Embed Size (px)
Citation preview

Farmacología básicaEquipo 3AntiviralesAntiparasitariosAntifúngicos

Fármacos antiparasitarios
Se describen los fármacos antiparasitarios internos (protozoos y helmintos) y externos (artrópodos).

Antiprotozoarios

Diloxánido
Derivado de la dicloroacetamida. Eficaz en la amebiasis intestinal asintomática, en la que destruye el parásito.
Vo en adultos es de 500 mg 3 veces al día/10 días. Niños 20 mg/kg/día.

Yodoquinol
Erradicar los quistes por protozoos en la amebiasis intestinal. Normalmente se administra después del tratamiento de las infecciones extraintestinales por amebas.
Vo adultos: 650 mg, 3 veces al día/20 días. Niños 30-40 mg/kg/día en 3 tomas.

Emetina y deshidroemetina
Eficaces en la amebiasis hepática (abscesos). Inhiben la síntesis proteica de los trofozoitos de E histolytica.
1 mg/kg/día por vía intramuscular. Deshidroemetina 1-1.5 mg/kg/día durante máximo 5 días. En niños 2 tomas misma dosis.

Paromomicina
Antibiótico aminoglucósido que no se absorbe por vía oral y que ejerce su acción amebicida en la luz intestinal.
25-30 mg/kg, en 3 tomas por 7 días (niños y adultos).

Tetraciclinas
Profilaxis y tratamiento de Plasmodium falciparum. Las tetraciclinas son amebicidas que se usan en combinación con otros amebicidas intestinales.La tiaciclina es el más potente.

Cloroquina
Eficaz en la amebiasis hepática, en la que se utiliza sola o combinada con emetina. Adultos dosis oral 1 gr/día por 2 días, seguida de 500 mg/día por 3 semanas. Niños 10 mg/kg/día.

Giardicidas
La giardiasis es una infección del intestino delgado producida por Giardia lambia. Se transmite por contagio directo e indirecto por los alimentos o el agua.

Quinacrina
Se intercala en el ADN e inhibe la ADN-polimerasa. Vo adultos 100 mg, 3 veces al día por 5-7 días y en niños 7 mg/kg.Tras su administración prolongada, causa una coloración amarilla.

Furazolidona
Ataca a G. lambia y bacterias entéricas grampositivas y gramnegativas, estafilococos, enterococos, Salmonella y E. coli.Dosis es de 100 mg, 4 veces al día por 7 días y para niños 5 mg/kg/día en 4 tomas.

Leishmanicidas
La enfermedad se inicia después de un periodo de incubación de 2-20 semanas con una mácula cutánea eritematosa y pruriginosa, que se transforma en pápula,
nódulo y finalmente úlcera, con costra y bordes elevados.

Antimoniales pentavalentes
Pentamidina

Hidroxistilbamidina
Segunda elección; 250 mg/día por vía intravenosa, 3 dosis en total con intervalos de 10 días.

Anfotericina B
Utilizado contra enfermedades fúngicas. Por vía oral apenas se absorbe. La semivida de eliminación, en adultos, es de 24 horas.
Dosis: 0.5-1 mg/kg/día por vía intravenosa.Dosis total acumulativa: 750-850 mg.

Alopurinol
Es de segunda elección en la leishmaniasis visceral. Se administra por vo en dosis de 7 mg/kg, 3 veces al día durante 2-10 semanas.

Ketoconazol e itraconazol
Altera las funciones de la membrana de Leishmania. Vo contra leishmaniasis cutánea. El ketoconazol 600 mg/día. Se recomienda monitorizar la función hepática durante la terapia.

Antipalúdicos
El paludismo o la malaria es una enfermedad adquirida de forma natural por la picadura de diferentes mosquitos vectores del género Anopheles, y el hombre constituye la única fuente de infección.
Infección protozoaria caracterizada por la aparición de fiebre, escalofríos y sudación, anemia, esplenomegalia y una evolución crónica.

Quinolinas
Cloroquina● Mecanismo de acción: interfiere en la síntesis de ácidos
nucleicos● Farmacocinética: Vo, vía intramuscular y subcutánea.
Se distribuye ampliamente en los tejidos y se elimina muy lentamente.
● Reacciones adversas: trastornos gastrointestinales, prurito, cefalea transitoria y alteraciones visuales.

● Indicaciones terapéuticas: en infecciones por P. falciparrum. Dosis oral de 1 g., seguido de 500 mg a las 6 horas, y posteriormente 500 mg/día por 2 días. En adultos la dosis inicial es de 2,5 g.

Amodiaquina
Análogo de la cloroquina, con una eficacia palúdica superior a ésta. Plasmodium falciparum resistentes a la cloroquina. Absorción digestiva rápida.

Derivados quinolinametanol
Quinina● Mecanismo de acción: unión a un componente del
pigmento palúdico hemozoína y a la interacción con el ADN.
● Farmacocinética: se absorbe en el aparato gastrointestinal y se distribuye en los tejidos. La concentración máxima plasmática se alcanza a las 3 horas. Semivida de 11 horas.

● Reacciones adversas: Síndrome cinconismo, que se manifiesta por náuseas, vértigo, cefalea y visión borrosa.
● Indicaciones terapéuticas: Vo 600 mg, 3 veces al día (en niños 25 mg/kg/día, en 3 tomas por 3 días.

Mefloquina
Se une al pigmento hemozoína, pero no se intercala con el ADN. Se administra sólo por vía oral. Semivida de 30 días.Dosis única de 1 g, o bien 500 mg cada 2 semanas para la profilaxis.

Proguanil
Primer antifolato usado en la profilaxis y el tratamiento del paludismo. Presenta una absorción oral lenta y una semivida de 16-20 horas; se excreta principalmente por la orina.Dosis en adultos 200 mg/día. Seguro para embarazadas.

Derivados 8-aminoquinolinas
Primaquina● Mecanismo de acción: interfiere en la cadena de
transporte electrónico en las mitocondrias de Plasmodium y en la síntesis de pirimidina.
● Farmacocinética: Vo., se metaboliza por completo en el hígado. Semivida de 3-6 horas.

● Reacciones adversas: molestias gastrointestinales.
● Indicaciones terapéuticas: dosis en adultos es de 15 mg/kg/día por 2 semanas, niños 0.3 mg/kg/día.

Atavaquona
Inhibe selectivamente el transporte de electrones de la mitocondria y, por consiguiente, la síntesis de pirimidinas. Está también indicada en el tratamiento de la neumonía leve o moderada por Pneumocystis jiroveci.

