Embed Size (px)
Citation preview

1
Acquired Resistance to Alectinib in ALK-Rearranged Lung Cancer Due to 1
ABCC11/MRP8 Overexpression in a Clinically Paired Resistance Model 2
3
Authors 4
Tomoko Yamamoto Funazo1, Takahiro Tsuji
1, Hiroaki Ozasa
1, Koh Furugaki
2, Yasushi 5
Yoshimura2, Tetsuya Oguri
3, Hitomi Ajimizu
1, Yuto Yasuda
1, Takashi Nomizo
1, Yuichi 6
Sakamori1, Hironori Yoshida
1, Young Hak Kim
1, and Toyohiro Hirai
1. 7
8
Affiliations 9
1Department of Respiratory Medicine, Kyoto University Graduate School of Medicine, 10
Kyoto, Japan. 11
2Product Research Department, Kamakura Research Laboratories, Chugai Pharmaceutical, 12
Kanagawa, Japan. 13
3Department of Education and Research Center for Community Medicine, Nagoya City 14
University Graduate School of Medical Sciences, Nagoya, Japan. 15
16
Running title 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

2
ABCC11/MRP8 May Confer Alectinib Resistance 1
2
Keywords 3
ABCC11, MRP8, alectinib, 4
5
Additional information 6
Corresponding Author: Hiroaki Ozasa, Department of Respiratory Medicine, Kyoto 7
University Graduate School of Medicine, 54, Shogoin-kawaharacho, Sakyo-ku, Kyoto 8
606-8507, Japan. Phone: +81-75-751-3830; Fax: +81-75-751-4643; E-mail: 9
Disclosure of Potential Conflicts of Interest: Tomoko Yamamoto Funazo has received 11
research grant from the Japan Society for the Promotion of Science (JSPS) KAKENHI 12
Grant Number JP18J14330. Hiroaki Ozasa has received research grant from JSPS 13
KAKENHI Grant Number JP19K08601. Tetsuya Oguri reports receiving honoraria from 14
the speakers' bureau of Chugai Pharmaceutical Co. Ltd. Young Hak Kim reports receiving 15
honoraria from the speakers' bureau of Chugai Pharmaceutical Co., Ltd., Pfizer, and 16
Novartis. Toyohiro Hirai reports receiving commercial research grant from Chugai 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

3
Pharmaceutical Co. Ltd. No potential conflicts of interest were disclosed by the other 1
authors. 2
3
Word count: 3482 4
Total No Figures: 4; Tables: 2 5
6
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics 7
Online (http://mct.aacrjournals.org/). 8
9
10
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

4
Abstract 1
Alectinib is used as a first-line treatment for anaplastic lymphoma kinase (ALK)-rearranged 2
non-small cell lung cancer (NSCLC). Whereas other ALK inhibitors have been reported to 3
be involved in resistance to ATP binding cassette (ABC) transporters, no data are available 4
regarding the association between resistance to alectinib and ABC transporters. To 5
investigate whether ABC transporters contribute to alectinib resistance, ABC transporter 6
expression in alectinib-resistant cell lines derived from a patient with ALK-rearranged 7
NSCLC and from H2228 lung cancer cells was evaluated and compared with that in each 8
parent cell type. ATP-binding cassette subfamily C member 11 (ABCC11) expression was 9
significantly increased in alectinib-resistant cell lines compared to that in alectinib-sensitive 10
lines. ABCC11 inhibition increased sensitivity to alectinib in vitro. 11
ABCC11-overexpressing cells were established by transfection of an ABCC11 expression 12
vector into H2228 cells, while control cells were established by transfecting H2228 cells 13
with an empty vector. ABCC11-overexpressing cells exhibited decreased sensitivity to 14
alectinib compared with that of control cells in vitro. Moreover, the tumor growth rate 15
following alectinib treatment was higher in ABCC11-overexpressing cells than that in 16
control cells in vivo. In addition, the intracellular alectinib concentration following 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

5
exposure to 100 nM alectinib was significantly lower in the ABCC11-overexpressing cell 1
line compared with that in control cells. This is the first preclinical evidence showing that 2
ABCC11 expression may be involved in acquired resistance to alectinib. 3
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

6
Introduction 1
In 2007, a novel driver oncogene EML4-ALK fusion gene, in which the echinoderm 2
microtubule-associated protein-like 4 (EML4) gene is fused to the anaplastic lymphoma 3
kinase (ALK) gene, was identified in patients with non-small cell lung cancer (NSCLC) (1, 4
2). Notably, the second-generation ALK inhibitor, alectinib, showed superior efficacy in 5
primary treatment of ALK-positive NSCLC compared with that of crizotinib, a 6
first-generation ALK inhibitor (3, 4). However, although alectinib is used as the first-line 7
treatment for ALK-positive NSCLC, approximately 30% of patients treated with alectinib 8
become refractory within a year of treatment owing to acquired resistance (4). 9
Several mechanisms of resistance to molecular-targeted therapeutic agents have been 10
reported including: mutations of the drug target; activation of bypass signaling pathways; 11
dysregulation of apoptosis; tumor microenvironment including the extracellular matrix, 12
immune and inflammatory cells, and blood vessels; and increased protein expression of 13
drug efflux pumps such as ATP binding cassette (ABC) transporters (5). Previous studies 14
showed that the presence of secondary mutations such as I1171T/N/S, V1180L, L1196M, 15
and G1202R in the kinase domain of ALK conferred resistance to alectinib (6, 7). Another 16
major resistance mechanism involves bypass salvage signaling that maintains downstream 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

7
signaling pathways to activate survival, anti-apoptotic, and proliferation signals. In 1
particular, activation of EGFR has reported to maintain survival signaling that induces 2
alectinib resistance, and we have reported that co-activation of c-Src and MET functions as 3
salvage signaling under alectinib exposure (8, 9). 4
ABC transporters transport various molecules and are involved in multidrug resistance 5
to anticancer agents. In particular, nine ABC transporters, viz., multidrug resistance protein 6
1/ABCB1 (ABCB1), multidrug resistance-associated protein 1 (MRP1)/ABCC1 (ABCC1), 7
MRP2/ABCC2 (ABCC2), MRP3/ABCC3 (ABCC3), MRP4/ABCC4 (ABCC4), 8
MRP5/ABCC5 (ABCC5), MRP7/ABCC10 (ABCC10), MRP8/ABCC11 (ABCC11), and 9
breast cancer resistance protein/ABCG2 (ABCG2) have been reported to be associated with 10
anticancer drug resistance (10, 11). Moreover, several tyrosine kinase inhibitors (TKIs) 11
(e.g., imatinib, nilotinib, dasatinib, ponatinib, gefitinib, erlotinib, lapatinib, vandetanib, 12
sunitinib, and sorafemib) were recently reported as substrates for ABC transporters 13
including ABCB1, ABCC1, ABCG2, and ABCC10 (12). Notably, overexpression of 14
ABCB1 was shown to be involved in the resistance to crizotinib and ceritinib, which 15
constitute other ALK-TKIs, but not to alectinib (13). In addition, it was demonstrated that 16
alectinib is not a substrate for ABCB1 and ABCG2 (14, 15). Thus, unlike other TKIs, the 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

