
HF/6-31G∗ Energy Surfaces forDisaccharide Analogs
ALFRED D. FRENCH,1 ANNE-MARIE KELTERER,2 GLENN P. JOHNSON,1
MICHAEL K. DOWD,1 CHRISTOPHER J. CRAMER3
1Southern Regional Research Center, Agricultural Research Service, U.S. Department of Agriculture,1100 Robt. E. Lee Blvd., P.O. Box 19687, New Orleans, Louisiana 70179-06872Institut für Physikalische und Theoretische Chemie, Technische Universität Graz, Technikerstrasse 4,A-8010 Graz, Austria3Department of Chemistry and Supercomputer Institute, 207 Pleasant St. SE, Minneapolis,Minnesota 55455-0431
Received 10 February 2000; accepted 20 June 2000
ABSTRACT: The HF/6-31G∗ level of theory was used to calculate relaxedpotential energy surfaces for 12 analogs of disaccharides. The analogs were madeby replacing glucose with tetrahydropyran and fructose with2-methyltetrahydrofuran. Molecules had zero, one or two anomeric carbonatoms, and di-axial, axial-equatorial, and di-equatorial linkages. Despite theabsence of hydroxyl groups, the surfaces account well for conformations that areobserved in crystals of the parent disaccharides. Thus, torsional energy and thesimple bulk of ring structures are major factors in determining disaccharideconformation. The contour shapes around the global minima depend on thenumber of anomeric carbons involved in the linkage, while the presence ofalternative minima that have relative energies less than 4 kcal/mol mostlyrequires equatorial bonds. However, molecules with two adjacent anomericcenters gave exceptions to these rules. Flexibility values related to a partitionfunction show that the di-axial trehalose analog is the most rigid. Thedi-equatorial pseudodisaccharide analog with no anomeric centers is mostflexible. Reproduction of these surfaces is proposed as a simple test of force fieldsfor modeling carbohydrates. Also, these surfaces can be used in a simple hybridmethod for calculating disaccharide energy surfaces. c© 2000 John Wiley &Sons, Inc.∗ J Comput Chem 22: 65–78, 2001
Dedicated to the memory of Professor Georgy A. Jeffrey, de-ceased February 13, 2000
Correspondence to: A. D. French; e-mail: [email protected] or to: A.-M. Kelterer; e-mail: [email protected]
This article includes Supplementary Material available fromthe author upon request or via the Internet at ftp.wiley.com/public/journals/jcc/suppmat/22/65 or http://journals.wiley.com/jcc
Journal of Computational Chemistry, Vol. 22, No. 1, 65–78 (2001)c© 2000 John Wiley & Sons, Inc. ∗This article is a US Government work and, as such, is in the public domain in the United Statesof America.

FRENCH ET AL.
Keywords: ab initio; anomeric effect; carbohydrate; conformational analysis;cellobiose; laminarabiose; galabiose; maltose; nigerose; quantum mechanics;sucrose; tetrahydrofuran; trehalose
Introduction
T he main descriptors of disaccharide shape arethe torsion angles (designated φ and ψ) for the
bonds that link the two monosaccharide residuestogether. Calculated relative energies for such mole-cules can be plotted on grids of φ and ψ , yieldingenergy surfaces that are often called Ramachandranplots.1 These maps predict the relative likelihood ofdifferent molecular conformations and characterizethe barriers among the various shapes. They alsopermit the assessment of the distortion of experi-mentally determined structures. The energies canbe calculated by methods of varying sophisticationand computational cost. Initial efforts used simpleallowed/disallowed “energy” functions, but weresoon improved to include van der Waals forces.2, 3
Those methods only allowed variation of the torsionangles for the linkage bonds.
The HSEA (Hard Sphere Exo-Anomeric Effect)program of Lemieux and coworkers4 – 6 used vander Waals energies and also incorporated tor-sional potentials to reproduce exo-anomeric effects.The exo-anomeric effect7 preferentially orients sub-stituents that are attached to the glycosidic oxy-gen atom to locations that are gauche to the ringoxygen. Thus, the magnitude and exact orienta-tion resulting from the exo-anomeric effect are ofdirect importance to the study of disaccharide con-formation. The torsional potentials for HSEA, onefor α-linkages and one for β-linkages, were pa-rameterized with single-point HF/4-31G ab initiocalculations on dimethoxymethane. Results fromHSEA sufficed to interpret some NMR spectra, butlimitations gradually became apparent.8 In partic-ular, the rigid linkage bond angle and geometriesof the monomeric residues led to strain energiesthat were much too high for some observed con-formations. Therefore, in the late 1980s, so-calledrelaxed-residue calculations9 – 12 were undertaken,in which energy minimization is employed at eachincrement of φ andψ . These more complex, relaxed-residue calculations can now be carried out withavailable molecular mechanics (MM) force fields.
For any given values of φ and ψ , however, rel-ative energies from various force fields often differby a few kcal (all energies herein are molar).13 This
degree of uncertainty for values near importantminima is large enough that important questionscannot be answered with confidence. In the questto obtain greater accuracy, the use of ab initio quan-tum mechanics (QM) is increasing. QM can be usedin studies of disaccharides in several ways besidessimply parameterizing the potential function forthe glycosidic torsion angle. Some MM force fieldsare now based exclusively on QM studies of smallmolecules.14 – 16 Alternatively, one might use QMto study disaccharides directly. However, such cal-culations are too time-consuming due to the largenumber of alternative orientations of the exo-cyclicgroups that must be considered to avoid biasing theanalysis.
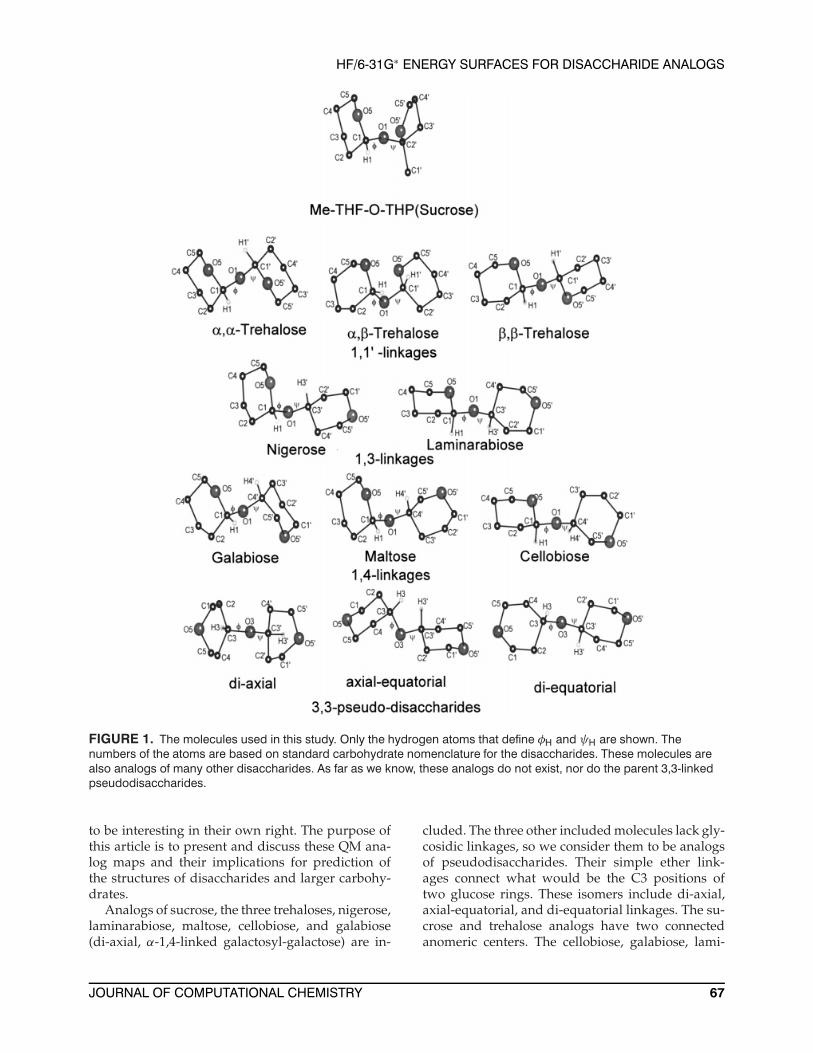
Another approach is to combine QM withMM. We have recently developed a simple hy-brid method for making Ramachandran plots.17 Itinvolves the separate calculation of the MM en-ergy surface for the complete disaccharide, with therequisite extensive variation of the exo-cyclic ori-entations. Also required are separate MM and QMenergy surfaces for the critical part of the disac-charide. The hybrid energy for the disaccharide atany φ,ψ point is the MM energy for the disaccha-ride, minus the MM energy for the critical part,plus the QM energy for the critical part. In thesestudies, the critical part is the disaccharide withall of the exo-cyclic hydroxyl and hydroxymethylgroups replaced by hydrogen. The resulting mole-cules (Fig. 1), except for the sucrose analog, aredimers of tetrahydropyran (THP). The analog of su-crose in the present work retains a methyl groupattached to its tetrahydrofuran (THF) ring at C2′.
