Embed Size (px)
Citation preview

CoagulopatCoagulopatíías adquiridas de origen inmune.as adquiridas de origen inmune.DiagnDiagnóóstico y actitudes terapstico y actitudes terapééuticasuticas
Mª Fernanda López FernándezUnidad de Hemostasia y Trombosis
Servicio de Hematología y HemoterapiaC. H. U. A Coruña
XXXIV Reunión de la AGHH
12-13 de Marzo 2010.
Vigo

CoagulopatCoagulopatíías adquiridas de origen inmune.as adquiridas de origen inmune.
Las coagulopatías adquiridas de etiología inmune son debidas a
autoanticuerpos que se desarrollan de forma espontánea en:
� Sujetos sin deficiencias previas de factores de la coagulación
� No expuestos previamente a antígenos externos.
Los autoanticuerpos neutralizan la función de un factor de la coagulación específico.
DiDiáátesis hemorrtesis hemorráágica gravegica grave
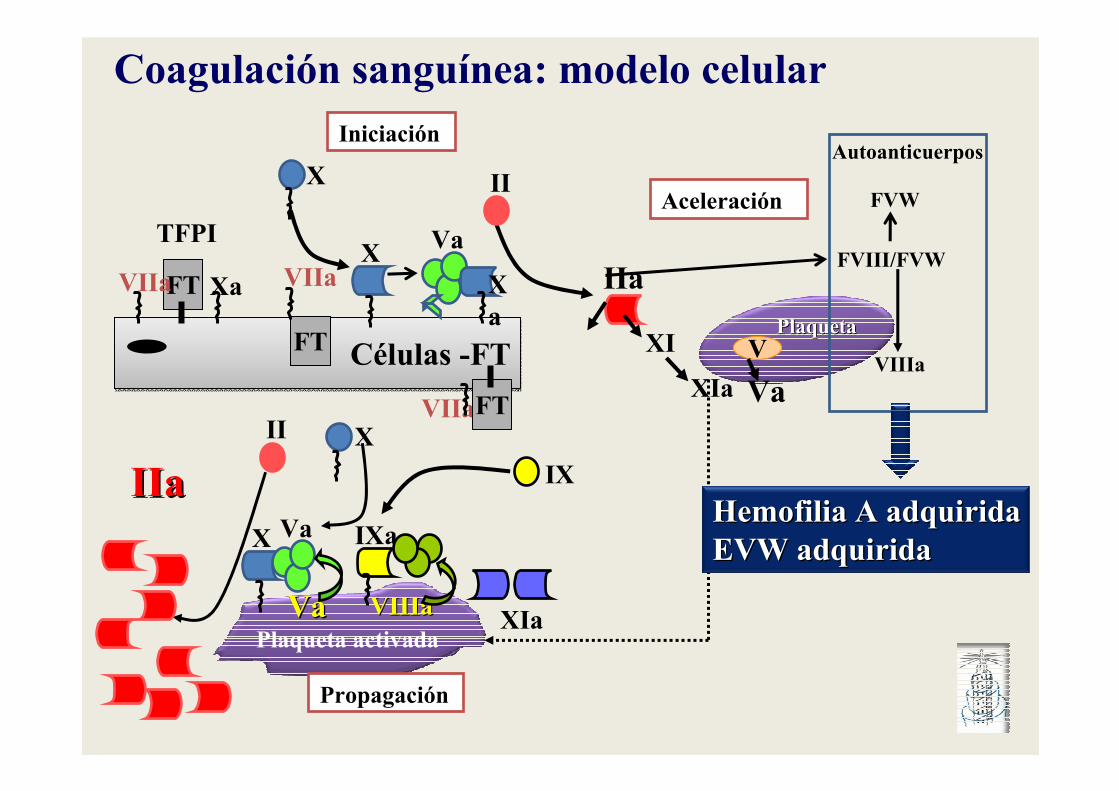
Coagulación sanguínea: modelo celular
VPlaquetaPlaqueta
Va
FVW
FVIII/FVW
VIIIaXI
XIaCélulas -FTCélulas -FT
FT XaVIIa
TFPI
VIIa
FT
Xa
X
IIaVa
Xa
II
IIaIIa
VIIa
IXa
Plaqueta activadaVIIIaVIIIaVaVa XIa
FT
IX
X
Xa
Va
II