Toxoplasmicidas
La toxoplasmosis es una enfermedad granulomatosa generalizada o del SNC causada por el protozoo intracelular T. gondii.Fármacos utilizados: pirimetamina-sulfadiacina, espiramicina y clindamicina.

Tricomonicidas
La tricomoniasis es una infección protozoaria que afecta al hombre, causada principalmente por T. vaginales. Este organismo habita en las vías urinarias del huésped parasitado, donde puede producir vaginitis en la mujer y uretritis en el hombre. Fármaco de elección es el metronidazol.

Tripanosomicidas
La tripanosomiasis africana o enfermedad del sueño se origina por las picaduras de la mosca tsetsé infectadas por dos subtipos de Trypanosoma brucei.Los fármacos usados son pentamidina, suramina y arsenicales orgánicos.

Suramina
Es una alternativa a la pentamidina en su etapa precoz.● Farmacocinética: intravenosa lenta. Semivida de 48
horas, semivida terminal de eliminación de 50 días.● Reacciones adversas: relativamente tóxica en especial
en pacientes desnutridos. Puede provocar lesión renal aguda.
● Indicaciones terapéuticas: 10% en adultos, dosis inicial de 100-200 mg cada 24-48 horas. Niños 20 mg/kg.

Melarsoprol
Actúa bloqueando el metabolismo energético del parásito. ● Farmacocinética: vía parenteral.● Reacciones adversas: muy tóxico. Fiebre, dolor torácico
y abdominal, diarrea, infecciones conjuntivales, vasodilatación y encefalopatía.
● Indicaciones terapéuticas: adultos 3,6 mg/kg/día por 3 días. Niños 18,8 mg/kg/día por 3 días.

Nifurtimox
Rotura del ADN del parásito. Se asocia a numerosas reacciones adversas; raras veces el paciente puede finalizar la terapia recomendada 3-4 meses.Eficaz en dosis orales de 8-10 mg/kg/día en adultos, y de 15 mg/kg/día en niños, en 4 tomas.

Benznidazol
Bloquea la síntesis de ácidos nucleicos y proteínas, pero se metaboliza en menor grado a radicales libres.
Vo 5 mg/kg/día en adultos y 10 mg/kg/día niños por 1-4 meses.

Antihelmínticos
Las infecciones por helmintos constituyen un problema para la salud pública por sus efectos sobre el estado nutricional e inmunológico. los parásitos helmintos que infectan al hombre incluyen las clases nematodos, cestodos y trematodos.

Dietilcarbamacina
Provoca la inmovilización del parásito.● Farmacocinética: se absorbe rápidamente por vo,
concentración plasmática máxima en 12 horas, semivida es de 2-10 horas.
● Reacciones adversas: cefaleas, mialgias, artralgia, anorexia, náuseas, vómitos.
● Indicaciones terapéuticas: niños 35 mg el primer día; 35 mg, 3 veces al día el segundo día; 75 mg 3 veces al día el tercer día y 2 mg 3 veces al día en los 18 días siguientes.

Mebendazol
Resulta útil en el tratamiento de infestaciones intestinales por vermes redondos. ● Mecanismo de acción: eliminación selectiva de los
microtúbulos citoplasmáticos en las células intestinales● Farmacocinética: la absorción aumenta con alimentos
ricos en grasas. Eliminación de primer paso a través del hígado, orina y heces.
● Reacciones adversas: dolor abdominal y diarrea.

Antivíricos.
★ La epidemia del SIDA ha constituido un estímulo importante para la investigación.
★ Virus: Parásitos intracelulares obligados que se aprovechan de las células que infectan para sobrevivir y replicarse.
➔ Interferencia en la adhesión del virus a la célula que infecta.
➔ Inhibición en procesos de transcripción y replicación.
➔ Interferencia en el ensamblaje vírico.


Análogos de los nucleósidos.
ACICLOVIR. Su actividad antivírica está limitada a algunos virus del herpes. (VHS1 y 2, VVZ, CMV, V Eipsten-Barr).Mecanismo de acción y actividad antivírica:● Inhibición de la síntesis del ADN del virus.● Tras penetrar por la célula infectada● Aciclovir trifosfato (forma activa)● Inhibe ADN polimerasa (actuando como un finalizador de
la cadena de ADN impidiendo l unión excesiva en su cadena lateral de varios nucleótidos.
aciclovir monofosfato timidincinasa


Farmacocinética: Vía oral, intravenosa o tópica.Su difusión tisular es excelente, capaz de atravesar membrana placentaria y alcanzar en el líquido cefalorraquídeo concentraciones de hasta el 50% de las plasmáticas.Reacciones adversas: Cuando se administra por vía intravenosa: flebitis en el punto de infusión, exantema cutáneo,diaforesis, náuseas y vómitos, puede alcanzar insuficiencia renal reversible.

VALACICLOVIR (Escasa absorción oral del aciclovir).
Se transforma en aciclovir tras su ingestión por vía oral. Mecanismo de acción y actividad antivírica: Es un profármaco del aciclovir, por lo que su mecanismo de acción y actividad son iguales a los expuestos para dicho fármaco.
Farmacocinética: Se absorbe rápidamente por vía oral. Biodisponibilidad de 3-5 veces superior a la del aciclovir, alcanza unos niveles séricos similares a los que alcanza el aciclovir por vía IV.

Reacciones adversas: Perfil de toxicidad similar al del aciclovir.

PENCICLOVIR-FAMCICLOVIR Análogo acíclico de la guanosina con actividad clínicamente relevante frente a algunos tipos de herpes. Mecanismo de acción y actividad antivírica: (Similar al del aciclovir). Tras su penetración en la célula sufre 3 fosforilaciones, una vez convertido en penciclovir-3-P, interrumpe la cadena de nucleótidos y aborta la replicación del virus.

Farmacocinética: Se absorbe por vía oral, las concentraciones intracelulares de forma activa se mantienen elevadas por mucho tiempo.Reacciones adversas: Cefalea, náuseas, vómitos.No se ha observado toxicidad hepática o hematológica. Indicaciones terapéuticas: Ha demostrado ser útil en herpes: zoster, genital y labial. Eficaz en la prevención del herpes genital recurrente.

GANCICLOVIR Análogo acíclico de la guanosina, con actividad frente al CMV 10 veces superior a la del aciclovir.Mecanismo de acción y actividad antivírica: Similar al del aciclovir, actúa como finalizador de la cadena de ADN tras ser convertido en ganciclovir-3-P. Es mucho más efectivo contra CMV y VH6 (mayor afinidad de la timidincinasa de estos virus por el fármaco).

