
Organic Synthesis
سنتز مواد آلی
به نام خدا
Dr Morteza MehrdadUniversity of Guilan, Department of Chemistry,
Rasht, [email protected]
2
C 1-1

2

Francis A. Carey
Department of Chemistry
Francis A. Carey is a native of Pennsylvania, educated in the public schools ofPhiladelphia, at Drexel University (B.S. in chemistry, 1959), and at Penn State (Ph.D.1963).Following postdoctoral work at Harvard and military service, he was appointed to thechemistry faculty of the University of Virginia in 1966.Prior to retiring in 2000, he regularly taught the two-semester lecture courses ingeneral chemistry and organic chemistry.With his students, Professor Carey has published over forty research papers insynthetic and mechanistic organic chemistry. In addition to this text, he is coauthor(with Robert C. Atkins) of Organic Chemistry: A Brief Course and (with Richard J.Sundberg) of Advanced Organic Chemistry, a two-volume treatment designed forgraduate students and advanced undergraduates. He was a member of the Committeeof Examiners of The Graduate Record Examination in Chemistry from 1993-2000.
University of Virginia

Richard J. Sundberg
Department of Chemistry
University of Virginia
Professor Sundberg is primarily engaged in teaching and chemical education.B.S. State University of Iowa, 1959Ph.D. University of Minnesota, 1962NIH Postdoctoral Fellow Stanford University, 1971-72Along with Francis A. Carey he is the author of “Advanced Organic Chemistry,” atwo-part text, which was recently published in its fifth edition.Professor Sundberg is also interested in synthetic methodology in heterocyclicchemistry and is the author of “Indoles” in the Best Synthetic Methods Series(Academic Press, 1996).

The focus of Part B is on the closely interrelated topics of reactions and synthesis.
We want to be able to answer questions such as:
What transformation does a reaction achieve?
What is the mechanism of the reaction?
What reagents and reaction conditions are typically used?
What substances can catalyze the reaction?
How sensitive is the reaction to other functional groups and the steric environment?
What factors control the stereoselectivity of the reaction?
Under what conditions is the reaction enantioselective?
5
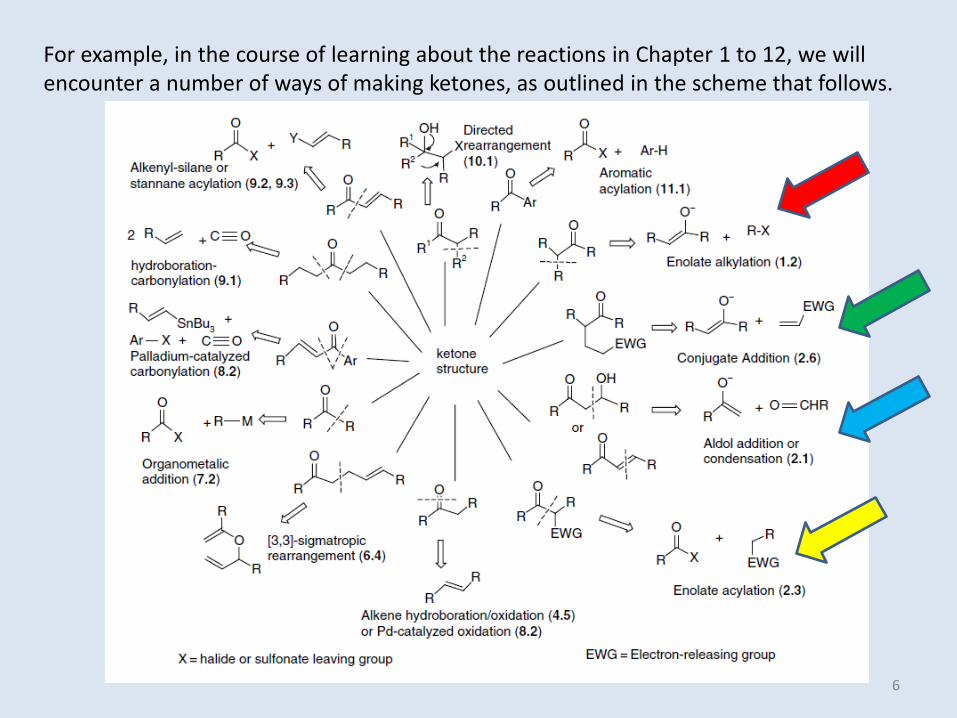
For example, in the course of learning about the reactions in Chapter 1 to 12, we will encounter a number of ways of making ketones, as outlined in the scheme that follows.
6

Part B emphasizes the most important reactions used in
organic synthesis. The material is organized by reaction
type.
Chapters 1 and 2 discuss the alkylation, conjugate
addition and carbonyl addition/condensation reactions of
enolates and other carbon nucleophiles.
7

8

Chapter 1. Alkylation of Enolates and Other Carbon Nucleophiles Introduction
1.1. Generation and Properties of Enolates and Other Stabilized Carbanions
1.1.1. Generation of Enolates by Deprotonation
1.1.2. Regioselectivity and Stereoselectivity in Enolate Formation from Ketone
and Esters
1.1.3. Other Means of Generating Enolates
1.1.4. Solvent Effects on Enolate Structure and Reactivity
1.2. Alkylation of Enolates
1.2.1. Alkylation of Highly Stabilized Enolates
1.2.2. Alkylation of Ketone Enolates
1.2.3. Alkylation of Aldehydes, Esters, Carboxylic Acids, Amides, and Nitriles
1.2.4. Generation and Alkylation of Dianions
1.2.5. Intramolecular Alkylation of Enolates
1.2.6. Control of Enantioselectivity in Alkylation Reactions
1.3. The Nitrogen Analogs of Enols and Enolates: Enamines and Imine Anions 9

Introduction
C-C bond formation is the basis for the construction of the molecular framework of organic molecules by synthesis.
One of the fundamental processes for C-Cbond formation is a reaction between a nucleophilic and an electrophilic carbon.
Reactions of C-nucleophile(enolates, imine anions, and enamines) with alkylating agents.
10

Crucial Factor for C-C bond formationby SN2 reaction
(1) the condition for generation of the carbonnucleophile
(2) the effect of the reaction conditions on thestructure and reactivity of the nucleophile
(3) the regio- and stereoselectivity of the alkylationreaction
11
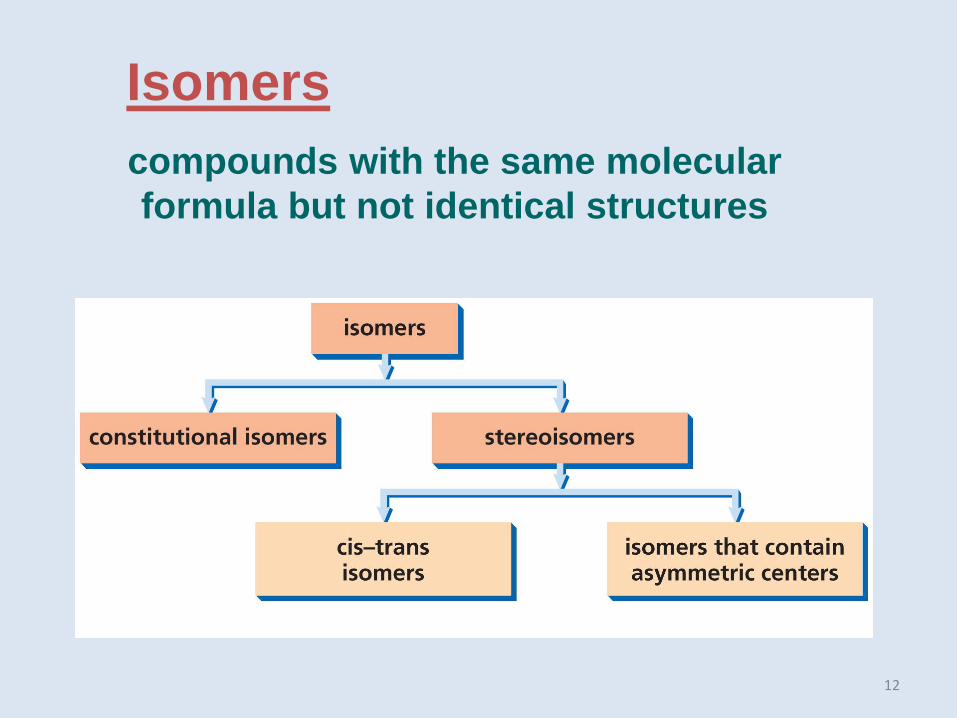
Isomers
compounds with the same molecular
formula but not identical structures
12

Constitutional Isomersایزومرهای ساختمانی
Different order of connections gives different carbon backboneand/or different functional groups
13

Stereochemistry of ReactionsRegioselective Reactions واکنشهای جهت گزین - reaction in which two constitutional
isomers can be obtained as products, but more of one is obtained than the other –
regioselectivity selects for a particular constitutional isomer
Stereoselective Reactions واکنشهای فضاگزین - reaction in which two stereoisomers
can be obtained as products, but more of one is obtained than the other –
stereoselectivity selects for a particular stereoisomer
Stereospecific Reactions واکنشهای فضاویژه- reaction in which each reactant stereo-
isomer forms a different stereoisomeric product or a different set of stereoisomeric
products
All stereospecific reactions are
stereoselective, but stereo-
selective reactions are not
necessarily stereospecific. 14
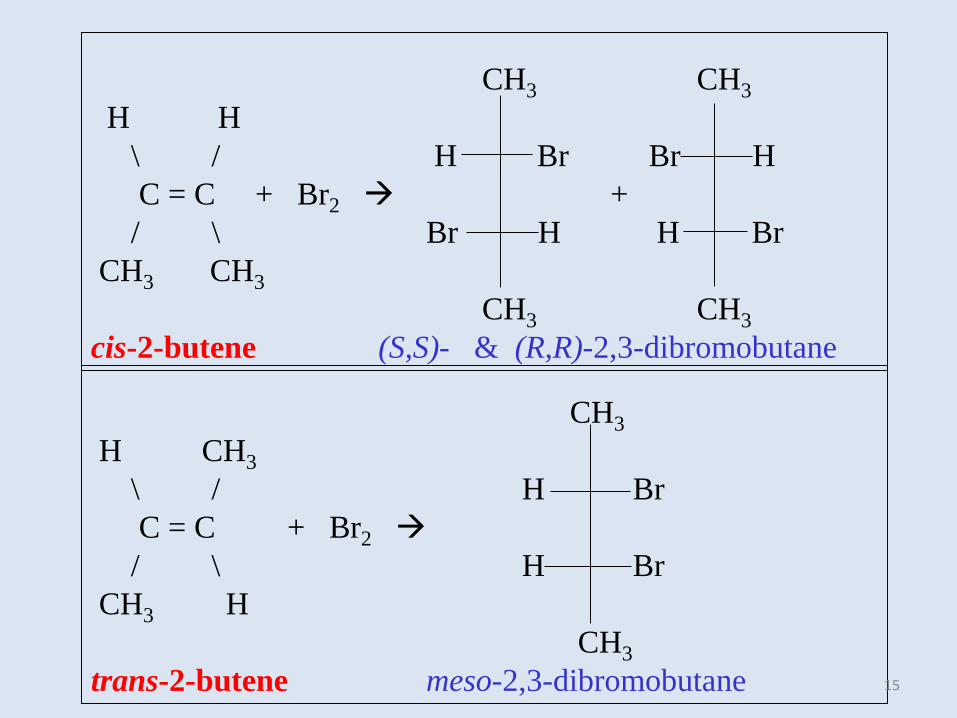
CH3
H CH3
\ / H Br
C = C + Br2
/ \ H Br
CH3 H
CH3
trans-2-butene meso-2,3-dibromobutane
CH3 CH3
H H
\ / H Br Br H
C = C + Br2 +
/ \ Br H H Br
CH3 CH3
CH3 CH3
cis-2-butene (S,S)- & (R,R)-2,3-dibromobutane
15

16
Addition of non-symmetrical reagent to a non-symmetrical
alkene, then two isomeric products that are constitutional
isomers can be obtained.
For example,
the reaction of HCl with propene gives 1-chloropropane
and 2-chloropropane.
Normally, 2-chloropropane is the major product.
Since one product is favoured over the other, the reaction
is said to be regioselective.
If 2-chloropropane were the only product then the reaction
is said to be regiospecific

