Embed Size (px)
DESCRIPTION
hematology
Citation preview

Blood and Blood Disorders made EZ
Comprehensive Hematology
Blood, the essence of life, can be a daunting subject to the typical nursing or medical student.
We are going to take a closer look at this subject to see if we can make it EZ for you.
Hematology: The study of blood and blood diseases
• Blood components
• Plasma
• Hematocrit
• Blood functions
• Respiration
• Immunity
• Hemostasis
• Lab Values
• Blood diseases
• Anemias
• Hemoglobinopathies
• Coagulopathies
• Decreased number of Cells
• Increased number of Cells

• Hematological Malignancies
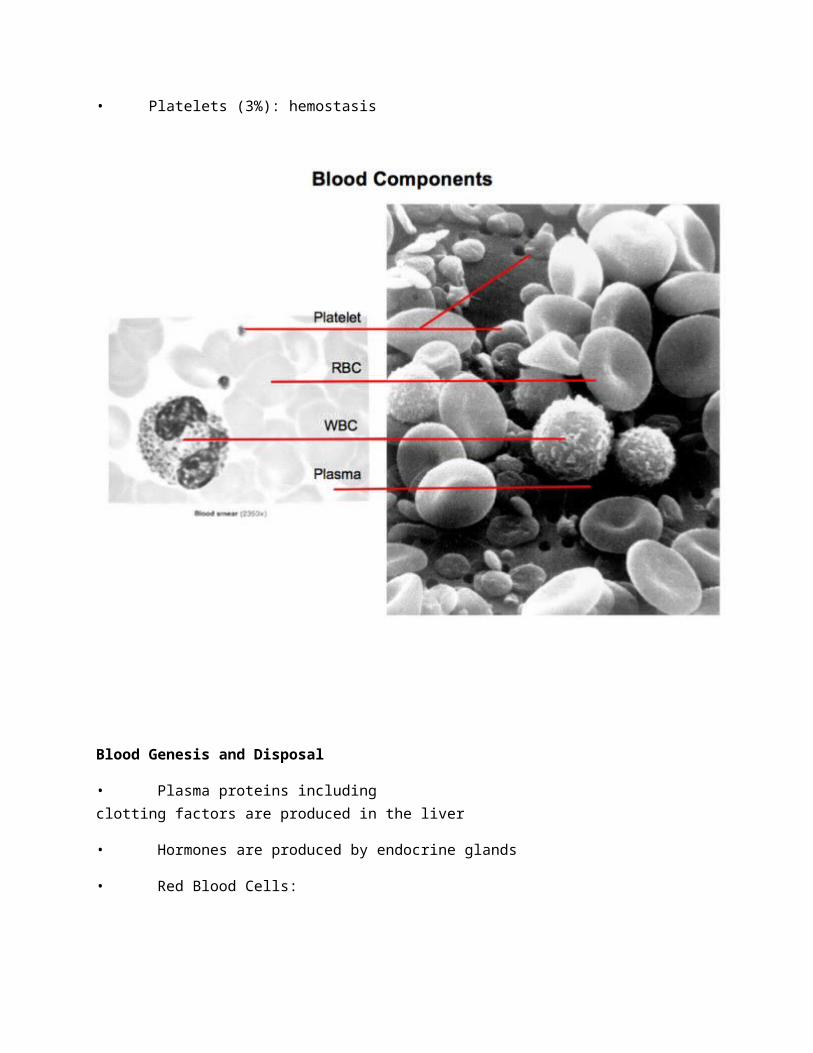
Blood components (and function)
• Plasma (55-60%)
• Water (92%) and Electrolytes (mostly Na, Cl): suspension of blood components, electrolytedelivery, waste disposal, pH regulation
• Proteins (8%)
• Fibrinogen and clotting factors: hemostasis
• Albumin: oncotic pressure, bind and deliver nutrients, transport thyroidhormone/fatty acids/bilirubin, binds Ca, buffers pH
• Globulins: immunity
• Hormones (trace): tissue signals
• Solids (Hematocrit) (40-45%)
• RBCs (95%): respiration
• WBCs (2%): immunity
• Platelets (3%): hemostasis
Blood Genesis and Disposal
• Plasma proteins including clotting factors are produced in the liver
• Hormones are produced by endocrine glands
• Red Blood Cells:
• Stem cells in the red bone marrow are stimulated by renal Erythropoietan to develop intoReticulocytes which enter the blood stream

