Embed Size (px)
Citation preview

Large-scale imputation of epigenetic datasets for systematic annotation
of diverse human tissues
2015/3/17 Epigenome Roadmap 輪読会
RIKEN ACCC BiT 露崎弘毅
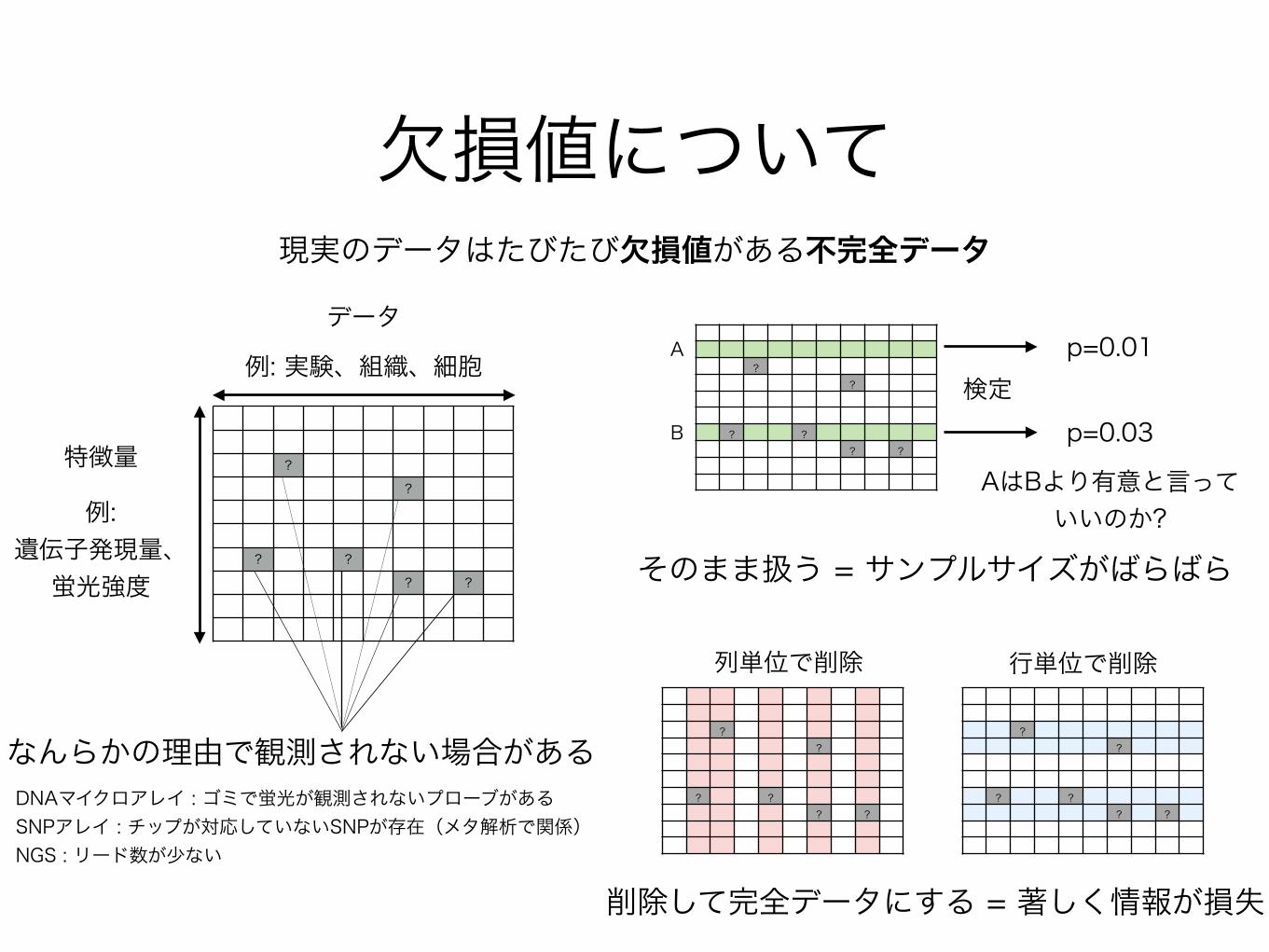
欠損値について
そのまま扱う = サンプルサイズがばらばら
削除して完全データにする = 著しく情報が損失
??
? ?? ?
なんらかの理由で観測されない場合がある
データ
特徴量
例:遺伝子発現量、
蛍光強度
例: 実験、組織、細胞
??
? ?? ?
??
? ?? ?
現実のデータはたびたび欠損値がある不完全データ
??
? ?? ?
AはBより有意と言っていいのか?
