Embed Size (px)
Citation preview

Tanaka et al. BMC Genomics 2012, 13:352http://www.biomedcentral.com/1471-2164/13/352
RESEARCH ARTICLE Open Access
A complete mitochondrial genome sequence ofOgura-type male-sterile cytoplasm and itscomparative analysis with that of normalcytoplasm in radish (Raphanus sativus L.)Yoshiyuki Tanaka, Mizue Tsuda, Keita Yasumoto, Hiroshi Yamagishi and Toru Terachi*
Abstract
Background: Plant mitochondrial genome has unique features such as large size, frequent recombination andincorporation of foreign DNA. Cytoplasmic male sterility (CMS) is caused by rearrangement of the mitochondrialgenome, and a novel chimeric open reading frame (ORF) created by shuffling of endogenous sequences is oftenresponsible for CMS. The Ogura-type male-sterile cytoplasm is one of the most extensively studied cytoplasms inBrassicaceae. Although the gene orf138 has been isolated as a determinant of Ogura-type CMS, no homologoussequence to orf138 has been found in public databases. Therefore, how orf138 sequence was created is a mystery.In this study, we determined the complete nucleotide sequence of two radish mitochondrial genomes, namely,Ogura- and normal-type genomes, and analyzed them to reveal the origin of the gene orf138.
Results: Ogura- and normal-type mitochondrial genomes were assembled to 258,426-bp and 244,036-bp circularsequences, respectively. Normal-type mitochondrial genome contained 33 protein-coding and three rRNA genes,which are well conserved with the reported mitochondrial genome of rapeseed. Ogura-type genomes containedsame genes and additional atp9. As for tRNA, normal-type contained 17 tRNAs, while Ogura-type contained 17tRNAs and one additional trnfM. The gene orf138 was specific to Ogura-type mitochondrial genome, and nosequence homologous to it was found in normal-type genome. Comparative analysis of the two genomes revealedthat radish mitochondrial genome consists of 11 syntenic regions (length >3 kb, similarity >99.9%). It was shownthat short repeats and overlapped repeats present in the edge of syntenic regions were involved in recombinationevents during evolution to interconvert two types of mitochondrial genome. Ogura-type mitochondrial genomehas four unique regions (2,803 bp, 1,601 bp, 451 bp and 15,255 bp in size) that are non-syntenic to normal-typegenome, and the gene orf138 was found to be located at the edge of the largest unique region. Blast analysisperformed to assign the unique regions showed that about 80% of the region was covered by short homologoussequences to the mitochondrial sequences of normal-type radish or other reported Brassicaceae species, althoughno homology was found for the remaining 20% of sequences.
Conclusions: Ogura-type mitochondrial genome was highly rearranged compared with the normal-type genomeby recombination through one large repeat and multiple short repeats. The rearrangement has produced fourunique regions in Ogura-type mitochondrial genome, and most of the unique regions are composed of knownBrassicaceae mitochondrial sequences. This suggests that the regions unique to the Ogura-type genomewere generated by integration and shuffling of pre-existing mitochondrial sequences during the evolution ofBrassicaceae, and novel genes such as orf138 could have been created by the shuffling process ofmitochondrial genome.
* Correspondence: [email protected] Laboratory, Faculty of Life Sciences, Kyoto Sangyo University, Motoyama,Kamigamo, Kita-ku, Kyoto 603-8555, Japan
© 2012 Tanaka et al.; licensee BioMed CentralCommons Attribution License (http://creativecreproduction in any medium, provided the or
Ltd. This is an Open Access article distributed under the terms of the Creativeommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andiginal work is properly cited.

Tanaka et al. BMC Genomics 2012, 13:352 Page 2 of 11http://www.biomedcentral.com/1471-2164/13/352
BackgroundCompared with animal mitochondrial genomes that arecompact and relatively uniform in size, plant mitochon-drial genomes are extremely complex and have charac-teristic features, including large genome size, frequentreorganization and incorporation of foreign DNA [1].Plant mitochondrial genome varies in size from 208 kbin Brassica hirta to 11.3 Mb in Silene conica [2,3], dueto the expansion of non-coding sequence and duplica-tion of a large segment. It is also well known that plantmitochondrial genome evolves rapidly in structure andslowly in sequence. Plant mitochondria contain a num-ber of repeats. The recombination via repeat sequencesis believed to be responsible for extensive structuralchange. Incorporation of foreign DNA fragments thatoriginated from plastid and nuclear genomes also can befound in plant mitochondrial genome. Because of fre-quent recombination and incorporation of foreign DNA,extensive genome reorganization and gene-order shuf-fling may occur. These changes in mitochondrial gen-ome can produce novel open reading frames, some ofwhich result in cytoplasmic male sterility (CMS) [4].CMS is a maternally inherited trait in which a plant is
unable to produce functional pollen. CMS is an econom-ically important trait for F1 hybrid seed production inmany crops. CMS has been widely observed in higherplants, and many reports show that CMS is caused by al-teration of the mitochondrial genome [5]. These altera-tions often create a novel open reading frame (ORF),which consists of a chimeric sequence generated by geneshuffling or by fusion between a native gene(s) and/oran unknown sequence(s).The Ogura male-sterile cytoplasm, which was origin-
ally found in an unknown variety of Japanese radish, isthe most widely studied cytoplasm in Brassicaceae [6].This cytoplasm is used in F1 seed production ofEuropean, Chinese and Japanese radishes. The cytoplasmwas introduced into Brassica crops by intergenerichybridization and somatic cell fusion, and has been uti-lized for F1 seed production in Brassica crops such asrapeseed worldwide [7,8].A mitochondrial gene, orf138, is responsible for Ogura
male sterility and specifically present in the mitochon-drial genome of various radishes with Ogura-type cyto-plasm [9]. The accumulation of ORF138 protein isobserved in plants expressing the CMS phenotype, andthe association of ORF138 with the inner mitochon-drial membrane of male-sterile plants was reported[10]. Although it appeared that expression of the nu-clear chalcone synthase gene, which is related to fla-vonoid biosynthesis, is inhibited in the anther of Oguraradish [11], the molecular mechanism underlyingOgura CMS by ORF138 protein is not known. The se-quence of the gene orf138 itself is also interesting;
there is no evidence that orf138 is a chimeric geneconsisting of native mitochondrial genes or sequence.No sequence homologous to orf138 is found in publicdatabases; therefore, the origin of orf138 is a mystery.In addition, it has been shown that the mitochondrialgenome of Ogura-type cytoplasm is highly rearrangedcompared with that of normal cytoplasm in radish.These rearrangements occurred in the loci atp1, atp6,cox1 and most notably orf138-atp8 [12-17]. The geneorf138 is specifically located in the 5’ upstream regionof the gene atp8 in Ogura radish, whereas the regionis occupied by the gene cox1 in normal radish [16]. Inthe evolution of Raphanus, how and when these exten-sive rearrangements occurred between two types ofmitochondrial genome are not known.In this study, in order to obtain a better understanding
of the origin of orf138 and Ogura-type mitochondrialgenome in radish, we determined the complete nucleo-tide sequences of two mitochondrial genomes of radish,that is, Ogura- and normal-type mitochondrial genomes,and compared their structures. Gene contents andunique regions for each genome were analyzed in detailat the sequence level. Furthermore, we focused on shortrepeated sequences that may be involved in the recom-bination, leading to extensive rearrangements of the twogenomes and to generation of the gene orf138.
