Embed Size (px)
Citation preview

Akademia Wiedzy Synevo Polska
zaprasza na szkolenie online …

Badania genetyczne
w niepłodności

Prelegent i autor prezentacji:
dr n. med. Agnieszka Sobczyńska-Tomaszewska
Pracownia Diagnostyki Genetycznej i Poradnia Genetyczna

Niepłodność
2. Ginekologia
3. Andrologia
. .
Program webinaru
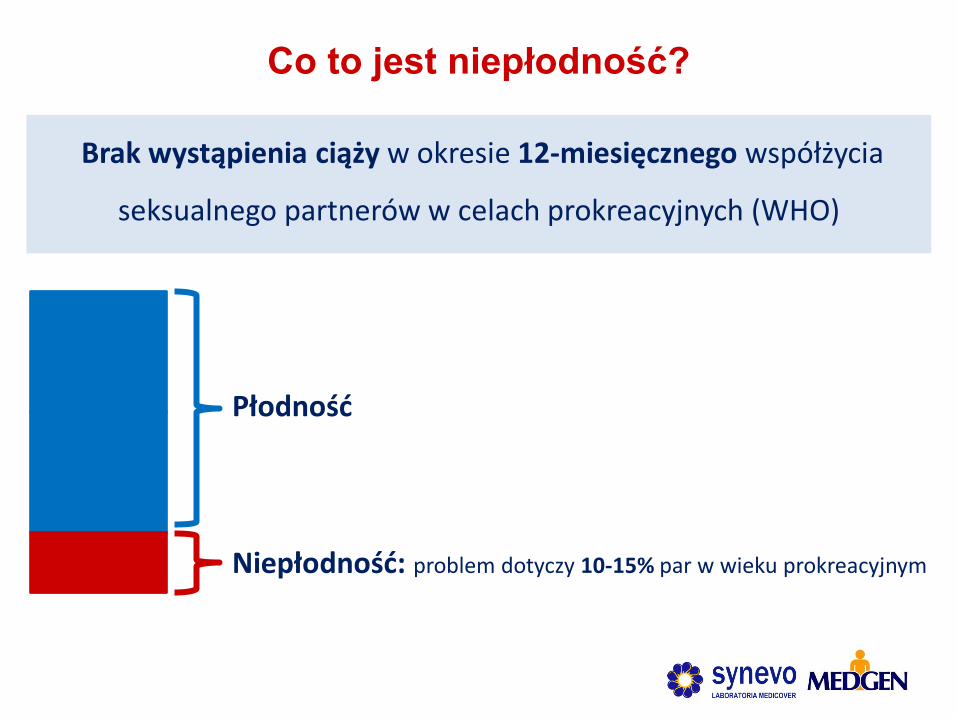
Brak wystąpienia ciąży w okresie 12-miesięcznego współżycia
seksualnego partnerów w celach prokreacyjnych (WHO)
Co to jest niepłodność?
Niepłodność: problem dotyczy 10-15% par w wieku prokreacyjnym
Płodność
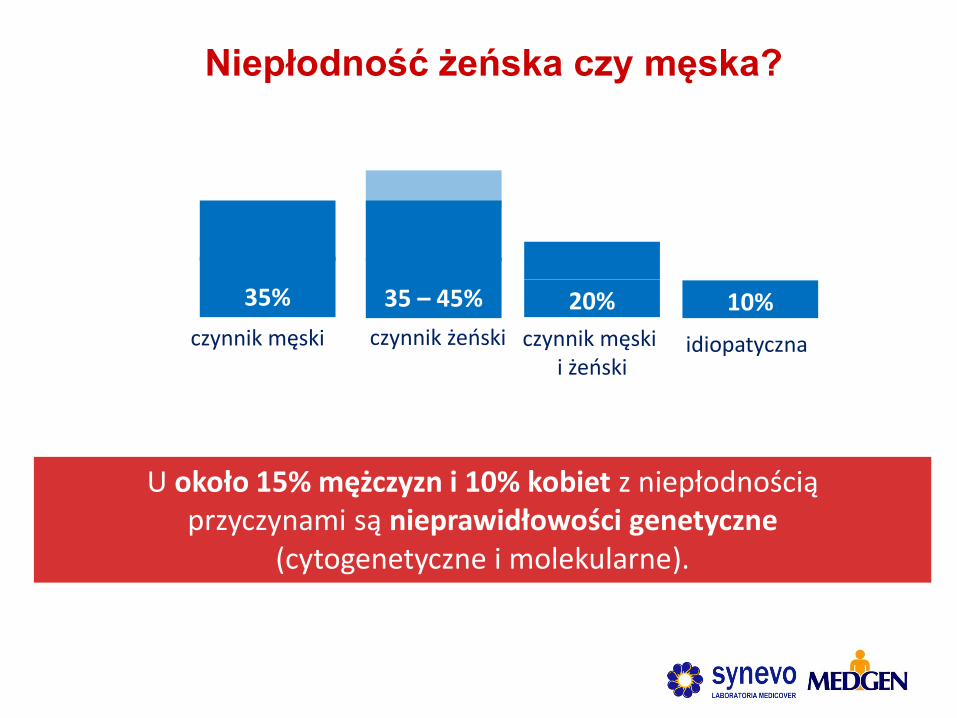
Niepłodność żeńska czy męska?
U około 15% mężczyzn i 10% kobiet z niepłodnością przyczynami są nieprawidłowości genetyczne
(cytogenetyczne i molekularne).
10% 35%
czynnik męski
35 – 45%
czynnik żeński
20%
czynnik męski i żeński
idiopatyczna

Ciężki, zazwyczaj przewlekły przebieg
Częste współistnienie upośledzenia umysłowego
i / lub fizycznego
Wysoki koszt diagnostyki i leczenia
Zwiększone ryzyko ponownego wystąpienia choroby w rodzinie
Choroba genetycznie uwarunkowana, to …
Choroba/predyspozycja przekazywana przez pokolenia,
spowodowana mutacją jednego lub kilku genów
lub też aberracją chromosomową.

Badanie cytogenetyczne (tzw. kariotyp)
Badanie CGH – porównawcza hybrydyzacja
genomowa (ang. comparative genomic
hybridization ) - umożliwia analizę całego genomu
w jednym badaniu z bardzo dużą rozdzielczością,
pozwala na identyfikację niezrównoważonych
aberracji chromosomowych (delecji/duplikacji),
które nie mogły by być rozpoznane klasycznymi
metodami cytogenetycznymi.
Aberracja chromosomowa
Nieprawidłowa liczba (47,XXY; 45,X0) lub
nieprawidłowa budowa chromosomów (delecje, translokacje)
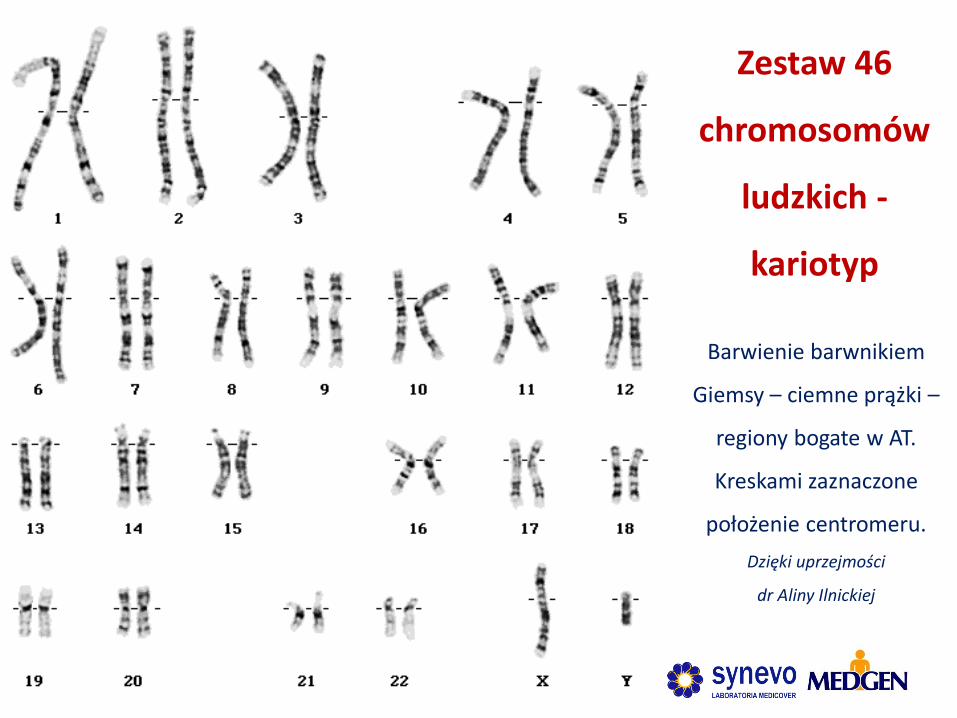
Barwienie barwnikiem
Giemsy – ciemne prążki –
regiony bogate w AT.
Kreskami zaznaczone
położenie centromeru.
Dzięki uprzejmości
dr Aliny Ilnickiej
Zestaw 46
chromosomów
ludzkich -
kariotyp

Badanie kariotypu = badanie cytogenetyczne
Ocena ilościowa i jakościowa garnituru chromosomalnego człowieka
Analiza cytogenetyczna chromosomów barwionych metodą GTG o rozdzielczości 550 prążków.
46XX - żeński
46XY - męski

pary z niepłodnością męską
dziewczynki z niskorosłością (podejrzenie zespołu Turnera)
chłopcy z zaburzeniami dojrzewania (podejrzenie zespołu
Klinefeltera)
dzieci z niepełnosprawnością intelektualną
dzieci z wadami wrodzonymi
Kiedy wykonujemy badanie kariotypu?