A
B
検定p=0.01
p=0.03
列単位で削除 行単位で削除
DNAマイクロアレイ : ゴミで蛍光が観測されないプローブがある SNPアレイ : チップが対応していないSNPが存在(メタ解析で関係) NGS : リード数が少ない
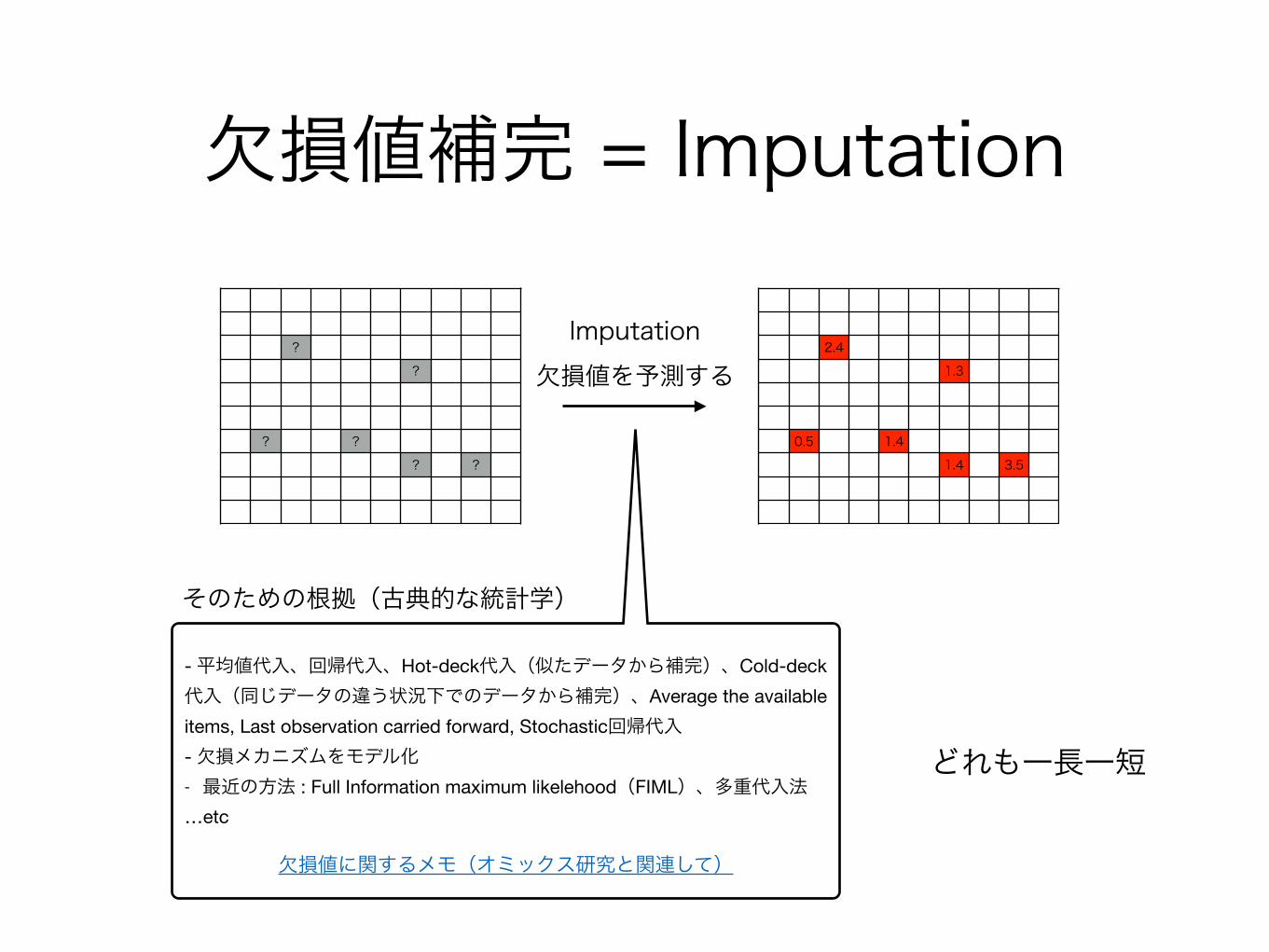
欠損値補完 = Imputation
??
? ?? ?
2.41.3
0.5 1.41.4 3.5
欠損値を予測するImputation
そのための根拠(古典的な統計学)
- 平均値代入、回帰代入、Hot-deck代入(似たデータから補完)、Cold-deck代入(同じデータの違う状況下でのデータから補完)、Average the available items, Last observation carried forward, Stochastic回帰代入- 欠損メカニズムをモデル化- 最近の方法 : Full Information maximum likelehood(FIML)、多重代入法…etc
欠損値に関するメモ(オミックス研究と関連して)
どれも一長一短
??
? ?? ?
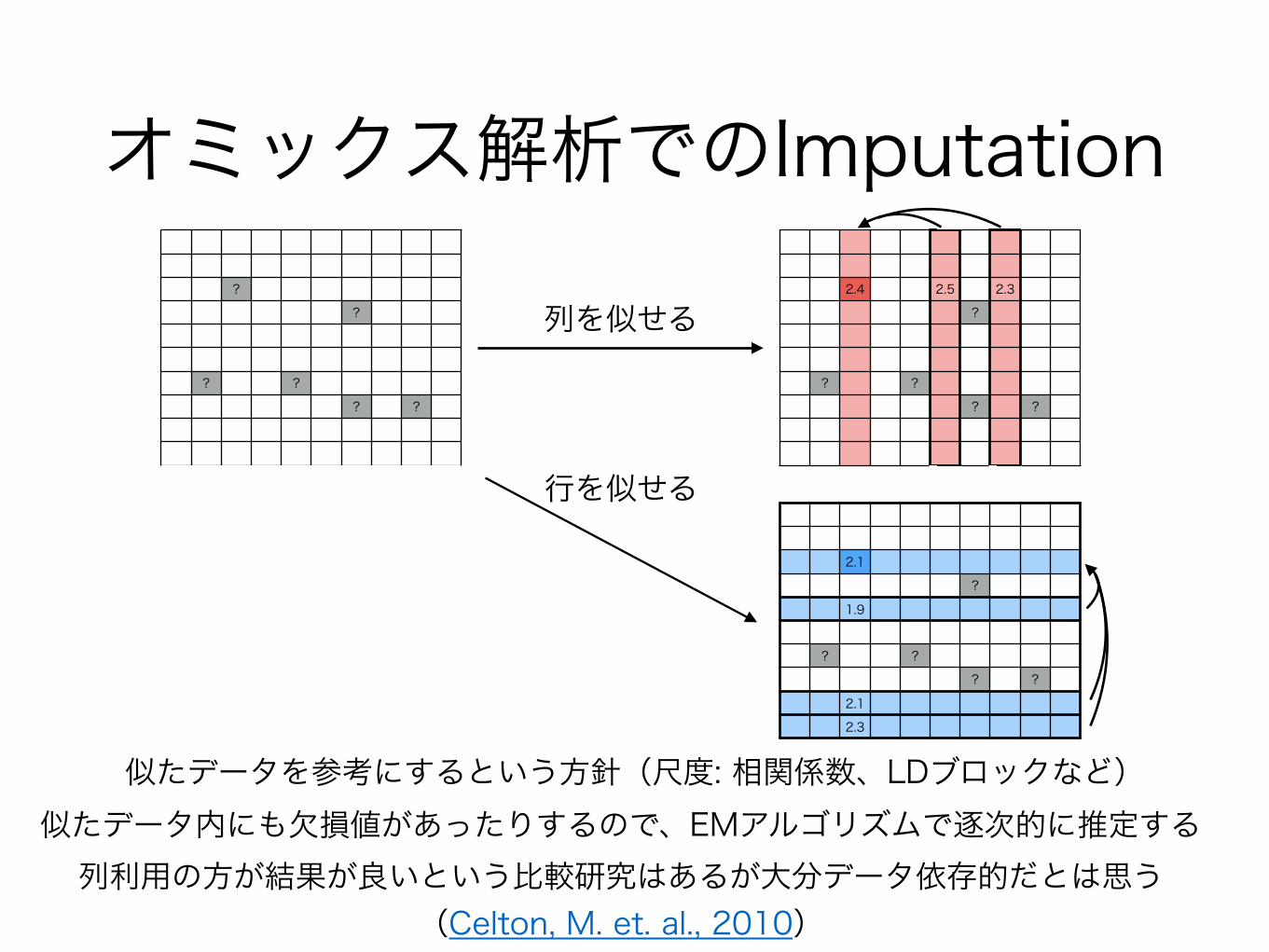
似たデータを参考にするという方針(尺度: 相関係数、LDブロックなど)
2.4 2.5 2.3?