ResultsRadish mitochondrial genomeThe Ogura and normal mitochondrial genomes areassembled as 258,426-bp and 244,036-bp circular mole-cules, respectively (Figure 1). The overall GC content ofboth mtDNAs is 45.2%, being comparable to those ofother mtDNAs of Brassicaceae. About 7% of Ogura and3% of normal sequences are unique to each mitochon-drial genome, and 80% of Ogura and 84% of normalsequences show high similarity to the sequence ofrapeseed (Brassica napus L.) mitochondrial genome(GenBank: AP006444.1) [18].
Gene organization of radish mitochondrial genomeNormal-type mitochondrial genome contained 33protein-coding and three rRNA genes, while Ogura-typegenomes contained same genes and one additional atp9(atp9-2). Gene content is identically conserved betweenthe two radish mitochondrial genomes and it is alsoidentical to that of rapeseed, except atp9-2 in Ogura-type genome. Normal-type genome contains similarsequence to atp9-2, but 3’ part of this sequence lackssimilarity to atp9-2. The atp9-2 like sequence innormal-type genome encodes 70 amino acid protein,which is lacking atp9 domain partially. Eighteen of theconserved 33 protein-coding genes produce compo-nents of the electron transport chain and ATP
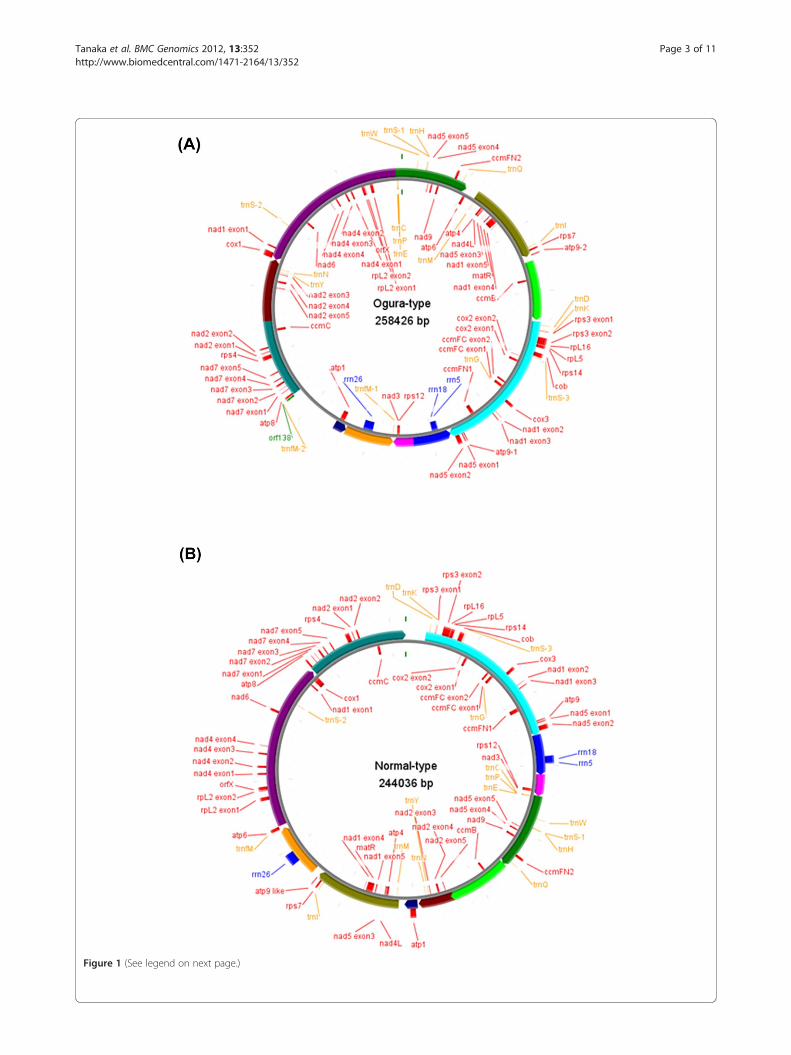
Figure 1 (See legend on next page.)
Tanaka et al. BMC Genomics 2012, 13:352 Page 3 of 11http://www.biomedcentral.com/1471-2164/13/352

(See figure on previous page.)Figure 1 The organization of Ogura-type (A) and normal-type (B) mitochondrial genomes represented as a "master circle". Features onforward and reverse strands are drawn on the outside and inside of the circles, respectively. Protein-coding genes are shown in red, rRNAs inblue, tRNAs in orange and orf138 in lime green. The arcs in the same colors indicate syntenic regions between Ogura- and normal-type genomes(refer to Figure 2). Genome maps were made with CGviewer [49].
Tanaka et al. BMC Genomics 2012, 13:352 Page 4 of 11http://www.biomedcentral.com/1471-2164/13/352
synthase: nine subunits of complex I(nad1, nad2, nad3,nad4, nad4L, nad5, nad6, nad7 and nad9), apocyto-chrome b (cob) of complex III, three subunits of com-plex IV (cox1, cox2 and cox3) and five subunits ofcomplex V (atp1, atp4, atp6, atp8 and atp9). Five add-itional proteins are involved in the biogenesis of cyto-chrome c (ccmB, ccmC, ccmFN1/ccmFN2 and ccmFC).Another eight genes encode ribosomal proteins (rpl2,rpl5 and rpl16, and rps3, rps4, rps7, rps12 and rps14).The two remaining genes encode maturase (matR) andorfX. The gene ccmFN is divided into two readingframes in mitochondrial genomes of radish like inthose of rapeseed and Arabidopsis [18,19]. Three rRNAgenes (rrn5, rrn18 and rrn26) are highly conserved be-tween the two mitochondrial genomes of radish.The 5’ region of the gene atp6 is highly diverged be-
tween the two radishes because of recombination thatoccurred in the locus [20]. The 3’ end of Ogura-typeorfX also lacks similarity to that of normal-type. This iscaused by a 48-bp repeated sequence that is regarded asa minisatellite. Eleven SNPs were identified in the ninegenes: four were synonymous and seven were non-synonymous (Table 1). As for tRNA, both Ogura and nor-mal mitochondrial genomes contain 17 tRNAs, whereasOgura genome has an additional trnfM (trnfM-2). ThetrnfM-2, which is located close to the gene orf138(Figure 1), has a specific SNP compared with othersequenced mitochondrial trnfMs in Brassicaceae.
Table 1 Difference in genes coding protein betweennormal-type and Ogura-type
Gene Nucleic acid Amino acidb
nad4 1010 T-C Non-synonymous (337 L-P)
cox1 117 G-T Synonymous
atp6 no homology in 5′ terminal
atp8 370 A-C Non-synonymous (124 I-L)
444 T-C Non-synonymous (150 V-A)
atp9 65 A-G Non-synonymous (22 I-V)
rpL2 840 C-T Synonymous
rpL16 492 G-A Synonymous
rps4 776 C-T Non-synonymous (259 S-F)
ccmC 351 G-A Synonymous
ccmFC 605 A-G Non-synonymous(202 E-G)
606 G-T
orfX no homology in 3′ terminal
a Location of SNP (Normal-Ogura).b Location of amino acid mutation (Normal-Ogura).
