Embed Size (px)
Citation preview

ORIGINAL ARTICLE
Caraparu virus induces damage and alterations in antioxidantdefenses in the liver of BALB/c mice after subcutaneous infection
Fernanda Caetano Camini • Letıcia Trindade Almeida • Carolina Silva Bernardes •
Maısa Silva • Maria Lucia Pedrosa • Daniela Caldeira Costa • Wanderson Geraldo de Lima •
Carla do Amaral Pinto • Paulo Cesar Peregrino Ferreira • Jose Carlos de Magalhaes •
Cintia Lopes de Brito Magalhaes
Received: 18 September 2013 / Accepted: 14 May 2014
� Springer-Verlag Wien 2014
Abstract Oxidative stress is a disturbance in the oxidant-
antioxidant balance leading to potential cellular damage.
Most cells can tolerate a mild degree of oxidative stress
because they have a system that counteracts oxidation that
includes antioxidant molecules such as glutathione (GSH)
and superoxide dismutase (SOD). Disruption of the host
antioxidant status has been recognized as an important
contributor to the pathogenesis of many viruses. Caraparu
virus (CARV) is a member of group C of the Bunyaviridae
family of viruses. In South American countries, group C
bunyaviruses are among the common agents of human
febrile illness and have caused multiple notable outbreaks
of human disease in recent decades; nevertheless, little is
known about the pathogenic characteristics of these viru-
ses. The purpose of this study was to examine the hepatic
pathogenesis of CARV in mice and the involvement of
oxidative stress and antioxidant defenses on this pathology.
Following subcutaneous infection of BALB/c mice, CARV
was detected in the liver, and histopathology revealed acute
hepatitis. Increased serum levels of aspartate and alanine
aminotransferases (AST/ALT) and greater hepatic expres-
sion of the proinflammatory cytokine tumor necrosis fac-
tor-a (TNF-a) were found in infected animals. CARV
infection did not alter the biomarkers of oxidative stress but
caused an increase in GSH content and altered the
expression and activity of SOD. This is the first report of an
alteration of oxidative homeostasis upon CARV infection,
which may, in part, explain the hepatic pathogenesis of this
virus, as well as the pathogenesis of other Bunyaviridae
members.
Introduction
Oxidative stress is an important contributor to pathogenesis
in many viral diseases, such as hepatitis B, hepatitis C, Rift
Valley fever, respiratory disease caused by respiratory
syncytial virus, and dengue [7, 8, 10, 15–18, 21, 24, 39].
Reactive oxygen species (ROS) are highly unstable mole-
cules that are involved in many forms of tissue damage,
including the damage caused to cellular components such
as lipids, proteins and DNA [14, 15]. However, cells are
protected against ROS and oxidative damage by well-
developed enzymatic and non-enzymatic antioxidant sys-
tems, including superoxide dismutase (SOD), catalase,
glutathione-dependent enzymes, thioredoxin and peroxire-
doxins [17]. Within cells, the redox potential is determined
primarily by the total content of glutathione (GSH) [26].
GSH is particularly important in the liver, where it serves
as the principal non-protein thiol involved in the cellular
F. C. Camini � M. L. Pedrosa � D. C. Costa �W. G. de Lima � C. L. de Brito Magalhaes
Nucleo de Pesquisas em Ciencias Biologicas, NUPEB,
Universidade Federal de Ouro Preto, Ouro Preto, Minas Gerais,
Brazil
F. C. Camini � L. T. Almeida � C. S. Bernardes � M. Silva �M. L. Pedrosa � D. C. Costa � W. G. de Lima �C. L. de Brito Magalhaes (&)
Departamento de Ciencias Biologicas, Universidade Federal de
Ouro Preto, Campus Universitario Morro do Cruzeiro,
Ouro Preto, Minas Gerais 35.400-000, Brazil
e-mail: [email protected]
C. do Amaral Pinto � P. C. P. Ferreira
Departamento de Microbiologia, Universidade Federal de Minas
Gerais, Belo Horizonte, Minas Gerais, Brazil
J. C. de Magalhaes
Departamento de Quımica, Biotecnologia e Engenharia de
Bioprocessos, Universidade Federal de Sao Joao del-Rei,
Ouro Branco, Minas Gerais, Brazil
123
Arch Virol
DOI 10.1007/s00705-014-2123-2

antioxidant defense mechanism. Additionally, antioxidant
enzymes (AOEs) can either directly decompose ROS (e.g.,
SOD and catalase) or facilitate these antioxidant reactions
(e.g., peroxidase using glutathione as a reducing agent).
The first ROS produced in the reduction pathway of oxy-
gen is the superoxide anion (O2-), which is metabolized to
hydrogen peroxide (H2O2) by enzymes of the SOD family.
Higher eukaryotes have three isoforms of SOD: cytoplas-
mic SOD1 (Cu/Zn-SOD), mitochondrial SOD2 (Mn-SOD)
and extracellular SOD3 (Cu/Zn-SOD) [23]. The glutathi-
one redox cycle is complementary to catalase in converting
H2O2 to water and oxygen [41]. Disruption of the host
antioxidant machinery is associated with many disease
states.
The family Bunyaviridae is composed of more than 350
RNA viruses that are classified into five distinct genera:
Orthobunyavirus, Hantavirus, Nairovirus, Phlebovirus and
Tospovirus [32]. Bunyavirus particles are approximately
100 nm in diameter and contain three single-stranded RNA
segments. All bunyaviruses encode four structural proteins:
the viral polymerase (L) on the large (L) segment, the
glycoproteins (Gn and Gc) on the medium (M) segment,
and the N protein on the small (S) segment. Viruses within
the genera Orthobunyavirus and Phlebovirus also encode
non-structural proteins, either on the M segment (termed
NSm) and/or on the S segment (NSs) [12]. With the
exception of the hantaviruses, these viruses are mainly
transmitted to mammals by arthropods and cause mild to
severe disease in humans, including fever, encephalitis and
hemorrhagic fever [32]. In tropical and subtropical areas,
bunyaviruses pose an increasing threat to human health and
are considered to be emerging pathogens [34]. In South
American countries, Oropouche (OROV), Guaroa (GROV)
and group C bunyaviruses are among the common agents
of human febrile illness and have caused multiple notable
outbreaks of human disease in recent decades [2, 11, 13,
37].
The genus Orthobunyavirus is subdivided into multiple
serological groups. Among these are the group C viruses,
which were initially discovered in the Brazilian Amazon
region during the 1950s [6, 9, 33]. A total of 14 distinct
group C viruses have been isolated from humans, mos-
quitoes, wild rodents, marsupials and bats [11, 38]. Cara-
paru virus (CARV) was first isolated in 1956 from a
sentinel monkey (Cebus apella) at the Utinga forest in
Belem, Para State, Brazil. Subsequently, CARV was iso-
lated from the blood of a febrile forest worker and from
arthropods in the same region [9]. In humans, the CARV
causes a self-limited disease lasting 2 to 5 days with
symptoms including fever, headache, myalgia, nausea,
vomiting and weakness [19, 25]. In experimentally infected
mice, CARV induces hepatitis [3]. In humans, there have
been no reports of CARV causing hepatitis; however,
autopsy or biomarkers of liver injury data are unavailable.
Interestingly, for another important bunyavirus in Brazil,
OROV, there have been no reports of clinical liver damage
in patients, but altered liver enzymes have been recorded,
and thus the liver is thought to be an important target organ
for human OROV infections [30].
Importantly, group C viruses of the genus Orthobunya-
virus are associated with febrile illness in humans. Because
of their nonspecific nature, they are usually not reported, or
are misdiagnosed, so their true incidence is unknown [2,
11, 13, 37]. Despite the public-health importance of the
group C viruses, little is known about their pathogenic
characteristics. A previous study demonstrated that intra-
peritoneal inoculation of CARV causes coagulative liver
necrosis in B6C3F1 mice; however, the mechanisms of
CARV-induced hepatic disease remain incompletely
defined [3]. Up to now, there have been no studies using
inoculation of experimental animals by subcutaneous
routes, which would more closely resemble the natural
means of CARV infection. Additionally, a non-lethal
mouse model of disease that more closely resembles the
disease in humans is not available. Thus, the aim of this
study was to characterize, in a non-lethal subcutaneous
model for CARV infection in BALB/c mice, the possible
involvement of oxidative homeostasis on hepatic disease.
