Infrared Spectra of CH2dM(H)NC, CH3-MNC, and η2-M(NC)-CH3 Produced byReactions of Laser-Ablated Group 5 Metal Atoms with Acetonitrile
Han-Gook Cho and Lester Andrews*Department of Chemistry, UniVersity of Incheon, 12-1 Songdo-dong, Yonsu-ku, Incheon, 406-840, South Korea,and Department of Chemistry, UniVersity of Virginia, P.O. Box 400319, CharlottesVille, Virginia 22904-4319
ReceiVed: February 9, 2010; ReVised Manuscript ReceiVed: March 24, 2010
Methylidene isocyanides, methyl isocyanides, and η2-nitrile-π-complexes are observed in the matrix IR spectrafrom reactions of Group 5 metals with acetonitrile isotopomers. The primary isocyanide products with notrace of cyanide complexes are consistent with the reaction path proposed in the analogous Zr study. Themajor products (CH2dTa(H)NC, CH3-NbNC, η2-Nb(NC)-CH3, and η2-V(NC)-CH3) after codepositionand reaction of metal with CH3CN clearly show the increasing preference for the higher oxidation-state complexon going down the group column, and the subsequent photochemistry provides further information for molecularrearrangements. The Group 5 metal methylidene isocyanides exhibit more agostic distortion than the Zrcounterparts and are comparable to the previously studied Group 5 metal methylidene hydrides and halides.The computed structures and observed frequencies indicate that the effects of metal conjugation (CdTa-NdC:)are minor.
Introduction
Coordination of metal atoms to electron-rich organic speciesplays a pivotal role for further rearrangements to more sophis-ticated end products.1-3 Metal interactions with lone electronpairs and π-electron systems provide driving forces for theformation of new metal bonds, leading to catalytic activitiesand further reactions such as C-H and C-C bond insertions.2-6
Metal-participating molecular rearrangements often consist ofmultistep configurational changes involving distinct stationarypoints and transition states.6 Therefore, investigation of theprocess for electrophilic coordination of metal atoms and theirsubsequent molecular rearrangements is essential to understand-ing the details of the reaction path and eventually to synthesizemore precious chemical reagents.
Recent studies have shown that reactions of laser-ablated metalatoms with small hydrocarbons and haloalkanes are efficient routesto produce characteristic complexes including carbenes, carbynes,π-complexes, and cyclic products.1-3,7-13 Although they are cousinsof the much larger complexes, their small sizes also allowopportunities to closely examine molecular processes involved inmetal coordination, bond insertion, H(X) migration, and photo-chemical reactions. Electronic structure calculations also offerhelpful information for spectral assignments and understanding thereaction mechanism.6,11,14 Studies show that Groups 3-10 metals,lanthanides, and actinides undergo C-H(X) bond insertion inreaction with small organic compounds and also produce highoxidation-state complexes15 either during reactions or upon pho-tolysis afterward.7-13
A more recent study shows that reactions of Zr with CH3CN,a well-known electron donor, yields methylidene isocyanide(CH2dZrHNC), methylisocyanide (CH3-ZrNC), and η2-nitrile-π-complex, with no trace of the corresponding cyanide products,and their energies are comparable.16 The primary productssuggest a reaction path that includes electrophilic coordinationof metal atom to the N-end of CH3CN, formation of the more
stable nitrile-π-complex, C-C bond insertion, and H migration.The observed products are the most stable computed speciesalong the reaction path, and the transition states between theproducts are examined. The extent of agostic distortion14 andthe observed frequencies indicate that the effects of the metalcontaining conjugation are minor.
In this investigation, reactions of Group 5 metal atoms withacetonitrile isotopomers are carried out in an effort to substanti-ate the previous Zr results. The primary products are identifiedthrough isotopic substitution and with helpful information fromDFT computations. The increasing preference for the higheroxidation-state product on going down the family column isclear, and the results are in line with the previously proposedreaction path for the Zr system.
Experimental and Computational Methods
Laser ablated Ta, Nb, and V atoms (Johnson-Matthey) werereacted with acetonitrile isotopomers (CH3CN, CD3CN, and13CH3
13CN) in excess argon during condensation at 10 K usinga closed-cycle refrigerator (Air Products Displex). Thesemethods have been described in detail in previous publications.17
Reagent gas mixtures are typically 0.5% in argon. The Nd:YAGlaser fundamental (1064 nm, 10 Hz repetition rate, 10 ns pulsewidth) was focused onto a rotating metal target using 5-10mJ/pulse. After initial reaction, infrared spectra were recordedat 0.5 cm-1 resolution using a Nicolet 550 spectrometer with aHg-Cd-Te range B detector. Then samples were irradiatedfor 20 min periods by a mercury arc street lamp (175 W) withthe globe removed using a combination of optical filters orannealed to allow further reagent diffusion.
To provide support for the assignment of new experimentalfrequencies and to correlate with related works,7-13 densityfunctional theory (DFT) calculations were performed using theGaussian 03 program system,18 the B3LYP density functional,19
the 6-311++G(3df,3pd) basis sets for H, C, N, and V20 andusing the SDD pseudopotential and basis sets21 for Nb and Tato provide vibrational frequencies for the reaction products.
* Author to whom correspondence should be addressed. E-mail: [email protected].
J. Phys. Chem. A 2010, 114, 5997–6006 5997
10.1021/jp1012686 2010 American Chemical SocietyPublished on Web 04/28/2010

Geometries were fully relaxed during optimization, and theoptimized geometry and transition-state structure were confirmedby vibrational analysis. The BPW9122 functional was alsoemployed to support the B3LYP results. The vibrationalfrequencies were calculated analytically, and zero-point energyis included in the calculation of binding and reaction energies.Previous investigations have shown that DFT calculated har-monic frequencies are usually slightly higher than observedfrequencies,7-13,23 and they provide useful predictions for theinfrared spectra of new molecules.
Results and Discussion
Reactions of Group 5 metal atoms with acetonitrile wereinvestigated and infrared spectra (Figures 1-6), and densityfunctional frequency calculations of the products and theirrelative energies (Figures 7-9) and structures (Figures 10-12)will be presented in turn.
Ta + Acetonitrile. Figures 1-3 show the product spectrafrom reactions of laser-ablated Ta atoms with acetonitrileisotopomers and their variation with subsequent photolysis and
annealing. Isomerization of acetonitrile due to the laser-plumeirradiation during ablation produces CH2CNH, CH2NCH, andCH3NC absorptions in the matrix IR spectra.24 The productabsorptions are all marked with “m” (m for methylidene), whichshow photoreversible intensity variations upon visible (λ > 420nm) and UV (240 < λ < 380 nm) irradiations. They are observedmostly in pairs 1.1-14.0 cm-1 apart as listed in Table 1, andthe intensity variations of the components for a pair are oppositeeach other. The ones shown with boldface letters in Table 1increase and decrease on UV and visible irradiations, and theother ones with plain letters show intensity changes in theopposite directions. This suggests that at least two competingsites for the Ta product exist in the matrix, and photolysisswitches from one to the other depending on the photon energy,leading to the considerable intensity alterations as shown inFigures 1-3. Similar photoreversible intensity variations dueto different matrix sites are also observed from Group 4 metalreactions with small alkanes and halomethanes.7
Excellent agreement between the observed and DFT com-puted frequencies shown in Table 1 substantiates generation ofthe small Ta methylidene isocyanide, CH2dTa(H)NC. Thecorresponding cyanide complex (CH2dTa(H)CN) would havesimilar frequencies except for the C-N stretching band expectedat about 2150 cm-1, which is not observed in this study as shownin Figures 1-3. The strongest m absorption at 2027.5 cm-1
(with a weak site absorption at 2024.2 cm-1) is assigned to theN-C stretching mode on the basis of the frequency and thenegligible D and substantial 13C (39.4 cm-1) shifts. On the otherhand, the m absorption observed at 1782.5 cm-1 (with a siteabsorption at 1788.7 cm-1) in the Ta-H stretching region showsa large D shift of 505.5 cm-1 (H/D ratio of 1.396) and anegligible 13C shift, leading to an assignment to the Ta-Hstretching mode. The observed Ta-H stretching frequency isalso compared with 1758.9 and 1732.9 cm-1 for TaH2
25 andthose for the previously studied Ta methylidenes (1753.8 and1731.9 cm-1 for CH2dTaH2, 1765.0 and 1759.3 cm-1 forCH2dTaHF, 1762.9 and 1759.6 cm-1 for CH2dTaHCl, and1760.3 cm-1 for CH2dTaHBr).7
A weak m absorption at 1012.9 cm-1 (with a site absorptionat 1005.3 cm-1) in the CD3CN spectra (Figure 2) has its 13Ccounterpart at 1297.3 cm-1, and it is assigned to the CD2
scissoring mode while its H counterpart is probably coveredby the common CH4 absorption center at ∼1305 cm-1. Anotherm absorption is observed at 819.6 cm-1 (with a site absorptionat 823.6 cm-1), has its D counterpart at 744.6 cm-1, and has13C counterparts at 798.2 cm-1 (with a site absorption at 800.7
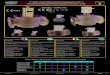
Figure 1. Infrared spectra in the 2200-1600 and 900-600 cm-1
regions for the reaction products of the laser-ablated tantalum atomwith CH3CN in excess argon at 10 K. (a) Ta and CH3CN (0.50% inargon) codeposited for 1 h, (b) as (a) after visible (λ > 420 nm)irradiation, (c) as (b) after UV (240-380 nm) irradiation, (d) as (c)after visible irradiation, (e) as (d) after UV irradiation, and (f) as (e)after annealing to 28 K. m designates the product absorption, and Pand c stand for the precursor and common in the CH3CN matrix spectra.CH2CNH, CH2NCH, and CH3NC absorptions are also indicated.
