Embed Size (px)
Citation preview
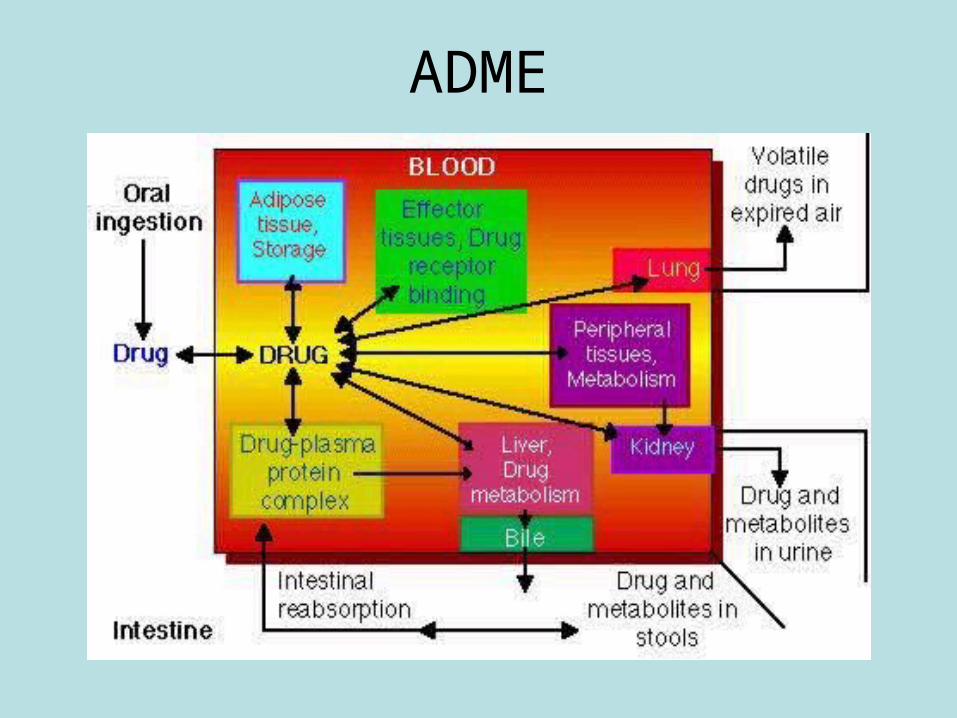
ADME

Absorção de fármacos
• Os fármacos devem atravessar as membranas biológicas para serem absorvidos.
• Os fármacos penetram as membranas biológicas por dois modos:
1. Difusão passiva.2. Mecanismos de transporte especializados (transporte
ativo e difusão facilitada).
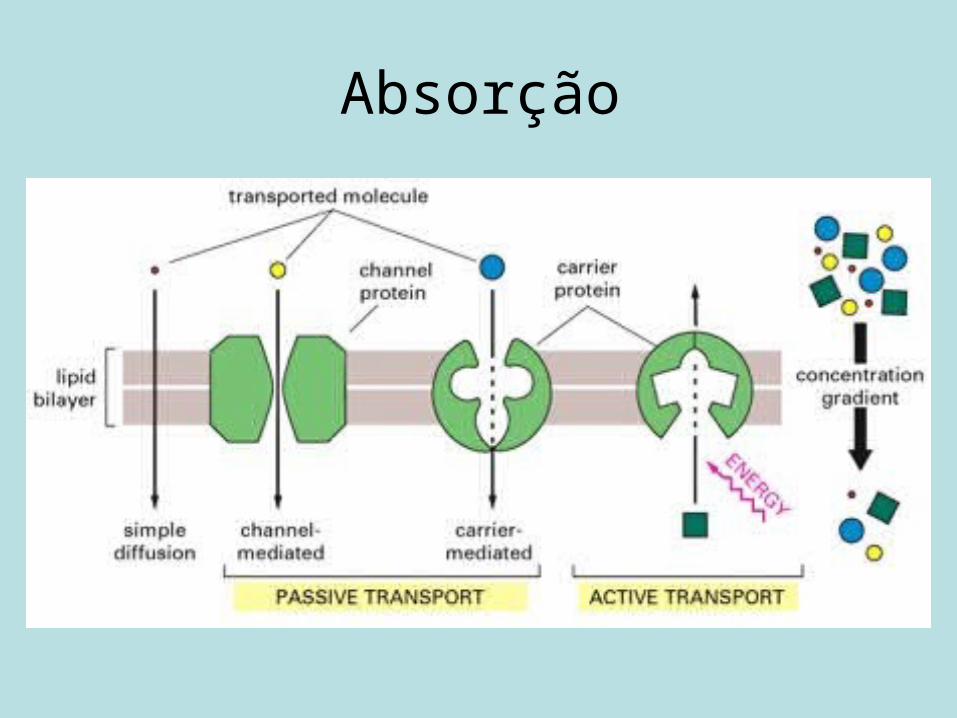
Absorção
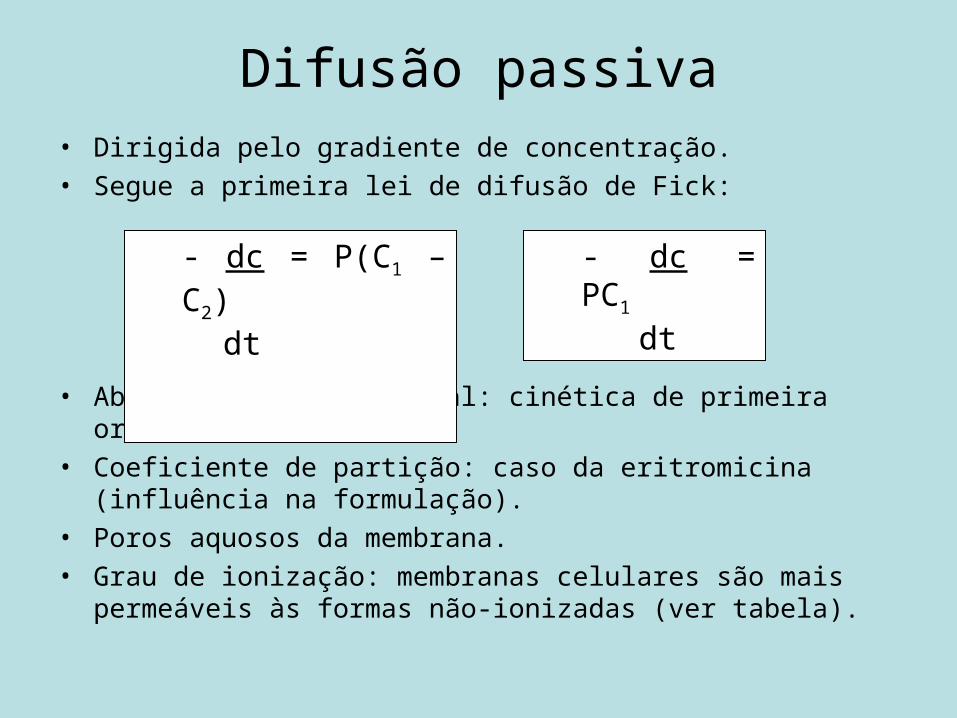
Difusão passiva• Dirigida pelo gradiente de concentração.• Segue a primeira lei de difusão de Fick:
• Absorção gastrintestinal: cinética de primeira ordem.• Coeficiente de partição: caso da eritromicina (influência na
formulação).• Poros aquosos da membrana.• Grau de ionização: membranas celulares são mais permeáveis às
formas não-ionizadas (ver tabela).
- dc = P(C1 – C2) dt
- dc = PC1
dt
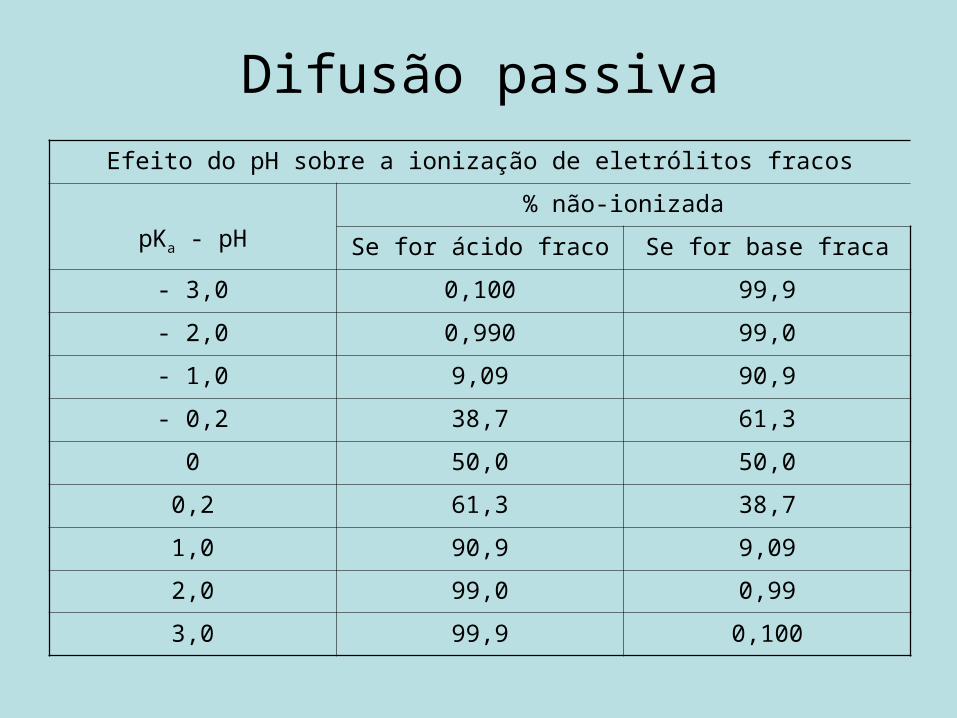
Difusão passivaEfeito do pH sobre a ionização de eletrólitos fracos
pKa - pH% não-ionizada
Se for ácido fraco Se for base fraca
- 3,0 0,100 99,9
- 2,0 0,990 99,0
- 1,0 9,09 90,9
- 0,2 38,7 61,3
0 50,0 50,0
0,2 61,3 38,7
1,0 90,9 9,09
2,0 99,0 0,99
3,0 99,9 0,100

Mecanismos de transporte especializado
• Moléculas muito insolúveis em lípides ou muito grandes para fluir ou filtrar pelos poros.
• Especificidade carreador-fármaco.• Transporte ativo: contra gradiente de conc. (ex.:
açúcares e aa no TGI, vitaminas, metildopa, 5-fluorouracil etc.).

