Embed Size (px)
Citation preview

Takeshi Matsubara,1 Makoto Araki,1 Hideharu Abe,2 Otoya Ueda,3 Kou-ichi Jishage,3
Akira Mima,2 Chisato Goto,4 Tatsuya Tominaga,2 Masahiko Kinosaki,3 Seiji Kishi,2
Kojiro Nagai,2 Noriyuki Iehara,1 Naoshi Fukushima,3 Toru Kita,5 Hidenori Arai,6 andToshio Doi2
Bone Morphogenetic Protein 4 andSmad1 Mediate Extracellular MatrixProduction in the Development ofDiabetic NephropathyDiabetes 2015;64:2978–2990 | DOI: 10.2337/db14-0893
Diabetic nephropathy is the leading cause of end-stagerenal disease. It is pathologically characterized by theaccumulation of extracellular matrix in the mesangium, ofwhich the main component is a1/a2 type IV collagen(Col4a1/a2). Recently, we identified Smad1 as a direct reg-ulator of Col4a1/a2 under diabetic conditions in vitro.Here, we demonstrate that Smad1 plays a key role in di-abetic nephropathy through bone morphogenetic protein4 (BMP4) in vivo. Smad1-overexpressing mice (Smad1-Tg)were established, and diabetes was induced by strepto-zotocin. Nondiabetic Smad1-Tg did not exhibit histologicalchanges in the kidney; however, the induction of diabetesresulted in an∼1.5-fold greater mesangial expansion, con-sistent with an increase in glomerular phosphorylatedSmad1. To address regulatory factors of Smad1, we de-termined that BMP4 and its receptor are increased in di-abetic glomeruli and that diabetic Smad1-Tg and wild-typemice treated with a BMP4-neutralizing antibody exhibitdecreased Smad1 phosphorylation and ∼40% lessmesangial expansion than those treated with controlIgG. Furthermore, heterozygous Smad1 knockout miceexhibit attenuated mesangial expansion in the diabeticcondition. The data indicate that BMP4/Smad1 signalingis a critical cascade for the progression of mesangial ex-pansion and that blocking this signal could be a noveltherapeutic strategy for diabetic nephropathy.
Diabetic nephropathy is a life-threatening complication ofdiabetes and the leading cause of end-stage renal disease
(1). The structural features of diabetic nephropathy in-clude thickening of the glomerular basement membrane(GBM) and mesangial matrix expansion (2,3). Mesangialmatrix expansion is pathologically important because itleads to glomerulosclerosis accompanied by various tubu-lointerstitial damages and subsequent nephron loss (4,5).In addition, the severity of mesangial matrix expansion isclinically important because it is closely associated withthe decline of the glomerular filtration rate (6).
Mesangial matrix expansion is characterized by in-creased amounts of extracellular matrix (7), particularlya1/a2 type IV collagen (Col4a1/a2) (8). Although variouspeptides or growth factors are shown to mediate the reg-ulation of this key component, the protein responsible forits direct regulation remains to be determined.
Because various injuries of epithelial, endothelial, andmesangial cells converge on the accumulation of Col4a1/a2in the mesangium, mesangial cells presumably play a centralrole for the regulation of Col4a1/a2, even if they are not theprimary target of injury (9). Therefore, we attempted toelucidate the direct regulation of Col4a1/a2 under diabeticconditions and demonstrate that Smad1 can transcription-ally regulate Col4a1/a2 in the presence of advanced glyca-tion end products in mesangial cells (10).
Smad1 is an intracellular molecule originally cloned asa signal transducer of the transforming growth factor(TGF)-b superfamily (11). In response to these stimuli,Smad1 is phosphorylated at the COOH-terminal SSXSmotif followed by accumulation in the nucleus where it
1Department of Nephrology, Kyoto University, Kyoto, Japan2Department of Nephrology, Tokushima University, Tokushima, Japan3Chugai Pharmaceutical Co., Ltd., Shizuoka, Japan4Chugai Research Institute for Medical Science, Inc., Shizuoka, Japan5Kobe City Medical Center General Hospital, Kyoto, Japan6National Center for Geriatrics and Gerontology, Aichi, Japan
Corresponding authors: Takeshi Matsubara, [email protected], andHideharu Abe, [email protected].
Received 7 June 2014 and accepted 12 April 2015.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0893/-/DC1.
T.M. and M.A. contributed equally to this work.
© 2015 by the American Diabetes Association. Readers may use this article aslong as the work is properly cited, the use is educational and not for profit, andthe work is not altered.
2978 Diabetes Volume 64, August 2015
COMPLIC
ATIO
NS

regulates the transcription of specific target genes (12). Invivo, Smad1 is essential for the development of the kidney(13), but it is not detected in adult murine glomeruli (14).Previously, we reported that Smad1 is induced and phos-phorylated by advanced glycation end products and binds tothe promoter of Col4a1/a2, thus upregulating its transcrip-tional activity in mesangial cells (10). We also found thatSmad1 is highly expressed in human diabetic glomeruli andthat glomerular expression of Smad1 is closely correlatedwith the severity of mesangial matrix expansion in a rodentmodel of diabetic nephropathy (15). However, the func-tional role of Smad1 in diabetic nephropathy in vivoremains unknown.
To this end, we sought to demonstrate that transgenicmice overexpressing Smad1 can accelerate mesangial matrixexpansion under diabetic conditions and to identify a regu-latory factor of Smad1. We also focused on bone morpho-genetic proteins (BMPs) because they are potent stimulatorsof Smad1. Of note, we demonstrate that BMP4 is increasedin diabetic glomeruli, so to prove the involvement of BMP4in diabetic nephropathy, we treated diabetic mice with ananti-BMP4 antibody and demonstrate that the neutraliza-tion of BMP4 prevents the phosphorylation of Smad1,accumulation of Col4a1/a2, and mesangial expansion inboth diabetic Smad1 transgenic (Smad1-Tg) mice and theirlittermates.
RESEARCH DESIGN AND METHODS
Generation of SMAD-Overexpressing MiceAll animal experiments in this study were performed inaccordance with institutional guidelines, and the reviewboard of Kyoto University granted ethical permission. ThepCAGGS-SMAD1 vector was constructed by inserting hu-man SMAD1 cDNA into the mammalian expression vectorpCAGGS (provided by J. Miyazaki, Osaka University). Al-though a previous report indicated that this promoter couldbe transactivated in glomerular epithelial cells (16), anotherreport showed that the transgene under this promoter isexpressed ubiquitously in glomeruli (17); therefore, it couldstill work in mesangial cells. Transgenic mice expressingSMAD1 were generated as described (18). Integration ofthe transgene into host genome was confirmed by South-ern blot analysis of DNA using a 32P-labeled SMAD1cDNA fragment as a probe (Fig. 1A).
Generation of Inducible Smad1-Tg MiceTo generate inducible Smad1-Tg mouse lines, we used thetamoxifen-regulated Cre-loxP system (TaconicArtemisGmbH, Köln, Germany). This system consists of two trans-genes. The first transgene is the inducible Smad1 expressioncassette pMacII-floxed GFP pA-BMP4 using expression vec-tor pMacII consisting of a cytomegalovirus enhancer andmouse b-actin promoter (Supplementary Fig. 2A). The sec-ond transgene is a construct for the expression of a fusionprotein of mutated murine estrogen receptor (Mer) and Crerecombinase (MerCreMer [MCM]), with the pCAGGS vec-tor (Supplementary Fig. 2A). MCM cDNA was a gift from
M. Reth (Max Planck Institute of Immunobiology and Epi-genetics) (19). Each transgene was microinjected into thepronuclei of C57BL/6J fertilized eggs to create a transgenicmouse line. For induction of SMAD1 gene expression,8-week-old transgenic mice were fed a diet containing tamox-ifen citrate 400 mg/kg.
Induction of DiabetesDiabetes was induced in 8-week-old mice by injectingstreptozotocin (STZ) 50 mg/kg i.p. (Sigma, St. Louis, MO)for 5 consecutive days. Control animals received0.1 mmol/L sodium citrate buffer (pH 4.5) alone.
Tissue PreparationThe right-side kidney was divided into fragments, each ofwhich was fixed with Carnoy solution, neutral bufferedformalin, and 2% glutaraldehyde or was frozen immedi-ately in optimal cutting temperature compound.
Antibody PreparationThe recombinant human BMP4 peptide was purchased fromR&D Systems (Minneapolis, MN). For immunization withthe peptide, 6-week-old mice were injected subcutaneouslywith 50 mg conjugated peptide once a week for 4 weeksfollowed by an interval of 2 weeks. Three days after finalimmunization, spleen cells were harvested for production ofhybridomas to recombinant human BMP4. Monoclonalantibodies were generated using established procedures.
Protocol for the Treatment With a BMP4-NeutralizingAntibody in MiceTen milligrams/kilogram of neutralizing anti-BMP4 anti-body was injected subcutaneously into each group of miceonce every 2 weeks from 24 until 36 weeks after theinduction of diabetes. As a negative control, mouse IgG(MP Biomedicals, Solon, OH) was injected at the sametime points.
Renal Histology and Morphometric AnalysesTwo-micrometer sections embedded in paraffin werecollected through the largest axial section and stainedwith periodic acid silver methenamine (PASM). To quantifymesangial expansion, all tissues were sectioned and stainedby one professional pathology technician (H. Uchiyama,Taigenkai Hospital) to control the thickness of sections andthe intensity of silver staining of the individual slides.Sections were further coded and read by an observer (M.A.)blinded to the experimental protocol applied (15).
