Embed Size (px)
Citation preview

9/27/2014
1
Sertifikat analizeSertifikat analize
doc. dr Biljana Stojanović
2

9/27/2014
2
“Dokument koji daje informaciju o kvalitetu ili čistoći nekog proizvoda”
“Sertifikat analize leka ili odgovarajućeg farmaceutskog proizvoda daje
tačne detalje o kvalitetu i o usaglašenosti proizvoda sa specifikacijom.
Dokument se odnosi na rezultate ispitivanja reprezentativnog uzorka
uzetog iz jedne serije proizvoda”
Sertifikat analize je dokumentovani dokaz o kontroli kvaliteta
urađenoj na nekom proizvodu.
Sertifikatanalize Papir koji nešto
sertifikuje...
4

9/27/2014
3
5
?Kvalitet lekova
�Proces proizvodnje
�Način pakovanja
�Način transporta
�Uslovi čuvanja
�Nedostatak terapijskog dejstva (produžetak bolesti, letalan ishod)�Toksične i neželjene reakcije�Finansijski gubici�Gubitak kredibiliteta
1. Testovi – ispitivanja koja je potrebno izvesti da bi se proverio kvalitet nekog preparata
2. Zahtevi za svaki od navedenih testova - granice u kojima rezultat treba da se nalazi
3. Rezultat ispitivanja – dobijeni rezultati (brojčane vrednosti) sa odgovarajućim zaključkom (odgovara/neodgovara)
6

9/27/2014
4
TESTOVI ZAHTEVI REZULTAT
Izgled No. 0 tvrde želatinske kapsule tamno zelene kapice i tela, sadrže žuto-bež sferne mikrogranule
odgovara
Prosečna masa punjenja kapsula 464 mg ± 7,5 % odgovara457,6 mg
Variranje mase Ph. Eur. 6 (2.9.5) odgovaraod - 2,1 % do + 1,9 %
Identifikacijaitrakonazola
HPLC odgovara
Semikvantitativno određivanje vode ≤ 2 % odgovara1,1 %
Raspadljivost moraju se raspasti za manje od 30 min. odgovara17 min.
Brzina rastvaranja Mora se osloboditi najmanje 75 % aktivne supstance za 60 minuta
odgovara98,6 %
Sadržajitrakonazola
od 95,0 mg/tbl. do 105,0 mg/tbl. (95 % – 105 %)
odgovara98,7 mg/tbl. (98,7 %)
Srodne supstancenečistoća Bnečistoća Fbilo koja druga nečistoćaukupne nečistoće
≤ 0,5 %≤ 0,5 %≤ 0,1 %≤ 1,5 %
odgovara0,07 %0,04 %0,03 %0,04 %
Mikrobiološka čistoća Ph. Eur 6 odgovara
Primer
7
Određeni su specifikacijom
8

9/27/2014
5
Sertifikat analize je odraz specifikacije...
Specifikaciju čini lista testova, zahteva i
metoda po kojima se vrši neko ispitivanje.
Sertifikat analize je rezultat primenjene
specifikacije na datu seriju.
9
10
Lista testova
Spisak kriterijuma
Spisak postupaka

9/27/2014
6
11
Šta se sve uzima u obzir pri određivanju ispitivanja koja će se naći u specifikaciji?
Farmakopeja
Regulatorni zahteviRazvoj proizvoda

9/27/2014
7
���� Pretkliničke studije
���� Testiranje prvih preparata razvijenih za kliničke studije
���� Faza II i faza III kliničkih studija
���� Priprema i podnošenje zahteva za registraciju i puštanje u promet
Faze
nas
taja
nja
leka
Faze značajne za nastajanje specifikacije13
���� Pretkliničke studije
U ovoj fazi vrše se ispitivanja za:
A) Aktivnu supstancu
B) Pretklinički dozirani oblik
C) Metabolite i nečistoće
14