Importância na cinética de absorção

Fatores físico-químicos do fármaco
• Área superficial.• Forma cristalina ou amorfa.• Tamanho de partícula.• Sais.• Estado de hidratação.• Interação fármaco-organismo biológico.

Vias de administraçãoOral Peroral, sublingual
Parenteral Intravenosa, intra-arterial, intracardíaca, intratecal, intra-óssea,
intra-articular, intrasinovial, intradérmica, subcutânea,
intramuscular
Epidérmica/transdérmica
Conjuntival
Intra-ocular/intra-auricular
Intranasal
Pulmonar
Retal
Vaginal
Uretral

Destino do fármacoDrug in dosage form
ReleaseRelease
Drug particles in body fluidsDissolution
Drug in solution Degradation
Absorption
Liver
Excretion
GI
Central Compartment
Free Bound
Distribution
PeripheralTissues
Pharmacologic effect
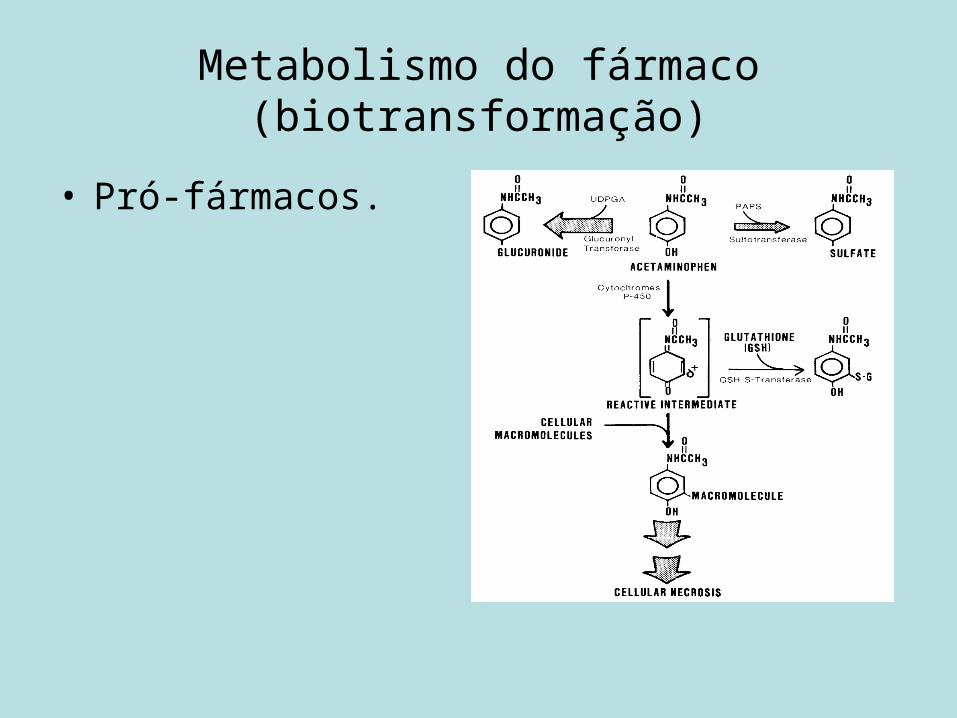
Metabolismo do fármaco (biotransformação)
• Pró-fármacos.

Liberação modificada de fármacos
• Expectativas da ação de um fármaco no organismo.
• Liberação modificada de fármacos.• Classificação dos sistemas de
liberação modificada.• Estratégias para formulação.• Vantagens e desvantagens.

Sistemas de liberação controlada
• Sistemas que mantêm algum controle temporal ou espacial, ou ambos, na liberação de fármacos no organismo.

Estratégias para formulação• Modificação química do fármaco.• Modificação da forma farmacêutica.
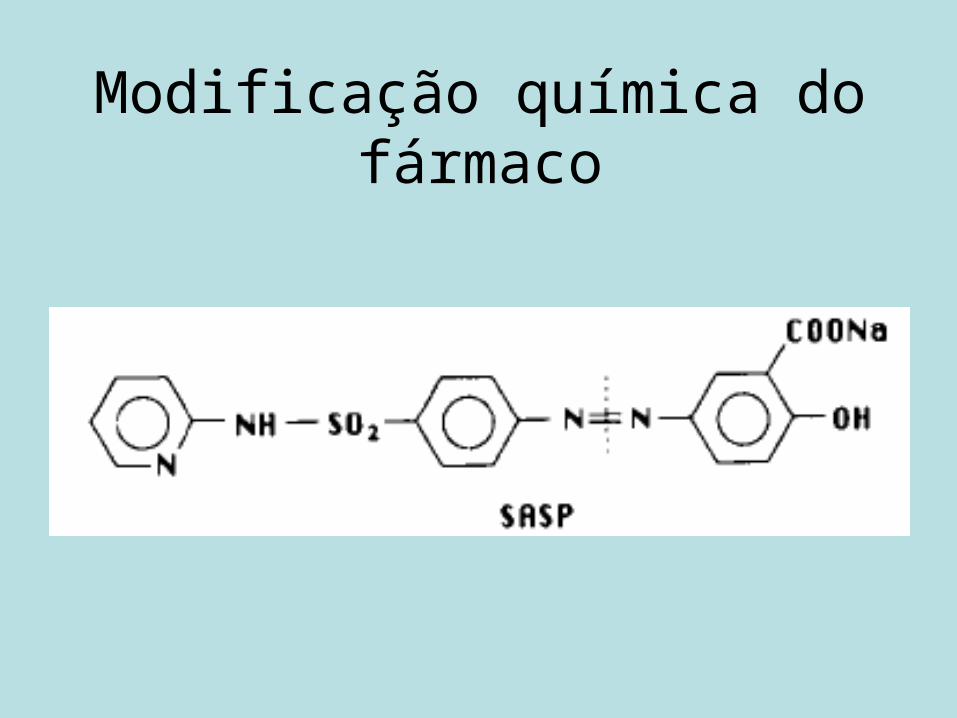
Modificação química do fármaco
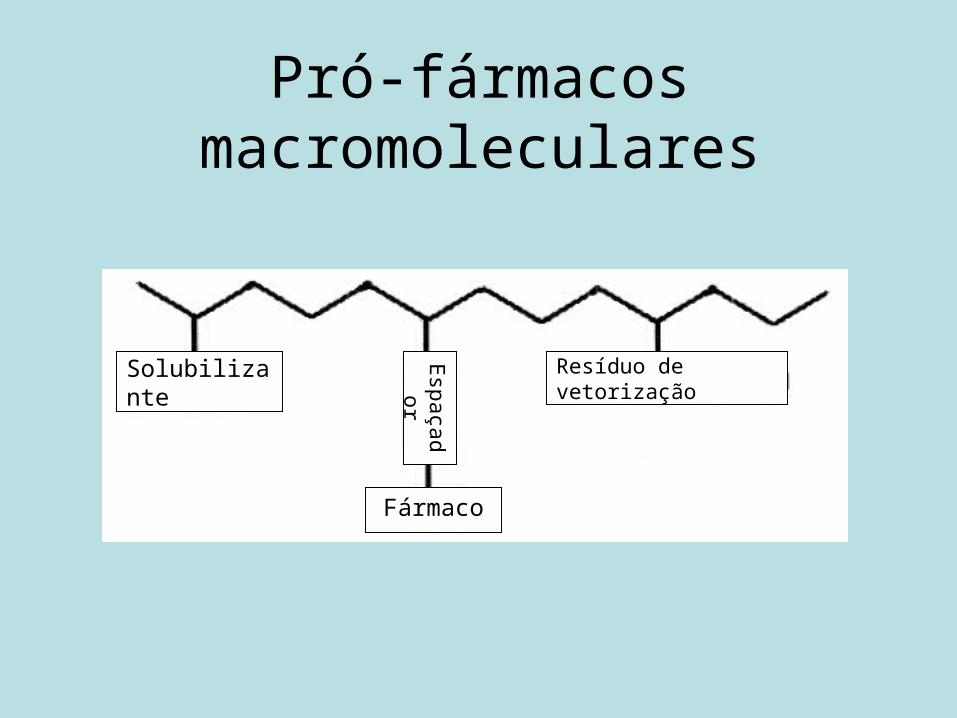
Pró-fármacos macromoleculares
Solubilizante
Espaçador
Fármaco
Resíduo de vetorização

Modificação da forma farmacêutica
• Microesferas.• Comprimidos.• Soluções.• Suspensões.• Cápsulas.• Nanopartículas.• Lipossomas.

Vantagens
• Adesão do paciente.• Redução nas variações das concentrações
plasmáticas.• Redução de efeitos colaterais sistêmicos e locais.• Minimização da acumulação do fármaco nos tecidos
corporais com terapia crônica.• Esquema posológico simplificado com menor
administração de ativo.• Controle mais adequado da absorção do fármaco.

Desvantagens• Impossibilidade da interrupção imediata da ação
terapêutica.• O médico perde a flexibilidade no ajuste dos regimes
posológicos, fixados durante a concepção da forma farmacêutica.
• Formas de liberação prolongada são concebidas para uma população normal: estados patológicos e variações interpacientes não são tidos em conta.
• Maior tamanho da maioria dos dispositivos. • Dor e rejeição no caso de implantes.• A produção pode envolver processos e equipamentos
mais caros.

Economia
• Custo inicial maior.• Custo médio do tratamento a longo prazo é
menor.• Diminuição com enfermagem/hospitalização.• Menos tempo de trabalho perdido.• Maior eficácia clínica.• Diminuição na freqüência ou no custo de
monitoração do paciente (incluindo visitas médicas).

