Embed Size (px)
Citation preview

Rychle progredující glomerulonefritidy
Josef ZadražilIII. Interní klinika FN a LF UP Olomouc

Rychle progredující glomerulonefritidy (RPGN)
Definice
Jde o heterogenní skupinu chorob, které mají
charakteristický histologický obraz a nejsou-li včas
rozpoznány a adekvátně léčeny obvykle rychle
(týdny až měsíce) progredují do terminálního selhání
ledvin
Incidence
2-5% bioptovaných nemocných
Při podezření na RPGN je biopsie ledvin indikována
okamžitě

Klinický obraz RPGN
únava, slabost, subfebrilie, bolesti v bedrech, makroskopická
hematurie
oligoanurie, otoky, kašel, dušnost
postižení horních dýchacích cest: epistaxe, chronická rýma,
recidivující sinusitídy
postižení dolních dýchacích cest: bolesti na hrudi,
hemoptýza, na rtg noduly, infiltráty, motýlovitá zastínění,
granulomy
postižení GIT: průjmy, bolesti břicha, krvácení do GIT
oční postižení: konjunktivitis, episkleritis, neuropatie optiku
srdeční postižení: perikarditida, dilatační kardiomyopatie,
arytmie
postižení periferních nervů: parézy, křeče, bolesti DKK
artralgie, myalgie, purpura

Podezření na RPGN
Progresivní renální selhávání během několika
týdnů až měsíců. Při každém nejasném
zvýšení kreatininu je nutno pomýšlet na
možnost RPGN a pátrat po dalších kritériích
Nefritický močový sediment (aktivní močový
sediment = hematurie, proteinurie, válce)
Nefrotický syndrom je přítomen u 10-30%
pacientů
Arteriální hypertenze není obvyklá a většinou
není závažná

Vaskulitidy – klasifikace
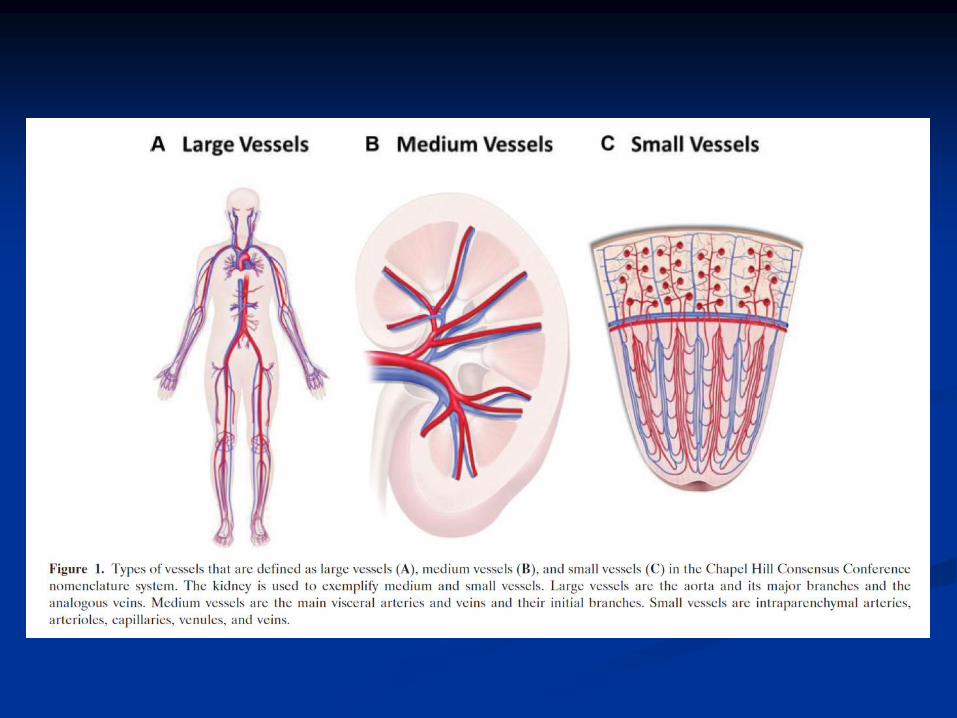

Klasifikace systémových vaskulitid
(Chapel Hill, 1993)
Velké cévyTemporální arteritis
Takayasuova arteritis
Cévy středního kalibruPolyarteritis nodosa
Kawasakiho syndrom
Cévy malého kalibruWegenerova granulomatosa*
Mikroskopická polyangiitis*
Churgův-Straussové syndrom*
Henochova-Schoenleinova purpura
Kryoglobulinemická vaskulitis
Kožní leukocytoklastická angiitis
*Lze prokázat přítomnost ANCA protilátek