Farmacocinética: Vía oral intravenosa. Alcanza LCR de 30% a 68%. Se elimina por leche materna, cruza barrera placentaria. Sin modificaciones al riñón.Reacciones adversas: Toxicidad medular reversible (efecto adverso más frecuente). Indicación terapéutica: Tratamiento de elección de todas las formas de enfermedad por CMV en pacientes inmunodeprimidos (incluidos pacientes con SIDA y trasplantados.

VALGANCICLOVIR Biodisponibilidad oral superior a la del ganciclovir, en el que se transforma completamente tras su absorción.
Mecanismo de acción y actividad antivírica: Es un profármaco del ganciclovir, su efecto y actividad son iguales.
Farmacocinética: Biodisponibilidad oral 60%, su absorción es mayor si se toma con la comida.

Reacciones adversas: Similares a las del ganciclovir oral.
Indicaciones terapéuticas: Enfermedades por CMV en pacientes con SIDA, tratamiento de infecciones con riesgo vital en pacientes trasplantados.

RIBAVIRINA Análogo de la guanosina que en su molécula presenta un anillo incompleto de purina y que tiene actividad in vitro frente muchos virus.
Mecanismo de acción y actividad antivírica: No se conoce con precisión, probablemente según el tipo de virus involucrado. Requiere fosforilarse en el interior de la célula para ejercer su acción.

Farmacocinética: Vía oral, intravenosa o aerosol.
Reacciones adversas: Ha demostrado ser teratogénica, por lo que su uso es contraindicado en el embarazo. Anemias (uso prolongado), náuseas, cefalea, insomnio.
Indicaciones terapéuticas:
Aerosol: VSR en niños, tratamiento de elección para VHC junto con interferón de 6-12 meses.

TRIFLURIDINA Análogo halogenado de la desoxiuridina con actividad in vitro frente a virus herpes y adenovirus que administrado por vía sistémica es muy tóxico.
“Queratinoconjuntivitis” por herpes simple, en la que se emplea por vía tópica.

LAMIVUDINA Fármaco que se describe en profundidad para el tratamiento con SIDA.
Además se utiliza como tratamiento por infección crónica con VHB.

ENTECAVIRAnálogo de la guanosina que inhibe de manera selectiva la replicación del VHB, la frecuencia con la que aparecen mutaciones resistentes es notable.

Análogos de los nucleótidos.
CIDOFOVIR Análogo acíclico fosforilado de la citosina, excelente actividad in vitro frente a todos los tipos de herpes.
Mecanismo de acción y actividad antivírica:
No requiere enzimas víricas para ejercer su acción antivírica, es fosforilado directamente por cinasas celulares y actúa como un inhibidor competitivo de la ADN-polimerasa del virus.

Farmacocinética: Solo se administra por vía intravenosa, no se absorbe por vía oral. Su semivida es de 20-30 hrs, lo que permite su administración semanal.
Reacciones adversas: Gravemente nefrotóxico, aunque se puede disminuir con la administración simultánea de probenecida.
Indicaciones terapéuticas: Tx de retinitis por CMV.

ADEFOVIR Análogo de la adenina con actividad in vitro frente al VHB y VIH y a algunos tipos de herpes. Mecanismo similar al de cidofovir.No se absorbe por vía oral, se utiliza como profármaco).Semivida de 16-18 hrs, se excreta por el riñón.E. adversos: cefalea y dispepsia.

TENOFOVIR Análogo acíclico de los nucleótidos con una actividad muy similar a la de adefovir frente al VHB y al VIH y que se ha demostrado eficaz en la coinfección de ambos virus.

Análogos de los pirofosfatos.
FOSCARNET Análogo orgánico del pirofosfato inorgánico con una actividad frente a una gran variedad de virus.
Mecanismo de acción y actividad antivírica:
No requiere fosforilación intracelular, ya que se une directamente a la ADN-polimerasa del virus en el lugar de unión del pirofosfato e impide la liberación de éste a partir de los desoxinucleósidos trifosfato y la elongación de ADN.

Farmacocinética: Debe administrarse por vía intravenosa, porque por vía oral no se absorbe. Se elimina de forma exclusiva por la orina.
Reacciones adversas: Nefrotoxicidad, generalmente reversible, aparece hasta en el 30% de los pacientes, es menor si se hidrata el paciente.
Indicaciones terapéuticas: TX de enfermedades por CMV (menos mielotóxica que el ganciclovir).

Aminas tricíclicas.
AMANTADINA. Amina tricíclica con actividad exclusiva frente al virus de la gripe A. Al parecer inhibe la descapsidación del virus de la gripe una vez que éste ha penetrado en la célula.Se absorbe bien por vía oral, no se metaboliza, semivida de 12-17 hrs. y se elimina por el riñón.“Toxicidad neurológica” (principal efecto adverso). Letargia, ansiedad, insomnio, confusión.

RIMANTADINA Fármaco similar a la amantadina con un importante metabolismo hepático y cuya principal ventaja es una menor incidencia de efectos adversos.
Indicaciones iguales a las de la amantadina.

Análogos del ácido siálico.
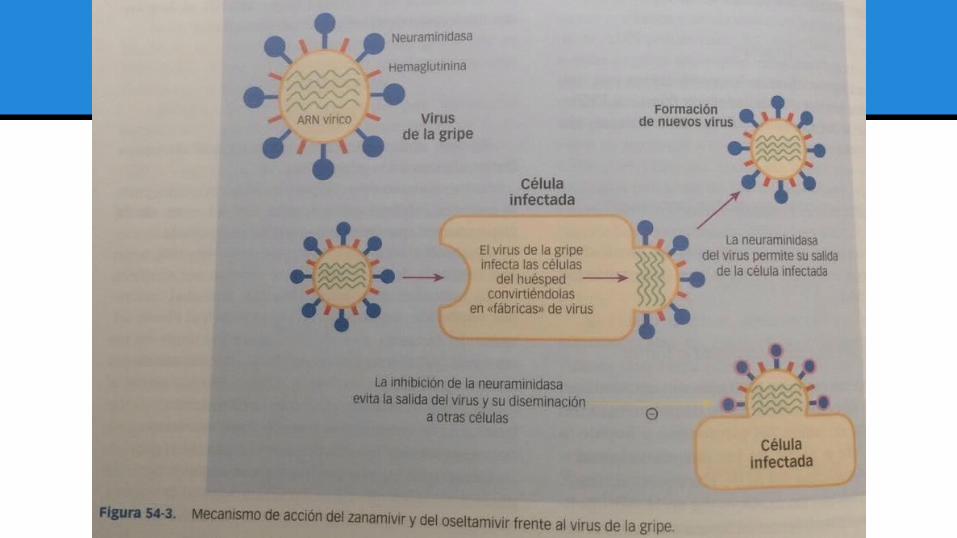
ZANAMIVIR Análogo del ácido siálico que inhibe específicamente la neuraminidasa del virus de la gripe.
Mecanismo de acción y actividad antivírica:
Inhibidor de la neuraminidasa del virus de la gripe, molécula que le permite entrar y salir de las células que infecta.
Actividad frente a los virus de la gripe A y B.

