
doi:10.1016/j.jmb.2005.02.059 J. Mol. Biol. (2005) 348, 335–347
The Native Energy Landscape for Interleukin-1b.Modulation of the Population Ensemble throughNative-state Topology
Melinda Roy1, Leslie L. Chavez2, John M. Finke2, David K. Heidary1
Jose N. Onuchic2 and Patricia A. Jennings1*
1Departments of Chemistry andBiochemistry, University ofCalifornia at San Diego, La JollaCA 92093-0359,USA
2Center for TheoreticalBiological Physics, University ofCalifornia at San Diego, La JollaCA 92093-0374, USA
0022-2836/$ - see front matter q 2005 E
Present address: D. K. Heidary, VAbbreviations used: IL-1b, interle
correlation; RMSD, root-mean-squarisomerase; MD, molecular dynamic
E-mail address of the correspond
A minimalist Go-model, with no energetic frustration in the nativeconformation, has been shown to describe accurately the folding pathwayof the b-trefoil protein, interleukin-1b (IL-1b). While it appears that thesemodels successfully model transition states and intermediates between theunfolded and native ensembles, it is unclear how accurately they capturesmaller, yet biologically relevant, structural changes within the nativeensemble after energetic perturbation. Here, we address the followingquestions. Can a simple Go-model of interleukin-1b, based on nativetopology, describe changes in structural properties of the native ensembleas the protein stability is changed? Or is it necessary to include a moreexplicit representation of atoms, electrostatic, hydrogen bonding, and vander Waals forces to describe these changes?
The native ensemble of IL-1b was characterized using a variety ofexperimental probes under native (0 M NaCl, guanidine hydrochloride(Gdn-HCl)), moderately destabilized (0 M NaCl, 0.8 M Gdn-HCl), and inmoderate salt concentration (0.8 M NaCl, 0 M Gdn-HCl). Heteronuclear1H–15N nuclear Overhauser effect spectroscopy (NOESY) and hetero-nuclear single quantum correlation (HSQC) NMR spectra confirmed thatthe b-trefoil global fold was largely intact under these three conditions.However, 25 of the 153 residues throughout the chain did demonstrate 13Cand 1H–15N chemical shifts when perturbed with 0.8 M NaCl or Gdn-HCl.Despite large differences in protection factors from solvent hydrogen-deuterium exchange for all residues between stable (0 M Gdn-HCl) anddestabilized (0.8 M Gdn-HCl) IL-1b, no difference in steady-state 15N–1HNOE enhancements were measured. Thus, the chemical shifts correlatewith a global but limited increase in residue flexibility in the presence ofGdn-HCl. Minimalist simulations highlight the regions of greatest positionshift between native and 0.8 M Gdn-HCl, which were determinedexperimentally. This correlation demonstrates that structural changeswithin the native ensemble of IL-1b are, at least partially, governed bythe principle of minimal energetic frustration.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: interleukin-1b; b-trefoil; topology; molecular dynamics; Go-model
*Corresponding authorlsevier Ltd. All rights reserved.
ertex Pharmaceuticals Inc., San Diego, CA 92121,USA.ukin-1b; Gdn-HCl, guanidine hydrochloride; HSQC, heteronuclear single quantume deviation; NOESY, nuclear Overhauser effect spectroscopy; TIM, triose phosphates; pdb, Protein Data Bank.ing author: [email protected]

336 Native Energy Landscape for IL-1b
Introduction
Simple theoretical models demonstrate that thefolding pathways of proteins with funneled energylandscapes are nearly completely determined bythe energetic requirements of adopting the finalnative state topology,1–8 with a few notable excep-tions in simulations of the folding of symmetricproteins.9 These models exploit topological infor-mation contained in the X-ray crystal structuresand are able to capture the dominant structuraland thermodynamic properties of the foldingmechanisms of both small proteins and morecomplex proteins such as interleukin-1b (IL-1b)and dihydrofolate reductase.4,10 These studies areespecially noteworthy, as both proteins are of asimilar size yet fold through different intermediatestructures.11,12 Thus, topological features of thenative protein are sufficient to predict the generalstructures and thermodynamic properties of thestates populated between distinct unfolded andfolded protein ensembles. Nonetheless, it remainsto be seen whether more subtle structural andthermodynamic changes, which occur during per-turbation of a single ensemble, i.e. the native stateensemble, can also be captured with these simplemodels. A recent study has shown that localperturbations induced by loop inserts can besimulated in a simple theoretical Go-model ofchymotrypsin inhibitor 2 (CI2) nearly exactly asobserved in experiments.13 Here, we addresswhether this agreement will hold for globalperturbation of the native IL-1b ensemble bychemical denaturants.
The inflammatory cytokine IL-1b has diverse andcomplex actions throughout the body and thebrain.14 IL-1b is partially unfolded during secretionfrom activated macrophages and monocytes,15 andis unfolded during in vitro aggregation16 and in vivoprecipitation in the brain during amyloid plaqueevolution in Alzheimer’s disease.17,18 Thus, theconformational dynamics that control the popu-lation of unfolded or partially unfolded stateswithin the native state ensemble are of growinginterest. The kinetic unfolding mechanism of IL-1bunder strongly denaturing conditions reveals apartially rugged landscape in which unfoldingseems to be best represented by the kineticprogression towards an ensemble of lessstructurally organized structures, rather thandiscrete unfolding intermediates.19
IL-1b displays the archetypal b-trefoil globalfold,20,21 where six b-strands form a b-barrel(b1, b4, b5, b8, b9, and b12) and six b-strandsform a three-hairpin cap (b2Cb3, b6Cb7, andb10Cb11).20,22,23 A discrete folding intermediatepopulates during the kinetic refolding of IL-1b,11,24,25
the structure of which has been captured withminimalist theoretical models.4 The equilibriumunfolding transition as monitored by CD spec-troscopy,26,27 Trp120 fluorescence average wave-length and anisotropy,28 and DSC29 is well fit by atwo-state unfolding model, which implies no
stable intermediates. In contrast, IL-1b exhibitshyperfluorescence, maximal near 0.8 M guanidinehydrochloride (Gdn-HCl), during the equilibriumGdn-HCl unfolding transition monitored withintrinsic tryptophan fluorescence and the transitioncannot be fit to either a two-state or a three-statefit.28 This hyperfluorescence can be explained byeither population of a metastable equilibriumintermediate and/or structural transitions withina conformationally flexible native state ensemble.30
Although not hyperfluorescent, another b-trefoilprotein, fibroblast growth factor 1 (FGF-1), alsopossesses a highly native-like intermediatepopulated during equilibrium Gdn-HClunfolding.31
In equilibrium titrations with a chemicaldenaturant, IL-1b exhibits hyperflourescence atlow denaturant concentrations, although thecircular dichroism signal remains largelyunchanged.16,26,27,32,35,36 Recent studies have usedH/2H exchange to probe proteins at low levels ofdenaturant, which destabilized the native ensemblebut did not produce a hyperfluorescent state.33,34 Inthe case of IL-1b, it remains unclear whether thehyperfluorescent ensemble populated at 0.8 MGdn-HCl is comprised of partially unfolded inter-mediates or slightly perturbed members of thenative ensemble. The present study investigates thestructural properties of the IL-1b hyperfluorescentstate populated at 0.8 M Gdn-HCl, revealing thestructural details of this ensemble. This study alsoemploys molecular dynamics simulations to testwhether a minimalist Ca-only protein model with afunneled energy landscape is sufficient to capturethe measured properties of the native ensemble.
Results
Experimental measurements of the native basin
Fluorescence spectroscopy
In IL-1b, the fluorescence intensity increases in alinear fashion with increasing concentrations ofNaCl, up to a maximum and constant value at andabove 3 M NaCl. In contrast, the equilibriumdenaturation profile of IL-1b exhibits an initialincrease in fluorescence signal that plateaus at 0.8 MGdn-HCl and then subsequently decreases between1 M and 2 M Gdn-HCl. The initial increase influorescence represents a hyperfluorescent ensem-ble populated at 0.8 M Gdn-HCl and is similar tothat populated at high NaCl concentration (data notshown). The decreasing signal between 1 M and2 M Gdn-HCl is coincident with the transition thatmonitors the cooperative unfolding of the protein asmeasured with a variety of probes.16,26,27,35,36 Con-trol experiments demonstrate that isolated tyrosineand tryptophan residue fluorescence increases withNaCl and Gdn-HCl concentration but can accountfor only half of the hyperfluorescence observed inIL-1b at the same NaCl and Gdn-HCl concentration.