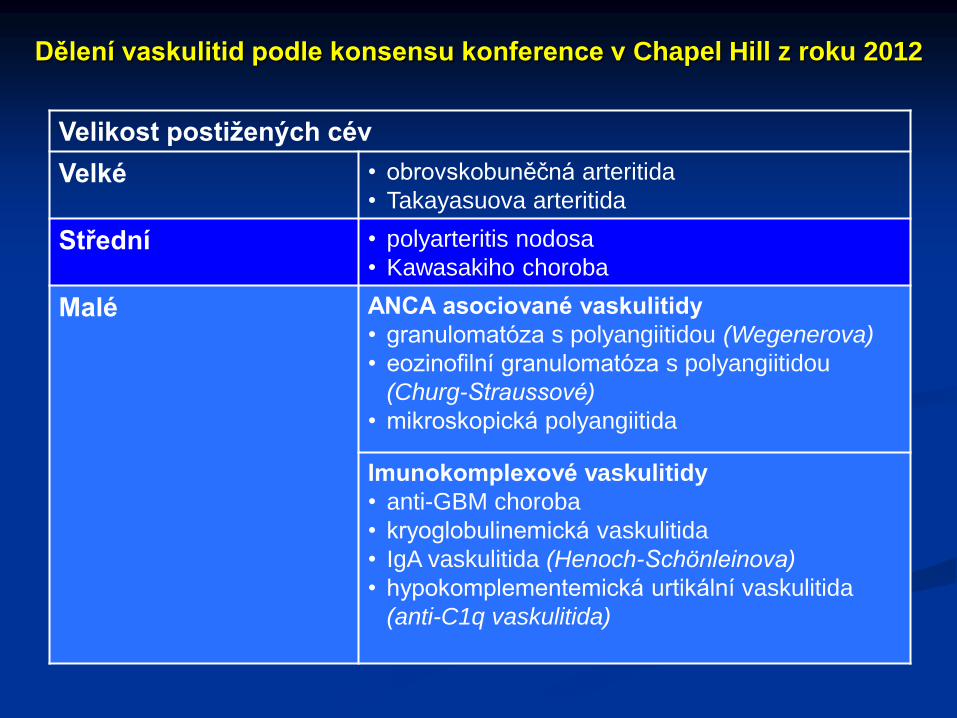
Dělení vaskulitid podle konsensu konference v Chapel Hill z roku 2012
Velikost postižených cév
Velké • obrovskobuněčná arteritida
• Takayasuova arteritida
Střední • polyarteritis nodosa
• Kawasakiho choroba
Malé ANCA asociované vaskulitidy
• granulomatóza s polyangiitidou (Wegenerova)
• eozinofilní granulomatóza s polyangiitidou
(Churg-Straussové)
• mikroskopická polyangiitida
Imunokomplexové vaskulitidy
• anti-GBM choroba
• kryoglobulinemická vaskulitida
• IgA vaskulitida (Henoch-Schönleinova)
• hypokomplementemická urtikální vaskulitida
(anti-C1q vaskulitida)



Laboratorní a pomocná vyšetření
urea, kreatinin, GF, mineralogram, Astrup, ostatní
biochem. vyšetření, KO (anemie, leukocytóza), vyšší FW,
vyšší CRP, hematurie, proteinurie,
hypergamaglobulinémie, protilátky proti BM, ANCA
(c-ANCA – proti proteináze 3, p-ANCA – proti
myeloperoxidáze)
sonografie ledvin, rtg S+P, EKG, echokardiografie, EMG,
gastroskopie, ORL a oční vyšetření

c-ANCA (proti proteináze 3)
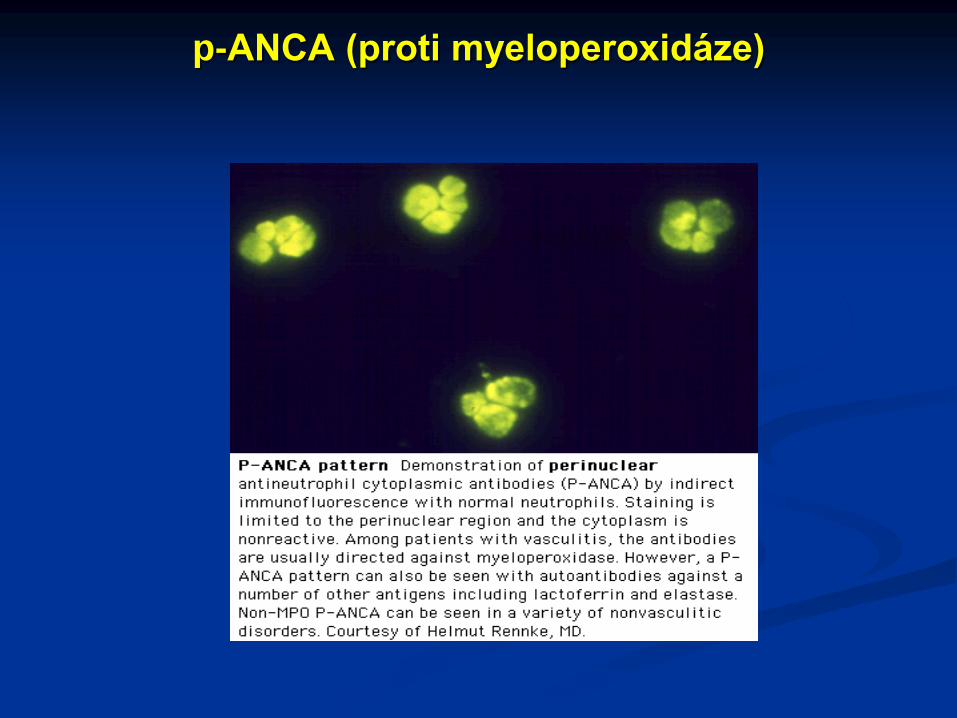
p-ANCA (proti myeloperoxidáze)

Pozitivita ANCA u AAV
Vaskulitida c-ANCA
(%)
p-ANCA
(%)
Negativní
(%)
Wegenerova
granulomatóza
70 25 5
Mikroskopická
polyangiitida
40 50 10
Syndrom Churga
a Straussové
10 60 30
Pauciimunitní
glomerulonefritida
20 70 10