These molecules include all the atoms used inconventional MM to define the torsion angles, andthus the torsional energies, of the glycosidic linkage.As discussed by Woods,8 calculation by the differentforce fields of glycosidic bond torsional energy is asource of variable modeling results, so a direct QMcalculation of all linkage torsional energies shouldbe advantageous. It also avoids the undesirable sit-uation implemented in the HSEA software whereinthe torsional parameters are different for axial andequatorial bonds.
While the hybrid energy surfaces for the variousdisaccharides are the finished product of this effort,we found the QM energy surfaces for the analogs
66 VOL. 22, NO. 1
HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
FIGURE 1. The molecules used in this study. Only the hydrogen atoms that define φH and ψH are shown. Thenumbers of the atoms are based on standard carbohydrate nomenclature for the disaccharides. These molecules arealso analogs of many other disaccharides. As far as we know, these analogs do not exist, nor do the parent 3,3-linkedpseudodisaccharides.
to be interesting in their own right. The purpose ofthis article is to present and discuss these QM ana-log maps and their implications for prediction ofthe structures of disaccharides and larger carbohy-drates.
Analogs of sucrose, the three trehaloses, nigerose,laminarabiose, maltose, cellobiose, and galabiose(di-axial, α-1,4-linked galactosyl-galactose) are in-
cluded. The three other included molecules lack gly-cosidic linkages, so we consider them to be analogsof pseudodisaccharides. Their simple ether link-ages connect what would be the C3 positions oftwo glucose rings. These isomers include di-axial,axial-equatorial, and di-equatorial linkages. The su-crose and trehalose analogs have two connectedanomeric centers. The cellobiose, galabiose, lami-
JOURNAL OF COMPUTATIONAL CHEMISTRY 67

FRENCH ET AL.
narabiose, maltose, and nigerose analogs have onlyone anomeric center, and the analogs of pseudo dis-accharides have no anomeric centers. The moleculesin Figure 1 are also analogs for many other mole-cules. For example, the analog of cellobiose is alsoan analog of lactose and of the β-1,4–linked dimersof mannose and xylose. It may also be suited forstudies of β-1,2–linked dimers.
This is not the first instance of studying confor-mations of disaccharide linkages by QM. In particu-lar, axial and equatorial 2-methoxytetrahydropyranhave been widely studied. Tvaroška and Carver18
have made especially thorough studies of variousmethoxytetrahydropyrans as models of disaccha-ride linkages. A few limited studies of disaccha-ride analogs based on cyclohexane, tetrahydropy-ran (THP), and tetrahydrofuran (THF) have beencarried out with ab initio QM theory.19 – 21 Full mapsfor trehalose analogs based on semiempirical QMtheory and partially minimized energies were con-structed more than a decade ago.22
Methods
Calculations on carbohydrates, which haveboth extensive hydrogen bonding possibilities andanomeric effects, are sensitive to the level of QMtheory.23 The HF/6-31G∗ level was chosen for sev-eral reasons. It has been used previously to studyanomeric effects in many molecules that are smallerthan our analogs.16, 24, 25 Because of fortuitous can-cellation of errors, HF/6-31G∗ relative energies forglucopyranose are similar to those produced by ad-vanced electronic structure theory.23, 26 Our workon calculated heats of formation27 showed that therelative electronic energies for the minimum en-ergy conformations of these analog molecules at theHF/6-31G∗ level of theory were similar to B3LYP/6-31G∗ calculations. That similarity is perhaps sur-prising because B3LYP/6-31G∗ and (MP2/6-31G∗)calculations for monosaccharides23, 26, 28 give rela-tive energies for different configurations and con-formations of carbohydrates that are quite differ-ent from energies from either HF/6-31G∗ or largecalculations. This was confirmed recently by Lii,Ma, and Allinger,29 who attributed the differencesto large Basis Set Superposition Errors (BSSE) inB3LYP/6-31G∗ calculations on hydrogen bonds. Be-cause there are no hydroxyl groups in our analogs,the relative energies should depend less on the levelof theory than would the relative energies of com-plete carbohydrates. Therefore, HF/6-31G∗ can bea representative, yet relatively economical, level oftheory for these models.
Energies for the analogs of sucrose, the tre-haloses, and the pseudodisaccharides were calcu-lated with GAMESS,30 and the other maps werecalculated with Gaussian 94.31 Starting modelsof sucrose and the trehalose analogs were basedon crystal structures of the actual disaccharideswith the exo-cyclic groups replaced with hydrogenatoms. These structures were then minimized withQM. Those molecules were then used to generate,by rigid rotations about the linkage bonds, the vari-ous initial structures. In the cases when the startingstructures were badly congested, with interpenetra-tion of the two monomeric residues, structures withthe required φ and ψ values were generated froma successfully optimized neighbor. A similar strat-egy was used for the pseudo disaccharide analogs.Analogs of galabiose, nigerose, maltose, laminara-biose, and cellobiose were sketched with CHEM-X32
and energy-minimized with MM3.33 The glycosidicangle was increased to 150◦ and MM3 was then usedto optimize individual starting structures at eachφ,ψ point for the QM calculations. The increase to150◦ avoided interpenetration of the atoms of thetwo rings in high-energy regions when starting eachMM3 minimization after rigid rotations about thelinkage bonds from the otherwise identical startingmolecule. All searches of conformation space werecarried out with 20◦ increments of φ and ψ . Thosetorsion angles were held fixed at each φ,ψ point,but all other internal coordinates were optimized.Structures at the lowest energy grid point on eachmap were subsequently minimized without φ,ψconstraints by computation of the Hessian matrixto precisely locate and confirm the minima. Furtherdetails of these methods are described elsewhere.34
We define the flexibility over the Ramachan-dran surface as the probability volume in degreessquared, with the probability calculated by a Boltz-mann relationship, pi = e−1E/RT. Therein, 1E is theelectronic energy relative to the global minimum de-termined by QM, R is the universal gas constant,and T is 298 K. This probability volume is relatedto the partition function, a summation of the proba-bility values over the surface. A completely flexiblemolecule would have a relative energy of 0 kcal (andpi = 1) everywhere on the surface. On a grid witha 1◦ spacing there are 360 × 360 = 129,600 φ,ψpoints. A completely rigid molecule would havea pi of 1 at only one point and a value of zeroat all others. Thus, the maximum possible valueof the partition function would be 129,600, andthe minimum on that basis would be 1. Similarly,the maximum and minimum probability volumeswould be 129,600 deg × deg, and 1 deg × deg, re-
68 VOL. 22, NO. 1

HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
FIGURE 2. Three-dimensional representation of theprobability volume from the QM surface for the maltoseanalog. The maximum, with a pi of 1, is at the globalminimum of φ = −57◦C, ψ = −33◦C. The outermostcontour line shown on this surface has a relativeprobability value of 0.00001. It correspondsapproximately to what would be the 7 kcal contour forthe maltose map shown in Figure 4b.
spectively. Numerical values of the dimensionlesspartition function were essentially the same as theprobability volume for the analog surfaces whenusing a 1◦ grid spacing and the Trapezoidal Rule,Simpson’s Rule, or Simpson’s 3/8 Rule. At largergrid increments, the probability volume representsa reasonably fine quadrature approach to the par-tition function calculated for a 1◦ grid, but valuesof the partition function for other grid incrementswere, of course, quite dependent on the numberof grid points in the summation. Figure 2 showsthe probability volume (2425 deg× deg), calculatedfor the maltose analog surface. The electronic en-ergy was interpolated to 1◦ intervals with the Gridfunction of Surfer 7.0,35 the program used to calcu-late the probability volume and produce the contourplots.