Electron Microscopy and Measuring GBM ThicknessPortions of the cortex were fixed in 2% glutaraldehydeand postfixed in 1% osmic acid. After embedding,ultrathin sections were stained (20). The average GBMthickness was measured using Image-Pro Plus software(Media Cybernetics, Bethesda, MD).
ImmunohistochemistryKidneys were processed as previously described (15). Theprimary antibodies used in this experiment are listed in
diabetes.diabetesjournals.org Matsubara and Associates 2979
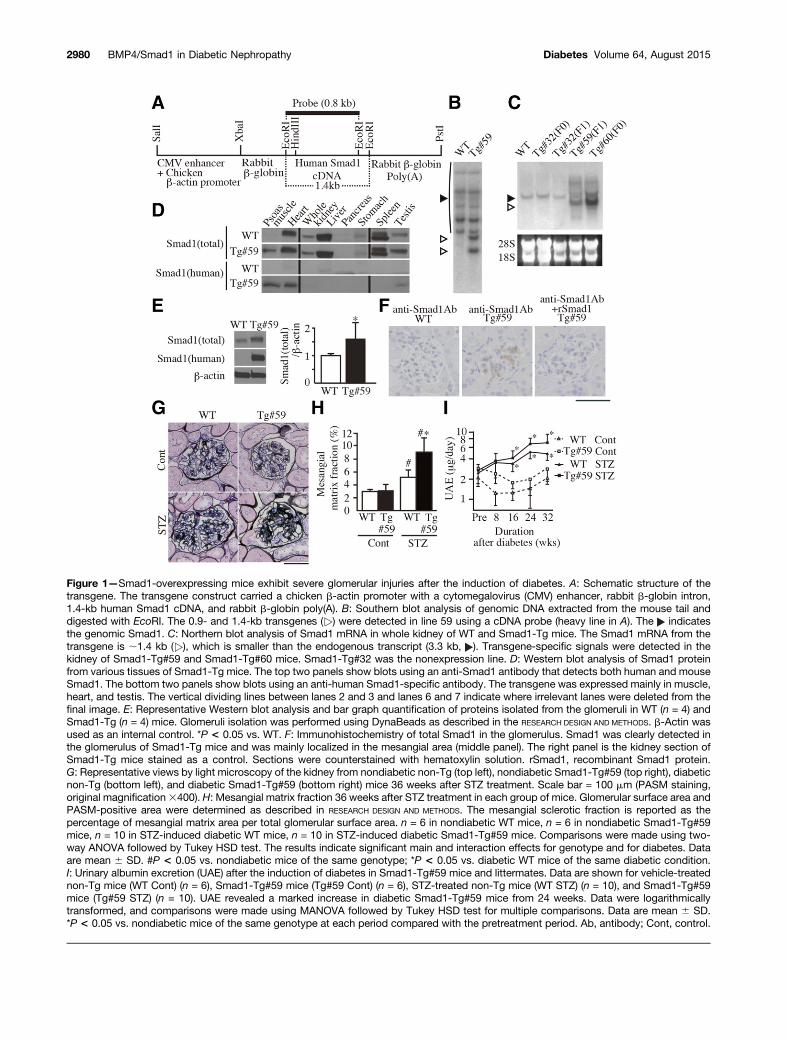
Figure 1—Smad1-overexpressing mice exhibit severe glomerular injuries after the induction of diabetes. A: Schematic structure of thetransgene. The transgene construct carried a chicken b-actin promoter with a cytomegalovirus (CMV) enhancer, rabbit b-globin intron,1.4-kb human Smad1 cDNA, and rabbit b-globin poly(A). B: Southern blot analysis of genomic DNA extracted from the mouse tail anddigested with EcoRI. The 0.9- and 1.4-kb transgenes ( ) were detected in line 59 using a cDNA probe (heavy line in A). The indicatesthe genomic Smad1. C: Northern blot analysis of Smad1 mRNA in whole kidney of WT and Smad1-Tg mice. The Smad1 mRNA from thetransgene is ;1.4 kb ( ), which is smaller than the endogenous transcript (3.3 kb, ). Transgene-specific signals were detected in thekidney of Smad1-Tg#59 and Smad1-Tg#60 mice. Smad1-Tg#32 was the nonexpression line. D: Western blot analysis of Smad1 proteinfrom various tissues of Smad1-Tg mice. The top two panels show blots using an anti-Smad1 antibody that detects both human and mouseSmad1. The bottom two panels show blots using an anti-human Smad1-specific antibody. The transgene was expressed mainly in muscle,heart, and testis. The vertical dividing lines between lanes 2 and 3 and lanes 6 and 7 indicate where irrelevant lanes were deleted from thefinal image. E: Representative Western blot analysis and bar graph quantification of proteins isolated from the glomeruli in WT (n = 4) andSmad1-Tg (n = 4) mice. Glomeruli isolation was performed using DynaBeads as described in the RESEARCH DESIGN AND METHODS. b-Actin wasused as an internal control. *P < 0.05 vs. WT. F: Immunohistochemistry of total Smad1 in the glomerulus. Smad1 was clearly detected inthe glomerulus of Smad1-Tg mice and was mainly localized in the mesangial area (middle panel). The right panel is the kidney section ofSmad1-Tg mice stained as a control. Sections were counterstained with hematoxylin solution. rSmad1, recombinant Smad1 protein.G: Representative views by light microscopy of the kidney from nondiabetic non-Tg (top left), nondiabetic Smad1-Tg#59 (top right), diabeticnon-Tg (bottom left), and diabetic Smad1-Tg#59 (bottom right) mice 36 weeks after STZ treatment. Scale bar = 100 mm (PASM staining,original magnification3400). H: Mesangial matrix fraction 36 weeks after STZ treatment in each group of mice. Glomerular surface area andPASM-positive area were determined as described in RESEARCH DESIGN AND METHODS. The mesangial sclerotic fraction is reported as thepercentage of mesangial matrix area per total glomerular surface area. n = 6 in nondiabetic WT mice, n = 6 in nondiabetic Smad1-Tg#59mice, n = 10 in STZ-induced diabetic WT mice, n = 10 in STZ-induced diabetic Smad1-Tg#59 mice. Comparisons were made using two-way ANOVA followed by Tukey HSD test. The results indicate significant main and interaction effects for genotype and for diabetes. Dataare mean 6 SD. #P < 0.05 vs. nondiabetic mice of the same genotype; *P < 0.05 vs. diabetic WT mice of the same diabetic condition.I: Urinary albumin excretion (UAE) after the induction of diabetes in Smad1-Tg#59 mice and littermates. Data are shown for vehicle-treatednon-Tg mice (WT Cont) (n = 6), Smad1-Tg#59 mice (Tg#59 Cont) (n = 6), STZ-treated non-Tg mice (WT STZ) (n = 10), and Smad1-Tg#59mice (Tg#59 STZ) (n = 10). UAE revealed a marked increase in diabetic Smad1-Tg#59 mice from 24 weeks. Data were logarithmicallytransformed, and comparisons were made using MANOVA followed by Tukey HSD test for multiple comparisons. Data are mean 6 SD.*P < 0.05 vs. nondiabetic mice of the same genotype at each period compared with the pretreatment period. Ab, antibody; Cont, control.
2980 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015

Supplementary Table 3. For immunostaining of Smad1,a mouse monoclonal antibody was used as described (21).
Immunofluorescence Staining and MorphometricAnalysis of Glomerular Type IV Collagen ExpressionImmunofluorescence staining was performed as described(15). Fluorescein isothiocyanate–labeled or biotinylatedsecondary antibody followed by avidin-labeled Alexa Fluor594 (Molecular Probes, Carlsbad, CA) was applied. Forimmunofluorescence staining of type IV collagen, 2-mmsections of formalin-fixed paraffin-embedded tissueblocks were used. The antitype IV collagen antibodyused in this experiment reacted mainly with mesangium,which includes Col4a1/a2, but not with the GBM, whichincludes a3/a4/a5 type IV collagen. The immunoreactiv-ity of type IV collagen was quantified as described (15).
Isolation of GlomeruliGlomeruli were isolated by DynaBeads (22). For the quanti-fication of a-smooth muscle actin (aSMA), laser-manipulatedmicrodissection was performed because some of the glo-meruli isolated by DynaBeads had the afferent and/orefferent arteriole still attached, which contained abun-dant aSMA (23).
RNA IsolationTotal RNA was extracted from isolated glomeruli usingthe guanidinium thiocyanate-phenol-chloroform method(TRIzol reagent, Invitrogen) in 20 mL RNase-free water orfrom microdissected glomeruli using a PicoPure RNA Iso-lation Kit (Arcturus, Foster City, CA) in 15 mL elutionbuffer.
cDNA Preparation and Quantification by Real-TimeRT-PCRFor RNA from isolated glomeruli, real-time RT-PCR wasperformed (24). Specific primers are listed in Supplemen-tary Table 2 except Col1a2, which was synthesized com-mercially (ABI primers and probes; Applied Biosystems).For RNA from microdissected glomeruli, the primers andprobes were obtained from Applied Biosystems.
Western BlottingTissues were homogenized in radioimmunoprecipitationassay buffer and subjected to immunoblotting (25). Theanti–b-actin antibody (#4967), anti-Smad1 antibody re-active only to human or monkey (#9512), and anti-Smad1antibody reactive to both human and mouse (#9743) wereobtained from Cell Signaling Technology (Danvers, MA).The antiphospho-Smad1/5/8 antibody was obtained fromChemicon (Millipore, Billerica, MA). The anti-aSMA anti-body was obtained from Sigma. The anti-GAPDH anti-body was obtained from BD Biosciences (San Jose, CA).
