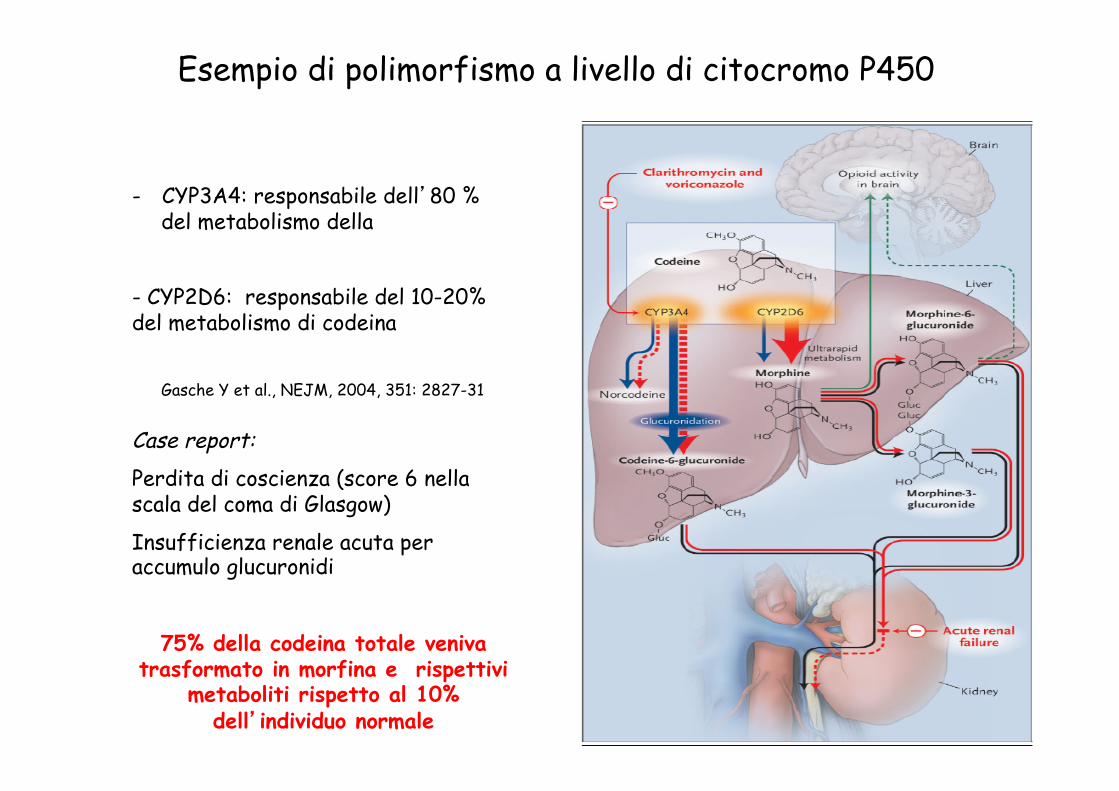
Gasche Y et al., NEJM, 2004, 351: 2827-31
- CYP3A4: responsabile dell’80 % del metabolismo della
- CYP2D6: responsabile del 10-20% del metabolismo di codeina
Esempio di polimorfismo a livello di citocromo P450
Case report: Perdita di coscienza (score 6 nella scala del coma di Glasgow)
Insufficienza renale acuta per accumulo glucuronidi
75% della codeina totale veniva
