Embed Size (px)
Citation preview

An Introduction to Bioinformatics
Protein Structure Prediction

Aims
• Understand the use of algorithms
• Recognize different approaches
• Understand the limitations
Objectives
• Predict occurrence of aspects of structure
• To select appropriate tools

Introduction
• Structure has several levels– 1 primary– 2 secondary– 3 tertiary– 4 quaternary

1 primary
• Amino acid sequence
NH2-MRLSWYDPDFQARLTRSNSKCQGQLEV YLKDGWHMVC SQSWGRSSKQWEDPSQASKVCQRLNCGVPLSLGPFLVTYTPQSSIICYGQLGSFSNCSHSRNDMCHSLGLTCLE-COOH
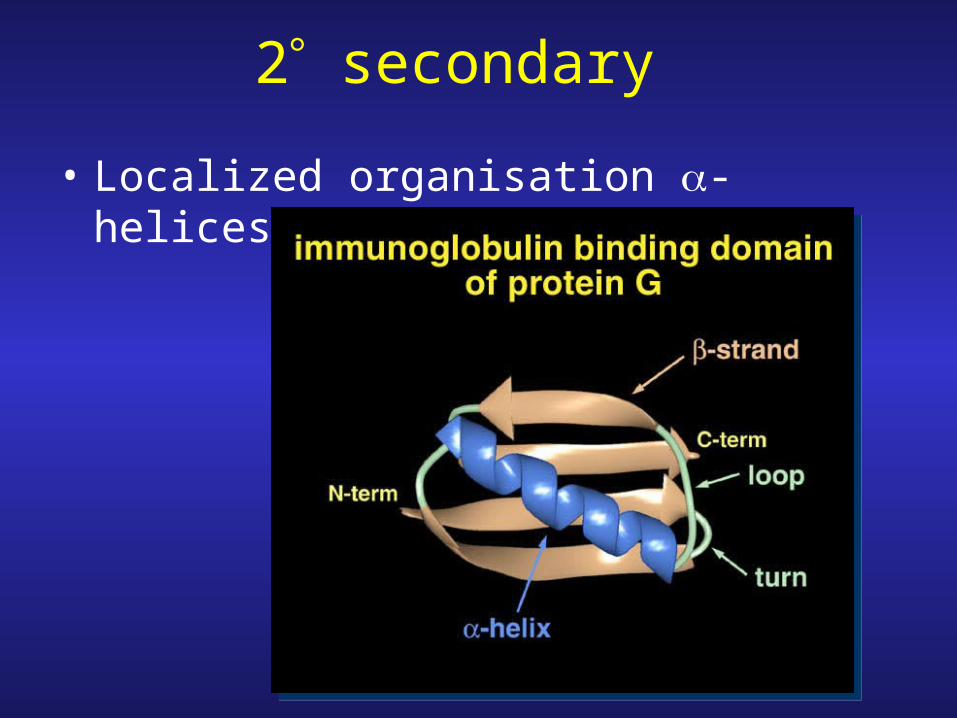
2 secondary
• Localized organisation -helices and -sheets