• These baby RBCs then become mature RBCs in 7 days, as they expel their nucleus andintracellular contents
• After 120 days the RBCs begin to lose their flexibility, and when they are unable to squeezethrough the RBC obstacle course in the spleen, they are consumed by Macrophage, with theirheme (Fe) being recycled and their globin being made into Bilirubin
• White Blood Cells:
• Both Granulocytes (neutrophils, basophils, eosinophils) and Agranulocytes (Macrophage, TCells, B Cells) are derived from Stem cells in the bone marrow; Live hours to days
• Platelets:
• Derived from the breakup of large Bone marrow cells called
Megakaryocytes, these important players in coagulation live about 10 days

Blood Functions
Respiration
• Respiration is the transport of oxygen from air to tissues, AND the transport of carbon dioxidein the opposite direction (Ventilation- like opening a window to vent smell if the dog poops on the rug)
• RBCs are the main player in the blood controlling respiration
• They contain hemoglobin which transports oxygen and carbon dioxide
• In the lungs, more oxygen is present in alveoli than in the blood, so it diffuses across the thinmembrane and attaches to the hemoglobin
• Conversely, in the lungs, more CO2 exists in the blood than in the alveoli so it diffuses into the alveoli and is cleared with exhalation
• In the tissue, more oxygen is present in the blood than in the interstitial space, so it diffusesinto tissue
• Conversely, in the tissue, more CO2 exists in the interstitial space, so it diffusesinto the blood and attaches to the hemoglobin
• Other factors influence the dissociation of O2 and hemoglobin, in an attempt to deliver moreO2 to the tissues (think of what happens when you exercise and need more oxygen)
• Increased acidity (lactic acid)
• Increased CO2 production in tissues forces exchange with O2

• Increased temperature
• Excess oxygen is normally stored in muscle by Myoglobin, which if it needs tocan forcibly pull oxygen from hemoglobin into tissue
Immunity
• Innate Immunity
• Non-specific response to any perceived threat
• Humoral components
• Complement: Lyse proteins that attack pathogens directly and activate othercomponents of immunity
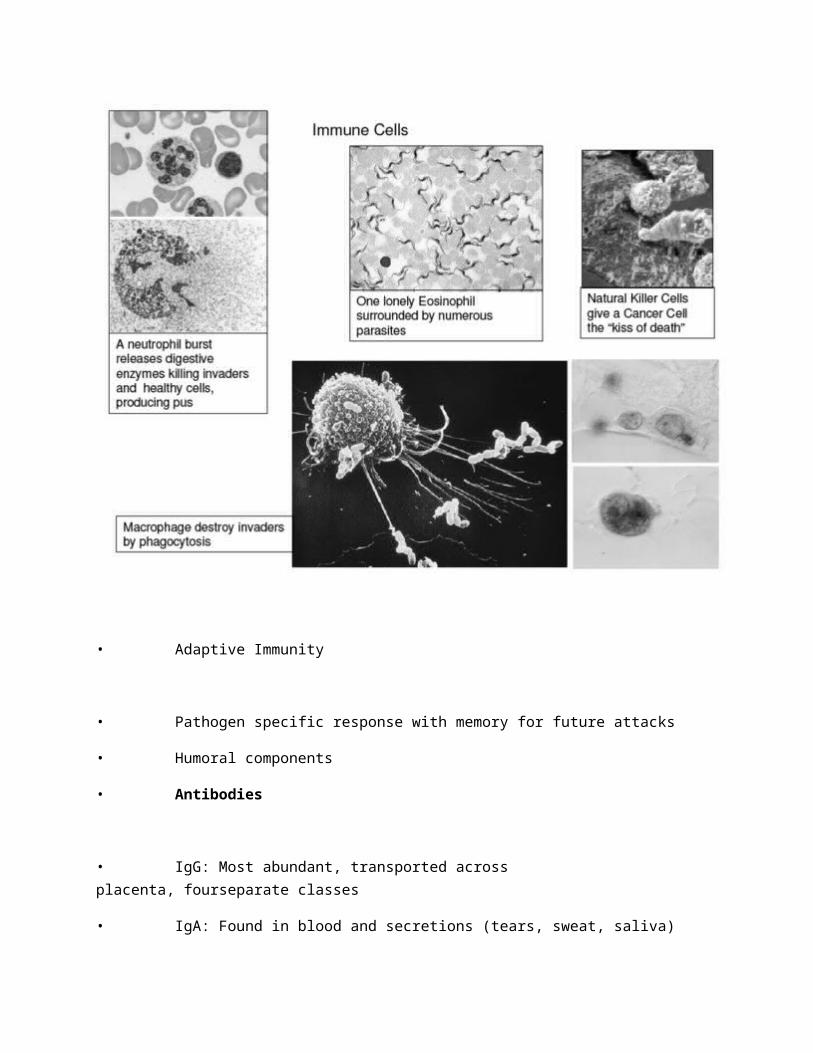
• Cell mediated components (For their order of abundance, remember Never Let Monkeys Eat Bananas: Neutrophils, Lymphocytes, Monocytes, Eosinophils, Basophils)
• Granulocytes
• Neutrophils: The suicide bomber of the immune system, they explode themselves and destroy anything in vicinity; Pus is the result ofNeutrophil activity
• Basophils: The alarm cell of Innate immunity, they releasechemical warning signals like histamine and cytokines
• Eosinophils: Specialized to fight parasites and viral infections; Thesecells are implicated in most allergic responses
• Agranulocytes