Iniciación
Propagación
Aceleración
Autoanticuerpos
Hemofilia A adquiridaHemofilia A adquiridaEVW adquiridaEVW adquirida
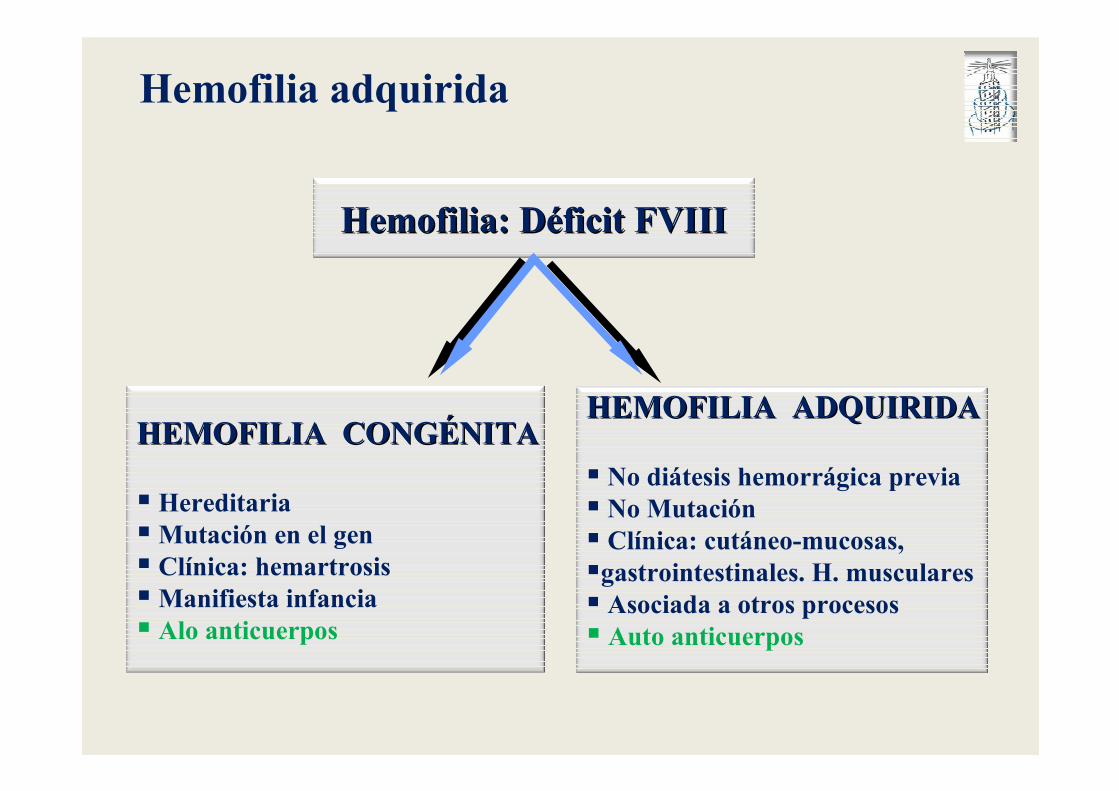
Hemofilia adquirida
HEMOFILIA CONGHEMOFILIA CONGÉÉNITANITA
� Hereditaria�Mutación en el gen � Clínica: hemartrosis�Manifiesta infancia� Alo anticuerpos
HEMOFILIA ADQUIRIDAHEMOFILIA ADQUIRIDA
� No diátesis hemorrágica previa� No Mutación� Clínica: cutáneo-mucosas, �gastrointestinales. H. musculares� Asociada a otros procesos� Auto anticuerpos
Hemofilia: DHemofilia: Dééficit FVIIIficit FVIII
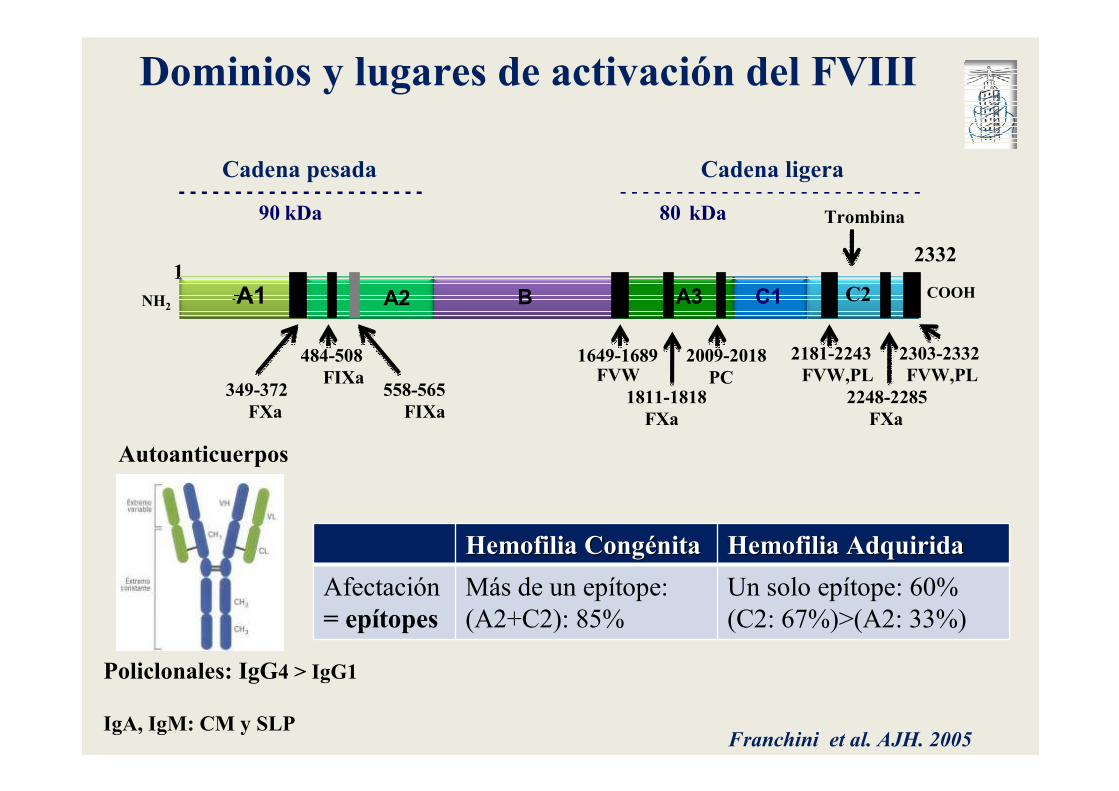
- - - - - - - - - - - - - - - - - - - - - -
90 kDa 80 kDa- - - - - - - - - - - - - - - - - - - - - - - - - - -
A1 A2 B A3 C1 C2 COOHNH2
12332
A1
2303-2332FVW,PL
2181-2243FVW,PL
Trombina
FXa
1649-1689FVW
2248-22851811-1818FXa
2009-2018PC
349-372FXa
484-508FIXa
558-565FIXa
Cadena pesada Cadena ligera
Dominios y lugares de activación del FVIII
Hemofilia CongHemofilia Congéénitanita Hemofilia AdquiridaHemofilia Adquirida
Afectación
= epítopesMás de un epítope:
(A2+C2): 85%