Following subcutaneous inoculation with CARV, the
animals were monitored for 14 days, and they displayed
conspicuous clinical signs two or three days postinfection
(pi). From the ninth day pi, no clinical signs were evident,
and none of the infected mice died. Aspartate and alanine
aminotransferases (AST/ALT) levels and the liver histo-
pathology of infected animals revealed hepatitis on days 3
and 7 pi. An increase in TNF-a mRNA expression was
observed in infected animals on day 3 pi. No involvement
of oxidative stress was observed in CARV-induced hepatic
injury, because the levels of oxidative stress markers were
similar between infected and control mice. However, the
levels of GSH were increased in infected animals on days
3, 7 and 14 pi. Concomitantly, levels of SOD changed on
different days following exposure to the virus. The SOD1
mRNA expression was downregulated on days 3 and 7 pi,
and SOD2 and SOD3 mRNA expression were upregulated
on day 14 pi. The total SOD activity showed a decrease on
day 3 pi, returned to control levels on day 7 pi, and
increased on day 14 pi.
Our results demonstrate that CARV infection in BALB/
c mice causes acute hepatitis, with strong positivity for
viral antigen, inflammation and liver damage. In this
pathology, we observed an increase in GSH content and an
early decrease followed by a late increase in SOD, but no
significant oxidative stress. These results suggest that
despite the decrease in SOD levels in the initial days pi,
GSH might prevent CARV-induced oxidative stress during
F. C. Camini et al.
123

liver injury in mice. One of the recurrent themes in recent
studies of many infectious diseases is the modulation of
host responses elicited by exposure to the infection.
Materials and methods
Virus stock preparation
CARV (BeAn3994) was obtained from the American Type
Culture Collection (ATCC) and propagated in Vero cell
culture (African green monkey kidney cell line). After viral
adsorption, the cells were washed twice with phosphate-
buffered saline (PBS) and fed with MEM (Cultilab, Brazil)
supplemented with 1 % fetal bovine serum (FBS). After
90 % of the cells exhibited cytopathogenic effects, the
supernatant was centrifuged at 3,000 g for 10 min at 4 �C and
the viral pools were aliquoted and stored at -80 �C. The
virus titer was 106 PFU/ml, as measured using a methylcel-
lulose plaque assay. Briefly, supernatants of virus-infected
Vero cells were diluted in PBS containing 2 % FBS and
subsequently used to infect confluent Vero cells grown in
six-well plates. After a 1-h incubation at 37 �C, the cells were
overlaid with MEM containing 2 % FBS and 1 % carboxy-
methylcellulose (Sigma-Aldrich, Brazil) and incubated at
37 �C for 5 days. Cells were fixed with 4 % formaldehyde in
PBS, and plaques were revealed with Giemsa stain.
Experimental infection of mice and sample collection
BALB/c mice aged 6 weeks were obtained from the Animal
Facility of Rene Rachou Research Center, Oswaldo Cruz
Foundation (Minas Gerais, Brazil), and were housed in filter-
top micro-isolator cages; they were provided with commercial
mouse feed and water ad libitum. Animal experimentation
was carried out in accordance with the regulations of the Ethics
Committee on Animal Research (CEUA) of Universidade
Federal de Ouro Preto (Minas Gerais, Brazil). Twenty-four
animals were infected via the subcutaneous route with 0.1 ml
of viral suspension containing 105 PFU of CARV, and twenty-
four animals were sham inoculated using the same volume of
control medium. Three groups containing sixteen animals each
(eight infected animals and eight uninfected controls) were
anesthetized with ketamine and xylazine and euthanized by
exsanguination on days 3, 7 and 14 pi. Blood samples were
collected and centrifuged for determination of the AST/ALT
serum biomarkers, and the livers were removed and immedi-
ately stored at -80 �C for subsequent analysis.
Study of hepatic function markers
Serum levels of AST and ALT were measured to determine
hepatic function using Labtest kits # 52 and 53 (Minas
Gerais, Brazil) according to the manufacturer’s
instructions.
Histopathology, immunohistochemistry
and morphometric analysis of inflammatory cells
After collection, the livers were fixed in 10 % buffered
formalin, dehydrated in increasing (70 to 100 %) ethanol
concentrations, cleared in xylene and embedded in paraffin.
The tissue blocks were cut into 4- to 5-lm sections with a
microtome, and the sections were stained with hematoxy-
lin/eosin. For viral antigen detection, 5- to 7-lm thick
tissue sections were deparaffinized and treated using the
heat-induced antigen retrieval technique (Target Retrieval
Solution, S1700, Dako Corporation, USA). Subsequently,
the tissue sections were treated with 3.5 % H2O2 in PBS,
blocked with 20 % normal goat serum in PBS for 30 min at
room temperature and incubated overnight at 4 �C with a
1:2,000 dilution of mouse immune ascitic fluid anti-arbo-
virus group C-I (NIH Research Reagent – Catalog No
G201-701-567, USA) in 2 % bovine serum albumin (BSA,
1870, Inlab, Brazil) in PBS. After incubation with the
primary antibody, the tissue sections were washed three
times with PBS and incubated for 30 min at room tem-
perature with the secondary, biotinylated antibody solution
(DAKO Kit K0675, USA) and then washed with PBS and
treated with the tertiary solution containing peroxidase-
conjugated streptavidin (DAKO Kit K0675, USA) for
30 min at room temperature. The sections were then rinsed
in PBS and incubated with 3,30-diaminobenzidine tetrahy-
drochloride (Sigma, USA) (0.05 %) and hydrogen peroxide
(0.03 %) for 5 min. The sections were then rinsed in PBS
and counterstained with hematoxylin (Reagen, Brazil).
Negative controls were prepared using PBS instead of the
primary antiserum. Morphometric measurements of the
inflammatory cells in liver sections (twenty sections/ani-
mal) were made using a light microscope (Leica DM5000)
and analyzed using Leica Qwin Image Processing and
Analysis Software (Germany).
Virus titration
The livers were macerated in MEM 0 % FBS (Cultilab,
Brazil) and centrifuged at 2,000 g for 3 min at 4 �C. The
supernatant was collected, and the viral titer (PFU/g of
liver) was determined using the methylcellulose plaque
assay in Vero cell culture as described above.
Measurement of the biomarkers of oxidative stress
and total glutathione content in liver homogenates
The level of thiobarbituric acid reactive substances
(TBARS) was estimated using the method described by
Changes in oxidative homeostasis in Caraparu virus infection
123

Buege and Aust [4]. Liver homogenate supernatants were
mixed with trichloroacetic acid (TCA 28 % w/v in 0.25 N
hydrochloric acid), thiobarbituric acid (TBA 1 % in
0.25 M acetic acid) and butylated hydroxytoluene (BHT
125 mM in ethanol); they were then heated for 15 min at
95 �C and then placed in an ice bath. The precipitated
material was removed by centrifugation, and the absor-
bance of the samples at 535 nm was determined. The
TBARS level was calculated using the molar absorption
coefficient of malondialdehyde (MDA). Carbonyl protein
levels were determined according to the method described
by Levine [20]. Each sample was precipitated with 10 %
(w/v) TCA. After centrifugation, the precipitate was treated
with 10 mmol of 2,4-dinitrophenylhydrazine (DNPH) in
2 M hydrochloric acid, incubated in the dark for 30 min
and treated with 10 % TCA. After centrifugation, the
precipitate was washed twice with ethanol/ethyl acetate
(1:1) and dissolved in 6 % sodium dodecyl sulfate (SDS).
The absorbance of the samples at 370 nm was determined.