Plasmid ConstructsThe reporter plasmid that contained the Smad1 respon-sive element (3GC2-Lux) was provided from K. Miyazono(University of Tokyo). The aSMA promoter reporter plas-mid (SMA-Luc) contains 219 base pairs of the proximal59-flanking region of the aSMA gene subcloned into the
luciferase reporter vector (Promega, Madison, WI). Ex-pression vectors for wild-type (WT) and mutant Smad1have been described previously (26).
Cell CulturesMurine mesangial cells were established as describedpreviously (25). After 12-h incubation, cells were starvedin DMEM containing 0.5% FCS followed by the stimuli.Treatment with dorsomorphin (Tocris Bioscience, Ellisville,MO), anti–BMP4-neutralizing antibody, or control IgG an-tibody was performed 30 min before the stimulation.
Plasmid Transfection and Reporter AssayMesangial cells or Cos7 cells (1.0 3 105/mL) were seededinto 12-well plates (Nunc). After 6 h, the cells were trans-fected with 375 ng SMA-Luc or 3GC2-Lux and 37.5 ngpRL-CMV (Promega) along with Smad1-DVD, Smad1-DCexpression vector, or a mock vector. Transfection was per-formed with FuGENE 6 (Roche Diagnostics, Indianapolis, IN).The medium was changed 12 h after transfection to 0.5%FCS in DMEM. Twenty-four hours after medium change, cellswere harvested and luciferase activity measured (15).
Immunostaining of Cultured CellsMesangial cells (1.0 3 105/mL) were seeded in chamberslides (Nalge Nunc, Roskilde, Denmark). Twenty-fourhours after transfection and medium change, cells werefixed in 4% paraformaldehyde and treated with anti-Smad1 antibody at 1:100 (T-20; Santa Cruz Biotechnology,Dallas, TX). An appropriate fluorescein isothiocyanate–conjugated secondary antibody was used.
Statistical AnalysisAll analyses were performed using JMP 11 software (SASInstitute, Cary, NC). Normal distribution assumptionswere verified using the Shapiro-Wilk test. For type II Bmpreceptor (BmprII), type I Bmp receptor (Alk3), Smad1,and albuminuria, logarithmically normal distributionassumptions were verified. Analyses were performedusing MANOVA for the time course of albuminuria andone-way or two-way ANOVA for other variables followedby Tukey honest significant difference (HSD) test formultiple comparisons. Data are presented as mean 6 SD.P , 0.05 was considered significant.
RESULTS
Establishment of Smad1-Overexpressing MiceWe constructed a transgene consisting of a fragment ofthe chicken b-actin promoter and human Smad1 cDNA(Fig. 1A). Transgenic founder lines carrying the humanSmad1 transgene were identified by Southern blot analy-sis (Fig. 1B). Human SMAD1 mRNA was detected in onlytwo founders (Tg#59 and Tg#60). Moreover, because oftheir poor fertilizing ability, only one line could be finallyestablished from the male founder (Tg#59) (Fig. 1C).Western blot analyses for Smad1 revealed that Smad1from the transgene was mainly expressed in skeletal mus-cle, heart, and testis and was slightly expressed in whole
diabetes.diabetesjournals.org Matsubara and Associates 2981

kidney, whereas endogenous Smad1 was expressed ubiqui-tously (Fig. 1D). On the other hand, Smad1 was increased;50% in Tg mice compared with their WT littermates inisolated glomeruli (Fig. 1E). Immunohistochemical analysisrevealed that Smad1 from the transgene was detectedmainly in the mesangial area (Fig. 1F). To confirm thatthe Smad1 transgene was expressed in mesangial cells,primary cultures were established from glomeruli isolatedfrom normal 4-week-old Smad1-Tg#59 mice. Smad1 fromthe transgene was detected in primary cultured mesangialcells from Tg#59 mice (Supplementary Fig. 1A). We alsoconfirmed no contamination of glomerular epithelial cellsin this primary culture from Tg#59 mice by demonstratingthat cell lysate from Tg#59 cells did not contain E-cadherin(Supplementary Fig. 1B). However, there were no signifi-cant histological changes in the kidney between Smad1-Tg#59 mice and their WT littermates. The glomerulardensity and glomerular surface area were also comparablebetween the two groups (10.8 6 1.9 vs. 11.3 6 2.2 mm2
and 1,980 6 290 vs. 1,910 6 260 mm2, respectively).Because we could obtain only one line using this
construct, we established another line of Smad1-Tg micewith inducible Smad1 expression using the tamoxifen-regulated Cre-loxP system (Supplementary Fig. 2A). Thetransgene was expressed mainly in epithelial cells (Sup-plementary Fig. 2B–J) and partly in the mesangial area(Supplementary Fig. 2K). After induction by tamoxifen,Smad1 was significantly increased in various tissues, in-cluding kidney (Supplementary Fig. 2L).
The Effect of Smad1 Overexpression on Albuminuriaand Mesangial Matrix Expansion After the Induction ofDiabetesTo examine the role of Smad1 overexpression in diabeticnephropathy, we induced diabetes by STZ. Body weights,HbA1c, and blood pressure in diabetic Smad1-Tg mice didnot differ from their diabetic littermates (Table 1).
First, we analyzed mesangial matrix expansion of eachgroup of mice 36 weeks after STZ treatment. Morphometricanalysis revealed a significant increase in the mesangialmatrix expansion of diabetic Smad1-Tg#59 mice (Fig. 1H)and inducible Smad1-Tg#5 mice (Supplementary Fig. 2M)
compared with their diabetic littermates. Histologically,most glomeruli exhibited a widespread increase in PASM-positive material within the mesangium, termed “diffuselesion” (Fig. 1G). Diabetic MCM-Tg mice, which did notexpress the Smad1 transgene, exhibited similar mesangialexpansion to their WT littermates (Supplementary Fig. 2M).
Next, we measured albuminuria in each group of mice.Both WT and Smad-Tg#59 diabetic mice exhibited morealbuminuria than nondiabetic mice at 24 weeks, and theseincreases were sustained through 32 weeks during theexperimental period (Fig. 1I). However, there was no differ-ence in albuminuria levels between WT and Smad1-Tg#59mice, although diabetic Smad1-Tg#59 mice tended to ex-hibit slightly more albuminuria than WT mice. InducibleSmad1-Tg#5 also exhibited a tendency toward slightly ex-tended albuminuria relative to their littermates or MCM-Tgmice 32 weeks after the induction of diabetes, but the trendwas not statistically significant (Supplementary Fig. 2N).
Effects of Smad1 Overexpression on DiabeticNephropathyPreviously, we reported that Smad1 transcriptionallyregulates Col4a1/a2 and other extracellular matrix proteins,such as type I collagen, in vitro (10). Because mesangialmatrix expansion was accelerated in diabetic Smad1-Tgmice compared with their diabetic littermates, we quantifiedthe glomerular expression of Col4a1/a2 and Col1a1/a2in these mice. As shown in Fig. 2A, the glomerularexpression of these molecules was increased in diabeticmice compared with nondiabetic mice. Moreover, the expres-sion was significantly increased in diabetic Smad1-Tg micerelative to their diabetic littermates. Immunohistochemistryrevealed that these molecules were accumulated mainly inthe mesangial area (Fig. 2B). aSMA is another key moleculein diabetic glomerulopathy and a marker of mesangial phe-notypic changes. Therefore, we examined the glomerular ex-pression of aSMA in each group of mice. In diabetic mice,the expression of aSMA was significantly increased after STZtreatment. Of note, Smad1-Tg mice exhibited significant ex-pression of aSMA at the same stage (Fig. 2A). Immunohis-tochemistry revealed that increased aSMA was also localizedmainly in the mesangial area (Fig. 2B).
Table 1—Characteristics of nondiabetic (control) and STZ-induced diabetic WT and Smad1-Tg mice
Control STZ
WT Smad1-Tg WT Smad1-Tg
n 6 6 10 10
BW (g) 34.0 6 3.1 34.2 6 2.0 28.2 6 1.6* 28.0 6 2.8*
HbA1c (NGSP) (%) 2.7 6 0.3 2.5 6 0.2 12.4 6 1.8* 11.0 6 1.8*
HbA1c (IFCC) (mmol/mol) 29.5 6 3.3 27.3 6 2.2 112 6 19.7* 97 6 19.7*
SBP (mmHg) 108.4 6 8.6 110.2 6 8.3 108.2 6 1.3 106.1 6 19.2
rKW/BW (mg/g) 6.6 6 1.1 7.6 6 1.2 11.1 6 0.9* 11.8 6 1.5*
CCr (mL/m/10 g BW) 0.12 6 0.07 0.10 6 0.04 0.50 6 0.13* 0.55 6 0.18*
Data are mean6 SD. BW, body weight; CCr, creatinine clearance; IFCC, International Federation of Clinical Chemistry and Laboratory Medicine;NGSP, National Glycohemoglobin Standardization Program; rKW, right kidney weight; SBP, systolic blood pressure. *P , 0.05 vs. control.