Un solo epítope: 60%
(C2: 67%)>(A2: 33%)
Franchini et al. AJH. 2005
Autoanticuerpos
Policlonales: IgG4 > IgG1
IgA, IgM: CM y SLP
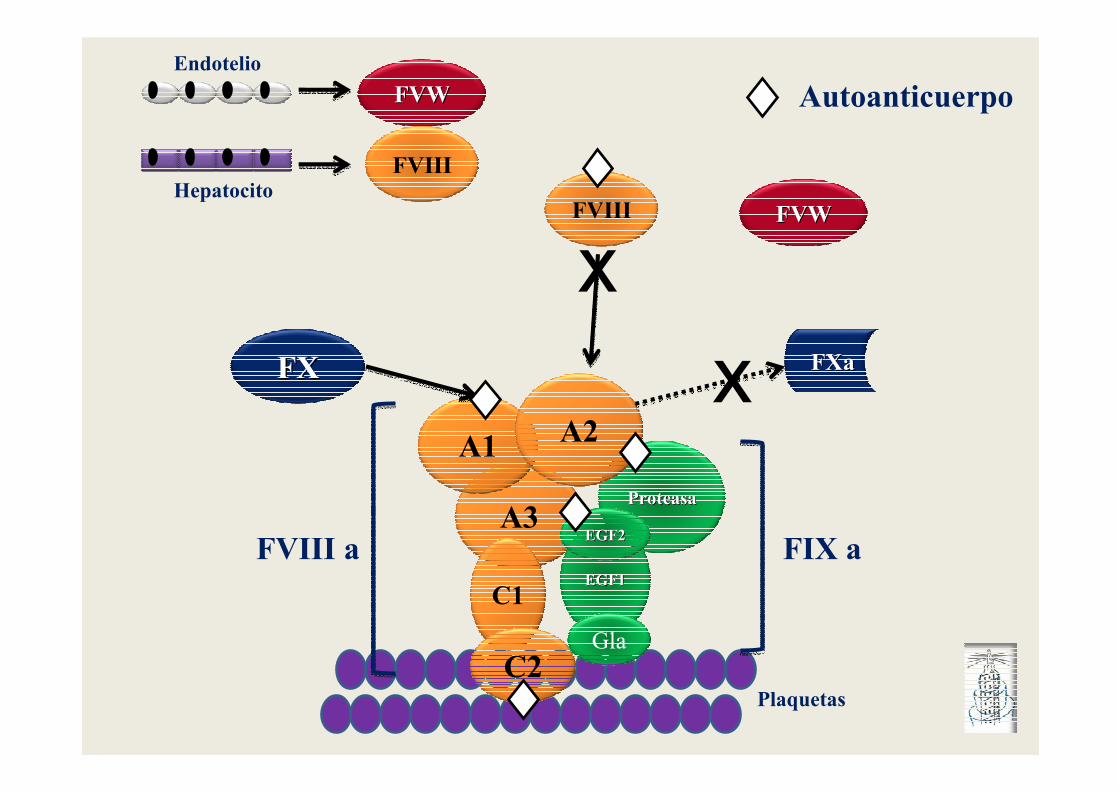
ProteasaProteasa
EGF1EGF1
A3
C1
PlaquetasC2
A1 A2
Gla
EGF2EGF2FIX aFVIII a
FXaFXaFXFX
Endotelio
FVWFVW
FVIIIHepatocito
FVWFVWFVIII
x
Autoanticuerpo
x
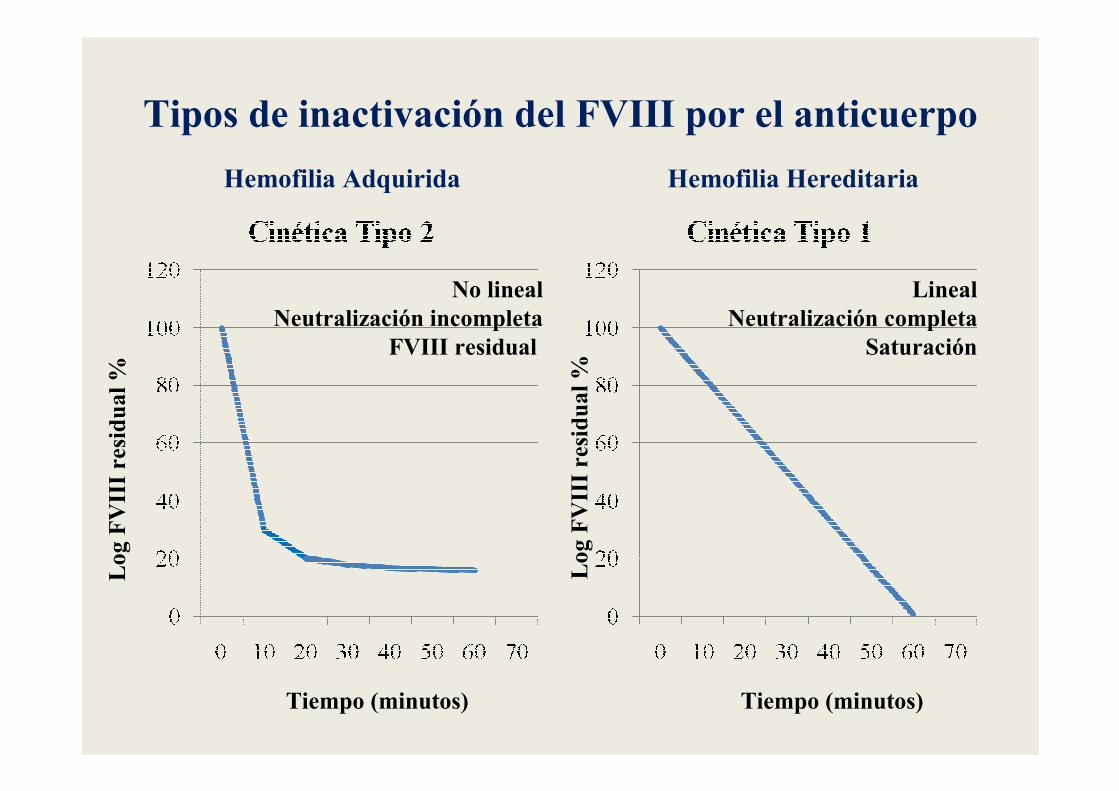
Tipos de inactivación del FVIII por el anticuerpo
Tiempo (minutos) Tiempo (minutos)
No linealNeutralización incompleta
FVIII residual
LinealNeutralización completa
Saturación
Log FVIII residual %
Log FVIII residual %
Hemofilia HereditariaHemofilia Adquirida
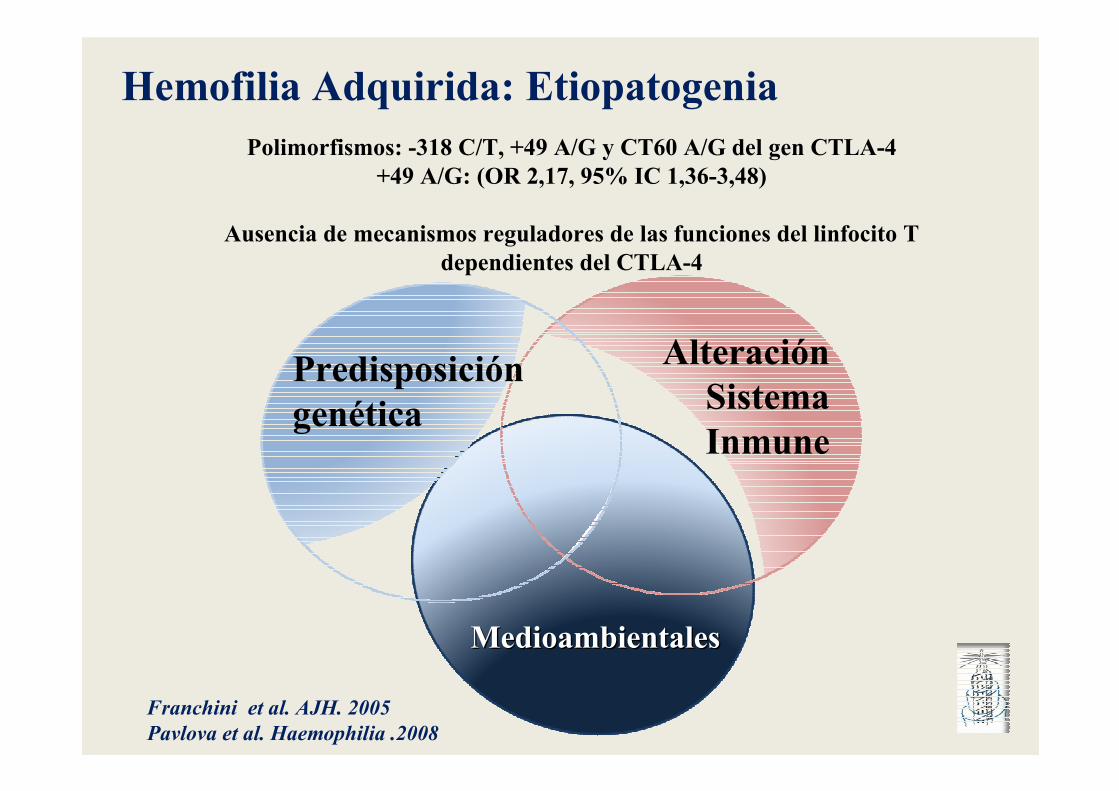
MedioambientalesMedioambientales
Alteración SistemaInmune
Predisposición genética
Franchini et al. AJH. 2005
Pavlova et al. Haemophilia .2008
Hemofilia Adquirida: EtiopatogeniaPolimorfismos: -318 C/T, +49 A/G y CT60 A/G del gen CTLA-4
+49 A/G: (OR 2,17, 95% IC 1,36-3,48)
Ausencia de mecanismos reguladores de las funciones del linfocito Tdependientes del CTLA-4
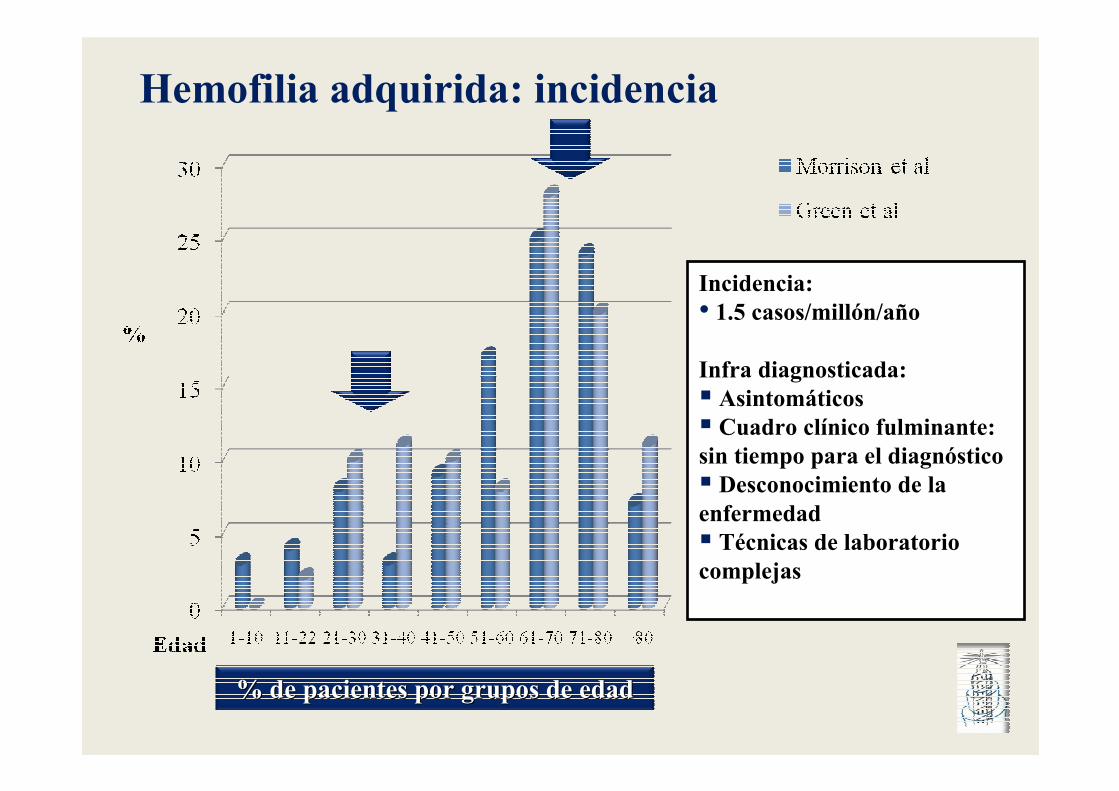