All maps herein are presented based on linkagetorsion angles defined by the exo-cyclic atoms (hy-drogen atoms except for C1′ on the sucrose analog)attached to the carbon atoms of the interring link-age. This emphasizes the differences resulting fromthe different linkage configurations. During calcu-lations for the trehaloses, the torsions were definedby φO5 (O5—C1—O1—C1′) and ψO5′ (O5′—C1′—O1—C1) and the sucrose torsions were defined byφO5 (O5—C1—O1—C2′) and ψO5′ (O5′—C2′—O1—C1). Calculations for the analogs of the di-axial andaxial-equatorial pseudo disaccharides were basedon φC2 (C2—C3—O—C3′) and ψC2′ (C2′—C3′—O—C3), and those for the di-equatorial analog were
based on φC4 (C4—C3—O—C3′) and ψC4′ (C4′—C3′—O—C3). To get the approximate appearance ofhydrogen-based maps, the values for the structureswith 2 or 0 anomeric centers were shifted by ±120◦(Table I gives the actual φH and ψH, and Supple-mentary Tables X–XXI show the original, unshifteddata.) Calculations for the structures with a singleanomeric center were based on torsions defined bythe hydrogen atoms.
Crystal structures for molecules having the samebackbones as the analogs were taken from the Cam-bridge Structural Database (CSD)36 and other litera-ture. Rather than rely on the experimental hydrogenatom positions, the φH and ψH values were ±120◦transformations of the torsions defined by heavyatoms. This introduces a small error for the calcula-tion of the corresponding energies for the reducingdisaccharides. The same shift was made for both theexperimental data and the models for the nonre-ducing disaccharides so they are not affected bythis minor problem. The energies corresponding tothe crystalline conformations were determined withthe “calculate residuals” function of Surfer. Struc-tures with 1,2-linkages were not used as analogsof 1,4-linkages. Molecules with additional bondsthat connect the two residues were not included inthese analyses because the covalent constraints maycause some deformation. However, cyclodextrinsand cycloamyloses are displayed on the maltoseanalog surface. The fit of the crystal structures tothe energy surfaces for each molecule will be dis-cussed in greater detail elsewhere (e.g., see ref. 17.)The crystal structures include molecules whose hy-droxyl groups have substantial substitution or aremissing, and molecules that form complexes withions are included. Corrections for incorrect chiralitywere made to structures identified by CSD refcodesJUMYUL, MCELOB,and WACHOX.
Results and Discussion
HF/6-31G∗ energy surfaces and the linkage con-formations from crystal structures are presented inFigures 3–5. Table I reports the positions of theglobal minima, along with the flexibility values.The crystal structure “refcodes,” φ and ψ values fortheir linkages, and corresponding relative energiesfrom our surfaces are in Supplementary Material,Tables S1–S9. Tables S10–S21 in Supplementary Ma-terial contain the absolute and relative energies ateach 20◦ increment of φ and ψ .
Global minima all have absolute φH values of<55◦. For the molecules with ψH, absolute val-ues are <40◦ unless the bond is glycosidic. Then
JOURNAL OF COMPUTATIONAL CHEMISTRY 69
FRENCH ET AL.
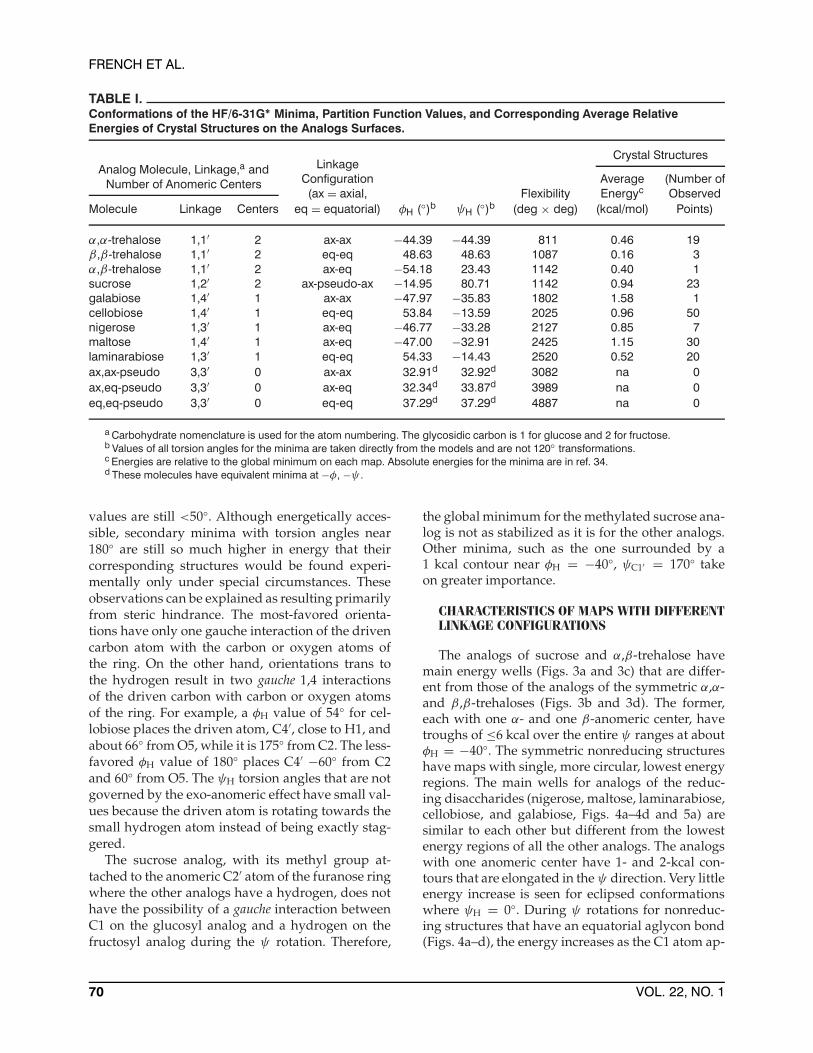
TABLE I.Conformations of the HF/6-31G∗ Minima, Partition Function Values, and Corresponding Average RelativeEnergies of Crystal Structures on the Analogs Surfaces.
Crystal StructuresAnalog Molecule, Linkage,a and
Number of Anomeric Centers
LinkageConfiguration Average (Number of(ax = axial, Flexibility Energyc Observed
Molecule Linkage Centers eq = equatorial) φH (◦)b ψH (◦)b (deg × deg) (kcal/mol) Points)
α,α-trehalose 1,1′ 2 ax-ax −44.39 −44.39 811 0.46 19β,β-trehalose 1,1′ 2 eq-eq 48.63 48.63 1087 0.16 3α,β-trehalose 1,1′ 2 ax-eq −54.18 23.43 1142 0.40 1sucrose 1,2′ 2 ax-pseudo-ax −14.95 80.71 1142 0.94 23galabiose 1,4′ 1 ax-ax −47.97 −35.83 1802 1.58 1cellobiose 1,4′ 1 eq-eq 53.84 −13.59 2025 0.96 50nigerose 1,3′ 1 ax-eq −46.77 −33.28 2127 0.85 7maltose 1,4′ 1 ax-eq −47.00 −32.91 2425 1.15 30laminarabiose 1,3′ 1 eq-eq 54.33 −14.43 2520 0.52 20ax,ax-pseudo 3,3′ 0 ax-ax 32.91d 32.92d 3082 na 0ax,eq-pseudo 3,3′ 0 ax-eq 32.34d 33.87d 3989 na 0eq,eq-pseudo 3,3′ 0 eq-eq 37.29d 37.29d 4887 na 0
a Carbohydrate nomenclature is used for the atom numbering. The glycosidic carbon is 1 for glucose and 2 for fructose.b Values of all torsion angles for the minima are taken directly from the models and are not 120◦ transformations.c Energies are relative to the global minimum on each map. Absolute energies for the minima are in ref. 34.d These molecules have equivalent minima at −φ, −ψ .