trasformato in morfina e rispettivi metaboliti rispetto al 10%
dell’individuo normale
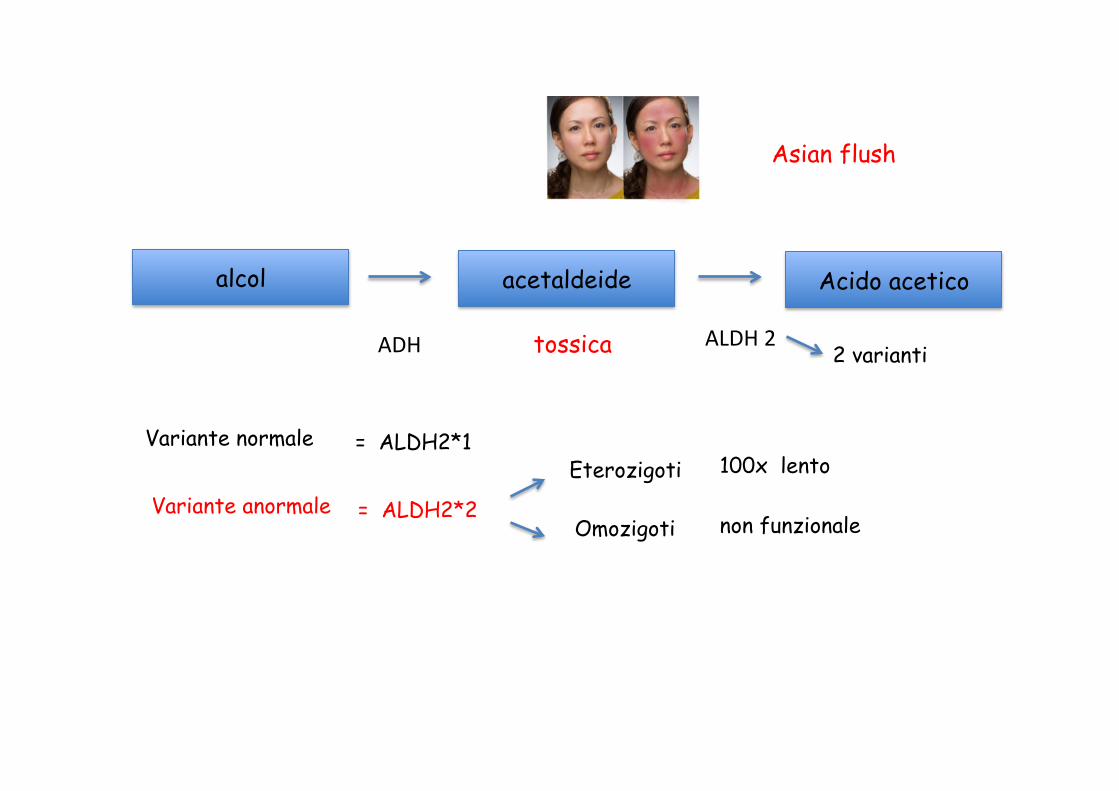
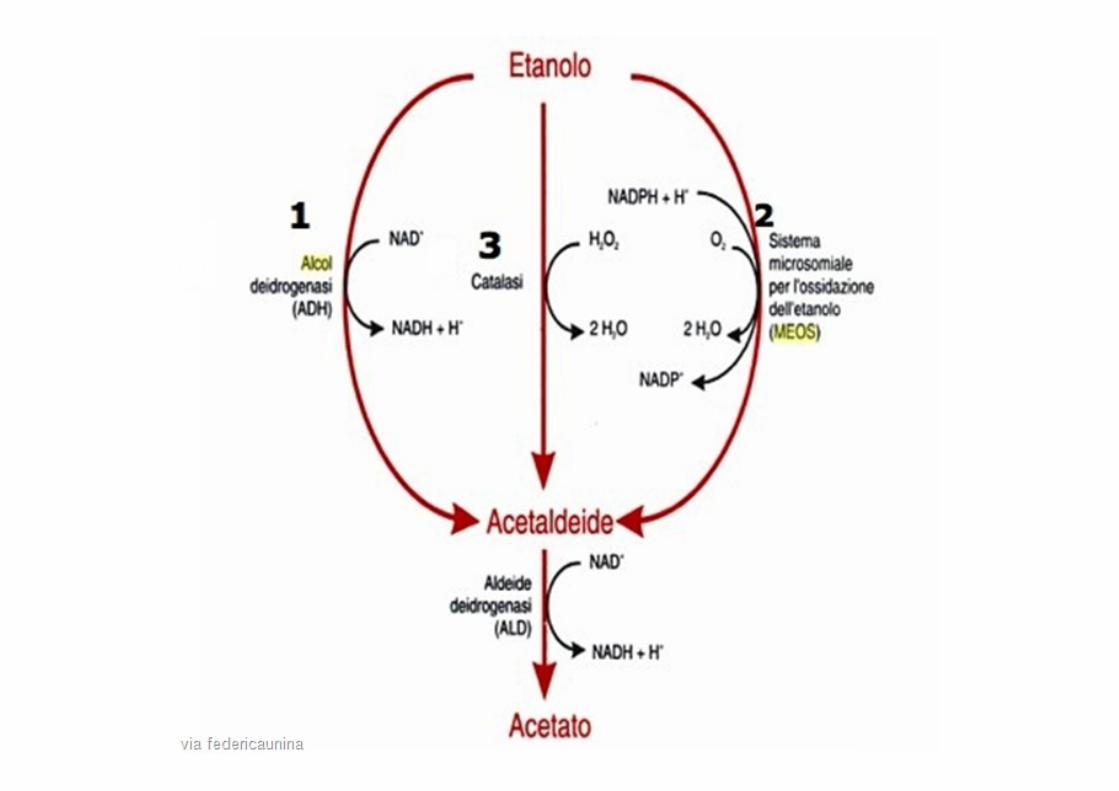
alcol acetaldeide Acido acetico
ADH ALDH 2 tossica
Asian flush
2 varianti
Variante normale
Variante anormale
= ALDH2*1
= ALDH2*2
Eterozigoti
Omozigoti
100x lento
non funzionale

Metabolizzatori Lenti (PM): incapacità di utilizzare la via del CYP Metabolizzatori Intermedi (IM): capacità fortemente