Dzika sekwencja ...AGACCATAGGCACTAGAACC...
Podstawienie ...AGACCATAGACACTAGAACC...
Wstawienie ...AGACCATAGCTGCACTAGAACC...
Delecja ...AGACCATAG ***CACTAGAACC...
Analiza molekularna
Mutacje to dziedziczne zmiany w genomie organizmu, które mogą
wywoływać określony efekt fenotypowy, na poziomie molekularnym
mutacje dotyczą zmiany w sekwencji lub organizacji DNA.
Np. mutacje w genie CFTR, mutacja Leiden, ...

Rodzaj diagnostyki genetycznej a źródło DNA
Diagnostyka
Preimplantacyjna Komórki zarodka, komórki linii płciowej
Diagnostyka
prenatalna
Komórki trofoblastu, komórki płynu owodniowego, leukocyty z krwi pępowinowej
Diagnostyka
postnatalna
Dowolne komórki jądrzaste, fragmenty tkanek

Badania molekularne: 1-4 ml krwi żylnej do probówki
morfologicznej (EDTA) lub kilka kropli na bibułę
Badania cytogenetyczne (kariotyp): 5 ml do probówki
z heparyną litową
Sposób pobrania

1. Niepłodność
Ginekologia
3. Andrologia
.
.

Około 30% wszystkich ciąż jest
tracona w trakcie poronień, większość
przed momentem klinicznego
rozpoznania ciąży.
8-15% klinicznie rozpoznanych ciąż
ulega poronieniu.
80% poronień przed 12 tyg. ciąży.
Poronienia
pozostałe przyczyny: infekcje,
nieprawidłowości dróg rodnych,
ekspozycja na toksyny, choroby
endokrynologiczne
i immunologiczne
50% poronień jest
wynikiem nieprawidłowości
chromosomowych

Poronienia - rekomendowane badania genetyczne:
badanie kariotypu u obydwojga rodziców – jeden
z partnerów spośród 3-5% par z poronieniami nawracającymi
jest nosicielem zrównoważonej strukturalnej anomalii
chromosomalnej (m.in. translokacja robertsonowska)
1.

Poronienia - rekomendowane badania genetyczne:
cytogenetyczne badanie zarodka lub kosmówki –
nieprawidłowy kariotyp poronionego płodu jest dobrą
prognozą dla kolejnej ciąży
2.

Poronienia - rekomendowane badania genetyczne:
wywiad w kier. trombofilii – badanie mutacji Leiden i genu
protrombiny
3.

Poronienia - rekomendowane badania genetyczne:
porada genetyczna – ewentualne wskazanie do diagnostyki
prenatalnej
4.

Badanie
materiału
poronnego
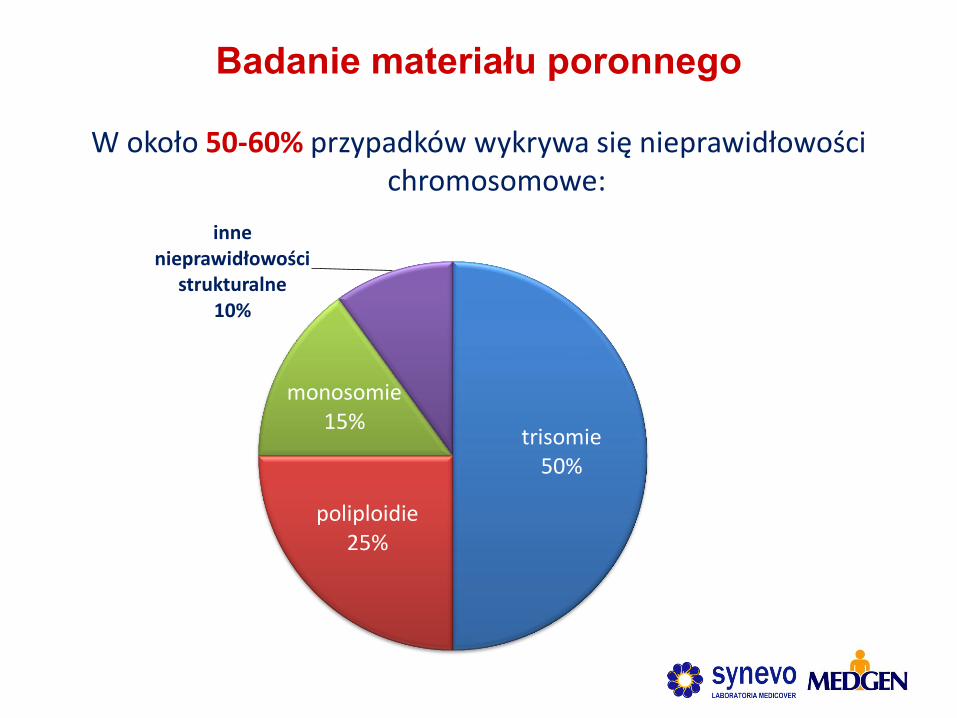
Badanie materiału poronnego
W około 50-60% przypadków wykrywa się nieprawidłowości chromosomowe:
trisomie 50%
poliploidie 25%
monosomie 15%
inne nieprawidłowości
strukturalne 10%

Metoda QF-PCR pozwala na szybkie, a zarazem wiarygodne
diagnozowanie najczęściej występujących zaburzeń genetycznych
będących głównymi przyczynami poronień, tj. aberracji liczbowych
chromosomów: 13, 15, 16, 18, 21 i 22 oraz aneuploidii
chromosomów płciowych (m. in. zespół Turnera).
Metoda QF-PCR
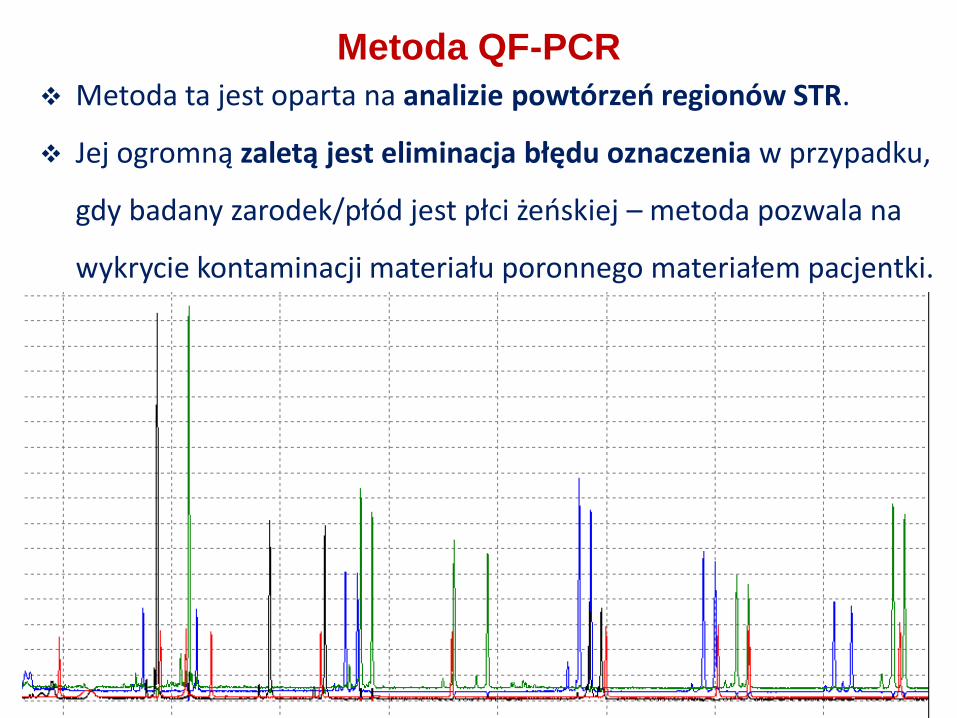
Metoda ta jest oparta na analizie powtórzeń regionów STR.
Jej ogromną zaletą jest eliminacja błędu oznaczenia w przypadku,
gdy badany zarodek/płód jest płci żeńskiej – metoda pozwala na
wykrycie kontaminacji materiału poronnego materiałem pacjentki.
Metoda QF-PCR

Zakrzepica żył
Spowolnienie przepływu krwi w naczyniach żylnych, zakrzepica żył głębokich
Wieloczynnikowa choroba wywołana interakcją czynników środowiskowych
(np.: siedzący tryb życia, otyłość, palenie tytoniu) i genetycznych
Objawy: ból i obrzęk kończyn, zaczerwienienie i zasinienie.

