Embed Size (px)
DESCRIPTION
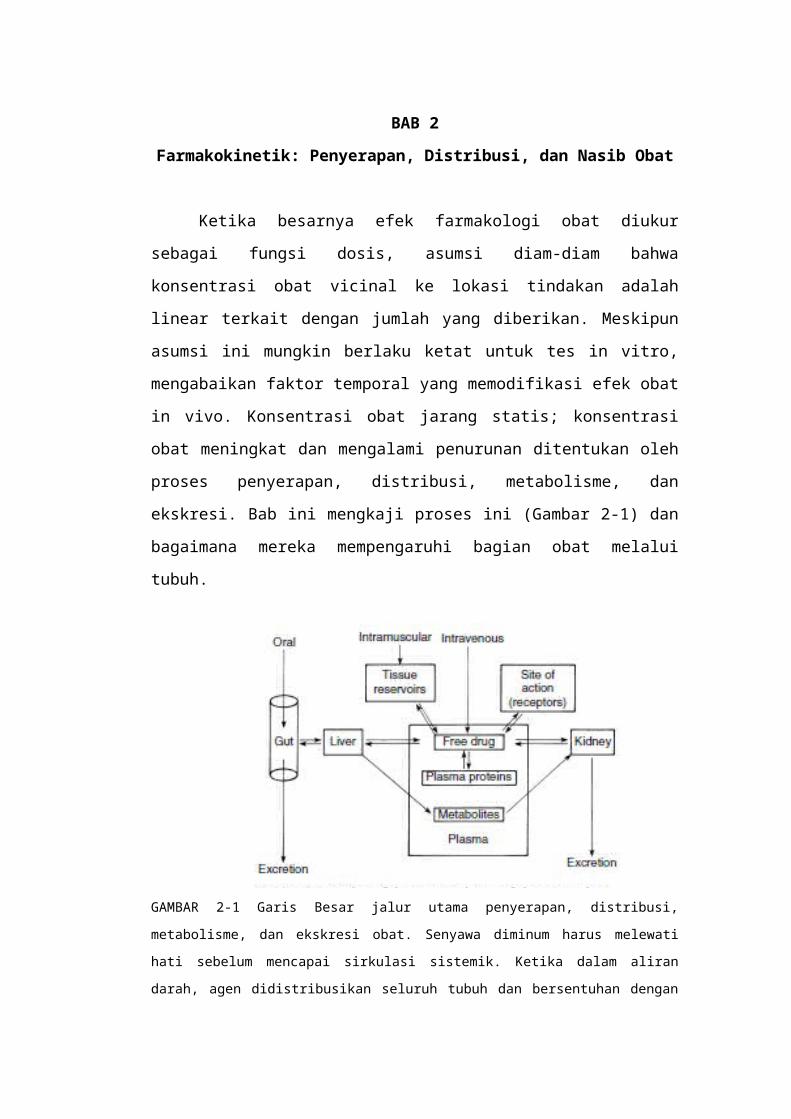
Ketika besarnya efek farmakologi obat diukur sebagai fungsi dosis, asumsi diam-diam bahwa konsentrasi obat vicinal ke lokasi tindakan adalah linear terkait dengan jumlah yang diberikan. Meskipun asumsi ini mungkin berlaku ketat untuk tes in vitro, mengabaikan faktor temporal yang memodifikasi efek obat in vivo. Konsentrasi obat jarang statis; konsentrasi obat meningkat dan mengalami penurunan ditentukan oleh proses penyerapan, distribusi, metabolisme, dan ekskresi. Bab ini mengkaji proses ini (Gambar 2-1) dan bagaimana mereka mempengaruhi bagian obat melalui tubuh.
Citation preview
BAB 2
Farmakokinetik: Penyerapan, Distribusi, dan Nasib Obat
Ketika besarnya efek farmakologi obat diukur sebagai fungsi dosis, asumsi
diam-diam bahwa konsentrasi obat vicinal ke lokasi tindakan adalah linear terkait
dengan jumlah yang diberikan. Meskipun asumsi ini mungkin berlaku ketat untuk
tes in vitro, mengabaikan faktor temporal yang memodifikasi efek obat in vivo.
Konsentrasi obat jarang statis; konsentrasi obat meningkat dan mengalami
penurunan ditentukan oleh proses penyerapan, distribusi, metabolisme, dan
ekskresi. Bab ini mengkaji proses ini (Gambar 2-1) dan bagaimana mereka
mempengaruhi bagian obat melalui tubuh.
GAMBAR 2-1 Garis Besar jalur utama penyerapan, distribusi, metabolisme, dan ekskresi obat.
Senyawa diminum harus melewati hati sebelum mencapai sirkulasi sistemik. Ketika dalam aliran
darah, agen didistribusikan seluruh tubuh dan bersentuhan dengan situs masing-masing tindakan.
Obat yang disaring oleh ginjal, hanya diserap kembali jika larut dalam lemak. Metabolisme banyak
obat terjadi terutama pada hati, setelah itu metabolit diekskresikan dalam empedu atau urin.
Beberapa agen dihilangkan dalam empedu tunduk pada reabsorpsi dan dapat berpartisipasi dalam
siklus enterohepatik. Bretscher MS: Molekul sel
2.1 Lintas Obat Melintasi Membran
Untuk obat yang akan diserap, mencapai tempat aksi dan akhirnya
dihilangkan, harus menyeberangi satu atau lebih hambatan membran biologis. Ini
dapat terdiri dari membran plasma tunggal atau merupakan lapisan sel yang

dikemas erat. Karena hambatan seperti obat-obatan berperilaku sama, membran
sel dapat berfungsi sebagai prototipe untuk semua. Membran plasma terdiri dari
lembaran Bimolekular lipid (terutama fosfolipid dan kolesterol) dengan protein
diselingi seluruh dan memperluas melampaui fase lipid dari membran (Gambar 2-
2). Kehadiran molekul protein yang mencakup seluruh ketebalan membran
menyediakan link yang diperlukan antara lingkungan ekstraseluler dan intraselular
sel, yang konsisten dengan konsep bahwa aktivasi obat reseptor membran-terikat
pada permukaan luar sel yang langsung dapat diterjemahkan ke dalam respon
intraseluler. Protein transmembran tertentu juga menyediakan jalan penting untuk
penyerapan dan ekstrusi obat.
GAMBAR 2-2 Membran plasma menggambarkan lapisan ganda lipid, terdiri dari
fosfolipid dan kolesterol, dan protein globular dan linier, yang berlabuh di dalam membran dengan
α-heliks-segmen KASIH dan melampaui bilayer tebal 40 Å pada permukaan ekstraseluler dan
sitoplasma. Untuk kejelasan, rasio lipid terhadap protein jauh lebih besar daripada yang ada di
membran alami. Komponen glikolipid membran dan sakarida polimer melekat pada protein juga
ditampilkan.
Difusi pasif
Bagian dari obat melintasi membran biologis dapat melibatkan beberapa
mekanisme yang berbeda. Dari jumlah tersebut, difusi pasif yang paling sering
ditemui. Karakteristik mendefinisikan difusi pasif adalah bahwa obat bergerak ke
bawah elektro nya gradien kimia ketika melintasi membran. sederhana difusi
Studi yang dilakukan oleh Overton dan Meyer lebih dari satu abad yang lalu
menunjukkan bahwa membran sel bertindak untuk sebagian besar sebagai sebuah

lipoid penghalang. Seperti yang ditunjukkan oleh Colander (Gambar 2-3), laju
perpindahan dari Nonelektrolit melintasi membran berbanding lurus dengan
koefisien partisi lipid / air. (Koefisien partisi adalah ukuran kelarutan relatif agen
di lemak pelarut, seperti minyak zaitun atau oktanol, dibandingkan kelarutannya
dalam air.) Sebuah obat dengan koefisien partisi yang tinggi (yaitu, lipofilik obat)
siap memasuki fase lipid membran dan melewati bawah gradien konsentrasi ke
fasa air pada sisi lain. Molekul yang lebih kemudian bebas untuk masuk membran
dan melanjutkan proses transfer. Dengan senyawa larut lipid buruk, namun,
beberapa molekul memasuki membran per unit waktu, dan tingkat bagian
tertekan.
GAMBAR 2-3 Hubungan antara permeabilitas membran dan lipid (minyak zaitun) /
koefisien partisi air di Chara ceratophylla. Setiap lingkaran merupakan nonelektrolit tunggal
dengan molekul radius seperti yang ditunjukkan dalam kunci. Senyawa kecil menyerap lebih
banyak mudah daripada koefisien partisi mereka akan menunjukkan; sebaliknya berlaku untuk
molekul besar.
Tidak adanya biaya ionik merupakan salah satu faktor utama mendukung
kelarutan lipid. Obat dengan biaya tetap, seperti obat yang mengandung atom
nitrogen kuaterner, menembus membran lambat jika sama sekali. Alasan untuk
kelarutan relatif molekul non terionisasi dalam lipid berhubungan dengan
pengecualian mereka dari kutub Media. Ion sederhana dan molekul bermuatan
yang stabil dalam air dengan kerang hidrasi yang mengelilingi mereka, akibat dari
kecenderungan spesies dibebankan pada molekul polar orientasi. Proses ini tidak
termasuk zat nonpolar, dan menghasilkan segregasi menyebabkan mereka

menyatu dengan cara yang analog dengan pembentukan tetesan minyak di
permukaan air. Ikatan hidrofobik istilah, diperkenalkan pada Bab 1, mengacu pada
kecenderungan untuk molekul air-larut menjadi ditarik bersama-sama; perilaku ini
bertanggung jawab untuk kecenderungan preferensial obat larut lemak untuk
menembus membran sel dengan cara komponen lipid. Senyawa terionisasi begitu
distabilkan oleh interaksi mereka dengan air yang bergerak ke fase lipid nyata
dibatasi. Banyak agen terapeutik weaelectrolytes; tergantung pada pH lingkungan
berair, mereka bisa eksis dalam terionisasi dan bentuk netral. Karena molekul
bermuatan menembus membran dengan kesulitan yang cukup besar, laju
pergerakan obat ini diatur oleh koefisien partisi spesies netral dan tingkat ionisasi.
Seperti diilustrasikan dalam Gambar 2-4, kondisi asam mendukung pengangkutan
asam lemah, dan sebaliknya berlaku untuk senyawa basa.
Konsep yang sama interaksi air yang digunakan untuk menjelaskan
kelarutan berair ion juga berlaku untuk banyak molekul nonionik. Meskipun
tersubstitusi hidrokarbon alifatik dan aromatik memiliki sedikit atau tidak ada
kecenderungan untuk bereaksi dengan air, afinitas untuk molekul air tidak terbatas
pada struktur dengan muatan formal. Residu organik yang memiliki atom
elektronegatif seperti oksigen, nitrogen, dan belerang dapat berinteraksi dengan
air melalui pembentukan ikatan hidrogen untuk memberikan beberapa derajat
kelarutan air.
Gambar 2-3 menunjukkan bahwa kelarutan lipid bukan satu-satunya faktor
mempengaruhi difusi sederhana obat bermuatan di sel membran; ukuran molekul
juga penting. Air, gliserol, dan beberapa molekul kecil lainnya menyerap jauh
lebih mudah daripada yang diprediksi dari koefisien partisi masing-masing.
Gambar 2-3 juga menunjukkan bahwa beberapa molekul organik yang besar
menyebar lebih lambat dari yang diharapkan. Nonelektrolit mengandung banyak
kelompok hidrofobik sering begitu larut dalam air yang transit mereka di seluruh
lipid antarmuka / air mungkin terbelakang meskipun koefisien partisi yang
menguntungkan. Temuan ini menunjukkan bahwa beberapa tingkat kelarutan air
diperlukan untuk difusi pasif obat melintasi membran.

GAMBAR 2-4 Membran penetrasi dengan elektrolit lemah. itu spesies ion non obat (HA,
B) menyerap membran banyak lebih efisien daripada bentuk-bentuk yang dibebankan (A- Kondisi
menggeser kurva disosiasi ke kiri, menguntungkan difusi asam lemah. Peningkatan pH nikmat
hilangnya hidrogen (H +) dan difusi basa lemah (H +).
Tidak peduli seberapa larut lemak agen, tidak akan pernah menyeberangi
membran jika tidak dapat larut dalam cairan ekstraseluler dan dibawa ke struktur.
Benzocaine, aktif lokal anestesi bila diterapkan secara langsung ke saraf, tidak
efektif setelah injeksi karena kelarutan air yang menghalangi signifikan difusi
jauh dari situs administrasi dan menuju nya lokus aksi dalam membran saraf.
ketika di dalam membran, obat dengan koefisien partisi yang sangat tinggi
mungkin begitu larut dalam fase lipid yang memiliki sedikit kecenderungan,
meskipun kelarutan moderat dalam air, untuk berdifusi keluar dari membran
bawah konsentrasi gradient. Sebuah tinjauan data klinis manusia yang melibatkan
lebih dari 2.400 senyawa menunjukkan bahwa difusi sederhana akan menjadi
miskin jika obat memiliki dua atau lebih dari karakteristik berikut: (1) lebih dari
lima Kelompok donor H-ikatan, (2) lebih dari lima H-ikatan kelompok akseptor,
(3) lebih dari 10 N dan O atom, (4) berat molekul lebih besar dari 500 Da, dan (5)
partisi Koefisien lebih besar dari 10.000: 1.28
Difusi sederhana di dinding kapiler menjamin khusus komentar. Selain
jalur transelular obat difusi hanya dijelaskan untuk agen larut-lipid, sebuah berair
jalur paracellular dibentuk oleh 10-nm ke celah 15-nm antara sel-sel endotel yang
paling kapiler memungkinkan difusi air obat larut dalam air antara plasma dan
ruang ekstraselular. Molekul hidrofilik hingga kecil protein dalam ukuran dapat
menggunakan rute ini; biaya tetap negatif bersama jalur difusi cenderung untuk

mempromosikan pergerakan bermuatan positif makromolekul sementara
membatasi move- ment dari mereka dengan muatan negatif bersih.
Menambah gerakan paracellular obat di seluruh kapiler adalah aliran
sebagian besar air yang bergerak dalam kaitannya dengan net keseimbangan
kekuatan hidrostatik dan osmotik antara vaskular dan kompartemen interstitial. Ini
transfer bersih cairan, konveksi diistilahkan, disertai dengan dibubarkan obat-
obatan dan lainnya zat terlarut. Gerakan konvektif dari kebanyakan obat secara
kuantitatif ngawur; Namun, hal itu mungkin memainkan peran utama dalam
pergerakan protein dan makromolekul lain yang menghindari filtrasi oleh
endothelium, terutama di jaringan yang meradang. Jumlah kecil albumin dan
protein plasma lain yang mencapai ruang ekstraselular (4% per jam untuk
albumin) yang sebagian besar kembali ke sirkulasi oleh konveksi limfatik.
Facilitated Difution, elektrolit kecil, dan hidrofilik molekul biologis
penting umumnya bergerak melintasi membran plasma jauh lebih mudah daripada
yang diperkirakan oleh difusi sederhana. Dalam hal ini, protein transmembran
yang menghindari lapisan ganda lipid memfasilitasi difusi. yang paling sederhana
Mekanisme melibatkan por transmembran, seperti aquaporin 1. Ditemukan pada
tahun 1991, aquaporin 1 adalah polipeptida 28-kDa yang membentuk saluran 3-Å
di mana air bisa masuk atau meninggalkan sel. Lebih dari 10 aquaporins telah
ditemukan di jaringan mamalia dan sangat menonjol dalam sel dan organ yang
terlibat dengan gerakan transelular air: ginjal, pembuluh kapiler, kelenjar sekresi,
sel-sel darah merah, koroid pleksus, glia otak, mata, dan lungs.1,24 Beberapa
aquaporins adalah selektif untuk air saja, meningkatkan permeabilitas membran
dengan faktor 10 sampai 100; lain memungkinkan perjalanan gliserol dan
beberapa molekul lain selain air.
Pergerakan ion tertentu (misalnya, Na +, Ca melintasi membran sel
difasilitasi oleh kehadiran saluran transmembran, seperti reseptor nicotinic
dijelaskan pada Gambar 1-2 dan Na + Gambar 16-4. Pembukaan ini gated
channels (kontras untuk porins, yang selalu terbuka) diatur oleh listrik potensial di
membran atau dengan kehadiran khusus ligan, seperti neurotransmitter. Ketika
saluran terbuka, difusi pasif dari ion yang mampu melintasi itu tergantung pada ,
K + dan Ca ++) saluran digambarkan dalam potensial listrik melintasi membran

dan singa sendiri gradien kimia. Meningkatkan gradien elektrokimia oleh
memanipulasi tegangan melintasi membran sel merupakan metode yang efektif
untuk meningkatkan aliran ion. Bahkan tanpa adanya saluran ion tertentu,
pengangkutan ion tetap dan lemah elektrolit seluruh hambatan jaringan dapat
difasilitasi oleh penggunaan yang tepat arus listrik (seperti dalam iontophoresis,
dibahas kemudian).
Banyak zat yang tidak larut-lipid yang shuttled di membran plasma dengan
membentuk kompleks dengan konstituen membran khusus yang disebut operator
atau transporter. Operator mirip dengan reseptor dalam banyak hal; mereka adalah
protein, seringkali cukup selektif dalam agen yang mereka menggabungkan, dan
tunduk pada penghambatan kompetitif. Karena jumlah molekul transporter
terbatas, pembawa dimediasi difusi dapat jenuh pada konsentrasi obat yang tinggi.
Keluarga GLUT transporter glukosa merupakan perwakilan dari protein pembawa
yang memfasilitasi pergerakan zat terlarut hidrofilik melintasi membran sel.
Langkah awal dalam difusi difasilitasi glukosa adalah mengikat ke situs aktif
terbuka dari protein transporter. Berurutan mengikat ini menyebabkan hambatan
eksternal atau gerbang untuk menutup dan pintu interior untuk membuka, setelah
glukosa dilepaskan ke dalam sel. Hilangnya glukosa menyebabkan gerbang
internal untuk menutup dan gerbang eksternal untuk membuka, memperlihatkan
sisi aktif dan menyelesaikan siklus.
Transportasi aktif Transpor aktif adalah istilah yang diberikan kepada
operator-dimediasi transfer obat terhadap gradien elektrokimia. dalam addi- tion
untuk menunjukkan selektivitas dan saturability, transpor aktif membutuhkan
pengeluaran energi dan dapat diblokir oleh inhibitor metabolisme sel. Izin
transportasi aktif penyerapan yang efisien zat penting untuk fungsi selular (dan
obat-obatan tertentu yang menyerupai mereka secara struktural) dan eliminasi
selektif produk limbah dan bahan kimia asing, termasuk banyak obat. Sekitar
2.000 gen-7% dari Total manusia genom-kode untuk transporter dan terkait
protein. Dua superfamili transporter yang penting khusus untuk farmakokinetik:
ATP-binding kaset (ABC) transporter, dan pembawa zat terlarut (SLC)
transporter.