diminuita Metabolizzatori Normali (EM): capacità normale Metabolizzatori Ultrarapidi (UM): capacità eccessiva
Farmacogenetica: basi ereditarie delle differenze tra gli individui
nell’azione dei farmaci
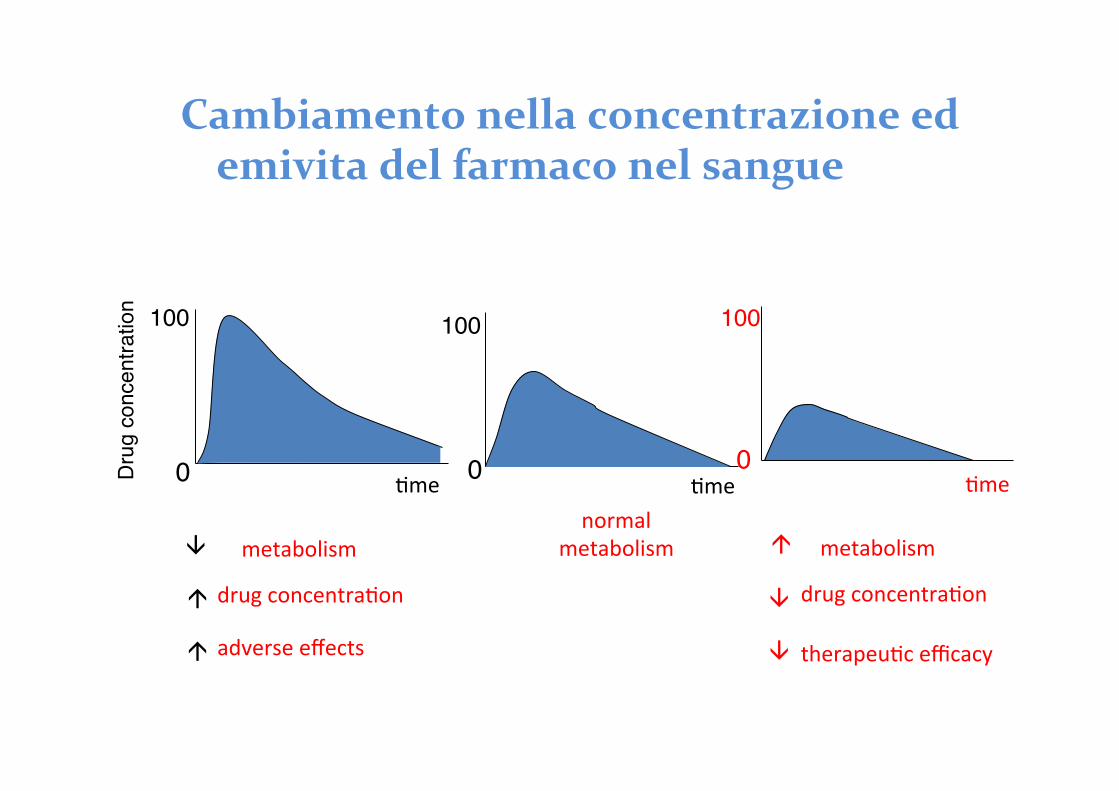
Cambiamento nella concentrazione ed emivita del farmaco nel sangue
100
normal metabolism
0 ^me Dru
g co
ncen
tratio
n 100
0 ^me
metabolism
à
drug concentra^on à
adverse effects à
100
^me 0
metabolism à