8
association between alectinib resistance and ABC transporter expression is not fully 1
understood. Therefore, the aim of the present study was to determine the ABC transporters 2
that are associated with alectinib resistance in lung cancer. 3
We previously reported the establishment of a clinical paired resistant model (CRPM) 4
comprised of patient-derived ALK-rearranged NSCLC cell lines from a treatment-naive and 5
subsequently, alectinib-refractory patient (9). In the present study, we evaluated the 6
expression of the nine chemoresistance-associated ABC transporters in these cell lines, 7
identifying that ABCC11 expression was significantly increased in alectinib-resistant cells 8
compared with that in alectinib-sensitive cells. Therefore, in the present study we 9
investigated whether increased ABCC11 expression may constitute a mechanism 10
underlying the acquired resistance to alectinib. 11
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

9
Materials and Methods 1
Clinical information and procedures for obtaining informed consent 2
The study protocol was prepared in accordance with the Declaration of Helsinki and 3
approved by the Kyoto University Graduate School and Faculty of Medicine Ethics 4
Committee (certification number: R0996). The patient provided written informed consent 5
to participate in this study. 6
7
Establishment of patient-derived lung cancer cells 8
KTOR1 and KTOR1-RE (EML4-ALK variant 1 E13; A20) cells were established from 9
a 29-year-old female patient with ALK-rearranged NSCLC, who regularly visited Kyoto 10
University Hospital. The method used to establish the patient-derived cells was described 11
previously (9). Briefly, 200 mL of pleural effusion was obtained from the patient with 12
ALK-rearranged lung cancer. Tumor cells were separated by centrifugation at 800 rpm for 5 13
min, and then cultured and maintained in alectinib-free RPMI 1640 medium (Nacalai 14
Tesque) supplemented with 8% heat-inactivated fetal bovine serum (Sigma-Aldrich) and 15
1% penicillin/streptomycin (Gibco) at 37.0°C in 5% CO2. Information about the patient 16
was obtained from electrical medical records at Kyoto University Hospital. 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

10
1
Cells and reagents 2
NCI-H2228 (EML4-ALK variant 3a/b E6; A20) lung cancer cells were purchased in 3
2016 from the American Type Culture Collection. The alectinib-resistant H2228-AR1S 4
cells were established by exposing NCI-H2228 cells to 300 nM alectinib for three months 5
in vitro. Similarly, KTOR1-AR was established from KTOR1 via exposure to 30 nM 6
alectinib for six months in vitro. All experiments were performed with cells that were 7
within 20 passages. In 2019, all cells were confirmed to be negative for mycoplasma using 8
the MycoAlert Mycoplasma Detection Kit (Lonza). Alectinib was kindly provided by 9
Chugai Pharmaceutical Co., Ltd. Crizotinib and ceritinib were purchased from LC 10
Laboratories (Woburn), and lorlatinib and brigatinib were purchased from ChemScene 11
(Monmouth Junction). Alectinib, crizotinib, ceritinib, and lorlatinib were dissolved in 12
dimethyl sulfoxide (DMSO) (Nacalai Tesque) at a concentration of 5 mmol/L and brigatinib 13
was dissolved in DMSO at a concentration of 0.5 mmol/L. DMSO was also used as a 14
vehicle control. 15
16
Cell viability and drug susceptibility assay 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

11
Cells were cultured overnight in 96-well plates at a density of 5000 cells/well and 1
treated with increasing concentrations of the reagents or vehicle control medium for 72–168 2
h. Subsequently, cells were incubated with CellTiter-Glo 2.0 Luminescent Cell Viability 3
Assay (Promega) for 20 min and luminescence was measured using an ARVO X3 plate 4
reader (PerkinElmer). The 50% inhibitory concentration (IC50) value was calculated using a 5
nonlinear regression model with a sigmoidal dose response using GraphPad Prism 8.0 6
(GraphPad software). Three independent experiments were conducted. 7
8
Immunoblotting 9
SDS-PAGE and immunoblotting were performed as described previously (16). Briefly, 10
antibodies against ABCC11 and GAPDH were purchased from Invitrogen. pALK (pY 11
1604) and secondary antibodies were purchased from Cell Signaling Technology. β-actin 12
was purchased from Sigma-Aldrich (details are provided in Supplementary Table S1). 13
Antibodies against ABCC11 (1:2000), GAPDH (1: 10,000), pALK (1:1000), and β-actin (1: 14
10,000), and secondary (1: 2000) antibodies were dissolved in 2.5% bovine serum albumin 15
(BSA)/Tris-buffered saline with Tween 20. BSA was purchased from Nacalai Tesque. 16
Quantification of protein bands was performed using ImageJ software 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

12
(https://imagej.nih.gov/ij/). 1
2
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) 3
Total RNA was extracted from cultured cells using the PureLink RNA mini kit (Thermo 4
Fisher Scientific). Gene expression was evaluated by qRT-PCR using 100 ng of total RNA, 5
the TaqMan RNA-to-Ct 1-Step Kit (Applied Biosystems), and a primer pair. A list of the 6
primers purchased from Thermo Fisher Scientific is shown in Supplementary Table S2. 7
Reactions were quantified using the 7300 Real-Time PCR System (Applied Biosystems). 8
9
Transfection with siRNA 10
ABCC11 siRNA oligonucleotides were purchased from Thermo Fisher Scientific 11
(Stealth RNAi siRNA; Supplementary Table S3). A total of 3.0 × 105 cells were transfected 12
with siRNA oligonucleotides at a final RNA concentration of 21 nM using Lipofectamine 13
RNAiMAX Transfection Reagent (Invitrogen). The reverse transfection method was 14
performed according to the manufacturer's protocol. RNA and total protein were extracted 15
at 30 and 54 h, respectively, following transfection. In the cell viability assay, transfected 16
cells (5000 cells/well) were cultured in a 96-well plate for 24 h and treated with increasing 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

13
concentrations of alectinib for 168 h. Cell viability was assessed as described above. 1
2
Establishment of ABCC11-overexpressing H2228 cells 3
The ABCC11 expression vector (ABCC11/pcDNA3.1/V5-His) and empty vector 4
(pcDNA3.1/V5-His C) were purchased from Invitrogen. The vector map of 5
ABCC11/pcDNA3.1/V5-His is shown in Supplementary Fig. S1; the ABCC11 sequence 6
inserted into the vector is shown in Supplementary file 2. H2228 cells were transfected with 7
the ABCC11 expression vector or empty vector using NEPA21 electroporator (Nepa Gene) 8
in serum-free Opti-MEM (Thermo Fisher Scientific) to generate stably expressing cells. At 9
24 h after transfection, cells were selected and cloned by limiting dilution in a medium 10
containing geneticin selective antibiotic (Wako) for two to three weeks. Proteins were 11
extracted from each clone and ABCC11 expression was analyzed by immunoblotting. Once 12
increased expression of ABCC11 was confirmed, two cell lines derived from the transfected 13
cells were selected as ABCC11-overexpressing cells (ABCC11a and ABCC11b). In the 14
same manner, cells transfected with the empty vector were cloned and one cell line that 15
exhibited similar ABCC11 expression as the parent cells was selected as the control cell 16
line (mock). 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