9/27/2014
8
15
Aktivna supstanca
� da bi izveli pretklinički i toksikološki testovi potrebno je poznavanje kvaliteta aktivne supstance
� da bi se izvele korektne studije nije neophodno da lek imavisok stepen čistoće
�u ovoj fazi nije potrebno imati formalnu specifikaciju
16
Uobičajeni testovi u ovoj fazi su:
Parametar Uobičajene metode ispitivanja
Identifikacija Infra crvena spektroskopijaTemperatura topljenja
Karakterizacija strukture Masena spektroskopijaNuklearna magnetna rezonanca
Određivanje sadržaja aktivnesupstance
HPLCTitracija
Srodne supstance Hromatografije metode(HPLC, GC, TLC)
Neorganske nečistoće Ostatak nakon žarenjaTeški metali
Rezidualni rastvarači Gasna hromatografija
Sadržaj vode Karl-Fišer titracija
pH rastvora merenje pH vrednosti rastvora

9/27/2014
9
17
Pretklinički dozirani oblik
� u pretkliničkim studijama lek se obično dozira u obliku
rastvora, suspenzija i hrane za životinje
� u ovoj fazi potrebno je znati:
a) koliki je sadržaj aktivne supstance u obliku koji se primenjuje
b) kakva je stabilnost aktivne supstance u primenjenomvehikulumu, kao i da li je supstanca stabilna tokom pretkliničkiheksperimenata
18
Uobičajeni testovi koji se izvode za rastvore i suspenzije u
ovoj fazi su:
1) izgled
2) sadržaj aktivne supstance
3) nivo nečistoća
4) pH vrednost rastvora
Formalna specifikacija još uvek ne postoji aliprimenjeni oblik mora pratiti odgovarajući sertifikat
analize.

9/27/2014
10
19
Nečistoće i metaboliti
� Kada god je to moguće potrebno je znati strukture i sadržaj
nečistoća u leku!
� Iako formalna specifikacija još nije razvijena potrebni su podaci
o količini i prirodi nečistoća, uključujući nečistoće iz procesa
sinteze kao i one koje nastaju kao posledica razgradnje leka.
� Metaboliti su potrebni za izvođenje i pretkliničkih i
kliničkih studija. Treba da budu određene, poznate čistoće.
20
���� Testiranje prvih preparata razvijenih za kliničkestudije
�Za male sintetske molekule definiše se tipična specifikacija bez
specifičnih testova
�Za složenije molekule kao što su peptidi, proteini ili smeše kao
proizvodi procesa fermentacije, specifikacije su znatno složenije
U ovoj fazi specifikacija ima probni karakter i podložna je daljim promenama
Kao kliničke formulacije koriste se uglavnom kapsule punjenelekom ili oblici koji se mogu injektovati. Specifikacije oblika
zasnovane su na specifikaciji aktivne supstance.

9/27/2014
11
Test Metoda Kriterujum za prihvatanjerezultata
Identifikacija IR Tt
odgovara standarduodgovara određenom intervalu
Razjašnjavanje strukture NMRMS
odgovara standardu
Određivanje sadržaja HPLC/GCTitracija
97,0 % – 102,0 %98,0 % – 102,0 %
Srodne supstance HPLC/GC ukupne: manje od 2,0 %pojedinačne: manje od 1,5 %
poznata nečistoća X: 1,0 %
Neorganske nečistoće ostatak nakon žarenjasulfatni ostatak
manje od 500 ppm
Rezidualni rastvarači gasna hromatografija rastvarač X: manje od 0,50 %rastvarač Y. manje od 0,20 %rastvarač Z: manje od 0,05 %
Sadržaj vode Karl-Fisher-ova titracija
manje od 0,5 %
pH 1 % – tnog rastvora pH merenje 4,6 – 5,0
Uobičajena specifikacija za male sintetske molekule
21
���� Faza II i faza III kliničkih studija
� Kada se pokaže da lek ima određenu podnošljivost i prihvatljiv
farmakokinetički profil u I fazi studija, u II fazi kliničkih studija vrši
se testiranje leka na malom broju pacijenata
� Sa ciljem testiranja proizvode se manje serije leka
Profil nečistoća treba da bude poznat što podrazumeva
poznavanje njihove strukture i tačne koncentracije
�Nakon faze II, lek prelazi u fazu III kliničkih studija
22