Figure 2. Infrared spectra in the 2150-1850 and 1300-450 cm-1
regions for the reaction products of the laser-ablated tantalum atomwith CD3CN in excess argon at 10 K. (a) Ta and CD3CN (0.50% inargon) codeposited for 1 h and (b-f) as (a) after visible, UV, visible,and UV irradiations and annealing to 28 K following the same sequencein Figure 1. m stands for the product absorption, and P and c designatethe precursor and common absorptions in the CH3CN matrix spectra.CD2CND and CD2NCD absorptions are also indicated.
Figure 3. Infrared spectra in the 2200-1700, 1350-1250, and850-450 cm-1 regions for the reaction products of the laser-ablatedtantalum atom with 13CH3
13CN in excess argon at 10 K. (a) Ta and13CH3
13CN (0.50% in argon) codeposited for 1 h and (b-f) as (a) aftervisible, UV, visible, and UV irradiations and annealing to 28 K followingthe same sequence in Figure 1. m stands for product absorption, and Pand c designate the product and common absorptions. 13CH2CNH,13CH2N13CH, and H13C13CNH2 absorptions are also indicated.
5998 J. Phys. Chem. A, Vol. 114, No. 19, 2010 Cho and Andrews
cm-1). Due to the modest H/D and relatively large 12/13 ratiosof 1.101 and 1.027, it is designated to be the C-Ta stretchingmode, and its relatively high carbon-metal stretching frequencyindicates that it is a carbon-tantalum double bond.
In the low frequency region, the weak product absorption at698.7 cm-1 and its 13C counterpart at 694.0 cm-1 are assignedto the Ta-H in-plane bending mode without observation of theD counterpart. Another m absorption is observed at 657.9 cm-1
(with site absorption at 643.9 cm-1) along with its D counterpartat 508.7 cm-1 and 13C counterpart at 652.6 cm-1 (with a siteabsorption at 638.9 cm-1) (H/D and 12/13 ratios of 1.293 and1.008). The large D and small 13C shifts leads to an assignmentto the CH2 wagging mode. The m absorption at 464.9 cm-1 inthe 13CH3
13CN spectra is tentatively designated to the CH2
rocking mode without observation of its 12C and D counterparts.Observation of the Ta methylidene isocyanide is parallel to
the previously studied CH2dZr(H)NC,16 reconfirming that thesmall conjugated high oxidation-state complexes can be providedin reactions of transition-metal atoms with a nitrile compound.However, unlike the previous Zr + acetonitrile study, the Tainsertion and π-complexes are not identified in the productspectra. The Ta methylidene isocyanide is the most stable amongthe plausible products. CH2dTa(H)NC, CH3-TaNC, η2-Ta(NC)-CH3, and CH3CNfTa in the doublet, quartet, doublet,and quartet ground states are 56, 48, 37, and 21 kcal/mol morestable than the reactants (Ta(4F) + CH3CN), respectively, andthe possible cyanide complexes, CH2dTa(H)CN(D) andCH3-TaCN(Q), are 52 and 47 kcal/mol more stable than thereactants.
Although the stability of CH2dTa(H)NC over other plausibleproducts is consistent with its exclusive generation,26 the absenceof CH2dTa(H)CN despite the comparable energy supports thepreviously proposed reaction path for the Zr system.
M + CH3CN f CH3CNfM f η2-Μ(ΝC)-CH3 f
CH3-MNC f CH2dM(H)NC (1)
Initial coordination of the metal atom to the electron-richN-end of acetonitrile is expected to form CH3CNfTa.27
Subsequent rearrangement will lead to the observed more stablenitrile π-complexes28 in the Nb and V systems (described below)and the previous Zr study. The observed isocyanide insertionproducts from the Nb, V, and Zr reactions with no trace of thecyanide counterparts, which are energetically comparable,indicates that during C-C bond insertion by the metal atom,
the N-Ta bond is preserved, leading to the C-Ta-N-Cbackbone. The most stable CH2dTa(H)NC is produced in theend via H migration.
Nb + Acetonitrile. The product absorptions from Nbreactions with CH3CN isotopomers are shown in Figures 4 and5. In contrast to the Ta system, three groups of productabsorptions are observed depending on their intensity variationupon photolysis and annealing, which are marked m, i, and π(for methylidene, insertion, and π-complexes). The m absorp-tions are almost invisible in the original spectrum after codepo-sition, remain as weak on visible irradiation. They emerge onUV photolysis and almost double on full arc (λ > 220 nm)irradiation. The i absorptions decrease ∼30% and disappear onvisible and UV irradiations. The π absorptions slightly increase,halve, and further decrease on visible, UV, and full arcirradiations, respectively. The observed intensity variations ofthe product absorption groups suggest that the products respon-sible for the i and π absorptions convert to another productresponsible for the m absorptions in the process of photolysis.
The m absorption at 2039.9 cm-1 (with site absorptions at2035.2 and 2031.0 cm-1) in the CD3CN spectra in Figure 5 hasits 13C counterpart at 1999.4 cm-1 (with site absorptions at1994.8 and 1990.8 cm-1). On the basis of the frequency andsignificant 13C shift, it is assigned to the NC stretching mode.24d
The H counterpart expected at the same frequency as that in
TABLE 1: Observed and Calculated Fundamental Frequencies of CH2dTa(H)NC Isotopomers in the Ground 2A′ Statea
CH2dTa(H)NC CD2dTa(D)NC 13CH2dTa(H)N13Capproximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′ CH2 as. str. 3226.3 7 3170.0 6 2389.4 7 2348.2 7 3215.2 6 3159.1 5A′ CH2 s. str. 2789.1 5 2706.1 4 2030.8 9 1696.9 9 2782.6 5 2699.8 4A′ NC str. 2027.5, 2024.2 2096.4 518 2016.2 462 2027.4 2095.1 472 2015.9 473 1988.1, covered 2054.1 510 1976.6 453A′ Ta-H str. 1788.7, 1782.5 1822.3 253 1791.8 215 1282.2, 1277.0 1293.2 102 1271.6 102 1788.5, 1782.4 1822.2 257 1791.6 219A′ CH2 scis. 1344.1 21 1307.0 20 1012.9, 1005.3 1048.1 18 1016.8 18 1297.3 1335.6 20 1299.2 20A′ C-Ta str. 823.6, 819.6 824.2 55 815.6 51 744.6 724.8 33 717.8 34 800.7, 798.2 802.1 54 793.5 50A′ Ta-H ip bend 698.7, 697.6 716.8 19 712.4 12 541.4 30 540.0 19 694.0 713.7 16 708.9 11A′′ CH2 wag 657.9, 643.9 703.6 76 668.2 68 508.7 549.6 51 521.5 46 652.6, 638.9 697.7 74 662.7 66A′ CH2 rock 474.7 25 488.2 11 344.7 8 348.5 11 464.9 468.7 20 483.3 8A′ Ta-NC str. 429.9 61 427.0 65 449.6 62 449.8 54 424.8 63 421.2 65A′′ CH2 twist 415.1 7 426.4 8 299.2 3 307.9 3 415.0 7 426.3 8A′ TaNC ip bend 210.0 1 215.5 2 195.4 1 201.8 2 208.6 1 214.0 1A′′ TaNC oop bend 120.5 14 116.4 6 112.8 1 108.5 2 119.5 14 115.1 6A′ Ta-H oop bend 107.0 12 87.3 4 83.7 4 82.3 4 106.5 11 84.8 3A′′ CTaN bend 89.0 3 55.6 15 82.6 15 41.7 11 86.4 3 55.5 15
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. The absorption that increases and decreases on UV andvisible irradiations is bold. c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computedwith BPW91/6-311++G(3df,3pd). CH2dTa(H)NC has a planar Cs structure.