Farmacocinética: Inhalación oral y alcanza concentraciones elevadas en la mucosa de las vías respiratorias superiores.
Reacciones adversas: Menos del 5% de los pacientes presentan efectos secundarios, la mayoría son leves, destacando el broncoespasmo.
Indicaciones terapéuticas: Acorta la duración d la gripe, se administra en las primeras 48 hrs. del comienzo del cuadro.

OLSETAMIVIR
Fármaco muy similar al zanamivir, con diferencia de que éste se administra por vía oral (biodisponibilidad del 70%). Muy seguro, solo del 5% - 10% de los pacientes presentan náuseas y vómito.

Interferones.
➔ Proteínas producidas por las células eucariotas, con especificidad de especie, poseen una amplia actividad antivírica.
➔ Se conocen tres clases (alfa, delta y gamma).

➔ Su mecanismo de acción no es completamente conocido, carecen de actividad antivírica directa. Al parecer, provocan en la célula infectada l producción de proteínas naturales que inhiben la replicación del virus.
➔ No se absorben por vía oral, se administran por vía intramuscular, subcutánea.
➔ Su actividad antivírica permanece durante varios días.



Fármacos antirretrovirales.
❖ Desde 1981 más de 60 millones de personas se han contagiado por el VIH y, actualmente es la cuarta causa de muerte en todo el mundo.
❖ Con el desarrollo del tx antirretroviral (TAR) se ha conseguido, sobre todo, en los últimos 10 años mejorar la supervivencia y la calidad de vida de las personas infectadas por el VIH.
❖ Desgraciadamente, no todos tienen esa accesibilidad.

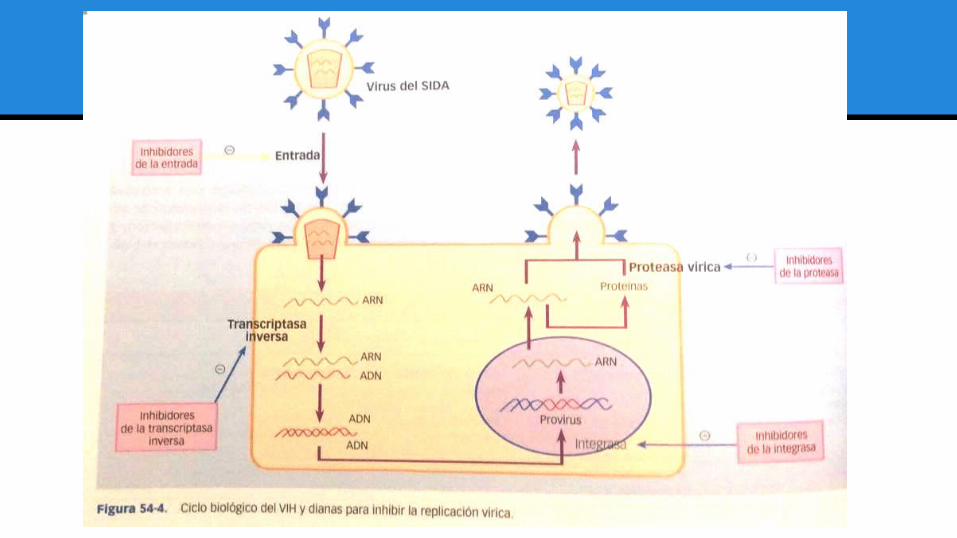
Ciclo biológico del VIH.
➢ Fases: Temprana y tardía. (Divididas por el estado de integración provírica).
➢ Etapas tempranas: ● Entrada en la célula Interacción con distintos receptores.● Fusión de membranas.● Desencapsidación del genoma del virus.● Retrotranscripción del ARN vírico en una doble hebra de ADN.● Transporte al núcleo y su integración en el genoma celular.
Latencia proviral, sin replicación detectable.

➢ Segunda fase:● Inicio de transcripción, síntesis y procesamiento del
ARN mensajero.● Expresión y procesamiento de las proteínas del virus.● Ensamblaje en partículas que salen de la célula
mediante un proceso de gemación.

Zidovudina
•Análogo estructural de la timidina.
•Zidovudina → monofosfato → difosfato → trifosfato.
•Trifosfato: sustrato e inhibidor de la TI del virus, bloqueando la formación de ADN provírico.
•Biodisponibilidad = 60-70%
•CPM = 30-190 min.
•Semivida plasmática = 1.1 hr
•Semivida intracelular = 3 hr
•Metabolismo = 75% en hígado, el resto en riñon sin metabolizar.

Zidovudina
•Reacciones adversas: anemia, leucopenia, neutropenia, hepatotoxicidad, náuseas, diarrea, miopatía, acidosis láctica, dispepsia, dolor abdominal.
•Dosis = 250-300 mg/12 hr.

Didanosina
•Inhibidor de la transcriptasa inversa, análogo de la base púrica inosina.
•Didanosina → ddATP: Inhibe la replicación y el TI del VIH.
•Libera sus microesferas en el estómago y se absorbe en el duodeno.
•CPM = 2 hr.
•Absorción máxima = 30-45 min.
•Biodisponibilidad = 30-40%
•Semivida intracelular = 25-40 hr.
•Metabolismo = renal.
•Reabsorción = 20% (administrado oral).

Didanosina
•Reacciones adversas: pancreatitis, neuropatía periférica, lipodistrofia, diarrea, náuseas, vómito, hiperuricemia.
•Dosis = ≥60 kg/400 mg/día <60 kg/250 mg/día

Zalcitabina
•Análogo sintético del nucleósido natural desoxicitina. Fase de replicación.
•Toxicidad
•Neuropatía periférica, pancreatitis, úlceras orales y esofágicas, esteatosis hepática, acidosis láctica lipodistrofia.