14
1
Intracellular concentration measurement 2
Cells were cultured overnight in a low attachment 24-well plate (Greiner Bio-One) at a 3
density of 2.5 × 105 cells/well and treated with 100 nM alectinib for 2 and 4 h. After 4
washing the cells twice with cold PBS, cells were lysed with 50 μL of 0.1 mol/L sodium 5
hydroxide solution (Nacalai Tesque) and homogenized. After incubating the cells at 37°C 6
for 1 h, cells were neutralized with 50 μL of 0.1 mol/L hydrochloric acid solution (Nacalai 7
Tesque). Alectinib concentration was measured using a Shimadzu Prominence HPLC 8
System (MD) and Qtrap 5500 quadrupole mass spectrometer (AB Sciex). Mass 9
spectrometry analysis was performed using electrospray ionization in positive ion mode. 10
The multiple reaction monitoring mode m/z transition values were set at 483.22–396.10 for 11
alectinib and 491.30–396.10 for d-alectinib as the internal standard. 12
13
Xenograft models 14
Six-week-old female BALB/c-nu mice (CAnN.Cg-Foxn1nu/CrlCrlj) were purchased 15
from Charles River Laboratories, Japan. Xenograft models were generated by suspending 16
1–3 × 106 cells in Matrigel (Corning), which was injected subcutaneously into the backs of 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

15
the mice. Mice with a tumor volume of 80–240 mm3 were randomly assigned to either 1
alectinib or vehicle groups (day 0). Mice were treated with alectinib (8 mg/kg/day) or 2
vehicle via oral gavage. Tumor volumes were evaluated using digital caliper measurements 3
and calculated using the formula (length × width × width) × 0.52, where length and width 4
represented the larger and smaller diameters, respectively. The mice were euthanized on 5
day 11. For immunoblotting, mice with a tumor volume of 500-1500 mm3 were treated with 6
alectinib (8 mg/kg/day) and were humanely sacrificed 24 hours after alectinib treatment. 7
Tumor tissues were collected, and immunoblotting was performed as mentioned above. All 8
animal experiments and protocols were approved by the Animal Research Committee of 9
Kyoto University (ID: MedKyo 18298) and implemented according to the ARRIVE 10
guidelines. 11
12
Statistical analysis 13
Continuous variable data are expressed as the means ± SEM. The significance of 14
differences was assessed using the Student’s t test. Sidak's multiple comparison test and 15
Holm–Sidak's multiple comparison test were used to compare the mean of more than three 16
groups. P-values <0.05 were defined as significant. All statistical analysis was performed 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

16
using JMP Pro version 12.0 (SAS Institute) and visualized by GraphPad Prism 8. 1
2
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

17
Results 1
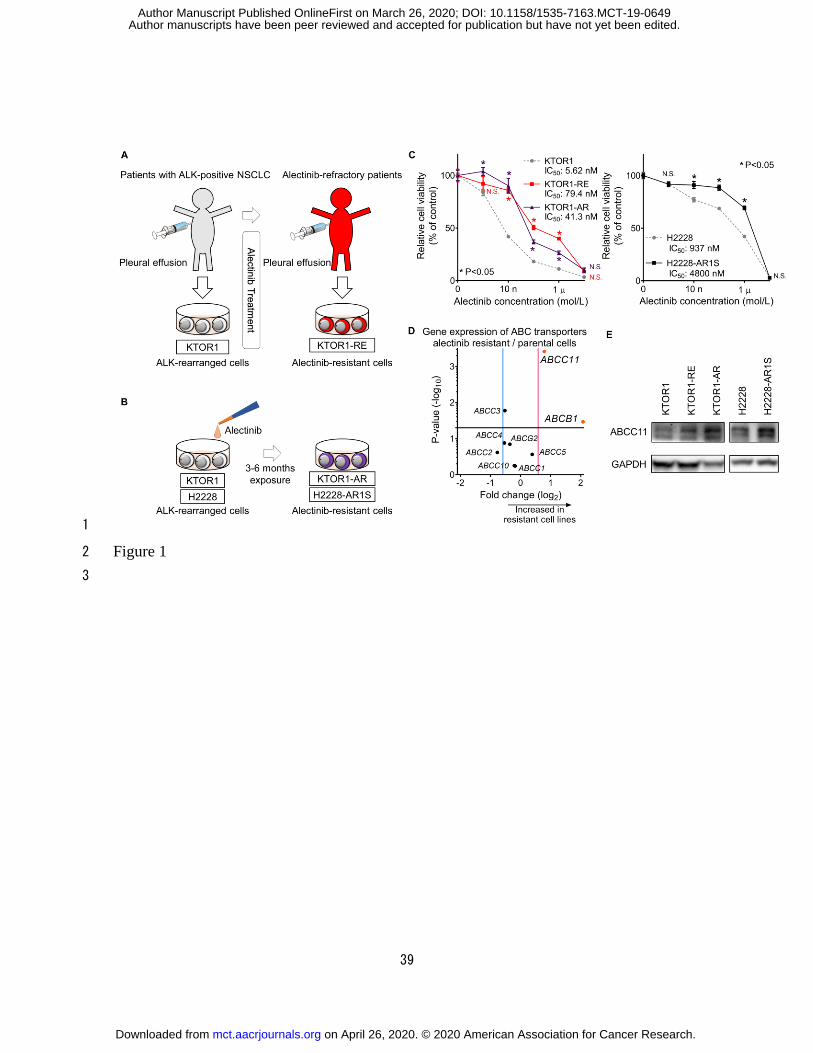
Establishment of alectinib-resistant cells 2
We established KTOR1 cells using cells derived from an alectinib-naïve patient with 3
ALK-rearranged NSCLC, and KTOR1-RE cells were established from the same 4
subsequently alectinib-refractory patient (9) (Fig. 1A). The H2228-AR1S and KTOR1-AR 5
cells were established from the H2228 and KTOR1 cells, respectively, through exposure to 6
alectinib to investigate the determinants of acquired resistance to alectinib in lung cancer. 7
KTOR1-RE, KTOR1-AR, and H2228-AR1S cells were more resistant to alectinib 8
compared with the parental cells (Fig. 1B). No secondary mutations (e.g., I1171N, V1180L, 9
L1196M, or G1202R) were detected in the ALK tyrosine kinase-coding regions of 10
KTOR1-RE, KTOR1-AR, or H2228-AR1S cells (9) (Supplementary Fig. S2) as determined 11
by ALK exon sequencing using ALK sequencing primers (Supplementary Table S4). The 12
IC50 values for alectinib treatment of KTOR1-RE, KTOR1-AR, and H2228-AR1S revealed 13
that the cells were approximately 14.1, 7.35, and 5.12-fold more resistant relative to the 14
parental cells, respectively (Fig. 1C, Table 1). We have previously reported that the IC50 of 15
H2228 is approximately 100 nM (9); notably, this was determined under low attachment 16
conditions, under which H2228 cells tend to exhibit greater sensitivity to alectinib 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