Importância para o mercado farmacêutico
• Sistemas de liberação tendem a crescer mais que o mercado farmacêutico como um todo.
• Mercado farmacêutico para 2009: U$ 900 bilhões.
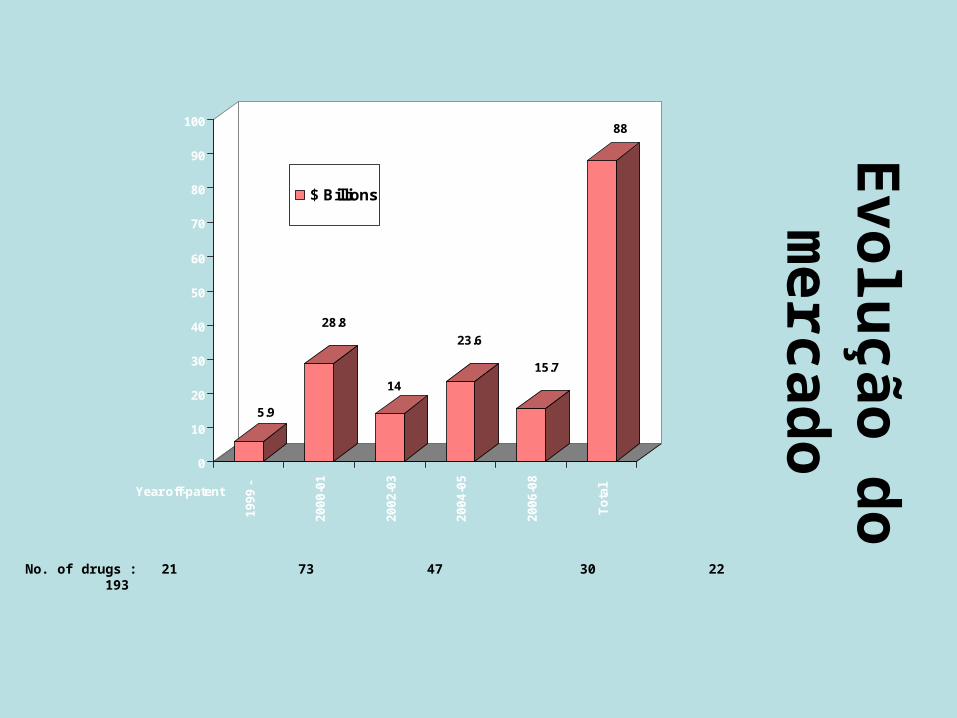
Evolução do mercado
5.9
28.8
14
23.6
15.7
88
0
10
20
30
40
50
60
70
80
90
100
1999
-
2000
-01
2002
-03
2004
-05
2006
-08
Tota
l
Year off-patent
$ Billions
No. of drugs : 21 73 47 30 22 193
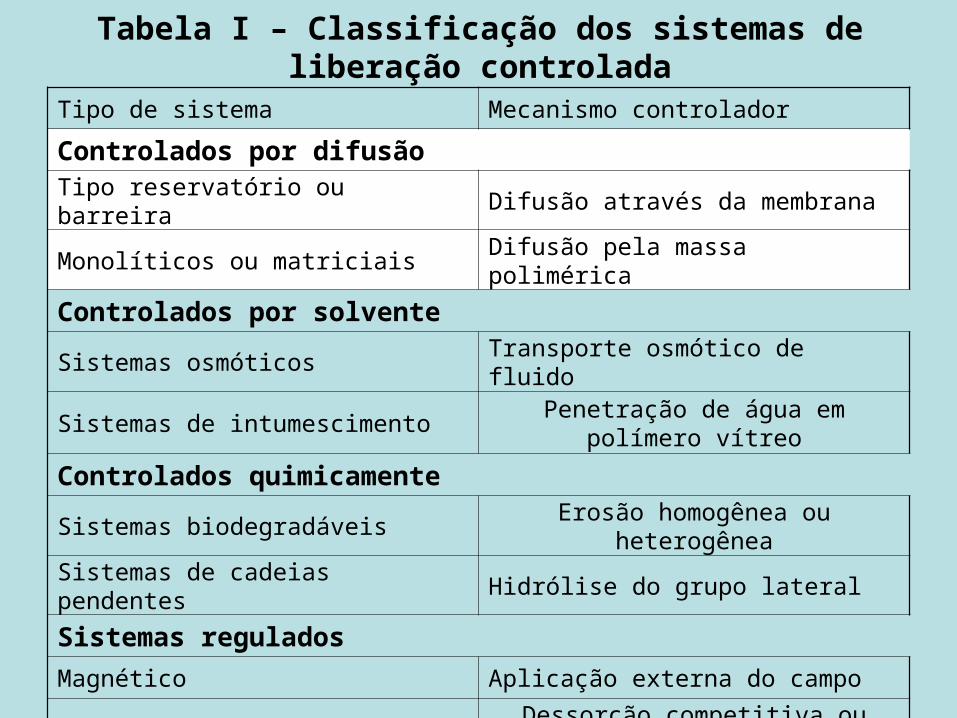
Tabela I – Classificação dos sistemas de liberação controlada
Tipo de sistema Mecanismo controlador
Controlados por difusãoTipo reservatório ou barreira Difusão através da membrana
Monolíticos ou matriciais Difusão pela massa polimérica
Controlados por solvente Sistemas osmóticos Transporte osmótico de fluido
Sistemas de intumescimento Penetração de água em polímero vítreo
Controlados quimicamenteSistemas biodegradáveis Erosão homogênea ou heterogênea
Sistemas de cadeias pendentes Hidrólise do grupo lateral
Sistemas reguladosMagnético Aplicação externa do campo
Ultra-som Dessorção competitiva ou reações de substrato enzimático

Sistemas controlados por difusão
• Os mais amplamente utilizados.• Dois tipos básicos: reservatório (barreira)
e matricial (monolítico).• Teoricamente, ambos controlados pela Lei
de Difusão de Fick.• Cineticamente, padrões mecanísticos
diferentes.

Difusividade polimérica (Dp)
• Composição polimérica.• Estrutura do fármaco.• Reticulações.• Cristalinidade do polímero.• Excipientes.
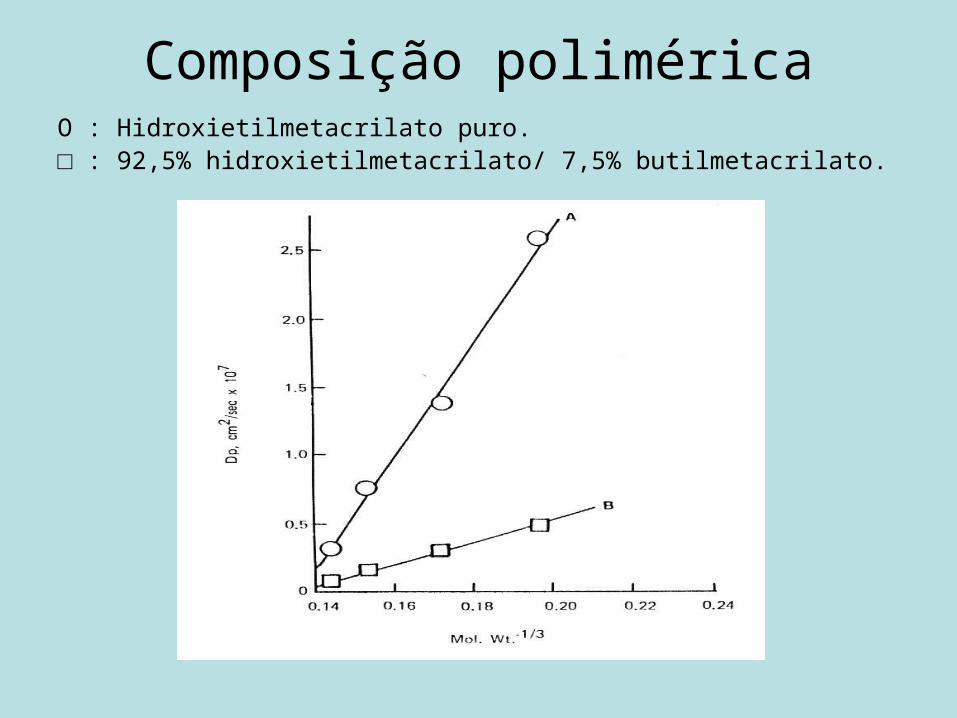
Composição poliméricaO : Hidroxietilmetacrilato puro.□ : 92,5% hidroxietilmetacrilato/ 7,5% butilmetacrilato.
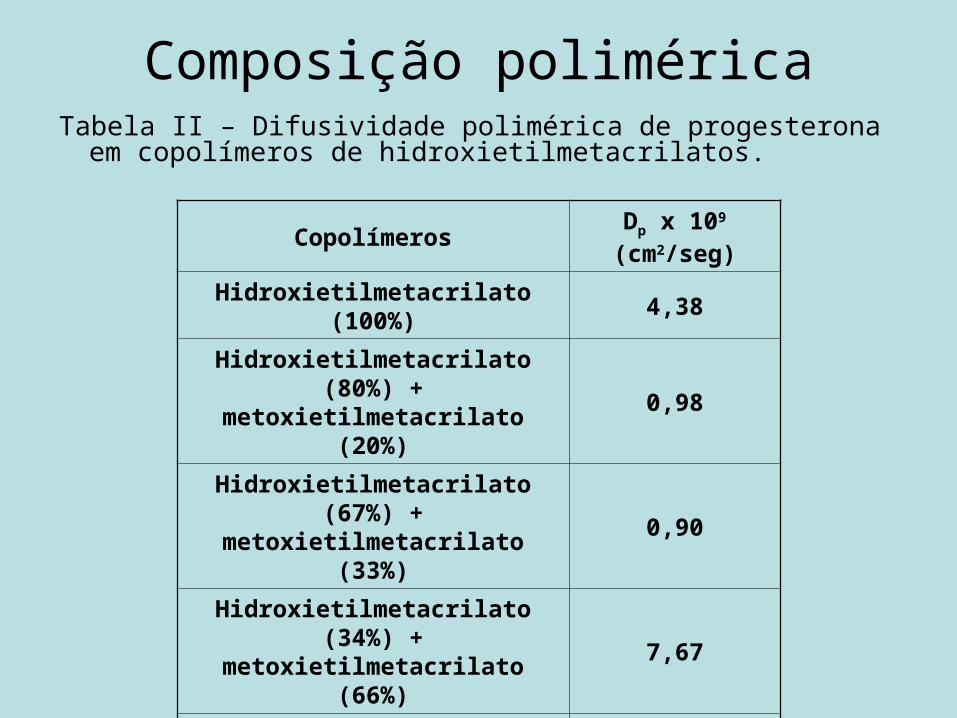
Composição poliméricaTabela II – Difusividade polimérica de progesterona em
copolímeros de hidroxietilmetacrilatos.
Copolímeros Dp x 109 (cm2/seg)
Hidroxietilmetacrilato (100%) 4,38
Hidroxietilmetacrilato (80%) + metoxietilmetacrilato (20%) 0,98
Hidroxietilmetacrilato (67%) + metoxietilmetacrilato (33%) 0,90
Hidroxietilmetacrilato (34%) + metoxietilmetacrilato (66%) 7,67
Metoxietilmetacrilato (100%) 12,70

Estrutura do fármacoTabela III – Difusividade polimérica em função da posição de grupos hidroxi em moléculas de progesterona.
Difusor Dp (cm2/seg)
Progesterona 6,34
21-Hidroxiprogesterona 4,51
11α-Hidroxiprogesterona 2,96
17α-Hidroxiprogesterona 1,82

