Embed Size (px)
Citation preview

九十一年六月分 CPC助猜三軍總醫院小兒科
林偉仁大夫 / 李春銘主治2002,JUN 18th

Present illness A female had been born to a 22-year-old women with birth history o
f G3P3A0, GA: 39+ wks, NSD, BBW: 3346 gm (75 percentile ), Apgar score: 79 at 15 min. BBL: 49.5CM (75 percentile) and HC: 33.4 CM (75 percentile ).
Because of congenital heart disease(CHD) suspected by fetal echocardiogram and uneventful pregnancy delivery with fetal distress, bradycardia and cyanosis, this newborn admitted to our NICU initially and the echocardiogram revealed large ASD(1.22 cm), muscular VSD(0.34 cm), R’t peripheral pulmonary artery stenosis (RPPS), pulmonary hypertension and r/o abnormal pulmonary venous return.

Present illness During the first year of life, she had 3 episodes of bronchi
olitis or bronchopneumonia and one gastroenteroritis. Due to failure to thrive BW: 5.8 kg(<3 percentile), BL: 67 cm(<3 percentile) when she was one-year-old), she was admitted for ASD repair on March 8, 2002.
Before the surgical procedure on March 11, 2002, her CV condition showed progressively severe pulmonary hypertension and cardiomegaly followed.

Hospital course However, a series of stressful, complicated episodes occur
red after operation. First of all, surgical wound infection with MRSA cultured was noted on March 23 after successful extubation. But the following respiratory distress then impending respiratory failure of her was present. Bronchoscope had found tracheal stenosis with bronchomalacia later. CXR showed bilateral exaggerative infiltration of lung, especially R’t upper lung and cardiomegaly was also revealed. The result of sputum culture was MRSA.

Hospital course Unfortunately, pulmonary hemorrhage and acute fulminen
t hepatic failure (AST/ALT revealed 8140/2840 U/L) occurred on April 7, and April 10 respectively. Meanwhile, DIC test had been shown positive finding. Repeated echocardiogram showed severe MR(4+), TR(4+); mild to moderate AR(2+), PR(2+); enlarged L’t atrium and R’t atrium RV-RA pressure gradient about 36 mmHg with normal LV ejection fraction. Due to unstable viral sign and occasional oliguria ( CCR: 57 ml/kg/1.73m2 pm March 24), dopamine and dobutamine were administered.

Hospital course
Moreover, Brain-echo showed generalized brain atrophy and benign subarachinoid fluid collection, coarse face and mild hypotonia were also present. Massive ascites and engorged hepatic vein, IVC and hepatosplenomegaly were scanned by abdominal sonogram.