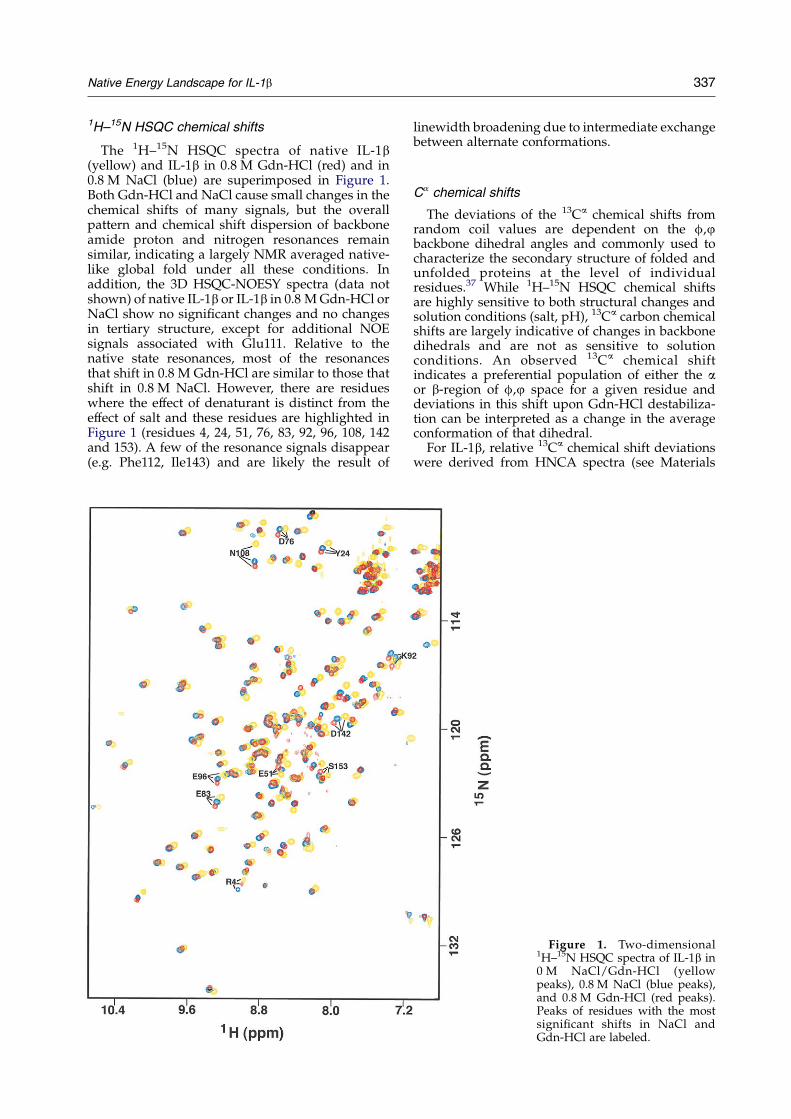
Native Energy Landscape for IL-1b 337
1H–15N HSQC chemical shifts
The 1H–15N HSQC spectra of native IL-1b(yellow) and IL-1b in 0.8 M Gdn-HCl (red) and in0.8 M NaCl (blue) are superimposed in Figure 1.Both Gdn-HCl and NaCl cause small changes in thechemical shifts of many signals, but the overallpattern and chemical shift dispersion of backboneamide proton and nitrogen resonances remainsimilar, indicating a largely NMR averaged native-like global fold under all these conditions. Inaddition, the 3D HSQC-NOESY spectra (data notshown) of native IL-1b or IL-1b in 0.8 M Gdn-HCl orNaCl show no significant changes and no changesin tertiary structure, except for additional NOEsignals associated with Glu111. Relative to thenative state resonances, most of the resonancesthat shift in 0.8 M Gdn-HCl are similar to those thatshift in 0.8 M NaCl. However, there are residueswhere the effect of denaturant is distinct from theeffect of salt and these residues are highlighted inFigure 1 (residues 4, 24, 51, 76, 83, 92, 96, 108, 142and 153). A few of the resonance signals disappear(e.g. Phe112, Ile143) and are likely the result of
linewidth broadening due to intermediate exchangebetween alternate conformations.
Ca chemical shifts
The deviations of the 13Ca chemical shifts fromrandom coil values are dependent on the f,4backbone dihedral angles and commonly used tocharacterize the secondary structure of folded andunfolded proteins at the level of individualresidues.37 While 1H–15N HSQC chemical shiftsare highly sensitive to both structural changes andsolution conditions (salt, pH), 13Ca carbon chemicalshifts are largely indicative of changes in backbonedihedrals and are not as sensitive to solutionconditions. An observed 13Ca chemical shiftindicates a preferential population of either the aor b-region of f,4 space for a given residue anddeviations in this shift upon Gdn-HCl destabiliza-tion can be interpreted as a change in the averageconformation of that dihedral.
For IL-1b, relative 13Ca chemical shift deviationswere derived from HNCA spectra (see Materials
Figure 1. Two-dimensional1H–15N HSQC spectra of IL-1b in0 M NaCl/Gdn-HCl (yellowpeaks), 0.8 M NaCl (blue peaks),and 0.8 M Gdn-HCl (red peaks).Peaks of residues with the mostsignificant shifts in NaCl andGdn-HCl are labeled.
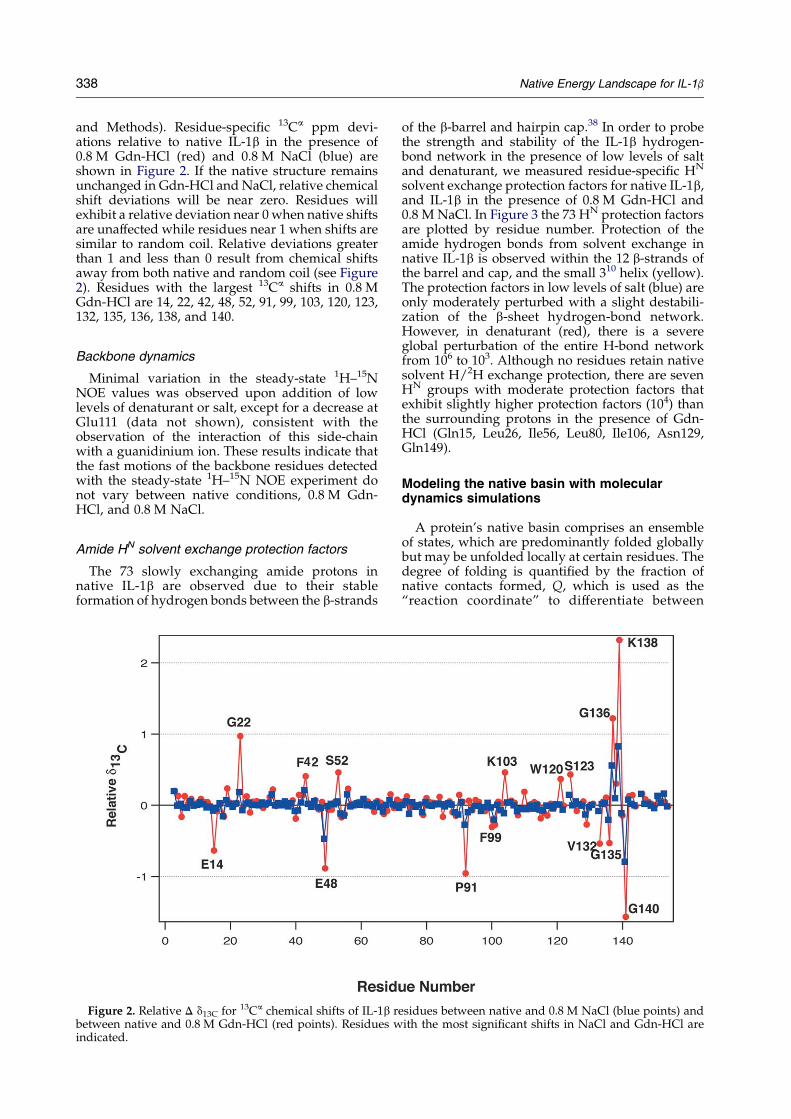
338 Native Energy Landscape for IL-1b
and Methods). Residue-specific 13Ca ppm devi-ations relative to native IL-1b in the presence of0.8 M Gdn-HCl (red) and 0.8 M NaCl (blue) areshown in Figure 2. If the native structure remainsunchanged in Gdn-HCl and NaCl, relative chemicalshift deviations will be near zero. Residues willexhibit a relative deviation near 0 when native shiftsare unaffected while residues near 1 when shifts aresimilar to random coil. Relative deviations greaterthan 1 and less than 0 result from chemical shiftsaway from both native and random coil (see Figure2). Residues with the largest 13Ca shifts in 0.8 MGdn-HCl are 14, 22, 42, 48, 52, 91, 99, 103, 120, 123,132, 135, 136, 138, and 140.