values are still <50◦. Although energetically acces-sible, secondary minima with torsion angles near180◦ are still so much higher in energy that theircorresponding structures would be found experi-mentally only under special circumstances. Theseobservations can be explained as resulting primarilyfrom steric hindrance. The most-favored orienta-tions have only one gauche interaction of the drivencarbon atom with the carbon or oxygen atoms ofthe ring. On the other hand, orientations trans tothe hydrogen result in two gauche 1,4 interactionsof the driven carbon with carbon or oxygen atomsof the ring. For example, a φH value of 54◦ for cel-lobiose places the driven atom, C4′, close to H1, andabout 66◦ from O5, while it is 175◦ from C2. The less-favored φH value of 180◦ places C4′ −60◦ from C2and 60◦ from O5. The ψH torsion angles that are notgoverned by the exo-anomeric effect have small val-ues because the driven atom is rotating towards thesmall hydrogen atom instead of being exactly stag-gered.
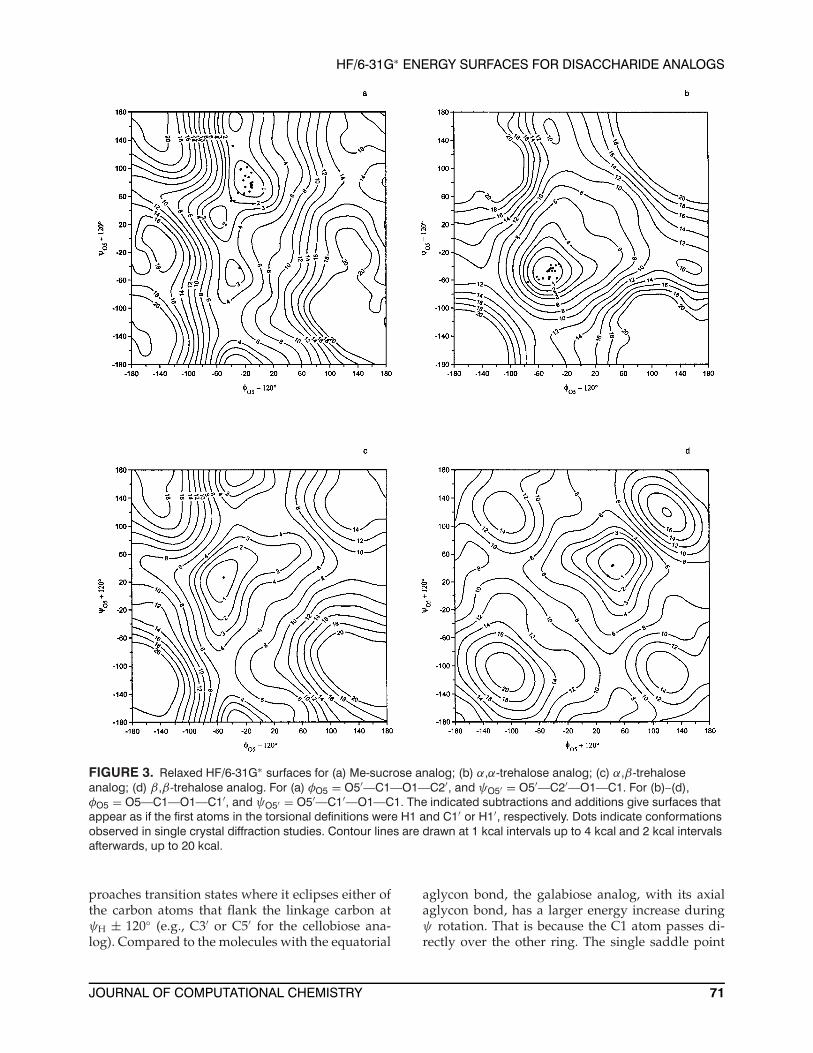
The sucrose analog, with its methyl group at-tached to the anomeric C2′ atom of the furanose ringwhere the other analogs have a hydrogen, does nothave the possibility of a gauche interaction betweenC1 on the glucosyl analog and a hydrogen on thefructosyl analog during the ψ rotation. Therefore,
the global minimum for the methylated sucrose ana-log is not as stabilized as it is for the other analogs.Other minima, such as the one surrounded by a1 kcal contour near φH = −40◦, ψC1′ = 170◦ takeon greater importance.
CHARACTERISTICS OF MAPS WITH DIFFERENTLINKAGE CONFIGURATIONS
The analogs of sucrose and α,β-trehalose havemain energy wells (Figs. 3a and 3c) that are differ-ent from those of the analogs of the symmetric α,α-and β,β-trehaloses (Figs. 3b and 3d). The former,each with one α- and one β-anomeric center, havetroughs of ≤6 kcal over the entire ψ ranges at aboutφH = −40◦. The symmetric nonreducing structureshave maps with single, more circular, lowest energyregions. The main wells for analogs of the reduc-ing disaccharides (nigerose, maltose, laminarabiose,cellobiose, and galabiose, Figs. 4a–4d and 5a) aresimilar to each other but different from the lowestenergy regions of all the other analogs. The analogswith one anomeric center have 1- and 2-kcal con-tours that are elongated in the ψ direction. Very littleenergy increase is seen for eclipsed conformationswhere ψH = 0◦. During ψ rotations for nonreduc-ing structures that have an equatorial aglycon bond(Figs. 4a–d), the energy increases as the C1 atom ap-
70 VOL. 22, NO. 1
HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
FIGURE 3. Relaxed HF/6-31G∗ surfaces for (a) Me-sucrose analog; (b) α,α-trehalose analog; (c) α,β-trehaloseanalog; (d) β,β-trehalose analog. For (a) φO5 = O5′—C1—O1—C2′, and ψO5′ = O5′—C2′—O1—C1. For (b)–(d),φO5 = O5—C1—O1—C1′, and ψO5′ = O5′—C1′—O1—C1. The indicated subtractions and additions give surfaces thatappear as if the first atoms in the torsional definitions were H1 and C1′ or H1′, respectively. Dots indicate conformationsobserved in single crystal diffraction studies. Contour lines are drawn at 1 kcal intervals up to 4 kcal and 2 kcal intervalsafterwards, up to 20 kcal.
proaches transition states where it eclipses either ofthe carbon atoms that flank the linkage carbon atψH ± 120◦ (e.g., C3′ or C5′ for the cellobiose ana-log). Compared to the molecules with the equatorial
aglycon bond, the galabiose analog, with its axialaglycon bond, has a larger energy increase duringψ rotation. That is because the C1 atom passes di-rectly over the other ring. The single saddle point
JOURNAL OF COMPUTATIONAL CHEMISTRY 71
FRENCH ET AL.