Hospital course
Finally, her general condition became worse day by day, and sinus bradycardia then frequent VPCs presented, urinary retention, imbalance electrolyte with hypokalemia, hypocalcaemia, and hypoglycemia were also noted but unresponsive to any correction. Due to ineffective resustation, P’t was expired on May 5, 2002.
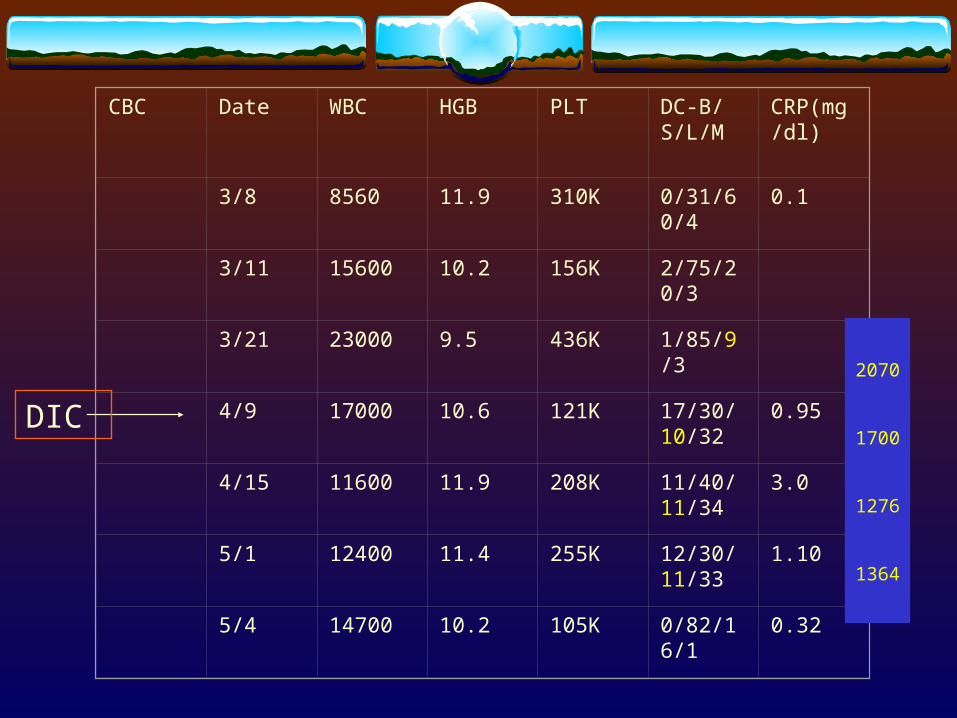
CBC Date WBC HGB PLT DC-B/S/L/M
CRP(mg/dl)
3/8 8560 11.9 310K 0/31/60/4 0.1
3/11 15600 10.2 156K 2/75/20/3
3/21 23000 9.5 436K 1/85/9/3
4/9 17000 10.6 121K 17/30/10/32
0.95
4/15 11600 11.9 208K 11/40/11/34
3.0
5/1 12400 11.4 255K 12/30/11/33
1.10
5/4 14700 10.2 105K 0/82/16/1 0.32
DIC
2070
1700
1276
1364
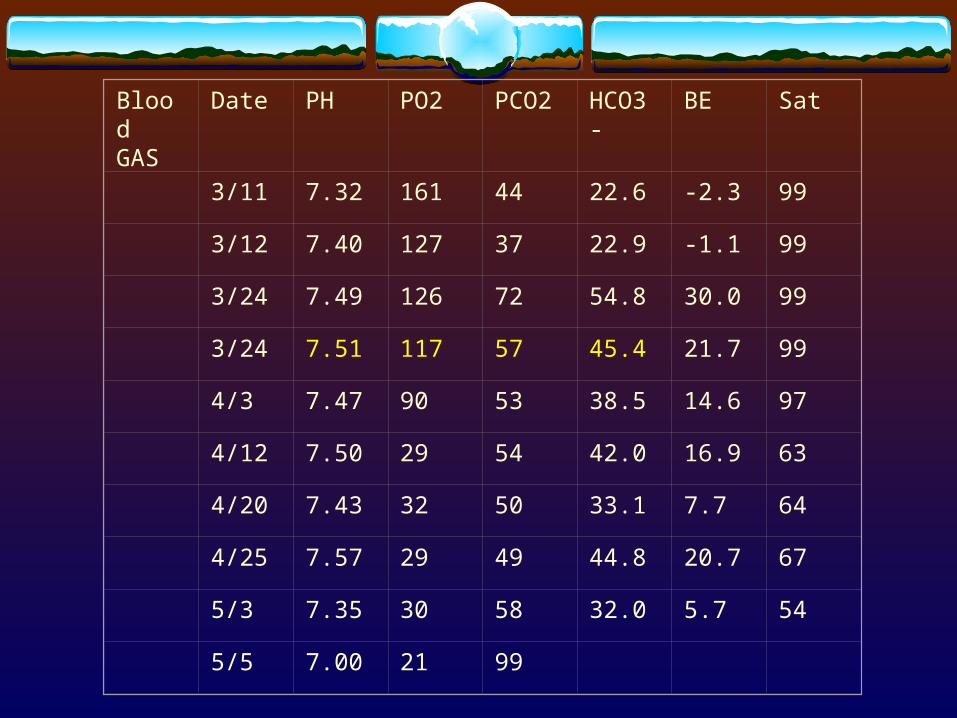
Blood GAS
Date PH PO2 PCO2 HCO3- BE Sat
3/11 7.32 161 44 22.6 -2.3 99
3/12 7.40 127 37 22.9 -1.1 99
3/24 7.49 126 72 54.8 30.0 99
3/24 7.51 117 57 45.4 21.7 99
4/3 7.47 90 53 38.5 14.6 97
4/12 7.50 29 54 42.0 16.9 63
4/20 7.43 32 50 33.1 7.7 64
4/25 7.57 29 49 44.8 20.7 67
5/3 7.35 30 58 32.0 5.7 54
5/5 7.00 21 99

