Embed Size (px)
Citation preview

www.elsevier.com/locate/ynbdi
Neurobiology of Disease 28 (2007) 3–15Cholinergic forebrain degeneration in the APPswe/PS1ΔE9transgenic mouse
Sylvia E. Perez,a Saleem Dar,a Milos D. Ikonomovic,b
Steven T. DeKosky,b and Elliott J. Mufsona,⁎
aDepartment of Neurological Sciences, Alla V. and Solomon Jesmer Chair in Aging, Rush University Medical Center, 1735 W. Harrison Street,Suite 300, Chicago, IL 60612, USA
bDepartments of Neurology and Psychiatry, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA
Received 10 April 2007; revised 5 June 2007; accepted 6 June 2007Available online 27 June 2007
The impact of Aβ deposition upon cholinergic intrinsic cortical andstriatal, as well as basal forebrain long projection neuronal systemswas qualitatively and quantitatively evaluated in young (2–6 months)and middle-aged (10–16 months) APPswe/PS1ΔE9 transgenic (tg)mice. Cholinergic neuritic swellings occurred as early as 2–3 months ofage in the cortex and hippocampus and 5–6 months in the striatum oftg mice. However, cholinergic neuron number or choline acetyltrans-ferase (ChAT) optical density measurements remained unchanged inthe forebrain structures with age in APPswe/PS1ΔE9 tg mice. ChATenzyme activity decreased significantly in the cortex and hippocampusof middle-aged tg mice. These results suggest that Aβ deposition hasage-dependent effects on cortical and hippocampal ChAT fibernetworks and enzyme activity, but does not impact the survival ofcholinergic intrinsic or long projection forebrain neurons in APPswe/PS1ΔE9 tg mice.© 2007 Elsevier Inc. All rights reserved.
Keywords: Alzheimer’s disease; Amyloid; Forebrain; Transgenics; Acetyl-choline; ChAT activity; AChE; Stereology; Interneurons; Degeneration
Introduction
Alzheimer’s disease (AD) is characterized by cognitive decline,which is accompanied by beta-amyloid (Aβ) plaque and neuro-fibrillary tangle formation in the diseased brain (Arnold et al.,1991; Braak and Braak, 1991). In early onset familial forms of AD(FAD) and in sporadic AD Aβ-deposits are composed of ∼4 kDaAβ-peptides derived from amyloid precursor proteins (APP). FADare autosomal dominant forms of AD caused by the expression ofmutant human genes encoding APP, presenilin 1 (PS1) orpresenilin 2 (PS2), that lead to increased production of highlypathogenic 42-amino-acid β-amyloid (Aβ1–42) peptides (Price and
⁎ Corresponding author. Fax: +1 312 563 3571.E-mail address: [email protected] (E.J. Mufson).Available online on ScienceDirect (www.sciencedirect.com).
0969-9961/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.nbd.2007.06.015
Sisodia, 1998). In addition to plaque and tangle formation there is areduction in subcortical cholinergic nucleus basalis neurons as wellas cortical acetylcholine in AD (Davies and Maloney, 1976;Davies, 1979; Richter et al., 1980; Whitehouse et al., 1981, 1985;Mufson et al., 1989a,b; DeKosky et al., 2002). Likewise,cholinergic interneurons and metabolic markers are reduced inthe striatum in the presence of Aβ and neurofibrillary tangles inAD (Davies, 1979; Oyanagi et al., 1989; Braak and Braak, 1990;Selden et al., 1994a,b; Scott et al., 1995; Boissiere et al., 1997;Klunk et al., 2004). Although Aβ aggregates/deposits are thoughtto be neurotoxic (Hartley et al., 1999; Selkoe, 2001), perhaps byspreading from the cortex to subcortical nuclei (Saper et al., 1987;Mufson et al., 1995; Yanker, 1996), the impact Aβ upon centralcholinergic neuron degeneration in AD remains unclear.
Various types of transgenic mice have been engineered to mimicdifferent aspects of AD neurodegeneration (Suh and Checler, 2002;Oddo et al., 2003; Götz et al., 2004). In terms of cholinergicpathology, most mice over-expressingmutant APP and/or PS1 genesdisplay an age-dependent Aβ deposition, cortical and hippocampalcholinergic fiber degeneration (Wong et al., 1999; Hernandez et al.,2001; Jaffar et al., 2001; Boncristiano et al., 2002; Buttini et al.,2002; German et al., 2003; Aucoin et al., 2005) and memory deficits(see Suh and Checler, 2002). However, these changes are notaccompanied by a loss of cholinergic basal forebrain neurons(Hernandez et al., 2001; Jaffar et al., 2001; Boncristiano et al., 2002;German et al., 2003). Therefore, none of these mutant mice fullyrecapitulates the robust cholinergic neurodegeneration seen as one ofthe hallmarks of human AD. It is interesting to note that the majorityof studies reporting cholinergic pathologies were performed in singleAPP or PS1 mutant mice, which have limited or no Aβ deposition(Hernandez et al., 2001; Jaffar et al., 2001; Boncristiano et al., 2002;Buttini et al., 2002; German et al., 2003; Aucoin et al., 2005; Bales etal., 2006) compared to APP/PS1 double transgenic mice (Wong etal., 1999; Hernandez et al., 2001; Jaffar et al., 2001). Moreover,Aβ1–42 disrupts normal cholinergic neurotransmission (Wang et al.,2000; Nagele et al., 2002; Chen et al., 2006), further suggesting thatAβ may precipitate cholinergic system dysfunction in AD.

4 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
The APPswe/PS1ΔE9 transgenic mouse displays an early andaggressive onset of neuritic Aβ deposition in the cortex, hippo-campus and striatum (Lazarov et al., 2002; Perez et al., 2005)together with memory impairment (Savonenko et al., 2005).Despite the report of alterations in cortical acetylcholinesteraseand ChAT activity seen in 19-month-old APPswe/PS1ΔE9transgenic mice (Savonenko et al., 2005), there are no detailedinvestigations of the impact of Aβ deposition upon cholinergiclocal and long projection neuronal systems in this transgenicmouse model of AD. Therefore, we performed quantitativemorphologic and biochemical analyses of cholinergic cortical,striatal, hippocampal, and nucleus basalis cholinergic systems inyoung (2–6 months) and middle-aged (10–16 months) amyloidover-expressing APPswe/PS1ΔE9 transgenic compared to age-matched non-transgenic mice.
Materials and methods
The present study used a total of 40 animals (both genders)consisting of young (2–6 months of age) and old (10–16 monthsof age) heterozygous transgenic (tg) mice harboring FAD-linkedmutant APPswe/PS1ΔE9 [co-expressing presenilin 1 (PS1) and achimerica mouse–human amyloid precursor protein (APP) 695with mutations (K595N, M596L) linked to Swedish FADpedigrees (APPswe) via the mouse prion protein promoter(Borchelt et al., 1996a,b, 1997; Lesuisse et al., 2001)] and age-matched non-transgenic (ntg) littermate mice. At least twofemale mice were included in each group examined. These micewere obtained by crossing single APPswe line C3-3 andPS1ΔE9 line S-9 tg mice and PS1ΔE9 line S-9 tg with ntglittermate mice. The background strains for APPswe are {C3H/HeJ×C57BL/6J F3}×C57BL/6J n1, and PS1ΔE9 are C3H/HeJ×C57BL/6J F3. Animal care and procedures were con-ducted according to the National Institutes of Health Guide forthe Care and Use of Laboratory Animals. All mice wereanesthetized with an injection of ketamine/xylazine (285 mg/kg/9.5 mg/kg) and perfused transcardially with ice-cold 0.9%sodium chloride (NaCl) solution. Brains were rapidly removedfrom the skull and hemisected in a frozen stainless steel brainblocker. One hemisphere was sectioned into 1 mm coronal slabson wet ice in the brain blocker. Tissue pieces were dissectedfrom the cortex, hippocampus and striatum using fiduciarylandmarks and frozen at −80 °C until processed for enzymaticassay. The other hemisphere was immersion-fixed in 4%paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphatebuffer for 12 h at 4 °C and cryoprotected in 30% sucrose at4 °C until the brain sank, and then cut on a freezing slidingknife microtome in the coronal plane at a thickness of 40 μm.The sections were stored at −4 °C in a cryoprotectant solution(30% glycerol, 30% ethylene glycol in 0.1 M phosphatebuffered saline) prior to use.
Immunohistochemistry, immunofluorescence and histochemistry
Fixed tissue was processed as free-floating sections andsingly labeled with a polyclonal antiserum raised against cholineacetyltransferase (ChAT) from human placenta (1:1000 dilution,Chemicon, CA, USA). The specificity of the antibody has beendescribed extensively (Mesulam et al., 1983; Mufson et al.,1989a,b). Sections were washed several times in Tris-bufferedsaline (TBS) before incubation with 0.1 M sodium periodate (to
inhibit endogenous peroxidase activity) in a TBS solution for20 min. After several rinses in a solution containing 0.25%Triton X-100 in TBS, the tissue was placed in a blockingsolution containing TBS with 0.25% Triton X-100 and 3%horse serum for 1 h. Sections were subsequently incubated ingoat anti-human ChAT for 48 h in a solution containing TBS,1% Triton X-100 and 1% horse normal serum. Followingwashes with 1% horse normal serum in TBS, sections wereincubated with biotinylated horse anti-goat secondary antibodies(Vector Laboratories, CA, USA) for 1 h. After several washes inTBS the tissue was incubated for 60 min with an avidin–biotincomplex (1:500; ‘Elite Kit’, Vector Laboratories, CA, USA).Tissue was rinsed in 0.2 M sodium acetate, 1.0 M imidazolbuffer (pH 7.4), and developed in an acetate–imidazol buffercontaining 2.5% nickel sulfate, 0.05% 3,3′-diaminobenzidinetetrahydrochochloride (DAB; Sigma-Aldrich, St. Louis, MO,USA) and 0.0015% H2O2. The reaction was terminated using anacetate–imidazol buffer solution. Finally, sections were mountedon glass slides, dehydrated in graded alcohols, cleared inxylenes and cover slipped with DPX (Biochemica Fluka,Switzerland). All sections were processed at the same timeusing the same chemical reagents to avoid batch-to-batchvariation during immunostaining. Additional sections weredual-labeled for ChAT and Aβ using the 10D5 monoclonalantibody raised against amino acids 1–16 of the human beta-amyloid protein (1:10,000 dilution, gift of Elan Pharmaceutics,San Francisco, CA, USA). For double immunostaining, ChATwas developed using a nickel chromagen followed by incubationwith the monoclonal 10D5 antibody and visualized using a novared substrate kit (Vector Laboratories, CA, USA). This dual-staining method results in a two-colored profile: dark-blue ChATpositive profiles, and red Aβ containing plaques. Immunofluor-escence was also used for Aβ visualization: these sections werestained with 10D5 antiserum (1:1000 dilution) and developedwith Cy3-conjugated donkey anti-mouse antibodies (1:300;Jackson ImmunoResearch Labs, West Grove, PA, USA). Inaddition sections were also double stained with 10D5 (nova redsubstrate kit) and/or histochemically reacted for acetylcholines-terase (AChE), the degrading enzyme for acetylcholine, usingthe Karnovsky and Roots method (1964) followed by silverintensification (Emre et al., 1993).