The results were expressed in nmol of DNPH incorporated/
mg of protein. The content of DNPH incorporated was
calculated using the molar absorption coefficient of DNPH
(22,000 M-1cm-1). Total protein content was determined
according to the method described by Lowry [22] using
bovine serum albumin (BSA) as the standard. The total
glutathione content of liver homogenates was determined
using a Glutathione Assay Kit (CS0260) from Sigma (St.
Louis, MO). This assay uses a kinetic method based on the
reduction of 5,50-dithiobis-(2-nitrobenzoic acid) (DTNB) to
5-thio-2-nitrobenzoic acid (TNB), which can be measured
spectrophotometrically at 412 nm.
Quantitative real-time reverse transcription PCR (q-RT-
PCR)
Total RNA was extracted from liver samples from control
and CARV-infected mice using an SV Total RNA Isolation
Kit (Promega, Brazil) according to the manufacturer’s
instructions. The concentration and purity of the RNA were
estimated spectrophotometrically using the A260/A280
ratio (NanoVue, GE Healthcare, Hertfordshere, UK). The
cDNA was synthesized from 2 lg of total RNA with ran-
dom primers using a High-Capacity cDNA Reverse Tran-
scription Kit (Applied Biosystems, Foster City, CA, USA)
according to the manufacturer’s instructions. The q-RT-
PCR was performed using SYBR Green PCR Master Mix
reagent (Applied Biosystems, Foster City, CA, USA) in a
final reaction volume of 12 lL. The reaction included 2 lL
of cDNA (100 ng) and 0.5 lL of each primer (forward and
reverse, 10 lM). The forward and reverse primers
sequences for TNF-a, SOD1, SOD2 and GAPDH were
obtained from the published nucleotide sequences [31, 40].
The primer sequences for SOD3 (forward primer 50-
TTACCACAAGGGACAGCCAA-30 and reverse primer
50-GTTATGTGAGCGAGCAGCAG-30) were derived
from the Mus musculus genome (National Center for
Biotechnology Information GenBank, accession number
NM_011435.3) and were constructed using the Primer-
BLAST program (http://www.ncbi.nlm.nih.gov/tools/pri
mer-blast/). The reactions were carried out under the fol-
lowing conditions: 50 �C for 2 min and 95 �C for 10 min,
followed by 40 cycles of 95 �C for 15 s and 60 �C for
1 min. Dissociation curves of the amplified product were
analyzed to confirm product specificity. The data obtained
were analyzed using the comparative CT method. Target
gene expression was determined relative to the endogenous
GAPDH gene. All analyses were performed in triplicate.
Biochemical assay for the total SOD activity
The activity of the total SOD was measured using a
Superoxide Dismutase Assay Kit (706002) from Cayman
Chemical Company (MI, USA). Briefly, 100 mg of liver
samples were homogenized in cold 20 mM HEPES, pH
7.2, containing 1 mM EGTA, 210 mM mannitol and
70 mM sucrose. Ten microliters of supernatant was used in
the assay. The reaction was initiated by adding xanthine
oxidase. The plate was incubated on a shaker for 20 min at
room temperature, and the absorbance at 450 nm was
measured using a plate reader (Biotek ELx808).
Statistical analysis
Data were analyzed using GraphPad Prism 3.0 software
and expressed as the mean ± standard deviation of eight
different animals. Student’s t-test at 95 % confidence was
used to determine the level of differences between the
CARV-infected groups and uninfected groups, with *, **
and *** representing significant differences at p \ 0.05,
p \ 0.005 and p \ 0.0005, respectively. The letters a, b,
and c represent differences between the groups of CARV-
infected animals, using one-way ANOVA and Tukey’s
post-test.
Results
Clinical findings and evaluation of hepatic function
markers
Control and CARV-infected BALB/c mice were examined
for clinical abnormalities twice daily for 14 days. All of
the infected mice were susceptible to infection and
developed conspicuous clinical signs within two or three
days pi, manifested by prostration, ruffled fur, shivering,
and a weight loss. This weight loss was more pronounced
F. C. Camini et al.
123
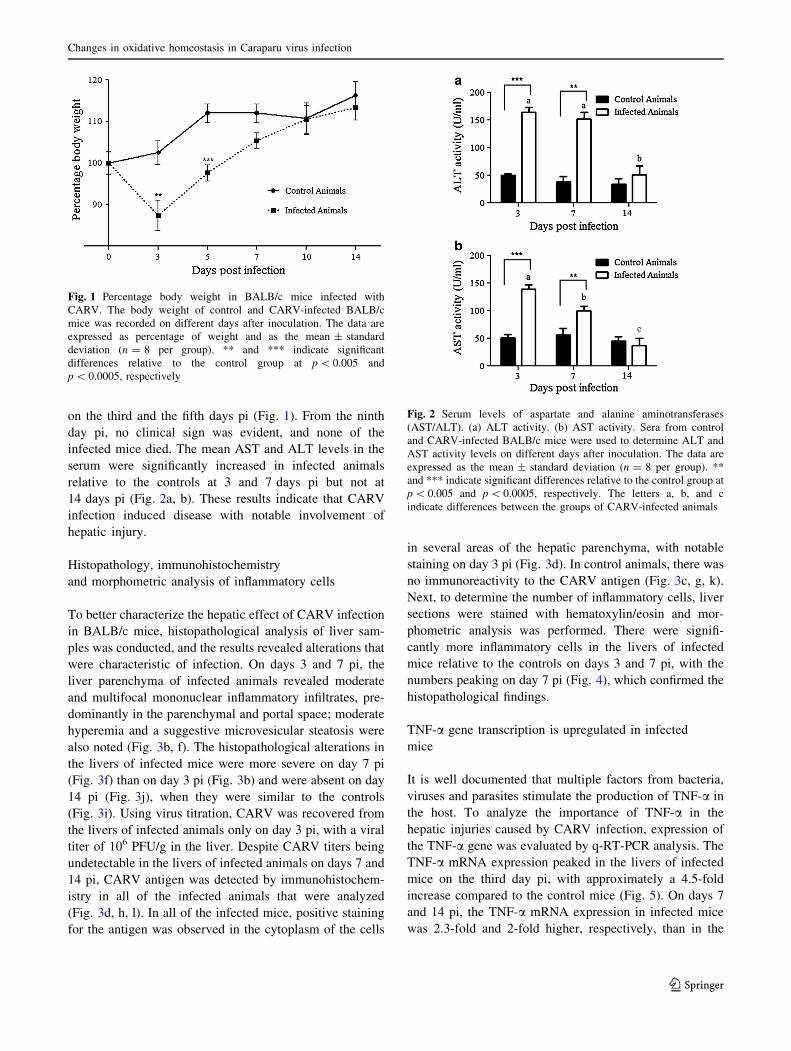
on the third and the fifth days pi (Fig. 1). From the ninth
day pi, no clinical sign was evident, and none of the
infected mice died. The mean AST and ALT levels in the
serum were significantly increased in infected animals
relative to the controls at 3 and 7 days pi but not at
14 days pi (Fig. 2a, b). These results indicate that CARV
infection induced disease with notable involvement of
hepatic injury.
Histopathology, immunohistochemistry
and morphometric analysis of inflammatory cells
To better characterize the hepatic effect of CARV infection
in BALB/c mice, histopathological analysis of liver sam-
ples was conducted, and the results revealed alterations that
were characteristic of infection. On days 3 and 7 pi, the
liver parenchyma of infected animals revealed moderate
and multifocal mononuclear inflammatory infiltrates, pre-
dominantly in the parenchymal and portal space; moderate
hyperemia and a suggestive microvesicular steatosis were
also noted (Fig. 3b, f). The histopathological alterations in
the livers of infected mice were more severe on day 7 pi
(Fig. 3f) than on day 3 pi (Fig. 3b) and were absent on day
14 pi (Fig. 3j), when they were similar to the controls
(Fig. 3i). Using virus titration, CARV was recovered from
the livers of infected animals only on day 3 pi, with a viral
titer of 106 PFU/g in the liver. Despite CARV titers being
undetectable in the livers of infected animals on days 7 and
14 pi, CARV antigen was detected by immunohistochem-
istry in all of the infected animals that were analyzed
(Fig. 3d, h, l). In all of the infected mice, positive staining
for the antigen was observed in the cytoplasm of the cells
in several areas of the hepatic parenchyma, with notable
staining on day 3 pi (Fig. 3d). In control animals, there was
no immunoreactivity to the CARV antigen (Fig. 3c, g, k).