We searched for a sequence homologous to the orf138in Ogura and normal mtDNA sequences. Only a 67-bpsequence, which includes the 3’ terminal of the orf138sequence and its 3’ UTR, was detected. This sequence ishomologous to 5’ flanking region of ccmFN1 in reportedBrassicaceae genomes. However, except for this short se-quence, no homology was found between the main partof the gene orf138 and other parts of the Brassicaceaemitochondrial genome.
Reorganization between two types of radishmitochondrial genomeThe bl2seq analysis suggested that the radish mitochon-drial genome consists of 11 syntenic regions (length>3 kb) (Figure 2). The sequence in the Ogura-typemitochondrial genome shows quite high homology(99.9%) to that of normal mitochondrial genome withinthese syntenic regions. This analysis revealed thatOgura-type mitochondrial genome has a large uniqueregion (15,255 bp) between syntenic regions 7 and 11.This unique region corresponds to the “region A” thatwas reported by Makaroff et al. [12]. The gene orf138 islocated at one of the terminals of this region (Figure 1).Repeats in two mitochondrial genomes have been
investigated. If a pair of sequences has over 90% similar-ity, the pair is defined as a repeat. A pair of large repeatsof 9,732 bp was found in Ogura-type mitochondrial gen-ome, while a pair of large repeats of 5,530 bp was identi-fied in normal-type. Even though the length of largerepeats is different, sequence of the large repeats arehighly conserved within 5,530 bp. In Ogura-type mito-chondrial genome, the large repeats are located in syn-tenic regions 5 and 11/6 (Figure 3, Ogura R1). A 9,732-bpsequence, corresponding to Ogura-type repeat, is con-served in syntenic region 5/6 of the normal-type gen-ome, but it is truncated in syntenic region 11 becausenormal-type genome has a specific sequence betweensyntenic regions 1 and 11. In rapeseed mitochondrialgenome, a 2,427-bp sequence that includes the gene cox2was reported as a large direct repeat. Large repeatedsequences found in Raphanus are completely differentfrom those reported in rapeseed [18]. These large re-peats are considered to be involved in the formation of atripartite structure; each radish mitochondrial genomecould recombine into two sub-genomic circles. Two ex-pected sub-genomic circles are 130,760 bp and 127,666 bpin Ogura-type, while they are 139,398 bp and 104,638 bpin normal-type. In addition, these repeats are also
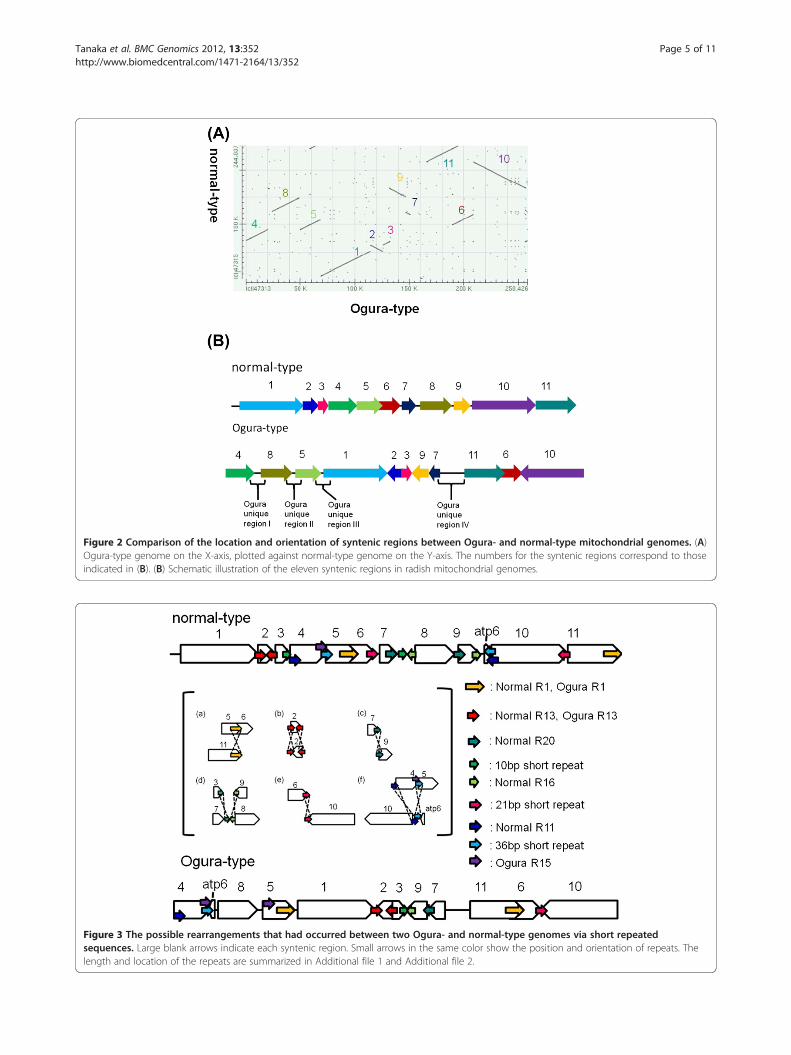
Figure 2 Comparison of the location and orientation of syntenic regions between Ogura- and normal-type mitochondrial genomes. (A)Ogura-type genome on the X-axis, plotted against normal-type genome on the Y-axis. The numbers for the syntenic regions correspond to thoseindicated in (B). (B) Schematic illustration of the eleven syntenic regions in radish mitochondrial genomes.
Figure 3 The possible rearrangements that had occurred between two Ogura- and normal-type genomes via short repeatedsequences. Large blank arrows indicate each syntenic region. Small arrows in the same color show the position and orientation of repeats. Thelength and location of the repeats are summarized in Additional file 1 and Additional file 2.