Radioenzymatic assay
Brain regions including cortex, hippocampus and striatumfrom 2–6 months old and 10–16 months old APPswe/PS1ΔE9 tgand age-matched ntg littermate mice were processed fordetermination of ChAT enzyme activity using a modification ofthe Fonnum method (Fonnum, 1975; DeKosky et al., 1985).Frozen tissue was homogenized using high frequency sonicationin a solution containing 0.5% Triton X-100 and 10 mM EDTA.Briefly, 5 μl of tissue homogenate was combined with C-14labeled acetyl Co-A (New England Nuclear, Boston, MA, USA),incubation buffer (100 mM sodium phosphate, 600 mM NaCl,20 mM choline chloride, 10 mM disodium EDTA, pH7.4), andphysostigmine (20 mM, Sigma-Aldrich, St. Louis, MO, USA).After 30 min of incubation at 37 °C in a water bath, the reactionwas stopped by the addition of 4 ml of 10 mM phosphate buffer(pH 7.4). Subsequently, 1.6 ml of acetonitrile/tetrephenalboronmixture and 8 ml of scintillation fluid were added to cause phaseseparation. After the samples stabilized for 24 h, they were

5S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
counted in a scintillation counter. Protein content of the sampleswas determined using BCA protein assay kits (Pierce, Rockford,IL, USA). ChAT activity was expressed as μmol/h/g protein.Samples were coded, and all assays performed in triplicate by atechnician blinded to experimental groups.
Stereologic analysis
The optical dissector method was used to determine thenumber of ChAT immunoreactive (-ir) neurons in the cerebralcortex (motor, cingulate and sensory cortices), striatum andnucleus basalis in 3- to 6-month-old and 10- to 16-month-oldAPPswe/PS1ΔE9 tg as well as age-matched ntg mice aspreviously described (Jaffar et al., 2001; Perez et al., 2005). Theregions were manually outlined under low magnification andsystematically analyzed using a random sampling design. Theestimated numbers of ChAT-ir neurons were performed usingMicroBrightField stereological software (Williston, VT) in anOlympus BX-60 microscope coupled with LEP MAC5000(BioVision Technologies; Exton, PA, USA). The coefficients oferror were calculated according to Gundersen et al. (1988) andvalues b0.10 were accepted (West, 1993). The ChAT-ir neurons inthe nucleus basalis were counted immediately caudal to thedecussation of the anterior commissure until the end of the globuspallidus (Figs. 1B–D). These neurons were located ventral to andwithin the globus pallidus as well as intermingled within theinternal capsule tracts. The ChAT-ir neurons in the sensory cortexwere counted until the appearance of cholinergic immunoreactivityin the interpeduncular nucleus (Figs. 1B–E), whereas in thecingulate and motor cortex as well as in the striatum they were
Fig. 1. (A–E) Rostral to caudal diagrams showing transverse sections delimitaticingulate and sensory cortices as well as the striatum and nucleus basalis processed fnucleus basalis; BST, bed nucleus of the stria terminalis; Cg, cingulate cortex; DGgeniculate nucleus; GP, globus pallidus; HB, horizontal limb of the diagonal bainterpeduncular nucleus; LH, lateral hypothalamic area; lo, lateral olfactory tract; LVstriatum; Pir, piriform cortex; R, red nucleus; RS, retrosplenial cortex, S, sensory corstria medullaris thalami; SN, substantia nigra; Th, thalamus.
counted throughout their entire extension (Fig. 1). Since hemi-secting a brain can result in asymmetrical midline cuts,quantification of ChAT-ir neurons in the medial septum andvertical limb of the diagonal band of Broca was not performed. Allof the ChAT-ir neuron counts were performed by an observerblinded to age and genotype.
Optical density and area measurements
Quantification of the relative optical density (OD) of neuronalChAT immunoreactivity in the cingulate, motor and sensorycortices, striatum and nucleus basalis from young (3–6 months)and middle-aged (10–16 months) APPswe/PS1ΔE9 tg and ntgmice was performed using a densitometry software program(Image 1.60, Scion 1.6) as previously described (Mufson et al.,1997; Ma et al., 1999; Jaffar et al., 2001; Perez et al., 2005). TheChAT-ir neurons of the different regions were outlined manually,and the OD and area measurements were automatically analyzedin gray-scale images. Background tissue levels were measured andthe average subtracted from the OD measurements of ChAT-ir.These measurements were performed by an observed blinded toage and genotype.
Statistical analysis
Data obtained from the ChAT enzymatic assay, stereologic andOD measurements were evaluated using the Mann–Whitney rank-sum test, a non-parametric test (SigmaStat 3.0; Aspire SoftwareInternational, Leesburg, VA, USA). This test is more powerful inevaluating small samples and minimizes the effect of the outliers
ng the level and borders (dotted-lines) outlining the regions of the motor,or stereology in APPswe/PS1ΔE9 tg and ntg mice. A, amygdala complex; B,, dentate gyrus; Ect, ectorhinal cortex; Ent, entorhinal cortex; fi, fimbria; G,nd of Broca; HC, hippocampus; I, insular cortex; ic, internal capsule; Ip,, lateral ventricle; M, motor cortex; ml, medial lemniscus; opt, optic tract; St,tex; SC, superior colliculus; SI/B, substantia innominata/nucleus basalis; sm,
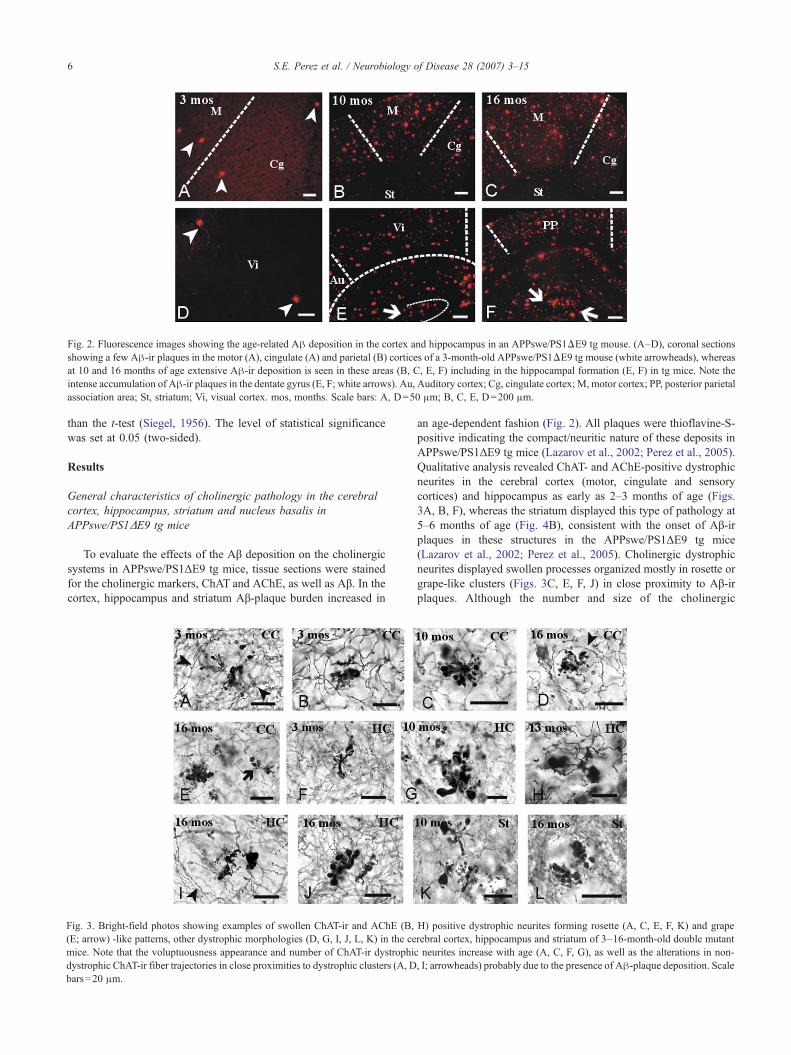
Fig. 2. Fluorescence images showing the age-related Aβ deposition in the cortex and hippocampus in an APPswe/PS1ΔE9 tg mouse. (A–D), coronal sectionsshowing a few Aβ-ir plaques in the motor (A), cingulate (A) and parietal (B) cortices of a 3-month-old APPswe/PS1ΔE9 tg mouse (white arrowheads), whereasat 10 and 16 months of age extensive Aβ-ir deposition is seen in these areas (B, C, E, F) including in the hippocampal formation (E, F) in tg mice. Note theintense accumulation of Aβ-ir plaques in the dentate gyrus (E, F; white arrows). Au, Auditory cortex; Cg, cingulate cortex; M, motor cortex; PP, posterior parietalassociation area; St, striatum; Vi, visual cortex. mos, months. Scale bars: A, D=50 μm; B, C, E, D=200 μm.
6 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
than the t-test (Siegel, 1956). The level of statistical significancewas set at 0.05 (two-sided).