Others Ascite culture, EBV, CMV, HBV, HCV, HAV stu
dy: Negative Urine culture (May 4): Yeast like pathogen and Fu
ngus culture: candida Antibiotics treatment: Cefazolin + Netromycin (March 11 – March 24) Vancomycin + Fortum ( March 24 – April 11) Vancomycin + Metropenem (April 11 – May 5)

Abnormal finding CV condition:
Congenital heart disease-- large ASD(1.22 cm), muscular VSD (0.34 cm), r’t peripheral pulmonary artery stenosis (RPPS), pulmonary hypertension and r/o abnormal pulmonary venous return.
Progressively severe pulmonary hypertension and cardiomegaly MR(4+), TR(4+); Mild to moderate AR(2+), PR(2+); Enlarged l’t atrium and r’t atrium RV-RA p
ressure gradient about 36 mmhg. Massive ascites and engorged hepatic vein, IVC and hepatosplenomegaly.
Infection: Frequent infection: 3 episodes of bronchiolitis or bronchopneumonia and one gastroen
teroritis during her first year. MRSA infection in surgical wound, sputum culture Urine culture: Yeast like pathogen and Fungus culture: candida DIC

Abnormal finding Immunodeficiency:
Absolute lymphocyte count< normal range (2180-8270 12mo-23mo) Frequent infection
Multiple organ failure: Oliguria--CCR: 57 ml/kg/1.73m Pulmonary hemorrhage and acute fulminent hepatic failure. Respiratory failure Flumient hepatic failure Generalized brain atrophy
Coarse face and mild hypotonia. Tracheal stenosis with bronchomalacia. Metabolic alkalosis Failure to thrive Hypokalemia, hypocalcaemia, and hypoglycemia unresponsive to any correction.
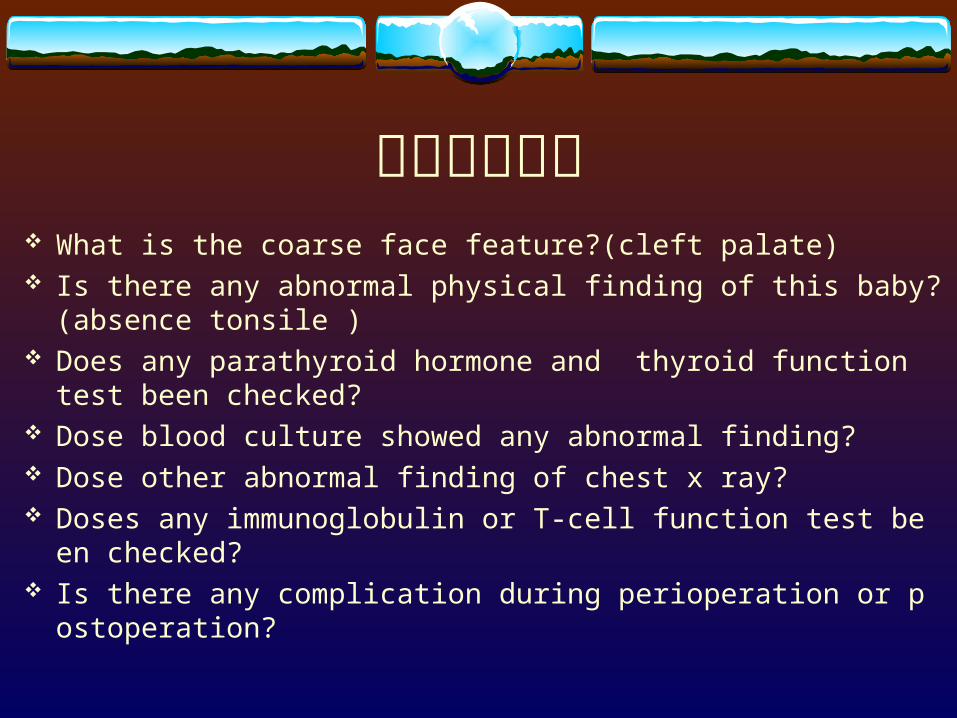
請教出題老師 What is the coarse face feature?(cleft palate) Is there any abnormal physical finding of this baby?(absence tonsile
) Does any parathyroid hormone and thyroid function test been chec
ked? Dose blood culture showed any abnormal finding? Dose other abnormal finding of chest x ray? Doses any immunoglobulin or T-cell function test been checked? Is there any complication during perioperation or postoperation?

