Embed Size (px)
Citation preview

Capitolo 5
CRIOPOROSIMETRIA
5.1 Introduzione
L’importante ruolo dei materiali porosi nelle industrie dell’olio,
chimica, della carta, tessile, costruzione, pelletteria, ecc. ha incentivato la
ricerca e l’applicazione di numerose tecniche mirate allo studio della
porosità.
Le tecniche porosimetriche possono essere classificate come segue:
• Metodi diretti; si basano sull’osservazione diretta
mediante l’uso di microscopi elettronici, tecniche
cristallografiche di diffrazione di raggi X, ecc.
Sfortunatamente, questi metodi pur permettendo
l’osservazione diretta del materiale non sono adatti allo
studio di materiali mesoporosi i cui raggi sono compresi tra i
2 e i 50 nm.
• Metodi indiretti; questi metodi si basano
sull’analisi del fenomeno di capillarità. I metodi più
frequentemente utilizzati sono la porimetria a mercurio per i
pori più grandi, mentre la porimetria a gas è utilizzata per la
misura dei pori più piccoli. Le trasformazioni di fase di una
sostanza che avvengono all’interno del materiale poroso che
la contiene, permettono la caratterizzazione della struttura
interna di quest’ultimo.
Ultimamente, un nuovo metodo indiretto sta conquistando
importanza: la crioporosimetria. La crioporosimetria si basa sulle
trasformazioni di fase solido – liquido in una sostanza che si trova
all’interno di una struttura porosa.

Crioporosimetria
118
Molti autori hanno osservato che le condizioni d’equilibrio di
temperatura e pressione delle fasi solida – liquida – gassosa di una
sostanza pura molto dispersa, dipendono dai raggi di curvatura delle
interfacce che si creano tra le fasi.
All’interno di un poro, la superficie dell’interfaccia solido – liquido
di una sostanza pura in esso contenuta, dipende strettamente dalla
dimensione del poro; quindi, la temperatura di solidificazione varierà per
ogni tipo di poro presente nel materiale.
Dunque, il tracciato di fusione (o solidificazione), che riporta
l’andamento del flusso di calore in funzione della temperatura, di una
sostanza conosciuta che fonde (solidifica) all’interno di un materiale
poroso di costituzione sconosciuta, permette di effettuare la seguente
affermazione: la dimensione del poro può essere determinata attraverso la
temperatura di fusione (solidificazione) ed il volume dei pori può essere
determinato attraverso la misura dell’energia coinvolta nella
trasformazione di fase.
Questo metodo è stato utilizzato, fino a non molto tempo fa, come
un metodo relativo per la calibratura di tracciati di campioni la cui
distribuzione del diametro dei pori era già conosciuta a priori.
L’obiettivo di questa trattazione, invece, è rendere questo metodo
assoluto per la determinazione della forma e della dimensione dei pori
attraverso relazioni teoriche.
La forma dei pori è una componente fondamentale che influisce
sulla permeabilità di un materiale, quest’ultima a sua volta, incide per
esempio, sulla cinetica di rilascio di farmaci.
Gli studi di crioporosimetria permettono di interpretare la forma dei
pori attraverso l’analisi del tracciato di fusione o solidificazione ottenuto
per mezzo di uno strumento denominato DSC (differential scanning
calorimeter). Pertanto, nei seguenti paragrafi saranno proposti due
modelli teorici che tengono conto di materiali costituiti da pori cilindrici o
sferici attraverso lo studio del tracciato DSC di fusione [1].

Capitolo 5
119
5.2 Crioporosimetria
La crioporosimetria o crioporimetria è un metodo calorimetrico che
può essere utilizzato per la caratterizzazione della struttura porosa dei
materiali a partire dalla valutazione del punto di fusione (o solidificazione)
di una sostanza in forma di nanocristalli all’interno dei pori di un
materiale.
Lo spostamento della temperatura T alla quale si verifica la
transizione di fase (fusione o solidificazione), dipende strettamente dai
raggi di curvatura delle interfacce delle fasi del sistema.
Un liquido contenuto all’interno di materiale poroso è distribuito e
suddiviso in moltissime parti; questo implica che il raggio di curvatura si
leghi strettamente alla dimensione del poro. Osservazioni sperimentali
sull’acqua, liquidi organici, ossigeno molecolare, ecc., hanno evidenziato
una dipendenza della diminuzione del punto di fusione al diminuire del
raggio nominale del poro.
I protocolli di preparazione dei materiali per le prove di
crioporosimetria, non sono complicati e l’acqua costituisce il liquido di
prova ideale. Dunque, questa tecnica assume ancora una maggior
importanza se i campioni da esaminare sono costituiti d’acqua. Il
vantaggio nell’utilizzo d’acqua, risiede nel proprio calore di fusione Hf =
334 J/gr, il quale è superiore di almeno un ordine di grandezza rispetto
alla maggior parte dei liquidi organici. Il valore del calore latente di
fusione dell’acqua, favorisce e di molto, la sensibilità degli sperimenti di
calorimetria a scansione differenziale per piccoli volumi di liquido.
Si ricorda che un calorimetro a scansione differenziale o differential
scanning calorimeter (DSC), rileva il segnale fornito da fenomeni
esotermici come la solidificazione o da fenomeni endotermici come la
fusione.
Lo svantaggio principale di questo metodo, è che si tratta di un
metodo non tradizionale, ed il suo impiego non è diffuso come i metodi

Crioporosimetria
120
porosimetrici ad azoto o altre tecniche che utilizzano liquidi penetranti
come il mercurio. Inoltre, l’effetto delle interazioni specifiche tra il liquido
di prova ed il mezzo poroso sono ancora incerte e possono dar luogo a
interpretazioni erronee del segnale.
Tuttavia, questo metodo può essere adottato per la determinazione
della dimensione dei pori qualora siano note a priori le seguenti
grandezze fisiche: tensione superficiale, l’angolo di contatto, il calore
latente di fusione ed i volumi specifici in funzioni della temperatura [2].
5.2.1 Determinazione della distribuzione delle
dimensioni dei pori
5.2.1.1 Considerazioni teoriche
Come è già stato suggerito nei paragrafi precedenti, i principi fisici
della porosimetria si basano sulle proprietà di un sistema all’equilibrio
termodinamico e le interazioni che si realizzano tra le interfacce delle fasi
solida, liquida e gassosa del sistema.
Le equazioni di Gibbs - Duhem, che descrivono il bilancio termico,
meccanico e di potenziale chimico di un sistema planare all’equilibrio in
cui coesistono le fasi solida, liquida e gassosa, sono le seguenti:
0=+− sssss dndPVdTS µ (5.1)
0=+− lllll dndPVdTS µ (5.2)
0=+− vvvvv dndPVdTS µ (5.3)
Tuttavia, per tenere conto dell’equilibrio meccanico, termico e di
potenziale chimico di una sostanza pura fortemente suddivisa e dispersa,
anche il contributo dell’energia o tensione superficiale deve essere preso
in considerazione. Quindi, in sistemi in cui le interfacce liquido/solido
(l/s), liquido/vapore (l/v), e solido/vapore (s/v) formino superfici curve, le
equazioni (5.1), (5.2) e (5.3) devono essere associate alle equazioni di
Laplace, le quali descrivono il bilancio per una superficie con raggio di
curvatura arbitrario jij dVdA / tra le fasi i e j :

Capitolo 5
121
=−
l
lvlvvl
dV
dAPP γ (5.4)
=−
v
vsvssv
dV
dAPP γ (5.5)
=−
s
slslls
dV
dAPP γ (5.6)
In cui:
lvγ
Tensione superficiale acqua liquida-vapore a
temperatura T.
vsγ
Tensione superficiale ghiaccio-vapore a temperatura T.
slγ
Tensione superficiale ghiaccio-acqua liquida a
temperatura T.
Quando un sistema chiuso è costituito da un componente puro
nelle sue tre forme fisiche: solida, liquida e gassosa, coesistenti e con
superfici planari, la regola delle fasi di Gibbs afferma che il sistema è
invariante, ovvero non vi sono gradi di libertà: il punto triplo può esistere
ad una ed una sola temperatura e pressione. Tuttavia per superfici curve,
la regola delle fasi di Defay stabilisce che esistono altri due gradi di
libertà indipendenti; questo significa che definendo due raggi di
curvatura indipendenti è possibile definire la temperatura e pressione del
punto triplo del sistema.
Al punto triplo, per un sistema fortemente disperso si hanno a
disposizioni due gradi di libertà, si considerino quindi le interfacce
“vapore – liquido” e “solido – liquido”. Si inizia dunque con la
differenziazione delle equazioni (5.4) e (5.6), i potenziali chimici delle fasi
all’equilibrio sono uguali:

Crioporosimetria
122
=−
v
vlvllv
dV
dAddPdP γ (5.7)
=−
s
slslls
dV
dAddPdP γ (5.8)
Si prosegue con la sottrazione dell’equazione (5.2) all’equazione
(5.1), e l’equazione (5.3) all’equazione (5.2), (siano iii nVv = , volume
molare ed iii nSs = entropia molare):
l
vl
lg
vl
v
vl
vl dPvv
vdP
vv
vdT
vv
ss
−+
−−=
−
− “vapore – liquido” (5.9)
s
ls
sl
ls
l
ls
ls dPvv
vdP
vv
vdT
vv
ss
−+
−−=
−
− “liquido – solido” (5.10)
Si sottrae successivamente l’equazione (5.9) all’equazione (5.10), e
sostituendo vdP e sdP con (5.7) e (5.8) rispettivamente, si ottiene:
−+
−=
−
−−
−
−
s
ssl
ls
s
v
vlv
vl
v
vl
vl
ls
ls
dV
dAd
vv
v
dV
dAd
vv
vdT
vv
ss
vv
ssγγ (5.11)
Questa equazione differenziale del punto triplo (5.11), dimostra
come la temperatura T dipenda dai raggi di curvatura di due interfacce.
Ovviamente, scegliendo in modo diverso le interfacce interagenti, altre
due equazioni molto simili alla (5.11) possono essere definite, tutto
dipende dal tipo di sistema fisico che venga a crearsi [1, 2].
Evidenze sperimentali dicono che nei nanopori riempiti di acqua,
esiste uno strato di spessore β, che non può solidificare a causa delle
interazioni tra le molecole di acqua e la parete solida del nanoporo. Per
distinguere i due tipi di stati d’aggregazione dell’acqua presenti nel

Capitolo 5
123
nanoporo, si parla di acqua solidificabile e acqua che non può
solidificare:
Figura 5.1. Tipi di acqua all’interno del poro: acqua non solidificabile “non
freezable water”, acqua solidificabile “freezable water”.
Sperimentalmente è stato accertato che β è costante, non è
funzione del raggio totale R del poro [3, 4, 5].
Per la determinazione dei diametri si comincia definendo le
seguenti relazioni:
nssp vvv += (5.12)
In cui, pv è il volume del nanoporo, sv è il volume dell’acqua
solidificabile o volume del cristallo d’acqua e nsv è il volume dell’acqua
non solidificabile.
Sulla base del tracciato DSC di melting o fusione, si procede alla
determinazione della distribuzione del raggio dei pori considerando
diverse geometrie e condizioni fisiche all’interno e fuori dei pori.
Ovviamente l’equazione (5.12) vale anche per il volume di tutti i
nanopori presenti, pV , il volume di tutti i cristalli di acqua,
sV ed il
volume di tutta l’acqua non solidificabile nsV :
nssp VVV += (5.13)

Crioporosimetria
124
Supponendo una distribuzione continua dei nanopori da minR a
maxR , ed indicando con N il numero di tutti i nanopori, differenziando
pV , sV e
nsV , per pori cilindrici si ha:
nssp dVdVdV += (5.14)
LdNRdV p
2π= (5.15)
LdNrdV s
2π= (5.16)
pdV indica il volume dei nanopori di raggio R , dN è il numero dei
nanopori di raggio R e sdV è il volume dei cristalli di raggio r .
Dunque:
2)(
2
2
2
β−==
R
RdVs
r
RdVsdVp (5.17)
Nel caso di pori sferici:
dNRdVp3
3
4π= (5.18)
dNrdVs 3
3
4π= (5.19)
Dunque:
3)(
3
3
3
β−==
R
RdVs
r
RdVsdVp (5.20)
Per i pori cubici:
dNrdV s
3= (5.21)
dNRdV p
3= (5.22)
Dunque:
3)(
3
β−=
R
RdVsdVp (5.23)