Zakrzepica żył
Ryzyko wystąpienia choroby dla:
heterozygot mutacji R506Q: 5-7x wyższe niż w populacji ogólnej,
heterozygot stosujących antykoncepcję doustną: 30-50x;
dla homozygot R506Q: ryzyko 80x
Dodatkowe badania: geny F2 (protrombina) i MTHFR (reduktaza
5,10- metylenotetrahydrofolianu)
Częstość występowania mutacji typu Leiden w genie czynnika V: 1-7%

Wskazania do wykonania badań (biochemicznych i genetycznych)
w kierunku trombofilii
zakrzepica żylna bez uchwytnej przyczyny przed 45-50. r. ż.
zakrzepica żylna u osoby obciążonym wywiadem rodzinnym (zakrzepica w rodzinie)
zakrzepica nawracająca
zakrzepica o nietypowej lokalizacji (np. żyły jamy brzusznej)
zakrzepica rozwijająca się w czasie ciąży
zakrzepica w trakcie stosowania doustnej antykoncepcji lub hormonalnej terapii
zastępczej
nawykowe poronienia lub urodzenie martwego płodu
badanie osoby asymptomatycznej z obciążonym wywiadem rodzinnym
Wskazania do wykonania badań w kierunku
trombofilii

Przedwczesne wygasanie funkcji jajników (POF)
a zespół łamliwego chromosomu X (FRAX)
ekson 1 intron 1
sekwencja prawidłowa (6-59 kopii CGG)
premutacja (60-220 kopii CGG)
pełna mutacja (ponad 220 kopii CGG)
niestabilna sekwencja (CGG)n
sekwencja eksonowa
1 2 3 4 5 6 7 8 9 10 11 1213 14 15 16 17
-(CGG)n- sekwencje eksonowe kodujące białko
sekwencje intronowe
sekwencja eksonowa niekodująca
Schemat genu FMR1

Najczęstsza dziedziczna przyczyna upośledzenia umysłowego: U chłopców i druga co do częstości wśród przyczyn
genetycznych (po zespole Downa).
W większości przypadków nie jest prawidłowo rozpoznawana, zwłaszcza u kobiet.
Częstość występowania FRAX, to …
1:1200-3600 u chłopców,
1:4000-6000 u dziewczynek
Nowotwór sutka
i/lub jajnika
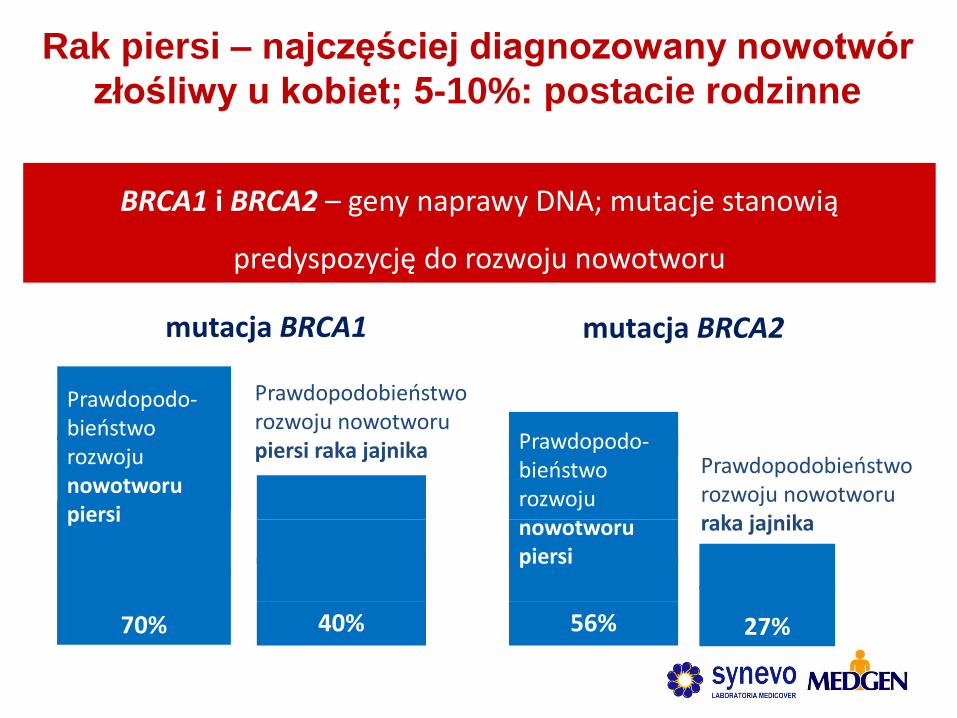
Rak piersi – najczęściej diagnozowany nowotwór
złośliwy u kobiet; 5-10%: postacie rodzinne
mutacja BRCA1
27% 70% 40%
mutacja BRCA2
Prawdopodo-bieństwo rozwoju nowotworu piersi
Prawdopodobieństwo rozwoju nowotworu piersi raka jajnika
Prawdopodobieństwo rozwoju nowotworu raka jajnika
56%
Prawdopodo-bieństwo rozwoju nowotworu piersi
BRCA1 i BRCA2 – geny naprawy DNA; mutacje stanowią
predyspozycję do rozwoju nowotworu

Nowotwór sutka i/lub jajnika
Rekomendacje EMQN:
W związku z heterogennością populacji europejskiej
należy badać cały region kodujący.
I etap diagnostyki
Identyfikacja mutacji: 5382insC,
C61G
i 4153delA + 150 mutacji rzadkich
Wykrycie mutacji:
profilaktyka przeciwnowotworowa, m.in.
chirurgiczna; przeciwwskazanie do stosowania
terapii hormonalnej, m.in. antykoncepcji

Rekomendacje Polskiego Towarzystwa
Ginekologicznego
Do grup wysokiego ryzyka zachorowania kwalifikuje się rodziny,
u których:
wystąpiły 3 lub więcej zachorowania na raka gruczołu sutkowego
lub/i jajnika u krewnych I i II stopnia (włączając w to pacjentkę)
u jednej i tej samej pacjentki lub u jej krewnej I lub II stopnia
wystąpiły jednoczasowo lub w różnym czasie zachorowania
na raka gruczołu sutkowego i jajnika
jeżeli u pacjentki stwierdzono wcześniej mutację genu BRCA1

Rekomendacje Polskiego Towarzystwa
Ginekologicznego c.d.
jeżeli wśród krewnych I i II stopnia (włączając w to pacjentkę)
wystąpiły 2 zachorowania na raka gruczołu sutkowego lub/i
jajnika – w tym 1 zachorowanie przed 50. rokiem życia
jeżeli pacjentka lub jedna z jej krewnych I stopnia zachorowała
na raka gruczołu sutkowego lub jajnika przed 40. rokiem życia

Rekomendacje Polskiego Towarzystwa
Ginekologicznego
Zalecenia dla kobiet nosicielek mutacji w genie BRCA1/BRCA2
Comiesięczna samokontrola piersi
Badanie palpacyjne piersi przez lekarza co 6 miesięcy
Badanie obrazowe piersi (USG, mammografia, rezonans
magnetyczny - w zależności od wieku i budowy piersi)
co 6 miesięcy (od 25 r. ż. USG, rezonans magnetyczny,
od 35 r. ż. mammografia)

Rekomendacje Polskiego Towarzystwa
Ginekologicznego
Badanie ginekologiczne co 6 miesięcy (od 30 r. ż.)
USG przezpochwowe co 6 miesięcy (od 30 r. ż.)
Oznaczenie poziomu markera CA 125 w surowicy krwi
co 6 miesięcy (od 30 r.ż.)
W oparciu o Rekomendacje PTG (2004) oraz Rekomendacje Europejskiego Towarzystwa
Onkologii Medycznej (ESMO 2011).