Pulmonary hypertension Secondary:
Increase pulmonary blood flow from CHD VSD AVSD PDA Isolated ASD, after many decades.
Irreversible pulmonary hypertension(with cyanosis) Pulmonary venous obstruction
Obstructed anomalous pulmonary venous return Pulmonary vneoocclusive disease
Left sided heart failure Dilated cardiomyopathy Mitral valve stenosis or severe incometence
Lung disease, airway obstruction, hypoventilation– cor pulmonale RV hypertrophy, right side heat failure secondary lung or airway disease

Atrial Septal Defect Ostium secundum defect: The most common yet least seri
ous type of ASD
Ostium primum defect: failure of the endocardial cushions to close the ostium primum. Because endocardial cushions also form the mitral and tricuspid valves, ostium primum defects virtually always are associated with a cleft in the anterior mitral valve leaflet.
Sinus venosus defect: This ASD is found in the posterior aspect of the septum near the superior vena cava
Coronary sinus septal defect: The least common type of ASD. A portion of the roof of the coronary sinus is missing.

Mortality/Morbidity In developed countries, the rate of mortality from ASD is very
low (ie, <1%). Morbidity secondary to ASD is unusual and is limited typically to the following 3 groups of patients:
Perhaps 1% of infants diagnosed with moderate or large (ie, nonrestrictive) ASDs but no ductus arteriosus have tachypnea and fail to thrive. Pulmonary artery pressure, is at or near systemic level in these individuals.
A second group of patients, in whom ASDs go unrecognized until later childhood, may develop arrhythmias (eg, atrial fibrillation) or pulmonary hypertension.
A third group of patients with ASDs has an embolic stroke as the initial presentation.

Surgical Care Not all children with an ASD are candidates for surgery, which is indicated only for
those children with clinically significant left-to-right shunting. Generally, a pulmonary-to-systemic flow ratio of 1.5:1 or more is considered the pri
ncipal indication for surgical repair. Because cardiac catheterization is rarely necessary, echocardiographic evidence o
f right atrial and right ventricular enlargement usually is considered evidence of a significant left-to-right shunt and an indication for surgical closure of the ASD.
Ideally, surgery is performed in children aged 2-4 years; however, surgery may be performed earlier if a child has evidence of CHF.
Operative mortality rate is low in patients with uncomplicated ASDs. Postoperative morbidity in individuals with ASDs almost exclusively is due to accu
mulation of pericardial fluid, which occurs in approximately one third of cases. Occasionally, tamponade occurs and requires pericardiocentesis.

Trabecular (muscular) VSD This form is the second most common type of VSD, occurring
in 5-20% of most series. Muscular VSD is also known as Swiss cheese VSD. Frequently, spontaneous closure of muscular VSD occurs in
the first 2 years of life, the majority by 6 months.

AVSDs Atrioventricular septal defects, characterized by a deficiency of the atrioventricular
septum, are a broad spectrum of malformations presumed to result from abnormal or inadequate fusion of the superior and inferior endocardial cushions with the mid-portion of the atrial septum and the muscular (trabecular) portion of the ventricular septum.
Pulmonary hypertension and an early tendency to increase pulmonary vascular resistance are common. AV valvular insufficiency increases the volume load on one or both vesicles. With time, progressive pulmonary vascular disease increases the right-left shunt so that clinical cyanosis develops.
With complete AVSD, CHF and intercurrent pulmonary infection usually appear in infancy. The liver is enlarged and the infant shows sings of failure to thrive.

Mortality/Morbidity: Prognosis depends on the magnitude of the left to right shunt, the degree
of elevation of pulmonary vascular resistance, and the severity of AV valve insufficiency.
Surgical for complete AVSD is more difficult, especially in infants with cardiac failure and pulmonary hypertension. The risk of developing pulmonary hypertension as early as 6-12 mo of age, surgical intervention must be performed during infancy.
Morbidity in infancy and early childhood is generally caused by mitral regurgitation (MR). Severe MR causes CHF, failure to thrive in infants and may result in death if left untreated.