Sekitar 49 ABC transporter menghidrolisis adenosin trifosfat (ATP) untuk
memberikan energi langsung dibutuhkan untuk transportasi molekul dan disebut
sebagai primary aktif transporter. P-glikoprotein (P untuk permeabilitas berubah),
juga dikenal sebagai resistensi multidrug protein-1 (MDR-1) dan diberi
penunjukan ABCB1 oleh Nomenklatur Human Gene Komite, adalah wakil yang
paling ekstensif diteliti. Awalnya diidentifikasi pada tahun 1976 karena
kemampuannya untuk mengusir berbagai obat antineoplastik dari sel-sel
bermutasi yang overexpress itu, P-glikoprotein adalah glikoprotein 170-kDa
terdiri dari dua subunit dalam susunan head-to-tail (Gambar 2-5).
Setiap subunit mengandung domain transmembran enam α-heliks yang
menjangkau membran plasma dan membantu membentuk pompa sendiri, dan
nukleotida-binding domain (juga dikenal sebagai ABC kaset) yang menghidrolisis
ATP untuk daya transportasi. Banyak transporter ABC disebut sebagai setengah
transporter karena mereka hanya terdiri dari subunit tunggal dan harus dimer- ize
untuk membuat pompa aktif. P-glikoprotein istimewa mempromosikan ekstrusi
seluler besar (300 Da sampai 2000 Da) zat hidrofobik dan netral atau bermuatan
positif molekul amphiphilic. Obat Diangkut meliputi banyak agen antikanker
(misalnya, doxorubicin, vinblastin, dan pacli- taxel), senyawa antiviral (misalnya,
ritonavir), Ca ++ - channel blocker (misalnya, diltiazem), digoxin, antibiotik dan
antijamur obat-obatan (misalnya, eritromisin dan ketoconazole), hormon
(misalnya, testosteron), dan imunosupresan (misalnya, siklosporin).
Obat mengikat terjadi dalam membran plasma dekat permukaan
sitoplasma, membatasi transportasi ke obat dengan baik kelarutan lipid atau cukup
panjang untuk mencapai lokasi aktif. P-glikoprotein dinyatakan dalam berbagai
sel, tetapi tertinggi Konsentrasi yang terletak di sel epitel usus; ginjal Sel-sel
tubular proksimal; membran canalicular hepatosit; endotelium kapiler otak,
pleksus koroid, testis, dan plasenta; trofoblas plasenta; Sel adrenocortical; dan
batang cells.30 transporter ABC lain yang penting dalam farmakokinetik meliputi
perlawanan terkait multidrug protein (MRP) keluarga. Secara kolektif, transporter
MRP adalah juga luas dan terlibat dalam vectorial (satu arah) pergerakan obat dan
xenobiotik lainnya. Berlawanan dengan P-glikoprotein, transporter MRP pompa
molekul amphiphilic dengan setidaknya satu muatan negatif. substrat termasuk

garam empedu, analog nukleotida, dan konjugat dari glutathione, asam
glukuronat, dan sulfat.
Transporter SLC yang dikenal mencakup 48 keluarga dikodekan dalam
gen 400. Karena transporter SLC tidak langsung menggunakan ATP sebagai
sumber energi untuk transportasi, mereka lebih tepat disebut transporter aktif
sekunder. pompa (atau Na + Na +keempat produksi ATP tubuh, adalah
mengemudi utama berlaku untuk transport aktif sekunder. Dengan
mempertahankan gradien elektrokimia besar untuk Na + melintasi membran
plasma, gerakan molekul yang penuh semangat digabungkan (atau ion lain dengan
potensi elektrokimia kuat untuk Na + perbedaan melintasi membran) dapat terjadi
terhadap gradien konsentrasi mereka sendiri. Transporter aktif sekunder yang
bergerak zat yang digabungkan dalam arah yang sama dengan ion terkait
diistilahkan cotransporters dan symporters. Sebaliknya, antiporters atau penukar
memindahkan zat digabungkan dalam arah yang berlawanan. Banyak transporter
SLC (termasuk keluarga GLUT dijelaskan sebelumnya) memungkinkan gerakan
transmembran bahan kimia tertentu ke gradien elektrokimia mereka sendiri dan
dukungan yang difasilitasi difusi. Berlawanan dengan transporter ABC,
transporter SLC dapat memfasilitasi pergerakan dua arah substrat berdasarkan
konsentrasi yang ada di membran sel.
Transporter anion organik (gandum) dan anion mengangkut polipeptida
organik (OATPs) adalah keluarga-keluarga penting transporter SLC terlibat dalam
pharmacokinetics.35 Sebagai sebuah kelompok, mereka mempromosikan ambilan
obat asam ke dalam hati, ginjal, usus, paru-paru, dan otak, dan ekskresi mereka
melalui empedu dan urin. Keluarga analog transporter kation organik (ACT)
memberikan penanganan yang sama bermuatan positif obat obatan.
Endositosis dan Eksositosis
Proses endositosis dan eksositosis bersama-sama dengan sebagian besar
metode yang kompleks transfer obat di seluruh biologis yang membran. The
endositosis merujuk pada serangkaian acara di substansi yang ditelan dan
diinternalisasi oleh sel. (Fagositosis, atau "makan sel," adalah varian dari

endositosis terkait lebih dengan penghapusan partikel oleh makrofag
dibandingkan dengan transportasi obat.)
Endositosis biasanya dimulai dengan pengikatan senyawa, biasanya
makromolekul, diserap oleh reseptor pada permukaan membran. Dua contoh yang
baik adalah lampiran low-density lipoprotein (LDL) dan insulin pada reseptor
masing-masing. Dengan waktu, kompleks reseptor agen terikat terkonsentrasi di
lekukan membran yang disebut lubang dilapisi. (Migrasi ini juga terjadi secara
spontan dengan reseptor LDL.) Clathrin, protein sitoplasma yang melekat pada
permukaan internal dari membran plasma, berfungsi untuk menangkap reseptor
dalam lubang sementara tidak termasuk permukaan proteins.49 penataan internal
lainnya strukturnya memperdalam pit, membentuk tunas dilapisi. Sebuah protein
kedua, disebut Dynamin, diyakini berkumpul di sekitar kerah tunas invaginated
dan memulai pemisahan dari membran. Ketika dirilis, vesikel kehilangan mantel
clathrin dan sekering dengan organel yang disebut endosome tersebut. Beberapa
isi ditangkap, seperti reseptor LDL, yang didaur ulang kembali ke membran
plasma oleh vesikel transportasi; sisanya menjalani proses lisosom dan
melepaskan ke dalam sitoplasma.
Sebuah metode alternatif endositosis melibatkan lekukan pada membran
plasma disebut caveolae. Caveolae mengandung banyak kolesterol kovalen terkait
dengan -caveolin 1, protein struktural utama dari struktur ini. Proses internalisasi
juga melibatkan pembentukan vesikel, tapi clathrin dan endosomes tidak
berpartisipasi dalam proses internalisasi.
Proses melengkapi eksositosis terjadi ketika vesikel, seperti yang
dihasilkan oleh aparat Golgi, sekering dengan membran plasma dan debit isinya di
luar sel. Eksositosis adalah metode utama yang digunakan produk seluler seperti
hormon peraturan yang dikeluarkan oleh sel. The transcytosis istilah deskriptif
bentuk digabungkan endositosis dan eksositosis yang mengarah ke transfer obat
capture, diangkut melintasi sel, dan sekering dengan membran plasma untuk
melepaskan isinya ekstrasel.
Sel umumnya mampu endositosis; Namun, eksositosis dan transcytosis
yang paling intensif dalam jaringan disesuaikan untuk penyerapan, distribusi, dan
ekspor bahan makanan penting, hormon regulasi, dan produk sekretorik.

Endositosis / transcytosis mungkin bertanggung jawab untuk penyerapan protein
antigenik dan racun tertentu dari usus kecil dan untuk transfer molekul besar
antara kompartemen jaringan. Hal ini memainkan peran kecil dalam transportasi
kebanyakan obat.
2.2 ABSORBSI
Penyerapan mengacu pada transfer obat dari situs nya administrasi ke
dalam aliran darah. Rute tertentu administrasi yang dipilih sangat mempengaruhi
tingkat dan mungkin tingkat penyerapan obat.
Konsumsi Obat Oral
Konsumsi oral adalah metode yang pertama dan masih yang paling umum
digunakan untuk administrasi agen terapi. Itu Keuntungan utama dari rute
kebohongan oral pada tiga bidang: kenyamanan, ekonomi, dan keamanan.
Penerimaan pasien oral obat yang baik karena teknik itu sendiri tidak
menimbulkan rasa sakit, dan personil terlatih tidak diperlukan untuk
pencapaiannya. Kenyamanan dan biaya rendah terhadap lainnya mode terapi
sangat menonjol untuk obat yang harus diberikan beberapa kali sehari secara
jangka panjang. Oral rute ini relatif aman karena penyerapan obat relative lambat.
Konsentrasi darah tinggi tiba-tiba tidak hamper mungkin untuk dicapai oleh
konsumsi obat-obatan karena mereka dengan injeksi parenteral. Reaksi alergi juga
kurang mungkin terjadi, reaksi sangat serius. Oral rute memang memiliki
beberapa kekurangan, namun. Karena diri administrasi adalah Aturan, kepatuhan
pasien diperlukan untuk terapi yang optimal. Obat penyerapan kemungkinan akan
tertunda (pada rata-rata klinis 30 60 menit) dan mungkin tidak lengkap. inaktivasi
metabolik atau pembentukan kompleks juga dapat terjadi sebelum obat memiliki
kesempatan untuk mencapai sirkulasi sistemik. keterbatasan ini dengan oral
menerjemahkan ke dalam variabilitas meningkat respon pasien. Akhirnya,
spektrum efek samping disebabkan oleh obat oral dapat memperpanjang dari satu
ujung saluran pencernaan yang lain.
Pengaruh PH

Seperti telah dibahas sebelumnya, penyerapan disukai ketika obat tertelan
adalah larut dalam lemak. Untuk elektrolit lemah, pH media sekitarnya
mempengaruhi tingkat ionisasi dan obat penyerapan. Karena konsentrasi H +
lambung dan usus halus berbeda secara luas, dua struktur tampaknya kualitatif
berbeda dalam pola masing-masing obat penyerapan. Gambar 2-6
mengilustrasikan perbedaan ini dan efeknya pada sebelumnya umum digunakan
kombinasi analgesik aspirin ditambah kodein. Aspirin adalah asam organik
dengan pKa (log negatif dari konstanta disosiasi) dari 3,49. dalam lambung jus
(pH 1 sampai 3), aspirin sebagian besar masih terionisasi, dan yang bagian di
mukosa lambung dan ke dalam aliran darah disukai. Plasma memiliki pH 7,4,
namun. memasuki lingkungan ini, aspirin menjadi terionisasi untuk seperti Sejauh
kembalinya obat untuk saluran pencernaan dicegah dengan kelarutan lipid rendah
dari spesies anionik.
Ketika cairan lambung masuk ke dalam usus kecil, cepat dinetralisir oleh
pankreas, empedu, dan sekresi usus. PH proksimal seperempat dari usus
bervariasi 3-6, tetapi mencapai netralitas lebih distal segmen. Dalam kondisi basa,
mengkonversi aspirin ke bentuk anionik, sedangkan fraksi yang signifikan dari
molekul codeine menyerah muatan positif mereka. meskipun obat dasar disukai
untuk penyerapan lebih asam di kecil usus, perangkap ion tidak begitu luas karena
diferensial pH di mukosa usus kecil. perbedaan penyerapan usus berdasarkan pH
lebih peduli dengan tingkat penyerapan dibandingkan dengan luasnya. Seperti
yang diharapkan, netralisasi isi lambung oleh administrasi antasida atau menelan
makanan sementara menghilangkan kualitatif disparitas penyerapan elektrolit
biasanya diamati antara lambung dan usus kecil.
Luas Permukaan Mukosa
Perbedaan utama kedua antara penyerapan di perut dan penyerapan di usus
kecil berhubungan dengan intraluminal yang area permukaan yang terlibat dalam
penyerapan obat. Selain tertentu penyimpangan mukosa (ruge), yang mendekati
lapisan perut bahwa dari kantong halus dengan lapisan lendir tebal. itu mukosa
usus kecil secara unik disesuaikan untuk penyerapan, Namun. Kontribusi oleh
lipatan Kerckring, vili, dan mikrovili bergabung untuk meningkatkan luas

permukaan efektif 600 kali lipat. Dengan asumsi usus kecil 280 cm dan 4 cm,
sekitar 200 m2 yang tersedia untuk obat penyerapan.
Pengosongan lambung
Karena hampir semua zat yang dapat menembus gastrointestinal epitel
yang terbaik diserap dalam usus kecil, tingkat pengosongan lambung dapat secara
signifikan mempengaruhi penyerapan obat, terutama untuk basa organik yang
tidak terserap sama sekali dari perut. Pengosongan lambung dilakukan dengan
kontraksi dari antrum lambung. Pola siklus aktivitas terjadi pada pasien puasa di
mana periode ketenangan (Sekitar 1 jam masing-masing) diikuti oleh kontraksi
yang meningkatkan intensitas selama 40 menit sebelum mengakhiri dalam
ledakan singkat kontraksi intens yang bermigrasi dari perut ke ileum distal.
Menelan tablet atau volume kecil cairan dapat menyebabkan retensi lambung obat
selama 1 jam atau lebih. Setelah makan makan, berkelanjutan antral dan pyloric
kontraksi membantu memecah makanan tertelan dan mengizinkan ekstrusi cairan
ke dalam duodenum sementara tetap mempertahankan partikel lebih dari 1 mm
dalam perut. Sebuah campuran makan padatan dan cairan biasanya mulai
memasuki duodenum dalam waktu sekitar 30 menit dan membutuhkan sekitar 4
jam untuk meninggalkan perut sepenuhnya.
Pengaruh Bentuk Sediaan
Meskipun waktu yang diperlukan untuk pengosongan lambung dan difusi
melintasi penghalang mukosa diragukan lagi berkontribusi untuk onset tertunda
aksi obat diambil secara lisan, situasi eksis di mana peristiwa ini tidak membatasi.
kebanyakan obat ditujukan untuk penggunaan oral yang dipasarkan dalam bentuk
kapsul atau tablet yang solid. Berbeda dengan solusi, persiapan ini harus pertama
larut dalam cairan pencernaan sebelum penyerapan bisa terjadi. Jika pembubaran
sangat lambat, hal itu dapat menjadi pengendali faktor dalam penyerapan obat.
Langkah pertama dalam proses pelarutan adalah disintegrasi tablet (atau
kapsul dan butiran-nya) untuk menghasilkan partikel obat utama. Berbagai
eksipien biasanya disertakan dalam persiapan obat kuat untuk mempromosikan
disintegrasi dan partikel dispersi. Jika disintegrasi terganggu, penyerapan obat

tertekan sesuai. Pembubaran partikel obat terjadi melalui mekanisme difusi
terbatas. Lapisan difusi pelarut sekitarnya setiap partikel menjadi jenuh sangat
cepat dengan molekul obat melarikan diri dari padat. karena kejenuhan lapisan
difusi terjadi jauh lebih cepat daripada melakukan difusi dari itu ke dalam larutan
massal, seluruh proses hasil tidak lebih cepat dari laju difusi obat. beberapa
metode dapat digunakan, namun, untuk mempercepat pembubaran tingkat. Karena
total luas permukaan partikel menentukan area yang tersedia untuk difusi,
mengurangi partikel rata-rata Ukuran melalui proses mikronisasi mempromosikan
solubilisasi. Penurunan ukuran partikel dari 85% dengan kompensasi yang
peningkatan jumlah partikel menggandakan tingkat dissolution.27 Pendekatan lain
yang berguna adalah untuk memproduksi obat dalam bentuk garam yang larut
dalam air. Konsentrasi obat dalam pembubaran Lapisan ditingkatkan (sering
berkali-kali), dan tingkat difusi meningkat.
Proses pelarutan dapat rate limiting dipertimbangkan setiap kali solusi
obat menghasilkan efek sistemik lebih cepat dari formulasi padat agen yang sama
tidak. kadang-kadang perbedaan dalam penyerapan antara bentuk sediaan yang
seperti besarnya bahwa perbedaan klinis dicatat. Dengan aspirin, konsentrasi obat
dalam plasma 30 menit setelah administrasi bisa dua kali lebih tinggi untuk solusi
seperti untuk padat tablet.27 Meskipun tidak jelas apakah hasil perbedaan ini
semata-mata dari pembubaran obat atau dari faktor lain, seperti lebih lambung
cepat pengosongan khas cairan, pembubaran adalah mungkin setidaknya sebagian
bertanggung jawab.
Proses pelarutan dapat tingkat dianggap membatasi setiap kali solusi obat
menghasilkan efek sistemik lebih cepat daripada formulasi padat agen yang sama
tidak. Kadang-kadang perbedaan dalam penyerapan antara bentuk sediaan yang
sebesar itu bahwa perbedaan klinis dicatat. Dengan aspirin, konsentrasi obat
dalam plasma 30 menit setelah pemberian dapat dua kali lebih tinggi untuk solusi
sebagai sebuah tablet yang solid. Meskipun tidak jelas apakah perbedaan hasil ini
semata-mata dari pembubaran obat atau dari faktor lain, seperti lambung lebih
cepat pengosongan khas cairan, pembubaran mungkin setidaknya sebagian
bertanggung jawab.

