Embed Size (px)
Citation preview

Universidad Nacional La Matanza
Ejercicio Muscular
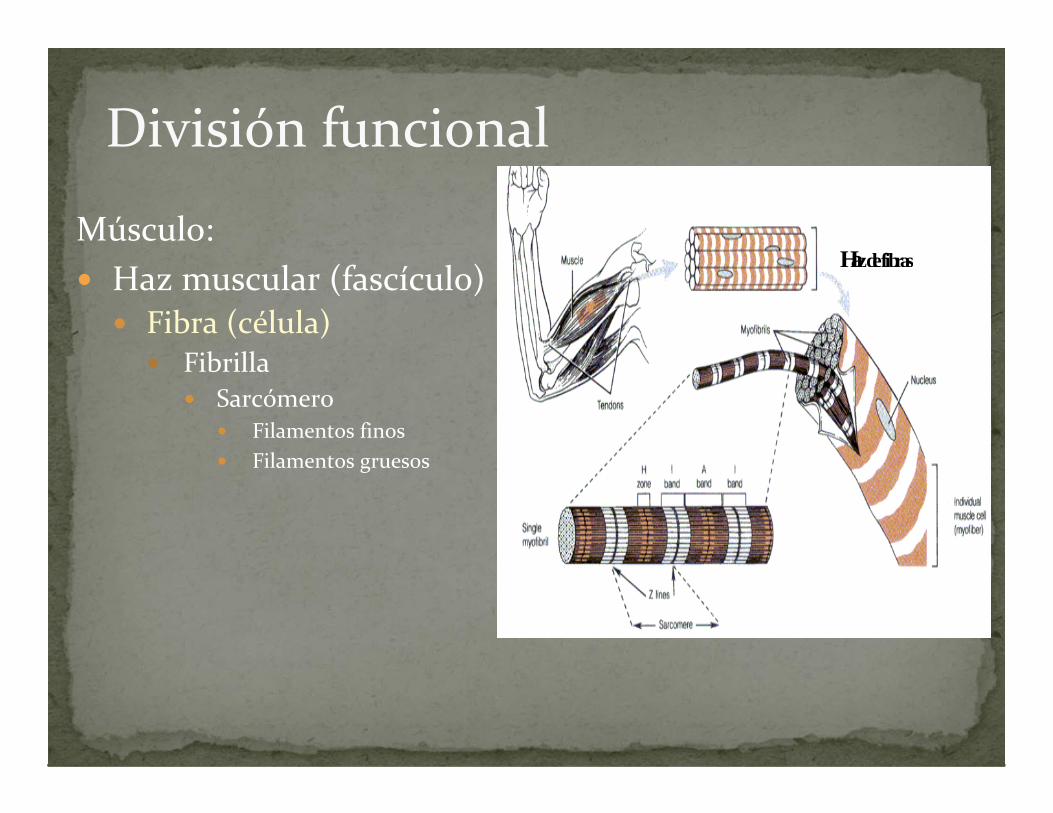
División funcional
Músculo: Haz muscular (fascículo)
Fibra (célula) Fibrilla
Sarcómero Filamentos finos Filamentos gruesos
Hazdefibras

