Embed Size (px)
Citation preview

1
ANEXO I
RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO

2
Este medicamento está sujeito a monitorização adicional. Isto irá permitir a rápida identificação
de nova informação de segurança. Pede-se aos profissionais de saúde que notifiquem quaisquer
suspeitas de reações adversas. Para saber como notificar reações adversas, ver secção 4.8.
1. NOME DO MEDICAMENTO
Benlysta 200 mg solução injetável em caneta pré-cheia.
Benlysta 200 mg solução injetável em seringa pré-cheia.
2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Caneta pré-cheia
Cada caneta pré-cheia de 1 ml contém 200 mg de belimumab.
Seringa pré-cheia
Cada seringa pré-cheia de 1 ml contém 200 mg de belimumab.
Belimumab é um anticorpo monoclonal recombinado IgG1λ humanizado, produzido numa linha
celular de mamíferos (CHO) por tecnologia de ADN recombinante.
Lista completa de excipientes, ver secção 6.1.
3. FORMA FARMACÊUTICA
Solução injetável em caneta pré-cheia (injetável).
Solução injetável em seringa pré-cheia (injetável).
Solução límpida a opalescente, incolor a amarelo pálido, com um pH de 6.
4. INFORMAÇÕES CLÍNICAS
4.1 Indicações terapêuticas
Benlysta é indicado como terapêutica adjuvante em doentes adultos com lúpus eritematoso sistémico
(LES) ativo, positivo para autoanticorpos, com um elevado grau de atividade da doença (por exemplo,
positivo, para anti-dsADN e complemento baixo) apesar de estarem a receber terapêutica padrão (ver
secção 5.1).
4.2 Posologia e modo de administração
O tratamento com Benlysta deve ser iniciado e supervisionado por um médico qualificado e
experiente no diagnóstico e tratamento de LES. Recomenda-se que a primeira injeção por via
subcutânea de Benlysta seja realizada sob supervisão de um profissional de saúde e num local
suficientemente habilitado para controlar reações de hipersensibilidade, se necessário. O profissional
de saúde deve providenciar treino adequado na técnica de administração subcutânea e educar sobre os
sinais e sintomas de reações de hipersensibilidade (ver secção 4.4). O doente pode autoinjetar-se ou o
cuidador do doente pode administrar Benlysta, após o profissional determinar que é apropriado.
Posologia

3
A dose recomendada é de 200 mg, uma vez por semana, administrada por via subcutânea. A dose não
é baseada no peso (ver secção 5.2).
O estado de saúde do doente deve ser avaliado continuamente. A descontinuação do tratamento com
Benlysta deve ser considerada se não existirem melhorias no controlo da doença após 6 meses de
tratamento.
Se falhar uma dose, esta deve ser administrada assim que possível. Depois, os doentes podem
recomeçar a administração da dose no dia habitual de administração, ou podem iniciar um novo
calendário semanal a partir do dia em que a dose em falta foi administrada. Não é necessário
administrar duas doses no mesmo dia.
Se os doentes desejarem alterar o dia da administração da sua dose semanal, pode ser administrada
uma nova dose no novo dia preferido da semana. Depois, o doente deve continuar com o novo
calendário semanal a partir desse dia, mesmo que o intervalo entre as doses seja temporariamente
inferior a menos de uma semana.
Transição da administração por via intravenosa para a via subcutânea
Se um doente está a transitar da administração de Benlysta por via intravenosa para a administração
por via subcutânea, a primeira injeção subcutânea deve ser administrada 1 a 4 semanas após a última
dose intravenosa (ver secção 5.2).
Populações especiais
Indivíduo idosos
A eficácia e segurança de Benlysta em idosos não foram estabelecidas. Os dados em doentes ≥65 anos
de idade estão limitados a <1,8% da população estudada. Consequentemente, não é recomendado o
uso de Benlysta em doentes idosos exceto se for esperado que os benefícios excedam os riscos. No
caso de se julgar necessária a administração de Benlysta a doentes idosos, não é necessário qualquer
ajuste da dose (ver secção 5.2).
Compromisso renal
Belimumab foi estudado num número limitado de doentes com LES com compromisso renal. Com
base na informação disponível, o ajuste de dose não é necessário em doentes com compromisso renal
ligeiro, moderado ou grave. Contudo, recomenda-se precaução em doentes com compromisso renal
grave devido à falta de dados (ver secção 5.2).
Compromisso hepático
Não foram realizados estudos específicos do uso de Benlysta em doentes com compromisso hepático.
É pouco provável que os doentes com compromisso hepático necessitem de um ajuste de dose (ver
secção 5.2).
População pediátrica
A segurança e eficácia de Benlysta em crianças e adolescentes (< 18 anos de idade) não foram
estabelecidas. Não existem dados disponíveis.
Modo de administração
A caneta pré-cheia ou a seringa pré-cheia devem ser utilizadas apenas para administração por via
subcutânea. Os locais de administração recomendados são o abdómem ou a coxa. Quando a
administração da injeção for na mesma região, os doentes devem ser aconselhados a usar um local de
injeção diferente, em cada semana. As injeções nunca devem ser administradas em zonas em que a
pele está sensível, ferida, vermelha ou dura.
Instruções detalhadas para a administração por via subcutânea de Benlysta em caneta pré-cheia ou
seringa pré-cheia são fornecidas no final do Folheto Informativo (Instruções passo a passo).

4
4.3 Contraindicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes mencionados na secção 6.1.
4.4 Advertências e precauções especiais de utilização
Benlysta não foi estudado nos seguintes grupos de doentes, e não é recomendado para:
• lúpus ativo grave com envolvimento do sistema nervoso central
• nefrite lúpica ativa grave (ver secção 5.1)
• VIH
• história ou diagnóstico atual de hepatite B ou C
• hipogamaglobulinemia (IgG <400 mg/dl) ou deficiência em IgA (IgA <10 mg/dl).
• história de transplante de órgão major ou transplante hematopoiético estaminal
celular/medula óssea ou transplante renal.
Uso concomitante com terapêuticas dirigidas às células B ou ciclofosfamida
Benlysta não foi estudado em combinação com outras terapêuticas dirigidas às células B ou
ciclofosfamida intravenosa. Recomenda-se precaução se Benlysta for administrado
concomitantemente com outras terapêuticas dirigidas às células B ou ciclofosfamida.
Reações de hipersensibilidade
A administração de Benlysta por via subcutânea ou intravenosa pode resultar em reações de
hipersensibilidade que podem ser graves e fatais. No caso de ocorrer uma reação grave, a
administração de Benlysta deve ser interrompida e administrada terapêutica médica apropriada (ver
secção 4.2). O risco de reações de hipersensibilidade é superior nas primeiras duas doses, contudo o
risco deve ser considerado para todas as administrações. Os doentes com história de alergias
medicamentosas múltiplas ou de hipersensibilidade significativa podem estar em maior risco. Foi
também observada recorrência de reações clinicamente significativas após o tratamento apropriado
inicial dos sintomas (ver secções 4.2 e 4.8).
Os doentes devem ser avisados de que são possíveis reações de hipersensibilidade no dia, ou nos
vários dias subsequentes, da administração e ser informados sobre os sinais e sintomas potenciais e da
possibilidade de recorrência. Os doentes devem ser instruídos a procurar ajuda médica imediata se
manifestarem algum desses sintomas. O folheto informativo deve ser entregue ao doente. As reações
de hipersensibilidade não agudas, do tipo retardado, foram também observadas e incluíram sintomas
como erupções cutâneas, náuseas, fadiga, mialgia, cefaleias e edema facial.
Nos ensaios clínicos por via intravenosa, as reações à perfusão ou de hipersensibilidade incluíram
reações anafiláticas, bradicardia, hipotensão, angioedema e dispneia. Ver o Resumo das
Características do Medicamento (RCM) de Benlysta pó para concentrado para solução para perfusão
(secção 4.4).
Infeções

5
O mecanismo de ação de belimumab pode aumentar o risco para o desenvolvimento de infeções,
incluindo infeções oportunistas. Foram notificadas infeções graves, incluindo casos fatais, em doentes
com LES a administrar terapêutica imunossupressora, incluindo belimumab (ver secção 4.8). Os
médicos devem tomar precauções ao considerar o uso de Benlysta em doentes com infeções graves ou
crónicas ou história de infeções recorrentes. Os doentes que desenvolvam uma infeção durante o
tratamento com Benlysta devem ser atentamente monitorizados e ser dada cuidadosa consideração
quanto à interrupção da terapêutica imunossupressora, incluindo o belimumab, até que a infeção seja
resolvida. O risco de utilizar Benlysta em doentes com tuberculose ativa ou latente é desconhecido.
Leucoencefalopatia multifocal progressiva (LMP)
A leucoencefalopatia multifocal progressiva (LMP) tem sido notificada com o tratamento com
Benlysta no LES. Os médicos devem estar particularmente atentos para os sintomas sugestivos de
LMP que os doentes possam não perceber (por exemplo, sinais ou sintomas cognitivos, neurológicos
ou psiquiátricos). Os doentes devem ser monitorizados para qualquer novo sintoma ou sinal ou seu
agravamento, e se ocorrerem estes sintomas/sinais serem referenciados para um neurologista e serem
consideradas medidas de diagnóstico apropriadas para a LMP. Se se suspeitar de LMP, deve ser
suspensa a administração de mais doses até ser excluída a LMP.
Imunização
As vacinas vivas não devem ser administradas durante os 30 dias antes ou concomitantemente com
Benlysta, uma vez que a segurança clínica não foi estabelecida. Não existem dados disponíveis sobre
a transmissão secundária da infeção em pessoas a quem foram administradas vacinas vivas para
doentes a quem está a ser administrado Benlysta.
Devido ao seu mecanismo de ação, belimumab pode interferir com a resposta a imunizações.
Contudo, num pequeno estudo que avaliou a resposta da vacina pneumocócica 23 valente,
globalmente as respostas imunitárias aos diferentes serotipos foram semelhantes nos doentes com
LES a administrar Benlysta, em comparação com os que estavam a administrar o tratamento
imunossupressor padrão à altura da vacinação. Os dados existentes são insuficientes para tirar
conclusões sobre a resposta a outras vacinas.
Dados limitados sugerem que Benlysta não afeta significativamente a capacidade de manter uma
resposta imunitária protetora referente a imunizações recebidas antes da administração de Benlysta.
Num subestudo, um pequeno grupo de doentes que tinha anteriormente recebido vacinas contra o
tétano, pneumococos ou gripe demonstrou manter títulos protetores após tratamento com Benlysta.
Neoplasias e doenças linfoproliferativas
Os medicamentos imunomodeladores, incluindo Benlysta, podem aumentar o risco de neoplasias.
Deve ser tomada precaução ao considerar a terapêutica com Benlysta em doentes com história de
neoplasias ou ao considerar a continuidade da terapêutica em doentes que desenvolvam neoplasias.
Não foram estudados doentes com neoplasias malignas nos últimos 5 anos, exceto aqueles com
carcinoma cutâneo basocelular ou das células escamosas ou com carcinoma do colo do útero, que
tenham sido completamente excisados cirurgicamente ou adequadamente tratados.
Conteúdo em sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose, ou seja, é praticamente
“isento de sódio”.
4.5 Interações medicamentosas e outras formas de interação
Não foram realizados estudos de interação in vivo. A formação de algumas enzimas CYP450 é
suprimida pelos níveis aumentados de certas citoquinas durante a inflamação crónica. Não é

6
conhecido se belimumab possa ser um modulador indireto destas citoquinas. Um risco para a redução
indireta da atividade CYP pelo belimumab, não pode ser excluído. Durante o início ou descontinuação
de belimumab, deve ser considerada a monitorização da terapêutica para os doentes em tratamento
com substratos CYP com um índice terapêutico estreito, em que a dose é ajustada individualmente
(por exemplo, varfarina).
4.6 Fertilidade, gravidez e aleitamento
Mulheres com potencial para engravidar/Contraceção em homens e mulheres
Mulheres em idade fértil têm de utilizar métodos contracetivos eficazes durante o tratamento com
Benlysta e até pelo menos 4 meses após o último tratamento.
Gravidez
Existe uma quantidade limitada de dados do uso de Benlysta em mulheres grávidas. Não foram
efetuados estudos formais. Para além de um efeito farmacológico espectável, i.e. depleção de células
B, estudos animais em macacos não indiciam efeitos prejudiciais diretos ou indiretos no que diz
respeito à toxicidade reprodutiva (ver secção 5.3).
Benlysta não deve ser utilizado durante a gravidez a menos que o potencial benefício justifique o
potencial risco para o feto.
Amamentação
Desconhece-se se Benlysta é excretado no leite materno ou se é absorvido sistemicamente após
ingestão. Contudo, belimumab foi detetado no leite de macacos fêmea às quais foram administrados
150 mg/kg a cada 2 semanas.
Dado que os anticorpos maternos (IgG) são excretados no leite materno, recomenda-se que a decisão
deva ser tomada entre descontinuar a amamentação ou descontinuar a terapêutica com Benlysta, tendo
em consideração o benefício da amamentação para a criança e o benefício da terapêutica para a
mulher.
Fertilidade
Não existem dados sobre os efeitos de belimumab na fertilidade humana. Os efeitos na fertilidade
masculina e feminina não foram formalmente avaliados durante os estudos animais (ver secção 5.3).
4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas
Não foram efetuados estudos sobre os efeitos na capacidade de conduzir e utilizar máquinas. Não se
preveem efeitos prejudiciais para estas atividades a partir da farmacologia de belimumab. O estado
clínico do indivíduo e o perfil de reações adversas de Benlysta deve estar presente ao considerar a
capacidade do doente desempenhar tarefas que exijam julgamento ou uso das capacidades motoras ou
cognitivas.
4.8 Efeitos indesejáveis
Resumo do perfil de segurança
A segurança de belimumab em doentes com LES foi avaliada em 3 estudos por via intravenosa,
controlados por placebo e em 1 estudo por via subcutânea, controlado por placebo.
Os dados apresentados na tabela em baixo refletem a exposição a Benlysta (10 mg/kg por via
intravenosa durante o período de 1 hora aos dias 0, 14, 28 e depois de 28 em 28 dias, durante até 52

7
semanas) administrado em 674 doentes com LES, incluindo 472 expostos durante um período de pelo
menos 52 semanas e 556 doentes expostos a 200 mg de Benlysta por via subcutânea uma vez por
semana, durante até 52 semanas. Os dados de segurança apresentados incluem dados para além da
semana 52 em alguns doentes. Também é incluída a informação proveniente de notificações após a
comercialização.
A maioria dos doentes estava também a receber uma ou mais das seguintes terapêuticas concomitantes
para LES: corticosteroides, medicamentos imunomoduladores, antimaláricos, medicamentos anti-
inflamatórios não esteroides.
Foram notificadas reações adversas em 87% dos doentes tratados com Benlysta e em 90% dos doentes
tratados com placebo. As reações adversas mais frequentemente notificadas (≥5% dos doentes com
LES tratados com Benlysta mais o tratamento padrão e a uma taxa ≥1% mais elevada que com
placebo) foram infeções víricas do trato respiratório superior, bronquite e diarreia. A proporção de
doentes que descontinuaram o tratamento devido a reações adversas foi de 7% para os doentes
tratados com Benlysta e de 7% dos doentes tratados com placebo.
Sumário tabelado das reações adversas
As reações adversas estão listadas abaixo de acordo com a classificação de sistemas de órgãos
MedDRA e por frequência. As categorias de frequência utilizadas são:
Muito frequente s > 1/10
Frequentes 1/100, <1/10
Pouco frequentes 1/1.000, <1/100
Raros >1/10.000, <1/1.000
Os efeitos indesejáveis são apresentados em ordem decrescente de gravidade dentro de cada classe de
frequência. A frequência incluída refere-se à superior observada com qualquer formulação.
Classificação de sistema de
órgãos
Frequência
Reações Adversas
Infeções e infestações Muito frequentes Infeções bacterianas, por exemplo
bronquite, infeção do trato urinário
Frequentes Gastroenterite vírica, faringite,
nasofaringite, infeção víricas do trato
respiratório superior
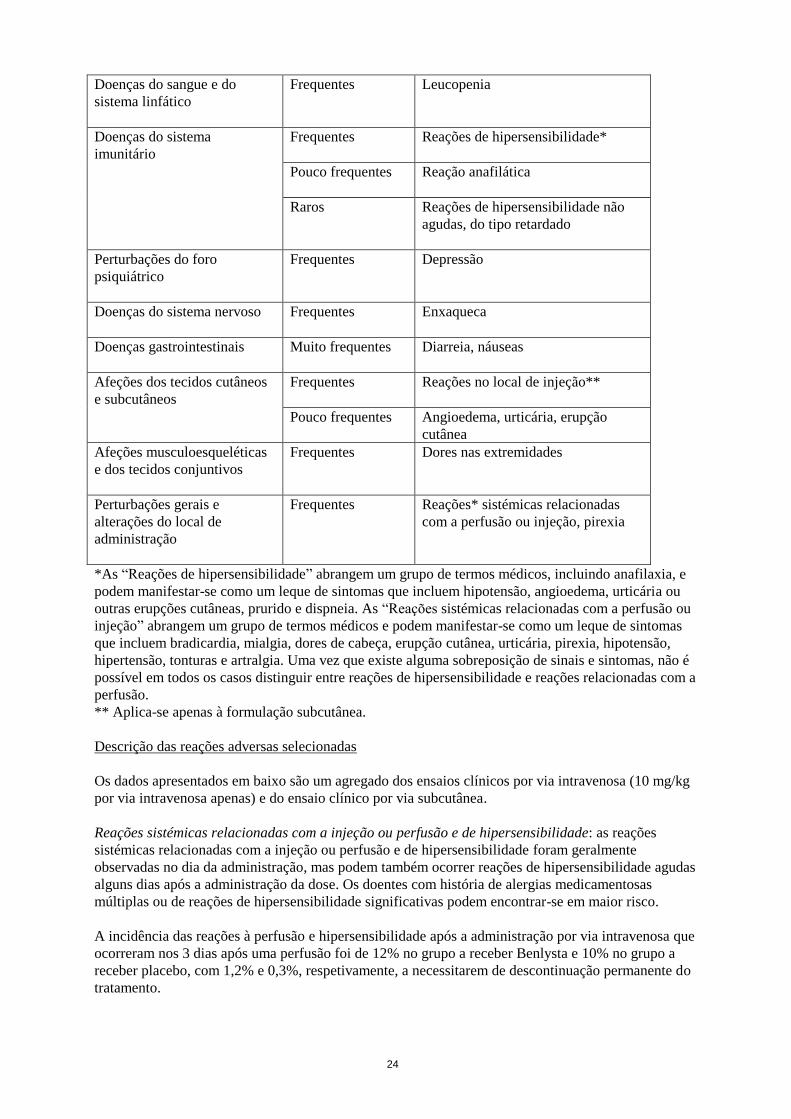
Doenças do sangue e do
sistema linfático
Frequentes Leucopenia
Doenças do sistema
imunitário
Frequentes Reações de hipersensibilidade*
Pouco frequentes Reação anafilática
Raros Reações de hipersensibilidade não
agudas, do tipo retardado
Perturbações do foro
psiquiátrico
Frequentes Depressão
Doenças do sistema nervoso
Frequentes Enxaqueca
Doenças gastrointestinais Muito frequentes Diarreia, náuseas

8
Afeções dos tecidos cutâneos
e subcutâneos
Frequentes
Reações no local de injeção**
Pouco frequentes Angioedema, urticária, erupção
cutânea
Afeções musculoesqueléticas
e dos tecidos conjuntivos
Frequentes Dores nas extremidades
Perturbações gerais e
alterações do local de
administração
Frequentes Reações* sistémicas relacionadas
com a perfusão ou injeção, pirexia
*As “Reações de hipersensibilidade” abrangem um grupo de termos médicos, incluindo anafilaxia, e
podem manifestar-se como um leque de sintomas que incluem hipotensão, angioedema, urticária ou
outras erupções cutâneas, prurido e dispneia. As “Reações sistémicas relacionadas com a perfusão ou
injeção” abrangem um grupo de termos médicos e podem manifestar-se como um leque de sintomas
que incluem bradicardia, mialgia, dores de cabeça, erupção cutânea, urticária, pirexia, hipotensão,
hipertensão, tonturas e artralgia. Uma vez que existe alguma sobreposição de sinais e sintomas, não é
possível em todos os casos distinguir entre reações de hipersensibilidade e reações relacionadas com a
perfusão.
** Aplica-se apenas à formulação subcutânea.
Descrição das reações adversas selecionadas
Os dados apresentados em baixo são um agregado dos ensaios clínicos por via intravenosa (10 mg/kg
por via intravenosa apenas) e do ensaio clínico por via subcutânea.
Reações sistémicas relacionadas com a injeção ou perfusão e de hipersensibilidade: as reações
sistémicas relacionadas com a injeção ou perfusão e de hipersensibilidade foram geralmente
observadas no dia da administração, mas podem também ocorrer reações de hipersensibilidade agudas
alguns dias após a administração da dose. Os doentes com história de alergias medicamentosas
múltiplas ou de reações de hipersensibilidade significativas podem encontrar-se em maior risco.
A incidência das reações à perfusão e hipersensibilidade após a administração por via intravenosa que
ocorreram nos 3 dias após uma perfusão foi de 12% no grupo a receber Benlysta e 10% no grupo a
receber placebo, com 1,2% e 0,3%, respetivamente, a necessitarem de descontinuação permanente do
tratamento.
A incidência de reações sistémicas após a injeção e reações de hipersensibilidade que ocorreram nos 3
dias após a administração por via subcutânea foi de 7% no grupo a receber Benlysta e 9% no grupo a
receber placebo. As reações de hipersensibilidade clinicamente significativas associadas com a
administração por via subcutânea e que necessitaram de descontinuação permanente do tratamento
foram notificadas em 0,2% dos doentes a receber Benlysta e em nenhum dos doentes a receber
placebo.
Infeções: A incidência global de infeções nos estudos por via intravenosa e subcutânea foi de 63% em
ambos os grupos que administraram Benlysta ou placebo. As infeções ocorreram em pelo menos 3%
dos doentes que administraram Benlysta, e pelo menos 1% mais frequentemente do que os doentes
que administraram placebo, foram infeção do trato respiratório superior, bronquite e infeção
bacteriana do trato urinário. As infeções graves ocorreram em 5% dos doentes em ambos os grupos
que administraram Benlysta ou placebo, as infeções oportunistas graves representaram 0,4% e 0%
destas, respetivamente. As infeções que originaram descontinuação do tratamento ocorreram em 0,7%
dos doentes a administrar Benlysta e 1,5% dos doentes a administrar placebo. Algumas infeções
foram graves ou fatais.