Estavudina
•Análogo de la timidina. Fase de replicación.
•Estavudina → estavudina-trifosfato → inhibe TI y síntesis de ADN vírico
•Biodisponibilidad = 100%
•Semivida plasmática = 1-1.6 hr
•Semivida intracelular = 3.5 hr
•Reacciones adversas: toxicidad mitocondrial, neuropatía periférica, lipodistrofia, esteatosis hepática, acidosis láctica, pancreatitis, vomito, hiperlipidemia, erupciones cutáneas.
•Dosis = > 60 kg/40 mg/12 hr <60 kg/30 mg/12 hr

Lamivudina
•Análogo de la citosina.
•Lamivudina → lamivudina-trifosfato → inhibe TI y elongación de la cadena de ADN
•Fase latente.
•Biodisponibilidad = 82%
•Semivida plasmática = 2.5 hr
•Semivida intracelular = 11-16 hr
•Dosis = 150 mg/12 hr 300 mg/24 hr
•Reacciones adversas: diarrea, astenia, cefalea, vómito.

Emtricitabina
•Análogo sintético de citidina.
•Emtricitabina → emtricitabina-5’-fosfato → interrumpe la cadena de ADN
•CPM = 1-2 hr
•Biodisponibilidad = 93%
•Excreción por orina 86% y en heces 14%
•Semivida intracelular = 39 hr

Emtricitabina
•Dosis = 200 mg/24 hr
•Reacciones adversas: cefalea, intolerancia digestiva, exantema cutáneo, lipodistrofia, neutropenia, anemia, esteatosis hepática, acidosis láctica.

Abacavir
•Análogo de la guanosina.
•Interrumpe la replicación celular.
•Abacavir → abacavir-monofosfato → carbovir-monofosfato → carbovir-trifosfato
•Semivida intracelular: 3.3 hr
•Biodisponibilidad = 76-100%
•

AbacavirDosis = 300 mg/12 hr 300 (2) mg/24 hr
•Reacciones adversas: hipersensibilidad con fiebre, letargo, erupciones cutáneas, síntomas respiratorios y gastrointestinales, edema, hipotensión, cefalea, conjuntivitis.

Inhibidores de la transcriptasa inversa análogos de los nucleótidos

Tenofovir
•Tenofovir → tenofovir disoproxil → tenofovir difosfato
•Semivida intracelular = 10 (cél. Activadas) y 50 (cél. En reposo) hr
•Termina de la cadena de ADN viral.
•CPM = 1 hr en ayunas y 2 hr con alimento.
•Dosis = 245 mg/24 hr
•Reacciones adversas: vómito, dolor abdominal, diarrea, astenia, cefalea.

Inhibidores de la transcriptasa inversa no análogos de nucleósidos

Efavirenz
•Inhibe la ADN-polimerasa.•Biodisponibilidad = 100%•CPM = 3-5 hr•Metabolizado por el sistema del citocromo P-450.•Dosis = 600 mg/24 hr•Reacciones adversas: síntomas del sistema nervioso, erupciones cutáneas.

Nevirapina
•Se une directamente a la TI.•CPM = 4 hr● Semivida plasmática = 12-24 hr● Metbolismo por el citocromo P-450● Dosis = 200 mg/24 hr/14 días → 200mg/12 hr● Reacciones adversas: hepatotoxicidad, exantema
cutáneo.

Inhibidores de la proteasa

Lopinavir
● biodisponibilidad = 70%● Metabolismo por citocromo P-450● Dosis = 400-100 mg/12 horas.● Reacciones adversas: diarrea, nauseas,
hiperglucemia, hipercolesterolemia, aumento de las transaminasas.

Fosamprenavir
● Fosamprenavir → hidrolisis en intestino → amprenavir y fosfato inorgánico
● CPM = 2 hr
● Semivida = 7.7 hr
● Metabolismo en CYP3A4.
● Dosis = 700 mg/12 hr con ritonavir 100/día

Atazanavir
● Previene la formación de viriones maduros.● administrar con alimentos.● Metabolismo por CYP3A4.● Dosis = 300 mg con ritonavir 100 mg / díaReacciones adversas: ictericia, vómito,
diarrea, dolor abdominal, dispepsia, astenia, fatiga, lipodistrofia.

Saquinavir
CPM = 3-4 hrBiodisponibilidad = 4% (gelatina dura) y 12-16% (gelatina blanda)Administrarse con alimentosEliminación en heces.Metabolismo por CYP3A4.Dosis = (2) 500 mg con 100 de ritonavir / 12 hrReacciones adversas: diarrea, nauseas, flatulencias
Indinavir
Biodisponibilidad = 30-60%CPM = <1 hrEliminacion en heces (83%) y orina (19%)Dosis = (2) 400 mg/8 hr Administrar en ayunas, hidratación abundanteReacciones adversas: hiperglucemia, hiperlipidemia, piel seca, erupciones cutáneas.

Tipranavir
Indicado en casos de resistencia.Inductor e inhibidor de CYP3A4.Unión alta a proteínas plasmáticas.Dosis = 500 mg con 200 mg de ritonavir / 12 hrReacciones adversas: hepatotoxicidad, vómito, flatulencias, hiperlipidemia, hemorragias, deposiciones blandas, dolor abdominal.

Darunavir
En pacientes que no han respondido a varios tratamientos.CPM = 2.5-4 hrBiodisponibilidad = 37%, 80% con 200 mg de ritonavir.Administración con alimentos.Dosis = 600 mg con 100 mg de ritonavir / 12 hrMetabolizado por CYP.Reacciones adversas: nausea, diarrea, cefalea, lipodistrofia.

Inhibidores de la entrada

Enfuvirtida
Inhibidor de la fusión.En pacientes con resistencia a varios tratamientos.Se cataboliza en los aminoácidos.Dosis = 90 microgramos/ 12 hr en inyección subcutánea en brazo, cara anterior del muslo o abdomenReacciones adversas: reación cutánea, dolor, eritema, hematomas, equimosis.

Antifúngicos

Fármacos Antifúngicos
Aumento en la frecuencia.Aumento en la morbilidad.Pacientes inmunodeprimidos.El pronóstico depende de:● Precocidad del diagnóstico.● Tratamiento.● Corrección de los factores de riesgo.
Infección Microorganismo
Candidiasis Cándida.● Candidiasis superficiales
(mucocutáneas)
● Candidiasis profunda.
Aspergilosis Aspergillus ● Mecanismos no infecciosos
(Aspergilosis broncopulmonar alérgica).
● De colonización (aspergiloma)
● Invasión tisular en pacientes inmunodeprimidos
Infección Microorganismo
Dermatofitos o tiñas. Hongos miceliales.Invaden los tejidos queratinizados.


Antibióticos
Anfotericina B.Actinomiceto Streptomyces nodosus.Infecciones fúngicas profundas.Amplio espectro fungicida in vitro e in vivo.