Backbone dynamics
Minimal variation in the steady-state 1H–15NNOE values was observed upon addition of lowlevels of denaturant or salt, except for a decrease atGlu111 (data not shown), consistent with theobservation of the interaction of this side-chainwith a guanidinium ion. These results indicate thatthe fast motions of the backbone residues detectedwith the steady-state 1H–15N NOE experiment donot vary between native conditions, 0.8 M Gdn-HCl, and 0.8 M NaCl.
Amide HN solvent exchange protection factors
The 73 slowly exchanging amide protons innative IL-1b are observed due to their stableformation of hydrogen bonds between the b-strands
Figure 2. Relative D d13C for 13Ca chemical shifts of IL-1b rbetween native and 0.8 M Gdn-HCl (red points). Residues windicated.
of the b-barrel and hairpin cap.38 In order to probethe strength and stability of the IL-1b hydrogen-bond network in the presence of low levels of saltand denaturant, we measured residue-specific HN
solvent exchange protection factors for native IL-1b,and IL-1b in the presence of 0.8 M Gdn-HCl and0.8 M NaCl. In Figure 3 the 73 HN protection factorsare plotted by residue number. Protection of theamide hydrogen bonds from solvent exchange innative IL-1b is observed within the 12 b-strands ofthe barrel and cap, and the small 310 helix (yellow).The protection factors in low levels of salt (blue) areonly moderately perturbed with a slight destabili-zation of the b-sheet hydrogen-bond network.However, in denaturant (red), there is a severeglobal perturbation of the entire H-bond networkfrom 106 to 103. Although no residues retain nativesolvent H/2H exchange protection, there are sevenHN groups with moderate protection factors thatexhibit slightly higher protection factors (104) thanthe surrounding protons in the presence of Gdn-HCl (Gln15, Leu26, Ile56, Leu80, Ile106, Asn129,Gln149).
Modeling the native basin with moleculardynamics simulations
A protein’s native basin comprises an ensembleof states, which are predominantly folded globallybut may be unfolded locally at certain residues. Thedegree of folding is quantified by the fraction ofnative contacts formed, Q, which is used as the“reaction coordinate” to differentiate between
esidues between native and 0.8 M NaCl (blue points) andith the most significant shifts in NaCl and Gdn-HCl are
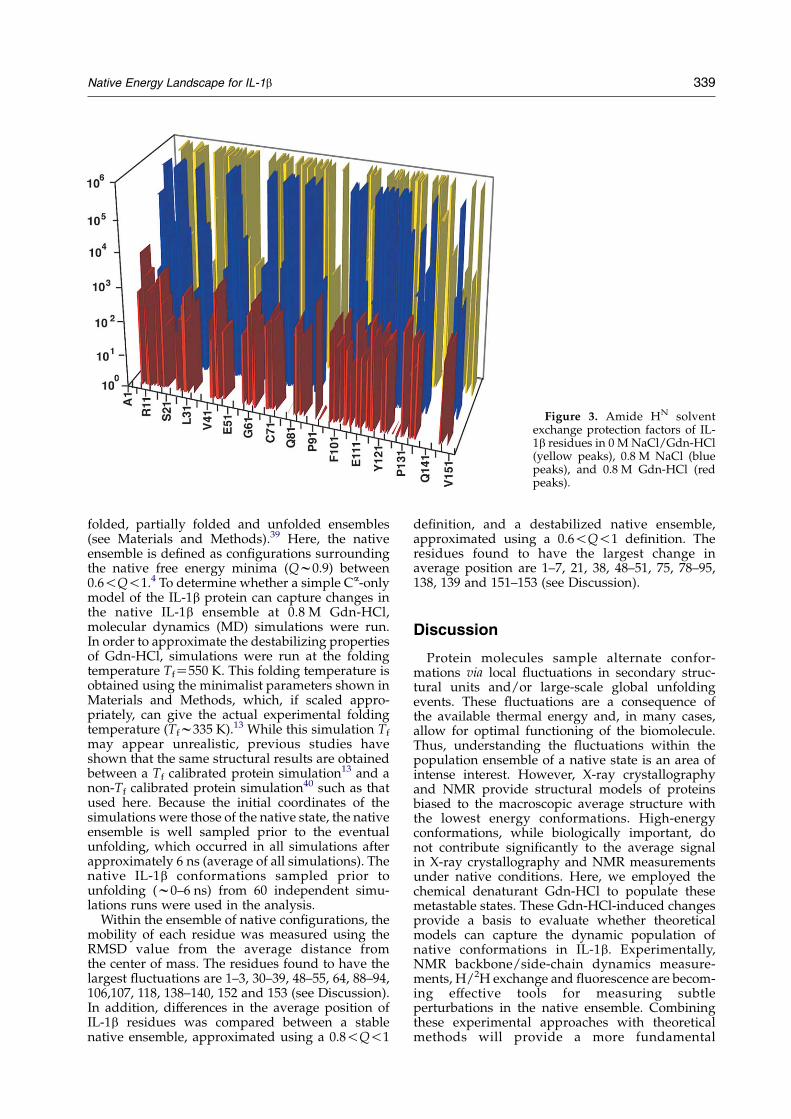
Figure 3. Amide HN solventexchange protection factors of IL-1b residues in 0 M NaCl/Gdn-HCl(yellow peaks), 0.8 M NaCl (bluepeaks), and 0.8 M Gdn-HCl (redpeaks).
Native Energy Landscape for IL-1b 339
folded, partially folded and unfolded ensembles(see Materials and Methods).39 Here, the nativeensemble is defined as configurations surroundingthe native free energy minima (Qw0.9) between0.6!Q!1.4 To determine whether a simple Ca-onlymodel of the IL-1b protein can capture changes inthe native IL-1b ensemble at 0.8 M Gdn-HCl,molecular dynamics (MD) simulations were run.In order to approximate the destabilizing propertiesof Gdn-HCl, simulations were run at the foldingtemperature TfZ550 K. This folding temperature isobtained using the minimalist parameters shown inMaterials and Methods, which, if scaled appro-priately, can give the actual experimental foldingtemperature (Tfw335 K).13 While this simulation Tf
may appear unrealistic, previous studies haveshown that the same structural results are obtainedbetween a Tf calibrated protein simulation13 and anon-Tf calibrated protein simulation40 such as thatused here. Because the initial coordinates of thesimulations were those of the native state, the nativeensemble is well sampled prior to the eventualunfolding, which occurred in all simulations afterapproximately 6 ns (average of all simulations). Thenative IL-1b conformations sampled prior tounfolding (w0–6 ns) from 60 independent simu-lations runs were used in the analysis.
Within the ensemble of native configurations, themobility of each residue was measured using theRMSD value from the average distance fromthe center of mass. The residues found to have thelargest fluctuations are 1–3, 30–39, 48–55, 64, 88–94,106,107, 118, 138–140, 152 and 153 (see Discussion).In addition, differences in the average position ofIL-1b residues was compared between a stablenative ensemble, approximated using a 0.8!Q!1
definition, and a destabilized native ensemble,approximated using a 0.6!Q!1 definition. Theresidues found to have the largest change inaverage position are 1–7, 21, 38, 48–51, 75, 78–95,138, 139 and 151–153 (see Discussion).
Discussion
Protein molecules sample alternate confor-mations via local fluctuations in secondary struc-tural units and/or large-scale global unfoldingevents. These fluctuations are a consequence ofthe available thermal energy and, in many cases,allow for optimal functioning of the biomolecule.Thus, understanding the fluctuations within thepopulation ensemble of a native state is an area ofintense interest. However, X-ray crystallographyand NMR provide structural models of proteinsbiased to the macroscopic average structure withthe lowest energy conformations. High-energyconformations, while biologically important, donot contribute significantly to the average signalin X-ray crystallography and NMR measurementsunder native conditions. Here, we employed thechemical denaturant Gdn-HCl to populate thesemetastable states. These Gdn-HCl-induced changesprovide a basis to evaluate whether theoreticalmodels can capture the dynamic population ofnative conformations in IL-1b. Experimentally,NMR backbone/side-chain dynamics measure-ments, H/2H exchange and fluorescence are becom-ing effective tools for measuring subtleperturbations in the native ensemble. Combiningthese experimental approaches with theoreticalmethods will provide a more fundamental

340 Native Energy Landscape for IL-1b
understanding of protein dynamics. While recentstudies have shown that simple theoretical modelscapture protein folding pathways accurately,4,10 thisstudy investigated whether these models can alsodescribe accurately the structural dynamics withinthe native ensemble of IL-1b.