1. Niepłodność
2. Ginekologia
Andrologia
.
.
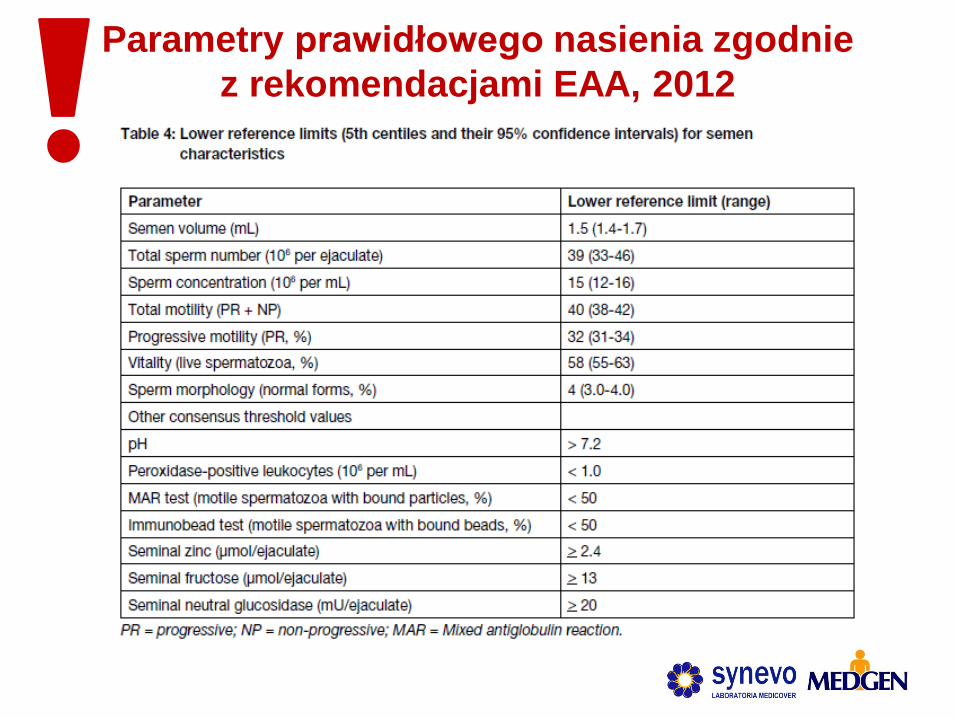
Parametry prawidłowego nasienia zgodnie
z rekomendacjami EAA, 2012
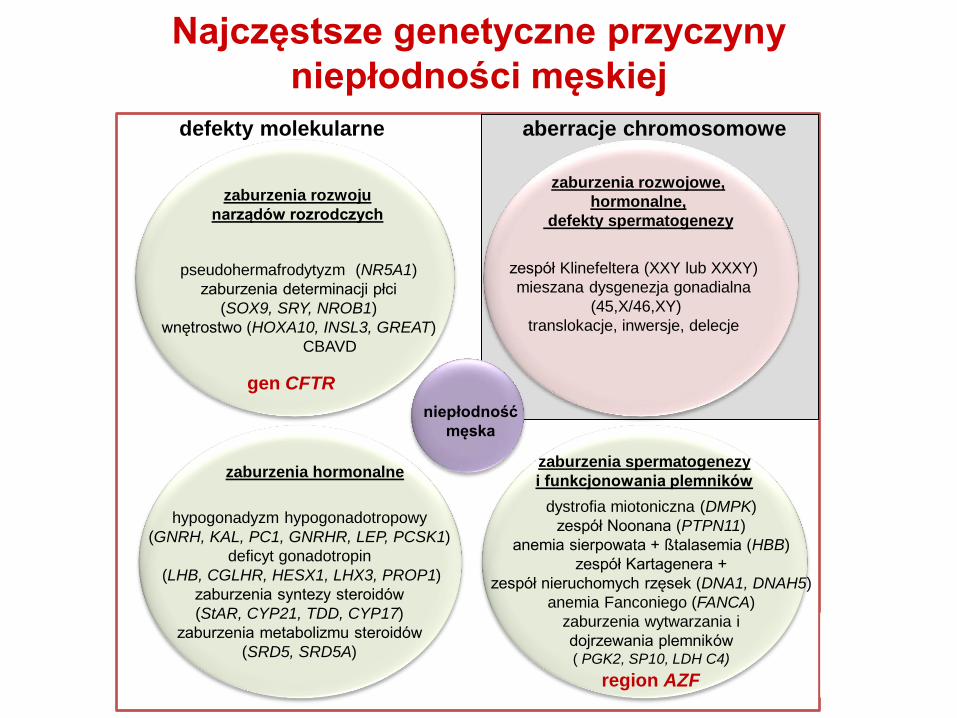
niepłodność męska
pseudohermafrodytyzm (NR5A1)
zaburzenia determinacji płci
(SOX9, SRY, NROB1)
wnętrostwo (HOXA10, INSL3, GREAT)
CBAVD
zaburzenia rozwoju
narządów rozrodczych
hypogonadyzm hypogonadotropowy
(GNRH, KAL, PC1, GNRHR, LEP, PCSK1)
deficyt gonadotropin
(LHB, CGLHR, HESX1, LHX3, PROP1)
zaburzenia syntezy steroidów
(StAR, CYP21, TDD, CYP17)
zaburzenia metabolizmu steroidów
(SRD5, SRD5A)
zaburzenia hormonalne
dystrofia miotoniczna (DMPK)
zespół Noonana (PTPN11)
anemia sierpowata + ßtalasemia (HBB)
zespół Kartagenera +
zespół nieruchomych rzęsek (DNA1, DNAH5)
anemia Fanconiego (FANCA)
zaburzenia wytwarzania i
dojrzewania plemników ( PGK2, SP10, LDH C4)
zaburzenia spermatogenezy
i funkcjonowania plemników
zespół Klinefeltera (XXY lub XXXY)
mieszana dysgenezja gonadialna
(45,X/46,XY)
translokacje, inwersje, delecje
defekty molekularne aberracje chromosomowe
niepłodność
męska
zaburzenia rozwojowe,
hormonalne,
defekty spermatogenezy
Najczęstsze genetyczne przyczyny
niepłodności męskiej
gen CFTR
region AZF
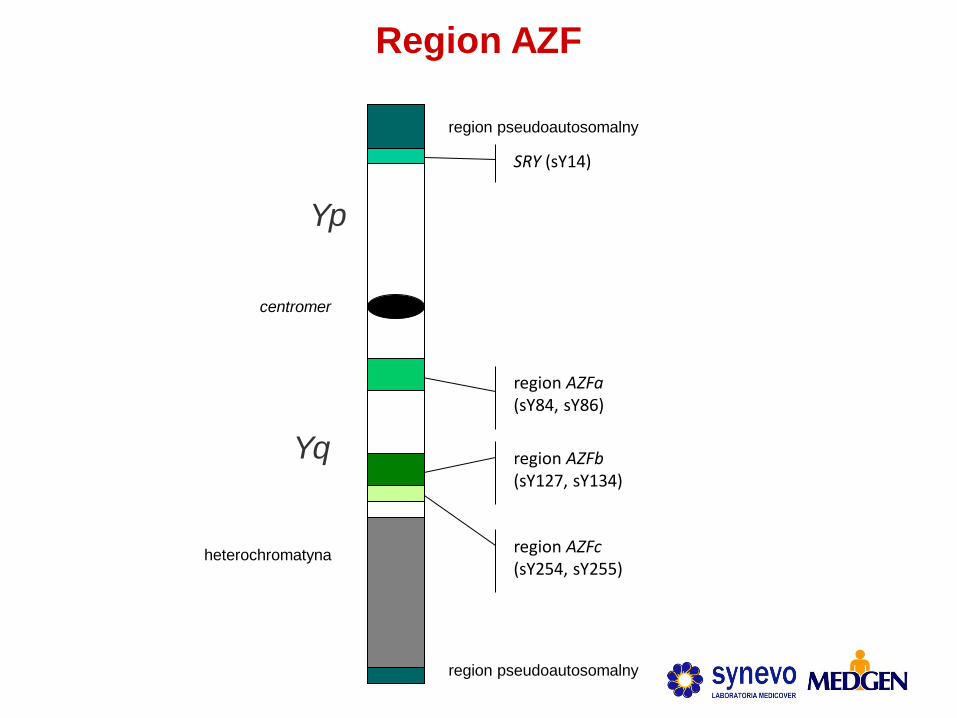
Region AZF
region AZFb (sY127, sY134)
Yp
Yq
SRY (sY14)
heterochromatyna
region pseudoautosomalny
centromer
region AZFa (sY84, sY86)
region AZFc (sY254, sY255)
region pseudoautosomalny