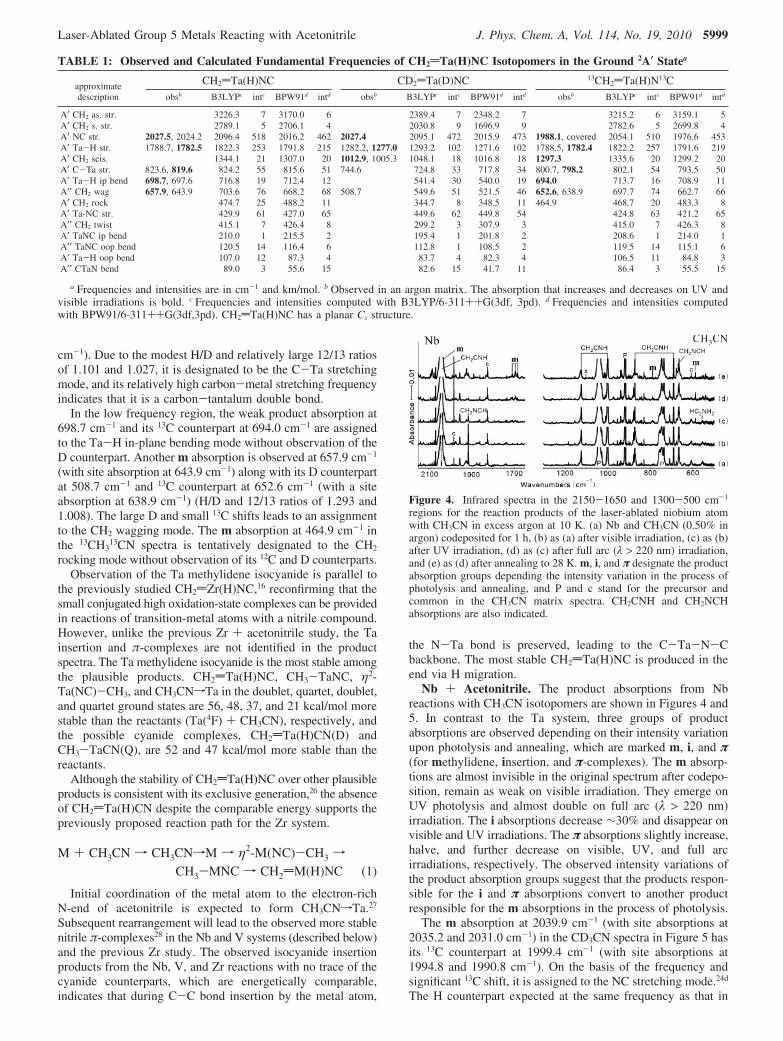
Figure 4. Infrared spectra in the 2150-1650 and 1300-500 cm-1
regions for the reaction products of the laser-ablated niobium atomwith CH3CN in excess argon at 10 K. (a) Nb and CH3CN (0.50% inargon) codeposited for 1 h, (b) as (a) after visible irradiation, (c) as (b)after UV irradiation, (d) as (c) after full arc (λ > 220 nm) irradiation,and (e) as (d) after annealing to 28 K. m, i, and π designate the productabsorption groups depending the intensity variation in the process ofphotolysis and annealing, and P and c stand for the precursor andcommon in the CH3CN matrix spectra. CH2CNH and CH2NCHabsorptions are also indicated.
Laser-Ablated Group 5 Metals Reacting with Acetonitrile J. Phys. Chem. A, Vol. 114, No. 19, 2010 5999
the CD3CN spectra is covered by CH2CNH absorption24 exceptfor a site absorption at 2031.0 cm-1. The possible nitrile (CN)stretching absorption expected at ∼2150 cm-1 is again notobserved. The m absorption at 1708.3 cm-1 (with site absorp-tions at 1714.2 and 1693.0 cm-1) in the Nb-H stretching regioncarries its D counterpart at 1228.0 cm-1 (with site absorptionsat 1238.2 and 1217.9 cm-1) and 13C counterpart at 1708.3 cm-1
(with site absorptions at 1714.2 and 1692.6 cm-1). The large D(H/D ratio of 1.391) and negligible 13C shifts lead to anassignment to the Nb-H stretching mode. The Nb-H stretchingabsorption is indicative of formation of a Nb high oxidation-state product, CH2dNb(H)NC, and its frequency is comparedwith 1611 and 1569 cm-1 for NbH2;25 1680.7 and 1652.5 cm-1
for CH2dNbH2; and 1698.7, 1697.8, and 1698.7 cm-1 forCH2dNbHF, CH2dNbHCl, and CH2dNbHBr.7
An m absorption is observed at 788.8 cm-1, and its D and13C counterparts at 691.8 and 770.5 cm-1 (H/D and 12/13 ratiosof 1.140 and 1.024 cm-1), and it is designated to the C-Nbdouble bond stretching mode on the basis of its frequency andthe modest D and relatively large 13C shifts. In the further lowfrequency region, the m absorption at 528.1 cm-1 in the CD3CNspectra is assigned to the CH2 wagging mode, and the H and13C counterparts are covered by precursor absorption. Anotherm absorption is observed at 587.4 cm-1, where 13C substitutionshifts it to 581.9 cm-1 and its D counterpart is too low infrequency to be observed, leading to an assignment to the CH2
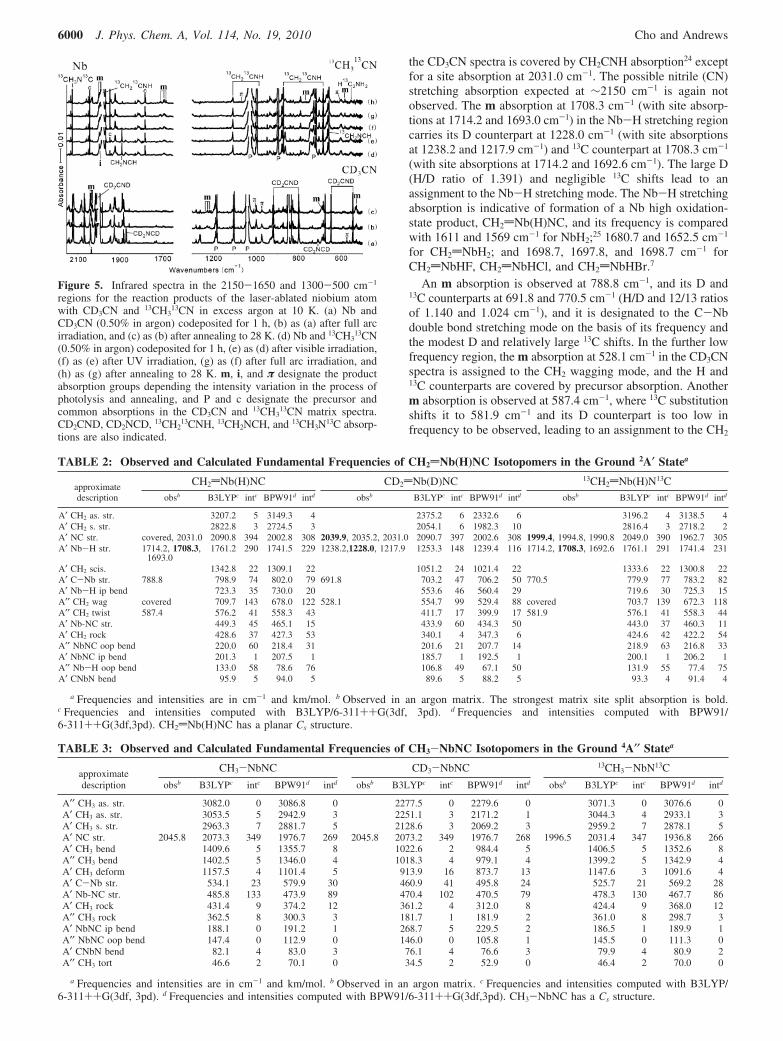
Figure 5. Infrared spectra in the 2150-1650 and 1300-500 cm-1
regions for the reaction products of the laser-ablated niobium atomwith CD3CN and 13CH3
13CN in excess argon at 10 K. (a) Nb andCD3CN (0.50% in argon) codeposited for 1 h, (b) as (a) after full arcirradiation, and (c) as (b) after annealing to 28 K. (d) Nb and 13CH3
13CN(0.50% in argon) codeposited for 1 h, (e) as (d) after visible irradiation,(f) as (e) after UV irradiation, (g) as (f) after full arc irradiation, and(h) as (g) after annealing to 28 K. m, i, and π designate the productabsorption groups depending the intensity variation in the process ofphotolysis and annealing, and P and c designate the precursor andcommon absorptions in the CD3CN and 13CH3
13CN matrix spectra.CD2CND, CD2NCD, 13CH2
13CNH, 13CH2NCH, and 13CH3N13C absorp-tions are also indicated.
TABLE 2: Observed and Calculated Fundamental Frequencies of CH2dNb(H)NC Isotopomers in the Ground 2A′ Statea
CH2dNb(H)NC CD2dNb(D)NC 13CH2dNb(H)N13Capproximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′ CH2 as. str. 3207.2 5 3149.3 4 2375.2 6 2332.6 6 3196.2 4 3138.5 4A′ CH2 s. str. 2822.8 3 2724.5 3 2054.1 6 1982.3 10 2816.4 3 2718.2 2A′ NC str. covered, 2031.0 2090.8 394 2002.8 308 2039.9, 2035.2, 2031.0 2090.7 397 2002.6 308 1999.4, 1994.8, 1990.8 2049.0 390 1962.7 305A′ Nb-H str. 1714.2, 1708.3,
1693.01761.2 290 1741.5 229 1238.2,1228.0, 1217.9 1253.3 148 1239.4 116 1714.2, 1708.3, 1692.6 1761.1 291 1741.4 231
A′ CH2 scis. 1342.8 22 1309.1 22 1051.2 24 1021.4 22 1333.6 22 1300.8 22A′ C-Nb str. 788.8 798.9 74 802.0 79 691.8 703.2 47 706.2 50 770.5 779.9 77 783.2 82A′ Nb-H ip bend 723.3 35 730.0 20 553.6 46 560.4 29 719.6 30 725.3 15A′′ CH2 wag covered 709.7 143 678.0 122 528.1 554.7 99 529.4 88 covered 703.7 139 672.3 118A′′ CH2 twist 587.4 576.2 41 558.3 43 411.7 17 399.9 17 581.9 576.1 41 558.3 44A′ Nb-NC str. 449.3 45 465.1 15 433.9 60 434.3 50 443.0 37 460.3 11A′ CH2 rock 428.6 37 427.3 53 340.1 4 347.3 6 424.6 42 422.2 54A′′ NbNC oop bend 220.0 60 218.4 31 201.6 21 207.7 14 218.9 63 216.8 33A′ NbNC ip bend 201.3 1 207.5 1 185.7 1 192.5 1 200.1 1 206.2 1A′′ Nb-H oop bend 133.0 58 78.6 76 106.8 49 67.1 50 131.9 55 77.4 75A′ CNbN bend 95.9 5 94.0 5 89.6 5 88.2 5 93.3 4 91.4 4
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. The strongest matrix site split absorption is bold.c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computed with BPW91/6-311++G(3df,3pd). CH2dNb(H)NC has a planar Cs structure.