3 tertiary
Three-dimensional organisation
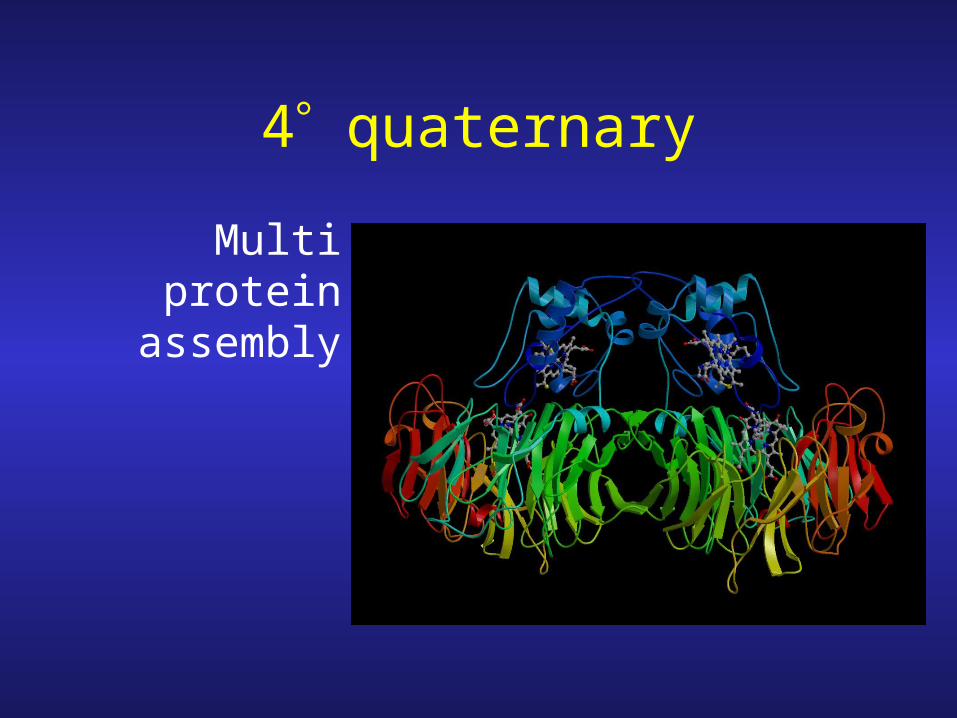
4 quaternary
Multi protein assembly

The problem…..
• The best way is by X-ray crystallography or NMR etc…
• Structure databases only hold about 10,000 + structures
• Therefore devise programs to deduce structural solutions
• Complex!
Secondary Structure prediction
• Signal peptides
• Intracellular targeting
•Trans-membrane -helices
• -helices and -sheets
•Super-secondary structure (motifs)