Fibra muscular (Célula muscular)
Diámetro 10-80 m; longitud la del músculo Sarcolema = membrana: se continúa con el tendón Sarcoplasma = citoplasma de la fibra muscular. Contiene
Glucógeno Mioglobina Miofibrillas
Núcleos numerosos, mitocondrias .... Retículo sarcoplásmico (retículo endoplásmico): contiene calcio. La
liberación de este catión dispara la contracción de las fibras. El calcio entra en el retículo contra gradiente por la acción de una bomba de calcio (ATPasa de Ca2+ ) y se une a una proteína, la calsecuestrina.
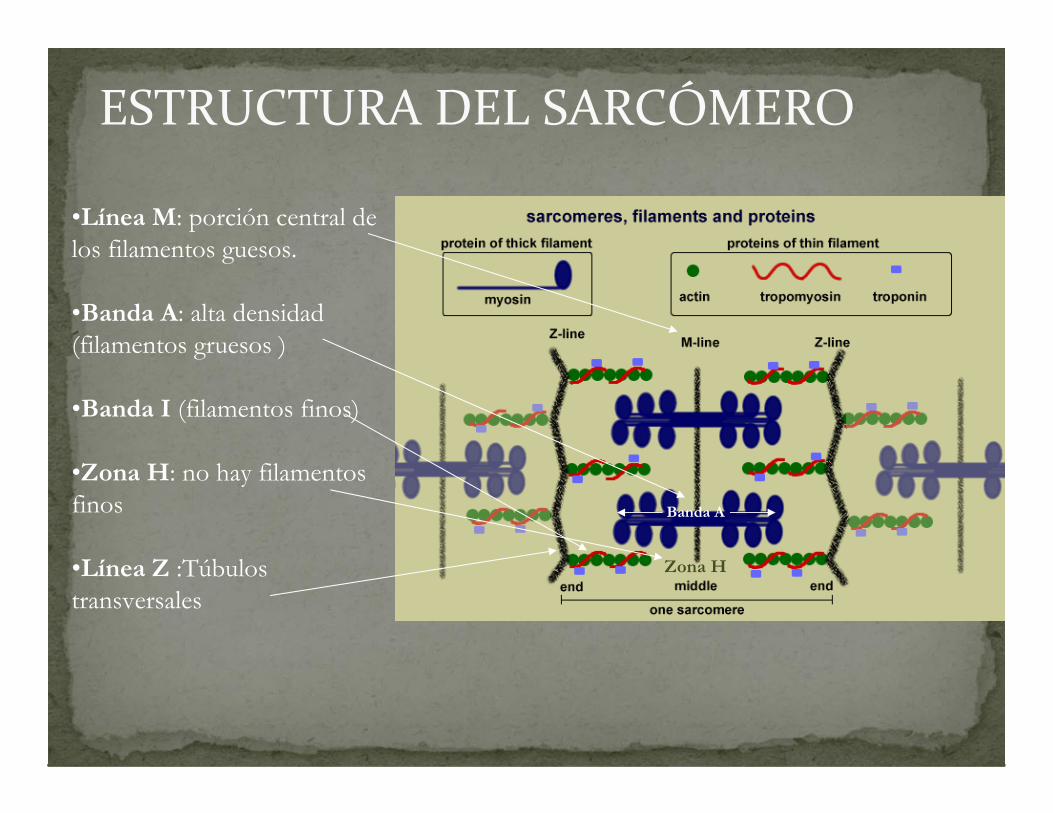
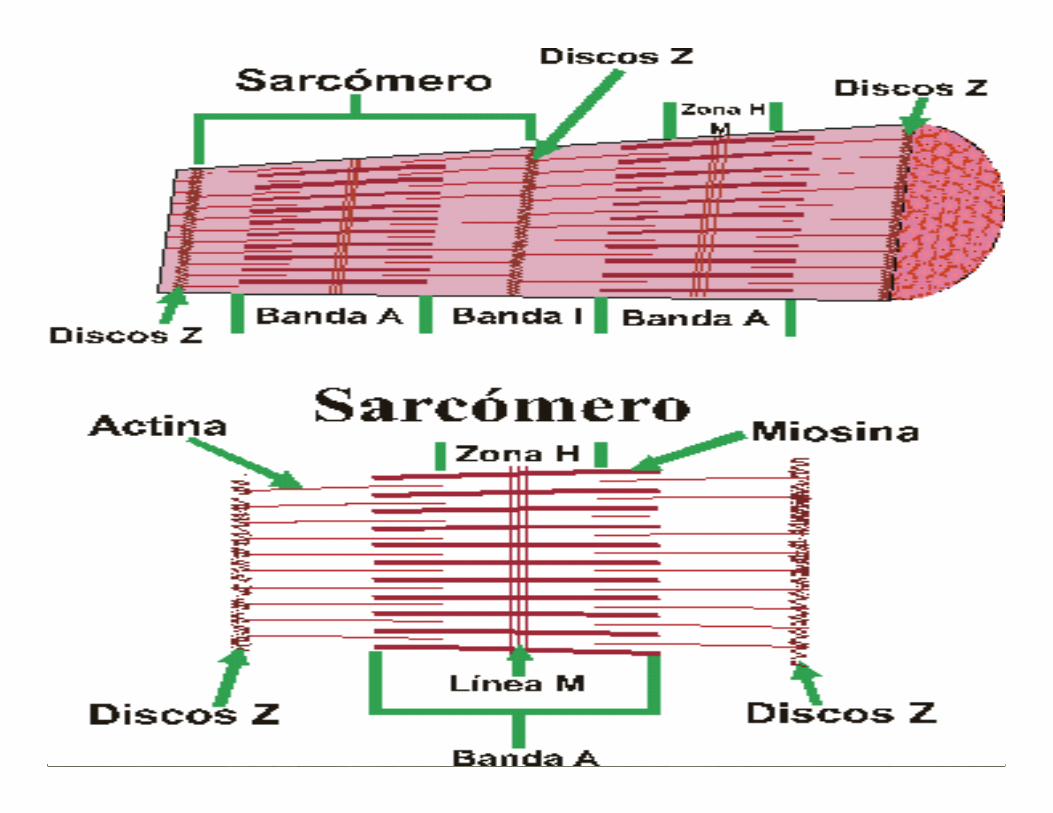
ESTRUCTURA DEL SARCÓMERO
•Línea M: porción central de los filamentos guesos.
•Banda A: alta densidad (filamentos gruesos )
•Banda I (filamentos finos)
•Zona H: no hay filamentos finos
•Línea Z :Túbulos transversales
Banda I
Banda A
Zona H
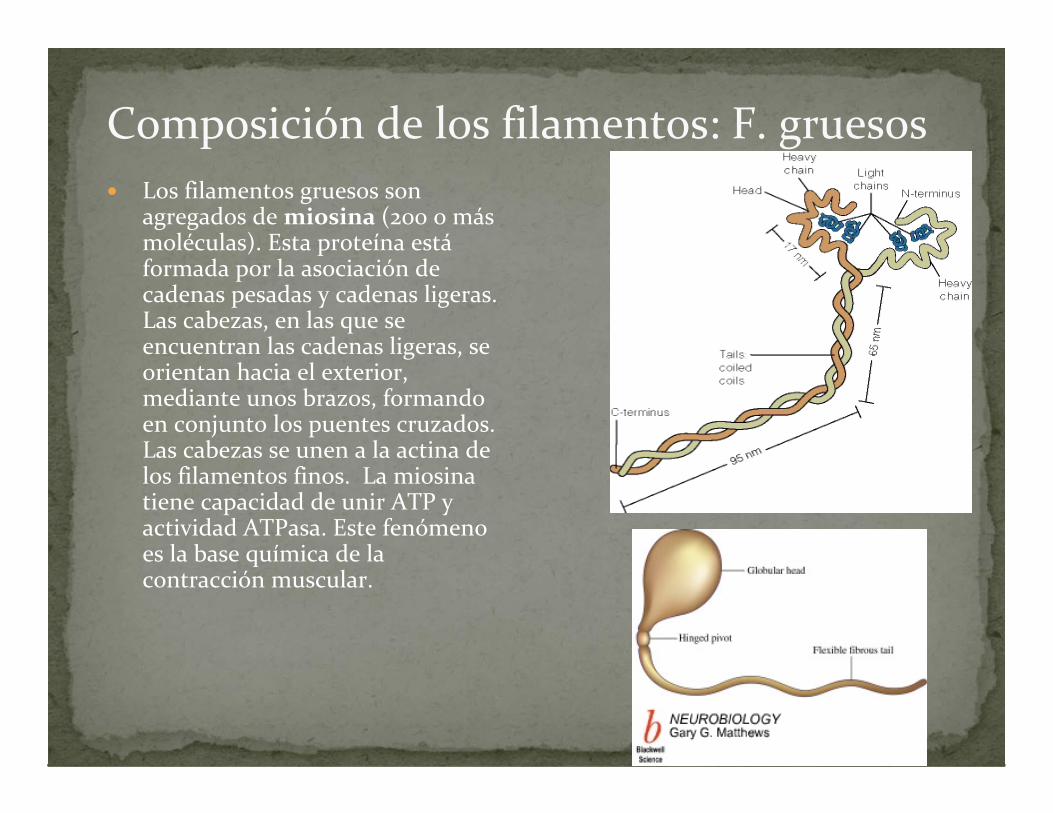
Composición de los filamentos: F. gruesos Los filamentos gruesos son
agregados de miosina (200 o más moléculas). Esta proteína estáformada por la asociación de cadenas pesadas y cadenas ligeras. Las cabezas, en las que se encuentran las cadenas ligeras, se orientan hacia el exterior, mediante unos brazos, formando en conjunto los puentes cruzados. Las cabezas se unen a la actina de los filamentos finos. La miosina tiene capacidad de unir ATP y actividad ATPasa. Este fenómeno es la base química de la contracción muscular.
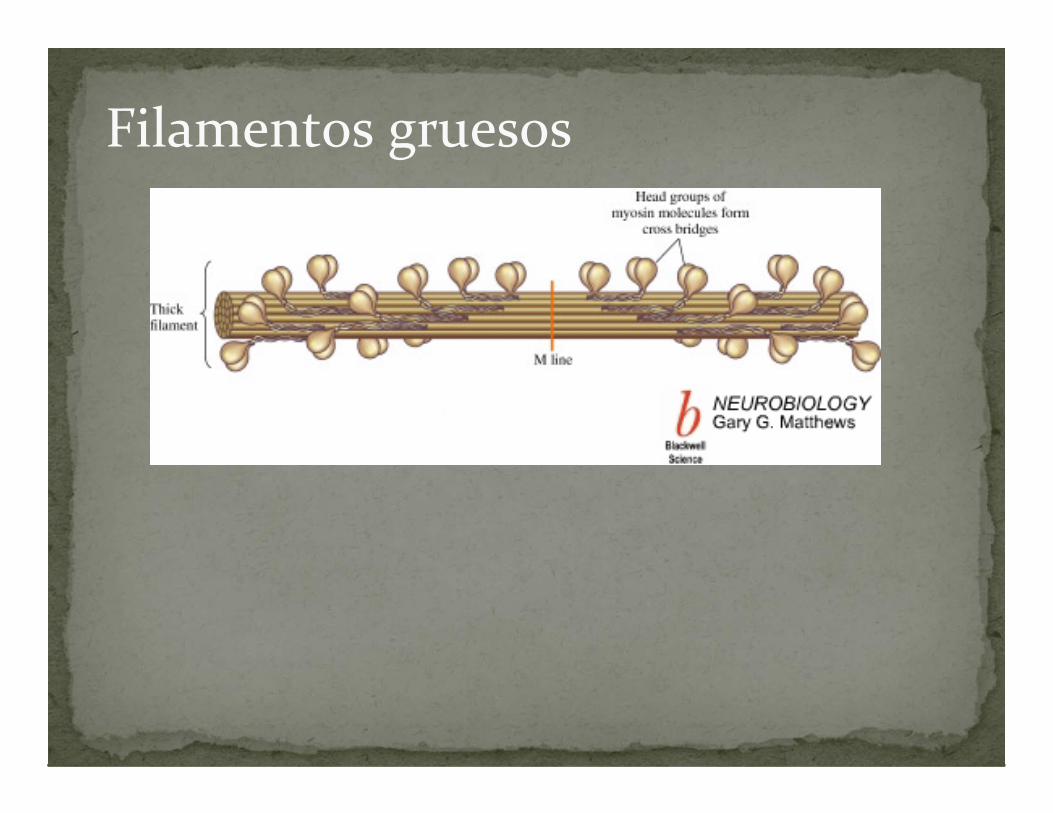
Filamentos gruesos

Filamentos finos
Actina (actina G, globular), forma agregados (actina F, fibrosa). Contiene un centro de unión para miosina. Cuando el músculo está en reposo este sitio estácubierto por la tropomiosina. (Alrededor de 13 agregados de actina por molécula de tropomiosina).