Next, to determine the number of inflammatory cells, liver
sections were stained with hematoxylin/eosin and mor-
phometric analysis was performed. There were signifi-
cantly more inflammatory cells in the livers of infected
mice relative to the controls on days 3 and 7 pi, with the
numbers peaking on day 7 pi (Fig. 4), which confirmed the
histopathological findings.
TNF-a gene transcription is upregulated in infected
mice
It is well documented that multiple factors from bacteria,
viruses and parasites stimulate the production of TNF-a in
the host. To analyze the importance of TNF-a in the
hepatic injuries caused by CARV infection, expression of
the TNF-a gene was evaluated by q-RT-PCR analysis. The
TNF-a mRNA expression peaked in the livers of infected
mice on the third day pi, with approximately a 4.5-fold
increase compared to the control mice (Fig. 5). On days 7
and 14 pi, the TNF-a mRNA expression in infected mice
was 2.3-fold and 2-fold higher, respectively, than in the
Fig. 1 Percentage body weight in BALB/c mice infected with
CARV. The body weight of control and CARV-infected BALB/c
mice was recorded on different days after inoculation. The data are
expressed as percentage of weight and as the mean ± standard
deviation (n = 8 per group). ** and *** indicate significant
differences relative to the control group at p \ 0.005 and
p \ 0.0005, respectively
Fig. 2 Serum levels of aspartate and alanine aminotransferases
(AST/ALT). (a) ALT activity. (b) AST activity. Sera from control
and CARV-infected BALB/c mice were used to determine ALT and
AST activity levels on different days after inoculation. The data are
expressed as the mean ± standard deviation (n = 8 per group). **
and *** indicate significant differences relative to the control group at
p \ 0.005 and p \ 0.0005, respectively. The letters a, b, and c
indicate differences between the groups of CARV-infected animals
Changes in oxidative homeostasis in Caraparu virus infection
123
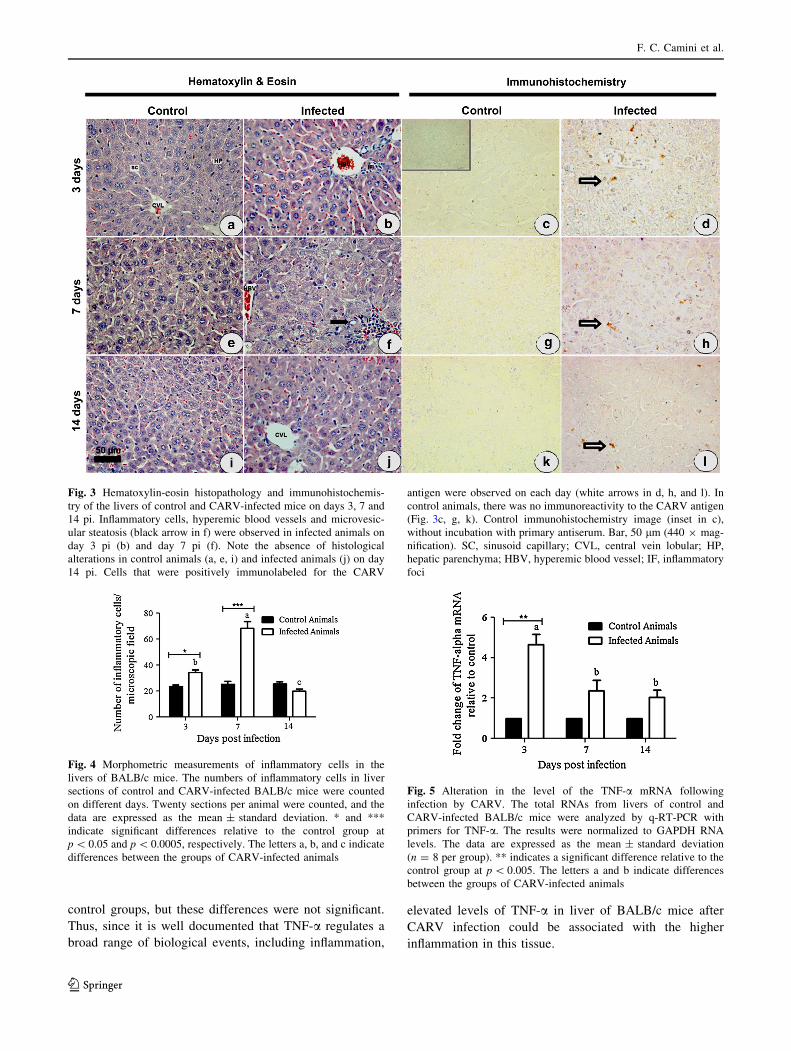
control groups, but these differences were not significant.
Thus, since it is well documented that TNF-a regulates a
broad range of biological events, including inflammation,
elevated levels of TNF-a in liver of BALB/c mice after
CARV infection could be associated with the higher
inflammation in this tissue.