Capitolo 5
125
Per la generica geometria si ha:
z
R
zRdVsdVp
)( β−= (5.24)
Ora, quello che interessa, è avere la distribuzione del volume dei
nanopori in funzione del loro raggio R, ovvero:
zR
zR
dR
dVs
dR
dVp
)( β−= (5.25)
Bisogna, dunque, valutare dR
dVs.
Vs è dato da:
ρh
HVs
∆
∆= (5.26)
In cui:
H∆ È l’entalpia di fusione di Vs [Joule].
h∆ È l’entalpia di fusione per unità di massa di Vs [J/kg].
ρ È la densità di Vs [kg/m3] .
La teoria di fusione dei nanocristalli dice che H∆ , h∆ , sono
funzioni della temperatura di fusione T , che a sua volta, è funzione del
raggio “ r ” del cristallo [6]. Ovviamente, essendo la densità ρ funzione
della temperatura, anche essa può essere vista come funzione del raggio
“ r ” del nanocristallo. Dunque, il volume dei cristalli di raggio “ r ” che
fonde a temperatura T, sarà dato da:
drdr
dVsdVs = (5.27)
Ricordando che β−= Rr , si ha che dRdr = , da cui:
dRdR
dVsdVs = (5.28)

Crioporosimetria
126
∆
∆=
))(())((
))((
rTrTh
rTH
dR
d
dR
dVs
ρ (5.29)
=∆
∆+
∆∆−−∆
∆
=))(())((
))((
22 rTrTh
dR
dh
dR
hdHrTh
dR
H
ρ
ρρρ
=∆
∆∆−
∆
∆
∆−
∆
∆=
dR
d
h
hH
dR
hd
h
H
hdR
Hd ρ
ρρ
ρ
ρ 2222
1
=∆
∆∆−
∆
∆
∆−
∆
∆=
dR
dT
dT
d
h
hH
dR
dT
dT
hd
h
H
hdR
dT
dT
Hd ρ
ρρρ 222
1
=
∆
∆−
∆
∆
∆−
∆
∆=
dT
d
h
H
dT
hd
h
H
hdT
Hd
dR
dT ρ
ρρρ 22
1
=
∆
∆−
∆
∆
∆−
∆
∆=
dT
dt
dt
d
h
H
dT
dt
dt
hd
h
H
hdT
dt
dt
Hd
dR
dT ρ
ρρρ 22
1
=
∆
∆−
∆
∆
∆−
∆
∆=
dT
dt
dt
d
h
H
dT
dt
dt
hd
h
H
hdT
dt
dt
Hd
dR
dT ρ
ρρρ 22
1
=
∆
∆−
∆
∆
∆−
∆
∆=
dt
d
h
H
dt
hd
h
H
hdt
Hd
dT
dt
dR
dT ρ
ρρρ 22
1
ρρ
ρ hv
Q
dR
dThd
hv
Q
dR
dT
initesimo∆
≅
∆−
∆=
&
43421
&
inf
)ln(1
Dunque dR
dVs è dato da:
ρhv
Q
dR
dT
dR
dVs
dr
dVs
∆==
& (5.30)
In cui:
dt
HdQ
∆=& È la potenza del tracciato DSC [ ]minJ .

Capitolo 5
127
dt
dTv = È la velocità di riscaldamento [ ]minC° .
Naturalmente, si sarebbe potuto giungere all’equazione (5.30)
dicendo che il volume dei nanocristalli di raggio “ r ” è dato da:
ρh
Hd
dr
dVs
∆
∆= (5.31)
In cui:
=∆
∆==
ρhdR
Hd
dR
dVs
dr
dVs 1
{=
∆
∆=
∆
∆=
ρρ hdR
dT
dT
dt
dt
Hd
hdR
dT
dT
Hd
vQ
11
1321&
dr
dVs
dR
dVs
dR
dT
hv
Q==
∆=
ρ
&.
Combinando le equazioni (5.25) e (5.30), si ottiene:
zR
zR
dR
dT
hv
Q
dR
dVp
)( βρ −∆=
&
(5.32)
In cui:
Q& È noto dal tracciato DSC.
v È la velocità di riscaldamento della DSC,
anch’essa nota.
dR
dTeh∆
Vanno valutati sulla base delle equazioni che
mettono in relazione T, il raggio del cristallo r e
l’entalpia specifica in funzione della T e di r.

Crioporosimetria
128
ρ La densità del ghiaccio, deve essere valutata
con apposite equazioni.
β Spessore dell’acqua non solidificabile, viene
valutata con una procedura iterativa.
Prima di continuare, bisogna procedere alla definizione di alcune
grandezze fisiche in funzione della temperatura.
Entalpia molare di Fusione h∆ [6]
∫∆−
−+−−∆=∆oT
T cp
ls
l
sl
s
svo dTcpcp
rThh
43421)()(
3)(
ρ
γ
ρ
γ [J/kg]
oT Temperatura di fusione del cristallo di raggio ∞.
sρ Densità del ghiaccio alla temperatura di fusione T.
lρ Densità del liquido alla temperatura di fusione T.
svγ Tensione superficiale ghiaccio-vapore a T.
lvγ Tensione superficiale acqua liquida-vapore a T
scp Calore specifico del solido (ghiaccio).
lcp Calore specifico del liquido (acqua).
L’entalpia specifica di fusione dell’acqua a 0 °C è pari a:
333797)0( ==∆ oTh ][ KgJ

Capitolo 5
129
Tensione Superficiale [1, 3, 7]
In letteratura si trovano diverse espressioni per la dipendenza delle
tensioni superficiale γsl e γlv dalla temperatura:
310)]20(138.088.72[ −∗−−= Tlvγ ][ 2mJ
310)]0(39.09.40[ −∗−+= Tslγ ][ 2mJ
Oppure:
][10)](102.08.23[ 23 mJCTsl
−∗°−=γ
][10)](283.05.30[ 23 mJCTsl
−∗°−=γ
][10)]0(323.036.20[ 23 mJTsl
−∗−−=γ “pori cilindrici”
][10)]0(188.043.20[ 23 mJTsl
−∗−−=γ “pori sferici”
La determinazione di γsv, si ottiene ricordando che l’acqua bagna
perfettamente il ghiaccio (angolo di contatto = 0 => 1cos =ϑ ) e
l’equazione di Young svsllv γγϑγ =+cos , diviene:
sllvsv γγγ +=
Dunque, noti γsl e γlv, si può determinare γsv.
Densità dell’acqua [3]
sghiaccio ρρ =
lOH ρρ =2
)1017.1032.1(917.0 4Ts
−∗−=ρ ][ 3cmg
3724 108649.3101959.30882.01114.7 TTTl
−− ∗+∗−+−=ρ ][ 3cmg
Le temperature delle equazioni di cui sopra vanno espresse in °K.
Calore specifico dell’acqua [1, 8]
sghiaccio cpcp =

Crioporosimetria
130
lOH cpcp =2
)107.3731(114.2 5−∗∗+= Tcp s ])([ CgJ °
)10541(222.4 5−∗∗−= Tcp l ])([ CgJ °
Oppure,
)10541(222.4 5−∗∗−= Tcp l ])([ KgJ °
Alla luce del fatto che BTAcp s += e FTEcp l += , si avrà
2
)()())(()()(
22
00
0 TTFBTTEAFTEBTAdTcpcp
T
T
T
T cp
ls
o −−+−−=−−+=− ∫∫
∆−
43421
Definizione di dR
dT
Per definire dR
dT è necessario rivedere l’equazione differenziale del
punto triplo per sistemi fortemente dispersi:
−+
−=
−
−−
−
−
s
ssl
ls
s
v
vlv
vl
v
vl
vl
ls
ls
dV
dAd
vv
v
dV
dAd
vv
vdT
vv
ss
vv
ssγγ (5.11)
In cui:
vls sss ,, Entropia molare fase solido, liquido, vapore
rispettivamente.
vls vvv ,, Volume molare fase solido, liquido, vapore
rispettivamente.
vV Volume fase vapore.
sV Volume fase solida.
vA Area interfacciale fase vapore.

Capitolo 5
131
sA Area interfacciale fase solida.
lvγ Tensione superficiale acqua liquida – vapore a T.
slγ Tensione superficiale ghiaccio-vapore a T.
Ipotizzando che vs vv << e vl vv << , si ha anche
vl
vl
ls
ls
vv
ss
vv
ss
−
−>>
−
−.
Dunque:
−+
−=
−
−
s
ssl
ls
s
v
vlv
ls
ls
dV
dAd
vv
v
dV
dAddT
vv
ssγγ (5.33)
Detto T
HssS
f
slf
∆=−=∆ , si ottiene:
∆−
∆
−=
s
ssl
f
s
v
vlv
f
ls
dV
dAd
S
v
dV
dAd
S
vvdT γγ (5.34)
fS∆ Entropia molare di fusione alla temperatura di
fusione T.
fH∆ Entalpia molare di fusione alla temperatura di
fusione T.
Si assume che la fusione avvenga all’equilibrio
0=∆−∆=∆ fff STHG
Arrivati a questo punto, bisogna valutare l’equazione (5.34) in base
alle diverse condizioni fisiche di fusione che si possono verificare nei
mezzi porosi che si stanno studiando, di conseguenza, è necessario
distinguere due casi:
a) l’acqua nel sistema poroso è in eccesso rispetto al
volume dei pori;

Crioporosimetria
132
b) l’acqua non è in eccesso.
La distinzione sperimentale dei due casi si opera semplicemente
guardando il tracciato DSC. Se compare il picco di fusione dell’acqua a
T=0°C, il sistema poroso contiene acqua che eccede il volume totale dei
pori, caso “a”; altrimenti, l’acqua si trova completamente all’interno dei
pori, caso “b”. Nelle figure successive, si possono apprezzare entrambe le
situazioni.
0
5
10
15
20
25
-4.50 -3.50 -2.50 -1.50 -0.50 0.50
T [°C]
Flusso di
Calore, Q
[mW]
Figura 5.2. Tracciato DSC di una membrana di idrogel di alginato di rame al
2% in peso. Il tracciato DSC di fusione denota l’esistenza di acqua strutturata in
nanopori che fonde a T < 0 °C. Non vi è acqua fuori dai pori che fonde a 0°C.
0.35422
-0.57466
0.033433
-2.5 -1.5 -0.5 0.50
2.5
5
7.5
10
12.5
0
2.5
5
7.5
10
12.5
2.5
5
7.5
10
12.5
15
2.5
5
7.5
10
12.5
15
Figura 5.3. Tracciato DSC di una membrana di idrogel di alginato di rame al
2% in peso invecchiata. Scomponendo la curva bianca come somma di tre curve,
la curva gialla denota l’esistenza d’acqua strutturata in pori più piccoli rispetto
alle altre due. Il resto dell’acqua fonde a 0 °C.

Capitolo 5
133
Pori cilindrici con acqua in eccesso rispetto al volume totale
dei pori
Aumentando la temperatura da valori molto bassi rispetto a 0°C, si
ha la fusione dei cristalli di acqua che si trovano confinati nei pori più
piccoli; mentre tutta l’altra acqua resterà allo stato solido. Dunque la
situazione che si verifica è quella disegnata in figura 5.4:
Figura 5.4. Condizione tipica negli sperimenti di termoporosimetria, in
questo caso, c’è un eccesso d'acqua nel materiale poroso. La fase solida in eccesso,
forma un superficie planare con la fase vapore.
slϑ , è l’angolo di contatto tra la fase liquida e quella solida. Poiché
vi è presenza di acqua non solidificabile adesa alle pareti del poro, si ha
che slϑ = 0.
Sulla base del disegno in figura 5.4, l’interfaccia solido – vapore è
piatta e dunque v
v
dV
dA= 0 , cioè, se parte del solido (ghiaccio) andasse in
fase vapore, causando un incremento di Vv, l’area interfacciale
rimarrebbe costante. Questo discende dal fatto che la curvatura prima e
seconda dell’interfaccia S-V sono uguali svR
1 e valgono zero, la superficie
non è curva ma piatta ∞→svR .