Pengaruh bentuk sediaan pada penyerapan obat sering dimanfaatkan oleh
produsen obat. Beberapa obat (misalnya, eritromisin) tidak stabil pada pH rendah,
dan lain-lain (misalnya, amonium klorida) mengiritasi mukosa lambung. Untuk
menghindari pelepasan obat ini dalam perut, mereka sering disiapkan dalam
bentuk tablet salut enterik. Mantel enterik terdiri dari film lak atau pengganti
polimer.
Meskipun persiapan ini sering menguntungkan, kegunaan mereka tetap
adalah negatif dipengaruhi oleh variabilitas meningkat sebagai respons pasien.
Karena penyerapan obat tidak bisa dimulai sampai tablet melewati ke duodenum,
waktu yang diperlukan untuk transit lambung menjadi variabel penting. Bagian
dari larut tunggal tablet dari perut ke dalam usus adalah random peristiwa yang
dapat memakan waktu beberapa menit untuk lebih dari 6 jam.
Persiapan lepas lambat merupakan metode lain memanfaatkan pengaruh
formulasi pada penyerapan obat. Produk-produk ini biasanya dirancang untuk
melepaskan sejumlah stabil obat dalam saluran pencernaan selama 12 sampai 24
jam. Beberapa persiapan juga menyediakan pemuatan dosis awal yang tersedia
untuk penyerapan. Rilis berkelanjutan dapat dicapai dengan menggunakan matriks
berpori, dengan obat terletak di ruang interior dan pada permukaan eksternal.
Sebuah alternatif adalah untuk membuat bola obat yang larut pada tingkat yang
berbeda karena berbagai lapisan.
Studi dengan beberapa persiapan telah mendokumentasikan variabilitas
yang lebih besar dalam kinerja, Namun, daripada yang biasanya ditemui dengan
konvensional bentuk sediaan. Karena produk berkelanjutan-release mengandung
beberapa dosis konvensional obat, bahaya ada yang rilis terlalu cepat obat dari
persiapan ini mungkin menyebabkan konsentrasi toksik yang tak terduga.
Sebaliknya, inordinately slow release atau tidak lengkap dapat menyebabkan tidak
memadai terapi obat. Ketidakpastian efek formulasi ini diakui oleh US Food and
Drug Administration (FDA), yang menganggap mereka sebagai obat baru dan
mengharuskan efikasi dan keamanan ditampilkan sebelum mereka dapat
dipasarkan.
Transpor Aktif

Kebanyakan obat ditujukan untuk penggunaan oral diserap oleh difusi
pasif. Sistem transportasi aktif memang ada, namun, untuk spesifik konstituen diet
yang kadang-kadang meningkatkan penyerapan obat-obatan tertentu. Penyerapan
levodopa dan baclofen dari usus ditingkatkan karena mereka amino analog asam
dan secara aktif diangkut ke dalam sel usus oleh besar netral transporter asam
amino (LNAT, sebuah transporter SLC). Valacyclovir adalah juga jauh lebih baik
diserap daripada asiklovir congener nya karena merupakan substrat untuk PepT-1,
transporter SLC lain.
Mekanisme transpor aktif juga dapat menghambat obat absorption.30 P-
glikoprotein sangat diekspresikan sepanjang permukaan luminal sel epitel usus, di
mana ekspor xenobiotik yang seharusnya dapat diserap. fungsi ini adalah dalam
konser dengan "chemoimmunity defensif" Peran Pglycoprotein bermain dalam
melindungi sel dari paparan potensial compounds beracun Meskipun P-
glikoprotein dapat menunda penyerapan banyak obat dan mencegah sama sekali
yang penyerapan obat-obatan potensi serap rendah, mungkin signifikansi kecil
mengenai tingkat penyerapan kebanyakan obat ditujukan untuk penggunaan oral,
yang konsentrasi di chyme yang cukup untuk membanjiri kapasitas P-glikoprotein
untuk mengekspor them. Gambar 2-7 menggambarkan aktif pengangkutan obat ke
dalam dan keluar dari sel-sel usus dan di lain situs penting.
Pengaruh dari bentuk dosis pada penyerapan obat cukup sering
dimanfaatkan oleh produsen obat. Misalnya, beberapa obat (misalnya, eritromisin)
tidak stabil pada pH rendah dan lain-lain (misalnya, Amonium klorida) adalah
mengiritasi mukosa lambung. Untuk menghindari pelepasan obat ini dalam perut,
mereka sering disiapkan dalam bentuk tablet salut enterik. Salut enterik terdiri
dari film shellacatau beberapa pengganti polimer. Penutup ini tidak mudah larut
dalam kondisi asam tetapi tidak pecah untuk mengizinkan tablet disintegrasi di
lingkungan yang lebih basa dari usus kecil. Meskipun dosis ini sering
menguntungkan, kegunaan mereka bagi penderita dari variabilitas peningkatan
respon pasien. Karena penyerapan obat tidak bisa dimulai sampai tablet masuk ke
dalam duodenum, waktu yang dibutuhkan untuk transit lambung menjadi variabel
penting. Bagian dari tablet larut tunggal dari perut ke dalam usus adalah peristiwa
acak yang bisa berlangsung dari beberapa menit sampai lebih dari 6 jam.

Persiapan lepas-lambat merupakan metode lain yang memanfaatkan
pengaruh formulasi pada absorpsi obat. Produk-produk ini biasanya dirancang
untuk melepaskan sejumlah tetap dari obat dalam saluran pencernaan untuk
jangka waktu ½ sampai 24 jam. Selain itu, beberapa persiapan juga menyediakan
pemuatan dosis awal yang tersedia untuk penyerapan. Rilis berkelanjutan dapat
dicapai dengan menggunakan matriks berpori, dengan obat yang terletak baik di
ruang interior dan pada permukaan eksternal. Sebuah alternatif adalah untuk
membuat bola obat yang larut pada tingkat yang berbeda karena berbagai
pelapisan. Bentuk menarik dari tablet lepas lambat adalah "pompa osmotik
elementer," di mana obat ini tertutup dalam membran semipermeabel yang
memungkinkan air tetapi membatasi jalan keluar obat. Rilis konstan melalui
lubang kecil di membran dicapai dengan tekanan osmotik yang terbentuk di dalam
tablet sebagai obat secara perlahan dibubarkan. Keuntungan diklaim untuk produk
obat ini termasuk kepatuhan pasien yang lebih besar dan fluktuasi kecil dalam
konsentrasi darah antara dosis. Studi dengan beberapa persiapan, bagaimanapun,
telah mendokumentasikan variabilitas yang lebih besar dalam kinerja dari
biasanya dijumpai dengan bentuk dosis konvensional. Karena produk lepas-
lambat berisi beberapa dosis konvensional obat, bahaya ada apabila rilis obat
terlalu cepat dari persiapan ini dapat menyebabkan konsentrasi beracun tak
terduga. Sebaliknya, rilis tidak biasa lambat atau tidak lengkap dapat
menyebabkan terapi obat yang tidak memadai. Ketidakpastian efek formulasi ini
diakui oleh Food and Drug Administration (FDA), yang menganggap mereka
sebagai obat baru dan membutuhkan baik keampuhan dan keamanan yang
didemonstrasikan sebelum mereka dapat dipasarkan. Secara umum, agen oral
diminum tiga atau empat kali setiap hari, jarang ada kesulitan untuk sebagian
besar pasien. Biaya tambahan persiapan lepas lambat juga dapat mempengaruhi
keputusan tentang penggunaan di tempat dari bentuk-bentuk yang lebih
konvensional.
Sensitivitas penyerapan gastrointestinal ke ragam formulasi obat yang
terbaik dicontohkan berkenaan dengan bioavailabilitas. Dalam beberapa kasus,
obat-obatan kimiawi identik telah terbukti tidak setara biologis. Dalam salah satu
penelitian terhadap hidroklorida tetrasiklin, misalnya, sembilan persiapan

pembuatan yang berbeda dibandingkan dengan larutan obat yang sama. Meskipun
tujuh merek yang diproduksi menghasilkan konsentrasi darah berkisar antara 70%
sampai 100% dari solusi referensi, dua produk yang menunjukkan bioavalabilitas
relatif hanya 20% sampai 30%. Perbedaan bioavailabilitas yang paling mungkin
secara klinis penting dengan obat yang kurang diserap, memiliki margin rendah
keselamatan, dan dinonaktifkan olehproses kapasitas terbatas. Undang-undang
Federal telah diperlukan sejak tahun 1977 bahwa tes bioekivalensi dilakukan pada
semua obat baru, dan FDA telah mengamanatkan pengujian tersebut pada produk
yang sudah ada dengan masalah ketidaksetaraan untuk diketahui ada.
Pertimbangan bioavailabilitas yang berkaitan dengan pemilihan obat yang
dianggap lebih lanjut dalam Bab 55.
Inaktivasi obat
Salah satu kekurangan dari konsumsi oral adalah inaktivasi obat sebelum
mereka mencapai sirkulasi sistemik. Perusakan beberapa agen (misalnya,
epinefrin dan insulin) adalah cukup besar untuk menghalangi administrasi mereka
lewat rute ini. Dengan obat lain (misalnya, penisilin G), kerugian mungkin lebih
kecil tapi masih cukup besar untuk membuat oral tidak efisien. Asam lambung
merupakan salah satu penyebab utama kerusakan obat dalam saluran pencernaan,
tetapi degradasi juga hasil dari aktivitas enzimatik. Misalnya, insulin vasopressir,
kalsitonin, dan polipeptida lainnya tunduk pada hidrolisis dengan peptidase
pankreas dan usus. Sel-sel usus juga mengandung enzim intraseluler untuk
memetabolisme obat-obatan. Yang paling penting adalah adanya monoamina
oksidase untuk inaktivasi amina biogenik sebuah enzim CYP3A4/5 (dijelaskan
kemudian) untuk berbagai senyawa oksidasi. Enzim bakteri enterik juga dapat
merusak agen tertelan tertentu, seperti klorpromazin. Akhirnya, isi usus dapat
mengubah efektivitas dari obat yang diberikan secara oral. Mengikat konstituen
dari chymochelation dengan divalen kation, atau pembentukan garam larut dapat
menurunkan jumlah obat yang tersedia untuk penyerapan.
Nasib khusus ada untuk zat-zat yang berhasil diserap dari saluran
pencernaan. Pengurasan vena dari lambung, usus kecil, dan usus besar disalurkan
lewat sistem portal hepatik ke hati.First pass dari konsentrasi obat tinggi melalui

organ sarat-enzim ini dapat signifikan] mengurangi jumlah agen mencapai
sirkulasi sistemik Lidocaine, pada kenyataannya, dimetabolisme begitu pesat
dalam hati hampir semua dosis oral hancur selama first pass.Meskipun kurang
jelas, kesenjangan opioid analgesik yang khasiat antibiotik diamati antara rute oral
dan moda lain dari administrasi adalah penting secara klinis untuk praktek
kedokteran gigi.
Rute enteral lainnya
Mukosa mulut dan dubur kadang-kadang digunakan sebagai tempat
penyerapan obat. Sublingual, di mana tablet, atau troche diperbolehkan untuk
larut sempurna dalam Cavit oral yang mengambil keuntungan dari permeabilitas
epitel mulut dan merupakan rute pilihan untuk beberapa obat lipofilik, ampuh,
seperti nitrogliserin dan oksitosin. Lapisan lendir mulut dan usus tidak berbeda
secara kualitatif seperti menyerap permukaan, yang penyerapan sebanding telah
terbukti untuk beberapa agen. Salah satu alasan untuk memilih rute sublingual
adalah untuk menghindari kerusakan obat. Karena asam lambung dan usus adalah
enzim-enzim hati yang dilewati, penyerapan sublingual bisa lebih efisien
keseluruhan untuk obat-obatan tertentu daripada penyerapan usus. Timbulnya efek
obat juga mungkin lebih cepat dari konsumsi oral.
Pemberian rektal dapat digunakan saat masuk rute yang menghalangi,
seperti pada pasien tidak sadar atau yang muntah. Meskipun sebagian besar obat
diserap masuk sirkulasi tanpa harus melewati penyerapan obat sering tak terduga.
Beberapa obat mengiritasi mukosa lambung (misalnya, xanthines) dapat diberikan
secara rektal; sensitivitas dubur melarang pemberian lewat rute ini.
Inhalasi
Membran alveolar merupakan jalur penting masuk beberapa obat dan
banyak zat-zat berbahaya. Meskipun lapisan alveolar sangat permeabel, dapat
diakses hanya untuk agen yang berada dalam keadaan gas atau dihirup dalam
bubuk halus atau tetesan untuk mencapai ujung terdalam pohon pernafasan.
Termasuk dalam kategori pertama adalah gas terapetik, karbon monoksida,
anestesi inhalasi, dan sejumlah pelarut organik yang mudah menguap. Kategori

kedua dari penetrants membran alveolar secara kolektif dikenal sebagai aerosol
Istilah ini mengacu pada partikel cair atau padat yang cukup kecil (biasanya 10
µm atau kurang diameter) untuk tetap tertahan di udara untuk waktu yang lama.
Partikel semacam ini termasuk bakteri, virus, asap, serbuk sari, semprotan, dan
debu. Setiap material halus yang terpisah tersebut, ketika dihirup, mencapai
beberapa bagian dari pohon pernapasan dan dipengaruhi oleh proses sedimentasi
dan endapan inersia. Kebanyakan aerosol mengandung campuran ukuran partikel.
Partikel yang relatif besar (lebih dari 6 µm) berdampak pada bronkiolus terminal
dan cabang besar pohon pernapasan dan dikeluarkan dari paru-paru dengan
selimut silia-didorong lendir mengalir terus menerus menuju faring. Partikel yang
lebih kecil, yang melakukan mencapai kantung alveolar, dapat diserap melalui sel-
sel lapisan ke dalam aliran darah, yang diambil oleh proses fagositosis, atau
dibawa oleh film berair meliputi sel alveolar bronkiolus terminal di mana mereka
bergabung dengan selimut lendir. Meskipun dua dari tiga hasil ini mungkin
melibatkan penyerapan partikel, mekanisme untuk menghilangkan padatan yang
sangat efisien. Hanya sebagian menit dari debu yang terhirup seumur hidup gagal
dihapus oleh transportasi silia.
Penggunaan terapi aerosol tidak meluas, tetapi beberapa obat darurat
disusun dalam bentuk ini. Karena timbulnya efek sangat cepat setelah menghirup
obat aerosol, dengan ini dapat memberikan sarana cepat pengobatan sendiri bagi
orang-orang dalam bahaya reaksi alergi akut pada venoms atau obat-obatan.
Epinefrin merupakan salah satu agen darurat seperti yang dipasarkan sebagai
aerosol. Banyak obat pernafasan juga disiapkan dalam bentuk aerosol karena
mereka sangat efektif dengan rute ini sambil meminimalkan paparan sistemik.
Namun, kecepatan dan efisiensi penyerapan membran alveolar dapat, kadang-
kadang, menimbulkan masalah untuk terapi, seperti yang digambarkan oleh
penggunaan aerosol bertekanan yang mengandung isoproterenol. Meskipun
hingga 97% dari semprotan isoproterenol ditelan dibawah kondisi normal dan
tidak diaktifkan oleh berbagai enzim, kelebihan obat dapat menghasilkan efek
toksik. Data yang dikumpulkan selama periode 7 tahun di Inggris menyarankan
bahwa penggunaan tanpa disiplin dari preparat ini menghasilkan angka kematian
meningkat pada pasien asma. Pembatasan penjualan obat non resep dan

peringatan kepada dokter yang disertai dengan penurunan tingkat kematian.
Temuan seperti ini mencerminkan bahaya aerosol ketika disalahgunakan dan
memberikan peringatan untuk pengobatan sendiri tak terkontrol dengan obat yang
berpotensi berbahaya. Keprihatinan atas aerosol juga terkait dengan pertanyaan
dari toksokologi, seperti penyerapan debu logam berat oleh pekerja industrial.
Injeksi parenteral
Obat sering diberikan melalui suntikan parenteral bila konsumsi oral
dilarang oleh kondisi pasien, ketika onset cepat dari efek diperlukan, atau ketika
konsentrasi darah lebih dari yang diperoleh dengan rute enteral diperlukan.
Metode injeksi yang dipilih beragam dengan obat tertentu dan kebutuhan terapetik
dari pasien.
Rute Intravenous
Pemberian obat melalui infus atau suntikan langsung ke dalam aliran darah
sangat berguna ketika efek langsung atau konsentrasi darah yang tepat yang
diinginkan. Karena penyerapan dilewati, injeksi intravena circumvents penundaan
dan variasi dalam respon obat biasanya berhubungan dengan moda administrasi.
Pengenceran cepat dalam aliran darah dan ketidakpekaan relatif dari endotelium
vena terhadap obat sering mengizinkan administrasi sukses dari senyawa atau
solusi terlalu mengiritasi untuk rute lainnya (misalnya, obat antikanker alkilasi
dan cairan hipertonik). Juga, melalui teknik titrasi, rute intravena menyediakan
sebuah jalan untuk administrasi obat dikendalikan yang memiliki margin
keselamatan yang sangat sempit antara konsentrasi terapeutik dan beracun. Infus
lidokain untuk mencegah aritmia ventricular dan suntikan tambahan obat
antiansietas selama sedasi intravena adalah dua contoh di mana titrasi digunakan
untuk mencapai efek yang diinginkan sementara menghindari efek samping.
Meskipun banyak agen intravena tidak memerlukan titrasi dan dapat diberikan
dalam dosis standar, mereka tetap harus disuntikkan perlahan-lahan. Jika
diberikan terlalu cepat, dosis bisa bergerak awalnya melalui jantung, paru-paru,
dan arteri utama sebagai bolus konsentrasi obat yang tinggi. Efek samping
cardiopulmonary nonspesifik tetapi berpotensi bencana bisa terjadi, bahkan dari

injeksi cepat larutan garam sederhana. Kebanyakan obat harus diberikan selama 1
menit, yang mendekati waktu sirkulasi darah melalui tubuh. Prosedur ini
menghindari konsentrasi tinggi, dan memungkinkan penghentian jika ada efek tak
diinginkan diamati selama injeksi.
Kelemahan utama dari rute intravena adalah bahwa setelah obat disuntikkan,
sangat sedikit yang bisa dilakukan untuk menghapusnya dari aliran darah. Ketika
respon yang merugikan dicatat dengan rute lain, penyerapan lanjut biasanya dapat
ditunda atau bahkan mungkin dicegah. Reaksi toksik terhadap obat yang diberikan
secara intravena sering seketika dan berat. Peristiwa anafilaksis mengancam jiwa
juga lebih mungkin karena kemungkinan dari reaksi antigen-antibodi besar.
Komplikasi lain injeksi intravena termasuk vasculitis dan emboli (baik dari iritasi
obat, partikel dalam larutan yang disuntikkan, atau trauma jarum), demam (dari
suntikan pirogen seperti lipopolisakarida bakteri), infeksi, dan pembentukan
hematoma.
Rute Intramuskular
Rute intramuskular yang sering dipilih untuk obat yang tidak dapat
diberikan secara oral karena lambat atau tidak menentu penyerapan, persentase
yang tinggi dari inaktivasi obat, atau kurangnya kerjasama pasien. Tingkat
penyerapan dari sebuah situs intramuskular diatur oleh faktor-faktor yang sama
yang mempengaruhi penyerapan gastrointestinal, seperti koefisien lipid/ partisi
air, derajat ionisasi, dan ukuran molekul. Banyak obat-obatan, namun diserap
mendekati tingkat yang sama, terlepas dari faktor-faktor ini. Satu-satunya
penghalang yang memisahkan obat disimpan intramuskuler dari aliran darah
adalah endotelium kapiler, membran multiseluler dengan kesenjangan antar besar.
Banyak zat yang tidak larut-lipid dapat memasuki kompartemen vaskular melalui
celah-celah tersebut, dan bahkan protein mampu diserap. Dalam keadaan ini,
aliran darah melalui jaringan sering penentu utama laju penyerapan obat. Dengan
demikian otot dengan arus darah tinggi (misalnya, deltoid) memberikan tingkat
penyerapan lebih cepat dari otot dengan arus yang lebih rendah (misalnya, gluteus
maximus). Secara umum, 5 sampai 30 menit diperlukan untuk timbulnya efek
obat, tetapi periode laten ini dapat dikontrol sampai batas tertentu. Latihan nyata