• Monocytes (Macrophage): Phagocytic cells with a voracious appetite; Thesecells consume invaders and present pieces of the invaders Antigens on their membrane to Lymphocytes
• Mast Cells: The combat engineers of the immune system, these cellsrelease histamine that vasodilates vascular tissue allowing blood cells tomigrate into tissues
• Natural Killer Cells: Less specific versions of the Killer T cells, thesecells recognize foreign or altered glycoproteins of cancerous or virallyinfected cells and kill the cell
• Adaptive Immunity
• Pathogen specific response with memory for future attacks
• Humoral components
• Antibodies
• IgG: Most abundant, transported across placenta, fourseparate classes
• IgA: Found in blood and secretions (tears, sweat, saliva)
• IgM: Largest; 1st antibody produced during primaryresponse; Present in fetus
• IgD: Located primarily on B lymphocyte membranes
• IgE: Least concentrated in blood; Mediates many allergicresponses; Defends against parasites by attracting eosinophils
• Cell mediated components
• T Cells: The General of the immune army
• Killer T Cells: Directly kill cancerous cells and virally infected cells byusing Perforins to pop the cell membrane
• Helper T Cells: Control the interaction between Innate and
Adaptive immune systems and direct specific attacks
• B Cells
• Plasma Cells: Activated B Cells that produce Antibodiesspecific to the invading Antigen

Hemostasis (covered in its own section in this blog)
Lab Note:
• Complete Blood Count (CBC) Respiration
• RBCs: 4-6 x1012/L
• Hematocrit: 35-50%
• Hemoglobin: 14-18 dg/L (male) or 12-16 dg/L (female)
• Reticulocytes: 10-100 x109/L
• Mean Cell Volume (MCV): 80-100 fL
• Mean Cell Hemoglobin: 26-34 pg
• Erythrocyte Sedimentation Rate (ESR): <20 mm/hr
• WBCs: 4-11 x109/L
• CBC with differential Immunity
• Absolute Neutrophil Count (aka polys, segs): 2-7.5 x109/L
• Lymphocytes: 1.3-4 x109/L
• Monocytes: 0.2-0.8 x109/L
• Eosinophils: 0.04-0.4 x109/L
• Basophils: 0.01-0.1 x109/L
• Coagulation Hemostasis

• Platelets: 150-400 x109/L
• PT: 10-13 sec
• INR: 0.9-1.2
• PTT: 30-40 sec
• Fibrinogen: 1.8-4 g/L

Blood Diseases
Anemias
• Normocytic
• Anemia of chronic disease
• Microcytic
• Iron deficiency
• Macrocytic
• B12 deficiency (Pernicious)
• Folate deficiency
Hemoglobinopathies
• Sickle Cell Anemia
• Thalassemia
Coagulopathies
• Hemophilia
• Thrombocytopenia
• Idiopathic Thrombocytopenic Purpura (ITP)
• Thrombotic Thrombocytopenic Purpura (TTP)
• Heparin Induced Thrombocytopenia (HIT)
• Disseminated Intravascular Coagulation (DIC)
Decreased number of Cells