9/27/2014
12
23
���� Priprema i podnošenje zahteva za registraciju ipuštanje leka u promet
� Kada se dođe do ove, poslednje faze, znači da se lek proizvodi poveć definisanom postupku
�Specifikacija je dostupna i uključuje fizičke osobine, nečistoće, stabilnost, itd
�Finalna specifikacija mora biti u skladu sa važećom regulativom
�Kako se ispitivanja vrše i dok je lek na tržištu, mogu se vršiti iodređene promene u specifikaciji
SMERNICA ICH Q6A
Kontrola novih farmaceutski
aktivnih supstanci
Kontrola novih farmaceutskih
oblika
Zajednička
ispitivanja
Specifična
ispitivanja
Zajednička
ispitivanja
Specifična
ispitivanja
24

9/27/2014
13
Kontrola novih farmaceutski aktivnih supstanciKontrola novih farmaceutski aktivnih supstanciBROJ ISPITIVANJE ZAHTEVI1 Izgled Beo do skoro beo prašak2 Rastvorljivost Umereno rastvoran u metanolu3 Identifikacija
A. IRB. HPLCC. hemijska
Odgovara spektru referentnog standarda.Retenciono vreme D i L izomera u uzorku mora odgovaratiradnom standardu tokom ispitivanja hiralne čistoće HPLC metodom.Odgovara testu za hloride.
4 Temperatura topljenja Između 223,0 °C i 230,0 °C5 Ostatak nakon sušenja Ne više 0,5 % w/w6 Specifična optička rotacija (računato na suvu bazu) Između -1,0 °C i +1,0 °C
7 Sulfatni ostatak Ne više od 0,1 %8 Teški metali Ne više od 10 ppm9 Sadržaj hlorida (% w/w računato na suvu bazu) Između 7,5 % i 8,5 %
10 Srodne supstance HPLC metodomA. Izomer AS na oko 0,88 RRTB. . Izomer AS izomer na oko 1,13 RRTC. Najveća pojedinačna nepoznata nečistoća D. Ukupne nečistoće
A. ne više od 0,15 %B. ne više od 0,10 %C. ne više od 0,10 %D. ne više od 0,60 %
11 Hiralna čistoća (HPLC)D-izomerL-izomerUkupno DL izomera
Između 48,5 % i 51,5 %Između 48,5 % i 51,5 %Ne manje od 99,0 %
12 Određivanje (potenciometrijski) 98,5 % – 101,5 % na osušenu bazu13 Rezidualni rastvarači (GC metoda)
(a) metanol(b) izopripil alkohol
(a) ne više od 1000 ppm(b) ne više od 1000 ppm
Dodatni testovi14 Veličina čestica (Malvern analizator) 50 % čestica treba da ima veličinu manju od 8 µm
15 Sadržaj paladijuma Ne više od 5 ppm 25
Specifična ispitivanja
� Opis
� Identifikacija aktivne
farmaceutske supstance
� Određivanje sadržaja
aktivne farmaceutske
supstance
� Određivanje nečistoća
���� fizičko−−−−hemijske karakteristike
���� distribucija veličina čestica
���� polimorfizam
���� testovi koji se izvode na novim
aktivnim supstancama sa
hiralnim karakteristikama
���� određivanje sadržaja vode
neorganske nečistoće
ispitivanje mikrobiološke čistoće
Zajednička ispitivanja
26

9/27/2014
14
Zajednička ispitivanja
1. OPIS – izgled, boja, miris
Ako se tokom proizvodnje ili čuvanja menjaju ove karakteristike, to
mora biti navedeno u specifikaciji.
Metoda: vizuelno, organoleptički
27
28
2. IDENTIFIKACIJA AKTIVNE FARMACEUTSKE SUPSTANCE
�Ako se koristi tečna hromatografija moraju biti najmanje dve metode i to biti kombinovane sa različitim tipovima detekcije: HPLC – DAD i HPLC –MS.
�Mora biti specifična i pouzdana metoda –najpouzdanija metoda za identifikaciju Infra crvena spektroskopija!
�Postupak mora biti takav da obezbedi jasno razlikovanje ispitivane susptance od strukturno sličnih supstanci
�Ako je supstanca u obliku soli – radi se specifična reakcija za so
�Za optički aktivne supstance metoda koja se primenjuje mora omogućiti razdvajanje i razlikovanje enantiomera