mempercepat penyerapan dengan merangsang sirkulasi lokal. Sebaliknya, serapan
dapat diminimalkan dengan penerapan paket es atau (dalam keadaan darurat)
torniket.
Dengan pengecualian dari beberapa obat yang relatif tidak larut pada pH
jaringan (misalnya, diazepam, phenytoin), penyerapan dari suntikan intramuskular
biasanya cepat dan lengkap. Formulasi itu telah dikembangkan untuk
menyediakan pelepasan obat lama dan mantap. Persiapan depot ini terdiri dari
obat yang diproduksi sebagai garam larut atau dibagikan dalam wahana pembawa
berminyak, atau keduanya, seperti penisilin prokain tersuspensi dalam minyak
kacang. Volume solusi yang relatif besar dapat diberikan oleh rute ini, tapi rasa
sakit di tempat suntikan adalah sering, dan beberapa obat (misalnya, doxycycline)
terlalu mengiritasi diberikan dengan cara ini.
Rute Subkutan
Injeksi obat ke dalam jaringan ikat subkutan adalah metode administrasi
yang digunakan secara luas untuk agen yang dapat diberikan dalam volume kecil
(2 ml atau kurang) dan tidak secara lokal merusak. Penyerapan subkutan mirip
dengan mengistirahatkan otot, dan waktu onset sering sebanding. Seperti dengan
rute intramuskular, penyerapan dapat ditunda dengan mengurangi aliran darah,
baik melalui penerapan tekanan atau pendinginan permukaan. Gangguan
farmakologi sirkulasi dengan vasokonstriktor juga merupakan strategi umum,
terutama dalam anestesi lokal. Karena kemudahan implantasi subkutan, pelet
terkompresi obat, kadang-kadang dicampur dengan bahan matriks yang tidak
larut, dapat dimasukkan untuk memberikan pelepasan obat hampir konstan selama
beberapa minggu atau bulan. Testosteron dan beberapa agen kontrasepsi
progestasional (misalnya, levonorgestrel) telah berhasil dikelola dengan
pendekatan ini. Penyerapan lambat dapat dicapai, juga, melalui penggunaan
bentuk depot seperti yang dijelaskan untuk suntikan intramuskular.
Ketika subkutan dipilih untuk efek sistemik, yang mempercepat penyerapan
obat kadang-kadang menguntungkan. Untuk itu, pemanasan jaringan
mempromosikan penyerapan obat dengan meningkatkan sirkulasi lokal. Memijat
tempat suntikan, selain menstimulasi aliran darah, membantu penyebaran obat dan

menyediakan peningkatan luas permukaan untuk penyerapan. Efek yang terakhir
ini juga dapat dicapai melalui pemberian bersamaan hyaluronidase, enzim yang
memecah matriks mukopolisakarida jaringan ikat. Penyebaran lateral larutan air
begitu ditingkatkan, pada kenyataannya, hyaluronidase yang kadang-kadang
digunakan untuk memungkinkan injeksi volume cairan yang besar dalam situasi
di mana infus intravena kontinu sulit atau tidak mungkin.
Rute Injeksi Parenteral Lainnya
Suntikan intraarterial kadang-kadang dilakukan ketika efek lokal pada organ
tertentu atau area tubuh yang diinginkan. Suntikan pewarna radiopaque untuk
tujuan diagnostik dan agen antineoplastik untuk mengendalikan tumor lokal
adalah contoh yang paling sering ditemui. Administrasi intratekal digunakan
ketika akses langsung dari obat SSP diperlukan. Indikasi untuk injeksi ke dalam
ruang subarachnoid meliputi produksi anestesi spinal dengan anestesi lokal dan
resolusi infeksi SSP akut dengan antibiotik. Infus intraperitoneal dari cairan
adalah pengganti berguna untuk hemodialisis dalam pengobatan keracunan obat.
Meskipun injeksi intraperitoneal umumnya digunakan dalam eksperimen hewan,
risiko infeksi biasanya termasuk menghalangi pada manusia. Terakhir,
intraosseous (anterior tibia) injeksi obat darurat dapat digunakan pada pasien anak
ketika akses intravena tidak dapat diperoleh dengan cepat.
Semua teknik injeksi khusus ini adalah berpotensi berbahaya untuk pasien.
Mereka harus dilakukan hanya bila secara tegas ditunjukkan dan hanya oleh
teknisi ahli.
Aplikasi Topikal
Obat-obatan yang sering diterapkan pada permukaan epitel untuk efek lokal dan
kurang sering untuk penyerapan sistemik. Penetrasi obat di seluruh epitel sangat
dipengaruhi oleh tingkat keratinisasi.
Kulit
Epidermis adalah jaringan yang sangat dimodifikasi yang mengisolasi tubuh
dari lingkungan eksternal. Lapisan luar kulit (stratum korneum) yang padat

dengan keratin protein. Lapisan ini tahan terhadap air dan obat-obatan karena itu
larut dalam air, dan ketebalan dan kurangnya lipid kontras dengan membran
biologis lainnya yang relatif menghambat bahkan difusi agen sangat lipofilik.
Sifat kulit terhadap obat yang larut dalam air sering mensyaratkan bahwa agen
(misalnya, antibiotik, fungisida) ditujukan untuk kondisi dermatologi] dikelola
oleh rute sistemik meskipun aksesibilitas dari kulit jelas. Untuk obat larut lipid,
rute perkutan sering berhasil untuk masalah-masalah lokal. Selain itu, ketika
lapisan keratin terganggu, penyerapan obat, terutama senyawa hidrofilik, secara
nyata ditingkatkan. TI jaringan ikat yang mendasari (dermis) cukup permeabel
banyak zat terlarut, meskipun berbeda dengan kebanyakan jaringan mempunyai
pasokan berlimpah shunts arteriovenosa, yang mungkin menyebabkan absorpsi
sistemik menjadi sangat sensitif terhadap perubahan suhu.
Hambatan umum kulit utuh terhadap obat tidak membatalkan perlunya
kehati-hatian ketika berhadapan dengan bahan kimia beracun yang potensial.
Dokumentasi yang cukup dari penyerapan epidermiszat-zat asing telah
menetapkan bahwa senyawa tertentu mungkin mudah menembus kulit
menyebabkan efek sistemik. Obat ini termasuk pelarut organik, organophospha
dan insektisida berbasis nikotin, dan beberapa gas saraf. Keracunan saraf juga
dihasilkan dari krim aplikasi tabir matahari yang berlebihan yang mengandung
anestesi lokal. Bahkan zat larut lipid seperti merkuri anorganik bisa menyebar
melintasi di kulit jika paparan berkepanjangan.
Manfaat yang jelas dari peningkatan dan cukup pengendalian penyerapan
perkutan untuk membuat pemberian obat dengan handal telah mendorong
beberapa strategi.Misalnya, " sistem terapi transdermal" telah dikembangkan
untuk memberikan penyerapan sistemik terus menerus dari skopolamin
nitroglyceri, fentanyl, dan nikotin, masing-masing, untuk profilaksis angina
pektoris dan mabuk, manajemen nyeri kronis, dan bantuan dengan penghentian
merokok. Sistem adalah patch kompleks yang terdiri dari impermeable backing,
cadangan yang mengandung obat dalam bentuk tertunda, membran
semipermeabel, dan perekat bagian dalam.
Pada awal 1960-an, ditemukan bahwa pelarut industri dimetil sulfoksida
mempromosikan percutanous penyerapan obat yang larut dalam air. Potensi

terapi yang disederhanakan (terapi bagi pasien rematik dan lainnya bahwa
pembawa obat ini disambut dengan penuh semangat. Laporan selanjutnya efek
samping pada hewan menyebabkan minat berkurang, namun sampai akhir 1970-
an ketika dipromosikan sebagai agen efektif untuk pereda gejala berbagai
gangguan muskuloskeletal dan kolagen. Meskipun banyak tersedia obat herbal,
dimetil sulfoksida saat ini disetujui oleh FDA hanya untuk pengobatan interstitial
cystitis.
Pendekatan lain untuk meningkatkan penetrasi obat melalui epidermis
adalah penggunaan dressing oklusif. Dressing mempertahankan kelembaban dan
memecah lapisan tanduk melalui proses maserasi. Sebuah teknik akhir,
iontophoresis, dibahas di bawah ini.
Membran Mukosa
Aplikasi topikal dari obat-obatan untuk selaput lendir menawarkan beberapa
keuntungan potensial untuk terapi lokal. Jaringan ini sering divisualisasikan
dengan dokter, memungkinkan penempatan akurat. Penggunaan jalur ini
umumnya meminimalkan sistem efek sambil memberikan konsentrasi optimal
obat pada daerah yang dirawat. Berbeda halnya dengan kulit, obat mempunyai
sedikit kesulitan menyerap membran mukosa uang mempengaruhi kondisi lokal.
Memang, penyerapan sistemik obat lipophilic dari selaput lendir mudah terjadi.
Sebelum hal ini secara luas dihargai, aplikasi topikal dari tetracairan ke faring dan
trakea mukosa adalah penyebab utama overdosis anestesi lokal. Dalam kedokteran
gigi, penggunaan kosteroid untuk memperbaiki kondisi peradangan memiliki
menaikkan respon sistemik, seperti menekan fungsi adrenokortikal oleh
triamcinolone. Meskipun efek umumnya ringan dan sementara, mereka dapat
menciptakan masalah bagi penderita hipertensi, diabetes mellitus, atau ulkus
peptikum. Terapi lokal juga dapat mempengaruhi kesehatan sistemik dengan
melayani sebagai stimulan antigenik dan, dalam kasus antibiotik, dengan
mengganggu ekologi mikroba normal dan mempromosikan munculnya
mikroorganisme resisten.
Obat kadang-kadang diterapkan secara mukosa untuk efek sistemik mereka.
Selain sublingual dibahas sebelumnya dan administrasi rute rektal, mukosa hidung

menawarkan jalan yang sesuai untuk penyerapan zat tertentu. Desmopresin,
digunakan dalam pengobatan diabetes insipidus, dan butorphanol, analgesik kuat,
adalah contoh obat yang dapat diberikan intranasal.
Iontophoresis
Iontophoresis adalah pengangkutan listrik obat positif atau negatif yang
dikenakan di jaringan permukaan. Teknik ini melibatkan melewati arus listrik
langsung dari polaritas yang tepat melalui solusi obat dan pasien. Perembesan
membran mukosa dan bahkan jaringan keras dan kulit mungkin dengan
pendekatan ini, namun dosis total disampaikan kecil, dan toksisitas sistemik
adalah tidak mungkin. Dalam terapi gigi, aplikasi iontophoretic obat telah
digunakan di beberapa situasi. Gigi sulung longgar telah diekstraksi berhasil
setelah pemberian intophoretic lidocaine dengan epinephrine untuk anestesi
jaringan lunak. Untuk pengobatan herpes orolabialis, arus galvanik meningkatkan
konsentrasi jaringan idoxuridine hingga tiga kali yang diperoleh dengan aplikasi
topikal saja. Mungkin penggunaan paling umum dari iontophoresis dalam
kedokteran gigi, bagaimanapun, adalah promosi penyerapan F- menjadi terpapar
dentin hipersensitif. Solusi 1% dari sodium fluoride diberikan dengan cara ini
menghasilkan hasil yang lebih baik daripada pasta 33%.
2.3 DISTRIBUSI
Distribusi mengacu pada pergerakan obat ke seluruh tubuh. Laju, urutan, dan
tingkat distribusi tergantung pada banyak faktor: sifat fisikokimia obat, cardiac
output dan aliran darah regional, karakteristik membran dari anatomi, listrik
transmembran dan gradient pH, ikatan protein plasma dan jaringan cadangan, dan
sesekali transportasi aktif, memfasilitasi difusi, atau endocytosis/transcytosis.
Untuk semua tapi sangat sedikit obat yang bekerja intravaskular, membran kapiler
merupakan penghalang jaringan pertama yang melintas dalam perjalanan obat dari
aliran darah ke situs kerjanya.
Penetrasi Kapiler

Setelah memperoleh akses obat ke sirkulasi sistemik, menjadi diencerkan
dengan volume plasma dari seluruh kompartemen vaskular. Untuk senyawa
intravena, proses ini hanya membutuhkan waktu beberapa menit untuk
menyelesaikan; untuk obat yang diberikan lewat rute lain, distribusi
intravascularberlangsung bersamaan dengan penyerapan. Transfer obat dari aliran
darah diatur oleh faktor-faktor yang sama yang mengontrol pintu masuknya. Obat
lipofilik, misalnya, berdifusi melintasi membran kapiler sangat cepat. Transfer ini
sangat cepat, pada kenyataannya, bahwa kesetimbangan dengan cairan interstitial
praktis seketika. Dengan kondisi tersebut, tingkat penyerapan obat ditentukan oleh
aliran darah melalui jaringan di bawah pertimbangan. Organ perfusi baik adalah
jenuh dengan obat jauh sebelum banyak jaringan lain memiliki kesempatan untuk
mencapai bahkan sebagian kecil dari keseimbangan konsentrasi. Obat yang larut
dalam air berdifusi melalui celah-celah yang terletak antara sel-sel endotel yang
berdekatan. Dengan agen ini, gerakan transkapiler lebih lambat dibandingkan obat
yang memiliki lipid yang tinggi/koefisien partisi air dan berbanding terbalik
dengan berat molekul. Ukuran obat meningkat, difusi air menjadi kurang penting,
dan filtrasi mengambil alih sebagai kekuatan motif utama di balik transportasi
obat. Zat dengan berat molekul yang lebih besar dari 60.000 lulus melintasi
membran kapiler sangat lambat. Bukti mikroskopis elektron menyarankan bahwa
transcytosis terlibat dalam distribusi mereka.
Masuknya obat ke dalam Sel
Seperti telah dibahas sebelumnya, membran sel bertindak sebagai
penghalang semipermeable, mengenali beberapa obat ke dalam sel sementara
mengeluarkan yang lain. Senyawa larut lipid, nonpolar, mendistribusikan merata
di seluruh membran plasma, namun distribusi elektrolit lemah pada
kesetimbangan agak lebih kompleks. PH intraseluler adalah sekitar 7.0, berbeda
sedikit dari 7.4 pH cairan ekstrasel. Asam dengan pK kurang dari 8.0 cenderung
tetap berada di luar sel, sedangkan obat dasar dengan pK lebih besar dari 6.0
cenderung menumpuk di dalam. Karena diferensial konsentrasi melintasi
membran sel berdasarkan gradien pH 0.4 setara 2.5:1, status asam-basa dari
pasien secara signifikan dapat mempengaruhi respon dosis elektrolit lemah

bertindak intraseluler. (Pengaruh pH pada distribusi anestesi lokal melintasi
membran saraf dijelaskan pada Bab 16.) Ion-ion, kecuali sangat kecil dalam
ukuran (berat molekul 60 atau kurang) atau diangkut oleh operator terikat-
membran, menembus membrane sel dengan kesulitan atau tidak sama sekali. Obat
membebani mendapatkan akses ke sel dengan cara difusi pasif didistribusikan di
kesetimbangan sesuai dengan gradien elektrokimia mereka melintasi membran.
2.4 Distribusi ke Kompartemen Cairan Khusus
Dalam beberapa jaringan atau organ, komponen anatomi memungkinkan
penyerapan cairan interstitial atau transelular dari ruang ekstraseluler umum.
Contoh yang paling penting bagi terapi melibatkan SSP, sirkulasi janin, dan,
sejauh kedokteran gigi yang bersangkutan, air liur.
Sistem saraf pusat
Masuknya obat ke dalam SSP adalah biasa tergantung pada kelarutan lipid.
Obat dengan koefisien partisi lipid /air yang tinggidiambil dengan sangat cepat,
seperti yang dicontohkan oleh onset segera anestesi umum setelah injeksi
intravena thiopental. Distribusi cepat obat lipofilik ke kabel otak dan tulang
belakang muncul dari fakta bahwa SSP menerima sekitar 15% dari output jantung
belum menyusun hanya 2% dari total berat badan. Meskipun suplai darah yang
menguntungkan ini, obat yang hemat larut lemak sebagian besar dikeluarkan dari
ruang ekstraselular otak. Berbeda dengan kapiler sebagian besar jaringan, sel-sel
endotel dari SSP bergabung bersama oleh persimpangan ketat yang membatasi
masuknya obat larut dalam air untuk mereka yang memiliki jari-jari molekul
efektif 8 Ǎ atau kurang. Sehingga molekul relatif besar (misalnya, inulin, dengan
berat molekul 5000) yang biasanya berlalu tanpa kesulitan ke dalam ruang
interstitial benar-benar dilarang, dan sebagian besar obat lain tergantung pada
saluran air untuk penetrasi dan beratnya lebih dari 100 sampai 200 dalton sangat
diperlambat. Adanya relatif endositosis/ transcytosis juga penting dalam kapiler
SSP.
Halangan kedua transfer ion dan zat yang larut dalam air lainnya adalah
selubung sel yang mengelilingi kapiler otak. Lapisan investasi ini terdiri dari

proses membentang dari astrosit jaringan ikat. Meskipun wilayah cakupan
permukaan kapiler tidak utuh, itu tetap cukup untuk menghambat difusi semua
kecuali senyawa lipid-mudah larut.
Faktor ketiga yang membatasi akses obat ke SSP adalah koleksi transporter
membran, seperti P-glikoprotein,yang secara efisien mengekspor obat
mendapatkan masuk ke sel-sel endotel. Bersama-sama, endothelium memodifikasi
kapiler, sarung astrositik, dan sistem pembawa ekspor merupakan penghalang
darah-otak.
Obat juga dapat memperoleh akses ke SSP dengan cara pleksus koroid.
Setiap pleksus koroid terdiri dari jaringan pembuluh darah kecil dan kapiler
memproyeksikan ke ruang ventrikel dan ditutupi oleh lapisan sel-sel epitel khusus
diadaptasi untuk sekresi cairan serebrospinal. Difusi obat di epitel koroid pleksus
dan ke dalam cairan cerebrospinal sebagian besar terbatas pada obat-obatan yang
sangat lipid-mudah larut, menunjukkan adanya fungsional analog dari penghalang
antara darah dan cairan serebrospinal. Koroid pleksus dan cairan serebrospinal
sebenarnya terlibat lebih dekat dengan penghapusan obat dari SSP dibandingkan
dengan masuknya mereka. Disekresi ke dalam ventrikel ketiga, keempat, dan
secara lateral, menggerakkan cairan serebrospinal oleh aliran massal melalui
sistem ventriculocisternal ke permukaan otak dan sumsum tulang belakang
sebelum keluar melalui vili arachnoid. Obat hadir dalam cairan ekstraselular SSP
bebas berdifusi ke dalam cairan cerebrospinal. Karena jumlah total cairan
serebrospinal (150 ml) mendekati volume ruang interstitial dan karena memiliki
tingkat moderat dengan cepat berubah (10% per jam), penghapusan obat oleh
aliran massal melalui viii arachnoid dapat mencegah agen di otak dari yang
pernah mencapai kesetimbangan dengan darah. Transpor aktif ion organik dari
cairan cerebrospinal kembali ke dalam sirkulasi sistemik oleh sel-sel lapisan
pleksus koroid juga mempromosikan penghapusan banyak obat dari otak.
Distribusi selektif senyawa-senyawa ke dalam SSP memiliki beberapa
konsekuensi terapi yang penting. Beberapa alkaloids ditujukan untuk efek sistem
saraf perifer, misalnya, dapat menyebabkan gangguan pada pusat masuk ke otak.
Konversi obat tersebut (misalnya, skopolamin) untuk secara positif dikenakan
turunan amonium kuaterner (misalnya, methscopolamine) mencegah pengaruh