• Neutropenia
• Agranulocytosis (granulocytopenia)
Increased number of Cells
• Polycythemia
• Leukocytosis
• Thrombocytosis
Hematological malignancies
• Lymphoma
• Hodgkins
• Non-Hodgkins
• Leukemia
• Acute Lymphoblastic Leukemia (ALL)
• Acute Myeloblastic Leukemia (AML)
• Chronic Lymphocytic Leukemia (CLL)
• Chronic Myeloid Leukemia (CML)

Anemia
• Defined as a deficiency in RBCs or Hemoglobin, occurring one of three ways:
1. Decreased RBC production: Deficiencies in Iron, Vitamin B12, or Folate, as well ascertain Chronic diseases can lead to less RBCs being made
2. Increased RBC destruction: Hemolysis can lead to anemias
3. Blood loss: Menses and GI bleeds are the most common causes
• S/S: All anemias share some common signs and symptoms: Weakness, Fatigue and
Dyspnea on exertion, Confusion
• Typically these vague symptoms occur around a Hct of 25 and Hgb of 8, but his dependsupon overall health of an individual (a healthy teen may not show symptoms until as low as 10/3)
• Some anemias have specific s/s that will be mentioned in the respective section
• Lab Data:
• Specific blood data is vital to determine type of anemia
• MCV (the RBC size) is the most important value to know, in fact anemias are classified according to this value
• Normocytic (MCV 80-100): Anemia of chronic disease
• Microcytic (MCV below 80): Fe deficiency, Thalassemia
• Macrocytic (MCV above 80): B12 deficiency (Pernicious), Folate deficiency
• Other data that help to differentiate anemias are:
• Serum Fe: Measures how much iron is bound to transferrin
• Transferrin: Iron transport protein,

• TIBC: Indicates the iron receptors on Transferrin that are not filled
• Ferritin: Iron storage protein, indicates the potential to hold iron

• Normocytic (MCV 80-100)
• Anemia of chronic disease
• Most common anemia in hospitalized patients
• Reduced stimulus to produce RBCS can be caused by infection, malignancies, kidneyfailure, or widespread inflammation
• Typically no iron is in the transportation phase (low serum Fe, low transferrin)
because no iron is being called to bone marrow for erythropoiesis
• The transferrin that is present is filled up with iron (low TIBC) because nothing is usingthe iron that is present
• Most iron is sitting around in storage (high ferritin)
• There is no deficiency in iron so do not treat with iron; Focus on treating the underlyingcause
• Microcytic (MCV <80)
• Fe deficiency anemia
• Most common anemia worldwide
• In women, menses and pregnancy are main causes
• In men, GI bleeds are the usual suspect
• Unique S/S:
• May be asymptomatic till severe
• May see spoon shaped fingernails, brittle hair, smooth tongue, stomatitis, orpica
• Since there is an overall low level of iron in the body (low Serum Fe), few Fe proteins are made by liver (low transferrin, low ferritin), and the transferrin that is present has little iron (high TIBC)
• Treat with Fe supplementation for months, but warn of probable constipation
• Thalassemia: Genetic absence of 1 to 4 of the 4 globins in each hemoglobin; NoFe deficiency so iron studies are normal

• Macrocytic (MCV above 100)
• Vitamin B12 deficiency
• The body has 3 years of B12 storage so this anemia develops over years
• B12 requires Intrinsic factor from the stomach parietal cells to be absorbed in the
Ileum, so any disruption = deficiency
• The most common cause is Pernicious anemia, an autoimmune attack onthe parietal cells
• Others causes include gastrectomy, ilectomy, celiac sprue, Crohns
• Unique s/s:
• Neurological defects due to Neuropathy is specific for B12 anemia and includenumbness in extremeties, loss of vibratory sense, ataxia, increased DTRs, +Babinski, Dementia
• Other s/s are similar to Folate deficiency anemia including beefy red tongue
• Treat with B12 injections that may be weekly or monthly for life
• Folate deficiency
• The body has 3 months of Folate storage, but if the diet is poor in greenvegetable a deficiency may develop
• Malabsorption may also be a cause but not near as common as diet related
• Typical anemia signs/symptoms with the beefy red tongue and stomatitis
• Treat with several weeks of Folate supplementation