drug concentra^on
à
therapeu^c efficacy
à

Per indurre un effetto farmacologico un agente deve:
ü interagire con siti specifici ad
appropriata concentrazione per tempo
sufficientemente lungo
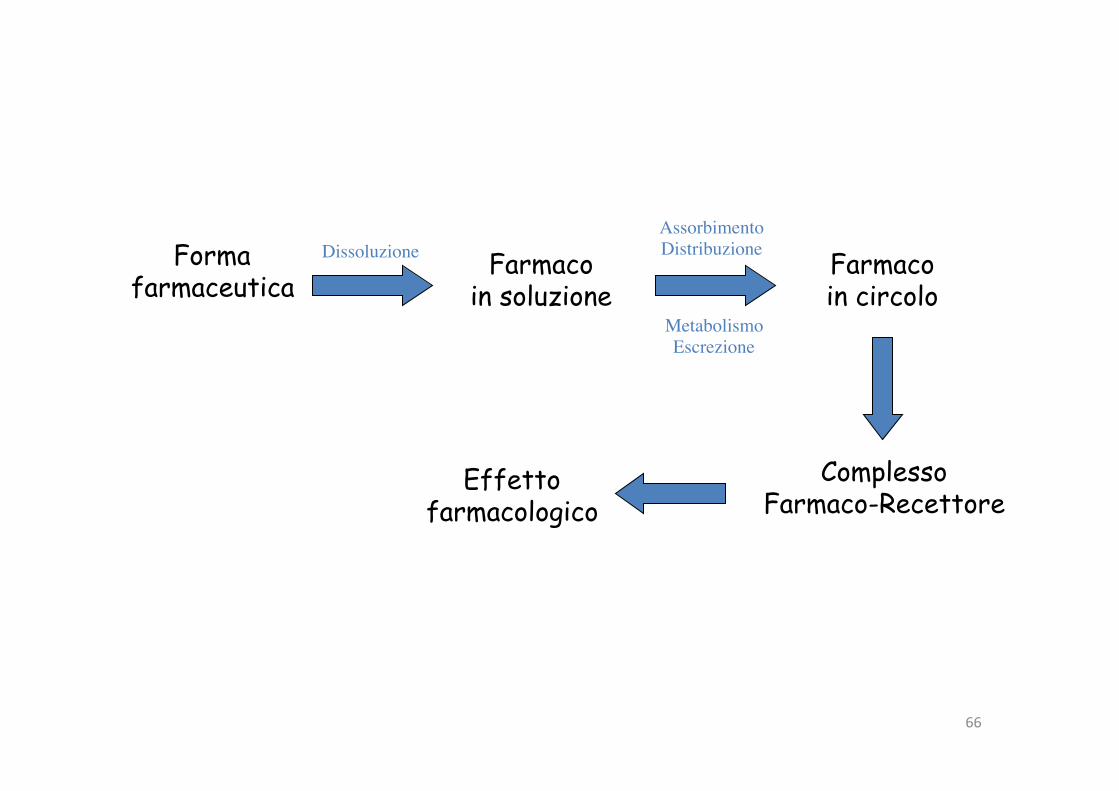
66
Forma farmaceutica
Dissoluzione Farmaco in soluzione
AssorbimentoDistribuzione Farmaco
in circoloMetabolismoEscrezione
Complesso Farmaco-Recettore
Effetto farmacologico
Recommended

