SSP belum memungkinkan aktivitas sistem saraf perifer penting. Sebaliknya, obat
yang digunakan untuk efek sentral mereka dapat merasakan manfaat dari
modifikasi molekul yang meningkatkan masuknya mereka ke dalam otak. Total
dosis rendah kemudian dapat diberikan dan efek perifer diminimalkan. Kadang-
kadang penghalang darah-otak adalah suatu halangan untuk terapi. Penisilin G,
asam organik larut dalam air dengan pK, 2.6, berdifusi perlahan ke dalam SSP dan
tunduk pada penghapusan aktif oleh pleksus koroid. Untuk orang dengan
ensefalitis bakteri, kurangnya penetrasi obat dapat mempersulit pengobatan.
(Permeabilitas kapiler di otak untungnya sering meningkat selama inflamasi
meningeal.) Pendekatan cerdas untuk mengelakkan penghalang darah-otak
diwujudkan dalam pengobatan parkinson. Kondisi ini dikaitkan dengan
kekurangan dopamin dalam bagian yang dipilih dari otak. Terapi penggantian
dengan dopamin tidak efektif, namun, karena obat ini dikecualikan oleh
penghalang darah-otak. Untuk menghindari masalah ini, levodopa, prekursor
asam amino dopamin, digunakan sebagai gantinya. Levodopa mudah memasuki
otak, di mana ia kemudian ber-dekarboksilasi untuk obat aktif. Sebuah metode
yang lebih drastis dan berpotensi berbahaya menembus penghalang darah-otak
adalah untuk mengganggu itu sementara dengan menanamkan solusi hipertonik ke
arteri karotis. Sebuah penyusutan osmotik diinduksi sel endotel serebrovaskular
menyebabkan persimpangan ketat untuk memisahkan dan memungkinkan
penyerapan obat yang larut dalam air. Strategi saat ini meliputi melampirkan obat
untuk zat pembawa, atau vektor, yang diangkut ke dalam otak. Vektor tersebut
dapat terdiri dari molekul alami yang diangkut atau melibatkan antibodi
monoklonal yang ditargetkan untuk molekul-molekul ini. Vektor peptida lainnya
telah diidentifikasi yang mempromosikan transcytosis. Kopling obat-obatan
seperti penisilin dan doxorubicin sangat mempertinggi serapan mereka di
penghalang darah-otak.
Transfer plasenta
Kelahiran obstetri dari bayi sadar dari anestesi, para ibu pernah
disalahartikan sebagai bukti penghalang plasenta unik termasuk obat bahkan lipid-
larut dari janin. Sekarang dipahami bahwa pengamatan tersebut sebagai hasil dari

terbatasnya tingkat transfer obat dari peredaran ibu ke jaringan janin. Pembuluh
darah janin memproyeksikan dalam sinus penuh dengan darah ibu yang ditutupi
oleh lapisan tunggal sel yang disebut trofoblas. Pergerakan obat dari plasenta
dibatasi oleh membran trofoblas, yang mana secara kualitatif mirip dengan
membran plasma dimanapun. Meskipun trofoblas diketahui aktif mengeluarkan
asam amina dan nutrisi lainnya ke dalam sirkulasi janin, masuk dari kebanyakan
obat tergantung pada difusi pasif yang melintasi penghalang lipofilik. Untuk obat
yang sangat lipofilik seperti thiopental, disibusi diperlambat hanya dengan tingkat
aliran darah ibu melalui plasenta dan dengan keanehan dalam sirkulasi janin yang
membatasi perfusi jaringan. Meski begitu, telah dihitung 40 menit yang
diperlukan untuk jaringan janin untuk mencapai kesetimbangan 90° dengan
konsentrasi konstan arteri ibu. Dibatasi oleh difusi transmembran lamban, transfer
senyawa larut dalam air sangat tidak efisien sehingga sebenarnya tidak ada obat
dari administrasi tunggal dapat memperoleh akses janin. Seperti di SSP, P-
glikoprotein dalam plasenta cenderung untuk mencegah zat yang berpotensi
berbahaya dari memasuki sirkulasi janin. Namun demikian, agen hemat lipid
mudah larut akhirnya akan terakumulasi pada janin yang diberikan kepada ibu
dalam beberapa dosis.
Menyangkut transfer plasenta dari obat-obatan muncul kemungkinan yang
mendorong manifestasi beracun di bayi baru lahir dan perkembangan cacat pada
embrio dan janin. Topik yang dibahas lebih lanjut dalam Bab 3.

Sekresi Air Liur
Distribusi obat ke air liur adalah kepentingan farmakologi dalam dua hal.
Pertama, obat mendapatkan akses ke lingkungan dari sirkulasi sistemik bisa
mempengaruhi mikroorganisme atau permukaan jaringan dalam mulut. Sekalipun
pengaruh ini biasanya tidak diinginkan, obat yang dikembangkan dengan efek
lokal, seperti pencegahan karies, dibayangkan bisa diberikan secara sistemik
untuk mencapai konsentrasi terapetik berkelanjutan dalam air liur sambil
menghindarkan kebutuhan berulang mulut pembilasan.Minat farmakologis pada
air liur yang berasal dari kenyataan bahwa penentuan obat air liur dapat
memberikan ukuran noninvasif konsentrasi plasma bebas dari obat. Karena
konsentrasi obat bebas dalam plasma biasanya penentu utama respon pasien,
manfaat jumlah obat air liur untuk terapetik peutics berpotensi besar. Namun,
penelitian klinis telah mendokumentasi hubungan kompleks antara plasma dan
titer air liur, salah satu obat yang harus dipahami sepenuhnya sebelum
pemantauan air liur dapat berhasil digunakan.

Obat bisa memasukkan cairan oral dari beberapa sumber:(1) difusi pasif di
alveolar dan sel-sel duktus kelenjar air liur, (2) difusi pasif seluruh epithelium oral
dan (3) aliran massal cairan dari celah gingiva. Kesempatan, yang pertama adalah
yang paling penting dan yang ketiga adalah kurang(kecuali untuk obat yang tidak
bisa mendapatkan masuk oleh salah satu dari dua rute lain). Seperti terlihat pada
Tabel 2-1, konsentrasi air ludah dari sebuah obat dipengaruhi oleh banyak faktor.
Agen yang relatif larut lemak (misalnya, diazepam) atau berukuran sangat kecil
(misalnya, etanol) mengalami sedikit kesulitan dalam kesetimbangan dengan air
liur. Karena hanya bagian tak terikat yang terlibat dalam distribusi di seluruh
membran dan kompartemen air liur cukup kecil terhadap total ruang intravaskular,
protein yang mengikat tidak mempengaruhi rasio air liur/plasma obat bebas
(misalnya, diazepam dan acetaminophe). Mengenai elektrolit lemah, perbedaan
pH antara plasma dan hasil air liur lebih asam dalam konsentrasi basa dengan pK,
lebih besar dari 5.5 dalam air liur (misalnya, quinidine) dan efek berlawanan pada
asam dengan pK, kurang dari 8.5 (misalnya, Sulfamerazin). Akhirnya, laju aliran
air liur dapat mengubah konsentrasi intraoral dalam setidaknya dua cara.
Peningkatan produksi air liur dapat melebihi laju difusi obat dengan moderat
untuk kelarutan lemak rendah (misalnya, acetaminophen), sehingga menurunkan
rasio air liur/plasma. Selain itu, pH air liur dirangsang cenderung mendekati7.4,
sehingga menghilangkan ketimpangan distribusi obat berdasarkan pH (misalnya,
quinidine). Dengan beberapa asam lemah, kedua pengaruh konsentrasi obat
mungkin cenderung membatalkan satu sama lain (misalnya, Sulfamerazin).
Volume Distribusi
Ini harus jelas dari pembahasan sebelumnya bahwa obat tidak terdistribusi
secara merata ke seluruh tubuh. Meskipun zat lipofilik cenderung menembus
semua kompartemen jaringan (asalkan mereka memiliki jumlah sedikit kelarutan
air), senyawa hidrofilik sering disebarkan lebih ketat. Volume distribusi (Vd)
adalah indikator yang berguna tentang bagaimana obat tersebar di antara tubuh
berbagai kompartemen. Dalam bentuk yang paling sederhana, Vd dihitung dari
persamaan Vd = Q/C, di mana Q adalah jumlah obat yang diberikan dan C adalah
konsentrasi plasma obat pada kesetimbangan. Oleh karena itu Vd adalah jumlah

air dimana dosis tertentu harus diencerkan untuk menghasilkan konsentrasi
plasma yang diberikan, asumsi bahwa tidak ada obat yang telah hilang melalui
penyerapan lengkap atau metabolisme atau ekskresi.
Evans pewarna biru khas dari beberapa obat yang didistribusikan hanya
dalam ruang vaskuler. Beberapa menit setelah injeksi intravena, Evans biru
seluruhnya dicampur dalam darah, dan Vd dari 3 L diperoleh. Nilai ini merupakan
total volume plasma seorang pria 70-kg rata-rata. Kebanyakan senyawa, namun,
lolos dengan mudah dari pohon vaskular ke dalam kompartemen interstitial. Pada
kesetimbangan, obat ini didistribusikan dalam volume ekstraseluler dari ½ L,
yang meliputi pembuluh darah dan cairan interstitial. Obat ionik (misalnya,
aminoglikosida) umumnya terkandung dalam Vd ini. Molekul yang dapat dengan
bebas menembus semua membran yang diencerkan dengan air seluruh tubuh,
sekitar 41 L. Gambar 2-6 menggambarkan volume cairan tubuh utama, dan Tabel
2-2 menyediakan daftar dari agen-agen dengan perwakilan nilai Vd.
Hal ini terlihat dari Tabel 2-2 bahwa Vddari banyak senyawa tidak sesuai
dengan setiap kompartemen cairan anatomi yang bisa didefinisikan. Menerima
bahwa pengukuran dilakukan dengan benar dan bahwa masalah dalam penyerapan
obat dan eliminasi berhasil dihindari, beberapa penjelasan tetap untuk hasil ini.
Persamaan Vd sebenarnya hanya menyediakan distribusi yang nampak, sebagian
karena menganggap bahwa obat telah disebar merata. Untuk menggambarkan hal
ini, Na+ hadir dalam semua cairan tubuh (dengan Vd sebenarnya 41 L), tapi jelas
(dihitung) Vd untuk Na+ hanya 18 L. Perbedaan ini timbul karena Na+ secara
aktif tapi tidak sempurna diekstrusi dari air intraseluler. Ketidakmiripan antara
volume yang benar dan kalkulasi distribusi berdasarkan konsentrasi kompartemen
yang tidak sama muncul setiap kali ion didistribusikan lintas

GAMBAR 2-½.Oksidasi mikrosomal.Obat bebas memasuki siklus (kanan atas) dan dikomplekskan dalam.Kehadiran fosfatidilkolin (PC) ke CYP dengan heme dalam teroksidasi (Fe +++) negara.The Fe +++ adalah berkurang (Fe ++) oleh elektron (e) yang dihasilkan dalam oksidasi NADPH ke NADP + oleh enzim NADPH-sitokrom P450 oksidoreduktase (CYP reduktase, kiri atas).Mengurangi menyerap kompleks molekul oksigen (O2, menengah bawah). Selain dari e kedua dan dua proton (2H +, kanan bawah) hasil dalam generasi satu molekul air (H2O), oksidasi obat (hidroksilasi dalam kasus ini),dan oksidasi Fe ++ untuk Fe +++. Siklus ini lengkap dengan pelepasan obat teroksidasi. (Diadaptasi dari Markey SP: Persiapan metabolisme obat. Dalam Atkinson AJ Jr, Abernethy DR, Daniels CE et al, editor:Prinsip farmakologi klinis, ed 2, Amsterdam, 2007, Elsevier.)
Oksidasi obat dapat saja mengarah untuk beberapa turunan yang berbeda. Oksigen mungkin dimasukkan dalam bentuk alkohol, aldehida, epoxide, keton, atau asam karboksilat sedemikian struktur sebagai residu alifatik , cincin aromatik gugus amino dan belerang moieties. Oksigen juga mungkin menggantikan seorang atom sulfur ( desulfuration ) atau gugus amino ( deaminasi ) atau mungkin tidak muncul dalam data metabolite sama sekali melainkan menjadi melekat pada sebuah hidrokarbon unit dirilis selama yang dealkylation dari nitrogen, oksigen atau belerang.

ReductionReduksi microsomal obat-obatan terbatas pada molekul dengan nitro atau carbonyl kelompok atau hubungan azo.Reaksi yang sama juga bisa diolah oleh enzim nonmicrosomal di dalam tubuh , akan tetapi kebanyakan berbagai pengurangan ini tampaknya hasil utama dari aksi bakteri enteric .Ketika terjadi reduksi di salah satu bagian di sebuah molekul , biasanya membutuhkan waktu oksidasi di tempat lain , dan produk final lebih polar meskipun ditambahkan atom hidrogen.
HydrolysisHidrolisis ester atau senyawa amida menyebabkan dua entitas yang lebih kecil , memiliki kutub di akhir setiap bagian, kadang dapat juga tergantung dari microsomal enzim .Hidrolisis dari meperidine ester dan pembelahan amida anestesi lokal dan metabolit oksidasinya adalah dua contoh penting dari microsomal hidrolisis. Epoxide hydrolase , bertanggung jawab atas biotransformasi yang sangat reaktif dan terbentuknya toksik selama reaksi oksidasi microsomal , dengan menghasilkan produk dihydrodiol tidak aktif .

DehalogenasiBerbagai senyawa, seperti chlorophenothane dan beberapa anestesi umum (uap), didehalogenasi oleh microsomal enzim.Reaksinya sangatlah kompleks, melibatkan kedua langkah oksidatif dan reductive sekaligus, dan dapat mengakibatkan pembentukan potensi metabolit toksik.
Konjugasi GlucoronideKombinasi senyawa dengan glucuronic asam adalah satu-satunya tahap II dari reaksi dikatalisis oleh enzim microsomal (dalam hal ini, dikarenakan dari sekelompok glucuronosyltransferases).Berasal dari glukosa, asam glucuronic ditransfer dari pendonor, diphosphate uridine, yang sesuai dengan reaksi pusat pada molekul obat.Hasil olahan konjugasi glucuronide aktif disekresikan melalui urin atau empedu.Berbeda dengan beberapa tahapan reaksi sebelumnya, konjugasi glucuronic asam hampir selalu mengakibatkan kerugian pada aktivitas farmakologis.Pengecualian terhadap morphine-6-glucuronide, yang 100 kali lebih ampuh sebagai analgesik daripada morfin saat disuntikkan ke dalam beberapa susunan saraf pusat. Beberapa glucuronides diekskresikan dalam empedu pada proses hidrolisis oleh bakteri usus dan β-glucuronidase enzim. Jika ini memiliki kelarutan lemak yang cukup , pelepasan obat dapat diserap kembali. Glucuronidation adalah sebuah jalur metabolisme yang signifikan secara kuantitas dari banyak obat dan metabolitnya, agen seperti morfin , ini merupakan metabolisme dasar.
Metabolisme NonmikrosomalPola metabolisme obat yang dimediasi oleh nonmicrosomal enzim berbeda
dari yang microsomal sistem.Meskipun vital, hati tidak selalu dominan dalam biotransformasi nonmikrosomal. Berbagai jenis utama reaksi nonsynthetic menggambarkan reaksi apa yang terjadi , tetapi relatif frekuensi yang terjadi berbeda. Umumnya, obat-obatan harus menyerupai substrat alam untuk dimetabolisme oleh nonmicrosomal enzim; yang menunjukkan kekhususan yang lebih microsomal oksidasi tidak ada di sini. Meskipun enzim cytosolic yang paling sering terlibat , enzim yang memiliki inti , mitokondria , membran plasma ini juga memiliki perananan yang terbatas. Plasma esterase adalah sebuah contoh penting dari ekstraselular sebuah enzim yang terlibat dalam metabolisme obat.
OksidasiNonmicrosomal enzim bertanggung jawab untuk oksidasi dari banyak macam senyawa.Dipilih alkohol dan aldehida yang teroksidasi oleh dehydrogenases di sitosol dalam hati. Reaksi oksidasi lain termasuk seperti deaminasi oksidatif obat seperti tyramine dan phenylephrine oleh enzim mitochondrial ditemukan di hati ,

ginjal , dan organ lainnya dari turunan purina hydroxylation dan teofilin allopurinol oleh xanthine oxidase.
Reduksi Enzim nonmicrosomal melipatgandakan hydrogenation dua kali lipat, melalui pembalikan dehydrogenase jalur normal , penghapusan atom oksigen. Pengurangan kloral hidrat untuk trichloroethanol oleh alkohol dehydrogenase adalah sebuah contoh hal yang sering disebut sebagai jenis reaksi terakhir.
HidrolysisKebanyakan reaksi hidrolisis zat asing tergantung pada esterase
mikrosomal non dan enzim amidase.Nonspesifik esterase yang ditemukan di seluruh tubuh, tetapi dua situs yang paling penting, berdasarkan kapasitas hidrolitik dan ketersediaan obat-obatan, adalah hati dan plasma.Ester anestesi lokal seperti prokain dan benzocaine yang dihidrolisis oleh enzim tersebut.Kecuali darah dan peptidase jaringan lainnya bertanggung jawab atas kerusakan farmakologipolipeptida aktif, sebagian besar kegiatan amidase berada di hati.
Reaksi KonjugasiSejumlah reaksi sintetik dikatalisis oleh non mikrosomalenzim transferase.
Seperti dengan sintesis mikrosomaldari glucuronides, tubuh biasanya memasok bagian asam(Misalnya, sulfat, asetat, sistein, glisin, glutamin, atau Ribosidefosfat) yang melekat pada kofaktor atau pembawa molekul tertentu. Penambahan kelompok metil untuk fenol, merkaptan,dan amina dapat menyebabkan senyawa kurang polar, tapi bahkan di sini oksidasi atau konjugasi reaksi selanjutnya menurunkan lipidkelarutan.