Morfologický obraz RPGN
Ledviny jsou obvykle zvětšené
Více než 70% glomerulů je postiženo extrakapilární proliferací
s tvorbou srpků (zprvu epiteliální, později vazivové). Na jejich tvorbě
se podílí proliferace epiteliálních buněk, monocyty (makrofágy) a
aktivace koagulace s tvorbou fibrinu. Velmi rychle dochází k tvorbě
kolagenu (okluzivní srpky, obsolescence glomerulů)
Proliferace, nekrózy a jizvení glomerulárního trsu (obraz fokálně
segmentální nekrózy)
Intersticiální infiltrát (neutrofily, T lymfocyty, monocyty)
Intersticiální fibróza a atrofie tubulů
Klasifikace
Základní dělení RPGN je založeno na imunofluorescenčním
vyšetření ledviny a na sérologickém vyšetření
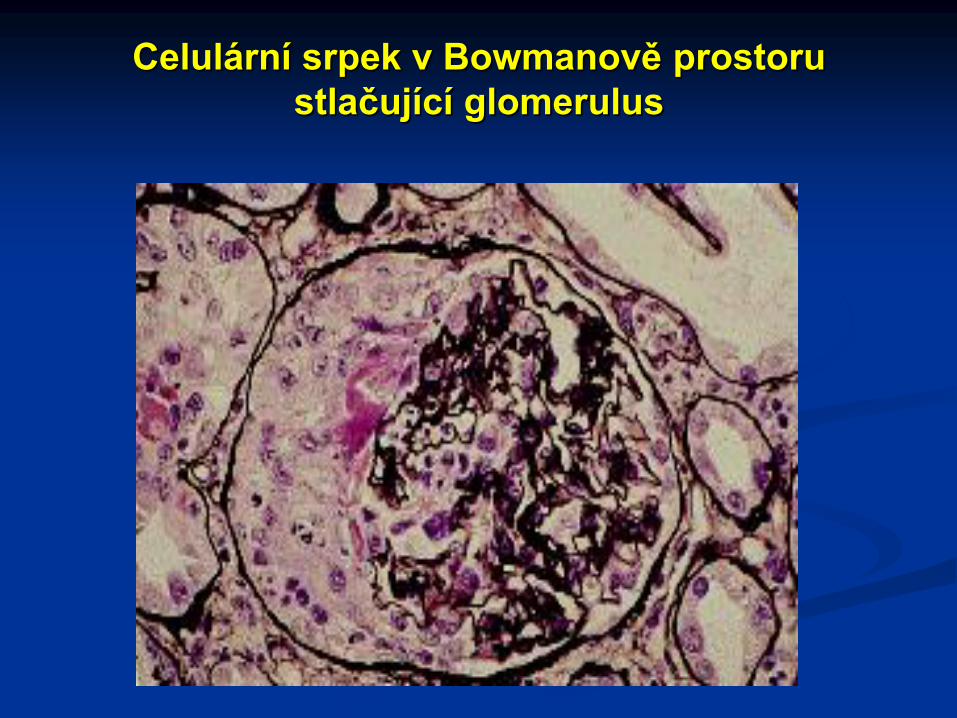
Celulární srpek v Bowmanově prostoru
stlačující glomerulus