TABLE 3: Observed and Calculated Fundamental Frequencies of CH3-NbNC Isotopomers in the Ground 4A′′ Statea
CH3-NbNC CD3-NbNC 13CH3-NbN13Capproximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′′ CH3 as. str. 3082.0 0 3086.8 0 2277.5 0 2279.6 0 3071.3 0 3076.6 0A′ CH3 as. str. 3053.5 5 2942.9 3 2251.1 3 2171.2 1 3044.3 4 2933.1 3A′ CH3 s. str. 2963.3 7 2881.7 5 2128.6 3 2069.2 3 2959.2 7 2878.1 5A′ NC str. 2045.8 2073.3 349 1976.7 269 2045.8 2073.2 349 1976.7 268 1996.5 2031.4 347 1936.8 266A′ CH3 bend 1409.6 5 1355.7 8 1022.6 2 984.4 5 1406.5 5 1352.6 8A′′ CH3 bend 1402.5 5 1346.0 4 1018.3 4 979.1 4 1399.2 5 1342.9 4A′ CH3 deform 1157.5 4 1101.4 5 913.9 16 873.7 13 1147.6 3 1091.6 4A′ C-Nb str. 534.1 23 579.9 30 460.9 41 495.8 24 525.7 21 569.2 28A′ Nb-NC str. 485.8 133 473.9 89 470.4 102 470.5 79 478.3 130 467.7 86A′ CH3 rock 431.4 9 374.2 12 361.2 4 312.0 8 424.4 9 368.0 12A′′ CH3 rock 362.5 8 300.3 3 181.7 1 181.9 2 361.0 8 298.7 3A′ NbNC ip bend 188.1 0 191.2 1 268.7 5 229.5 2 186.5 1 189.9 1A′′ NbNC oop bend 147.4 0 112.9 0 146.0 0 105.8 1 145.5 0 111.3 0A′ CNbN bend 82.1 4 83.0 3 76.1 4 76.6 3 79.9 4 80.9 2A′′ CH3 tort 46.6 2 70.1 0 34.5 2 52.9 0 46.4 2 70.0 0
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computed with BPW91/6-311++G(3df,3pd). CH3-NbNC has a Cs structure.
6000 J. Phys. Chem. A, Vol. 114, No. 19, 2010 Cho and Andrews
twisting mode. Therefore, the primary product responsible forthe m absorptions carries Nb-H, CdNb, CH2, and NC moieties,most probably CH2dNb(H)NC. The observed frequencies arewell reproduced as shown in Table 2, within the limits ofDFT,7-13,23 which substantiate formation of the small Nbmethylidene isocyanide.
Only one i absorption is observed at 2045 cm-1, whichdisappears on UV irradiation, and deuteration does not changeits frequency, but 13C substitution shifts it to 1996.5 cm-1. Weassign the NC stretching absorption to the insertion complex,CH3-NbNC. Table 3 shows that the NC stretching band is thestrongest one, and the predicted D and 13C shifts of 0.1 and41.9 cm-1 correlate excellently with the observed values.Unfortunately, the second strong Nb-N stretching band withabout a third the NC stretching absorption intensity expectedat ∼470 cm-1 is not observed in the noisier low frequencyregion.
The strongest π absorption is observed at 1101.6 cm-1, andits D and 13C counterparts at 973.1 and 1069.0 cm-1 (H/D and12/13 ratios of 1.132 and 1.031). It is assigned to the A′ CH3
rocking mode, which is the strongest for the nitrile π-complex,η2-Nb(NC)-CH3, on the basis of the frequency and goodcorrelation with the DFT values (Table 4). For example, theobserved frequencies for the isotopomers are compared withthe B3LYP frequencies of 1122.6, 993.9, and 1097.8 cm-1. Inthe low frequency region, another π absorption is observed at606.4 cm-1, and the D and 13C counterparts are at 572.5 and600.2 cm-1. The frequency, relatively small isotopic shifts, andreasonable correlation with the DFT values lead to an assign-ment to the N-Nb stretching mode. The weak π absorptionobserved at 1000.1 cm-1 in the CD3CN spectra is tentativelydesignated to the A′ CH3 bending mode. Unfortunately, otherabsorptions from the Nb nitrile π-complex (η2-Nb(NC)-CH3)are expected to be too weak to be observed or to be covered byprecursor absorption in the congested areas.
CH2dNb(H)NC, CH3-NbNC, η2-Nb(NC)-CH3, andCH3CNfNb in the doublet, quartet, quartet, and sextet groundstates are 42, 48, 35, and 27 kcal/mol more stable than thereactants (Nb(6D) + CH3CN), respectively, and CH2dNb(H)CNand CH3-NbCN in the ground doublet and quartet states are43 and 48 kcal/mol more stable. The observed CH3-NbNC andη2-Nb(NC)-CH3Nb in the original spectra and CH2dNb(H)CNupon photolysis are the most stable products in the proposedreaction path 1 whereas the cyanide counterparts have essentiallythe same energy.
V + CH3CN. Figure 6 shows the product absorptions formreactions of V with acetonitrile isotopomers and their variationupon photolysis and annealing. Two groups of product absorp-tions marked i and π are observed depending on the intensityvariation upon photolysis and annealing. The i absorptions arealmost invisible in the original spectra, emerge on visibleirradiation, and dramatically increase (∼400%) on UV irradia-tion. They sharpen up in the early stage of annealing and latergradually decrease. The π absorptions, in contrast, are strongestin the original spectra, slightly decrease on visible irradiation,and almost disappear on UV irradiation. The intensity variationsof the i and π absorptions suggest that the primary productresponsible for the π absorptions from V reaction with aceto-nitrile converts to a different product responsible for the iabsorptions in the process of photolysis.
The i absorption at 2058.8 cm-1 shows a negligible D shift;and following the Zr, Ta, and Nb cases, it is assigned to the
TABLE 4: Observed and Calculated Fundamental Frequencies of the η2-π-Complex Nb(NC)-CH3 Isotopomers in the Ground4A′′ Statea
η2-Nb(NC)-CH3 Nb(NC)-CD3 Nb(N13C)-13CH3approximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′ CH3 as. str. 3094.7 20 3042.0 19 2293.9 10 2254.4 10 3083.5 19 3031.0 18A′′ CH3 as. str. 3089.9 1 3036.9 2 2285.5 2 2245.2 1 3079.1 1 3026.3 2A′ CH3 s. str. 3020.2 34 2962.0 30 2168.1 13 2126.2 11 3017.0 35 2958.9 31A′ CCN as. str. 1635.8 36 1561.6 14 1632.1 39 1556.7 17 1595.9 38 1523.0 14A′′ CH3 bend 1468.3 16 1420.5 14 1058.8 6 1023.9 6 1466.0 16 1418.4 14A′ CH3 bend 1467.4 15 1420.4 17 1000.1 1055.4 11 1022.7 14 1464.9 16 1417.6 18A′ CH3 deform 1386.2 7 1335.2 8 1131.8 62 1103.2 59 1375.6 6 1324.9 7A′ CH3 rock 1101.6 1122.6 120 1101.0 92 973.1 993.9 93 973.2 51 1069.0 1097.8 107 1075.1 84A′′ CH3 rock 1001.1 17 964.5 4 807.4 21 780.8 7 988.9 15 952.5 3A′ C-C str. 942.0 26 917.0 15 792.7 5 765.9 4 925.5 30 902.8 18A′ N-Nb str. 606.4 640.8 76 630.5 58 572.5 617.8 65 610.9 54 600.2 637.4 76 627.3 58A′ C-Nb str. 496.4 4 505.7 5 467.0 2 473.3 2 485.3 4 494.3 4A′′ CCNNb deform 311.7 17 308.0 10 282.6 13 278.5 7 302.7 16 299.2 9A′ CCNb bend 244.5 14 243.4 13 224.1 13 222.6 12 240.0 14 239.0 13A′′ CH3 tort 48.6 1 26.9 0 35.4 1 17.2 1 48.6 1 26.9 0
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computed with BPW91/6-311++G(3df,3pd). The Nb nitrile-π-complex has a Cs structure.