Results
General characteristics of cholinergic pathology in the cerebralcortex, hippocampus, striatum and nucleus basalis inAPPswe/PS1ΔE9 tg mice
To evaluate the effects of the Aβ deposition on the cholinergicsystems in APPswe/PS1ΔE9 tg mice, tissue sections were stainedfor the cholinergic markers, ChAT and AChE, as well as Aβ. In thecortex, hippocampus and striatum Aβ-plaque burden increased in
Fig. 3. Bright-field photos showing examples of swollen ChAT-ir and AChE (B,(E; arrow) -like patterns, other dystrophic morphologies (D, G, I, J, L, K) in the cemice. Note that the voluptuousness appearance and number of ChAT-ir dystrophidystrophic ChAT-ir fiber trajectories in close proximities to dystrophic clusters (A, Dbars=20 μm.
an age-dependent fashion (Fig. 2). All plaques were thioflavine-S-positive indicating the compact/neuritic nature of these deposits inAPPswe/PS1ΔE9 tg mice (Lazarov et al., 2002; Perez et al., 2005).Qualitative analysis revealed ChAT- and AChE-positive dystrophicneurites in the cerebral cortex (motor, cingulate and sensorycortices) and hippocampus as early as 2–3 months of age (Figs.3A, B, F), whereas the striatum displayed this type of pathology at5–6 months of age (Fig. 4B), consistent with the onset of Aβ-irplaques in these structures in the APPswe/PS1ΔE9 tg mice(Lazarov et al., 2002; Perez et al., 2005). Cholinergic dystrophicneurites displayed swollen processes organized mostly in rosette orgrape-like clusters (Figs. 3C, E, F, J) in close proximity to Aβ-irplaques. Although the number and size of the cholinergic
H) positive dystrophic neurites forming rosette (A, C, E, F, K) and graperebral cortex, hippocampus and striatum of 3–16-month-old double mutanc neurites increase with age (A, C, F, G), as well as the alterations in non, I; arrowheads) probably due to the presence of Aβ-plaque deposition. Scale
t-

Fig. 4. Photographs of the striatum and nucleus basalis showing the normal morphology of the ChAT-ir neurons and ChAT-ir dystrophic neurites in 3-, 6- and16-month-old APPswe/PS1ΔE9 tg and ntg mice. (A–F), Coronal sections of the striatum illustrating the presence of ChAT-ir dystrophic neurites at 6- and16-month-old tg mice (arrowheads) compared with their absence in age-matched ntg mice (A, D). (C, F), Insets showing detail of ChAT-ir dystrophic neuritesfrom B and E, respectively. (G–I), Coronal sections of the nucleus basalis showing the absence of dystrophic neurites in a 3-month-old double mutant mouse (H)as well as in 3- and 16-month-old ntg mice (G, I). (J), Coronal section of the nucleus basalis showing a few Aβ-ir plaques (red; arrows) surrounded by ChAT-irdystrophic neurites (dark blue) in a 16-month-old double mutant mice. (K), Inset illustrating detail of swollen ChAT-ir dystrophic neurites (dark blue) in thevicinities of Aβ-ir plaques (red) in the nucleus basalis of a 16-month-old double tg mouse. ntg, non-transgenic mice; tg, APPswe/PS1ΔE9 transgenic mice; mos,months. Scale bars: A, B, D, E, G–J=50 μm; C, F, K=20 μm.
7S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
dystrophic neurites were variable, they increased in parallel withthe increase in plaque load seen in the cortex, hippocampus andstriatum (Figs. 3A, C, F, J). However, within the region containingthe cholinergic nucleus basalis neurons only occasional Aβ-irplaques and cholinergic dystrophic neurites were seen mainly inthe oldest APPswe/PS1ΔE9 tg mice (Figs. 4J, K). Furthermore,cholinergic processes arising from the neurons of nucleus basalis in16 months old tg mice showed larger axonal varicosities comparedwith age-matched ntg mice. ChAT and AChE staining revealedsimilar alterations in the laminar organization of cholinergic fibersin the cortex and the hippocampus of young and middle-agedAPPswe/PS1ΔE9 tg mice (Figs. 5B, D, F, H, J, L). In youngAPPswe/PS1ΔE9 tg mice, there was virtually no disruption ofcortical cholinergic fiber innervation compared to the dramaticdisturbance of these fibers, which occurs in association with thepresence of amyloid plaques in older mutant mice (Figs. 5D, H, L,Q and 6). However the older transgenic animals did not display alayer-specific alteration in cortical cholinergic fiber innervation(Fig. 6D). Aged tg mice (16 months) also showed a strikingreduction in the density of cholinergic fibers staining revealed byChAT immuno- or AChE histochemical staining (Figs. 5D, H, O,Q and 6D). Cholinergic fiber trajectory was altered near as wellbeyond the borders of the Aβ plaque in mutant, but was morepronounced in aged tg mice (Figs. 3A, D and 5B, D, L, Q).
Cholinergic neuronal number in the cerebral cortex, striatum andnucleus basalis of APPswe/PS1ΔE9 tg mice
To examine the effects of Aβ-plaques upon local and longprojection cholinergic neurons in young and middle-aged APPswe/PS1ΔE9 tg mice (Table 1), we quantified the number of ChAT-ircortical, striatal and nucleus basalis neurons using an unbiased
stereological counting method. Cortical and striatal ChAT-irneurons are implicated in the modulation of local neuronal circuitry,whereas ChAT-ir neurons in the nucleus basalis have longprojections and are the main source of acetylcholine in the cerebralcortex (Wainer et al., 1984). Cortical small and mainly bipolarChAT-ir neurons were counted in the motor, cingulate and sensorycortices (Figs. 5A–L and 6). ChAT-ir neurons in the cingulate andmotor cortex were found mainly in layers II–III, whereas in thesensory cortex they were seen in layers II to VI. Despite thepresence of Aβ-ir plaques in these cortical areas, as well as thestriatum there was not a significant difference in the number ofChAT-ir interneurons in these structures, except in the motor cortex(Fig. 7A). In this cortical area the number of ChAT-ir neurons weresignificantly lower in the APPswe/PS1ΔE9 tg compared to ntgmice, independent of the age-group examined (Fig. 7A; Mann–Whitney rank-sum test; p=0.033). No differences were detected inthe number of ChAT-ir neurons in the nucleus basalis between theyoung and middle-aged double mutant compared to the age-matched ntg mice (Mann–Whitney rank-sum test; pN0.05). Thesedata suggest that Aβ deposition may not lead to frank degene-ration of the cholinergic local and long projection neurons in thecingulate and sensory cortices, striatum and nucleus basalis.
Optical density measurements of cortical, striatal and nucleusbasalis neurons in APPswe/PS1ΔE9 tg mice
Levels of neuronal ChAT-ir were evaluated in neurons of themotor, cingulate and sensory cortices, striatum as well as in thenucleus basalis in young and middle-aged tg mice (Table 1), usingOD measurements (Scion image system). OD measurements ofthe ChAT-ir neurons in the motor, cingulate and sensory corticesas well as the striatum and nucleus basalis did not show
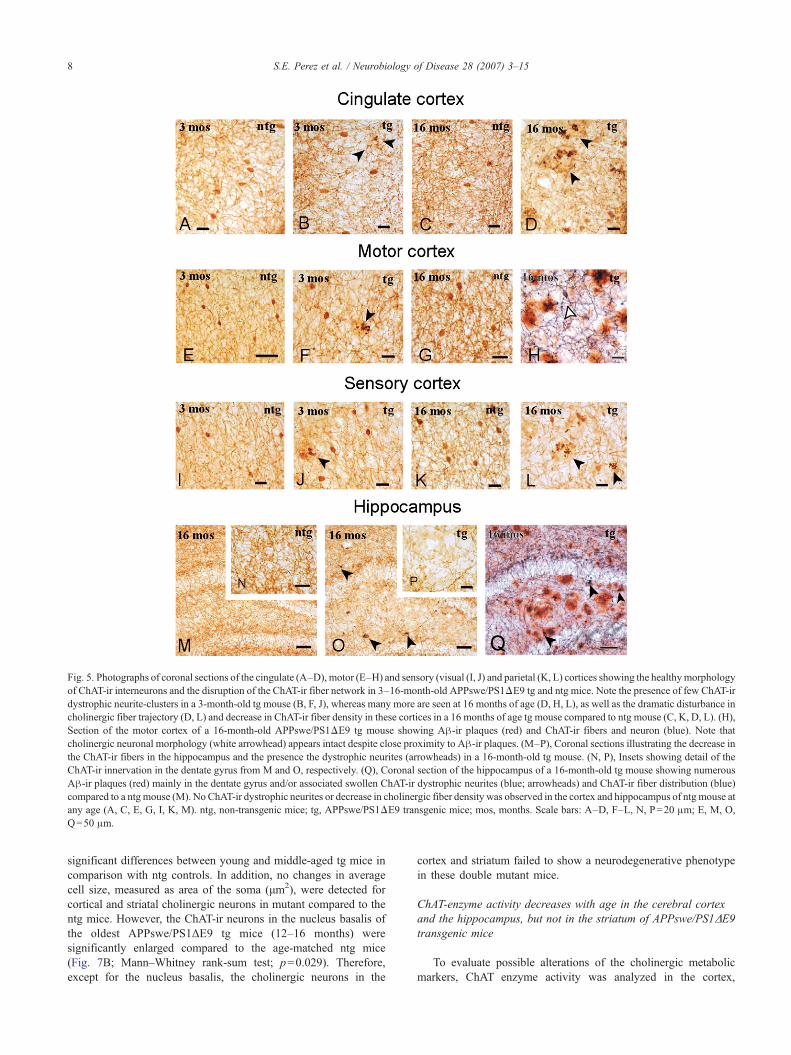
Fig. 5. Photographs of coronal sections of the cingulate (A–D), motor (E–H) and sensory (visual (I, J) and parietal (K, L) cortices showing the healthymorphologyof ChAT-ir interneurons and the disruption of the ChAT-ir fiber network in 3–16-month-old APPswe/PS1ΔE9 tg and ntg mice. Note the presence of few ChAT-irdystrophic neurite-clusters in a 3-month-old tg mouse (B, F, J), whereas many more are seen at 16 months of age (D, H, L), as well as the dramatic disturbance incholinergic fiber trajectory (D, L) and decrease in ChAT-ir fiber density in these cortices in a 16 months of age tg mouse compared to ntg mouse (C, K, D, L). (H),Section of the motor cortex of a 16-month-old APPswe/PS1ΔE9 tg mouse showing Aβ-ir plaques (red) and ChAT-ir fibers and neuron (blue). Note thatcholinergic neuronal morphology (white arrowhead) appears intact despite close proximity to Aβ-ir plaques. (M–P), Coronal sections illustrating the decrease inthe ChAT-ir fibers in the hippocampus and the presence the dystrophic neurites (arrowheads) in a 16-month-old tg mouse. (N, P), Insets showing detail of theChAT-ir innervation in the dentate gyrus from M and O, respectively. (Q), Coronal section of the hippocampus of a 16-month-old tg mouse showing numerousAβ-ir plaques (red) mainly in the dentate gyrus and/or associated swollen ChAT-ir dystrophic neurites (blue; arrowheads) and ChAT-ir fiber distribution (blue)compared to a ntg mouse (M). No ChAT-ir dystrophic neurites or decrease in cholinergic fiber density was observed in the cortex and hippocampus of ntg mouse atany age (A, C, E, G, I, K, M). ntg, non-transgenic mice; tg, APPswe/PS1ΔE9 transgenic mice; mos, months. Scale bars: A–D, F–L, N, P=20 μm; E, M, O,Q=50 μm.