Immunodeficiency Most immunodeficiency diseases lead to repeated or chronic infections.
Two or more systemic bacterial infection(sepsis, osteomyelitis or meningitis) Three or more serious respiratory or documented bacterial infecions(cellulitis, drainin
g OM, lymphadenitis within 1 yr) Infctions occureing at unusual sited Unusual pathogens
Patients with defects in immunoglobulins (B cells), complement proteins or phagocytes are very susceptible to recurrent pyogenic infections;
Screening tests for T-cell defectsAbsolute lymphocyte count(normal result indicates T-c
ell defect unlikely)

Immunodeficiency
T cell deficiencies are usually marked by repeated opportunistic infections caused by viruses, fungi, and protozoans. Since B cell function is largely T cell-dependent, T cell deficiency also results in humoral deficiency. Thus, T cell deficiency leads to a combined deficiency of both humoral and cell-mediated immunity.
Infections can be treated by administration of the appropriate anti-microbial agents. Some T cell deficiencies can be corrected by bone marrow transplantation or fetal thymus graft.

Infective endocarditis Native valve endocarditis
Congenital heart disease (15% of NVE) - Underlying etiologies include a patent ductus arteriosus, ventricular septal defect, tetralogy of Fallot, or any native or surgical high-flow lesion.
Approximately 70% of cases are caused by Streptococcus species including Streptococcus viridans, Streptococcus bovis, and enterococci. Staphylococcus species cause 25% of cases and generally demonstrate a more aggressive acute course.
Mortality/Morbidity: Increased mortality rates are associated with increased age, infection involving the
aortic valve, development of congestive heart failure, central nervous system (CNS) complications, and underlying disease. Mortality rates also vary with the infecting organism.
Mortality rates in native valve disease range from 16-27%. Mortality rates in patients with prosthetic valve infections are higher. More than 50% of these infections occur within 2 months after surgery.

Fungal endocarditis
Fungal endocarditis is found in intravenous drug users and intensive care unit patients who receive broad-spectrum antibiotics.
Blood cultures are often negative, and diagnosis frequently is made after microscopic examination of large emboli.
Mortality/Morbidity: The mortality rate remains 75-90% because of difficulty making the diagnosis, lack of effective antifungal antibiotics, need for surgical intervention in most cases, presence of underlying or predisposing conditions, and frequent comorbid conditions in these typically critically ill neonates and children.
Although a small number of patients have survived with medical therapy alone, the majority of survivors have required both medical and surgical treatment. Operative intervention is almost always required.

Fungal endocarditis
fewer than half of candidal endocarditis cases yield positive blood cultures and other causative organisms are even less frequently identified in blood
Culture of urine, sputum, cerebrospinal fluid, synovial fluid, lymph node, and/or bone marrow may offer the only evidence of systemic fungal infection.
Chest radiography A chest radiograph may reveal cardiomegaly.
Echocardiography Transthoracic echocardiography is less sensitive than transesophageal echocardiograph
y but is also less invasive. Vegetations and intracardiac thrombi are the most common types seen but are rare non
etheless. Echocardiography may demonstrate pericardial effusion. Normal valves are rarely involved. Echocardiography may suggest myocardial abscesses. Echocardiography may demonstrate associated myocarditis or pericarditis.

Metabolic alkalosis Causes:
Chloride-responsive type Gastric fluid loss (vomiting, NG drainage) Volume contraction -- stimulates release of renin-angiotensin Chronic furosemide therapy Congenital chloride diarrhea Posthypercapnia syndrome -- Chronic CO2 retention causes a compensatory increase in HCO3 l
evels. Hypoparathyroidism
Chloride-resistant type Primary aldosteronism Bartter syndrome –urine Cl loss, plasma renin and aldosterone increase. Liddle syndrome Excessive ingestion of licorice Chronic potassium depletion Primary reninism Hyperglucocorticoidism Milk-alkali syndrome

Severe metabolic alkalosis Is associated with increased morbidity and mortality, probably due to its pr
ofound influences on multiple organ systems and, more importantly, to tissue anoxia caused by hypoventilation and shift of the oxygen-dissociation curve to the left.
Cardiovascular system: Life-threatening arrhythmias are the most significant adverse effect of metabolic alkalosis. Because of heightened electrical excitability, ventricular arrhythmias can occur, which often are unresponsive to antiarrhythmic agents.
Neuromuscular system: Patients with severe metabolic alkalosis can develop obtundation and marked muscle weakness that resolve only with correction of the pH.
Ionized calcium concentration: Metabolic alkalosis may cause a decrease in ionized calcium because of increased binding of calcium to plasma proteins; consequences include tetany and seizures.