18
(Supplementary Fig. S3). In the present study, we continued the experiments under 1
attachment conditions to facilitate siRNA knockdown experiments. 2
3
Expression levels of ABC transporters in alectinib-resistant cells 4
To identify transporters that conferred resistance to alectinib, ABC transporters 5
exhibiting increased expression in alectinib-resistant cells were explored. In particular, gene 6
expression of the nine ABC transporters previously reported to be related to anticancer drug 7
resistance was screened in the five cell types, and the expression ratio (resistant 8
cells/parental cells) of the transporters was calculated (10, 11, 12). For each ABC 9
transporter, the fold change in gene expression (horizontal line) and significance between 10
parental and alectinib- resistant cells calculated using the t-test [−log(P-value); vertical line] 11
was plotted (Fig. 1D; Supplementary Fig. S4). Among the transporters, the expression of 12
ABCB1 and ABCC11 genes was significantly increased in the resistant cells (fold change 13
>1.5, P < 0.05). We focused on ABCC11 as a potential transporter candidate for alectinib 14
resistance, as previous studies suggested that ABCB1 was not involved in the extracellular 15
efflux of alectinib (14,15). 16
17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

19
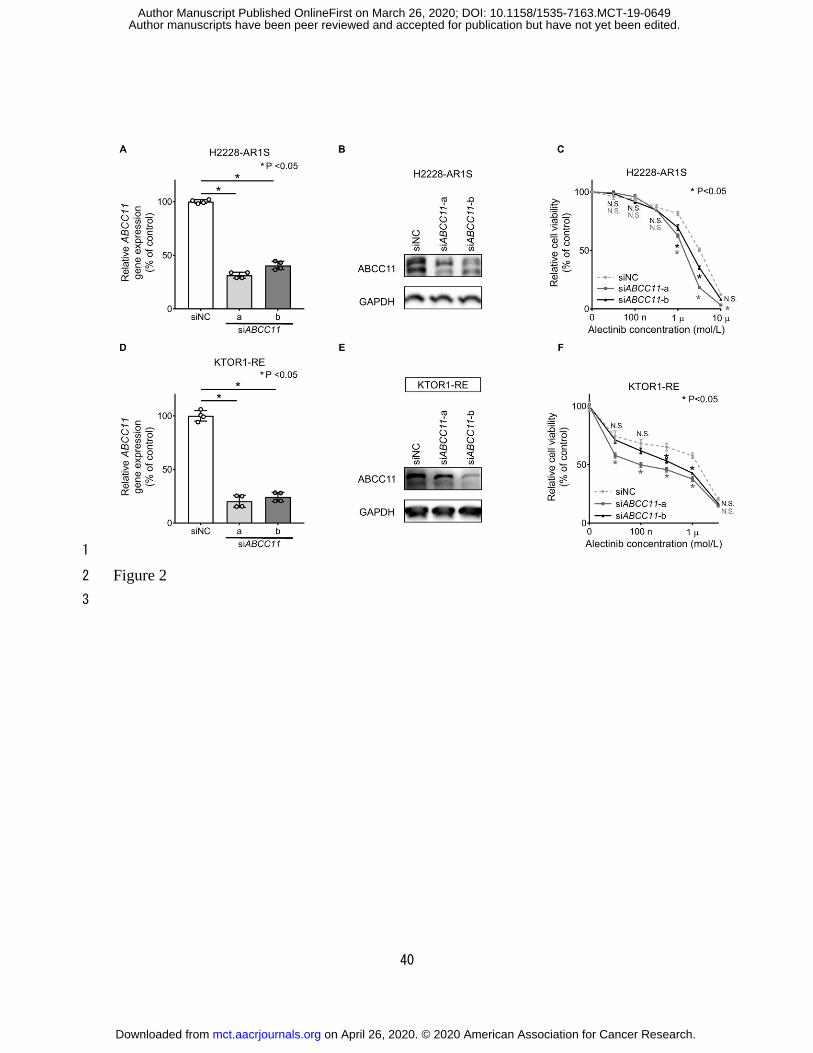
ABCC11 inhibition sensitizes H2228-AR1S and KTOR1-RE cells to alectinib 1
Cell viability in the presence of alectinib during siRNA inhibition of the ABCC11 gene 2
was measured to evaluate the functional role of ABCC11 overexpression in alectinib 3
resistance. H2228-AR1S cells transfected with siRNA targeting the ABCC11 gene exhibited 4
reduced cell viability of alectinib compared with that of cells transfected with negative 5
control siRNA (Fig. 2C; Supplementary Table S5). KTOR1-RE cells transfected with 6
siRNA targeting the ABCC11 gene also demonstrated reduced cell viability following 7
alectinib exposure (Fig. 2F; Supplementary Table S6). 8
9
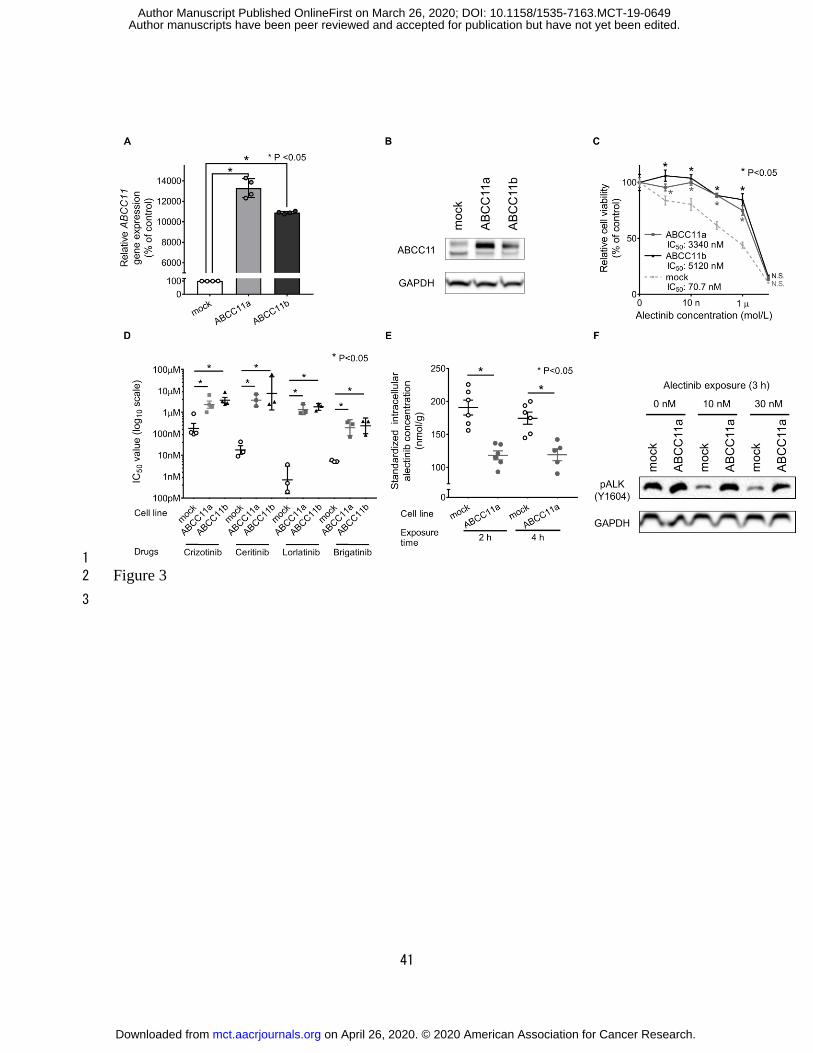
ABCC11 overexpression confers alectinib resistance 10
To investigate whether ABCC11 overexpression induced alectinib resistance, the 11
ABCC11 expression vector (ABCC11/pcDNA3.1/V5-His) was transfected into H2228 cells 12
to establish two ABCC11-overexpressing cell lines, ABCC11a and ABCC11b. H2228 cells 13
were also transfected with an empty vector (mock). Expression levels of the ABCC11 gene 14
and protein were increased in both ABCC11a and ABCC11b cells compared with those in 15
mock cells (Fig. 3A and B). The negative impact of alectinib on cell viability was decreased 16
in ABCC11a and ABCC11b cells compared with that on mock cells (Fig. 3C, Table 2). The 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