Efeito do grau de reticulação
• Dp = D . ε ∂ D = difusividade intrínseca
ε = porosidade ∂ = tortuosidade
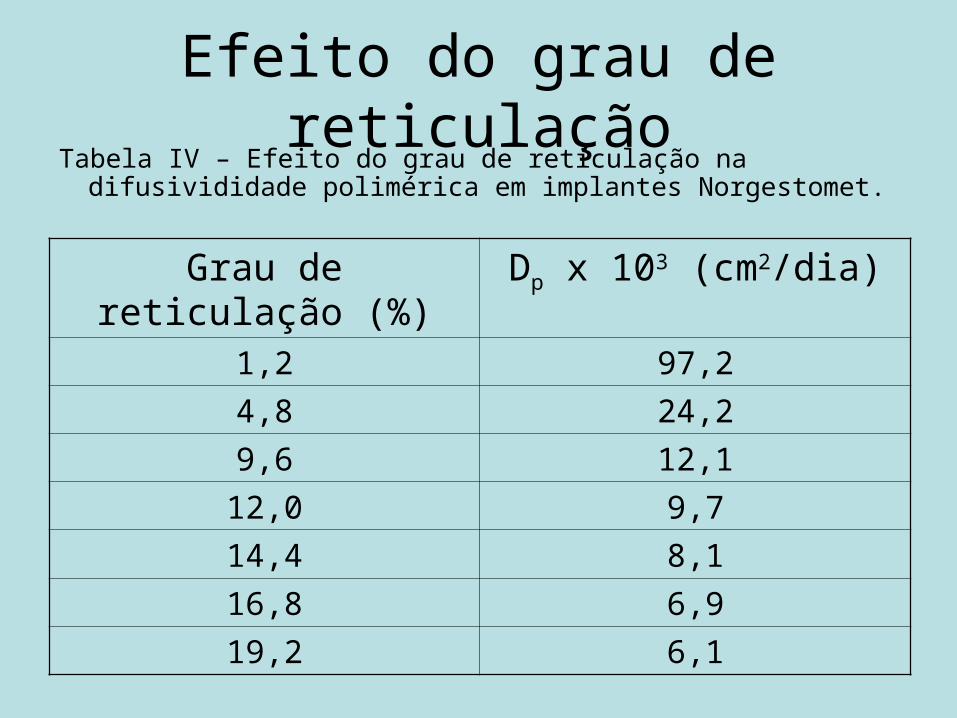
Efeito do grau de reticulaçãoTabela IV – Efeito do grau de reticulação na difusivididade
polimérica em implantes Norgestomet.
Grau de reticulação (%) Dp x 103 (cm2/dia)1,2 97,24,8 24,29,6 12,1
12,0 9,714,4 8,116,8 6,919,2 6,1
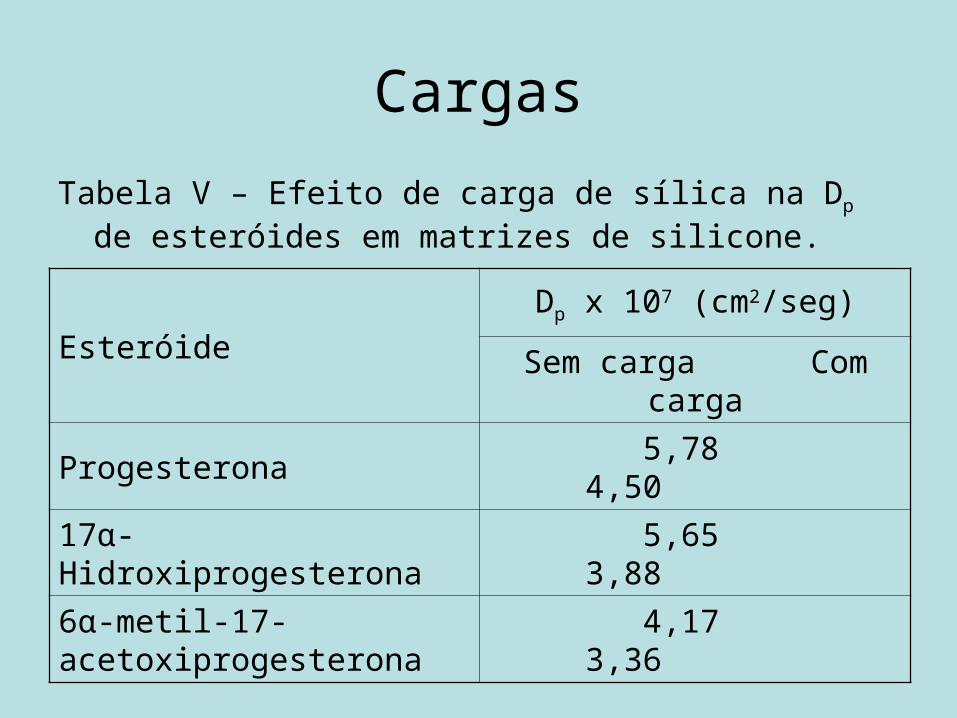
CargasTabela V – Efeito de carga de sílica na Dp de esteróides
em matrizes de silicone.
EsteróideDp x 107 (cm2/seg)
Sem carga Com carga
Progesterona 5,78 4,50
17α-Hidroxiprogesterona 5,65 3,88
6α-metil-17-acetoxiprogesterona 4,17 3,36
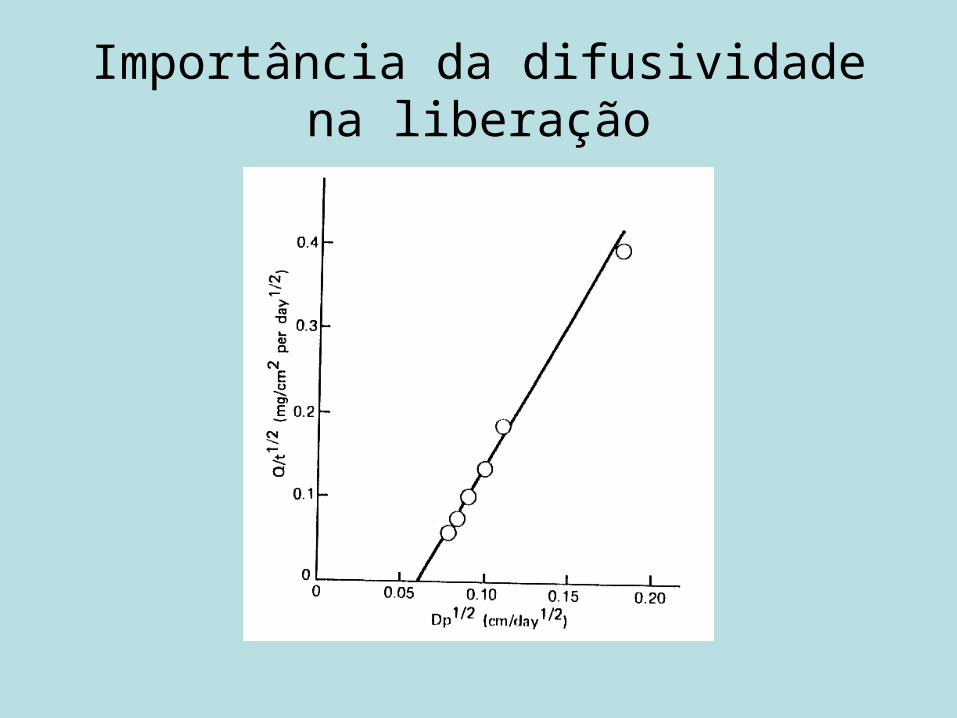
Importância da difusividade na liberação

Sistemas de barreira
• Um núcleo (sólido ou solução) de fármaco é envolvido por um filme polimérico inchável ou não-inchável.
• Fator limitante: difusão do fármaco através do polímero.
• Membranas, cápsulas, micro- e nanocápsulas, lipossomas.
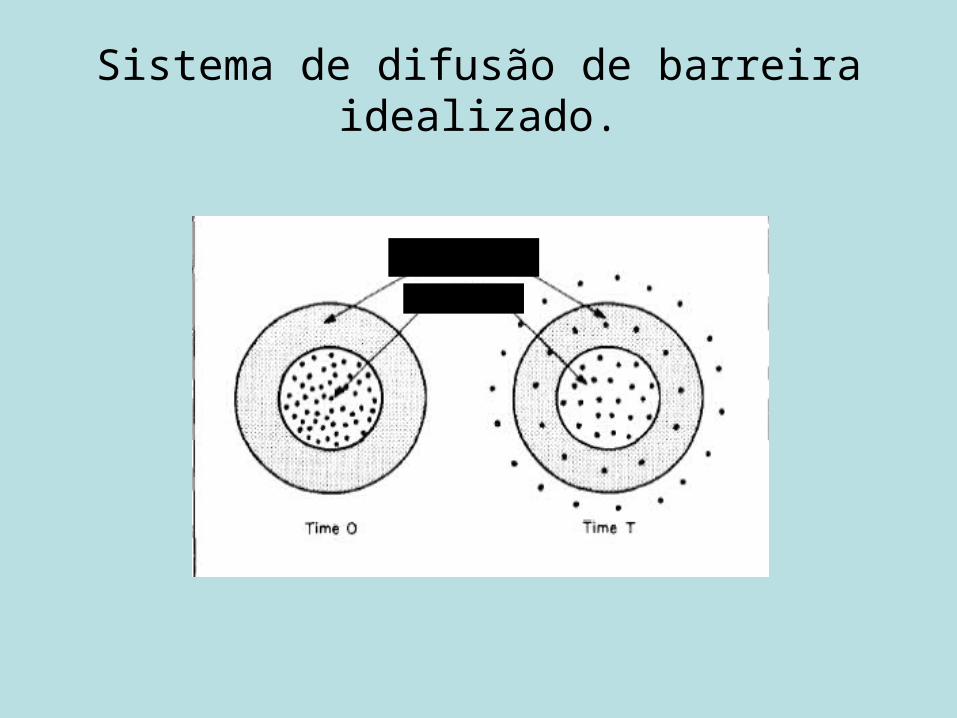
Sistema de difusão de barreira idealizado.
PolímeroFármaco

Controle da liberação
• Difusão controlada pela Primeira Lei de Fick:
J = DKΔC L
• O formato do dispositivo altera a equação: esfera, cilindro ou placa.

Vantagem
• Facilidade para produzir cinéticas de liberação próximas à ordem zero.