Dengan amina, metilasi dapat meningkatkan polaritas, sepertidalam pembentukan amonium kuaterner kation.Itukontribusi kuantitatif dari berbagai tahap II reaksidiilustrasikan pada Gambar2-10.
Konjugasi dengan glutation tidak biasa karenaditujukan terhadap metabolit yang sangat reaktif, seperti epoksida dan kuinon, dan dapat terjadi dengan atau tanpa dukungan enzimatik.Meskipun jalur kuantitatif kecil, glutathione konjugasi sering major pentingnya dalam mencegahmetabolisme yang disebabkan keracunan obat.Tahap II reaksi dapat diharapkan setiap kali obat membawa satu atau lebih dari pusat reaktif yang tercantum dalam Tabel 2-5. Demikiankonjugasi umumnya mengakibatkan penghentian efek obat,pembatasan dalam jelas Vd, dan percepatan ekskresi obatmelalui proses sekresi aktif.
Metabolisme Nonhepatik
Meskipun berfokus pada hati ketika mempertimbangkan biotransformasi
secara umum adalah sesuai, jaringan lain juga mengandung enzim metabolisme-
obat, termasuk anggota keluarga CYP, dan berkontribusi terhadap mikrosomal
dan metabolisme nonmicrosomal dari obat-obatan. Kemampuan ini kadang-

kadang diambil dari keuntungan dengan mempersiapkan prodrugs yang menjadi
metabolik diaktifkan dalam jaringan target. Penggunaan levodopa untuk
menghindari penghalang darah-otak adalah contoh dari pendekatan ini;
administrasi asiklovir, sebuah prodrug antivirus yang dikonversi ke bentuk
nukleotida aktif dalam sel yang sakit (lihat Bab 40), adalah hal lain. Berdasarkan
lokasi dan suplai darah, organ-organ tertentu memainkan peran khusus dalam
metabolisme obat. Seperti dijelaskan sebelumnya dalam konteks bioavailabilitas,
usus, bekerja sendiri atau bersama dengan hati, dapat memetabolisme beberapa
obat sehingga benar-benar lengkap bahwa rute oral tidak dapat digunakan untuk
administrasi mereka. CYP3A4 adalah utama enzim yang terlibat. Ginjal cocok
untuk metabolisme obat karena memiliki sistem enzim mikrosomal yang
berkembang dengan baik dan menerima suplai darah berlimpah. Glucuronidation
merupakan kegiatan sangat menonjol.
Dalam beberapa tahun terakhir, peran paru-paru pada disposisi obat telah
menjadi area aktif penyelidikan. Dengan cara sirkulasi paru, hampir semua darah
mengekspos jaringan paru-paru dengan masing-masing sirkulasi. Penelitian telah
menunjukkan bahwa paru-paru adalah situs utama untuk metabolisme dari
senyawa endogen melalui darah seperti bradikinin, angiotensin I, prostaglandin,
dan amina biogenik. Perannya dalam biotransformasi senyawa eksogenus murni
adalah dikurangi karena hati memiliki konten tinggi enzim metabolisme obat.
Namun, alasan ini gagal untuk memperhitungkan pengaruh penting dari aliran
darah atau pemberian obat pada metabolisme beberapa obat. Jadi sementara
aktivitas arylhydrocarbon hidroksilase dalam hati lebih dari 1000 kali dari paru-
paru, metabolisme paru dari benzo[a]pyrene oleh enzim in vivo ini dapat
mendekati atau bahkan melebihi laju hati.
Faktor-Faktor yang Mempengaruhi Metabolisme Obat
Tingkat biotransformasi obat tergantung pada banyaknya variabel. Ini
termasuk akses ke situs metabolisme, konsentrasi dan fenotip dari enzim yang
ada, dan efek dari agen tertentu pada aktivitas enzimatik. Karena sebagian besar
obat dimetabolisme di hati, perhatian dipusatkan pada faktor-faktor yang
mempengaruhi biotransformasi obat hati.

Masuk ke dalam hati
Seperti yang dinyatakan sebelumnya, ikatan protein plasma secara
signifikan dapat mengurangi tingkat penyerapan dan metabolisme obat oleh hati.
Korelasi terbalik antara tingkat biotransformasi dan tingkat protein mengikat telah
dilaporkan untuk sulfonamides, warfarin, dan fenitoin, diantaranya. Hubungan
serupa juga memegang, untuk obat terikat reservoirs ekstravaskuler. Untuk
beberapa senyawa, ikatan protein plasma yang tidak menghalangi metabolisme
dan bahkan dapat mempertinggi itu. Lidocaine dan propranolol begitu efektif
diserap oleh jaringan hati, bahkan dengan mengikat signifikan, jarak obat ini dari
tubuh terutama dibatasi oleh aliran darah hati. Karena protein mengikat tetap obat
ekstra dengan kompartemen vaskular, banyak dihadirkan ke hati 1 unit waktu
untuk metabolisme.
Beberapa kondisi penyakit dan interaksi obat dapat mempengaruhi
aksesibilitas enzim hati untuk agen farmakologis. Uremia, dengan mengurangi
kapasitas pengikatan albumin, mempromosikan biotransformasi beberapa ikatan
yang sangat tinggi, Karena peradangan dan stres meningkatkan konsentrasi
plasma dari asam α1-glikoprotein, efek sebaliknya dapat terjadi dengan beberapa
obat dasar. Kerusakan hepatik dapat mempengaruhi pengiriman ke hati dalam
beberapa cara. Misalnya, mungkin mengurangi konsentrasi ikatan protein plasma
dan diubah. Penurunan metabolisme bilirubin dan substrat lainnya mungkin juga
mengubah distribusi obat dan ketersediaannya untuk penyerapan hati. Masuk ke
hati lebih lanjut dapat diperlambat oleh penurunan transportasi dimediasi-
pembawa. Sirosis akhir, insufisiensi jantung, dan kondisi lainnya dapat
mengurangi aliran darah hati secara signifikan dapat memperlambat metabolisme
dari lidocaine dan agen seperti agen yang mempunyai biotransformasi biasanya
dibatasi oleh tingkat pengiriman obat ke hati.
Penghambatan enzim
Enzim memetabolisme-obat tunduk pada antagonisme kompetitif daripada
non kompetitif. Karena begitu banyak obat yang bertindak oleh sistem CYP,
penghambatan kompetitif dari oksidasi mikroskop mudah dibuktikan di

laboratorium. Untungnya interaksi obat jenis ini biasanya tidak secara klinis
penting. Dalam banyak kasus, tingkat biotransformasi terbatas bukan oleh rantai
transpor elektron CYP tapi oleh pergerakan obat ke dalam endoplasma reticulum
halus. Beberapa senyawa, namun, menunjukkan kinetika jenuh dan dibatasi dalam
metabolisme oleh laju mengikat untuk enzim C tertentu. Kompetisi yang
melibatkan agen ini (misalnya, phenytonin, dan dicumarol bersaing untuk
CYP2C9) adalah sangat penting.
Berbagai racun metabolik – karbonmonoksida, sianida, logam berat -
nonkompetitif menghambat biotransformasi mikroson. Tindakan ini hanya dari
kepentingan percobaan, meskipun, karena efek pada respirasi dan proses lainnya
diutamakan in vivo. Yang jauh lebih spesifik dalam inhibisi oksidasi mikrosomal
dicapai dengan proadif yang rajin mengikat besi heme dari CYP.Senyawa ini
memblokir metabolisme sejumlah besar agen tergantung pada enzim CYP; juga
dapat menghambat glucuronidation. Efek pada kebanyakan obat adalah
perpanjangan aksi, tetapi senyawa membutuhkan aktivasi mikrosomal mungkin
memiliki potensi kerugian. Banyaknya zat yang dipengaruhi oleh proadifen
menghambat penggunaannya pada manusia; namun, senyawa yang mirip
menemukan aplikasi sebagai potensiator insektisida yang diaktifkan oleh
mikrosomal biotransformasi. Penggunaan obat-obatan klinis yang menghambat
metabolisme berbagai usia lainnya dengan menonaktifkan berbagai enzim CYP
termasuk antibiotik macrol (selain azitromisin), chloramphenic turunan imidazol
tertentu (simetidin dan antijamur azole), dan amiodaron (lihat Tabel 2-3). Obat-
obat ini - metabolit mereka -bereaksi kovalen atau sebaliknya sangat kuat dengan
situs-situs tertentu pada molekul CYP. St. John’s wort dan jus buah anggar adalah
herbal dan makanan pokok, masing-masing sangat kuat menghambat kelas
tertentu enzim CYP.
Beberapa obat yang digunakan khusus sebagai inhibitor selektif dari enzim
nonmicrosomal. Ketika enzim terpapar terjadi, untuk bertanggung jawab untuk
inaktivasi dari agen terapetik lainnya, interaksi obat yang mungkin untuk
mengembang. Contoh enzim tersebut monoamine oxidase, pseudocholinesten dan
xantin oksidase. Penghambatan aldehida dehydrogenase oleh disulfiram adalah

pengecualian karena indikasi utama obat adalah untuk mengganggu metabolisme
senyawa asing lainnya, etanol (lihat Bab 43).
Induksi Enzim
Enzim metabolisme-obat mikrosomal adalah bisa diinduksi; di bawah
stimulus kimia yang tepat, aktivitas katalitik akan meningkat. Banyak bahan
kimia, termasuk agen terapeutik, obat"sosial", dan racun lingkungan, mampu
merangsang biotransformasi mereka sendiri dan senyawa terkait erat. Selain itu,
beberapa bahan kimia dapat meningkatkan pemecahan berbagai macam zat yang
beragam. Phenobarbital menggambarkan jenis induksi berikutnya. Saat mencapai
sitosol hepatosit, fenobarbital mengikat faktor transkripsi disebut constitutive
androstane receptor, yang kemudian bermigrasi ke inti untuk mengaktifkan gen
dengan elemen respons yang tepat. Beberapa jam setelah itu, ketinggian dalam
sintesis protein hati menjadi jelas. Penurunan metabolisme paruh obat yang
terkena dampak disejajarkan dengan peningkatan berat mikrosomal dan dalam
konsentrasi NADPH-sitokrom P450 reduktase dan beberapa enzim CYP (paling
penting CYP2B6, 2C8/9, 2C18 /19, dan 3A4/5). Hati akhirnya hipertropi, dan
darah hati mengalir dan sekresi empedu juga ditingkatkan. Rifampisin, inducer
spektrum luas lain, mengikat protein terkait erat disebut pregnane X
receptoruntuk memulai respons yang sama.
Dengan cara sebaliknya, benzo[a]pyrene mencontohkan agen dengan bentuk
yang lebih ketat dari induksi. Meskipun benzo[a]pyrene memerlukan
pembentukan enzim baru untuk stimulasi metabolismenya (inhibitor sintesis
protein memblokir aksinya), perubahan struktural dalam retikulum endoplasma
halus tidak menonjol dan mungkin tidak terdeteksi. Induksi enzim dalam hal ini
terutama melibatkan famili gen CYP1 (CYP1A1/2 dan CYP1B1). Faktor
transkripsi untuk benzo[a]pyrene dan banyak hidrokarbon aromatik lainnya dan
heterocyclics adalah reseptor aril hidrokarbon.
Tidak peduli pola induksi, tingkat metabolismedari senyawa yang terkena
dapat mempertinggi eksperimen sebanyak tujuh kali baseline. Stimulasi biasanya
kurang ditegaskan secara klinis; Namun demikian, induksi enzim memiliki
banyak konsekuensi terapi yang penting. Hal ini, misalnya, penyebab utama

interaksi obat. Sebuah contoh klasik dari bentuk interaksi obat adalah stimulasi
dengan fenobarbital dari metabolisme dicumarol antikoagulan, yang
menyebabkan dosis standar antikoagulan untuk menjadi tidak efektif. Induksi
enzim mikrosomal menyebabkan hilangnya respon farmakologis disebut toleransi
farmakokinetik. Akhirnya, induksi enzim dapat mempengaruhi fungsi bahan
kimia endogen dimetabolisme secara microsomal. Akselerasi dari oksidasi
vitamin D untuk menghasilkan produk-produk yang tidak aktif adalah penyebab
utama dari rakhitis dan osteomalacia pada pasien epilepsi yang menerima obat-
obatan seperti fenitoin dan phenobarbital.
Tampaknya hasil jelas bahwa induksi enzim harus mengurangi toksisitas
obat dalam perencanaan dengan pengurangan dalam potensi obat. Anehnya, hal
ini tidak selalu terjadi. Perhatian yang kuat di bidang toksikologi adalah potensi
bahaya yang ditimbulkan oleh zat-zat perantara yang sangat reaktif yang
dihasilkan selama oksidasi mikrosomal obat-obatan seperti acetaminophen,
halotan, dan benzo[a]pyrene.Zat ini biasanya disintesis dalam jumlah terbatas
sehingga reaksi berikutnya, termasuk: hidrolisis dan konjugasi glutathione,
menonaktifkan senyawa sebelum cedera selular dapat terjadi. Induksi Enzim
mikrosomal selektif, bagaimanapun, jadi dapat meningkatkan sintesis mereka
bahwa reaksi protektif selanjutnya menjadi kewalahan. Dalam perjanjian dengan
tesis ini adalah estimasi bahwa perokok yang menunjukkan induktibilitas tinggi
dari aktivitas arylhydrocarbon hidroksilase, yang mengubah benzo[a]pyrene dan
hidrocarbons polycyclic terkait ke intermediet epoksida, diperkirakan memiliki 36
kali lipat peningkatan risiko karsinoma bronkogenik daripada orang yang
memiliki induksibilitas rendah.
Faktor genetik
Individu bervariasi dalam kemampuan mereka untuk memetabolisme obat.
Meskipun perbedaan dapat hasil dari induksi lingkungan enzim mikrosomal
(seperti yang terlihat pada pekerja pabrik kimia dan perokok), studi yang
membandingkan kembar identik dan fraternal telah meyakinkan didirikan pada
pengaruh unggul hereditas pada tingkat biotransformasi. Untuk beberapa obat,
kisaran waktu paruh metabolik dapat melebihi urutan besaran, tetapi biasanya

angka ini terbatas pada nilai dua atau tiga. Kemampuan untuk memetabolisme
jenis senyawa tertentu pada tingkat normal biasanya tidak menunjukkan apa-apa
tentang biotransformasi zat yang tidak terkait. Namun, orang normal
menunjukkan tingkat metabolisme microsomal terendah adalah yang paling
mungkin untuk menjalani induksi enzim yang mendalam setelah perawatan
fenobarbital.
Pengaruh genetik pada metabolisme yang paling mudah dicirikan ketika gen
tunggal yang terlibat. Sebuah contoh yang baik dari prinsip ini disediakan oleh
pseudocholinesterase enzim plasma. Sekitar satu orang dari 3000 adalah
homozigot untuk - gen atipikal yang produknya enzim memetabolisme ester-
sangat lambat. Dosis konvensional dari succinylcholine relaksan otot
menghasilkan apnea yang berkepanjangan pada pasien ini. Mereka dengan
kombinasi gen yang khas dan atipikal (heterozigot) masih memiliki cukup enzim
normal menghidrolisis obat cukup cepat untuk menghindari manifestasi klinis
yang tidak biasa. Sejak penemuan gen atipikal untuk pseudokolinesterase,
genotipe baru lainnya telah dijelaskan, termasuk salah satu yang "diam" (produk
tidak memiliki aktivitas enzimatik apapun) dan salah satu yang menghasilkan
enzim sangat efektif dalam katalisis bahwa pasien dengan itu memperlihatkan
resistensi bawaan yang luar biasa untuk efek melumpuhkan succinylcholine.
Pharmacogenetics dari metabolisme obat dieksplorasi lebih lengkap dalam Bab 4.
Usia
Neonatus, terutama bayi prematur, sering kekurangan sistem metabolisme-
obat fungsional tertentu. Ketidakmampuan relatif terhadap konjugasi bilirubin
dengan asam glukuronat dan pengembangan yang dihasilkan dari
hiperbilirubinemia adalah contoh yang biasa terlihat dari kekurangan ini dalam
biotransformasi. Kegagalan untuk memperhitungkan perbedaan yang nyata secara
kuantitatif dalam metabolisme neonatal tragis disorot oleh "sindrom abu-abu" dan
kematian bayi yang berhubungan dengan chloramphenicol. Tidak seperti bayi
yang baru lahir, anak-anak sering lebih mahir memetabolisasi obat berdasarkan
berat daripada orang dewasa muda. Setelah itu, kapasitas biotransformasi muncul

berkurang bersama dengan usia, orang tua mungkin sering menunjukkan tingkat
terlambat dalam metabolisme obat.
Patologi
Jelas, kerusakan yang signifikan pada parenkim hati dengan hilangnya
enzim metabolisme-obat dapat langsung menekan biotransformasi banyak agen.
Efek klinis, bagaimanapun, mungkin cukup kecil karena kapasitas cadangan
metabolisme hati dan karena induksi enzim dalam jaringan tidak terpengaruh.
(Lihat Bab 3 untuk pembahasan lebih lanjut dari disfungsi hati dan respon pasien)
Efek lebih halus dari patologi dicontohkan oleh pengaruh infeksi pada
metabolism hati.Sakit virus telah dikaitkan dengan depresi aktivitas CYP dan
oksidasi mikrosomal teofilin dan beberapa
obat-obatan. Interferon diproduksi dalam menanggapi penyakit-penyakit ini
atauvaksin dipersiapkan dari virion yang terganggu dapat benar-benar
menyebabkan penghambatan. Beberapa infeksi nonviral, seperti malaria, kusta,
dan berbagai bentuk pneumonia, juga telah dikaitkan dengan gangguan
biotransformasi obat.
Akhirnya, gangguan endokrin dapat mengubah metabolisme obat.
Hypothyroidism dapat memperlambat biotransformasi obat-obatan tertentu;
hipertiroidisme cenderung memiliki efek sebaliknya. Pada hewan, gangguan
kelenjar hipofisis, adrenal korteks, dan gonad telah terbukti mempengaruhi
metabolisme obat; apakah efek yang sama terjadi pada manusia tidak diketahui.
EKSKRESI
Zat-zat asing, termasuk obat-obatan terapi, dicegah dari membangun dalam
tubuh dengan aksi gabungan metabolisme dan ekskresi. Obat-obatan dan
metabolites mereka dapat dihilangkan dengan sejumlah rute: urin, empedu,
keringat, air liur dan sekresi gastrointestinal lainnya, pernafasan paru, air mata,
dan air susu ibu.Pertimbangan kuantitatif membuat ginjal organ utama ekskresi
obat.
Ekskresi ginjal