? ?? ?
2.1?
1.9
? ?? ?
2.12.3
列を似せる
行を似せる
オミックス解析でのImputation
列利用の方が結果が良いという比較研究はあるが大分データ依存的だとは思う(Celton, M. et. al., 2010)
似たデータ内にも欠損値があったりするので、EMアルゴリズムで逐次的に推定する
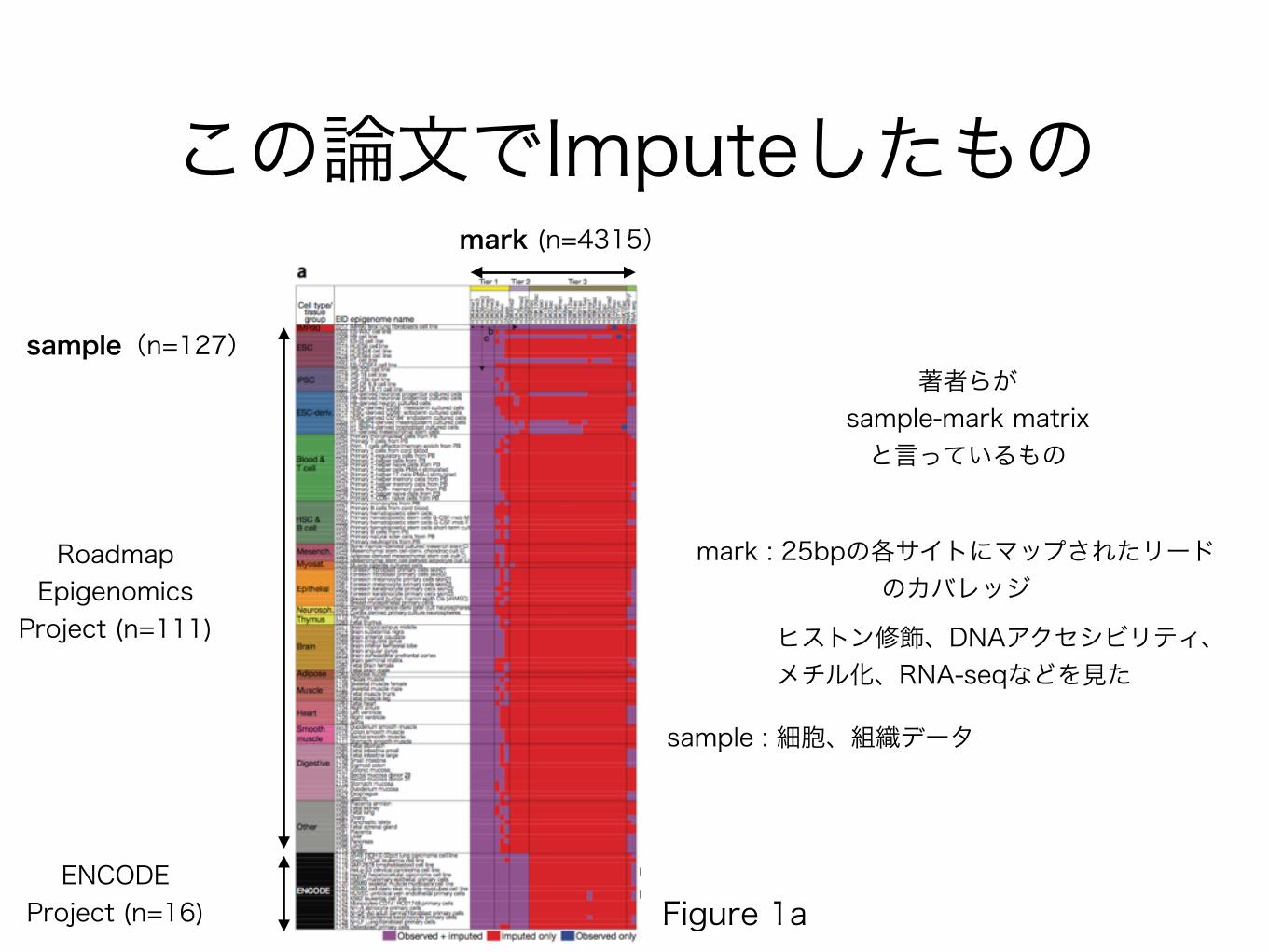
この論文でImputeしたもの
sample(n=127)
mark (n=4315)
Figure 1a
Roadmap Epigenomics
Project (n=111)
ENCODE Project (n=16)
著者らが sample-mark matrix と言っているもの
mark : 25bpの各サイトにマップされたリードのカバレッジ
sample : 細胞、組織データ