% de pacientes por grupos de edad% de pacientes por grupos de edad
Incidencia: • 1.5 casos/millón/año
Infra diagnosticada:� Asintomáticos � Cuadro clínico fulminante: sin tiempo para el diagnóstico� Desconocimiento de la enfermedad� Técnicas de laboratorio complejas
Hemofilia adquirida: incidencia
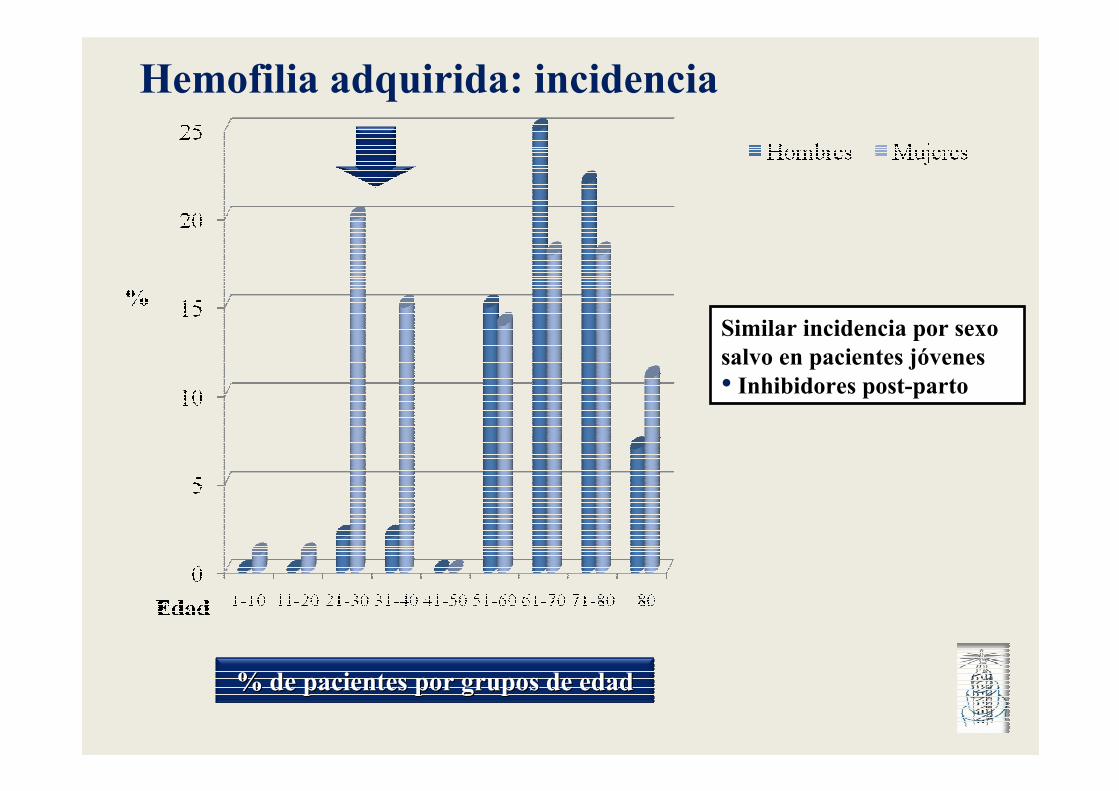
% de pacientes por grupos de edad% de pacientes por grupos de edad
Similar incidencia por sexo salvo en pacientes jóvenes• Inhibidores post-parto
Hemofilia adquirida: incidencia
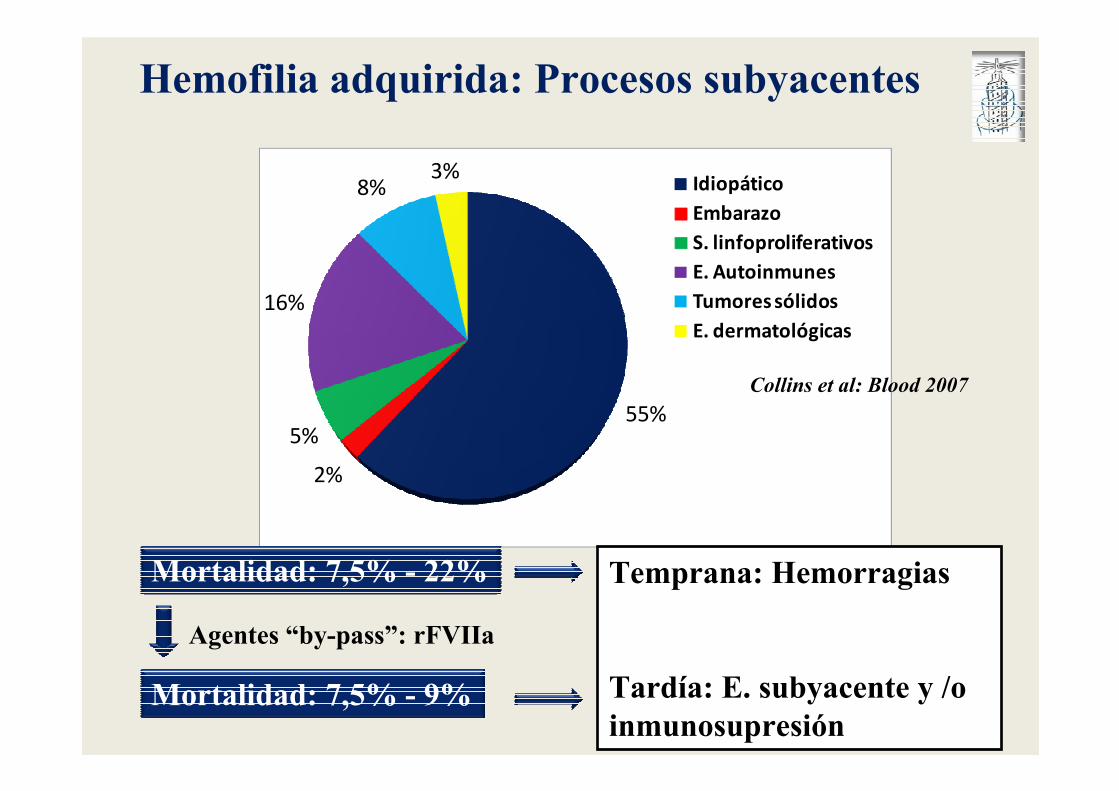
55%
2%
5%
16%
8%3%
Idiopático
Embarazo
S. linfoproliferativos
E. Autoinmunes
Tumores sólidos
E. dermatológicas
Hemofilia adquirida: Procesos subyacentes
Mortalidad: 7,5% - 22%
Mortalidad: 7,5% - 9%
Agentes “by-pass”: rFVIIa
Temprana: Hemorragias
Tardía: E. subyacente y /o inmunosupresión
Collins et al: Blood 2007
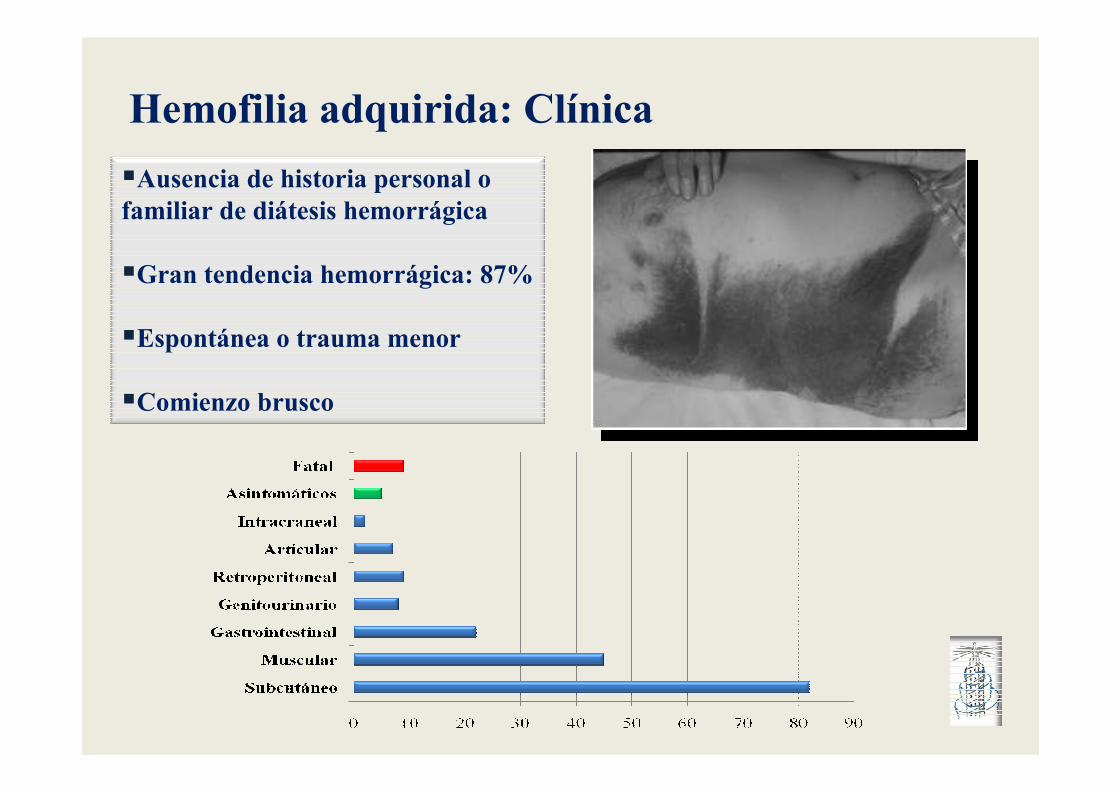
Hemofilia adquirida: Clínica
�Ausencia de historia personal o familiar de diátesis hemorrágica
�Gran tendencia hemorrágica: 87%
�Espontánea o trauma menor
�Comienzo brusco
