Embed Size (px)
Citation preview
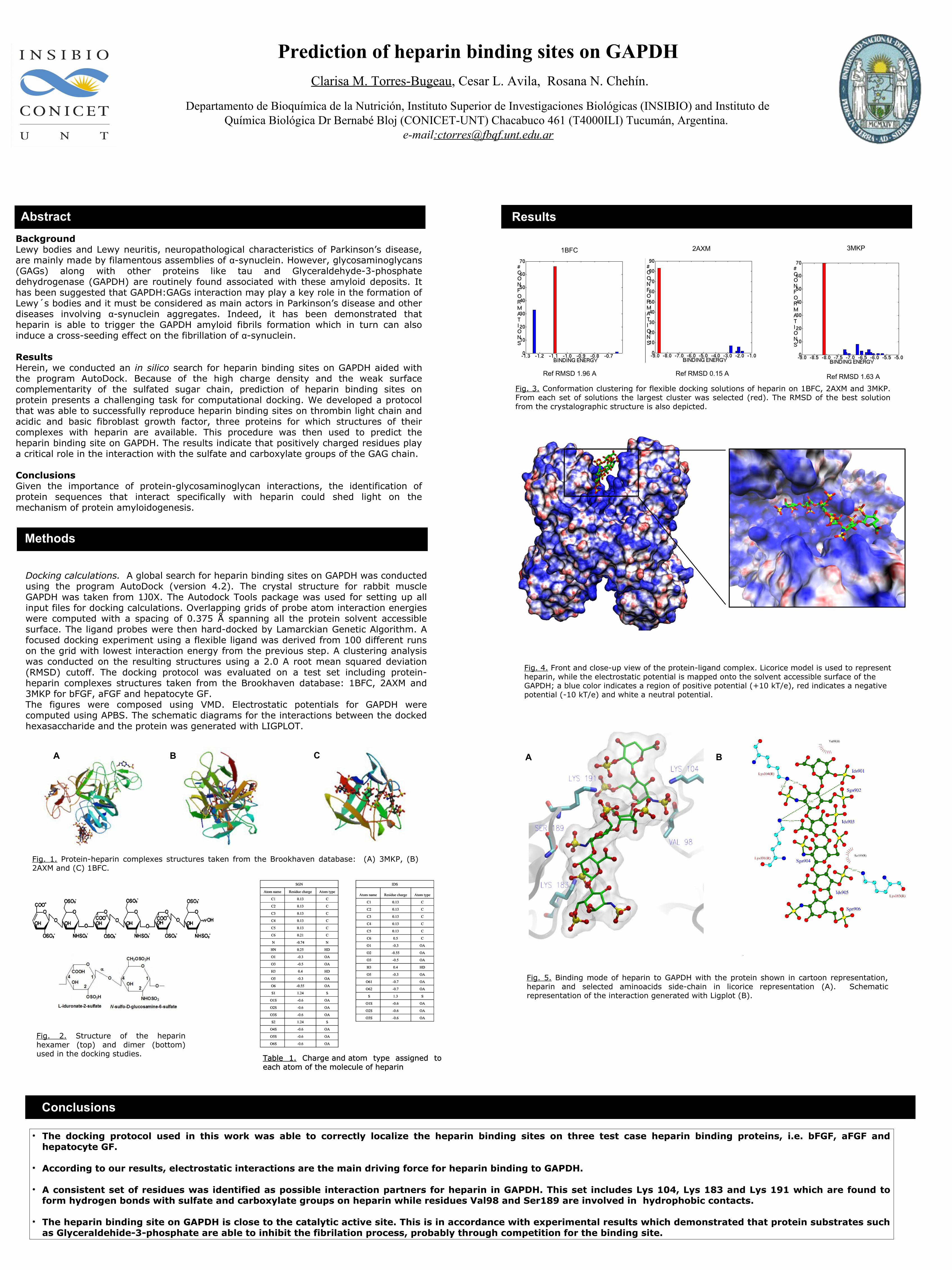
A B C
BackgroundLewy bodies and Lewy neuritis, neuropathological characteristics of Parkinson’s disease, are mainly made by filamentous assemblies of α-synuclein. However, glycosaminoglycans (GAGs) along with other proteins like tau and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are routinely found associated with these amyloid deposits. It has been suggested that GAPDH:GAGs interaction may play a key role in the formation of Lewy´s bodies and it must be considered as main actors in Parkinson’s disease and other diseases involving α-synuclein aggregates. Indeed, it has been demonstrated that heparin is able to trigger the GAPDH amyloid fibrils formation which in turn can also induce a cross-seeding effect on the fibrillation of α-synuclein.
ResultsHerein, we conducted an in silico search for heparin binding sites on GAPDH aided with the program AutoDock. Because of the high charge density and the weak surface complementarity of the sulfated sugar chain, prediction of heparin binding sites on protein presents a challenging task for computational docking. We developed a protocol that was able to successfully reproduce heparin binding sites on thrombin light chain and acidic and basic fibroblast growth factor, three proteins for which structures of their complexes with heparin are available. This procedure was then used to predict the heparin binding site on GAPDH. The results indicate that positively charged residues play a critical role in the interaction with the sulfate and carboxylate groups of the GAG chain.
ConclusionsGiven the importance of protein-glycosaminoglycan interactions, the identification of protein sequences that interact specifically with heparin could shed light on the mechanism of protein amyloidogenesis.
Abstract Results
Conclusions
• The docking protocol used in this work was able to correctly localize the heparin binding sites on three test case heparin binding proteins, i.e. bFGF, aFGF and hepatocyte GF.