Mecanismo de acciónActividad antimicótica mediante la creación de poros en la membrana plasmática del hongo que alteran su función.
Se une al ergosterol y el centro hidrófilo forma un conducto transmembrana que interfiere en la permeabilidad y función de barrera osmótica.
Pérdida de iones que conducen a la muerte celular.
Diana terapéutica debe atravesar la pared celular del hongo.
Puede desempeñar un papel en los mecanismos de resistencia y sinergía con otros antifúngicos que actuén sobre la pared.

Actividad y resistencia.
Efecto antifúngico de la anfotericiba B puede ser:Fungistático o fungicida.Depende de la sensibilidad del microorganismo .Influido In vitro por PH (max actividad 6- 7.5). La mayoría de levaduras y hongos que causan infecciones en el ser humano son sensibles a la anfotericina B.


La resistencia a la anfotericina no es habitual.Puede ser por:Disminución en la cantidad de ergosterol de la membrana.Alteración en su composición que la hace menos afín.

Farmacocinética:

Anfotericina B desoxiclonato.
Mínima absorción en el tubo digestivo.Tratarse por vía intravenosa.
Concentraciones más altas Hígado bazo, pulmón y riñones.
50-60% de las concentraciones plasmáticas mínimas.
Líquido pleural, peritoneal, sinovial y el humor acuoso inflamados
2% Penetración en el líquido cefalorraquídeo .Aumenta en caso de inflamación meníngea.
Mínimo El paso al humor vítreo y al líquido amniótico.Cruza barrera placentaría.

No se conocen metabolitos.
Eliminación muy lenta por vía biliar < 15%
RenalSe detectó en orina hasta siete semanas después del tratamiento
3%
Perfil farmacocinético en niños Menor volumen de distribución.Mayor aclaramiento.
Pico sérico Se consigue con la mitad de la dosis de un adulto.

Anfotericina B Complejo lipídico
Picos séricos y áreas bajo la curva ● Menores que la anfotericina B desoxicolato.
● Por su rápida distribución tisular y el aclaramiento total.
Distribución tisular ● No es plenamente conocida.● Se acomula en el sistema monocito-
macrófago.● Alcanza concentraciones elevadas
en pulmón, hígado y bazo.● Concentraciones bajas en ganglios
linfáticos, riñón, corazón y cerebro.

Anfotericina B liposomal.
Picos séricos y áreas bajo la curva Menores que la anfotericina B desoxicolato y que el complejo lipídico.
Volumen de distribución y aclaramiento Son menores debido a que los liposomas no pueden eliminarse por la filtración glomerular.
Concentraciones más altas
Concentraciones más bajas
● En órganos ricos en células del sistema reticuloendotelial.Hígado y bazo.
● Riñón y pulmón.

Anfotericina B en dispersión coloidal
Concentraciones plasmáticas más bajas y mayores concentraciones hepáticas.

Reacciones adversasFiebre y escalofríos Pre Medicación con fármacos que evitan la aparición de estos efectos.Paracetamol 30 min antes de la infusión
En la primera semana y posteriormente disminuyen.Fiebre 40 ºC
Arritmias ventriculares Infusión rápida
Náuseas y vómitos Frecuente, disminuye con el tiempo
TromboflebitisExtravasación
Asociada a la infusión intravenosaNecrosis tisular.
Reacciones anafilácticas son raras
Insuficiencia respiratoria y síndrome de distrés respiratorio del adulto con infiltrados intersticiales
Transfusión de leucocitos de forma concomitante con la anfotericina B

Reacciones adversas.Toxicidad renal80 % de los pacientes aproximadamente
La anfotericina B disminuye el flujo sanguíneo renal y la filtración glomerular, afectando la reabsorción de electrolitos en los túbulos proximal y distal.
Acidosis tubular En px que reciben dosis de 0.5-1g y suelen acompañarse de hipopotasemia e hipomagnasemia.Se revierte cuando se interrumpe la terapia
Anemia normocítica normocrómica Consecuencia de la inhibición de la síntesis de eritropoyetina y por acción directa sobre la médula ósea
Insuficiencia renal transitoria en el feto
Toxicidad hepática
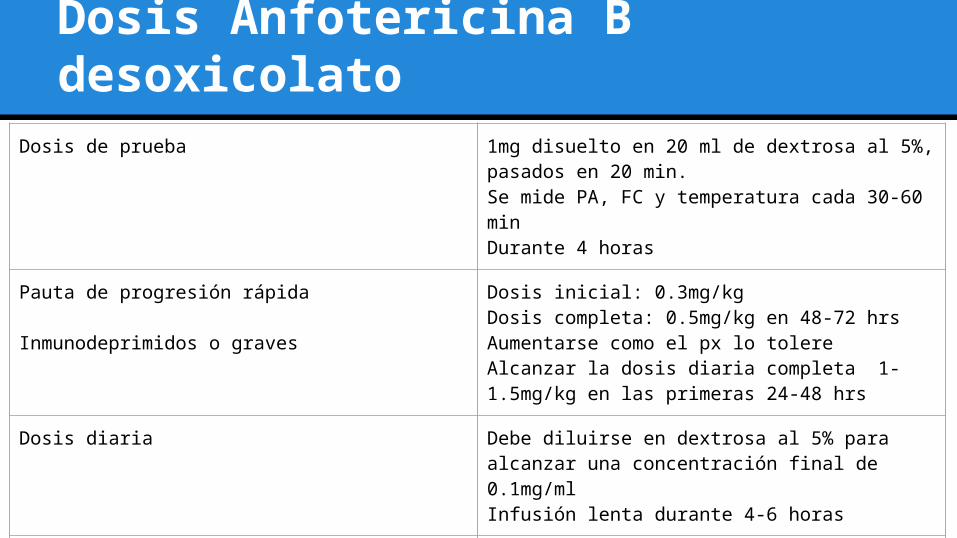
Dosis Anfotericina B desoxicolatoDosis de prueba 1mg disuelto en 20 ml de dextrosa al 5%, pasados en
20 min.Se mide PA, FC y temperatura cada 30-60 minDurante 4 horas
Pauta de progresión rápida
Inmunodeprimidos o graves
Dosis inicial: 0.3mg/kgDosis completa: 0.5mg/kg en 48-72 hrsAumentarse como el px lo tolereAlcanzar la dosis diaria completa 1-1.5mg/kg en las primeras 24-48 hrs
Dosis diaria Debe diluirse en dextrosa al 5% para alcanzar una concentración final de 0.1mg/mlInfusión lenta durante 4-6 horas
Administración tópica Por la escasa penetración o llegada del antifúngico sistémico a los focos de infección fúngica.