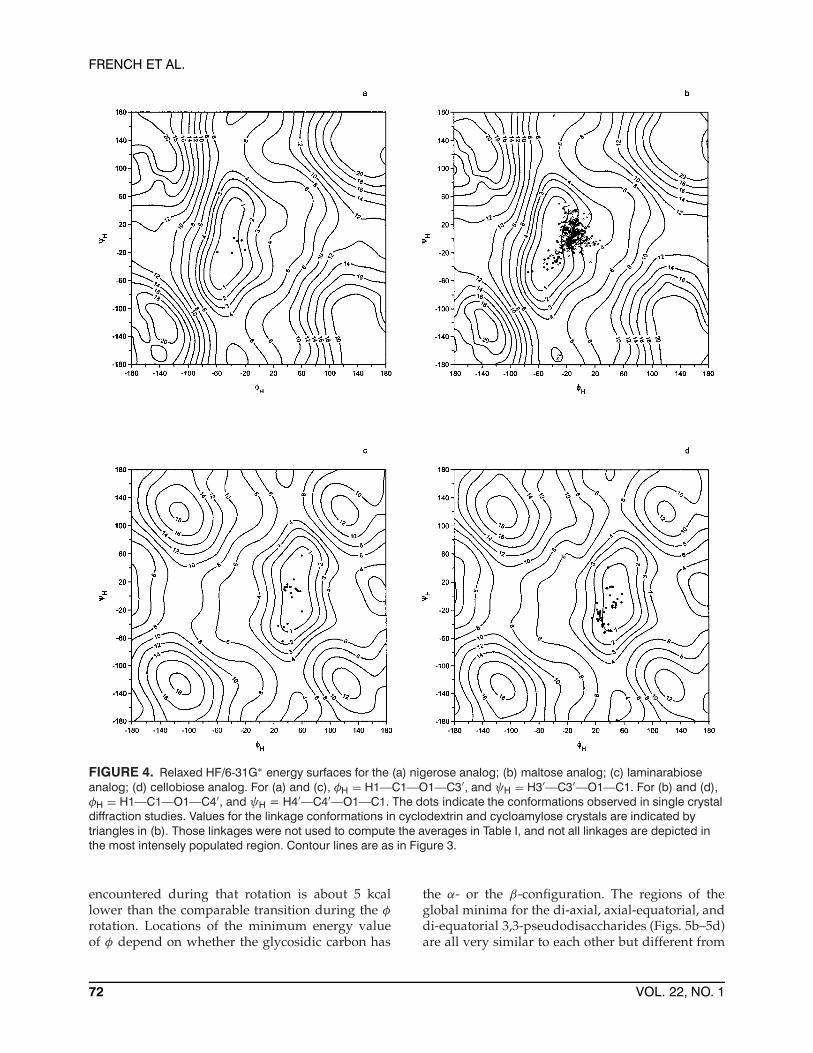
FIGURE 4. Relaxed HF/6-31G∗ energy surfaces for the (a) nigerose analog; (b) maltose analog; (c) laminarabioseanalog; (d) cellobiose analog. For (a) and (c), φH = H1—C1—O1—C3′, and ψH = H3′—C3′—O1—C1. For (b) and (d),φH = H1—C1—O1—C4′, and ψH = H4′—C4′—O1—C1. The dots indicate the conformations observed in single crystaldiffraction studies. Values for the linkage conformations in cyclodextrin and cycloamylose crystals are indicated bytriangles in (b). Those linkages were not used to compute the averages in Table I, and not all linkages are depicted inthe most intensely populated region. Contour lines are as in Figure 3.
encountered during that rotation is about 5 kcallower than the comparable transition during the φrotation. Locations of the minimum energy valueof φ depend on whether the glycosidic carbon has
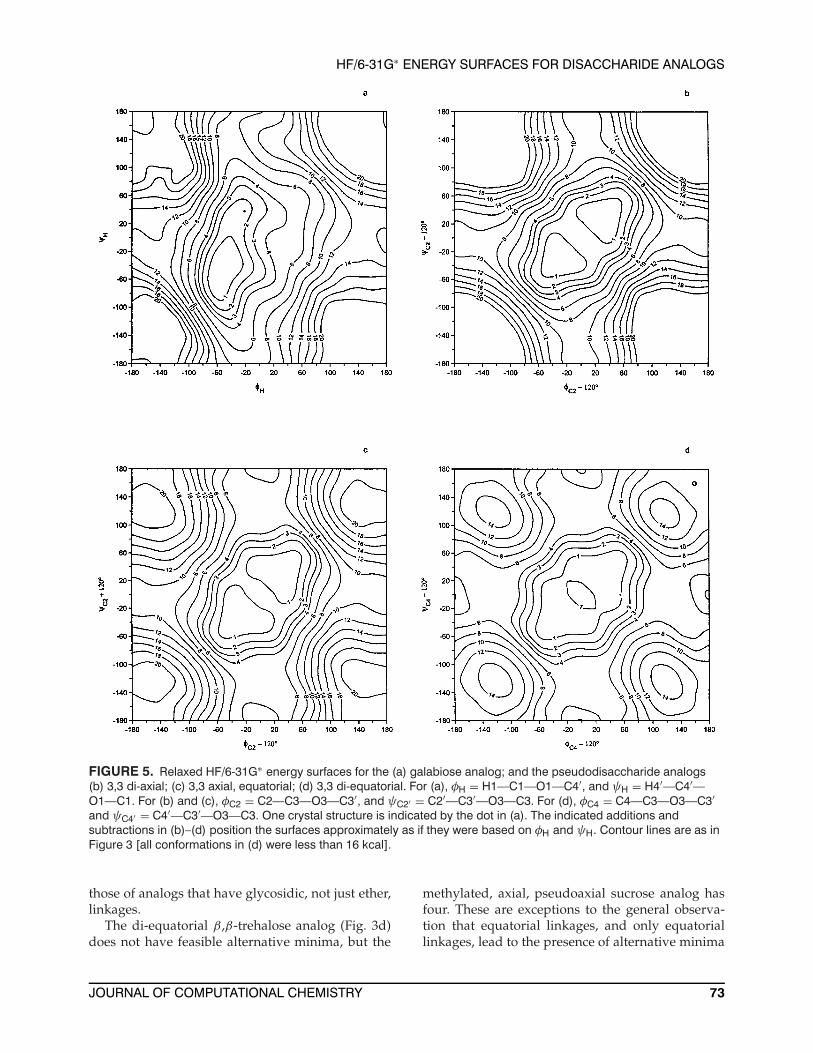
the α- or the β-configuration. The regions of theglobal minima for the di-axial, axial-equatorial, anddi-equatorial 3,3-pseudodisaccharides (Figs. 5b–5d)are all very similar to each other but different from
72 VOL. 22, NO. 1
HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
FIGURE 5. Relaxed HF/6-31G∗ energy surfaces for the (a) galabiose analog; and the pseudodisaccharide analogs(b) 3,3 di-axial; (c) 3,3 axial, equatorial; (d) 3,3 di-equatorial. For (a), φH = H1—C1—O1—C4′, and ψH = H4′—C4′—O1—C1. For (b) and (c), φC2 = C2—C3—O3—C3′, and ψC2′ = C2′—C3′—O3—C3. For (d), φC4 = C4—C3—O3—C3′and ψC4′ = C4′—C3′—O3—C3. One crystal structure is indicated by the dot in (a). The indicated additions andsubtractions in (b)–(d) position the surfaces approximately as if they were based on φH and ψH. Contour lines are as inFigure 3 [all conformations in (d) were less than 16 kcal].
those of analogs that have glycosidic, not just ether,linkages.
The di-equatorial β,β-trehalose analog (Fig. 3d)does not have feasible alternative minima, but the
methylated, axial, pseudoaxial sucrose analog hasfour. These are exceptions to the general observa-tion that equatorial linkages, and only equatoriallinkages, lead to the presence of alternative minima
JOURNAL OF COMPUTATIONAL CHEMISTRY 73

FRENCH ET AL.
of <4 kcal. For example, the regions at φH = 180◦are feasible for the di-equatorial laminarabiose ana-log (Fig. 4c) but not for the axial-equatorial nigeroseanalog (Fig. 4a). The same applies to the maltoseand cellobiose analogs (Figs. 4b and 4d). Neitherof the alternative conformations of the di-axial gal-abiose analog (Fig. 5a) with φH or ψH = 180◦ arefeasible. The allowed secondary minima also ben-efit from gauche OCOC torsions (the exo-anomericeffect). In the case of the maltose and nigerose sec-ondary minima, φH values are about the same asfor the global minimum. The global and secondaryminima for laminarabiose and cellobiose have φH
torsion angles of about +60◦ and 180◦, and thesecorrespond to O—C—O—C (φO5) torsion angles of−60◦ and +60◦. For the secondary minima withφH = 180◦, the energy is higher than for φH tor-sions of +60◦. This is perhaps partly because whenφH = 60◦, the C2—C1—O1—Cn′ (φC2) torsion an-gle is about −60◦, likely to be of higher energy thanwhen φC2 is 180◦. Arguments regarding gauche,steric interactions at the beginning of Results andDiscussion also apply. Alternative minima with φ
or ψ = 180◦ also occur for the pseudodisaccharideanalogs if there is an equatorial bond.