2982 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015
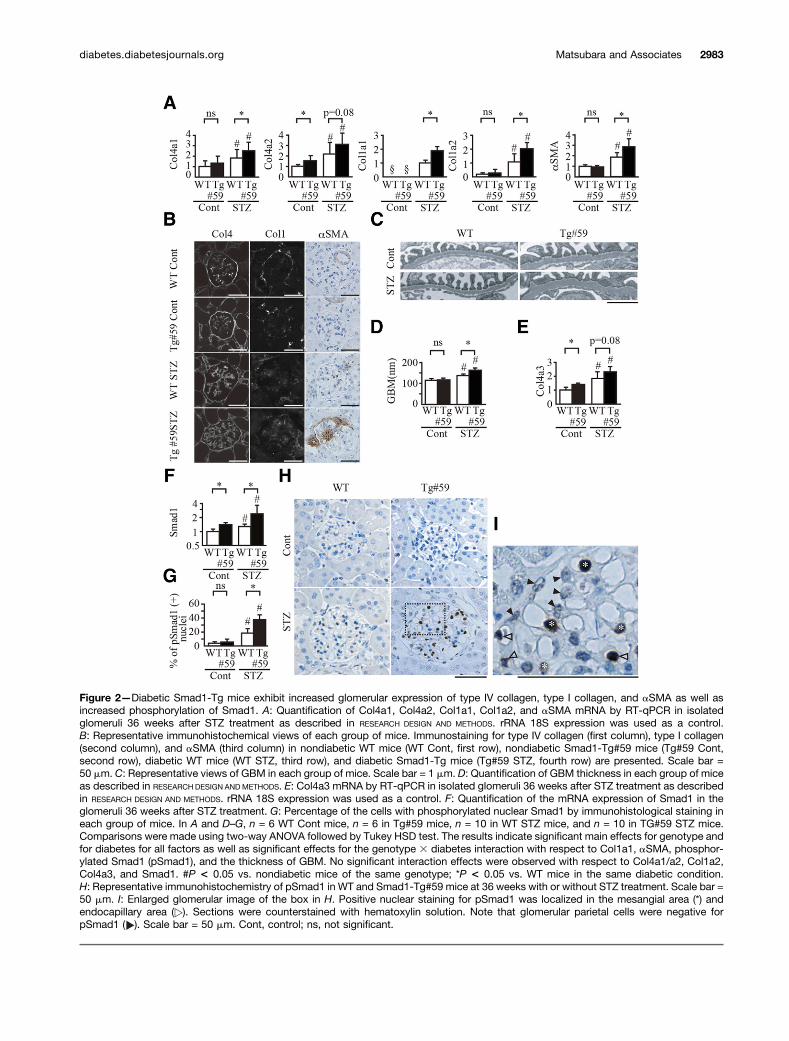
Figure 2—Diabetic Smad1-Tg mice exhibit increased glomerular expression of type IV collagen, type I collagen, and aSMA as well asincreased phosphorylation of Smad1. A: Quantification of Col4a1, Col4a2, Col1a1, Col1a2, and aSMA mRNA by RT-qPCR in isolatedglomeruli 36 weeks after STZ treatment as described in RESEARCH DESIGN AND METHODS. rRNA 18S expression was used as a control.B: Representative immunohistochemical views of each group of mice. Immunostaining for type IV collagen (first column), type I collagen(second column), and aSMA (third column) in nondiabetic WT mice (WT Cont, first row), nondiabetic Smad1-Tg#59 mice (Tg#59 Cont,second row), diabetic WT mice (WT STZ, third row), and diabetic Smad1-Tg mice (Tg#59 STZ, fourth row) are presented. Scale bar =50 mm. C: Representative views of GBM in each group of mice. Scale bar = 1 mm. D: Quantification of GBM thickness in each group of miceas described in RESEARCH DESIGN ANDMETHODS. E: Col4a3 mRNA by RT-qPCR in isolated glomeruli 36 weeks after STZ treatment as describedin RESEARCH DESIGN AND METHODS. rRNA 18S expression was used as a control. F: Quantification of the mRNA expression of Smad1 in theglomeruli 36 weeks after STZ treatment. G: Percentage of the cells with phosphorylated nuclear Smad1 by immunohistological staining ineach group of mice. In A and D–G, n = 6 WT Cont mice, n = 6 in Tg#59 mice, n = 10 in WT STZ mice, and n = 10 in TG#59 STZ mice.Comparisons were made using two-way ANOVA followed by Tukey HSD test. The results indicate significant main effects for genotype andfor diabetes for all factors as well as significant effects for the genotype 3 diabetes interaction with respect to Col1a1, aSMA, phosphor-ylated Smad1 (pSmad1), and the thickness of GBM. No significant interaction effects were observed with respect to Col4a1/a2, Col1a2,Col4a3, and Smad1. #P < 0.05 vs. nondiabetic mice of the same genotype; *P < 0.05 vs. WT mice in the same diabetic condition.H: Representative immunohistochemistry of pSmad1 in WT and Smad1-Tg#59 mice at 36 weeks with or without STZ treatment. Scale bar =50 mm. I: Enlarged glomerular image of the box in H. Positive nuclear staining for pSmad1 was localized in the mesangial area (*) andendocapillary area ( ). Sections were counterstained with hematoxylin solution. Note that glomerular parietal cells were negative forpSmad1 ( ). Scale bar = 50 mm. Cont, control; ns, not significant.
diabetes.diabetesjournals.org Matsubara and Associates 2983
We further investigated the thickness of the GBM byelectron microscopy. Diabetic Smad1-Tg mice exhibitedsignificant GBM thickening relative to their diabetic litter-mates at the same stage (Fig. 2C and D). Glomerular expres-sion of Col4a3, a major molecular component of the GBM,was also increased in diabetic Smad1-Tg in parallel withGBM thickening (Fig. 2E).
Phosphorylation of Smad1 After the Induction ofDiabetes in Smad1-Tg MiceThese data demonstrate that the overexpression ofSmad1 per se does not exacerbate nephropathy. Smad1
is activated by the phosphorylation of its carboxylterminus. Therefore, we quantified Smad1 activation bycounting cells with positive staining of phosphorylatedSmad1. Although glomerular expression of Smad1 wasincreased in Smad1-Tg mice relative to their littermates(Fig. 2F), phosphorylated Smad1 was barely detectable inboth Smad1-Tg mice and their littermates before STZtreatment. After STZ treatment, however, Smad1 wasphosphorylated and translocated into the nucleus inboth Smad1-Tg mice and their littermates (Fig. 2G andH). Of note, nuclear translocation of phosphorylatedSmad1 was more evident in diabetic Smad1-Tg mice
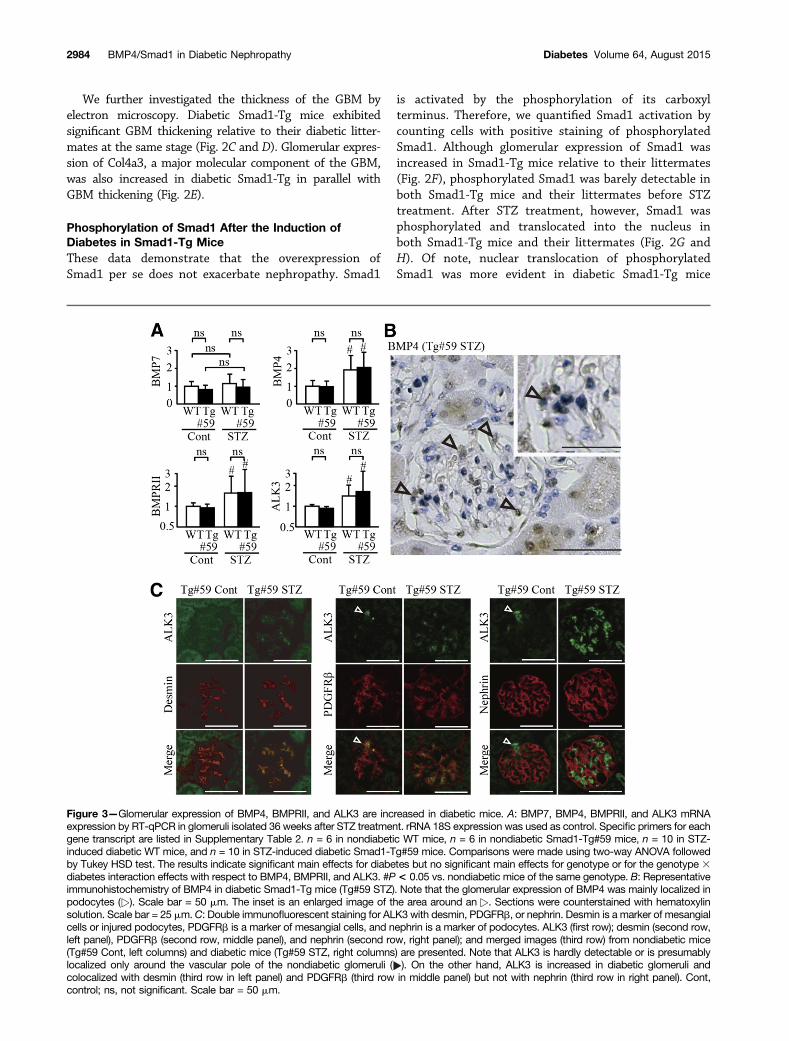
Figure 3—Glomerular expression of BMP4, BMPRII, and ALK3 are increased in diabetic mice. A: BMP7, BMP4, BMPRII, and ALK3 mRNAexpression by RT-qPCR in glomeruli isolated 36 weeks after STZ treatment. rRNA 18S expression was used as control. Specific primers for eachgene transcript are listed in Supplementary Table 2. n = 6 in nondiabetic WT mice, n = 6 in nondiabetic Smad1-Tg#59 mice, n = 10 in STZ-induced diabetic WT mice, and n = 10 in STZ-induced diabetic Smad1-Tg#59 mice. Comparisons were made using two-way ANOVA followedby Tukey HSD test. The results indicate significant main effects for diabetes but no significant main effects for genotype or for the genotype 3diabetes interaction effects with respect to BMP4, BMPRII, and ALK3. #P< 0.05 vs. nondiabetic mice of the same genotype. B: Representativeimmunohistochemistry of BMP4 in diabetic Smad1-Tg mice (Tg#59 STZ). Note that the glomerular expression of BMP4 was mainly localized inpodocytes ( ). Scale bar = 50 mm. The inset is an enlarged image of the area around an . Sections were counterstained with hematoxylinsolution. Scale bar = 25 mm.C: Double immunofluorescent staining for ALK3 with desmin, PDGFRb, or nephrin. Desmin is a marker of mesangialcells or injured podocytes, PDGFRb is a marker of mesangial cells, and nephrin is a marker of podocytes. ALK3 (first row); desmin (second row,left panel), PDGFRb (second row, middle panel), and nephrin (second row, right panel); and merged images (third row) from nondiabetic mice(Tg#59 Cont, left columns) and diabetic mice (Tg#59 STZ, right columns) are presented. Note that ALK3 is hardly detectable or is presumablylocalized only around the vascular pole of the nondiabetic glomeruli ( ). On the other hand, ALK3 is increased in diabetic glomeruli andcolocalized with desmin (third row in left panel) and PDGFRb (third row in middle panel) but not with nephrin (third row in right panel). Cont,control; ns, not significant. Scale bar = 50 mm.