زاشیمیدانهاهورمونیادارویکسنتزبرایStereoselectiveفضاگزینواکنشهای Reactionsاستفاده
هکآورندبدستراایزومریفقطکهکنندمیخودازبیولوژیکیسیستمدرایزومرآن
.باشدداشتهStereospecificityفضاویژگیفعالیت
17
Stereospecific: A term indicating that only a
single stereoisomer is produced in a given
reaction rather than a mixture.
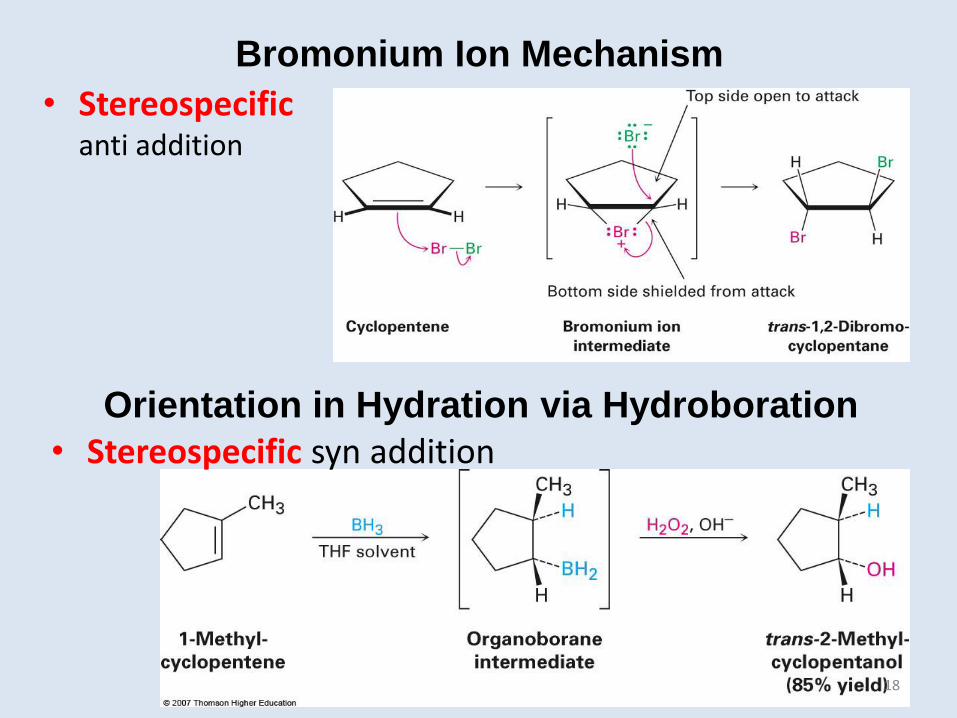
Bromonium Ion Mechanism
• Stereospecific anti addition
18
Orientation in Hydration via Hydroboration
• Stereospecific syn addition
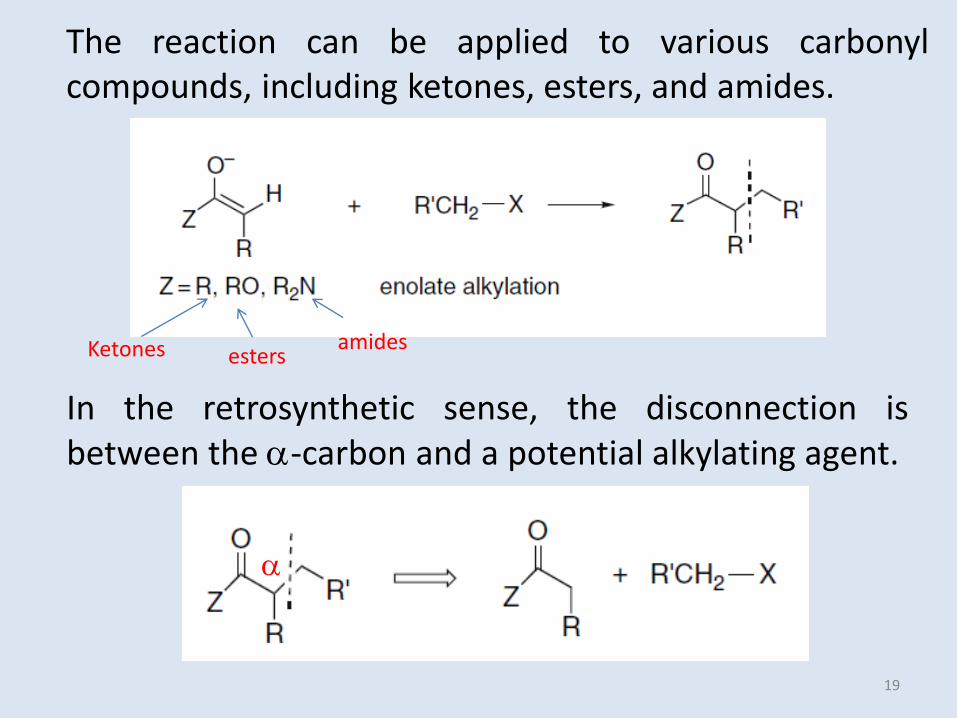
The reaction can be applied to various carbonylcompounds, including ketones, esters, and amides.
In the retrosynthetic sense, the disconnection isbetween the -carbon and a potential alkylating agent.
amidesKetones esters
19
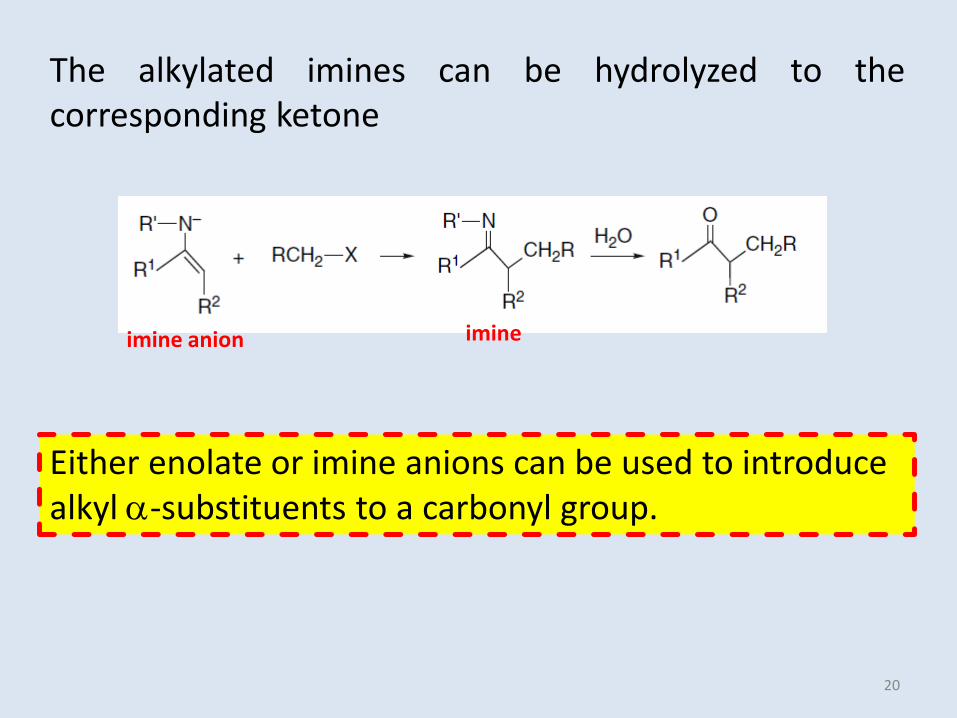
imine anion
The alkylated imines can be hydrolyzed to thecorresponding ketone
Either enolate or imine anions can be used to introduce alkyl -substituents to a carbonyl group.
imine
20

In the present chapter we relate the properties and
reactivity of carbanions stabilized by carbonyl and
other EWG substituents to their application as
nucleophiles in synthesis
there is a fundamental relationship between the
stabilizing functional group and the acidity of the
C−H groups, as illustrated by the pK data
21

1) pK data
the stability and reactivity of carbanions.
(The acidity of the reactant determines which bases can
be used for generation of the anion.)
2) distinction between
kinetic or thermodynamic control
of enolate formation by deprotonation
which determines the enolate composition.
22

Generation of an enolate or other stabilizedcarbanion by deprotonation
• under conditions in which the enolate is in
equilibrium with its conjugate acid or under
which the reactant is completely converted to its
conjugate base
(The key determinant is the amount and strength
of the base)
23

The base must be derived from a substantiallyweaker acid than the reactant.
Or the reagent must be a stronger base thanthe anion of the reactant.
Most current procedures for alkylation ofenolates and other carbanions involvecomplete conversion to the anion.
The solvent and other coordinating orchelating additives also have strong effects onthe structure and reactivity of carbanionsformed by deprotonation.
24
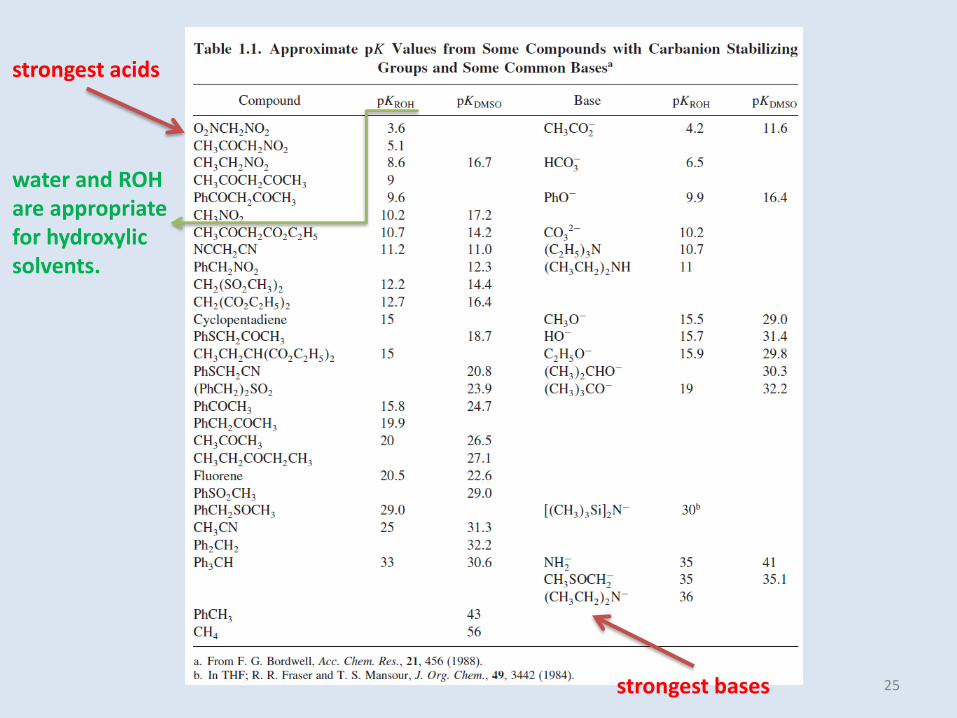
strongest acids
strongest bases
water and ROH are appropriate for hydroxylicsolvents.
25