20
loss of cell viability from crizotinib, ceritinib, lorlatinib, and brigatinib exposure was also 1
significantly lower in ABCC11a and ABCC11b cells compared with that in mock cells (Fig. 2
3D). 3
4
ABCC11 overexpression decreases the intracellular alectinib concentration 5
To obtain evidence supporting that ABCC11 effluxes alectinib, we measured the 6
intracellular accumulation of alectinib in ABCC11a or mock cells using liquid 7
chromatography-mass spectrometry. Following treatment with 100 nM alectinib for 2 and 4 8
hours, the intracellular concentration of alectinib was significantly lower in ABCC11a cells 9
compared with that in mock cells (Fig. 3E; Supplementary Fig. S5). To evaluate the effect 10
of alectinib treatment on ABCC11-overexpressing cells, ALK phosphorylation was 11
evaluated by immunoblotting in cells treated with increasing concentrations of alectinib. 12
Treatment of cells with 10 and 30 nM alectinib suppressed ALK phosphorylation in mock 13
but not ABCC11a cells (Fig. 3F). 14
15
In vivo effect of ABCC11 overexpression on alectinib response 16
To determine whether ABCC11 overexpression might alter sensitivity to alectinib in 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

21
vivo, xenograft models using H2228 cells transfected with the ABCC11 expression vector 1
and cells transfected with empty vector were established and randomized into two groups: 2
alectinib treatment and vehicle. The dosage of alectinib was selected as 8 mg/kg (10 days) 3
in accordance with the human dose (600 mg/day/body) (17). The tumor growth rate 4
following alectinib treatment was higher with H2228 cells transfected with ABCC11 5
expression vector compared with that from cells containing empty vector (−34% for 6
ABCC11a, −24% for ABCC11b, and −77% for mock; Fig. 4A; Supplementary Fig. S6) and 7
did not affect body weight (Fig. 4B). ALK phosphorylation after alectinib treatment in 8
tumors established from ABCC11a and ABCC11b cells was not suppressed ,but those in 9
mock cells was suppressed (Fig. 4C). These results suggested that ABCC11 overexpression 10
could reduce the tumor response to alectinib treatment in vivo. 11
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

22
Discussion 1
The results of the present study showed that ABCC11 expression was increased in 2
alectinib-resistant cells compared with that in the parental cells. We also revealed that 3
altered ABCC11 expression affected the cell viability following alectinib exposure. This is 4
the first report to demonstrate that ABCC11 overexpression may represent a mechanism 5
involved in acquired resistance to alectinib. 6
ABC transporters transport various molecules from the interior to the exterior of the cell. 7
ABCC11 was identified as an ABC transporter in 2001 (18-20) and is expressed in various 8
tissues including the brain, lung, liver, kidney, and apocrine glands (21-24). ABCC11 is 9
also involved in the intracellular to extracellular efflux of dehydroepiandrosterone 3-sulfate, 10
methotrexate, and tenofovir (25, 26), and is a substrate for cytotoxic anticancer agents 11
including 5-fluorouracil and pemetrexed (19, 20, 27). However, to the best of our 12
knowledge, molecular targeted therapeutics have not previously been described as potential 13
substrates for ABCC11. In the present study, we showed that H2228 cells transfected with 14
an ABCC11 expression vector exhibited a lower intracellular alectinib concentration 15
compared with that of cells transfected with empty vector, and exposure to alectinib 16
suppressed ALK phosphorylation in cells transfected with empty vector but not in cells 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

23
transfected with the ABCC11 expression vector. These data suggested that ABCC11 may be 1
involved in intracellular to extracellular alectinib efflux (Fig. 4D). 2
In the present study, ABCC11 expression was increased in alectinib-resistant cells 3
relative to the level in their parental cells. The cell viability of H2228-AR1S cells 4
transfected with siRNA targeting ABCC11 treated with alectinib was significantly 5
decreased compared with that of cells transfected with negative control siRNA, whereas 6
sensitivity to alectinib in H2228 cells transfected with the ABCC11 expression vector was 7
lower compared with that of cells transfected with empty vector. In addition, ABCC11 8
expression was significantly lower in cells derived from the alectinib-naïve patient than in 9
those from the same alectinib-refractory patient. These results suggested that increased 10
expression of ABCC11 may be involved in acquired resistance to alectinib. 11
However, the clinical relevance of ABCC11 in alectinib resistance remains unclear. 12
ABCC11 expression in other patients treated with alectinib could not be evaluated. 13
Although several antibodies were evaluated to assess ABCC11 expression in tumors using 14
immunostaining, no useful antibodies could be obtained. We evaluated ABC transporters 15
using a clinically paired resistance model (CPRM) that compares patient-derived resistant 16
cells with patient-derived treatment-sensitive cells to provide information regarding 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

