Embed Size (px)
Citation preview

1
Eduardo Aceves RMI
Esclerosis tuberosa Revisión bibliográfica
El complejo de Esclerosis Tuberosa (TSC) es un desorden multisistémico,
autosómico, dominante que afecta a niños y adultos. Se da a partir de mutaciones en uno de dos genes reguladores del crecimiento y diferenciación celular, el TSC1 (codifica para hamartina) ó TSC2 (codifica para tuberina).Se han encontrado dos regiones cromosómicas para la génesis de TSC, en el cromosoma 9q y en el 16p. Se encuentra descrito por primera vez por Bourneville en 1880 y se caracteriza por la aparición de hamartomas diseminados, generalmente en cerebro, ojos, piel, riñones, hígado, corazón y pulmones. La incidencia aproximada es de 1 en 10,000.
Frecuentemente causa desórdenes neurológicos incapacitantes incluyendo epilepsia, retraso mental y autismo. Otras manifestaciones frecuentes son angiofibromas faciales, angiomiolipomas renales y linfangiomatosis pulmonar.
Los criterios diagnósticos consisten en mayores y menores (tabla 1). Los hallazgos de genética molecular actualmente se utilizan únicamente para corroborar el diagnóstico. Es importante tener en cuenta que las manifestaciones clínicas aparecen en distintos puntos de desarrollo y tienen penetrancia variable. Previamente el diagnóstico se asociaba a la triada de retraso mental, epilepsia y angiofibromas faciales aunque la triada completa se presenta únicamente en 1/3 de pacientes. Tabla 1. Criterios diagnósticos para TSC Criterios mayores Criterios menores
Angiofibromas faciales Fibromas ungueales Parches de Shagreen Máculas hipomelanóticas Tuberosidades corticales Nódulos subependimarios Tumores subependimarios de células gigantes Hamartomas renales Rabdomiolipoma renal Linfangiomatosis
Múltiples criptas en piezas dentarias Pólipos rectales hamartomatosos Quístes óseos Líneas radiales de migración de materia blanca Fibromas gingivales Parches retinales acrómicos Lesiones cutáneas en confeti Quístes renales múltiples
Dos criterios mayores ó un criterio mayor más uno menor hacen el diagnóstico de TSC. Para un diagnóstico probable, se necesita un mayor y uno menor. Para diagnóstico posible, uno mayor ó 2 menores.
Manifestaciones dermatológicas Las manifestaciones cutáneas más frecuentes son los angiofibromas faciales, máculas hipomelanóticas, fibromas subungueales múltiples, parches en piel de zapa, placas frontales y lesiones “de confeti”. Es importante recordar de nuevo, que también las lesiones dermatológicas se presentan en distintos puntos del desarrollo por lo que algunas pueden aparecer y desaparecer al paso del tiempo. Jozwiak et al realizaron un estudio con 106 niños con diagnóstico de TSC con edades entre 1 mes y 18 años y de 1984 y 1995. El diagnóstico de TSC fue realizado basado en los criterios utilizados en 1992 (muy similares a los actuales). Todos los pacientes fueron revisados por un pediatra y un dermatólogo además de asistir a una clínica de neurología. Estos fueron sus resultados (tabla 2):
2
Tabla 2. Lesiones cutáneasen 106 pacientes con diagnóstico de TSC Lesión cutánea n %
Máculas hipomelanóticas Angiofibromas faciales Parches en piel de zapa Manchas café con leche Molusco fibroso péndulo Placa fibrosa frontal Fibromas periungueales Lesiones en confeti