2984 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015
than in their littermates (Fig. 2H) and was largely local-ized in mesangial and/or endocapillary cells (Fig. 2I).
Diabetic Changes in the Regulatory Factors of Smad1PhosphorylationThe data demonstrate that Smad1 is phosphorylated underdiabetic conditions. BMPs/BMPRs are generally accepted aspotent stimulators of Smad1. Among the BMPs, BMP2, -4,and -7 are expressed at various sites in different embryonicstages of renal development (27–29). We observed that
Bmp7 was abundantly expressed both in nondiabetic anddiabetic glomeruli, but there was no difference in its expres-sion between the nondiabetic and diabetic groups of mice(Fig. 3A). Of note, glomerular expression of Bmp4 was in-creased approximately twofold after STZ treatment in bothWT and Smad1-Tg mice (Fig. 3B), although Bmp2 was notdetected by quantitative RT-PCR (RT-qPCR) from the samealiquot (data not shown). These data suggest that BMP4 isinvolved in the progression of diabetic nephropathy. BMPsinduce Smad1 phosphorylation by forming heterotetrameric
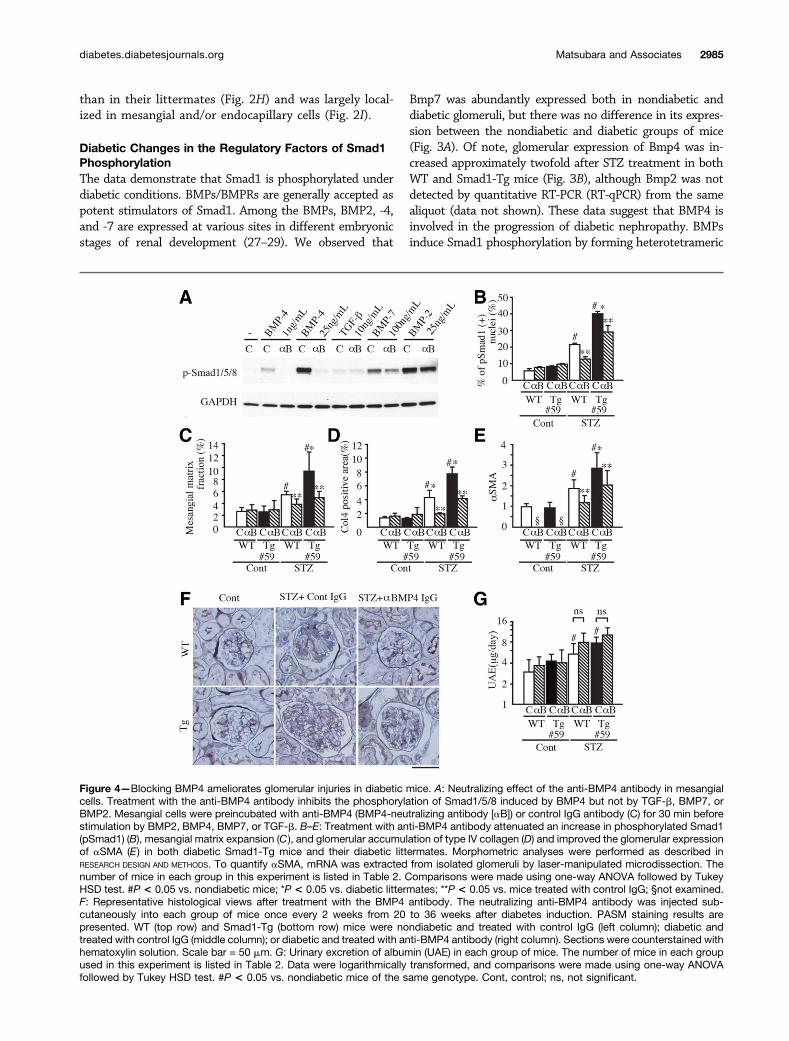
Figure 4—Blocking BMP4 ameliorates glomerular injuries in diabetic mice. A: Neutralizing effect of the anti-BMP4 antibody in mesangialcells. Treatment with the anti-BMP4 antibody inhibits the phosphorylation of Smad1/5/8 induced by BMP4 but not by TGF-b, BMP7, orBMP2. Mesangial cells were preincubated with anti-BMP4 (BMP4-neutralizing antibody [aB]) or control IgG antibody (C) for 30 min beforestimulation by BMP2, BMP4, BMP7, or TGF-b. B–E: Treatment with anti-BMP4 antibody attenuated an increase in phosphorylated Smad1(pSmad1) (B), mesangial matrix expansion (C ), and glomerular accumulation of type IV collagen (D) and improved the glomerular expressionof aSMA (E) in both diabetic Smad1-Tg mice and their diabetic littermates. Morphometric analyses were performed as described inRESEARCH DESIGN AND METHODS. To quantify aSMA, mRNA was extracted from isolated glomeruli by laser-manipulated microdissection. Thenumber of mice in each group in this experiment is listed in Table 2. Comparisons were made using one-way ANOVA followed by TukeyHSD test. #P < 0.05 vs. nondiabetic mice; *P < 0.05 vs. diabetic littermates; **P < 0.05 vs. mice treated with control IgG; §not examined.F: Representative histological views after treatment with the BMP4 antibody. The neutralizing anti-BMP4 antibody was injected sub-cutaneously into each group of mice once every 2 weeks from 20 to 36 weeks after diabetes induction. PASM staining results arepresented. WT (top row) and Smad1-Tg (bottom row) mice were nondiabetic and treated with control IgG (left column); diabetic andtreated with control IgG (middle column); or diabetic and treated with anti-BMP4 antibody (right column). Sections were counterstained withhematoxylin solution. Scale bar = 50 mm. G: Urinary excretion of albumin (UAE) in each group of mice. The number of mice in each groupused in this experiment is listed in Table 2. Data were logarithmically transformed, and comparisons were made using one-way ANOVAfollowed by Tukey HSD test. #P < 0.05 vs. nondiabetic mice of the same genotype. Cont, control; ns, not significant.
diabetes.diabetesjournals.org Matsubara and Associates 2985

complexes with two major types of membrane-bound ser-ine/threonine kinase receptors: the type I ALK receptors andthe type II receptors (30). In vitro binding assays suggestthat ALK3/6 are type I receptors for BMP4 (31,32). There-fore, we examined the glomerular expression of BMPRII,Alk3, and Alk6 in these mice. The glomerular expressionof BmprII and Alk3 but not Alk6 was increased after STZtreatment. Next, we studied the localization of BMP4 andALK3 by immunohistochemistry. In nondiabetic mice,BMP4 was barely detected in the glomeruli (data notshown). In contrast, 36 weeks after diabetes induction,BMP4 was extensively expressed in the podocytes and partlyin the mesangium (Fig. 3B). Double immunostaining forALK3 and desmin, platelet-derived growth factor (PDGF)receptor b (PDGFRb), or nephrin revealed that increasedALK3 in diabetic glomeruli was mainly localized in themesangium (Fig. 3C). These data suggest that BMP4/ALK3/Smad1 signaling contributes to the progression of di-abetic nephropathy.