Hemoglobinopathies
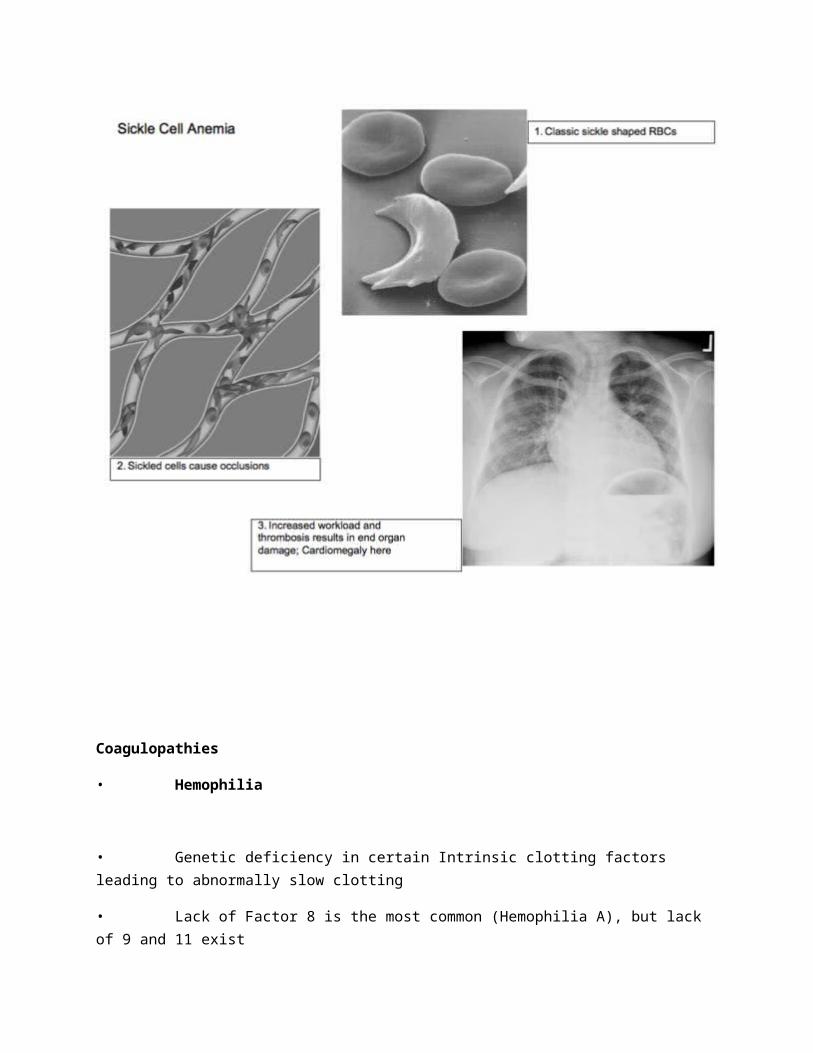
• Sickle Cell Anemia (SCA)
• Autosomal recessive disease characterized by a defect in the hemoglobin structure,caused by one abnormal amino acid
• The defect causes cells to sickle when oxygen levels or pH is low
• Almost exclusively in people of African descent (1 in 400 have disease)
• The defect leads to near immunity against Malaria as the invader can not penetrate the altered cell membrane
• Can carry only one copy of the defect and be a carrier with few symptoms or may carrytwo copies and have full blown Sickle Cell Disease
• Sickle Cell Disease: Periodic flare ups often caused by low oxygen or dehydration
• Hemolysis: Sickle cells can not pass inspection in the spleen and are destroyed byMacrophage leading to increased levels of bilirubin and Gallstones; This increasedbilirubin overloads the Liver from doing its job so Jaundice and Scleral Icterus may beseen; Iron overload may also occur
• Splenomegaly: Due to overuse the spleen becomes enlarged and is at risk for infections or rupture
• Vaso-occlusive crisis: Sickle cells obstruct small vasculature leading to ischemic painin bones, hands, legs, chest, etc; Strokes are common; Priapism can occur and is anemergency; Leg ulcers are common
• Treat infections promptly (fever or leukocytosis = antibiotics); Vaccinate against
S.Pneumoniae and H.Influenzae
• Acute treatment of acute crisis include hydration, analgesics, and oxygen
• Transfusion reserved for refractory crisis, or chest ischemia, strokes and priapism