Fulminant Hepatic Failure
Infectious agents: In approximately 50% of patients, FHF is caused by acute viral hepatitis, commonly caused by hepatitis viruses A; B; non-A, non-B; D; or E. Many viruses other than hepatitis also are recognized causes of FHF in childhood, including Epstein- Barr virus; cytomegalovirus (CMV); paramyxovirus; varicella-zoster virus; herpesvirus types 1, 2, and 6; parvovirus; and adenovirus.
Circulatory causes: Circulatory causes are uncommon in FHF. They include congestive heart failure, cardiomyopathy, sepsis, shock, cyanotic heart disease, obstructive lesions of the aorta, vascular occlusions, myocarditis, and severe asphyxia.

Fulminant Hepatic Failure
In the US:In the pediatric age group, exact incidence of FHF remains unknown, but it is estimated to be approximately 50 cases per year.
Mortality/Morbidity: The mortality rate may reach 80-90% in the absence of liver
transplantation. In some pediatric series, survival rates of 50-75% have been reported.
Complications: Infections: Bacterial and fungal infections commonly occur, leading to the
development of peritonitis, pneumonia, urinary tract infections, or septicemia.

Ascites, splenomegaly, hepatomegaly The ascites frequently is a component or a complication of hepatic sirrhosis, cong
estive heart failure, nephrosis, or carcinoma. (transudate) Obstructed venous blood flow from intrahepatic or extrahepatic etiologies
can cause splenomegaly. The most common causes include portal vein thrombosis, hepatic cirrhosis, and congestive heart failure.
Excessive antigenic stimulation from infection causes most splenomegaly in children. Viral infections are the most frequent culprits, and the associated splenomegaly usually is transient and only mild to moderate in severity.
Other common infectious etiologies include bacterial, protozoal, and fungal infections. Concomitant generalized lymphadenopathy is common in many of these infectious etiologies.
Hepatomegaly with underlying systemic disorder present--- cardiovascular disease( right heart failure)

Hypokalemia may be due to a total body deficit of potassiu
m, which may occur due to chronic inadequate intake, long-term diuretic or laxative use, and chronic diarrhea, hypomagnesemia, or hyperhidrosis.
Acute causes of potassium depletion include diabetic ketoacidosis, severe gastrointestinal losses from vomiting and diarrhea, dialysis, and diuretic therapy.

Hypokalemia
Mortality/Morbidity: Mortality is rare, except when hypokalemia is
severe or occurs in a patient with underlying heart disease requiring digoxin therapy, following cardiac surgery, or when accompanied by arrhythmia.

Hypocalcemia The concentration of calcium in the serum is critical t
o many important biological functions, including the following:
Calcium messenger system by which extracellular messengers regulate cell function
Activation of several cellular enzyme cascades Smooth muscle and myocardial contraction Nerve impulse conduction Secretory activity of exocrine glands

HypoparathyroidismAplasia or hypoplasia - DiGeorge syndrome, velocardiofacial syndrome, maternal diab
etes mellitus or treatment with retinoic acid, VATER complex (vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, radial and renal abnormalities), CHARGE association (coloboma, heart defects, choanal atresia, renal abnormalities, growth retardation, male genital anomalies, ear abnormalities)
Symptoms:weakness , muscle cramps, abnormal sensations such as tingling, burning and nu
mbness (paresthesias) of the hands excessive nervousness, loss of memory, headaches and uncontrollable cramping muscle movements of the wrists and feet.
Other symptoms may be spasms of the facial muscles (Chvostek Sign), the contraction of muscles produced by mild compression of nerves (Trousseaus Sign), malformations of the teeth, including enamel and roots of the teeth; and malformed finger nails.