9
Leucopenia: A incidência de leucopenia notificada como acontecimento adverso foi de 3% no grupo a
administrar Benlysta e de 2% no grupo a administrar placebo.
Perturbações do foro psiquiátrico: A incidência de depressão notificada como acontecimento
adverso foi de 3% em ambos os grupos a administrar Benlysta ou placebo.
Reações no local de injeção: No ensaio clínico por via subcutânea, a frequência de reações no local
de injeção foi de 6,1% (34/556) e 2,5% (7/280) para os doentes a administrar Benlysta e placebo,
respetivamente. Estas reações no local de injeção (mais frequentemente dor, eritema, hematoma,
prurido e endurecimento) foram de gravidade ligeira a moderada. A maioria não requereu a
descontinuação do medicamento.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas após a autorização do medicamento é importante, uma
vez que permite uma monitorização contínua da relação benefício-risco do medicamento. Pede-se aos
profissionais de saúde que notifiquem quaisquer suspeitas de reações adversas através do sistema
nacional de notificação mencionado no Apêndice V.
4.9 Sobredosagem
A experiência clínica de sobredosagem com Benlysta é limitada. As reações adversas notificadas em
associação com casos de sobredosagem foram consistentes com as esperadas para belimumab.
Duas doses até 20 mg/kg administradas com 21 dias de intervalo por perfusão intravenosa foram
administradas a seres a humanos sem aumento na incidência ou gravidade das reações adversas
quando comparadas com doses de 1, 4 ou 10 mg/kg.
No caso de sobredosagem não intencional, os doentes devem ser cuidadosamente observados e, se
apropriado, devem ser administrados cuidados de suporte.
5. PROPRIEDADES FARMACOLÓGICAS
5.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Imunossupressores seletivos, código ATC: L04AA26
Mecanismo de ação
Belimumab é um anticorpo monoclonal IgG1 humano, específico para a proteína humana solúvel
Estimuladora dos Linfócitos B (BLyS, também referida como BAFF e TNFSF13B). Belimumab
bloqueia a ligação do BlyS solúvel, um fator de sobrevivência celular, aos seus recetores nas células
B. Belimumab não se liga às células B diretamente, mas ao ligar-se ao BLyS, Belimumab inibe a
sobrevivência das células B, incluindo as células B autorreactivas, e reduz a diferenciação das células
B em células plasmáticas produtoras de imunoglobulinas.
Os níveis de BLyS estão elevados em doentes com LES e outras doenças autoimunes. Existe uma
associação entre os níveis plasmáticos de BLyS e a atividade da doença LES. A contribuição relativa
dos níveis de BLyS para a patofisiologia do LES não está completamente compreendida.
Efeitos farmacodinâmicos
A mediana dos níveis IgG à semana 52 foi reduzida em 11% nos doentes a receberem Benlysta, em
comparação com o aumento em 0,7% nos doentes a receber placebo.

10
Em doentes com anticorpos anti ds-ADN na linha de base inicial, a mediana dos níveis de anticorpos
anti ds-ADN à semana 52 foi reduzida em 56% nos doentes a receber Benlysta, em comparação com
41% nos doentes a receber placebo. Nos doentes com anticorpos anti-dsADN na linha de base inicial,
à semana 52, 18% dos doentes tratados com Benlysta tinham convertido para anti-dsADN negativos,
em comparação com 15% dos doentes a receber placebo.
Em doentes com níveis de complemento baixos, foi observada à semana 52 a normalização de C3 e
C4 em 42% e 53% dos doentes a receberem Benlysta e em 21% e 20% dos doentes a receberem
placebo, respetivamente.
Benlysta reduziu significativamente a circulação global das células B, transitórias, naive (não
ativadas) e LES bem como células plasmáticas, à semana 52. As reduções nas células B naive e
transitórias bem como no subgrupo das células B LES, foram observadas tão cedo como à semana 8.
As células de memória aumentaram inicialmente e diminuíram lentamente em direção à linha de base
inicial à semana 52.
A resposta das células B e IgG ao tratamento a longo prazo com Benlysta por via intravenosa foi
avaliada num estudo de extensão, não controlado. Após 7 anos e meio de tratamento (incluindo o
estudo principal de 72 semanas), foi observada uma diminuição substancial e sustentada em vários
subtipos de células B, conduzindo a uma redução mediana de 87% nas células B não ativadas (naive),
de 67% nas células B de memória, de 99% nas células B ativadas e uma redução mediana de 92% dos
níveis das células plasmáticas após mais de 7 anos de tratamento. Após cerca de 7 anos, foi observada
uma redução mediana de 28% dos níveis de IgG, com 1,6% dos indivíduos a terem uma diminuição
nos níveis de IgG para abaixo de 400 mg/dl. Ao longo do estudo, a incidência de notificações de
acontecimentos adversos, em geral, manteve-se estável ou diminuiu.
Imunogenicidade
No estudo por via subcutânea foram analisadas amostras séricas de mais de 550 doentes com LES,
não foram detetados anticorpos anti-belimumab durante o tratamento ou após o tratamento com 200
mg de belimumab por via subcutânea.
Eficácia e segurança clínicas
Injeção subcutânea
A eficácia de Benlysta administrado por via subcutânea foi avaliada num estudo de Fase III com
aleatorização, em dupla ocultação, controlado por placebo, de 52 semanas (HGS1006-C1115;
BEL112341), realizado em 836 doentes adultos com um diagnóstico clínico de LES de acordo com os
critérios de classificação do Colégio Americano de Reumatologia. Os doentes elegíveis tinham
doença LES ativa, definida como um resultado 8 na escala SELENA-SLEDAI e resultados positivos
no teste para deteção de anticorpos antinucleares (ANA ou anti-dsADN; títulos de ANA 1:80 e/ou
um resultado positivo anti-dsADN [30 unidades/ml]) durante avaliação inicial. Os doentes estavam a
receber um regime terapêutico estável (cuidados padrão) para LES, que consistia num dos seguintes
(isolados ou em combinação): corticosteroides, antimaláricos, anti-inflamatórios não esteroides
(AINEs) ou outros imunossupressores. Os doentes eram excluídos do estudo se tinham lúpus ativo
grave com envolvimento do sistema nervoso central ou nefrite lúpica ativa grave.
Este estudo foi realizado nos EUA, América do Sul, Europa e Ásia. A mediana das idades dos doentes
foi de 37 anos (intervalo de 18 a 77 anos) e a maioria (94%) era do género feminino. A medicação
prévia concomitante incluiu corticosteroides (86%; >7,5 mg/dia de prednisolona equivalente a 60%),
imunossupressores (46%) e antimaláricos (69%). Os doentes foram aleatorizados numa proporção de
2:1 para a administração de 200 mg de belimumab ou placebo, por via subcutânea, uma vez por
semana durante 52 semanas.
Na linha de base inicial, 62,2% dos doentes tinha atividade elevada da doença (resultado SELENA
SLEDAI de >10), 88% dos doentes tinha envolvimento mucocutâneo, 78% dos doentes tinham

11
envolvimento musculoesquelético, 8% dos doentes tinham envolvimento hematológico, 12% dos
doentes tinham envolvimento renal e 8% dos doentes tinham envolvimento vascular dos órgãos.
O objetivo primário de eficácia foi um objetivo composto (Índice de Resposta LES) que definia a
resposta como o atingir cada um dos seguintes critérios à semana 52 quando comparado com a linha
de base inicial:
• redução de 4 pontos na escala SELENA-SLEDAI e
• sem novos resultados de envolvimento de órgãos na escala British Isles Lupus Assessment
Group (BILAG) ou 2 novos resultados de envolvimento orgânico BILAG B, e
• sem agravamento (aumento <0,30 pontos) na escala de Avaliação Médica Global (PGA)
O Índice de Resposta LES mede a melhoria da atividade da doença LES, sem agravamento de
qualquer sistema de órgãos ou na condição geral do doente.
Tabela 1: Taxa de resposta à semana 52
Resposta
Placebo
(n=279)
Benlysta
200 mg
semanalmente
(n=554)
Índice de resposta LES
Diferença observada vs
placebo
Odds Ratio (OR) (IC 95%)
vs placebo
48,4%
61,4%
(p=0,0006)
12,98%
1,68
(1,25, 2,25)
Componentes do Índice de resposta LES
Percentagem de doentes com
redução na escala SELENA-
SLEDAI 4
49,1%
62,3%
(p=0,0005)
Percentagem de doentes sem
agravamento medido pelo
índice BILAG
74,2% 80,9%
(p=0,0305)
Percentagem de doentes sem
agravamento medido por
PGA
72,8% 81,2%
(p=0,0061)
Todos os doentes receberam a terapêutica padrão
As diferenças entre os grupos de tratamento foram aparentes pela semana 16 e sustentadas até à
semana 52 (Figura 1).
Figura 1. Proporção de respondedores SRI por consulta

12
Os picos (flares) no LES foram definidos pelo Índice de Picos SELENA SLEDAI no LES. O risco do
primeiro pico foi reduzido em 22% durante as 52 semanas de observação no grupo a administrar
Benlysta, em comparação com o grupo a administrar placebo (Hazard Ratio=0,78, p=0,0061). A
mediana do tempo para o primeiro pico, entre os doentes que tiveram um pico, foi atrasado no grupo a
administrar Benlysta quando comparado com placebo (190 dias vs 141 dias). Foram observados picos
graves em 10,6% dos doentes no grupo a administrar Benlysta em comparação com 18,2% dos
doentes no grupo a administrar placebo ao longo das 52 semanas de observação (diferença observada
entre tratamentos =-7,6%). O risco de picos graves foi reduzido em 49% durante as 52 semanas de
observação no grupo a administrar Benlysta, em comparação com o grupo a administrar placebo (risco
relativo=0,51, p=0,0004). A mediana do tempo para o primeiro pico grave, entre os doentes que
tiveram um pico grave, foi atrasada nos doentes a administrar Benlysta, em comparação com o
placebo (171 dias vs 118 dias).
A percentagem de doentes a administrar mais de 7,5 mg de prednisolona (ou equivalente) na linha de
base inicial, aos quais a média de corticosteroides foi reduzida em pelo menos 25% deste a linha basal
inicial para a dose equivalente de prednisolone ≤ 7,5 mg/dia durante as semanas 40 e 52, foi de 18,2%
no grupo a administrar Benlysta e 11,9%b no grupo a administrar placebo (p=0,0732).
Benlysta demonstrou melhorias na fadiga quando comparado com placebo, medido pela escala de
fadiga FACIT. A alteração média ajustada do resultado à Semana 52 a partir da linha de base inicial é
significativamente maior com Benlysta quando comparado com placebo (4,4 vs 2,67, p=0,0130).
A análise do subgrupo do objetivo primário demonstrou que o maior benefício foi observado em
doentes com atividade mais elevada da doença na linha de base inicial incluindo doentes com
resultados SELENA SLEDAI ≥ 10 ou doentes que necessitavam de esteroides para controlar a sua
doença ou doentes com níveis de complemento baixos.
Adicionalmente, no grupo previamente identificado serologicamente ativo, tal como doentes com
níveis de complemento baixos e resultados positivos para anti-dsADN na linha de base inicial,
também demonstrou uma resposta relativa superior, ver Tabela 2 para os resultados deste exemplo de
um grupo com atividade da doença elevada.
Tabela 2: Doentes com níveis de complemento baixos e resultados positivos anti-dsADN na linha
de base inicial

13
Subgrupo Anti-dsADN positivo E complemento
baixo
Placebo Benlysta
200 mg
semanalmente
Taxa de resposta SRI à semana 52 (%)
Diferença entre tratamentos observada vs placebo
(%)
(n=108)
47,2
(n=246)
64,6 (p=0,0014)
17,41
Picos graves ao longo das 52 semanas:
Doentes que sofreram um pico grave (%)
Diferença entre tratamentos observada vs placebo
(%)
Tempo até um pico grave [HR (95% CI)]
(n=108)
31,5
(n=248)
14,1
17,4
0,8 (0,24, 0,61)
(p<0,0001)
Redução de prednisolona em ≥25% desde a linha de
base inicial até ≤7,5 mg/dia durante as semanas 40
até 52* (%)
Diferença entre tratamentos observada vs placebo
(diferença média)
(n=70)
11,4
(n=164)
20,7 (p=0,0844)
9,3
Melhoria na escala de fadiga FACIT desde a linha de
base inicial até à semana 52 (média):
Diferença entre tratamentos observada vs placebo
(diferença média)
(n=108)
2,4
(n=248)
4,6 (p=0,0324)
2,1
* Entre doentes com dose de prednisolona >7,5 mg/dia na linha de base inicial
Idade e etnia
Não foram incluídos nos ensaios clínicos controlados doentes suficientes ≥ 65 anos de idade, ou
doentes de etnia negra/afro-americana, para se poderem retirar conclusões significativas sobre os
efeitos da idade ou etnia nos resultados clínicos.
População pediátrica
A Agência Europeia de Medicamentos diferiu a obrigação de submissão dos resultados dos estudos
com Benlysta num ou mais sub-grupos da população pediátrica com LES (ver secção 4.2 para
informação sobre utilização pediátrica).
5.2 Propriedades farmacocinéticas
Os parâmetros farmacocinéticos subcutâneos seguintes são baseados em estimativas dos parâmetros
da população de 661 doentes, incluindo 554 doentes com LES e 107 indivíduos saudáveis, que
administraram Benlysta por via subcutânea.
Absorção

14
Benlysta em caneta pré-cheia ou seringa pré-cheia destina-se administração por injeção por via
subcutânea.
Após a administração por via subcutânea, a biodisponibilidade de belimumab foi aproximadamente
74%. A exposição no estado de equilíbrio foi atingida após aproximadamente 11 semanas de
administração subcutânea. A concentração sérica máxima (Cmáx) de belimumab no estado de
equilíbrio foi de 108 µg/ml.
Distribuição
Belimumab distribuiu-se pelos tecidos com um volume de distribuição no estado de equilíbrio (Vss)
de aproximadamente 5 litros.
Biotransformação
Belimumab é uma proteína para a qual a via metabólica expectável é a degradação em pequenos
péptidos e aminoácidos individuais por enzimas proteolíticas largamente disseminadas. Não foram
realizados estudos clássicos de biotransformação.
Eliminação
Após a administração subcutânea, belimumab apresentou um tempo de semi-vida de eliminação de
18,3 dias. A depuração sistémica foi de 204 ml/dia.
Populações Especiais de Doentes
População pediátrica: Não existem dados farmacocinéticos em doentes pediátricos.
Indivíduos idosos: Benlysta foi estudado num número limitado de doentes idosos. A idade não afetou
a exposição ao belimumab na população farmacocinética subcutânea em análise. Contudo, dado o
reduzido número de indivíduos com ≥65 anos de idade, um efeito relacionado com a idade não pode
ser conclusivamente excluído.
Compromisso renal: Não foram realizados estudos específicos para examinar os efeitos do
compromisso renal na farmacocinética de belimumab. Durante o desenvolvimento clínico, Benlysta
foi estudado num número limitado de doentes com LES com compromisso renal ligeiro (depuração da
creatinina [CrCl] 60 e <90 ml/min), moderado (CrCl e <60 ml/min) ou grave (CrCl 15 e
<30 ml/min): 121 doentes com compromisso renal ligeiro e 30 doentes com compromisso renal
moderado administraram Benlysta por via subcutânea; 770 doentes com compromisso renal ligeiro,
261 doentes com compromisso renal moderado e 14 doentes com compromisso renal grave
administraram Benlysta por via intravenosa.
Não foi observada uma redução clínica significativa na depuração sistémica em resultado do
compromisso renal. Consequentemente, não é recomendado qualquer ajuste de dose para doentes com
compromisso renal.
Compromisso hepático: Não foram realizados estudos específicos para examinar os efeitos do
compromisso hepático na farmacocinética do belimumab. As moléculas IgG como o belimumab são
catabolizadas por enzimas proteolíticas largamente distribuídas, que não estão restritas ao tecido
hepático, e alterações da função hepática não irão provavelmente ter qualquer efeito na eliminação do
belimumab.
Peso corporal/Índice de Massa Corporal (IMC):

15
Os efeitos do peso corporal e do IMC na exposição de belimumab após a administração por via
subcutânea não foram considerados com significado clínico. Não houve impacto significativo na
eficácia e segurança com base no peso. Consequentemente, não é recomendado ajuste da dose.
Transição da administração por via intravenosa para a via subcutânea
Os doentes com LES em transição da administração de 10 mg/kg por via intravenosa, de 4 em 4
semanas, para 200 mg por via subcutânea semanalmente, utilizando o intervalo de permutação de 1 a
4 semanas, tiveram uma concentração sérica de belimumab pré-dose na sua primeira dose por via
subcutânea próximo da sua eventual concentração subcutânea no estado de equilíbrio (ver secção 4.2).
Com base em simulações com parâmetros farmacocinéticos da população, as concentrações médias de
belimumab no estado de equilíbrio para 200 mg por via subcutânea, todas as semanas, foram
semelhantes a 10 mg por via intravenosa, de 4 em 4 semanas.
5.3 Dados de segurança pré-clínica
Os dados não clínicos não revelam riscos especiais para o ser humano, segundo estudos de toxicidade
de dose repetida e toxicidade reprodutiva.
A administração intravenosa e subcutânea em macacos resultou numa redução esperada do número de
células B periféricas e nos tecidos linfoides sem achados toxicológicos associados.
Foram realizados estudos reprodutivos em macacos fêmea cinomolgos grávidas a receber 150 mg/kg
de belimumab por perfusão intravenosa (aproximadamente 9 vezes a exposição clínica máxima
antecipada em seres humanos) a cada 2 semanas durante 21 semanas, e o tratamento com belimumab
não foi associado com efeitos negativos diretos ou indiretos relacionados com toxicidade materna,
toxicidade do desenvolvimento ou teratogenicidade.
Os resultados relacionados com o tratamento foram limitados à redução das células B, esperada e
reversível, tanto nas progenitoras como nas crias e à redução reversível dos níveis de IgM nas crias de
macacos. As contagens de células B recuperaram após a cessação do tratamento com belimumab cerca
de 1 ano após parto em macacos adultos e perto dos 3 meses de vida nas crias de macacos; os níveis
de IgM em crias expostas in utero a belimumab recuperaram por volta dos 6 meses de idade.
Os efeitos na fertilidade masculina e feminina em macacos foram determinados em 6 meses de
estudos de toxicidade de dose repetida, em doses de até 50 mg/kg inclusive. Não foram notadas
alterações relacionadas com o tratamento nos órgãos reprodutivos masculinos e femininos de animais
sexualmente maduros. Uma avaliação informal do ciclo menstrual em fêmeas não demonstrou
alterações relacionadas com o belimumab.
Uma vez que belimumab é um anticorpo monoclonal não foram realizados estudos de genotoxicidade.
Não foram efetuados estudos de carcinogenicidade ou estudos de fertilidade (masculinos ou
femininos).
6. INFORMAÇÕES FARMACÊUTICAS
6.1. Lista dos excipientes
Cloridrato de arginina
Histidina
Monocloridrato de histidina
Polissorbato 80
Cloreto de sódio
Água para preparações injetáveis

16
6.2 Incompatibilidades
Não conhecidas.
6.3 Prazo de validade
3 anos.
6.4 Precauções especiais de conservação
Conservar no frigorífico (2ºC a 8ºC).
Não congelar.
Manter o frasco para injetáveis dentro da embalagem exterior para proteger da luz.
6.5 Natureza e conteúdo do recipiente
Caneta pré-cheia
1 ml de solução numa seringa de vidro tipo I com uma agulha fixa (aço inoxidável) numa caneta pré-
cheia.
Disponível em embalagens de 1 ou 4 canetas pré-cheias e embalagens múltiplas com 12 canetas pré-
cheias de dose única (3 embalagens de 4 canetas pré-cheias).
É possível que não sejam comercializadas todas as apresentações.
Seringa pré-cheia
1 ml de solução numa seringa de vidro tipo I com uma agulha fixa (aço inoxidável) e uma cápsula de
fecho da agulha.
Disponível em embalagem de 1 seringa pré-cheia ou embalagem de 4 seringas pré-cheias.
É possível que não sejam comercializadas todas as apresentações.
6.6 Precauções especiais de eliminação e manuseamento
Instruções detalhadas para administração por via subcutânea de Benlysta em caneta pré-cheia ou
seringa pré-cheia são fornecidas no final do Folheto Informativo (ver Instruções passo a passo).
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências
locais.
7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Reino Unido
8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO

17
EU/1/11/700/003: 1 caneta pré-cheia
EU/1/11/700/004: 4 canetas pré-cheias
EU/1/11/700/005: 12 (3x4) canetas pré-cheias (embalagem múltipla)
EU/1/11/700/006: 1 seringa pré-cheia
EU/1/11/700/007: 4 seringas pré-cheias
9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE
INTRODUÇÃO NO MERCADO
Data da primeira autorização: 13 de julho de 2011
Data da última renovação: 18 de fevereiro de 2016
10. DATA DA REVISÃO DO TEXTO
Está disponível informação pormenorizada sobre este medicamento no sítio da internet da Agência
Europeia de Medicamentos: http://www.ema.europa.eu.