Tanaka et al. BMC Genomics 2012, 13:352 Page 5 of 11http://www.biomedcentral.com/1471-2164/13/352

Tanaka et al. BMC Genomics 2012, 13:352 Page 6 of 11http://www.biomedcentral.com/1471-2164/13/352
related to reorganization among syntenic regions 5, 6 and11 (Figure 3a).We defined 22 and 23 short repeats in Ogura- and
normal-type genomes, respectively (Additional file 1 andAdditional file 2). These repeats can account forreorganization of mitochondrial genome via homologousrecombination. We have identified repeats that can beinvolved in reorganization from normal- to Ogura-typemitochondrial genomes, although the direction ofchanges cannot be fixed. The pair of inverted repeats(Normal R13) is located on either side of syntenic region2 in normal-type genome. These inverted repeats maychange the orientation of syntenic region 2 in Ogura-type genome (Figure 3b). In normal-type mitochondrialgenome, a pair of direct repeats (Normal R20) is locatedat the edge of syntenic regions 7 and 9. These repeatscan be related to generation of a linkage between 7 and9 in Ogura-type genome (Figure 3c). In normal-typegenome, Normal R16 is present at the edge of syntenicregion 9 and in the unique sequence located betweensyntenic regions 7 and 8 in the inverted orientation. A10-bp short repeat is also present in this unique se-quence, and it exists at the edge of syntenic region 3 inthe direct orientation. These repeats can be associatedwith the generation of a linkage between syntenicregions 3 and 9 in Ogura-type genome (Figure 3d).Syntenic regions 6 and 10 in normal-type genome havea 21-bp short inverted repeat. This inverted repeat canbe involved in inversion and recombination betweensyntenic regions 6 and 10 in Ogura-type genome(Figure 3e). Finally, in normal-type genome, syntenic re-gion 4 has Normal R11 and a short 36-bp repeat at eachedge. Normal R11 is also present at one edge of syntenicregion 10, being overlapped with a 36-bp repeat. InOgura-type genome, a linkage between syntenic regions10 and 4 is generated via these two sequences. This re-combination event also changes the location of the geneatp6 (Figure 3f ). Moreover, in Ogura-type genome, twocopies of a short direct repeat (Ogura R15) are present
Table 2 Mitotype specific open-reading frames caused by rec
ORF Similarity of predicted protein
Ogura-type specific orf122 No similarity
orf154 AAG51754.1 reverse transcriptase, pu
orf117 YP_717156.1 hypothetical protein Br
orf138 No similarity
orf102 NP_085574.1 hypothetical protein Ar
orf344 YP_717145.1 hypothetical protein Br
normal-type specific
orf322 YP_717145.1 hypothetical protein Br
orf150 No similarity
orf145 NP_085575.1 hypothetical protein Ar
at the edge of syntenic regions 4 and 5, between whichsyntenic region 8 is inserted with a novel sequence(Figure 3f ).
Open reading frames that are unique to eachmitochondrial genome typeA comprehensive comparison of predicted ORFs wasconducted between Ogura- and normal-type mitochon-drial genomes. We picked up unique ORFs that can en-code over 100 amino acids and were generated byrecombination. As a result, six ORFs including orf138were specifically detected in Ogura-type mitochondrialgenome (Table 2). The five Ogura-type ORFs werelocated between syntenic regions 7 and 11, whereasorf344 was in syntenic region 3. With the exception oforf138, the nucleotide sequences of the five ORFs showedhigh similarity to the reported sequences in plant mito-chondrial genomes of other species. As for the normal-type genome, three unique ORFs were detected. Theorf322 has strong similarity to the orf344 in Ogura-type.The extension of orf344 was caused by a new initiationcodon that was created in 5’ region of orf322 through arecombination between syntenic regions 3 and 9. Thetwo remaining ORFs were located within the regionunique to the normal-type genome located between syn-tenic regions 11 and 1. Protein Blast analysis was con-ducted to characterize unique ORFs. Even thoughhomologous amino acid sequences were not found fororf150 in normal-type and for orf122 and orf138 inOgura-type, other unique ORFs encoded similar proteinsthat have been reported in the mitochondrial genome ofrapeseed or Arabidopsis.
Regions unique to the Ogura-type mitochondrial genomeOgura-type mitochondrial genome has four uniqueregions that are non-syntenic to normal-type mitochon-drial genome. Unique region I is 2,802 bp, which islocated between syntenic regions 4 and 8 (Figure 2B).Unique regions II and III are 1,601-bp and 451-bp
ombination
Location
between syntenic regions 7 and 11
tative[Arabidopsis thaliana] between syntenic regions 7 and 11
napMp059 [Brassica napus] between syntenic regions 7 and 11
between syntenic regions 7 and 11
thMp044 [Arabidopsis thaliana] between syntenic regions 7 and 11
napMp048 [Brassica napus] the edge of syntenic region 3
napMp048 [Brassica napus] the edge of syntenic region 3
between syntenic regions 1 and 10
thMp101 [Arabidopsis thaliana] between syntenic regions 1 and 10

Tanaka et al. BMC Genomics 2012, 13:352 Page 7 of 11http://www.biomedcentral.com/1471-2164/13/352
sequences that are present between syntenic regions 8and 5, and between syntenic regions 5 and 1, respect-ively. The largest unique region IV is located betweensyntenic regions 7 and 11 (Figure 2B). Total length ofthe unique regions in Ogura-type genome is 20,110 bp,which occupies 7% of the whole Ogura-type mitochon-drial genome. The largest unique region contains fiveunique ORFs including orf138.To obtain more information on the sequence unique
to Ogura-type genome, nucleotide Blast search wasconducted to assign the sequences. The search showedthat over 80% of these regions consist of sequenceshomologous to those reported for Brassicaceae or radishmitochondrial genomes. Bl2seq analyses were furtherconducted between the sequence unique to Ogura-typeand each of five mitochondrial genomes in Brassicaceae(normal and Ogura-type radish, A. thaliana and twotypes of Brassica napus). Seventy-two percent of uniqueregion I (2,044 bp in a total of 2,802 bp) and 100% ofunique region II (1,601 bp) were occupied by five mito-chondrial sequences of Brassicaceae (Additional file 3).As for the smallest unique region III, 97% of the se-quence (440 bp in a total of 451 bp) was shown to havesimilarity to rapeseed mtDNAs. As for the largest uniqueregion IV, sequences homologous to the five Brassica-ceae mitochondrial genomes covered 80% of this region(12,187 bp in a total of 15,255 bp), even though orf138sequence itself had no similarity. Homologous sequencesdetected by bl2seq were plotted on unique region IV inOgura-type mitochondrial genome (Figure 4). Homolo-gous sequences were scattered throughout this region.Insertion of plastid or nuclear sequence has not beendetected in this region. It seems, therefore, that the re-gion unique to Ogura-type mitochondrial genome was
Figure 4 The sequences homologous to Brassicaceae mitochondrial gindicates the region specific to the Ogura-type mitochondrial genome (15,orf138 is located at the edge of this region. Black boxes indicate the sequegenomes.
created by frequent recombination and shuffling ofmitochondrial DNA sequences during the evolution ofBrassicaceae.