Crioporosimetria
134
Per quanto riguarda s
s
dV
dA, si ha che
s
s
dV
dA=
slR
2. Infatti
s
s
dV
dA è uguale
alla somma della curvatura prima e seconda che, in questo caso, sono
uguali in quanto si è implicitamente assunto che l’interfaccia S-L abbia
forma di una calotta sferica, poiché slϑ = 0 : siamo in presenza di una
mezza sfera. Inoltre, le due curvature sono > 0 in quanto la superficie
interfacciale S-L è concava rispetto alla fase solida. In altre parole, un
aumento di Vs comporta un aumento di As :
→
2
1 14
slRAS π=
< 2
2 24
slRAS π=
Allora l’equazione (5.34) diventa:
∆−=
∆−
∆
−=
sl
sl
f
s
R
s
ssl
f
s
v
vlv
f
ls
Rd
S
v
dV
dAd
S
v
dV
dAd
S
vvdT
sl
γγγ 2
2
0
0
321
444 3444 21
321
(5.35)
Integrando tra T0 , temperatura di fusione a 0 °C e T , temperatura
di fusione del nanocristallo:
∫∫
∆−=−=
slR
sl
sl
f
s
T
TR
dS
vTTdT
1
0
0
12
0
γ (5.36)

Capitolo 5
135
Si assumono sv , slγ e fS∆ costanti. Sebbene questo non sia della
tutto vero, data la tecnica numerica che verrà adottata, tale
approssimazione risulta ragionevole.
Dunque, la soluzione dell’equazione (5.34) è:
slf
sls
RS
vTT
120
∆−=−
γ (5.37)
TRH
vTT
slf
sls 120
∆−=−
γ (5.38)
Se, nell’integrazione della (5.36), avessimo supposto ∆Sf = T*∆Hf,
avremmo avuto:
sl
slsf
R
vHTT
γ2)ln( 0 −=∆ (5.39)
Con passi di integrazione < di 0.5 °C, la (5.38) e la (5.39) sono
assolutamente equivalenti.
In termini di unità di massa:
TRH
TTsslf
sl
ρ
γ 120
∆−=− (5.40)
)12
1(0
sslf
sl
RHTT
ρ
γ
∆+= (5.41)
ssl
slf
RHTT
ρ
γ2)ln( 0 −=∆ (5.42)
Pori cilindrici senza acqua in eccesso rispetto al volume totale
dei pori
Aumentando la temperatura da valori ben inferiori a quella di
fusione dell’acqua, T = 0 °C, si avrà la fusione dei nanocristalli a partire
da quelli più piccoli. La situazione fisica che si realizza è quella

Crioporosimetria
136
evidenziata in figura 5.5, ( slϑ , è l’angolo di contatto solido - liquido; lvϑ , è
l’angolo di contatto liquido – vapore, entrambi si assumono uguali a zero)
Figura 5.5. Condizione tipica negli sperimenti di crioporosimetria, in
questo caso non c’è un eccesso d'acqua nel poro. La fase liquida forma una
superficie non planare con la fase gassosa.
In questo caso abbiamo v
v
dV
dA=
lvR
2, la fase vapore prevede
un’interfaccia concava, quindi con curvatura prima e seconda positive ed
uguali a lvR
1 e
s
s
dV
dA=
slR
2, anche in questo caso la fase solida prevede
un’interfaccia concava e dunque, positiva.
Pertanto la (5.34) diventa:
∆−
∆
−=
s
ssl
f
s
v
vlv
f
ls
dV
dAd
S
v
dV
dAd
S
vvdT γγ =
∆−
∆
−=
sl
sl
f
s
lv
lv
f
ls
Rd
S
v
Rd
S
vvdT
22γγ (5.43)

Capitolo 5
137
Integrando tra T e T0, mantenendo fS∆ sv , lv slγ e lvγ costanti, si
ha:
∫∫∫
∆−
∆
−=−=
sllv R
sl
sl
f
s
R
lv
lv
f
ls
T
TR
dS
v
Rd
S
vvTTdT
1
0
1
0
0
1212
0
γγ (5.44)
sl
slf
s
lv
lvf
ls
RS
v
RS
vvTT γγ
∆−
∆
−=−
220 (5.45)
sl
slf
slv
lvf
ls
RH
vT
RH
vvTTT γγ
∆−
∆
−=−
220 (5.46)
sl
slf
slv
lvf
ls
RH
vT
RH
vvTTT γγ
∆−
∆
−=−
220 (5.47)
( )( )ssllslv
f
vvvRH
TTT γγ −−
∆=−
20 (5.48)
( )( )
−−
∆+= lslvssl
f
vvvRH
TT γγ2
10 (5.49)
Anche in questo caso, se, nell’integrazione della (5.44), avessimo
supposto ∆Sf = T*∆Hf, avremmo avuto:
( ) ( )( )lslvsslf vvvR
HTT −−−=∆ γγ2
ln 0 (5.50)
Le eq.(5.49) e (5.50) sono equivalenti per intervalli di T < 0.5°C.
E’ interessante notare che, essendo vs > vl, il termine
( ) 0>− lslv vvγ . Ovviamente R = Rsl = Rlv.
L’equazione (5.49), si può esprimere in termini di massa:
−−
∆+=
ls
lv
s
sl
f RHTT
ρργ
ρ
γ 11210 (5.51)
Essendo 011
≅
−
ls ρρ,
La (5.50), invece, diviene:

Crioporosimetria
138
( ) ( )( )lslvsslfR
HTT ρργργ 112
ln 0 −−−=∆ (5.52)
Pori Sferici
Per i pori sferici l’unico modello adattabile è il semplice sistema a
due fasi mostrato in figura 5.1, (non prevede acqua in eccesso, e quindi
non è previsto un sistema trifasico in equilibrio), per tale sistema bifasico
fortemente disperso all’equilibrio occorre definire le equazioni di equilibrio
accoppiate alle equazioni di Laplace, si ha perciò:
0=+−− SLSLSSLL dAdVPdVP γ (5.53)
Essendo SL dVdV −= , si ha dunque:
SLSLSSSL dAdVPdVP γ−=− (5.54)
SLSLSLS dAPPdV γ−=− )( (5.55)
SL
SL
S
SLSLLS
RdV
dAPP
γγ
2)( ==− (5.56)
SL
SLLS
RdPdP
γ2+= (5.57)
Le due equazioni di Gibbs - Duhem, per un sistema bifasico sono:
0=−+ SSS dPVdudTs (5.58)
0=−+ LLL dPVdudTs (5.59)
Sottraendo la (5.59) alla (5.58):
0)( =+−− LLSSLS dPVdPVdTss (5.60)
Sostituendo SL
SLLS
RddPdP
γ2+= alla (5.60) si ottiene:
0)2
()( =++−−LL
SL
SLLSLS
dPVR
ddPVdTssγ
(5.61)

Capitolo 5
139
02)()( =−−+−SL
SLS
SLLLSR
VdVVdPdTss
γ (5.62)
0)1
(2)()( =−−+−SL
SLSLSLLSR
dVdPVVdTss γ (5.63)
Integrando tra T e T0 la (5.63):
01
2)(000
=−−+∆− ∫∫∫SLR
SL
SLS
P
P
LSL
T
T
fR
dVdPVVT
dTH γ (5.64)
02
))(()ln( 00 =−−−+∆−SL
SLS
SLfR
VPPVVTTH
γ (5.65)
La pressione della fase liquida non varia al variare del raggio del
nanocristallo, pertanto 0)( 0 =− PP pertanto l’equazione (5.65) diventa:
SL
SLSf
R
VTTH
γ2)ln( 0 −=∆ (5.66)
Un'altra via consiste nell’arrangiare diversamente l’equazione
(5.63):
0))1(2()(
)1(2)(
=−+−−
−−
444444 3444444 21SLSLLSLS RdVVVdP
SLSLSLSSLS RddPVdPVdTss
γ
γ (5.67)
Integrando tra gli estremi d’integrazione corrispondenti:
0)1(2)()(
1
00
=−−+− ∫∫∫SLS
L
R
SLLSL
P
P
SSL
T
T
LS RdVdPVVdTss γ (5.68)
02
))(()ln( 0 =−−−+∆−SL
LSLLSSLf
R
VPPVVTTH
γ (5.69)
Dall’equazione di Laplace (5.56) si ha cheSL
SLLS
RPP
γ2)( =− , pertanto
l’equazione (5.69) diventa:
022
)()ln( 0 =−−+∆−SL
LSL
SL
SLSLf
R
V
RVVTTH
γγ (5.70)

Crioporosimetria
140
In termini molari:
SL
SLSf
RVTTH
γ2)ln( 0 −=∆ (5.71)
In termini di massa:
SLS
SLf
RTTH
ρ
γ2)ln( 0 −=∆ (5.71’)
5.2.1.2 Considerazioni Matematiche
Una volta definite tutte le equazioni necessarie si procede al calcolo
delle grandezze.
Valutazione di β [1]
La procedura è iterativa. Preventivamente si valutano le seguenti
grandezze:
∫°=
∆=
CT
T
fp dTThv
QW
00
)(
1& (5.72)
∫°=
∆=
CT
T ICE
fpThT
dT
v
QV
00
)()(ρ
& (5.73)
∫>
∆=
0
0
2 )(
1
0
0
TT
T
H dTThv
QW
& (5.74)
02Hfptnfp WWWW −−= (5.75)
In cui:
tW Massa di acqua totale presente nel sistema dentro e
fuori dai pori. Si misura sperimentalmente. [Kg]
fpW Massa di acqua che solidifica nei pori. Si valuta dal
tracciato sperimentale DSC. [Kg]
fpV Volume occupato dall’acqua ghiacciabile. Si valuta
dal tracciato sperimentale DSC. [m3]

Capitolo 5
141
02HW Massa di acqua che fonde a 0°C. si valuta dal
tracciato sperimentale DSC. [Kg]
nfpW Massa di acqua contentua nei pori che non fonde
poiché aderisce alla parete del poro. [Kg]
h∆ È l’entalpia di fusione per unità di massa di [J/kg]
ICEρ È la densità del ghiaccio di Vfp a T<0°C, e non
solidificabile perché aderito alle pareti. [kg/m3]
Il volume dei pori può essere valutato in questo modo:
fp
ICE
nfp
p VC
WV +
°=
)0(ρ (5.75)
Procedura iterativa per la stima di β
1) assumere β e assumere la geometria dei pori, z
2) calcolare dRR
R
dR
dT
RRhv
QV
ZCTR
R ICE
calcp
Max
Min
−∆= ∫
°=
βρ
)0(
.,)()(
& (5.76)
3) valutare ε<−
=∆
p
pcalcp
p V
VV
V
V , (5.77)
In cui ε è la tolleranza.
Se il requisito non viene soddisfatto, allora si pone βββ ∆+= , e
si ripetono i passi iniziando dal punto 2.
Altrimenti il valore di β è stato determinato. La procedura di
calcolo è arrivata alla FINE.
Noto β , si può finalmente avere la distribuzione di probabilità
delle dimensioni dei pori cercata:

Crioporosimetria
142
dRdR
dV
dR
dV
dR
dMax
Min
R
R
p
p
∫=
φ (5.78)
Ponendo dR
dRf
φ=)( , si ottiene:
1)( ==
=
∫
∫∫∫
dRdR
dV
dRdR
dV
dRdR
ddRRf
Max
Min
Max
Min
Max
Min
Max
Min
R
R
p
R
R
p
R
R
R
R
φ (5.79)
Determinazione della funzione dR
dT
Poter risolvere l’integrale (5.76), è necessario calcolare dR
dT.
A tale fine conviene procedere come segue:
I° intervallo
1
01 )(
00R
ATTSdT
T
HSdT
T
T
T
T
−=−∆≅∆
=∆ ∫∫ (5.80)
SSLA ργ /2=
Pori sferici.
Pori cilindrici con acqua in
eccesso.
))11
((2LS
LV
S
SLAρρ
γρ
γ−−=
Pori cilindrici senza acqua in
eccesso.
Dunque, )( 011
1TTS
AR
−∆−= (5.81)
Ma deve anche risultare:

Capitolo 5
143
)(3 1001 TTcHH p
l
lv
s
sv −∆−
−−∆=∆
ρ
γ
ρ
γ (5.82)
)(3
10
1
0011 TTcR
STST p
l
lv
s
sv −∆−
−−∆=∆
ρ
γ
ρ
γ (5.83)
Riarrangiando la (5.83) si ottiene:
−
−∆−∆−∆=
l
lv
s
sv
p TTcSTST
R
ρ
γ
ρ
γ3
)(1 101100
1
(5.84)
)(
3
100011
1TTcSTST
Rp
l
lv
s
sv
−∆+∆−∆
−−
=ρ
γ
ρ
γ
(5.85)
Uguagliando la (5.85) alla (5.81) si ottiene:
)(
3
)( 100011011 TTcSTSTTTS
A
p
l
lv
s
sv
−∆+∆−∆
−
=−∆
ρ
γ
ρ
γ
(5.86)
Determinazione di 1S∆ :
( ) )()(3
011100011 TTSTTcSTSTA
p −∆=−∆+∆−∆β
(5.87)
[ ])(3
)(3
1000011
1 TTcSTA
TTAT
S p −∆−∆=
−−∆
ββ (5.88)
β
β
3)(
)(
3 101
0010
1 ATTT
STTTcAS
p
−−
∆−−∆=∆ (5.89)
111 STH ∆=∆ (5.90)
)(
3
)( 100011011
1TTcSTSTTTS
AR
p −∆+∆−∆
−=
−∆
−=
β (5.91)
In cui
−=
l
lv
s
sv
ρ
γ
ρ
γβ .