18
Este medicamento está sujeito a monitorização adicional. Isto irá permitir a rápida identificação
de nova informação de segurança. Pede-se aos profissionais de saúde que notifiquem quaisquer
suspeitas de reações adversas. Para saber como notificar reações adversas, ver secção 4.8.
1. NOME DO MEDICAMENTO
Benlysta 120 mg pó para concentrado para solução para perfusão.
Benlysta 400 mg pó para concentrado para solução para perfusão.
2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Benlysta 120 mg pó para concentrado para solução para perfusão
Cada frasco para injetáveis contém 120 mg de belimumab. Após reconstituição, a solução contém 80
mg de belimumab por ml.
Benlysta 400 mg pó para concentrado para solução para perfusão
Cada frasco para injetáveis contém 400 mg de belimumab. Após reconstituição, a solução contém 80
mg de belimumab por ml.
Belimumab é um anticorpo monoclonal recombinado IgG1λ humanizado, produzido numa linha
celular de mamíferos (CHO) por tecnologia de ADN recombinante.
Lista completa de excipientes, ver secção 6.1.
3. FORMA FARMACÊUTICA
Pó para concentrado para solução para perfusão.
Pó branco a esbranquiçado.
4. INFORMAÇÕES CLÍNICAS
4.1 Indicações terapêuticas
Benlysta é indicado como terapêutica adjuvante em doentes adultos com lúpus eritematoso sistémico
(LES) ativo, positivo para autoanticorpos, com um elevado grau de atividade da doença (por exemplo,
positivo, para anti-dsADN e complemento baixo) apesar de estarem a receber terapêutica padrão (ver
secção 5.1).
4.2 Posologia e modo de administração
O tratamento com Benlysta deve ser iniciado e supervisionado por um médico qualificado e
experiente no diagnóstico e tratamento de LES. As perfusões de Benlysta devem ser administradas
por um profissional de saúde qualificado, treinado na administração de terapêuticas por perfusão.
A administração de Benlysta pode resultar em reações de hipersensibilidade graves ou que coloquem
a vida em risco e reações à perfusão. Foram notificados casos de doentes que desenvolveram sintomas
de hipersensibilidade aguda várias horas após a perfusão ter sido administrada. Foi também observada
recorrência de reações clinicamente significativas após o tratamento apropriado inicial dos sintomas
(ver secções 4.4 e 4.8). Consequentemente, Benlysta deve ser administrado num local onde estejam
imediatamente disponíveis recursos para o tratamento deste tipo de reações. Os doentes devem

19
permanecer sobre supervisão clínica durante um período de tempo prolongado (durante várias horas),
após pelo menos a administração das 2 primeiras perfusões, tendo em consideração a possibilidade de
uma reação de início retardado.
Os doentes tratados com Benlysta devem estar informados sobre o potencial risco de
hipersensibilidade grave ou ameaçadora da vida e do potencial início retardado ou de recorrência dos
sintomas. O folheto informativo deve ser entregue ao doente de cada vez que Benlysta é administrado
(ver secção 4.4).
Posologia
Pode ser administrada medicação prévia antes da perfusão com Benlysta, incluindo um anti-
histamínico, com ou sem antipirético (ver secção 4.4).
O regime posológico recomendado é de 10 mg/kg de Benlysta nos dias 0, 14 e 28, e seguidamente a
cada intervalo de 4 semanas. O estado de saúde do doente deve ser avaliado continuamente. A
descontinuação do tratamento com Benlysta deve ser considerada se não existirem melhorias no
controlo da doença após 6 meses de tratamento.
Transição da administração por via intravenosa para a via subcutânea
Se um doente está a transitar da administração de Benlysta por via intravenosa para a administração
por via subcutânea, a primeira injeção subcutânea deve ser administrada 1 a 4 semanas após a última
dose intravenosa (ver secção 5.2).
Populações especiais
Indivíduo idosos
A eficácia e segurança de Benlysta em idosos não foram estabelecidas. Os dados em doentes ≥65 anos
de idade estão limitados a <1,8% da população estudada. Consequentemente, não é recomendado o
uso de Benlysta em doentes idosos exceto se for esperado que os benefícios excedam os riscos. No
caso de se julgar necessária a administração de Benlysta a doentes idosos, não é necessário qualquer
ajuste da dose (ver secção 5.2).
Compromisso renal
Belimumab foi estudado num número limitado de doentes com LES com compromisso renal. Com
base na informação disponível, o ajuste de dose não é necessário em doentes com compromisso renal
ligeiro, moderado ou grave. Contudo, recomenda-se precaução em doentes com compromisso renal
grave devido à falta de dados (ver secção 5.2).
Compromisso hepático
Não foram realizados estudos específicos do uso de Benlysta em doentes com compromisso hepático.
É pouco provável que os doentes com compromisso hepático necessitem de um ajuste de dose (ver
secção 5.2).
População pediátrica
A segurança e eficácia de Benlysta em crianças e adolescentes (< 18 anos de idade) não foram
estabelecidas. Não existem dados disponíveis.
Modo de administração
Benlysta é administrado intravenosamente por perfusão, e deve ser reconstituído e diluído antes da
administração. Para instruções acerca da reconstituição, diluição e conservação do medicamento antes
da administração, ver secção 6.6.
Benlysta deve ser perfundido durante um período de 1 hora.

20
Benlysta não pode ser administrado como um bólus intravenoso.
O ritmo de perfusão pode ser diminuído ou interrompido se o doente desenvolver uma reação à
perfusão. A perfusão deve ser descontinuada imediatamente se o doente sofrer uma reação adversa
que coloque a vida em risco (ver secções 4.4 e 4.8).
4.3 Contraindicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes mencionados na secção 6.1.
4.4 Advertências e precauções especiais de utilização
Benlysta não foi estudado nos seguintes grupos de doentes, e não é recomendado para:
• lúpus ativo grave com envolvimento do sistema nervoso central
• nefrite lúpica ativa grave (ver secção 5.1)
• VIH
• história ou diagnóstico atual de hepatite B ou C
• hipogamaglobulinemia (IgG <400 mg/dl) ou deficiência em IgA (IgA <10 mg/dl).
• história de transplante de órgão major ou transplante hematopoiético estaminal
celular/medula óssea ou transplante renal.
Uso concomitante com terapêuticas dirigidas às células B ou ciclofosfamida
Benlysta não foi estudado em combinação com outras terapêuticas dirigidas às células B ou
ciclofosfamida intravenosa. Recomenda-se precaução se Benlysta for administrado
concomitantemente com outras terapêuticas dirigidas às células B ou ciclofosfamida.
Reações à perfusão e hipersensibilidade
A administração de Benlysta pode resultar em reações de hipersensibilidade e reações à perfusão que
podem ser graves e fatais. No caso de ocorrer uma reação grave, a administração de Benlysta deve ser
interrompida e administrada terapêutica médica apropriada (ver secção 4.2). O risco de reações de
hipersensibilidade é superior nas primeiras duas perfusões, contudo o risco deve ser considerado para
todas as perfusões administradas. Os doentes com história de alergias medicamentosas múltiplas ou
de hipersensibilidade significativa podem estar em maior risco.
A medicação prévia incluindo um anti-histamínico, com ou sem um antipirético, pode ser
administrada antes da perfusão com Benlysta. Não existe conhecimento suficiente para determinar se
a medicação prévia pode diminui a frequência ou a gravidade das reações à perfusão.
Nos ensaios clínicos, as reações graves à perfusão e de hipersensibilidade afetaram aproximadamente
0,9% dos doentes, e incluíram reações anafiláticas, bradicardia, hipotensão, angioedema e dispneia.
As reações à perfusão ocorreram mais frequentemente na administração das 2 primeiras perfusões e
tendencialmente diminuíram nas perfusões subsequentes (ver secção 4.8). Foram notificados casos de
doentes que desenvolveram sintomas de hipersensibilidade aguda várias horas após a perfusão ter sido
administrada. Foi também observada recorrência de reações clinicamente significativas após o
tratamento apropriado inicial dos sintomas (ver secções 4.2 e 4.8). Consequentemente, Benlysta deve

21
ser administrado num local onde estejam imediatamente disponíveis recursos para o tratamento deste
tipo de reações. Os doentes devem permanecer sobre supervisão clínica durante um período de tempo
prolongado (durante várias horas), após pelo menos a administração das 2 primeiras perfusões, tendo
em consideração a possibilidade de uma reação de início retardado. Os doentes devem ser avisados de
que são possíveis reações de hipersensibilidade no dia, ou nos vários dias subsequentes, da
administração da perfusão e ser informados sobre os sinais e sintomas potenciais e da possibilidade de
recorrência. Os doentes devem ser instruídos a procurar ajuda médica imediata se manifestarem algum
desses sintomas. O folheto informativo deve ser entregue ao doente de cada vez que Benlysta é
administrado (ver secção 4.2).
As reações de hipersensibilidade não agudas, do tipo retardado, foram também observadas e incluíram
sintomas como erupções cutâneas, náuseas, fadiga, mialgia, cefaleias e edema facial.
Infeções
O mecanismo de ação de belimumab pode aumentar o risco para o desenvolvimento de infeções,
incluindo infeções oportunistas. Foram notificadas infeções graves, incluindo casos fatais, em doentes
com LES a administrar terapêutica imunossupressora, incluindo belimumab (ver secção 4.8). Os
médicos devem tomar precauções ao considerar o uso de Benlysta em doentes com infeções graves ou
crónicas ou história de infeções recorrentes. Os doentes que desenvolvam uma infeção durante o
tratamento com Benlysta devem ser atentamente monitorizados e ser dada cuidadosa consideração
quanto à interrupção da terapêutica imunossupressora, incluindo o belimumab, até que a infeção seja
resolvida. O risco de utilizar Benlysta em doentes com tuberculose ativa ou latente é desconhecido.
Leucoencefalopatia multifocal progressiva (LMP)
A leucoencefalopatia multifocal progressiva (LMP) tem sido notificada com o tratamento com
Benlysta no LES. Os médicos devem estar particularmente atentos para os sintomas sugestivos de
LMP que os doentes possam não perceber (por exemplo, sinais ou sintomas cognitivos, neurológicos
ou psiquiátricos). Os doentes devem ser monitorizados para qualquer novo sintoma ou sinal ou seu
agravamento, e se ocorrerem estes sintomas/sinais serem referenciados para um neurologista e serem
consideradas medidas de diagnóstico apropriadas para a LMP. Se se suspeitar de LMP, deve ser
suspensa a administração de mais doses até ser excluída a LMP.
Imunização
As vacinas vivas não devem ser administradas durante os 30 dias antes ou concomitantemente com
Benlysta, uma vez que a segurança clínica não foi estabelecida. Não existem dados disponíveis sobre
a transmissão secundária da infeção em pessoas a quem foram administradas vacinas vivas para
doentes a quem está a ser administrado Benlysta.
Devido ao seu mecanismo de ação, belimumab pode interferir com a resposta a imunizações.
Contudo, num pequeno estudo que avaliou a resposta da vacina pneumocócica 23 valente,
globalmente as respostas imunitárias aos diferentes serotipos foram semelhantes nos doentes com
LES a administrar Benlysta, em comparação com os que estavam a administrar o tratamento
imunossupressor padrão à altura da vacinação. Os dados existentes são insuficientes para tirar
conclusões sobre a resposta a outras vacinas.
Dados limitados sugerem que Benlysta não afeta significativamente a capacidade de manter uma
resposta imunitária protetora referente a imunizações recebidas antes da administração de Benlysta.
Num subestudo, um pequeno grupo de doentes que tinha anteriormente recebido vacinas contra o
tétano, pneumococos ou gripe demonstrou manter títulos protetores após tratamento com Benlysta.
Neoplasias e doenças linfoproliferativas

22
Os medicamentos imunomodeladores, incluindo Benlysta, podem aumentar o risco de neoplasias.
Deve ser tomada precaução ao considerar a terapêutica com Benlysta em doentes com história de
neoplasias ou ao considerar a continuidade da terapêutica em doentes que desenvolvam neoplasias.
Não foram estudados doentes com neoplasias malignas nos últimos 5 anos, exceto aqueles com
carcinoma cutâneo basocelular ou das células escamosas ou com carcinoma do colo do útero, que
tenham sido completamente excisados cirurgicamente ou adequadamente tratados.
Conteúdo em sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose, ou seja, é praticamente
“isento de sódio”.
4.5 Interações medicamentosas e outras formas de interação
Não foram realizados estudos de interação in vivo. A formação de algumas enzimas CYP450 é
suprimida pelos níveis aumentados de certas citoquinas durante a inflamação crónica. Não é
conhecido se belimumab possa ser um modulador indireto destas citoquinas. Um risco para a redução
indireta da atividade CYP pelo belimumab, não pode ser excluído. Durante o início ou descontinuação
de belimumab, deve ser considerada a monitorização da terapêutica para os doentes em tratamento
com substratos CYP com um índice terapêutico estreito, em que a dose é ajustada individualmente
(por exemplo, varfarina).
4.6 Fertilidade, gravidez e aleitamento
Mulheres com potencial para engravidar/Contraceção em homens e mulheres
Mulheres em idade fértil têm de utilizar métodos contracetivos eficazes durante o tratamento com
Benlysta e até pelo menos 4 meses após o último tratamento.
Gravidez
Existe uma quantidade limitada de dados do uso de Benlysta em mulheres grávidas. Não foram
efetuados estudos formais. Para além de um efeito farmacológico espectável, i.e. depleção de células
B, estudos animais em macacos não indiciam efeitos prejudiciais diretos ou indiretos no que diz
respeito à toxicidade reprodutiva (ver secção 5.3).
Benlysta não deve ser utilizado durante a gravidez a menos que o potencial benefício justifique o
potencial risco para o feto.
Amamentação
Desconhece-se se Benlysta é excretado no leite materno ou se é absorvido sistemicamente após
ingestão. Contudo, belimumab foi detetado no leite de macacos fêmea às quais foram administrados
150 mg/kg a cada 2 semanas.
Dado que os anticorpos maternos (IgG) são excretados no leite materno, recomenda-se que a decisão
deva ser tomada entre descontinuar a amamentação ou descontinuar a terapêutica com Benlysta, tendo
em consideração o benefício da amamentação para a criança e o benefício da terapêutica para a
mulher.
Fertilidade
Não existem dados sobre os efeitos de belimumab na fertilidade humana. Os efeitos na fertilidade
masculina e feminina não foram formalmente avaliados durante os estudos animais (ver secção 5.3).
4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas

23
Não foram efetuados estudos sobre os efeitos na capacidade de conduzir e utilizar máquinas. Não se
preveem efeitos prejudiciais para estas atividades a partir da farmacologia de belimumab. O estado
clínico do indivíduo e o perfil de reações adversas de Benlysta deve estar presente ao considerar a
capacidade do doente desempenhar tarefas que exijam julgamento ou uso das capacidades motoras ou
cognitivas.
4.8 Efeitos indesejáveis
Resumo do perfil de segurança
A segurança de belimumab em doentes com LES foi avaliada em 3 estudos por via intravenosa,
controlados por placebo e em 1 estudo por via subcutânea, controlado por placebo.
Os dados apresentados na tabela em baixo refletem a exposição a Benlysta (10 mg/kg por via
intravenosa durante o período de 1 hora aos dias 0, 14, 28 e depois de 28 em 28 dias, durante até 52
semanas) administrado em 674 doentes com LES, incluindo 472 expostos durante um período de pelo
menos 52 semanas e 556 doentes expostos a 200 mg de Benlysta por via subcutânea, uma vez por
semana, durante até 52 semanas. Os dados de segurança apresentados incluem dados para além da
semana 52 em alguns doentes. Também é incluída a informação proveniente de notificações após a
comercialização.
A maioria dos doentes estava também a receber uma ou mais das seguintes terapêuticas concomitantes
para LES: corticosteroides, medicamentos imunomoduladores, antimaláricos, medicamentos anti-
inflamatórios não esteroides.
Foram notificadas reações adversas em 87% dos doentes tratados com Benlysta e em 90% dos doentes
tratados com placebo. As reações adversas mais frequentemente notificadas (≥5% dos doentes com
LES tratados com Benlysta mais o tratamento padrão e a uma taxa ≥1% mais elevada que com
placebo) foram infeções víricas do trato respiratório superior, bronquite e diarreia. A proporção de
doentes que descontinuaram o tratamento devido a reações adversas foi de 7% para os doentes
tratados com Benlysta e de 7% dos doentes tratados com placebo.
Sumário tabelado das reações adversas
As reações adversas estão listadas abaixo de acordo com a classificação de sistemas de órgãos
MedDRA e por frequência. As categorias de frequência utilizadas são:
Muito frequente s > 1/10
Frequentes 1/100, <1/10
Pouco frequentes 1/1.000, <1/100
Raros >1/10.000, <1/1.000
Os efeitos indesejáveis são apresentados em ordem decrescente de gravidade dentro de cada classe de
frequência. A frequência incluída refere-se à superior observada com qualquer formulação.
Classificação de sistema de
órgãos
Frequência
Reações Adversas
Infeções e infestações Muito frequentes Infeções bacterianas, por exemplo
bronquite, infeção do trato urinário
Frequentes Gastroenterite vírica, faringite,
nasofaringite, infeção víricas do trato
respiratório superior
24
Doenças do sangue e do
sistema linfático
Frequentes Leucopenia
Doenças do sistema
imunitário
Frequentes Reações de hipersensibilidade*
Pouco frequentes Reação anafilática
Raros Reações de hipersensibilidade não
agudas, do tipo retardado
Perturbações do foro
psiquiátrico
Frequentes Depressão
Doenças do sistema nervoso
Frequentes Enxaqueca
Doenças gastrointestinais
Muito frequentes Diarreia, náuseas
Afeções dos tecidos cutâneos
e subcutâneos
Frequentes
Reações no local de injeção**
Pouco frequentes Angioedema, urticária, erupção
cutânea
Afeções musculoesqueléticas
e dos tecidos conjuntivos
Frequentes Dores nas extremidades
Perturbações gerais e
alterações do local de
administração
Frequentes Reações* sistémicas relacionadas
com a perfusão ou injeção, pirexia
*As “Reações de hipersensibilidade” abrangem um grupo de termos médicos, incluindo anafilaxia, e
podem manifestar-se como um leque de sintomas que incluem hipotensão, angioedema, urticária ou
outras erupções cutâneas, prurido e dispneia. As “Reações sistémicas relacionadas com a perfusão ou
injeção” abrangem um grupo de termos médicos e podem manifestar-se como um leque de sintomas
que incluem bradicardia, mialgia, dores de cabeça, erupção cutânea, urticária, pirexia, hipotensão,
hipertensão, tonturas e artralgia. Uma vez que existe alguma sobreposição de sinais e sintomas, não é
possível em todos os casos distinguir entre reações de hipersensibilidade e reações relacionadas com a
perfusão.
** Aplica-se apenas à formulação subcutânea.
Descrição das reações adversas selecionadas
Os dados apresentados em baixo são um agregado dos ensaios clínicos por via intravenosa (10 mg/kg
por via intravenosa apenas) e do ensaio clínico por via subcutânea.
Reações sistémicas relacionadas com a injeção ou perfusão e de hipersensibilidade: as reações
sistémicas relacionadas com a injeção ou perfusão e de hipersensibilidade foram geralmente
observadas no dia da administração, mas podem também ocorrer reações de hipersensibilidade agudas
alguns dias após a administração da dose. Os doentes com história de alergias medicamentosas
múltiplas ou de reações de hipersensibilidade significativas podem encontrar-se em maior risco.
A incidência das reações à perfusão e hipersensibilidade após a administração por via intravenosa que
ocorreram nos 3 dias após uma perfusão foi de 12% no grupo a receber Benlysta e 10% no grupo a
receber placebo, com 1,2% e 0,3%, respetivamente, a necessitarem de descontinuação permanente do
tratamento.