Signal peptides
• Short N-terminal amino acid sequences
• Direct to membrane
• Cleaved after translocation
• SignalP – Nobel Prize 1999 Günter Blobel
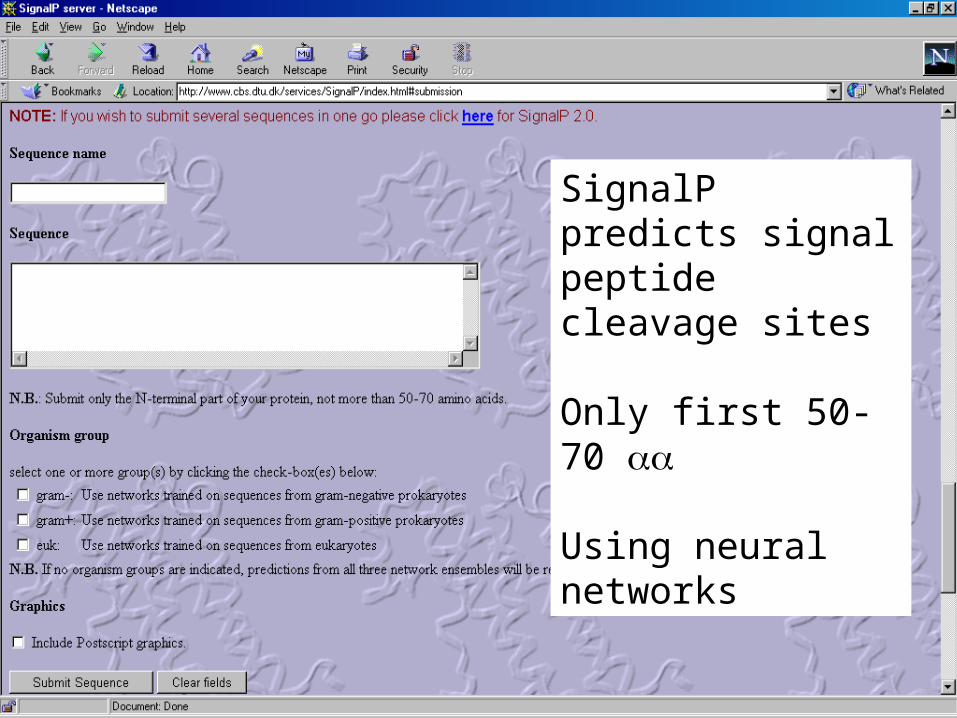
SignalP predicts signal peptide cleavage sites
Only first 50-70
Using neural networks
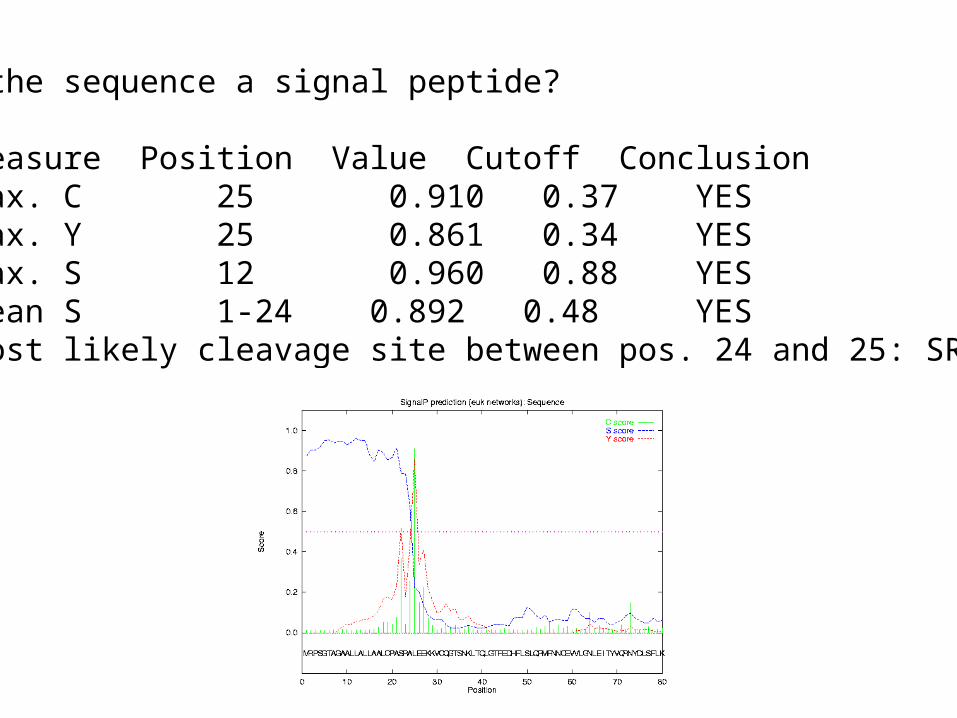
Is the sequence a signal peptide?
# Measure Position Value Cutoff Conclusion max. C 25 0.910 0.37 YES max. Y 25 0.861 0.34 YES max. S 12 0.960 0.88 YES mean S 1-24 0.892 0.48 YES# Most likely cleavage site between pos. 24 and 25: SRA-LE

Intracellular targeting
• TargetP
• Predict subcellular location of eukaryotic protein
• Presequences – Chloroplasts– Mitochondria– signal peptide