Complications of hypoparathyroidism
Are largely due to hypocalcemia. Neurologic: Hypocalcemia leads to neuromuscular irritability. Affected pati
ents may experience paresthesias, muscle cramping, tetany, or seizures. However, patients can be asymptomatic. Neck muscle cramping can cause dystoniclike neck movements.
Cardiac: Hypocalcemia causes prolongation of the QTc interval. Affected individuals may be asymptomatic or suffer syncope, seizure, or death due to arrhythmias, such as polymorphic ventricular tachycardia.
Respiratory: Laryngospasm, a form of tetany, can lead to stridor and significant airway obstruction.

Bronchoscopy Tracheo/broncho malacia is the most common cause of expiratory stridor.
It is caused by a defect on the cartilage resulting in the loss of rigidity necessary to maintain the tracheal lumen patent or by an extrinsic compression of the trachea.
Tracheal stenosis can be congenital or secondary to extrinsic compression. Congenital stenosis is usually in the for on complete tracheal rings, characterized by a persistent stridor, and requires surgery based on severity of symptoms. The most common extrinsic causes of stenosis include vascular rings, slings, and a double aortic arch that encircles the trachea and esophagus. External compression could result in tracheomalacia as well. Patients usually present during the first year of life with noisy breathing, intercostal retractions, and a prolonged expiratory phase.

DiGeorge Syndrome Hx. DiGeorge syndrome (DGS), which was described by Angelo DiGeorge, MD, in 1965, ---- con
genital cardiac anomalies, craniofacial dysmorphology, and learning dysfunction, all of which were traced to a defect in the third and fourth pharyngeal pouches during embryogenesis.
10 years later, in Japan, Kinouchi et al described the conotruncal anomalies face (CTAF) syndrome, composed of congenital conotruncal cardiac anomalies, characteristic facies, learning dysfunction, and developmental delay
Shprintzen et al described their experiences in a craniofacial clinic with a syndrome of congenital conotruncal cardiac anomalies, characteristic facies, velopharyngeal dysfunction with or without cleft palate, and learning dysfunction, which they termed the velocardiofacial syndrome (VCFS).
Genetic support for this came in 1981 A common area of deletion, known as the DiGeorge syndrome critical region (DGCR), was fo
und on band 22q11.2. As a result, these 3 syndromes have been combined into one genetic entity with variable phenotypic presentations.
CATCH 22 be used for this group, since it describes the findings of cardiac anomalies, abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia on chromosome 22

DiGeorge Syndrome A variable degree of hypoplasia of the thymus and parathyroid glan
ds is more frequent than total aplasia. (partial DiGeorge syndrome– they may have little trouble with infections and grow normally. )
Complete DiGeorge syndrome resemble patients with severe combined immunodeficiency (SCID) in their susceptibility to infections with low grade or opportunistic pathogens, including fungi, viruses an
d Pneumocystis carinii, and to graft versus host disease(GVHD)

Severe combined immunodeficiency disease
(SCID) Failure of stem cells to differentiate into T and/or B cells. Infants with SCID have very few lymphocytes in their blood (<3000/ml), and the
re are no or few lymphocytes in their lymphoid tissue (including the thymus, which has a fetal appearance).
SCID patients are susceptible to all microbial infections, most notably rotavirus, cytomegalovirus, Candida albicans and Pneumocystis carinii.
Symptoms occur in early infancy and prove fatal within the first year of life if left untreated. They have prolonged diarrhea, develop pneumonia, and may die of progressive infection if immunized with live organisms.
Bone marrow transplants are the treatment of choice.