ヒストン修飾、DNAアクセシビリティ、メチル化、RNA-seqなどを見た
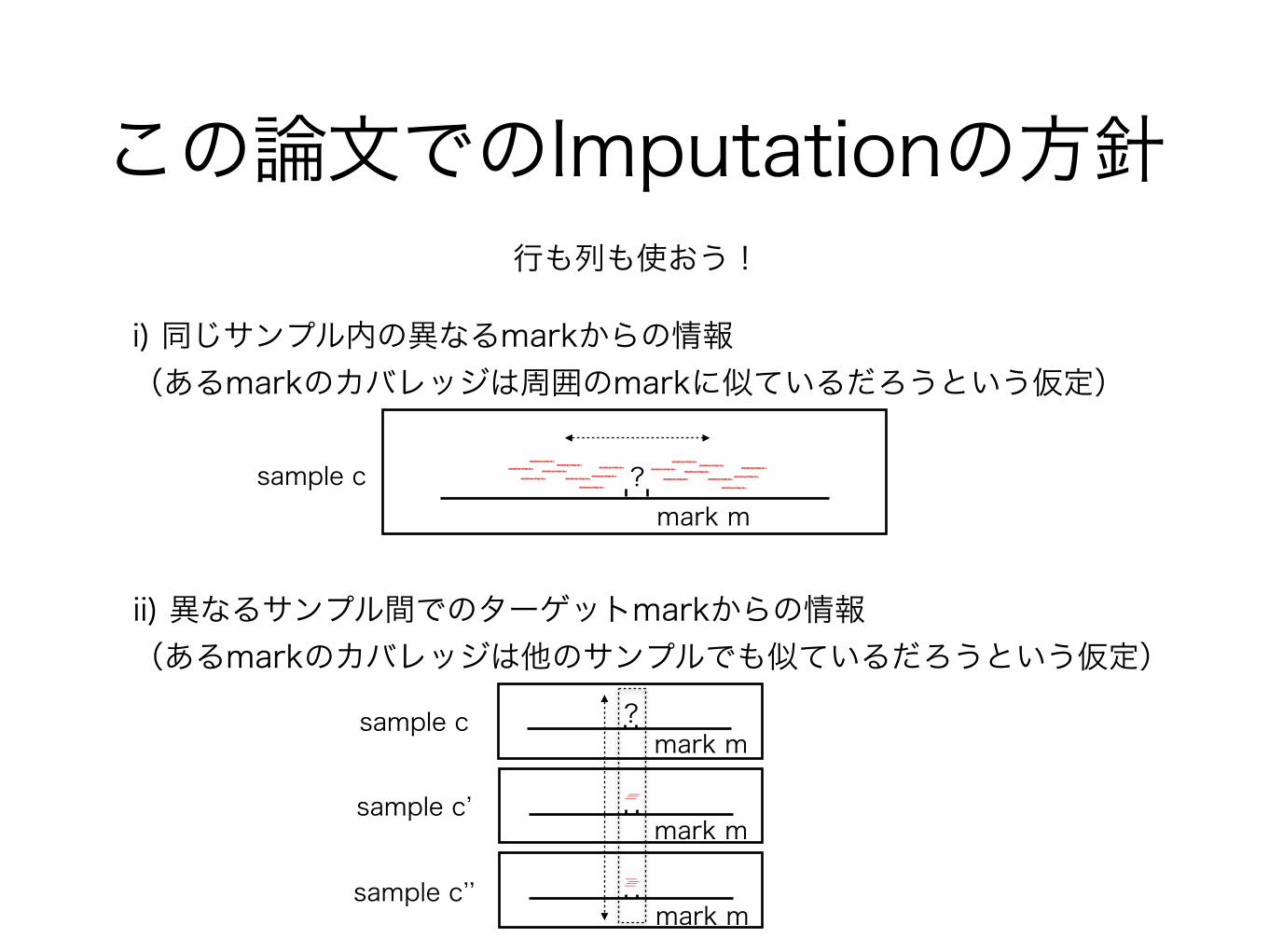
この論文でのImputationの方針行も列も使おう!
i) 同じサンプル内の異なるmarkからの情報 (あるmarkのカバレッジは周囲のmarkに似ているだろうという仮定)
ii) 異なるサンプル間でのターゲットmarkからの情報(あるmarkのカバレッジは他のサンプルでも似ているだろうという仮定)
sample c ?mark m
sample c ?mark m
mark m
mark m
sample c’
sample c’’
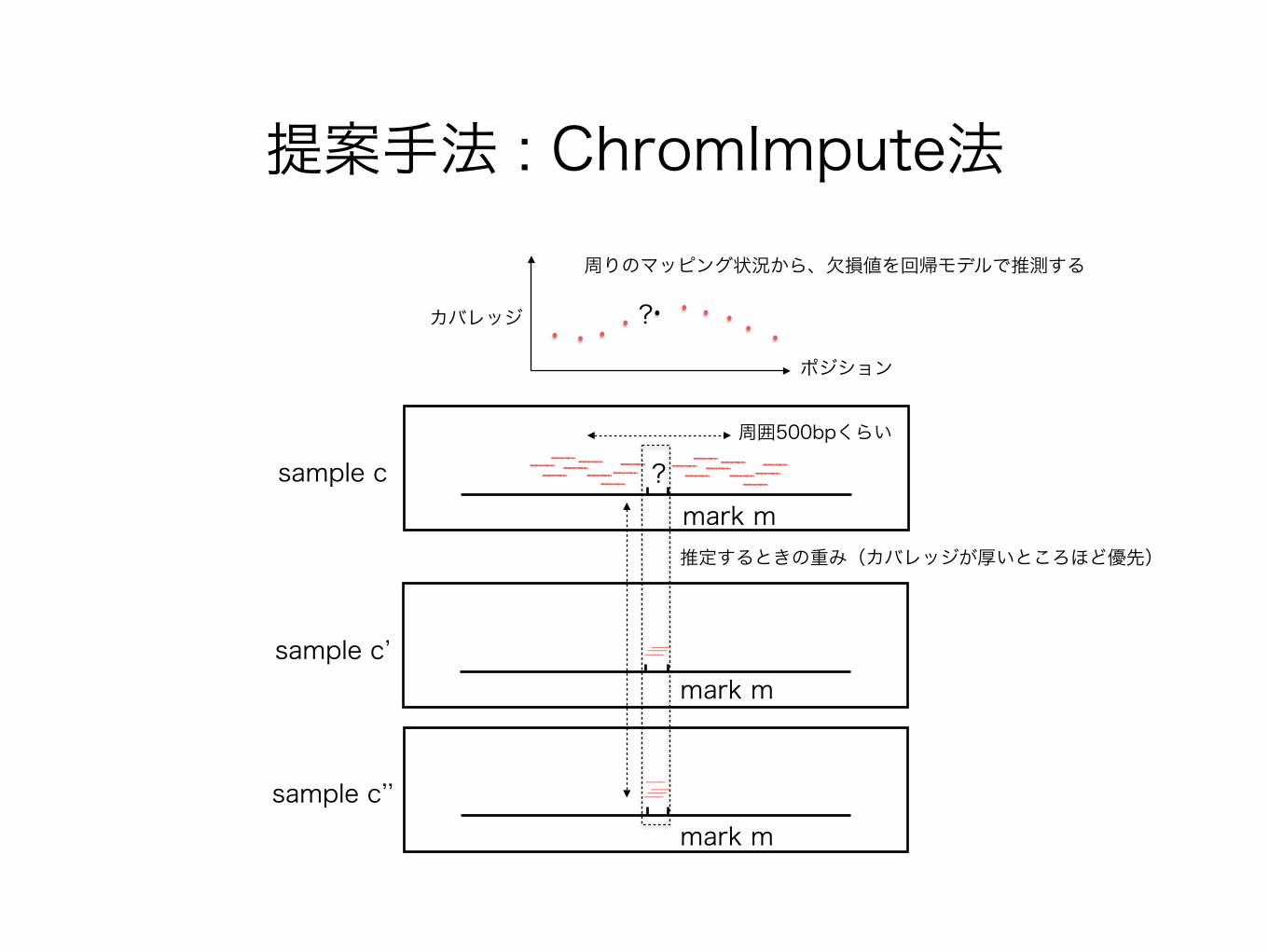
提案手法 : ChromImpute法
sample c ?mark m
sample c’mark m
sample c’’mark m
?