Transmembrane Domains
• Lots of programs
• TMHMM -helices– hydrophobic – helix topology– R or K +ve charge cytoplasmic
side– Hidden Markov Modelling
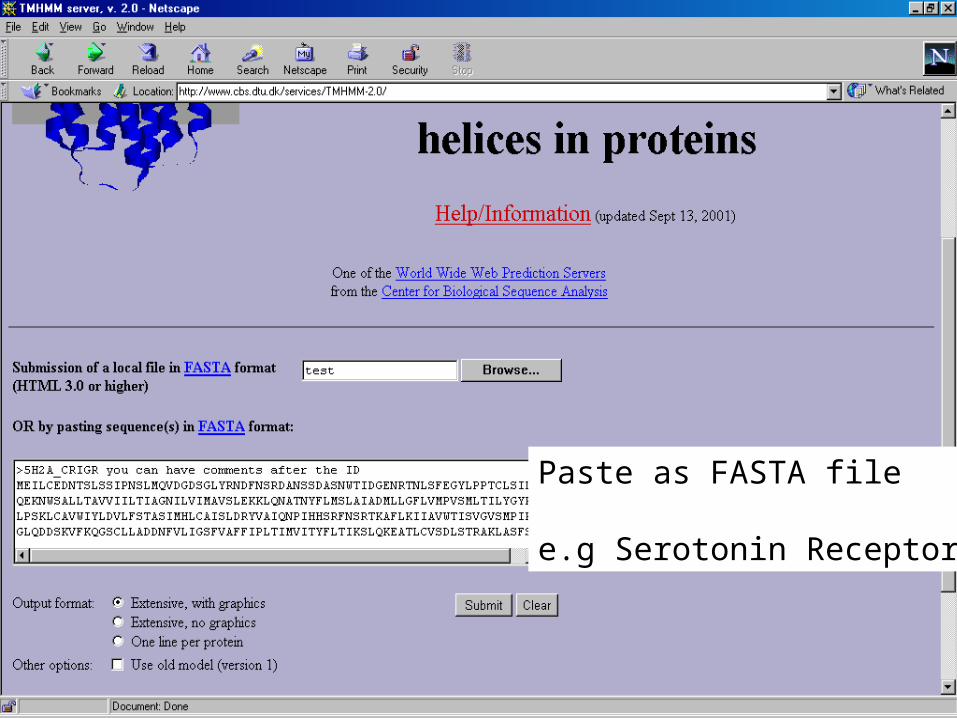
Paste as FASTA file
e.g Serotonin Receptor
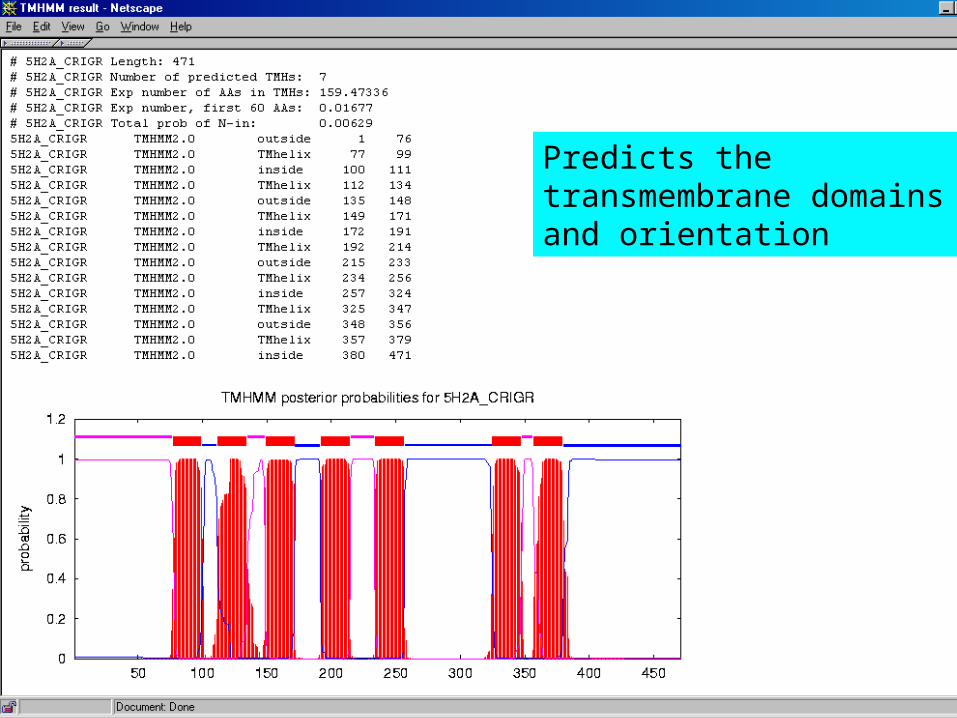
Predicts the transmembrane domains and orientation
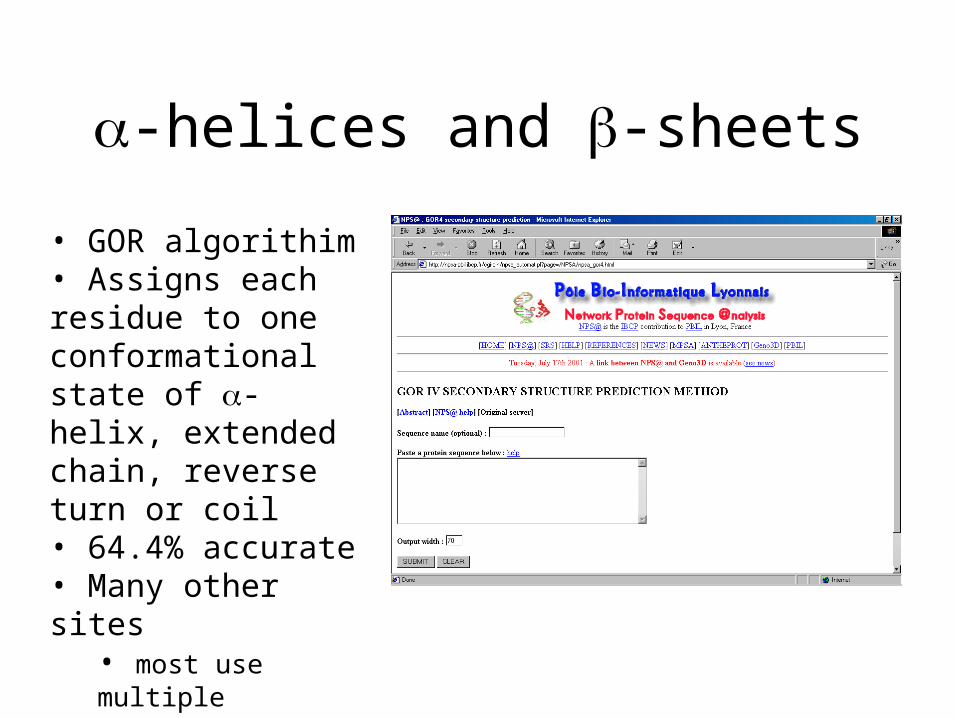
-helices and -sheets
• GOR algorithim• Assigns each residue to one conformational state of -helix, extended chain, reverse turn or coil• 64.4% accurate• Many other sites
• most use multiple alignments
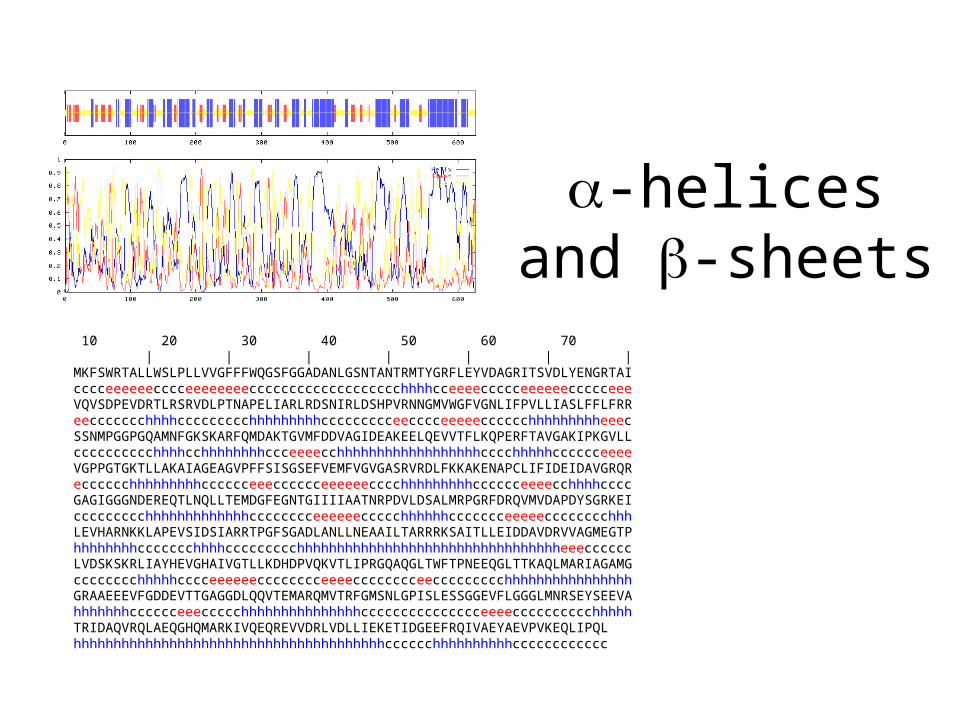
-helices and -sheets