• According to our results, electrostatic interactions are the main driving force for heparin binding to GAPDH.
• A consistent set of residues was identified as possible interaction partners for heparin in GAPDH. This set includes Lys 104, Lys 183 and Lys 191 which are found to form hydrogen bonds with sulfate and carboxylate groups on heparin while residues Val98 and Ser189 are involved in hydrophobic contacts.
• The heparin binding site on GAPDH is close to the catalytic active site. This is in accordance with experimental results which demonstrated that protein substrates such as Glyceraldehide-3-phosphate are able to inhibit the fibrilation process, probably through competition for the binding site.
Fig. 4. Front and close-up view of the protein-ligand complex. Licorice model is used to represent heparin, while the electrostatic potential is mapped onto the solvent accessible surface of the GAPDH; a blue color indicates a region of positive potential (+10 kT/e), red indicates a negative potential (-10 kT/e) and white a neutral potential.
Prediction of heparin binding sites on GAPDH Clarisa M. Torres-Bugeau, Cesar L. Avila, Rosana N. Chehín.
Departamento de Bioquímica de la Nutrición, Instituto Superior de Investigaciones Biológicas (INSIBIO) and Instituto de Química Biológica Dr Bernabé Bloj (CONICET-UNT) Chacabuco 461 (T4000ILI) Tucumán, Argentina.
e-mail:[email protected]
Methods
Fig. 5. Binding mode of heparin to GAPDH with the protein shown in cartoon representation, heparin and selected aminoacids side-chain in licorice representation (A). Schematic representation of the interaction generated with Ligplot (B).
Docking calculations. A global search for heparin binding sites on GAPDH was conducted using the program AutoDock (version 4.2). The crystal structure for rabbit muscle GAPDH was taken from 1J0X. The Autodock Tools package was used for setting up all input files for docking calculations. Overlapping grids of probe atom interaction energies were computed with a spacing of 0.375 Å spanning all the protein solvent accessible surface. The ligand probes were then hard-docked by Lamarckian Genetic Algorithm. A focused docking experiment using a flexible ligand was derived from 100 different runs on the grid with lowest interaction energy from the previous step. A clustering analysis was conducted on the resulting structures using a 2.0 A root mean squared deviation (RMSD) cutoff. The docking protocol was evaluated on a test set including protein-heparin complexes structures taken from the Brookhaven database: 1BFC, 2AXM and 3MKP for bFGF, aFGF and hepatocyte GF.The figures were composed using VMD. Electrostatic potentials for GAPDH were computed using APBS. The schematic diagrams for the interactions between the docked hexasaccharide and the protein was generated with LIGPLOT.
Ref RMSD 0.15 A
2AXM1BFC
Ref RMSD 1.96 A Ref RMSD 1.63 A
3MKP
Fig. 3. Conformation clustering for flexible docking solutions of heparin on 1BFC, 2AXM and 3MKP. From each set of solutions the largest cluster was selected (red). The RMSD of the best solution from the crystalographic structure is also depicted.
Fig. 1. Protein-heparin complexes structures taken from the Brookhaven database: (A) 3MKP, (B) 2AXM and (C) 1BFC.
Fig. 2. Structure of the heparin hexamer (top) and dimer (bottom) used in the docking studies.
BA
SGN
Atom name Residue charge Atom type
C1 0.13 C
C2 0.13 C
C3 0.13 C
C4 0.13 C
C5 0.13 C
C6 0.21 C
N -0.74 N
HN 0.25 HD
O1 -0.3 OA
O3 -0.5 OA
H3 0.4 HD
O5 -0.3 OA
O6 -0.55 OA
S1 1.24 S
O1S -0.6 OA
O2S -0.6 OA
O3S -0.6 OA
S2 1.24 S
O4S -0.6 OA
O5S -0.6 OA
O6S -0.6 OA
IDS
Atom name Residue charge Atom type
C1 0.13 C
C2 0.13 C
C3 0.13 C
C4 0.13 C
C5 0.13 C
C6 0.5 C
O1 -0.3 OA
O2 -0.55 OA
O3 -0.5 OA
H3 0.4 HD
O5 -0.3 OA
O61 -0.7 OA
O62 -0.7 OA
S 1.3 S
O1S -0.6 OA
O2S -0.6 OA
O3S -0.6 OA
Table 1. Charge and atom type assigned to each atom of the molecule of heparin
SGN
Atom name Residue charge Atom type
C1 0.13 C
C2 0.13 C
C3 0.13 C
C4 0.13 C
C5 0.13 C
C6 0.21 C
N -0.74 N
HN 0.25 HD
O1 -0.3 OA
O3 -0.5 OA
H3 0.4 HD
O5 -0.3 OA
O6 -0.55 OA
S1 1.24 S
O1S -0.6 OA
O2S -0.6 OA
O3S -0.6 OA
S2 1.24 S
O4S -0.6 OA
O5S -0.6 OA
O6S -0.6 OA
IDS
Atom name Residue charge Atom type
C1 0.13 C
C2 0.13 C
C3 0.13 C
C4 0.13 C
C5 0.13 C
C6 0.5 C
O1 -0.3 OA
O2 -0.55 OA
O3 -0.5 OA
H3 0.4 HD
O5 -0.3 OA
O61 -0.7 OA
O62 -0.7 OA
S 1.3 S
O1S -0.6 OA
O2S -0.6 OA
O3S -0.6 OA
Table 1. Charge and atom type assigned to each atom of the molecule of heparin





