Ability to stablize carbanion:
26
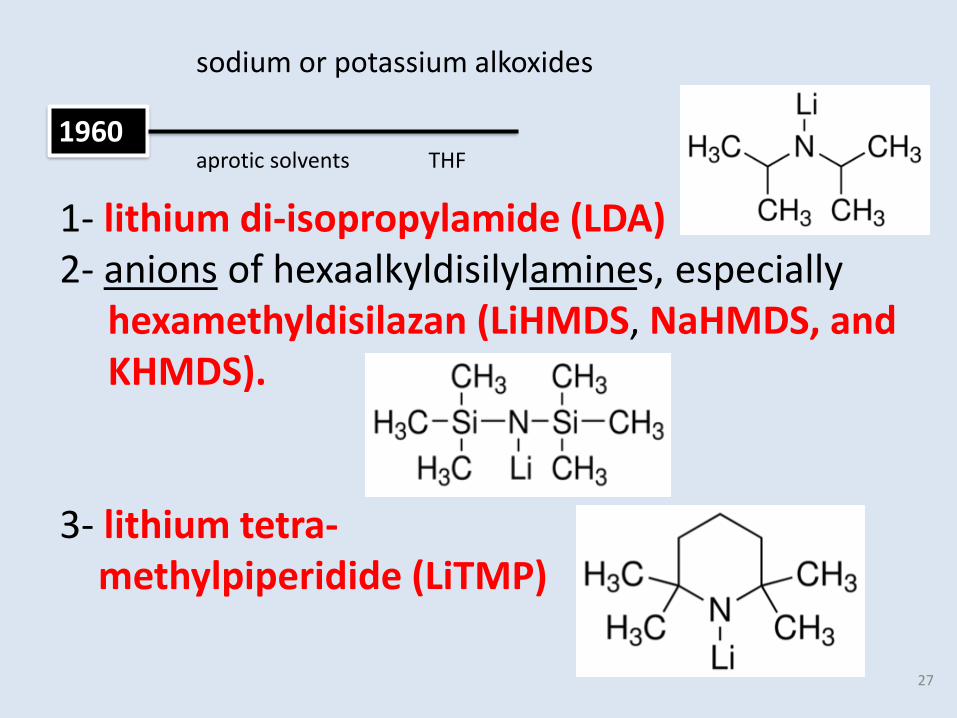
1960
sodium or potassium alkoxides
aprotic solvents
1- lithium di-isopropylamide (LDA)2- anions of hexaalkyldisilylamines, especially
hexamethyldisilazan (LiHMDS, NaHMDS, andKHMDS).
3- lithium tetra-methylpiperidide (LiTMP)
THF
27
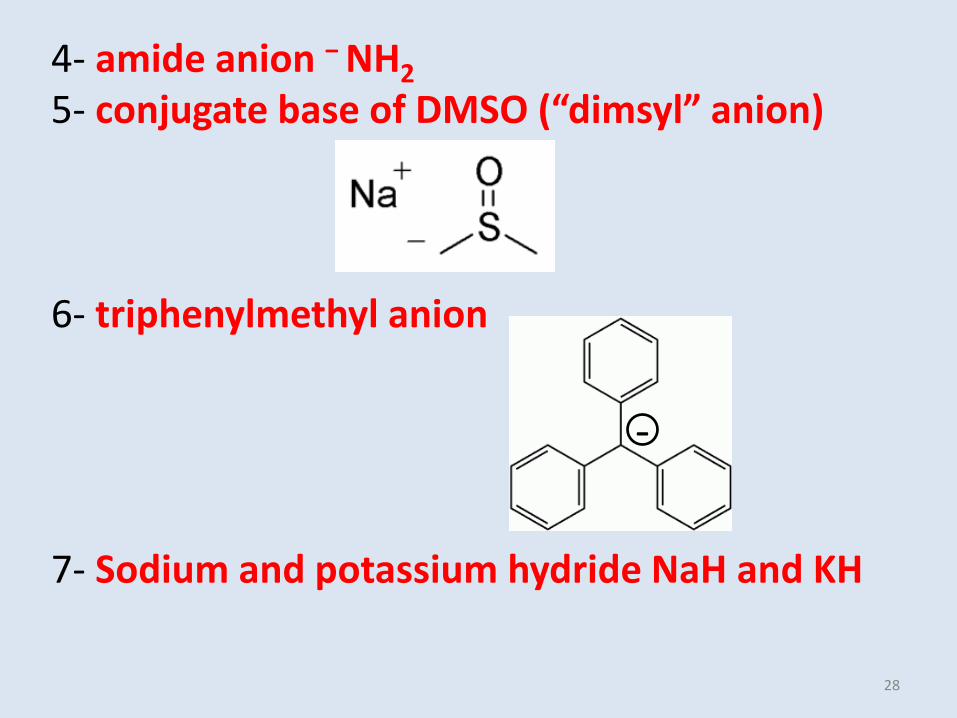
4- amide anion − NH2
5- conjugate base of DMSO (“dimsyl” anion)
6- triphenylmethyl anion
7- Sodium and potassium hydride NaH and KH
28
-

For a carbon acid C−H and a base B−H,
29
By comparing the approximate pK values of the bases with those of the carbon acid of interest,
it is possibleto estimate the position of the acid-base equilibrium for a given
reactant-base combination
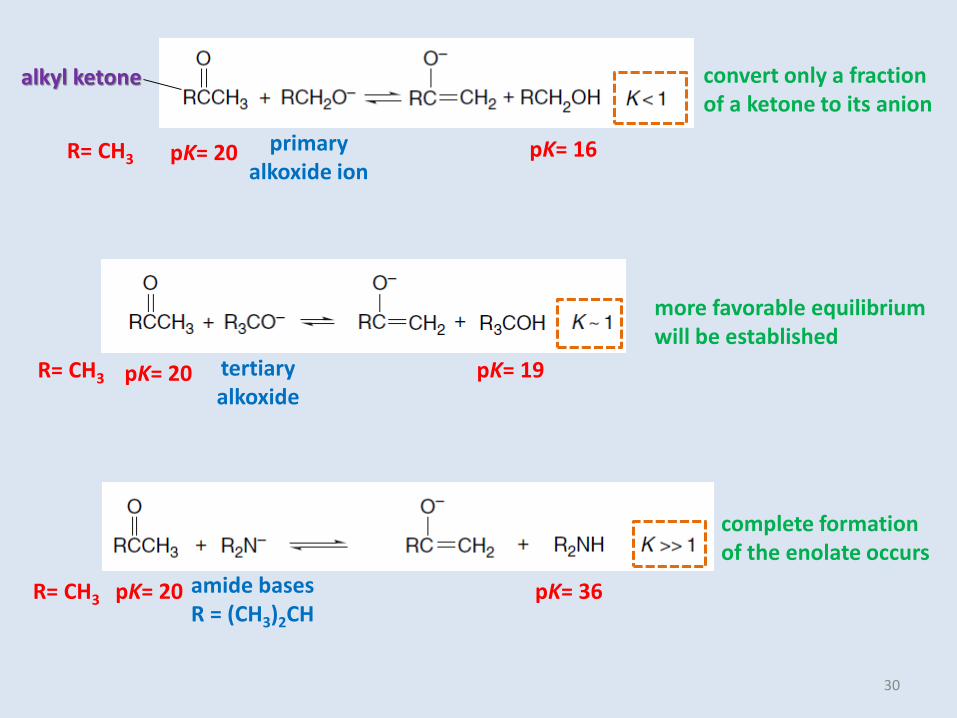
R= CH3
alkyl ketone
primary alkoxide ion
convert only a fraction of a ketone to its anion
pK= 20 pK= 16
tertiary alkoxide
more favorable equilibrium will be established
R= CH3 pK= 20 pK= 19
amide basesR = (CH3)2CH
complete formation of the enolate occurs
R= CH3 pK= 20 pK= 36
30
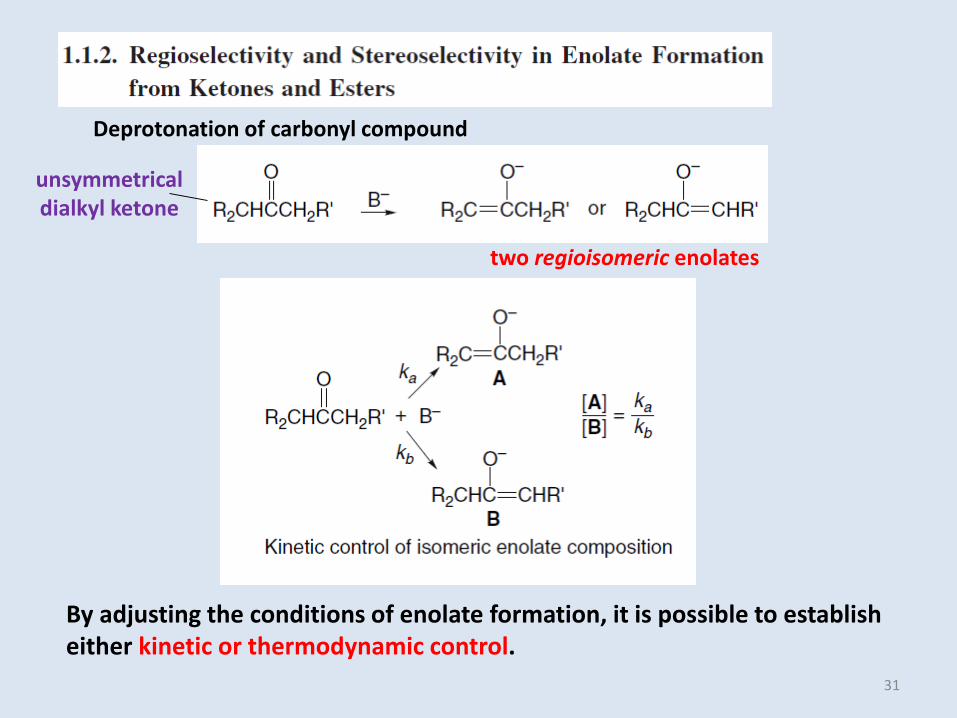
Deprotonation of carbonyl compound
unsymmetrical dialkyl ketone
two regioisomeric enolates
By adjusting the conditions of enolate formation, it is possible to establisheither kinetic or thermodynamic control.
31

Conditions for kinetic control of enolate formation are those in
which deprotonation is:
- rapid,
- quantitative,
- and irreversible
This requirement is met experimentally by using:
- a very strong base such as LDA or LiHMDS
- in an aprotic solvent
- in the absence of excess ketone.
32

Lithium is a better counterion than sodium or potassium forregioselective generation of the kinetic enolate,as it maintains a tighter coordination at oxygen and reduces therate of proton exchange.
Use of an aprotic solvent is essentialbecause protic solvents permit enolate equilibration by reversibleprotonation-deprotonation,which gives rise to the thermodynamically controlled enolatecomposition.
Excess ketone also catalyzes the equilibration by protonexchange.
33