9/27/2014
15
29
3. ODREĐIVANJE SADRŽAJA AKTIVNE FARMACEUTSKE SUPSTANCE
Ista metoda može se koristiti za određivanje sadržaja aktivne
farmaceutske supstance i za određivanje prisustva srodnih supstanci
Metoda: HPLC
Nespecifične metode – titrimetrija, spektrofotometrija
Za određivanje sadržaja nečistoća – HPLC metoda
30
4. ISPITIVANJE NEČISTOĆA
organskih, neorganskih i rezidualnih rastvarača
Organske nečistoće :
� polazne supstance i sporedni proizvodi postupka sinteze
� degradacioni proizvodi
Neorganske nečistoće:
� elementi u tragovima (katalizatori)
Rezidualni rastvarači:
� koriste se u različitim fazama sinteze aktivne farmaceutske
supstance

9/27/2014
16
Metode koje se koriste za ispitivanje
Organske nečistoće :
� Metoda tečne hromatografije sa različitim tipovima detekcije (UV,
Diode Array, MS...)
Neorganske nečistoće:
� različiti limit testovi, atomska apsorpciona spektroskopija
Rezidualni rastvarači:
� Metoda gasne hromatografije sa različitim tipovima detekcije
31
Značaj ispitivanja nečistoća
32
9/27/2014
17
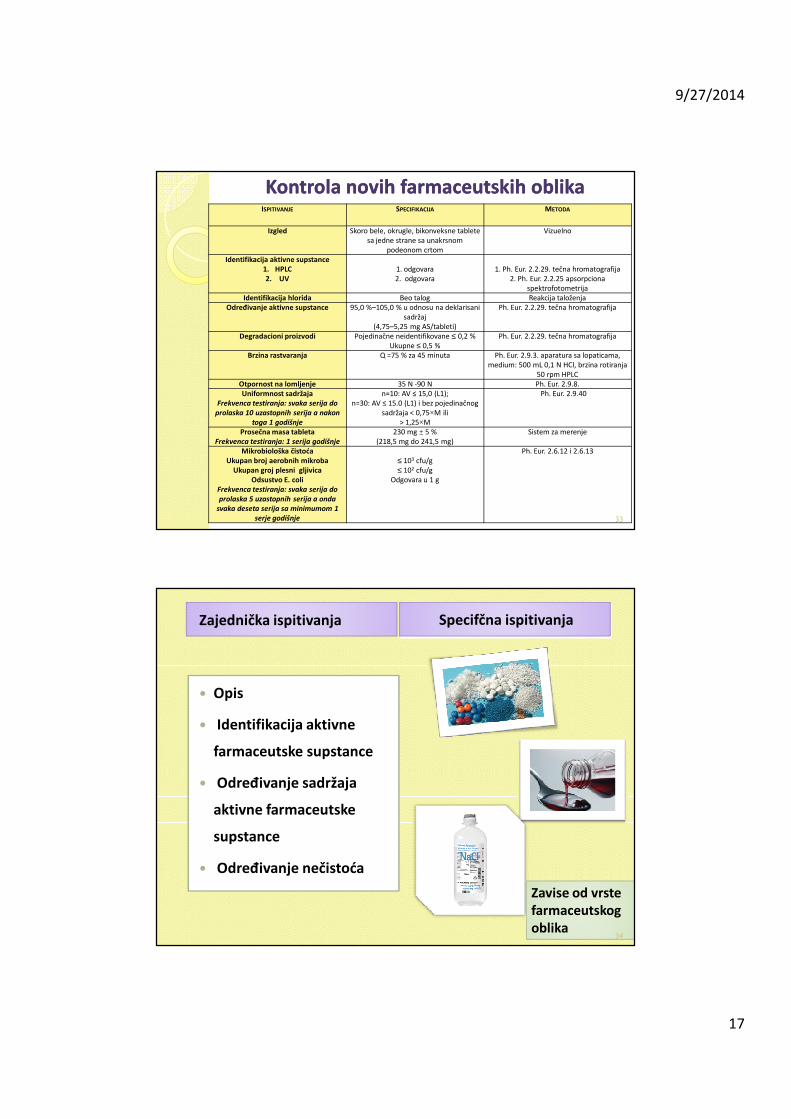
Kontrola novih farmaceutskih oblikaKontrola novih farmaceutskih oblikaISPITIVANJE SPECIFIKACIJA METODA
Izgled Skoro bele, okrugle, bikonveksne tablete sa jedne strane sa unakrsnom
podeonom crtom
Vizuelno
Identifikacija aktivne supstance1. HPLC2. UV
1. odgovara2. odgovara