25
Infeções: A incidência global de infeções nos estudos por via intravenosa e subcutânea foi de 63% em
ambos os grupos que administraram Benlysta ou placebo. As infeções ocorreram em pelo menos 3%
dos doentes que administraram Benlysta, e pelo menos 1% mais frequentemente do que os doentes
que administraram placebo, foram infeção do trato respiratório superior, bronquite e infeção
bacteriana do trato urinário. As infeções graves ocorreram em 5% dos doentes em ambos os grupos
que administraram Benlysta ou placebo, as infeções oportunistas graves representaram 0,4% e 0%
destas, respetivamente. As infeções que originaram descontinuação do tratamento ocorreram em 0,7%
dos doentes a administrar Benlysta e 1,5% dos doentes a administrar placebo. Algumas infeções
foram graves ou fatais.
Leucopenia: A incidência de leucopenia notificada como acontecimento adverso foi de 3% no grupo a
administrar Benlysta e de 2% no grupo a administrar placebo.
Perturbações do foro psiquiátrico: A incidência de depressão notificada como acontecimento
adverso foi de 3% em ambos os grupos a administrar Benlysta ou placebo.
Doenças gastrointestinais: Doentes obesos [Índice de Massa Corporal (IMC)>30 kg/m2] tratados com
Benlysta administrado por via intravenosa notificaram taxas mais elevadas de náuseas, vómitos e
diarreia relativamente ao placebo, e quando comparados aos doentes com peso normal (IMC≥18,5
kg/m2 a IMC≤30 kg/m2). Nenhum destes acontecimentos gastrointestinais em doentes obesos foi
grave.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas após a autorização do medicamento é importante, uma
vez que permite uma monitorização contínua da relação benefício-risco do medicamento. Pede-se aos
profissionais de saúde que notifiquem quaisquer suspeitas de reações adversas através do sistema
nacional de notificação mencionado no Apêndice V.
4.9 Sobredosagem
A experiência clínica de sobredosagem com Benlysta é limitada. As reações adversas notificadas em
associação com casos de sobredosagem foram consistentes com as esperadas para belimumab.
Duas doses até 20 mg/kg administradas com 21 dias de intervalo por perfusão intravenosa foram
administradas a seres a humanos sem aumento na incidência ou gravidade das reações adversas
quando comparadas com doses de 1, 4 ou 10 mg/kg.
No caso de sobredosagem não intencional, os doentes devem ser cuidadosamente observados e, se
apropriado, devem ser administrados cuidados de suporte.
5. PROPRIEDADES FARMACOLÓGICAS
5.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Imunossupressores seletivos, código ATC: L04AA26
Mecanismo de ação
Belimumab é um anticorpo monoclonal IgG1 humano, específico para a proteína humana solúvel
Estimuladora dos Linfócitos B (BLyS, também referida como BAFF e TNFSF13B). Belimumab
bloqueia a ligação do BlyS solúvel, um fator de sobrevivência celular, aos seus recetores nas células
B. Belimumab não se liga às células B diretamente, mas ao ligar-se ao BLyS, Belimumab inibe a
sobrevivência das células B, incluindo as células B autorreactivas, e reduz a diferenciação das células
B em células plasmáticas produtoras de imunoglobulinas.

26
Os níveis de BLyS estão elevados em doentes com LES e outras doenças autoimunes. Existe uma
associação entre os níveis plasmáticos de BLyS e a atividade da doença LES. A contribuição relativa
dos níveis de BLyS para a patofisiologia do LES não está completamente compreendida.
Efeitos farmacodinâmicos
Foram observadas alterações nos biomarcadores em ensaios clínicos com Benlysta administrado por
via intravenosa. Em doentes com hipergamaglobulinemia, foi observada a normalização dos níveis de
IgG à semana 52 em 49% e 20% dos doentes a receberem Benlysta e placebo, respetivamente.
Em doentes com anticorpos anti-dsADN, 16% dos doentes tratados com Benlysta reverteram para
anti-dsADN negativos, comparativamente a 7% dos doentes a receber placebo, à semana 52.
Em doentes com níveis de complemento baixos, foi observada à semana 52 a normalização de C3 e
C4 em 38% e 44% dos doentes a receberem Benlysta e em 17% e 18% dos doentes a receberem
placebo, respetivamente.
Dos anticorpos antifosfolipídicos apenas foi medido o anticorpo anticardiolipina. Para o anticorpo
anticardiolipina IgA foi observada uma redução de 37% à semana 52 (p=0,0003), para o anticorpo
anticardiolipina IgG foi observada uma redução de 26% à semana 52 (p=0,0324) e para o anticorpo
anticardiolipina IgM foi observada uma redução de 25% à semana 52 (p=NS 0,46).
As alterações nos níveis de células B (incluindo células B não ativadas (naive), de memória e ativadas
e células plasmáticas) e de IgG que ocorreram nos doentes durante o decurso do tratamento com
belimumab por via intravenosa foram seguidas num estudo de extensão, não controlado e de longo
prazo. Após 7 anos e meio de tratamento (incluindo o estudo principal de 72 semanas), foi observada
uma diminuição substancial e sustentada em vários subtipos de células B, conduzindo a uma redução
mediana de 87% nas células B não ativadas (naive), de 67% nas células B de memória, de 99% nas
células B ativadas e uma redução mediana de 92% dos níveis das células plasmáticas após mais de 7
anos de tratamento. Após cerca de 7 anos, foi observada uma redução mediana de 28% dos níveis de
IgG, com 1,6% dos indivíduos a terem uma diminuição nos níveis de IgG para abaixo de 400 mg/dl.
Ao longo do estudo, a incidência de notificações de acontecimentos adversos, em geral, manteve-se
estável ou diminuiu.
Imunogenicidade
A sensibilidade da determinação de anticorpos neutralizantes e de anticorpos antimedicamento não
específicos (ADA) está limitada pela presença do fármaco ativo nas amostras recolhidas.
Consequentemente, não se conhece a verdadeira ocorrência de anticorpos neutralizantes e de
anticorpos anti medicamento não específicos nas populações em estudo.
Em dois estudos de Fase III, 4 dos 563 doentes (0,7%) no grupo de 10 mg/kg e 27 dos 559 doentes
(4,8%) no grupo de 1 mg/kg tiveram um resultado positivo no teste para a presença persistente de
anticorpos antibelimumab.
Entre os indivíduos persistentemente positivos nos estudos de Fase III, 1/10 (10%), 2/27 (7%) e 1/4
(25%) indivíduos nos grupos a tomar placebo, 1 mg/kg e 10 mg/kg, respetivamente, sofreram reações
relacionadas com a perfusão num dia de administração; todas estas reações relacionadas com a
perfusão não foram graves e foram de gravidade ligeira a moderada. Poucos doentes com ADA
notificaram acontecimentos adversos sérios/graves. As taxas de reação relacionada com a perfusão
entre indivíduos persistentemente positivos foram comparáveis às taxas para os doentes ADA
negativos de 75/552 (14%), 78/523 (15%) e 83/559 (15%) nos grupos a tomar placebo, 1 mg/kg e 10
mg/kg, respetivamente.
Eficácia e segurança clínicas

27
Perfusão intravenosa
A eficácia de Benlysta administrado por via intravenosa foi avaliada em 2 estudos com aleatorização,
em dupla ocultação, controlados por placebo em 1.684 doentes com um diagnóstico clínico de LES de
acordo com os critérios de classificação do Colégio Americano de Reumatologia. Os doentes tinham
doença LES ativa, definida como um resultado 6 na escala SELENA-SLEDAI (SELENA=Safety of
Estrogens in Systemic Lupus Erythematosus National Assessment; SLEDAI=Systemic Lupus
Erythematosus Disease Activity Index) e resultados positivos no teste para deteção de anticorpos
antinucleares (ANA; títulos de ANA 1:80 e/ou um resultado positivo anti-dsADN
[30 unidades/ml]) durante avaliação inicial. Os doentes estavam a receber um regime terapêutico
estável para LES, que consistia em (isolados ou em combinação): corticosteroides, antimaláricos, anti-
inflamatórios não esteroides (AINEs) ou outros imunossupressores. Os dois estudos foram
semelhantes quanto ao seu desenho, exceto pelo facto do BLISS-76 ser um estudo de 76 semanas
enquanto que o BLISS-52 ser de 52 semanas. Em ambos os estudos o objetivo primário de eficácia foi
avaliado às 52 semanas.
Os doentes com nefrite lúpica ativa grave e os doentes com lúpus ativo grave com envolvimento do
sistema nervoso central (SNC) foram excluídos.
O BLISS-76 foi realizado principalmente na América do Norte e na Europa Ocidental. A medicação
prévia concomitante incluiu corticosteroides (76%; 46% >7,5 mg/dia), imunossupressores (56%) e
antimaláricos (63%).
O BLISS-52 foi realizado na América do Sul, Europa de Leste, Ásia e Austrália. A medicação prévia
concomitante incluiu corticosteroides (96%; 69% >7,5 mg/dia), imunossupressores (42%) e
antimaláricos (67%).
Na linha de base inicial, 52% dos doentes tinha atividade elevada da doença (resultado SELENA
SLEDAI de >10), 59% dos doentes tinha envolvimento mucocutâneo, 60% dos doentes tinham
envolvimento musculoesquelético, 16% dos doentes tinham envolvimento hematológico, 11% dos
doentes tinham envolvimento renal e 9% dos doentes tinham envolvimento vascular dos órgãos
(BILAG A ou B na linha de base inicial).
O objetivo primário de eficácia foi um objetivo composto (Índice de Resposta LES) que definia
resposta como o atingir cada um dos seguintes critérios à semana 52 quando comparado com a linha
de base inicial:
• redução de 4 pontos na escala SELENA-SLEDAI e
• sem novos resultados de envolvimento de órgãos na escala British Isles Lupus Assessment Group
(BILAG) ou 2 novos resultados de envolvimento orgânico BILAG B, e
• sem agravamento (aumento <0,30 pontos) na escala de Avaliação Médica Global (PGA)
O Índice de Resposta LES mede a melhoria da atividade da doença LES, sem agravamento de
qualquer sistema de órgãos ou na condição geral do doente.

28
Tabela 1: Taxa de resposta à semana 52
Resposta
BLISS-76 BLISS-52 BLISS-76 e BLISS-52
Combinados
Placebo*
(n=275)
Benlysta
10 mg/kg*
(n=273)
Placebo*
(n=287)
Benlysta
10 mg/kg*
(n=290)
Placebo*
(n=562)
Benlysta
10 mg/kg*
(n=563)
Índice de
resposta LES
33,8% 43,2%
(p=0,021)
43,6% 57,6%
(p=0, 0006)
38,8% 50,6%
(p<0,0001)
Diferença
observada vs
placebo
9,4% 14,0% 11,8%
Odds Ratio
(OR) (IC 95%)
vs placebo
1,52
(1,07, 2,15)
1,83 (1,30,
2,59)
1,68
(1,32, 2,15)
Componentes do Índice de resposta LES
Percentagem
de doentes
com redução
na escala
SELENA-
SLEDAI 4
35,6% 46,9%
(p=0,006)
46,0% 58,3%
(p=0,0024)
40,9% 52,8%
(p<0,0001)
Percentagem
de doentes
sem
agravamento
medido pelo
índice BILAG
65,1% 69,2%
(p=0,32)
73,2% 81,4%
(p=0,018)
69,2% 75,5%
(p=0,019)
Percentagem
de doentes
sem
agravamento
medido por
PGA
62,9% 69,2%
(p=0,13)
69,3% 79,7%
(p=0,0048)
66,2% 74,6%
(p=0,0017)
* Todos os doentes receberam a terapêutica padrão
Numa análise combinada dos dois estudos, a percentagem de doentes a receber prednisolona >7,5
mg/dia (ou equivalente) na linha de base inicial, cuja dose média de corticosteroides foi reduzida em
pelo menos 25% para uma dose equivalente a prednisolona ≤7,5 mg/dia durante o intervalo entre a
Semana 40 e 52 foi de 17,9% no grupo a receber Benlysta e 12,3% no grupo a receber placebo
(p=0,0451).
Os picos (flares) no LES foram definidos pelo Índice de Picos SELENA SLEDAI no LES. O tempo
médio para o primeiro pico foi atrasado no grupo combinado a receber Benlysta quando comparado
com o grupo a receber placebo (110 vs 84 dias, Hazard Ratio (HR)=0,84, p=0,012). Foram
observados picos graves em 15,6% do grupo de Benlysta em comparação com 23,7% no grupo de
placebo ao longo das 52 semanas de observação (diferença observada entre tratamentos =-8,1%: risco
relativo=0,64, p=0,0011).

29
Benlysta demonstrou melhorias na fadiga quando comparado com placebo, medido pela escala de
fadiga FACIT na análise combinada. Esta alteração média do resultado à Semana 52 a partir da linha
de base inicial é significativamente maior com Benlysta quando comparado com placebo (4,70 vs
2,46, p=0,0006).
Análises univariadas e multivariadas do objetivo primário em subgrupos pré-especificados
demonstraram que o maior benefício foi observado em doentes com atividade mais elevada da doença
incluindo doentes com resultados SELENA SLEDAI ≥ 10 ou doentes que necessitavam de esteroides
para controlar a sua doença ou doentes com níveis de complemento baixos.
Uma análise subsequente identificou subgrupos de resposta elevada, tais como doentes com níveis de
complemento baixos e resultados positivos para anti ds-ADN na linha de base inicial, ver Tabela 2
para os resultado deste exemplo de um grupo com atividade da doença elevada. Destes doentes,
64,5% tinham resultados SELENA SLEDAI ≥ 10 na linha de base inicial.
Tabela 2: Doentes com níveis de complemento baixos e resultados positivos anti ds-ADN na
linha de base inicial
Subgrupo Anti-dsADN positivo E complemento
baixo
BLISS-76 e BLISS-52 dados combinados Placebo
(n=287)
Benlysta
10 mg/kg
(n=305)
Taxa de resposta systemic lupus erythematosus responder
index (SRI) à semana 52 (%)
31,7 51,5 (p<0,0001)
Diferença entre tratamentos observada vs placebo (%) 19,8
Taxa de resposta SRI à semana 52 (excluindo alterações
de complemento e anti ds-ADN) (%)
28,9 46,2 (p<0,0001)
Diferença entre tratamentos observada vs placebo (%) 17,3
Picos graves ao longo das 52 semanas
Doentes que sofreram um pico grave (%)
29,6 19,0
Diferença entre tratamentos observada vs placebo (%) 10,6
Tempo até um pico grave [HR (95% CI)] 0,61 (0,44, 0,85)
(p=0,0038)
Redução de prednisolona em ≥25% desde a linha de base
inicial até ≤7,5 mg/dia durante as semanas 40 até 52* (%)
(n=173)
12,1
(n=195)
18,5 (p=0,0964)
Diferença entre tratamentos observada vs placebo (%) 6,3
Melhoria na escala de fadiga FACIT desde a linha de base
inicial até à semana 52 (média)
1,99 4,21 (p=0,0048)
Diferença entre tratamentos observada vs placebo
(diferença média)
2,21
Apenas Estudo BLISS-76 Placebo
(n=131)
Benlysta
10 mg/kg
(n=134)
Taxa de resposta SRI à Semana 76 (%)
27,5 39,6 (p=0,0160)
Diferença entre tratamentos observada vs placebo (%) 12,1

30
* Entre doentes com dose de prednisolona >7,5 mg/dia na linha de base inicial
Idade e etnia
Não foram incluídos nos ensaios clínicos controlados doentes suficientes ≥ 65 anos de idade, ou
doentes de etnia negra/afro-americana, para se poderem retirar conclusões significativas sobre os
efeitos da idade ou etnia nos resultados clínicos.
População pediátrica
A Agência Europeia de Medicamentos diferiu a obrigação de submissão dos resultados dos estudos
com Benlysta num ou mais sub-grupos da população pediátrica com LES (ver secção 4.2 para
informação sobre utilização pediátrica).
5.2 Propriedades farmacocinéticas
Os parâmetros farmacocinéticos descritos abaixo são baseados em estimativas dos parâmetros da
população para os 563 doentes que receberam 10 mg/kg de Benlysta nos dois estudos de Fase III.
Absorção
Benlysta é administrado por perfusão intravenosa. As concentrações séricas máximas de belimumab
foram geralmente observadas no final da perfusão ou pouco depois. A concentração máxima sérica foi
313 µg/ml (intervalo: 173-573 µg/ml) baseado na simulação do perfil concentração-tempo utilizando
os parâmetros típicos para um modelo farmacocinético populacional.
Distribuição
Belimumab distribuiu-se pelos tecidos com um volume de distribuição no estado de equilíbrio (Vss)
de aproximadamente 5 litros.
Biotransformação
Belimumab é uma proteína para a qual a via metabólica expectável é a degradação em pequenos
péptidos e aminoácidos individuais por enzimas proteolíticas largamente disseminadas. Não foram
realizados estudos clássicos de biotransformação.
Eliminação
As concentrações séricas de belimumab diminuíram de forma bi-exponencial, com um tempo de
semivida de distribuição de 1,75 dias e um tempo de semivida de eliminação de 19,4 dias. A
depuração sistémica foi de 215 ml/dia (intervalo: 69-622 ml/dia).
Populações especiais de doentes
População pediátrica: Não existem dados farmacocinéticos em doentes pediátricos.
Indivíduos idosos: Benlysta foi estudado num número limitado de doentes idosos. Dentro da
população total com LES nos estudos com administração intravenosa, a idade não afetou a exposição
ao belimumab na população farmacocinética em análise. Contudo, dado o reduzido número de
indivíduos com ≥65 anos de idade, um efeito relacionado com a idade não pode ser conclusivamente
excluído.
Compromisso renal: Não foram realizados estudos específicos para examinar os efeitos do
compromisso renal na farmacocinética de belimumab. Durante o desenvolvimento clínico, Benlysta

31
foi estudado em doentes com LES e compromisso renal (261 indivíduos com compromisso renal
moderado, depuração da creatinina 30 e <60 ml/min; 14 indivíduos com compromisso renal grave,
depuração da creatinina 15 e <30 ml/min). A redução na depuração sistémica estimada por modelo
farmacocinético populacional para doentes nos objetivos médios das categorias de compromisso renal
relativamente a doentes com depuração média da creatinina na população farmacocinética (79,9
ml/min) foi de 1,4% para compromisso renal ligeiro (75 ml/min), 11,7% para moderado (45 ml/min) e
24,0% para grave (22,5 ml/min). Apesar da proteinúria (≥ 2 g/dia) ter aumentado a depuração do
belimumab e diminuições na depuração da creatinina ter diminuído a depuração do belimumab, estes
efeitos estavam dentro do intervalo de variabilidade esperado. Consequentemente, não é recomendado
qualquer ajuste de dose para doentes com compromisso renal.
Compromisso hepático: Não foram realizados estudos específicos para examinar os efeitos do
compromisso hepático na farmacocinética do belimumab. As moléculas IgG como o belimumab são
catabolizadas por enzimas proteolíticas largamente distribuídas, que não estão restritas ao tecido
hepático, e alterações da função hepática não irão provavelmente ter qualquer efeito na eliminação do
belimumab.
Peso corporal/Índice de Massa Corporal (IMC):
O doseamento de belimumab ajustado ao peso leva a uma exposição diminuída para indivíduos com
baixo peso (IMC<18,5) e a uma exposição aumentada em indivíduos obesos (IMC≥30). As alterações
na exposição dependentes do IMC não levaram a alterações correspondentes na eficácia. A exposição
aumentada em indivíduos obesos a receberem 10 mg/kg belimumab não levou a um aumento global
das taxas de acontecimentos adversos ou de acontecimentos adversos graves quando comparados com
indivíduos obesos a receber placebo. Contudo, foram observadas taxas mais elevadas de náuseas,
vómitos e diarreia em doentes obesos. Nenhum destes efeitos gastrointestinais em doentes obesos foi
grave. Não é recomendado nenhum ajuste de dose para indivíduos de baixo peso ou obesos.
Transição da administração por via intravenosa para a via subcutânea
Os doentes com LES em transição da administração de 10 mg/kg por via intravenosa, de 4 em 4
semanas, para 200 mg por via subcutânea semanalmente, utilizando o intervalo de permutação de 1 a
4 semanas, tiveram uma concentração sérica de belimumab pré-dose na sua primeira dose por via
subcutânea próximo da sua eventual concentração subcutânea no estado de equilíbrio (ver secção 4.2).
Com base em simulações com parâmetros farmacocinéticos da população, as concentrações médias de
belimumab no estado de equilíbrio para 200 mg por via subcutânea, todas as semanas, foram
semelhantes a 10 mg por via intravenosa, de 4 em 4 semanas.
5.3 Dados de segurança pré-clínica
Os dados não clínicos não revelam riscos especiais para o ser humano, segundo estudos de toxicidade
de dose repetida e toxicidade reprodutiva.
A administração intravenosa e subcutânea em macacos resultou numa redução esperada do número de
células B periféricas e nos tecidos linfoides sem achados toxicológicos associados.
Foram realizados estudos reprodutivos em macacos fêmea cinomolgos grávidas a receber 150 mg/kg
de belimumab por perfusão intravenosa (aproximadamente 9 vezes a exposição clínica máxima
antecipada em seres humanos) a cada 2 semanas durante 21 semanas, e o tratamento com belimumab
não foi associado com efeitos negativos diretos ou indiretos relacionados com toxicidade materna,
toxicidade do desenvolvimento ou teratogenicidade.
Os resultados relacionados com o tratamento foram limitados à redução das células B, esperada e
reversível, tanto nas progenitoras como nas crias e à redução reversível dos níveis de IgM nas crias de
macacos. As contagens de células B recuperaram após a cessação do tratamento com belimumab cerca