8 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
significant differences between young and middle-aged tg mice incomparison with ntg controls. In addition, no changes in averagecell size, measured as area of the soma (μm2), were detected forcortical and striatal cholinergic neurons in mutant compared to thentg mice. However, the ChAT-ir neurons in the nucleus basalis ofthe oldest APPswe/PS1ΔE9 tg mice (12–16 months) weresignificantly enlarged compared to the age-matched ntg mice(Fig. 7B; Mann–Whitney rank-sum test; p=0.029). Therefore,except for the nucleus basalis, the cholinergic neurons in the
cortex and striatum failed to show a neurodegenerative phenotypein these double mutant mice.
ChAT-enzyme activity decreases with age in the cerebral cortexand the hippocampus, but not in the striatum of APPswe/PS1ΔE9transgenic mice
To evaluate possible alterations of the cholinergic metabolicmarkers, ChAT enzyme activity was analyzed in the cortex,

Fig. 6. Photographs showing lamina disturbances of cholinergic fiber inner-vation of the somatosensory cortex in 3- and 16-month-old tg mice (B, D)compared to ntg mice (A, C). There was no age-related alteration in thelaminar distribution of cholinergic fibers in the somatosensory cortexbetween young and aged ntg mice. By contrast, there was an extensivedisruption of the cholinergic fiber network and the presence of dystrophicneurites (arrowheads) in the somatosensory cortex between young and agedmutant mice. Cortical layers are indicated by the roman numerals I–VI;ntg, non-transgenic mice; tg, APPswe/PS1ΔE9 transgenic mice. Scalebar=150 μm.
9S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
hippocampus and striatum of young (2–6 months) and middle-aged(10–16 months) APPswe/PS1ΔE9 tg mice as well as in age-matched ntg mice (Table 2). ChAT enzyme activity in the cerebralcortex and hippocampus of middle-aged APPswe/PS1ΔE9 wassignificantly decreased compared to young tg mice (Figs. 8A, B;Mann–Whitney rank-sum test; cerebral cortex p=0.048, hippo-campus p=0.035). A comparison of ChATenzyme activity betweenyoung and middle-aged tg mice revealed a greater reduction in thehippocampus (∼30%) than in the cerebral cortex (∼15%). CorticalChAT enzyme activity in middle-aged tg mice was significantlyreduced compared to the middle-aged ntg mice, but not in thehippocampus (Fig. 8A; Mann–Whitney rank-sum test; p=0.018).ChAT enzyme activity in the cerebral cortex and hippocampus wasnot statistically different between young and middle-aged ntg mice(Figs. 8A, B). Striatal ChAT enzyme activity did not differ betweenthe young and the middle-aged APPswe/PS1ΔE9 tg mice incomparison with ntg mice (Fig. 8C; Mann–Whitney rank-sum test,pN0.05).
Discussion
The present study evaluated the impact of Aβ deposition uponcholinergic local and long projection forebrain neuronal systems inAPPswe/PS1ΔE9 tg mice. Swollen dystrophic cholinergic neuriteswere seen in close association with Aβ plaques throughout thecortex and hippocampus at 2–3 months and in the striatum at5–6 months in these mutant tg mice. In addition, we also observedthat the trajectory of non-dystrophic cholinergic fibers was
displaced beyond plaque borders in young and middle-agedmutant mice (D’Amore et al., 2003, Knowles et al., 1998, 1999).ChAT positive neurite size and number increased in parallel withplaque load and was more conspicuous in older mutant tg mice.These findings support and expand other investigations showingthat the formation of cholinergic neurites in the cortex, hippocam-pus and in the striatum are associated with compact (thioflavine-S-positive) rather than diffuse plaques described in other over-expressing APP mice (Hernandez et al., 2001; Boncristiano et al.,2002; Brendza et al., 2003; D’Amore et al., 2003; German et al.,2003; Hu et al., 2003). These observations suggest that the toxicproperties of Aβ proteins are dictated by its conformationalfeatures as reported for AD (Probst et al., 1983; Masliah et al.,1990; Knowles et al., 1998). Recently, it was shown that corticalcholinergic neurites are the first to form in APP over-expressedmice, followed by glutamatergic and GABA-ergic neurites (Huet al., 2003; Bell et al., 2003, 2005). Stokin et al. (2005) showedthat cholinergic axonal swellings arising from nucleus basalisneurons precede plaque formation in single APPswe tg miceleading to the suggestion that their presence is related to axonaltransport deficits (Pigino et al., 2003). Other studies suggest thatAβ deposition has neurotrophic effects (Jaffar et al., 2001), whichinduce aberrant neuritic sprouting in both, animal models of AD aswell as in the disease itself (Masliah et al., 1991, 2003; Koo et al.,1993; Phinney et al., 1999; Hernandez et al., 2001). Studies alsodemonstrate that dystrophic neurites are secondary to the presenceof Aβ and are only partially reversed by anti-Aβ antibodytreatments (Lombardo et al., 2003; Brendza et al., 2005). Theseobservations lend support to the concept that the very earlymorphological changes seen in cholinergic projections in APPswe/PS1ΔE9 tg mice parallels early changes in cholinergic transmissionin these same tg mice assessed by neuropharmacologicaltechniques (Machová et al., in press). Ultrastructural analysis ofdystrophic swellings in APPswe/PS1ΔE9 (Perez et al., 2004) andother over-expressing APP tg mice reveal the presence of vesicularorganelles (Masliah et al., 1996; Phinney et al., 1999; Luth et al.,2003), which are part of lysosomal systems implicated in endo-cytic (Cataldo et al., 2000, 2004; Mathews et al., 2002) and/orautophagic (Wang et al., 2006) mechanisms. Autophagia may bean early remodeling response to aberrant neuritic processes(Wang et al., 2006), and therefore, an attempt by the brain atself-repair.
In addition to the cholinergic dystrophic pathology, we found asignificant decrease in ChAT enzyme activity in the cerebral cortexand hippocampus of 10–16 months old compared with 2–6 monthsold APPswe/PS1ΔE9 tg mice, but not in the striatum. Similar toprevious studies in aged APPswe/PS1ΔE9 tg mice (19 months;Savonenko et al., 2005) and homozygous PDAPP tg mice(24 months; German et al., 2003), we found significant differencesin ChAT enzyme activity in the cerebral cortex between middle-aged tg and ntg mice, but not in the hippocampus. This discrepancymay be due to the pattern of reduction depicted for hippocampalChAT enzyme activity in tg mice, and in the fact that young doublemutant mice showed higher levels of ChAT enzyme activity thanthe age-matched control mice. These findings are similar to thosereported in end-stage of AD, where there is marked decrease ofChAT enzyme activity in the cortex, but not in the striatum(Davies, 1979; Araujo et al., 1988; DeKosky et al., 2002).Interestingly, ChAT activity was reported to be increased in corticalareas in middle-aged APP and PS1 single tg mice (Hernandezet al., 2001), perhaps resembling the up-regulation of cortical and
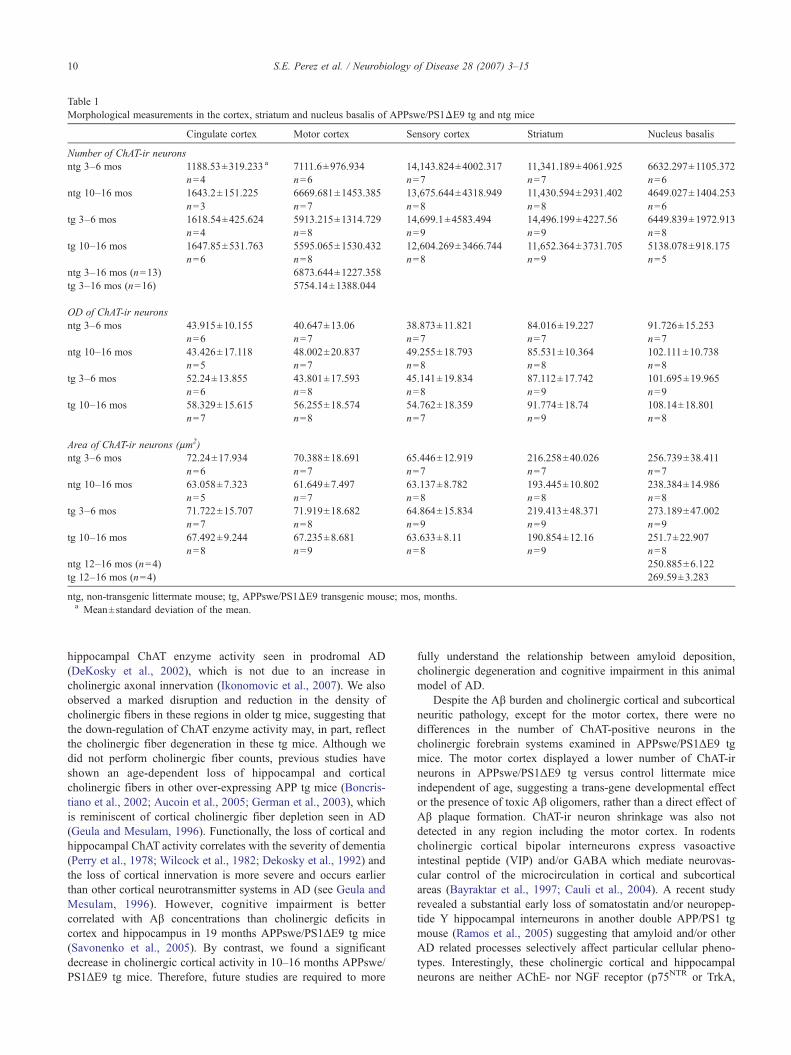
Table 1Morphological measurements in the cortex, striatum and nucleus basalis of APPswe/PS1ΔE9 tg and ntg mice
Cingulate cortex Motor cortex Sensory cortex Striatum Nucleus basalis
Number of ChAT-ir neuronsntg 3–6 mos 1188.53±319.233 a 7111.6±976.934 14,143.824±4002.317 11,341.189±4061.925 6632.297±1105.372
n=4 n=6 n=7 n=7 n=6ntg 10–16 mos 1643.2±151.225 6669.681±1453.385 13,675.644±4318.949 11,430.594±2931.402 4649.027±1404.253
n=3 n=7 n=8 n=8 n=6tg 3–6 mos 1618.54±425.624 5913.215±1314.729 14,699.1±4583.494 14,496.199±4227.56 6449.839±1972.913
n=4 n=8 n=9 n=9 n=8tg 10–16 mos 1647.85±531.763 5595.065±1530.432 12,604.269±3466.744 11,652.364±3731.705 5138.078±918.175
n=6 n=8 n=8 n=9 n=5ntg 3–16 mos (n=13) 6873.644±1227.358tg 3–16 mos (n=16) 5754.14±1388.044
OD of ChAT-ir neuronsntg 3–6 mos 43.915±10.155 40.647±13.06 38.873±11.821 84.016±19.227 91.726±15.253
n=6 n=7 n=7 n=7 n=7ntg 10–16 mos 43.426±17.118 48.002±20.837 49.255±18.793 85.531±10.364 102.111±10.738
n=5 n=7 n=8 n=8 n=8tg 3–6 mos 52.24±13.855 43.801±17.593 45.141±19.834 87.112±17.742 101.695±19.965
n=6 n=8 n=8 n=9 n=9tg 10–16 mos 58.329±15.615 56.255±18.574 54.762±18.359 91.774±18.74 108.14±18.801
n=7 n=8 n=7 n=9 n=8
Area of ChAT-ir neurons (μm2)ntg 3–6 mos 72.24±17.934 70.388±18.691 65.446±12.919 216.258±40.026 256.739±38.411
n=6 n=7 n=7 n=7 n=7ntg 10–16 mos 63.058±7.323 61.649±7.497 63.137±8.782 193.445±10.802 238.384±14.986
n=5 n=7 n=8 n=8 n=8tg 3–6 mos 71.722±15.707 71.919±18.682 64.864±15.834 219.413±48.371 273.189±47.002
n=7 n=8 n=9 n=9 n=9tg 10–16 mos 67.492±9.244 67.235±8.681 63.633±8.11 190.854±12.16 251.7±22.907
n=8 n=9 n=8 n=9 n=8ntg 12–16 mos (n=4) 250.885±6.122tg 12–16 mos (n=4) 269.59±3.283
ntg, non-transgenic littermate mouse; tg, APPswe/PS1ΔE9 transgenic mouse; mos, months.a Mean±standard deviation of the mean.