Delecje regionu AZF
Nie wykryto ich u pacjentów z normozoospermią ->
potwierdzenie zależności przyczynowo-skutkowej
Wykryto u pacjentów z azoospermią (8-12%) oraz
oligozoospermią (3-7%)
• Bardzo rzadko występują u pacjentów > 5 mln plemników/ml (0,7%)
• Częstość występowania poszczególnych delecji:
• AZFc 65-70%
• AZFb, AZFb+c lub AZFa+b+c 25-30%
• AZFa 5%
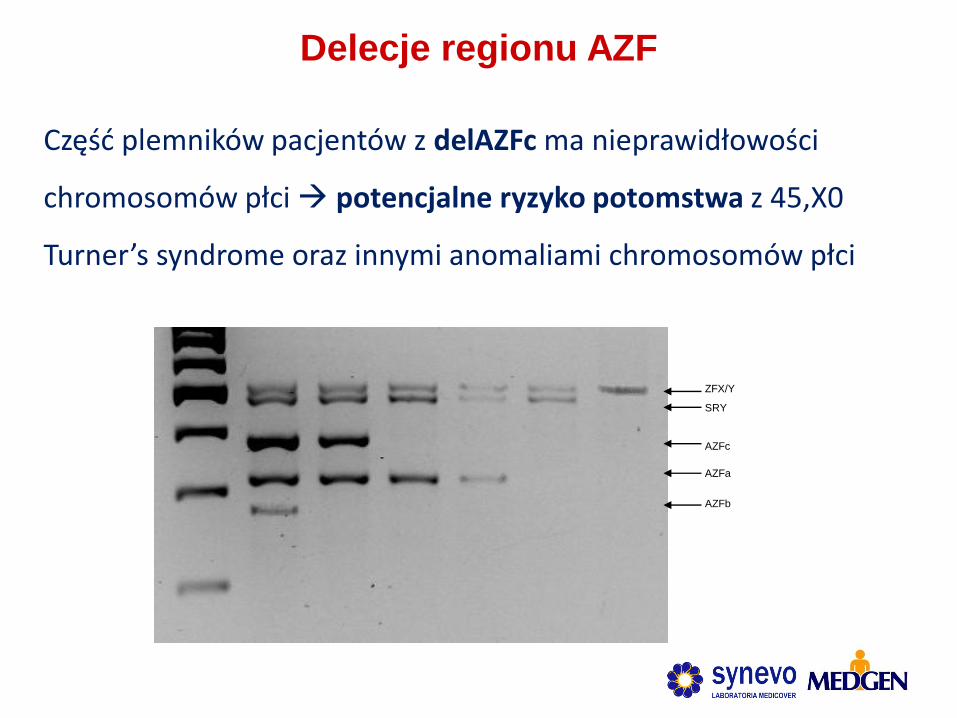
Delecje regionu AZF
Część plemników pacjentów z delAZFc ma nieprawidłowości
chromosomów płci potencjalne ryzyko potomstwa z 45,X0
Turner’s syndrome oraz innymi anomaliami chromosomów płci
ZFX/Y
SRY
AZFc
AZFa
AZFb
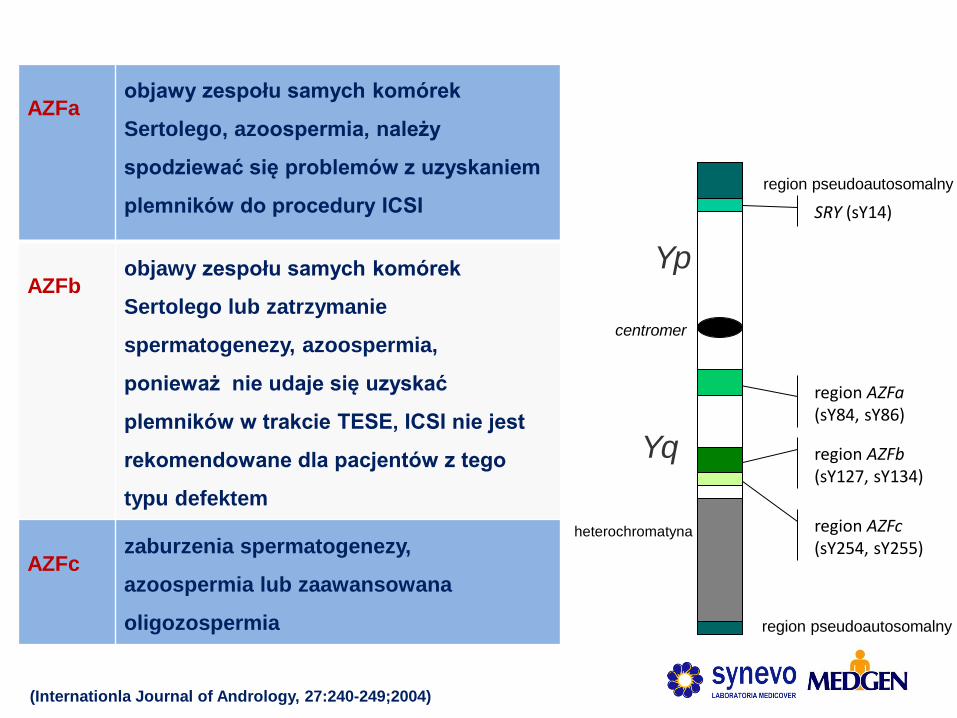
(Internationla Journal of Andrology, 27:240-249;2004)
AZFa objawy zespołu samych komórek
Sertolego, azoospermia, należy
spodziewać się problemów z uzyskaniem
plemników do procedury ICSI
AZFb objawy zespołu samych komórek
Sertolego lub zatrzymanie
spermatogenezy, azoospermia,
ponieważ nie udaje się uzyskać
plemników w trakcie TESE, ICSI nie jest
rekomendowane dla pacjentów z tego
typu defektem
AZFc zaburzenia spermatogenezy,
azoospermia lub zaawansowana
oligozospermia
region AZFb (sY127, sY134)
Yp
Yq
SRY (sY14)
heterochromatyna
region pseudoautosomalny
centromer
region AZFa (sY84, sY86)
region AZFc (sY254, sY255)
region pseudoautosomalny

Fertil Steril. 2006 Feb;85(2):441-5. „Y-chromosome microdeletions and recurrent pregnancy loss”.
OBJECTIVE: To determine the prevalence of Y-chromosome microdeletions in recurrent pregnancy
loss (RPL) couples as compared with couples with male factor infertility and fertile couples.
PATIENT(S): 17 men from RPL couples, 18 men from couples with a live birth and no history of
miscarriages, and 10 men from couples with male factor infertility.
RESULT(S): 14 of the 17 men (82%) tested had microdeletions in one or more of the four segments
studied. Two of the 10 male factor infertility patients (20%) had microdeletions in 2 different
segments. None of the 18 fertile men had any microdeletions in the 4 segments of the proximal AZFc
region studied.
CONCLUSION(S): The prevalence of the Y-chromosome microdeletions in the proximal AZFc region
was much higher in men from RPL couples than from fertile or infertile couples.
Wysoka częstość AZFc delecji u partnerów kobiet z nawracającymi poronieniami

Delecja gr/gr jest również potencjalnym czynnikiem ryzyka raka
jąder.
Pierwotne guzy jąder (ang. testicular germ cell tumor) są jednym
z najczęstszych nowotworów złośliwych u młodych mężczyzn
w wieku 20-44 lata.
W 2006 r. ten typ nowotworu stwierdzono u 21% młodych
mężczyzn chorych na nowotwory złośliwy (wg danych Krajowego Rejestru
Nowotworów, Centrum Onkologii-Instytut, Warszawa).
Delecja gr/gr – subregion AZFc
Pacjenci z delecją gr/gr mają 7-8-krotnie wyższe ryzyko rozwoju
oligozoospermii (OR = 7.9, 95%, CI: 1.8-33.8; p < 0.001)