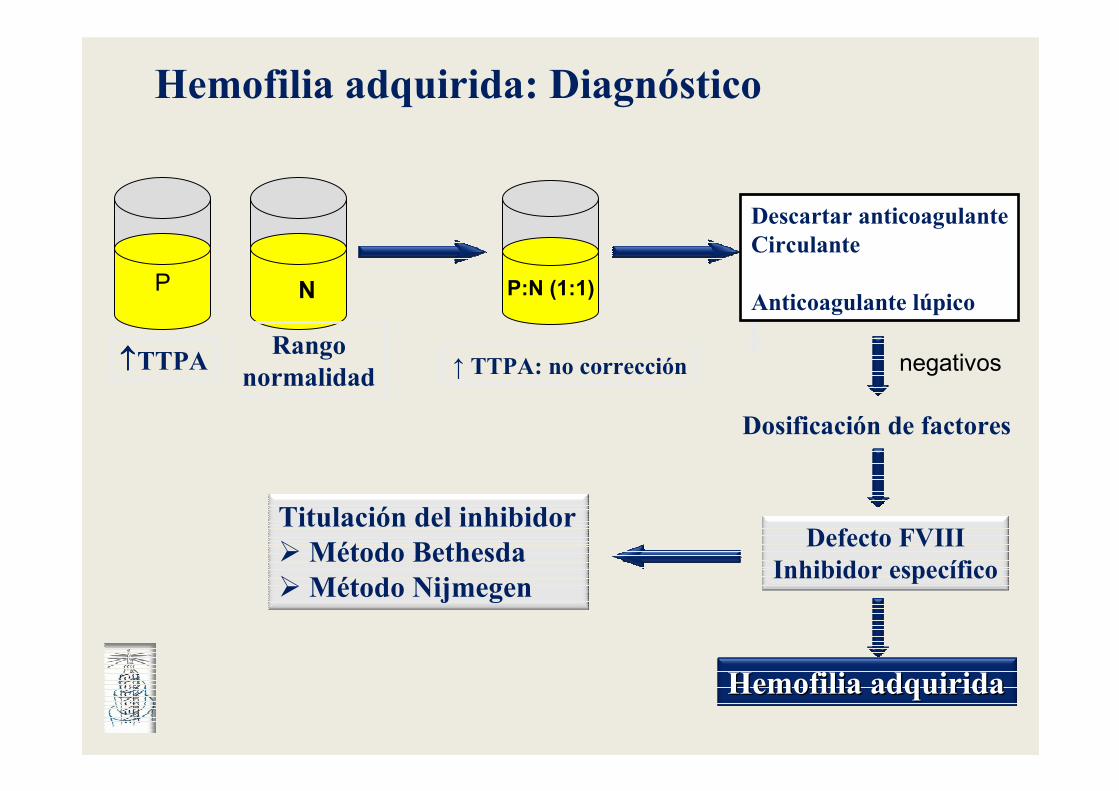
↑↑↑↑TTPA
P N
Rangonormalidad
P:N (1:1)
↑ TTPA: no corrección
Descartar anticoagulanteCirculante
Anticoagulante lúpico
Dosificación de factores
negativos
Defecto FVIIIInhibidor específico
Hemofilia adquiridaHemofilia adquirida
Titulación del inhibidor�Método Bethesda�Método Nijmegen
Hemofilia adquirida: Diagnóstico
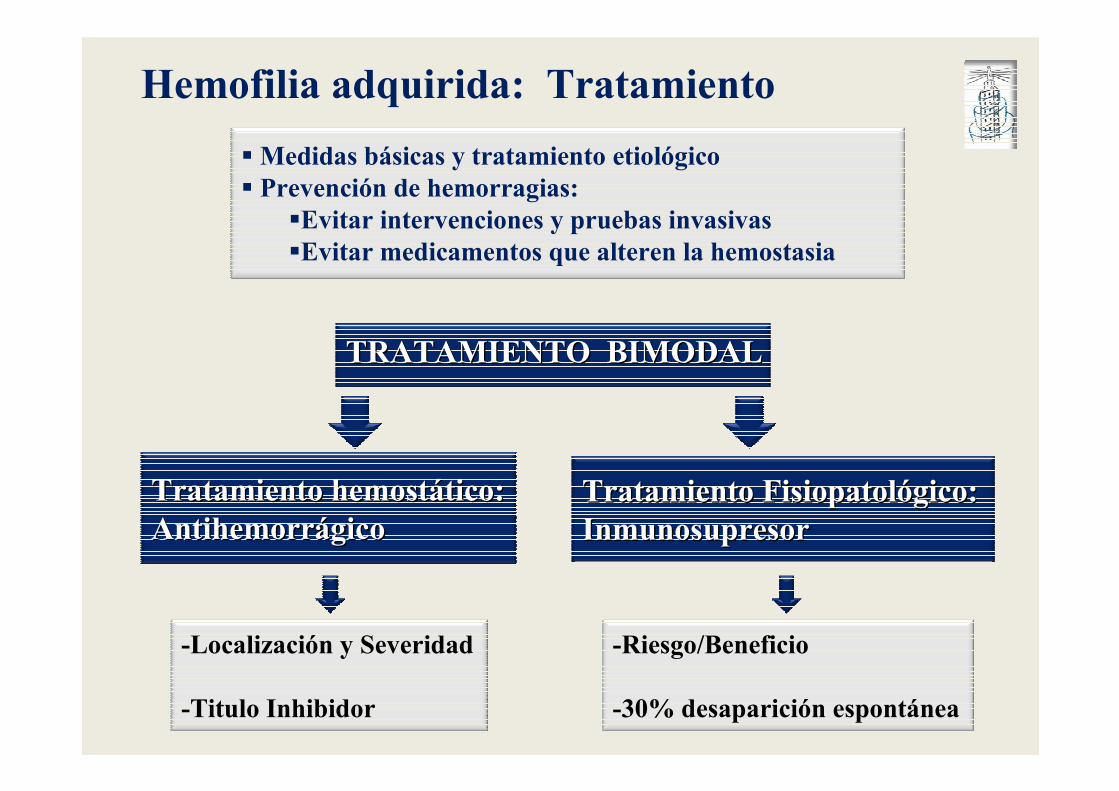
Hemofilia adquirida: Tratamiento
Tratamiento hemostTratamiento hemostáático:tico:AntihemorrAntihemorráágicogico
Tratamiento FisiopatolTratamiento Fisiopatolóógico:gico:InmunosupresorInmunosupresor
�Medidas básicas y tratamiento etiológico� Prevención de hemorragias:
�Evitar intervenciones y pruebas invasivas�Evitar medicamentos que alteren la hemostasia
TRATAMIENTO BIMODALTRATAMIENTO BIMODAL
-Localización y Severidad
-Titulo Inhibidor
-Riesgo/Beneficio
-30% desaparición espontánea

Hemofilia adquirida: Tratamiento hemostático
Aumentar Niveles FVIII:�Desmopresina (DDAVP)
�Concentrados de Factor VIII�Factor VIII:C