Crioporosimetria
144
Intervallo i-esimo
i
i
j
iiijjj
T
TR
ATTSTTSSdT
i
−=−∆+−∆=∆ ∑∫−
=
−−
1
1
11 )()(
0
(5.92)
)(
3
)()( 000
1
1
11ipii
i
j
iiijjj
iTTcSTST
TTSTTS
AR
−∆+∆−∆
−=
−∆+−∆
−=
∑−
=
−−
β (5.93)
[ ])(3
)()( 000
1
1
11 ipii
i
j
iiijjj TTcSTSTA
TTSTTS −∆+∆−∆=−∆+−∆∑−
=
−−β
(5.94)
[ ] ∑−
=
−− −∆−−∆+∆−∆=
−−∆
1
1
10001 )()(33
)(i
j
jjjipii
i
iii TTSTTcSTSTAAT
TTSββ
(5.95)
[ ]
−−
−∆−−∆+∆−∆
=∆
−
−
=
−∑
β
β
3)(
)()(3
1
1
1
1000
i
ii
i
j
jjjipii
iAT
TT
TTSTTcSTSTA
S (5.96)
iii STH ∆=∆ (5.97)
Infine:
)(
3
000 ipii
iTTcSTST
R−∆+∆−∆
−=
β (5.98)
Poiché questa trattazione è termodinamica, T deve essere espressa
in gradi Kelvin (K), infatti se si usassero i gradi Celsius (°C), si avrebbe
∞→∆ iS per CTi °→ 0 , essendo i
i
iT
HS
∆=∆ .
Si potrebbe calcolare l’integrale ∫∫∆
=∆T
T
T
T
dTT
HSdT
00
anche in
maniera diversa. Ritenendo costante ∆Hf e non ∆Sf. In tal caso avremmo
( )01ln
0
TTHSdT
T
T
∆=∆∫ . Infatti, procedendo in maniera analoga, si arriva
allo stesso risultato.

Capitolo 5
145
I° Intervallo
1
011 )ln(1
0R
ATTHdT
T
HT
T
−=∆=∆∫ (5.99)
)ln( 011
1TTH
AR
∆−= (5.100)
In cui 0T è la temperatura di fusione del ghiaccio a 0°C e 01 TT < .
))/1/1(/(2 LSLVSSLA ρργργ −−= Pori cilindrici senza acqua
in eccesso.
SSLA ργ /2=
Pori sferici.
Pori cilindrici con acqua in
eccesso.
Valutazione di 1H∆ , deve anche risultare:
∫∆
−−
−−∆=∆
0
1
)(3
1
01
T
Tc
pspl
l
lv
s
sv
p
dTccR
HH4434421
43421β
ρ
γ
ρ
γ (5.101)
dTcHHR
T
T
p∫ ∆−∆−∆=0
1
10
1
3β (5.102)
dTcHH
RT
T
p∫ ∆+∆−∆
−=
0
1
01
1
3β (5.103)
Uguagliando la (5.100) alla (5.103), si ha:
)ln(
3
011
01
0
1
TTH
A
dTcHH
T
T
p
∆=
∆+∆−∆ ∫
β

Crioporosimetria
146
A
TTHdTcHH
T
T
p
)ln(
3
011
01
0
1∆
=
∆+∆−∆ ∫
β
)ln(3
01101
0
1
TTHdTcHHA
T
T
p ∆=
∆+∆−∆ ∫β
−∆=
∆−∆∫ ββ 3
)ln(3
0110
0
1
ATTHHdTc
AT
T
p
β
β
3)ln(
301
0
1
0
1
ATT
HdTcA
H
T
T
p
−
∆−∆
=∆
∫ (5.104)
Vediamo ora gli altri intervalli:
i
i
j
iiijjj
T
TR
ATTHTTHdT
T
Hi
−=∆+∆=∆
∑∫−
=
−−
1
1
11 )ln()ln(
0
(5.105)
∑∫−
=
−− ∆+∆=∆
−=
1
1
11 )ln()ln(
0
i
j
iiijjj
T
T
i
TTHTTHdTT
H
AR
i (5.106)
Ma deve anche valere:
dTcR
HH
T
T
p
i
i ∫ ∆−−∆=∆0
1
30
β (5.107)
dTcHHR
T
T
pi
i
∫ ∆−∆−∆=0
1
0
3β (5.108)
dTcHH
RT
T
pi
i
∫ ∆+∆−∆
−=
0
1
0
3β (5.109)
Uguagliando la (5.106) alla (5.109):

Capitolo 5
147
dTcHHTTHTTH
AT
T
pi
i
j
iiijjj ∫∑ ∆+∆−∆
=
∆+∆−
=
−−
0
1
0
1
1
11
3
)ln()ln(
β
β3
)ln()ln(
0
1
0
1
1
11dTcHH
A
TTHTTH
T
T
pi
i
j
iiijjj ∫∑ ∆+∆−∆
=
∆+∆−
=
−−
∑∫−
=
−− ∆−
∆−∆=
−∆
1
1
101 )ln(33
)ln(0
1
i
j
jjj
T
T
piii TTHHdTcAA
TTHββ
β
β
3)ln(
)ln(3
1
1
1
10
0
1
ATT
TTHHdTcA
H
ii
i
j
jjj
T
T
p
i
−
∆−
∆−∆
=∆
−
−
=−∑∫
(5.110)
In cui:
( ) ( )[ ]dTTBFAEdTc
T
T
i
T
T
p ∫∫ −+−=∆0
1
0
1
(5.111)
( )( ) ( )( )22
00
0
1
iii
T
T
p TTBFTTAEdTc −−+−−=∆∫ (5.112)
( ) ( )( )[ ]( )ii
T
T
p TTBFTTAEdTc −−++−=∆∫ 00
0
1
(5.113)
( )ip
T
T
p TTCdTc −∆=∆∫ 0
*0
1
(5.114)
Dove ( ) ( )( )BFTTAEC ip −++−=∆ 0
* (5.115)
5.3 Porosimetria a Gas
5.3.1 Determinazione della distribuzione delle
dimensioni dei pori
5.3.1.1 Considerazioni Teoriche
La porosimetria a gas si basa sul principio di condensazione
capillare e sulle interazioni che si stabiliscono tra le fasi vapore - liquida
all’equilibrio all’interno dei pori [2]. La Figura 5.6 mostra l’esempio di un

Crioporosimetria
148
liquido all’equilibrio con la propria fase gassosa, confinato all’interno di
un poro cilindrico. Il liquido bagna la parete con un angolo di contatto Θ.
Questa semplice figura rappresenta la regione ad alta pressione in una
condizione d’assorbimento isotermo. Gli effetti gravitazionali non sono
considerati.
Per un fluido con bagnabilità perfetta, i.e. Θ =0, il raggio di
curvatura è identico al raggio del poro. Il liquido è dalla parte convessa
dell’interfaccia. Prendendo in considerazione l’equazione di Laplace,
secondo lo schema rappresentato in figura 5.6, si nota che la tensione di
vapore della fase vapore è maggiore rispetto alla pressione idrostatica
esercitata dal liquido. La figura 5.6 rappresenta il modello fisico che si
utilizzerà per la determinazione della dimensione dei pori.
In genere, la fase dal lato concavo esercita una pressione maggiore
rispetto alla fase che si trova dalla parte convessa dell’interfaccia.
Figura 5.6. Liquido all'interno di un poro cilindrico in equilibrio con la fase
vapore. Il liquido bagna le pareti del poro con un angolo di contatto Θ, il raggio di
curvatura risultante è più grande del raggio del poro rp.
Un approccio per la determinazione del raggio del poro, utilizza le
equazione di Gibbs- Duhem per descrivere le equazioni di stato
termodinamiche per la coesistenza delle fasi liquide e gassose. Le
equazioni di Laplace sono state utilizzate per la descrizione dell’equilibrio
meccanico all’ interfaccia gas-liquido con tensione interfacciale γgl.
Per poter determinare il raggio del poro attraverso la tecnica di
porosimetria a gas, bisogna introdurre le equazione di Gibbs – Duhem.

Capitolo 5
149
0=+− lllll dndPVdTS µ (5.2)
0=+− vvvvv dndPVdTS µ (5.3)
Le equazioni (5.2) e (5.3) riferiscono il bilancio termodinamico tra la
fase vapore e la fase liquida all’esterno del poro quando le superfici o aree
interfacciali tra le fasi sono piatte. Le equazioni mettono in relazione, i
contributo chimici, termici e meccanici, all’equilibrio, su variazioni
infinitesime del potenziale chimico δµi , temperatura δT, e pressione δPi.
Le sottoscritture i,j = g,l si riferiscono alla fase vapore e liquida
rispettivamente, Si, Vi ed ni sono la entropia, il volume ed il numero di
moli della fase i o j, rispettivamente.
Lo sperimento di porimetria ad azoto è condotto a temperatura
costante (δT = 0) e sotto condizioni tali per cui la fase gassosa dell’azoto è
in equilibrio con la propria fase liquida, pertanto i potenziali chimici δµg
= δµl = 0. E quindi, eguagliando le equazioni di Gibbs–Duhem (5.2) e
(5.3), si ottiene:
llvg dPvdPv = (5.116)
I volumi totali sono stati sostituiti dai volumi molari per ogni fase
rispettivamente (vi = Vi/ni). L’equazione di Laplace (Eq. (5.4)), per
superfici semisferiche, soggette ad un incremento di pressione δP,
relativa al sistema fisico rappresentato in figura 5.6:
=−
l
lvlvlv
dV
dAPP γ (5.4)
Derivando (5.4), si giunge a:
=−
rddPdP lv
lv
γ2 (5.117)
Per convenzione, la fase con la pressione maggiore si trova dalla
parte convessa dell’interfaccia. Combinando le equazioni (5.117) e
(5.116), si ottiene,
v
v
l
v
l
vllv
P
dP
v
RTdP
v
vv
rd −=
−=
γ2 (5.118)

Crioporosimetria
150
v
v
l
lv
P
dP
v
RT
rd −=
γ2 (5.118’)
Essendo vl vv << , e assumendo che la fase vapore si comporti
come gas ideale, gv PRTv = . Integrando l’equazione (5.118’) da 0
vP che
corrisponde a r =∞, a vP che corrisponde a r = r , si ottiene:
rRT
v
P
P llv
v
v γ2ln
0−= (5.119)
L’equazione risultante è conosciuta come equazione di Kelvin.
L’equazione (5.119), mette in relazione la tensione di vapore
all’equilibrio vP , di un liquido con volume specifico vl e raggio del menisco
r, con la tensione di vapore 0
vP dello stesso liquido su una superficie
planare; incrementando la pressione del gas avviene la condensazione
capillare nei pori più grandi. La maggior parte dei fluidi sonda inclusi
l’azoto, hanno un angolo di contatto Θ con la parete del poro.
Mettendo in relazione la (5.119) con l’equazione che lega il raggio
del poro pr all’angolo di contatto, Θ= cosrrp , si perviene a:
Θ−= cos2
ln0
RTr
v
P
P
p
llv
v
v γ (5.120)
In conclusione, una volta che si ha a disposizione la distribuzione
di 0
v
v
P
P, note che siano le altre grandezze definite nella (5.120), si può
risalire alla distribuzione dei raggi dei pori [2].
5.3.1.2 Considerazioni Sperimentali: Il metodo BET
Il metodo principale di misura della superficie specifica di un
materiale microposo è per adsorbimento dei gas (metodo BET) sviluppato
da Brunauer, Emmett e Teller.
La misura si effettua a basse temperature (-200 °C) e pressioni e
consiste nella valutazione del volume di gas (N2) adsorbito dal solido a
varie temperature a varie pressioni inferiori alle pressioni di saturazione,

Capitolo 5
151
da cui si calcola il volume teorico di uno strato monomolecolare
adsorbito.
Figura 5.7. Schema semplificato dell’apparecchiatura per lo sperimento di
porosimetria ad azoto.
L'isoterma di Stephen Brunauer, Paul H. Emmett ed Edward Teller
o isoterma BET (dalle iniziali dei loro cognomi), sviluppata nel 1938, è
utilizzata per descrivere l'adsorbimento su superfici da fase vapore. In
tale modello è contemplato l'adsorbimento multistrato e si distinguono
due possibili interazioni fondamentali: adsorbato-superficie e adsorbato-
adsorbato "verticale" (interazione attrattiva). L'interazione adsorbato-
adsorbato verticale è indipendente dallo strato considerato, mentre
l'interazione adsorbato-adsorbato nello stesso monostrato (orizzontale) è
considerata trascurabile rispetto all'interazione adsorbato-superficie. Il
sistema è composto quindi da infiniti monostrati che seguono il modello
di Langmuir.
L'equazione BET è:
( )[ ] cvP
P
cv
c
PPv mm
11
1
1
00
+
−=
− (5.121)
in cui P e P0 sono le pressioni all'equilibrio nel sistema poroso e la
pressione di saturazione degli adsorbati alla temperatura di