Rychle progredující glomerulonefritis
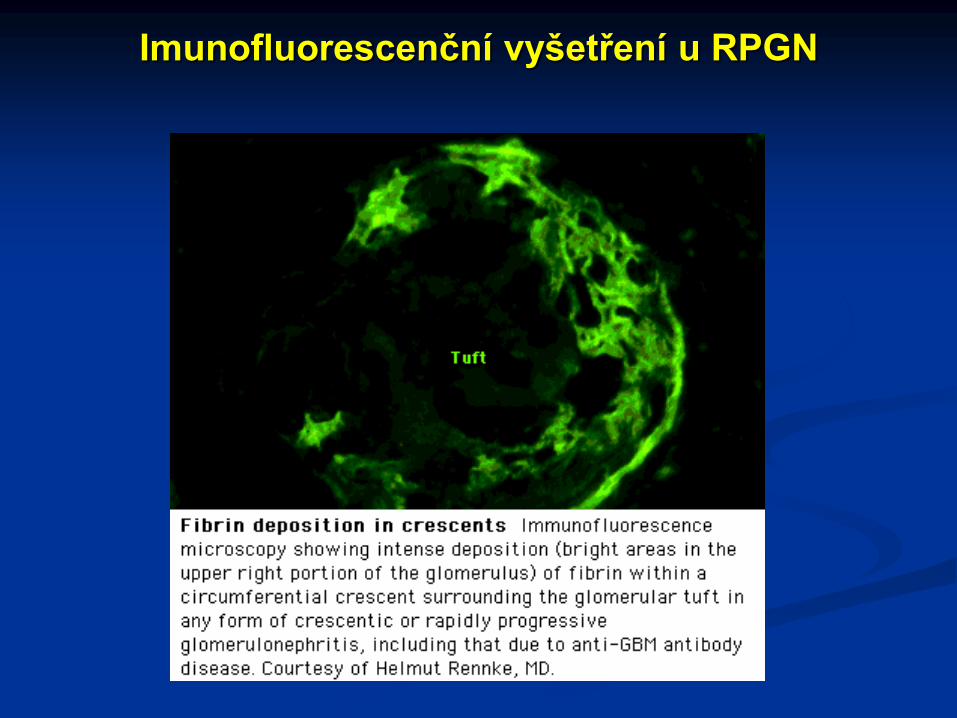
Imunofluorescenční vyšetření u RPGN
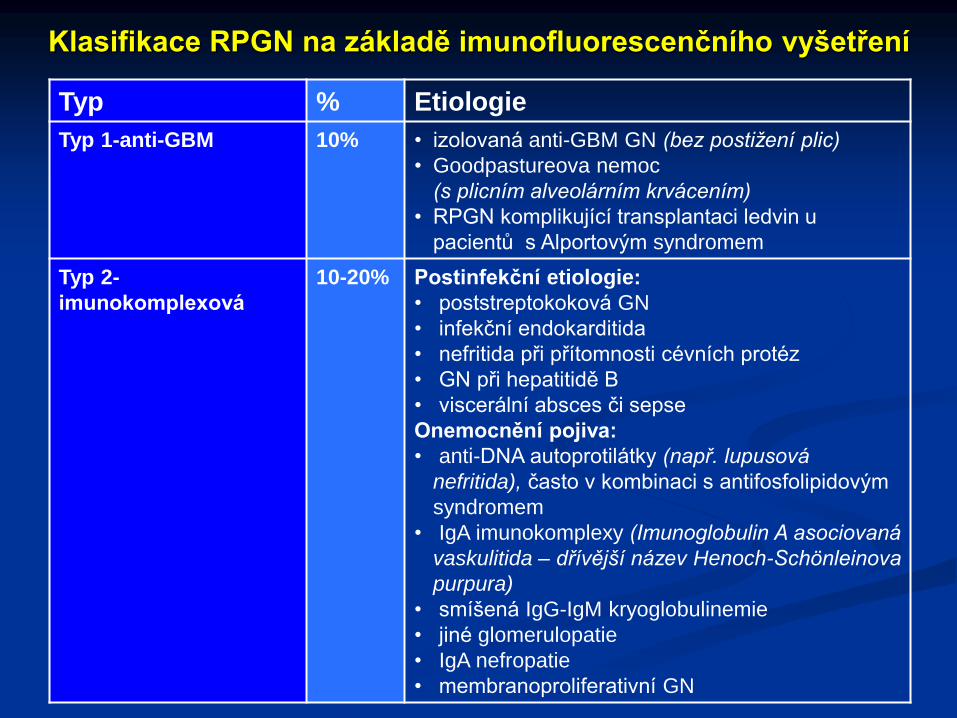
Klasifikace RPGN na základě imunofluorescenčního vyšetření
Typ % Etiologie
Typ 1-anti-GBM 10% • izolovaná anti-GBM GN (bez postižení plic)
• Goodpastureova nemoc
(s plicním alveolárním krvácením)
• RPGN komplikující transplantaci ledvin u
pacientů s Alportovým syndromem
Typ 2-
imunokomplexová
10-20% Postinfekční etiologie:
• poststreptokoková GN
• infekční endokarditida
• nefritida při přítomnosti cévních protéz
• GN při hepatitidě B
• viscerální absces či sepse
Onemocnění pojiva:
• anti-DNA autoprotilátky (např. lupusová
nefritida), často v kombinaci s antifosfolipidovým
syndromem
• IgA imunokomplexy (Imunoglobulin A asociovaná
vaskulitida – dřívější název Henoch-Schönleinova
purpura)
• smíšená IgG-IgM kryoglobulinemie
• jiné glomerulopatie
• IgA nefropatie
• membranoproliferativní GN
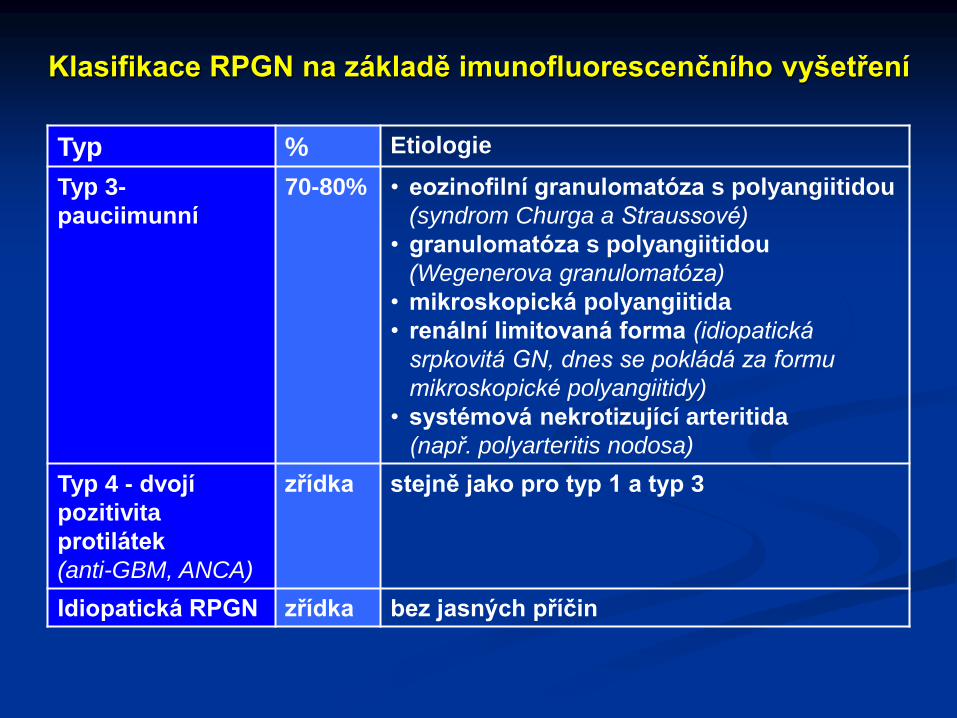
Klasifikace RPGN na základě imunofluorescenčního vyšetření
Typ % Etiologie
Typ 3-
pauciimunní
70-80% • eozinofilní granulomatóza s polyangiitidou
(syndrom Churga a Straussové)
• granulomatóza s polyangiitidou
(Wegenerova granulomatóza)
• mikroskopická polyangiitida
• renální limitovaná forma (idiopatická
srpkovitá GN, dnes se pokládá za formu
mikroskopické polyangiitidy)
• systémová nekrotizující arteritida
(např. polyarteritis nodosa)
Typ 4 - dvojí
pozitivita
protilátek
(anti-GBM, ANCA)
zřídka stejně jako pro typ 1 a typ 3
Idiopatická RPGN zřídka bez jasných příčin