Filamentos finos
Tropomiosina: proteína filamentosa que se asocia a la actina. Troponina: proteína reguladora asociada a la tropomiosina. Es
un complejo formado por tres proteínas globulares (troponina T, I y C). T: unión a la tropomiosina. I: inhibidora de la unión de la miosina a la actina. C: une calcio. Está unión dispara la contracción.
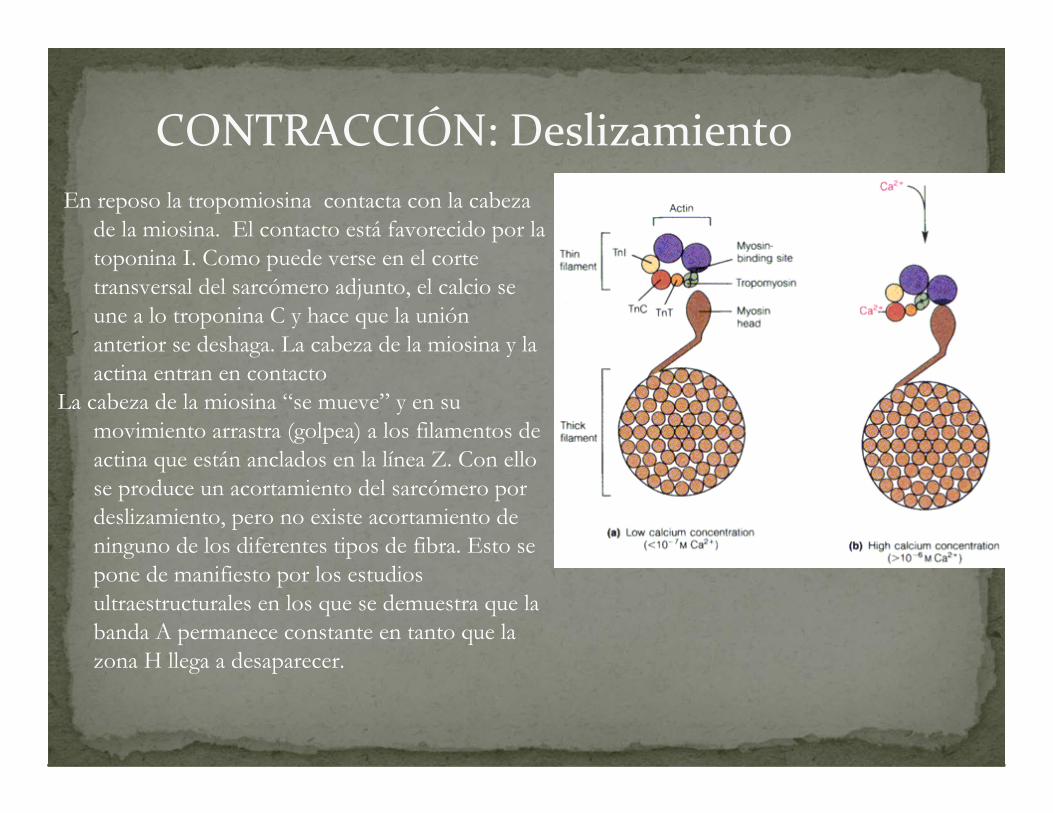
CONTRACCIÓN: Deslizamiento
En reposo la tropomiosina contacta con la cabeza de la miosina. El contacto está favorecido por la toponina I. Como puede verse en el corte transversal del sarcómero adjunto, el calcio se une a lo troponina C y hace que la unión anterior se deshaga. La cabeza de la miosina y la actina entran en contacto
La cabeza de la miosina “se mueve” y en su movimiento arrastra (golpea) a los filamentos de actina que están anclados en la línea Z. Con ello se produce un acortamiento del sarcómero por deslizamiento, pero no existe acortamiento de ninguno de los diferentes tipos de fibra. Esto se pone de manifiesto por los estudios ultraestructurales en los que se demuestra que la banda A permanece constante en tanto que la zona H llega a desaparecer.
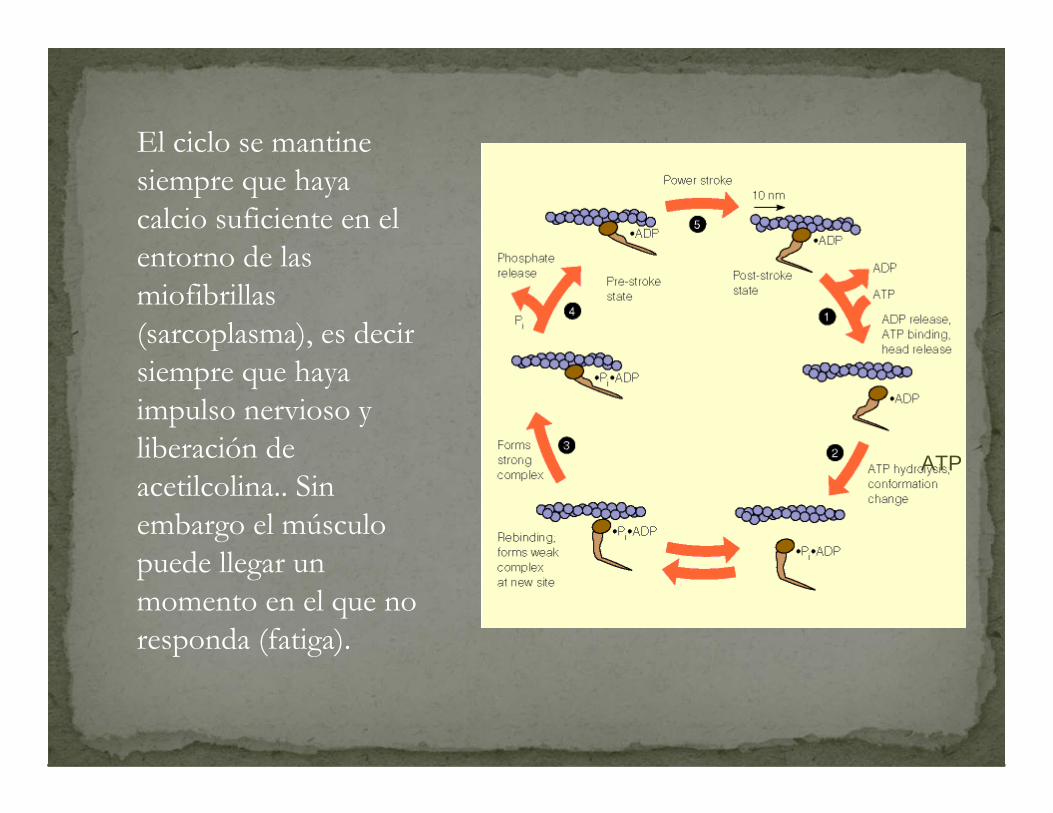
El ciclo se mantinesiempre que haya calcio suficiente en el entorno de las miofibrillas(sarcoplasma), es decir siempre que haya impulso nervioso y liberación de acetilcolina.. Sin embargo el músculo puede llegar un momento en el que no responda (fatiga).
ATP
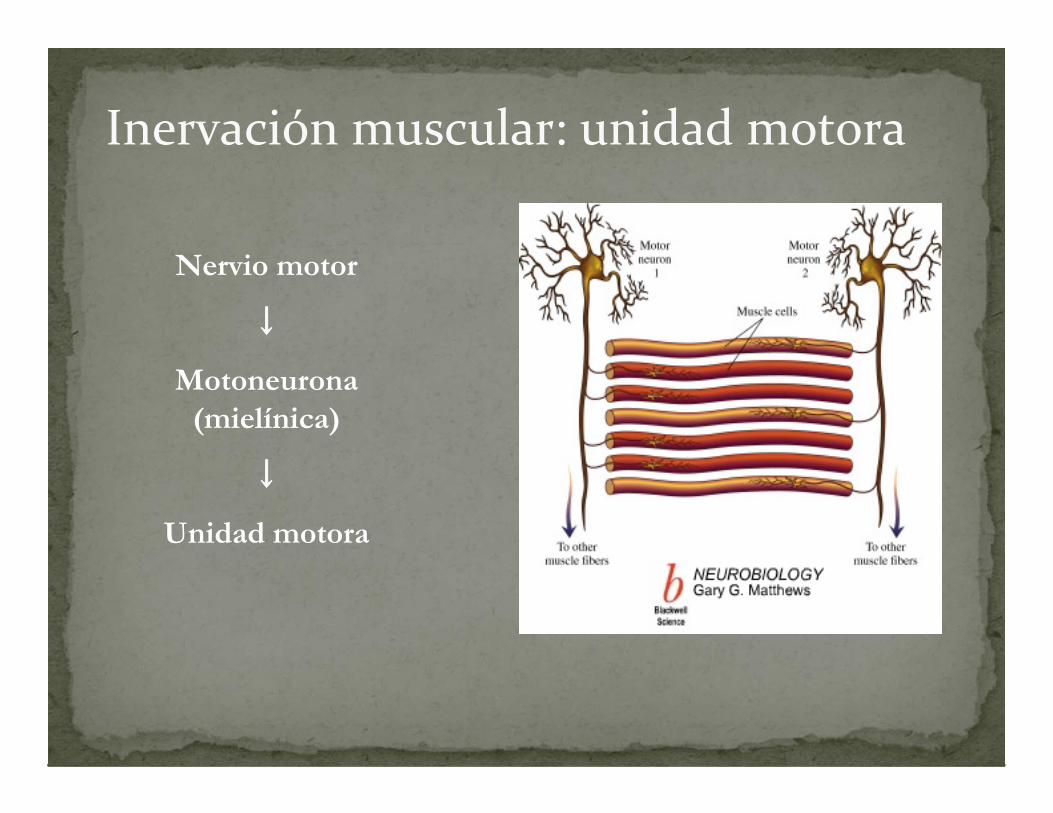
Inervación muscular: unidad motora
Nervio motor
↓
Motoneurona(mielínica)
↓
Unidad motora
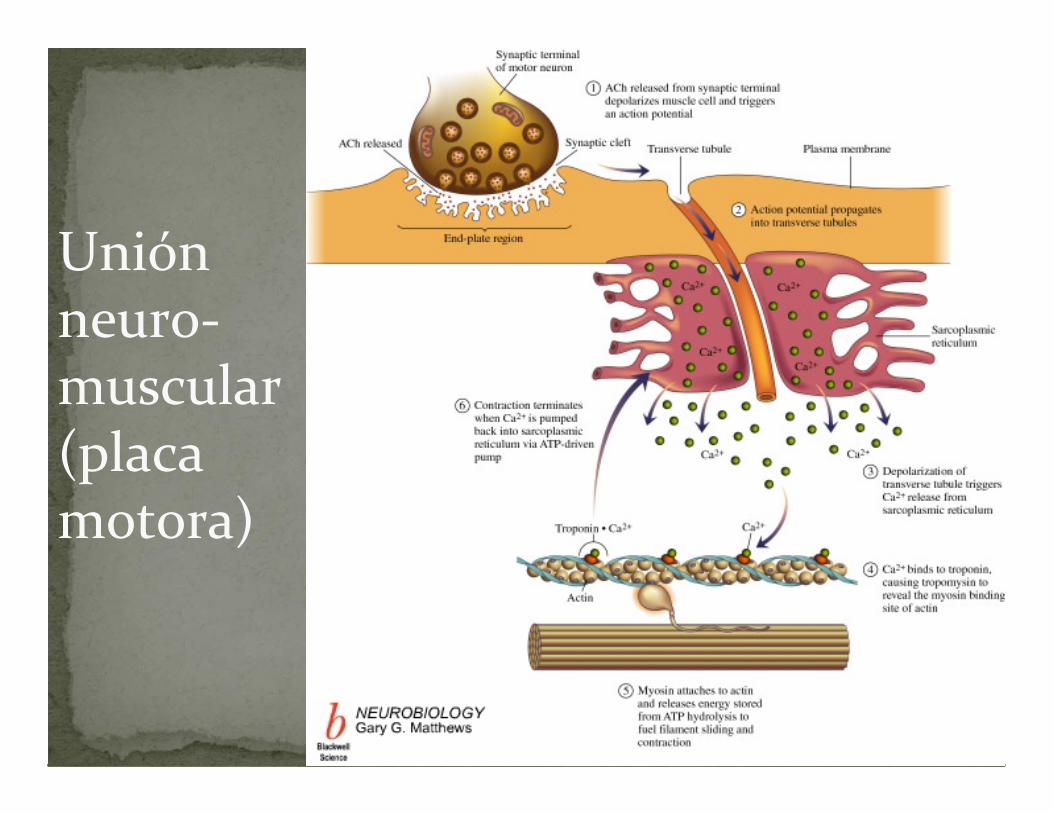
Unión neuro‐muscular (placa motora)