DiscussionComparative sequence analysis of Ogura- and normal-type mitochondrial genomesOgura-type mitochondrial genome was 14 kb larger thanthat of normal type. Region IV that is unique to Ogura-type mitochondrial genome mainly contributes to thissize difference. Comparative analysis has been performedbetween mitochondrial genomes of CMS and normallines in other higher plant species. In sugar beet, Owen-type CMS line has a significantly larger mitochondrialgenome than fertile line because the Owen-type mito-chondrial genome contains one 86-kb large repeat [21].In wheat, the mitochondrial genome of the CMS lineKs3 was reported to be 192 kb larger than that of com-mon wheat [22]. This difference is also caused by add-itional large repeats, including a 98-kb repeat. Incontrast, a rice (Oryza sativa) LD-CMS line has a smal-ler mitochondrial genome than fertile line, whileCW-CMS derived from wild rice (O. rufipogon) has alarger genome than fertile line [23]. The difference inmitochondrial genome size within the Oryza genus isalso attributable to large repeats. It should be noted thatthe size difference between two radish mitochondrialgenomes mainly arises from a novel sequence of15,255 bp, not from a large repeated sequence.The reported Brassicaceae species including Brassica and
Arabidopsis have small mitochondrial genomes amonghigher plants [24]. It is also known that Brassicaceae mito-chondrial genomes reported so far do not contain muchnuclear sequence. The number of large repeats (>1 kb) is
enome plotted on Ogura-type specific region IV. A black line255 bp in size) located between syntenic regions 7 and 11. The genence homologous to radish and/or other Brassicaceae mitochondrial

Tanaka et al. BMC Genomics 2012, 13:352 Page 8 of 11http://www.biomedcentral.com/1471-2164/13/352
less in reported Brassicaceae (Arabidopsis and Brassica)than in other plants species [25]. While mitochondrial gen-omes of rice or wheat have more than ten large repeats,Brassica and Raphanus have only two large repeats. Inaddition, sequences derived from nuclear or plastid gen-omes were rarely observed. This is one of the reasons whymitochondrial genomes of Brassica and Raphanus aresmall. Because of the small genome size and few largerepeats, we can consider Brassica and Raphanus as speciesthat have the “simple” mitochondrial genomes in higherplants. An understanding of the remarkable reorganizationevents that occurred in the simple radish genome will pro-vide us with a clear picture of mitochondrial genome evo-lution, such as insertion of a 15-kb large novel sequence inOgura-type.As expected, gene contents and their sequences were
highly conserved between the two mitochondrial gen-omes in radish. The 5’ sequence of the atp6 in Ogura-type mitochondrial genome is, however, different fromthat of normal-type. Recombination had occurred at thesite of syntenic region 4 and atp6, which produced anew initiation codon for atp6 in Ogura-type genome.According to the genome sequence, Ogura-type atp6encodes a different polypeptide from that of normal-typeatp6. However, the N-terminal of ATP6 polypeptide ofOgura-type is processed to produce a protein identicalto normal-type ATP6 [20]. The 3’ end of orfX also lackssimilarity to the sequence of normal-type. This is causedby a 48-bp repeated sequence regarded as a minisatellite.This minisatellite will be a useful marker to detect poly-morphism among radish mitochondrial genomes includ-ing Ogura and normal types [26]. A total of 11 SNPswere detected in nine protein-coding genes betweenOgura and normal types. The number of SNPs in radishwas similar to that in sugar beet; the sugar beet Owen-CMS line contains 24 SNPs in 11 protein-coding genescompared with the fertile lines [21]. As for tRNA, 17tRNAs were highly conserved between two radish mito-chondrial genome types, but Ogura-type contains oneadditional trnfM (trnfM-2). This additional trnfM-2 islocated near orf138, and it is interesting to note that thistrnfM-2 has a unique SNP that has not been found inother reported trnfMs in Brassicaceae. As for uniqueORFs, Ogura-type has six unique ORFs including orf138.These ORFs can be related to unique biological charac-teristics in Ogura-type cytoplasm. In particular, tran-scription of four ORFs (orf122, orf154, orf117 andorf102) that are specific to Ogura-type genome shouldbe investigated in future study.
The origin of Ogura-type unique sequenceCMS-related ORFs have been isolated in various plantspecies, such as T-urf13 in maize [27], orf522 in sun-flower [28], pcf in petunia [29], orf456 in pepper [30],
orf107 in sorghum [31] and orf79 in rice [32]. In Brassi-caceae crops, in addition to orf138 in radish, threeCMS-related ORFs have been reported: orf224 andorf222 in rapeseed [33], and orf267 in Brassica tournefor-tii [34]. With the exception of orf138, all these CMS-related ORFs have a chimeric structure that contains thesequence of known mitochondrial genes. The orf138,however, does not contain any conserved mitochondrialsequence. The sequence of orf138 is totally novel, and isnot present in normal-type mitochondrial genome ofradish. It is quite interesting to reveal how the orf138 se-quence was created during the evolution of radish mito-chondrial genome. However, it now appears difficult toinfer the origin of the gene orf138, since no sequencehomologous to the main part of orf138 is present in ei-ther Ogura- or normal-type mitochondrial genome.In this study, we have determined the nucleotide se-
quence for unique region IV (15,255 bp in size) inOgura-type mitochondrial genome. The gene orf138 islocated in this region. Blast analysis using the sequencein region IV as a query showed that 80% of this regionhas similarity to the mitochondrial sequences in otherBrassicaceae plants, including rapeseed and Arabidopsis.The analysis also showed that the homologoussequences are scattered throughout unique region IV(Figure 4). Therefore, region IV is a mosaic of mitochon-drial sequences from Brassicaceae, suggesting that thesequence was generated through shuffling and fusion ofpre-existing mitochondrial sequences in ancestral Brassi-caceae. The sequence of which the gene orf138 consistsmay be one of the products of this shuffling process. Itis unlikely that the sequence of orf138 is derived fromforeign DNA, such as a nuclear or plastid sequence, be-cause no sequence with clear homology to foreign DNAis found within this unique region. However the possibil-ity remains that they may be derived from horizontalgene transfer from unidentified organism.This composition of novel sequence in radish con-
trasts to that reported for sugar beet. In sugar beet mito-chondrial genome, 7.6% of the unique regions showedsignificant homology to previously determined mito-chondrial sequences, 17.9% to nuclear DNA, 4.6% tomitochondrial episome and 0.1% to plastid DNA [35].
The emergence of Ogura-type cytoplasmThe sequence of Ogura-type unique region IV can bederived from a mixture of sequences in the reportedBrassicaceae mitochondrial genome. The next questionrelates to how this unique region IV was generated orintegrated into the radish mitochondrial genome. Wesearched for repeated sequences around this region.Ogura-type unique region IV was shown to be sand-wiched between syntenic regions 7 and 11 (Figure 5).There were identical 176-bp inverted repeats at the edges

Figure 5 A possible model for integration of the sequence (unique region IV) in Ogura-type mitochondrial genome. Yellow small arrowsindicate identical 176-bp inverted repeats at the edges of regions 7 and 11. On the insides of these repeated sequences, another pair of 28-bpinverted repeats is present (purple small arrows). Unique region IV had been integrated into radish mitochondria genome via these repeats whenrecombination between syntenic regions 7 and 11 occurred.
Figure 6 An atp8 promoter sequence affected byrearrangement among syntenic regions. Alignment of nucleotidesequences shows the comparison of predicted promoters for thegene atp8 among three mitochondrial genomes includingArabidopsis, and Ogura and normal radish. In Ogura-type genome, apromoter core (in a dotted box) is lost. This nucleotide change canaffect transcription of the gene atp8.