10 20 30 40 50 60 70 | | | | | | |MKFSWRTALLWSLPLLVVGFFFWQGSFGGADANLGSNTANTRMTYGRFLEYVDAGRITSVDLYENGRTAIcccceeeeeecccceeeeeeeeccccccccccccccccccchhhhcceeeeccccceeeeeeccccceeeVQVSDPEVDRTLRSRVDLPTNAPELIARLRDSNIRLDSHPVRNNGMVWGFVGNLIFPVLLIASLFFLFRReeccccccchhhhccccccccchhhhhhhhhccccccccceecccceeeeecccccchhhhhhhhheeecSSNMPGGPGQAMNFGKSKARFQMDAKTGVMFDDVAGIDEAKEELQEVVTFLKQPERFTAVGAKIPKGVLLcccccccccchhhhcchhhhhhhhccceeeecchhhhhhhhhhhhhhhhhhcccchhhhhcccccceeeeVGPPGTGKTLLAKAIAGEAGVPFFSISGSEFVEMFVGVGASRVRDLFKKAKENAPCLIFIDEIDAVGRQRecccccchhhhhhhhhcccccceeecccccceeeeeecccchhhhhhhhhcccccceeeecchhhhccccGAGIGGGNDEREQTLNQLLTEMDGFEGNTGIIIIAATNRPDVLDSALMRPGRFDRQVMVDAPDYSGRKEIccccccccchhhhhhhhhhhhhcccccccceeeeeeccccchhhhhhccccccceeeeecccccccchhhLEVHARNKKLAPEVSIDSIARRTPGFSGADLANLLNEAAILTARRRKSAITLLEIDDAVDRVVAGMEGTPhhhhhhhhccccccchhhhccccccccchhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhheeeccccccLVDSKSKRLIAYHEVGHAIVGTLLKDHDPVQKVTLIPRGQAQGLTWFTPNEEQGLTTKAQLMARIAGAMGcccccccchhhhhcccceeeeeecccccccceeeecccccccceeccccccccchhhhhhhhhhhhhhhhGRAAEEEVFGDDEVTTGAGGDLQQVTEMARQMVTRFGMSNLGPISLESSGGEVFLGGGLMNRSEYSEEVAhhhhhhhcccccceeeccccchhhhhhhhhhhhhhhccccccccccccccceeeecccccccccchhhhhTRIDAQVRQLAEQGHQMARKIVQEQREVVDRLVDLLIEKETIDGEEFRQIVAEYAEVPVKEQLIPQLhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhcccccchhhhhhhhhhcccccccccccc
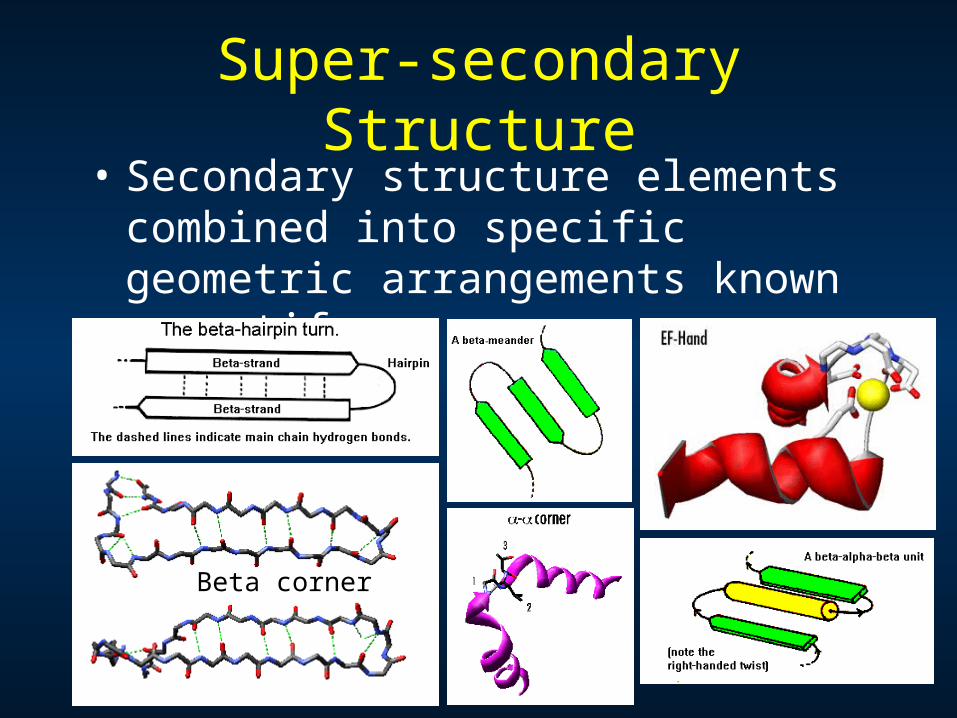
Super-secondary Structure• Secondary structure elements
combined into specific geometric arrangements known as motifs
Beta corner