Neutralizing BMP4 Ameliorates the Exacerbation ofGlomerular Injuries in Diabetic MiceTo further delineate the role of BMP4 in the developmentof diabetic nephropathy, we administered a neutralizingantibody against BMP4 to both diabetic Smad1-Tg miceand their diabetic littermates. First, we evaluated thespecificity of the neutralizing activity of the antibodyusing an assay measuring Smad1 phosphorylation inmesangial cells induced by BMPs and TGF-b. The additionof the antibody completely inhibited the phosphorylationof Smad1/5/8 induced by BMP4 but not by BMP2, BMP7,or TGF-b (Fig. 4A), indicating its specificity. Next, weadministered the neutralizing antibody or control IgG toeach group of mice (Table 2). The administration of theneutralizing antibody attenuated the nuclear translocationof phosphorylated Smad1 (Fig. 4B), ameliorated the glomer-ular accumulation of type IV collagen (Fig. 4D), and inhibitedmesangial matrix expansion (Fig. 4C and F) in both di-abetic Smad1-Tg mice and littermates. Furthermore, RT-qPCR of RNA from glomeruli isolated by laser-manipulatedmicrodissection revealed that treatment with the BMP4-neutralizing antibody also improved glomerular expression
of aSMA in both diabetic Smad1-Tg mice and littermates(Fig. 4E). However, the BMP4-neutralizing antibody didnot have any effect on albuminuria (Fig. 4G).
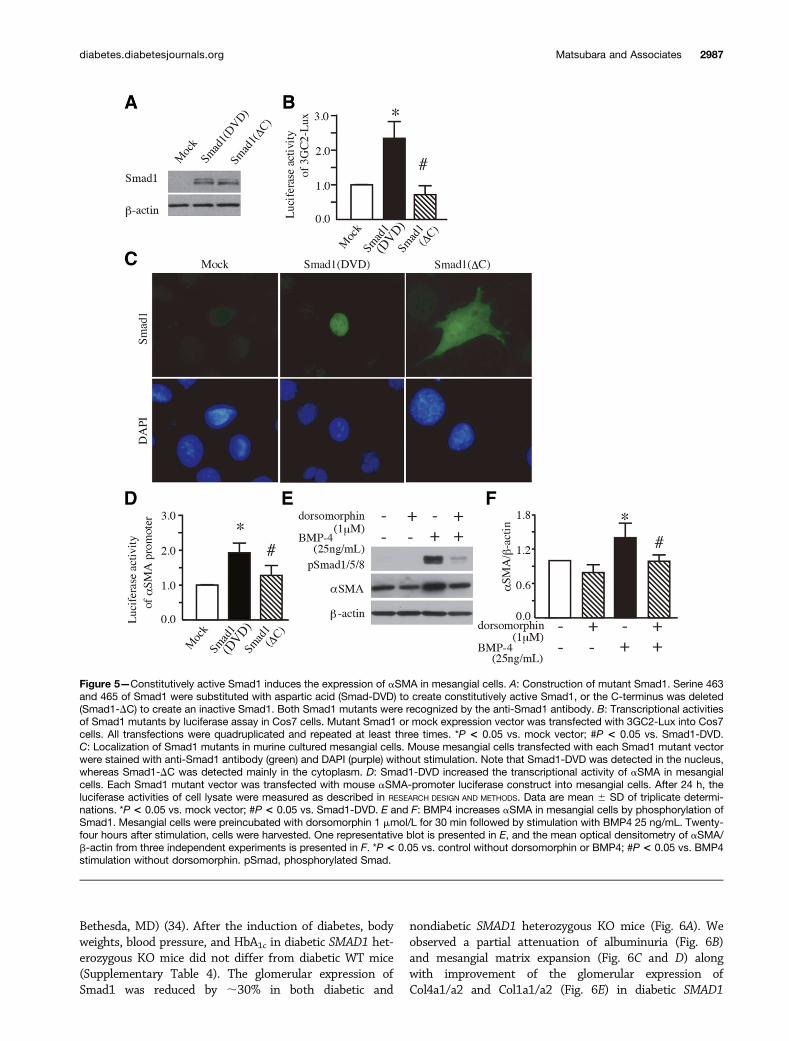
Role of BMP4 in Mesangial Cell aSMA ExpressionIn this study, we demonstrate that overexpression andsubsequent phosphorylation of Smad1 results in anincrease in the glomerular expression of aSMA, whereasthe inhibition of Smad1 phosphorylation using an anti-BMP4 antibody leads to the improvement of glomerularexpression of aSMA. To further elucidate the relationshipbetween Smad1 phosphorylation and aSMA, we generatedSmad1 mutants: a constitutively active mutant in whichtwo serine residues at the carboxyl termini were substitutedwith aspartic acid (Smad1-DVD) (26) and a dominant neg-ative mutant in which the carboxyl termini were lacking(Smad1-DC) (Fig. 5A). The expression of Smad1-DVD in-creased the transcriptional activity of 3GC2-Lux, a Smad1-dependent reporter, whereas the expression of Smad1-DCdid not (Fig. 5B). In mesangial cells, Smad1-DVD was local-ized in the nuclei, whereas Smad1-DC was localized mainlyin the cytoplasm (Fig. 5C). Therefore, we asked whether thetranscriptional activity of aSMA is modulated by the con-stitutive activation of Smad1 in mesangial cells. Asexpected, the expression of Smad1-DVD but not Smad1-DC increased the transcriptional activity of aSMA inmesangial cells (Fig. 5D). Finally, we examined whetherdorsomorphin, a small molecule inhibitor of BMP signaling(33), can affect the expression of aSMA by inhibiting thephosphorylation of Smad1 in mesangial cells. As shown inFig. 5E and F, BMP4 induced the phosphorylation of Smad1along with increased aSMA expression, which was blockedby dorsomorphin. These data indicate that BMP4 mediatesaSMA expression through the phosphorylation of Smad1 inmesangial cells.
Heterozygous SMAD1 Knockout Mice ExhibitAttenuated Mesangial Sclerosis in Diabetes
We further investigated whether the reduction of Smad1expression improves diabetic glomerular changes by usingheterozygous SMAD1 knockout (KO) mice, which were pro-vided from Anita B. Roberts (National Cancer Institute,
Table 2—Characteristics of each group of mice
Control STZ
WT Smad1-Tg WT Smad1-Tg
IgG aBMP4 IgG IgG aBMP4 IgG IgG aBMP4 IgG IgG aBMP4 IgG
n 5 5 5 5 9 9 9 9
BW (g) 38.6 6 3.9 37.4 6 4.3 37.3 6 2.2 38.4 6 4.6 30.8 6 2.9* 31.3 6 2.2* 30.3 6 2.3* 29.3 6 3.2*
HbA1c (NGSP) (%) 2.7 6 0.4 2.6 6 0.3 2.3 6 0.5 2.4 6 0.4 9.9 6 0.6* 10.1 6 0.7* 10.2 6 0.9* 9.8 6 2.0*
HbA1c (IFCC) (mmol/mol) 29.5 6 4.4 28.4 6 3.3 25.2 6 5.5 26.2 6 4.4 85 6 6.6* 87 6 7.7* 84 6 9.8* 84 6 21.9*
rKW/BW (mg/10 g BW) 5.2 6 0.5 5.2 6 0.6 5.8 6 1.1 5.6 6 0.6 9.7 6 0.9* 9.8 6 1.0* 9.5 6 0.9* 9.0 6 1.4*
Data are mean 6 SD. aBMP4 IgG, mice treated with BMP4-neutralizing antibody; BW, body weight; IFCC, International Federation ofClinical Chemistry and Laboratory Medicine; IgG, mice treated with control IgG antibody; NGSP, National Glycohemoglobin Standard-ization Program; rKW, right kidney weight. *P , 0.05 vs. control.
2986 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015
Bethesda, MD) (34). After the induction of diabetes, bodyweights, blood pressure, and HbA1c in diabetic SMAD1 het-erozygous KO mice did not differ from diabetic WT mice(Supplementary Table 4). The glomerular expression ofSmad1 was reduced by ;30% in both diabetic and
nondiabetic SMAD1 heterozygous KO mice (Fig. 6A). Weobserved a partial attenuation of albuminuria (Fig. 6B)and mesangial matrix expansion (Fig. 6C and D) alongwith improvement of the glomerular expression ofCol4a1/a2 and Col1a1/a2 (Fig. 6E) in diabetic SMAD1
Figure 5—Constitutively active Smad1 induces the expression of aSMA in mesangial cells. A: Construction of mutant Smad1. Serine 463and 465 of Smad1 were substituted with aspartic acid (Smad-DVD) to create constitutively active Smad1, or the C-terminus was deleted(Smad1-DC) to create an inactive Smad1. Both Smad1 mutants were recognized by the anti-Smad1 antibody. B: Transcriptional activitiesof Smad1 mutants by luciferase assay in Cos7 cells. Mutant Smad1 or mock expression vector was transfected with 3GC2-Lux into Cos7cells. All transfections were quadruplicated and repeated at least three times. *P < 0.05 vs. mock vector; #P < 0.05 vs. Smad1-DVD.C: Localization of Smad1 mutants in murine cultured mesangial cells. Mouse mesangial cells transfected with each Smad1 mutant vectorwere stained with anti-Smad1 antibody (green) and DAPI (purple) without stimulation. Note that Smad1-DVD was detected in the nucleus,whereas Smad1-DC was detected mainly in the cytoplasm. D: Smad1-DVD increased the transcriptional activity of aSMA in mesangialcells. Each Smad1 mutant vector was transfected with mouse aSMA-promoter luciferase construct into mesangial cells. After 24 h, theluciferase activities of cell lysate were measured as described in RESEARCH DESIGN AND METHODS. Data are mean 6 SD of triplicate determi-nations. *P < 0.05 vs. mock vector; #P < 0.05 vs. Smad1-DVD. E and F: BMP4 increases aSMA in mesangial cells by phosphorylation ofSmad1. Mesangial cells were preincubated with dorsomorphin 1 mmol/L for 30 min followed by stimulation with BMP4 25 ng/mL. Twenty-four hours after stimulation, cells were harvested. One representative blot is presented in E, and the mean optical densitometry of aSMA/b-actin from three independent experiments is presented in F. *P < 0.05 vs. control without dorsomorphin or BMP4; #P < 0.05 vs. BMP4stimulation without dorsomorphin. pSmad, phosphorylated Smad.