The greatest range in relative energy (about35 kcal) is found for the analog of di-axial α,α-trehalose. That molecule is thought to be very ster-ically hindered. The flattest surfaces are for theanalog of the di-equatorial 3,3-pseudodisaccharide(range <16 kcal), and other di-equatorial maps arealso fairly flat (range <23 kcal). Maps for the 1,3-and 1,4-linked, axial-equatorial nigerose and mal-tose analogs are very similar to each other, whilethe surfaces for the comparable di-equatorial pair ofanalogs of laminarabiose and cellobiose are some-what different from each other.
THE INDIVIDUAL ANALOG SURFACES
The surface for the methylated THF-O-THP su-crose analog (Fig. 3a) is fairly different from ourpreviously published17 HF/6-31G∗ surface for theanalog that lacks the methyl group. There is a con-siderably better fit of the crystal structures on thesurface of this more complete methylated model.The outlier on the 3 kcal contour is the 4,1′,6′-tri-chlorogalactosucrose structure.37 Structures nearthe 2 kcal contour are from NaI sucrose38 andtwo molecules having the alternative, southern ringshape for the furanose ring.39, 40 The low barrierto complete the ψ rotation is remarkable consider-ing the pseudoaxial orientation of the linkage fromthe furanosyl ring. The barrier for the ψ rotation of
α,β-trehalose, with an equatorial bond, is slightlyhigher.
The surface for the α,α-trehalose analog (Fig. 3b)locates the observed crystal structures on or withinthe 1-kcal contour. The exception is an ionic complexof calcium with an allopyranosyl analog of trehaloseat more than 2 kcal.41 The presence of the ions mayalter the torsional potential.42
The surface for the analog of α,β-trehalose(Fig. 3c) is nominally similar to that of the su-crose analog, but there are three important struc-tural differences between the two molecules. Themethyl group on the furanose ring is over the in-terresidue linkage on the present sucrose analog,and the map for the α,β-trehalose analog is moresimilar to the map for the sucrose analog withoutthe methyl group.17 Secondly, the β-fructose ring insucrose (α-D-glucopyranosyl-1,2′-β-D-fructofurano-side) has five atoms. That flatter, more flexible ringwould be expected to have different steric interac-tions with the α-glucose analog ring than wouldthe six-membered β-ring in the α,β-trehalose ana-log. Last, the glycosidic bond from the fructose ringis usually pseudoaxial, as preferred by the anomericeffect for D-fructose. In α,β-trehalose, the bond fromthe β-pyranose ring is equatorial. The few availablecrystal structures are found on or within the 1-kcalcontours on the α,β- and β,β-trehalose analog sur-faces (Figs. 3c and 3d.)
Only seven structures have the nigerose (Fig. 4a)backbone. In contrast, more than 1000 linkages areavailable for maltose (Fig. 4b). Many of the maltoselinkages do not coincide well with the global mini-mum. One reason is that most linkages are from cy-clodextrins having six to eight glucose residues. In-terresidue linkages in these macrocyclic moleculesare apparently somewhat distorted when the macroring is formed. Linkage conformations from cy-cloamyloses having 10, 14, and 26 glucose residues43
give six points with ψ values near ±180◦, the re-gion of the secondary minimum. Those alternativeconformations allow the cycloamyloses to avoid agreat deal of steric strain that would occur if allof their linkages were restricted to φ and ψ valuesnear the global minimum. Another important rea-son for the relatively poor fit of the crystal data onthe maltose analog map is that C6, adjacent to thelinkage, is not included in the present models. Thesubstantial effect of C6 on the energy surface can beseen in the MM3 maps published for nigerose andmaltose analogs that included a methyl group at-tached to C5.44 Those maps are quite different fromeach other, while our QM surfaces for the analogsmissing the C6 groups are very similar. Fits of the
74 VOL. 22, NO. 1

HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
maltosyl linkages from nonmacrocyclic moleculeswere considerably improved on our hybrid surfacesfor maltose.34 In particular, the 1-kcal contour onthe hybrid surface with ε = 7.5 surrounds 27 ofthe 30 observations. Although the hydroxymethyland hydroxyl groups are important for preciselydefining the low energy region for maltose, the 1-kcal contour for the present analog still surrounds agood number of observed structures.
Figure 4c and 4d, the surfaces for the laminara-biose and cellobiose analogs, are both successfulat predicting the locations of the crystal structuresof the disaccharide molecules. A heavily substi-tuted cellobiose derivative45 is near the alternativeminimum with ψH = −180◦. The 1-kcal contour en-compasses more area on the laminarabiose analogsurface than on the map for the cellobiose analog, al-though the two surfaces are generally quite similar.On the surface for the cellobiose analog, a numberof structures are close to a line (not shown) passingthrough the lower-right and upper-left corners. Theline corresponds approximately to a twofold screwaxis, and there may be a crystal-packing bias in fa-vor of such arrangements.
Despite the di-axial linkage of galabiose, the2-kcal contour of its analog (Fig. 5a) is roughly com-parable to the 2-kcal contours on the nigerose andmaltose analog surfaces. The linkage conformationfrom the sole crystal structure46 is at 1.58 kcal. Thatcrystal structure has an O3′—O5 hydrogen bond ofnearly ideal geometry.
Figure 5b–d shows the energy surfaces for theanalogs of the pseudodisaccharides. The centralminima are very similar for these molecules. Also,there is a mirror plane of symmetry through theatom on each ring that is connected to the centraloxygen atom. There is some enlargement of the 1-kcal contour on the surfaces for the molecules withequatorial linkages. The individual values of the φand ψ torsion angles for the freely minimized struc-tures of these di-axial and di-equatorial structures,as well as for the α,α- and β,β-trehalose analogstructures are very similar (Table I). This indicatessymmetric structures and successful minimization.
FLEXIBILITIES
The flexibility values in Table I show the impactof the exo-anomeric effect on the flexibilities of theseanalogs. Structures with two adjacent anomeric cen-ters have the smallest values and are more rigidthan those with one anomeric center. Those withno exo-anomeric sequences are least rigid. Group-ings of flexibility values that arise based on the
number of anomeric centers are similar to thosefound for calculated heats of formation.27 That workshowed the lowest heats of formation for moleculeswith two anomeric centers. A second group withone anomeric center had heats about 8 kcal higher,and then came the analogs of the pseudodisaccha-rides, about 8 kcal higher still. The largest calculatedvalue of the flexibility, 4887 deg × deg for the di-equatorial, pseudodisaccharide analog, is 3.8% ofthe theoretical maximum, whereas the most rigidmolecule, the α,α-trehalose analog, has a flexibilityonly 0.63% of the maximum.
The linkage of the cellobiose analog is calculatedto be 20% less flexible than the linkage of the lam-inarabiose analog. It is also less flexible than themaltose and nigerose structures, which have axial-equatorial configurations. This is borne out by thesmaller 1-kcal contour on its energy surface, com-pared to the other structures with one anomericcenter. We can offer no explanation for why it isso much less flexible than the similar analog oflaminarabiose. We attempted to gain insight by car-rying out calculations for these two analogs withMM3(96).33 The magnitude of the flexibility wassimilar, but MM3 gave a different assessment of theorder of flexibility, with values of 2170 deg × degfor the cellobiose analog and 1782 deg× deg for thelaminarabiose analog. These discrepancies are themotivation for recourse to higher level QM calcu-lations.
The flexibilities for the α,β-trehalose and themethylated sucrose analogs were almost identical,despite surfaces that are fairly different in appear-ance. Also, they are a little greater than for the α,α-and β,β-trehaloses, despite the axial-equatorial andaxial-pseudoaxial dispositions of their glycosidicbonds. The minimum-energy conformations of theα,β-trehalose and the methylated sucrose analogshave adjacent, nearly parallel dipoles for the bondsthat connect the anomeric carbon and the ring oxy-gen atoms (Fig. 1). This may destabilize the globalminima in these molecules and cause the slightlyhigher electronic energy for the α,β-trehalose com-pared to the other trehaloses.27 A flatter energyminimum would allow greater flexibility than forthe other trehaloses.