24
acquired resistance. The present study revealed that ABCC11 expression was increased in 1
KTOR1-RE compared with KTOR1 cells. Although the relative intracellular alectinib 2
concentration in these cells was not evaluated, ALK phosphorylation was upregulated in 3
KTOR1-RE cells compared with that in KTOR1 cells following treatment with 20 nM 4
alectinib (Supplementary Fig. S7). This result suggested that the intracellular alectinib 5
concentration of KTOR1-RE cells might be lower than that of KTOR1 cells following 6
alectinib exposure. Moreover, inhibition of ABCC11 in KTOR1-RE cells increased 7
sensitivity to alectinib (Fig. 2F). These results indicated that increased ABCC11 expression 8
may contribute to efflux the alectinib from the interior to the exterior of the cells in the 9
evaluated patient. Further studies are required to evaluate whether high expression of 10
ABCC11 may be associated with acquired resistance to alectinib in other patients. We aim 11
to continue to accumulate evidence to support our model using CPRM. 12
ABC transporters are known to cause multidrug resistance (12, 19, 20). In the present 13
study, the sensitivity of crizotinib, ceritinib, lorlatinib, and brigatinib was lower in 14
H2228-AR1S cells compared with that in H2228 cells (Supplementary Fig. S8). These 15
results revealed that H2228-AR1S cells exhibit cross-resistance to other ALK inhibitors. In 16
addition, as with alectinib, the cell viability impairment upon exposure to ALK inhibitors 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

25
was decreased in H2228 cells transfected with the ABCC11 expression vector compared 1
with that of cells transfected with empty vector (Fig. 3D). Although we were unable to 2
measure the intracellular concentration of ALK inhibitors other than alectinib in cells 3
transfected with the ABCC11 expression vector, these findings suggest that ABCC11 4
expression may be involved in the acquired resistance to multiple ALK inhibitors. 5
As we were unable to clarify the mechanism underlying ABCC11 overexpression in 6
resistant cells, it is unclear whether exposure to alectinib selects cells with high expression 7
of ABCC11 prior to treatment or induces increased expression of ABCC11 in these cells. 8
Previous studies showed that cancer stem cells (CSCs), which are innately resistant to 9
many standard therapies, are associated with overexpression of ABCB1 and ABCG2 (5, 10, 10
28, 29). In particular, CD44 constitutes a CSC marker that exhibits strong negative 11
correlations with patient survival and has been associated with the expression of ABC 12
transporters, most notably ABCG2 (5, 28). ABCB1 overexpression was also reported to be 13
associated with short survival in stage 1 lung adenocarcinoma and rendered CSC-like 14
properties (30, 31). These reports indicated that cancer cells may highly express ABC 15
transporters prior to treatment and that clonal selection of cells with high expression of 16
ABC transporters might be associated with acquired resistance to anticancer agents. 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

26
Although an association between ABCC11 and CSC-like properties has not been reported, 1
clonal selection of ALK fusion gene-positive lung cancer cells with ABCC11 2
overexpression may represent a mechanism of acquired resistance to alectinib. 3
Overall, our findings provide new insight into the mechanisms underlying alectinib 4
resistance in ALK-rearranged NSCLC. We found that ABCC11 expression was 5
significantly increased in cells derived from an alectinib-refractory patient using CPRM. 6
We propose that alectinib may constitute a substrate for ABCC11 and that overexpression 7
of ABCC11 may represent an important determinant of acquired resistance to alectinib, 8
although other resistance mechanisms, including activation of salvage signaling pathways, 9
may also be involved. Further studies are required to elucidate the mechanisms of alectinib 10
resistance using CPRM to overcome the problem of acquired resistance in lung cancer. 11
12
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

27
Acknowledgments 1
The authors would like to thank Editage (https://www.editage.jp/) for the English language 2
review. 3
4
Author contributions 5
Conception and design: T. Y. Funazo, T. Tsuji, H. Ozasa 6
Development of methodology: T. Tsuji, H. Ozasa 7
Acquisition of data: T. Y. Funazo, T. Tsuji, H. Ozasa, K. Furugaki, Y. Yoshimura, H 8
Ajimizu, Y.H. Kim, 9
Analysis and interpretation of data: T. Y. Funazo, T. Tsuji, H. Ozasa, Y. Yasuda 10
Writing, review, and/or revision of the manuscript: T. Y. Funazo, T. Tsuji, H. Ozasa, K. 11
Furugaki, Y. Yoshimura, T. Oguri, Y. Yasuda, T Nomizo, Y Sakamori, H Yoshida, T. 12
Hirai. 13
Administrative, technical, and material support: T. Y. Funazo, T. Tsuji, H. Ozasa, H 14
Ajimizu, Y. Yasuda 15
Study supervision: H. Ozasa, T. Oguri, T. Hirai 16
17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

28
Reference 1
1. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification 2
of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 3
2007;448(7153):561-6 doi 10.1038/nature05945. 4
2. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma 5
kinase inhibition in non-small-cell lung cancer. The New England journal of medicine 6
2010;363(18):1693-703 doi 10.1056/NEJMoa1006448. 7
3. Nishio M, Nakagawa K, Mitsudomi T, Yamamoto N, Tanaka T, Kuriki H, et al. Analysis 8
of central nervous system efficacy in the J-ALEX study of alectinib versus crizotinib 9
in ALK-positive non-small-cell lung cancer. Lung cancer (Amsterdam, Netherlands) 10
2018;121:37-40 doi 10.1016/j.lungcan.2018.04.015. 11
4. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus 12
Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. The New England 13
journal of medicine 2017;377(9):829-38 doi 10.1056/NEJMoa1704795. 14
5. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an 15
evolving paradigm. Nature Reviews Cancer 2013;13:714 doi 10.1038/nrc3599. 16
6. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular 17
Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in 18
ALK-Rearranged Lung Cancer. Cancer discovery 2016;6(10):1118-33 doi 19
10.1158/2159-8290.Cd-16-0596. 20
7. Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, et al. Two novel 21
ALK mutations mediate acquired resistance to the next-generation ALK inhibitor 22
alectinib. Clinical cancer research : an official journal of the American Association 23
for Cancer Research 2014;20(22):5686-96 doi 10.1158/1078-0432.Ccr-14-1511. 24
8. Tani T, Yasuda H, Hamamoto J, Kuroda A, Arai D, Ishioka K, et al. Activation of EGFR 25
Bypass Signaling by TGFalpha Overexpression Induces Acquired Resistance to Alectinib 26
in ALK-Translocated Lung Cancer Cells. Molecular cancer therapeutics 27
2016;15(1):162-71 doi 10.1158/1535-7163.Mct-15-0084. 28
9. Tsuji T, Ozasa H, Aoki W, Aburaya S, Funazo T, Furugaki K, et al. Alectinib Resistance 29
in ALK-Rearranged Lung Cancer by Dual Salvage Signaling in a Clinically Paired 30
Resistance Model. Molecular cancer research : MCR 2019;17(1):212-24 doi 31
10.1158/1541-7786.Mcr-18-0325. 32
10. Dean M. ABC transporters, drug resistance, and cancer stem cells. Journal of mammary 33
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