1. Llegada del potencial de acción al terminal del nervio motor : se abren canales para calcio dependientes de voltaje en la membrana presináptica, aumenta el calcio y esto estimula la liberación de acetil‐colina (AC) en la hendidura sináptica.
ACOPLAMIENTO EXCITACIACOPLAMIENTO EXCITACIÓÓNN--CONTRACCICONTRACCIÓÓN : Placa motora.N : Placa motora.
2. La AC liberada se une a receptores (receptoresnicotínicos) en la membrana postsináptica (membrana de la célula muscular). Este receptor es un canal de cationes (Na, K) que se abre por la AC produciéndose la despolarización local de la membrana

3. La despolarización local de la membrana abre nuevos canales dependientes de voltaje, propagándose el potencial de acción por toda la membrana, incluyendo los túbulos T
ACOPLAMIENTO EXCITACIACOPLAMIENTO EXCITACIÓÓNN--CONTRACCICONTRACCIÓÓN : Placa motora.N : Placa motora.

ACOPLAMIENTO EXCITACIÓN‐CONTRACCIÓN : Placa motora.4. Los túbulos T conectan directamente con el retículo
sarcoplásmico, de forma que cuando los primeros se despolarizan se abren canales de Ca+ dependientes de voltaje del segundo, esto provoca que el Ca2+ salga del retículo sarcoplásmico al sarcoplasma. Esto dispara la contracción. Como la señal (potencial de acción) se propaga en milisegundos a través de los túbulos T, a cada sarcómero de la célula, todas las miofibrillas se contraen al mismo tiempo
5. La acetilcolina es degradada en la hendidura sináptica por la acción de la acetilcolina esterasa
6. El calcio es devuelto al retículo sarcoplásmico por la ATPasa de Ca2+.

Metabolismo muscular
Fuente inmediata de E para la contracción musc: ATP
1° ATP ADP + Pi
Creatina-P-quinasa2° Creatina-P + ADP Creatina + ATP
3º Vía del ácido láctico
4º Vía aeróbica
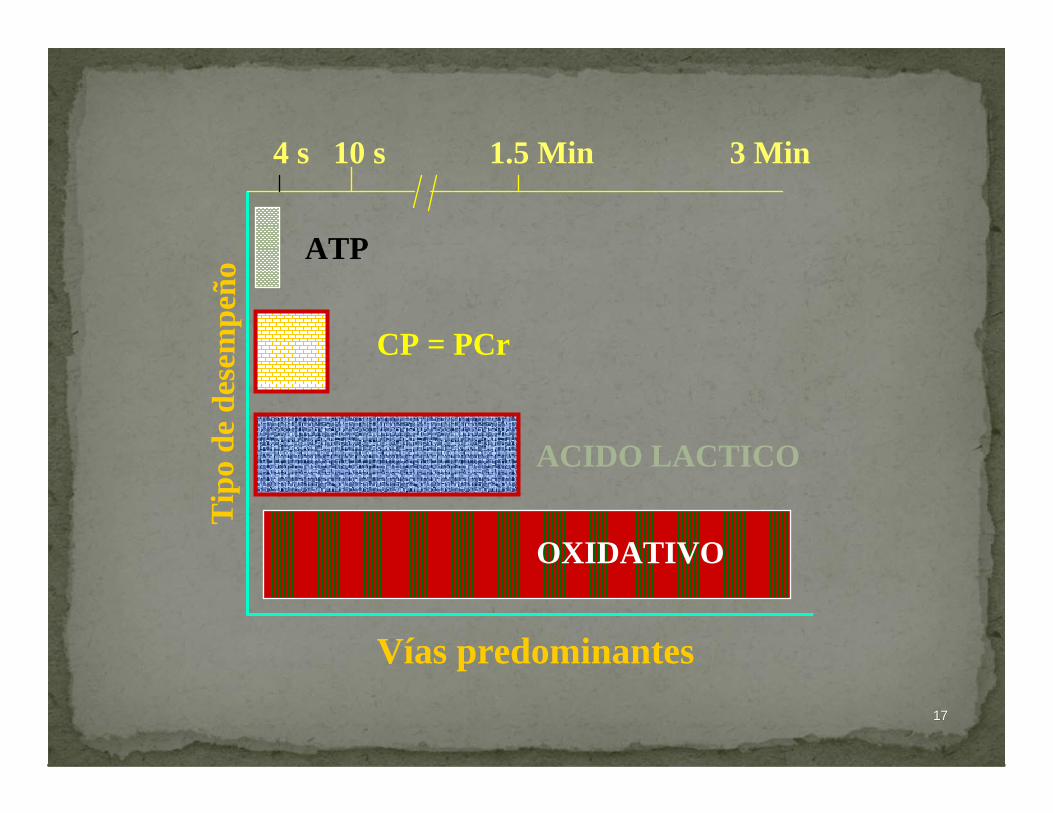
1717
ATP
CP = PCr
ACIDO LACTICO
OXIDATIVO
Vías predominantes
Tip
o de
des
emp e
ñ o
4 s 10 s 1.5 Min 3 Min