Tanaka et al. BMC Genomics 2012, 13:352 Page 9 of 11http://www.biomedcentral.com/1471-2164/13/352
of regions 7 and 11. On the insides of these repeatedsequences, another pair of 28-bp repeated sequences wasfound in inverted orientation. These 28-bp sequences arespecific to the Ogura-type mitochondrial genome, andnot found in normal-type genome. This characteristic ar-rangement of repeats indicates that unique region IV wasintegrated into the radish mitochondrial genome whenrecombination between syntenic regions 7 and 11occurred.Previously, we conducted sequence analysis on the
atp8 locus in normal- and Ogura-type cytoplasm fromwild and cultivated radishes [36]. While plants withnormal-type cytoplasm contained several types of se-quence, plants with Ogura-type cytoplasm had only onetype of sequence. These results suggested that a muta-tional event, which created linkage of atp8 and orf138,occurred only once in the history of radish [36]. Thepresent data indicate that this event probably occurredat the same time as when recombination between syn-tenic regions 7 and 11 occurred via 176-bp repeats andthe unique region IV was integrated into the Ogura-typemitochondrial genome (Figure 5). Interestingly, the rad-ish mitochondrial genome may have lost one putative pro-moter region for atp8 by this recombination (Figure 6). InArabidopsis, the promoters of mitochondrial genes havebeen determined [37]. A promoter sequence of atp8(CTATCAATCTCATAAGAGAAGAAAT) is almost con-served at the edge of syntenic region 11 of normal radish.The position of the promoter exactly matched the size ofatp8 transcript (ca. 600 bp in size) [16]. The recombin-ation of syntenic regions 7 and 11 would destroy the pro-moter sequence. Therefore, recombination betweensyntenic regions 7 and 11 in normal radish can lead to loss
of biological function in mitochondria. A reasonable hy-pothesis to explain the generation of Ogura-type mito-chondrial genome is that the mitochondrial genomerearrangement that could compensate for the loss of bio-logical function had been selected among many rear-ranged genomes. Integration of unique region IV canprovide a new promoter for the gene atp8. Actually, inOgura-type genome, atp8 is co-transcribed with orf138and trnfM, while in normal-type, atp8 is transcribed fromits own promoter.Another possibility to explain the emergence of Ogura-
type mitochondrial genome is substoichiometric shift(SSS). SSS is a phenomenon by which copy number of

Tanaka et al. BMC Genomics 2012, 13:352 Page 10 of 11http://www.biomedcentral.com/1471-2164/13/352
the substoichiometric molecule dramatically increasesover generations [38]. Research in some plants hasshown that SSS is related to the occurrence of CMS[39,40]. Recently, it has been reported that two mito-chondrial genome types, pol -type and nap-type, co-existamong many rapeseed cultivars, and SSS can explain theemergence of these CMS [41]. In radish with normal-type cytoplasm, orf138 was reported to be present insubstoichiometric molecules with low copy number bygenomic PCR [42]. However, our previous analyses failedto detect the gene orf138 in various normal radishes. Thefact that no sequence homologous to orf138 was found inthe current pyrosequencing data (20,646,046 bp in total)from a normal radish suggests that substoichiometricmolecules containing orf138 are absent in normal radish.The discrepancy between our results and a previousstudy [42] can be attributed to the differences of materi-als used and to the conditions of PCR including primers,type of DNA polymerase and particularly amplificationcycles (30 cycles [43,44] vs. 40 cycles in ref. [42]). How-ever, our PCR conditions can amplify two different con-figurations of orf138 regarded as subliminal fragments invarious radishes with Ogura-type cytoplasm. This indi-cates that, if there were orf138 sequences in normal rad-ish, they would be at quite a low level, and thecontribution of SSS as a possible mechanism to explaingeneration of the Ogura-type mitochondrial genomefrom normal-type genome remains to be elucidated.Previously, we conducted a large-scale sequence analysis
of orf138 in 107 Japanese wild radishes, 29 cultivatedradishes and seven R. raphanistrum [43]. On the basis ofthe pattern of mutation and the distribution of orf138variants, we inferred the mechanism behind the differenti-ation of Ogura-type cytoplasm. These studies showed thatthe original Ogura-type radish is derived from Japanesewild radish and that orf138 sequence of Japanese wildradish was introduced from R. raphanistrum [44-46].However, it was still unclear when Ogura-type cytoplasmwas initially generated. This is still an open question, andcomparative sequence analysis among more mitochon-drial genomes in Raphanus and molecular evolutionarystudies of the data should provide clues to resolve thisissue.
ConclusionsThe complete nucleotide sequence of the mitochondrialgenome from Ogura male-sterile radish reveals that thegenome has been highly rearranged compared with thatof normal radish. The radish mitochondrial genome con-sists of 11 syntenic regions. The reorganization of thegenome occurred via a pair of large repeats and multiplepairs of short repeats, and produced four regions uniqueto the Ogura-type mitochondrial genome. Most of theseunique regions consist of sequences homologous to
known Brassicaceae mitochondrial genomes. Insertionsof sequences derived from plastid or nuclear genomewere not identified. This suggested that the uniqueregions were generated by integration and shuffling ofpre-existing mitochondrial sequences during the evolu-tion of Brassicaceae, and novel genes such as orf138could have been created by the shuffling process.
MethodsPlant materialsA cultivar 'Uchiki-Gensuke' (UC-G) and its male-sterileline 'MS-Gensuke' (MS-G) were used in this study.These two lines are in the category of Japanese culti-vated radish (Raphanus sativus var. hortensis) witha long white root. UC-G and MS-G have normalcytoplasm and Ogura-type cytoplasm, respectively. TheMS-G was developed by introducing Ogura-type male-sterile cytoplasm into UC-G and maintained by repeatedbackcrosses.
Mitochondrial DNA extractionMitochondrial DNA was isolated from leaves of MS-Gand UC-G, according to the method of Bonen and Gray[47] using discontinuous (1.15 M, 1.30 M and 1.45 Msucrose) density gradient centrifugation with minormodifications. DNaseI-treated mitochondria were col-lected from the interface between 1.30 M and 1.45 Msucrose. MtDNA was purified by EtBr/CsCl centrifuga-tion and ethanol precipitation.
Sequence assemblyFor normal-type mitochondrial genomePyrosequencing using the GS-FLX system (Roche) andde novo assembly were conducted by Hokkaido SystemScience (Sapporo, Japan). A nucleotide sequence of20,646,046 bp in total was obtained. Sequences wereassembled to 105 contigs (4,414 bp long on average). Onthe basis of the physical map for the normal-type mito-chondrial genome, we connected contigs to develop amaster circle. If contigs had similarity to plant plastidgenome and their depth value was low, we ignored them.All linkages between contigs were confirmed by genomicPCR (Additional file 4 and Additional file 5). By se-quencing genomic-PCR product, we determined thesequences between contigs. Sequences were assembledusing the software Sequencher ver. 4.9 (Gene CodesCorporation, Ann Arbor, MI, USA), and deposited in adatabase (DDBJ) under accession No. AB694743.
For Ogura-type mitochondrial genomePyrosequencing was conducted using the GS-FLX sys-tem (Roche) by Takara-Bio (Ohtsu, Japan). A nucleotidesequence of 12,059,770 bp in total for Ogura-type wasobtained. Sequences were assembled to 147 contigs

Tanaka et al. BMC Genomics 2012, 13:352 Page 11 of 11http://www.biomedcentral.com/1471-2164/13/352
(4,402 bp long on average), using GS De Novo Assem-bler version 2.0 (Roche). The average sequence depth,which was defined as the total nucleotide number usedfor assembly divided by the total length of contigs, was36. On the basis of linkage information of the contigsand the physical map of radish mitochondria, we con-nected contigs to develop a master circle by a parsimo-nious method, so that each contig appeared at least onceto construct the smallest genome [23]. If the depth oflinkage was under 10, we did not use the linkages tomake the master circle. When it was difficult to judgewhether a given contig really appears in the master cir-cle, we checked the existence of the contig in all possibleregions by genomic PCR and sequencing (Additional file 6).Sequences were assembled using the software Sequencherver. 4.9, and deposited in a database (DDBJ) under acces-sion No. AB694744.
