Embed Size (px)
Citation preview

LAS LAS TALASEMIASTALASEMIAS

TALASEMIASTALASEMIAS
Las talasemias son trastornos hereditarios en la sangre, lo cual causa que el cuerpo
produzca menos glóbulos rojos y sanos y menos hemoglobina de lo normal.
La hemoglobina se compone de
dos proteínas:
La globina alfa y la globina beta.
La talasemia ocurre cuando hay
un defecto en un gen que ayuda a
controlar la producción de una
de estas proteínas.

ETIOLOGIAETIOLOGIA
Genética:
HERENCIA AUTOSOMICA RECESIVA
Se manifiesta al momento que ambos miembros del par génico afectados (genes
recesivos). Homocigotos para alelo recesivo son encontrados en el genotipo del
individuo.
Los portadores sanos poseen un
alelo recesivo del par de alelos en su
genotipo, el cual no es manifestado
hasta el cruce de transmisión a su
siguiente generación

FISIOPATOLOGÍA
La talasemia alfa ocurre cuando
un gen o los genes relacionados
con la proteína globina alfa
faltan o han cambiado (mutado).
La talasemia beta ocurre
cuando defectos genéticos
similares afectan la producción
de la proteína globina beta.

CLASIFICACIÓNCLASIFICACIÓN
Talasemia Alfa : hay
disminución o ausencia
de la globina alfa
Talasemia Beta : hay
disminución o ausencia
de la globina beta.
Talasemia Delta: rara.
Talasemia gamma: rara.
La alfa y la beta son las
más importantes.
Las talasemias también se
categorizan por el número de
alelos que están defectuosos:
•Talasemia Menor - un alelo
anormal
(portador / rasgo talasémico /
heterocigoto)
•Talasemia Mayor - dos alelos
anormales
(Enfermo / homocigoto)

TALASEMIA MAYOR (BETA) (ANEMIA DE COOLEY)
Es la forma más severa de anemia y es una forma hereditaria de la anemia
hemolítica.
Caracterizada por anomalías en la producción de los glóbulos rojos
sanguíneos (hemoglobina).
Se pueden observar los glóbulos rojos sanguíneos de forma anormal, pálidos
(hipocrómicos) y pequeños asociados con esta enfermedad.
Es la forma más severa de anemia y es una forma hereditaria de la anemia
hemolítica.
Caracterizada por anomalías en la producción de los glóbulos rojos
sanguíneos (hemoglobina).
Se pueden observar los glóbulos rojos sanguíneos de forma anormal, pálidos
(hipocrómicos) y pequeños asociados con esta enfermedad.