1. Ph. Eur. 2.2.29. tečna hromatografija2. Ph. Eur. 2.2.25 apsorpciona
spektrofotometrijaIdentifikacija hlorida Beo talog Reakcija taloženja
Određivanje aktivne supstance 95,0 %–105,0 % u odnosu na deklarisani sadržaj
(4,75–5,25 mg AS/tableti)
Ph. Eur. 2.2.29. tečna hromatografija
Degradacioni proizvodi Pojedinačne neidentifikovane ≤ 0,2 %Ukupne ≤ 0,5 %
Ph. Eur. 2.2.29. tečna hromatografija
Brzina rastvaranja Q =75 % za 45 minuta Ph. Eur. 2.9.3. aparatura sa lopaticama, medium: 500 mL 0,1 N HCl, brzina rotiranja
50 rpm HPLCOtpornost na lomljenje 35 N -90 N Ph. Eur. 2.9.8.Uniformnost sadržaja
Frekvenca testiranja: svaka serija do
prolaska 10 uzastopnih serija a nakon
toga 1 godišnje
n=10: AV ≤ 15,0 (L1);n=30: AV ≤ 15.0 (L1) i bez pojedinačnog
sadržaja < 0,75×M ili> 1,25×M
Ph. Eur. 2.9.40
Prosečna masa tabletaFrekvenca testiranja: 1 serija godišnje
230 mg ± 5 %(218,5 mg do 241,5 mg)
Sistem za merenje
Mikrobiološka čistoćaUkupan broj aerobnih mikroba
Ukupan groj plesni gljivicaOdsustvo E. coli
Frekvenca testiranja: svaka serija do
prolaska 5 uzastopnih serija a onda
svaka deseta serija sa minimumom 1
serje godišnje
≤ 103 cfu/g≤ 102 cfu/g
Odgovara u 1 g
Ph. Eur. 2.6.12 i 2.6.13
33
Specifčna ispitivanja
� Opis
� Identifikacija aktivne
farmaceutske supstance
� Određivanje sadržaja
aktivne farmaceutske
supstance
� Određivanje nečistoća
Zajednička ispitivanja
Zavise od vrste farmaceutskog oblika
34