29
gland biology and neoplasia 2009;14(1):3-9 doi 10.1007/s10911-009-9109-9. 1
11. Kathawala RJ, Gupta P, Ashby CR, Jr., Chen ZS. The modulation of ABC 2
transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug 3
resistance updates : reviews and commentaries in antimicrobial and anticancer 4
chemotherapy 2015;18:1-17 doi 10.1016/j.drup.2014.11.002. 5
12. Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L. Overcoming ABC 6
transporter-mediated multidrug resistance: The dual role of tyrosine kinase 7
inhibitors as multitargeting agents. European journal of medicinal chemistry 8
2017;142:271-89 doi 10.1016/j.ejmech.2017.07.062. 9
13. Katayama R, Sakashita T, Yanagitani N, Ninomiya H, Horiike A, Friboulet L, et al. 10
P-glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma 11
Kinase-rearranged Non-small Cell Lung Cancer. EBioMedicine 2016;3:54-66 doi 12
10.1016/j.ebiom.2015.12.009. 13
14. Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity 14
of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer 15
chemotherapy and pharmacology 2014;74(5):1023-8 doi 10.1007/s00280-014-2578-6. 16
15. Yang K, Chen Y, To KK, Wang F, Li D, Chen L, et al. Alectinib (CH5424802) antagonizes 17
ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. 18
Experimental & molecular medicine 2017;49(3):e303 doi 10.1038/emm.2016.168. 19
16. Ozasa H, Oguri T, Maeno K, Takakuwa O, Kunii E, Yagi Y, et al. Significance of c-MET 20
overexpression in cytotoxic anticancer drug-resistant small-cell lung cancer cells. 21
Cancer science 2014;105(8):1032-9 doi 10.1111/cas.12447. 22
17. Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) 23
for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP 24
study): a single-arm, open-label, phase 1-2 study. The Lancet Oncology 25
2013;14(7):590-8 doi 10.1016/s1470-2045(13)70142-6. 26
18. Gottesman MM. Mechanisms of Cancer Drug Resistance. 2002;53(1):615-27 doi 27
10.1146/annurev.med.53.082901.103929. 28
19. Oguri T, Bessho Y, Achiwa H, Ozasa H, Maeno K, Maeda H, et al. MRP8/ABCC11 directly 29
confers resistance to 5-fluorouracil. Molecular cancer therapeutics 2007;6(1):122-7 30
doi 10.1158/1535-7163.Mct-06-0529. 31
20. Uemura T, Oguri T, Ozasa H, Takakuwa O, Miyazaki M, Maeno K, et al. ABCC11/MRP8 confers 32
pemetrexed resistance in lung cancer. Cancer science 2010;101(11):2404-10 doi 33
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

30
10.1111/j.1349-7006.2010.01690.x. 1
21. Bera TK, Lee S, Salvatore G, Lee B, Pastan I. MRP8, a new member of ABC transporter 2
superfamily, identified by EST database mining and gene prediction program, is highly 3
expressed in breast cancer. Molecular medicine (Cambridge, Mass) 2001;7(8):509-16. 4
22. Bortfeld M, Rius M, Konig J, Herold-Mende C, Nies AT, Keppler D. Human multidrug 5
resistance protein 8 (MRP8/ABCC11), an apical efflux pump for steroid sulfates, is 6
an axonal protein of the CNS and peripheral nervous system. Neuroscience 7
2006;137(4):1247-57 doi 10.1016/j.neuroscience.2005.10.025. 8
23. Yabuuchi H, Shimizu H, Takayanagi S, Ishikawa T. Multiple splicing variants of two 9
new human ATP-binding cassette transporters, ABCC11 and ABCC12. Biochemical and 10
biophysical research communications 2001;288(4):933-9 doi 10.1006/bbrc.2001.5865. 11
24. Toyoda Y, Takada T, Gomi T, Nakagawa H, Ishikawa T, Suzuki H. Clinical and Molecular 12
Evidence of ABCC11 Protein Expression in Axillary Apocrine Glands of Patients with 13
Axillary Osmidrosis. International journal of molecular sciences 2017;18(2) doi 14
10.3390/ijms18020417. 15
25. Chen ZS, Guo Y, Belinsky MG, Kotova E, Kruh GD. Transport of bile acids, sulfated 16
steroids, estradiol 17-beta-D-glucuronide, and leukotriene C4 by human multidrug 17
resistance protein 8 (ABCC11). Molecular pharmacology 2005;67(2):545-57 doi 18
10.1124/mol.104.007138. 19
26. Tun-Yhong W, Chinpaisal C, Pamonsinlapatham P, Kaewkitichai S. Tenofovir Disoproxil 20
Fumarate Is a New Substrate of ATP-Binding Cassette Subfamily C Member 11. 21
Antimicrobial agents and chemotherapy 2017;61(4) doi 10.1128/aac.01725-16. 22
27. Oba T, Izumi H, Ito KI. ABCB1 and ABCC11 confer resistance to eribulin in breast cancer 23
cell lines. Oncotarget 2016;7(43):70011-27 doi 10.18632/oncotarget.11727. 24
28. Bhatavdekar JM, Patel DD, Chikhlikar PR, Trivedi TI, Gosalia NM, Ghosh N, et al. 25
Overexpression of CD44: a useful independent predictor of prognosis in patients with 26
colorectal carcinomas. Annals of surgical oncology 1998;5(6):495-501. 27
29. Shervington A, Lu C. Expression of multidrug resistance genes in normal and cancer 28
stem cells. Cancer investigation 2008;26(5):535-42 doi 10.1080/07357900801904140. 29
30. Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, et al. Acquired resistance to 30
EGFR inhibitors is associated with a manifestation of stem cell-like properties in 31
cancer cells. Cancer research 2013;73(10):3051-61 doi 10.1158/0008-5472.Can-12-4136. 32
31. Zou F, Seike M, Noro R, Kunugi S, Kubota K, Gemma A. Prognostic significance of ABCB1 33
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

31
in stage I lung adenocarcinoma. Oncology letters 2017;14(1):313-21 doi 1
10.3892/ol.2017.6145. 2
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

32
Table 1. IC50 values for alectinib treatment of H2228, H2228-AR1S, KTOR1, KTOR1-RE, 1
and KTOR1-AR cells. 2
3
Cells H2228 H2228-AR1S KTOR1 KTOR1-RE KTOR1-AR
Alectinib
IC50 (95% CI)
IC50 (95% CI) RR
IC50 (95% CI) IC50 (95% CI) RR
IC50 (95% CI) RR
(nmol/L)
937 (577–1450)
4800 (3340–7900) 5.12
5.62 (4.27–7.36) 79.4 (35.0–185) 14.1
41.3 (30.1–56.3) 7.35
95% CI, 95% confidence interval; RR, relative resistance [(IC50 in resistant subline) ⁄ (IC50 in 4
parental cells)]. 5
6
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