32
de 1 ano após parto em macacos adultos e perto dos 3 meses de vida nas crias de macacos; os níveis
de IgM em crias expostas in utero a belimumab recuperaram por volta dos 6 meses de idade.
Os efeitos na fertilidade masculina e feminina em macacos foram determinados em 6 meses de
estudos de toxicidade de dose repetida, em doses de até 50 mg/kg inclusive. Não foram notadas
alterações relacionadas com o tratamento nos órgãos reprodutivos masculinos e femininos de animais
sexualmente maduros. Uma avaliação informal do ciclo menstrual em fêmeas não demonstrou
alterações relacionadas com o belimumab.
Uma vez que belimumab é um anticorpo monoclonal não foram realizados estudos de genotoxicidade.
Não foram efetuados estudos de carcinogenicidade ou estudos de fertilidade (masculinos ou
femininos).
6. INFORMAÇÕES FARMACÊUTICAS
6.1. Lista dos excipientes
Ácido cítrico mono-hidratado (E330)
Citrato de sódio (E331)
Sacarose
Polissorbato 80
6.2 Incompatibilidades
Benlysta não é compatível com glucose a 5%.
Este medicamento não deve ser misturado com outros medicamentos, exceto os mencionados na
secção 6.6.
6.3 Prazo de validade
Frascos para injetáveis não abertos
5 anos.
Solução reconstituída
Após reconstituição com água para preparações injetáveis, a solução reconstituída, se não utilizada
imediatamente, deve ser protegida da luz solar direta e armazenada refrigerada de 2ºC a 8ºC.
Solução reconstituída e diluída para perfusão
A solução de Benlysta diluída em solução para injetáveis de cloreto de sódio 9 mg/ml (0,9%), cloreto
de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer pode ser armazenada de 2ºC a 8ºC ou à temperatura
ambiente (15ºC a 25ºC).
O tempo total entre a reconstituição de Benlysta até à conclusão da perfusão não deve exceder 8
horas.
6.4 Precauções especiais de conservação
Conservar no frigorífico (2ºC a 8ºC).
Não congelar.
Manter o frasco para injetáveis dentro da embalagem exterior para proteger da luz.
Condições de conservação do medicamento após reconstituição e diluição, ver secção 6.3.
6.6 Natureza e conteúdo do recipiente

33
Benlysta 120 mg pó para concentrado para solução para perfusão
Frascos para injetáveis de vidro tipo I (5 ml), selados com uma rolha de clorobutilo siliconizada e um
selo de abertura fácil de alumínio contendo 120 mg de pó.
Tamanho da embalagem: 1 frasco para injetáveis
Benlysta 400 mg pó para concentrado para solução para perfusão
Frascos para injetáveis de vidro tipo I (20 ml), selados com uma rolha de clorobutilo siliconizada e
um selo de abertura fácil de alumínio contendo 400 mg de pó.
Tamanho da embalagem: 1 frasco para injetáveis
6.6 Precauções especiais de eliminação e manuseamento
Preparação da solução para perfusão de 120 mg
Reconstituição
A reconstituição e diluição devem ser executadas em condições assépticas.
Aguardar 10-15 minutos para que o frasco para injetáveis aqueça até à temperatura ambiente (15ºC-
25ºC).
Recomenda-se a utilização de uma agulha de 21-25 gauge para furar a rolha do frasco para injetáveis
na reconstituição e diluição.
O frasco para injetáveis de uso único de 120 mg de belimumab é reconstituído em 1,5 ml de água para
preparações injetáveis para obter uma concentração final de 80 mg/ml de belimumab.
O fluxo de água para preparações injetáveis deve ser dirigido para a face lateral do frasco para
injetáveis de forma a minimizar a formação de espuma. Rodar suavemente o frasco para injetáveis
durante 60 segundos. Deixar o frasco para injetáveis em repouso à temperatura ambiente (15ºC a
25ºC) durante a reconstituição, rodar suavemente o frasco para injetáveis durante 60 segundos a cada
5 minutos até que o pó esteja dissolvido. Não agitar. A reconstituição normalmente está completa em
10 a 15 minutos após a adição da água, mas poderá demorar até 30 minutos.
Proteger a solução reconstituída da luz solar.
Se for utilizado um aparelho de reconstituição mecânico para reconstituir Benlysta, não se deve
exceder as 500 rpm e o frasco para injetáveis não deve ser agitado durante mais do que 30 minutos.
Quando a reconstituição estiver completa, a solução deve apresentar-se opalescente e incolor a
amarelo pálido e sem partículas. Contudo, são aceitáveis e expectáveis pequenas bolhas de ar.
Após a reconstituição, um volume de 1,5 ml (correspondente a 120 mg de belimumab) pode ser
retirado de cada frasco para injetáveis.
Diluição
O medicamento reconstituído é diluído para 250 ml com uma solução para injetáveis de cloreto de
sódio 9 mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer.

34
Não devem ser utilizadas soluções intravenosas de glucose a 5% porque são incompatíveis com
Benlysta.
A partir de um saco de perfusão ou frasco de 250 ml de solução para injetáveis de cloreto de sódio 9
mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer retirar e eliminar um volume
igual ao volume da solução reconstituída de Benlysta necessário para a dose do doente.
Seguidamente, adicionar o volume necessário da solução reconstituída de Benlysta ao saco ou frasco
de perfusão. Inverter suavemente o saco ou frasco para homogeneizar a solução. Toda a solução não
utilizada nos frascos para injetáveis deve ser eliminada.
Inspecionar visualmente a solução de Benlysta para a presença de partículas e descoloração antes da
administração. Eliminar a solução se forem observadas partículas ou descoloração.
O tempo total a partir da reconstituição de Benlysta até ao final da perfusão não deve exceder 8 horas.
Preparação da solução para perfusão de 400 mg
Reconstituição
A reconstituição e diluição devem ser executadas em condições assépticas.
Aguardar 10-15 minutos para que o frasco para injetáveis aqueça até à temperatura ambiente (15ºC a
25ºC).
Recomenda-se a utilização de uma agulha de 21-25 gauge para furar a rolha do frasco para injetáveis
na reconstituição e diluição.
O frasco para injetáveis de uso único de 400 mg de belimumab é reconstituído em 4,8 ml de água para
preparações injetáveis para obter uma concentração final de 80 mg/ml de belimumab.
O fluxo de água para preparações injetáveis deve ser dirigido para a face lateral do frasco para
injetáveis de forma a minimizar a formação de espuma. Rodar suavemente o frasco para injetáveis
durante 60 segundos. Deixar o frasco para injetáveis em repouso à temperatura ambiente (15ºC a
25ºC) durante a reconstituição, rodar suavemente o frasco para injetáveis durante 60 segundos a cada
5 minutos até que o pó esteja dissolvido. Não agitar. A reconstituição normalmente está completa em
10 a 15 minutos após a adição da água, mas poderá demorar até 30 minutos.
Proteger a solução reconstituída da luz solar.
Se for utilizado um aparelho de reconstituição mecânico para reconstituir Benlysta, não se deve
exceder as 500 rpm e o frasco para injetáveis não deve ser agitado durante mais do que 30 minutos.
Quando a reconstituição estiver completa, a solução deve apresentar-se opalescente e incolor a
amarelo pálido e sem partículas. Contudo, são aceitáveis e expectáveis pequenas bolhas de ar.
Após a reconstituição, um volume de 5 ml (correspondente a 400 mg de belimumab) pode ser retirado
de cada frasco para injetáveis.
Diluição
O medicamento reconstituído é diluído para 250 ml com uma solução para injetáveis de cloreto de
sódio 9 mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer.
Não devem ser utilizadas soluções intravenosas de glucose a 5% porque são incompatíveis com
Benlysta.

35
A partir de um saco de perfusão ou frasco de 250 ml de solução para injetáveis de cloreto de sódio 9
mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer retirar e eliminar um volume
igual ao volume da solução reconstituída de Benlysta necessário para a dose do doente.
Seguidamente, adicionar o volume necessário da solução reconstituída de Benlysta ao saco ou frasco
de perfusão. Inverter suavemente o saco ou frasco para homogeneizar a solução. Toda a solução não
utilizada nos frascos para injetáveis deve ser eliminada.
Inspecionar visualmente a solução de Benlysta para a presença de partículas e descoloração antes da
administração. Eliminar a solução se forem observadas partículas ou descoloração.
O tempo total a partir da reconstituição de Benlysta até ao final da perfusão não deve exceder 8 horas.
Modo de administração
Benlysta é perfundido durante um período de 1 hora.
Benlysta não deve ser perfundido concomitantemente na mesma linha intravenosa com outros
fármacos. Não foram realizados estudos de compatibilidade físicos ou bioquímicos para avaliar a
administração concomitante de Benlysta com outros fármacos.
Não foram observadas incompatibilidades entre Benlysta e sacos de cloreto de polivinilo ou
poliolefina.
Eliminação
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências
locais.
7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Reino Unido
8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/001: 1 frasco para injetáveis – 120 mg
EU/1/11/700/002: 1 frasco para injetáveis – 400 mg
9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE
INTRODUÇÃO NO MERCADO
Data da primeira autorização: 13 de julho de 2011
Data da última renovação: 18 de fevereiro de 2016
10. DATA DA REVISÃO DO TEXTO

36
Está disponível informação pormenorizada sobre este medicamento no sítio da internet da Agência
Europeia de Medicamentos: http://www.ema.europa.eu.

37
ANEXO II
A. FABRICANTE DA SUBSTÂNCIA ATIVA DE ORIGEM
BIOLÓGICA E FABRICANTE(S) RESPONSÁVEL(VEIS)
PELA LIBERTAÇÃO DO LOTE
B. CONDIÇÕES OU RESTRIÇÕES RELATIVAS AO
FORNECIMENTO E UTILIZAÇÃO
C. OUTRAS CONDIÇÕES E REQUISITOS DA AUTORIZAÇÃO
DE INTRODUÇÃO NO MERCADO
D. CONDIÇÕES OU RESTRIÇÕES RELATIVAS À UTILIZAÇÃO
SEGURA E EFICAZ DO MEDICAMENTO

38
A FABRICANTE DA SUBSTÂNCIA ATIVA DE ORIGEM BIOLÓGICA FABRICANTE
RESPONSÁVEL PELA LIBERTAÇÃO DO LOTE
Nome e endereço do fabricante da substância ativa de origem biológica
Human Genome Sciences, Inc.
Belward Large Scale Manufacturing (LSM) Facility
9911 Belward Campus Drive
Rockville, MD 20850
Estados Unidos da América
Nome e endereço do fabricante responsável pela libertação do lote
GlaxoSmithKline Manufacturing S.P.A
Strada Provinciale Asolana No. 90
I-43056 San Polo di Torrile, Parma
Itália
Ou
Glaxo Operations UK Ltd
Harmire Road
Barnard Castle
County Durham, DL12 8DT
Reino Unido
B. CONDIÇÕES OU RESTRIÇÕES RELATIVAS AO FORNECIMENTO E
UTILIZAÇÃO
Medicamento de receita médica restrita, de utilização reservada a certos meios especializados (ver
anexo I: Resumo das Características do Medicamento, secção 4.2).
C. OUTRAS CONDIÇÕES E REQUISITOS DA AUTORIZAÇÃO DE INTRODUÇÃO
NO MERCADO
• Relatórios Periódicos de Segurança
Os requisitos para a apresentação de relatórios periódicos de segurança para este medicamento estão
estabelecidos na lista Europeia de datas de referência (lista EURD), tal como previsto nos termos do
n.º 7 do artigo 107.º-C da Diretiva 2001/83/CE e quaisquer atualizações subsequentes publicadas no
portal europeu de medicamentos.
D. CONDIÇÕES OU RESTRIÇÕES RELATIVAS À UTILIZAÇÃO SEGURA E EFICAZ
DO MEDICAMENTO
• Plano de Gestão do Risco (PGR)
O Titular da AIM deve efetuar as atividades e as intervenções de farmacovigilância requeridas e
detalhadas no PGR apresentado no Módulo 1.8.2. da Autorização de Introdução no Mercado, e
quaisquer atualizações subsequentes do PGR que sejam acordadas.
Deve ser apresentado um PGR atualizado:

39
• A pedido da Agência Europeia de Medicamentos
• Sempre que o sistema de gestão do risco for modificado, especialmente como resultado da
receção de nova informação que possa levar a alterações significativas no perfil benefício-risco
ou como resultado de ter sido atingido um objetivo importante (farmacovigilância ou
minimização do risco).
• Obrigação de concretizaras medidas de pós-autorização
O Titular da Autorização de Introdução no Mercado deverá completar, dentro dos prazos indicados,
as seguintes medidas:
Descrição Data Limite
O Titular da AIM deve providenciar um relatório com os dados referentes a 5
anos de um estudo alargado de segurança com aleatorização controlado por
placebo, duplamente cego, baseado num protocolo acordado com o CHMP. O
estudo irá avaliar, ao longo de um período mínimo de 1 ano, a incidência de
mortalidade por todas as causas e os acontecimentos adversos de especial
interesse em doentes com lúpus eritematoso sistémico. Estes acontecimentos
adversos de especial interesse abrangem infeções graves (incluindo infeções
oportunistas graves e não graves e leucoencefalopatia multifocal progressiva
(LMP)), neoplasias (incluindo cancro da pele não-melanoma), reações de
hipersensibilidade e à perfusão, e acontecimentos psiquiátricos graves incluindo
perturbações de humor, ansiedade e suicídio.
31 de
dezembro de
2019
O Titular da AIM deve também de providenciar um relatório com os dados
referentes à base de dados de segurança controlada a longo prazo onde todos os
doentes são seguidos durante um período mínimo de 5 anos, com base no
protocolo acordado com o CHMP. A base de dados de segurança irá avaliar a
incidência da mortalidade por qualquer causa e acontecimentos adversos de
especial interesse em doentes com lúpus eritematoso sistémico. Estes
acontecimentos adversos de especial interesse incluem infeções graves (incluindo
infeções oportunistas e leucoencefalopatia multifocal progressiva (LMP)),
acontecimentos psiquiátricos graves selecionados e neoplasias (incluindo cancro
da pele não-melanoma).
28 de fevereiro
de 2026

40
ANEXO III
ROTULAGEM E FOLHETO INFORMATIVO

41
A. ROTULAGEM

42
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
CARTONAGEM – CANETA(S) PRÉ-CHEIA(S)
1. NOME DO MEDICAMENTO
Benlysta 200 mg solução injetável em caneta pré-cheia
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada caneta pré-cheia de 1ml contém 200 mg de belimumab
3. LISTA DOS EXCIPIENTES
Também contém cloridrato de arginina, histidina, monocloridrato de histidina, polissorbaro 80,
cloreto de sódio e água para preparações injetáveis.
4. FORMA FARMACÊUTICA E CONTEÚDO
Solução injetável em caneta pré-cheia.
1 caneta pré-cheia.
4 canetas pré-cheias.
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Consultar o folheto informativo antes de utilizar.
Via subcutânea.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP

43
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
Conservar no frigorífico.
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/003: 1 caneta pré-cheia
EU/1/11/700/004: 4 canetas pré-cheias
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
caneta de benlysta
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
Código de barras 2D com identificador único incluído.
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA
PC:
SN:
NN:

44
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
ROTULAGEM SECUNDÁRIA – Embalagem múltipla com 12 canetas pré-cheias (3
embalagens de 4 canetas pré-cheias) - com blue-box
1. NOME DO MEDICAMENTO
Benlysta 200 mg solução injetável em caneta pré-cheia
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada caneta pré-cheia de 1 ml contém 200 mg de belimumab
3. LISTA DOS EXCIPIENTES
Também contém cloridrato de arginina, histidina, monocloridrato de histidina, polissorbaro 80,
cloreto de sódio e água para preparações injetáveis.
4. FORMA FARMACÊUTICA E CONTEÚDO
Solução injetável em caneta pré-cheia.
Embalagem múltipla: 12 canetas pré-cheias (3 embalagens de 4 canetas pré-cheias).
Não vender separadamente.
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Consultar o folheto informativo antes de utilizar.
Via subcutânea.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP

45
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
Conservar no frigorífico.
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford
Middlesex
TW8 9GS
Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/005
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
caneta de benlysta
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
Código de barras 2D com identificador único incluído.
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA

46
PC:
SN:
NN:

47
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
CARTONAGEM – CANETA PRÉ-CHEIA - Embalagem múltipla com 12 canetas pré-cheias (3
embalagens de 4 canetas pré-cheias) - sem blue-box
1. NOME DO MEDICAMENTO
Benlysta 200 mg solução injetável em caneta pré-cheia
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada caneta pré-cheia de 1 ml contém 200 mg de belimumab
3. LISTA DOS EXCIPIENTES
Também contém cloridrato de arginina, histidina, monocloridrato de histidina, polissorbaro 80,
cloreto de sódio e água para preparações injetáveis.
4. FORMA FARMACÊUTICA E CONTEÚDO
Solução injetável em caneta pré-cheia.
4 canetas pré-cheias. Componente de uma embalagem múltipla.
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Consultar o folheto informativo antes de utilizar.
Via subcutânea.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP

48
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
Conservar no frigorífico.
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford
Middlesex
TW8 9GS
Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
Medicamento sujeito a receita médica.
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
caneta de benlysta
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA

49
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE
ACONDICIONAMENTO PRIMÁRIO
RÓTULO DA CANETA PRÉ-CHEIA
1. NOME DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO
Benlysta 200 mg injetável
belimumab
SC
Via subcutânea
2. MODO DE ADMINISTRAÇÃO
3. PRAZO DE VALIDADE
EXP
4. NÚMERO DO LOTE
Lot
5. CONTEÚDO EM PESO, VOLUME OU UNIDADE
1 ml
6. OUTRAS

50
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
CARTONAGEM – SERINGA(S) PRÉ-CHEIA(S)
1. NOME DO MEDICAMENTO
Benlysta 200 mg solução injetável em seringa pré-cheia
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada seringa pré-cheia de 1ml contém 200 mg de belimumab
3. LISTA DOS EXCIPIENTES
Também contém cloridrato de arginina, histidina, monocloridrato de histidina, polissorbaro 80,
cloreto de sódio e água para preparações injetáveis.
4. FORMA FARMACÊUTICA E CONTEÚDO
Solução injetável em seringa pré-cheia.
1 seringa pré-cheia.
4 seringas pré-cheias.
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Consultar o folheto informativo antes de utilizar.
Via subcutânea.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP

51
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
Conservar no frigorífico.
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/006: 1 seringa pré-cheia
EU/1/11/700/007: 4 seringas pré-cheias
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
seringa de benlysta
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
Código de barras 2D com identificador único incluído.
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA
PC:
SN:
NN:

52
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE
ACONDICIONAMENTO PRIMÁRIO
RÓTULO DA SERINGA PRÉ-CHEIA
1. NOME DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO
Benlysta 200 mg
belimumab
SC
2. MODO DE ADMINISTRAÇÃO
3. PRAZO DE VALIDADE
EXP
4. NÚMERO DO LOTE
Lot
5. CONTEÚDO EM PESO, VOLUME OU UNIDADE
1 ml
6. OUTRAS

53
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
CARTONAGEM – FRASCO PARA INJETÁVEIS
1. NOME DO MEDICAMENTO
Benlysta 120 mg pó para concentrado para solução para perfusão
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada frasco para injetáveis contém 120 mg de belimumab (80 mg/ml após reconstituição)
3. LISTA DOS EXCIPIENTES
Ácido cítrico mono-hidratado (E330), citrato de sódio (E331), sacarose, polissorbato 80
4. FORMA FARMACÊUTICA E CONTEÚDO
Pó para concentrado para solução para perfusão
1 frasco para injetáveis
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Para perfusão intravenosa após reconstituição e diluição.
Consultar o folheto informativo antes de utilizar.
Via intravenosa.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO

54
Conservar no frigorífico.
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/001
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
Foi aceite a justificação para não incluir a informação em Braille
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
Código de barras 2D com identificador único incluído.
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA
PC:
SN:
NN:

55
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO
CARTONAGEM – FRASCO PARA INJETÁVEIS
1. NOME DO MEDICAMENTO
Benlysta 400 mg pó para concentrado para solução para perfusão
belimumab
2. DESCRIÇÃO DA(S) SUBSTÂNCIA(S) ATIVA(S)
Cada frasco para injetáveis contém 400 mg de belimumab (80 mg/ml após reconstituição)
3. LISTA DOS EXCIPIENTES
Ácido cítrico mono-hidratado (E330), citrato de sódio (E331), sacarose, polissorbato 80
4. FORMA FARMACÊUTICA E CONTEÚDO
Pó para concentrado para solução para perfusão
1 frasco para injetáveis
5. MODO E VIA(S) DE ADMINISTRAÇÃO
Para perfusão intravenosa após reconstituição e diluição.
Consultar o folheto informativo antes de utilizar.
Via intravenosa.
Para uma única utilização.
6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DA VISTA E DO ALCANCE DAS CRIANÇAS
Manter fora da vista e do alcance das crianças.
7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO
8. PRAZO DE VALIDADE
EXP
9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
Conservar no frigorífico.