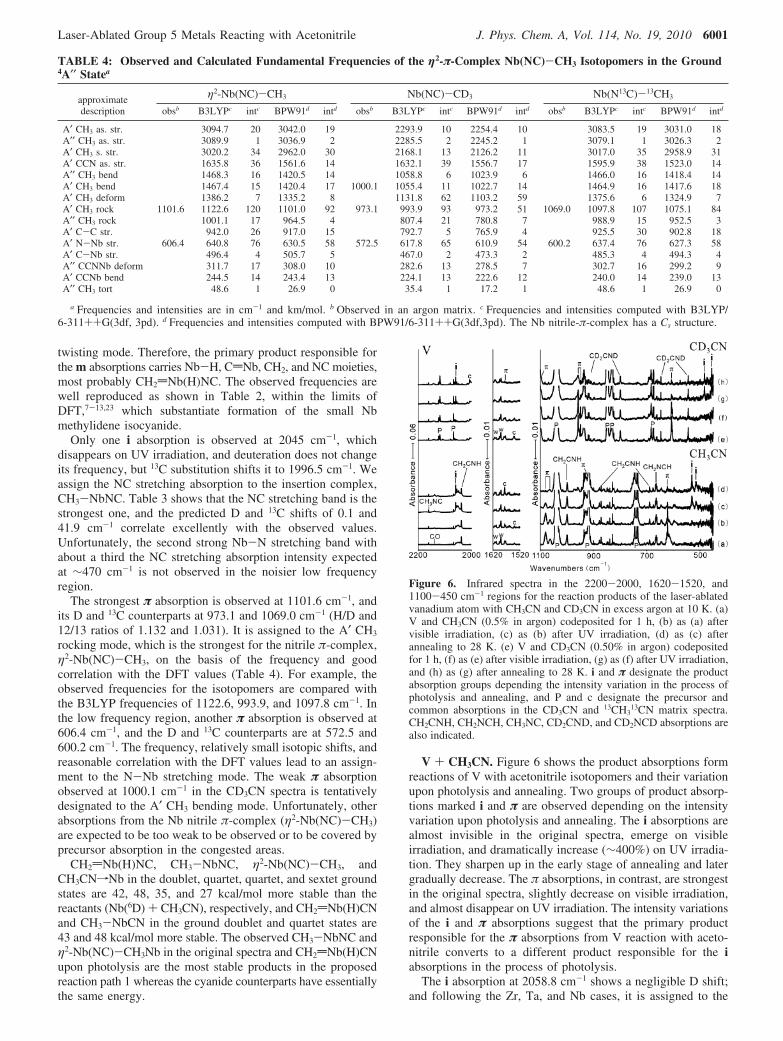
Figure 6. Infrared spectra in the 2200-2000, 1620-1520, and1100-450 cm-1 regions for the reaction products of the laser-ablatedvanadium atom with CH3CN and CD3CN in excess argon at 10 K. (a)V and CH3CN (0.5% in argon) codeposited for 1 h, (b) as (a) aftervisible irradiation, (c) as (b) after UV irradiation, (d) as (c) afterannealing to 28 K. (e) V and CD3CN (0.50% in argon) codepositedfor 1 h, (f) as (e) after visible irradiation, (g) as (f) after UV irradiation,and (h) as (g) after annealing to 28 K. i and π designate the productabsorption groups depending the intensity variation in the process ofphotolysis and annealing, and P and c designate the precursor andcommon absorptions in the CD3CN and 13CH3
13CN matrix spectra.CH2CNH, CH2NCH, CH3NC, CD2CND, and CD2NCD absorptions arealso indicated.
Laser-Ablated Group 5 Metals Reacting with Acetonitrile J. Phys. Chem. A, Vol. 114, No. 19, 2010 6001
NC stretching mode. In the low frequency region, the iabsorptions at 530.9 and 515.2 cm-1 along with its D counterpartat 483.1 and 455.6 cm-1 (H/D ratios of 1.099 and 1.131) areassigned to the C-V and V-N stretching modes on the basisof the frequencies and modest D shifts. Table 5 shows that theyare the strongest bands for CH3-VNC, and the observedfrequencies are 0.981, 0.953, and 1.011 of the B3LYP values.Mode analysis shows substantial coupling between the C-Vand V-N stretching modes, which might be the source of theslight under- and overestimation of the C-V and V-Nstretching frequencies. The other bands of the insertion complexare too weak to be observed, as shown in Table 5.
A strong and relatively broad π absorption is observed at1080.0 cm-1 (with a site absorption at 1066.5 cm-1), and its Dcounterpart is at 957.8 cm-1 (with a site absorption at 948.6cm-1). The only plausible product from reaction of V withCH3CN that shows a strong absorption in the frequency regionis the nitrile η2-π-complex, η2-(NC)-CH3. It is assigned to theA′ CH3 rocking mode (Table 6), and the relatively small D shift(H/D ratio of 1.128) for a rocking mode originates from couplingwith the C-C and C-V stretching modes. The weak A′ CCNsymmetric stretching absorption is observed at 923.4 cm-1, and
the even weaker D counterpart is not observed. The uniquelystrong π absorption in the low frequency region at 622.5 cm-1
has its D counterpart at 607.0 cm-1 (H/D ratio of 1.026) and isassigned to the N-V stretching mode (Table 6). The πabsorption at 1573.4 and 1097 cm-1 in the CD3CN spectra aredesignated to the CCN antisymmetric stretching and A′ CH3
bending modes, but their H counterpart expected at ∼1577 and∼1445 cm-1 are believed to be covered in the congested areas.Table 6 shows very good agreement with the observed andpredicted values, where the observed frequencies are 0.954-1.006of the B3LYP values.
Generation of CH3-VNC and η2-V(NC)-CH3 is also con-sistent with their relative stabilities. CH2dV(H)NC, CH3-VNC,η2-V(NC)-CH3, and CH3CNfV in their doublet, quartet,quartet, and quartet ground states are 7, 41, 21, 17 kcal/molmore stable than the reactants (V(4F) + CH3CN), respectively,whereas CH2dV(H)CN and CH3-VCN in the doublet andquartet ground states are 3 and 40 kcal/mol more stable.Although CH3-VNC is more stable than η2-V(NC)-CH3, theprimary product in the original spectra after codeposition of Vand CH3CN is the nitrile π-complex. The present results areindicative of a low barrier between η2-V(NC)-CH3 and
TABLE 5: Observed and Calculated Fundamental Frequencies of CH3-VNC Isotopomers in the Ground 4A′′ Statea
CH3-VNC CD3-VNCapproximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′ CH3 as. str. 3109.3 2 3079.2 3 2298.0 0 2275.1 1A′′ CH3 as. str. 3053.7 3 2991.4 2 2258.0 0 2215.1 0A′ CH3 s. str. 2981.1 6 2898.2 60 2137.5 1 2078.6 31A′ NC str. 2058.8 2099.2 363 2001.2 287 2058.7 2099.2 364 2001.2 282A′′ CH3 bend 1426.9 4 1375.6 7 1037.0 4 1002.2 5A′ CH3 bend 1410.7 3 1340.3 35 1024.2 3 972.4 26A′ CH3 deform 1142.4 0 1075.4 10 896.1 6 851.5 4A′ C-V str. 530.9 556.9 44 559.5 42 483.1 507.9 124 503.0 65A′′ CH3 rock 545.1 39 613.1 19 408.6 24 465.3 11A′ V-NC str. 515.2 509.7 162 504.2 95 455.6 471.9 54 482.7 48A′ CH3 rock 413.5 7 381.3 20 341.1 12 309.2 22A′ VNC ip bend 179.9 2 148.1 8 175.3 1 143.4 6A′′ VNC oop bend 174.3 1 181.2 1 168.0 2 168.1 2A′ CVN bend 79.8 8 81.1 5 75.0 8 75.9 5A′′ CH3 tort 52.6 1 84.8 0 41.1 2 66.8 1
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computed with BPW91/6-311++G(3df,3pd). CH3VNC has a Cs structure.
TABLE 6: Observed and Calculated Fundamental Frequencies of the η2-π-Complex V(NC)-CH3 Isotopomers in the Ground3A′′ Statea
η2-V(NC)-CH3 V(NC)-CD3approximatedescription obsb B3LYPc intc BPW91d intd obsb B3LYPc intc BPW91d intd
A′ CH3 as. str. 3087.9 29 3037.7 22 2288.1 15 2250.8A′′ CH3 as. str. 3087.8 6 3035.8 6 2284.0 3 2244.3A′ CH3 s. str. 3014.1 57 2957.9 41 2163.9 20 2123.2A′ CCN as. str. covered 1652.5 252 1586.8 51 1573.4 1649.6 253 1583.0 54A′′ CH3 bend 1468.3 10 1420.5 8 1058.8 5 1023.8 6A′ CH3 bend covered 1466.9 16 1419.7 18 1097 1116.3 91 1088.5 61A′ CH3 deform 1382.3 5 1331.1 6 1054.2 11 1020.3 14A′ CH3 rock 1080.0, 1066.5 1082.4 226 1072.6 126 957.8, 948.6 951.8 245 950.9 93A′′ CH3 rock 1013.3 0 983.6 6 821.9 0 803.8 3A′ CCN s. str. 923.4 928.7 95 910.3 24 791.4 8 764.8 6A′ N-V str. 622.5 635.3 48 636.6 62 607.0 614.1 28 622.2 53A′ C-V str. 471.7 23 506.9 3 446.6 19 473.4 1A′′ CCNV deform 323.8 2 326.7 0 292.1 2 292.8 0A′ CCV bend 247.2 31 248.7 23 227.9 28 228.7 21A′′ CH3 tort 75.7 0 50.3 0 57.4 1 38.6 0
a Frequencies and intensities are in cm-1 and km/mol. b Observed in an argon matrix. The strongest matrix site split absorption is bold.c Frequencies and intensities computed with B3LYP/6-311++G(3df, 3pd). d Frequencies and intensities computed with BPW91/6-311++G(3df,3pd). The V nitrile-π-complex has a Cs structure.