Crioporosimetria
152
adsorbimento, v è la quantità di vapore adsorbita (in unità di volume), vm
è la quantità adsorbita in un monostrato e c è la costante BET, pari a:
−=
RT
EEc L1exp (5.122)
In cui E1 è l'entalpia di adsorbimento del primo strato (quello che
interagisce con la superficie) e EL rappresenta l'entalpia per tutti gli altri
strati ed equivale all'entalpia di fusione.
Se si considera un grafico con 1 / v[(P0 / P) − 1] sull'asse delle
ordinate e P / P0 sull'asse delle ascisse (il cosiddetto grafico BET)
l'equazione (5.121) corrisponde ad una funzione lineare (cioè una retta).
In particolare, si osserva sperimentalmente che la relazione è lineare
nell'intervallo 0.05 < P / P0 < 0.35. A partire dalla pendenza della retta e
dal valore dell'intercetta sull'asse delle ordinate è possibile stimare la
quantità di vapore adsorbita per monostrato vm e la costante BET c.
Utilizzando opportune molecole-sonda (solitamente azoto o gas rari)
di cui si conosce l’area di ingombro e determinando m0 (quantità
adsorbita dal monostrato), si ottiene così l’area superficiale del solido
(solitamente normalizzata a massa unitaria di adsorbente).
5.4 Le Zeoliti
Le zeoliti sono dei materiali speciali. Esse formano una classe
affascinante di minerali microporosi [9]. Vengono largamente utilizzate in
applicazioni di scambio ionico, hanno proprietà uniche come assorbenti e
setacci molecolari e giocano un ruolo dominante in catalisi eterogenea. Le
proprietà delle zeoliti derivano direttamente dalle caratteristiche
particolari delle loro strutture cristalline, e la chimica dello stato solido
conosce pochi altri esempi in cui la relazione tra struttura e proprietà
macroscopiche può essere osservata così direttamente. Gli interessi sia
teorici che applicativi per le zeoliti sono in continua crescita. Ciò riflette
l’espansione delle procedure sintetiche di nuove specie e gli ulteriori

Capitolo 5
153
sviluppi negli utilizzi commerciali. Una più approfondita conoscenza della
chimica delle zeoliti offre la possibilità di un più diretto controllo delle
sintesi e di una migliore capacità previsionale nella selezione in vista di
specifiche applicazioni.
Per molti anni le zeoliti sono state utilizzate come scambiatori
cationici per addolcire l’acqua (e sono tuttora utilizzate nei detergenti per
questo scopo al posto dei fosfati, eliminati per via dei problemi ambientali
che essi causavano). Esistono in natura almeno 40 forme di zeoliti ma il
loro numero è stato di molto aumentato per via sintetica.
Il nome zeolite fu coniato nel 1756 dal mineralogista svedese A. F.
Cronstedt che osservò che questi minerali (in particolare la stilbite)
quando riscaldati emettevano bolle per via del rilascio di acqua
interstiziale. Da questo il nome zeo lithos da ‘bollire’ e ‘pietra’ in greco.
5.4.1 Struttura e composizione
Le zeoliti rappresentano una particolare classe di minerali collegati
ai feldspati e feldspatoidi. Sono dei tetroalluminosilicati e hanno strutture
cristalline costruite da tetraedri TO4 (T = specie tetraedrica, Si, Al, etc.), i
cui atomi di ossigeno sono scambiati con tetraedri adiacenti (vedi figura
5.8).
Figura 5.8. Strutture tetraedriche.
Le zeoliti, per definizione, si distinguono per avere strutture più
aperte, in grado di poter assorbide e disassorbire reversibilmente
molecole d’acqua o molecole più grandi, e che contengono grandi cationi

Crioporosimetria
154
non legati al network che possono essere facilmente scambiati. La
formula generale delle zeoliti è:
Mx/n[(AlO2)x(SiO2)y].mH2O
dove i cationi M, di valenza n, neutralizzano le cariche negative sul
reticolo di allumosilicato.
I mattoni costituenti le zeoliti sono unità tetraedriche [SiO4]4- e
[AlO4]5- legate insieme dalla condivisione di un vertice per ogni coppia di
tetraedri, a formare dei ponti di ossigeno non-lineari. Una unità di base
composta da due tetraedri uniti tra loro, è mostrata in figura 5.9.
Figura 5.9. Unità base composta da due tetraedri uniti tra loro.
Mentre i tetraedri TO4 nelle strutture zeolitiche sono in genere
regolari, i valori degli angoli T-O-T sono distribuiti in un range tra circa
125° e 180°. I tetraedri si possono quindi combinare a dare una varietà di
strutture. I tetraedri SiO4 sono elettricamente neutri quando legati tra
loro in un reticolo tridimensionale come il quarzo. La sostituzione di
Si(IV) con Al(III) nella struttura provoca uno squilibrio di carica e, per
conservare la elettroneutralità, ogni tetraedro AlO4 deve essere
controbilanciato da una carica positiva. La carica proviene da cationi
legati in modo elettrostatico alla zeolite. Il fatto che le zeoliti siano
cristalline, con una microporosità che è una intrinseca caratteristica della
struttura cristallina, le differenzia da molti altri materiali microporosi
come i setacci molecolari di carbone, il gel di silice o certe argille
colonnari.

Capitolo 5
155
Figura 5.10. Strutture molto comuni nelle zeoliti: cicli a 4, 5, 6 membri che
generano tipi diversi di catene.
I tetraedri TO4 formano anelli: unità molto comuni nelle zeoliti sono
cicli a 4, a 5 e a 6 membri (figura 5.10). Le configurazioni che può
adottare un anello a 4 membri illustrano la flessibilità dei tetraedri come
building blocks strutturali. Gli apici dei quattro tetraedri TO4 possono
puntare in su (U, up) o in giù (D, down), generando tipi diversi di catene.
Tutti questi modi di connessione si osservano nelle strutture zeolitiche.
In genere i tetraedri uniti in questo modo sono rappresentati
disegnando solo le linee congiungenti i centri di tetraedri adiacenti (Si-Si,
Si-Al etc.). In questo modo la rappresentazione della struttura si
semplifica, ma va sempre ricordato che i legami Al-O-Si etc. non sono
lineari (vedi anello esagonale in figura 5.11). La struttura di molte zeoliti è
basata sull’ unità di 24 tetraedri di Si o Al uniti insieme, l’unità sodalitica
(gabbia β), illustrata in figura 5.12.
Figura 5.11. Esempio di come la rapresentazione della struttura
semplificata dei tetraedri adiacenti (sinistra) non sia lineare come un anello
esagonale (destra).

Crioporosimetria
156
Figura 5.12. L’unità sodalitica (gabbia β), struttura di molte zeoliti, è basata
sull’ unità di 24 tetraedri di Si o Al uniti insieme.
Si possono riconoscere anelli a sei e a quattro tetraedri uniti tra
loro a formare un ottaedro troncato. Questa, come abbiamo visto, è
l’unità base della sodalite. Molte delle zeoliti più comuni sono basate
sull’unità sodalitica (figura 5.13). Si noti che nella sodalite la ‘cavità
interna’ definita dalle otto unità sodalitiche è anch'essa una unità
sodalitica. Come sappiamo, infatti, l’ottaedro troncato è uno dei poliedri
che riempiono completamente lo spazio. La struttura risultante è
altamente simmetrica e contiene canali che viaggiano paralleli ai tre assi
del sistema cubico.
La figura 5.13 mostra oltre alla sodalite, una zeolite sintetica, la
zeolite-A o LTA. Questa è correlata alla struttura della sodalite, ma con le
unità sodalitiche di base unite mediante ponti ad ossigeno tra gli anelli a
4 membri. In questo modo si forma un nuovo reticolo di cavità più grandi
connesse tra loro. La formula della zeolite-A è Na12[(SiO2)12(AlO2)12].27
H2O. Il rapporto Si/Al è quindi 1:1 e gli atomi dei due elementi si
alternano regolarmente nel reticolo. La struttura della faujasite (FAU), un
minerale, è mostrata nella stessa figura 5.13. Le unità sodalitiche sono
legate da ponti a ossigeno tra quattro degli otto anelli a 6 membri, in
arrangiamento tetraedrico, formando prismi esagonali. Le zeoliti
sintetiche X e Y hanno lo stesso framework, ma nella zeolite X il rapporto
Si/Al è tra 1 e 1.5 mentre nella zeolite Y è tra 1.5 e 3. Queste zeoliti sono
caratterizzate dalla comparsa di una supercavità a cui si accede tramite
finestre che sono anelli a 12 membri. Il framework emt è una variante
esagonale della FAU, presente nella zeolite EMC-2.

Capitolo 5
157
Figura 5.13. Esempio di alcune zeoliti che derivano dall’unità sodalitica.
Come detto la zeolite-A ha un rapporto Si/Al unitario. In alcune
zeoliti questo rapporto può essere molto alto. La ZK-4, con la stessa
struttura della zeolite-A, ha un rapporto Si/Al di 2.5. Molte delle zeoliti
sintetizzate in anni recenti per scopi catalitici sono altamente silicee. La
ZSM-5 (zeolite Socony-Mobil 5) può avere un rapporto Si/Al tra 20 e ∞
(cioè SiO2 pura). Chiaramente, la variazione del rapporto Si/Al comporta
anche una variazione del contenuto di cationi nel reticolo. Un più basso
numero di atomi di Al significa un minor numero di cationi scambiabili.
Le zeoliti ad alto contenuto di Si sono maggiormente idrofobiche e hanno
una maggiore affinità per gli idrocarburi. Alcune strutture zeolitiche
contenenti anelli a 5 membri di tetraedri sono mostrate in Figura 5.14.
Oltre alle zeoliti convenzionali, diverse nuove classi di materiali
zeolitici sono stati preparati come gli AlPO (alluminofosfati), i SAPO
(alluminofosfati Si sostituiti) etc. Il reticolo Si-O-Al nelle zeoliti è
relativamente rigido; i cationi non fanno parte integrante della struttura e
sono spesso chiamati cationi intercambiabili. Sono relativamente mobili e
possono essere sostituiti da altri cationi. La presenza e posizione dei
cationi nella zeolite è importante per varie ragioni. Le sezioni degli anelli e

Crioporosimetria
158
dei canali nella zeolite possono venire modificate cambiando la carica (e
quindi il numero) dei cationi.
Figura 5.14. Alcune strutture zeolitiche contenenti anelli a 5 membri di
tetraedri.
Oltre alle zeoliti convenzionali, diverse nuove classi di materiali
zeolitici sono stati preparati come gli AlPO (alluminofosfati), i SAPO
(alluminofosfati Si sostituiti) etc. Il reticolo Si-O-Al nelle zeoliti è
relativamente rigido; i cationi non fanno parte integrante della struttura e
sono spesso chiamati cationi intercambiabili. Sono relativamente mobili e
possono essere sostituiti da altri cationi. La presenza e posizione dei
cationi nella zeolite è importante per varie ragioni. Le sezioni degli anelli e
dei canali nella zeolite possono venire modificate cambiando la carica (e
quindi il numero) dei cationi.
Questo ha una grande importanza per il tipo di molecole che
possono venire assorbite nella zeolite. Una variazione nella occupazione
dei siti da parte dei cationi porta anche a cambiare le proprietà catalitiche
del sistema. Per questa ragione è diventato molto importante determinare
la posizione dei cationi all'interno della zeolite. I cationi all’interno della
zeolite possono avere più di una possibile collocazione. Nella figura 5.15
sono mostrati alcuni siti di assorbimento di ioni K+ nella zeolite-A. Alcuni
cationi occupano il centro degli anelli a 6, mentre altri sono all’interno
degli anelli ad 8.

Capitolo 5
159
Figura 5.15. Alcuni siti d’assorbimento di ioni K+ nella zeolite-A. Alcuni
cationi occupano il centro degli anelli a 6, mentre altri sono all’interno degli anelli
ad 8.
La presenza dei cationi in queste posizioni riduce la dimensione del
canale e impedisce l’entrata di altre molecole. Se, ad esempio, si vuole
introdurre una molecola organica come l’etano, si possono sostituire gli
ioni K+ con ioni divalenti in modo da dimezzare il numero di cationi
presenti nella zeolite.
Figura 5.16. I principali siti cationici nel minerale faujasite.