1818
PRODUCCION ANAEROBICA DE ATP
La forma mas simple y rápida envuelve la donación de ungrupo fosforico (P) y del
enlace energetico de la CP para el ADP y formar ATP.
CP + ADP ATP + CCREATIN-
QUINASA
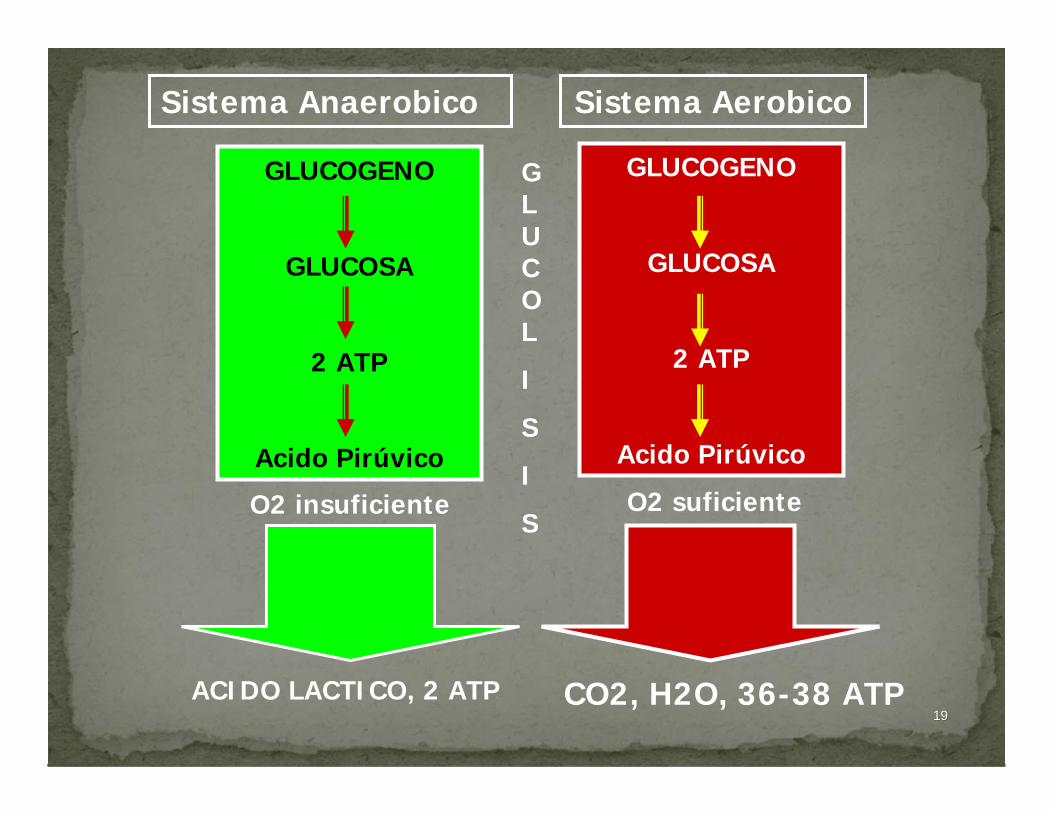
1919
Sistema AerobicoSistema Anaerobico
GLUCOGENO
GLUCOSA
2 ATP
Acido Pirúvico
GLUCOGENO
GLUCOSA
2 ATP
Acido Pirúvico
O2 insuficiente O2 suficiente
CO2, H2O, 36-38 ATPACIDO LACTICO, 2 ATP
GLUCOL
I
S
I
S
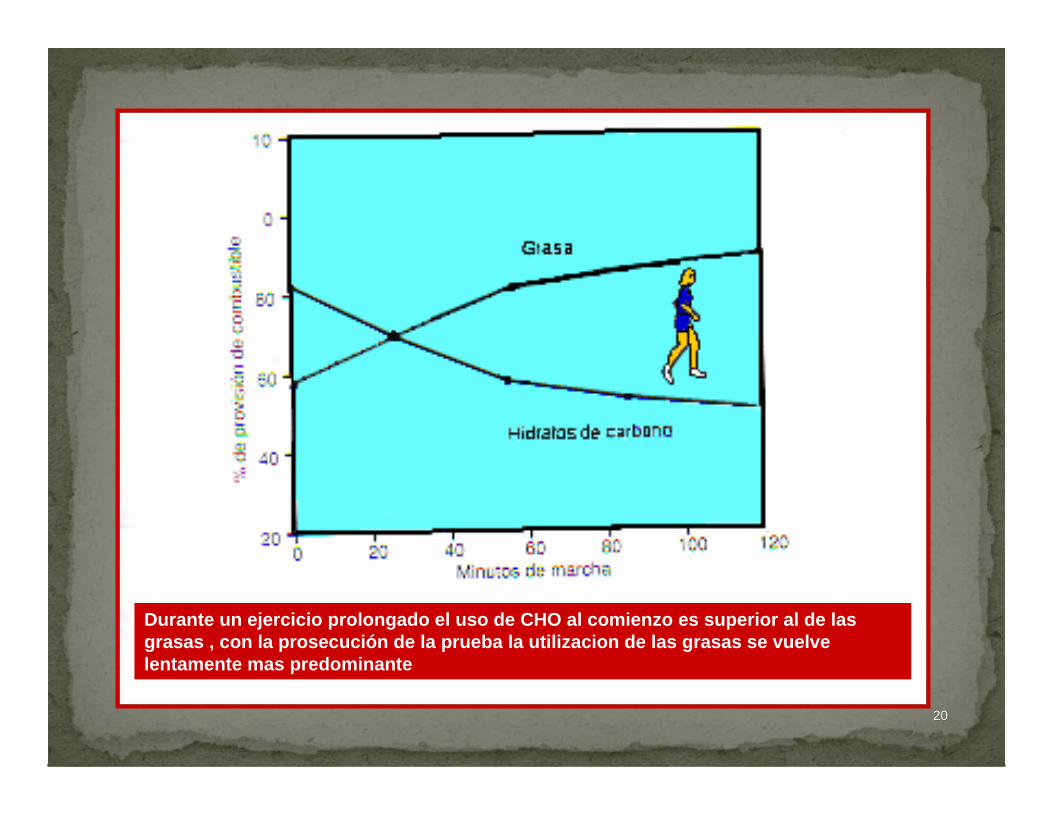
2020
Du
Durante un ejercicio prolongado el uso de CHO al comienzo es superior al de las grasas , con la prosecución de la prueba la utilizacion de las grasas se vuelve lentamente mas predominante
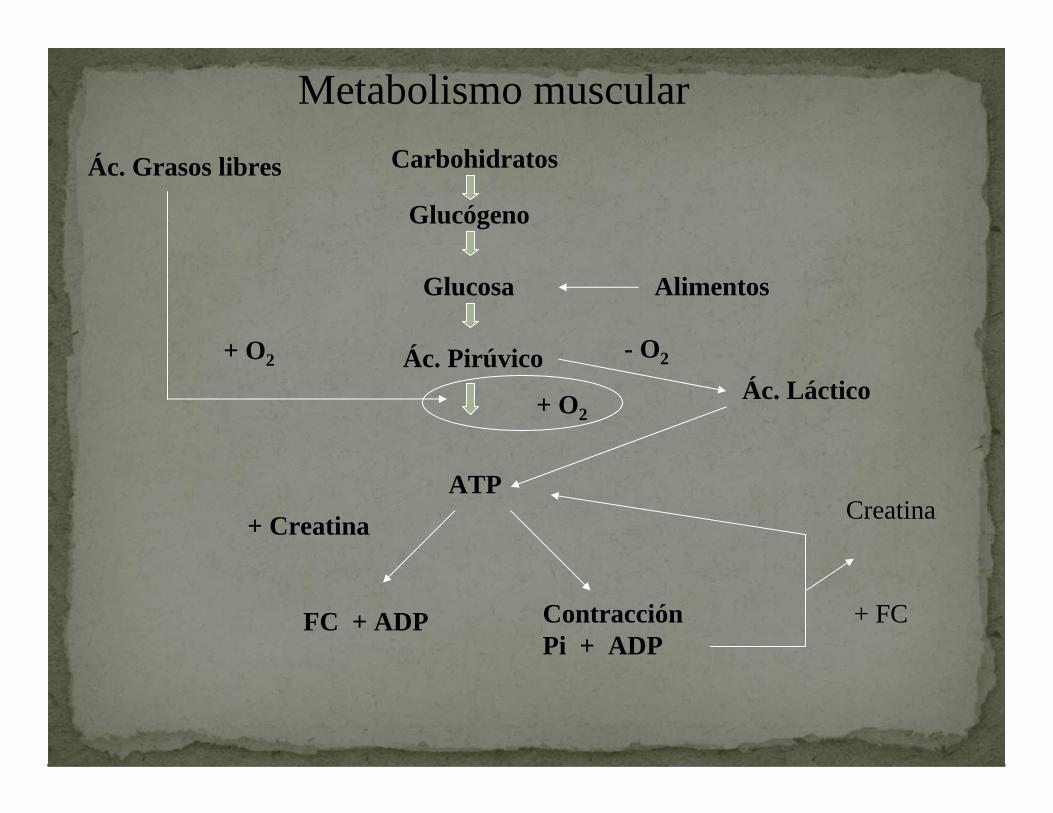
Metabolismo muscularCarbohidratos
Glucógeno
Glucosa Alimentos
Ác. Pirúvico
ATP
+ O2
Ác. Grasos libres
+ O2
Ác. Láctico- O2
FC + ADP ContracciónPi + ADP
+ Creatina Creatina
+ FC

En la contracción el Ca se une a troponina C y a la CALMODULINA
Estimula
Ca-Calmodulina Fosforilasa quinasa
GLUCOGENÓLISIS
Inicia
De esta manera el Calcio provoca la contracción muscular y estimula la provisión del sustrato necesario.