Pharm Note: Hydroxyurea is a chemotherapy drug that destroys sickle cells, and may help to makecrisis less common; Caution is used as the drug can be toxic to bone marrow and causepancytopenia

Coagulopathies
• Hemophilia
• Genetic deficiency in certain Intrinsic clotting factors leading to abnormally slow clotting
• Lack of Factor 8 is the most common (Hemophilia A), but lack of 9 and 11 exist
• Hemophilia A only effects males
• Many hemophiliacs contracted HIV due to multiple transfusions prior to screening

• Due to normal platelet plug formation and extrinsic coagulation, all cuts do not abnormally bleed, especially those on the surface. However, certain areas such as muscles, joints, the digestivetract, and brain are very prone to bleed for prolonged time
• PTT is abnormal in the presence of a normal PT
• TX: factor replacement (one specific factor or FFP)
• Disseminated Intravascular Coagulation (DIC)
• DIC is caused by an abnormal and massive activation of the coagulation cascade to thepoint that all factors are used up
• Remember that tissue factor activated the cascade? Well bacterial endotoxinslook enough like tissue factor to do the same. Therefore, Sepsis is a commoncause of DIC
• Other causes include Obstetric complications, Trauma, and Malignancy
• Bleeding:
• Tends to occur in DIC that develops rapidly as factors are used up and not replenished
• Bleed tends to occur both superficially (ecchymoses) and in GI/GU (melena, hematuria)
• PT/PTT and D-dimer are all increased; Fibrinogen and Platelets are decreased
• TX: replace factors (FFP), platelets, and fibrinogen (cryoprecipitate)
• Thrombosis
• Tends to occur in DIC that develops more slowly
• Mottling and tachycardia may be indications that organ infarctions are occuring, mostoften in CNS and Kidney
• PT/PTT may not be significantly elevated as factors have had time to be replenished