Fig. 3 Hematoxylin-eosin histopathology and immunohistochemis-
try of the livers of control and CARV-infected mice on days 3, 7 and
14 pi. Inflammatory cells, hyperemic blood vessels and microvesic-
ular steatosis (black arrow in f) were observed in infected animals on
day 3 pi (b) and day 7 pi (f). Note the absence of histological
alterations in control animals (a, e, i) and infected animals (j) on day
14 pi. Cells that were positively immunolabeled for the CARV
antigen were observed on each day (white arrows in d, h, and l). In
control animals, there was no immunoreactivity to the CARV antigen
(Fig. 3c, g, k). Control immunohistochemistry image (inset in c),
without incubation with primary antiserum. Bar, 50 lm (440 9 mag-
nification). SC, sinusoid capillary; CVL, central vein lobular; HP,
hepatic parenchyma; HBV, hyperemic blood vessel; IF, inflammatory
foci
Fig. 4 Morphometric measurements of inflammatory cells in the
livers of BALB/c mice. The numbers of inflammatory cells in liver
sections of control and CARV-infected BALB/c mice were counted
on different days. Twenty sections per animal were counted, and the
data are expressed as the mean ± standard deviation. * and ***
indicate significant differences relative to the control group at
p \ 0.05 and p \ 0.0005, respectively. The letters a, b, and c indicate
differences between the groups of CARV-infected animals
Fig. 5 Alteration in the level of the TNF-a mRNA following
infection by CARV. The total RNAs from livers of control and
CARV-infected BALB/c mice were analyzed by q-RT-PCR with
primers for TNF-a. The results were normalized to GAPDH RNA
levels. The data are expressed as the mean ± standard deviation
(n = 8 per group). ** indicates a significant difference relative to the
control group at p \ 0.005. The letters a and b indicate differences
between the groups of CARV-infected animals
F. C. Camini et al.
123
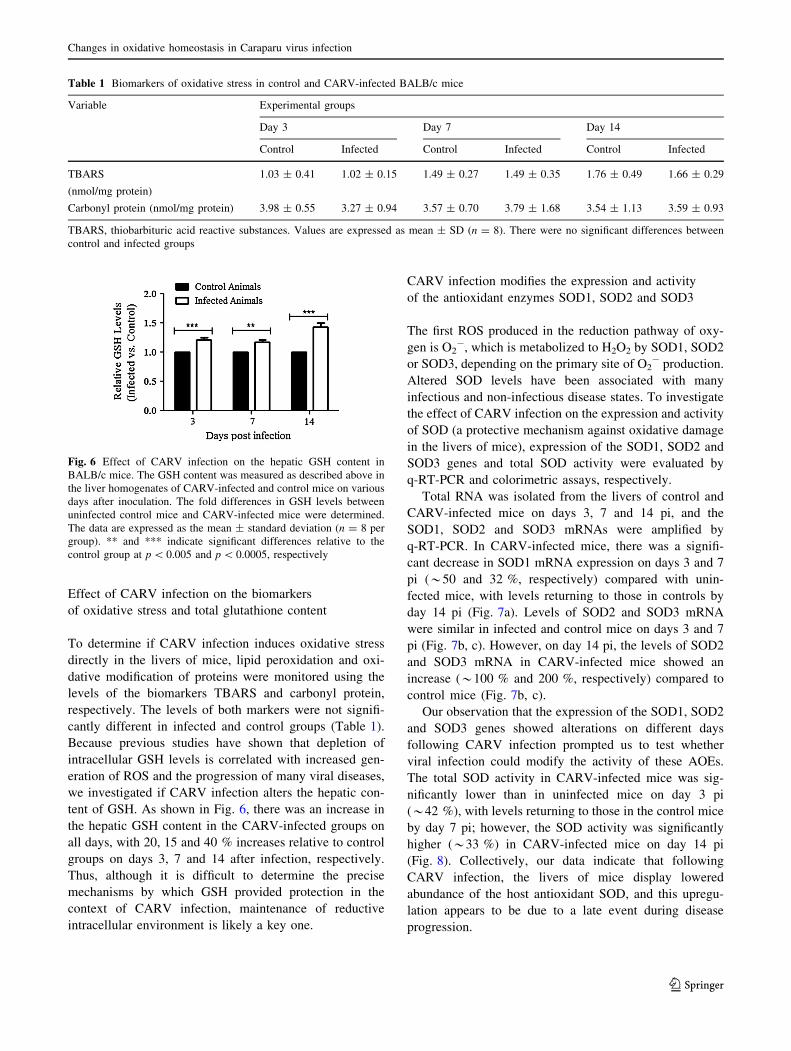
Effect of CARV infection on the biomarkers
of oxidative stress and total glutathione content
To determine if CARV infection induces oxidative stress
directly in the livers of mice, lipid peroxidation and oxi-
dative modification of proteins were monitored using the
levels of the biomarkers TBARS and carbonyl protein,
respectively. The levels of both markers were not signifi-
cantly different in infected and control groups (Table 1).
Because previous studies have shown that depletion of
intracellular GSH levels is correlated with increased gen-
eration of ROS and the progression of many viral diseases,
we investigated if CARV infection alters the hepatic con-
tent of GSH. As shown in Fig. 6, there was an increase in
the hepatic GSH content in the CARV-infected groups on
all days, with 20, 15 and 40 % increases relative to control
groups on days 3, 7 and 14 after infection, respectively.
Thus, although it is difficult to determine the precise
mechanisms by which GSH provided protection in the
context of CARV infection, maintenance of reductive
intracellular environment is likely a key one.
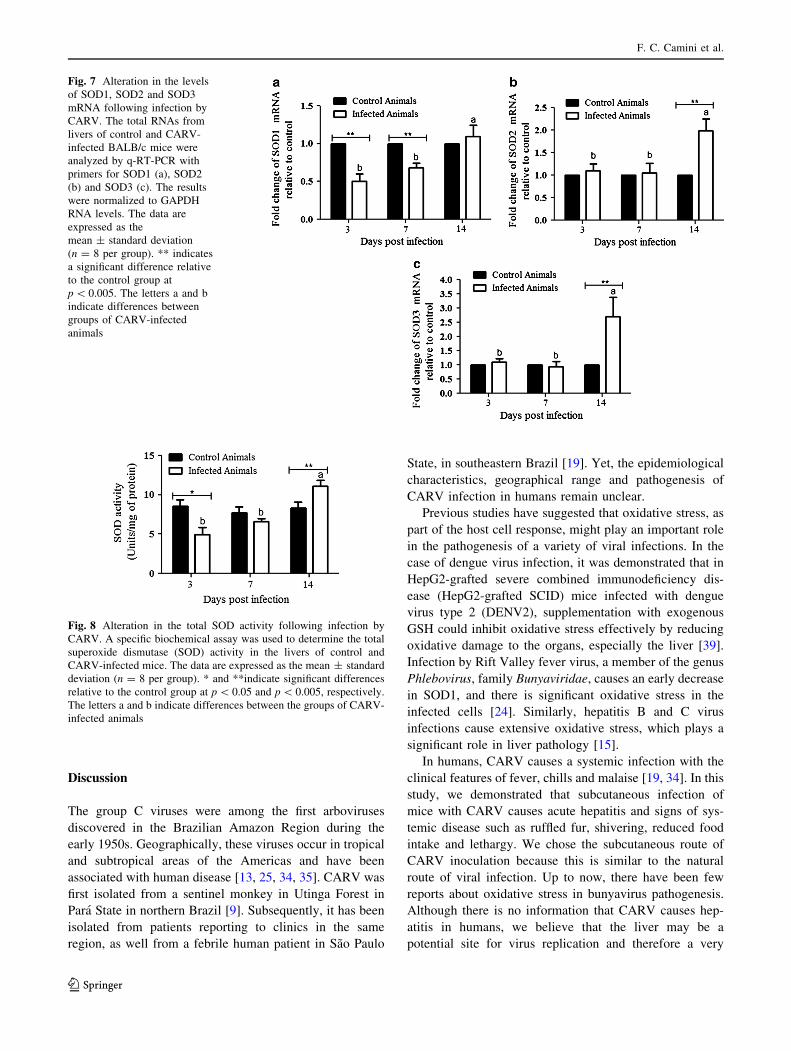
CARV infection modifies the expression and activity
of the antioxidant enzymes SOD1, SOD2 and SOD3
The first ROS produced in the reduction pathway of oxy-
gen is O2-, which is metabolized to H2O2 by SOD1, SOD2
or SOD3, depending on the primary site of O2- production.
Altered SOD levels have been associated with many
infectious and non-infectious disease states. To investigate
the effect of CARV infection on the expression and activity
of SOD (a protective mechanism against oxidative damage
in the livers of mice), expression of the SOD1, SOD2 and
SOD3 genes and total SOD activity were evaluated by
q-RT-PCR and colorimetric assays, respectively.
Total RNA was isolated from the livers of control and
CARV-infected mice on days 3, 7 and 14 pi, and the
SOD1, SOD2 and SOD3 mRNAs were amplified by
q-RT-PCR. In CARV-infected mice, there was a signifi-
cant decrease in SOD1 mRNA expression on days 3 and 7
pi (*50 and 32 %, respectively) compared with unin-
fected mice, with levels returning to those in controls by
day 14 pi (Fig. 7a). Levels of SOD2 and SOD3 mRNA
were similar in infected and control mice on days 3 and 7
pi (Fig. 7b, c). However, on day 14 pi, the levels of SOD2
and SOD3 mRNA in CARV-infected mice showed an
increase (*100 % and 200 %, respectively) compared to
control mice (Fig. 7b, c).
Our observation that the expression of the SOD1, SOD2
and SOD3 genes showed alterations on different days
following CARV infection prompted us to test whether
viral infection could modify the activity of these AOEs.
The total SOD activity in CARV-infected mice was sig-
nificantly lower than in uninfected mice on day 3 pi
(*42 %), with levels returning to those in the control mice
by day 7 pi; however, the SOD activity was significantly
higher (*33 %) in CARV-infected mice on day 14 pi
(Fig. 8). Collectively, our data indicate that following
CARV infection, the livers of mice display lowered
abundance of the host antioxidant SOD, and this upregu-
lation appears to be due to a late event during disease
progression.
Table 1 Biomarkers of oxidative stress in control and CARV-infected BALB/c mice
Variable Experimental groups
Day 3 Day 7 Day 14
Control Infected Control Infected Control Infected
TBARS
(nmol/mg protein)
1.03 ± 0.41 1.02 ± 0.15 1.49 ± 0.27 1.49 ± 0.35 1.76 ± 0.49 1.66 ± 0.29
Carbonyl protein (nmol/mg protein) 3.98 ± 0.55 3.27 ± 0.94 3.57 ± 0.70 3.79 ± 1.68 3.54 ± 1.13 3.59 ± 0.93
TBARS, thiobarbituric acid reactive substances. Values are expressed as mean ± SD (n = 8). There were no significant differences between
control and infected groups
Fig. 6 Effect of CARV infection on the hepatic GSH content in
BALB/c mice. The GSH content was measured as described above in
the liver homogenates of CARV-infected and control mice on various
days after inoculation. The fold differences in GSH levels between
uninfected control mice and CARV-infected mice were determined.