Desvantagens
• Geralmente não-biodegradáveis (implantes necessitam de remoção cirúrgica).
• Apenas fármacos de baixa massa molar.
• Criação de orifícios com risco de liberação total.
• Equipamentos/processo mais caros.

Tipos
• Filmes poliméricos homogêneos não-porosos.
• Membranas microporosas.

Membranas não-porosas (homogêneas)
• Etapas básicas na liberação:1) Partição entre o núcleo e a membrana.2) Difusão do fármaco em solução através
da membrana.3) Partição para o meio aquoso.

Membranas porosas
• Difusão através de poros hidrossaturados.
• Partículas incorporadas são solubilizadas quando do contanto com o meio, criando os poros.

Sistemas matriciais (monolíticos)
• Fármaco uniformemente distribuído por todo o polímero sólido.
• Matrizes hidrofílicas: absorção de água pelo polímero, gelificação e difusão do fármaco pelo gel viscoso.

Representação
Fármaco no polímero

Vantagens e desvantagens
• Facilidade de fabricação.• Custo de produção.
• Dificuldade em atingir liberação de ordem zero.• Solubilidades relativamente baixas de fármacos em
polímeros apropriados limitam a quantidade total de fármaco que pode ser incorporada e, assim, limita o tempo útil de tais sistemas.

Sistemas osmóticos
• Alza Corporation: 90% das patentes.• Bomba osmótica de Theeuwes de 1974.• Normalmente para fármacos bastante
hidrossolúveis, em virtude da densidade das membranas.
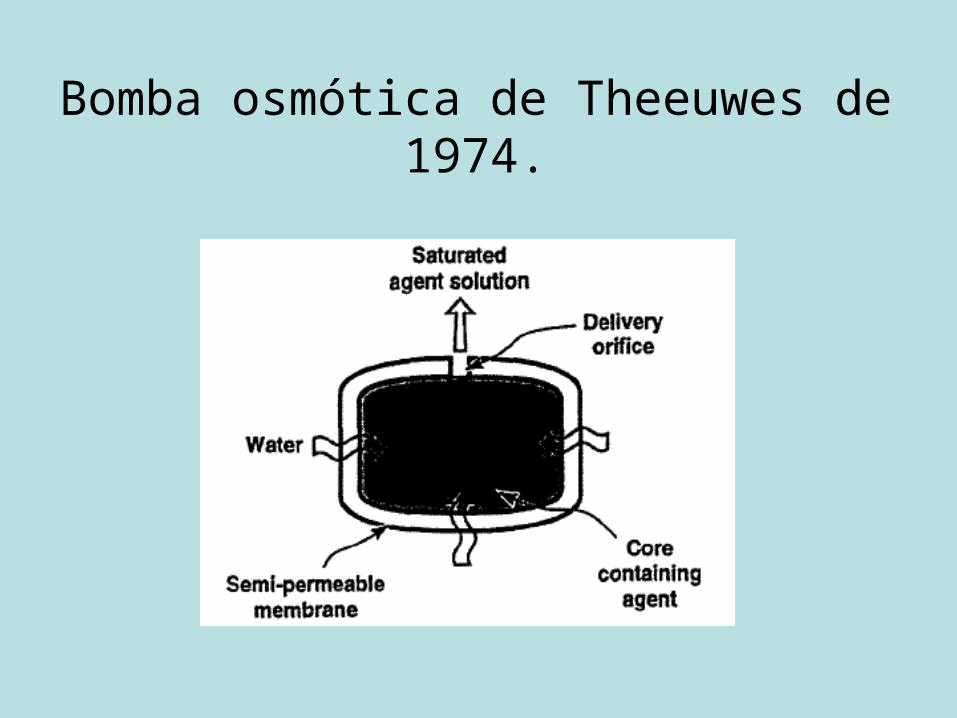
Bomba osmótica de Theeuwes de 1974.

Sistemas biodegradáveis• Biodegradação: processo químico de quebra das
cadeias. Agente biológico responsável pela degradação (enzima, microorganismo, célula).
• Bioerosão: processo físico (como dissolução) e químico (como quebra de cadeia, reticulações ou grupos laterais) de esgotamento do material. Um polímero hidro-insolúvel torna-se hidrossolúvel sob condições fisiológicas.
• Bioabsorção: o polímero ou seu produto de degradação é removido por atividade celular (como fagocitose).
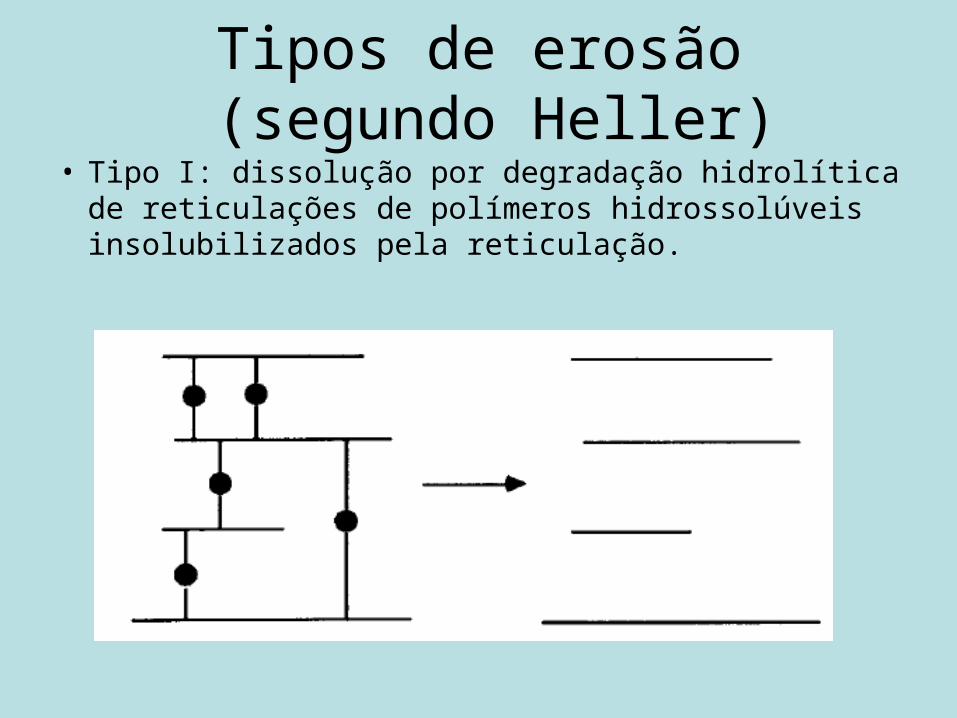
Tipos de erosão (segundo Heller)
• Tipo I: dissolução por degradação hidrolítica de reticulações de polímeros hidrossolúveis insolubilizados pela reticulação.

Tipos de erosão (segundo Heller)
• Tipo II: Dissolução aquosa de polímeros hidrossolúveis por hidrólise, ionização ou protonação de um grupo pendente na cadeia.
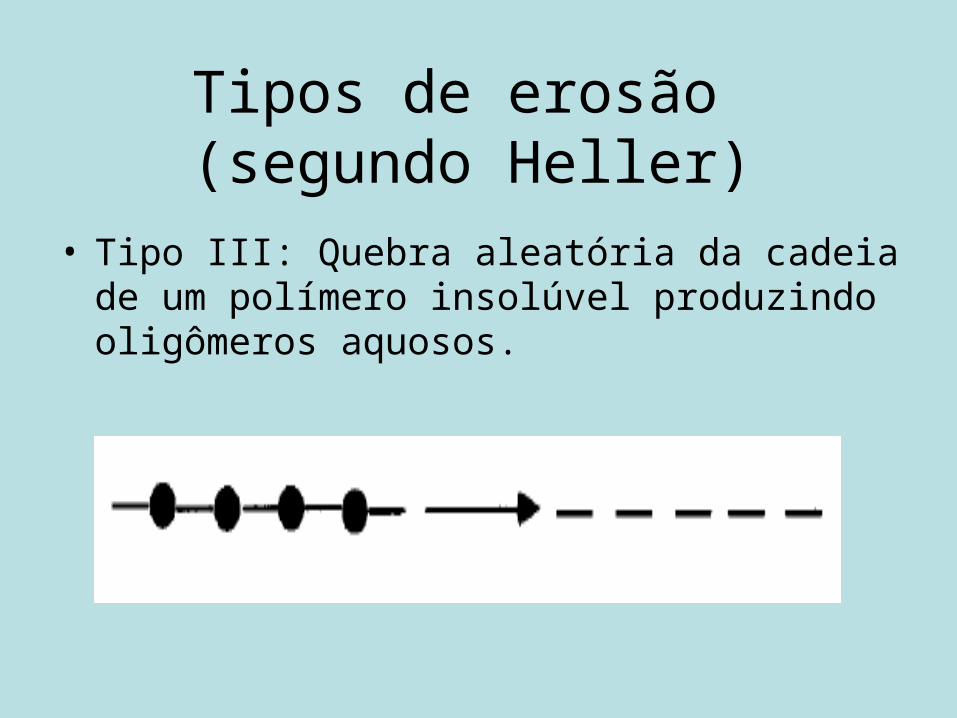
Tipos de erosão (segundo Heller)
• Tipo III: Quebra aleatória da cadeia de um polímero insolúvel produzindo oligômeros aquosos.
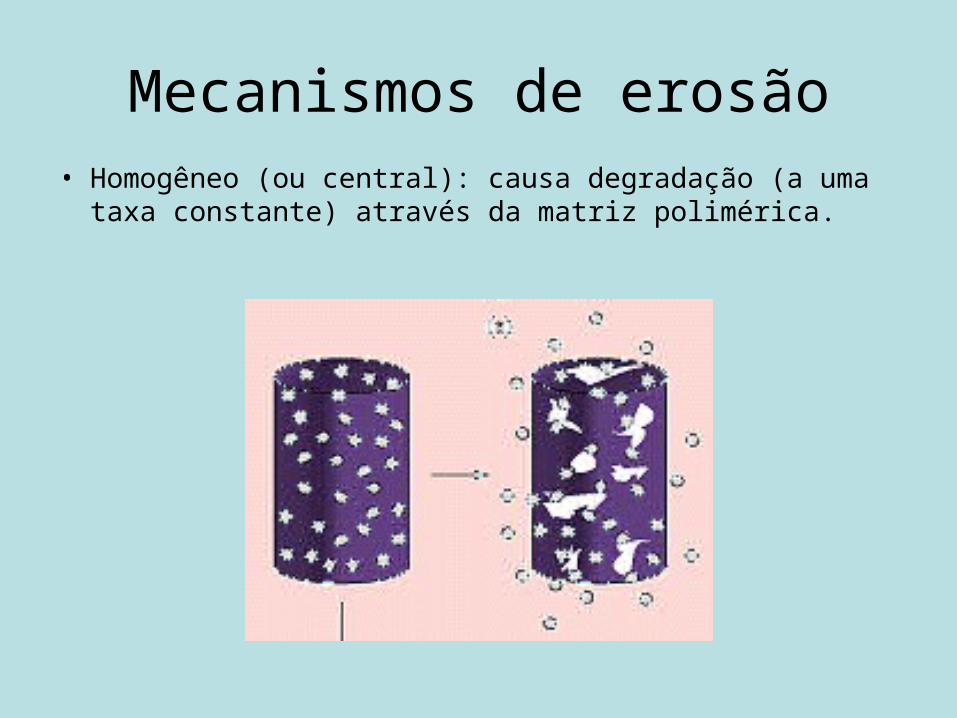
Mecanismos de erosão• Homogêneo (ou central): causa degradação (a uma taxa
constante) através da matriz polimérica.