ADRENALINA Se une a receptores de membrana
+ Adenilato Ciclasa
Si AMPc + PKA Fosforilasa quinasa(a) Fosforila
Glucógeno fosforilasa
Fosforila
Glucogenólisis
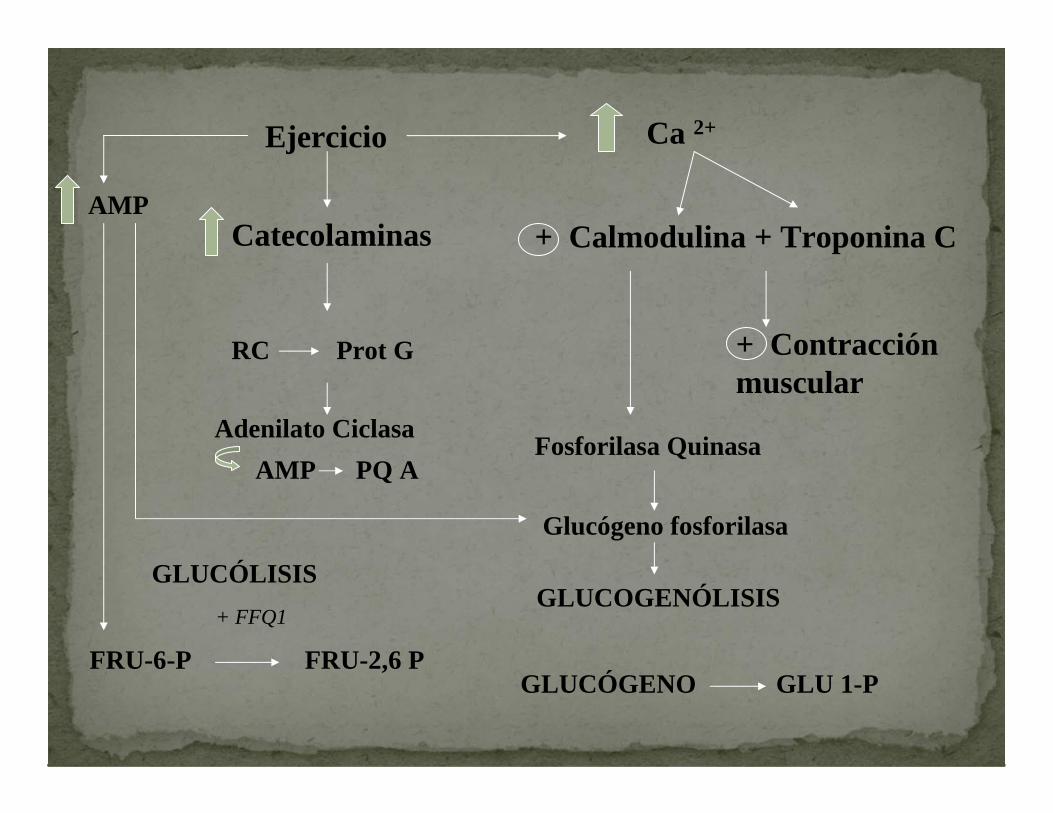
Ejercicio Ca 2+
AMPCatecolaminas + Calmodulina + Troponina C
RC Prot G
Adenilato Ciclasa
+ Contracción muscular
Fosforilasa QuinasaAMP PQ A
Glucógeno fosforilasa
GLUCOGENÓLISISGLUCÓLISIS
FRU-6-P FRU-2,6 P
+ FFQ1
GLUCÓGENO GLU 1-P

Universidad Nacional La Matanza
Miopatías Metabólicas
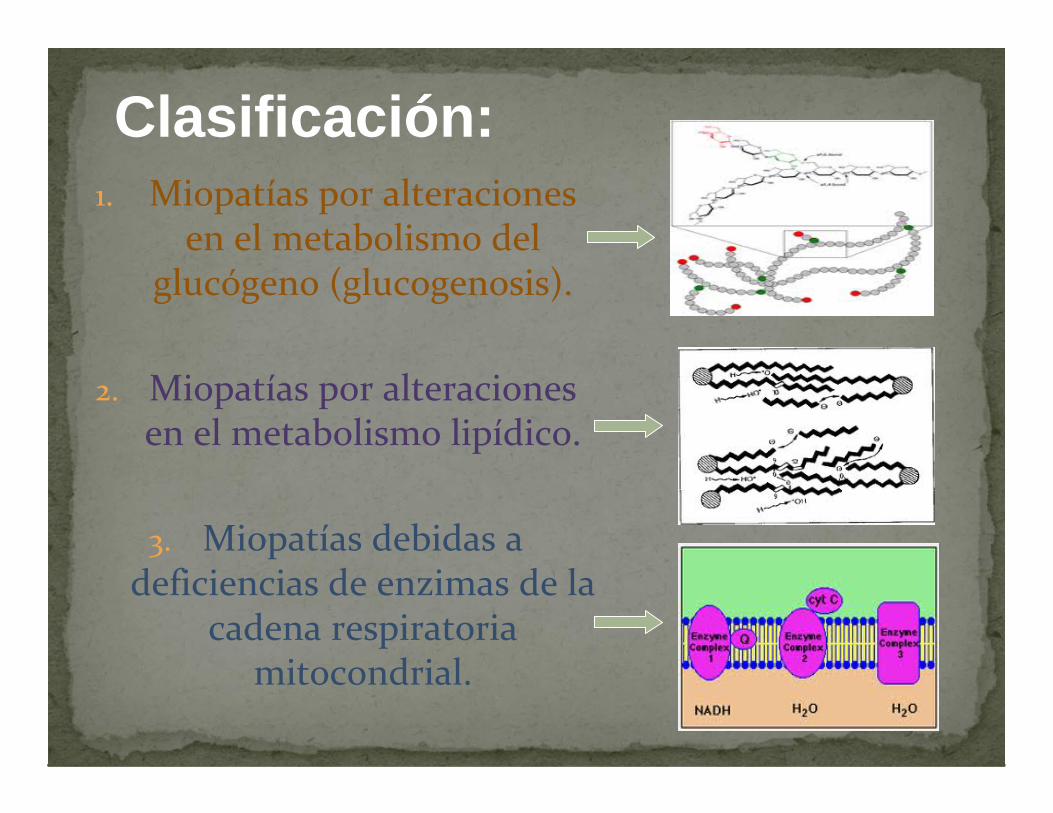
Clasificación:1. Miopatías por alteraciones
en el metabolismo del glucógeno (glucogenosis).
2. Miopatías por alteraciones en el metabolismo lipídico.
3. Miopatías debidas a deficiencias de enzimas de la
cadena respiratoria mitocondrial.
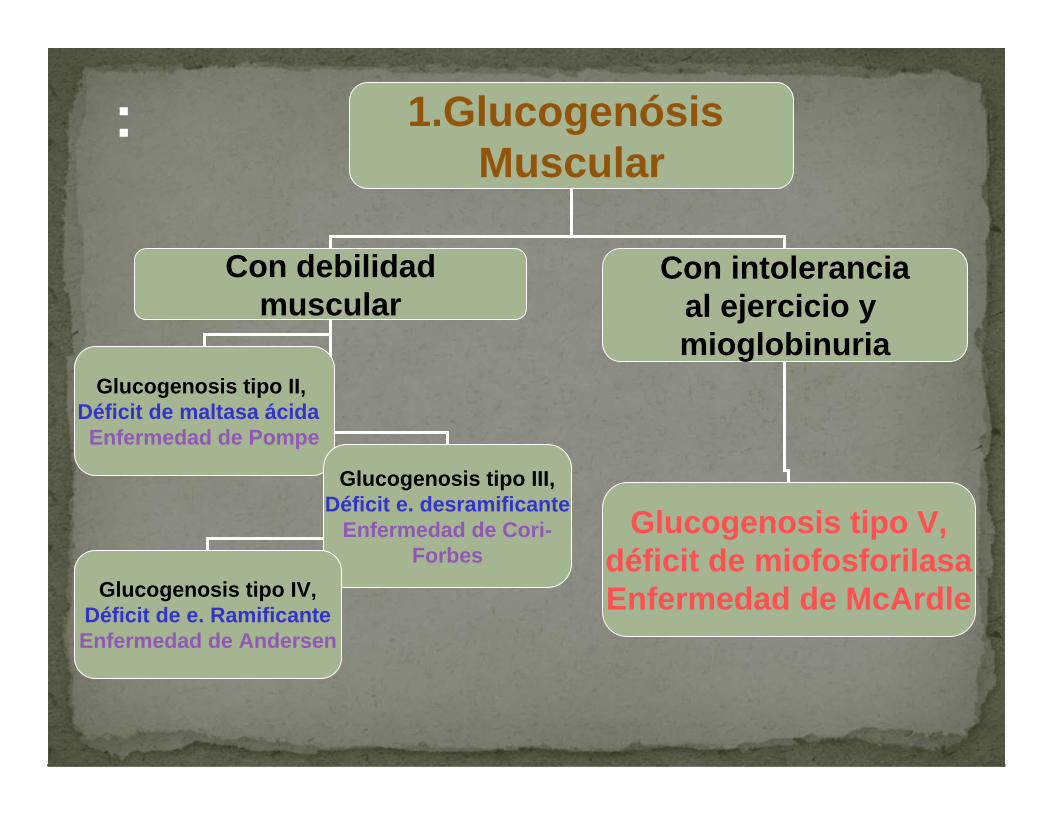
: 1.GlucogenósisMuscular
Con debilidadmuscular
Con intoleranciaal ejercicio y mioglobinuria
Glucogenosis tipo II,Déficit de maltasa ácidaEnfermedad de Pompe
Glucogenosis tipo III,Déficit e. desramificante
Enfermedad de Cori-Forbes
Glucogenosis tipo IV,Déficit de e. RamificanteEnfermedad de Andersen
Glucogenosis tipo V,déficit de miofosforilasaEnfermedad de McArdle

Repaso de la acción de la enzima: FOSFORILASA. Rompe uniones alfa 1‐‐‐4 por inserción de un grupo fosfato en el C1 de la glucosa.
Producto: Glucosa‐1‐P No hay gasto de ATP. El Pi proviene del medio.
Déficit de miofosforilasaEnfermedad de McArdle

Déficit de miofosforilasaEnfermedad de McArdle
Bioquímica: disminución notable o ausencia de la actividad de la miofosforilasa en músculo.
se acumula glucógeno el cual puede ser demostrado mediante tinciones histoquímicas en biopsias musculares
Síntomas aparecen en la adolescencia o juventud: intolerancia al ejercicio, mialgias, calambres musculares rigidez muscular.
un 50% de los casos presenta crisis de mioglobinuriatras el ejercicio intenso

Déficit de miofosforilasaEnfermedad de McArdle
Diagnóstico:
La biopsia muscular es siempre
diagnóstica.
Esta reacción se puede determinar
histoquímicamente.