6002 J. Phys. Chem. A, Vol. 114, No. 19, 2010 Cho and Andrews
CH3CNfV, which allows the swift conversion to the π complexduring codeposition and much higher barrier betweenCH3-VNC and η2-(NC)-CH3. Formation of CH3-VNC fromthe nitrile π-complex requires photolysis (particularly with UV)following codeposition as shown in Figures 6.
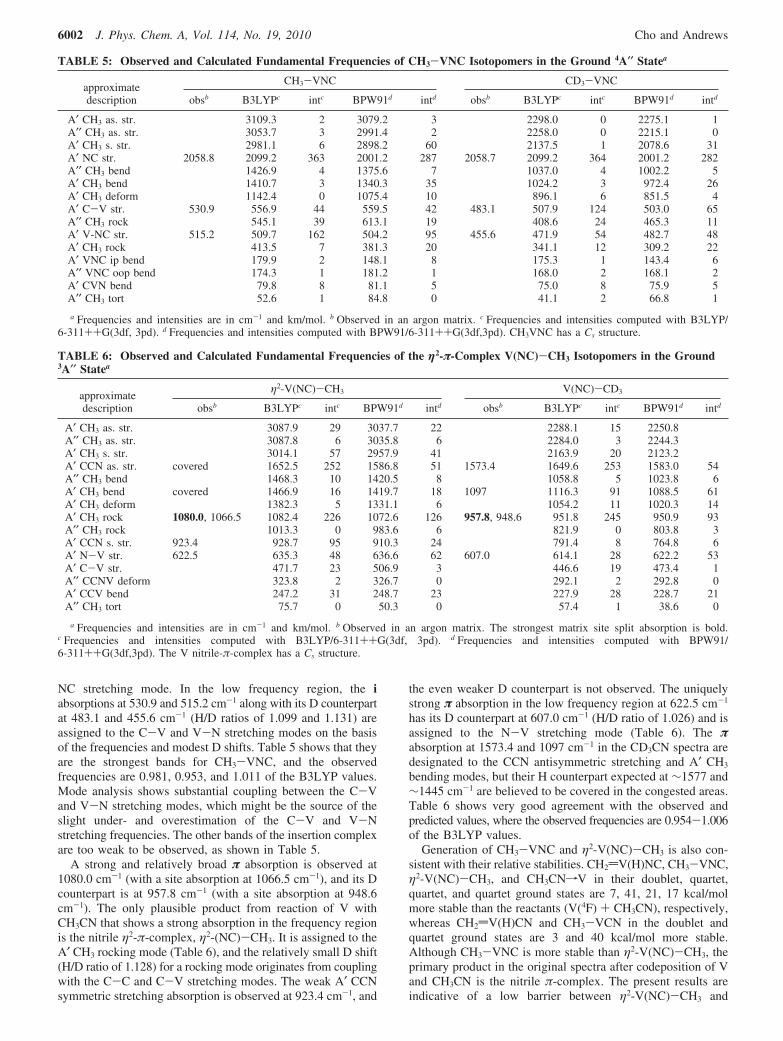
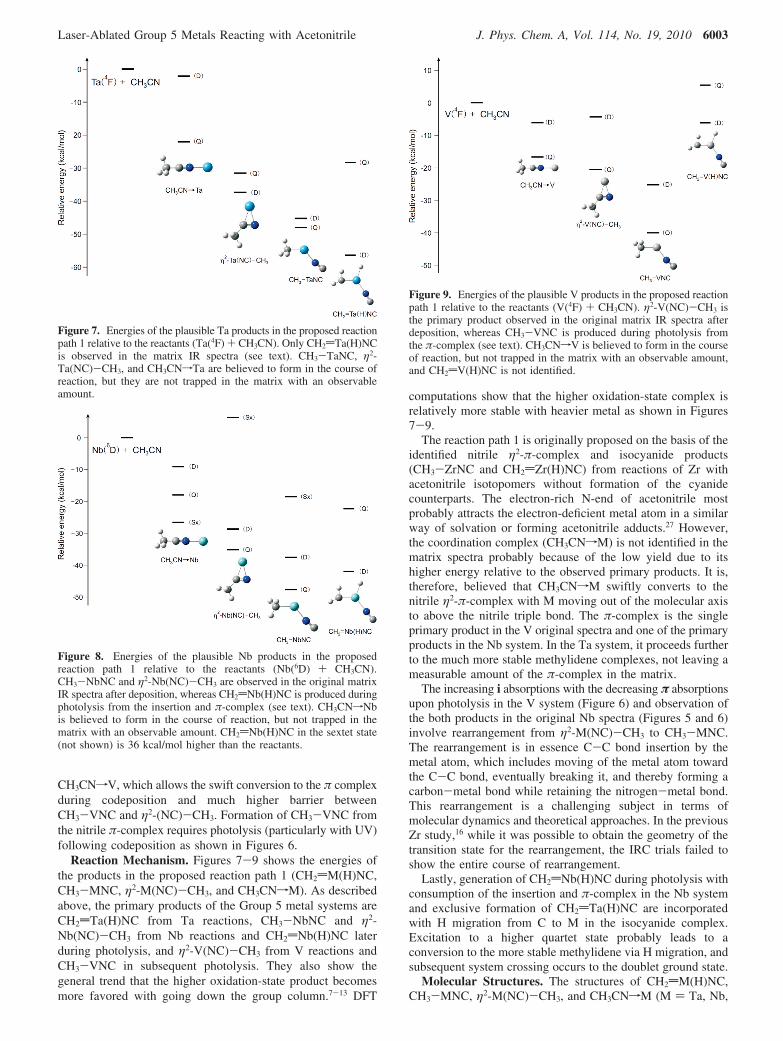
Reaction Mechanism. Figures 7-9 shows the energies ofthe products in the proposed reaction path 1 (CH2dM(H)NC,CH3-MNC, η2-M(NC)-CH3, and CH3CNfM). As describedabove, the primary products of the Group 5 metal systems areCH2dTa(H)NC from Ta reactions, CH3-NbNC and η2-Nb(NC)-CH3 from Nb reactions and CH2dNb(H)NC laterduring photolysis, and η2-V(NC)-CH3 from V reactions andCH3-VNC in subsequent photolysis. They also show thegeneral trend that the higher oxidation-state product becomesmore favored with going down the group column.7-13 DFT
computations show that the higher oxidation-state complex isrelatively more stable with heavier metal as shown in Figures7-9.
The reaction path 1 is originally proposed on the basis of theidentified nitrile η2-π-complex and isocyanide products(CH3-ZrNC and CH2dZr(H)NC) from reactions of Zr withacetonitrile isotopomers without formation of the cyanidecounterparts. The electron-rich N-end of acetonitrile mostprobably attracts the electron-deficient metal atom in a similarway of solvation or forming acetonitrile adducts.27 However,the coordination complex (CH3CNfM) is not identified in thematrix spectra probably because of the low yield due to itshigher energy relative to the observed primary products. It is,therefore, believed that CH3CNfM swiftly converts to thenitrile η2-π-complex with M moving out of the molecular axisto above the nitrile triple bond. The π-complex is the singleprimary product in the V original spectra and one of the primaryproducts in the Nb system. In the Ta system, it proceeds furtherto the much more stable methylidene complexes, not leaving ameasurable amount of the π-complex in the matrix.
The increasing i absorptions with the decreasing π absorptionsupon photolysis in the V system (Figure 6) and observation ofthe both products in the original Nb spectra (Figures 5 and 6)involve rearrangement from η2-M(NC)-CH3 to CH3-MNC.The rearrangement is in essence C-C bond insertion by themetal atom, which includes moving of the metal atom towardthe C-C bond, eventually breaking it, and thereby forming acarbon-metal bond while retaining the nitrogen-metal bond.This rearrangement is a challenging subject in terms ofmolecular dynamics and theoretical approaches. In the previousZr study,16 while it was possible to obtain the geometry of thetransition state for the rearrangement, the IRC trials failed toshow the entire course of rearrangement.
Lastly, generation of CH2dNb(H)NC during photolysis withconsumption of the insertion and π-complex in the Nb systemand exclusive formation of CH2dTa(H)NC are incorporatedwith H migration from C to M in the isocyanide complex.Excitation to a higher quartet state probably leads to aconversion to the more stable methylidene via H migration, andsubsequent system crossing occurs to the doublet ground state.
Molecular Structures. The structures of CH2dM(H)NC,CH3-MNC, η2-M(NC)-CH3, and CH3CNfM (M ) Ta, Nb,
Figure 7. Energies of the plausible Ta products in the proposed reactionpath 1 relative to the reactants (Ta(4F) + CH3CN). Only CH2dTa(H)NCis observed in the matrix IR spectra (see text). CH3-TaNC, η2-Ta(NC)-CH3, and CH3CNfTa are believed to form in the course ofreaction, but they are not trapped in the matrix with an observableamount.