Mecanismos de erosão
• Heterogêneo (superficial): ocorre apenas na superfície polimérica. Acarreta uma liberação mais constante.

Fatores que afetam a degradação
• Tg.
• Cristalinidade.
• Estabilidade química da cadeia.
• Hidrofobicidade do monômero.

Cinética de degradação
• Depende da posição da ligação dentro da cadeia e/ou do tamanho total da cadeia.
• Ligações centrais quebram preferencialmente em relação às das pontas.

Vantagens e desvantagens
• Não há necessidade de remoção cirúrgica no caso de implantes.
• Reposição por tecido regenerado à medida que o implante degrada.
• Os produtos de degradação podem ser tóxicos, imunogênicos ou carcinogênicos.

Sistemas controlados por inchamento
• Polímeros hidrofílicos absorvedores de água.
• Inchamento modifica a liberação.• Hidrogel: matriz inchável.
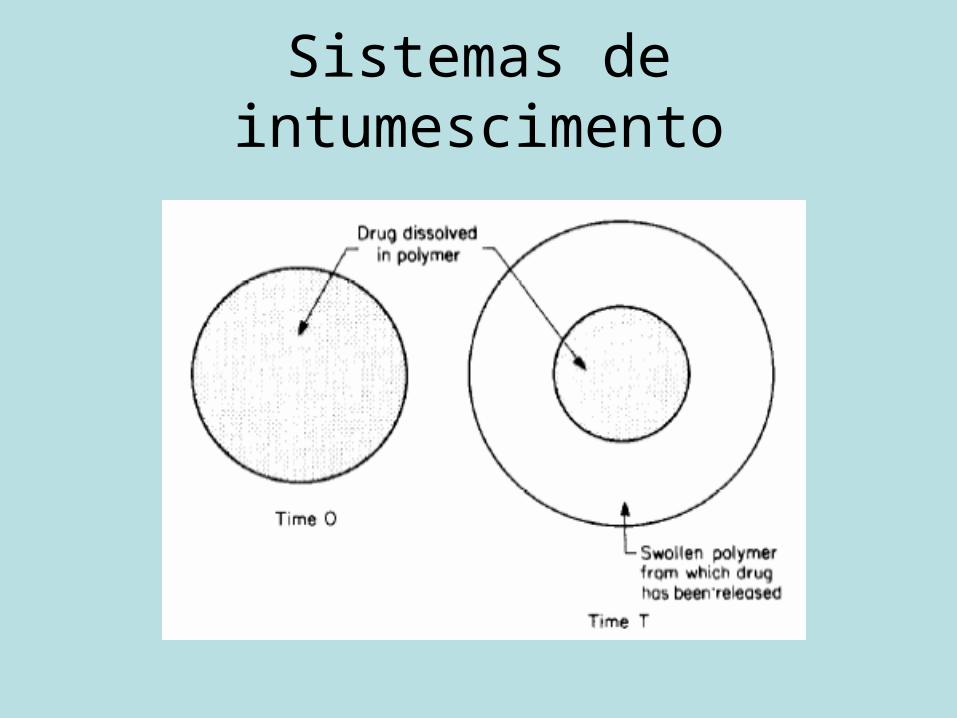
Sistemas de intumescimento

Mecanismo molecular• Relaxação macromolecular (Tg).• Difusão.
• Dependência da solubilidade do fármaco e dos excipientes incorporados.
• Variáveis: quantidade de polímero, tamanho de partícula (fármaco e polímero), pressão de compactação, razão fármaco/polímero.
• Elemento central da liberação: camada de gel envolvendo a matriz.

Cinética de liberação• Mt = K . tn
M∞
Expoente difusional, nMecanismo de
liberaçãoFilme fino Cilindro Esfera
0,5 0,45 0,43 Difusão Fickiana
0,5 < n < 1,0 0,45 < n < 0,89 0,43 < n < 0,85Transporte
anômalo (não-Fickiano)
1,0 0,89 0,85 Transporte caso II

Top 100 (fármacos) - valor de mercado nos EUA
Não - Oral24% Oral
76%
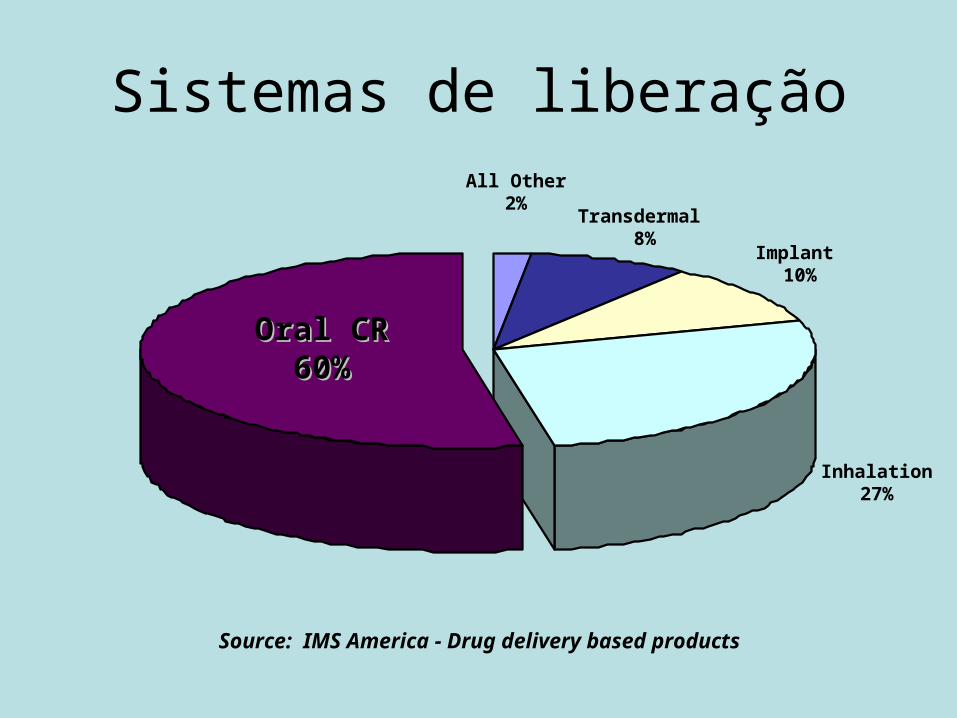
Sistemas de liberação
Source: IMS America - Drug delivery based products
Oral CROral CR60%60%
Implant 10%
Inhalation27%
Transdermal 8%
All Other2%

Segmentos de liberação de fármacosMercado mundial / Crescimento ($ em bilhões)
95%16.08.2TRANSNASAL DELIVERY
171%6.52.4TRANSMUCOSAL
106%104.350.6TOTAL67%2.51.5MISCELLANEOUS0%50CELL/GENE THERAPY
175%3.31.2LIPOSOMAL140%1.20.5RECTAL150%10.4NEEDLE-LESS INJECTION89%7.23.8INJECTABLE/IMPLANTABLE90%12.76.7TRANSDERMAL DELIVERY
93%22.611.7PULMONARY, INHALATION85%26.314.2CONTROLLED RELEASE
GROWTH20052000TECHNOLOGY

Lipossomas• Vesículas fosfolipídicas.• Introdução como sistemas de liberação de fármacos nos anos de
1970.• 1999: vendas de US$ 250 milhões.• Impedimento: alto custo das matérias-primas (maior para
cosméticos do que medicamentos).• Medicamentos (DOXIL (Caelyx), Daunosomes, Ableet, Amphotech
e AmbiSome) e cosméticos (Niosome e Capture).• Maior parte da pesquisa é acadêmica.
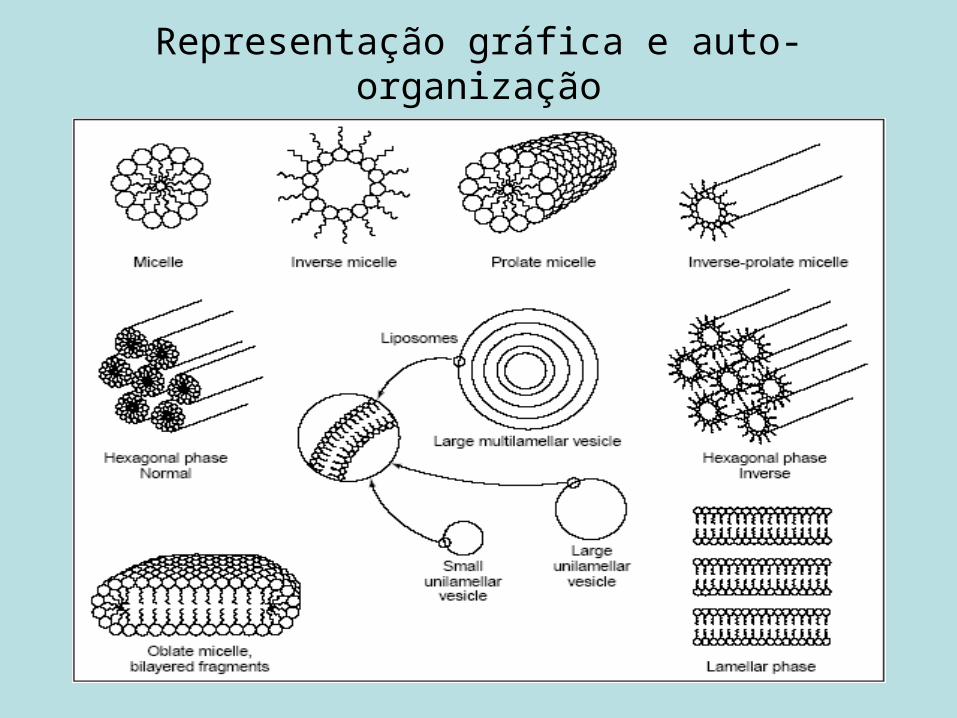
Representação gráfica e auto-organização

Esquemas lipossomais principais
• Natural: Viral: Retrovirus. Adenovirus.
• Synthetic: Vesicles. Complexes.