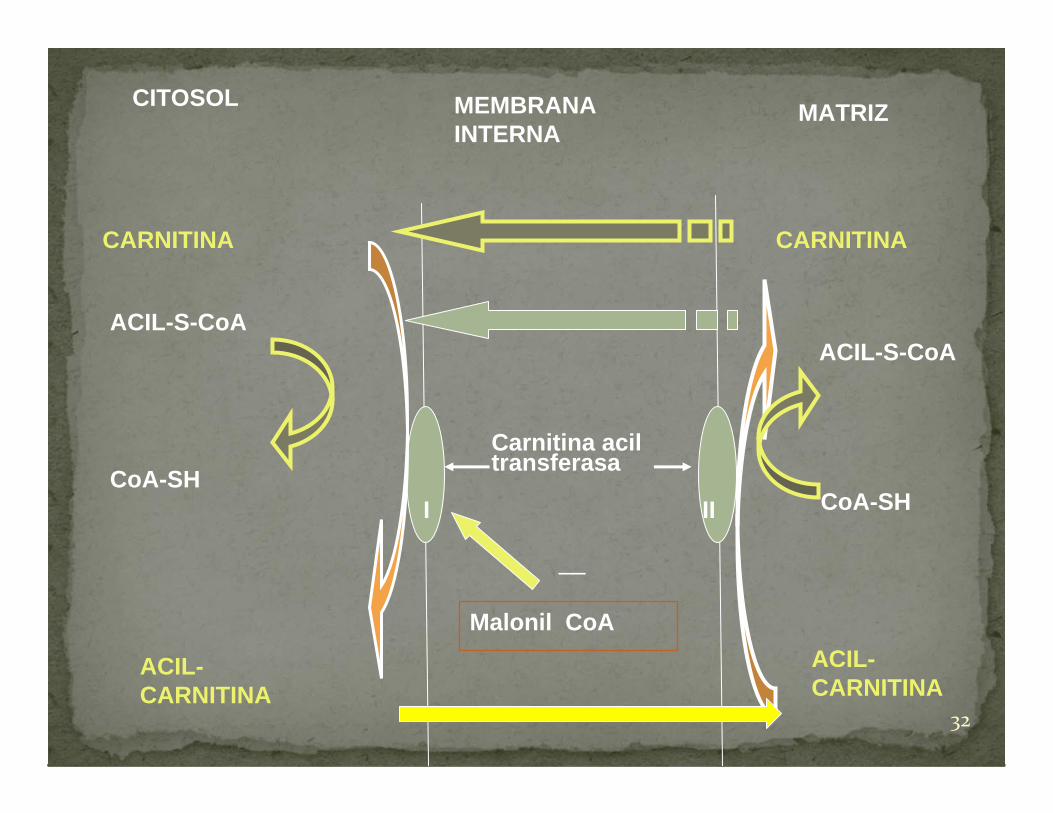
Alteración en el transporte de los ácidos
grasos de cadena larga al interior de la
mitocondria donde son metabolizados
mediante la ß‐ oxidación, proporcionando
la energía necesaria al músculo.
Miopatías por alteraciones en el metabolismo lipídico.
Déficit muscular de Carnitina Palmitoil Transferasa
32
CITOSOL MEMBRANA INTERNA
MATRIZ
CARNITINA CARNITINA
I II
Carnitina aciltransferasa
ACIL-S-CoAACIL-S-CoA
CoA-SHCoA-SH
ACIL-CARNITINA
ACIL-CARNITINA
Malonil CoA
__

Se acumulan lípidos bajo la forma de grasas neutras.
• Los síntomas se desencadenan por el ejercicio prolongado.
• consisten en:• crisis de mialgias, • calambres, • debilidad muscular y • rigidez con mioglobinuria, • y en casos severos fracaso renal agudo. • Las crisis se favorecen con el ayuno, la fiebre,
el frío o el «stress».
Miopatías por alteraciones en el metabolismo lipídico.

Miopatías debidas a deficiencias de enzimas de la cadena respiratoria
mitocondrialMIOPATIA MITOCONDRIAL
Se presenta: Desde la infancia temprana hasta la edad adulta. Síntomas:
Debilidad generalizada, Flacidez en los músculos del cuello e imposibilidad para caminar. El cerebro frecuentemente se ve afectado y pueden presentar
ataques, pérdida de la consciencia, la visión, el oído o el equilibrio, así como signos de retardo mental.
Progresión: La enfermedad puede presentar distintos grados de severidad y progresión. Herencia: Gen materno mitocondrial (mtADN).

Diferencias en el músculo según la deficiencia

DISTROFIA MUSCULAR DE DUCHENNE

DISTROFIA MUSCULAR DE DUCHENNE
Produce degeneración muscularAfecta a todas las razasEl gen anormal en el locus Xp21Codifica para la distrofina

La distrofia muscular de Duchenne, es una de las distrofias más comunes y más graves que afectan al ser humano. Se hereda de forma recesiva ligada al sexo, el gen que la determina está ubicada en el brazo corto del cromosoma X, este gen es incapaz de codificar la proteína distrofina, lo que se traduce en un perdida progresiva de las fibras musculares.
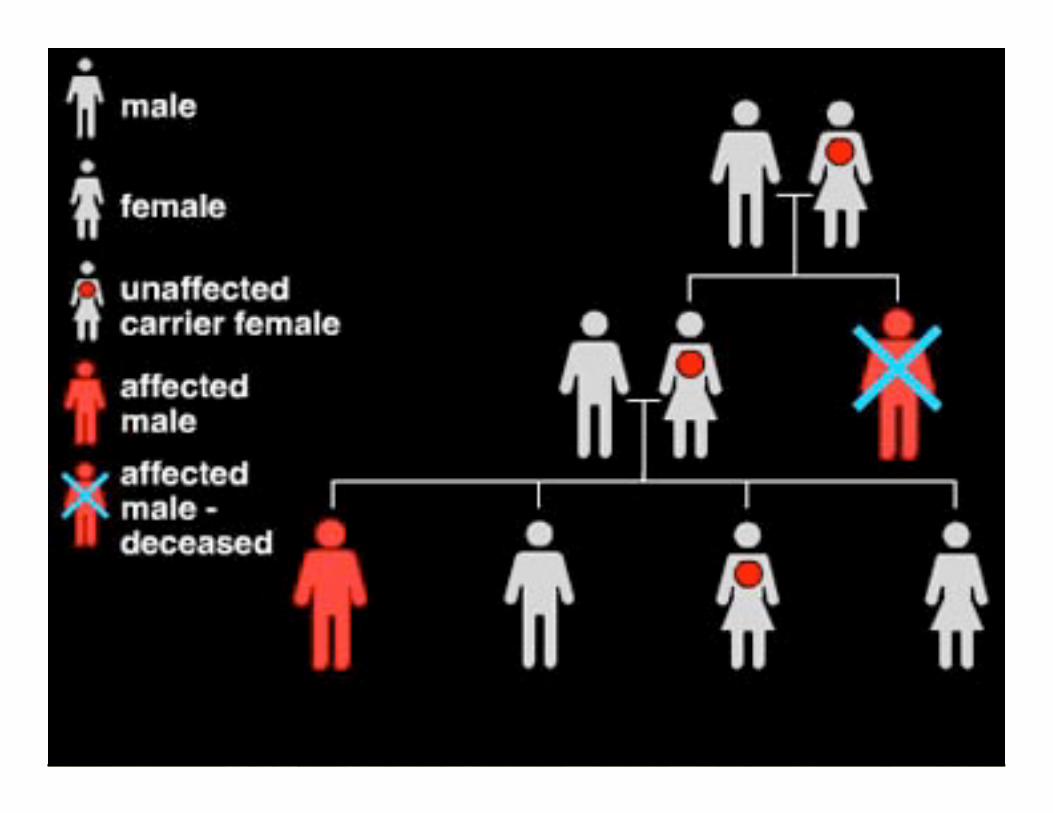