AVERAGE CORRESPONDING ENERGIES OFTHE CRYSTALLINE CONFORMATIONS ON THEANALOG SURFACES
The average energies in Table I for the conforma-tions observed in crystal structures are all less than1.2 kcal, except for the single available structure
JOURNAL OF COMPUTATIONAL CHEMISTRY 75

FRENCH ET AL.
of galabiose (1.58 kcal). No observed conformationcorresponds to an energy higher than 4 kcal, evenincluding the cyclodextrins. Of the nonmacrocyclicmolecule conformations, only a heavily substitutedcellobiose derivative exceeded 3 kcal. Only 16 moreof the 154 structures exceeded 2 kcal.
Although the various energy surfaces provide amostly satisfactory accounting for the crystal con-formations, we did not a priori expect this to bethe case. In one view, the analogs would be moreflexible than the actual disaccharides because ofless steric hindrance. Also, the lack of specific hy-drogen bonds might avoid rigidity. On the otherhand, disaccharide energy surfaces are complex,with multiple contributions. It is possible that the in-creased steric hindrance from the exo-cyclic groupswould occur in the areas of lowest torsional energy,and optimal hydrogen bonding would occur some-where else. This would make minima shallowerand broaden the potential distribution of crystalstructures, increasing the molecular flexibility of theactual disaccharides. Besides changing the flexibil-ity, the substituents could cause the relocation of theminima.
Apparently the increased flexibility of theanalogs and their failure to exactly locate the min-ima because of missing substituents are mostlycounterbalancing factors. The overall average cor-responding energy for the crystalline disaccharideson the QM analog surfaces is 0.86 kcal. This valuecan be compared with the average of correspond-ing energies on the QM/MM hybrid energy surfacesfor these disaccharides (except galabiose, which hasnot yet been calculated). At dielectric constants of1.5, 3.5, and 7.5, the overall average energies were1.80, 0.91, and 0.83 kcal, respectively.34 Therefore,it appears that the QM plots for the analogs ac-count better for the crystal structures than do theQM/MM surfaces for the full disaccharides thatwere computed at a dielectric constant of 1.5 (rec-ommended for use in MM3 for modeling moleculesin the gas phase). For the higher dielectric hybridsurfaces (which have diminished hydrogen bond-ing stabilization), the averages (0.91 and 0.83 kcal)are similar to the average for the QM surfaces forthe analogs (0.86 kcal), which cannot form hydrogenbonds at all.
Although hydrogen bonding dominates theenergy hypersurface for vacuum-phase (isolatedmolecule) calculations on carbohydrates,29 intra-molecular hydrogen bonds do not occur as often asthe intermolecular kind in most condensed phases.The overall relative importance of hydrogen bond-ing and the exo-anomeric effect on preferred con-
formations may even be reversed in condensedphases. Studies with the MM3 force field require in-creased dielectric constants to best account for theobserved crystal structures.47 Several factors thatmight be responsible for the need for weakenedhydrogen bond strengths in models of condensedphase systems were discussed in ref. 17. In partic-ular, the electronic energy for the hydrogen bondin the water dimer is about −4.94 kcal,48 but thezero-point vibrational energy correction yields afree energy of about −2.86 kcal.49 That suggeststhat prediction by potential energy calculations ofcondensed phase structures with isolated modelswould be done best by using a reduced strength,compared to the enthalpy, for hydrogen bondingcalculations.
Conclusions
We have prepared relaxed HF/6-31G∗ energysurfaces for analogs of sucrose and a variety ofdimers of glucose that have a wide range of linkageconfigurations. All the analogs of glucose dimershad small absolute (14–55◦) values of φH and ψH atthe global minima, presumably for steric reasons.In all analogs having glycosidic linkages, the loca-tions of the global, minimum-energy regions wereconsistent with exo-anomeric effects. Further, thecrystallographically observed conformations of theparent disaccharide molecules were almost entirelyclustered near the global minima.
In a way, we have confirmed Lemieux’s hypoth-esis that the primary factors that determine con-densed phase conformations are the exo-anomerictorsional energies and simple nonbonded interac-tions. The use of QM analog maps to account fordisaccharide conformations is somewhat similar tothe HSEA approach. Each takes less time than acomplete treatment of all interactions in a disac-charide. Both approaches ignore conventional hy-drogen bonding. Our analogs lack the requisite hy-droxyl groups, while the HSEA potential lacks anyterm that alters the calculated energy in the event ofa hydrogen bond.
The main goal of our work is to learn more aboutthe factors that determine carbohydrate conforma-tion. The present work shows that the regions ofthe global minima for disaccharides of numerousconfigurations were reliably located by these HF/6-31G∗ calculations on analogs, as confirmed by thecrystal structure geometries. The simplest interpre-tation is that the forces inherent in our models havemajor roles in determining disaccharide conforma-tion. If so, then it is reasonable to ask that MM force
76 VOL. 22, NO. 1

HF/6-31G∗ ENERGY SURFACES FOR DISACCHARIDE ANALOGS
fields reproduce such analog surfaces. To facilitatequantitative comparison, we have included the mapdata in the Supplementary Material. These isomericanalogs offer a rich variety of energy surfaces and,therefore, offer a good test of the balance of the var-ious terms in an MM force field. As computationalpower continues to increase, it will be interesting tosee the effects of the addition of electron correlationand larger basis sets (especially diffuse functions)on the analog surfaces.
Given this set of energy surfaces, we were ableto describe some inherent characteristics of disac-charide surfaces that will be modified to varyingdegrees when the model is made more complete byaddition of the particular substituents. One charac-teristic is that the shape of the region of the globalminimum depends on the number of anomeric cen-ters. When two anomeric centers are present, theshape depends on whether the centers have thesame or opposite configuration. Whether the con-figuration results in axial or equatorial geometryis of lesser importance in the region of the globalminimum. The number of anomeric centers wasalso important in determining whether viable alter-native minima would be found. For the linkageswith no anomeric centers, or those with one, anequatorial bond led to an alternative minimum.The di-equatorial β,β-trehalose analog has only asingle likely minimum, but the β-linkages in thesucrose and in α,β-trehalose analogs gave alter-native minima, despite the pseudoaxial linkage inthe sucrose analog. The number of anomeric cen-ters in the linkage was also an important factorin the calculated flexibility. Two anomeric centersin the linkage gave the most rigid models, andmodels with no anomeric centers had the leastrigidity.
In the case of sucrose, there was a large differencebetween the MM3 and QM results for the analogsurface in the region of the crystal structures.17, 19
Therefore, we wanted to make use of the QM en-ergies in a study of the whole disaccharide. Thiswas accomplished with our hybrid method.17 Thereare also differences between the other QM ana-log surfaces and their MM3 counterparts, exceptfor the 3,3-linked pseudo disaccharides. However,those differences were not as large in the areas ofthe observed structures.
Acknowledgments
Professor John Brady encouraged us to addressthe issue of the effect of linkage configuration on
the overall surface characteristics. He also providedhelpful comments on the article, as did Dr. WilliamFranklin and Professor Gabor Csonka. C.J.C. thanksthe Alfred P. Sloan Foundation. Members of Profes-sor Wolfrom Saenger’s group supplied cycloamy-lose coordinates before publication.
Supplementary Material
The Cambridge Structural Database Refcodes,the translated φH andψH, and the corresponding en-ergies are supplied for the experimental structures(Supplementary Tables I–IX). Tables of φ, ψ , andrelative energy are supplied for each of the analogsurfaces (Supplementary Tables X–XXI).
References
1. (a) Sasisekharan, V. Collagen Proc Symp, Madras, India,1962, p. 39, vol. 1060; (b) Rao, V. S. R.; Sundararajan, P. R.;Ramakrishnan, C.; Ramachandran, G. N. Conformation ofBiopolymers; Academic: London, 1967, vol. 2.