56
Não congelar.
Manter na embalagem exterior para proteger da luz.
10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE
APLICÁVEL
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido
12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU/1/11/700/002
13. NÚMERO DO LOTE
Lot
14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO
15. INSTRUÇÕES DE UTILIZAÇÃO
16. INFORMAÇÃO EM BRAILLE
Foi aceite a justificação para não incluir a informação em Braille
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
Código de barras 2D com identificador único incluído.
18 IDENTIFICADOR ÚNICO - DADOS PARA LEITURA HUMANA
PC:
SN:
NN:

57
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE
ACONDICIONAMENTO PRIMÁRIO
RÓTULO DO FRASCO PARA INJETÁVEIS
1. NOME DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO
Benlysta 120 mg pó para concentrado para solução para perfusão
belimumab
IV
2. MODO DE ADMINISTRAÇÃO
3. PRAZO DE VALIDADE
EXP
4. NÚMERO DO LOTE
Lot
5. CONTEÚDO EM PESO, VOLUME OU UNIDADE
120 mg
6. OUTRAS

58
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE
ACONDICIONAMENTO PRIMÁRIO
RÓTULO DO FRASCO PARA INJETÁVEIS
1. NOME DO MEDICAMENTO
Benlysta 400 mg pó para concentrado para solução para perfusão
belimumab
IV
2. MODO DE ADMINISTRAÇÃO
3. PRAZO DE VALIDADE
EXP
4. NÚMERO DO LOTE
Lot
5. CONTEÚDO EM PESO, VOLUME OU UNIDADE
400 mg
6. OUTRAS

59
B. FOLHETO INFORMATIVO

60
Folheto informativo: Informação para o utilizador
Benlysta 200 mg solução injetável em caneta pré-cheia
belimumab
Este medicamento está sujeito a monitorização adicional. Isto irá permitir a rápida identificação de
nova informação de segurança. Poderá ajudar, comunicando quaisquer efeitos secundários que tenha.
Para saber como comunicar efeitos secundários, veja o final da secção 4.
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém
informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode
ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados
neste folheto, fale com o seu médico. Ver secção 4.
O que contém este folheto
1. O que é Benlysta e para que é utilizado
2. O que precisa de saber antes de utilizar Benlysta
3. Como utilizar Benlysta
4. Efeitos secundários possíveis
5. Como conservar Benlysta
6. Conteúdo da embalagem e outras informações
Instruções passo a passo para utilização da caneta pré-cheia
1. O que é Benlysta e para que é utilizado
Benlysta é um medicamento utilizado para o tratamento do lúpus (lúpus eritematoso sistémico,
LES) em adultos (com e acima de 18 anos de idade) cuja doença esteja ainda altamente ativa apesar
do tratamento padrão.
O lúpus é uma doença na qual o sistema imunitário (o sistema que combate infeções) ataca as suas
próprias células e tecidos, provocando inflamação e danos nos órgãos. Pode afetar qualquer órgão do
corpo e pensa-se que envolve um tipo de células brancas sanguíneas chamado células B.
Benlysta contém belimumab (um anticorpo monoclonal). Diminui o número de células B no seu
sangue bloqueando a ação do BLyS, uma proteína que ajuda as células B a viverem mais tempo e que
se encontra em níveis aumentados nas pessoas com lúpus.
Ser-lhe-á administrado Benlysta bem como o seu tratamento habitual para o lúpus.
2. O que precisa de saber antes de utilizar Benlysta
Não utilize Benlysta:
• se tem alergia ao belimumab ou a qualquer outro componente deste medicamento (indicados
na secção 6).
Consulte o seu médico para saber se isto se pode aplicar a si.

61
Advertências e precauções
Fale com o seu médico antes de utilizar Benlysta:
• se tiver uma infeção atual ou crónica (permanente) ou se tiver infeções repetidamente. O seu
médico irá decidir se lhe pode ser dado Benlysta
• se estiver a planear ser vacinado ou tiver sido vacinado nos últimos 30 dias. Algumas vacinas não
devem ser administradas imediatamente antes ou durante o tratamento com Benlysta
• se o seu lúpus afetar os seus rins ou sistema nervoso
• se é VIH positivo(a) ou tem baixos níveis de imunoglobulinas
• se tem ou tiver tido hepatite B ou C
• se tiver tido um transplante de órgãos ou um transplante de medula óssea ou de células
estaminais
• se tiver tido cancro.
Informe o seu médico se alguma destas condições se aplicar a si.
Tenha atenção para sintomas importantes
Os indivíduos a tomar medicamentos que afetem o seu sistema imunitário podem ter um risco maior
para infeções, incluindo uma rara, mas grave, infeção do cérebro chamada de leucoencefalopatia
multifocal progressiva (LMP).
Leia a informação “Risco aumentado de infeção do cérebro” na secção 4 deste
folheto.
Crianças e adolescentes
Não administrar este medicamento a crianças e adolescentes com idade inferior a 18 anos.
Outros medicamentos e Benlysta
Informe o seu médico se estiver a tomar, tiver tomado recentemente, ou se vier a tomar outros
medicamentos.
Em especial, informe o seu médico se estiver a ser tratado com medicamentos que afetem o seu
sistema imunitário:
• ciclofosfamida (um medicamento para tratar alguns cancros e doenças autoimunes)
• qualquer medicamento que afete as células B (para tratar o cancro ou doenças inflamatórias).
Informe o seu médico. Utilizar estes medicamentos em associação com Benlysta pode
tornar o seu sistema imunitário menos efetivo. Isto pode aumentar o seu risco de sofrer uma
infeção grave.
Gravidez e aleitamento
Contraceção na mulher com potencial para engravidar
• Utilize um método contracetivo efetivo enquanto estiver a ser tratada com Benlysta e durante
pelo menos 4 meses após a última dose.
Gravidez
Benlysta não é normalmente recomendado se estiver grávida.

62
• Informe o seu médico se está grávida, se pensa estar grávida, ou planeia engravidar. O seu
médico irá decidir se pode utilizar Benlysta.
• Se engravidar enquanto estiver a ser tratada com Benlysta, informe o seu médico.
Amamentação
Informe o seu médico se estiver a amamentar. É provável que Benlysta possa passar para o leite
materno. O seu médico irá discutir consigo sobre se deve parar o tratamento com Benlysta enquanto
estiver a amamentar ou se deve parar de amamentar.
Condução de veículos e utilização de máquinas
Benlysta pode ter efeitos secundários que podem diminuir a sua capacidade de conduzir ou operar
máquinas.
Informação importante sobre os componentes de Benlysta
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose, é praticamente “isento de
sódio”.
3. Como utilizar Benlysta
Qual a quantidade a utilizar
A dose recomendada é de 200 mg (totalidade do conteúdo de uma caneta), uma vez por semana,
administrada (injetada) por baixo da pele, no mesmo dia de cada semana.
Se pretender alterar o dia da administração da dose (injeção):
Administre a dose no novo dia (mesmo que seja menos de uma semana depois da sua última dose).
Continue com o novo calendário semanal a partir desse dia.
Injeção de Benlysta
O médico ou enfermeiro irão mostrar a si ou ao seu cuidador como injetar Benlysta. A sua primeira
injeção com a caneta pré-cheia de Benlysta será supervisionada por um médico ou enfermeiro. Após
ter sido treinado(a) em como utilizar a caneta, o médico ou enfermeiro podem decidir que pode
administrar a injeção a si próprio(a), ou o seu cuidador pode administrá-la a si. O seu médico ou
enfermeiro vão também informar sobre os sinais e sintomas a que deve estar atento(a) quando utilizar
Benlysta, porque podem ocorrer reações alérgicas graves (ver “Reações alérgicas” na secção 4).
Irá administrar (injetar) Benlysta por baixo da pele na zona do estômago (abdómen) ou na parte
superior da perna (coxa).
A injeção de Benlysta subcutâneo não deve ser injetada numa veia (por via intravenosa).
As instruções sobre como utilizar a caneta pré-cheia encontram-se no final deste folheto.
Se utilizar mais Benlysta do que deveria
Se isto acontecer, contacte o médico ou enfermeiro imediatamente, que o(a) irão monitorizar para
quaisquer sinais ou sintomas de efeitos secundários, e tratar estes sintomas se necessário. Se possível,
mostre-lhes a embalagem ou este folheto.
Caso se tenha esquecido de administrar Benlysta
Administre (injete) a dose em falta assim que se lembrar. Depois continue com o calendário semanal
como habitualmente ou inicie um novo calendário semanal a partir do dia da injeção da dose em falta.
Se não notou que se tinha esquecido de uma dose até ser o momento da dose seguinte, então
administre (injete) apenas a dose seguinte como planeado. Não é necessário administrar (injetar) duas
doses no mesmo dia.

63
Se parar o tratamento com Benlysta
O seu médico irá decidir se necessita que lhe seja interrompido o tratamento com Benlysta.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não
se manifestem em todas as pessoas.
Reações alérgicas – procure ajuda médica imediatamente
Benlysta pode causar uma reação à injeção ou uma reação alérgica (hipersensibilidade).
Estas são efeitos secundários frequentes (podem afetar até 1 em 10 pessoas). Podem ocasionalmente
ser graves (pouco frequente, afetando até 1 em 100 pessoas) e podem colocar a vida em risco. Estas
reações graves são mais prováveis de ocorrer no dia do primeiro ou do segundo tratamento com
Benlysta, mas podem ser do tipo retardado e podem ocorrer vários dias depois.
Se manifestar algum dos seguintes sintomas de uma reação alérgica ou relacionada com a
injeção, informe o seu médico ou enfermeiro imediatamente ou dirija-se à urgência médica do
hospital mais próximo:
• inchaço da face, lábios, boca ou língua
• pieira, dificuldades em respirar ou falta de ar
• erupção na pele
• altos com comichão ou urticária
Raramente, as reações do tipo retardado menos graves a Benlysta podem também ocorrer, geralmente
5 a10 dias após uma injeção. Estas incluem sintomas como erupção na pele, sensação de mal-estar,
cansaço, dores musculares, dores de cabeça ou inchaço da face.
Se manifestar estes sintomas, especialmente se manifestar uma associação de dois ou mais destes
sintomas:
Informe o seu médico ou enfermeiro.
Infeções
Benlysta pode levar a que seja mais provável que tenha infeções, incluindo infeção do trato urinário e
das vias respiratórias. Estas são muito frequentes e podem afetar mais de 1 em 10 pessoas. Algumas
infeções podem ser graves e podem, pouco frequentemente, causar a morte.
Se manifestar alguns destes sintomas de uma infeção:
• febre
• tosse, problemas de respiração
• diarreia, vómitos
• sensação de ardor ao urinar
Informe o seu médico ou enfermeiro imediatamente.
Risco aumentado de infeção do cérebro
Os medicamentos que enfraquecem o sistema imunitário, tais como Benlysta, podem colocá-lo em
maior risco de ter uma infeção do cérebro rara, mas grave e que podem colocar a vida em perigo,
chamada de leucoencefalopatia multifocal progressiva (LMP).
Os sintomas de LMP incluem:
• perda de memória
• problemas em pensar

64
• dificuldade em falar ou em andar
• perda de visão.
Informe o seu médico imediatamente se tiver algum destes sintomas ou problemas semelhantes
que se tenham prolongado por vários dias.
Se já tinha estes sintomas antes de ter iniciado o tratamento com Benlysta:
Informe o seu médico imediatamente se notar qualquer alteração nestes sintomas.
Outros efeitos secundários possíveis:
Efeitos secundários muito frequentes
Estes podem afetar mais de 1 em 10 pessoas:
• infeções bacterianas (ver “Infeções” em cima)
• sentir-se maldisposto, diarreia.
Efeitos secundários frequentes
Estes podem afetar até 1 em 10 pessoas:
• temperatura alta ou febre
• contagem baixa de células sanguíneas brancas (pode ser observada nas análises sanguíneas)
• infeção no nariz, garganta ou estômago
• dor nas mãos ou pés
• enxaqueca
• depressão
• reações no local de injeção, por exemplo, erupção na pele, vermelhidão, prurido ou inchaço
da pele no local onde administrou (injetou) Benlysta.
Efeitos secundários pouco frequentes
Estes podem afetar até 1 em 100 pessoas:
• erupção na pele irregular e com prurido (urticária), erupção na pele.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste
folheto, fale com o seu médico ou enfermeiro. Também poderá comunicar efeitos secundários
diretamente através do sistema nacional de notificação mencionado no Apêndice V. Ao comunicar
efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste
medicamento.
5. Como conservar Benlysta
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no rótulo e na embalagem exterior,
após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conservar no frigorífico (2ºC a 8ºC).
Não congelar.
Conservar na embalagem original para proteger da luz.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu
farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger
o ambiente.

65
6. Conteúdo da embalagem e outras informações
Qual a composição de Benlysta
A substância ativa é belimumab.
Cada caneta pré-cheia de 1 ml contém 200 mg de belimumab.
Os outros componentes são cloridrato de arginina, histidina, monocloridrato de histidina, polissorbato
80, cloreto de sódio, água para preparações injetáveis. Ver “Informação importante sobre os
componentes de Benlysta” na secção 2 para mais informação.
Qual o aspeto de Benlysta e conteúdo da embalagem
Benlysta é fornecido numa caneta pré-cheia de utilização única com 1 ml de solução incolor a
ligeiramente amarelada.
Disponível em embalagens de 1 ou 4 canetas pré-cheias por embalagem e em embalagens múltiplas
com 12 canetas pré-cheias (3 embalagens de 4 canetas pré-cheias).
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Reino Unido
Fabricante
Glaxo Operations UK Ltd
Harmire Road
Barnard Castle
County Durham, DL12 8DT
Reino Unido
Para quaisquer informações sobre este medicamento, queira contactar o representante local do Titular
da Autorização de Introdução no Mercado:
België/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Tél/Tel: + 32 (0) 10 85 52 00
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
България
ГлаксоСмитКлайн ЕООД
Teл.: + 359 2 953 10 34
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111
Malta
GlaxoSmithKline (Malta) Limited
Tel: + 356 21 238131
Danmark
GlaxoSmithKline Pharma A/S
Tlf: + 45 36 35 91 00
Nederland
GlaxoSmithKline BV
Tel: + 31 (0)30 6938100

66
Deutschland
GlaxoSmithKline GmbH & Co. KG
Tel.: + 49 (0)89 36044 8701
Norge
GlaxoSmithKline AS
Tlf: + 47 22 70 20 00
Eesti
GlaxoSmithKline Eesti OÜ
Tel: + 372 6676 900
Österreich
GlaxoSmithKline Pharma GmbH
Tel: + 43 (0)1 97075 0
Ελλάδα
GlaxoSmithKline A.E.B.E.
Τηλ: + 30 210 68 82 100
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
España
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700
Portugal
GlaxoSmithKline – Produtos Farmacêuticos, Lda.
Tel: + 351 21 412 95 00
France
Laboratoire GlaxoSmithKline
Tél.: + 33 (0)1 39 17 84 44
România
GlaxoSmithKline (GSK) S.R.L.
Tel: + 4021 3028 208
Hrvatska
GlaxoSmithKline d.o.o.
Tel:+ 385 1 6051 999
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00
Ireland
GlaxoSmithKline (Ireland) Limited
Tel: + 353 (0)1 4955000
Slovenská republika
GlaxoSmithKline Slovakia s. r. o.
Tel: + 421 (0)2 48 26 11 11
Ísland
Vistor hf.
Sími: +354 535 7000
Suomi/Finland
GlaxoSmithKline Oy
Puh/Tel: + 358 (0)10 30 30 30
Italia
GlaxoSmithKline S.p.A.
Tel: + 39 (0)45 9218 111
Sverige
GlaxoSmithKline AB
Tel: + 46 (0)8 638 93 00
Κύπρος
GlaxoSmithKline (Cyprus) Ltd
Τηλ: + 357 22 39 70 00
United Kingdom
GlaxoSmithKline UK Ltd
Tel: + 44 (0)800 221441
Latvija
GlaxoSmithKline Latvia SIA
Tel: + 371 67312687

67
Lietuva
GlaxoSmithKline Lietuva UAB
Tel: + 370 5 264 90 00
Este folheto foi revisto pela última vez em
Está disponível informação pormenorizada sobre este medicamento no sítio da internet da Agência
Europeia de Medicamentos: http://www.ema.europa.eu.