Figure 8. Energies of the plausible Nb products in the proposedreaction path 1 relative to the reactants (Nb(6D) + CH3CN).CH3-NbNC and η2-Nb(NC)-CH3 are observed in the original matrixIR spectra after deposition, whereas CH2dNb(H)NC is produced duringphotolysis from the insertion and π-complex (see text). CH3CNfNbis believed to form in the course of reaction, but not trapped in thematrix with an observable amount. CH2dNb(H)NC in the sextet state(not shown) is 36 kcal/mol higher than the reactants.
Figure 9. Energies of the plausible V products in the proposed reactionpath 1 relative to the reactants (V(4F) + CH3CN). η2-V(NC)-CH3 isthe primary product observed in the original matrix IR spectra afterdeposition, whereas CH3-VNC is produced during photolysis fromthe π-complex (see text). CH3CNfV is believed to form in the courseof reaction, but not trapped in the matrix with an observable amount,and CH2dV(H)NC is not identified.
Laser-Ablated Group 5 Metals Reacting with Acetonitrile J. Phys. Chem. A, Vol. 114, No. 19, 2010 6003

and V), the plausible products along the reaction path 1, areillustrated in Figures 10-12. Unlike previously studied non-planar CH2dZr(H)NC,16 the Group 5 metal-conjugated meth-ylidene complexes all have planar Cs structures. Their CH2
moieties are more agostically distorted than the Zr methylideneisocyanide.16 The H-C-M angles of 88.8, 90.0, and 85.3° forCH2dTa(H)NC, CH2dNb(H)NC, and CH2dV(H)NC are smallerthan that of 91.3° for CH2dZr(H)NC. They are also comparedwith those of 92.7, 92.4, and 88.2° for CH2dTaHF,CH2dNbHF, and CH2dVHF and those of 88.7, 88.9, 88.2, and
87.9° for CH2dTaHCl, CH2dNbHCl, CH2dTaHBr, andCH2dNbHBr, respectively.7 It is reasonable that more-electron-deficient (higher electronegativity) Group 5 metals lead tostronger agostic interactions, but the conjugation effects on themagnitude of the distortion are apparently not noticeable.
The observed C-Ta and C-Nb stretching frequencies of819.6 and 788.8 cm-1 are also compared with those of 809.8,816.7, 816.6, 777.7, 794.3, and 796.4 cm-1 for CH2dTaHF,CH2dTaHCl, CH2dTaHBr, CH2dNbHF, CH2dNbHCl, andCH2dNbHBr, respectively. The CH2 wagging frequencies of657.9 cm-1 for CH2dTa(H)NC are also comparable with thoseof 672.9, 656.3, and 653.6 cm-1 for CH2dTaHF, CH2dTaHCl,and CH2dTaHBr, respectively.7 Evidently, weakening of theC-M bond by conjugation is not observable in the Group 5metal methylidene cyanides, reconfirming the previous resultthat the conjugation effects of CdM-N′C: is minor.16
The insertion complex, on the other hand, has a Cs structureand the M-N′C moiety is slightly bent (167.6, 169.4, and 167.7°for the Ta, Nb, and V insertion products). The π-complex alsohas a Cs structure, and the C-M and N-M bonds are shorterthan those in CH3-MNC and CH3CNfM but are longer thanthe C-M bond in the methylidene complex as shown in Figures10-12. This indicates that binding of the Group 5 metal atomsto the nitrile group is weaker than in the Zr system, where theC-Zr and N-Zr bonds of η2-Zr(NC)-CH3 are the shortestamong those of the Zr products including CH2dZr(H)NC.
The back-donation from the Group 5 metal center to the π*-orbitals of the CN triple bond is evidently less effective thanthe exceptionally strong interaction in the corresponding Zrsystem.16 The binding energy for the Ta, Nb, and V nitrileπ-complexes of 48, 48, and 21 kcal/mol are compared with thatfor the Zr π-complex of 69 kcal/mol. The present results arealso in line with the previously reported exceptionally strongback-donation from the Group 4 metals to the π*-orbital of theCC triple bond in η2-M-C2H2.29 The shorter CN bonds for theTa, Nb, and V η2-π-complexes (1.278, 1.269, and 1.256 Å) thanthat of η2-Zr(NC)-CH3 (1.280 Å) are also consistent with theweaker back-donations, resulting in the stronger CN bonds.
Figure 10. The B3LYP structures of the plausible Ta products in theproposed reaction path 1 with the 6-311++G(3df,3pd) basis sets forH, C, and N, and SDD pseudopotential and basis for Ta. Bond distancesand angles are in Å and degrees, respectively. The Ta methylideneisocyanide has a doublet ground state, and the other ones have quartetground states. The molecular symmetry is denoted under the structure.Notice the agostic distortion of the methylidene isocyanide complex.The C-Ta and N-Ta bonds in the nitrile π-complex (2.045 and 1.986Å) are shorter than those in CH3-TaNC and CH3CNfTa but are muchlonger than the C-Ta bond in the Ta methylidene isocyanide (1.896 Å).
Figure 11. The B3LYP structures of the plausible Nb products in theproposed reaction path 1 with the 6-311++G(3df,3pd) basis sets forH, C, and N, and SDD pseudopotential and basis for Nb. Bond distancesand angles are in Å and degrees, respectively. The Nb methylideneisocyanide has a doublet ground state, and the other ones have quartetground states. The molecular symmetry is denoted under the structure.Notice the agostic distortion of the methylidene isocyanide complex.The C-Nb and N-Nb bonds in the nitrile π-complex (2.042 and 1.997Å) are shorter than those in CH3-NbNC and CH3NCfNb but are muchlonger than the C-Nb bond in the Nb methylidene isocyanide (1.892 Å).
Figure 12. The B3LYP structures of the plausible V products in theproposed reaction path 1 with the 6-311++G(3df,3pd) basis sets forH, C, N, and V. Bond distances and angles are in Å and degrees,respectively. The V methylidene isocyanide has a doublet ground state,and the other ones have quartet ground states. The molecular symmetryis denoted under the structure. Notice the agostic distortion of themethylidene isocyanide complex. The C-V and N-V bonds in thenitrile π-complex (1.967 and 1.889 Å) are shorter than those inCH3-VNC and CH3CNfV but are longer than the C-V bond in theV methylidene isocyanide (1.757 Å).
6004 J. Phys. Chem. A, Vol. 114, No. 19, 2010 Cho and Andrews

On the other hand, the coordination complexes with the N-endof CH3CN (CH3CNfM), which is believed to form first inreactions of the metal atoms with acetonitrile although notobserved in the matrix spectra, largely retain the structure offree acetonitrile. They all have C3V structures computed withthe DFT methods used in this study.
Conclusions
Laser-ablated Group 5 metal atoms react with acetonitrile,and the products are identified on the basis of isotopic shiftsand correlation with the DFT results. Ta exclusively producesCH2dTa(H)NC in the process of both codeposition andsubsequent photolysis, whereas Nb generates CH3-NbNCand η2-Nb(NC)-CH3 in reaction with CH3CN and theytransform to CH2dNb(H)NC upon subsequent photolysis. Co-deposition of V with acetonitrile leads to almost exclusiveproduction of η2-V(NC)-CH3, and following photolysistransforms the nitrile π-complex to CH3-VNC. Productionof the Group 5 metal isocyanide and π-complexes duringcodeposition and subsequent photolysis and absence of theenergetically comparable cyanide counterparts are anotherevidence for the proposed reaction path 1.
The Group 5 metal methylidene isocyanides are more agosticthan the Zr methylidene, but the extents of distortion arecomparable to those for the previously studied Group 5 metalmethylidene halides. The observed C-M stretching and CH2
wagging frequencies of the methylidene isocyanides are alsosimilar to those of the halide analogues, indicating that theeffects of the metal containing conjugation system are minor,parallel to the recent results for the Zr methylidene cyanide.The C-M and N-M bonds are shorter than those ofCH3CNfM and CH3-MNC but are longer than the C-M bondof CH2dM(H)NC, indicating that binding of the Group 5 metalatoms to the nitrile triple bond is considerably weaker than inthe previously studied Zr system,16 where the C-M and N-Mbonds are the shortest. It is also consistent with the recent reportthat the Group 4 metals form exceptionally strong bonds withacetylene due to the unusually strong back-donation from themetal center to the π*-orbitals.29
Acknowledgment. We gratefully acknowledge financialsupport from National Science Foundation (U.S.) Grant CHE03-52487 to L.A., and support from the Korea ResearchFoundation (KRF) grant funded by the Korean government(MEST) (No. 2009-0075428).
References and Notes
(1) (a) Chang, S.-C.; Hauge, R. H.; Kafafi, Z. H.; Margrave, J. L.;Billups, W. E. J. Am. Chem. Soc. 1988, 110, 7975. (b) de Almeida, K. J.;Cesar, A. Organometallics 2006, 25, 3407. (c) Bihlmeier, A.; Greene, T. M.;Himmel, H.-J. Organometallics 2004, 23, 2350.