Imunopatologická klasifikace RPGN (1)
1. Antirenální RPGN s protilátkami proti bazální
membráně glomerulů (10% všech RPGN)
- RPGN bez plicní hemoragie
- RPGN s plicní hemoragií (Goodpastureův syndrom)
- RPGN komplikující transplatnaci ledviny u nemocných
s Alportovým syndromem

Imunopatologická klasifikace RPGN (2)
2. Imunokomplexové RPGN s granulárními depozity(10-20% všech RPGN)
- RPGN u preexistující primární glomerulopatie(IgA nefropatie, proliferativní GN, membranózní GN)
- RPGN u systémových chorob (Lupus, esenciální kryoglobulinémie)
- RPGN u infekčních onemocnění (infekční endokarditída, viscerální abscesy)

Imunopatologická klasifikace RPGN (3)
3. RPGN s žádnými nebo minimálními
imunodepozity – pauciimunní
(70-80% všech RPGN)
Spojené s průkazem ANCA
- glomerulonefritis bez systémového poškození
- mikroskopická polyarteritis nodosa (p-ANCA)
- Wegenerova granulomatóza (c-ANCA)
V patogenezi ANCA - asociované vaskulitity byla nově
popsána velmi významná role protilátek proti lysosomálnímu
membránovému proteinu -2 (LAMP-2)
Bez průkazu ANCA

Jednoduchá klinicko-patologická klasifikace ANCA-pozitivní
vaskulitidy (podle Jennetta a Falka)
Vaskulitida bez astmatu a granulomů
Mikroskopická polyangitida
Vaskulitida s granulomy a bez astmatu
Wegenerova granulomatóza
Vaskulitida s eozinofilií, astmatem a granulomy
Syndrom Churga a Straussové

Granulomatóza s polyangiitidou (1)
Systémové onemocnění charakterizované nekrózami, tvorbou granulomů a vaskulitídou horních a dolních dýchacích cest, nekrotizující glomerulonefritidou se srpky a možným postižením řady dalších orgánů.
Postižení horních dýchacích cest nekrotizujícími granulomy se projevuje epistaxí, chronickou rýmou, chronickou sinusitídou až destrukcí nosních chrupavek
Plicní postižení se projevuje kašlem, bolestmi na hrudníku, hemoptýzou. Rentgenologicky lze prokázat různě velké granulomy (až 10 cm), krvácení do plic se projevuje typickým motýlovitým zastřením