Tiga proses filtrasi glomerular, penyerapan tubular, dan transportasi aktif -
mengontrol eliminasi urin dari obat. Meskipun semua obat tunduk pada filtrasi,
persentase disaring berbanding terbalik sesuai dengan tingkat mengikat plasma
protein dan ke Vd. Setelah disaring, agen cenderung diserap dalam kaitannya
dengan koefisien partisi lipid/air mereka. Pertimbangan ini mendukung ekskresi
ginjal dari senyawa yang sangat polar, tetapi tingkat yang tepat dari eliminasi juga
tergantung pada apakah transpor aktif masuk (atau, jarang, dari) cairan tubular
juga terjadi.
Filtrasi glomerulus
Setiap hari ginjal menyaring sekitar 180 L plasma. Darah arteri memasuki
kapsul Bowman disalurkan melalui seberkas kapiler secara kolektif disebut
sebagai glomerulus. Kapiler ini unik dimodifikasi untuk filtrasi, memiliki
sejumlah besar pori-pori dengan diameter efektif untuk 80 A menembus melalui
endotel. Sekitar seperlima dari plasma masuk aparat glomerular sebenarnya
disaring, sisanya keluar dengan cara arterioles eferen untuk memasok bagian-
bagian lain dari nefron. Secara umum, molekul kecil dari albumin (berat molekul
69.000) muncul dalam cairan tubulus. Karena protein plasma hampir sepenuhnya
dipertahankan dalam aliran darah, obat terikat tidak dikenakan filtrasi.
Reabsorpsi tubular
Karena hanya sekitar 1,5 L urin sebenarnya dikeluarkan setiap 24 jam
(kurang dari 1% dari beban disaring setiap hari), ginjal harus memiliki sistem
reabsorpsi efisien. Memang, adalah hal ini tidak terjadi, seseorang akan
kehilangan cairan dan nutrisi yang berharga dan cepat mati. Sekitar 80% filtrat
glomerular yang direklamasi oleh tubulus proksimal konvulated. Sebuah pompa
berkapasitas tinggi aktif mengangkut Na+ kembali ke dalam aliran darah, dengan
anion (terutama Cl-) dan air mengikuti secara pasif. Proses ini berlanjut sepanjang
nefron dan dibantu oleh sistem transportasi tertentu, seperti dalam lingkaran
menaik dari Henle, di mana transportasi penyerta dari Na+, K+, dan Cl- terjadi.
Selain itu, reabsorpsi Na+ dibantu oleh pertukaran dengan H+ dan, dalam tubulus
konvulated distal, KK. Konsentrasi yang dihasilkan dari cairan tubular

menciptakan gradien kimia untuk difusi obat kembali ke dalam sirkulasi sistemik.
Agen dengan koefisien partisi lipid/air yang menguntungkan maka mudah
melintasi epithelium tubular dan melarikan diri dari urin.
Faktor utama yang mempengaruhi reabsorpsi elektrolit lemah dari cairan
tubulus ginjal adalah pH. Tergantung pada tingkat sekresi H+, pH urin dapat
bervariasi 4.5-8.Asam lemah seperti aspirin dan fenobarbital yang menyerap lebih
efektif di bawah kondisi asam; sebaliknya adalah basa lemah seperti amphetamine
dan ephedrine.Kadang-kadang, pengaruh pH pada ekskresi obat digunakan untuk
keunggulan klinis.Jadi strategi umum dalam menghadapi toksitas aspirin adalah
untuk mempromosikan penghapusan salisilat melalui alkalizati dari urin dengan
administrasi sistemik natrium bikarbonat. Untuk sulfonamid (juga asam lemah),
alkalisasi urin dapat mengurangi waktu paruh plasma sebesar 50% dan mencegah
pengembangan kristaluria dengan meningkatkan solubilitas air. Tentu saja, upaya
untuk meningkatkan ekskresi ginjal bernilai kecil untuk agen yang inaktivasi
tergantung sebagian besar pada biotransformasi.
Sekresi aktif
Banyak kation dan anion organik secara aktif mensekresi sel-sel tubulus
proksimal konvulated. Sistem transportasi anionik, yang bertanggung jawab untuk
sekresi amphipathic dan metabolit konjugasi (misalnya, glucuronides, sulfat
melibatkan transporter seperti P-glikoprotein dan resistensi multi-drugsterkait
jenis protein. Mekanisme transportasi kationik menggerakkan senyawa dengan
dibebani grup amino. Karena setiap transportasi pembawa agak tidak selektif,
persaingan untuk situs mengikat kadang-kadang diamati. Probenesid, anion
keasaman, telah digunakan untuk memblokir sekresi aktif asam lain, Penisilin G.
Ekskresi Penghambatan penisilin adalah menguntungkan pada saat obat itu
menunjukkan pasokan dan masih menemukan kegunaan ketika diperlukan untuk
mempertahankan konsentrasi tinggi antibiotik untuk waktu yang lama.
Sistem pembawa spesifik dari ginjal, ditemukan terutama dalam tubulus
distal konvulated, aktif menyerap agen tertentu. Resorpsi aktif yang paling
penting dari organik adalah dengan mekanisme ini melibatkan asam urat. Karena

probenesid bersaing dengan ion urateyang telah ada bersama dengan probenesid
penicillin menemukan aplikasi dalam gout sebagai promotor ekskresi asam urat.
Sekresi aktif dari zat-zat ke dalam urin adalah sebaliknya dipengaruhi oleh
ikatan protein plasma. Transportasi sering sangat efektif sehingga disosiasi obat
berlangsung instan membuat ketersediaan obat lebih banyak untuk sekresi,
sampai semua obat telah dibersihkan dari suplai darah lokal. Mengikat jaringan
ekstravaskular, bagaimanapun, tidak mengurangi tingkat eliminasi terlepas dari
mekanisme yang terlibat.
Pembersihan
Jumlah obat yang dihapus oleh ginjal per unit sering dievaluasi sebagai
fungsi dari air plasma pembersihan obat. Secara matematis, volume plasma
dibersihkan setiap menit (CL) dapat ditulis sebagai CL = U x V/P , di mana U
adalah konsentrasi urin dari obat, V adalah volume urin p: diproduksi per menit,
dan P adalah konsentrasi plasma. Pembersihan dan Vd terkait dengan rumus
sederhana CL=k Vd, di mana k, adalah penghapusan. konstanta laju. Agen-agen
yang disaring tetapi tidak diserap atau dikeluarkan, seperti inulin, menghasilkan
pembersihan dari 130 ml/menit (dengan asumsi tidak ada ikatan protein plasma)
dan berfungsi sebagai ukuran tingkat filtrati glomerulus. Dengan Vd dari ½ L, laju
pembersihan diterjemahkan menjadi waktu paruh plasma untuk inulin dari 64
menit. Aktivitas senyawa yang disekresikan ke dalam urin dan tidak diserap,
seperti pensilin G dan p-aminohippurate, mungkin mendekati pembersihan 650
ml/menit, yang merupakan laju aliran plasma melalui ginjal. Dengan asumsi Vd
dari ½ L, seperti obat memiliki waktu paruh plasma sekitar 13 menit. Sebaliknya,
obat-obatan yang sangat terikat dan tunduk pada pass reabsorpsi mungkin
menunjukkan laju pembersihan mendekati 0.
Ekskresi Empedu
Sejumlah kationik, anionik, dan molekul seperti steroidsecara selektif
dihapus dari darah untuk ekskresi dalam empedu dan akhirnya kotoran. Secara
umum, zat ini memiliki berat molekul melebihi 500 dalton. Proses transportasi
merupakan salah satu yang aktif di mana bahan terlarut ditransfer dari plasma ke

hepatosit dan kemudian ke empedu. Seperti dalam semua situasi semacam ini,
mekanisme yang membutuhkan energi khusus untuk transfer seperti itu ada.
Seperti yang telah disebutkan anion organik mengangkut famili polipeptida dari
transporter termasuk anggota yang memfasilitasi penyerapan ke dalam bahan
kimia sitoplasma hati yang beragam seperti asam empedu, digoxin, dan
methotrexate. Sistem operator lainada untuk molekul kationik tapi belum ditandai.
P-glycoprotein dan multidrug resistance-associated protein tipe 2 transporter
penting untuk memindahkan obat dari sel hati ke empedu. Protein ini juga terlibat
dalam transfer metabolit asam yang dihasilkan oleh CYP3A hati dan reaksi fase II
ke dalam empedu, plasma, atau keduanya.
Ekskresi bilier bertanggung jawab untuk semua tapi sebagian kecil dari
penghapusan kotoran obat. Sisanya dihasilkan dari bagian transmucosal langsung
ke saluran pencernaan dari aliran darah atau merupakan senyawa dilarutkan dalam
satu atau lebih sekresi saluran pencernaan. Tentu saja, kotoran juga mengandung
variabel jumlah obat yang tidak diserap juga. Reabsorpsi molekul diekskresikan
melalui empedu dapat terjadi, seperti dengan fenolftalein obat pencahar. Entero-
hepatic daur ulang tersebut dapat memperpanjang durasi kerja dan akan terus tak
terhingga sampai sistem terganggu (misalnya, oleh metabolisme, pembatasan
aliran empedu, atau menelan obat chelator).
Rute lain dari Ekskresi
Ekskresi paru adalah rute utama untuk penghapusan gas dan beberapa
senyawa volatil. Kecuali untuk anestesi inhalasi, namun, ekskresi bahan kimia ke
dalam pohon pernafasan dapat menjadi perhatian lebih estetik dari kepentingan
farmakokinetik. Sebagai contoh, halitosis yang dihasilkan oleh agen bau-bauan
(misalnya, Paraldehid) dapat mengganggu kesesuaian klinis.
Penghapusan obat oleh ASI penting, bukan karena signifikansi kuantitatif, tetapi
karena itu merupakan potensi berbahaya untuk bayi menyusui. Obat-obatyang
mendapat perhatian khusus termasuk lithium, berbagai agen antikanker, dan
isoniazid. Variabel utama yang mempengaruhi bagian dari obat ke dalam susu
adalah kelarutan lipid.

Rute kecil lainnya dari ekskresi termasuk; keringat, air mata; air liur; dan
lambung, pankreas, dan sekresi usus. Dalam semua kasus, ekskresi dibatasi oleh
koefisien partisi lipid/air. Untuk air liur dan cairan pencernaan yang terkait, obat
disetorkan ke saluran pencernaan setelah sekresi dan tersedia untuk reabsorpsi ke
sirkulasi sistemik.
WAKTU JALAN DARI AKSI OBAT
Hubungan erat antara konsentrasi plasma dari agen dan besarnya efek telah
ditekankan. Karena administrasi obatbiasanya mencakup midrange linear dari
kurva log dosis-respons, hubungan antara plasma dan titer reaksi pasien sering
mudah. Jadi deskripsi temporal konsentrasi obat atas dasar prinsip-prinsip
farmakokinetik berguna dalam menggambarkan bagaimana penyerapan,
distribusi, metabolisme, danekskresi mempengaruhi efek obat dalam
penyelenggaran dan memberikan bimbingan untuk menyesuaikan jadwal dosis
untuk mencapai hasil-hasil terapetik dengan toksisitas obat minimal.
Kinetika dari Absorpsi dan Penghapusan
Kebanyakan peristiwa biologis yang melibatkan nasib obat dapat
digambarkan secara sederhana kinetik: zero-order, first order, atau kapasitas
terbatas (kombinasi dari keduanya).
Kinetika Zero-order
Kinetika zero-ordermendefinisikan proses yang terjadi dengan laju yang
konstan per unit waktu. Secara matematis ini dapat ditulis sebagai dC/dt = k0,
dimana dC/dt adalah laju perubahan konsentrasi dan k0 adalah konstanta dalam
satuan jumlah per waktu. Sebuah contoh yang baik dari absorpsi obat zero-order
adalah infus intravena kontinu di mana kuantitas senyawa memasuki aliran darah
setiap menit tetap konstan (misalnya, 5 mg/menit). Contoh lain dari absorpsi zero-
order disediakan oleh suntikan intramuskular atau subkutan bentuk depot obat.
Kelarutan air yang buruk dari persiapan memungkinkan tingkat pelepasan obat
konstan selama berjam-jam. Dengan pemberian oral, pada dasarnya penyerapan
zero-order menyadari setiap kali, gerbang-faktor pembatas adalah pembubaran

partikel obat utama. Akhirnya, terapi topikal sering menyebabkan penyerapan
zero-order. Selama agen adalah lebih besar, jumlah yang relatif tetap obat
menembus kulit per unit waktu.
Kinetika First-order
Kinetika first-order berhubungan dengan peristiwa yang terjadi pada tingkat
pecahan konstan per unit waktu (misalnya, 5%/menit). Berikut dC/dt = k1C,
dengan k1 mewakili fraksi tingkat konstan dalam satuan waktu-dan C mewakili
konsentrasi obat. Penyerapan, distribusi, dan eliminasi senyawa umumnya
menunjukkan jenis kinetika karena mereka umumnya mengandalkan proses
karakter first-order : difusi pasif, filtrasi, aliran darah, atau transportasi obat atau
metabolisme beroperasi di bawah saturasi. Karena fraksi obat yang terkena per
unit waktu adalah independen dari konsentrasi, mengacu pada laju reaksi dengan
waktu paruhnya (t½) , periode yang diperlukan untuk proses untuk mencapai 50%
penyelesaian, sering berguna. Waktu paruh adalah terkait dengan pecahan laju
konstan oleh rumus t½= 0.693/k1. Dengan demikian semakin besar tingkat
konstan, semakin pendek waktu paruh dan lebih cepat reaksi. Hal ini mudah
menunjukkan bahwa proses first-order pada dasarnya lengkap (94%) setelah
empat waktu paruh hidup. Gambar 2-10 memberikan contoh penghapusan first-
order dari obat dengan t½ dari 2 jam, dan Tabel 2-6 mendaftar eliminasi waktu
paruh hidup beberapa kategori obat umum digunakan dalam kedokteran gigi.
Reaksi kapasitas terbatas
Reaksi kapasitas terbatas melibatkan enzim yang bertanggung jawab untuk
metabolisme obat dan molekul pembawa berkaitan dengan transportasi obat
melintasi membran. Proses semacam ini awalnya menampilkan kinetika zero-
order ketika faktor endogen (enzim atau carrier) yang jenuh dengan obat; secara
bertahap mengambil fitur reaksi first-order saat konsentrasi obat jatuh. Seperti
telah disebutkan, dosis yang digunakan secara klinis biasanya di bawah yang
dibutuhkan untuk saturasi. Beberapa pengecualian ada, namun, di mana kinetika
saturasi yang jelas. Alkohol, bahkan dalam dosis cukup memabukkan,
dimetabolisme dengan laju konstan sekitar 8 g/jam. Hanya ketika konsentrasi

jatuh jauh di bawah memproduksi efek yang bisa diamati apakah dehidrogenasi
alkohol mengasumsikan tingkat first order.
GAMBAR 2-13 Pertama-order penghapusan obat yang diberikan sebagai bolus
intravena. Dalam contoh ini plasma kurva konsentrasi-waktu, diasumsikan bahwa
tubuh berperilaku sebagai kompartemen tunggal dan bahwadistribusi obat pada
dasarnya seketika. A, konsentrasi plasma diplot pada aritmatikaskala. B, A skala
logaritmik digunakan untuk menghasilkan garis lurus. Penghapusan paruh (t ½)
Ditentukanoleh interval waktu yang diperlukan (2 jam dalam kasus ini) untuk
konsentrasi plasma menurun50%. Cp o menunjukkan konsentrasi interpolasi obat
segera setelah penyuntikan obat.
Contoh lain yang penting dari kapasitas terbatas biotransformasi melibatkan
aspirin. Aspirin cepat deasetilasi menjadi salisilat, anion bertanggung jawab untuk
banyak kegiatan farmakologis obat. Salisilat tersebut kemudian dihilangkan
melalui beberapa jalur metabolisme dan ekskresi oleh ginjal, menghasilkan
eliminasi paruh keseluruhan sekitar 3 jam. Beberapa rute inaktivasi, meskipun,
mudah jenuh, sehingga ketika overdosis tertelan masalah toksisitas diperparah
oleh kerugian relatif efisiensi eliminasi. Penghapusan paruh dapat dihitung untuk
obat menampilkan kinetika kapasitas terbatas,tetapi nilai-nilai yang diperoleh
bervariasi terus menerus sesuai dengankonsentrasi obat (lihat Tabel 2-6). Salisilat

memiliki plasmat ½ dari 20 jam saat konsentrasi yang sangat tinggi hadir dialiran
darah. Sebagai titer salisilat menurun keberbagai terapi, t eliminasi ½
menurun hingga konstandari 3 jam.
GAMBAR 2-14Model single-kompartemen kinetika obat.Penyerapan dalam dan
eliminasi dari tubuh masing-masing ditugaskantingkat konstan orde pertama
tunggal. Distribusi, diasumsikancepat mengenai penyerapan dan eliminasi, tidak
dianggap.
Model Single-CmpartmentSecara keseluruhan, disposisi tubuh dari obat yang diberikan melibatkan suatu
interaksi sementara kompleks biokimia dan proses fisiologis, masing-masing
dengan unik sendiriparameter kinetik, bahwa deskripsi kuantitatif penuhTentu
saja waktu kerja obat mungkin mustahil untuk dicapai. Untuktujuan praktis,
bagaimanapun, pemukiman banyak agen bisadijelaskan oleh model sistem yang
sederhana (Gambar 2-14) di mana tubuh digambarkan sebagai kompartemen
tunggal yang ukurannya bersesuaiandengan Vd dan yang eliminasi didasarkan
pada urutan pertamakinetika. Dalam model ini, yang mengasumsikan distribusi