Przyczyn rozwoju guzów jąder jest wiele.
Istotną rolę odgrywają tu różne czynniki genetyczne – ryzyko
rozwoju pierwotnego guza jąder jest 4-6-krotnie wyższe u synów
osób chorych i 8-10-krotnie u braci osób chorych.
Obecność deletions gr/gr jest predyspozycją do wystąpienia
delecji AZFc w następnym pokoleniu.
Delecja gr/gr – subregion AZFc

Badanie nieprawidłowości chromosomalnych
w nasieniu
Badanie z zastosowaniem metody FISH
Aneuploidie, szczególnie dotyczące chromosomów płci
są związane z ciężkimi zaburzeniami spermatogenezy
(3,6-10 mln plemników/ml) i są identyfikowane często
u mężczyzn z translokacjami
Badanie cytogenetyczne plemników NIE JEST BADANIEM
DIAGNOSTYCZNYM, jest wciąż badaniem naukowym.
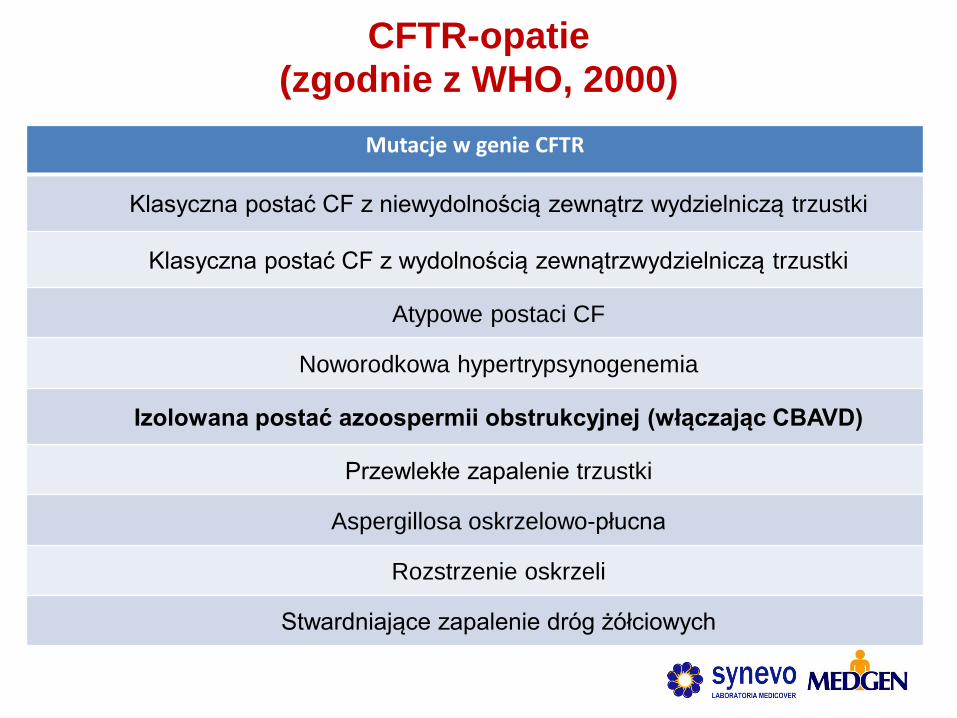
CFTR-opatie (zgodnie z WHO, 2000)
Mutacje w genie CFTR
Klasyczna postać CF z niewydolnością zewnątrz wydzielniczą trzustki
Klasyczna postać CF z wydolnością zewnątrzwydzielniczą trzustki
Atypowe postaci CF
Noworodkowa hypertrypsynogenemia
Izolowana postać azoospermii obstrukcyjnej (włączając CBAVD)
Przewlekłe zapalenie trzustki
Aspergillosa oskrzelowo-płucna
Rozstrzenie oskrzeli
Stwardniające zapalenie dróg żółciowych

CBAVD
50-82% pacjentów ma mutację CFTR w co najmniej jednej kopii genu (w jednym allelu genu)
Azoospermia, objętość nasienia < 1,5ml, pH <7,0
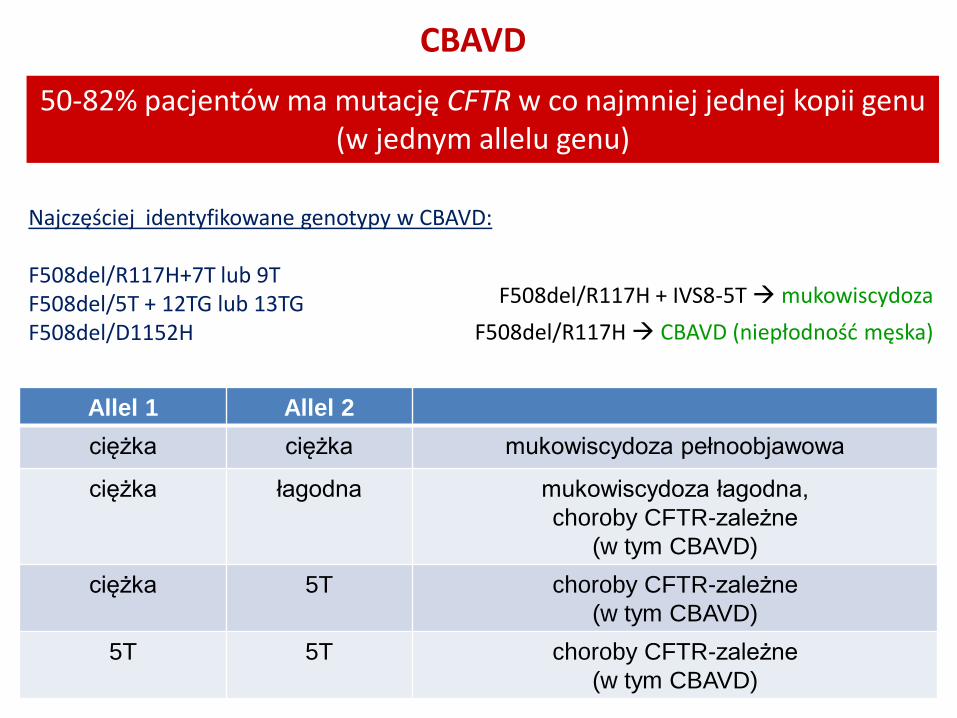
F508del/R117H + IVS8-5T mukowiscydoza
F508del/R117H CBAVD (niepłodność męska)
Allel 1 Allel 2
ciężka ciężka mukowiscydoza pełnoobjawowa
ciężka łagodna mukowiscydoza łagodna,
choroby CFTR-zależne
(w tym CBAVD)
ciężka 5T choroby CFTR-zależne
(w tym CBAVD)
5T 5T choroby CFTR-zależne
(w tym CBAVD)
Najczęściej identyfikowane genotypy w CBAVD: F508del/R117H+7T lub 9T F508del/5T + 12TG lub 13TG F508del/D1152H
CBAVD
50-82% pacjentów ma mutację CFTR w co najmniej jednej kopii genu (w jednym allelu genu)
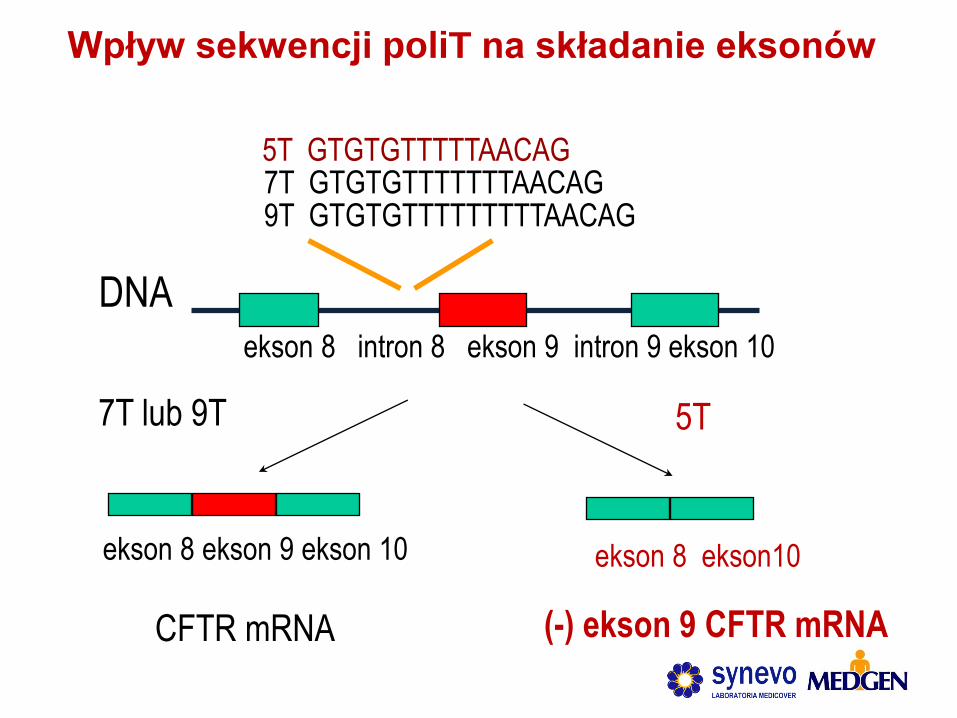
ekson 8 intron 8 ekson 9 intron 9 ekson 10
DNA
5T GTGTGTTTTTAACAG
9T GTGTGTTTTTTTTTAACAG 7T GTGTGTTTTTTTAACAG
ekson 8 ekson 9 ekson 10
CFTR mRNA
7T lub 9T
ekson 8 ekson10
5T
(-) ekson 9 CFTR mRNA
Wpływ sekwencji poliT na składanie eksonów
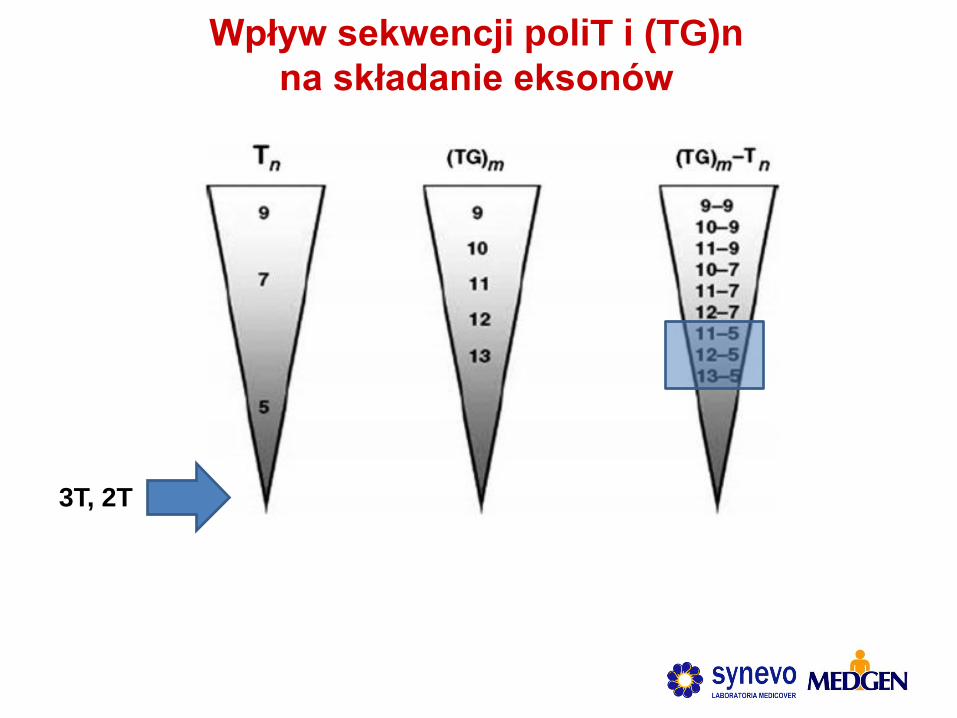
3T, 2T
Wpływ sekwencji poliT i (TG)n
na składanie eksonów