Retention of native-like structure in 0.8 MGdn-HCl
The consensus of the experimental measurementsindicates the ensemble of IL-1b in 0.8 M Gdn-HCl islargely native-like (Figures 1 and 2). Althoughclearly less stable to solvent intrusion than that in0 M Gdn-HCl (Figure 3), no increases in backbonedynamics are detected (data not shown), whichwould be expected if a partially folded intermediatewas populated. Thus, the hyperfluorescenceobserved at 0 and 0.8 M Gdn-HCl must stem fromrelatively subtle structural changes between differ-ent native ensembles and not from population of apartially folded intermediate.30
In Figure 1, most peaks in the 1H–15N HSQCspectra IL-1b are shifted slightly by either 0.8 MGdn-HCl or NaCl. The pattern of chemical shiftdispersion is nonetheless quite similar to that of thenative state. Since 0.8 M Gdn-HCl elicits changes inthe electronic anisotropy surrounding each residuesimilar to those of 0.8 M NaCl, many of thedeviations from native resonances may result fromchanges in solvent electrostatics and not fromstructural changes.
In Figure 2, most (w90%) of the 13Ca resonancesare unperturbed by the presence of 0.8 M Gdn-HCl,indicating the presence of mostly native dihedralsin the destabilized ensemble. However, it is clearfrom Figure 2 that the dihedrals near specificresidues do exhibit changes in 0.8 M Gdn-HCl thatcannot be explained simply from the effects ofincreased ionic strength produced with 0.8 M NaCl.Thus, while the IL-1b ensemble in 0.8 M Gdn-HClretains most of its native dihedrals, local deviationsfrom the native conformation are observed nearresidues 14, 22, 42, 48, 52, 91, 99, 103, 120, 123, and inthe region between 135 and 140. Thus, the 0.8 MGdn-HCl ensemble is a native-like ensemble withlocal deviations at select residues.
Residue flexibility does not increase between 0 Mand 0.8 M Gdn-HCl to the extent that the backbonedynamics are altered (data not shown). However,Figure 3 shows that H/2H protection factors aredecreased by at least two orders of magnitude forall residues. Thus, the population of locallyunfolded or exposed residue backbones mustincrease to account for these decreased protectionfactors, which suggests a more flexible ensemble at0.8 M Gdn-HCl. With the exception of the sevenresidues showing a higher protection factor (104),most other residues are decreased to a similardegree (103) in 0.8 M Gdn-HCl. Despite thisdecrease in global stability observed across theentire IL-1b chain in 0.8 M Gdn-HCl (Figure 3), the
resulting increase in residue flexibility is notsufficient alter backbone dynamics.
The location of both 1H–15N HSQC and 13Ca
resonance changes between 0 M and 0.8 MGdn-HCl in the two-dimensional schematic ofIL-1b structure are shown in Figure 4 where, bluecircles indicate shifts in 1H–15N HSQC resonancesand red circles indicate shifts in 13Ca resonances.Contrary to conventional wisdom, the regions ofgreatest perturbation are not always in loop andturn regions. While most residue perturbations dooccur in loop/turn regions (residues 14, 22, 24, 48,51, 52, 76, 91, 92, 96, 99, 108, 123, 132, 136, 138, 140,142 and 153), some do occur within structuredregions of the b-strands in the barrel (residues 4, 42,and 103) and caps (residues 83, 120 and 135).Furthermore, certain loop/turn regions appear tobe unperturbed in the presence of 0.8 M Gdn-HCl(residues 30–39, 63–67 and 114–119). Thus, regionsof IL-1b are stabilized by a more complex networkof interactions than simply nearby contact partners.Molecular dynamics simulations were used toinvestigate whether a simple Ca-only topology-based model produces the experimental pattern ofresidue perturbation in the presence of denaturants.
Comparison of experimental native ensembleperturbations with experimental metrics and afunneled landscape model
Figure 5 shows the experimental 1H–15N HSQC(short blue lines) and 13Ca (long red lines) reson-ances, which are clearly shifted as shown inFigures 1 and 2. To predict these shifts, fourdifferent methods are shown for each residue.(1) Experimental B-factors determined from theProtein Data Bank (pdb) file 2I1B (Figure 5(b)). (2)Number of native contacts determined from 2I1B(Figure 5(c)). (3) RMSD values of residue positionsdetermined from MD simulation (Figure 5(d)). (4)Normalized change in average position from thecenter of mass (DD) between native conformationswithin 0.6!Q!1 and conformations within 0.8!Q!1 in the MD simulation (Figure 5(e)).
In Figure 5(a), the residues with the largest shiftin 13Ca resonance between 0 M and 0.8 M Gdn-HClare highlighted with red lines so as to compare withthe predictive methods in Figure 5(b)–(e). Forclarity, the highlighted residues are also codedalong the top axis with red letters: (a) Glu14, (b)Gly22, (c) Phe42, (d) Glu48, (e) Ser52, (f) Pro91, (g)Phe99, (h) Lys103, (i) Trp120, (j) Ser123, (k) Val132,(l) Gly135CGly136, (m) Lys138, and (n) Gly140.These residues all clearly show significant deviationfrom native backbone dihedrals given in Figure 2.Also shown for comparison are blue lines indicatingthe residue peaks that show significant shifts in the1H–15N HSQC spectra (Figure 1). Since 1H–15NHSQC peak shifts can result from many localchanges in residue environment, interpretingthese 1H–15N HSQC peak shifts as a loss of localnative structure is less straightforward than those of13Ca resonances and is not used for direct

Figure 4. Indication of IL-1b residues with significant 1H–15N HSQC peaks shifts (blue circles) and 13Ca chemical shiftsD d13C (red circles) in 0.8 M Gdn-HCl overlayed on a two-dimensional representation of the IL-1b structure.
Native Energy Landscape for IL-1b 341
comparison. Nonetheless, the 1H–15N data arepresented as complementary to the 13Ca shift data,although of a lesser significance.
The experimental results in Figure 5(a) arecompared to MD simulations of a minimalistmodel of IL-1b containing only Ca atoms. Residue-specific properties of the simulation are quantifiedand are compared to the experimental results.However, the Ca B-factors (Figure 5(b)) and thenumber of native residue contacts (Figure 5(c)) arealso shown to demonstrate whether the simulationmodel provides information above and beyond asimple analysis of the X-ray structural data.
In Figure 5(b), Ca B-factors provided in pdb file2I1B are shown. A periodic increase in the B-factorsis observed for each turn and loop region of theprotein. If used in a predictive manner, the higherB-factors correctly predict local unfolding inresidues (b) 22, (e) 52, (m) 138, and (n) 140 but donot predict (a) 14, (c) 42, (d) 48, (f) 91, (g) 99, (h) 103,(i) 120, (j) 123, (k) 132, and (l) 135C136.
In Figure 5(c), the number of native contacts isshown for each residue, as determined using CSUanalysis on pdb file 2I1B.41 A periodic decrease innative contacts is observed for each turn and loop
region of the protein. If used in a predictive manner,the lower native contacts correctly predict localunfolding in residues (a) 14, (b) 22, (e) 52, (l) 135C136, (m) 138, and (n) 140 but do not pick out (c) 42,(d) 48, (f) 91, (g) 99, (h) 103, (i) 120, (j) 123, and (k)132.
In Figure 5(d), the RMSD fluctuations for each Ca
atom in the simulated IL-1b native state model isshown (see Results). A periodic increase in RMSDvalue is observed for each turn and loop region ofthe protein. If used in a predictive manner, thehigher RMSD values correctly predict localunfolding in residues (d) 48, (e) 52, (f) 91, (m)138, and (n) 140 but do not pick out (a) 14, (b) 22, (c)42, (g) 99, (h) 103, (i) 120, (j) 123, (k) 132, and (l)135C136.