33
Table 2. IC50 values for alectinib treatment of mock, ABCC11a, and ABCC11b cells. 1
Cells Mock ABCC11a ABCC11b
Alectinib
IC50 (95% CI) IC50 (95% CI) RR IC50 (95% CI) RR
(nmol/l)
70.7 (25.9–172) 3340 (2260–5430) 47.2 5120 (3200–14400) 72.4
95% CI, 95% confidence interval; RR, relative resistance [(IC50 in resistant subline) ⁄ (IC50 in parental 2
cells)] 3
4
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

34
Figure legends 1
Figure 1. 2
ABCC11 as a potential candidate transporter associated with alectinib resistance. A, 3
Schematic representation of establishment of patient-derived alectinib-resistant tumor cells 4
(KTOR1-RE). Pleural effusion from a patient with ALK-rearranged NSCLC was obtained 5
at disease progression and collected cancer cells were cultured in vitro. B, Schematic 6
representation of conventional alectinib-resistant cells (H2228-AR1S and KTOR1-AR). 7
H2228-AR1S cells were established by exposing parental H2228 cells to 300 nM alectinib 8
for 3 months. The KTOR1-AR cells were established by exposure of KTOR1 cells to 30 9
nM alectinib for 6 months. C, Cell viability assay of KTOR1, KTOR1-RE, and KTOR1-AR 10
(left) or H2228 and H2228-AR1S (right) cells treated with alectinib for 72 hours. P-values 11
were calculated using two-way ANOVA, Sidak's multiple comparisons test (n = 5). D, 12
Integrated results of the qRT-PCR analysis in the five cell types. Volcano plot representing 13
gene expression of the ABC transporters. For each gene, the fold change of 14
alectinib-resistant cells (KTOR1-RE, KTOR1-AR, and H2228-AR1S) compared with that 15
of parental cells (KTOR1 and H2228) (horizontal line) and significance between 16
alectinib-resistant (n = 3) and parental cells (n = 2) calculated using the t-test 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

35
[-log(P-value); vertical line] was plotted. Expression of ABCB1 and ABCC11 was 1
significantly increased in resistant cells (plotted in red). E, ABCC11 protein expression of 2
the five cell types was confirmed by immunoblotting. 3
4
Figure 2. 5
Improved alectinib sensitivity in H2228-AR1S cells transfected with ABCC11 siRNA. A, D, 6
Gene expression of ABCC11 following siRNA knockdown of ABCC11 using two different 7
ABCC11-directed RNAi sequences (ABCC11-a and ABCC11-b) in H2228-AR1S (A) and 8
KTOR1-RE cells (D). P-values were calculated using one-way ANOVA and the Dunnett 9
test (n = 4). B, E, ABCC11 protein expression detected using immunoblotting in 10
H2228-AR1S (B) and KTOR1-RE cells (E) transfected with ABCC11 siRNA was 11
decreased compared with that in cells transfected with negative control siRNA. C, F, Cell 12
viability assay of H2228-AR1S (C) and KTOR1-RE cells (F) transfected with ABCC11 13
siRNA and negative control siRNA then treated with alectinib for 168 hours following 14
24-hour transfection. Statistical significance was calculated using two-way ANOVA 15
followed by Sidak's multiple comparisons test (n = 5). 16
17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

36
Figure 3. 1
ABCC11 overexpression leads to decreased alectinib intracellular concentration and low 2
sensitivity to alectinib. ABCC11 expression vectors were introduced into H2228 cells to 3
establish ABCC11-overexpressing cell lines (ABCC11a, ABCC11b). Empty vector was 4
transfected into H2228 cells to establish control cells (mock). A, Gene expression levels of 5
ABCC11 in ABCC11a, ABCC11b, and mock cell lines. P-values were calculated using 6
one-way ANOVA followed by Dunnett’s multiple comparison test (n = 4). B, Protein 7
expression of ABCC11 and GAPDH in mock, ABCC11a, and ABCC11b cell lines 8
determined by immunoblotting. C, Cell viability assay of ABCC11a, ABCC11b, and mock 9
cell lines treated with alectinib for 72 hours. P-values were calculated using two-way 10
ANOVA and post-hoc Sidak’s method (n = 5). D, IC50 value of mock, ABCC11a, and 11
ABCC11b cell lines in the presence of crizotinib, ceritinib, lorlatinib, and brigatinib. 12
P-values were calculated using two-way ANOVA and post-hoc Sidak’s multiple 13
comparisons test. E, Intracellular alectinib concentrations of mock and ABCC11a cells 14
exposed to 100 nM alectinib. Raw data for intracellular concentrations prior to 15
standardization using intracellular protein are shown in Supplementary Fig. S4. P-values 16
were calculated using two-way ANOVA and post-hoc Sidak’s multiple comparisons test. F, 17
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

37
ALK phosphorylation in mock and ABCC11a cell lines exposed to alectinib for 3 hours as 1
determined using immunoblotting. 2
3
Figure 4. 4
ABCC11 overexpression limits the effect of alectinib. Xenograft models of mock, 5
ABCC11a, and ABCC11b cell lines were established and randomized into two groups: 6
alectinib treatment and vehicle. The alectinib dose was 8 mg/kg/day. A, Tumor volume 7
following alectinib or vehicle treatment. In the alectinib group, tumor growth rate was 8
significantly higher in the ABCC11a and ABCC11b groups compared with that in the mock 9
group. P-values were calculated using two-way ANOVA and Holm–Sidak's multiple 10
comparisons test. B, Body weights of the mice. C, ALK phosphorylation in tumors 11
established from ABCC11a, ABCC11b, and mock cell lines treated with alectinib for 24 12
hours as determined using immunoblotting. Quantification of protein bands was performed 13
using ImageJ software. Quantification values for each band were firstly normalized to 14
corresponding values of β-actin. Then, the ratio between normalized values for 15
ABCC11-overexpressing cells and mock cells was calculated. D, Schematic of ABCC11 16
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

38
role in alectinib resistance. ABCC11 overexpression reduces alectinib intracellular 1
concentrations and attenuates the effects of alectinib. 2
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649
39
1
Figure 1 2
3
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649
40
1
Figure 2 2
3
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649
41
1
Figure 3 2
3
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

42
1
Figure 4 2
3
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649

Published OnlineFirst March 26, 2020.Mol Cancer Ther Tomoko Yamamoto Funazo, Takahiro Tsuji, Hiroaki Ozasa, et al. Paired Resistance ModelCancer Due to ABCC11/MRP8 Overexpression in a Clinically Acquired Resistance to Alectinib in ALK-Rearranged Lung
Updated version
10.1158/1535-7163.MCT-19-0649doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2020/03/26/1535-7163.MCT-19-0649.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/early/2020/03/26/1535-7163.MCT-19-0649To request permission to re-use all or part of this article, use this link
on April 26, 2020. © 2020 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 26, 2020; DOI: 10.1158/1535-7163.MCT-19-0649





