68
Instruções passo a passo para utilização da caneta pré-cheia
Uma vez por semana
Siga estas instruções sobre como utilizar a caneta pré-cheia corretamente. O não cumprimento destas
instruções pode afetar o funcionamento adequado da caneta pré-cheia. Deve também receber treino em
como utilizar a caneta pré-cheia.
Benlysta destina-se a ser administrado por baixo da pele apenas (por via subcutânea).
Conservação
• Manter no frigorífico até 30 minutos antes da utilização.
• Manter na cartonagem para proteger da luz.
• Manter fora da vista e do alcance das crianças.
• Não congelar.
• Não utilizar se deixado à temperatura ambiente por mais de 12 horas.
Advertências
• A caneta pré-cheia deve apenas ser utilizada uma vez e depois deve ser eliminada.
• Não partilhar a sua caneta pré-cheia de Benlysta com outra pessoa.
• Não agitar.
• Não utilizar se tiver caído numa superfície dura.
• Não remover a cápsula de fecho do anel até antes da injeção.
Componentes da caneta pré-cheia de Benlysta
Janela para inspeção
Rolha cinzenta
Cápsula de fecho do anel
Proteção da
agulha dourada
Prazo de validade
Exp: Mês - Ano

69
Elementos que precisa para a injeção
1. Juntar e verificar todos os elementos
Junte os elementos
• Retire uma bandeja selada com a caneta pré-cheia do frigorífico.
• Encontre uma superfície confortável, bem iluminada e limpa e coloque os seguintes elementos
ao seu alcance:
• caneta pré-cheia de Benlysta
• toalhete com álcool (não incluído na embalagem)
• compressa de gaze ou bola de algodão (não incluído na embalagem)
• recipiente vazio com tampa ajustada para eliminação da caneta (não incluído na
embalagem)
• Não realizar a injeção se não tiver todos os elementos listados.
Verifique o prazo de validade
• Remova a pelicula da bandeja e retire a caneta pré-cheia.
• Verifique o prazo de validade na caneta pré-cheia. (Figura 1)
Caneta pré-cheia de Benlysta
Toalhete com
álcool
(não incluído)
Compressa de gaze ou bola de
algodão
(não incluído)

70
Figura 1
Não utilizar após o prazo de validade
2. Preparar e inspecionar a caneta pré-cheia
Permitir que atinja a temperatura ambiente
• Deixe a caneta à temperatura ambiente durante 30 minutos. (Figura 2) Injetar Benlysta frio
pode demorar mais tempo e pode ser desconfortável.
Figura 2
• Não aquecer a caneta de outra forma. Por exemplo, não aqueça num micro-ondas, com água
quente ou à luz solar direta.
• Não remover a cápsula de fecho do anel durante este passo.
Inspecionar a solução de Benlysta
• Olhe pela janela de inspeção para verificar se a solução de Benlysta é incolor a ligeiramente
amarelada. (Figura 3)
É normal observar uma ou mais bolhas de ar na solução.
Exp: Mês - Ano
Exp: Mês - Ano
Esperar 30
minutos
min

71
Figura 3
• Não utilizar se a solução estiver enevoada, com cor alterada ou com partículas.
3. Escolher e limpar um local de injeção
Escolha um local de injeção
• Escolha um local de injeção (abdómen ou coxa) como indicado na Figura 4.
Figura 4
• Não injetar exatamente no mesmo local todas as vezes. Isto é para evitar que a pele fique
endurecida.
• Não injetar em zonas que a pele está sensível, ferida, vermelha ou dura.
• Não injetar numa área de 5 cm do umbigo.
Limpe o local de injeção
• Lave as mãos.
• Limpe o local de injeção, limpando-o com um toalhete com álcool. (Figura 5). Deixe a pele
secar ao ar.
Solução de Benlysta

72
Figura 5
Não tocar nesta área novamente antes de administrar a injeção.
4. Preparar-se para a injeção
Remover a cápsula de fecho do anel.
• Não remova a cápsula de fecho do anel até ao momento imediatamente antes da administração
da injeção.
• Remova a cápsula de fecho do anel puxando ou torcendo-a. A cápsula de fecho do anel pode ser
rodada no sentido dos ponteiros do relógio ou no sentido contrário aos ponteiros do relógio.
(Figura 6)
Figura 6
• Não colocar a cápsula de fecho do anel de novo na caneta.
Posição da caneta
• Segure a caneta confortavelmente de modo a que possa ver a janela de inspeção. Isto é
importante para que possa confirmar a administração de uma dose completa. (Figura 7)
Figura 7

73
• Se necessário, firme o local de injeção, puxando ou esticando a pele.
• Posicione a caneta diretamente sobre o local de injeção (com um ângulo de 90º). Certifique-se
de que a proteção da agulha dourada está assente na pele.
5. Administrar (injetar) Benlysta
Iniciar a injeção
Isto irá introduzir a agulha e iniciar a injeção.
Figura 8
Pode ouvir um primeiro “click” ao inicio da injeção. Irá ver o indicador roxo começar a mover-se na
janela de inspeção. (Figura 9)
• Pressione com firmeza a caneta completamente para baixo no local de injeção e segure-a no
local. (Figura 8)

74
Figura 9
Completar a injeção
Continue a segurar a caneta até o indicador roxo deixar de se mover. Pode ouvir um segundo “click”
uns segundos antes do indicador roxo parar. (Figura 10)
Figura 10
• Quando a injeção estiver finalizada, levante a caneta do local de inspeção.
6. Eliminar e inspecionar
Eliminar a caneta usada
• Não colocar a cápsula de fecho do anel de novo na caneta.
• Eliminar a caneta usada e a cápsula de fecho do anel num recipiente vazio com uma tampa
ajustada.
• Pergunte ao médico ou farmacêutico instruções de como eliminar adequadamente a caneta
usada ou o recipiente com as canetas usadas
A injeção pode demorar até 15 segundos para estar finalizada.
Primeiro “click”
Indicador roxo
Segundo “click” Despois espere
até…
… o indicador roxo
parar

75
• Não reciclar ou deitar fora a caneta usada, ou os recipientes com as canetas usadas, no lixo
doméstico.
Inspecionar o local de injeção
Pode haver uma pequena quantidade de sangue no local de injeção.
• Se necessário, pressione a bola de algodão ou a compressa de gaze no local de injeção.
• Não esfregue o local de injeção.

76
Folheto informativo: Informação para o utilizador
Benlysta 200 mg solução injetável em seringa pré-cheia
belimumab
Este medicamento está sujeito a monitorização adicional. Isto irá permitir a rápida identificação de
nova informação de segurança. Poderá ajudar, comunicando quaisquer efeitos secundários que tenha.
Para saber como comunicar efeitos secundários, veja o final da secção 4.
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém
informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode
ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados
neste folheto, fale com o seu médico. Ver secção 4.
O que contém este folheto
1. O que é Benlysta e para que é utilizado
2. O que precisa de saber antes de utilizar Benlysta
3. Como utilizar Benlysta
4. Efeitos secundários possíveis
5. Como conservar Benlysta
6. Conteúdo da embalagem e outras informações
Instruções passo a passo para utilização da seringa pré-cheia
1. O que é Benlysta e para que é utilizado
Benlysta é um medicamento utilizado para o tratamento do lúpus (lúpus eritematoso sistémico,
LES) em adultos (com e acima de 18 anos de idade) cuja doença esteja ainda altamente ativa apesar
do tratamento padrão.
O lúpus é uma doença na qual o sistema imunitário (o sistema que combate infeções) ataca as suas
próprias células e tecidos, provocando inflamação e danos nos órgãos. Pode afetar qualquer órgão do
corpo e pensa-se que envolve um tipo de células brancas sanguíneas chamado células B.
Benlysta contém belimumab (um anticorpo monoclonal). Diminui o número de células B no seu
sangue bloqueando a ação do BLyS, uma proteína que ajuda as células B a viverem mais tempo e que
se encontra em níveis aumentados nas pessoas com lúpus.
Ser-lhe-á administrado Benlysta bem como o seu tratamento habitual para o lúpus.
2. O que precisa de saber antes de utilizar Benlysta
Não utilize Benlysta:
• se tem alergia ao belimumab ou a qualquer outro componente deste medicamento (indicados
na secção 6).
Consulte o seu médico para saber se isto se pode aplicar a si.

77
Advertências e precauções
Fale com o seu médico antes de utilizar Benlysta:
• se tiver uma infeção atual ou crónica (permanente) ou se tiver infeções repetidamente. O seu
médico irá decidir se lhe pode ser dado Benlysta
• se estiver a planear ser vacinado ou tiver sido vacinado nos últimos 30 dias. Algumas vacinas não
devem ser administradas imediatamente antes ou durante o tratamento com Benlysta
• se o seu lúpus afetar os seus rins ou sistema nervoso
• se é VIH positivo(a) ou tem baixos níveis de imunoglobulinas
• se tem ou tiver tido hepatite B ou C
• se tiver tido um transplante de órgãos ou um transplante de medula óssea ou de células
estaminais
• se tiver tido cancro.
Informe o seu médico se alguma destas condições se aplicar a si.
Tenha atenção para sintomas importantes
Os indivíduos a tomar medicamentos que afetem o seu sistema imunitário podem ter um risco maior
para infeções, incluindo uma rara, mas grave, infeção do cérebro chamada de leucoencefalopatia
multifocal progressiva (LMP).
Leia a informação “Risco aumentado de infeção do cérebro” na secção 4 deste
folheto.
Crianças e adolescentes
Não administrar este medicamento a crianças e adolescentes com idade inferior a 18 anos.
Outros medicamentos e Benlysta
Informe o seu médico se estiver a tomar, tiver tomado recentemente, ou se vier a tomar outros
medicamentos.
Em especial, informe o seu médico se estiver a ser tratado com medicamentos que afetem o seu
sistema imunitário:
• ciclofosfamida (um medicamento para tratar alguns cancros e doenças autoimunes)
• qualquer medicamento que afete as células B (para tratar o cancro ou doenças inflamatórias).
Informe o seu médico. Utilizar estes medicamentos em associação com Benlysta pode
tornar o seu sistema imunitário menos efetivo. Isto pode aumentar o seu risco de sofrer uma
infeção grave.
Gravidez e aleitamento
Contraceção na mulher com potencial para engravidar
• Utilize um método contracetivo efetivo enquanto estiver a ser tratada com Benlysta e durante
pelo menos 4 meses após a última dose.
Gravidez
Benlysta não é normalmente recomendado se estiver grávida.

78
• Informe o seu médico se está grávida, se pensa estar grávida, ou planeia engravidar. O seu
médico irá decidir se pode utilizar Benlysta.
• Se engravidar enquanto estiver a ser tratada com Benlysta, informe o seu médico.
Amamentação
Informe o seu médico se estiver a amamentar. É provável que Benlysta possa passar para o leite
materno. O seu médico irá discutir consigo sobre se deve parar o tratamento com Benlysta enquanto
estiver a amamentar ou se deve parar de amamentar.
Condução de veículos e utilização de máquinas
Benlysta pode ter efeitos secundários que podem diminuir a sua capacidade de conduzir ou operar
máquinas.
Informação importante sobre os componentes de Benlysta
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose, é praticamente “isento de
sódio”.
3. Como utilizar Benlysta
Qual a quantidade a utilizar
A dose recomendada é de 200 mg (totalidade do conteúdo de uma seringa), uma vez por semana,
administrada (injetada) por baixo da pele, no mesmo dia de cada semana.
Se pretender alterar o dia da administração da dose (injeção):
Administre a dose no novo dia (mesmo que seja menos de uma semana depois da sua última dose).
Continue com o novo calendário semanal a partir desse dia.
Injeção de Benlysta
O médico ou enfermeiro irão mostrar a si ou ao seu cuidador como injetar Benlysta. A sua primeira
injeção com a seringa pré-cheia de Benlysta será supervisionada por um médico ou enfermeiro. Após
ter sido treinado(a) em como utilizar a seringa, o médico ou enfermeiro podem decidir que pode
administrar a injeção a si próprio(a), ou o seu cuidador pode administrá-la a si. O seu médico ou
enfermeiro vão também informar sobre os sinais e sintomas a que deve estar atento(a) quando utilizar
Benlysta, porque podem ocorrer reações alérgicas graves (ver “Reações alérgicas” na secção 4).
Irá administrar (injetar) Benlysta por baixo da pele na zona do estômago (abdómen) ou na parte
superior da perna (coxa).
A injeção de Benlysta subcutâneo não deve ser injetada numa veia (por via intravenosa).
As instruções sobre como utilizar a seringa pré-cheia encontram-se no final deste folheto.
Se utilizar mais Benlysta do que deveria
Se isto acontecer, contacte o médico ou enfermeiro imediatamente, que o(a) irão monitorizar para
quaisquer sinais ou sintomas de efeitos secundários, e tratar estes sintomas se necessário. Se possível,
mostre-lhes a embalagem ou este folheto.
Caso se tenha esquecido de administrar Benlysta
Administre (injete) a dose em falta assim que se lembrar. Depois continue com o calendário semanal
como habitualmente ou inicie um novo calendário semanal a partir do dia da injeção da dose em falta.
Se não notou que se tinha esquecido de uma dose até ser o momento da dose seguinte, então
administre (injete) apenas a dose seguinte como planeado. Não é necessário administrar (injetar) duas
doses no mesmo dia.

79
Se parar o tratamento com Benlysta
O seu médico irá decidir se necessita que lhe seja interrompido o tratamento com Benlysta.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não
se manifestem em todas as pessoas.
Reações alérgicas – procure ajuda médica imediatamente
Benlysta pode causar uma reação à injeção ou uma reação alérgica (hipersensibilidade).
Estas são efeitos secundários frequentes (podem afetar até 1 em 10 pessoas). Podem ocasionalmente
ser graves (pouco frequente, afetando até 1 em 100 pessoas) e podem colocar a vida em risco. Estas
reações graves são mais prováveis de ocorrer no dia do primeiro ou do segundo tratamento com
Benlysta, mas podem ser do tipo retardado e podem ocorrer vários dias depois.
Se manifestar algum dos seguintes sintomas de uma reação alérgica ou relacionada com a
injeção, informe o seu médico ou enfermeiro imediatamente ou dirija-se à urgência médica do
hospital mais próximo:
• inchaço da face, lábios, boca ou língua
• pieira, dificuldades em respirar ou falta de ar
• erupção na pele
• altos com comichão ou urticária
Raramente, as reações do tipo retardado menos graves a Benlysta podem também ocorrer, geralmente
5 a10 dias após uma injeção. Estas incluem sintomas como erupção na pele, sensação de mal-estar,
cansaço, dores musculares, dores de cabeça ou inchaço da face.
Se manifestar estes sintomas, especialmente se manifestar uma associação de dois ou mais destes
sintomas:
Informe o seu médico ou enfermeiro.
Infeções
Benlysta pode levar a que seja mais provável que tenha infeções, incluindo infeção do trato urinário e
das vias respiratórias. Estas são muito frequentes e podem afetar mais de 1 em 10 pessoas. Algumas
infeções podem ser graves e podem, pouco frequentemente, causar a morte.
Se manifestar alguns destes sintomas de uma infeção:
• febre
• tosse, problemas de respiração
• diarreia, vómitos
• sensação de ardor ao urinar
Informe o seu médico ou enfermeiro imediatamente.
Risco aumentado de infeção do cérebro
Os medicamentos que enfraquecem o sistema imunitário, tais como Benlysta, podem colocá-lo em
maior risco de ter uma infeção do cérebro rara, mas grave e que podem colocar a vida em perigo,
chamada de leucoencefalopatia multifocal progressiva (LMP).
Os sintomas de LMP incluem:
• perda de memória
• problemas em pensar
• dificuldade em falar ou em andar

80
• perda de visão.
Informe o seu médico imediatamente se tiver algum destes sintomas ou problemas semelhantes
que se tenham prolongado por vários dias.
Se já tinha estes sintomas antes de ter iniciado o tratamento com Benlysta:
Informe o seu médico imediatamente se notar qualquer alteração nestes sintomas.
Outros efeitos secundários possíveis:
Efeitos secundários muito frequentes
Estes podem afetar mais de 1 em 10 pessoas:
• infeções bacterianas (ver “Infeções” em cima)
• sentir-se maldisposto, diarreia.
Efeitos secundários frequentes
Estes podem afetar até 1 em 10 pessoas:
• temperatura alta ou febre
• contagem baixa de células sanguíneas brancas (pode ser observada nas análises sanguíneas)
• infeção no nariz, garganta ou estômago
• dor nas mãos ou pés
• enxaqueca
• depressão
• reações no local de injeção, por exemplo, erupção na pele, vermelhidão, prurido ou inchaço
da pele no local onde administrou (injetou) Benlysta.
Efeitos secundários pouco frequentes
Estes podem afetar até 1 em 100 pessoas:
• erupção na pele irregular e com prurido (urticária), erupção na pele.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste
folheto, fale com o seu médico ou enfermeiro. Também poderá comunicar efeitos secundários
diretamente através do sistema nacional de notificação mencionado no Apêndice V. Ao comunicar
efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste
medicamento.
5. Como conservar Benlysta
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no rótulo e na embalagem exterior,
após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conservar no frigorífico (2ºC a 8ºC).
Não congelar.
Conservar na embalagem original para proteger da luz.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu
farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger
o ambiente.

81
6. Conteúdo da embalagem e outras informações
Qual a composição de Benlysta
A substância ativa é belimumab.
Cada seringa pré-cheia de 1 ml contém 200 mg de belimumab.
Os outros componentes são cloridrato de arginina, histidina, monocloridrato de histidina, polissorbato
80, cloreto de sódio, água para preparações injetáveis. Ver “Informação importante sobre os
componentes de Benlysta” na secção 2 para mais informação.
Qual o aspeto de Benlysta e conteúdo da embalagem
Benlysta é fornecido numa seringa pré-cheia com cápsula de fecho da agulha de utilização única com
1 ml de solução incolor a ligeiramente amarelada.
Disponível em embalagens de 1 ou 4 seringas pré-cheias por embalagem.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Reino Unido
Fabricante
Glaxo Operations UK Ltd
Harmire Road
Barnard Castle
County Durham, DL12 8DT
Reino Unido
Para quaisquer informações sobre este medicamento, queira contactar o representante local do Titular
da Autorização de Introdução no Mercado:
België/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Tél/Tel: + 32 (0) 10 85 52 00
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
България
ГлаксоСмитКлайн ЕООД
Teл.: + 359 2 953 10 34
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111
Malta
GlaxoSmithKline (Malta) Limited
Tel: + 356 21 238131
Danmark
GlaxoSmithKline Pharma A/S
Tlf: + 45 36 35 91 00
Nederland
GlaxoSmithKline BV
Tel: + 31 (0)30 6938100

82
Deutschland
GlaxoSmithKline GmbH & Co. KG
Tel.: + 49 (0)89 36044 8701
Norge
GlaxoSmithKline AS
Tlf: + 47 22 70 20 00
Eesti
GlaxoSmithKline Eesti OÜ
Tel: + 372 6676 900
Österreich
GlaxoSmithKline Pharma GmbH
Tel: + 43 (0)1 97075 0
Ελλάδα
GlaxoSmithKline A.E.B.E.
Τηλ: + 30 210 68 82 100
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
España
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700
Portugal
GlaxoSmithKline – Produtos Farmacêuticos, Lda.
Tel: + 351 21 412 95 00
France
Laboratoire GlaxoSmithKline
Tél.: + 33 (0)1 39 17 84 44
România
GlaxoSmithKline (GSK) S.R.L.
Tel: + 4021 3028 208
Hrvatska
GlaxoSmithKline d.o.o.
Tel:+ 385 1 6051 999
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00
Ireland
GlaxoSmithKline (Ireland) Limited
Tel: + 353 (0)1 4955000
Slovenská republika
GlaxoSmithKline Slovakia s. r. o.
Tel: + 421 (0)2 48 26 11 11
Ísland
Vistor hf.
Sími: +354 535 7000
Suomi/Finland
GlaxoSmithKline Oy
Puh/Tel: + 358 (0)10 30 30 30
Italia
GlaxoSmithKline S.p.A.
Tel: + 39 (0)45 9218 111
Sverige
GlaxoSmithKline AB
Tel: + 46 (0)8 638 93 00
Κύπρος
GlaxoSmithKline (Cyprus) Ltd
Τηλ: + 357 22 39 70 00
United Kingdom
GlaxoSmithKline UK Ltd
Tel: + 44 (0)800 221441
Latvija
GlaxoSmithKline Latvia SIA
Tel: + 371 67312687

83
Lietuva
GlaxoSmithKline Lietuva UAB
Tel: + 370 5 264 90 00
Este folheto foi revisto pela última vez em
Está disponível informação pormenorizada sobre este medicamento no sítio da internet da Agência
Europeia de Medicamentos: http://www.ema.europa.eu.

84
Instruções passo a passo para utilização da seringa pré-cheia
Uma vez por semana
Siga estas instruções sobre como utilizar a seringa pré-cheia corretamente. O não cumprimento destas
instruções pode afetar o funcionamento adequado da seringa pré-cheia. Deve também receber treino
em como utilizar a seringa pré-cheia.
Benlysta destina-se a ser administrado por baixo da pele apenas (por via subcutânea).
Conservação
• Manter no frigorífico até 30 minutos antes da utilização.
• Manter na cartonagem para proteger da luz.
• Manter fora da vista e do alcance das crianças.
• Não congelar.
• Não utilizar se deixado à temperatura ambiente por mais de 12 horas.
Advertências
• A seringa pré-cheia deve apenas ser utilizada uma vez e depois deve ser eliminada.
• Não partilhar a sua seringa pré-cheia de Benlysta com outra pessoa.
• Não agitar.
• Não utilizar se tiver caído numa superfície dura.
• Não remover a cápsula de fecho da agulha até antes da injeção.
Componentes da seringa pré-cheia de Benlysta

85
Elementos que precisa para a injeção
1. Juntar e verificar todos os elementos
Junte os elementos
Antes da utilização
Cápsula de fecho da agulha
Agulha
Corpo
Êmbolo
Janela de
inspeção
Suporte dos dedos
Após a utilização – a agulha é coberta pela proteção da agulha
Proteção da
agulha ativada
Seringa pré-cheia de Benlysta
Toalhete com álcool
(não incluído)
Compressa de gaze ou
bola de algodão
(não incluído)

86
• Retire uma bandeja selada com a seringa pré-cheia do frigorífico.
• Encontre uma superfície confortável, bem iluminada e limpa e coloque os seguintes elementos
ao seu alcance:
• seringa pré-cheia de Benlysta
• toalhete com álcool (não incluído na embalagem)
• compressa de gaze ou bola de algodão (não incluído na embalagem)
• recipiente vazio com tampa ajustada para eliminação da seringa (não incluído na
embalagem)
• Não realizar a injeção se não tiver todos os elementos listados.
Verifique o prazo de validade
• Remova a pelicula da bandeja e retire a seringa pré-cheia, segurando pelo meio do corpo.
• Verifique o prazo de validade na seringa pré-cheia. (Figura 1)
Figura 1
•
• Não utilizar após o prazo de validade.
2. Preparar e inspecionar a seringa pré-cheia
Permitir que atinja a temperatura ambiente
• Deixe a seringa à temperatura ambiente durante 30 minutos. (Figura 2) Injetar Benlysta frio
pode demorar mais tempo e pode ser desconfortável.
Exp: Mês - Ano
Exp: Mês - Ano

87
Figura 2
• Não aquecer a seringa de outra forma. Por exemplo, não aqueça num micro-ondas, com água
quente ou à luz solar direta.
• Não remover a cápsula de fecho da agulha durante este passo.
Inspecionar a solução de Benlysta
• Olhe pela janela de inspeção para verificar se a solução de Benlysta é incolor a ligeiramente
amarelada. (Figura 3)
Figura 3
• Não utilizar se a solução estiver enevoada, com cor alterada ou com partículas.
3. Escolher e limpar um local de injeção
Escolha um local de injeção
• Escolha um local de injeção (abdómen ou coxa) como indicado na Figura 4.
É normal observar uma ou mais bolhas de ar na solução.
Esperar 30
minutos
min
Solução de Benlysta

88
Figura 4
• Não injetar exatamente no mesmo local todas as vezes. Isto é para evitar que a pele fique
endurecida.
• Não injetar em zonas que a pele está sensível, ferida, vermelha ou dura.
• Não injetar numa área de 5 cm do umbigo.
Limpe o local de injeção
• Lave as mãos.
• Limpe o local de injeção, limpando-o com um toalhete com álcool. (Figura 5). Deixe a pele
secar ao ar.
Figura 5
• Não tocar nesta área novamente antes de administrar a injeção.
4. Preparar-se para a injeção
• Não remova a cápsula de fecho da agulha até ao momento imediatamente antes da
administração da injeção.
• Segure a seringa pré-cheia pelo corpo e com a agulha afastada de si. (Figura 6a)
• Remova a cápsula de fecho da agulha puxando-a. (Figura 6b)

89
Figura 6
Pode ver uma gota de líquido na ponta da agulha. Isto é normal.
• Não deixar a agulhar tocar em nenhuma superfície.
• Não expelir nenhuma bolha de ar da seringa.
• Não colocar a cápsula de fecho da agulha de novo na seringa.
5. Administrar (injetar) Benlysta
Inserir a agulha
• Segure a seringa com uma mão.
• Utilize a mão livre para gentilmente apertar a pele à volta do local de injeção. (Figura 7)
• Inserir a agulha completamente na área da pele apertada num ligeiro ângulo de (45°), através um
movimento de dardo.
Figura 7
a
b
90
• Após a agulha estar completamente inserida, libertar a pele apertada.
Completar a injeção
• Empurrar todo o êmbolo para baixo até toda a solução ser injetada. (Figura 8)
Figura 8
Mantendo-se a segurar na seringa, retire devagar o polegar, permitindo que o êmbolo suba (Figura 9).
A agulha vai automaticamente entrar na proteção da agulha.
Figura 9
6. Eliminar e inspecionar
Eliminar a seringa usada
• Eliminar a seringa usada e a cápsula de fecho da agulha num recipiente vazio com uma tampa
ajustada.
Empurrar todo o
êmbolo para baixo
Retirar devagar o
polegar

91
• Pergunte ao médico ou farmacêutico instruções de como eliminar adequadamente a seringa
usada ou o recipiente com as seringas usadas
• Não reciclar ou deitar fora a seringa usada, ou os recipientes com as seringas usadas, no lixo
doméstico.
Inspecionar o local de injeção
Pode haver uma pequena quantidade de sangue no local de injeção.
• Se necessário, pressione a bola de algodão ou a compressa de gaze no local de injeção.
• Não esfregue o local de injeção.