Conditions of kinetic control usually favor formation of the less
substituted enolate, especially for methyl ketones.
The main reason for this result is that
removal of a less hindered hydrogen is faster, for steric reasons,
than removal of a more hindered hydrogen.
Steric factors in ketone deprotonation are accentuated by using
bulky bases
34
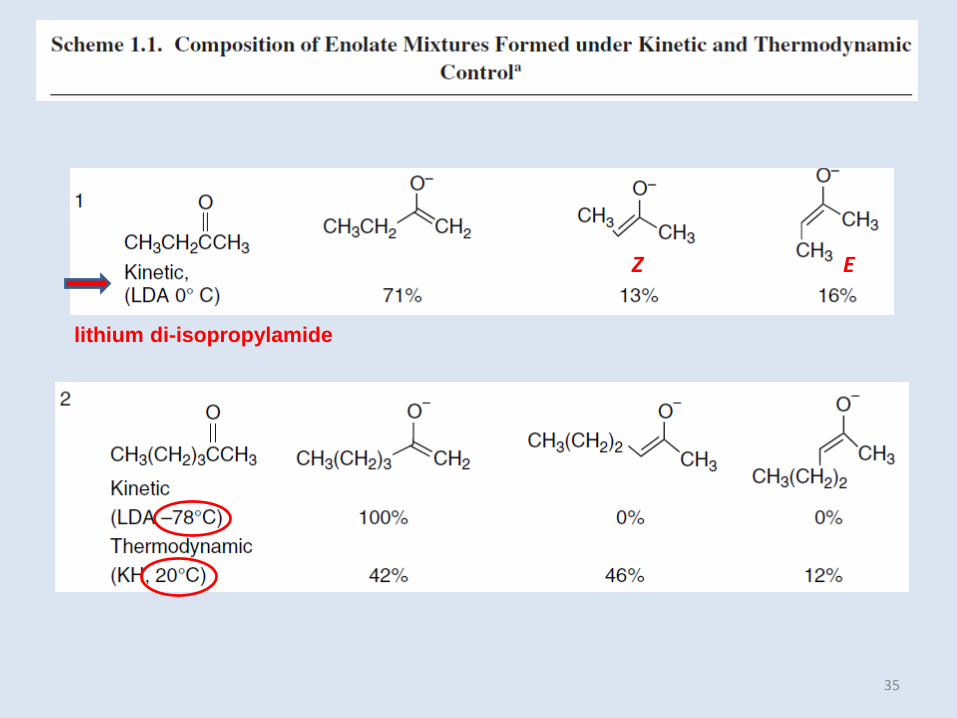
35
lithium di-isopropylamide
EZ
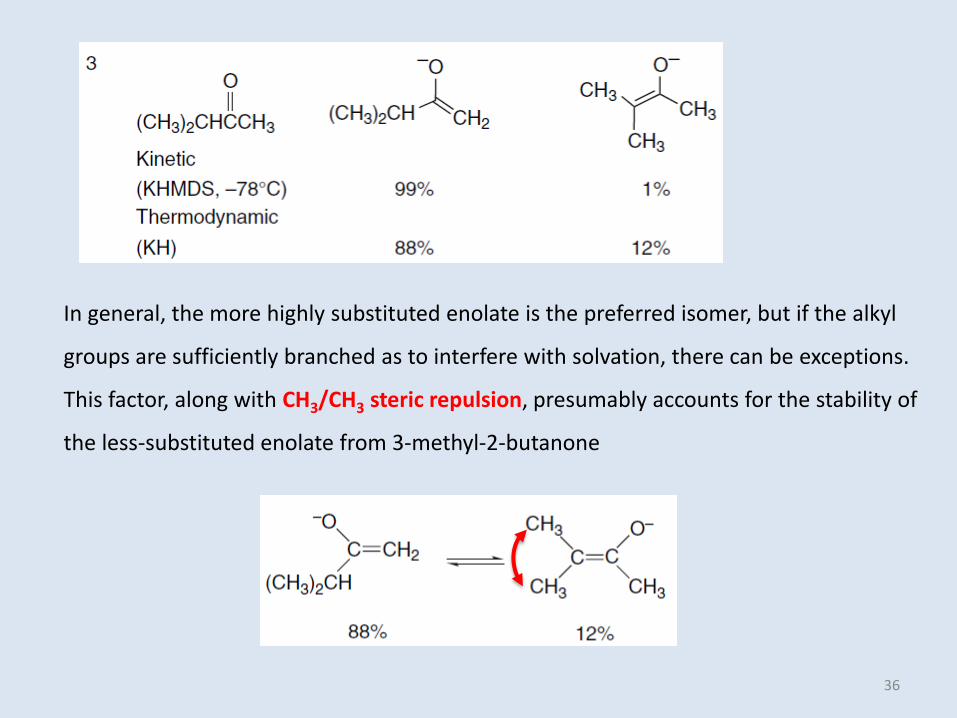
In general, the more highly substituted enolate is the preferred isomer, but if the alkyl
groups are sufficiently branched as to interfere with solvation, there can be exceptions.
This factor, along with CH3/CH3 steric repulsion, presumably accounts for the stability of
the less-substituted enolate from 3-methyl-2-butanone
36
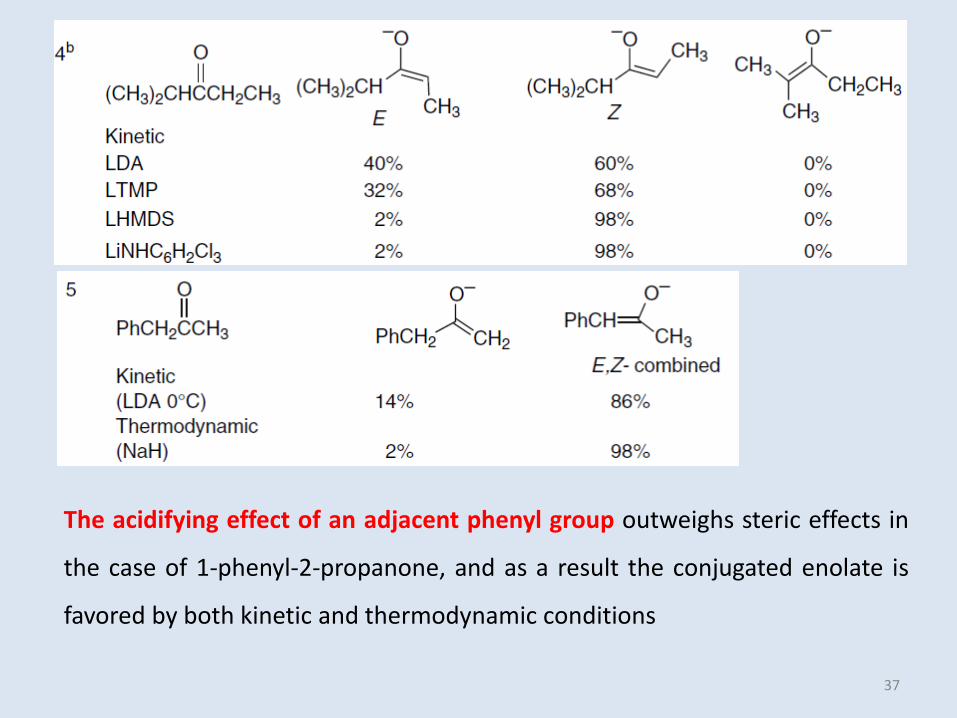
The acidifying effect of an adjacent phenyl group outweighs steric effects in
the case of 1-phenyl-2-propanone, and as a result the conjugated enolate is
favored by both kinetic and thermodynamic conditions
37
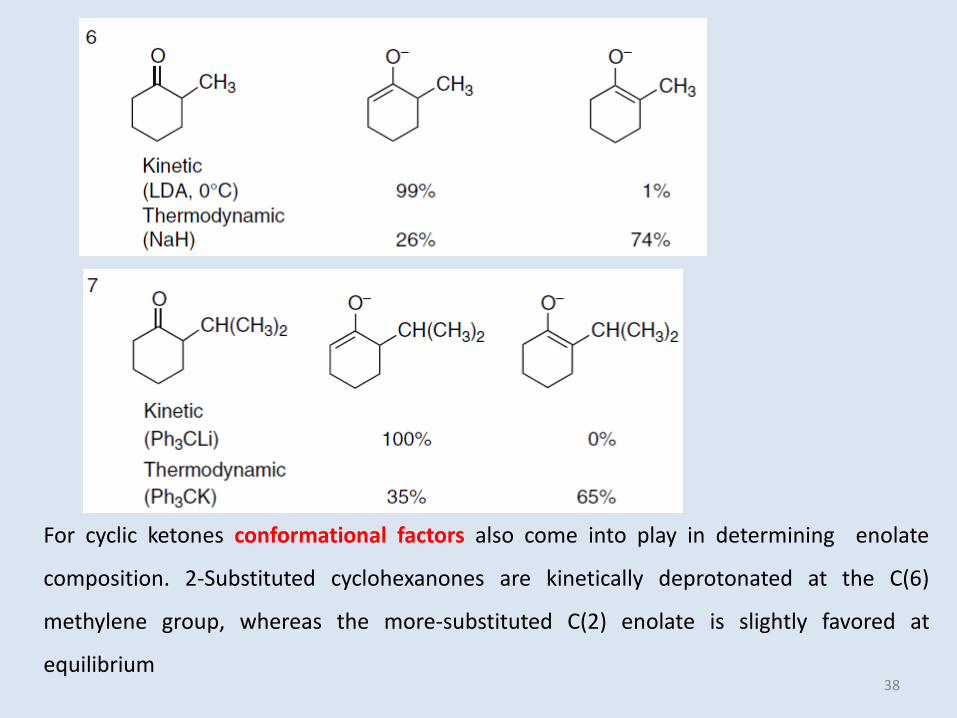
For cyclic ketones conformational factors also come into play in determining enolate
composition. 2-Substituted cyclohexanones are kinetically deprotonated at the C(6)
methylene group, whereas the more-substituted C(2) enolate is slightly favored at
equilibrium38
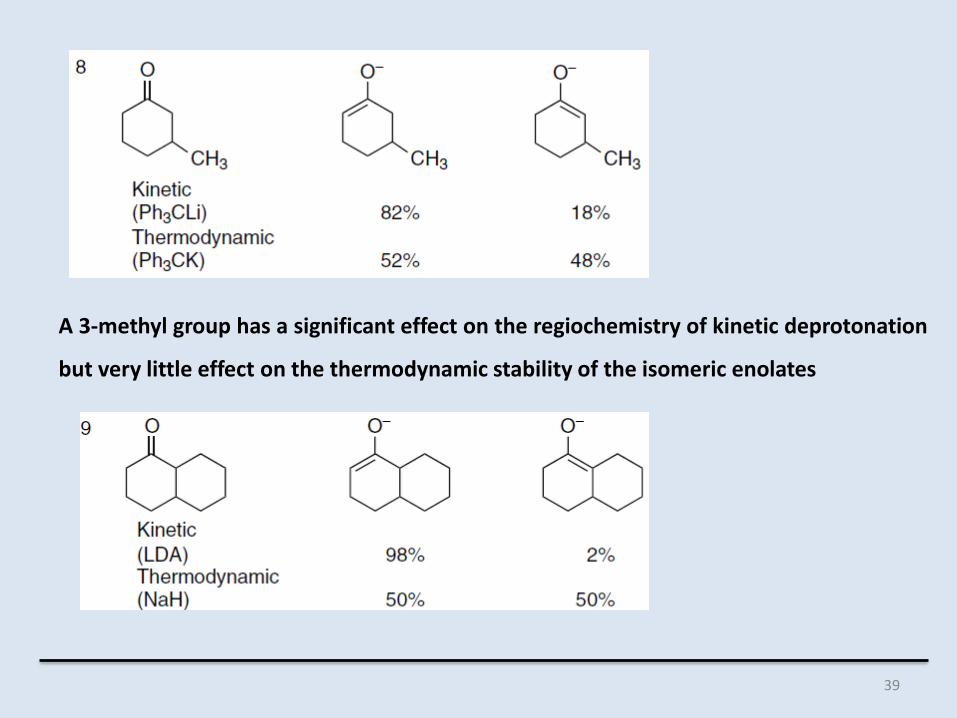
A 3-methyl group has a significant effect on the regiochemistry of kinetic deprotonation
but very little effect on the thermodynamic stability of the isomeric enolates
39
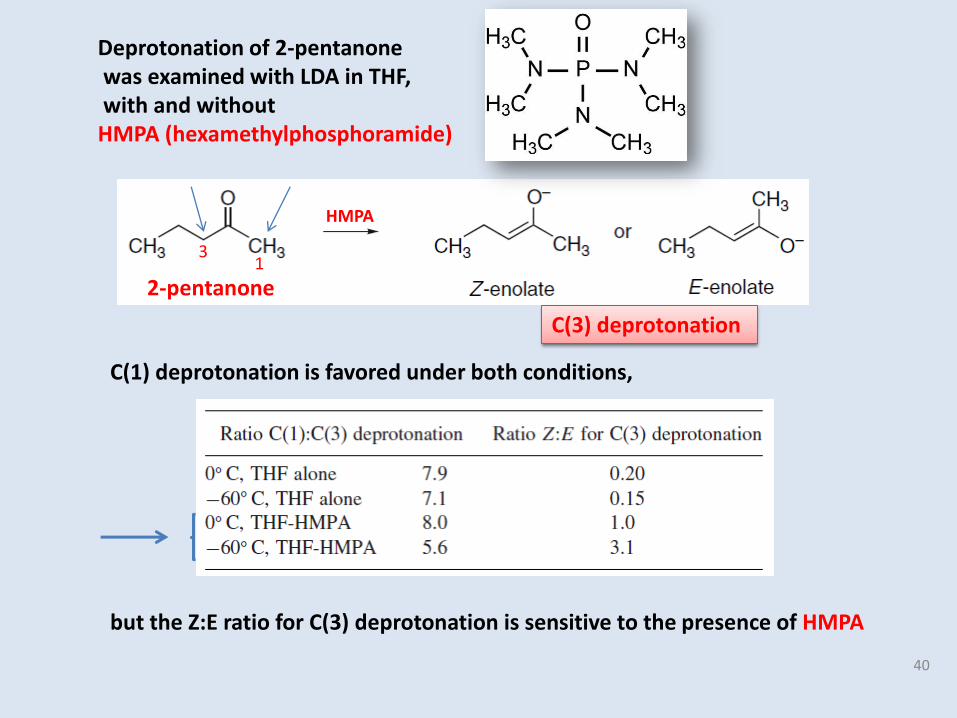
C(1) deprotonation is favored under both conditions,
Deprotonation of 2-pentanonewas examined with LDA in THF,with and without
HMPA (hexamethylphosphoramide)