In Figure 5(e), the normalized change betweeneach Ca atom and the IL-1b center of mass (DD),using native definitions 0.6!Q!1 and 0.8!Q!1,in the simulated IL-1b native state model is shown(see Results). Increased values of DD do notcorrelate with loops and turns and are observed atthe N terminus, near residues 20, 40, 50, 80–95, 140and the C terminus. If used in a predictive manner,the higher DD values correctly predict local
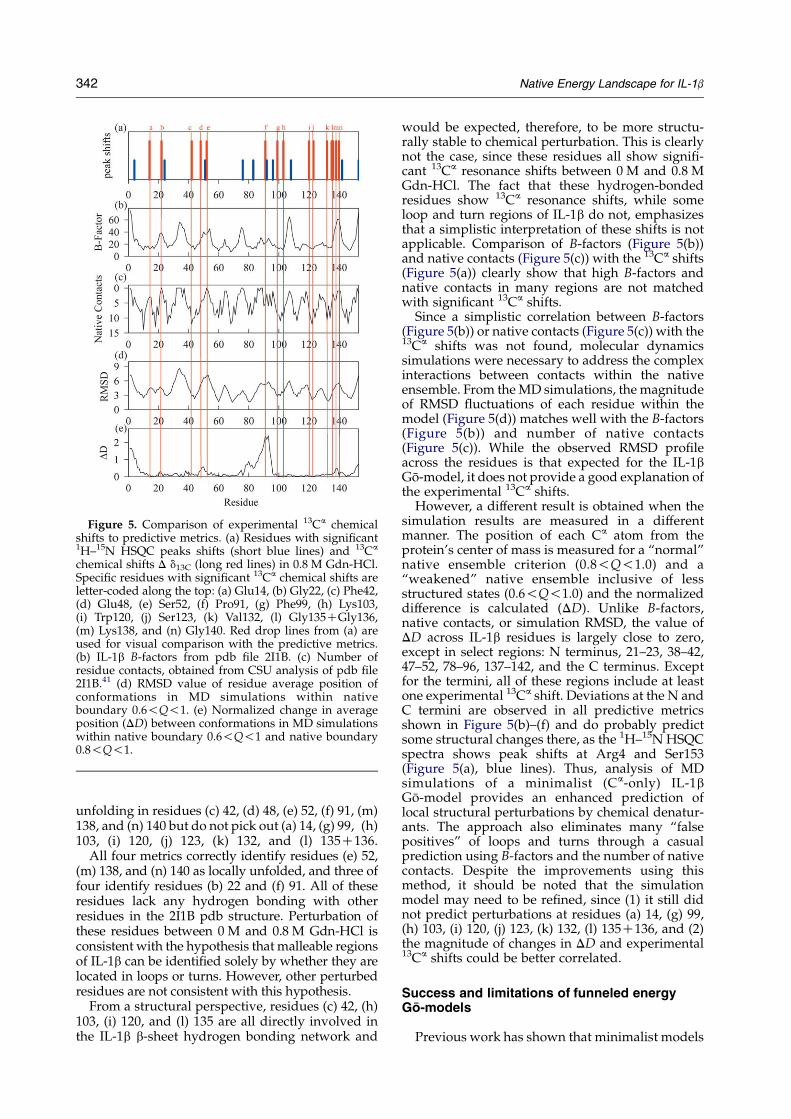
Figure 5. Comparison of experimental 13Ca chemicalshifts to predictive metrics. (a) Residues with significant1H–15N HSQC peaks shifts (short blue lines) and 13Ca
chemical shifts D d13C (long red lines) in 0.8 M Gdn-HCl.Specific residues with significant 13Ca chemical shifts areletter-coded along the top: (a) Glu14, (b) Gly22, (c) Phe42,(d) Glu48, (e) Ser52, (f) Pro91, (g) Phe99, (h) Lys103,(i) Trp120, (j) Ser123, (k) Val132, (l) Gly135CGly136,(m) Lys138, and (n) Gly140. Red drop lines from (a) areused for visual comparison with the predictive metrics.(b) IL-1b B-factors from pdb file 2I1B. (c) Number ofresidue contacts, obtained from CSU analysis of pdb file2I1B.41 (d) RMSD value of residue average position ofconformations in MD simulations within nativeboundary 0.6!Q!1. (e) Normalized change in averageposition (DD) between conformations in MD simulationswithin native boundary 0.6!Q!1 and native boundary0.8!Q!1.
342 Native Energy Landscape for IL-1b
unfolding in residues (c) 42, (d) 48, (e) 52, (f) 91, (m)138, and (n) 140 but do not pick out (a) 14, (g) 99, (h)103, (i) 120, (j) 123, (k) 132, and (l) 135C136.
All four metrics correctly identify residues (e) 52,(m) 138, and (n) 140 as locally unfolded, and three offour identify residues (b) 22 and (f) 91. All of theseresidues lack any hydrogen bonding with otherresidues in the 2I1B pdb structure. Perturbation ofthese residues between 0 M and 0.8 M Gdn-HCl isconsistent with the hypothesis that malleable regionsof IL-1b can be identified solely by whether they arelocated in loops or turns. However, other perturbedresidues are not consistent with this hypothesis.
From a structural perspective, residues (c) 42, (h)103, (i) 120, and (l) 135 are all directly involved inthe IL-1b b-sheet hydrogen bonding network and
would be expected, therefore, to be more structu-rally stable to chemical perturbation. This is clearlynot the case, since these residues all show signifi-cant 13Ca resonance shifts between 0 M and 0.8 MGdn-HCl. The fact that these hydrogen-bondedresidues show 13Ca resonance shifts, while someloop and turn regions of IL-1b do not, emphasizesthat a simplistic interpretation of these shifts is notapplicable. Comparison of B-factors (Figure 5(b))and native contacts (Figure 5(c)) with the 13Ca shifts(Figure 5(a)) clearly show that high B-factors andnative contacts in many regions are not matchedwith significant 13Ca shifts.
Since a simplistic correlation between B-factors(Figure 5(b)) or native contacts (Figure 5(c)) with the13Ca shifts was not found, molecular dynamicssimulations were necessary to address the complexinteractions between contacts within the nativeensemble. From the MD simulations, the magnitudeof RMSD fluctuations of each residue within themodel (Figure 5(d)) matches well with the B-factors(Figure 5(b)) and number of native contacts(Figure 5(c)). While the observed RMSD profileacross the residues is that expected for the IL-1bGo-model, it does not provide a good explanation ofthe experimental 13Ca shifts.
However, a different result is obtained when thesimulation results are measured in a differentmanner. The position of each Ca atom from theprotein’s center of mass is measured for a “normal”native ensemble criterion (0.8!Q!1.0) and a“weakened” native ensemble inclusive of lessstructured states (0.6!Q!1.0) and the normalizeddifference is calculated (DD). Unlike B-factors,native contacts, or simulation RMSD, the value ofDD across IL-1b residues is largely close to zero,except in select regions: N terminus, 21–23, 38–42,47–52, 78–96, 137–142, and the C terminus. Exceptfor the termini, all of these regions include at leastone experimental 13Ca shift. Deviations at the N andC termini are observed in all predictive metricsshown in Figure 5(b)–(f) and do probably predictsome structural changes there, as the 1H–15N HSQCspectra shows peak shifts at Arg4 and Ser153(Figure 5(a), blue lines). Thus, analysis of MDsimulations of a minimalist (Ca-only) IL-1bGo-model provides an enhanced prediction oflocal structural perturbations by chemical denatur-ants. The approach also eliminates many “falsepositives” of loops and turns through a casualprediction using B-factors and the number of nativecontacts. Despite the improvements using thismethod, it should be noted that the simulationmodel may need to be refined, since (1) it still didnot predict perturbations at residues (a) 14, (g) 99,(h) 103, (i) 120, (j) 123, (k) 132, (l) 135C136, and (2)the magnitude of changes in DD and experimental13Ca shifts could be better correlated.
Success and limitations of funneled energyGo-models
Previous work has shown that minimalist models

Native Energy Landscape for IL-1b 343
with funneled energy landscape can captureprotein folding pathways accurately, in which thereaction coordinate comprises very disorderedunfolded conformations (Qw0.1) as well as highlyordered conformations (Qw0.9).4,40,42 However,biological activity can depend on relatively subtlestructural changes between two different orderedprotein structures.43 It is important to test whethersimple models are capable of capturing these subtlechanges if we are to extend these models to manybiological interactions involving different nativestate conformations. The present work shows thatsimulations of a simple IL-1b model offers clearadvantages over non-simulation approaches inpredicting the regions most susceptible to structuralchange under chemical stress. However, the presentsimulation approach does not completely capturethe regions and magnitudes of local residueunfolding and may require more atomic complexityin future studies. Nonetheless, it is important todemonstrate that improvements can be gained froma simple model before moving to more complexatomic models.
Conclusion
Under destabilizing conditions, the population ofthe IL-1b protein ensemble retains a macroscopicnative-state structural average although lowdenaturant levels modulate the populations ofconformational states within the native-stateensemble. In the presence of 0.8 M Gdn-HCl, thereare region-specific sites of local perturbation withinboth loop/turn regions as well as the b-sheethydrogen bond network. Previously, engineeringthe topological features of native IL-1b in asimulation model was sufficient to capture itsprotein folding landscape with an experimentallydetermined folding intermediate.4,11,28,44 This fold-ing landscape is similar to that seen in anotherb-trefoil protein, hisactophilin, suggesting thatother b-trefoil proteins may also have funneledenergy landscape where topological, not energetic,frustration presents the dominant free energyfolding barriers.45 The agreement in the presentstudy between the IL-1b theoretical model andNMR experiments solution results suggest thattransitions between native IL-1b ensemble confor-mations are, at least partially, determined bytopological frustration.