10 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
hippocampal ChAT enzyme activity seen in prodromal AD(DeKosky et al., 2002), which is not due to an increase incholinergic axonal innervation (Ikonomovic et al., 2007). We alsoobserved a marked disruption and reduction in the density ofcholinergic fibers in these regions in older tg mice, suggesting thatthe down-regulation of ChAT enzyme activity may, in part, reflectthe cholinergic fiber degeneration in these tg mice. Although wedid not perform cholinergic fiber counts, previous studies haveshown an age-dependent loss of hippocampal and corticalcholinergic fibers in other over-expressing APP tg mice (Boncris-tiano et al., 2002; Aucoin et al., 2005; German et al., 2003), whichis reminiscent of cortical cholinergic fiber depletion seen in AD(Geula and Mesulam, 1996). Functionally, the loss of cortical andhippocampal ChAT activity correlates with the severity of dementia(Perry et al., 1978; Wilcock et al., 1982; Dekosky et al., 1992) andthe loss of cortical innervation is more severe and occurs earlierthan other cortical neurotransmitter systems in AD (see Geula andMesulam, 1996). However, cognitive impairment is bettercorrelated with Aβ concentrations than cholinergic deficits incortex and hippocampus in 19 months APPswe/PS1ΔE9 tg mice(Savonenko et al., 2005). By contrast, we found a significantdecrease in cholinergic cortical activity in 10–16 months APPswe/PS1ΔE9 tg mice. Therefore, future studies are required to more
fully understand the relationship between amyloid deposition,cholinergic degeneration and cognitive impairment in this animalmodel of AD.
Despite the Aβ burden and cholinergic cortical and subcorticalneuritic pathology, except for the motor cortex, there were nodifferences in the number of ChAT-positive neurons in thecholinergic forebrain systems examined in APPswe/PS1ΔE9 tgmice. The motor cortex displayed a lower number of ChAT-irneurons in APPswe/PS1ΔE9 tg versus control littermate miceindependent of age, suggesting a trans-gene developmental effector the presence of toxic Aβ oligomers, rather than a direct effect ofAβ plaque formation. ChAT-ir neuron shrinkage was also notdetected in any region including the motor cortex. In rodentscholinergic cortical bipolar interneurons express vasoactiveintestinal peptide (VIP) and/or GABA which mediate neurovas-cular control of the microcirculation in cortical and subcorticalareas (Bayraktar et al., 1997; Cauli et al., 2004). A recent studyrevealed a substantial early loss of somatostatin and/or neuropep-tide Y hippocampal interneurons in another double APP/PS1 tgmouse (Ramos et al., 2005) suggesting that amyloid and/or otherAD related processes selectively affect particular cellular pheno-types. Interestingly, these cholinergic cortical and hippocampalneurons are neither AChE- nor NGF receptor (p75NTR or TrkA,
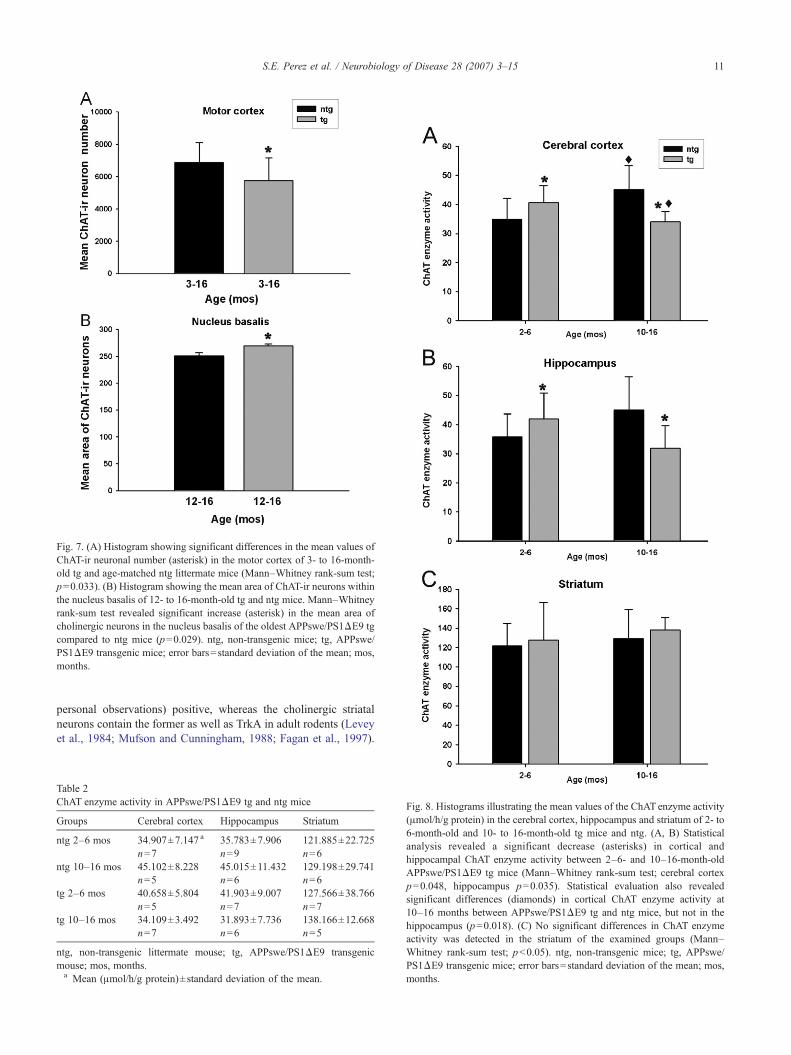
Fig. 7. (A) Histogram showing significant differences in the mean values ofChAT-ir neuronal number (asterisk) in the motor cortex of 3- to 16-month-old tg and age-matched ntg littermate mice (Mann–Whitney rank-sum test;p=0.033). (B) Histogram showing the mean area of ChAT-ir neurons withinthe nucleus basalis of 12- to 16-month-old tg and ntg mice. Mann–Whitneyrank-sum test revealed significant increase (asterisk) in the mean area ofcholinergic neurons in the nucleus basalis of the oldest APPswe/PS1ΔE9 tgcompared to ntg mice (p=0.029). ntg, non-transgenic mice; tg, APPswe/PS1ΔE9 transgenic mice; error bars=standard deviation of the mean; mos,months.
11S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
personal observations) positive, whereas the cholinergic striatalneurons contain the former as well as TrkA in adult rodents (Leveyet al., 1984; Mufson and Cunningham, 1988; Fagan et al., 1997).
Table 2ChAT enzyme activity in APPswe/PS1ΔE9 tg and ntg mice
Groups Cerebral cortex Hippocampus Striatum
ntg 2–6 mos 34.907±7.147 a 35.783±7.906 121.885±22.725n=7 n=9 n=6
ntg 10–16 mos 45.102±8.228 45.015±11.432 129.198±29.741n=5 n=6 n=6
tg 2–6 mos 40.658±5.804 41.903±9.007 127.566±38.766n=5 n=7 n=7
tg 10–16 mos 34.109±3.492 31.893±7.736 138.166±12.668n=7 n=6 n=5
ntg, non-transgenic littermate mouse; tg, APPswe/PS1ΔE9 transgenicmouse; mos, months.a Mean (μmol/h/g protein)±standard deviation of the mean.
Fig. 8. Histograms illustrating the mean values of the ChAT enzyme activity(μmol/h/g protein) in the cerebral cortex, hippocampus and striatum of 2- to6-month-old and 10- to 16-month-old tg mice and ntg. (A, B) Statisticalanalysis revealed a significant decrease (asterisks) in cortical andhippocampal ChAT enzyme activity between 2–6- and 10–16-month-oldAPPswe/PS1ΔE9 tg mice (Mann–Whitney rank-sum test; cerebral cortexp=0.048, hippocampus p=0.035). Statistical evaluation also revealedsignificant differences (diamonds) in cortical ChAT enzyme activity at10–16 months between APPswe/PS1ΔE9 tg and ntg mice, but not in thehippocampus (p=0.018). (C) No significant differences in ChAT enzymeactivity was detected in the striatum of the examined groups (Mann–Whitney rank-sum test; pb0.05). ntg, non-transgenic mice; tg, APPswe/PS1ΔE9 transgenic mice; error bars=standard deviation of the mean; mos,months.