Super-secondary Structure
Several programs/websites for specific domains e.g.
• PAIRCOIL and MULTICOIL - detect coiled-coiled regions– regions separating domains
• TRESPASSER - detects Leucine Zippers– Leu-X6-Leu-X6-Leu-X6-Leu protein interaction domain
• NPS@nalysis Helix-Turn-Helix– Protein interaction/DNA binding

Integrated stucture prediction
• One stop shop!• Predict Protein at EBI
– secondary structure
– solvent accessibility globular regions
– transmembrane helices coiled-coil regions
– a multiple sequence alignment ProSite sequence motifs
– low-complexity retions
– ProDom domain assignments
Tertiary Structure Prediction
• Homology modelling
• Fold recognition
• Threading
• Model building
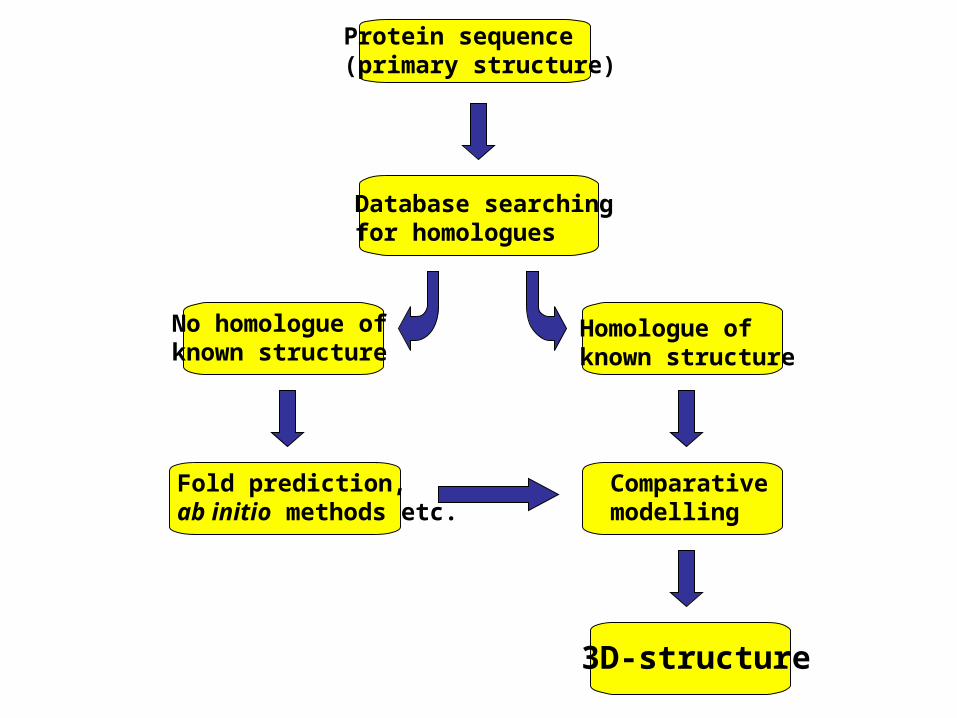
Protein sequence(primary structure)
Database searchingfor homologues
Homologue ofknown structure
No homologue ofknown structure
Comparativemodelling
3D-structure
Fold prediction,ab initio methods etc.