Crioporosimetria
160
Inoltre, gli ioni divalenti preferiscono posizionarsi all'interno degli
anelli a 6, lasciando liberi i canali di accesso alle cavità zeolitiche (figura
5.16). Questi siti sono collocati :
• Nei prismi esagonali, S(I), occupati da cationi che
preferiscono alte coordinazioni;
• Immediatamente adiacenti ai precedenti nelle gabbie β,
S(I’), occupati solo in alternativa ai siti S(I);
• Sulle pareti della supergabbia, S(II), quasi tutti
occupati;
• Nella parte centrale delle gabbie β, S(II’), quasi sempre
vuoti.
Le zeoliti cristalline contengono molecole d’acqua che sono
coordinate agli ioni scambiabili. Le strutture possono essere disidratate
mediante riscaldamento sotto vuoto, con la conseguenza che anche i
cationi si spostano e in genere si collocano in siti con basso numero di
coordinazione. Le zeoliti disidratate sono ottimi agenti essiccanti.
5.4.2 Cavità e canali
L’aspetto strutturale di maggior importanza nelle zeoliti è la
presenza di cavità e pori collegati tra loro mediante dei canali a formare
una vera e propria rete di canali all'interno della struttura. Queste cavità
hanno dimensioni molecolari e possono assorbire specie chimiche
abbastanza piccole da passare attraverso i canali. Un fattore che
controlla la possibilità o meno di assorbire molecole nella zeolite è la
dimensione della finestra o apertura del poro (figura 5.17).

Capitolo 5
161
Figura 5.17. Finestre o aperture dei pori di alcuni tipi di zeoliti.
Questa finestra a sua volta dipende dal numero di tetraedri, atomi
T, e di ossigeni, O, uniti tra loro e quindi dalla dimensione degli anelli. Le
dimensioni delle finestre sono ottenute usando i raggi di van der Waals di
O, 1.35 Å, e Si, 1.40 Å (figura 5.18).
Figura 5.18. Diverse dimensioni delle finestre ottenute utilizzando i raggi di
van der Waals.
Una cavità nella sodalite ha aperture costituite da anelli a 4 tetraedri con
un diametro di circa 260 pm; questo diametro è abbastanza piccolo e
permette solo l’ingresso di molecole d’acqua. L’apertura dei pori nella
zeolite-A è di 410 pm, ed è determinato dalla dimensione degli anelli a 8.

Crioporosimetria
162
La cavità interna però misura 1140 pm in diametro. La faujasite ha
aperture costituite da anelli a 12 con diametro di 740 pm e una
supercavità di 1180 pm in diametro.
Le dimensioni delle finestre dei pori variano quindi tra 300 e 1000
pm, da cui il nome di setaccio molecolare dato a questi allumosilicati. Di
conseguenza, le zeoliti hanno una elevatissima area superficiale, con la
possibilità di adsorbire grandi quantità di specie chimiche, una
caratteristica di grande importanza in catalisi. Valori tipici di area
superficiale per le zeoliti sono 300-700 m2 g-1 e in cristalliti della
dimensione di 0.1-5 mm più del 98% dell'area superficiale totale è
interna.
Le zeoliti sono suddivise in tre grandi categorie. I canali possono
essere paralleli a: (1) una singola direzione (si parla di zeoliti fibrose); (2)
due direzioni distribuite su dei piani (zeoliti lamellari); (3) tre direzioni
(zeoliti a framework). Come sempre accade, esistono strutture che non
appartengono in modo netto a nessuna di queste categorie. Un tipico
esempio di zeolite fibrosa è la edingtonite, Ba[(AlO2)2(SiO2)3].4H2O, che ha
una caratteristica forma a catena legata alla ripetizione regolare di cinque
tetraedri (figura 5.19).
Figura 5.19. L’edingtonite, Ba[(AlO2)2(SiO2)3].4H2O, ha una caratteristica
forma a catena legata alla ripetizione regolare di cinque tetraedri.
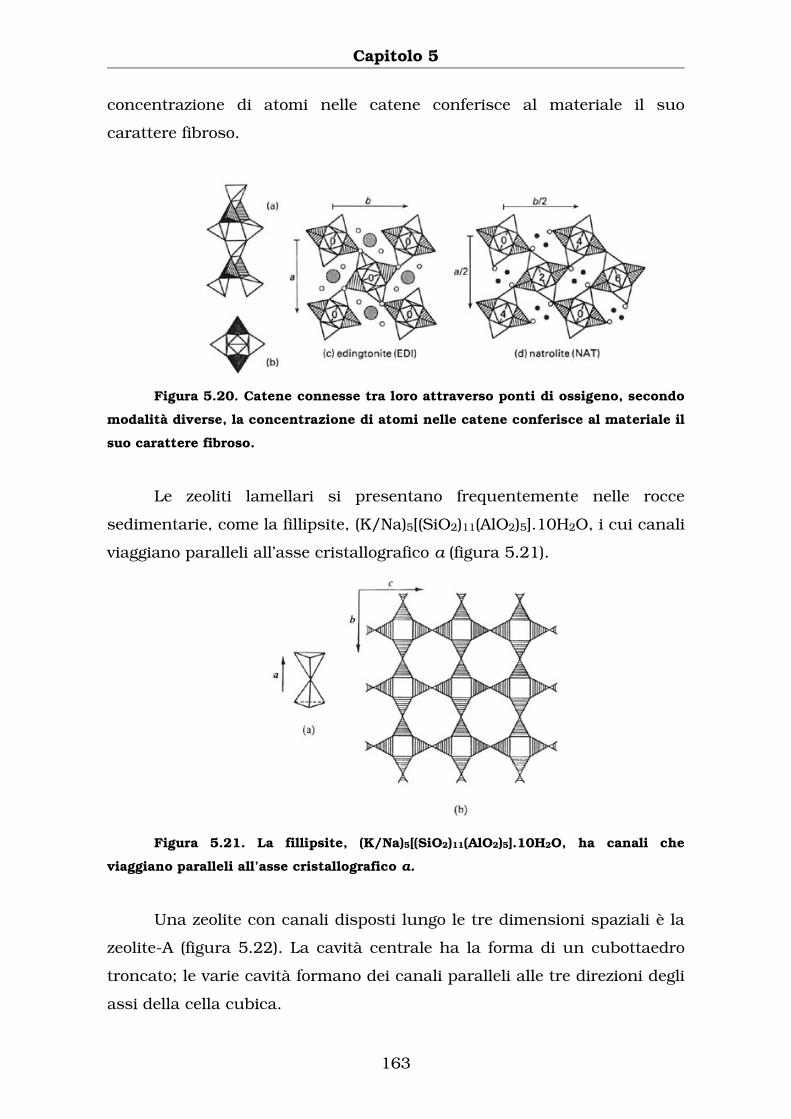
Le catene sono connesse tra loro attraverso ponti di ossigeno,
secondo modalità diverse (vedi figura 5.20, i tetraedri ombreggiati
contengono Al, cationi e acque sono disposti tra le catene), ma la
Capitolo 5
163
concentrazione di atomi nelle catene conferisce al materiale il suo
carattere fibroso.
Figura 5.20. Catene connesse tra loro attraverso ponti di ossigeno, secondo
modalità diverse, la concentrazione di atomi nelle catene conferisce al materiale il
suo carattere fibroso.
Le zeoliti lamellari si presentano frequentemente nelle rocce
sedimentarie, come la fillipsite, (K/Na)5[(SiO2)11(AlO2)5].10H2O, i cui canali
viaggiano paralleli all’asse cristallografico a (figura 5.21).
Figura 5.21. La fillipsite, (K/Na)5[(SiO2)11(AlO2)5].10H2O, ha canali che
viaggiano paralleli all’asse cristallografico a.
Una zeolite con canali disposti lungo le tre dimensioni spaziali è la
zeolite-A (figura 5.22). La cavità centrale ha la forma di un cubottaedro
troncato; le varie cavità formano dei canali paralleli alle tre direzioni degli
assi della cella cubica.

Crioporosimetria
164
Figura 5.22. La zeolite-A ha canali disposti lungo le tre dimensioni spaziali.
Nella metà degli anni '70 furono sintetizzate alcune zeoliti con
struttura completamente nuova, che hanno portato a significativi sviluppi
nell’area. Si tratta di una famiglia di zeoliti a framework 3D, che
includono le specie sintetizzate presso i laboratori della compagnia
petrolifera Mobil, note come ZSM-5 e ZSM-11 (dette anche silicaliti 1 e 2,
rispettivamente) e alcune altre zeoliti naturali, che sono chiamate con il
nome generale di pentasil.
La struttura della ZSM-5 (ZSM = Zeolite Socony Mobil), un
catalizzatore utilizzato industrialmente in tutto il mondo, è riportata in
figura 5.23. L’unità di base pentasil è mostrata a sinistra (si noti che può
essere descritta come un poliedro con 8 facce pentagonali). Queste unità
di base sono collegate in catene unite tra loro a formare degli strati. La
sovrapposizione appropriata di questi strati genera poi le diverse
strutture pentasil.

Capitolo 5
165
Figura 5.23. La struttura della ZSM-5 (ZSM = Zeolite Socony Mobil). E’ un
catalizzatore utilizzato industrialmente in tutto il mondo.
Sia la ZSM-5 che la ZSM-11 sono caratterizzate da canali controllati da
anelli a 10, con diametro di circa 550 pm (figura 5.24).
Figura 5.24. La ZSM-5 e la ZSM-11 sono caratterizzate da canali controllati
da anelli a 10, con diametro di circa 550 pm.
Il sistema dei pori in queste zeoliti non unisce grandi cavità ma
contiene zone di intersezione dove lo spazio disponibile permette che
possano manifestarsi interazioni molecolari. La struttura del sistema dei
pori della ZSM-5 con canali circolari a zigzag che si intersecano con
canali lineari a sezione ellittica è mostrata in figura 5.24. Nella ZSM-11
invece i canali che si intersecano hanno sezioni praticamente circolari.

Crioporosimetria
166
Il sistema di canali della mordenite è mostrato in figura 5.25. Si
distinguono due tipi di canali, governati da anelli a 8 e 12 membri
interconnessi da piccoli anelli a 5 e 6 membri.
Figura 5.25. Il sistema di canali della mordenite.
5.4.3 Preparazione e caratterizzazione delle zeoliti
Le zeoliti vengono preparate da soluzioni contenenti silicati e
alluminati, [Al(OH)4]-, di sodio, ad alti pH realizzati usando l’idrossido di
un metallo alcalino o una base organica. Si forma un gel attraverso un
processo di copolimerizzazione degli ioni silicato e alluminato. Il gel viene
poi riscaldato moderatamente a 60-100 °C in un recipiente chiuso per
circa 2 giorni, producendo una zeolite condensata (si parla di condizioni
idrotermali). Il prodotto ottenuto è determinato dalle condizioni di sintesi:
temperatura, tempo, pH e movimento meccanico. Alcuni microcristalli di
zeoliti diverse osservati al SEM sono mostrati in figura 5.26.
La presenza di basi organiche favorisce la formazione di fasi ricche
di silicio.
La formazione di nuove fasi sintetiche ricche di Si è stata facilitata
dall’uso di templati. Questo metodo è un caso particolare di
precipitazione molto usato per la sintesi di zeoliti. Il processo implica la
cristallizzazione da una soluzione acquosa basica contente gli ioni
costituenti più una sostanza detta templato, generalmente uno ione a
base organica. La forma della molecola di templato dirige la
cristallizzazione dei tetraedri di alluminato e silicato e determina la

Capitolo 5
167
struttura del prodotto finale. Tipicamente vengono usati sali di ammonio
quaternario a grandi dimensioni, come tetrapropilammonio. Il catione
tetrametilammonio viene usato nella sintesi di ZK-4.
Figura 5.26. Alcuni microcristalli di zeoliti diverse osservati al SEM.
La figura 5.27 illustra i cationi tetra-alchilammonio occlusi nelle
cavità della sodalite (a) e tetrapropilammonio in un canale di intersezione
nella zeolite sintetica ZSM-5 (b).
Figura 5.27. Cationi tetra-alchilammonio occlusi nelle cavità della sodalite
(a) e tetrapropilammonio in un canale di intersezione nella zeolite sintetica ZSM-5
(b).
Una volta terminata la sintesi, il templato viene rimosso per
decomposizione termica o per via chimica.