diabetes.diabetesjournals.org Matsubara and Associates 2987
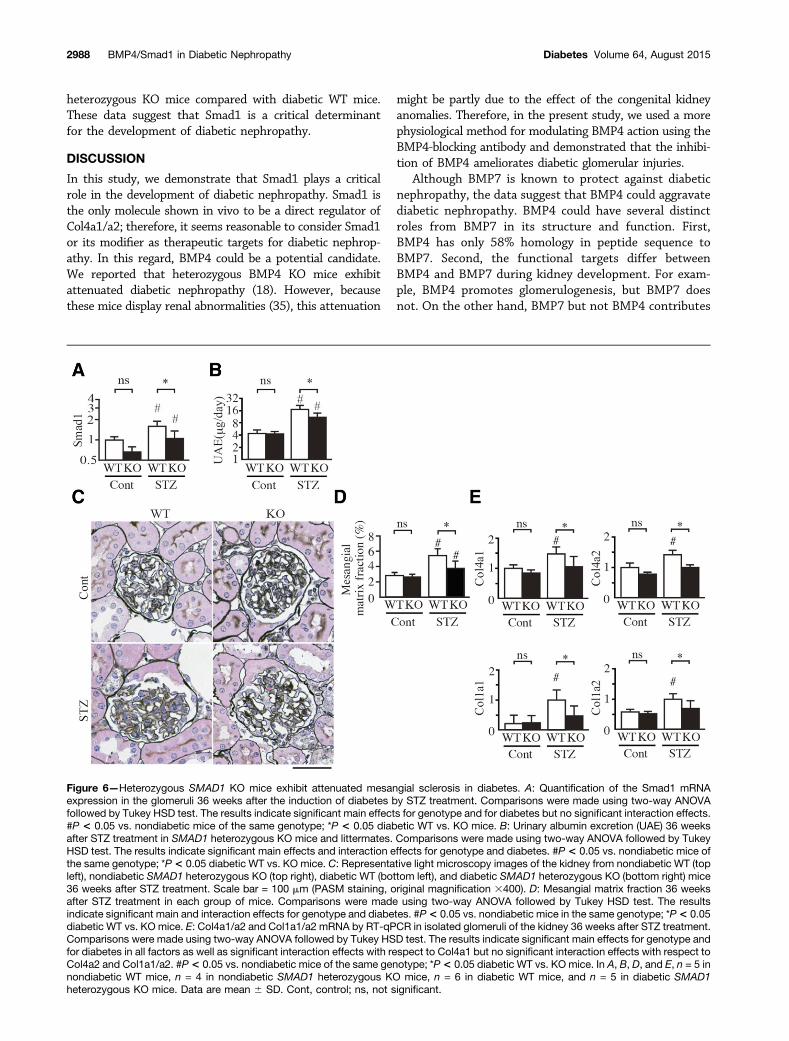
heterozygous KO mice compared with diabetic WT mice.These data suggest that Smad1 is a critical determinantfor the development of diabetic nephropathy.
DISCUSSION
In this study, we demonstrate that Smad1 plays a criticalrole in the development of diabetic nephropathy. Smad1 isthe only molecule shown in vivo to be a direct regulator ofCol4a1/a2; therefore, it seems reasonable to consider Smad1or its modifier as therapeutic targets for diabetic nephrop-athy. In this regard, BMP4 could be a potential candidate.We reported that heterozygous BMP4 KO mice exhibitattenuated diabetic nephropathy (18). However, becausethese mice display renal abnormalities (35), this attenuation
might be partly due to the effect of the congenital kidneyanomalies. Therefore, in the present study, we used a morephysiological method for modulating BMP4 action using theBMP4-blocking antibody and demonstrated that the inhibi-tion of BMP4 ameliorates diabetic glomerular injuries.
Although BMP7 is known to protect against diabeticnephropathy, the data suggest that BMP4 could aggravatediabetic nephropathy. BMP4 could have several distinctroles from BMP7 in its structure and function. First,BMP4 has only 58% homology in peptide sequence toBMP7. Second, the functional targets differ betweenBMP4 and BMP7 during kidney development. For exam-ple, BMP4 promotes glomerulogenesis, but BMP7 doesnot. On the other hand, BMP7 but not BMP4 contributes
Figure 6—Heterozygous SMAD1 KO mice exhibit attenuated mesangial sclerosis in diabetes. A: Quantification of the Smad1 mRNAexpression in the glomeruli 36 weeks after the induction of diabetes by STZ treatment. Comparisons were made using two-way ANOVAfollowed by Tukey HSD test. The results indicate significant main effects for genotype and for diabetes but no significant interaction effects.#P < 0.05 vs. nondiabetic mice of the same genotype; *P < 0.05 diabetic WT vs. KO mice. B: Urinary albumin excretion (UAE) 36 weeksafter STZ treatment in SMAD1 heterozygous KO mice and littermates. Comparisons were made using two-way ANOVA followed by TukeyHSD test. The results indicate significant main effects and interaction effects for genotype and diabetes. #P < 0.05 vs. nondiabetic mice ofthe same genotype; *P< 0.05 diabetic WT vs. KO mice. C: Representative light microscopy images of the kidney from nondiabetic WT (topleft), nondiabetic SMAD1 heterozygous KO (top right), diabetic WT (bottom left), and diabetic SMAD1 heterozygous KO (bottom right) mice36 weeks after STZ treatment. Scale bar = 100 mm (PASM staining, original magnification 3400). D: Mesangial matrix fraction 36 weeksafter STZ treatment in each group of mice. Comparisons were made using two-way ANOVA followed by Tukey HSD test. The resultsindicate significant main and interaction effects for genotype and diabetes. #P< 0.05 vs. nondiabetic mice in the same genotype; *P< 0.05diabetic WT vs. KO mice. E: Col4a1/a2 and Col1a1/a2 mRNA by RT-qPCR in isolated glomeruli of the kidney 36 weeks after STZ treatment.Comparisons were made using two-way ANOVA followed by Tukey HSD test. The results indicate significant main effects for genotype andfor diabetes in all factors as well as significant interaction effects with respect to Col4a1 but no significant interaction effects with respect toCol4a2 and Col1a1/a2. #P< 0.05 vs. nondiabetic mice of the same genotype; *P< 0.05 diabetic WT vs. KO mice. In A, B, D, and E, n = 5 innondiabetic WT mice, n = 4 in nondiabetic SMAD1 heterozygous KO mice, n = 6 in diabetic WT mice, and n = 5 in diabetic SMAD1heterozygous KO mice. Data are mean 6 SD. Cont, control; ns, not significant.
2988 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015

to mesenchymal survival (36). Finally, the downstream tar-get of BMP4 is Smad1 in mesangial cells, as demonstratedin the present study, whereas the downstream target ofBMP7 in mesangial cells is Smad5 (37). Thus, we sug-gest that each BMP has its own function and signalingtargets in kidney development and disease progression.
It remains to be determined whether BMP4/Smad1signaling is specific for diabetic nephropathy. We pre-viously reported increased glomerular expression ofBMP4/Smad1 in another diabetic mouse model (38) andidentified PDGF-b as another activator of Smad1 inmesangial cells through Src in murine experimental glo-merulonephritis (39). Smad1 also operates during chronicstages of fibrosis in scleroderma (40). Thus, we speculatethat the Smad1 signaling is a common pathway amongvarious glomerular injuries and in organ fibrosis. To de-termine the specificity of BMP4 in diabetic nephropathy,the glomerular expression of BMP4 should be tested inother disease models in future experiments.
In this study, albuminuria did not reflect mesangialexpansion in BMP4 antibody–treated diabetic mice anddiabetic Smad1-Tg mice, although diabetic Smad1 hetero-zygous KO mice exhibited slightly improved albuminuria.The mechanisms responsible for these results remain un-known. Previous reports have indicated that albuminuriais not correlated with the severity of mesangial expansionin incipient diabetic nephropathy both in humans (41)and in a rodent model (15). Treatment with an anti–TGF-b antibody does not attenuate albuminuria in db/dbmice despite its beneficial effects on glomerular matrixexpansion (42). These data suggest that distinct mecha-nisms may underlie albuminuria and mesangial matrixexpansion in diabetic nephropathy.
In conclusion, this study of Smad1-overexpressingmice reveals that both the induction and the phosphor-ylation of Smad1 play critical roles in the development ofdiabetic glomerulopathy in vivo. BMP4 might be re-sponsible for the phosphorylation of Smad1 and repre-sents a novel therapeutic target for diabetic nephropathy.