92
Folheto informativo: Informação para o utilizador
Benlysta 120 mg pó para concentrado para solução para perfusão
Benlysta 400 mg pó para concentrado para solução para perfusão
belimumab
Este medicamento está sujeito a monitorização adicional. Isto irá permitir a rápida identificação de
nova informação de segurança. Poderá ajudar, comunicando quaisquer efeitos secundários que tenha.
Para saber como comunicar efeitos secundários, veja o final da secção 4.
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém
informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados
neste folheto, fale com o seu médico. Ver secção 4.
O que contém este folheto
1. O que é Benlysta e para que é utilizado
2. O que precisa de saber antes de utilizar Benlysta
3. Como utilizar Benlysta
4. Efeitos secundários possíveis
5. Como conservar Benlysta
6. Conteúdo da embalagem e outras informações
1. O que é Benlysta e para que é utilizado
Benlysta é um medicamento utilizado para o tratamento do lúpus (lúpus eritematoso sistémico,
LES) em adultos (com e acima de 18 anos de idade) cuja doença esteja ainda altamente ativa apesar
do tratamento padrão.
O lúpus é uma doença na qual o sistema imunitário (o sistema que combate infeções) ataca as suas
próprias células e tecidos, provocando inflamação e danos nos órgãos. Pode afetar qualquer órgão do
corpo e pensa-se que envolve um tipo de células brancas sanguíneas chamado células B.
Benlysta contém belimumab (um anticorpo monoclonal). Diminui o número de células B no seu
sangue bloqueando a ação do BLyS, uma proteína que ajuda as células B a viverem mais tempo e que
se encontra em níveis aumentados nas pessoas com lúpus.
Ser-lhe-á administrado Benlysta bem como o seu tratamento habitual para o lúpus.
2. O que precisa de saber antes de utilizar Benlysta
Não administrar Benlysta:
• se tem alergia ao belimumab ou a qualquer outro componente deste medicamento (indicados
na secção 6)
Consulte o seu médico para saber se isto se pode aplicar a si.
Advertências e precauções

93
Fale com o seu médico antes de Benlysta lhe ser administrado:
• se tiver uma infeção atual ou crónica (permanente) ou se tiver infeções repetidamente. O seu
médico irá decidir se lhe pode ser dado Benlysta
• se estiver a planear ser vacinado ou tiver sido vacinado nos últimos 30 dias. Algumas vacinas
não devem ser administradas imediatamente antes ou durante o tratamento com Benlysta
• se o seu lúpus afetar os seus rins ou sistema nervoso
• se é VIH positivo(a) ou tem baixos níveis de imunoglobulinas
• se tem ou tiver tido hepatite B ou C
• se tiver tido um transplante de órgãos ou um transplante de medula óssea ou de células
estaminais
• se tiver tido cancro.
Informe o seu médico se alguma destas condições se aplicar a si.
Tenha atenção para sintomas importantes
Os indivíduos a tomar medicamentos que afetem o seu sistema imunitário podem ter um risco maior
para infeções, incluindo uma rara, mas grave, infeção do cérebro chamada de leucoencefalopatia
multifocal progressiva (LMP)
Leia a informação “Risco aumentado de infeção do cérebro” na secção 4 deste
folheto.
Outros medicamentos e Benlysta
Informe o seu médico se estiver a tomar, tiver tomado recentemente, ou se vier a tomar outros
medicamentos.
Em especial, informe o seu médico se estiver a ser tratado com medicamentos que afetem o seu
sistema imunitário:
• ciclofosfamida (um medicamento para tratar alguns cancros e doenças autoimunes)
• qualquer medicamento que afete as células B (para tratar o cancro ou doenças inflamatórias).
Informe o seu médico. Utilizar estes medicamentos em associação com Benlysta pode
tornar o seu sistema imunitário menos efetivo. Isto pode aumentar o seu risco de sofrer uma
infeção grave.
Gravidez e aleitamento
Contraceção na mulher com potencial para engravidar
• Utilize um método contracetivo efetivo enquanto estiver a ser tratada com Benlysta e durante
pelo menos 4 meses após a última dose.
Gravidez
Benlysta não é normalmente recomendado se estiver grávida.
• Informe o seu médico se está grávida, se pensa estar grávida, ou planeia engravidar. O seu
médico irá decidir se Benlysta lhe pode ser administrado.
• Se engravidar enquanto estiver a ser tratada com Benlysta informe o seu médico.
Amamentação

94
Informe o seu médico se estiver a amamentar. É provável que Benlysta possa passar para o leite
materno. O seu médico irá discutir consigo sobre se deve parar o tratamento com Benlysta enquanto
estiver a amamentar ou se deve parar de amamentar.
Condução de veículos e utilização de máquinas
Benlysta pode ter efeitos secundários que podem diminuir a sua capacidade de conduzir ou operar
máquinas.
Informação importante sobre os componentes de Benlysta
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose, é praticamente “isento de
sódio”.
3. Como utilizar Benlysta
Um enfermeiro ou um médico irão administrar-lhe Benlysta sob a forma de um líquido gota a gota
numa veia (perfusão intravenosa) ao longo de uma hora.
O seu médico irá decidir quanto à dose correta dependendo do seu peso corporal. A dose
recomendada é 10 mg por cada quilograma (kg) do seu peso corporal.
Normalmente, é-lhe administrado Benlysta no primeiro dia de tratamento e depois novamente 14 e 28
dias mais tarde. Após isso, Benlysta é normalmente administrado uma vez a cada 4 semanas.
Medicamentos administrados antes de uma perfusão
O seu médico poderá decidir dar-lhe medicamentos que podem ajudar a diminuir qualquer reação à
perfusão (administração gota a gota) antes de lhe ser administrado Benlysta. Estes medicamentos
podem incluir um tipo de medicamentos chamados anti-histamínicos e um medicamento para evitar a
subida da sua temperatura. Irá ser acompanhado atentamente e se, de facto, tiver quaisquer reações
estas serão tratadas.
Se parar o tratamento com Benlysta
O seu médico irá decidir se necessita que lhe seja interrompido o tratamento com Benlysta.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não
se manifestem em todas as pessoas.
Reações alérgicas – procure ajuda médica imediatamente
Benlysta pode causar uma reação à perfusão ou uma reação alérgica (hipersensibilidade).
Estas são efeitos secundários frequentes (podem afetar até 1 em 10 pessoas). Podem ocasionalmente
ser graves (pouco frequente, afetando até 1 em 100 pessoas) e podem colocar a vida em risco. Estas
reações graves são mais prováveis de ocorrer no dia do primeiro ou do segundo tratamento com
Benlysta, mas podem ser do tipo retardado e podem ocorrer vários dias depois.
Se manifestar algum dos seguintes sintomas de uma reação alérgica ou de perfusão, informe o
seu médico ou enfermeiro imediatamente ou dirija-se à urgência médica do hospital mais
próximo:
• inchaço da face, lábios, boca ou língua
• pieira, dificuldades em respirar ou falta de ar
• erupção na pele

95
• altos com comichão ou urticária
Raramente, as reações do tipo retardado menos graves a Benlysta podem também ocorrer, geralmente
5 a10 dias após uma perfusão. Estas incluem sintomas como erupção na pele, sensação de mal-estar,
cansaço, dores musculares, dores de cabeça ou inchaço da face.
Se manifestar estes sintomas, especialmente se manifestar uma associação de dois ou mais destes
sintomas:
Informe o seu médico ou enfermeiro.
Infeções
Benlysta pode levar a que seja mais provável que tenha infeções, incluindo infeção do trato urinário e
das vias respiratórias. Estas são muito frequentes e podem afetar mais de 1 em 10 pessoas. Algumas
infeções podem ser graves e podem, pouco frequentemente, causar a morte.
Se manifestar alguns destes sintomas de uma infeção:
• febre
• tosse, problemas de respiração
• diarreia, vómitos
• sensação de ardor ao urinar
Informe o seu médico ou enfermeiro imediatamente.
Risco aumentado de infeção do cérebro
Os medicamentos que enfraquecem o sistema imunitário, tais como Benlysta, podem colocá-lo em
maior risco de ter uma infeção do cérebro rara, mas grave e que podem colocar a vida em perigo,
chamada de leucoencefalopatia multifocal progressiva (LMP).
Os sintomas de LMP incluem:
• perda de memória
• problemas em pensar
• dificuldade em falar ou em andar
• perda de visão.
Informe o seu médico imediatamente se tiver algum destes sintomas ou problemas semelhantes
que se tenham prolongado por vários dias.
Se já tinha estes sintomas antes de ter iniciado o tratamento com Benlysta:
Informe o seu médico imediatamente se notar qualquer alteração nestes sintomas.
Outros efeitos secundários possíveis:
Efeitos secundários muito frequentes
Estes podem afetar mais de 1 em 10 pessoas:
• infeções bacterianas (ver “Infeções” em cima)
• sentir-se maldisposto, diarreia.
Efeitos secundários frequentes
Estes podem afetar até 1 em 10 pessoas:
• temperatura alta ou febre
• contagem baixa de células sanguíneas brancas (pode ser observada nas análises sanguíneas)
• infeção no nariz, garganta ou estômago
• dor nas mãos ou pés
• enxaqueca
• depressão.

96
Efeitos secundários pouco frequentes
Estes podem afetar até 1 em 100 pessoas:
• erupção na pele irregular e com prurido (urticária), erupção na pele.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste
folheto, fale com o seu médico ou enfermeiro. Também poderá comunicar efeitos secundários
diretamente através do sistema nacional de notificação mencionado no Apêndice V. Ao comunicar
efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste
medicamento.
5. Como conservar Benlysta
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no rótulo e na embalagem exterior,
após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conservar no frigorífico (2ºC a 8ºC).
Não congelar.
Conservar na embalagem original para proteger da luz.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências
locais.
6. Conteúdo da embalagem e outras informações
Qual a composição de Benlysta
• A substância ativa é belimumab.
Cada frasco para injetáveis de 5 ml contém 120 mg de belimumab.
Cada frasco para injetáveis de 20 ml contém 400 mg de belimumab.
Após reconstituição, a solução contém 80 mg de belimumab por ml.
• Os outros componentes são ácido cítrico mono-hidratado (E330), citrato de sódio (E331),
sacarose e polissorbato 80. Ver “Informação importante sobre os componentes de Benlysta” na
secção 2 para mais informação.
Qual o aspeto de Benlysta e conteúdo da embalagem
Benlysta é fornecido como um pó branco a esbranquiçado para solução para perfusão, num frasco
para injetáveis de vidro com uma rolha de borracha siliconizada e um selo de alumínio de abertura
fácil.
Cada embalagem contém 1 frasco para injetáveis.
Titular da Autorização de Introdução no Mercado
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex TW8 9GS
Reino Unido
Fabricante

97
GlaxoSmithKline Manufacturing S.P.A.
Strada Provinciale Asolana No. 90
I-43056 San Polo di Torrile
Parma
Itália
Para quaisquer informações sobre este medicamento, queira contactar o representante local do Titular
da Autorização de Introdução no Mercado:
België/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Tél/Tel: + 32 (0) 10 85 52 00
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
България
ГлаксоСмитКлайн ЕООД
Teл.: + 359 2 953 10 34
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111
Malta
GlaxoSmithKline (Malta) Limited
Tel: + 356 21 238131
Danmark
GlaxoSmithKline Pharma A/S
Tlf: + 45 36 35 91 00
Nederland
GlaxoSmithKline BV
Tel: + 31 (0)30 6938100
Deutschland
GlaxoSmithKline GmbH & Co. KG
Tel.: + 49 (0)89 36044 8701
Norge
GlaxoSmithKline AS
Tlf: + 47 22 70 20 00
Eesti
GlaxoSmithKline Eesti OÜ
Tel: + 372 6676 900
Österreich
GlaxoSmithKline Pharma GmbH
Tel: + 43 (0)1 97075 0
Ελλάδα
GlaxoSmithKline A.E.B.E.
Τηλ: + 30 210 68 82 100
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
España
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700
Portugal
GlaxoSmithKline – Produtos Farmacêuticos, Lda.
Tel: + 351 21 412 95 00
France
Laboratoire GlaxoSmithKline
Tél.: + 33 (0)1 39 17 84 44
România
GlaxoSmithKline (GSK) S.R.L.
Tel: + 4021 3028 208
Hrvatska
GlaxoSmithKline d.o.o.
Tel:+ 385 1 6051 999
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00

98
Ireland
GlaxoSmithKline (Ireland) Limited
Tel: + 353 (0)1 4955000
Slovenská republika
GlaxoSmithKline Slovakia s. r. o.
Tel: + 421 (0)2 48 26 11 11
Ísland
Vistor hf.
Sími: +354 535 7000
Suomi/Finland
GlaxoSmithKline Oy
Puh/Tel: + 358 (0)10 30 30 30
Italia
GlaxoSmithKline S.p.A.
Tel: + 39 (0)45 9218 111
Sverige
GlaxoSmithKline AB
Tel: + 46 (0)8 638 93 00
Κύπρος
GlaxoSmithKline (Cyprus) Ltd
Τηλ: + 357 22 39 70 00
United Kingdom
GlaxoSmithKline UK Ltd
Tel: + 44 (0)800 221441
Latvija
GlaxoSmithKline Latvia SIA
Tel: + 371 67312687
Lietuva
GlaxoSmithKline Lietuva UAB
Tel: + 370 5 264 90 00
Este folheto foi revisto pela última vez em
Está disponível informação pormenorizada sobre este medicamento no sítio da internet da Agência
Europeia de Medicamentos: http://www.ema.europa.eu.
<-----------------------------------------------------------------------------------------------------------------------------

99
A informação que se segue destina-se apenas a profissionais de saúde:
Instruções para utilização e manipulação - reconstituição, diluição e administração
1) Como reconstituir Benlysta
A reconstituição e diluição necessitam de ser executadas sob condições assépticas.
Aguardar 10 a15 minutos para que o frasco para injetáveis aqueça até à temperatura ambiente (15ºC a
25ºC).
Recomenda-se a utilização de uma agulha de 21-25 gauge para furar a rolha do frasco para injetáveis
na reconstituição e diluição.
AVISO: Os frascos para injetáveis de 5 ml e 20 ml são reconstituídos com volumes diferentes de
solvente, ver abaixo:
Frasco para injetáveis de 120 mg
O frasco para injetáveis de uso único de 120 mg de Benlysta é reconstituído com 1,5 ml de água para
preparações injetáveis para obter uma concentração final de 80 mg/ml de belimumab.
Frasco para injetáveis de 400 mg
O frasco para injetáveis de uso único de 400 mg de Benlysta é reconstituído com 4,8 ml de água para
preparações injetáveis para obter uma concentração final de 80 mg/ml de belimumab.
Quantidade de Benlysta
Tamanho do frasco para injetáveis
Volume de solvente Concentração final
120 mg 5 ml 1,5 ml 80 mg/ml
400 mg 20 ml 4,8 ml 80 mg/ml
O fluxo de água para preparações injetáveis deve ser dirigido para a face lateral do frasco para
injetáveis de forma a minimizar a formação de espuma. Rodar suavemente o frasco para injetáveis
durante 60 segundos. Deixar o frasco para injetáveis em repouso à temperatura ambiente (15ºC a
25ºC) durante a reconstituição, rodar suavemente o frasco para injetáveis durante 60 segundos a cada
5 minutos até que o pó esteja dissolvido. Não agitar. A reconstituição normalmente está completa em
10 a 15 minutos após a adição da água, mas pode demorar até 30 minutos. Proteja a solução
reconstituída da luz solar.
Se for utilizado um aparelho de reconstituição mecânico para reconstituir Benlysta, não se deve
exceder as 500 rpm e o frasco para injetáveis não deve ser agitado durante mais do que 30 minutos.
2) Antes de diluir Benlysta
Quando a reconstituição estiver completa, a solução deve apresentar-se opalescente e incolor a
amarelo pálido e sem partículas. Contudo, são expectáveis e aceitáveis pequenas bolhas de ar.
Frasco para injetáveis de 120 mg
Após a reconstituição, um volume de 1,5 ml (correspondente a 120 mg de belimumab) pode ser
retirado de cada frasco para injetáveis de 5 ml.

100
Frasco para injetáveis de 400 mg
Após a reconstituição, um volume de 5 ml (correspondente a 400 mg de belimumab) pode ser retirado
de cada frasco para injetáveis de 20 ml.
3) Como diluir a solução para perfusão
O medicamento reconstituído é diluído para 250 ml com uma solução para injetáveis de cloreto de
sódio a 9 mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer.
Não devem ser utilizadas soluções intravenosas de glucose a 5% porque são incompatíveis com
Benlysta.
A partir de um saco ou frasco de perfusão de 250 ml de solução para injetáveis de cloreto de sódio 9
mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer retirar e eliminar um volume
igual ao volume de solução de Benlysta reconstituída necessário para a dose do doente.
Seguidamente, adicionar o volume necessário da solução reconstituída de Benlysta ao saco ou frasco
de perfusão. Inverter suavemente o saco ou frasco para homogeneizar a solução. Toda a solução não
utilizada nos frascos deve ser eliminada.
Inspecionar visualmente a solução de Benlysta para a presença de partículas e descoloração antes da
administração. Eliminar a solução se forem observadas partículas ou descoloração.
Se não for utilizada imediatamente, a solução reconstituída deve ser protegida da luz solar direta e
armazenada de forma refrigerada a 2ºC-8ºC. As soluções diluídas em solução para injetáveis de
cloreto de sódio 9 mg/ml (0,9%), cloreto de sódio 4,5 mg/ml (0,45%) ou Lactato de Ringer podem ser
armazenadas a 2ºC-8ºC ou a temperatura ambiente (15ºC a 25ºC).
O tempo total a partir da reconstituição de Benlysta até ao final da perfusão não deve exceder 8 horas.
4) Como administrar a solução diluída
Benlysta é perfundido durante um período de 1 hora.
Benlysta não deve ser perfundido concomitantemente na mesma linha intravenosa com outros
fármacos.
Não foram observadas incompatibilidades entre Benlysta e sacos de cloreto de polivinilo (PVC) ou
poliolefina.
----------------------------------------------------------------------------------------------------------------------------------





