2-pentanone1
3
40
C(3) deprotonation
HMPA
but the Z:E ratio for C(3) deprotonation is sensitive to the presence of HMPA
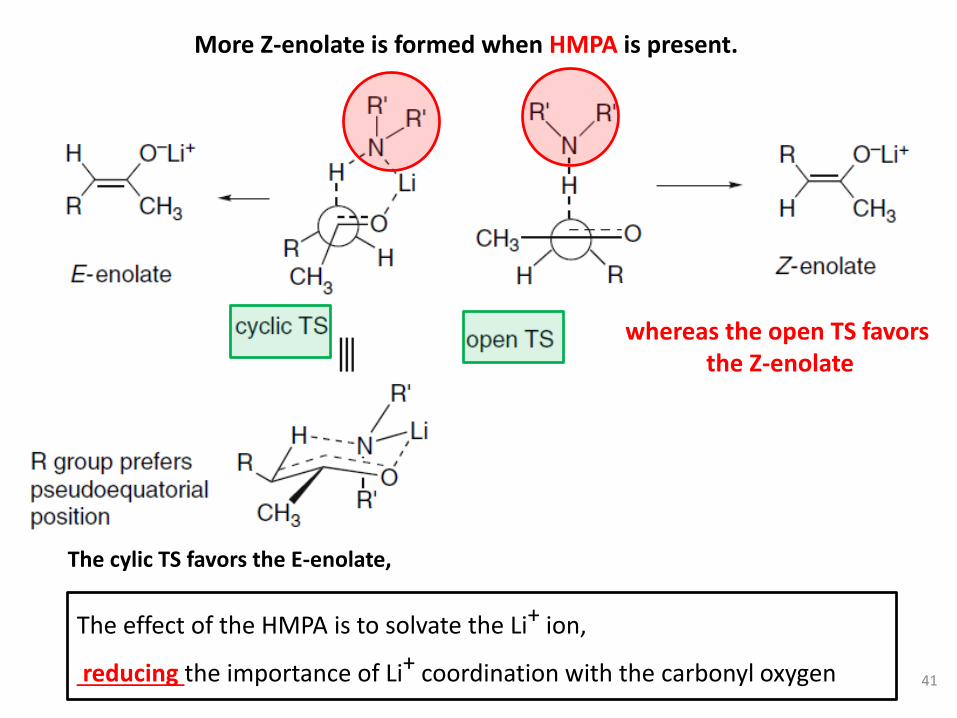
whereas the open TS favorsthe Z-enolate
The effect of the HMPA is to solvate the Li+ ion,
reducing the importance of Li+ coordination with the carbonyl oxygen
More Z-enolate is formed when HMPA is present.
The cylic TS favors the E-enolate,
41
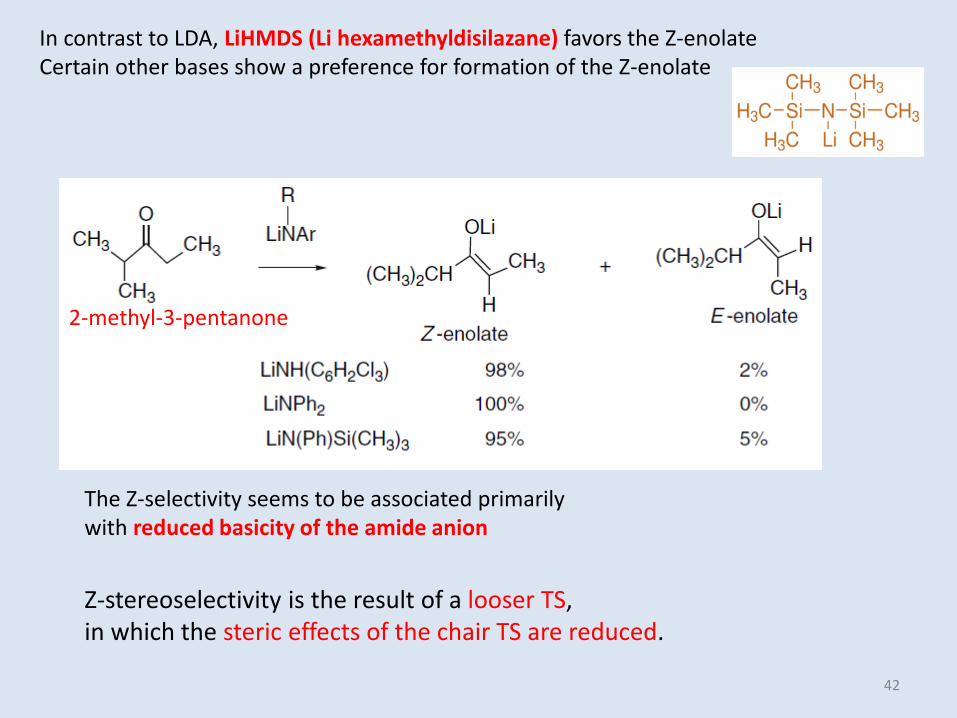
In contrast to LDA, LiHMDS (Li hexamethyldisilazane) favors the Z-enolateCertain other bases show a preference for formation of the Z-enolate
The Z-selectivity seems to be associated primarily with reduced basicity of the amide anion
2-methyl-3-pentanone
Z-stereoselectivity is the result of a looser TS,in which the steric effects of the chair TS are reduced.
42
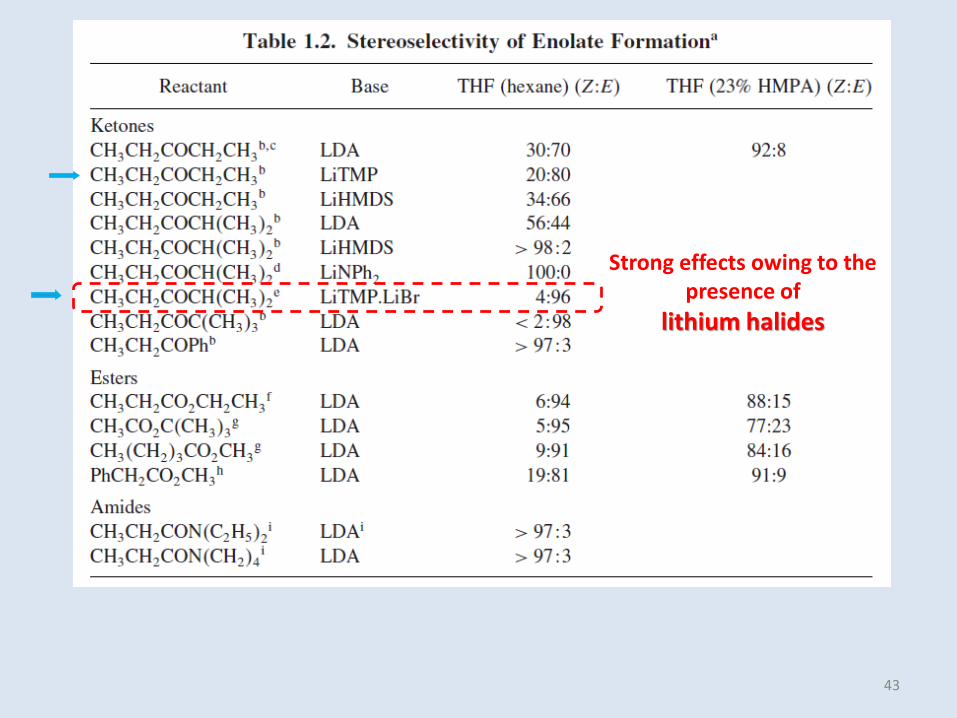
Strong effects owing to the presence of
lithium halides
43
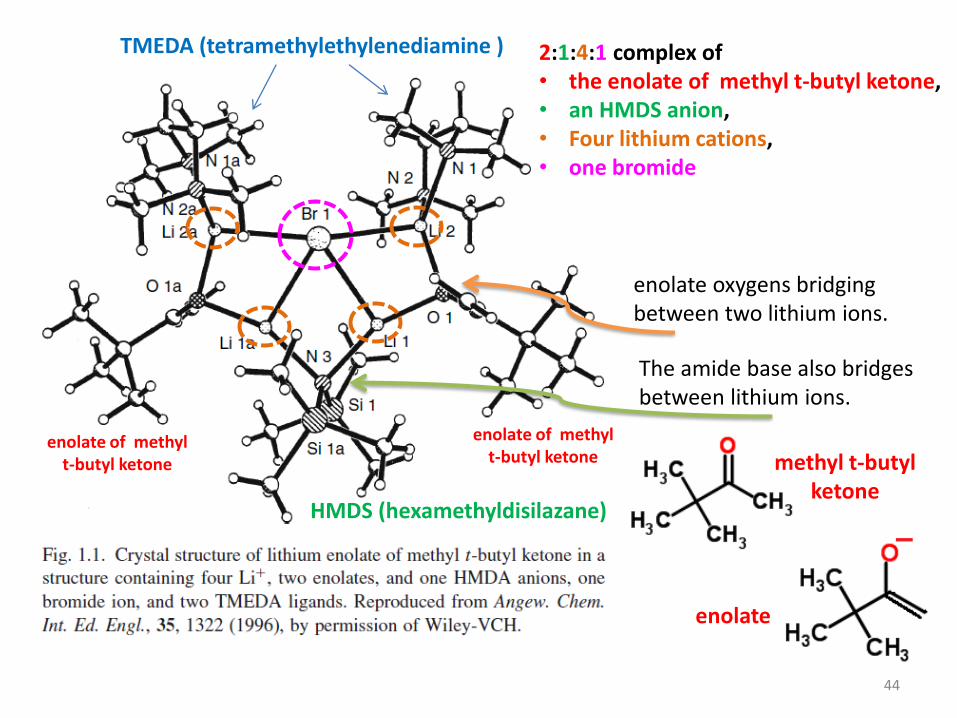
2:1:4:1 complex of• the enolate of methyl t-butyl ketone, • an HMDS anion, • Four lithium cations, • one bromide
HMDS (hexamethyldisilazane)
TMEDA (tetramethylethylenediamine )
enolate oxygens bridging between two lithium ions.
enolate of methyl t-butyl ketone
enolate of methyl t-butyl ketone
The amide base also bridges between lithium ions.
44
methyl t-butyl ketone
enolate
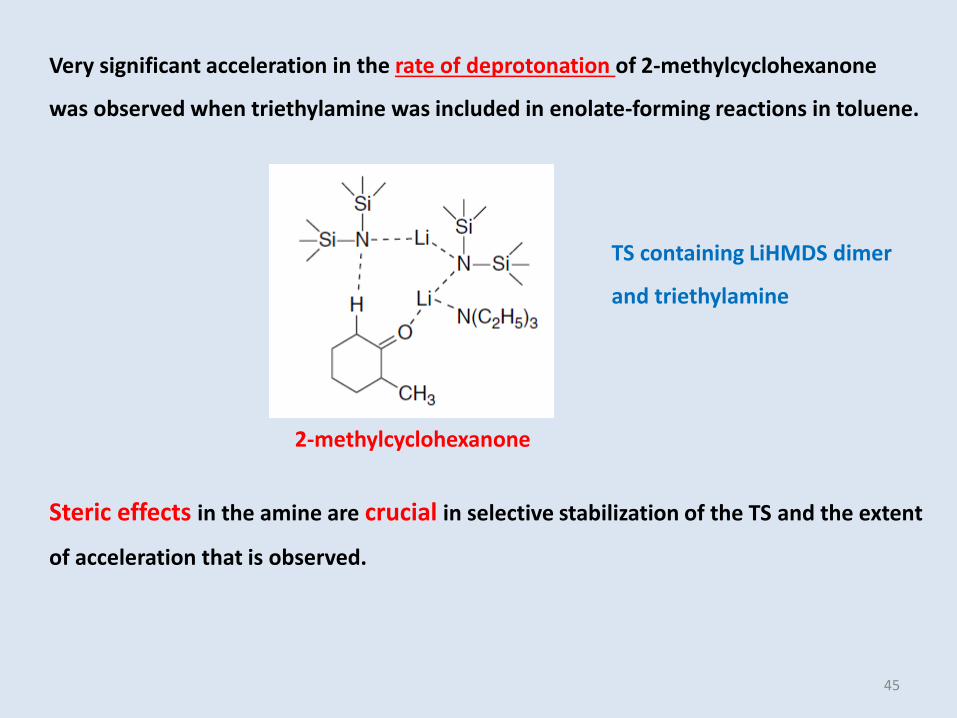
Very significant acceleration in the rate of deprotonation of 2-methylcyclohexanone
was observed when triethylamine was included in enolate-forming reactions in toluene.
Steric effects in the amine are crucial in selective stabilization of the TS and the extent
of acceleration that is observed.
2-methylcyclohexanone
TS containing LiHMDS dimer
and triethylamine
45