Anfotericina B complejo lipídico
Suspensión en viales de 20 ml (100 mg de anfotericina B)
Deben diluirse en dextrosa al 5% antes de la administración intravenosa.
Dosis diaria recomendadaRitmo
5mg/kg/día2,5mg/kg/dia
Duración del tratamiento y dosis acuminada
Depende del tipo de infección, del microorganismo involucrado y de la respuesta clínica.

Anfotericina B liposomal
ReconstituyeDiluyeInfusión intravenosa
Agua esteril Dextrosa al 5% 30-60 min
Se inicia conSe incrementa
1mg/kg/día3-5mg/kg/hora
Dosis toleradaRiesgo de nefrotoxicidad
15mg/kg/día10mg/kg/día

Interacciones farmacológicas
Pacientes VIH positivos Incremento en la nefrotoxicidad y la mielotoxicidad con el uso concomitante de zidovudina, e insuficiencia renal.
Hipopotasemia por anfotericina B toxicidad de los digitálicos y relajantes musculares
Combinación de arabinósido de citosina con anfotericina B
Pakinson

NistatinaProducida Streptomyces noursei
Antibiótico macrólido poliénico Mecanismo de acciónEspectro de actividadSimilares a anfotericina B
Puede ser FungostáticoFungicidaSegún la concentración
Administración
Cutánea
Vaginal
Candidiasis mucocutánea: oral, esofágica vaginal y cutánea. vía oral 100 mil y 1 millón U Pomada cada 8-12 hrs durante 2-4 semanasComprimidos vaginales 12-24 hrs14 semanas
Reacciones adversas Via oral: náuseas vómitos y diarrea.Tópico Irritación cutánea.

GriseofulvinaEfecto Producida por griseofulvum.
inhibe la mitosis actuando sobre los microtúbulos del huso mitótico.Tiene afinidad por las células productoras de queratina, se fija a esta, ejerciendo acción protectora.Activo frente a las especies de dermatofitos Trichophylon, Epidermophyton y Microsporum
Mecanismo de acción
Se absorbe por vía oral 4-5 hrs. Absorción facilitada por las grasas. La absorción depende del tamaño de partícula; existen dos preparados: la microcristalina y la ultramicrocristalina.Se distribuye por todo el organismo, con especial tropismo por la piel.Se metaboliza en el hígado a 6 metilgriseofulvina.Semivida de 24-30 horas.

Dosis
Infecciones extensas
Duración
Adultos: 0,1g Niños: 10-15mg/kg microcristalina 330g ultramicrocristalina en un única dosis después de la comida.
1g repartido en varias tomas.
Tiña carpitis 6-12 semanas.onicomicosis 12 meses.
Reacciones adversas
50% cefaleas desaparecen en la primera semanamareos, parestesias, polineuropatia, pérdida de memoria, confusión e insomnio.Náuseas, vómitos, cambios de sabor, pérdida temporal de sabor.Reacciones cutáneas: fotosensibilidad en px con lupus eritematosa sistémica.

Azoles
Mecanismo de acción Actúan inhibiendo la enzima 14 alpha desmetilasa.Forma un complejo de azol con una parte del citocromo p-450.Impide la conversión del lanosterol en ergosterol.Produciendo una alteración de la permeabilidad de dicha membranay acomolación de períxidos que la dañan.

Actividad antifúngica.
Itraconazol CMI excelente frente a los hongos dermatofitos.Actividad frente a las levaduras: mayor.Activo frente algunas especies de Candida
Fluconazol Activo frente a levaduras (criptococcus spp)
Voriconazol Activo frente especies de Candida y Aspergillus.
Posaconazol Activo frente especies de Candida y Aspergillus.Activo frente a Zygomicetales, Fusarium, coccidioides y Blastomyces.

FarmacocinéticaItraconazol Insoluble en agua.
Solución oral se absorbe mejor que los comprimidos gracias a su unión con un oligosacárido.Alcanza pequeñas porciones en el LCR y saliva.Elevadas concentraciones en el cerebro. Biodisponibilidad oral excelente.Administración una hora antes o después de la comida.
Fluconazol HidrosolubleAbsorción oral muy rápida y completa, no modificada por alimentación.Equilibrio se alcanza 4-5 días.LCR de pacientes con meningitis se alcanzan niveles del 90% con respecto a los alcanzados en el suero.Insuficiencia renal altera la excreción y obliga a reducir las dosis.Los pacientes con hemodiálisis deben recibir una dosis diaria por cada sesión

Voriconazol Biodisponibilidad oral excelente.Administración una hora antes o una hora después de la comida.Forma intravenosa lleva ciclodextrina como excipiente.En niños es necesario usar dosis mayores para obtener los niveles séricos del adultoEquilibrio tras 5- 6 días.Si se administra una dosis de carga de 6mg/kg cada 12 hrs el equilibrio puede alcanzarse el primer día.Se metaboliza en el hígado a través del citocromo p-450.
Pozaconazol Se absorbe y se elimina lentamente.Farmacocinética lineal tras la administración de dosis únicas y múltiples hasta 800mg cuando se toma con comida ricas en grasas.Debe administrarse con alimentos o suplemento alimenticio.

Reacciones adversasItraconazol Intolerancias digestivas, prurito, cefalea y vértigo,
diarrea y náuseas.
Fluconazol Alteraciones de la funcionalidad hepática leves, En enfermos con SIDA la afectación hepática puede ser grave.
Voriconazol Trastorno reversible de la visión 30% de los pacientes.Trastornos de la percepción del color, visión borrosa y percepción de manchas brillantes.Manifestaciones cutáneas
Posaconazol Náuseas y cefaleas.

Indicaciones TerapeúticasFluconazol Puede utilizarse por vía oral o vía intravenosa.
Candidiasis orofaríngea, esofágica,urinaria y la invasora crónica.Meningitis criptocócica solo o combinado con flucitosina.Profilaxis de un subgrupo de pacientes oncohematológicos neutropénicos.200-400 mg/día vía oral o intravenosa.En casos graves o en meningitis puede usarse el doble.
Itraconazol En comprimidos, como solución oral asociada a la ciclodextrina o por vía intravenosa.Blastomicosis, histoplasmosis, coccidioidomicosis, paracoccidioidomicosis, tiña y onicomicosis.Solución oral: candidiasis oral y esofágica.Dosis: 100 y 400 mg/día

Voriconazol Mayor eficacia en el tratamiento inicial de aspergilosis invasora.Micosis emergentes debidas a Fusarium y Scedosporium.Especies de Candida KruseiActividad in vitro frente a Criptococcus.Dosis por vía intravenosa: 6mg/kg cada 12 horas inicialmente.Para disminuir a 4mg/kg cada 12 horas.Vía oral: 200 a 400mg/kg cada 12 horas en niños.
Posaconazol Tratamiento de rescate de la aspergilosis invasiva en px con enfermedad resistente a la anfotericina B o al itroconazol.Prevención de invasión fúngica en px con quimioterapia de remisión inducción para leucemia mieloide aguda o síndromes mielodisplásicos.En receptores de trasplante de progenitores hematopoyéticos con enfermedad del injerto contra el huésped grave.