2. Rees, D. A.; Skerrett, R. J. Carbohydr Res 1968, 7, 334.
3. Rao, V. S. R.; Qasba, P. K.; Balaji, P. V.; Chandrasekaran, R.Conformation of Carbohydrates; Harwood Academic: Am-sterdam, 1998.
4. Lemieux, R. U.; Koto, S. Tetrahedron 1974, 30, 1933.
5. Bock, K. Pure Appl Chem 1983, 55, 605.
6. Thogersen, H.; Lemieux, R. U.; Bock, K.; Meyer, B. CanJ Chem 1982, 60, 44.
7. (a) Szarek, W. A.; Horton, D. ACS Symp Ser 1979, 87; (b)Tvaroška, I.; Bleha, T. Adv Carbohydr Chem Biochem 1989,47, 45; (c) Thatcher, G. R. J. ACS Symp Ser 1994, 539.
8. Woods, R. Carbohydrate Force Fields. Encyclopedia of Com-putational Chemistry; Schleyer, P. v. R.; Allinger, N. L.;Clark, T.; Gasteiger, J.; Kollman, P. A.; Schaefer, H. F., III;Schreiner, P. R., Eds.; John Wiley & Sons: Chichester, 1998,p. 220, vol. 1.
9. Jimenez–Barbero, J.; Noble, O.; Pfeffer, C.; Pérez, S. NewJ Chem 1988, 12, 941.
10. French, A. D. Biopolymers 1988, 27, 1519.
11. Ha, S. N.; Madsen, L.; Brady, J. W. Biopolymers 1988, 27,1927.
12. Tran, V.; Buleon, A.; Imberty, A.; Pérez, S. Biopolymers 1989,28, 679.
13. Pérez, S.; Imberty, A.; Engelsen, S. B.; Gruza, J.; Mazeau, K.;Jimenez–Barbero, J.; Poveda, A.; Espinosa, J. F.; van Eyck,B. P.; Johnson, G. P.; French, A. D.; Kouwijzer, M. L.C. E.; Grootenhuis, P. D. J.; Bernardi, A; Raimondi, L.;Senderowitz, H.; Durier, V.; Vergoten, G.; Rasmussen, K.Carbohydr Res 1998, 314, 141.
14. Halgren, T. A. J Comput Chem 1996, 17, 490.
15. Hwang, M.-J.; Ni, X.; Waldman, M.; Ewig, C. S.; Hagler, A.T. Biopolymers 1998, 45, 435.
JOURNAL OF COMPUTATIONAL CHEMISTRY 77

FRENCH ET AL.
16. Damm, W.; Frontera, A.; Tirado–Rives, J.; Jorgensen, W. L.J Comput Chem 1997, 18, 1955.
17. French, A. D.; Kelterer, A.-M.; Johnson, G. P.; Cramer, C. J.;Dowd, M. K. Carbohydr Res 2000, 326, 305.
18. Tvaroška, I.; Carver, J. P. Carbohydr Res 1998, 309, 1; andtheir previous work cited therein.
19. Van Alsenoy, C.; French, A. D.; Cao, M.; Newton, S. Q.;Schäfer, L. J Am Chem Soc 1994, 116, 9590.
20. Odelius, M.; Laaksonen, A.; Widmalm, G. J Phys Chem 1995,99, 12686.
21. Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P.;Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A. J Am ChemSoc 1998, 120, 11158.
22. Tvaroška, I.; Váklavík, L. Carbohydr Res 1987, 160, 137.
23. Barrows, S. E.; Dulles, F. J.; Cramer, C. J.; French, A. D.; Truh-lar, D. G. Carbohydr Res 1995, 276, 219.
24. (a) Salzner, U.; Schleyer, P. v. R. J Org Chem 1994, 59, 2138;(b) Salzner, U.; Schleyer, P. v. R. J Org Chem 1995, 60, 8430.
25. Cramer, C. J. J Org Chem 1992, 57, 7034.
26. Barrows, S. E.; Storer, J. W.; Cramer, C. J.; Truhlar, D. G.;French, A. D. J Comput Chem 1998, 19, 1111.
27. French, A. D.; Kelterer, A.-M.; Johnson, G. P.; Dowd, M. K.J Mol Struct, to appear.
28. (a) Csonka, G. I.; Éliás, K.; Csizmadia, I. G. Chem PhysLett 1996, 257, 49; (b) Csonka, G. I.; Éliás, K.; Kolossváry, I.;Sosa, C. P.; Csizmadia, I. G. J Phys Chem A 1998, 102, 1219;(c) Csonka, G. I.; Éliás, K.; Csizmadia, I. G. J Comput Chem1997, 18, 330.
29. Lii, J.-H.; Ma, B.; Allinger, N. L. J Comput Chem 1999, 20,1593.
30. Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.;Gordon, M. S.; Jensen, J.; Koseki, H. S.; Matsunaga, N.;Nguyen, K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Mont-gomery, J. A. J Comput Chem 1993, 14, 1347.
31. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.;Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.;Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Oritz, J. V.; Foresman,J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challa-combe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.;Andres, J. L.; Replogle, E. S.; Bomperts, R.; Martin, R. L.; Fox,D. J.; Binkley, J. S.; DeFrees, D. J.; Baker, J.; Stewart, J. P.;
Head–Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian 94;Gaussian, Inc.: Pittsburgh, PA, 1995.
32. Chem-X, version 1999.1; Oxford Molecular Ltd., 1999.33. Allinger, N. L.; Rahman, M.; Lii, J.-H. J Am Chem Soc 1990,
112, 8293. MM3 is available to academic users from theQuantum Chemistry Program Exchange, Creative Arts, In-diana University, Bloomington, IN. Others may obtain itfrom Tripos Associates, St. Louis, MO.
34. French, A. D.; Kelterer, A.-M.; Johnson, G. P.; Cramer, C. J.;Dowd, M. K. J Mol Graph Model, 2000, 18, 95.accepted.
35. Surfer, version 7.0.0, Aug 25 1999; Golden Software, Inc.:Golden, CO. http://www.goldensoftware.com/frames/surferframe.htm.
36. Allen, F. H.; Kennard, O. Chem Design Automat News 1993,8, 1, 31.
37. Kanters, J. A.; Scherrenbert, R. L.; Leeflang, B. R.; Kroon, J.;Mathlouthi, M. Carbohydr Res 1988, 180, 175.
38. Accorsi, C. A.; Bertolasi, B.; Ferretti, V.; Gilli, G. CarbohydrRes 1989, 191, 91.
39. Jeffrey, G. A.; Park, Y. J. Acta Crystallogr B 1972, 28, 257.40. Sachinvala, N. D.; Chen, H.; Niemczura, W. P.; Furusawa, E.;
Cramer, R. E.; Rup, J. J.; Ganjian, I. J Med Chem 1993, 36,1791.
41. Ollis, J.; James, V. J.; Angyal, S. J.; Pojer, P. M. Carbohydr Res1978, 60, 219.
42. Tvaroška, I.; Carver, J. J Phys Chem 1995, 99, 6234.43. Saenger, W.; Jacob, J.; Gessler, K.; Steiner, T.; Hoffamnn, D.;
Sanbe, H.; Koizumi, K.; Smith, S. M.; Takaha, T. Chem Rev1998, 98, 1787. Coordinates were furnished by the authors.
44. Dowd, M. K.; Zeng, J.; French, A. D.; Reilly, P. J. CarbohydrRes 1992, 230, 223.
45. Ernst, A.; Vasella, A. Helv Chim Acta 1996, 79, 1279.46. Svensson, G.; Albertsson, J.; Svensson, C.; Magnusson, G.;
Dahmen, J. Carbohydr Res 1986, 146, 29.47. French, A. D.; Rowland, R. S.; Allinger, N. L. ACS Symp Ser
1990, 340, 120.48. Xantheas, S. S. J Chem Phys 1996, 104, 8821.49. Xantheas, S. S. Personal communication. The difference in
the zero-point energies at the MP2/aug-cc-pVTZ level forthe water dimer and two separated water molecules is2.12 kcal/mol.
78 VOL. 22, NO. 1
Recommended

