9/27/2014
18
Ispitivanja specifična za tablete
1. Određivanje brzine rastvaranja
2. Raspadljivost
3. Ujednačenost mase/sadržaja
4. Određivanje sadržaja vode
5. Čvrstina i/ili frijabilnost
6. Ispitivanje mikrobiološke čistoće
35
ISPITIVANJE ZAHTEVI REZULTATI
Izgled Skoro bele, okrugle, bikonveksne tablete sa jedne strane sa unakrsnom podeonom crtom
Odgovara
Identifikacija aktivne supstance1. HPLC2. UV
1. odgovara2. odgovara
Odgovara
Identifikacija hlorida Beo talog OdgovaraOdređivanje aktivne supstance
95,0 %–105,0 % u odnosu na deklarisani sadržaj(4,75–5,25 mg AS/tableti)
Odgovara100,2 %
(5,01 mg AS/tbl)Degradacioni proizvodi
Pojedinačne neidentifikovane ≤ 0,2 %Ukupne ≤ 0,5 %
Odgovara0,05 %0,14 %
Brzina rastvaranja Q =75 % za 45 minuta Odgovara92,8 %
Otpornost na lomljenje 35 N -90 N Odgovara80 N
Uniformnost sadržaja n=10: AV ≤ 15,0 (L1);n=30: AV ≤ 15,0 (L1) i bez pojedinačnog sadržaja < 0,75×M ili
> 1,25×M
Odgovara
Prosečna masa tableta 230 mg ± 5 %(218,5 mg do 241,5 mg)
Odgovara231,0 mg
Mikrobiološka čistoćaUkupan broj aerobnih mikroba
Ukupan groj plesni gljivicaOdsustvo E. coli
≤ 103 cfu/g≤ 102 cfu/g
Odgovara u 1 g
Odgovara
Sertifikat analize tableta
36

9/27/2014
19
Ispitivanja specifična za tečne farmaceutske oblike za per os primenu
1. pH rastvora
2. Određivanje brzine rastvaranja
3. Ujednačenost mase/sadržaja
4. Određivanje sadržaja vode
5. Određivanje sadržaja alkohola
6. Ispitivanje materija koje se mogu ekstrahovati iz pakovanja
7. Distribucija veličine čestica
8. Redisperzibilnost
9. Reološka svojstva
10. Vreme potrebno za rekonstituisanje
11. Ispitivanje mikrobiološke čistoće 37
Ispitivanja specifična za parenteralne preparate1. pH vrednost rastvora
2. Sterilnost
3. Endotoksini/pirogeni
4. Ujednačenost mase/sadržaja
5. Određivanje sadržaja vode
6. Vidljive i subvidljive čestice
7. Ispitivanje materija koje se mogu ekstrahovati iz pakovanja ili
sistema zatvaranja kontejnera
8. Distribucija veličine čestica
9. Redisperzibilnost
10. Osmolarnost
11. Vreme potrebno za rekonstituisanje
12. Ispitivanje funkcionalnosti sistema 38

9/27/2014
20
39
Na koji način može da se obezbedi odgovarajući kvalitet lekova?
� Izbor odgovarajućeg proizvoda� Dug rok upotrebe� Prihvatljiva stabilnost� Prihvatljiva bioraspoloživost
� Izbor odgovarajućih dobavljača� Po potrebi izmena dobavljača� Uzimanje uzoraka od novih dobavljača� Postavljanje specifičnih zahteva za dobavljača (na primer o
bioraspoloživosti supstance i stabilnosti)
� Sertifikovanje proizvodnje� GMP sertifikat proizvođača� Sertifikovanje proizvoda i serije proizvoda� Nasumična testiranja
40
�Upotreba farmakopejskih referentnih standarda u
ispitivanju
�Za označavanje proizvoda koristiti jezik zemlje u
kojoj se lek izdaje
�Koristiti odgovarajuće pakovanje koje će zadovoljiti
kriterijume za čuvanje i transport
� Odgovarajući način čuvanja, transporta,
distribucije i upotrebe
Pored toga i :
...

9/27/2014
21
41
Ko je sve odgovoran za očuvanje kvaliteta proizvodaKo je sve odgovoran za očuvanje kvaliteta proizvoda??
Kvalitet lekova
Agencija za lekove
Lekari, stomatolozi,
svi koji propisuju
lekove
Službe koje učestvuju u
nabavci lekova
Apoteke Pacijenti
Broj koji je karakterističan za datu analizu i datum izdavanja sertifikata
Podaci o uzorku koji se ispituje
Ispitivan
ja i d
ob
ijeni
rezultati
Konačan zaključak o kvalitetu ispitivanog proizvoda
Potpis odgovornih osoba
42

9/27/2014
22
+ Serifikat analizePuštanje
leka u
promet
43





