in Table 1.2, The switch from E to Z in the presence of HMPA (hexamethylphosphoramide)is particularly prominent for ester enolates
2-methyl-3-pentanone and ethyl propanoate, good selectivity is possible for both stereoisomers.
There are several important factors in determining regio- and stereoselectivity in enolate formation:- The strength of the base, - The identity of the cation, - The nature of the solvent and additives.
46
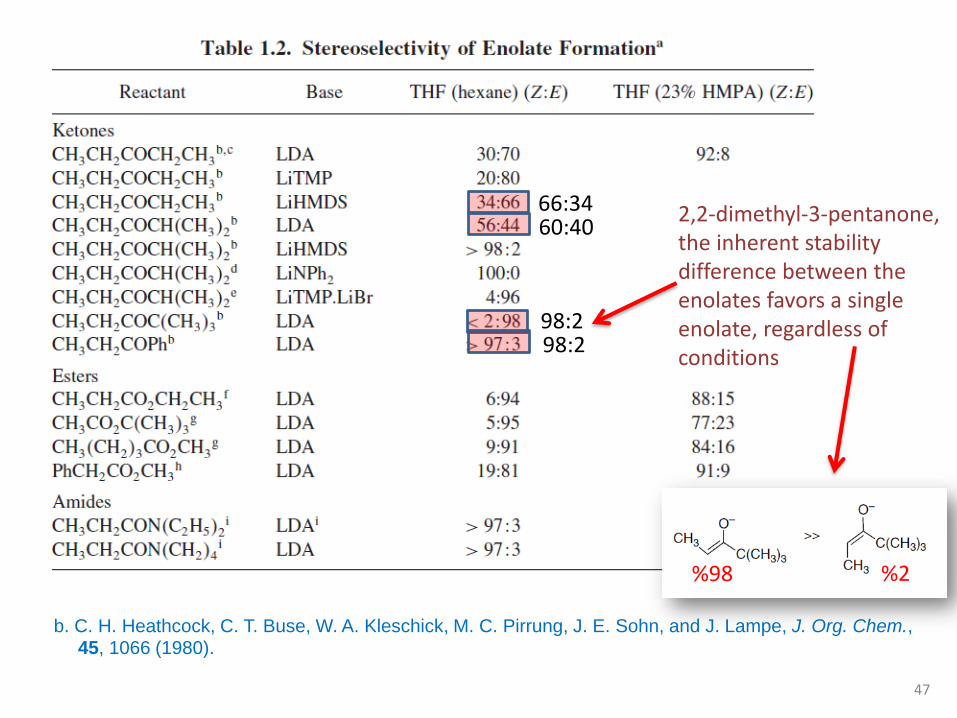
2,2-dimethyl-3-pentanone, the inherent stability difference between the enolates favors a single enolate, regardless of conditions
47
b. C. H. Heathcock, C. T. Buse, W. A. Kleschick, M. C. Pirrung, J. E. Sohn, and J. Lampe, J. Org. Chem., 45, 1066 (1980).
%98 %2
66:3460:40
98:298:2
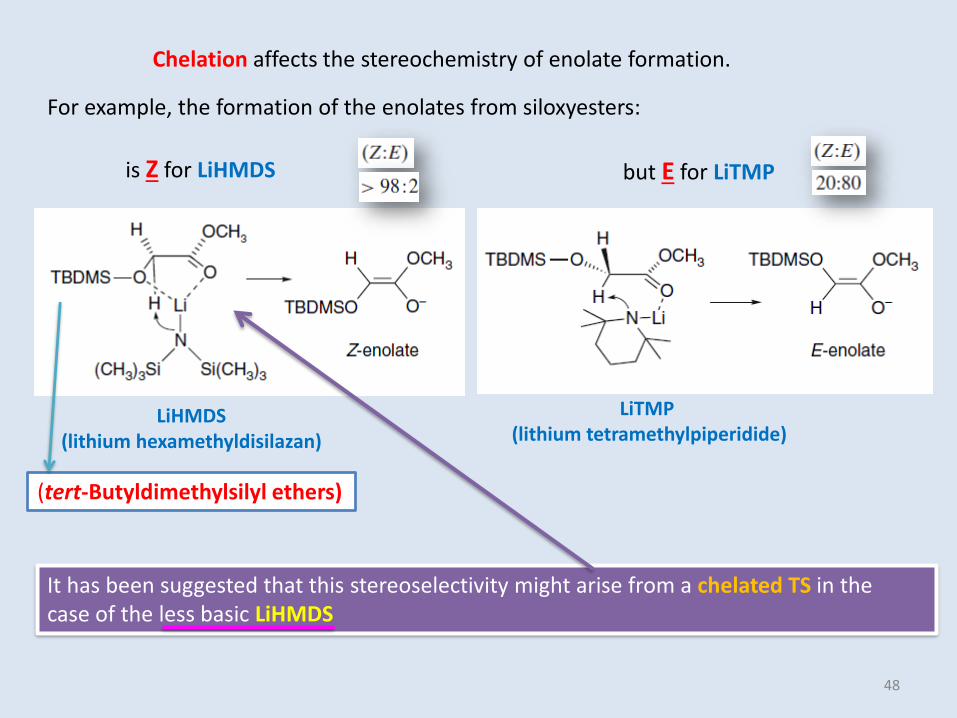
For example, the formation of the enolates from siloxyesters:
It has been suggested that this stereoselectivity might arise from a chelated TS in thecase of the less basic LiHMDS
(tert-Butyldimethylsilyl ethers)
Chelation affects the stereochemistry of enolate formation.
is Z for LiHMDS but E for LiTMP
LiTMP(lithium tetramethylpiperidide)
LiHMDS(lithium hexamethyldisilazan)
48
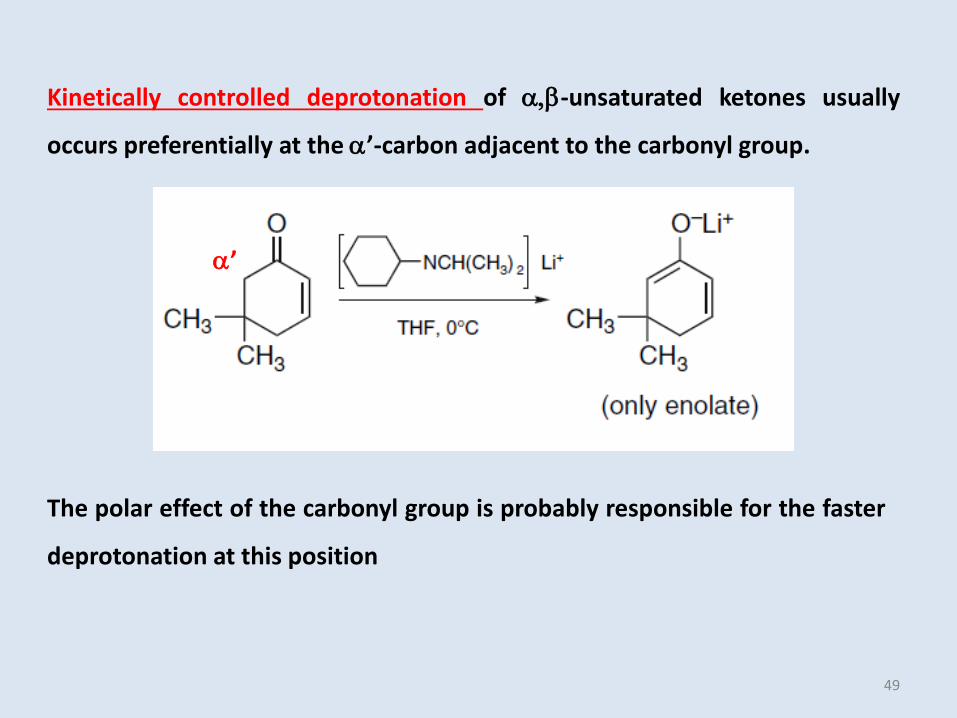
Kinetically controlled deprotonation of ,b-unsaturated ketones usually
occurs preferentially at the ’-carbon adjacent to the carbonyl group.
’
The polar effect of the carbonyl group is probably responsible for the faster
deprotonation at this position
49
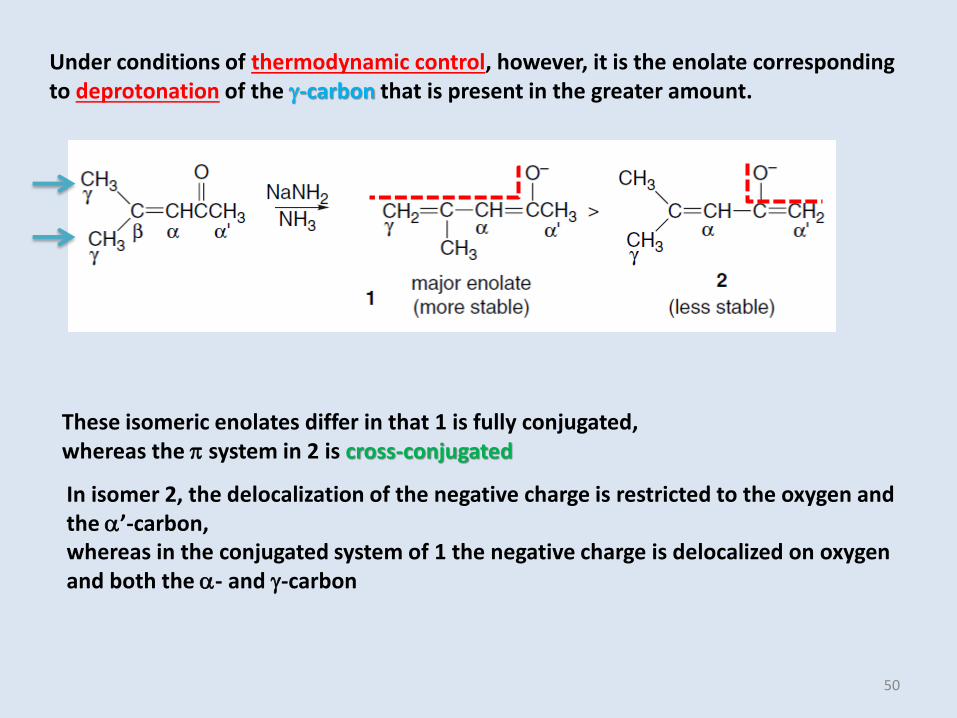