Materials and Methods
Purification of interleukin 1b
Expression and purification of IL-1b followed apublished protocol.19 IL-1b isoforms and the extent ofisotope enrichment were identified by electrosprayionization mass spectrometric analysis.
NMR spectroscopy
Protein samples were 0.8 mM IL-1b (the 1-Met isoform)in 100 mM sodium acetate-d3 (pH 5.4), 10%/90%2H2O/H2O at 36 8C. All NMR experiments wereperformed on a Bruker DMX500 spectrometer, using atriple-resonance gradient probe. The 1H chemical shiftwas referenced directly to sodium 2,2-dimethyl-2-sila-pentane-5-sulfonate (DSS), and the 13C and 15N chemicalshifts indirectly referenced to DSS.46 Assignments for 13C,15N and 1H resonances were based on comparison withpublished work.47,48 Water suppression was achievedusing a 3-9-19 train pulse sequence with gradients. Whenrequired, broadband 15N decoupling during acquisitionwas accomplished using a WALTZ16 decoupling scheme.All experiments were processed using Felix 95.0 software(MSI, San Diego, CA). Two-dimensional homonuclear 1HNOESY spectra of IL-1b were collected at 0 M salt, 0.8 MNaCl, and 0.8 M Gdn-HCl.
1H–15N HSQC and NOESY-HSQC spectra
Two-dimensional 1H–15N heteronuclear single quan-tum coherence (HSQC)49,50 and 3D NOESY-1H–15NHSQC51 spectra were acquired for IL-1b at 0 M NaCl/Gdn-HCl, 0.8 M NaCl, and 0.8 M Gdn-HCl. A mixingtime of 100 ms was used for the NOESY-HSQCexperiment.
Residue-specific 13Ca chemical shifts
HNCA spectra52 were collected of IL-1b at 0 M salt,0.8 M NaCl, and 0.8 M Gdn-HCl with constant time in t1and aliasing in the 15N (t1) dimension. A total of 16complex t1 (15N), 70 complex t2 (13C), and 512 complex t3(1H) points were collected. The spectral widths were810 Hz (16 ppm) (t1), 3017 Hz (24 ppm) (t2), and3004.808 Hz (6 ppm) (t3), respectively. Linear predictionwas performed in the indirectly detected dimensions andzero-filling in all dimensions. The relative perturbation in13Ca ppm for each specific amino acid residue in IL-1b in0.8 M Gdn-HCl was then calculated from the followingequation:
Relative Dd13C Z
ðd13CnativeKd13Cgdn-HClÞ=ðd13CnativeKd13CÞ(1)
where d13C refers to the chemical shift of the 13Ca of theindividual amino acid in a random coil, d13Cgdn-HCl isthe chemical shift of the specific amino acid residue inIL-1b in 0.8 M Gdn-HCl, and d13Cnative is the chemicalshift of the specific amino acid residue in native IL-1b. Forthe perturbation in 13Ca ppm for each specific amino acidresidue in IL-1b in 0.8 M NaCl, the following equation isused:
Relative Dd13C Z
ðd13CnativeKd13CNaClÞ=ðd13CnativeKd13CÞ(2)
where d13CNaCl is the chemical shift of a specific aminoacid residue in Il-1b in 0.8 M NaCl. Random coil 13Cchemical shifts for Ca atoms were taken from theliterature.37,53 When the relative perturbation in 13Cchemical shift is near 0G0.2, chemical shifts of specificresidues are taken to be native-like. When the relativedeviation is close to 1, this implies that the chemical shiftof the specific residues is close to the random coil value inthe presence of the chaotropic agent.

344 Native Energy Landscape for IL-1b
Heteronuclear steady-state NOE
Steady-state 2D 1H–15N NOE experiments54 wereacquired for IL-1b at 0 M salt, 0.8 M NaCl, and 0.8 MGdn-HCl in order to examine the backbone dynamics.Water suppression was achieved by application of a 1H908 pulse followed by a gradient pulse immediatelyprior to the start of the experiment. Backbonedynamics of native IL-1b measurements have beenstudied,55 and the results given here agree well with theprevious ones.
Amide HN solvent exchange protection factors
The rates of H/2H exchange of specific amide protonswere measured at 0 M salt, 0.8 M NaCl, and 0.8 M Gdn-HCl by following the change in intensity of cross-peaks inthe 1H–15N-HSQC spectra as a function of time upon achange of buffer from H2O to 2H2O. IL-1b was applied to aQuick-Spin buffer-exchange column (BoehringerMannheim) that was equilibrated in one of the followingthree buffers: 100 mM sodium acetate-d3 in 99.99% 2H2O(pD 5.0); 0.8 M guanidine-d6 in 100 mM sodium acetate-d3 in 99.99% 2H2O (pD 5.0); 0.8 M NaCl in 100 mMsodium acetate-d3 in 99.99% 2H2O. Elution by centri-fugation at 600g took three minutes. The first HSQCexperiment for each H/2H exchange experiment wasstarted uniformly at 30 minutes for 50 time-points(25 hours). The decay in intensity of individual aminoacid cross-peaks over time in 0 M salt and 0.8 M NaCl wasfit to a single-exponential function. In 0.8 M Gdn-HCl, theexchange was rapid enough that instead, a minimum orthreshold rate k was determined using the followingequation:
I1=I2 ¼ eKkðt1Kt2Þ (3)
where I1 is the cross-peak intensity at time t1 and I2 is thecross-peak intensity at time t2. Intrinsic rates of solventexchange for individual residues in IL-1b were calculatedwith in-house software based on values incorporatingnearest-neighbor effects, and activation energies.56
Protection factors for individual residues were thencalculated using the following equation:
PZ kint=kobs (4)
where P is the protection factor, kint is the intrinsicexchange rate and kobs is the observed rate. The relativestability of amide protons in the presence of denaturantwas then determined by:
Relative protection ZPgdnKHCl=Pnative (5)
Go-model
To model IL-1b, each amino acid is approximated withits single backbone Ca atom from pdb file 2I1B. The modelparameters used are the same as those used in ourprevious studies.4,10 The overall potential energy for agiven protein conformation is given by equation (6):
Etotal ZEbond CEangle CEdihedral CELJ CErep (6)
Consistent with the original Go-model,57 the minimumenergy of each energy term is obtained when the proteinis in the native folded state.
For covalent bond distance terms:
Ebond ZX
bonds
1
23rðrKr0Þ
2 (7)
where 3rZ100 kcal/mol is the bond energy, r is the bonddistance in the simulation, and r0 is the native Ca/Ca
bond distance, summed over each consecutive Ca/Ca
bond pair in pdb structure 2ILB.For the bond angle term:
Eangle ZX
angles
1
23qðqKq0Þ
2 (8)
where 3qZ20 kcal/mol is the bond angle energy, q is thebond angle in the simulation, and q0 is the Ca–Ca–Ca
native bond angle, summed over all Ca–Ca–Ca bondangles in pdb structure 2ILB.
For dihedral energies:
Edihedral ZX
dihedrals
½31f½1KcosðfKf0Þ�
C32f½1Kcosð3ðfKf0ÞÞ��
(9)
where 31fZ1:0 kcal=mol, 32
fZ0:5 kcal=mol, f is thedihedral angle in the simulation, and f0 is the CaCaCaCa
native dihedral angle, summed over all CaCaCaCa
dihedral angles in the pdb structure 2I1B.For long-range attractive interactions in the Go-model,
two Ca atoms in a protein were selected as attractive ifthey are separated by four or more residues and have anyheavy atoms less than 4 A apart. Each attractive Ca–Ca
contact is described by an attractive Lennard-Jones (LJ)potential:
ELJ ZX
jiKjjO3
3LJ 5sij
rij
!12
K6sij
rij
!10" #(10)
where 3LJZ1.0 kcal/mol is the contact energy, sij is thenative distance between the two contact atoms, i and j,given from the crystal structure, and rij is the distancebetween the two contact atoms i and j determined for agiven iteration of the simulation.
If any two atoms are not determined to be attractive orfall within three residues of each other (i,iC3), then theirinteraction is defined by a repulsive term:
Erep ZXi;j
3reps12
r12
� �12
(11)
where 3repZ1.0 kcal/mol is the repulsive energy, sij is thehard-sphere distance between the two repulsive atoms iand j (3.81 A), and rij is the distance between the tworepulsive atoms i and j determined for a given iteration ofthe simulation.