The data are expressed as the mean ± standard deviation (n = 8 per
group). ** and *** indicate significant differences relative to the
control group at p \ 0.005 and p \ 0.0005, respectively
Changes in oxidative homeostasis in Caraparu virus infection
123
Discussion
The group C viruses were among the first arboviruses
discovered in the Brazilian Amazon Region during the
early 1950s. Geographically, these viruses occur in tropical
and subtropical areas of the Americas and have been
associated with human disease [13, 25, 34, 35]. CARV was
first isolated from a sentinel monkey in Utinga Forest in
Para State in northern Brazil [9]. Subsequently, it has been
isolated from patients reporting to clinics in the same
region, as well from a febrile human patient in Sao Paulo
State, in southeastern Brazil [19]. Yet, the epidemiological
characteristics, geographical range and pathogenesis of
CARV infection in humans remain unclear.
Previous studies have suggested that oxidative stress, as
part of the host cell response, might play an important role
in the pathogenesis of a variety of viral infections. In the
case of dengue virus infection, it was demonstrated that in
HepG2-grafted severe combined immunodeficiency dis-
ease (HepG2-grafted SCID) mice infected with dengue
virus type 2 (DENV2), supplementation with exogenous
GSH could inhibit oxidative stress effectively by reducing
oxidative damage to the organs, especially the liver [39].
Infection by Rift Valley fever virus, a member of the genus
Phlebovirus, family Bunyaviridae, causes an early decrease
in SOD1, and there is significant oxidative stress in the
infected cells [24]. Similarly, hepatitis B and C virus
infections cause extensive oxidative stress, which plays a
significant role in liver pathology [15].
In humans, CARV causes a systemic infection with the
clinical features of fever, chills and malaise [19, 34]. In this
study, we demonstrated that subcutaneous infection of
mice with CARV causes acute hepatitis and signs of sys-
temic disease such as ruffled fur, shivering, reduced food
intake and lethargy. We chose the subcutaneous route of
CARV inoculation because this is similar to the natural
route of viral infection. Up to now, there have been few
reports about oxidative stress in bunyavirus pathogenesis.
Although there is no information that CARV causes hep-
atitis in humans, we believe that the liver may be a
potential site for virus replication and therefore a very
Fig. 7 Alteration in the levels
of SOD1, SOD2 and SOD3
mRNA following infection by
CARV. The total RNAs from
livers of control and CARV-
infected BALB/c mice were
analyzed by q-RT-PCR with
primers for SOD1 (a), SOD2
(b) and SOD3 (c). The results
were normalized to GAPDH
RNA levels. The data are
expressed as the
mean ± standard deviation
(n = 8 per group). ** indicates
a significant difference relative
to the control group at
p \ 0.005. The letters a and b
indicate differences between
groups of CARV-infected
animals
Fig. 8 Alteration in the total SOD activity following infection by
CARV. A specific biochemical assay was used to determine the total
superoxide dismutase (SOD) activity in the livers of control and
CARV-infected mice. The data are expressed as the mean ± standard
deviation (n = 8 per group). * and **indicate significant differences
relative to the control group at p \ 0.05 and p \ 0.005, respectively.
The letters a and b indicate differences between the groups of CARV-
infected animals
F. C. Camini et al.
123

useful model for evaluating the involvement of oxidative
homeostasis on CARV pathology as well as the pathology
of other members of the family Bunyaviridae.
In the present study, CARV-infected mice developed
significant hepatic damage on the third and seventh days pi,
confirmed by histopathological studies and changes in the
serum levels of ALT/AST. CARV antigen was detected in
all infected animals on different days pi, in several areas of
the hepatic parenchyma and in the cytoplasm of the cells.
However, stronger staining was observed in the paren-
chyma of infected mice on day 3 pi when the CARV
progeny titer was 106 PFU per gram of liver, indicating that
CARV replication is highly efficient in the liver. Despite
the accentuated inflammation and liver damage observed
on the seventh day after CARV infection, the virus was not
recovered from the liver by titration. It is possible that the
extremely high level of inflammatory response observed on
day 7 pi is sufficient to inhibit viral replication. The
observed hepatotropic nature of CARV in our study sub-
stantiates earlier findings obtained upon intraperitoneal
inoculation of mice (B6C3F1), where the animals devel-
oped liver disease [3].
Next, we investigated whether CARV infection in mice
led to cellular oxidative stress in the liver. Elevation of the
levels of TBARS and carbonyl protein is an indicator of
oxidative damage, which is usually caused by an increase
in intracellular ROS. In this study, the levels of TBARS
and carbonyl protein in infected animals were similar to
those in control mice on different days, indicating that
oxidative-stress-induced damage of the liver does not
necessarily occur in CARV infection. Because GSH is the
major endogenous antioxidant produced by cells, we
investigated if CARV infection alters the GSH content in
the liver. There was an increase in hepatic GSH content in
the CARV-infected groups relative to the control groups on
all days pi. Thus, our study demonstrated that in CARV
infection, endogenous GSH might be involved in neutral-
izing ROS and constitute an important cellular system that
counteracts oxidation and plays a significant role in
maintaining a reductive intracellular environment.
Because it is essential to proper liver function, exoge-
nous GSH has been used for the inhibition of oxidative
stress. In a previous study using HepG2 cells, DENV2
infection resulted in decreased intracellular GSH levels,
and supplementation with GSH significantly decreased the
production of DENV2 [36]. Additionally, in mice infected
with DENV2, supplementation with GSH inhibited hepatic
oxidative stress [39]. In other studies, treatment with GSH
had an inhibitory effect on the production of influenza A
virus or herpes simplex virus type 1 (HSV-1), suppressed
rhinovirus-induced superoxide radical production, and
reduced the weights of the spleen and lymph node in HIV-
infected mice [5, 27–29]. These studies suggest that the
inhibition of oxidative pathways by antioxidants such as
GSH has therapeutic potential for viral diseases.
Because the other component of the cellular antioxidant
machinery is the SOD family of enzymes, we investigated
the roles of the intracellular isoforms SOD1/SOD2 and of
the extracellular isoform SOD3 in CARV infection. Levels
of SOD1 mRNA were decreased in CARV-infected mice
on days 3 and 7 pi, and levels of SOD2 and SOD3 mRNA
were higher in the infected mice on day 14 pi. Consistent
with the observed levels of SOD1, SOD2 and SOD3
mRNA, the total SOD activity in CARV-infected mice
relative to control mice was lower on day 3 pi and higher
on day 14 pi. Despite the reduction of cellular SOD activity
3 days after infection, there was an increase of approxi-
mately 20 % in GSH content on this day. SOD enzymes
convert O2- to H2O2, and the glutathione redox cycle is
complementary to catalase in converting H2O2 to water and
oxygen. In the presence of H2O2, reduced GSH can be
oxidized to its dimeric oxidized form, GSSH, protecting
cells from oxidative stress. The decrease in total SOD
activity 3 days after infection suggests that livers of
infected mice display lower levels of H2O2 at early time
points of infection. Then, when the amount of H2O2 is low,
the cells maintain high GSH content in part by preventing
the oxidation of GSH to GSSG.