12 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
These findings indicate differences in cholinergic phenotypebetween cortical, hippocampal, striatal as opposed to nucleusbasalis neurons which display AChE and both NGF receptors(Sobreviela et al., 1994). It is important to note that cortical orhippocampal cholinergic neurons have not been reported in theprimate brain (Wainer et al., 1984) suggesting a species andperhaps functional difference between rodents and non-humanprimates which needs to be considered when modeling AD.
Although the present study failed to reveal a loss of cholinergicstriatal neurons in APPswe/PS1ΔE9 tg mice, it should be noted thatthe prion protein promoter drives the highest trans-gene expressionin the cerebral cortex and hippocampus. This may possibly be afactor underlying the lower amyloid load and cholinergic pathologyseen in the striatum (Borchelt et al., 1996a). Although dystrophicneurites and plaques are found in the AD striatum (Rudelli et al.,1984; Suenaga et al., 1990; Brilliant et al., 1997), it remainscontroversial whether there is a loss of cholinergic neurons in theneostriatum (Oyanagi et al., 1989; Selden et al., 1994b) as comparedto the reduction seen in the ventral striatum in AD (Lehericy et al.,1989; Selden et al., 1994b). These observations suggest that thecurrent model of AD is not a complete replication of the diseaseinduced pathology. Similarly, the present and other studies havefailed to show cholinergic neuronal loss in the nucleus basalis (Jaffaret al., 2001; Hernandez et al., 2001; Boncristiano et al., 2002) or inthe medial septum/vertical and horizontal limb of the band of Brocain animal models of amyloidosis (Boncristiano et al., 2002; Germanet al., 2003) as compared to the dramatic loss of these neurons inadvancedAD (Whitehouse et al.,1981). Interestingly, we reported anincrease in p75NTR/cholinergic neuron number in the medial septumin APPswemice (Jaffar et al., 2001). In the present study, we found asignificant increase in nucleus basalis cholinergic neuron cell size inaged APPswe/PS1ΔE9 tg mice unlike that seen in APP23 mice(Boncristiano et al., 2002). The increase in nucleus basalis cell sizemay be indicative of cytoskeletal changes and/or axonal transportdefects, which have been implicated in cellular neurodegeneration inAD and in transgenic animal models of cholinergic neuronaldysfunction (Gajdusek, 1985; Morfini et al., 2002; Pigino et al.,2003; Stokin et al., 2005). The differential effect of amyloid upondifferent cholinergic neurons may also be related to a particularcholinergic phenotype and hodology, which confers resistance to Aβtoxicity or to trans-gene expression.
In summary, the present findings indicate that Aβ plaques do notdirectly impact the survival of cholinergic neurons in APPswe/PS1ΔE9 tg mice. It is possible that if our APP over-expressing micehad lived longer or expressed tangle bearing material (Oddo et al.,2003), a necessary precondition for neuronal death in AD, theywould have displayed frank neuronal loss. However, the increase ofcholinergic dystrophic neuritic and Aβ pathology with age in thecortex, striatum and hippocampus, together with the reduction ofcortical ChAT enzyme activity in the absence of cholinergicneuronal death in the APPswe/PS1ΔE9 tg mouse suggest that Aβmay affect cholinergic neuronal transport processes withoutimpacting upon cortical or subcortical cholinergic neuronal viability.
Acknowledgments
The authors thank M. Nadeem and W.R. Paljug for technicalassistance and K. Schafernak for editing the manuscript. We alsothank the Shapiro Foundation and a UCR grant from RushUniversity Medical Center.
References
Araujo, D.M., Lapchak, P.A., Robitaille, Y., Gauthier, S., Quirion, R., 1988.Differential alteration of various cholinergic markers in corticaland subcortical regions of human brain in Alzheimer’s disease.J. Neurochem. 50, 1914–1923.
Arnold, S.E., Hyman, B.T., Flory, J., Damasio, A.R., Van Hoesen, G.W.,1991. The topographical and neuroanatomical distribution ofneurofibrillary tangles and neuritic plaques in the cerebral cortex ofpatients with Alzheimer’s disease. Cereb. Cortex 1, 103–116.
Aucoin, J.S., Jiang, P., Aznavour, N., Tong, X.K., Buttini, M., Descarries,L., Hamel, E., 2005. Selective cholinergic denervation, independentfrom oxidative stress, in a mouse model of Alzheimer’s disease.Neuroscience 132, 73–86.
Bales, K.R., Tzavara, E.T., Wu, S., Wade, M.R., Bymaster, F.P., Paul, S.M.,Nomikos, G.G., 2006. Cholinergic dysfunction in a mouse model ofAlzheimer disease is reversed by an anti-A beta antibody. J. Clin. Invest.116, 825–832.
Bayraktar, T., Staiger, J.F., Acsady, L., Cozzari, C., Freund, T.F., Zilles, K.,1997. Co-localization of vasoactive intestinal polypeptide, gamma-aminobutyric acid and choline acetyltransferase in neocorticalinterneurons of the adult rat. Brain Res. 757, 209–217.
Bell, K.F., de Kort, G.J., Steggerda, S., Shigemoto, R., Ribeiro-da-Silva, A.,Cuello, A.C., 2003. Structural involvement of the glutamatergicpresynaptic boutons in a transgenic mouse model expressing earlyonset amyloid pathology. Neurosci. Lett. 353, 143–147.
Bell, K.F., Ducatenzeiler, A., Ribeiro-da-Silva, A., Duff, K., Bennett, D.A.,Cuello, A.C., 2005. The amyloid pathology progresses in a neuro-transmitter-specific manner. Neurobiol. Aging 27, 1644–1657.
Boissiere, F., Faucheux, B., Agid, Y., Hirsch, E.C., 1997. Cholineacetyltransferase mRNA expression in the striatal neurons of patientswith Alzheimer’s disease. Neurosci. Lett. 225, 169–172.
Boncristiano, S., Calhoun, M.E., Kelly, P.H., Pfeifer, M., Bondolfi, L.,Stalder, M., Phinney, A.L., Abramowski, D., Sturchler-Pierrat, C., Enz,A., Sommer, B., Staufenbiel, M., Jucker, M., 2002. Cholinergic changesin the APP23 transgenic mouse model of cerebral amyloidosis.J. Neurosci. 22, 3234–3243.
Borchelt, D.R., Davis, J., Fischer, M., Lee, M.K., Slunt, H.H., Ratovitsky,T., Regard, J., Copeland, N.G., Jenkins, N.A., Sisodia, S.S., Price, D.L.,1996a. A vector for expressing foreign genes in the brains and hearts oftransgenic mice. Genet. Anal. 13, 159–163.
Borchelt, D.R., Thinakaran, G., Eckman, C.B., Lee, M.K., Davenport, F.,Ratovitsky, T., Prada, C.M., Kim, G., Seekins, S., Yager, D., Slunt, H.H.,Wang, R., Seeger, M., Levey, A.I., Gandy, S.E., Copeland, N.G.,Jenkins, N.A., Price, D.L., Younkin, S.G., Sisodia, S.S., 1996b. FamilialAlzheimer’s disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013.
Borchelt, D.R., Ratovitski, T., van Lare, J., Lee, M.K., Gonzales, V.,Jenkins, N.A., Copeland, N.G., Price, D.L., Sisodia, S.S., 1997.Accelerated amyloid deposition in the brains of transgenic mice co-expressing mutant presenilin 1 and amyloid precursor proteins. Neuron19, 939–945.
Braak, H., Braak, E., 1990. Alzheimer’s disease: striatal amyloid depositsand neurofibrillary changes. J. Neuropathol. Exp. Neurol. 49,215–224.
Braak, H., Braak, E., 1991. Neuropathological stageing of Alzheimer-relatedchanges. Acta Neuropathol. 82, 239–259.
Brendza, R.P., O’Brien, C., Simmons, K., McKeel, D.W., Bales, K.R., Paul,S.M., Olney, J.W., Sanes, J.R., Holtzman, D.M., 2003. PDAPP; YFPdouble transgenic mice: a tool to study amyloid-beta associated changesin axonal, dendritic, and synaptic structures. J. Comp. Neurol. 456,375–383.
Brendza, R.P., Bacskai, B.J., Cirrito, J.R., Simmons, K.A., Skoch, J.M.,Klunk, W.E., Mathis, C.A., Bales, K.R., Paul, S.M., Hyman, B.T.,Holtzman, D.M., 2005. Anti-Abeta antibody treatment promotes therapid recovery of amyloid-associated neuritic dystrophy in PDAPPtransgenic mice. J. Clin. Invest. 115, 428–433.

13S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
Brilliant, M.J., Elble, R.J., Ghobrial, M., Struble, R.G., 1997. Thedistribution of amyloid beta protein deposition in the corpus striatumof patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 23,322–325.
Buttini, M., Yu, G.Q., Shockley, K., Huang, Y., Jones, B., Masliah, E.,Mallory, M., Yeo, T., Longo, F.M., Mucke, L., 2002. Modulation ofAlzheimer-like synaptic and cholinergic deficits in transgenic mice byhuman apolipoprotein E depends on isoform, aging, and overexpressionof amyloid beta peptides but not on plaque formation. J. Neurosci. 22,10539–10548.
Cataldo, A.M., Peterhoff, C.M., Troncoso, J.C., Gomez-Isla, T., Hyman,B.T., Nixon, R.A., 2000. Endocytic pathway abnormalities precedeamyloid beta deposition in sporadic Alzheimer’s disease and Downsyndrome: differential effects of APOE genotype and presenilinmutations. Am. J. Pathol. 157, 277–286.
Cataldo, A.M., Peterhoff, C.M., Schmidt, S.D., Terio, N.B., Duff, K., Beard,M., Mathews, P.M., Nixon, R.A., 2004. Presenilin mutations in familialAlzheimer disease and transgenic mouse models accelerate neuronallysosomal pathology. J. Neuropathol. Exp. Neurol. 63, 821–830.
Cauli, B., Tong, X.K., Rancillac, A., Serluca, N., Lambolez, B., Rossier, J.,Hamel, E., 2004. Cortical GABA interneurons in neurovascularcoupling: relays for subcortical vasoactive pathways. J. Neurosci. 24,8940–8949.
Chen, L., Yamada, K., Nabeshima, T., Sokabe, M., 2006. Alpha7 Nicotinicacetylcholine receptor as a target to rescue deficit in hippocampal LTPinduction in beta-amyloid infused rats. Neuropharmacology 50,254–268.