Azoles de aplicación tópica.Bifonazol Dermatofitos candidiasis
Dermatomicetos Malassezia fur furCorunebacterium minutissium
Clotrimazol Infecciones dermatofíticas: tiña versicolor candidiasis cutánea, candidiasis mucosa y mucocutánea.
Econazol Dermatofitosis: tiña de los pies, inguinal, del cuerpo, pitiriasis versicolor, y candidiasis cutánea superficial
Sulconazol Dermatofitosis, infecciones por candida y M furfur.
Tioconazol Trichophyton, Epidermophyton, M Furfur y C. AlbicansClamidias, Tricomonas y bacterias Grampositivas.
Ketoconazol Dermatomicosis

Pirimidinas Fluoradas:flucitosinaMecanismo de acción Impide la síntesis de ADN en el hongo.
Sufre una desanimación y se transforma en 5-fluorouracilo, un inhibidor no competitivo de la timidilato-sintetasa que interfiere en la síntesis de ADN.Se lleva a cabo en el interior del hongo.
Actividad antifúngicaAntifúngico de espectro reducido.Con actividad exclusiva frente a Candida y Cryptococcus.Algunas cepas de Aspergillus.

Farmacocinética Absorción gastrointestinal y distribución excelentes.Concentraciones elevadas en la mayoría de los tejidos incluyendo:LCR.Buena penetración en el humor acuoso, articulaciones, secreciones bronquiales, líquido peritoneal, cerebro, bilis y hueso.Eliminación sin transformación.90% por orina.Puede depurarse por hemodiálisis y diálisis peritoneal
Reacciones adversas e interacciones
Toxicidad sobre la médula ósea y gastrointestinal.Depresión medular relacionada con los niveles séricos máximos del fármaco se produce cuando son superiores a 100mg/mlToxicidad hematológica se potencia al asociar con otros fármacos mielotóxicos
Indicaciones En pacientes con criptococosis y candidiasis.Dosis 150mg/kg/día por vía oral repartida en 4 tomas. En caso de insuficiencia renal hay q ajustar las dosis.

Equinocandinas
Nueva familia de antifúngicos:Caspofungina.Anidulafungina.Próximamente:micafungina.

Mecanismo de acción Inhibe la síntesis del 1,3 b-D-glucano, componente clave de la pared celular, provoca inestabilidad osmótica de las células fúngicas.Impide crecimiento y replicación
Farmacocinética Se une a las proteínas en más del 95%.La distribución a la mayoría de los tejidos es buena.Eliminación del plasma es lenta.Se metaboliza en el hígado.Semivida plasmática prolongadaPequeña cantidad de caspofungina se excreta sin cambio en la orina.Anidulafungina Semivida más prolongada y mayor volumen.No son dializables.

Reacciones adversas muy bien toleradas
Indicaciones terapeúticas Candidiasis invasiva y candidemia.Aspergilosis invasiva resistente.Caspofungina se administra por vía intravenosa en intravenosa en infusión salina al 0.9% no glucosada durante 60 minutos en dosis de 70 mg en el primer día.50mg/día hasta finalizar el tratamiento

Aplicación sistémica: terbinafinaEfecto Alilamina,fungicida queratinofila y muy lipófila, se usa por vía sistémica y tópica.
Actúa sobre la enzima escualeno-epoxidasa
Mecanismo de acción Se absorbe por vía oral .Por vía tópica menos de 5%.Se acomula en tejido graso y se fija en la queratina de la piel, pelo y uñas.Se metaboliza en el hígado Metabolitos inactivos se excretan por la orina.
Indicaciones Vía oral 250mg/día en una o dos tomas2-4 semanas en tiña del cuerpo 2-6 semanas tiña del pie2-12 semanas onicomicosis
Reacciones adversas Leves y autolimitadasGastrointestinalesCutáneasHepatotoxicidad
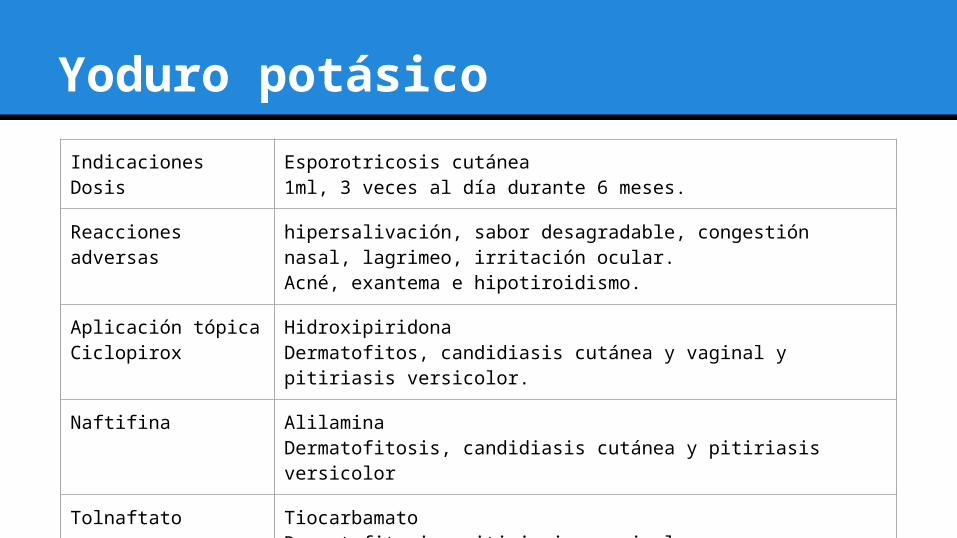
Yoduro potásico
IndicacionesDosis
Esporotricosis cutánea1ml, 3 veces al día durante 6 meses.
Reacciones adversas hipersalivación, sabor desagradable, congestión nasal, lagrimeo, irritación ocular.Acné, exantema e hipotiroidismo.
Aplicación tópicaCiclopirox
HidroxipiridonaDermatofitos, candidiasis cutánea y vaginal y pitiriasis versicolor.
Naftifina Alilamina Dermatofitosis, candidiasis cutánea y pitiriasis versicolor
Tolnaftato TiocarbamatoDermatofitosis, pitiriasis versicolor.