Next, since recent studies have demonstrated that an
increase in TNF-a causes downregulation of SOD1
expression [1, 24], we investigated the relationship
between TNF-a and SOD1 after CARV infection. In
infected animals, there is a significant increase in TNF-amRNA levels 3 days after infection and a concomitant
decrease in SOD1 expression. Thus, our data suggest that
upregulated TNF-a could contribute to lowered SOD1
mRNA levels in CARV-infected mice during the early
stages of infection. TNF-a is a pleiotropic cytokine that
regulates a broad range of biological events, including cell
differentiation, proliferation, tissue development and death,
as well as inflammation and innate and adaptive immune
responses. The increase in the levels of TNF-a mRNA on
the third day after CARV infection was associated with
severity of clinical signs and inflammation in the liver of
BALB/c mice. Yet, this increase on the levels of TNF-apossibly activates different pathways of the innate immune
response, favoring the control of viral load, which may
result in resistance to CARV in BALB/c mice. However,
further studies are needed to better determine the role of
TNF-a in this model of CARV-induced liver disease.
Although our study was performed only on days 3, 7 and
14 after infection, it included a balanced representation of
the progression of the disease caused by CARV in the
livers of BALB/c mice from acute hepatitis until the
recuperation of the animals. However, assessing the effect
of the infection within 3 days could contribute to a better
Changes in oxidative homeostasis in Caraparu virus infection
123

understanding of the relationships between endogenous
GSH, ROS detoxification, other AOEs, and anti-inflam-
matory mediators on CARV-induced hepatic disease.
In relation to possible involvement of brain damage after
CARV infection, there are no data in the literature showing
that CARV is neurotropic after inoculation in adult mice.
According to Briton et al. [3], newborn mice develop
hepatitis and encephalitis after intracerebral injection of
CARV, but adult mice inoculated with virus by the intra-
peritoneal route develop only hepatic disease. Here, ani-
mals infected by the subcutaneous route with CARV
developed clinical signs and liver disease, without notice-
able neurological manifestations; however, possible dam-
age to the brain has not been investigated. It would be
interesting to examine, in future studies, if subcutaneous
infection by CARV in adult mice causes neurologic injury
and to investigate its relationship to oxidative homeostasis.
Based on our findings, we propose a model considering
the possible factors that might contribute to acute hepatitis
and disease resolution in CARV-infected BALB/c mice
(Fig. 9). Briefly, following subcutaneous infection with
105 PFU of CARV, the virus exhibits hepatic tropism.
Then, there is an increase in TNF-a levels at the early
stages of infection, which possibly activates the inflam-
matory response, leading to downregulation of SOD1
expression and favoring acute inflammation in the tissue
(with an increase in serum levels of AST/ALT), as well as
reduced SOD activity. However, despite the reduction in
SOD activity, no evident oxidative stress was observed. As
the infection progresses, an effective host response is
elicited in which TNF-a levels, inflammatory cells and
AST/ALT levels return to normal. There is also an increase
in SOD2 and SOD3 expression and total SOD activity,
which might help to control both virus load and disease
resolution. Furthermore, the increase in both the total SOD
activity and GSH content corroborate the hypothesis that
CARV infection may lead to increased levels of intracel-
lular O2-, which is metabolized to H2O2 by SOD, and GSH
detoxifies H2O2 by generating water and oxygen, main-
taining the oxidant/antioxidant balance and preventing
oxidative stress during CARV liver injury. However, fur-
ther work is needed to confirm this hypothesis, since levels
of ROS were not tested here.
The development of experimental models is a necessity
for virologists to understand pathogenesis. This report
describes a subcutaneous experimental infection of BALB/
c mice with CARV, resulting in hepatic disease with
alterations in antioxidant defenses, but no obvious oxida-
tive stress. Other important aspects to be investigated in
future studies comparing infections caused by CARV to
those caused by other hepatotropic pathogens include the
profiles of AOEs that are altered in severe versus mild
forms of hepatitis, the ability of each pathogen to trigger
the generation of ROS, and the extent and type of oxidative
damage. Collectively, our data shed light on some early
mechanisms that are operational in the murine host fol-
lowing exposure to CARV.
Acknowledgements This work received financial support from
Fundacao de Amparo a Pesquisa do Estado de Minas Gerais (FAP-
EMIG) – Process APQ-04125-10, Brazil. We thank Universidade
Federal de Ouro Preto (UFOP) and the Research Center in Biological
Sciences (NUPEB/UFOP), Brazil. The authors are grateful to col-
leagues from the Virus Laboratory (UFMG), Laboratory of Metabolic
Biochemistry (UFOP) and Experimental Nutrition (UFOP) for their
technical and scientific support. We are also grateful to Maria
Terezinha Bahia from Chagas’ Disease Laboratory (UFOP) for the
use of the real-time PCR ABI 7300 equipment (Applied Biosystems)
Subcutaneous infection (105 PFU)
CARV
ACUTE HEPATITIS
Effective host response
Virus replication
TNF- mRNA
AST/ALT
Inflammation
DISEASE RESOLUTION
No obvious oxidative damage
SOD1 mRNA
SOD activityLiver injuryGSH
Viral clearance
TNF- mRNA
AST/ALT
Inflammation
No obvious oxidative damage
SOD1/2/3 mRNA
SOD activityLiver recovery
LIVER
Hepatic tropism
H2O2(?)
H2O
GSH
Fig. 9 Schematic
representation of possible
factors associated with CARV
infection that contribute to the
acute hepatitis in BALB/c mice
and disease resolution
F. C. Camini et al.
123

and Jaquelline G. de Oliveira, Marcelo Eustaquio Silva, Melina Oli-
veira de Souza and Joamyr Victor Rossoni Junior for help with certain
experiments.
References
1. Afonso V, Santos G, Collin P, Khatib AM, Mitrovic DR, Lomri
N, Leitman DC, Lomri A (2006) Tumor necrosis factor-alpha
down-regulates human Cu/Zn superoxide dismutase 1 promoter
via JNK/AP-1 signaling pathway. Free Radic Biol Med
41:709–721
2. Bernardes-Terzian AC, de-Moraes-Bronzoni RV, Drumond BP,
da Silva-Nunes M, da Silva NS, Urbano-Ferreira M, Speranca
MA, Nogueira ML (2009) Sporadic oropouche virus infection,
Acre, Brazil. Emerg Infect Dis 15:348–350
3. Brinton MA, Gavin EI, Lo WK, Pinto AJ, Morahan OS (1993)
Characterization of murine Caraparu Bunyavirus liver infection
and imunomodulator-mediated antiviral protection. Antiviral Res
20:155–171
4. Buege JA, Aust SD (1978) Microsomal lipid peroxidation.
Methods Enzymol 52:302–310
5. Cai J, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP
(2003) Inhibition of influenza infection by glutathione. Free
Radic Biol Med 34:928–936
6. Casals J, Whitman L (1961) Group C, a new serological group of
hitherto undescribed arthropod-borne viruses. Immunological
studies. Am J Trop Med Hyg 10:250–258
7. Casola A, Burger N, Liu T, Jamaluddin M, Brasier AR, Garofalo
RP (2001) Oxidant tone regulates RANTES gene expression in
airway epithelial cells infected with respiratory syncytial virus.
Role in viral-induced interferon regulatory factor activation.
J Biol Chem 276:19715–19722
8. Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA,
Colasurdo GN, Khan AM, Garofalo RP, Casola A (2006) Anti-
oxidant treatment ameliorates respiratory syncytial virus-induced
disease and lung inflammation. Am J Respir Crit Care Med
174:1361–1369
9. Causey OR, Causey CE, Maroja OM, Macedo DG (1961) The
isolation of arthropod-born viruses including members of two
hitherto undescribed serological groups, in the Amazon Region of
Brazil. Am J Trop Med Hyg 10:227–249
10. Chen TH, Tang P, Yang CF, Kao LH, Lo YP, Chuang CK, Shih
YT, Chen WJ (2011) Antioxidant defense is one of the mecha-
nisms by which mosquito cells survive dengue 2 viral infection.