TALASEMIA MENOR(ALFA TALASEMIA)
Es un tipo hereditario de anemia
hemolítica menos severo que la
talasemia mayor. Se pueden
observar glóbulos rojos
sanguíneos de varias formas
(poiquilocitosis), pálidos
(hipocrómicos) y pequeños
(microcíticos), los cuales tienen
menor capacidad para transportar
el oxígeno que los glóbulos rojos
sanguíneos normales.
Es un tipo hereditario de anemia
hemolítica menos severo que la
talasemia mayor. Se pueden
observar glóbulos rojos
sanguíneos de varias formas
(poiquilocitosis), pálidos
(hipocrómicos) y pequeños
(microcíticos), los cuales tienen
menor capacidad para transportar
el oxígeno que los glóbulos rojos
sanguíneos normales.
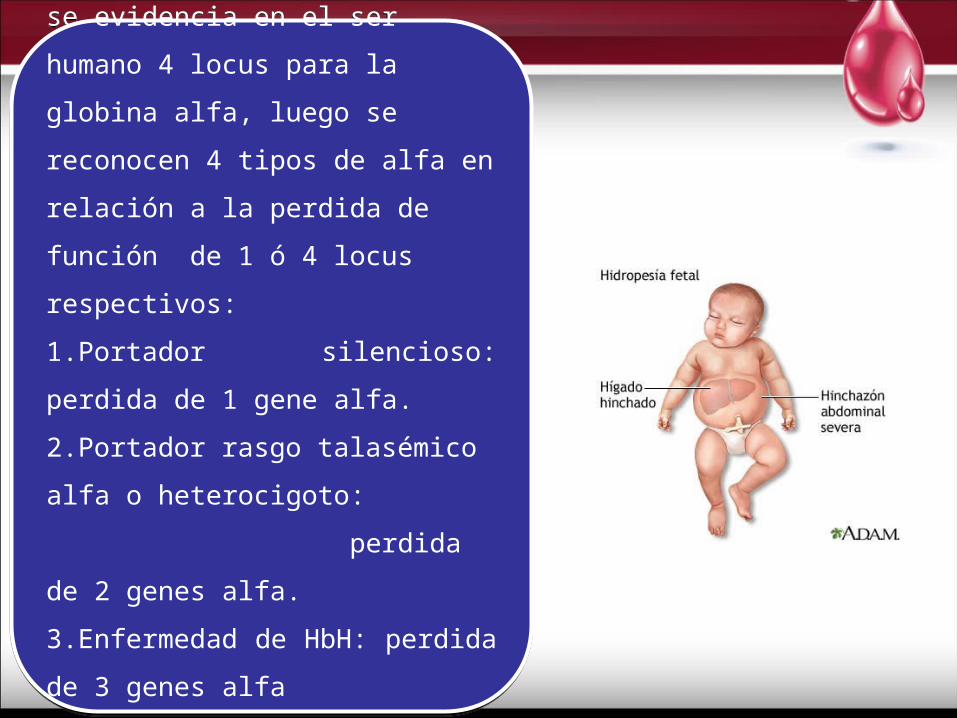
Como genética y químicamente se
evidencia en el ser humano 4 locus para
la globina alfa, luego se reconocen 4 tipos
de alfa en relación a la perdida de función
de 1 ó 4 locus respectivos:
1.Portador silencioso: perdida de 1 gene
alfa.
2.Portador rasgo talasémico alfa o
heterocigoto: perdida de
2 genes alfa.
3.Enfermedad de HbH: perdida de 3
genes alfa
4.Hidropesía fetal: no hay síntesis de
cadena alfa.
Como genética y químicamente se
evidencia en el ser humano 4 locus para
la globina alfa, luego se reconocen 4 tipos
de alfa en relación a la perdida de función
de 1 ó 4 locus respectivos:
1.Portador silencioso: perdida de 1 gene
alfa.
2.Portador rasgo talasémico alfa o
heterocigoto: perdida de
2 genes alfa.
3.Enfermedad de HbH: perdida de 3
genes alfa
4.Hidropesía fetal: no hay síntesis de
cadena alfa.

SIGNOS Y SINTOMASSIGNOS Y SINTOMAS
Talasemia Alfa: Benigna
•Microcitosis e hipocromía
eritrocitaria.
•Disminución mínima de la
hemoglobina.
•Disminución de HCM
(hemoglobina corpuscular media).
•Disminución de HCM
(hemoglobina corpuscular media).
Talasemia Alfa: Severa
•Anemia grave con una tasa en la
síntesis de globinas irregular.
•Vida fetal exceso de globina alfa
(Hemoglobina Bart).
•Vida adulta deficiencia de globina
alfa y exceso de globinas beta
(Hemoglobina H).

SIGNOSSIGNOS Y SINTOMAS Y SINTOMAS
Talasemia Beta: Menor
•Anemia moderada (No tienen
indicada transfusión sanguínea).
•Aumento de glóbulos rojos con
hipocromía y Microcitosis.
•Asintomático en la mayoría de
los casos.
•Aumento en los niveles de
hemoglobina A.
Talasemia Beta: Mayor
•Primeros meses de vida anemia hemolítica y hepatoesplenomegalia (Indicación de transfusión sanguínea).
•Prominencia del cráneo y sobre crecimiento de la región maxilar con facies mongoloides.
•Aumento en los niveles de hierro sérico lo cual produce afecciones cardiaca como ICC, endocrinas como hipotiroidismo e hipoparatiroidismo y afección hepática (Cirrosis).