周囲500bpくらい
推定するときの重み(カバレッジが厚いところほど優先)
カバレッジ
ポジション
周りのマッピング状況から、欠損値を回帰モデルで推測する
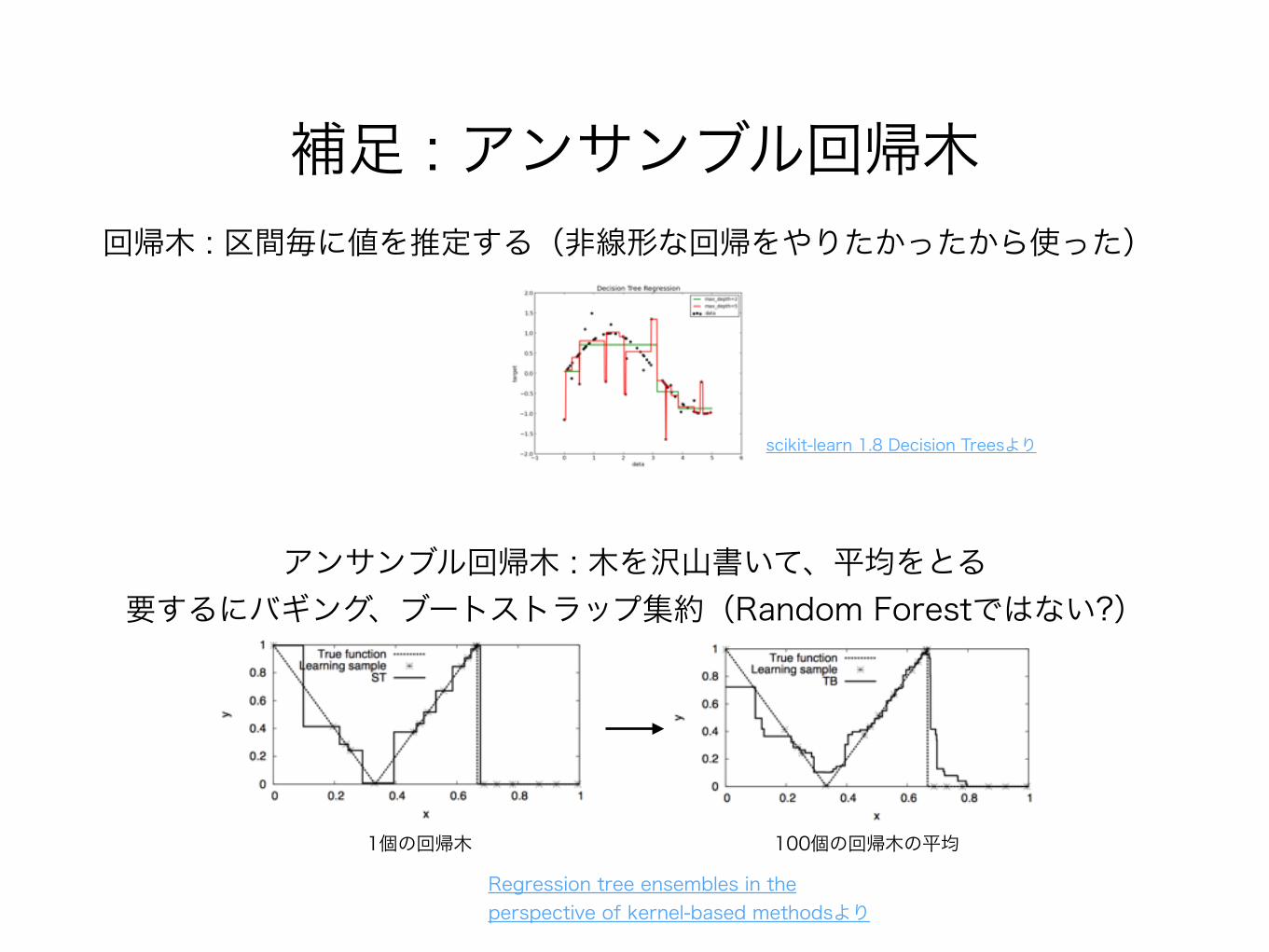
補足 : アンサンブル回帰木回帰木 : 区間毎に値を推定する(非線形な回帰をやりたかったから使った)
scikit-learn 1.8 Decision Treesより
アンサンブル回帰木 : 木を沢山書いて、平均をとる 要するにバギング、ブートストラップ集約(Random Forestではない?)