• TX: possibly with Heparin to avoid infarcts but is controversial
• What do I do? I/Os, Vitals, Pulses qhr; Check for volume overload; Abdominal girth q4hr
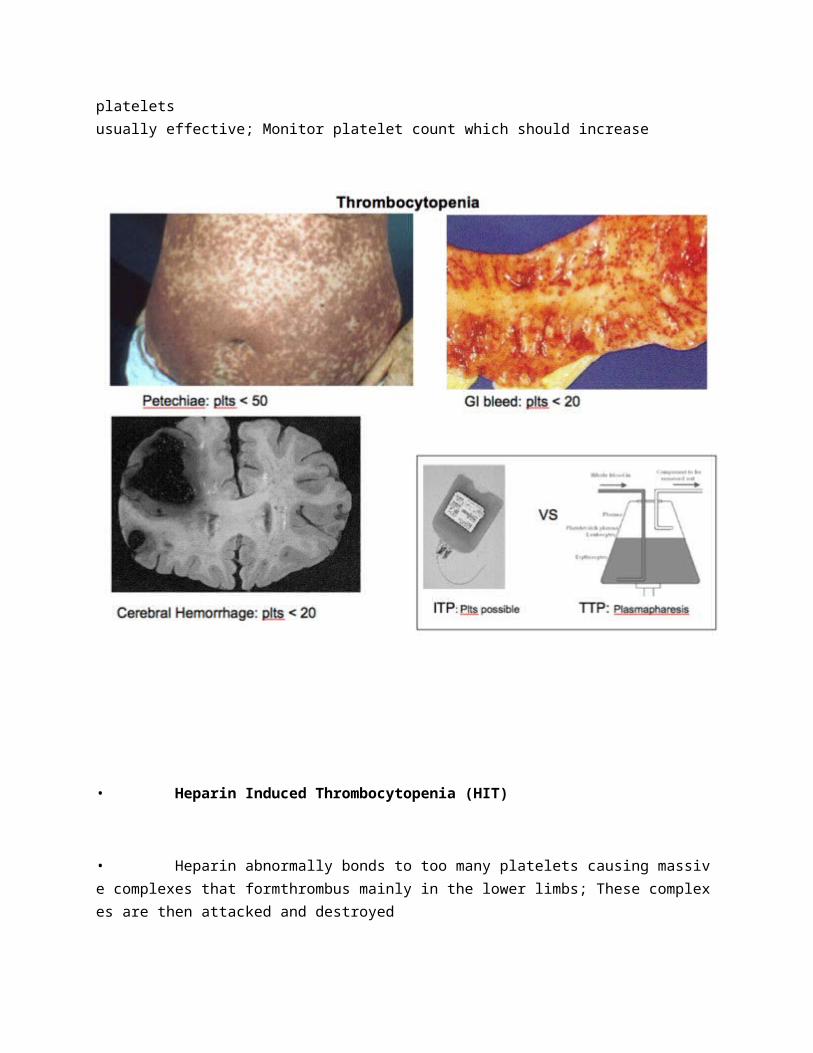
• Thrombocytopenia
• Defined as a decreased number of platelets below 150
• Severity
• 100-150: Abnormal bleeding is unusual
• 20-70: Increased bleeding during surgery or trauma
• < 50: Minor spontaneous bleeding: easy bruising, petechiae, epistaxis,menorrhagia, bleeding gums
• < 20: Major spontaneous bleeding such as intracranial and GI
• Causes
• Decreased production: Bone marrow invasion (mets) or injury (drugs likealcohol, chemo, radiation); often in the presence of pancytopenia
• Increased destruction: Immune mediated (ITP) or nonimmune causes (TTP, HIT)
• Immune Thrombocytopenic Purpura (ITP) – often referred to as Idiopathic TP
• Autoimmune antibody production against platelets, typically after viral infection, leadsto consumption by macrophage in the spleen; Usually in children and women 20-40yo
• S/S: Petichiae, ecchymoses, and mucosal bleeding are most common
• DX: Plt < 20, remainder of CBC usually normal unless significant bleeding (low H/H)
• TX:
• Steroids reduce Macrophage activity
• IV immune globulins saturate Macrophage receptors so less ability torecognize the marked platelets
• Splenectomy reduces recurrences of ITP in 80%
• Platelet transfusions for serious hemorrhagic episodes, but new plts aretargeted
• Thrombotic Thrombocytopenic Purpura (TTP)
• Idiopathic d/o causing platelets to collect in small vessels and are thendestroyed, similar to DIC, except instead of factors, platelets are involved
• S/S: Hemolytic anemia, Neuro problems (AMS, hemiplegia), Fever, Renal insufficiency
PT/PTT normal
• TX: Do not give platelets! Plasmapheresis (removal, cleansing, and reinfusion)of platelets usually effective; Monitor platelet count which should increase