Barreiras para a liberação lipossomal in vivo
• Filtração (tamanho molecular e carga): fígado e baço.• Leucócitos.• Defesas naturais da célula (membranas).

Filtração
• Lipossomas pequenos neutros e positivamente carregados são eliminados menos rapidamente que os negativos.
• Interação de lipossomas negativados com proteínas plasmáticas
pode promover rápida eliminação do sangue. • Lipossomas grandes negativados são incorporados por monócitos
sangüíneos mais eficientemente que os compostos de lipídeos positivados ou neutros.
• Lipossomas grandes negativados têm maior tendência de serem incorporados pelo pulmão que os positivados ou neutros.
• Lipossomas carregando um ligante específico na superfície tendem
a ser mais rapidamente eliminados do sangue que os negativados.
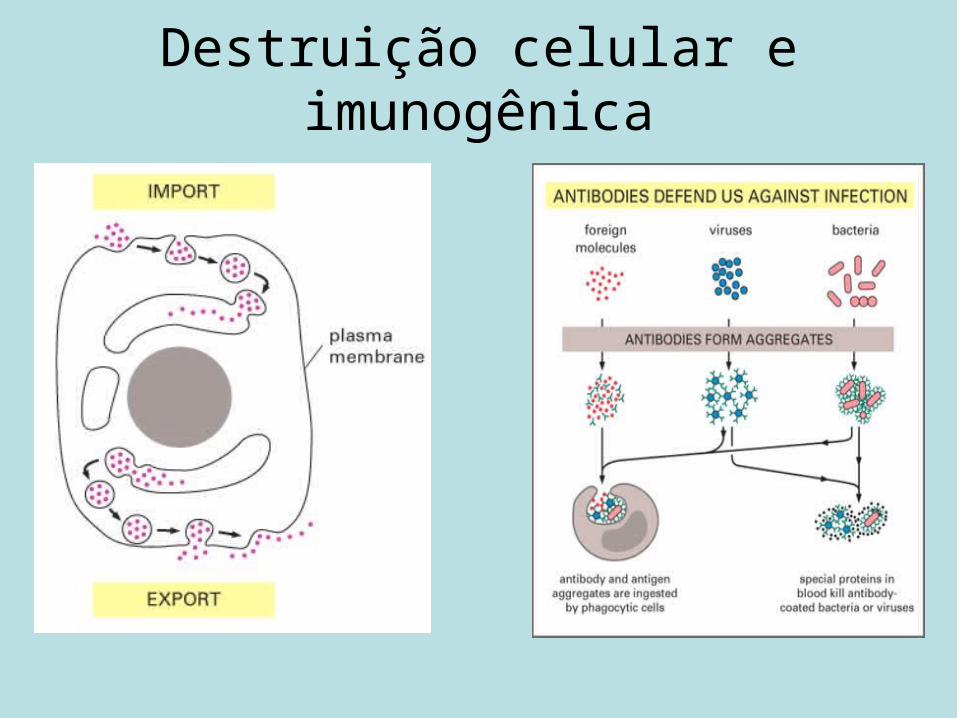
Destruição celular e imunogênica

Avanços na área• Os três principais avanços na área são:
1. Desenvolvimento de lipossomas estericamente estabilizados ou revestidos: mimetizar a estrutura superficial de células sangüíneas.
2. Aquisição de uma razão fármaco:lipídeo alta e estável por métodos de incorporação remotos direcionados por gradiente.
3. Introdução de lipídeos catiônicos e lipossomas catiônicos para formar lipoplexos (complexos com proteínas e ácidos nucléicos carregados anionicamente).

Lipossomas revestidos – estericamente estabilizados
• A qtd de PEG incluída na bicamada lipídica diminui com o aumento na massa molar do PEG.
• Baixa rigidez da bicamada → desestabilização da membrana do lipossoma → liberação do fármaco encapsulado.
• Melhores condições: cadeias de PEG longas e alta densidade superficial.
• Impedimento da opsonização pelas proteínas plasmáticas.• Estrutura transiente, flexível e de mudança rápida: dificuldade do
sistema imune em modelar um anticorpo ao redor do sistema.• Outros candidatos: PVP, poliacrilamida.• Polímeros protetores não deve apresentar grupos hidroxila (como
polissacarídeos), vetores para o complemento C3, ou amina (como polilisina), vetores para C4.

Lipossomas revestidos – estericamente estabilizados
• Revestimento de PEG: Aumenta a estabilidade in vivo. Reduz a incorporação pelo SRE.
• Baixa permeabilidade da matriz lipídica e composição intralipossomal aquosa selecionada:
Alta carga de fármaco. Encapsulação estável (para retenção do fármaco durante a residência).
• Diâmetro médio (100 nm): Grande o suficiente para carrear uma qtd razoável de fármaco. Pequeno bastante para permitir extravasamento eficiente através de
defeitos do endotélio vascular em tecidos tumorais.

Doxil
• Principal objetivo: vetorização (tumores e inflamações).• DOXIL: vetorização passiva.• Lançado em 1995.• U$ 100 milhões/ano, nos EUA.

Ambisome – Anfotericina B lipossomal liofilizada
• Frasco ampola 10mL.• 50 mg anfotericina.• Lipossomas: 213 mg fosfatidilcolina de soja
hidrogenada, 52 mg colesterol, 84 mg distearoilfosfatidilglicerol, 0,64 mg alfa tocoferol.
• 900 mg sacarose, 27 mg succinato dissódico 7.H2O.• Lipossoma com monolamela dupla.

Nanopartículas• Desenvolvidas inicialmente nos anos 70.• Incorporadas pelo fígado (mais as hidrofílicas), baço e outras partes
do RES.• Partículas < 100nm (evitar o clearance).• Revestimento com polímeros hidrofílicos (repelir proteínas
plasmáticas).• EPR (enhanced permeation and retention effect): efeito de
permeação e penetração aumentada.

Vantagens e desvatagens
• Proteção do fármaco contra degradação in vivo.• Estabilidade.• Habilidade de controlar a liberação do fármaco.• Alta eficiência de encapsulação.• Habilidade na internacionalização celular.
• Interação não-específica com células e proteínas, levando à acumulação em tecidos não-específicos.

Revestimento com PEG• Evitar adsorção protéica e seqüestro pelo RES.• Retarda a depuração e reduzir a imunogenicidade das proteínas.• Falta de grupos ligantes na superfície de carreadores PEGlados:
dificuldade de vetorização ativa.• Redução da degradação por enzimas proteolíticas.• Aumento no tamanho aparente do polipeptídeo redução na
filtração renal alteração da biodistribuição.• Fatores importantes: Número de cadeias de PEG ligadas ao polipeptídeo. Massa molar e estrutura da cadeia de PEG. Localição do PEG no polipeptídeo. Química usada para ligar o PEG ao polipeptídeo.

Propriedades do PEG
• Linear: mais comum.• Solúvel em soluções aquosas e solventes orgânicos.• Ligação de 2-3 moléculas de água por unidade de óxido de etileno.• Pela água ligada e flexibilidade da cadeia: PEG age como se fosse
5-10 vezes maior que uma proteína solúvel de massa comparável.• Oligômeros de PEG (<400 Da): degradação in vivo por álcool
desidrogenase a metabólitos tóxicos.• PEG (>1000 Da): não-tóxico (anos de uso em alimentos,
cosméticos e produtos farmacêuticos).• Eliminação rápida in vivo: <20 Kda: excreção na urina. >20 Kda: excreção na urina e nas fezes.• Pouco imunogênico.

Química da PEGlação
• Normalmente derivatização do PEG para ligação a lisina ou um grupo N-terminal.
• Heterogeneidade na substituição da lisina e na MM do PEG: necessidade de reprodutibilidade.
• Ligações estáveis: Melhor na estocagem. Purificação mais fácil. Disponibilidade em seringas prefilled.• Ligações mais instáveis: Podem melhorar a atividade (facilitando a ligação da proteína ao
seu sítio, sem impedimento direto ou estérico)

Características para liberação• Circulação pelos menores capilares: < 5μm.• Prevenção de efeitos de filtragem no baço: < 200nm.• Allen: “se você quiser ser invisível, pareça água”: partículas
hidrofílicas ou revestimento das lipofílicas.

Avanço metodológico• Tecnologia inicial: PEGs pequenos (12 Kda). Múltiplos PEGs por fármaco. Ligações instáveis. Baixa bioatividade. Pureza variável do produto. Adagen®, Oncospar®, PEG-
Intron®.
• PEGlação avançada: PEGs grandes. Único PEG por fármaco. Produto estável. Alta bioatividade. Alta pureza do produto.