D’Amore, J.D., Kajdasz, S.T., McLellan, M.E., Bacskai, B.J., Stern, E.A.,Hyman, B.T., 2003. In vivo multiphoton imaging of a transgenic mousemodel of Alzheimer disease reveals marked thioflavine-S-associatedalterations in neurite trajectories. J. Neuropathol. Exp. Neurol. 62,137–145.
Davies, P., 1979. Neurotransmitter-related enzymes in senile dementia of theAlzheimer’s type. Brain Res. 171, 319–327.
Davies, P., Maloney, A.J., 1976. Selective loss of central cholinergic neuronsin Alzheimer’s disease. Lancet 2–1403.
DeKosky, S.T., Scheff, S.W., Markesbery, W.R., 1985. Laminar organizationof cholinergic circuits in human frontal cortex in Alzheimer’s diseaseand aging. Neurology 35, 1425–1431.
DeKosky, S.T., Harbaugh, R.E., Schmitt, F.A., Bakay, R.A., Chui, H.C.,Knopman, D.S., Reeder, T.M., Shetter, A.G., Senter, H.J., Markesbery,W.R., 1992. Cortical biopsy in Alzheimer’s disease: diagnosticaccuracy and neurochemical, neuropathological, and cognitive cor-relations. Intraventricular Bethanecol Study Group. Ann. Neurol. 32,625–632.
DeKosky, S.T., Ikonomovic, M.D., Styren, S.D., Beckett, L., Wisniewski,S., Bennett, D.A., Cochran, E.J., Kordower, J.H., Mufson, E.J., 2002.Upregulation of choline acetyltransferase activity in hippocampus andfrontal cortex of elderly subjects with mild cognitive impairment. Ann.Neurol. 51, 145–155.
Emre, M., Heckers, S., Mash, D.C., Geula, C., Mesulam, M.M., 1993.Cholinergic innervation of the amygdaloid complex in the human brainand its alterations in old age and Alzheimer’s disease. J. Comp. Neurol.336, 117–134.
Fagan, A.M., Garber, M., Barbacid, M., Silos-Santiago, I., Holtzman, D.M.,1997. A role for TrkA during maturation of striatal and basal forebraincholinergic neurons in vivo. J. Neurosci. 17, 7644–7654.
Fonnum, F., 1975. A rapid radiochemical method for the determination ofcholine acetyltransferase. J. Neurochem. 24, 407–409.
Gajdusek, D.C., 1985. Hypothesis: interference with axonal transport ofneurofilament as a common pathogenetic mechanism in certaindiseases of the central nervous system. N. Engl. J. Med. 312,714–719.
German, D.C., Yazdani, U., Speciale, S.G., Pasbakhsh, P., Games, D., Liang,C.L., 2003. Cholinergic neuropathology in a mouse model ofAlzheimer’s disease. J. Comp. Neurol. 462, 371–381.
Geula, C., Mesulam, M.M., 1996. Systematic regional variations in the loss
of cortical cholinergic fibers in Alzheimer’s disease. Cereb. Cortex 6,165–177.
Götz, J., Schild, A., Hoerndli, F., Pennanen, L., 2004. Amyloid-inducedneurofibrillary tangle formation in Alzheimer’s disease: insight fromtransgenic mouse and tissue-culture models. Int. J. Dev. Neurosci. 22,453–465.
Gundersen, H.J., Bagger, P., Bendtsen, T.F., Evans, S.M., Korbo, L.,Marcussen, N., Moller, A., Nielsen, K., Nyengaard, J.R., Pakkenberg,B., et al., 1988. The new stereological tools: dissector, fractionator,nucleator and point sampled intercepts and their use in pathologicalresearch and diagnosis. APMIS 96, 857–881.
Hartley, D.M., Walsh, D.M., Ye, C.P., Diehl, T., Vasquez, S., Vassilev, P.M.,Teplow, D.B., Selkoe, D.J., 1999. Protofibrillar intermediates of amyloidbeta-protein induce acute electrophysiological changes and progressiveneurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884.
Hernandez, D., Sugaya, K., Qu, T., McGowan, E., Duff, K., McKinney, M.,2001. Survival and plasticity of basal forebrain cholinergic systems inmice transgenic for presenilin-1 and amyloid precursor protein mutantgenes. NeuroReport 12, 1377–1384.
Hu, L., Wong, T.P., Cote, S.L., Bell, K.F., Cuello, A.C., 2003. The impact ofAbeta-plaques on cortical cholinergic and non-cholinergic presynapticboutons in Alzheimer’s disease-like transgenic mice. Neuroscience 121,421–432.
Ikonomovic, M.D., Abrahamson, E.E., Isanski, B.A., Wuu, J., Mufson, E.J.,DeKosky, S.T., 2007. Superior frontal cortex cholinergic axon density inmild cognitive Impairment and early Alzheimer’s Disease. Arch. Neurol.64, 1–6.
Jaffar, S., Counts, S.E., Ma, S.Y., Dadko, E., Gordon, M.N., Morgan, D.,Mufson, E.J., 2001. Neuropathology of mice carrying mutant APP(swe) and/or PS1 (M146L) transgenes: alterations in the p75(NTR)cholinergic basal forebrain septohippocampal pathway. Exp. Neurol.170, 227–243.
Karnovsky, M.J., Roots, L., 1964. “A direct-coloring” thiocholine methodfor cholinesterases. J. Histochem. Cytochem. 12, 219–232.
Klunk, W.E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D.P.,Bergstrom, M., Savitcheva, I., Huang, G.F., Estrada, S., Ausen, B.,Debnath, M.L., Barletta, J., Price, J.C., Sandell, J., Lopresti, B.J., Wall,A., Koivisto, P., Antoni, G., Mathis, C.A., Langstrom, B., 2004. Imagingbrain amyloid in Alzheimer’s disease with the Pittsburgh compound-B.Ann. Neurol. 55, 306–319.
Knowles, R.B., Gomez-Isla, T., Hyman, B.T., 1998. Abeta associatedneuropil changes: correlation with neuronal loss and dementia.J. Neuropathol. Exp. Neurol. 57, 1122–1130.
Knowles, R.B., Wyart, C., Buldyrev, S.V., Cruz, L., Urbanc, B., Hasselmo,M.E., Stanley, H.E., Hyman, B.T., 1999. Plaque-induced neuriteabnormalities: implications for disruption of neural networks inAlzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 96, 5274–5279.
Koo, E.H., Park, L., Selkoe, D.J., 1993. Amyloid beta-protein as a substrateinteracts with extracellular matrix to promote neurite outgrowth. Proc.Natl. Acad. Sci. U.S.A. 90, 4748–4752.
Lazarov, O., Lee, M., Peterson, D.A., Sisodia, S.S., 2002. Evidence thatsynaptically released beta-amyloid accumulates as extracellulardeposits in the hippocampus of transgenic mice. J. Neurosci. 22,9785–9793.
Lehericy, S., Hirsch, E.C., Cervera, P., Hersh, L.B., Hauw, J.J., Ruberg, M.,Agid, Y., 1989. Selective loss of cholinergic neurons in the ventralstriatum of patients with Alzheimer disease. Proc. Natl. Acad. Sci.U. S. A. 86, 8580–8584.
Lesuisse, C., Xu, G., Anderson, J., Wong, M., Jankowsky, J., Holtz, G.,Gonzalez, V., Wong, P.C., Price, D.L., Tang, F., Wagner, S., Borchelt,D.R., 2001. Hyper-expression of human apolipoprotein E4 in astrogliaand neurons does not enhance amyloid deposition in transgenic mice.Hum. Mol. Genet. 10, 2525–2537.
Levey, A.I., Wainer, B.H., Rye, D.B., Mufson, E.J., Mesulam, M.M., 1984.Choline acetyltransferase-immunoreactive neurons intrinsic to rodentcortex and distinction from acetylcholinesterase-positive neurons.Neuroscience 13, 341–353.

14 S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
Lombardo, J.A., Stern, E.A., McLellan, M.E., Kajdasz, S.T., Hickey, G.A.,Bacskai, B.J., Hyman, B.T., 2003. Amyloid-beta antibody treatmentleads to rapid normalization of plaque-induced neuritic alterations.J. Neurosci. 23, 10879–10883.
Luth, H.J., Apelt, J., Ihunwo, A.O., Arendt, T., Schliebs, R., 2003.Degeneration of beta-amyloid-associated cholinergic structures intransgenic APP SW mice. Brain Res. 977, 16–22.
Ma, S.Y., Ciliax, B.J., Stebbins, G., Jaffar, S., Joyce, J.N., Cochran, E.J.,Kordower, J.H., Mash, D.C., Levey, A.I., Mufson, E.J., 1999. Dopaminetransporter-immunoreactive neurons decrease with age in the humansubstantia nigra. J. Comp. Neurol. 409, 25–37.
Machová, E., Jakubik, J., Michal, P., Oksman, M., Iivonen, H., Tanila, H.,Dolezal, V., in press. Impairment of muscarinic transmission intransgenic APPswe/PS1dE9 mice Neurobiol. Aging.
Masliah, E., Terry, R.D., Mallory, M., Alford, M., Hansen, L.A., 1990.Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease.Am. J. Pathol. 137, 1293–1297.
Masliah, E., Mallory, M., Hansen, L., Alford, M., Albright, T., DeTeresa, R.,Terry, R., Baudier, J., Saitoh, T., 1991. Patterns of aberrant sprouting inAlzheimer’s disease. Neuron 6, 729–739.
Masliah, E., Sisk, A., Mallory, M., Mucke, L., Schenk, D., Games, D., 1996.Comparison of neurodegenerative pathology in transgenic mice over-expressing V717F beta-amyloid precursor protein and Alzheimer’sdisease. J. Neurosci. 16, 5795–5811.
Masliah, E., Alford, M., Adame, A., Rockenstein, E., Galasko, D., Salmon,D., Hansen, L.A., Thal, L.J., 2003. Abeta1–42 promotes cholinergicsprouting in patients with AD and Lewy body variant of AD. Neurology61, 206–211.
Mathews, P.M., Guerra, C.B., Jiang, Y., Grbovic, O.M., Kao, B.H., Schmidt,S.D., Dinakar, R., Mercken, M., Hille-Rehfeld, A., Rohrer, J., Mehta, P.,Cataldo, A.M., Nixon, RA., 2002. Alzheimer’s disease-relatedoverexpression of the cation-dependent mannose 6-phosphate receptorincreases Abeta secretion: role for altered lysosomal hydrolasedistribution in beta-amyloidogenesis. J. Biol. Chem. 277, 5299–5307.