• Heparin Induced Thrombocytopenia (HIT)
• Heparin abnormally bonds to too many platelets causing massive complexes that formthrombus mainly in the lower limbs; These complexes are then attacked and destroyed
• Platelet counts can range from 10-200, but classic definition is a reduction of 50% from baseline, with recent Heparin use and other causes (hypervolemia) ruled out
• Despite low platelet counts, HIT is mainly a thrombotic, not a bleeding disorder

• HIT develops in 3% of patients on Unfractionated Heparin, but only 0.1% onlow molecular weight heparins (Lovenox)
• 40% of those who develop HIT will develop significant thrombosis 3-12 daysafter starting Heparin infusion
• S/S: Skin necrosis, septic appearance from massive immune response
• End result can be MI, Stroke, DVT/PE and limb amputations
• TX: Prompt withdrawal of Heparin and replacement with an alternative such as
Argatroban (fda approved), Fondaparinux (Similar to Lovenox w/0% chance of HIT)
Decreased number of cells
• Neutropenia
• Abnormally low number of Neutrophils
• Since these cells are the main defense against bacteria, infections are common

• Similar to Agranulocytosis (Granulocytopenia) except these terms refer to low
Basophils and Eosinophils
• Often caused (75%) by drugs that suppress the bone marrow
• S/S: Fever/Chills, Mouth ulcers, Diarrhea, Dysuria, Dysphagia, Dyspnea
• TX: Mainly preventative; Maintain strict precautions with these patients
Increased number of cells
• Polycythemia
• Increase in the number of RBCs
• Can be a congenital problem, termed Polycythemia Vera
• Increased Hct, Hypertension, HSM, Pruritis, H/A, Vertigo, Tinnitus
• Can be due to high altitudes, or illegal blood doping with EPO
• Leukocytosis
• Increase in the number of WBCs
• Often an indication of an acute bacterial infection, but may also be caused by Steroids as thesedrugs force WBCs to leave tissues (lower immunity) and reside in blood
• Thrombocytosis
• Increase in the number of Platelets
• Can occur with Polycythemia Vera, or during acute inflammation states (Surgery)
Hematological malignancies
• Lymphoma:
• Cancers of the lymphatic system, usually involving abnormal growth of B or T cells
• Because it is a cancer of WBCs, leukocytosis is present
• Hodgkins
• This form of lymphoma is characterized clinically as the orderly spread of the disease from one lymph node to another
• The current cure rate is 93% making it one of the most treatable cancers, and leading to its TV name, the good Hodgkins (Larry David)
• Most common symptom is painless, rubbery, lymphadenopathy (LAN)
• Systemic symptoms may or may not be seen, including fever, night sweats,weight loss, pruritus, and cough
• Bimodal age distribution occurring most frequently between 15-30 and above 50
• Lymph Node biopsy needed for diagnosis, revealing Reed-Sternberg cells
• Radiation and Chemotherapy effective agents in treatment, though S/E oftreatment include sterility, pulmonary fibrosis, and peripheral neuropathy
• Non-Hodgkins
• Twice as common as Hodgkins
• The distinguishing characteristic of Non-Hodgkins is the lack of Reed Sternberg cells on biopsy; If these cells are absent, the predictability of spreading is very low as thistype of Lymphoma often spreads outside of the lymph system
• Due to the likelihood of spreading outside of lymph, there is a much higher rate ofmortality and failure of treatment; High grade lives a few months while low grade livesaround 5 years
• Most common early symptom is also painless LAN, Weight loss, reddened skin
• May invade Liver (LFTs, bilis elevated)or Bone marrow (Thrombocytopenia, Anemia); Hilar LAN may cause Vena Cava compression syndrome
• TX: Chemo and Radiation may slow progres

• Leukemia:
• Cancers of blood or bone marrow, resulting in a proliferation of immature WBCs, and an underproduction of RBCs (Anemia) and Platelets (Thrombocytopenia)
• Symptoms for all types may include anemic s/s, abnormal bleeding, bone pain andfrequent infections
• 4 main types divided into Acute (very immature cells) and Chronic (relatively immature, yetabnormal) & Myeloid (Granulocytes-Neutrophils) and Lymphocytic (T and B cells)
• Acute Myeloblastic Leukemia (AML):

• Occurs mainly in adults, with WBC widely varied from 1-100
• Very unresponsive to treatment if over 60, and often rapidly fatal
• Acute Lymphoblastic Leukemia (ALL):
• Most common cancer under the age of 15
• Most responsive leukemia to treatment, but if untreated can kill in months
• 75% remission and 50% completely cured
• Chronic Myeloid Leukemia (CML):
• Often asymptomatic until discovered on routine CBC that reveals a WBC
count of 50-200
• Typically occurs over 40 years of age and may remain indolent untilprogressing to aggressive Acute Leukemia with total life span after diagnosis averaging 3 years
• Treatment ineffective once the Acute blast stage has been reached
• Chronic Lymphocytic Leukemia (CLL):
• Often asymptomatic until discovered on routine CBC that reveals a WBC
count of 50-200, almost completely Lymphocytes
• Most common leukemia after the age of 60
• Treatment has little effect on survival times, but typically CLLprogresses slow

![Euphorbia-Derived Natural Products with Potential for Use ...€¦ · warts [47], while in China, it is used to treat blood disorders (e.g., haematuria, haemoptysis, epistaxis, and](https://img.pdfslide.tips/doc/110x75/609d654cf6ad966e433f5913/euphorbia-derived-natural-products-with-potential-for-use-warts-47-while.jpg)



