Homology Modelling
• Method of choice following BLAST search
• SWISSModel is agood WWWInterface
URL: http://www.expasy.ch/swissmod/SWISS-MODEL.html

• Requires at least one sequence of known 3D-structure with significant similarity to the target sequence.
• Compare the target sequence with database - FastA and BLAST.
• Sequences with a FastA score 10.0 standard deviations above the mean of the random scores or a P(N) lower than 10-5 (BLAST) considered for the model building
• Restrict to those which share at least 30% residue identity
Homology Modelling

Homology Modelling
• Framework construction– compare atom positions - Cs
• Build non-conserved loops
• Complete backbone - add other atoms
• Add side chains
• Refine
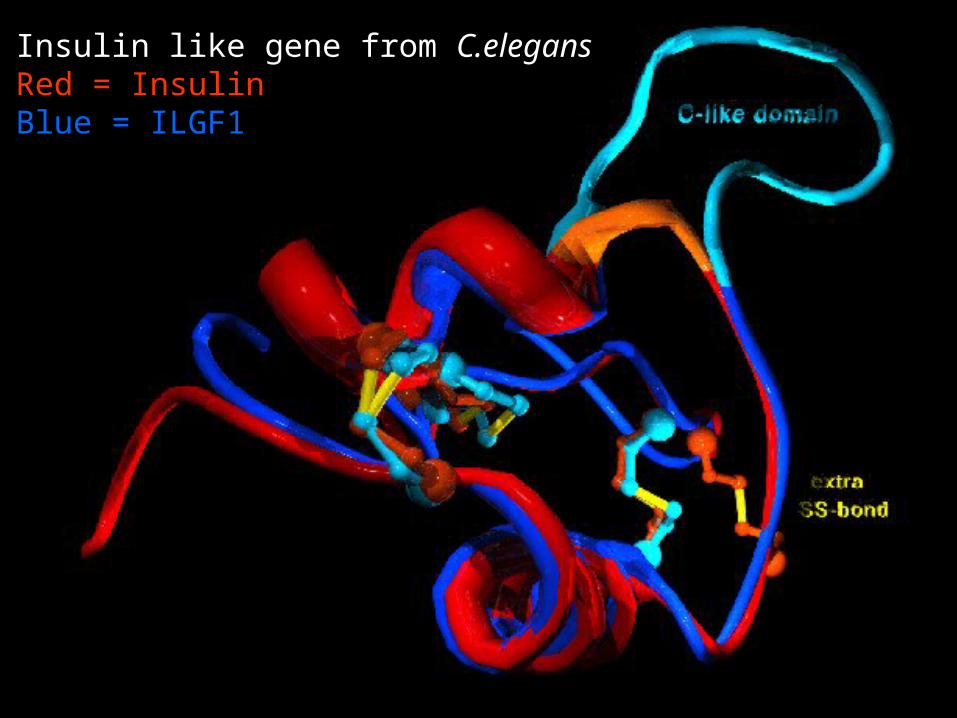
Insulin like gene from C.elegansRed = InsulinBlue = ILGF1

What if I have no homologue?
Ab initio methods - Threading
• Sequence of unknown structure
• Thread through a through a sequence of known structure
• Move query sequence through residue by resudue and compare computationally
– include thermodynamic criteria, solvent accessibility, secondary structure information
• Computing intensive
http://www.cs.bgu.ac.il/~bioinbgu/form.html