103 79 51 30 24 20 16 3
97.2 74.5 48.1 28.3 22.6 18.9 15.1 2.8
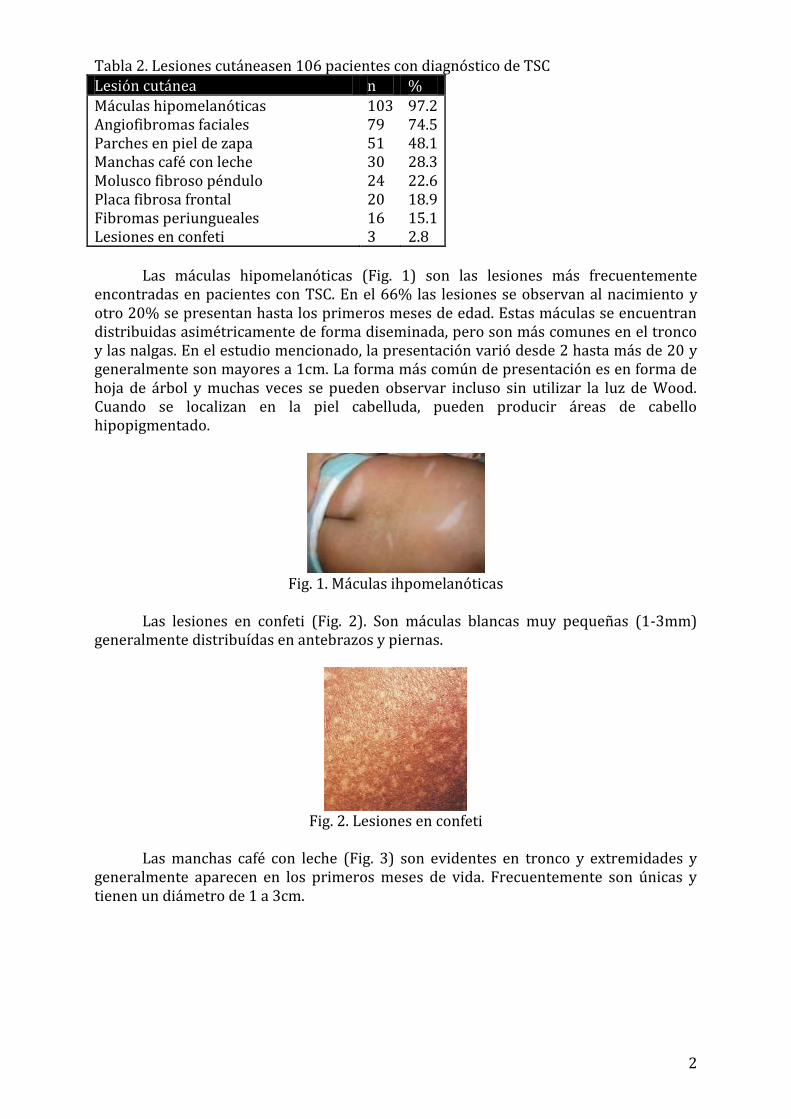
Las máculas hipomelanóticas (Fig. 1) son las lesiones más frecuentemente encontradas en pacientes con TSC. En el 66% las lesiones se observan al nacimiento y otro 20% se presentan hasta los primeros meses de edad. Estas máculas se encuentran distribuidas asimétricamente de forma diseminada, pero son más comunes en el tronco y las nalgas. En el estudio mencionado, la presentación varió desde 2 hasta más de 20 y generalmente son mayores a 1cm. La forma más común de presentación es en forma de hoja de árbol y muchas veces se pueden observar incluso sin utilizar la luz de Wood. Cuando se localizan en la piel cabelluda, pueden producir áreas de cabello hipopigmentado.
Fig. 1. Máculas ihpomelanóticas
Las lesiones en confeti (Fig. 2). Son máculas blancas muy pequeñas (1-3mm) generalmente distribuídas en antebrazos y piernas.
Fig. 2. Lesiones en confeti
Las manchas café con leche (Fig. 3) son evidentes en tronco y extremidades y generalmente aparecen en los primeros meses de vida. Frecuentemente son únicas y tienen un diámetro de 1 a 3cm.

3
Fig. 3. Manchas café con leche
Los angiofibromas papulonodulares (Fig. 4) faciales son nódulos rosados-rojos con superficie suave. Se distribuyen de forma bilateral en áreas centrofaciales, particularmente en los pliegues nasolabiales (en mariposa), hacia las mejillas y barbilla. En la mayoría de pacientes, las lesiones se hacen evidentes en 2-5 años. Estas lesiones presentan crecimiento progresivo, especialmente en la pubertad.
Fig. 4. Angiofibromas faciales
La placa fibrosa frontal (Fig. 5) son placas amarillentas-cafés, levemente elevadas, de consistencia variable, crecen muy lentamente al correr de los años.
Fig. 5. Placa fibrosa frontal
Parches en piel de zapa (Fig. 6). Se localizan asimétricamente en superficies dorsales y de forma particular en la región lumbosacra. Se encuentran levemente elevados, son de color amarillo-café o rosadas con una textura similar a piel de naranja. La frecuencia de estas lesiones aumentan con la edad. Son múltiples y varían en tamaño de pequeños milímetros a varios centímetros.
Fig. 6. Parches en piel de zapa

4
Los fibromas periungueales o subungueales (tumores de Koenen) se localizan alrededor o debajo de los lechos ungueales (Fig. 7).
Fig. 7. Fibromas periungueales
Molusco fibroso péndulo (Fig. 8). Son crecimientos pedunculados solitarios o múltiples particularmente en el cuello y rara vez en axilas e ingles.
Fig. 8. Molusco fibroso péndulo
Lesiones renales
Los angiomiolipomas renales son tumores benignos compuestos de vasos anormales, células inmaduras de músculo liso y células lipídicas. Son bilaterales y en muchas ocasiones se presentan de forma múltiple en cada riñón. Tienen vasculatura anormal y muy frecuentemente aneurismas por lo que una complicación importante es el sangrado que pone en riesgo la vida. La remoción quirúrgica es evitada en la medida de lo posible tratando de preservar la función renal. Los angiomiolipomas que son mayores de 3 a 4cm de diámetro se pueden tratar con embolización de forma efectiva.