Sequence analysisThe genes encoding known mitochondrial proteins andthose for rRNAs, as well as repeated sequences, wereidentified using the Basic Local Alignment Search Tool(http://blast.ncbi.nlm.nih.gov/Blast.cgi). A tRNA genesearch was conducted with the tRNA scan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) [48]. ORFs encodinghypothetical proteins were identified using Getorf soft-ware (http://emboss.dbcls.jp/). ORFs predicted to encodeproteins longer than 100 amino acids were picked up inthe current analysis.
Additional files
Additional file 1: List of repeated sequences in normal-typegenome. Repeated sequences(>80 bp, >90% homology) in normal-typegenome were listed.
Additional file 2: List of repeated sequences in Ogura-typegenome. Repeated sequences(>80 bp, >90% homology) in Ogura-typegenome were listed.
Additional file 3: The sequences homologous to Brassicaceaemitochondrial genome plotted on Ogura-type specific region I -III. Ablack line indicates the region specific to the Ogura-type mitochondrialgenome. Black boxes indicate the sequence homologous to radish and/or other Brassicaceae mitochondrial genomes.
Additional file 4: Validation of contig linkage by PCR analysis. Theprimer information used for this PCR analysis is described in Additionalfile 5.
Additional file 5: The primers used for the PCR analysis to validatethe linkage between contigs of normal-type genome. Information onthe primers used for the PCR analysis in Additional file 4.
Additional file 6: The primers used for PCR analysis to validate theshort repeats. Information on the primers used for PCR and sequencing,in order to confirm short repeats in Ogura-type genome.
AbbreviationsCMS, Cytoplasmic male sterility; mtDNA, Mitochondrial DNA; ORF, Openreading frame; SNP, Single nucleotide polymorphism; SSS, Substoichiometricshifting.
Competing interestsThe authors declare that they have no competing interests.
Authors’ contributionsYT carried out the sequence data analysis, and drafted the manuscript. MTisolated mitocnodrial DNA from radish. KY participated in the sequenceanalysis. HY prepared plant materials. TT supervised the work and edited themanuscript. All authors read and approved the final manuscript.
AcknowledgementThis study was supported in part by Private University Strategic ResearchFoundation Support Program, Grants-in-Aid for Scientific Research, ScientificResearch (B) (No. 22380008), and the Program for Promotion of Basic andApplied Researches for Innovations in Bio-oriented Indurstry (BRAIN), JAPAN.
Received: 14 February 2012 Accepted: 20 July 2012Published: 31 July 2012
References1. Kubo T, Newton KJ: Angiosperm mitochondrial genomes and mutations.
Mitochondrion 2008, 8(1):5–14.2. Palmer JD, Herbo LA: Unicircular structure of the Brassica hirta
mitochondrial genome. Curr Genet 1987, 11(6–7):565–570.3. Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD,
Taylor DR: Rapid Evolution of Enormous, Multichromosomal Genomes inFlowering Plant Mitochondria with Exceptionally High Mutation Rates.PLoS Biol 2012, 10(1):e1001241.
4. Schnable PS, Wise RP: The molecular basis of cytoplasmic male sterilityand fertility restoration. Trends Plant Sci 1998, 3(5):175–180.
5. Hanson MR, Bentolila S: Interactions of mitochondrial and nuclear genesthat affect male gametophyte development. Plant Cell 2004,16(SUPPL):S154–S169.
6. Ogura H: Studies on the new male sterility in Japanese radish, withspecial references to the utilization of this sterility towards the practicalraising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ. 1968, 6:39–78.
7. Sakai T, Imamura J: Intergeneric transfer of cytoplasmic male sterilitybetween Raphanus sativus (cms line) and Brassica napus throughcytoplast-protoplast fusion. Theor Appl Genet 1990, 80(3):421–427.
8. Bannerot H, Boulidard L, Chupeau Y: Unexpected difficulties met with theradish cytoplasm in Brassica oleracea. Eucarpia Cruciferae Newsletter1977, 2:16.
9. Krishnasamy S, Makaroff CA: Organ-specific reduction in the abundance ofa mitochondrial protein accompanies fertility restoration in cytoplasmicmale-sterile radish. Plant Mol Biol 1994, 26(3):935–946.
10. Duroc Y, Gaillard C, Hiard S, Defrance MC, Pelletier G, Budar F: Biochemicaland functional characterization of ORF138, a mitochondrial proteinresponsible for Ogura cytoplasmic male sterility in Brassiceae. Biochimie2005, 87(12):1089–1100.
11. Yang S, Terachi T, Yamagishi H: Inhibition of chalcone synthase expressionin anthers of Raphanus sativus with ogura male sterile cytoplasm. AnnBot 2008, 102(4):483–489.
12. Makaroff CA, Palmer JD: Mitochondrial DNA rearrangements andtranscriptional alterations in the male-sterile cytoplasm of Ogura radish.Mol Cell Biol 1988, 8(4):1474–1480.
13. Makaroff CA, Apel IJ, Palmer JD: The atp6 coding region has beendisrupted and a novel reading frame generated in the mitochondrialgenome of cytoplasmic male-sterile radish. J Biol Chem 1989,264(20):11706–11713.
14. Makaroff CA, Apel IJ, Palmer JD: Characterization of radish mitochondrialatpA: influence of nuclear background on transcription of atpA-associated sequences and relationship with male sterility. Plant Mol Biol1990, 15(5):735–746.
15. Makaroff CA, Apel IJ, Palmer JD: The role of coxI-associated repeatedsequences in plant mitochondrial DNA rearrangements and radishcytoplasmic male sterility. Curr Genet 1991, 19(3):183–190.
16. Krishnasamy S, Makaroff CA: Characterization of the radish mitochondrialorfB locus: Possible relationship with male sterility in Ogura radish. CurrGenet 1993, 24(1–2):156–163.
17. Rankin CT, Cutright MT, Makaroff CA: Characterization of the radishmitochondrial nad3/rps12 locus: Analysis of recombination repeats andRNA editing. Curr Genet 1996, 29(6):564–571.

Tanaka et al. BMC Genomics 2012, 13:352 Page 12 of 11http://www.biomedcentral.com/1471-2164/13/352
18. Handa H: The complete nucleotide sequence and RNA editing content ofthe mitochondrial genome of rapeseed (Brassica napus L.): Comparativeanalysis of the mitochondrial genomes of rapeseed and Arabidopsisthaliana. Nucleic Acids Res 2003, 31(20):5907–5916.
19. Unseld M, Marienfeld JR, Brandt P, Brennicke A: The mitochondrial genomeof Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. NatGenet 1997, 15(1):57–61.
20. Krishnasamy S, Grant RA, Makaroff CA: Subunit 6 of the Fo-ATP synthasecomplex from cytoplasmic male-sterile radish: RNA editing and NH2-terminal protein sequencing. Plant Mol Biol 1994, 24(1):129–141.
21. Satoh M, Kubo T, Nishizawa S, Estiati A, Itchoda N, Mikami T: Thecytoplasmic male-sterile type and normal type mitochondrial genomesof sugar beet share the same complement of genes of known functionbut differ in the content of expressed ORFs. Mol Genet Genomics 2004,272(3):247–256.