Molecular dynamics
MD simulations were carried out using AMBER 6software, compiled on a Linux platform, employing thesander_classic program as an integrator for initial energyminimization and subsequent molecular dynamics.58 Thefollowing describes the AMBER sander_classic moleculardynamics parameters used here. The specific parametervalues are listed in parentheses. The time-step was0.001 ps (DTZ0.001). Translational and rotational motionwas removed at the beginning of each run and every 1000time-steps thereafter (NTCMZ1, NSCMZ1000,NDFMINZ0). Initial velocities were randomly selected(INITZ3, IGZrandom). If the absolute value of the

Native Energy Landscape for IL-1b 345
velocity of any atom exceeded 500 A/time-step, velocitiesare scaled such that the absolute value of the velocity ofthat atomZ500 A/time-step (VLIMITZ500). Tempera-ture was maintained with an external bath using themethod of Berendsen59 with a coupling constant of 0.2 ps(NTTZ5, TAUTPZ0.2, TAUTSZ0.2). If the simulationtemperature Tsim exceeded the average temperature T bygreater than 10 K, velocities were scaled such that TsimZT. SHAKE was not used. A constant dielectric was used(IDIELZ1) with a delectric constant of 1 (DIELCZ1). Theparticle mesh ewald method was not used (IEWALDZ0).During each integration step, interactions between allatom pairs were calculated and this contact pair-list wasupdated only once at the beginning of the simulation(CUTZ9999, NSNBZ9999). No periodic boundary orpressure regulations were used (NTBZ0, NTPZ0).Structures and energies were saved every 1 ps (NTPRZ1000, NTWRZ1000, NTWXZ1000, NTWVZ1000,NTWEZ1000).
Sixty simulations were run independently for 10 nseach near the folding temperature Tf (550 K for thepresent model). For each structure sampled in thesimulations, Q, the fraction of native contacts, wascalculated where each native contact was determined tobe formed if it falls within 1.5 times the native distance.4,39
The Tf value of IL-1b was determined by the temperatureat which the simulated protein’s specific heat wasmaximal.40 This peak is typically found at a temperaturewhere the protein spends half its simulation time infolded conformations (Qw0.9) and the other half inunfolded conformations (Qw0.1). It should be noted that,here, the Tf value of 550 K is much greater than that ofproteins. This does not affect the interpretation, sincesimilar folding pathways have been determined formodels where parameters have been calibrated toproduce an experimentally matched folding Tf
13 valueand those that have not.40
Acknowledgements
We thank Dimitri Morikis for the program tocalculate intrinsic rates of solvent exchange, LarryA. Gross for mass spectrometric analyses, and theKeck Institute for computational support. Forfinancial support, we acknowledge, NIH grantGM54038 (to P.A.J.), NIH grant CA09523 (to M.R.),NSF grant MCB-0084797 (to J.N.O.), NIH Post-doctoral Fellowship GM064936-01 (to J.M.F.), the LaJolla Interfaces in Science Interdisciplinary TrainingProgram and the Burroughs Welcome Fund (toJ.M.F.) with additional support from PHY-0216576(to J.N.O.), PHY-0225630 (to J.N.O.), and the MARCpredoctoral fellowship to L.L.C. from NIGMS (adivision of NIH).
References
1. Bryngelson, J. D. & Wolynes, P. G. (1987). Spin glassesand the statistical mechanics of protein folding. Proc.Natl Acad. Sci. USA, 84, 7524–7528.
2. Bryngelson, J. D., Onuchic, J. N., Socci, N. D. &Wolynes, P. G. (1995). Funnels, pathways, and theenergy landscape of protein folding: a synthesis.Proteins: Struct. Funct. Genet. 21, 167–195.
3. Chiti, F., Taddei, N., White, P. M., Bucciantini, M.,Magherini, F., Stefani, M. & Dobson, C. M. (1999).Mutational analysis of acylphosphatase suggests theimportance of topology and contact order in proteinfolding. Nature Struct. Biol. 6, 1005–1009.
4. Clementi, C., Jennings, P. A. & Onuchic, J. N. (2000).How native-state topology affects the folding ofdihydrofolate reductase and interleukin-1beta. Proc.Natl Acad. Sci. USA, 97, 5871–5876.
5. Koga, N. & Takada, S. (2001). Roles of native topologyand chain-length scaling in protein folding: a simu-lation study with a Go-like model. J. Mol. Biol, 313,171–180.
6. Onuchic, J. N., Nymeyer, H., Garcia, A. E., Chahine, J.& Socci, N. D. (2000). The energy landscape theory ofprotein folding: insights into folding mechanisms andscenarios. Advan. Protein Chem. 53, 87–152.
7. Plaxco, K. W., Simons, K. T. & Baker, D. (1998).Contact order, transition state placement and therefolding rates of single domain proteins. J. Mol. Biol.277, 985–994.
8. Riddle, D. S., Grantcharova, V. P., Santiago, J. V., Alm,E., Ruczinski, I. & Baker, D. (1999). Experiment andtheory highlight role of native state topology in SH3folding. Nature Struct. Biol. 6, 1016–1024.
9. Clementi, C., Garcia, A. E. & Onuchic, J. N. (2003).Interplay among tertiary contacts, secondary struc-ture formation and side-chain packing in the proteinfolding mechanism: all-atom representation study ofprotein L. J. Mol. Biol. 326, 933–954.
10. Clementi, C., Jennings, P. A. & Onuchic, J. N. (2001).Prediction of folding mechanism for circular-permuted proteins. J. Mol. Biol. 311, 879–890.
11. Heidary, D. K., Gross, L. A., Roy, M. & Jennings, P. A.(1997). Evidence for an obligatory intermediate in thefolding of interleukin-1b. Nature Struct. Biol. 4, 1–10.
12. Heidary, D. K., O’Neill, J. C., Jr, Roy, M. & Jennings,P. A. (2000). An essential intermediate in the folding ofdihydrofolate reductase. Proc. Natl Acad. Sci. USA, 97,5866–5870.
13. Finke, J. M., Cheung, M. S. & Onuchic, J. N. (2004). Astructural model of polyglutamine determined from ahost-guest method combining experiments andlandscape theory. Biophys. J. 87, 1900–1918.
14. Rothwell, N. J. & Luheshi, G. N. (2000). Interleukin 1in the brain: biology, pathology and therapeutictarget. Trends Neurosci. 23, 618–625.
15. Rubartelli, A., Cozzolino, F., Talio, M. & Sitia, R.(1990). A novel secretory pathway for interleukin-1beta, a protein lacking a signal sequence. EMBO J. 9,1503–1510.
16. Finke, J. M., Roy, M., Zimm, B. H. & Jennings, P. A.(2000). Aggregation events occur prior to stableintermediate formation during refolding of inter-leukin 1beta. Biochemistry, 39, 575–583.
17. Griffin, W. S., Sheng, J. G., Roberts, G. W. & Mrak, R. E.(1995). Interleukin-1 expression in different plaquetypes in Alzheimer’s disease: significance in plaqueevolution. J. Neuropathol. Expt. Neurol. 54, 276–281.
18. Griffin, W. S., Nicoll, J. A., Grimaldi, L. M., Sheng, J. G.& Mrak, R. E. (2000). The pervasiveness of inter-leukin-1 in alzheimer pathogenesis: a role for specificpolymorphisms in disease risk. Expt. Gerontol. 35,481–487.
19. Roy, M. & Jennings, P. A. (2003). Real-time NMRkinetic studies provide global and residue-specificinformation on the non-cooperative unfolding of thebeta-trefoil protein, interleukin-1beta. J. Mol. Biol. 328,693–703.

346 Native Energy Landscape for IL-1b
20. Murzin, A. G., Lesk, A. M. & Chothia, C. (1992).b-Trefoil fold: patterns of structure and sequence inthe Kunitz inhibitors interleukins-1b and 1a andfibroblast growth factors. J. Mol. Biol. 223, 531–543.
21. Salem, G. M., Hutchinson, E. G., Orengo, C. A. &Thornton, J. M. (1999). Correlation of observed foldfrequency with the occurrence of local structuralmotifs. J. Mol. Biol. 287, 969–981.
22. Finzel, B. C., Clancy, L. L., Holland, D. R., Muchmore,S. W., Watenpaugh, K. D. & Einspahr, H. M. (1989).Crystal structure of recombinant human interleukin-1beta at 2.0 A resolution. J. Mol. Biol. 209, 779–791.
23. Clore, G. M., Wingfield, P. T. & Gronenborn, A. M.(1991). High-resolution three-dimensional structureof interleukin 1b in solution by three- and four-dimensional nuclear magnetic resonance spec-troscopy. Biochemistry, 30, 2315–2323.
24. Varley, P., Gronenborn, A. M., Christensen, H.,Wingfield, P. T., Pain, R. H. & Clore, G. M. (1993).Kinetics of folding of the all-beta sheet proteininterleukin-1 beta. Science, 260, 1110–1113.