Rekomendacje American College
of Medical Genetics
Badanie powinno być proponowane:
Populacji kaukaskiej (rasa biała) pochodzenia
nie-żydowskiego
Żydom Aszkenazyjskim
Badanie nosicielstwa mutacji CFTR
test prekoncepcyjny
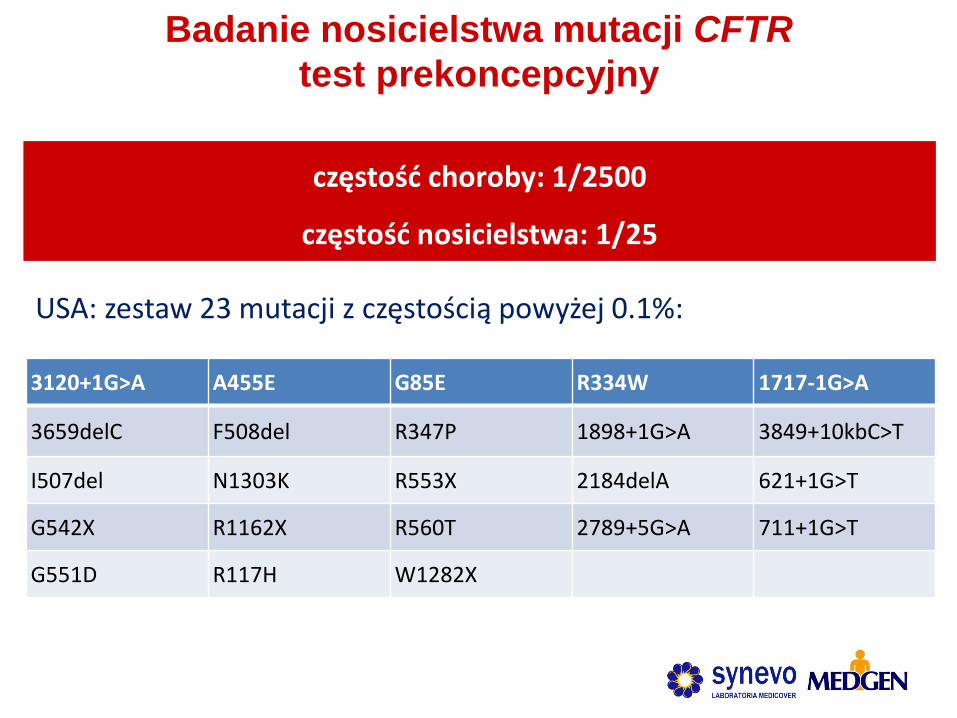
Badanie nosicielstwa mutacji CFTR
test prekoncepcyjny
3120+1G>A A455E G85E R334W 1717-1G>A
3659delC F508del R347P 1898+1G>A 3849+10kbC>T
I507del N1303K R553X 2184delA 621+1G>T
G542X R1162X R560T 2789+5G>A 711+1G>T
G551D R117H W1282X
USA: zestaw 23 mutacji z częstością powyżej 0.1%:
częstość choroby: 1/2500
częstość nosicielstwa: 1/25