Under conditions of thermodynamic control, however, it is the enolate corresponding to deprotonation of the g-carbon that is present in the greater amount.
These isomeric enolates differ in that 1 is fully conjugated, whereas the p system in 2 is cross-conjugated
In isomer 2, the delocalization of the negative charge is restricted to the oxygen and the ’-carbon, whereas in the conjugated system of 1 the negative charge is delocalized on oxygen and both the - and g-carbon
50
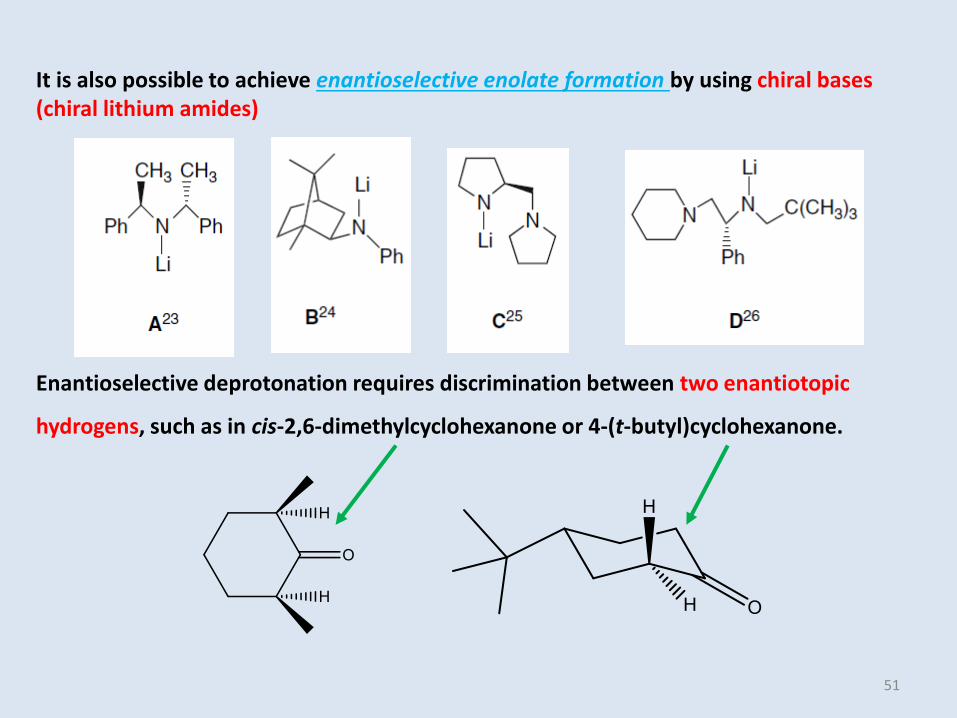
It is also possible to achieve enantioselective enolate formation by using chiral bases(chiral lithium amides)
Enantioselective deprotonation requires discrimination between two enantiotopic
hydrogens, such as in cis-2,6-dimethylcyclohexanone or 4-(t-butyl)cyclohexanone.
51
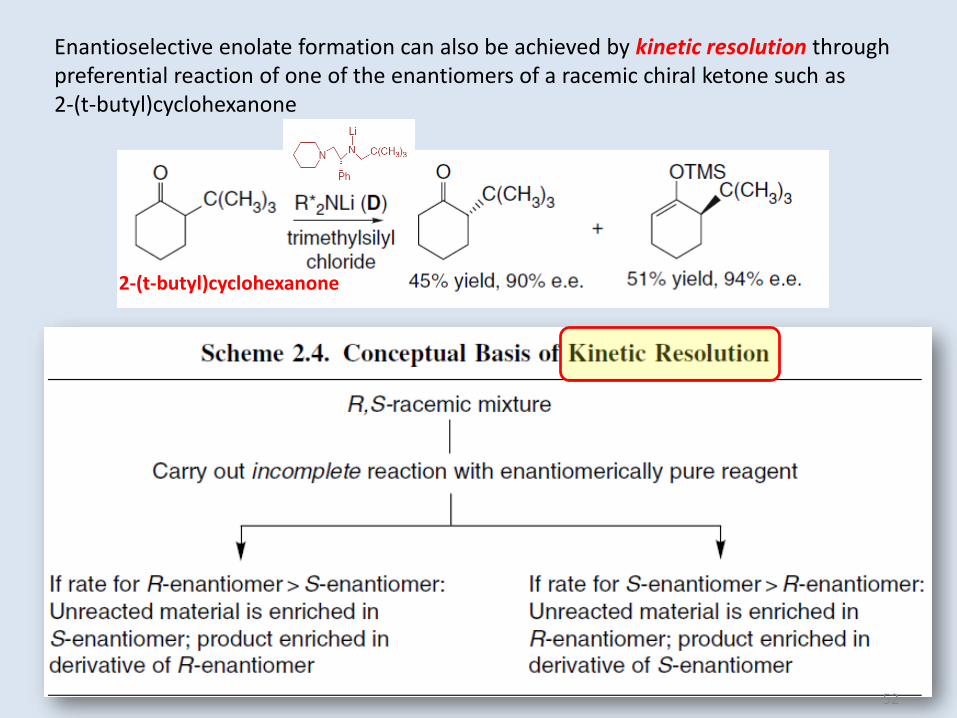
Enantioselective enolate formation can also be achieved by kinetic resolution throughpreferential reaction of one of the enantiomers of a racemic chiral ketone such as2-(t-butyl)cyclohexanone
52
2-(t-butyl)cyclohexanone
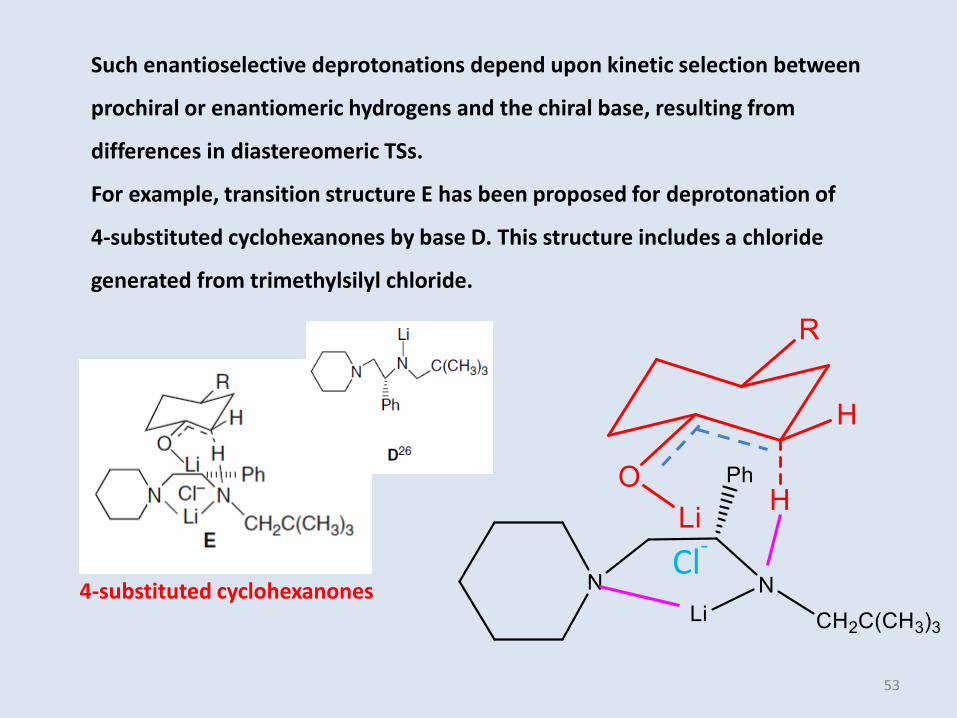
Such enantioselective deprotonations depend upon kinetic selection between
prochiral or enantiomeric hydrogens and the chiral base, resulting from
differences in diastereomeric TSs.
For example, transition structure E has been proposed for deprotonation of
4-substituted cyclohexanones by base D. This structure includes a chloride
generated from trimethylsilyl chloride.
Cl-
53
4-substituted cyclohexanones
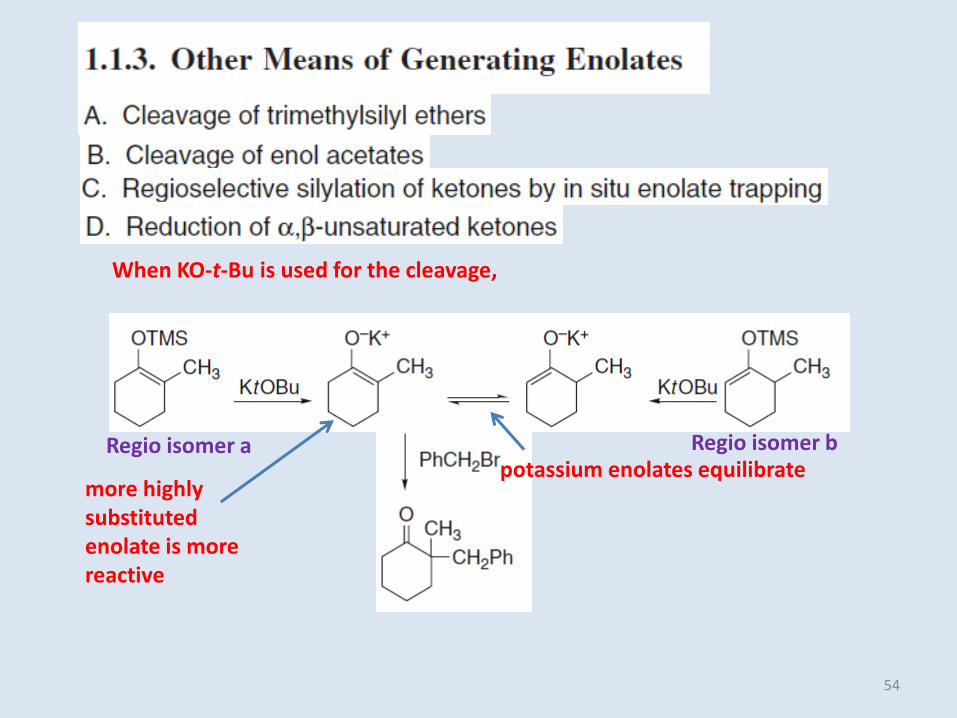
potassium enolates equilibratemore highly substituted enolate is more reactive
When KO-t-Bu is used for the cleavage,