25. Finke, J. M., Gross, L. A., Ho, H. M., Sept, D., Zimm,B. H. & Jennings, P. A. (2000). Commitment to foldedand aggregated states occurs late in interleukin- 1betafolding. Biochemistry, 39, 15633–15642.
26. Covalt, J. C., Jr, Roy, M. & Jennings, P. A. (2001). Coreand surface mutations affect folding kinetics, stabilityand cooperativity in IL-1 beta: does alteration inburied water play a role? J. Mol. Biol. 307, 657–669.
27. Heidary, D. K. & Jennings, P. A. (2002). Threetopologically equivalent core residues affect thetransition state ensemble in a protein folding reaction.J. Mol. Biol. 316, 789–798.
28. Finke, J. M. & Jennings, P. A. (2002). Interleukin-1 betafolding between pH 5 and 7: experimentalevidence for three-state folding behavior and robusttransition state positions late in folding. Biochemistry,41, 15056–15067.
29. Makhatadze, G. I., Clore, G. M., Gronenborn, A. M. &Privalov, P. L. (1994). Thermodynamics of unfoldingof the all b-sheet protein interleukin-1b. Biochemistry,33, 9327–9332.
30. Ervin, J., Larios, E., Osvath, S., Schulten, K. &Gruebele, M. (2002). What causes hyperfluorescence:folding intermediates or conformationally flexiblenative states? Biophys. J. 83, 473–483.
31. Srimathi, T., Kumar, T. K., Chi, Y. H., Chiu, I. M. & Yu,C. (2002). Characterization of the structure anddynamics of a near-native equilibrium intermediatein the unfolding pathway of an all beta-barrel protein.J. Biol. Chem. 277, 47507–47516.
32. Finke, J. M. & Jennings, P. A. (2001). Early aggregatedstates in the folding of interleukin-1b. J. Biol. Phys. 27,119–131.
33. Bai, Y., Sosnick, T. R., Mayne, L. & Englander, S. W.(1995). Protein folding intermediates: native-statehydrogen exchange. Science, 269, 192–197.
34. Chamberlain, A. K., Handel, T. M. & Marqusee, S.(1996). Detection of rare partially folded molecules inequilibrium with the native conformation of RNase H.Nature Struct. Biol. 3, 782–787.
35. Craig, S., Schmeissner, U., Wingfield, P. & Pain, R. H.(1987). Conformation, stability, and folding of inter-leukin 1b. Biochemistry, 26, 3570–3576.
36. Chrunyk, B. A., Evans, J. & Wetzel, R. (1993). Probingthe role of protein folding in inclusion body for-mation. In Protein Folding In Vivo and In Vitro(Cleland, J. L., ed.), pp. 46–58, American ChemicalSociety, Washington, DC.
37. Wishart, D. S. & Sykes, B. D. (1994). The 13C chemical-shift index: a simple method for the identification ofprotein secondary structure using 13C chemical-shiftdata. J. Biomol. NMR, 4, 171–180.
38. Driscoll, P. C., Gronenborn, A. M., Wingfield, P. T. &Clore, G. M. (1990). Determination of the secondarystructure and molecular topology of interleukin-1beta by use of two- and three-dimensional hetero-nuclear 15N-1H NMR spectroscopy. Biochemistry, 29,4668–4682.
39. Nymeyer, H., Garcia, A. E. & Onuchic, J. N. (1998).Folding funnels and frustration in off-latticeminimalist protein landscapes. Proc. Natl Acad. Sci.USA, 95, 5921–5928.
40. Cheung, M. S., Finke, J. M., Callahan, B. & Onuchic,J. N. (2003). Exploring the interplay between topologyand secondary structural formation in the proteinfolding problem. J. Phys. Chem. B, 107, 11193–11200.
41. Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E. &Edelman, M. (1999). Automated analysis of inter-atomic contacts in proteins. Bioinformatics, 15,327–332.
42. Nymeyer, H., Socci, N. D. & Onuchic, J. N. (2000).Landscape approaches for determining the ensembleof folding transition states: success and failure hingeon the degree of frustration. Proc. Natl Acad. Sci. USA,97, 634–639.
43. McCammon, J. A., Gelin, B. R., Karplus, M. &Wolynes, P. G. (1976). The hinge-bending mode inlysozyme. Nature, 262, 325–326.
44. Varley, P., Gronenborn, A. M., Christensen, H.,Wingfield, P. T., Pain, R. H. & Clore, G. M. (1993).Kinetics of folding of the all-b sheet protein inter-leukin-1b. Science, 260, 1110–1113.
45. Liu, C., Gaspar, J. A., Wong, H. J. & Meiering, E. M.(2002). Conserved and nonconserved features of thefolding pathway of hisactophilin, a beta-trefoilprotein. Protein Sci. 11, 669–679.
46. Wishart, D. S., Bigam, C. G., Holm, A., Hodges, R. S. &Sykes, B. D. (1995). 1H, 13C and 15N random coil NMRchemical shifts of the common amino acids. I.Investigations of nearest-neighbor effects. J. Biomol.NMR, 5, 67–81.
47. Clore, G. M., Bax, A., Driscoll, P. C., Wingfield, P. T. &Gronenborn, A. M. (1990). Assignment of the side-chain 1H and 13C resonances of interleukin-1 betausing double- and triple-resonance heteronuclearthree-dimensional NMR spectroscopy. Biochemistry,29, 8172–8184.
48. Driscoll, P. C., Clore, G. M., Marion, D., Wingfield,P. T. & Gronenborn, A. M. (1990). Complete resonanceassignment for the polypeptide backbone of Inter-leukin 1b using three-dimensional heteronuclearNMR spectroscopy. Biochemistry, 29, 3542–3556.
49. Bodenhausen, G. & Ruben, D. J. (1980). Naturalabundance nitrogen-15 NMR by enhanced hetero-nuclear spectroscopy. Chem. Phys. Letters, 69, 185–189.
50. Mori, S., Abeygunawardana, C., O’Neill Johnson, M.& Van Zul, P. C. M. (1995). Improved sensitivity ofHSQC spectra of exchanging protons at short inter-scan delays using a new fast HSQC (FHSQC)detection scheme that avoids water saturation.J. Magn. Reson. B, 108, 94–98.
51. Marion, D., Driscoll, P. C., Kay, L. E., Wingfield, P. T.,Bax, A., Gronenborn, A. M. & Clore, G. M. (1989).Overcoming the overlap problem in the assignmentof 1H NMR spectra of larger proteins by use ofthree-dimensional heteronulcear 1H–15N Hartman-Hahn-multiple quantum coherence and nuclear

Native Energy Landscape for IL-1b 347
Overhauser-multiple quantum coherence spec-troscopy: application to interleukin 1b. Biochemistry,28, 6150–6156.
52. Grzesiek, S. & Bax, A. (1992). Correlating backboneamide and side chain resonances in larger proteins bymultiple relayed triple resonance NMR. J. Am. Chem.Soc. 114, 6291–6293.
53. Wishart, D. S., Bigam, C. G., Yao, J., Abildgaard, F.,Dyson, H. J., Oldfield, E. et al. (1995). 1H, 13C and 15Nchemical shift referencing in biomolecular NMR.J. Biomol. NMR, 6, 135–140.
54. Farrow, N. A., Muhandiram, R., Singer, A. U., Pascal,S. M., Kay, C. M., Gish, G. et al. (1994). Backbonedynamics of a free and phosphopeptide-complexedSrc homology 2 domain studied by 15N NMRrelaxation. Biochemistry, 33, 5984–6003.
55. Clore, G. M., Driscoll, P. C., Wingfield, P. T. &Gronenborn, A. M. (1990). Analysis of the backbone
dynamics of interleukin-1b using two-dimensionalinverse detected heteronuclear 15N–1H NMR spec-troscopy. Biochemistry, 29, 7387–7401.
56. Bai, Y., Milne, J. S., Mayne, L. & Englander, S. W.(1993). Primary structure effects on peptide grouphydrogen exchange. Proteins: Struct. Funct. Genet. 17,75–86.
57. Go, N. (1983). Theoretical studies of protein folding.Annu. Rev. Biophys. Bioeng. 12, 183–210.
58. Pearlman, D. A., Case, D. A., Caldwell, J. W., Ross,W. R., Cheatham, T. E., III, DeBolt, S. et al. (1995).AMBER, a computer program for applying molecularmechanics, normal mode analysis, moleculardynamics and free energy calculations to elucidatethe structures and energies of molecules. Comput.Phys. Commun. 91, 1–41.
59. Berendsen, H. J. (1984). Molecular dynamics withcoupling to an external bath. J. Chem. Phys. 81,3684–3690.
Edited by C. R. Matthews
(Received 8 September 2004; received in revised form 2 February 2005; accepted 21 February 2005)
Recommended

