Virology 410:410–417
11. de Brito Magalhaes CL, Drumond BP, Novaes RF, Quinan BR,
de Magalhaes JC, dos Santos JR, Pinto CA, Assis MT, Bonjardim
CA, Kroon EG, Ferreira PC (2011) Identification of a phyloge-
netically distinct orthobunyavirus from group C. Arch Virol
156:1173–1184
12. Elliott RM, Weber F (2009) Bunyaviruses and the Type I inter-
feron system. Viruses 1:1003–1021
13. Forshey BM, Guevara C, Laguna-Torres VA, Cespedes M,
Vargas J, Gianella A, Vallejo E, Madrid C, Aguayo N, Gotuzzo
E, Suarez V, Morales AM, Beingolea L, Reyes N, Perez J,
Negrete M, Rocha C, Morrison AC, Russell KL, Blair PJ, Olson
JG, Kochel TJ (2010) Arboviral etiologies of acute febrile ill-
nesses in Western South America, 2000–2007. PLoS Negl Trop
Dis 4:787
14. Gabbita SP, Robinson KA, Stewart CA, Floyd RA, Hensley K
(2000) Redox regulatory mechanisms of cellular signal trans-
duction. Arch Biochem Biophys 376:1–13
15. Ha HL, Shin HJ, Feitelson MA, Yu DY (2010) Oxidative stress
and antioxidants in hepatic pathogenesis. World J Gastroenterol
16:6035–6043
16. Hosakote YM, Jantzi PD, Esham DL, Spratt H, Kurosky A, Ca-
sola A, Garofalo RP (2011) Viral-mediated inhibition of antiox-
idant enzymes contributes to the pathogenesis of severe
respiratory syncytial virus bronchiolitis. Am J Respir Crit Care
Med 183:1550–1560
17. Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A (2009)
Respiratory syncytial virus induces oxidative stress by modulat-
ing antioxidant enzymes. Am J Respir Cell Mol Biol 41:348–357
18. Huang SH, Cao XJ, Liu W, Shi XY, Wei W (2010) Inhibitory
effect of melatonin on lung oxidative stress induced by respira-
tory syncytial virus infection in mice. J Pineal Res 48:109–116
19. Iversson LB, Travassos da Rosa APA, Coimbra TLM, Ferreira
IB, Nassar ES (1987) Human disease in Ribeira Valley, Brazil
caused by Caraparu, a group C arbovirus- report of a case. Rev
Inst Med Trop 29:112–117
20. Levine RL, Williams JA, Stadtman ER, Shacter E (1994) Car-
bonyl assays for determination of oxidatively modified proteins.
Methods Enzymol 233:346–357
21. Liu T, Castro S, Brasier AR, Jamaluddin M, Garofalo RP, Casola
A (2004) Reactive oxygen species mediate virus-induced STAT
activation: role of tyrosine phosphatases. J Biol Chem
279:2461–2469
22. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein
measurement with the Folin phenol reagent. J Biol Chem
193:265–275
23. Miao L, St Clair DK (2009) Regulation of superoxide dismutase
genes: implications in disease. Free Radic Biol Med 47:344–356
24. Narayanan A, Popova T, Turell M, Kidd J, Chertow J, Popov SG,
Bailey C, Kashanchi F, Kehn-Hall K (2011) Alteration in
superoxide dismutase 1 causes oxidative stress and p38 MAPK
activation following RVFV infection. PLoS One 6:e20354
25. Nunes MRT, Travassos da Rosa APA, Weaver SC, Tesh RB,
Vasconcelos PF (2005) Molecular epidemiology of group C
viruses (Bunyaviridae, Orthobunyavirus) isolated in the Ameri-
cas. J Virol 79:10561–10570
26. Ostergaard H, Tachibana C, Winther JR (2004) Monitoring
disulfide bond formation in the eukaryotic cytosol. J Cell Biol
166:337–345
27. Palamara AT, Garaci E, Rotilio G, Ciriolo MR, Casabianca A,
Fraternale A, Rossi L, Schiavano GF, Chiarantini L, Magnani M
(1996) Inhibition of murine AIDS by reduced glutathione. AIDS
Res Hum Retroviruses 12:1373–1381
28. Palamara AT, Perno CF, Ciriolo MR, Dini L, Balestra E,
D’Agostini C, Di Francesco P, Favalli C, Rotilio G, Garaci E
(1995) Evidence for antiviral activity of glutathione: in vitro
inhibition of herpes simplex virus type 1 replication. Antiviral
Res 27:237–253
29. Papi A, Contoli M, Gasparini P, Bristot L, Edwards MR, Chicca
M, Leis M, Ciaccia A, Caramori G, Johnston SL, Pinamonti S
(2008) Role of xanthine oxidase activation and reduced gluta-
thione depletion in rhinovirus induction of inflammation in
respiratory epithelial cells. J Biol Chem 283:28595–28606
30. Pinheiro FP, Travassos da Rosa AP, Travassos da Rosa JF, Ishak
R, Freitas RB, Gomes ML, LeDuc JW, Oliva OF (1981) Oro-
pouche virus. I. A review of clinical, epidemiological, and eco-
logical findings. Am J Trop Med Hyg 30:149–160
31. Qin Y, Tian Y (2010) Protective effects of total glucosides of
paeony and the underlying mechanisms in carbon tetrachloride-
induced experimental liver injury. Arch Med Sci 7:604–612
32. Schmaljohn CS, Nichol ST (2007) Bunyaviridae. In: Fields BN,
Knipe DM, Howley PM, Griffin DE (eds) Fields Virology, 5th
edn. Lippincott Williams and Wilkins, Philadelphia,
pp 1741–1778
33. Shope RE, Whitman L (1966) Nepuyo virus, a new group C agent
isolated in Trinidad and Brazil. II Serological studies. Am J Trop
Med Hyg 15:772–774
Changes in oxidative homeostasis in Caraparu virus infection
123

34. Shope RE, Woodall JP, Travassos da Rosa APA (1988) The
epidemiology of disease caused by viruses in group C and Guama
(Bunyaviridae). In: Monath TP (ed) The arboviruses: epidemi-
ology and ecology. CRC Press Inc, Boca Raton, pp 37–52
35. Soldan SS, Gonzalez-Scarano F (2005) Emerging infectious
diseases: the Bunyaviridae. J Neurovirol 11:412–423
36. Tian Y, Jiang W, Gao N, Zhang J, Chen W, Fan D, Zhou D, An J
(2010) Inhibitory effects of glutathione on dengue virus produc-
tion. Biochem Biophys Res Commun 397:420–424
37. Vasconcelos HB, Azevedo RS, Casseb SM, Nunes-Neto JP,
Chiang JO, Cantuaria PC, Segura MN, Martins LC, Monteiro
HA, Rodrigues SG, Nunes MR, Vasconcelos PF (2009) Or-
opouche fever epidemic in Northern Brazil: epidemiology and
molecular characterization of isolates. J Clin Virol
44:129–133
38. Vasconcelos PFC, Da Rosa JF, Da Rosa AP, Degallier N, Pin-
heiro FP, Sa Filho GC (1991) Epidemiology of encephalitis
caused by arbovirus in the Brazilian Amazonia. Rev Inst Med
Trop Sao Paulo 33:465–476
39. Wang J, Chen Y, Gao N, Wang Y, Tian Y, Wu J, Zhang J, Zhu J,
Fan D, An J (2013) Inhibitory effect of glutathione on oxidative
liver injury induced by dengue virus serotype 2 infections in
mice. PLoS One 8:e55407
40. Xiong Q, Xie P, Li H, Hao L, Li G, Qiu T, Liu Y (2010) Acute
effects of microcystins exposure on the transcription of antioxi-
dant enzyme gene in three organs (liver, kidney, and testis) of
male Wistar rats. J Biochem Mol Toxicol 24:361–367
41. Zamocky M, Furtmuller PG, Obinger C (2008) Evolution of
catalases from bacteria to humans. Antioxid Redox Signal
10:1527–1548
F. C. Camini et al.
123





![Case Report Subcutaneous Emphysema, …downloads.hindawi.com/journals/criem/2015/134816.pdfpneumothorax, pneumomediastinum, pneumopericardium, or subcutaneous emphysema [ ]. Diagnosis](https://img.pdfslide.tips/doc/110x75/5f4072ff5627821a5534fd08/case-report-subcutaneous-emphysema-pneumothorax-pneumomediastinum-pneumopericardium.jpg)






