Crioporosimetria
168
Un esempio importante di zeolite sintetica è la ZMS-5. Si parte da una
miscela di acido silicico (SiO2. nH2O), idrossido di sodio, solfato di
alluminio, acqua, n-propilammina e bromuro di tetrapropilammonio che
viene scaldata a 160 °C per parecchi giorni. Il templato organico dirige la
crescita con i gruppi alchilici che riempiono le cavità della zeolite.
La preparazione di zeoliti ricche in silicio, come la zeolite-Y, può essere
effettuata variando la composizione del materiale di partenza, ma anche
per rimozione successiva degli ioni Al3+ dal reticolo della zeolite mediante
acidi minerali o agenti complessanti.
Il pH è un fattore molto importante nella sintesi. Piccole variazione di pH
infatti possono variare sensibilmente la quantità di prodotto ottenuto.
5.4.4 Applicazioni delle zeoliti
Agenti deidratanti. Le zeoliti contengono sempre molecole di acqua
coordinate agli ioni incorporati nella struttura. Le strutture possono
essere disidratate per riscaldamento sotto vuoto. Le zeoliti così ottenute
sono dei buoni agenti essiccanti.
Zeoliti come scambiatori di ioni. Le zeoliti hanno la capacità di
scambiare in parte o tutti i loro cationi (non di reticolo) per trattamento
con un soluzione o un sale fuso. Il carattere dell’equilibrio di scambio
ionico tra la soluzione e la zeolite dipende da vari fattori tra cui il tipo di
catione scambiabile e i possibili siti di coordinazione cationica presenti
nella zeolite. La massima capacità di scambio è determinata dal rapporto
Si/Al.
La zeolite-A nella forma con Na+ è usata come additivo per addolcire le
acque; gli ioni Na+ vengono rilasciati e sostituiti dagli ioni Ca2+
provenienti dall’acqua dura. L’additivo può essere rigenerato facendovi
passare una soluzione salina molto pura di NaCl. Questo processo è
familiare a chi usa la lavastoviglie. Attualmente le zeoliti-A sono aggiunte
a tutti i detersivi per lavatrice al posto dei polifosfati. Annualmente si
producono più di 250000 tonnellate di zeolite-A per uso nei detergenti. E'
anche possibile produrre acqua dolce per desalinazione dell’acqua di
mare usando zeoliti che contengono una miscela di ioni Ag e Ba. Il

Capitolo 5
169
processo però è talmente costoso da essere utile solo in caso di
emergenza.
Alcune zeoliti hanno una affinità per un catione in particolare. E' il caso
della clinoptilolite, una zeolite naturale, che scambia facilmente con il Cs.
Può quindi essere usata per separare il 137Cs da rifiuti radioattivi
scambiando ioni Na+ con ioni Cs+. In modo simile la zeolite-A è usata per
recuperare lo Sr radioattivo.
Zeoliti come adsorbenti. Dato che le zeoliti deidratate hanno
strutture porose aperte, possiedono alte aree superficiali e sono in grado
di adsorbire grandi quantità di sostanze oltre all’acqua. In questo modo le
zeoliti possono essere utilizzate come setacci molecolari per purificare o
separare sostanze.
Ad esempio la chabazite (figura 5.28) è nota per la sua capacità di
assorbire piccole molecole come acido formico e metanolo ma non
benzene e molecole più grandi.
La chabazite è stata usata industrialmente per rimuovere la SO2 dalle
emissioni inquinanti di impianti di combustione industriali. Similmente i
pori di apertura 410 pm nella zeolite-A, associati a finestre costituite da
anelli a 8 membri (si noti però che la cavità interna ha un diametro molto
maggiore, di 1140 pm), lasciano passare molecole di metano ma
escludono molecole più grandi come il benzene.
Figura 5.28. La chabazite è nota per la sua capacità di assorbire piccole
molecole come acido formico e metanolo ma non benzene e molecole più grandi.

Crioporosimetria
170
La computer graphic può essere molto utile nell’illustrare se una
molecola è in grado o meno di passare attraverso una finestra o un
canale. Gli atomi vengono rappresentati coi loro raggi di van der Waals.
La selettività di forma della ZSM-5 è illustrata in figura 5.29 (è mostrata
la cross-section del canale lineare, a confronto con dimensioni e forma di
alcune molecole: metanolo, 2,2-dimetilpentano, p-xilene).
Figura 5.29. Cross-section del canale lineare della ZSM-5, a confronto con
dimensioni e forma di alcune molecole: metanolo, 2,2-dimetilpentano, p-xilene,
ottenuta mediante la computer graphic. Gli atomi vengono rappresentati coi loro
raggi di van der Waals.
Le zeoliti sono particolarmente utili per questi processi dato che la
loro struttura non cambia durante la deidratazione e che il reticolo non si
decompone sino a temperature dell’ordine dei 700 °C. In una zeolite-A
dopo deidratazione il volume delle cavità rappresenta circa il 50% del
volume totale del materiale. Dopo il loro utilizzo come setacci molecolari,
le zeoliti possono essere ripristinate mediante riscaldamento o riflusso
con gas puri.
Zeoliti come catalizzatori. La applicazione forse più importante delle
zeoliti è in catalisi. E' nota la necessità di ottenere catalizzatori ad elevata
area superficiale e le zeoliti da questo punto di vista sono uniche.
Permettono infatti di trattare sino a 100 volte la quantità di molecole che

Capitolo 5
171
si possono assorbire su un catalizzatore amorfo tradizionale. Le zeoliti
possono essere ottenute cristalline e la loro sintesi è altamente
riproducibile: non mostrano quindi tendenza a variare la attività
catalitica in funzione della preparazione del catalizzatore. Infine, la
possibilità di agire da setacci molecolari consente una selezione delle
molecole che possono accedere ad un sito attivo. Quest’ultima proprietà è
particolarmente importante ed è nota come selettività di forma (shape
selectivily). L’attività catalitica di zeoliti prive di cationi è attribuita alla
presenza di siti acidi che derivano dalle unità tetraedriche [AlO4] nella
struttura. Queste possono comportarsi da siti acidi di Brönsted o di
Lewis. Le zeoliti sono sintetizzate di solito con ioni Na+ per bilanciare le
cariche negative del reticolo; gli ioni Na+ possono facilmente venire
sostituiti da protoni per reazione di scambio diretto con una soluzione
acquosa, dando luogo alla formazione di siti acidi di Brönsted. In
alternativa, se la zeolite non è stabile in soluzione acida, si prepara il sale
della zeolite con lo ione ammonio, NH4+, e si scalda in modo da rimuovere
l’ammoniaca lasciando il protone all’interno. Se si riscalda ulteriormente
si rimuove acqua dal sito acido di Brönsted lasciando uno ione Al3+ tri-
coordinato con tipiche proprietà di accettore di coppie elettroniche (acido
di Lewis). Lo schema di formazione di queste specie è mostrato in figura
5.30.
La superficie di una zeolite può quindi presentare siti acidi sia di
Brönsted che di Lewis o entrambi a seconda della preparazione. I siti di
Brönsted sono convertiti in quelli di Lewis a temperature superiori ai 600
°C. Non tutte le zeoliti sono usate in forma priva dei cationi o in forma
acida. E' comune sostituire gli ioni Na+ con ioni La3+ o Ce3+. Questi ioni si
dispongono nella struttura in modo da neutralizzare tre cariche negative
di altrettanti tetraedri [AlO4]-. La separazione di carica genera un forte
campo elettrostatico nella cavità. Questo può risultare abbastanza forte
da polarizzare o addirittura ionizzare il legame C-H di una molecola
adsorbita.
Una delle primi applicazioni delle zeoliti nel campo della catalisi
eterogenea è stata legata all’uso di zeoliti sostituite da terre rare per

Crioporosimetria
172
processi di cracking del petrolio negli anni ‘60. Il petrolio viene prima
separato per distillazione in frazioni leggere e pesanti di idrocarburi e le
frazioni pesanti sono ulteriormente trattate in processi di cracking su
catalizzatori per dare benzine.
Figura 5.30. Schema di formazione di una zeolite come catalizzatore.
Uno schema di processo in un impianto di cracking di idrocarburi a
letto catalitico fluido è mostrato in figura 5.31.
Figura 5.31. Schema di processo in un impianto di cracking di idrocarburi a
letto catalitico fluido.

Capitolo 5
173
Un terzo uso delle zeoliti in catalisi si basa sulla sostituzione degli
ioni Na+ con ioni di metalli di transizione come Ni2+, Pd2+ o Pt2+. Questi
vengono poi ridotti in sito in modo da formare atomi neutri depositati nel
reticolo. Il materiale risultante mostra le proprietà di un catalizzatore
metallico supportato, ma con una enorme dispersione del metallo.Ci sono
tre tipi di catalisi selettiva per forma (Figura 5.32):
Figura 5.32. Tre tipi di catalisi per forma del catalizzatore.
a) selettività sui reagenti: solo le molecole con dimensioni più
piccole di un valore critico possono entrare nei pori e raggiungere i
siti attivi. Ad esempio, un idrocarburo lineare può venire assorbito
e uno ramificato no.
• b) selettività sui prodotti: solo i prodotti di una certa
dimensione
• possono lasciare i siti attivi e diffondere attraverso i
canali come illustrato nella Figura seguente per il caso dello xilene.
Si possono formare tre isomeri nelle cavità zeolitiche, ma solo la
forma para- è in grado di uscire.

Crioporosimetria
174
c) selettività sullo stato di transizione: alcune reazioni non avvengono
in quanto lo stato di transizione (complesso attivato) richiede più
spazio di quanto non sia disponibile nelle cavità.
Uno degli usi industriali della ZSM-5 sta nella produzione di 1,4 (para)
xilene. Si tratta di un esempio tipico di selettività sui prodotti. Gli xileni
sono ottenuti per alchilazione del toluene con metanolo (figura 5.33).
Figura 5.33. Gli xileni ottenuti per alchilazione del toluene con metanolo.
La selettività della ZSM-5 è dovuta alle diverse cinetiche di
diffusione degli isomeri attraverso i canali. La diffusione del p-xilene è
circa 1000 volte superiore a quella degli altri isomeri. Va ribadito che un
ausilio molto importante nella progettazione di nuove zeoliti e nello studio
della loro attività catalitica è quello della grafica al computer e dei
programmi di dinamica molecolare. Esiste attualmente una intensa
attività in questo campo.
5.5 Parte Sperimentale
Lo scopo di questa sezione è caratterizzare la struttura interna di
campioni di zeoliti mediante studi di crioporosimetria e di porosimetria a
gas (N2). Si metteranno a confronto i risultati ottenuti da entrambi i
metodi, prendendo come riferimento dati reperibili in letteratura
specializzata [5]. In questo modo potrà essere valutata la bontà
dell’utilizzo della crioporosimetria per la determinazione della forma e

Capitolo 5
175
della dimensione dei pori per poter estenderla allo studio di sistemi in cui
le tecniche tradizionali non sono in grado di arrivare.
I campioni oggetto di questo studio comprendono due tipi di zeoliti
denominate commercialmente Si 60 e Si 100. Le caratteristiche fornite dal
produttore sono le seguenti: Si 60 ha un diametro medio del poro di 3,3
nm, una massa di 1.0612 g ed una densità di 2.091 g/cm3; Si 100 è
caratterizzato da un diametro medio di 7,7 nm, una massa di 0.878 g ed
una densità di 2.126 g/cm3.
5.5.1 Preparazione dei Materiali
5.5.1.1 Crioporosimetria
I campioni sono stati preparati immergendoli dapprima in acqua
distillata, dopodichè, il sistema è stato mantenuto a pressione ridotta, o
sottovuoto per un paio di ore in modo di eliminare l’eventuale aria
contenuta nei pori e riempire i pori di acqua.
Il sistema risultante si trova in una condizioni fisica tale per cui si
ha sia acqua all’interno dei pori che al di fuori di esso, infatti, dal
tracciato DSC si può apprezzare dell’acqua che solidifica attorno a 0°C
(figura 5.34).
Caratteristiche dei sistemi sottoposti all’esame DSC:
Silica 60
Peso totale del campione = 20.093 mg,
Peso della zeoliti a secco = 10.477 mg,
Peso totale dell’acqua = 9.616 mg.
Silica 100
Peso totale del campione = 23.981 mg,
Peso della zeoliti a secco = 10.264 mg,
Peso totale dell’acqua = 3.717 mg.

Crioporosimetria
176
Tracciati DSC
Temperature (°C)
-40 -30 -20 -10 0 10
He
at F
low
(m
W)
0
10
20
30
40
50
60 water in silica 60
water in silica 100
Figura 5.34. Tracciati DSC per i campioni di Zeoliti Si 60 e Si 100.
Entrambi i tracciati DSC evidenziano la presenza di due tipi di acqua: strutturata
all’interno dei pori (T di fusione < 0°C) ed acqua in eccesso che fonde a 0°C.
Le misure sono state condotte con un apparecchio DSC. Sono stati
rilevati i dati relativi al fenomeno di fusione dell’acqua durante il
riscaldamento.
La procedura effettuata è la seguente:
1- Raffreddamento da 25°C a -50°C a 10 °C/min
2- Stazionamento a -50 °C per un minuto
3- Riscaldamento da -50 °C a 25 °C ad una velocità di 3
°C/min per Si 100 ed a 10 °C/min per Si 60 . Acquisizione dati.
I calcoli sono stati eseguiti accuratamente con l’utilizzo di un
programma informatico scritto in linguaggio Fortran ed eseguito in
Windows XP.