Chromosome 22q11 deletion syndrome
The estimates are now closer to 1:4,000. The characteristic facies of this syndrome are often subtle in i
nfancy and not fully manifested until the child is older; therefore, they are not common causes of genetic investigation. The same is true of developmental delays, which often are not noticed until the child is of school age. A recent review of a large series reported involvement as follows: cardiac system (49%), developmental delay (16%), behavioral disturbance (7%), otolaryngologic system (6%), psychiatric symptoms (3%), and mental retardation (2%).
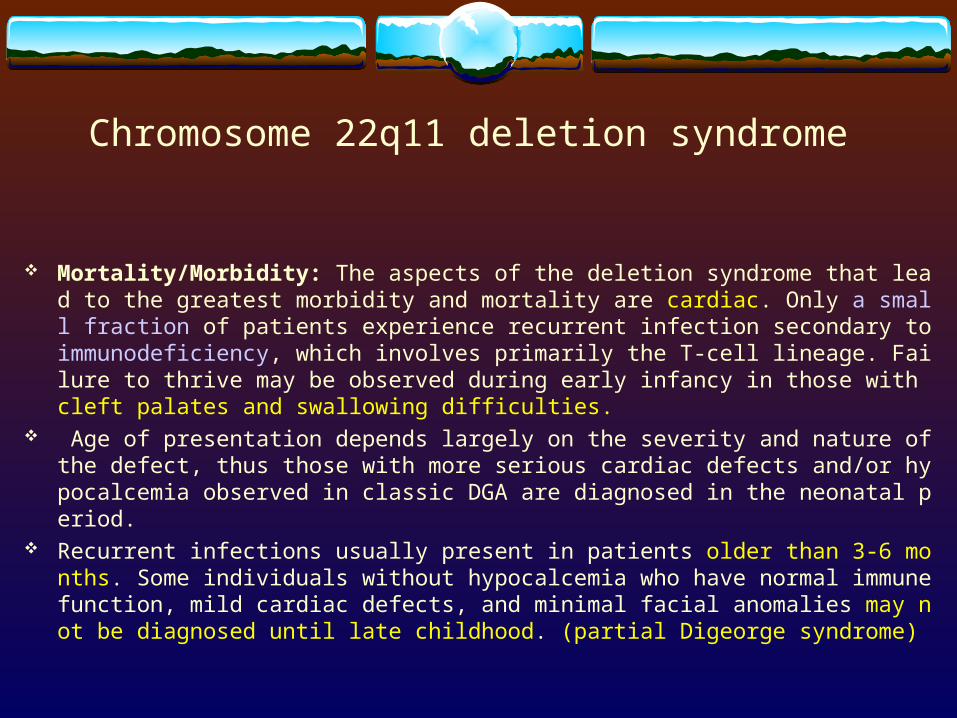
Chromosome 22q11 deletion syndrome
Mortality/Morbidity: The aspects of the deletion syndrome that lead to the greatest morbidity and mortality are cardiac. Only a small fraction of patients experience recurrent infection secondary to immunodeficiency, which involves primarily the T-cell lineage. Failure to thrive may be observed during early infancy in those with cleft palates and swallowing difficulties.
Age of presentation depends largely on the severity and nature of the defect, thus those with more serious cardiac defects and/or hypocalcemia observed in classic DGA are diagnosed in the neonatal period.
Recurrent infections usually present in patients older than 3-6 months. Some individuals without hypocalcemia who have normal immune function, mild cardiac defects, and minimal facial anomalies may not be diagnosed until late childhood. (partial Digeorge syndrome)

Chromosome 22q11 deletion syndrome
interrupted aortic arch, followed by a right aortic arch. bicuspid aortic valve, membranous ventricular septal defects, aberrant right subcla
vian artery, and atrial septal defects. Patients usually have characteristic facies, which become more pronounced as the
children grow into the second decade. Many affected children present when aged 1-2 months with poor suck as a result of the velopharyngeal insufficiency alone and/or in combination with the generalized hypotonia that sometimes is observed in these infants.
IMAGE: Thymus: Chest radiographs can demonstrate a decreased thymic silhouette bu
t are unreliable. MRI is slightly better; however, thymic size is a poor predictor of immune function.
Chromosome 22q11 deletion syndrome
Alcohol consumption during the early stages of fetal development may be one of many environmental explanations for the microdeletion.
Prenatal testing for DiGeorge syndrome is widely available and is recommended for fetuses that have been detected as having cleft palate or heart malformation through ultrasound, and have at least one parent testing positive for the 22q11 microdeletion.
Infants with the 22q11 microdeletion should be monitored for hypocalcemia, renal function and low lymphocyte count. Babies with low lymphocyte counts should be seen by an immunologist and should not be given live vaccine immunizations.

Diagnosis of mortality:
Sepsis with DIC (MRSA), R/O Fungal endocarditis Congestive heart failure with pulmonary hypertension. Congenital heart disease with AVSD S/P repair with Postoperative mediastinitis Arrythemia R/o hypoparathyrodism R/O Chromosome 22q11 deletion syndrome Fulminant Hepatic Failure Acute renal failure Acute respiratory failure Metabolic alkalosis Hypocalcemia





