Granulomatóza s polyangiitidou (2)
Pacienti mívají často nespecifické příznaky systémového onemocnění, tj. teploty, artralgie, myalgie, únavu atd.
Postižení ledvin je časté a během několika týdnů vede k selhání ledvin vyžadující hemodialyzační léčbu
K měně častým projevům patří oční postižení, srdeční postižení a postižení GIT s krvácením do trávicího ústrojí
Je o systémovou vaskulitidu a postižen může být jakýkoli orgán, kde jsou cévy
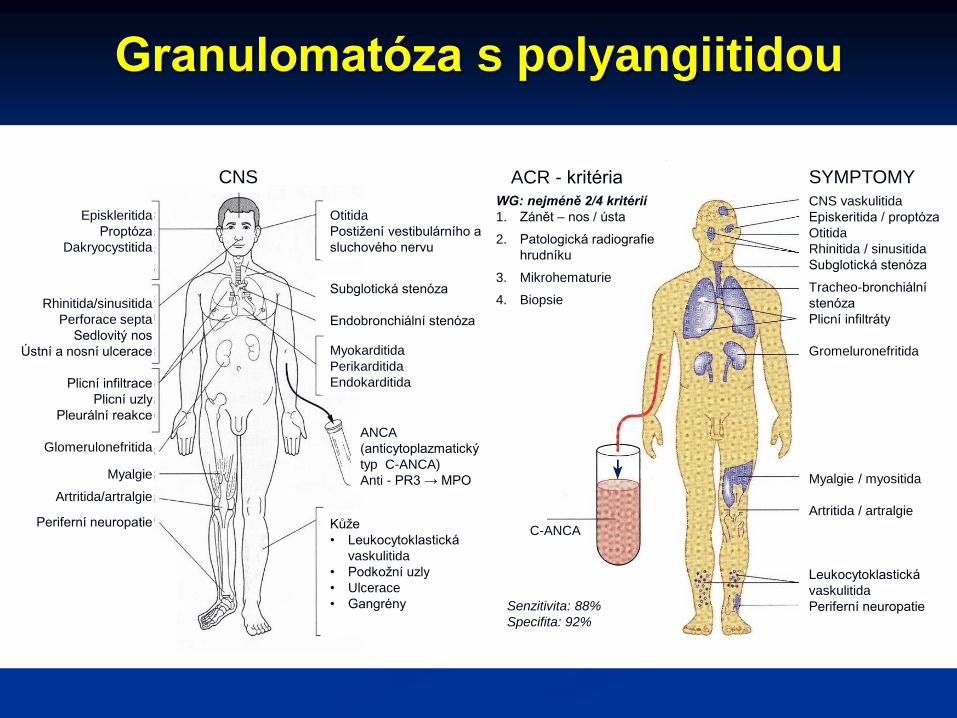
Granulomatóza s polyangiitidou
CNS
Otitida
Postižení vestibulárního a
sluchového nervu
Myokarditida
Perikarditida
Endokarditida
Subglotická stenóza
Endobronchiální stenóza
ANCA
(anticytoplazmatický
typ C-ANCA)
Anti - PR3 → MPO
Kůže
• Leukocytoklastická
vaskulitida
• Podkožní uzly
• Ulcerace
• Gangrény
C-ANCA
Episkleritida
Proptóza
Dakryocystitida
Rhinitida/sinusitida
Perforace septa
Sedlovitý nos
Ústní a nosní ulcerace
Plicní infiltrace
Plicní uzly
Pleurální reakce
Glomerulonefritida
Myalgie
Artritida/artralgie
Periferní neuropatie
CNS vaskulitida
Episkeritida / proptóza
Otitida
Rhinitida / sinusitida
Subglotická stenóza
Tracheo-bronchiální
stenóza
Plicní infiltráty
Gromeluronefritida
Myalgie / myositida
Artritida / artralgie
Leukocytoklastická
vaskulitida
Periferní neuropatie
SYMPTOMYACR - kritériaWG: nejméně 2/4 kritérií
1. Zánět – nos / ústa
2. Patologická radiografie
hrudníku
3. Mikrohematurie
4. Biopsie
Senzitivita: 88%
Specifita: 92%
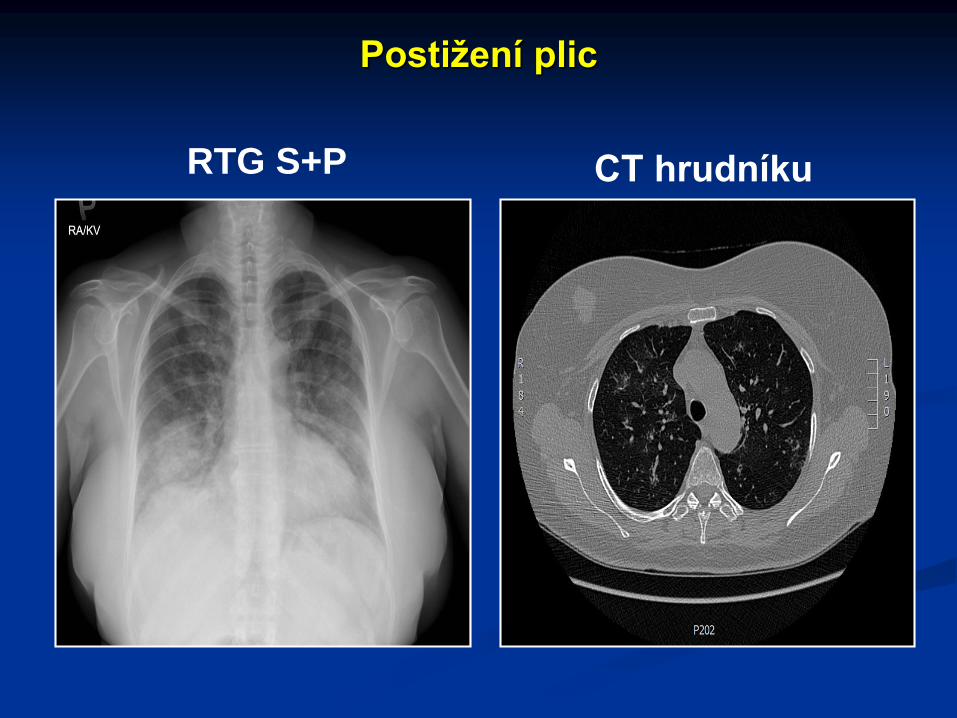
Postižení plic
RTG S+P CT hrudníku
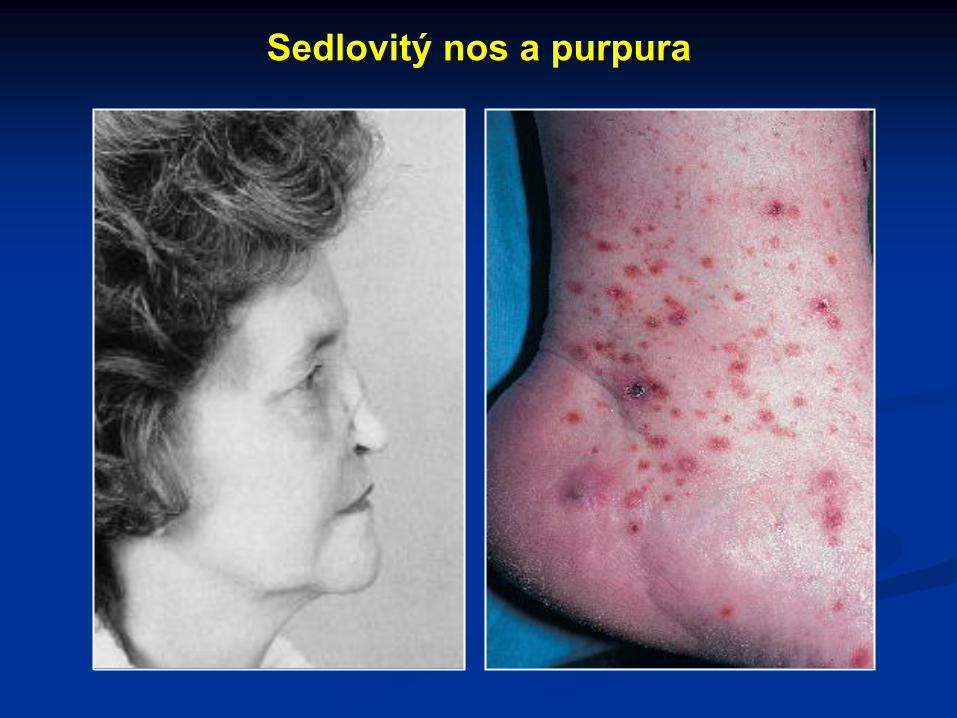
Sedlovitý nos a purpura