Capitolo 5
177
5.5.1.2 Porosimetria a Gas
La porosimetria a gas (N2) è stata condotta con l’apparecchiatura
Sorptomatic 1990, che utilizza il metodo di BET per la determinazione
della distribuzione dei diametri dei pori. Si opera a temperatura costante
a -200 ° C e basse pressioni.
Sotto vengono riportate le condizioni analitiche corrispondenti ai
due campioni.
Si 60
Condizioni analitiche relative all’azoto
Peso molecolare (g/mol): 28.01
Area molecolare (A²) : 16.2
Spessore del monostrato (Å) : 3.54
Risultati del calcolo BET
Valori iniziali e finali di P/P0 : .0486 - .326
Volume specifico del monostrato vm (cm3/g) 88.83
Superficie specifica (m²/g) : 386.68
Valore di C (equazione BET) : 122.95
Volume Specifico del Poro (cm3/g): 0.78
Si 100
Condizioni analitiche relative all’azoto
Peso molecolare (g/mol): 28.01
Area molecolare (A²) : 16.2
Spessore del monostrato (Å) : 3.54
Risultati del calcolo BET
Valori iniziali e finali di P/P0 : 0.0954 - 0.311
Volume specifico del monostrato vm (cm3/g) : 68.43
Superficie specifica (m²/g) : 297.88
Valore di C (equazione BET): 323.51
Volume Specifico del Poro (cm3/g) : 1.16

Crioporosimetria
178
5.5.2 Risultati e Discussione
Le distribuzioni di probabilità dei raggi dei pori delle zeoliti ottenute
mediante porosimetria ad azoto e crioporosimetria, assumendo una
configurazione cilindrica z = 2 e sferica z = 3, sono mostrate nelle figure
5.35 e 5.36, rispettivamente per Si 60 e Si 100.
Si 60
0
0.05
0.1
0.15
0.2
0.25
0.3
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17R [nm]
Distribuzione
Normale
Differenziata
[1/nm]N2
Crioporimetria Z=3
Criporimetria Z=2
Figura 5.35. Distribuzioni di probabilità dei raggi dei pori per Si 60,
ottenute mediante porosimetria ad azoto (linea rossa) e crioporosimetria,
assumendo una configurazione cilindrica z = 2 (linea verde) e sferica z = 3 (linea
blu).
In linea di massima, le distribuzioni dei raggi dei pori in relazione
ad una funzione di probabilità ottenute mediante crioporosimetria e
porosimetria a gas sono risultate concordi tra di loro.
È fondamentale fare una precisazione, per Si 100 è stato
necessario ricorrere ad una approssimazione della distribuzione
normalizzata della curva di porosimetria a gas: in conseguenza alla
mancanza di dati, si è dovuto ipotizzare l’estremo inferiore dell’insieme
delle variabili ed assegnarne una probabilità pari a zero, in sostanza si è
dovuto chiudere la curva. Per insieme delle variabili vengono intesi i
valori dei raggi che possono assumere i campioni di zeoliti ed ai quali

Capitolo 5
179
viene assegnato una probabilità di frequenza. Essendo la funzione di
probabilità una distribuzione normalizzata che dipende quindi dall’area
sottesa dalla curva, è facile intuire che maggiore è l’area, più si abbassa
l’altezza della funzione.
Si 100
0
0.05
0.1
0.15
0.2
0 5 10 15 20 25 30
R [nm]
Distribuzione
Normale
Differenziata
[1/nm]N2
Criporimetria Z=3
Crioporimetria Z=2
Figura 5.36. Distribuzioni di probabilità dei raggi dei pori per Si 100
ottenute mediante porosimetria ad azoto (linea rossa) e crioporosimetria,
assumendo una configurazione cilindrica z = 2 (linea verde) e sferica z = 3 (linea
blu).
Dai risultati ottenuti si osserva che Si 60 ha raggi minori rispetto a
Si 100.
Non sono state rilevate grosse differenze tra i raggi calcolati
relativamente alla configurazione cilindrica e sferica, in quanto i
campione esaminati avevano acqua in eccesso rispetto al volume totale
dell’acqua interna ai pori, in questa condizione fisica il modello cilindrico
è equivalente a quello sferico. Ciò che cambia invece, è il valore calcolato
per lo strato d’acqua non solidificabile β che interagisce con le pareti
del poro.
Prima di continuare, si definisce la seguente funzione per valutare
la deviazione dei raggi dei modelli cilindrico e sferico rispetto a quello
calcolato col metodo BET a parità di probabilità:

Crioporosimetria
180
( )
i
i
CrioiNi
N
RpRp∑ −
=
2
,, 2
σ (5.123)
In cui:
2,NiRp Raggio del poro calcolato mediante porosimetria ad
azoto.
CrioiRp , Raggio del poro calcolato mediante crioporosimetria
con il modello sferico o cilindrico.
iN Numero totale dei dati.
i Dato i-esimo.
Nelle figure successive vengono riportati i valori dei raggi calcolati
mediante crioporosimetria con una configurazione cilindrica e sferica in
funzione dei raggi ottenuti mediante porosimetria ad azoto, in questo
modo si possono apprezzare le deviazioni di un modello rispetto all’altro.
3
5
7
9
11
13
15
3 5 7 9 11 13 15
RpN2 [nm]
Rp [nm]
N2
Z=3 (sferico)
Z=2 (cilindrico)
Figura 5.37. Deviazioni dei valori dei raggi calcolati mediante
crioporosimetria con una configurazione cilindrica (pallini verdi) e sferica (pallini
blu) dai raggi ottenuti mediante porosimetria ad azoto (linea rossa) per Si 60.
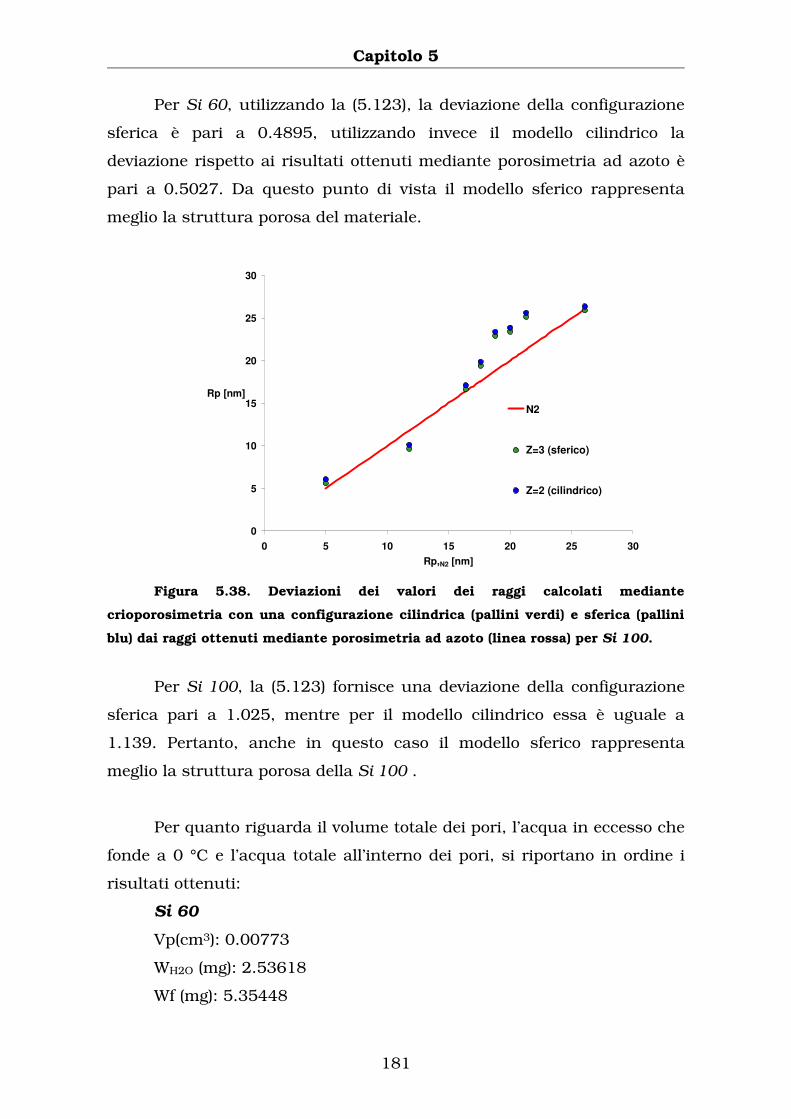
Capitolo 5
181
Per Si 60, utilizzando la (5.123), la deviazione della configurazione
sferica è pari a 0.4895, utilizzando invece il modello cilindrico la
deviazione rispetto ai risultati ottenuti mediante porosimetria ad azoto è
pari a 0.5027. Da questo punto di vista il modello sferico rappresenta
meglio la struttura porosa del materiale.
0
5
10
15
20
25
30
0 5 10 15 20 25 30
Rp,N2 [nm]
Rp [nm]
N2
Z=3 (sferico)
Z=2 (cilindrico)
Figura 5.38. Deviazioni dei valori dei raggi calcolati mediante
crioporosimetria con una configurazione cilindrica (pallini verdi) e sferica (pallini
blu) dai raggi ottenuti mediante porosimetria ad azoto (linea rossa) per Si 100.
Per Si 100, la (5.123) fornisce una deviazione della configurazione
sferica pari a 1.025, mentre per il modello cilindrico essa è uguale a
1.139. Pertanto, anche in questo caso il modello sferico rappresenta
meglio la struttura porosa della Si 100 .
Per quanto riguarda il volume totale dei pori, l’acqua in eccesso che
fonde a 0 °C e l’acqua totale all’interno dei pori, si riportano in ordine i
risultati ottenuti:
Si 60
Vp(cm3): 0.00773
WH2O (mg): 2.53618
Wf (mg): 5.35448

Crioporosimetria
182
Si 100
Vp(cm3): 0.01164
WH2O (mg): 3.06265
Wf (mg): 8.56158
Determinazione dello strato β
I risultati ottenuti dello spessore dello strato d’acqua non
solidificabile all’interno del poro β , determinato attraverso una
procedura iterativa, è stato confrontato coi risultati riportati in tabella 5.1
[5].
Si 60
Per quanto riguarda la geometria sferica, lo spessore β individuato
corrisponde a 0,56 nm, mentre per quanto riguarda la geometria
cilindrica β , esso assume un valore leggermente superiore, 0.86 nm. I
risultati ottenuti con entrambi in modelli rientrano nel set di dati forniti
in tabella 5.1[5].
Tabella 5.1. Spessore degli strati di acqua non solidificabile β , determinati
mediante prove di crioporosimetria. mβ e
fβ corrispondono ai valori ottenuti dai
tracciati DSC di fusione e solidificazione, rispettivamente.

Capitolo 5
183
Si 100
Assumendo una geometria sferica, il valore ottenuto per β è pari a
0.85 nm, mentre se si assume una geometria cilindrica il valore di β
risultante è di 1.31 nm. Anche in questo caso i valori ottenuti sono in
accordo con quelli riportati in tabella 5.1 [5].
5.5.3 Conclusioni
La distribuzione dei raggi in relazione ad una funzione di
probabilità, il volume dei pori e la loro forma, ottenuti dallo studio dei
tracciati DSC di fusione dell’acqua medianti studi di crioporosimetria
sono risultati conformi ai risultati ottenuti mediante porosimetria ad
azoto. Pertanto, la crioporosimetria può essere considerata un metodo
ragionevole per lo studio della porosità dei materiali, soprattutto per lo
studio di quei composti in cui le tecniche tradizionali non sono in grado
di riuscire. I materiali più complessi sia per struttura chimica che fisica,
che hanno come uno dei costituenti principali l’acqua, gli idrogel
polimerici per esempio, rappresentano un buon candidato per
l’applicazione di questa innovativa tecnologia.
Bibliografia
[1] M. Brun, A. Lallemand, J.-F. Quinson, C. Eyraud, Thermochim.
Acta 21, (1977) 59.
[2] M. Landry, Thermochim. Acta 433 , (2005) 27-50.
[3] K. Ishikiriyama, M. Todoki, K. Motomura, J. Colloid Interface Sci.
171 , (1995), 92 -102.
[4] T. Yamamoto, S.R. Mukaib, K. Nitta, H. Tamonb, A. Endo, T.
Ohmoria, M. Nakaiwa, Thermochimica Acta 439, (2005) 74–79
[5] K. Ishikiriyama, M. Todoki, K. Motomura, J. Colloid Interface Sci.
171, (1995), 103 -111.
[6] Zhang M. et al., Physical review B, 62, 15, 10548-10557, 2000

Crioporosimetria
184
[7] Gerosa, N. P., Determinazione della distribuzione volumetrica
dei cristalli da calorimetria differenziale a scansione, tesi triennale,
Politecnico di Milano, Dipartimenti d’Ingegneria Chimica, A.A. 2004/2005
[8] K. Ishikiriyama and M. Todoki, Thermochim. Acta, 256 (1995),
213.
[9] D. M. Proserpio, G. Ciani, Appunti del corso di Chimica dello
Stato Solido: Zeoliti, Dipartimento di Chimica Strutturale e Stereochimica
Inorganica, Università degli Studi di Milano, via Venezian 21, 20133
Milano, ITALIA.