La enfermedad de duchenne se debe a la falta de DISTROFINA en la célula muscular.
La cual constituye una unión elástica entre las fibras de actina del citoesqueleto y la matriz extracelular que permite disipar la fuerza contráctil. Fundamental para el mantenimiento del tejido muscular
Lo que significa que sin distrofina las células musculares se degeneran gradualmente y mueren . Luego son sustituidas por tejido graso.
CPK elevada: diagnostico diferencial.
Al ser esta una enfermedad degenerativa la fuerza muscular de lapersona va disminuyendo al pasar los años afectando así los distintos segmentos musculares del cuerpo tales como musculaturacardiaca , esquelética y musculatura lisa
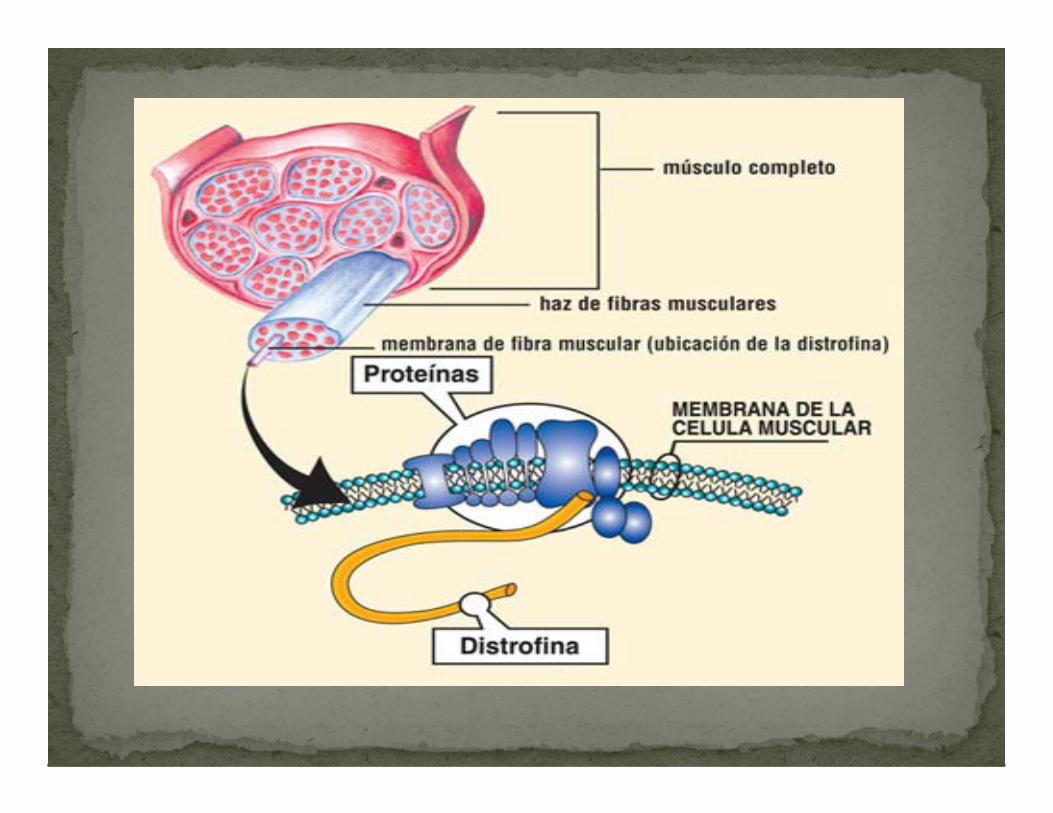
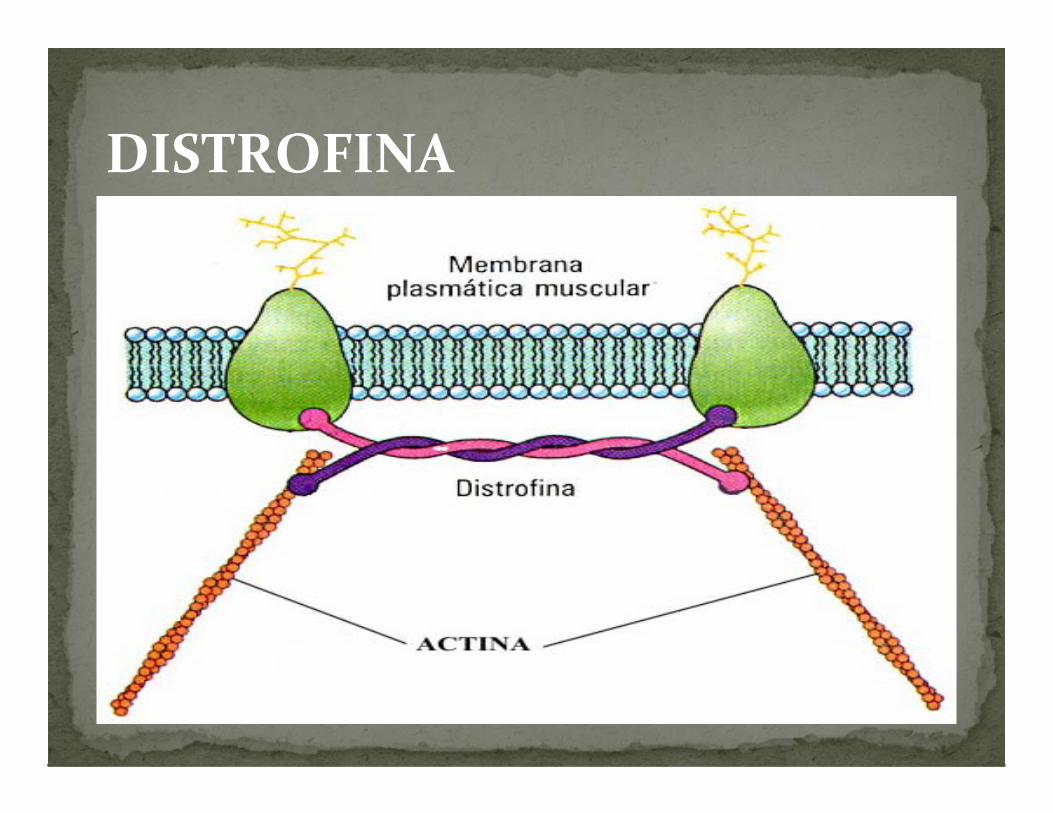
DISTROFINA

Evolución de la enfermedad
En los primeros meses se muestra una leve hipotonía (afecta el desarrollo psicomotor del lactante y del niño)
A los 3 años aparecen signos de alteraciones al inclinarse y caminar, alcanzando su máxima expresión alrededor de los 5 años
Entre 7 y 12 años aparece debilidad progresiva hasta el punto de precisar silla de ruedas.
La enfermedad progresa imparablemente, la afectación de la musculatura respiratoria. Comienza a manifestarse, infecciones pulmonares frecuentes y disminución de la capacidad respiratoria. Las contracturas y la escoliosis comprometen aún más la mecánica pulmonar e incluso comprimen el corazón.

La muerte puede sobrevenir, en algunos casos, alrededor de los 18 años de edad por insuficiencia respiratoria durante el sueño, insuficiencia cardiaca congestiva intratable, neumonía, aspiración y obstrucción de la vía respiratoria.

ENFERMEDAD NEUROMUSCULAR
DEBILIDAD MUSCULAR
DIFICULTAD MOVIMIENTOS DE EXTREMIDADES Y HABILIDAD
FUNCIONAL
DIFICULTAD EN DEGLUCIÓN, MASTICACÓN, HABLA,
RESPIRACIÓN
complicaciones secundarias
contracturas y escoliosis


Ejercicios: fortalecer la musculatura y dan aprendizaje motor.
Elongaciones: el desarrollo de contracturas y retracciones tienden a fijar articulaciones
produciendo deformidades, dolor y disminución en ciertas funciones. el objetivo de las elongaciones es retrasar las retracciones, ya que las mismas dificultan la marcha, haciéndose necesario a veces la cirugía ortopédica.
Postura: los pacientes que han perdido la función de la marcha, la postura en la
silla de ruedas debe ser cuidada al máximo, ya que tienden a hacer deformaciones escolióticas, con las consiguientes complicaciones, principalmente respiratorias es recomendable el uso de almohadones adecuados para una correcta base de apoyo,
Ejercicios respiratorios: los pacientes se ven complicados respiratoriamente en estadios
terminales,o como consecuencia de deformaciones torácicas graves. Presentando dificultades para toser y movilizar secreciones, haciéndose necesario la asistencia kinésica respiratoria.

Universidad Nacional La Matanza
Càtedra BioquímicaKinesiología UNLaM


![UNIVERSIDADE FFEDERAL DDE UUBERLÂNDIA ......Com exceção das miosinas I, II e V pouco se conhece das propriedades bioquímicas dessas enzimas [24]. Miosina II muscular ou miosina](https://img.pdfslide.tips/doc/110x75/5e6951af49f0237a9606ebb5/universidade-ffederal-dde-uuberlndia-com-exceo-das-miosinas-i-ii.jpg)


