Agentes ByAgentes By--PassPass��Feiba (CCPa)Feiba (CCPa)��Factor VII activado recombinanteFactor VII activado recombinante

Hemofilia Adquirida: Tratamiento hemostático
Desmopresina (DDAVP):Desmopresina (DDAVP):
��Dosis:0.3Dosis:0.3µµg/Kg (i.v.)g/Kg (i.v.)��PrecauciPrecaucióón: n:
��HTAHTA��Alteraciones CardAlteraciones Cardííacasacas��Antecedentes de trombosisAntecedentes de trombosis
Factor VIII:Factor VIII:��Dosis: Muy AltasDosis: Muy Altas��Problemas:Problemas:
��Respuesta anamnRespuesta anamnéésicasica��Mala correlaciMala correlacióón n
�� Regimenes de inmunotoleranciaRegimenes de inmunotolerancia
�Hemorragias leves
�Título Inhibidor bajo <3 UB
�Nivel Factor VIII>5%
Utilidad muy limitadaUtilidad muy limitada
AntifibinolAntifibinolííticos:ticos:
��CoadyuvantesCoadyuvantes

Hemofilia Adquirida: CCPA (FEIBA®)
Contiene : FII, VIIa, IXa, Xa, PC, PSContiene : FII, VIIa, IXa, Xa, PC, PSy pequey pequeññas cantidades de FVIIIas cantidades de FVIII
�Dosis: *50-100 U/Kg /8-12 h�(no>200 U/Kg en 24h)
�Contraindicados: antifibrinolíticos
�Problemas:�Trombogénico�Plasmático : (Calor seco)� No Monitorización�Respuesta anamnésica