Regression tree ensembles in the perspective of kernel-based methodsより
1個の回帰木 100個の回帰木の平均
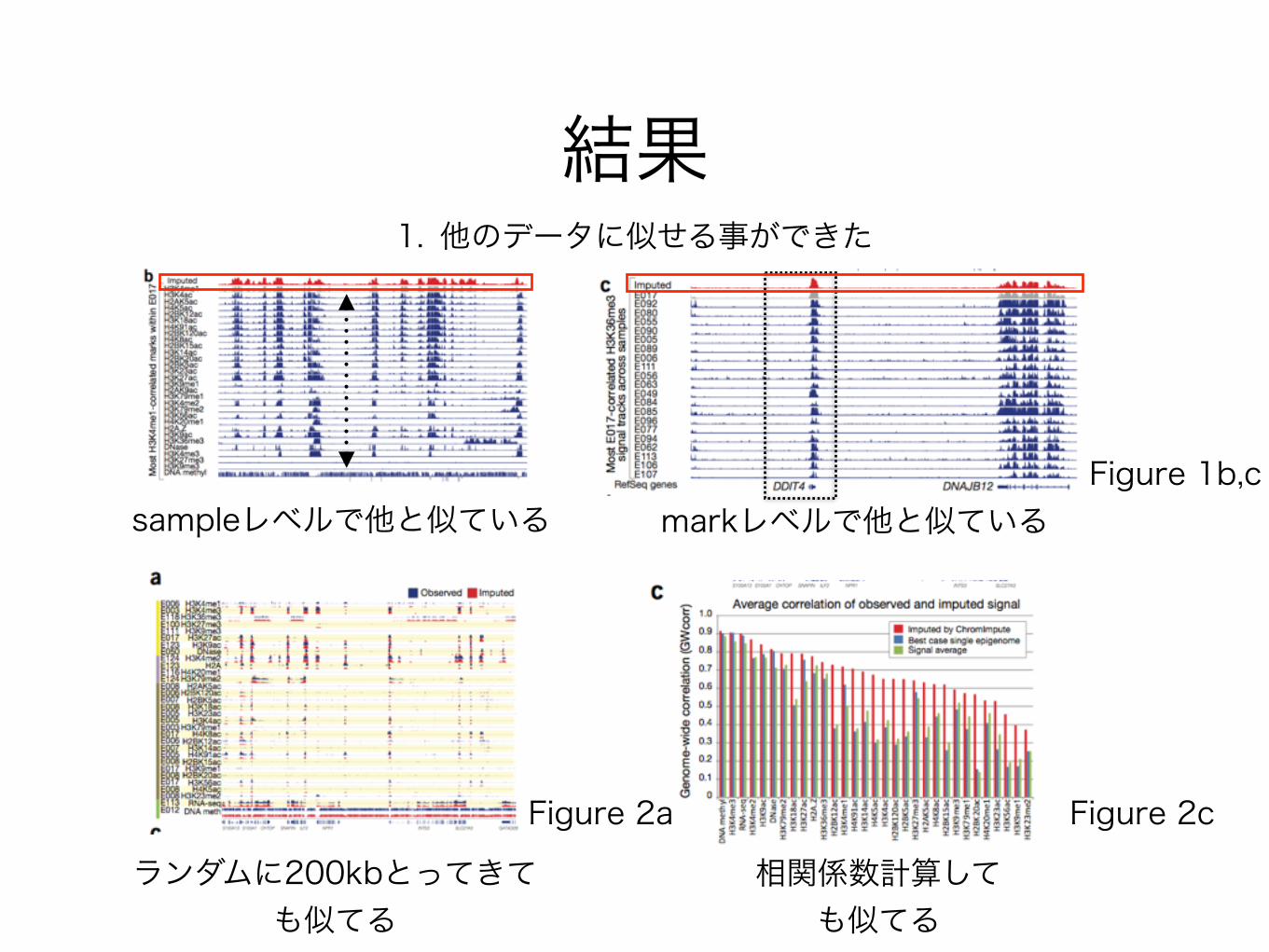
結果
sampleレベルで他と似ている markレベルで他と似ているFigure 1b,c
1. 他のデータに似せる事ができた
ランダムに200kbとってきても似てる
Figure 2a Figure 2c
相関係数計算しても似てる
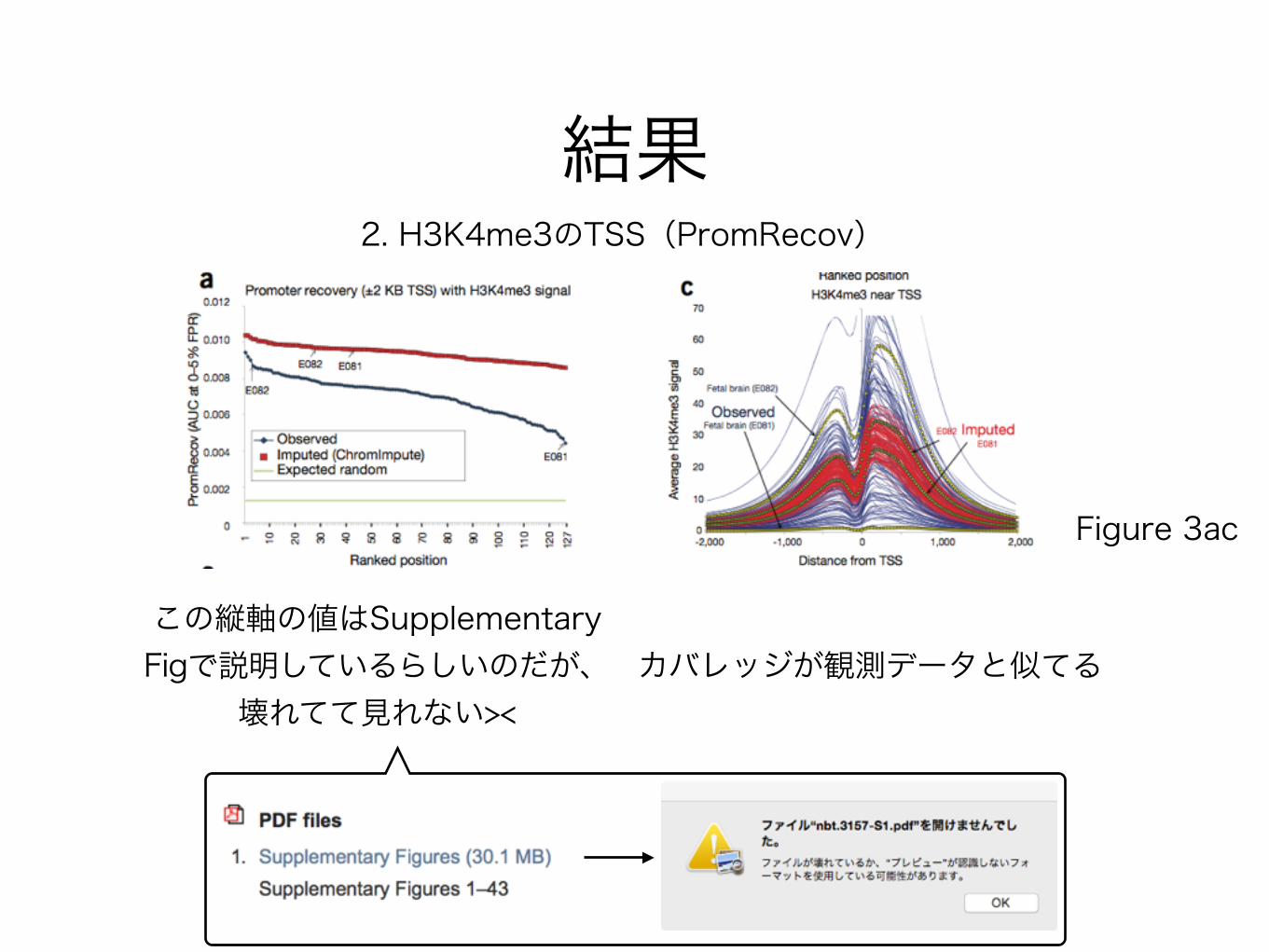
結果2. H3K4me3のTSS(PromRecov)
Figure 3ac
この縦軸の値はSupplementary Figで説明しているらしいのだが、
壊れてて見れない><カバレッジが観測データと似てる