Regio isomer a Regio isomer b
54
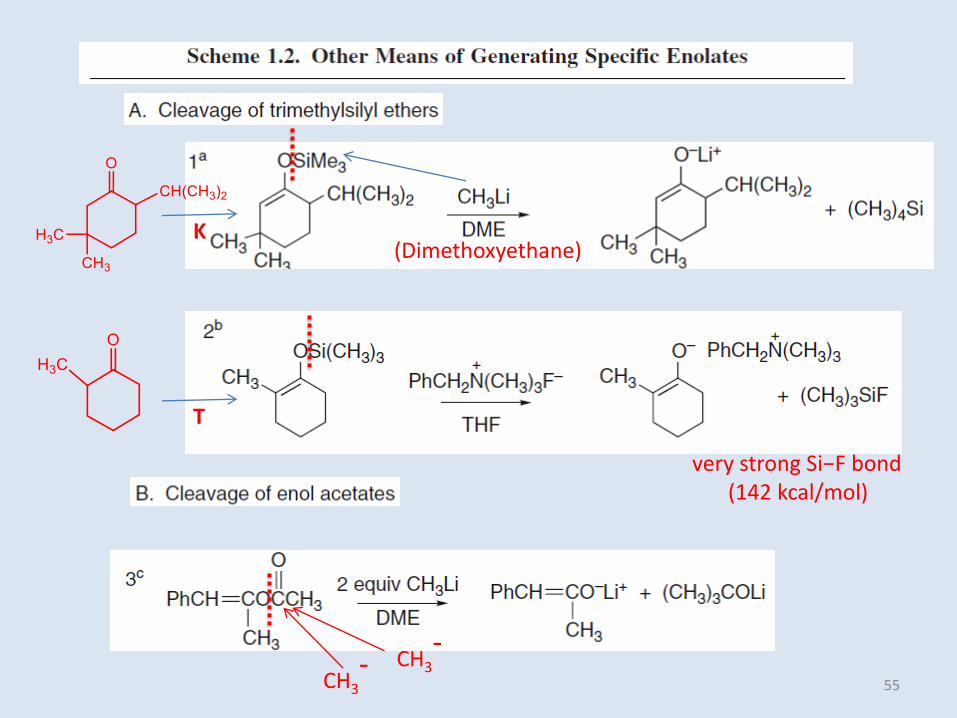
(Dimethoxyethane)K
very strong Si−F bond(142 kcal/mol)
CH3
-CH3
-
T
55
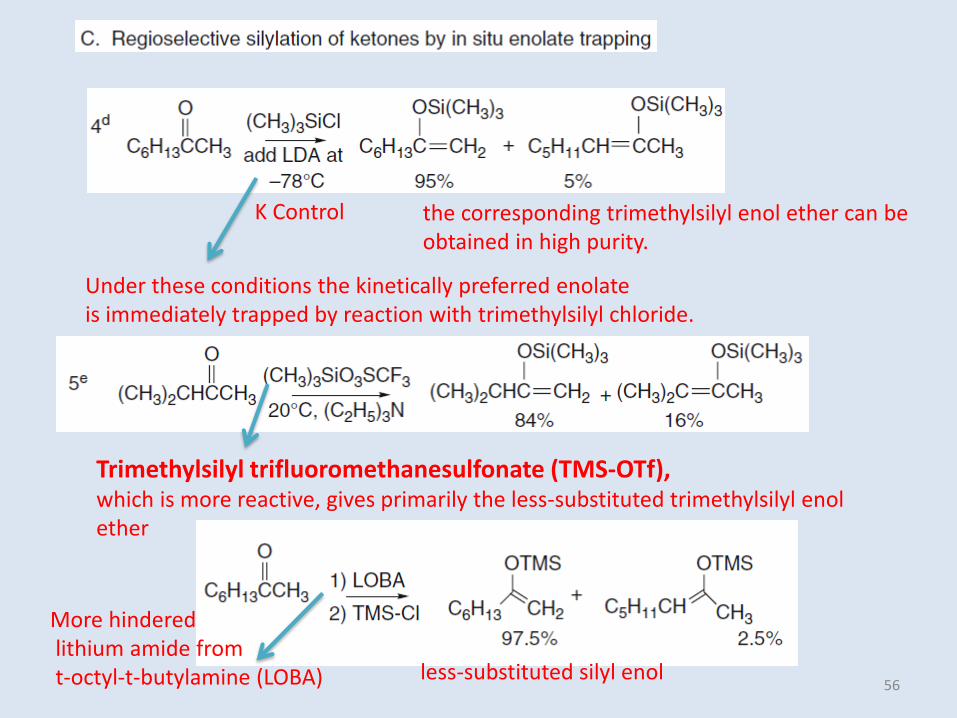
K Control the corresponding trimethylsilyl enol ether can be obtained in high purity.
Trimethylsilyl trifluoromethanesulfonate (TMS-OTf), which is more reactive, gives primarily the less-substituted trimethylsilyl enolether
Under these conditions the kinetically preferred enolateis immediately trapped by reaction with trimethylsilyl chloride.
less-substituted silyl enol
More hinderedlithium amide fromt-octyl-t-butylamine (LOBA) 56
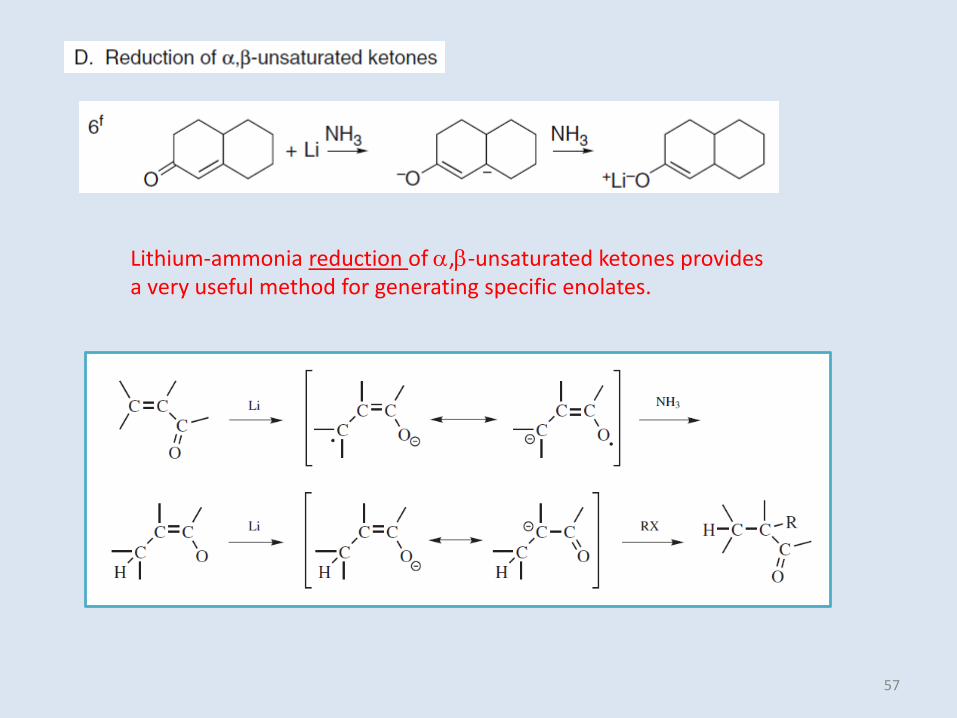
Lithium-ammonia reduction of ,b-unsaturated ketones provides a very useful method for generating specific enolates.
57
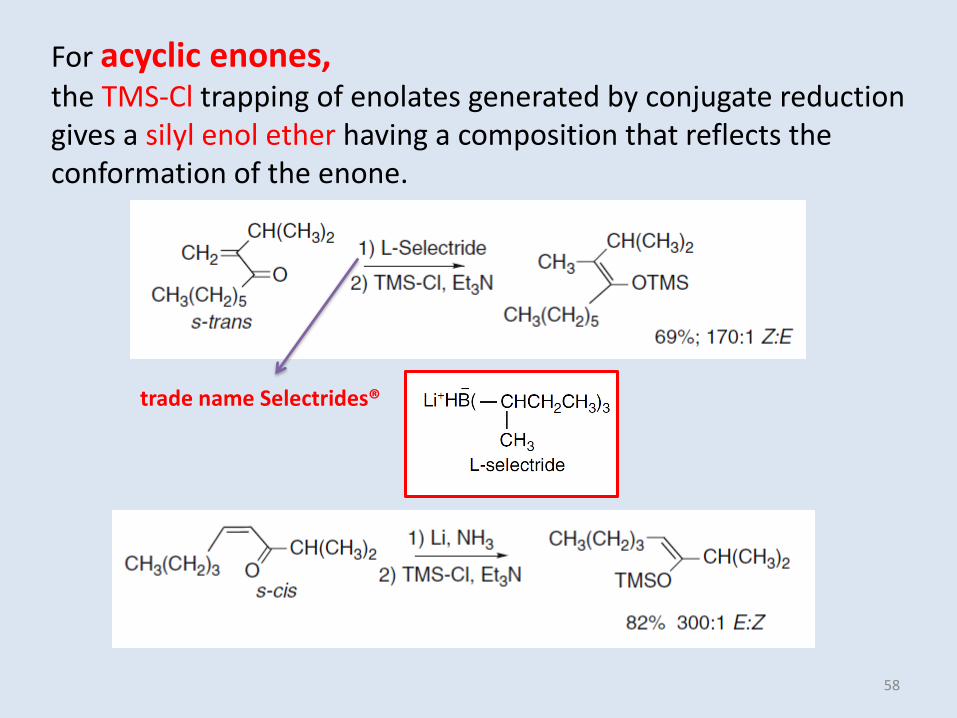
For acyclic enones, the TMS-Cl trapping of enolates generated by conjugate reduction gives a silyl enol ether having a composition that reflects the conformation of the enone.
trade name Selectrides®
58
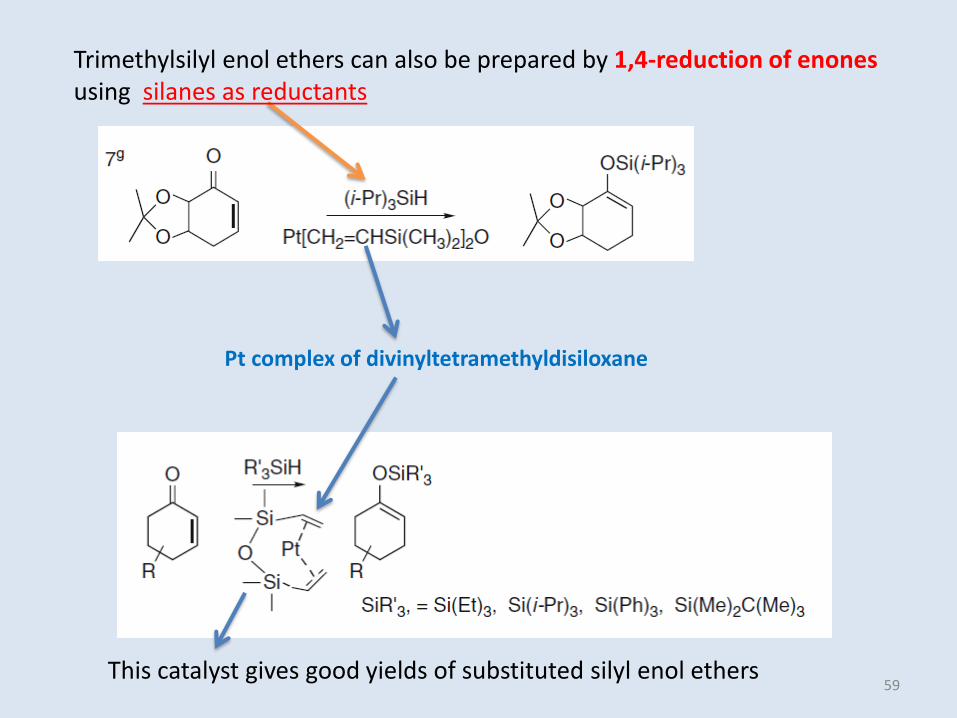
Trimethylsilyl enol ethers can also be prepared by 1,4-reduction of enonesusing silanes as reductants
Pt complex of divinyltetramethyldisiloxane
This catalyst gives good yields of substituted silyl enol ethers59
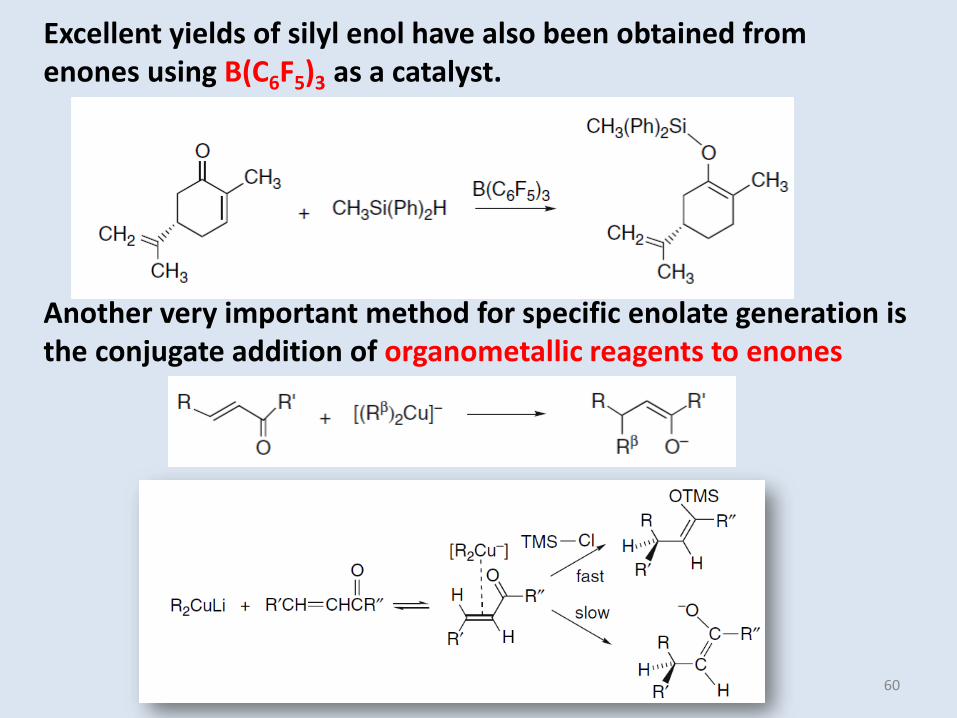
Excellent yields of silyl enol have also been obtained from enones using B(C6F5)3 as a catalyst.
Another very important method for specific enolate generation is the conjugate addition of organometallic reagents to enones
60
Recommended

