Vantagens da PEGlação• Aumenta a solubilidade.• Diminui a qtd de proteína para manter a eficácia
terapêutica.• Diminui a freqüência de doses pela menor eliminação.
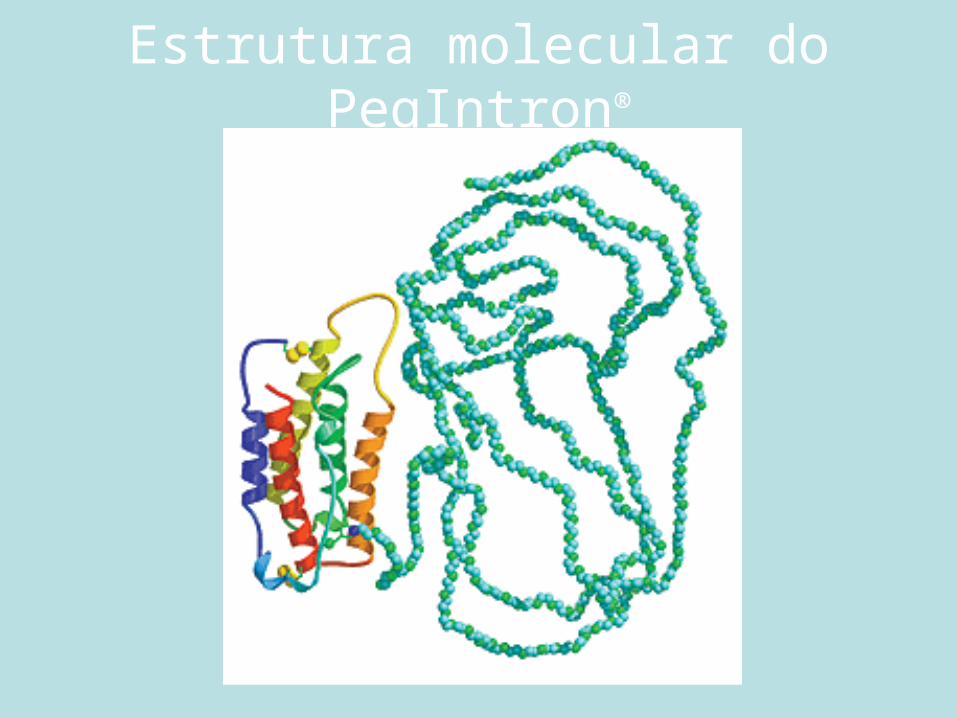
Estrutura molecular do PegIntron®

Clearance renal de PEG

Oncaspar®
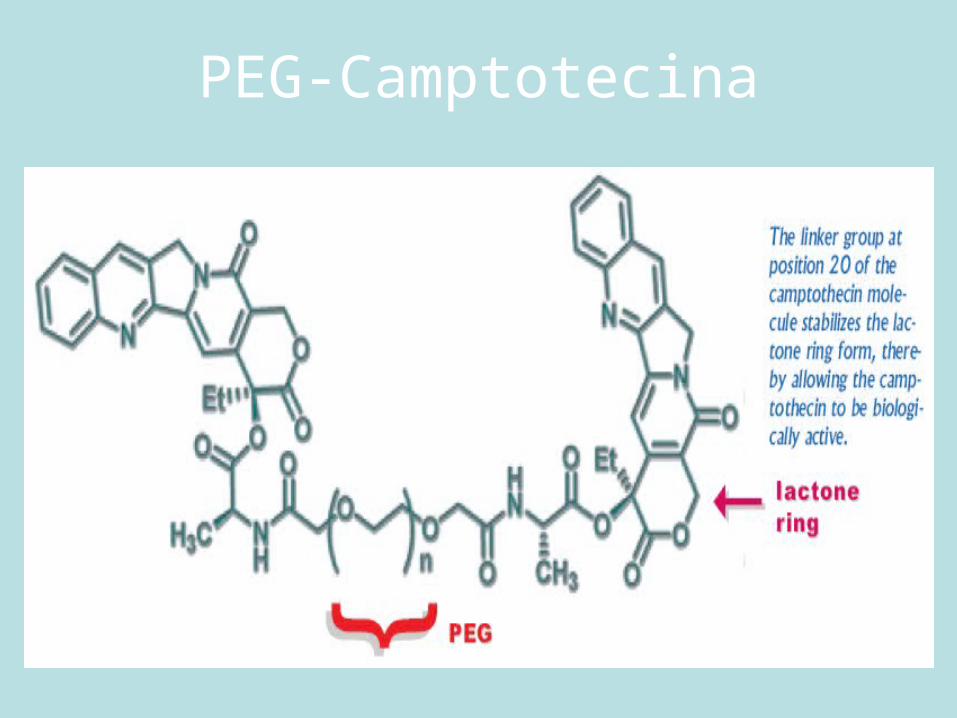
PEG-Camptotecina

Proteínas PEGladas no mercado

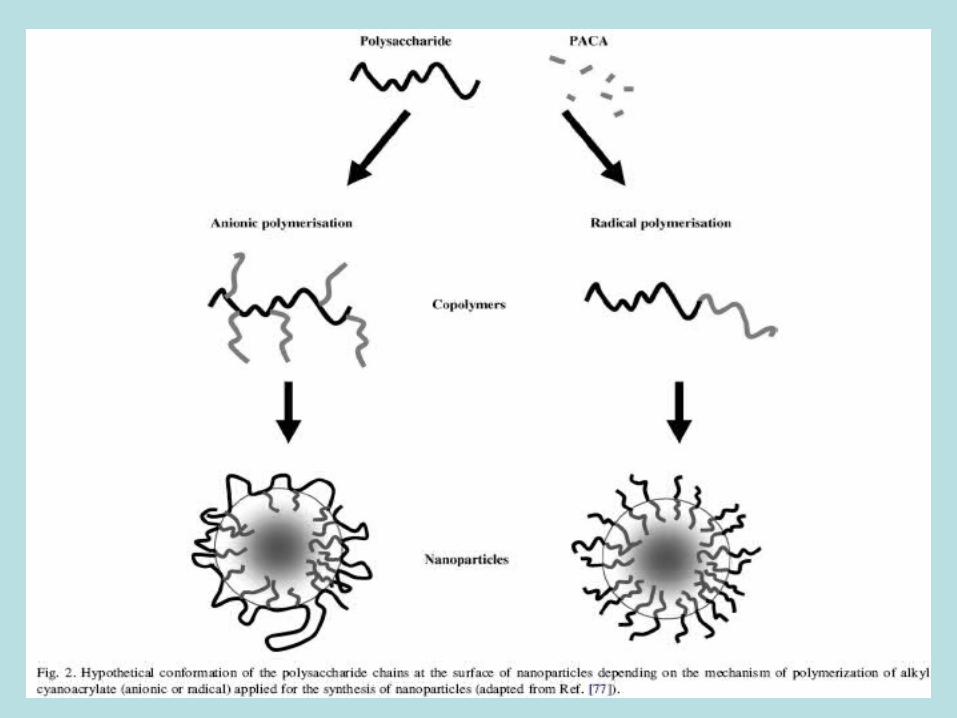
Revestimento polissacarídeo
• Vetorização ativa: receptores específicos em células e tecidos.• Propriedades antivirais, antibacterianas e antitumorais próprias.• Ácido siálico: revestimento de células vermelhas (biomimetismo).• Propriedades mucoadesivas: quitosano e ácido hialurônico.

Diagnóstico • SPIO (óxido de ferro super-paramagnético): partículas de ~150nm
envoltas por uma camada de dextrana.• Primeira aplicação (Endorem®): agente de contraste em órgãos
SRE.• USPIO (óxido de ferro paramagnético ultra-pequeno): evita a
incorporação pelo SER. Incorporação também por células tumorais. Sinerem®. Linfografia de linfonodos hiperplásicos ou metastático. Tumores cerebrais.

Terapia oncológica
• Quimioterapia/radioterapia.
• Melhoria na qualidade de vida / aumento da expectativa com o tratamento.

Vetorização tumor-específica
• Ligantes com clivagem por peptidase ou ácida.• Falta de estabilidade in vivo.• Menor potência do fármaco quando clivado erradamente.• Anticorpos monoclonais: ligação a antígenos tumorais (1975).
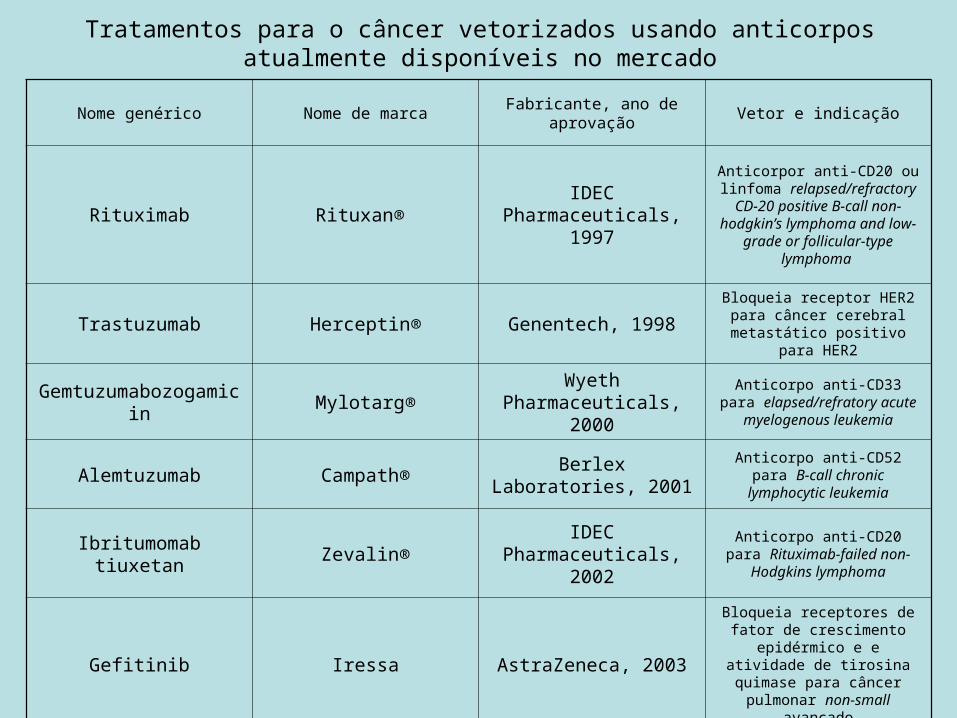
Tratamentos para o câncer vetorizados usando anticorpos atualmente disponíveis no mercado
Nome genérico Nome de marca Fabricante, ano de aprovação Vetor e indicação
Rituximab Rituxan® IDEC Pharmaceuticals, 1997
Anticorpor anti-CD20 ou linfoma relapsed/refractory CD-20 positive B-call non-hodgkin’s
lymphoma and low-grade or follicular-type lymphoma
Trastuzumab Herceptin® Genentech, 1998Bloqueia receptor HER2 para câncer cerebral metastático
positivo para HER2
Gemtuzumabozogamicin Mylotarg® Wyeth Pharmaceuticals, 2000
Anticorpo anti-CD33 para elapsed/refratory acute myelogenous leukemia
Alemtuzumab Campath® Berlex Laboratories, 2001
Anticorpo anti-CD52 para B-call chronic lymphocytic leukemia
Ibritumomab tiuxetan Zevalin® IDEC Pharmaceuticals, 2002
Anticorpo anti-CD20 para Rituximab-failed non-Hodgkins
lymphoma
Gefitinib Iressa AstraZeneca, 2003
Bloqueia receptores de fator de crescimento epidérmico e e
atividade de tirosina quimase para câncer pulmonar non-
small avançado


















![6. LTM_Grau de Liberação[2].pdf](https://img.pdfslide.tips/doc/110x75/577c7ba41a28abe054981bb4/6-ltmgrau-de-liberacao2pdf.jpg)