yang cepatsehubungan dengan penyerapan dan eliminasi, hubunganantara
eliminasi t ½, jumlah izin tubuh (CL, atauVolume darah "dibersihkan" dari obat
per unit waktu olehproses gabungan metabolisme dan ekskresi), dan Vd adalah
langsung, sebagai berikut:
Yang tidak diketahui dari persamaan ini sebaiknya ditentukan
olehmenyuntikkan obat intravena (menghilangkan penyerapanvariabel) dan
mengukur konsentrasi plasma di regulerinterval yang cukup untuk membangun
plasma konsentrasi-waktukurva, seperti yang ditunjukkan pada Gambar 2-13.
Mengingat dosis awal500 mg (Q) dan konsentrasi plasma awal 6,4 mg / mL(Cpo),
ditentukan dengan ekstrapolasi konsentrasi plasmakurva kembali ke saat injeksi,
Vd sama Q Cpo, atausekitar 78 L. Dengan t ½dari 2 jam, pembukaan mendekati
27 L / jam atau 450 mL / menit.
Penghapusan t ½ yang tidak tergantung pada variabel independen dari dua
atribut, obt dan vd.Obat menunjukkan peningkatan jika tidak dalam 2, 1. Ini bisa
berarti mengikat jaringan obat yang lebih besar seperti daripada yang mudah
untuk menunjukkan penurunan tingkat metabolisme atau ekskresi dari agen.Sama,
pengurangan yang signifikan dalam vd. Penyakit yang dapat terjadi, hal ini dapat
mengurangi tidak ingin satu dari dua obat bahkan tidak akan terganggu.
Konsentrasi Plasma-Dosis Tunggal
Agen terapeutik sering diberikan dalam dosis tunggal oleh dokter gigi. Baik
obat-obatan seperti lidokain yang disuntikkan untuk anestesi regio, atropin untuk
mengendalikan air liur, atau midazolam untuk memberikan efek sedasi,
konsentrasi plasma yang meningkat hingga puncak selama tahapan haustoriumnya
dan kemudian menurun akhirnya ke nol, sehingga obat dihilangkan dari aliran
darah. Dengan menggunakan model single-compartment, hal ini memungkinkan
untuk membangun teori konsentrasi plasma kurva dan mengamati bagaimana
modifikasi dosis, penyerapan, atau bebasnya obat dapat mengubah konsentrasi
dan, mungkin, efek obat.Seperti terlihat pada gambar 2-15, konsentrasi plasma
adalah sama sekali kali langsung sebanding dengan dosis. Hubungan ini tidak ada
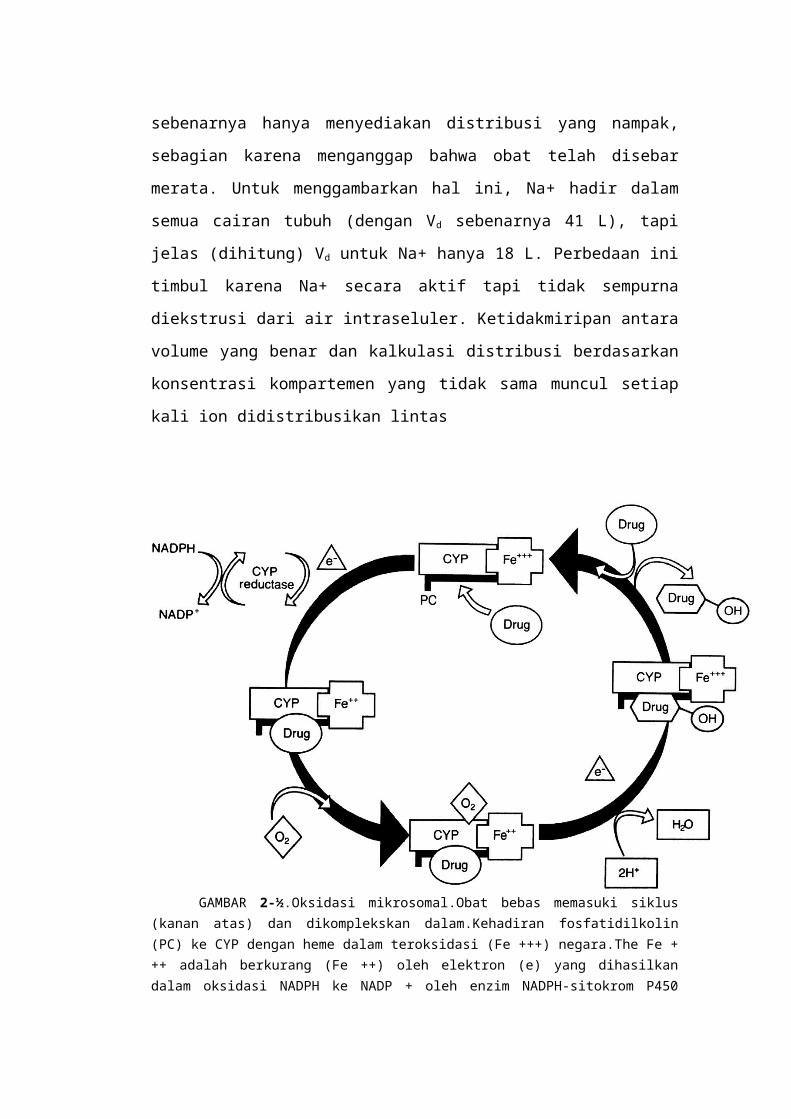
untuk agen yang kapasitas terbatas dalam penyerapan, mengikat, metabolisme,
atau ekskresi.
Selama penyerapannya beberapa kali lebih cepat dari sebelumnya ,
perubahan dalam tingkat penyerapan obat lain yang memiliki sedikit efek dari
konsentrasi untuk mengubah puncak.Durasi tindakan yang tidak terpengaruh sama
sekali.Muncul pola yang berbeda , akan tetapi , dalam kasus di mana tingkat
penyerapan anggaran yang mendekati eliminasi ( tidak ditampilkan pada gambar
2-15), baik karena timed-release sebuah rumusan yang digunakan untuk
memperlambat penyerapan atau karena obat tersebut dengan cepat mengalami
atau dibuang.Seperti dicontohkan oleh penisilin g (t 1 2 dari 30 menit), lambatnya
penyerapan dicapai dengan lisan konsumsi relatif terhadap hasil ekskresi yang
cepat di puncak konsentrasi yang jauh berkurang dan jauh dibandingkan dengan
suntikan intravena tertunda. Di sisi positif, lisan administrasi dapat menghasilkan
durasi efek yang secara signifikan berkepanjangan.
Variasi dalam hilangnya rate sangat mempengaruhi postabsorptive fase
obat.Seperti terlihat pada gambar 2-15, tiga kali lipat penurunan eliminasi dapat
lebih efektif daripada yang sama peningkatan dosis dalam memberikan durasi
efek.Karena puncak titer umumnya hampir sama tidak peka terhadap perubahan
kondisi seperti ini sebelumnya adalah perubahan dalam dosis , memperlambat
penghapusan mungkin kamu lebih baik untuk memperpanjang durasi efek dengan
senyawa yang rendah atau margin yang cukup aman .
Untuk penisilin g, perlambatan eliminasi dapat dicapai dengan menghambat
ekskresi kemih melalui coadministration dari probenecid .Penisilin g adalah luar
biasa , namun dalam antibiotik yang memiliki beberapa sebuah toksisitas yang
rendah yang cepat eliminasi dapat mengimbangi dengan aman dengan hanya
mengalikan jumlah dosis beberapa kali.
Konsentrasi Plasma Dosis Berulang
Setiap kali obat yang diberikan lebih dari sekali dalam empat half-lives
sebelumnya , senyawa tersebut terjadi penumpukan dalam tubuh .2-16 gambar
menunjukkan hasil dari terus menggunakan obat yang diberikan infus intravena
baik oleh ( zeroorder repetitif atau proses administrasi (first-order penyerapan

dosis masing-masing , tapi dalam agregat ) zero-order.Terlepas dari format
administrasi , konsentrasi adalah sebuah dataran tinggi yang dicapai di sekitar
empat half-lives sebelumnya. Fluktuasi berselang periodik yang diperoleh dengan
administrasi adalah fungsi dari penyerapan umum dari interval dosis. Mendekati
50% dari puncak konsentrasi ketika penyerapannya sangat cepat dan dosis periode
sama dengan nasional anti t½, seperti variasi dapat diminimalkan dengan
meningkatkan frekuensi pemerintahan atau perlambatan tingkat penyerapan.
Rata-rata steady-state konsentrasi relatif terhadap nilai puncak didapat
setelah awal dosis dapat ditentukan oleh memperbanyak jumlah dosis dikelola per
eliminasi t½ oleh 1.44.Konsentrasi obat diberikan sekali setiap t½ sama dengan
144% konsentrasi awal puncak. Untuk konsumsi diazepam tiga kali per hari, rata-
rata kesetimbangan konsentrasi mendekati (dengan asumsi t 2 ½ hari) 1.44x6, atau
8,6 kali konsentrasi puncak setelah dosis tunggal.Sedikitnya dibutuhkan delapan
hari ( 4 ½ t ) untuk mencapai titer akhir ini obat.
Bertahap steady-state yang berhubungan dengan konsentrasi pendekatan
untuk dihilangkan perlahan-lahan bisa saja menghambat manfaat obat atau
terapi .Di sisi positif , lama t ½ yang memungkinkan untuk mengelola obat ini di
interval yang nyaman, mungkin sekali dalam sehari, tanpa harus terpengaruhi
dengan ayunan konsentrasi dalam plasma.Jika monitor pasien mengungkap
penumpukan obat yang tidak biasa karena gangguan metabolisme atau ekskresi
atau beberapa penyebab, masih tersedia waktu untuk menyesuaikan dosis sebelum
efek toksik berlangsung. Di sisi debet, pencapaian terapi efek tertunda karena
perlu waktu untuk mengumpulkan obat yang sama. Yang terjadi jika efek
farmakologis yang diperlukan, komposisi sebuah dosis obat harus diulang. Sebuah
muatan dosis yang cukup besar, kuantitas obat-obatan awal diganti kejumlah
normal cepat menghasilkan konsentrasi mendekati kesetimbangan.Bagi seorang
agen yang diberikan pada setiap t ½ , pengangkutan dosis dua kali lipat sama
dengan pemeliharaan dan dosis obat yang diberikan lebih sering, dari
pengangkutan dosis lebih besar.Membagi dosis yang memuat menjadi beberapa
pecahan kecil. Korban diharapkan mencapai kecepatan terapi yang beratnya lebih
dari biasanya lebih sesuai dengan kemampuan pasien mengevaluasi respon selama
tahap awal dari terapi. Fakta bahwa tingkat eliminasi, yang membantu mengatur

steady-state konsentrasi, dapat sangat bervariasi antara individu-individu sehingga
harus berhati-hati pada setiap kali kumulatif efek obat yang diberikan.
GAMBAR 2-16 Waktu tentu konsentrasi plasma yang melibatkan akumulasi
obat. Garis bergerigi mencerminkan pola akumulasi diamati selama pemberian
ulang obat pada intervalsama dengan eliminasi paruhnya, ketika penyerapan obat
adalah 10 kali lebih cepat sebagai eliminasi. Sebagai relatif tingkat kenaikan
penyerapan, maksimal konsentrasi mendekati dua kali minimum selamasteady
state. Garis halus menggambarkan akumulasi obat selama pemerintahan dosis
setaradengan infus intravena kontinu. (Diadaptasi dari Benet LZ, Kroetz DR,
Sheiner LB: Farmakokinetik:dinamika penyerapan obat, distribusi, dan eliminasi.
Di Hardman JG, Limbird LE, GilmanAG, editor: Goodman & Gilman yang dasar
farmakologi terapi, ed 9, New York, tahun 1996,McGraw-Hill.)
Model Multiple-Compartment
Kebanyakan obat, single-compartment model yang sederhana tidak
cukup menggambarkan konsentrasi plasma dari waktu awal saja.Perbedaan sangat
besar kemungkinan besar akan diamati ketika waktu yang relatif berupa obat yang
diberikan secara intravena, seperti dalam penggunaan untuk obat penenang sadar,
ssp depressants.Dalam soal tersebut, anggapan bahwa tubuh yang bertindak
sebagai satu kompartemen tidak pasti, dan satu atau lebih obat tambahan harus
diperhitungkan.Gambar 2-17 menggambarkan sebuah two-compartment model di
mana obat dikelola menjadi sebuah pusat kompartemen kecil.Agen mungkin

meninggalkan pusat kompartemen baik oleh distribusi ke yang lebih besar
kompartemen perifer atau oleh proses eliminasi. Dengan waktu, keadaan kuasi-
stabil didirikan antara wadah pusat dan perifer yang bersihredistribusi kembali ke
dalam kompartemen sentral terjadi sebagaiobat dimetabolisme atau diekskresi.
Dalam contoh yang diberikan, yang analog dengan skenario pada
Gambar 2-13, awalkonsentrasi tinggi (Cpo) setelah 500 mg dosis
mencerminkanVd kecil dari kompartemen sentral (Vc = 9,8 L).Pusat
kompartemen terdiri dari organ (termasuk otak, jantung, paru-paru, dan ginjal)
yang menerima pasokan darah besar. Sebuah terminal t ½ ditentukan dari log-
linear bagian dari kurva, tapi dalam kasus ini istilahnya mencerminkan distribusi
dan pemberantasan fungsi. Dan parameter lain seperti total vd. Yang sama dalam
makna dan derivation yang lebih kompleks daripada rekan-rekan mereka, pada
seluruh single-compartment.Kompleksitas ini masih ditambah dengan jumlah dari
kompartemen dalam model yang akan meningkat. Namun, multicompartment
model yang berguna dalam memahami bagaimana durasi dari efek obatsetelah
satu injeksi mungkin sebagian besar independen dari tingkat memungkinkan atau
penghapusan t ½. Pada gambar 2-17, jika konsentrasi di ambang untuk sebuah
akibat sedative obat adalah 10 µg/ml, seorang pasien akan pulih dari efek sedasi
30 menit, jika metabolisme dan pengeluaran sepenuhnya menutup, hanya dengan
distribusi ke dalam jaringan penyerapan-baik yang lebih kecil.
Setengah-Hidup Konteks-Sensitif
Banyaknya variabel model yang membuat multicompartment itu tidak
mungkin untuk membuat prediksi pengaruh pharmacokinetic dari tiap tiap seperti
parameter half-lives, nilai vd, dan kliring pada tingkat profil plasma-concentration
yang sangat lipid-soluble diberikan obat atau secara terus menerus berulang kali
untuk jangka waktu tertentu. Situasi ini menimbulkan masalah jika agen intravena
dikelola dengan terus-menerus infus untuk anestesi atau sedasi.Sebagian solusi
melibatkan penggunaan context-sensitive pemodelan komputer untuk
memperkirakan halflives. Para context-sensitive t ½ adalah waktu yang
dibutuhkan untuk konsentrasi plasma obat untuk mengurangi oleh 50% ketika
tidak diberikan pertimbangan untuk berapa lama obat ini telah dimasukkan.Seperti

yang digambarkan pada gambar 2-18, fentanyl menunjukkan peningkatan yang
signifikan dalam hal ini sebagai parameter infus yang melebihi durasi 2
jam.Fenomena ini adalah hasil dari kejenuhan dari lokasi terjadinya redistribusi.
Sementara itu, propofol, dengan kapasitas yang besar untuk terjadinya
redistribusi,hanya tumbuh di atas waktu yang lambat.Informasi tersebut sangat
bermanfaat secara klinis dalam penetapan agen yang sesuai untuk digunakan dan
di memperkirakan jangka waktu yang berubah tersebut efek obat.Mirip context-
sensitive kurva yang bisa diperoleh untuk pemulihan ke berbagai persentase
(misalnya, 25%) dari plasma konsentrasi, bergantung pada apa yang memprediksi
pemulihan fungsi nilai terbaik.Kemajuan lebih lanjut dalam pemodelan komputer
tidak diragukan lagi akan membantu mengatasi keterbatasan multicompartment
model yang lain, antara lain osilasi dalam arteri yang terjadi dengan jumlah
plasma bolus obat suntik dan kesalahan yang terkait dengan fakta bahwa beberapa
obat yang mengalami di lebih dari satu kompartemen.
GAMBAR 2-17 Model Dua-kompartemen kinetika obat. A, Dalam model ini,
obat-obatan yang diserap ke dalam dantersingkir dari kompartemen sentral yang
dihubungkan oleh proses distribusi (memiliki konstanta lajukd dan kr) untuk
kedua, kompartemen perifer. Kompartemen pusat termasuk darah, dariyang
penentuan obat yang diambil. B, The konsentrasi plasma-waktu kurva terdiri dari
dua tahap:distribusi awal atau fase α, di mana konsentrasi menurun terutama
sebagai akibat dari distribusidari kompartemen sentral, dan eliminasi akhir atau

fase β, di mana metabolisme danekskresi mendominasi. Terminal paruh (t ½ b)
dihitung dari bagian log-linear darikurva eliminasi.
GAMBAR 2-18 Konteks-sensitif setengah kali. (Digambar ulang dari Hughes
MA, Kaca PSA, Jacobs JR: Konteks Sensitifbabak pertama dalam model multi-
kompartemen farmakokinetik obat bius intravena,Anestesiologi 76: 334-341,
1992.)
PEMODELAN FARMAKOKITENIK-FARMAKODINAMIK
Dua dasar asumsi yang mendasari pharmacokinetic mempelajari bahwa
plasma konsentrasi obat yang diprediksi dari konsentrasi di sekitar lokasi tindakan
obat dan besarnya efek obat tergantung pada konsentrasi ini.Meskipun hal-hal itu
asumsi umum, perkecualian penting untuk mereka memang ada.Sebelumnya,
seperti, untuk mengikat dengan obat yang menghasilkan efek covalently reseptor
yang jauh melampauiobat yang terjadi di dalam darah.Obat yang mengandalkan
transcription dalam sintesis protein efek yang tertunda karena waktu yang
dibutuhkan untuk proses ini bisa terjadi. Tambahan kesenjangan antara plasma
konsentrasi dan efek obat timbul karena penundaan yang dalam menjangkau

masyarakat dari tindakan dan temporal perubahan yang terjadi direseptor
ketanggapan .Pharmacokinetic-pharmacodynamic pemodelan berusaha untuk
menjelaskan perbedaan ini.
GAMBAR 2-19 distorsi temporal antara konsentrasi plasmalorazepam dan
kognisi yang diukur dengan digitUji substitusi simbol. The hysteresis loop
berlawananmenunjukkan keterlambatan dalam distribusi lorazepam ke situs
nyatindakan dalam otak. (Diadaptasi dari Gupta SK, Ellinwood EH,Nikaido AM,
et al: pemodelan simultan farmakokinetik yangdan sifat farmakodinamik
benzodiazepin. 1.Lorazepam, J Pharmacokinet Biopharm 18: 89-102, 1990.)

GAMBAR 2-20 toleransi akut dengan dosis tunggal intranasal kokain. Searah
jarum jam hysteresis loop menunjukkan hilangnya subjektifefek obat sebagai
fungsi dari konsentrasi plasma ataswaktu. (Data dari Van Dyke C, Jatlow P, J
Ungerer, et al: Oralkonsentrasi plasma dan efek sentral, Sains: kokain200: 211-
213, 1978.)
Lorazepam memberikan contoh yang baik karena efeknya temporal
(gambar 2-19) tertunda. Lorazepam, benzodiazepin yang digunakan untuk
menghilangkan kecemasan, harus mendapatkan akses ke ssp dengan reseptor
untuk merangsang dan menghasilkan efek sebagai karakteristik efek ssp.Kelarutan
lipid sederhana obat memastikan,konsentrasi puncak plasma setelah pemberian
secara oralterjadi sebelum obat memiliki efek yang signifikandi otak.
Kokain menghasilkan kebalikan hubungan di yang maksimal obat efek
setelah lisan mendahului puncak konsentrasi administrasi plasma (gambar 2-
20).Dalam hal ini, yang mengatur reseptor yang pharmacologic efek kokain
mengalami desensitization.Desensitization sering melibatkan perubahan dalam
reseptor sensitivitas, baik dengan pengaturan yang phosphorylation dari subunit
dari reseptor atau penerima uncoupling dari sistem respon dari intraseluler.Lebih
tahan lama kerugian respon obat mungkin termasuk downregulation dari reseptor,
di mana jumlah reseptor menurun pada terus-menerus paparan obat.Lihat gambar
1-12 sebagai contoh dari desensitization dan downregulation.





