22. Liu H, Cui P, Zhan K, Lin Q, Zhuo G, Guo X, Ding F, Yang W, Liu D, Hu S, YuJ, Zhang A: Comparative analysis of mitochondrial genomes between awheat K-type cytoplasmic male sterility (CMS) line and its maintainerline. BMC Genomics 2011, 12:163.
23. Fujii S, Kazama T, Yamada M, Toriyama K: Discovery of global genomic re-organization based on comparison of two newly sequenced ricemitochondrial genomes with cytoplasmic male sterility-related genes.BMC Genomics 2010, 11:209.
24. Palmer JD, Herbon LA: Plant mitochondrial DNA evolved rapidly instructure, but slowly in sequence. J Mol Evol 1988, 28(1–2):87–97.
25. Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD: The mitochondrialgenome of the legume vigna radiata and the analysis of recombinationacross short mitochondrial repeats. PLoS One 2011, 6(1):e16404.
26. Honma Y, Yoshida Y, Terachi T, Toriyama K, Mikami T, Kubo T: Polymorphicminisatellites in the mitochondrial DNAs of Oryza and Brassica. CurrGenet 2011, 57(4):261–270.
27. Rhoads DM, Levings Iii CS, Siedow JN: URF13, a ligand-gated, pore-forming receptor for T-toxin in the inner membrane of cms-Tmitochondria. J Bioenerg Biomembr 1995, 27(4):437–445.
28. Gagliardi D, Leaver CJ: Polyadenylation accelerates the degradation of themitochondrial mRNA associated with cytoplasmic male sterility insunflower. EMBO J 1999, 18(13):3757–3766.
29. Young EG, Hanson MR: A fused mitochondrial gene associated withcytoplasmic male sterility is developmentally regulated. Cell 1987,50(1):41–49.
30. Kim DH, Kang JG, Kim BD: Isolation and characterization of thecytoplasmic male sterility-associated orf456 gene of chili pepper(Capsicum annuum L.). Plant Mol Biol 2007, 63(4):519–532.
31. Tang HV, Chen W, Pring DR: Mitochondrial orf107 transcription, editing,and nucleolytic cleavage conferred by the gene Rf3 are expressed insorghum pollen. Sex Plant Reprod 1999, 12(1):53–59.
32. Wang Z, Zou Y, Li X, Zhang Q, Chen L, Wu H, Su D, Chen Y, Guo J, Luo D,et al: Cytoplasmic male sterility of rice with Boro II cytoplasm is causedby a cytotoxic peptide and is restored by two related PPR motif genesvia distinct modes of mRNA silencing. Plant Cell 2006, 18(3):676–687.
33. L'Homme Y, Stahl RJ, Li XQ, Hameed A, Brown GG: Brassica napcytoplasmic male sterility is associated with expression of a mtDNAregion containing a chimeric gene similar to the pol CMS- associatedorf224 gene. Curr Genet 1997, 31(4):325–335.
34. Landgren M, Zetterstrand M, Sundberg E, Glimelius K: Alloplasmic male-sterile Brassica lines containing B. tournefortii mitochondria express anORF 3′ of the atp6 gene and a 32 kDa protein. Plant Mol Biol 1996,32(5):879–890.
35. Satoh M, Kubo T, Mikami T: The Owen mitochondrial genome in sugarbeet (Beta vulgaris L.): Possible mechanisms of extensiverearrangements and the origin of the mitotype-unique regions. TheorAppl Genet 2006, 113(3):477–484.
36. Terachi T, Yamaguchi K, Yamagishi H: Sequence analysis on themitochondrial orfB locus in normal and Ogura male-sterile cytoplasmsfrom wild and cultivated radishes. Curr Genet 2001, 40(4):276–281.
37. Kuhn K, Weihe A, Borner T: Multiple promoters are a common feature ofmitochondrial genes in Arabidopsis. Nucleic Acids Res 2005,33(1):337–346.
38. Woloszynska M: Heteroplasmy and stoichiometric complexity of plantmitochondrial genomes-though this be madness, yet there's methodin't. J Exp Bot 2010, 61(3):657–671.
39. Janska H, Sarria R, Woloszynska M, Arrieta-Montiel M, Mackenzie SA:Stoichiometric shifts in the common bean mitochondrial genomeleading to male sterility and spontaneous reversion to fertility. Plant Cell1998, 10(7):1163–1180.
40. Feng X, Kaur AP, MacKenzie SA, Dweikat IM: Substoichiometric shifting inthe fertility reversion of cytoplasmic male sterile pearl millet. Theor ApplGenet 2009, 118(7):1361–1370.
41. Chen J, Guan R, Chang S, Du T, Zhang H, Xing H: Substoichiometricallydifferent mitotypes coexist in mitochondrial genomes of Brassica napusl. PLoS One 2011, 6(3).
42. Kim S, Lim H, Park S, Cho KH, Sung SK, Oh DG, Kim KT: Identification of anovel mitochondrial genome type and development of molecularmarkers for cytoplasm classification in radish (Raphanus sativus L.).Theor Appl Genet 2007, 115(8):1137–1145.
43. Yamagishi H, Terachi T: Intra- and inter-specific variations in themitochondrial gene orf138 of Ogura-type male-sterile cytoplasm fromRaphanus sativus and Raphanus raphanistrum. Theor Appl Genet 2001,103(5):725–732.
44. Yamagishi H, Terachi T: Molecular and biological studies on male-sterilecytoplasm in the Cruciferae. IV. Ogura-type cytoplasm found in the wildradish, Raphanus raphanistrum. Plant Breeding 1997, 116(4):323–329.
45. Yamagishi H, Terachi T: Molecular and biological studies on male-sterilecytoplasm in the Cruciferae. 1.The origin and distribution of Ogura male-sterile cytoplasm in Japanese wild radishes (Raphanus sativus L.)revealed by PCR-aided assay of their mitochondrial DNAs. Theor ApplGenet 1994, 87(8):996–1000.
46. Yamagishi H, Terachi T: Molecular and biological studies on male-sterilecytoplasm in the cruciferae. III. Distribution of Ogura-type cytoplasmamong Japanese wild radishes and Asian radish cultivars. Theor ApplGenet 1996, 93(3):325–332.
47. Bonen L, Gray MW: Organization and expression of the mitochondrialgenome of plants. I. The genes for wheat mitochondrial ribosomal andtransfer RNA: Evidence for an unusual arrangement. Nucleic Acids Res1980, 8(2):319–335.
48. Lowe TM, Eddy SR: tRNAscan-SE: A program for improved detection oftransfer RNA genes in genomic sequence. Nucleic Acids Res 1997,25(5):955–964.
49. Stothard P, Wishart DS: Circular genome visualization and explorationusing CGView. Bioinformatics 2005, 21(4):537–539.
doi:10.1186/1471-2164-13-352Cite this article as: Tanaka et al.: A complete mitochondrial genomesequence of Ogura-type male-sterile cytoplasm and its comparativeanalysis with that of normal cytoplasm in radish (Raphanus sativus L.).BMC Genomics 2012 13:352.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit





