Mesulam, M.M., Mufson, E.J., Wainer, B.H., Levey, A.I., 1983. Centralcholinergic pathways in the rat: an overview based on an alternativenomenclature (Ch1–Ch6). Neuroscience 10, 1185–1201.
Morfini, G., Pigino, G., Beffert, U., Busciglio, J., Brady, S.T., 2002. Fastaxonal transport misregulation and Alzheimer’s disease. Neuromol.Med. 2, 89–99.
Mufson, E.J., Cunningham, M.G., 1988. Observations on cholineacetyltransferase containing structures in the CD-1 mouse brain.Neurosci. Lett. 84, 7–12.
Mufson, E.J., Bothwell, M., Hersh, L.B., Kordower, J.H., 1989a. Nervegrowth factor receptor immunoreactive profiles in the normal, agedhuman basal forebrain: colocalization with cholinergic neurons. J. Comp.Neurol. 285, 196–217.
Mufson, E.J., Bothwell, M., Kordower, J.H., 1989b. Loss of nerve growthfactor receptor-containing neurons in Alzheimer’s disease: a quantitativeanalysis across subregions of the basal forebrain. Exp. Neurol. 105,221–232.
Mufson, E.J., Conner, J.M., Kordower, J.H., 1995. Nerve growth factor inAlzheimer’s disease: defective retrograde transport to nucleus basalis.NeuroReport 9, 1063–1066.
Mufson, E.J., Lavine, N., Jaffar, S., Kordower, J.H., Quirion, R., Saragovi,H.U., 1997. Reduction in p140-TrkA receptor protein within the nucleusbasalis and cortex in Alzheimer’s disease. Exp. Neurol. 146, 91–103.
Nagele, R.G., D’Andrea, M.R., Anderson, W.J., Wang, H.Y., 2002.Intracellular accumulation of beta-amyloid (1–42) in neurons is facili-tated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’sdisease. Neuroscience 110, 199–211.
Oddo, S., Caccamo, A., Shepherd, J.D., Murphy, M.P., Golde, T.E., Kayed,R., Metherate, R., Mattson, M.P., Akbari, Y., LaFerla, F.M., 2003.Triple-transgenic model of Alzheimer’s disease with plaques andtangles: intracellular Abeta and synaptic dysfunction. Neuron 39,409–421.
Oyanagi, K., Takahashi, H., Wakabayashi, K., Ikuta, F., 1989. Correlative
decrease of large neurons in the neostriatum and basal nucleus ofMeynert in Alzheimer’s disease. Brain Res. 504, 354–357.
Perez, S.E., Lazarov, O., Rodriguez, V., Sisodia, S.S., Mufson, E.J., 2004.Amyloidosis induces nigro-striatal degeneration in APPswe/PS1ΔE9transgenic mice: A model for Alzheimer’s/Parkinson’s Disease. AbstractSFN, 34th Annual Meeting in San Diego, CA.
Perez, S.E., Lazarov, O., Koprich, J.B., Chen, E.Y., Rodriguez-Menendez,V., Lipton, J.W., Sisodia, S.S., Mufson, E.J., 2005. Nigrostriataldysfunction in familial Alzheimer’s disease-linked APPswe/PS1ΔE9transgenic mice. J. Neurosci. 25, 10220–10229.
Perry, E.K., Tomlinson, B.E., Blessed, G., Bergmann, K., Gibson, P.H.,Perry, R.H., 1978. Correlation of cholinergic abnormalities with senileplaques and mental test scores in senile dementia. Br. Med. J. 2,1457–1459.
Phinney, A.L., Deller, T., Stalder, M., Calhoun, M.E., Frotscher, M.,Sommer, B., Staufenbiel, M., Jucker, M., 1999. Cerebral amyloidinduces aberrant axonal sprouting and ectopic terminal formation inamyloid precursor protein transgenic mice. J. Neurosci. 19, 8552–8559.
Pigino, G., Morfini, G., Pelsman, A., Mattson, M.P., Brady, S.T., Busciglio,J., 2003. Alzheimer’s presenilin 1 mutations impair kinesin-basedaxonal transport. J. Neurosci. 23, 4499–4508.
Price, D.L., Sisodia, S.S., 1998. Mutant genes in familial Alzheimer’sdisease and transgenic models. Annu. Rev. Neurosci. 21, 479–505.
Probst, A., Basler, V., Bron, B., Ulrich, J., 1983. Neuritic plaques in seniledementia of Alzheimer type: a Golgi analysis in the hippocampal region.Brain Res. 268, 249–254.
Ramos, B., Baglietto-Vargas, D., del Rio, J.C., Moreno-Gonzalez, I.,Santa-Maria, C., Jimenez, S., Caballero, C., Lopez-Tellez, J.F., Khan,Z.U., Ruano, D., Gutierrez, A., Vitorica, J., 2005. Early neuropathologyof somatostatin/NPY GABAergic cells in the hippocampus of aPS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging27, 1658–1672.
Richter, J.A., Perry, E.K., Tomlinson, B., 1980. Acetylcholine and cholinelevels in post-mortem human brain tissue: preliminary observations inAlzheimer’s disease. Life Sci. 26, 1683–1689.
Rudelli, R.D., Ambler, M.W., Wisniewski, H.M., 1984. Morphology anddistribution of Alzheimer neuritic (senile) and amyloid plaques instriatum and diencephalon. Acta Neuropathol. 64, 273–281.
Saper, C.B., Wainer, B.H., German, D.C., 1987. Axonal and transneuronaltransport in the transmission of neurological disease: potential role insystem degenerations, including Alzheimer’s disease. Neuroscience 23,389–398.
Savonenko, A., Xu, G.M., Melnikova, T., Morton, J.L., Gonzales, V., Wong,M.P., Price, D.L., Tang, F., Markowska, A.L., Borchelt, D.R., 2005.Episodic-like memory deficits in the APPswe/PS1(E9 mouse model ofAlzheimer’s disease: relationships to beta-amyloid deposition andneurotransmitter abnormalities. Neurobiol. Dis. 18, 602–617.
Scott, S.A., Mufson, E.J., Weingartner, J.A., Skau, K.A., Crutcher, K.A.,1995. Nerve growth factor in Alzheimer’s disease: increased levelsthroughout the brain coupled with declines in nucleus basalis.J. Neurosci. 15, 6213–6221.
Selden, N., Mesulam, M.M., Geula, C., 1994a. Human striatum: thedistribution of neurofibrillary tangles in Alzheimer’s disease. Brain Res.648, 327–331.
Selden, N., Geula, C., Hersh, L., Mesulam, M.M., 1994b. Human striatum:chemoarchitecture of the caudate nucleus, putamen and ventral striatumin health and Alzheimer’s disease. Neuroscience 60, 621–636.
Selkoe, D.J., 2001. Alzheimer’s disease: genes, proteins, and therapy.Physiol. Rev. 81, 741–766.
Siegel, S., 1956. Non-Parametric Statistics: for the Behavioral Sciences.McGrawn-Hill, New York, pp. 116–127.
Sobreviela, T., Clary, D.O., Reichardt, L.F., Brandabur, M.M., Kordower,J.H., Mufson, E.J., 1994. TrkA immunoreactive profiles in the centralnervous system: colocalization with neurons containing p75 nervegrowth factor receptor, choline acetyltransferase and serotonin. J. Comp.Neurol. 350, 587–611.
Stokin, G.B., Lillo, C., Falzone, T.L., Brusch, R.G., Rockenstein, E., Mount,

15S.E. Perez et al. / Neurobiology of Disease 28 (2007) 3–15
S.L., Raman, R., Davies, P., Masliah, E., Williams, D.S., Goldstein, L.S.,2005. Axonopathy and transport deficits early in the pathogenesis ofAlzheimer’s disease. Science 307, 1282–1288.
Suenaga, T., Hirano, A., Llena, J.F., Yen, S.H., Dickson, D.W., 1990.Modified Bielchowsky stain and immunohistochemical studies onstriatal plaques in Alzheimer’s disease. Acta Neuropathol. 80, 280–286.
Suh, Y.H., Checler, F., 2002. Amyloid precursor protein, presenilins, andalpha-synuclein: molecular pathogenesis and pharmacological appli-cations in Alzheimer’s disease. Pharmacol. Rev. 54, 469–525.
Wainer, B.H., Levey, A.I., Mufson, E.J., Mesulam, M.M., 1984. Cholinergicsystems in mammalian brain identified with antibodies against cholineacetyltransferase. Neurochem. Int. 6, 163–182.
Wang, H.Y., Lee, D.H., D’Andrea, M.R., Peterson, P.A., Shank, R.P., Reitz,A.B., 2000. Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholinereceptor with high affinity. Implications for Alzheimer’s diseasepathology. J. Biol. Chem. 275, 5626–5632.
Wang, Q.J., Ding, Y., Kohtz, D.S., Mizushima, N., Cristea, I.M., Rout, M.P.,Chait, B.T., Zhong, Y., Heintz, N., Yue, Z., 2006. Induction of autophagyin axonal dystrophy and degeneration. J. Neurosci. 26, 8057–8068.
West, M.J., 1993. New stereological methods for counting neurons.Neurobiol. Aging 14, 275–285.
Whitehouse, P.J., Price, D.L., Clark, A.W., Coyle, J.T., DeLong, M.R., 1981.Alzheimer disease: evidence for selective loss of cholinergic neurons inthe nucleus basalis. Ann. Neurol. 10, 122–126.
Whitehouse, P.J., Struble, R.G., Hedreen, J.C., Clark, A.W., Price, D.L.,1985. Alzheimer’s disease and related dementias: selective involvementof specific neuronal systems. CRC Crit. Rev. Clin. Neurobiol. 1,319–339.
Wilcock, G.K., Esiri, M.M., Bowen, D.M., Smith, C.C., 1982. Alzheimer’sdisease. Correlation of cortical choline acetyltransferase activity with theseverity of dementia and histological abnormalities. J. Neurol. Sci. 57,407–417.
Wong, T.P., Debeir, T., Duff, K., Cuello, A.C., 1999. Reorganization ofcholinergic terminals in the cerebral cortex and hippocampus intransgenic mice carrying mutated presenilin-1 and amyloid precursorprotein transgenes. J. Neurosci. 19, 2706–2716.
Yanker, B.A., 1996. Mechanisms of neuronal degeneration in Alzheimer’sdisease. Neuron 16, 921–932.





