Mikroskopická polyangiitida (polyarteridita)
V klinickém obraze dominuje poškození
ledvin, mikroskopická hematurie, proteinurie
a různý stupeň renální insuficience s tendencí
k rychlé progresi do terminálního selhání
Průběh onemocnění je méně akutní než
u Wegenerovy granulomatózy, postižení plic
je vzácné, sklon k relapsům je malý
Z dalších projevů se může vyskytnout
purpura, myalgie, artralgie, periferní
neuropatie nebo krvácení do trávícího ústrojí


Indukční léčba RPGN
Cíl: Zastavit imunopatologický proces vedoucí k tvorbě srpků, potlačit akutní zánět v glomerulech
omezit nebo zabránit jizvení glomerulů
Metylprednisolon 0.25-05 g i.v. 3-5 dnů, dále Prednison
1 mg/kg/den po dobu 6-ti měsíců
Cyklofosfamid 15mg/kg/měsíc 6 x (1,5-2 mg/kg/den p.o.)
Mykofenolát mofetil jako alternativa CFA
Plazmaferézy u pacientů s plicním krvácením a selháním
funkce ledvin (kreatinin >500 µmol/l )
[výměny 3-4 l plazmy denně, případně ob den, až 14dnů
podle klin.vývoje nebo titru protilátek]
Vysoké dávky imunoglobulinů (2g/kg hmotnosti)
Biologická léčba
protilátky proti antigenu CD 20 (rituximab)

Udržovací léčba RPGN
Cíl: Zabránit relapsu onemocnění a stabilizovat GF
Kortikosteroidy se snahou o podání co nejnižších účinných dávek
Azathioprin, alternativně mykofenolát mofetil
Metotrexát
Deoxysperqualin
U vybraných pacientů rituximab
Podpůrná a doplňující medikace (vitamin D, vápník, cotrimoxazol, antimykotika, antivirotika)
Léčba se řídí klinickým stavem, laboratorními nálezy a opakovaným sledováním protilátek GBM, případně titru ANCA (vzestup titru ANCA předchází u většiny nemocných o více než měsíc rozvoj klinických známek relapsu)

Granulomatóza s polyangiitidou
Prognóza
Střední doba přežití u pacientů s neléčeným
onemocněním byla 5 měsíců, 80% pacientů
zemřelo do 1 roku od stanovení diagnózy
Vysoké dávky steroidů prodloužily střední dobu
přežití na 12,5 měsíce
Indukční léčba CFA + steroidy navodí remisi
onemocnění ve více než 90%
U značné části dialyzovaných pacientů
(dle různých studií až u 60%) se podaří obnovit
renální funkce

Antirenální glomerulonefritída
Goodpastureův syndrom – RPGN a krvácení do plic
Goodpastureova nemoc – anti GBM nefritída s plicním krvácením
Pulmorenální syndrom – jakékoli součas. závažné poškození ledvin a plic
Krvácení do plic se objevuje asi u 2/3 nemocných s antirenálníglomerulonefritídou. Hemoptýza může být minimální nebo masivnía není v přímém vztahu k rozsahu plicního krvácení. Je častopředcházena prohlubující se dušností. Fyzikální nález na plicíchmůže být normální nebo minimální. Důležité je opakované rtgvyšetření plic (symetrické postižení středních plicních polí nodulyaž splývajícími rozsáhlými infiltráty). Postižení plic špatně korelujes titrem protilátek proti bazální membráně glomerulů, což jezpůsobeno zejména rozdíly v dostupnosti antigenu pro cirkulujícíautoprotilátky (alveolární endotelie nejsou fenestrovány).

Diferenciální diagnostika pulmo-renálního syndromu
Wegenerova granulomatóza
Mikroskopická polyarteritída
Polyarteritis nodosa
Imunokomplexová RPGN
Goodpastureův syndrom
Schoenleinova-Henochova purpura
Smíšená kryoglobulinémie
Hypersenzitivní angiitis
Poléková reakce (např. Penicilamin)
Bakteriální endokarditída postihující pravé srdce
Systémový lupus erythematodes
Legionářšká nemoc
Kongestivní srdeční selhání s urémií
Trombóza renální žíly s plicní embolizací
Sarkoidóza