DIAGNOSTICODIAGNOSTICO
Homocigoto de beta talasemia: es fácil el
diagnostico porque desde temprana edad
tiene hepato y esplenomegalia, anemia
hemolítica intensa y aumento de
hemoglobina fetal.
Homocigoto de alfa talasemia: se hace en
el mortinato con hidropesía fetal que tiene
cifra aumentada de Hb Bart´s (4 gamma).
En todas las talasemias hay microcitosis e
hipocromías.
DIAGNOSTICO DIAGNOSTICO PRENATALPRENATAL
Es útil en la beta talasemia pero no en la alfa, se puede hacer en
sangre del cordón umbilical desde la 12 semanas y determinar
la síntesis alfa y no alfa, diagnosticar anemia falciforme

Es una enfermedad hereditaria causada por una deficiencia enzimática y se manifiesta con incapacidad de utilizar el azúcar simple galactosa, lo cual provoca una acumulación de éste dentro del organismo, produciendo lesiones en el hígado y el sistema nervioso central.
Galactosemia

Etiología
La galactosemia sigue un patrón de herencia autosómico recesivo en sus tres tipologías. Generalmente los padres portadores asintomáticos tendrán un 25% de los hijos sanos, un 50% de los hijos portadores asintomáticos y un 25% de los hijos con galactosemia.

Metabolismo La metabolización de la galactosa a glucosa se realiza a través de la vía Leloir, la cual implica una serie de reacciones enzimáticas realizadas por diferentes enzimas:
Galactitol. Galactosa + ATP ---
(Galactoquinasa) ---> Galactosa 1-fosfato + ADP.
Galactosa 1-fosfato + UDP-glucosa ---(Galactosa 1-fosfato uridiltransferasa) ---> Glucosa 1-fosfato + UDP-galactosa.
UDP-galactosa --- (UDP-galactosa 4-epimerasa) ---> UDP-glucosa.
Vía Leloir

Si la galactosa no es metabolizada por la vía Leloir puede metabolizarse por dos vías alternativas:En la primera la galactosa es reducida por una aldosa reductasa a galactitol, mientras que en la segunda la galactosa es oxidada por una galactosa deshidrogenasa a galactonato.
Si la galactosa no es metabolizada por la vía Leloir puede metabolizarse por dos vías alternativas:En la primera la galactosa es reducida por una aldosa reductasa a galactitol, mientras que en la segunda la galactosa es oxidada por una galactosa deshidrogenasa a galactonato.
TiposLas deficiencias de cada uno de estos enzimas (aldosa reductasa, galactosa deshidrogenasa) producen los diferentes tipos de galactosemia:

Deficiencia de galactoquinasa (GALK). En este tipo de galactosemia la galactosa no puede ser fosforilada a galactosa 1-fosfato, por lo que se acumula en los tejidos y se metaboliza por las vías alternativas citadas anteriormente.
Deficiencia de UDP-galactosa 4-
epimerasa (GALE). La reacción que transforma la
UDP-galactosa en UDP-glucosa y viceversa no se realiza.
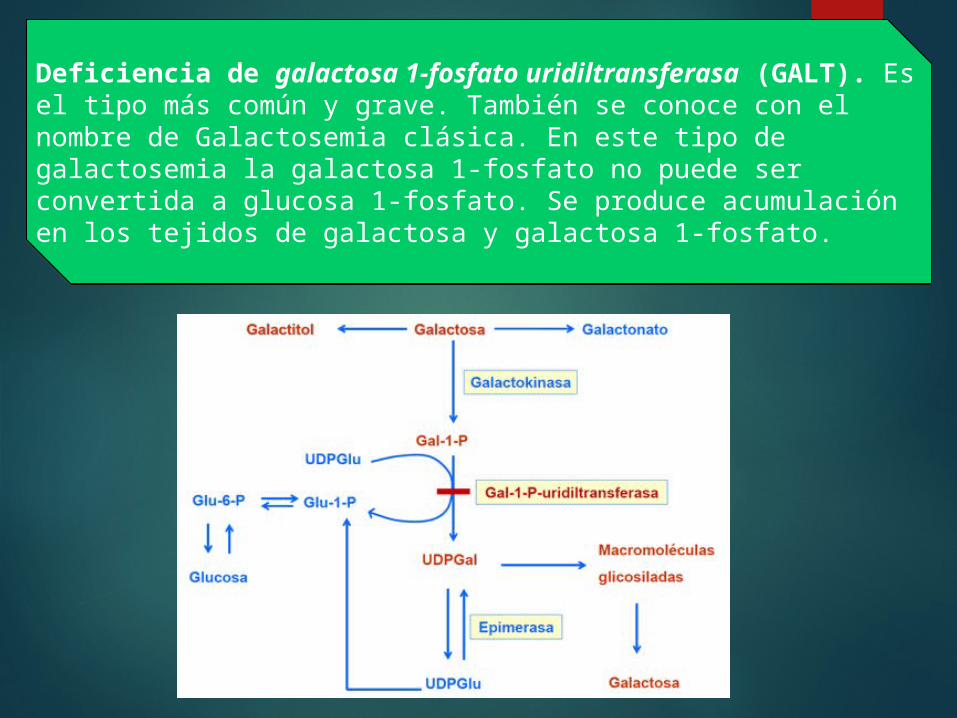
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Es el tipo más común y grave. También se conoce con el nombre de Galactosemia clásica. En este tipo de galactosemia la galactosa 1-fosfato no puede ser convertida a glucosa 1-fosfato. Se produce acumulación en los tejidos de galactosa y galactosa 1-fosfato.
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Es el tipo más común y grave. También se conoce con el nombre de Galactosemia clásica. En este tipo de galactosemia la galactosa 1-fosfato no puede ser convertida a glucosa 1-fosfato. Se produce acumulación en los tejidos de galactosa y galactosa 1-fosfato.

Cuadro clínicoCuadro clínico
La sintomatología y su intensidad está determinada por el tipo de deficiencia enzimática que se presente.
Convulsiones Irritabilidad Letargo Alimentación
deficiente (el bebé se niega a tomar fórmula que contenga leche)
Poco aumento de peso
Coloración amarillenta de la piel y de la esclerótica (ictericia)
Vómitos

Los bebés con galactosemia pueden desarrollar síntomas en los primeros días de vida si consumen leche artificial o leche materna que contengan lactosa. Los síntomas pueden deberse a una infección grave en la sangre con la bacteria E. coli.

Deficiencia de galactoquinasa (GALK). Únicamente se presenta la formación de cataratas debido a la acumulación de galactitol en el cristalino. No hay afectación de hígado, riñones o cerebro. Aumento de galactosa y galactitol en plasma y galactosuria.
Deficiencia de UDP-galactosa 4-epimerasa (GALE). Se pueden no mostrar síntomas o presentar síntomas parecidos a los de la galactosemia clásica. En ambos casos se produce una acumulación de UDP-Galactosa y Galactosa 1-fosfato.
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Se presentan letargo, rechazo al alimento y manifestaciones tóxicas generales, incluyendo vómitos y diarreas, pérdida de peso, ictericia, hepatomegalia, ascitis y la formación de cataratas entre otros debido a la acumulación de galactosa, galactitol y galactosa 1-fosfato en los tejidos. Aumento de galactosa y galactitol en plasma, galactosuria e hiperaminoaciduria.

Diagnostico
El diagnóstico se realiza por:•Cuantificación de galactosa y galactitol en plasma.•Cuantificación de galactosa 1-fosfato, galactitol, galactonato y actividad enzimática GALK, GALE y GALT en glóbulos rojos.•Presencia de sustancias reductoras en orina.
• Aminoácidos presentes en la orina y/o en el plasma sanguíneo (aminoaciduria)
• Agrandamiento del hígado (hepatomegalia)• Acumulación de líquido en el abdomen
(ascitis)• Azúcar bajo en la sangre (hipoglucemia)• Las pruebas de detección en recién nacidos
en muchos estados evalúan esta afección.





