(2) (a) Kline, E. S.; Kafafi, Z. H.; Hauge, R. H.; Margrave, J. L. J. Am.Chem. Soc. 1987, 109, 2402. (b) Horacek, M. H.; Hiller, J.; Thewalt, U.;Stepnicka, P.; Mach, K. J. Oranomet. Chem. 1998, 571, 77. Michelini,M. D. C.; Russo, N.; Alikhani, M. E.; Silvi, B. J. Comput. Chem. 2004,25, 1647; M + C2H2.
(3) (a) Siegbahn, P. E. M.; Blomberg, M. R. A.; Svensson, M. J. Am.Chem. Soc. 1993, 115, 1952. (b) Alikhani, M. E.; Hannachi, Y.; Manceron,L.; Bouteiller, Y. Chem. Phys. 1995, 103, 10128. (c) Cho, H.-G.; Andrews,L. J. Phys. Chem. A 2008, 112, 12071; M + C2H4.
(4) (a) Prusse, T.; Drewello, T.; Lebrilla, C. B.; Schwarz, H. J. Am.Chem. Soc. 1989, 111, 2857. (b) Legon, A. C.; Lister, D. G.; Warner, H. E.Angew. Chem., Int. Ed. Engl. 1992, 31, 202. (c) Purcell, K. F.; Drago, R. S.J. Am. Chem. Soc. 1966, 88, 919.
(5) (a) Wells, N. P.; Phillips, J. A. J. Phys. Chem. A 2002, 106, 1518.(b) Hattori, R.; Suzuki, E.; Shimizu, K. J. Mol. Struct. 2005, 750, 123.
(6) (a) Porembski, M.; Weisshaar, J. C. J. Phys. Chem. A 2000, 104,1524. (b) Willis, P. A.; Stauffer, H. U.; Hinrichs, R. Z.; Davis, H. F. J.Phys. Chem. A 1999, 103, 3706.
(7) (a) Andrews, L.; Cho, H.-G. Organometallics 2006, 25, 4040, andreferences therein (Review article, Groups 4-6. (b) Lyon, J. T.; Cho, H.-G.; Andrews, L. Organometallics 2007, 26, 2519; Ti, Zr, Hf + CHX3,CX4(c) Lyon, J. T.; Cho, H.-G.; Andrews, L. Organometallics 2007, 26,6373; Cr, Mo, W + CHX3, CX4(d) Cho, H.-G.; Andrews, L. J. Phys. Chem.A 2006, 110, 10063; Group 5 + CH3X.
(8) (a) Cho, H.-G.; Andrews, L. J. Phys. Chem. A 2007, 111, 2480.(b) Cho, H.-G.; Andrews, L. Organometallics 2007, 26, 633; Group 3.
(9) (a) Cho, H.-G.; Andrews, L. Organometallics 2007, 26, 4096. (b)Cho, H.-G.; Andrews, L. Inorg. Chem. 2008, 47, 1653; Re.
(10) (a) Cho, H.-G.; Lyon, J. T.; Andrews, L. Organometallics 2008,27, 5241. (b) Cho, H.-G.; Andrews, L. Eur. J. Inorg. Chem. 2008, 2537.(c) Cho, H.-G.; Andrews, L. Organometallics 2008, 27, 1786; Group 8.
(11) Cho, H.-G.; Andrews, L. Reactions of Group 9 Metals withHalomethanes, unpublished data.
(12) (a) Cho, H.-G.; Andrews, L. J. Am. Chem. Soc. 2008, 130, 15836.(b) Cho, H.-G.; Andrews, L. J. Phys. Chem. A 2008, 112, 12293. (c) Cho,H.-G.; Andrews, L. Organometallics 2009, 28, 1358. (d) Cho, H.-G.;Andrews, L.; Vlaisavljevich, B.; Gagliardi, L. Organometallics 2009, 28,5623; Group 10.
(13) (a) Andrews, L.; Cho, H.-G. J. Phys. Chem. A 2005, 109, 6796.(b) Cho, H.-G.; Lyon, J. T.; Andrews, L. J. Phys. Chem. A 2008, 112, 6902.(c) Lyon, J. T.; Cho, H.-G.; Andrews, L. Eur. J. Inorg. Chem. 2008, 1047;Actinides. (d) Cho, H.-G.; Andrews, L. Reactions of Lanthanides withMethane and Halomethanes, unpublished data.
(14) (a) von Frantzius, G.; Streubel, R.; Brandhorst, K.; Grunenberg, J.Organometallics 2006, 25, 118, and references therein. (b) Berkaine, N.;Reinhardt, P.; Alikhani, M. E. Chem. Phys. 2008, 343, 241. (c) Roos, B. O.;Lindh, R.; Cho, H.-G.; Andrews, L. J. Phys. Chem. A 2007, 111, 6420.Clot, E. ; Eisenstein in O. Computational Inorganic Chemistry; Kaltzoyannis,N.; McGrady, J. E. Eds.; Structure and Bonding; Springer: Heidelberg, 2004;pp 1-36. Aubert, C.; Buisine, O.; Malacria, M. Chem. ReV 2002, 102, 813,and references therein. (agostic structures).
(15) (a) Crabtree, R. H. Chem. ReV. 1995, 95, 987, and referencestherein. (b) Wada, K.; Craig, B.; Pamplin, C. B.; Legzdins, P.; Patrick,B. O.; Tsyba, I.; Bau, R. J. Am. Chem. Soc. 2003, 125, 7035. (c) Ujaque,G.; Cooper, A. C.; Maseras, F.; Eisenstein, O.; Caulton, K. G. J. Am. Chem.Soc. 1998, 120, 361.
(16) Cho, H.-G.; Andrews, L. J. Phys. Chem. A 2010, 114, 891.(17) (a) Andrews, L.; Citra, A. Chem. ReV 2002, 102, 885, and references
therein. (b) Andrews, L. Chem. Soc. ReV. 2004, 33, 123, and referencestherein. .
(18) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.;Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.;Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson,G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.;Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai,H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo,C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin,A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma,K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.;Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.;Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui,Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu,G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe,M.; Gill, P. M. W.; Johnson, vB.; Chen, vW.; Wong, M. W.; Gonzalez,vC.; Pople, J. A. Gaussian 03, ReVision C.02; Gaussian, Inc.: Walling-ford, CT, 2004.
(19) (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Lee, C.; Yang,Y.; Parr, R. G. Phys. ReV. B 1988, 37, 785.
(20) Raghavachari, K.; Trucks, G. W. J. Chem. Phys. 1989, 91, 1062.(21) Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H.
Theor. Chim. Acta 1990, 77, 123.(22) (a) Becke, A. D. Phys. ReV. A 1988, 38, 3098. (b) Burke, K.;
Perdew, J. P.; Wang, Y. In Electronic Density Functional Theory. RecentProgress and New Directions; Dobson, J. F., Vignale, G., Das, M. P., Ed.s;Plenum: 1998.
(23) (a) Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502. (b)Andersson, M. P.; Uvdal, P. L. J. Phys. Chem. A 2005, 109, 2937.
(24) (a) Maier, G.; Schmidt, C.; Reisenauer, H. P.; Endlein, E.;Becker, D.; Eckwert, J.; Hess, B. A.; Schaad, L. J. Chem. Ber. 1993,126, 2337. (b) Jacox, M. E. Chem. Phys. 1979, 43, 157. (c) Yang, X.;Maeda, S.; Ohno, K. J. Phys. Chem. A 2005, 109, 7319; Photolysis ofCH3CN. (d) Hattori, R.; Suzuki, E.; Shimizu, K. J. Mol. Struct. 2005,738, 165; CH3NC. (e) Deng, R.; Trenary, M. J. Phys. Chem. C 2007,111, 17088; HCCNH2.
(25) (a) Van Zee, R. J.; Weltner, W., Jr. J. Chem. Phys. 1995, 102,4367. (b) Wang, X.; Andrews, L. unpublished data. (Nb and Ta + H2).
Laser-Ablated Group 5 Metals Reacting with Acetonitrile J. Phys. Chem. A, Vol. 114, No. 19, 2010 6005

(26) (a) Petrie, S. Phys. Chem. Chem. Phys. 1999, 1, 2897. (b) Rayon,V. M.; Redondo, P.; Valdes, H.; Barrientos, C.; Largo, A. J. Phys. Chem.A 2007, 111, 6334. (c) Kafafi, Z. A.; Hauge, R. H.; Margrave, J. L.Polyhedron 1983, 2, 167; M(CN) isomerism.
(27) (a) Balasubrahmanyam, K.; Janz, G. J. J. Am. Chem. Soc. 1970,92, 4189. (b) Jamros, D.; Wojcik, M.; Lindgren, J.; Stangret, J. J. Phys.Chem. B 1997, 101, 6758. (c) Cha, J.-N.; Cheong, B.-S.; Cho, H.-G. J.Phys. Chem. A 2001, 105, 1789; solvation in CH3CN.
(28) (a) Garcia, J. J.; Brunkan, N. M.; Jones, W. D. J. Am. Chem. Soc.2002, 124, 9547. (b) Favero, G.; Morvillo, A.; Turco, A. J. Organomet.Chem. 1983, 241, 251; CN π-complex.
(29) Cho, H.-G.; Kushto, G. P.; Andrews, L.; Bauschlicher, C. W., JrJ. Phys. Chem. A 2008, 112, 6295.
JP1012686
6006 J. Phys. Chem. A, Vol. 114, No. 19, 2010 Cho and Andrews
Recommended