Acknowledgments. The authors thank Hideo Uchiyama and KazumasaUsami (Taigenkai Hospital) for preparing tissue sections and PASM staining andNorihiko Suzuki (Nagoya University) for electron microscope sample preparation.They also thank Ayumi Hosotani and Maki Watanabe (Kyoto University) andS. Hayashi and A. Sakurai (Tokushima University) for excellent technical assistance.The authors are grateful to H. Kanamori (Fukuchiyama City Hospital);M. Matsuura, T. Murakami, and T. Araoka (Tokushima University); K. Torikoshi(Kitano Hospital); A. Fukatsu (Yachiyo Hospital); M. Yanagita (Kyoto University);and T. Kimura (Kyoto University) for helpful discussion. Finally, they thank ChugaiResearch Institute for Medical Science, Inc., colleagues Yousuke Kawase, TakanoriTachibe, Toshio Hani, and Hiromi Tateishi for manipulation of mouse embryos;Toshio Mori for technical support in pathological examination; and Satomi Uchidaand Yumiko Nakajima for excellent technical assistance.Funding. This study was supported by Grants-in-Aid for Young Scientists (B)(21790808), Grants-in-Aid for Scientific Research (21591033), Grants-in-Aid forScientific Research (C) (25461221), the Kidney Foundation of Japan (JKFB09-41), and Takeda Science Foundation.
Duality of Interest. Funding was provided in part by an AstraZenecaVirtual Research Institute research grant. T.D. received collaborative researchfunds from Chugai Pharmaceutical Co., Ltd. No other potential conflicts of interestrelevant to this article were reported.Author Contributions. T.M. contributed to the data analysis and writingof the manuscript. M.A. contributed to the experiments and data analysis. H.Ab.contributed to the experiments and writing of the manuscript. O.U. contributed toestablishing the transgenic mice and to the experiments. K.-i.J. contributed toestablishing the transgenic mice. A.M. contributed to the data analysis. C.G.contributed to the experiments. T.T., S.K., K.N., N.I., and T.K. contributed tothe data analysis. M.K. established the neutralizing antibody and contributed tothe data analysis. N.F. contributed to the study concept and design, experiments,and data analysis. H.Ar. and T.D. contributed the study concept and design andwriting of the manuscript. T.M. and H.Ab. are the guarantors of this work and, assuch, had full access to all the data in the study and take responsibility for theintegrity of the data and the accuracy of the data analysis.Prior Presentation. Parts of this study were presented at the 71st Sci-entific Sessions of the American Diabetes Association, San Diego, CA, 24–28June 2011.
References1. Bojestig M, Arnqvist HJ, Hermansson G, Karlberg BE, Ludvigsson J. De-clining incidence of nephropathy in insulin-dependent diabetes mellitus. N Engl JMed 1994;330:15–182. Kanwar YS, Wada J, Sun L, et al. Diabetic nephropathy: mechanisms ofrenal disease progression. Exp Biol Med (Maywood) 2008;233:4–113. Yamamoto Y, Kato I, Doi T, et al. Development and prevention of advanceddiabetic nephropathy in RAGE-overexpressing mice. J Clin Invest 2001;108:261–2684. Kriz W, LeHir M. Pathways to nephron loss starting from glomerular dis-eases-insights from animal models. Kidney Int 2005;67:404–4195. Striker GE, Schainuck LI, Cutler RE, Benditt EP. Structural-functional cor-relations in renal disease. I. A method for assaying and classifying histopathologicchanges in renal disease. Hum Pathol 1970;1:615–6306. Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC.Structural-functional relationships in diabetic nephropathy. J Clin Invest 1984;74:1143–11557. Roberts AB, McCune BK, Sporn MB. TGF-beta: regulation of extracellularmatrix. Kidney Int 1992;41:557–5598. Striker LJ, Peten EP, Elliot SJ, Doi T, Striker GE. Mesangial cell turnover:effect of heparin and peptide growth factors. Lab Invest 1991;64:446–4569. Fogo AB. Mesangial matrix modulation and glomerulosclerosis. Exp Nephrol1999;7:147–15910. Abe H, Matsubara T, Iehara N, et al. Type IV collagen is transcriptionallyregulated by Smad1 under advanced glycation end product (AGE) stimulation.J Biol Chem 2004;279:14201–1420611. Liu F, Hata A, Baker JC, et al. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature 1996;381:620–62312. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membraneto nucleus through SMAD proteins. Nature 1997;390:465–47113. Tremblay KD, Dunn NR, Robertson EJ. Mouse embryos lacking Smad1signals display defects in extra-embryonic tissues and germ cell formation.Development 2001;128:3609–362114. Huang S, Flanders KC, Roberts AB. Characterization of the mouse Smad1gene and its expression pattern in adult mouse tissues. Gene 2000;258:43–5315. Matsubara T, Abe H, Arai H, et al. Expression of Smad1 is directly asso-ciated with mesangial matrix expansion in rat diabetic nephropathy. Lab Invest2006;86:357–36816. Akagi Y, Isaka Y, Akagi A, et al. Transcriptional activation of a hybridpromoter composed of cytomegalovirus enhancer and beta-actin/beta-globingene in glomerular epithelial cells in vivo. Kidney Int 1997;51:1265–1269
diabetes.diabetesjournals.org Matsubara and Associates 2989

17. Miyata T, Inagi R, Nangaku M, et al. Overexpression of the serpin megsininduces progressive mesangial cell proliferation and expansion. J Clin Invest2002;109:585–59318. Tominaga T, Abe H, Ueda O, et al. Activation of bone morphogenetic protein4 signaling leads to glomerulosclerosis that mimics diabetic nephropathy. J BiolChem 2011;286:20109–2011619. Zhang Y, Riesterer C, Ayrall AM, Sablitzky F, Littlewood TD, Reth M. In-ducible site-directed recombination in mouse embryonic stem cells. NucleicAcids Res 1996;24:543–54820. Tanaka M, Asada M, Higashi AY, et al. Loss of the BMP antagonist USAG-1ameliorates disease in a mouse model of the progressive hereditary kidneydisease Alport syndrome. J Clin Invest 2010;120:768–77721. Suganami T, Mukoyama M, Mori K, et al. Prevention and reversal of renalinjury by leptin in a new mouse model of diabetic nephropathy. FASEB J 2005;19:127–12922. Takemoto M, Asker N, Gerhardt H, et al. A new method for large scaleisolation of kidney glomeruli from mice. Am J Pathol 2002;161:799–80523. Ohashi S, Abe H, Takahashi T, et al. Advanced glycation end products in-crease collagen-specific chaperone protein in mouse diabetic nephropathy. J BiolChem 2004;279:19816–1982324. Yanagita M, Okuda T, Endo S, et al. Uterine sensitization-associated gene-1(USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubularinjury. J Clin Invest 2006;116:70–7925. Nagai K, Arai H, Yanagita M, et al. Growth arrest-specific gene 6 is involvedin glomerular hypertrophy in the early stage of diabetic nephropathy. J Biol Chem2003;278:18229–1823426. Qin BY, Chacko BM, Lam SS, de Caestecker MP, Correia JJ, Lin K.Structural basis of Smad1 activation by receptor kinase phosphorylation. Mol Cell2001;8:1303–131227. Dudley AT, Robertson EJ. Overlapping expression domains of bone mor-phogenetic protein family members potentially account for limited tissue defectsin BMP7 deficient embryos. Dev Dyn 1997;208:349–36228. Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic pro-tein-4 is required for mesoderm formation and patterning in the mouse. GenesDev 1995;9:2105–211629. Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and havedefects in amnion/chorion and cardiac development. Development 1996;122:2977–2986
30. Liu F, Ventura F, Doody J, Massagué J. Human type II receptor for bonemorphogenic proteins (BMPs): extension of the two-kinase receptor model to theBMPs. Mol Cell Biol 1995;15:3479–348631. Koenig BB, Cook JS, Wolsing DH, et al. Characterization and cloning ofa receptor for BMP-2 and BMP-4 from NIH 3T3 cells. Mol Cell Biol 1994;14:5961–597432. ten Dijke P, Yamashita H, Sampath TK, et al. Identification of type I re-ceptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem1994;269:16985–1698833. Anderson GJ, Darshan D. Small-molecule dissection of BMP signaling. NatChem Biol 2008;4:15–1634. Huang S, Tang B, Usoskin D, et al. Conditional knockout of the Smad1 gene.Genesis 2002;32:76–7935. Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogeneticprotein 4 regulates the budding site and elongation of the mouse ureter. J ClinInvest 2000;105:863–87336. Cain JE, Hartwig S, Bertram JF, Rosenblum ND. Bone morphogeneticprotein signaling in the developing kidney: present and future. Differentiation2008;76:831–84237. Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposingtransforming growth factor beta in mesangial cells. J Biol Chem 2004;279:23200–2320638. Kishi S, Abe H, Akiyama H, et al. SOX9 protein induces a chondrogenicphenotype of mesangial cells and contributes to advanced diabetic nephropathy.J Biol Chem 2011;286:32162–3216939. Mima A, Abe H, Nagai K, et al. Activation of Src mediates PDGF-inducedSmad1 phosphorylation and contributes to the progression of glomerulosclerosisin glomerulonephritis. PLoS One 2011;6:e1792940. Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforminggrowth factor-beta receptor type I-dependent fibrogenic gene program is mediatedvia activation of Smad1 and ERK1/2 pathways. J Biol Chem 2007;282:10405–1041341. Chavers BM, Bilous RW, Ellis EN, Steffes MW, Mauer SM. Glomerular le-sions and urinary albumin excretion in type I diabetes without overt proteinuria.N Engl J Med 1989;320:966–97042. Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renalinsufficiency, excess matrix gene expression, and glomerular mesangial matrixexpansion by treatment with monoclonal antitransforming growth factor-betaantibody in db/db diabetic mice. Proc Natl Acad Sci U S A 2000;97:8015–8020
2990 BMP4/Smad1 in Diabetic Nephropathy Diabetes Volume 64, August 2015