結果3. H3K36me3のGeneBody(GeneRecov)
Figure 3bd
よくわからない>< カバレッジが観測データと似てる
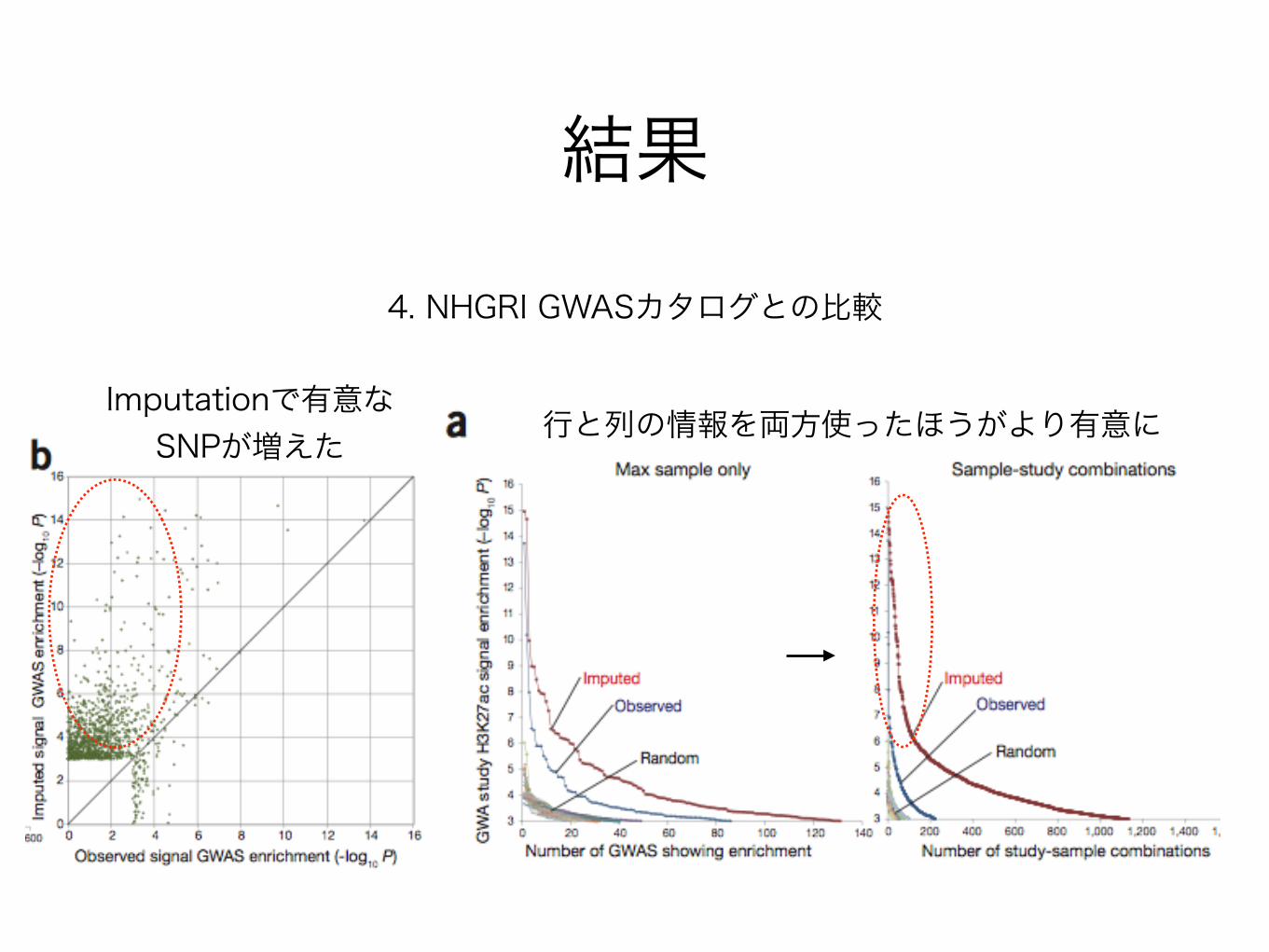
4. NHGRI GWASカタログとの比較
結果
Imputationで有意なSNPが増えた 行と列の情報を両方使ったほうがより有意に
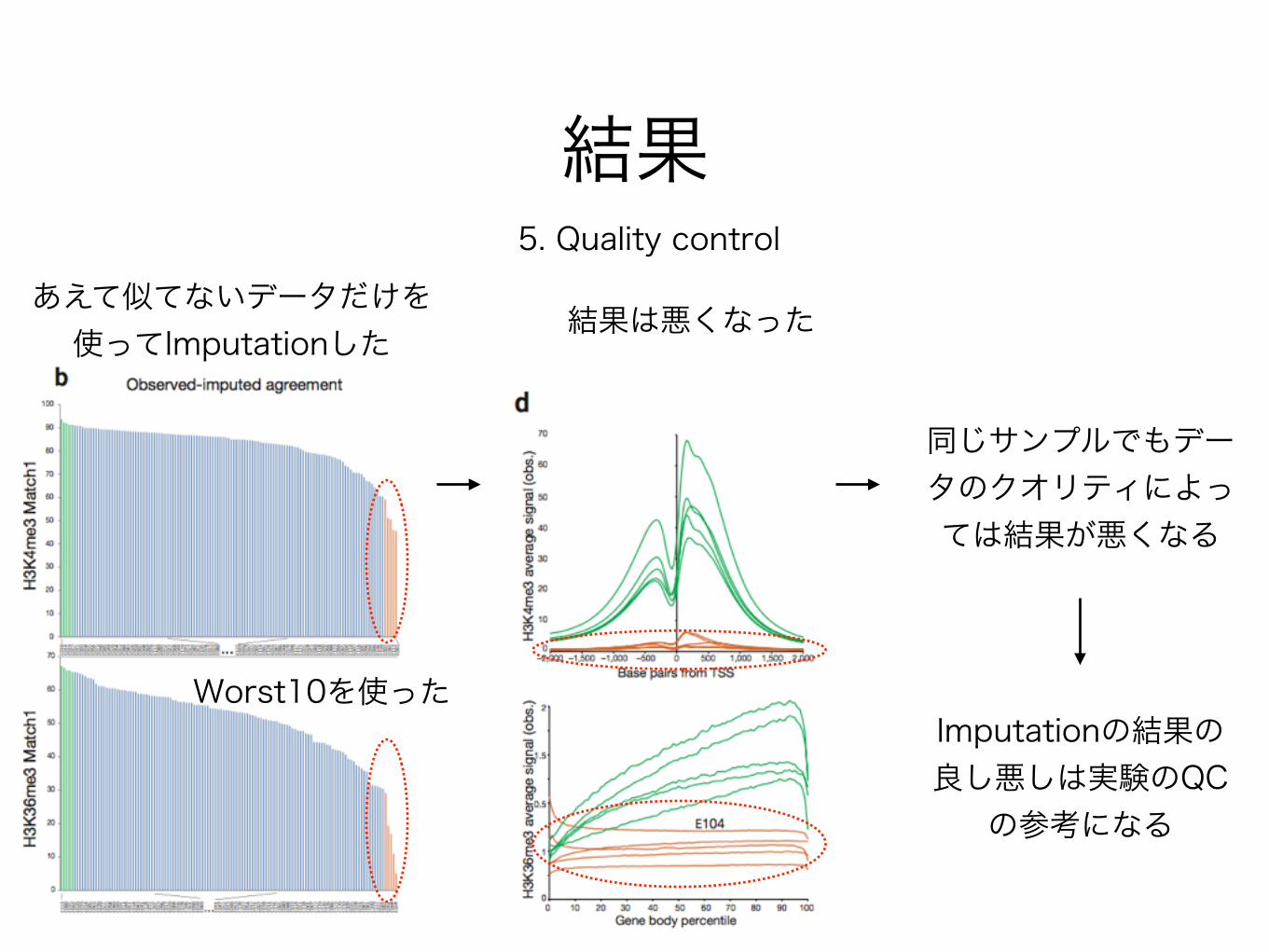
結果5. Quality control
あえて似てないデータだけを 使ってImputationした
Worst10を使ったImputationの結果の 良し悪しは実験のQC
の参考になる
結果は悪くなった
同じサンプルでもデータのクオリティによっては結果が悪くなる
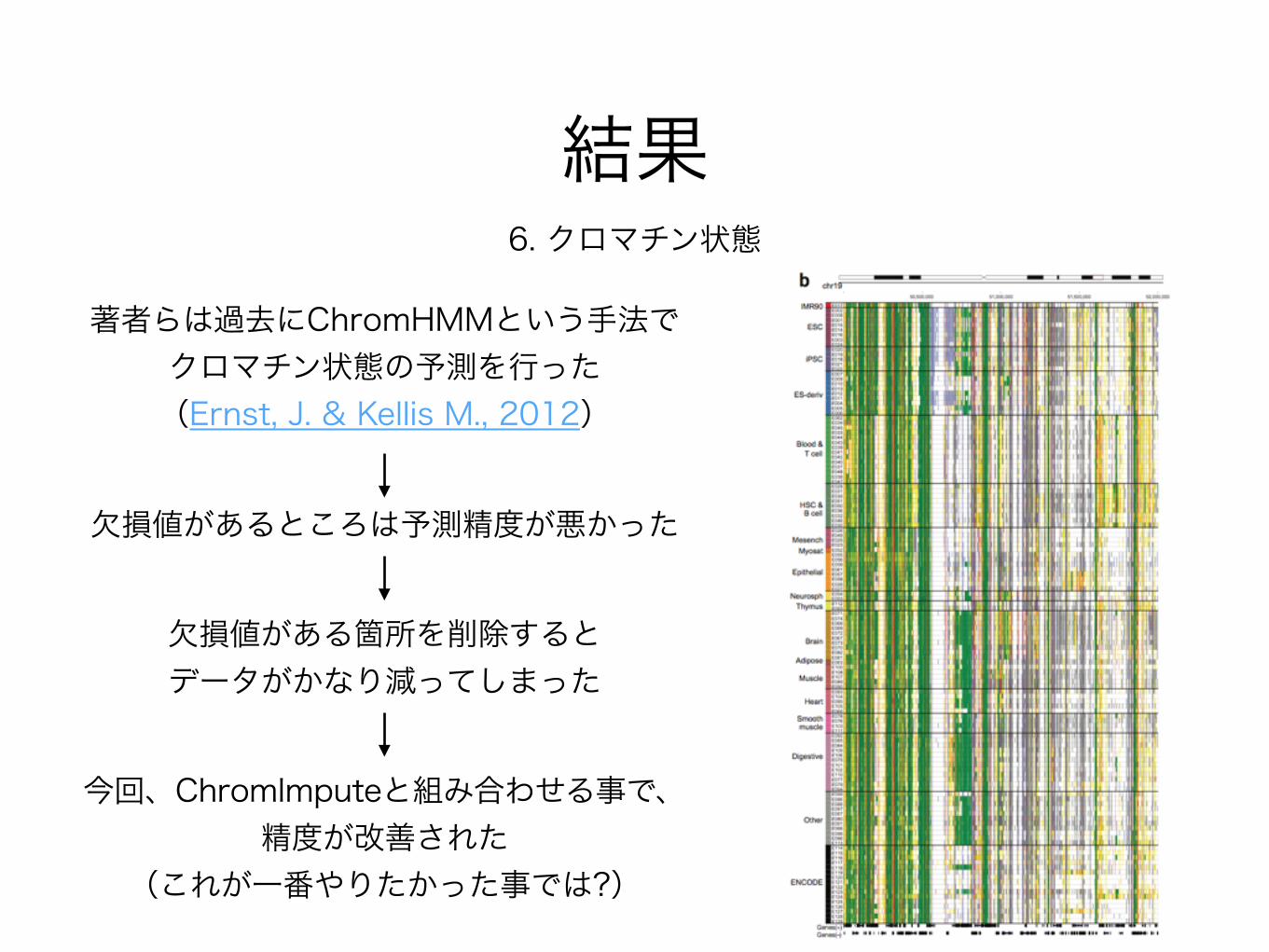
結果6. クロマチン状態
著者らは過去にChromHMMという手法でクロマチン状態の予測を行った(Ernst, J. & Kellis M., 2012)
欠損値があるところは予測精度が悪かった
欠損値がある箇所を削除すると データがかなり減ってしまった
今回、ChromImputeと組み合わせる事で、精度が改善された
(これが一番やりたかった事では?)

まとめ・Epigenomics RoadmapとENCODEデータのImputationを行った
・sampleレベル、markレベルで類似度が高くなるように欠損値を推定するChromImpute法を提案した
・他のデータに似せることができた
・評価が難しい話しだけど、色々なデータから察するに多分良い推定ができた




