Test prekoncepcyjny w kierunku
mukowiscydozy w Polsce - ???
Zakres badania zgodny z zaleceniami
Polskiego Towarzystwa Mukowiscydozy
F508del R334W 1717-1G>A 2143delT
G542X R347P R117H+IVS8-T dele2,3(21kb)
R553X W1282X 3849+10kbC>T 2184insA
G551D N1303K 2183delAA>G K710X
Min.16 mutacji:
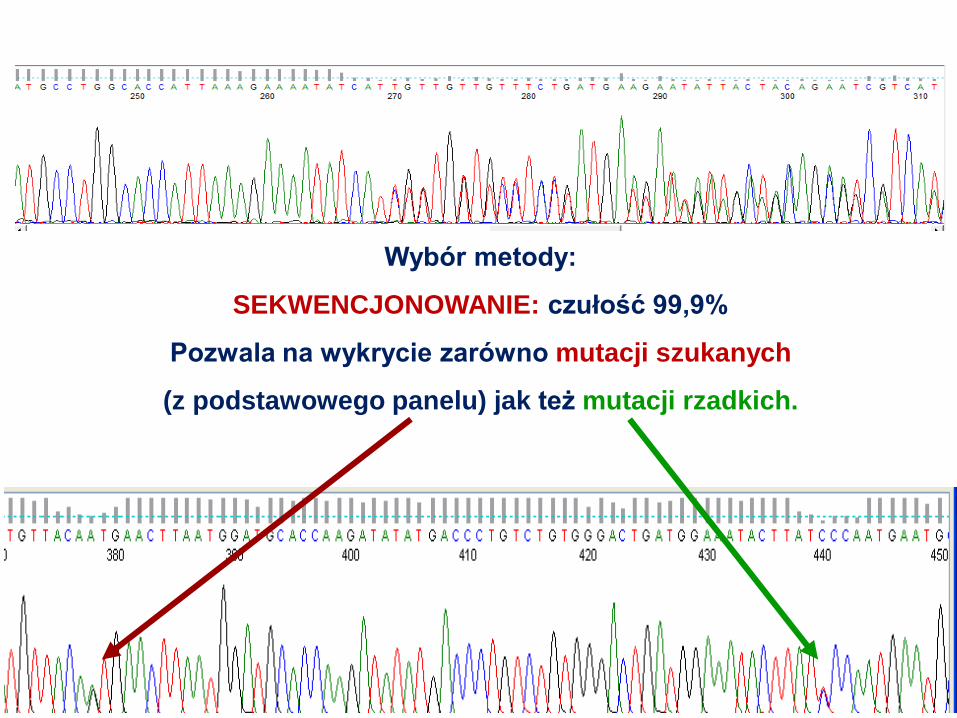
Wybór metody:
SEKWENCJONOWANIE: czułość 99,9%
Pozwala na wykrycie zarówno mutacji szukanych
(z podstawowego panelu) jak też mutacji rzadkich.
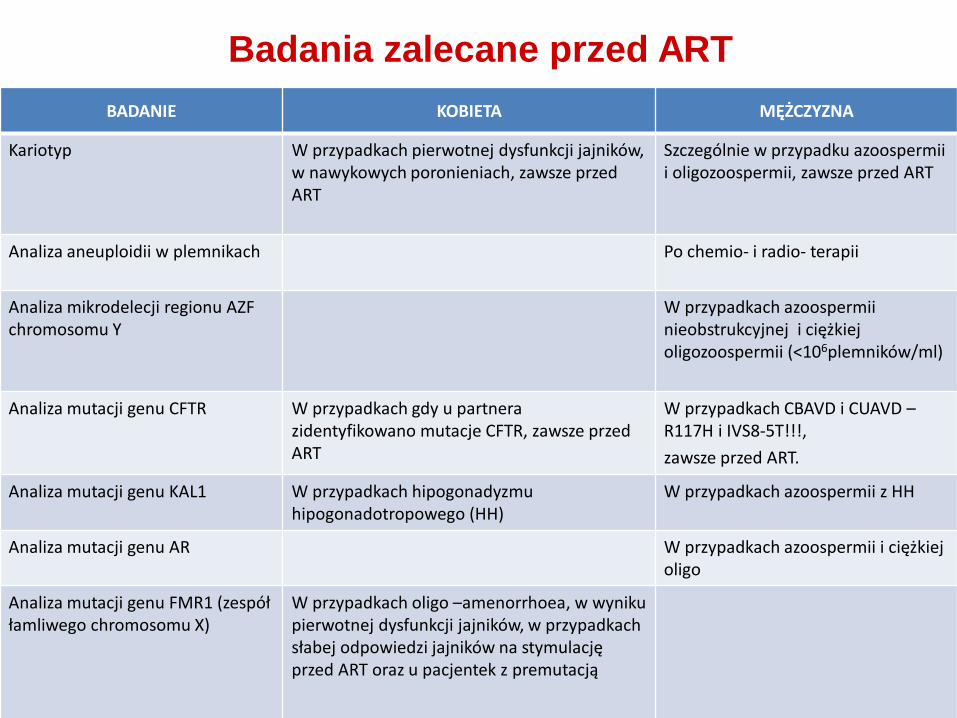
Badania zalecane przed ART
BADANIE KOBIETA MĘŻCZYZNA
Kariotyp W przypadkach pierwotnej dysfunkcji jajników, w nawykowych poronieniach, zawsze przed ART
Szczególnie w przypadku azoospermii i oligozoospermii, zawsze przed ART
Analiza aneuploidii w plemnikach Po chemio- i radio- terapii
Analiza mikrodelecji regionu AZF chromosomu Y
W przypadkach azoospermii nieobstrukcyjnej i ciężkiej oligozoospermii (<106plemników/ml)
Analiza mutacji genu CFTR W przypadkach gdy u partnera zidentyfikowano mutacje CFTR, zawsze przed ART
W przypadkach CBAVD i CUAVD – R117H i IVS8-5T!!!,
zawsze przed ART.
Analiza mutacji genu KAL1 W przypadkach hipogonadyzmu hipogonadotropowego (HH)
W przypadkach azoospermii z HH
Analiza mutacji genu AR W przypadkach azoospermii i ciężkiej oligo
Analiza mutacji genu FMR1 (zespół łamliwego chromosomu X)
W przypadkach oligo –amenorrhoea, w wyniku pierwotnej dysfunkcji jajników, w przypadkach słabej odpowiedzi jajników na stymulację przed ART oraz u pacjentek z premutacją

Rekomendacje EAA, 2012
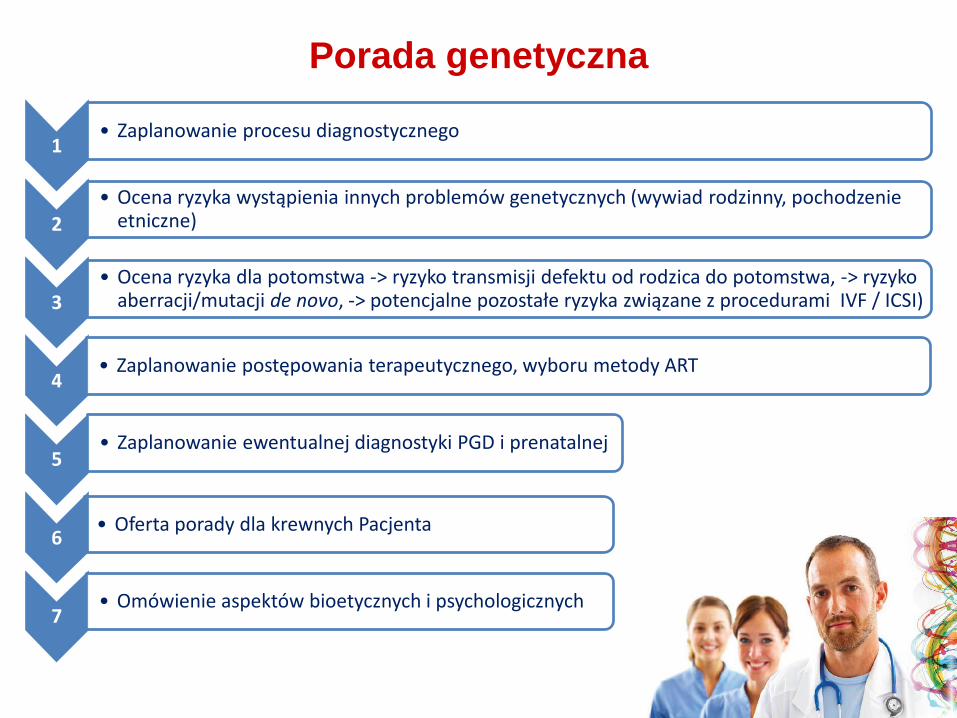
Porada genetyczna
1 • Zaplanowanie procesu diagnostycznego
2
• Ocena ryzyka wystąpienia innych problemów genetycznych (wywiad rodzinny, pochodzenie etniczne)
3
• Oferta porady dla krewnych Pacjenta
4 • Zaplanowanie postępowania terapeutycznego, wyboru metody ART
5
• Ocena ryzyka dla potomstwa -> ryzyko transmisji defektu od rodzica do potomstwa, -> ryzyko aberracji/mutacji de novo, -> potencjalne pozostałe ryzyka związane z procedurami IVF / ICSI)
6
• Zaplanowanie ewentualnej diagnostyki PGD i prenatalnej
7 • Omówienie aspektów bioetycznych i psychologicznych

Diagnostyka prenatalna
Materiał do badań genetycznych:
- trofoblast:11-14 Hbd
- komórki płynu owodniowego:15-17 Hbd
Molekularna diagnostyka prenatalna jest możliwa wyłącznie po wcześniejszym
określeniu defektu genetycznego u rodziców!
Wskazania do diagnostyki cytogenetycznej (kariotyp):
wiek matki > 35 lat; wiek ojca > 55 lat;
nieprawidłowy obraz USG;
aberracje chromosomowe w rodzinie.
Możliwość wykonania szybkiego testu MLPA w kierunku najczęstszych wad
płodu: zespół Downa, Edwardsa i Pataua – wynik po 3 dniach.

Kontakt:
MEDGEN Pracownia Badań Genetycznych i Poradnia Genetyczna ul. Orzycka 27, 02-695 Warszawa www.medgen.pl [email protected] dr Kamila Czerska 515-14-14-14; [email protected] dr Agnieszka Sobczyńska-Tomaszewska 506-069-568; [email protected]
SYNEVO Laboratoria Medicover Ul. Dzika 4, 00-194 Warszawa dr Andrzej Marszałek 604-172-806; andrzej.marszał[email protected]

Dziękujemy





