Además, puede haber lesiones epiteliales como quístes, enfermedad renal poliquística y carcinomas de células renales. En una serie de pacientes con TSC, el carcinoma renal tuvo un desarrollo para los 28 años, 25 años antes de lo que se espera en pacientes sin TSC. Manifestaciones pulmonares
La linfangiomatosis afecta a mujeres y se caracteriza por proliferación diseminada de células de músculo liso y cambios quísticos en el parénquima pulmonar. Durante la edad adulta temprana, se manifiesta como disnea o neumotórax. La incidencia en mujeres con TSC es de entre el 26 y 39%; la mayoría son asintomáticas. Manifestaciones neurológicas
Las manifestaciones neurológicas, que incluyen epilepsia, retraso cognitivo y anormalidades de comportamiento aparentemente están relacionadas a las tuberosidades corticales cerebrales que se encuentran presentes en hasta el 80% de los pacientes. Las tuberosidades son anormalidades de desarrollo en la corteza caracterizadas histológicamente por la pérdida de estructura, neuronas dismórficas,

5
astrocitos grandes y células gigantes. Estas lesiones persisten durante toda la vida pero no tienen el riesgo de volverse malignas. Los pacientes con TSC, hasta en el 70-80% de los casos presentan epilepsia. Los espasmos infantiles ocurren del 20-30% de los niños con TSC. Estudios clínicos sugieren que un número más alto de tuberosidades (más de 7) se relacionan con el desarrollo de espasmos infantiles y epilepsia intratable. En aproximadamente el 10% de los pacientes con TSC, el crecimiento de tumores subependimarios de células gigantes puede causar obstrucción de flujo de LCR, hidrocefalia, hipertensión intracraneal e incluso muerte. Estos tumores, se cree, derivan de nódulos subependimarios, que son hamartomas asintomáticos que protruyen de las paredes de los ventrículos laterales y tercero. Lesiones cardiacas
Los rabdomiomas cardiacos se presentan en el 50 al 70% de los niños con TSC y son el principal tumor cardiaco diagnosticado in utero. Estos tumores pueden estar asociados a falla cardiaca en la infancia y el 47% de los pacientes pueden tener asociadas arritmias cardicas como taquicardia auricular, taquicardia ventricular, bloqueo AV completo y síndrome de Wolff-Parkinson-White. Muy frecuentemente los rabdomiomas desaparecen espontáneamente en la vida adulta. Tratamiento
Una vez hecho el diagnóstico, lo más importante es el envío a centros especialistas en esta enfermedad. En caso de no tener la posibilidad de hacerlo, se recomienda la interconsulta a especialistas en los órganos probablemente afectados para tener un manejo global del paciente.
Las afecciones dermatológicas pueden ser manejadas con fines cosméticos. En cuanto al seguimiento a largo plazo, lo más importante es el monitoreo del
crecimiento de lesiones. Lo que se recomienda es la obtención de imágenes radiológicas de encéfalo y abdomen por lo menos cada 3 años y de forma más temprana en pacientes con lesiones cerebrales ó renales con crecimiento progresivo demostrado. Se recomienda una RM de encéfalo en pacientes hasta por lo menos los 21 años de edad y posteriormente cada 2 ó 3 años. En pacientes con angiomiolipomas múltiples ó una lesión única que es progresiva, esta indicado un US, RM ó TC de forma anual. En pacientes con linfangiomatosis, se recomiendan pruebas de función pulmonar anuales.
El tercer punto es el consejo genético para ayudar a los pacientes a la planeación familiar. Debido a que es un desorden autosómico dominante, se debe explicar que el riesgo de tener hijos afectados es de aproximadamente 50%.
Actualmente se esta investigando el uso de sirolimus (inhibidor de mTOR) como un potencial agente terapéutico. Bibliografía Borkowska J, Schwartz R, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and
tumorigenesis. International Journal of Dermatology 2011;50:13-20 Crino P, Nathanson K, Henski E. The Tuberous Sclerosis Complex. N Engl J Med
2006;355(13):1345-1356 Jozwiak S, Schwartz R, Janniger C, Michalowics R, Chmielik J. Skin lesions in children with
tuberous sclerosis complex: their prevalence, natural course and diagnostic significance. International Journal of Dermatology 1998;37:911-917
Webb D, Clarke A, Fryer A, Osborne J. The cutaneous features of tuberous sclerosis: a population study. British Jorunal of Dermatology 1996;135:1-5





