Diagnostika antirenální RPGN
Průkaz cirkulujících protilátek proti GBM
Protilátky proti GBM jsou většinou třídy IgG1 nebo IgG4
a reagují s tzv. Goodpastureovým antigenem, který je
lokalizován do C-terminální nekolagenní globulární
domény molekuly α3 řetězce kolagenu IV.
Imunofluorescenční průkaz fixace protilátek ke GBM
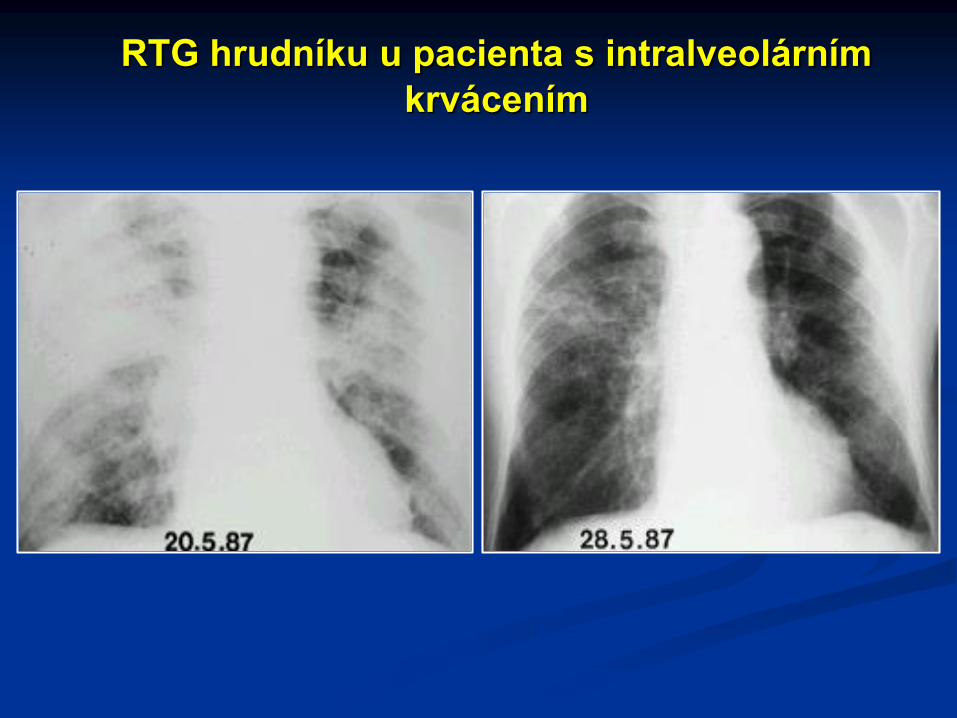
RTG hrudníku u pacienta s intralveolárním
krvácením
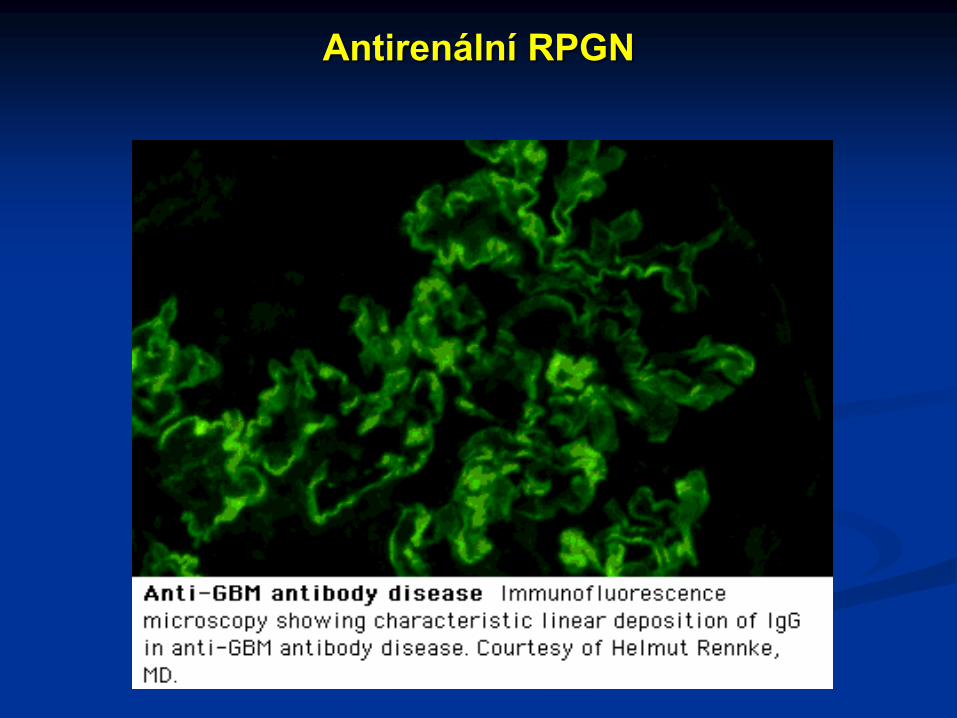
Antirenální RPGN

Goodpasterova nemoc
Goodpastureův antigen je přítomen:
glomerulární bazální membrána
tubulární bazální membrána
bazální membrána Bowmanova pouzdra
alveolární bazální membrána
bazální membrána rohovky, retiny, kochleárního aparátu, chorioidálního plexu a některých endokrinních orgánů
V diagnostice Goodpastureova sy (nemoci) nemůže plicní biopsie nahradit biopsii ledviny
Prognóza neléčených nemocných je velmi špatná (roční úmrtnost 77-96%)
Léčba – kombinace plazmaferéz a podávání kortikosteroidů a cyklofosfamidu
Relapsy Goodpastureovy nemoci nejsou časté
Rekurence Goodpastureovy nemoci po transplantaci je vzácná (nutno však dodržet interval 6 měs. po vymizení protilátek proti GBM)



