Sallah et al: Haemophilia 2004
34 pacientes: 75 U/Kg/8-12 horas� 86% respuestas
•Episodios Graves:10 dosis•Episodios Leves: 6 dosis
Goudemand et al: WFH 2004
55 episodios hemorrágicos17 pacientes Dosis media; 68 U/Kg/8-24 horasDurante una media de 3,5 días� 89% respuestas Evidencia limitadaEvidencia limitada
No estudios prospectivosNo estudios prospectivos
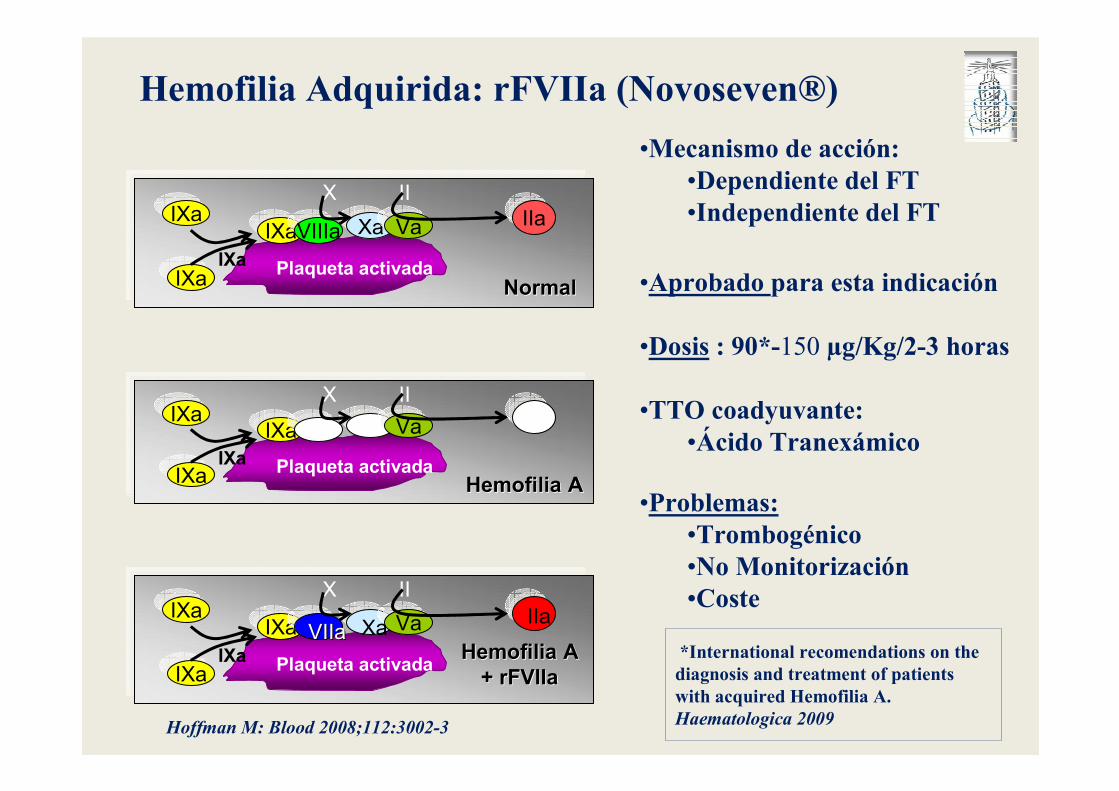
Plaqueta activada
IXaVIIIa Xa Va
IXa
IXa
IXa
X II
IIa
NormalNormal
Plaqueta activada
IXa Va
IXa
IXa
IXa
X II
Hemofilia AHemofilia A
Plaqueta activada
IXa Va
IXa
IXa
IXa
X II
Hemofilia AHemofilia A
+ rFVIIa+ rFVIIa
XaVIIaVIIaIIa
Hoffman M: Blood 2008;112:3002-3
Hemofilia Adquirida: rFVIIa (Novoseven®)
•Mecanismo de acción:•Dependiente del FT•Independiente del FT
•Aprobado para esta indicación
•Dosis : 90*-150 µg/Kg/2-3 horas
•TTO coadyuvante: •Ácido Tranexámico
•Problemas: •Trombogénico•No Monitorización•Coste
*International recomendations on the diagnosis and treatment of patients with acquired Hemofilia A. Haematologica 2009

Hemofilia Adquirida: rFVIIa (Novoseven®)
•Hay et al 1997•Estudio multicéntrico•74 episodios hemorrágicos•38 pacientes
TTO 2ª línea ó rescate (60 casos):� 75% buenas respuestas� 17% respuestas parciales� 8% pobre respuesta
TTO Primera línea (14 Casos)� 100% Buenas respuestas
�Nº Dosis medias 6
•Baudo et al 1997•Registro italiano•Prospectivo•20 episodios hemorrágicos•14 pacientes
TTO 1 línea (19 casos):� 86,6% control de la hemorragia
*International recomendations on the diagnosis and treatment of patients with acquired Hemofilia A. Haematologica 2009
*Primera l*Primera líínea de tratamientonea de tratamiento

Hemofilia Adquirida: Tratamiento recomedaciones
� El tratamiento hemostático en pacientes con hemorragia grave debe iniciarse lo antes posible, independientemente del título del inhibidor y el
FVIII residual.
� Se recomienda el uso de rFVIIa o CCPA en hemorragias graves�Concentrados de FVIII recombinantes o hemoderivados deben utilizarse
sólo si no se dispone de agentes “by-pass”
� En caso de fallo terapéutico deben buscarse estrategias alternativas � Incremento dosis rFVIIa, cambio agente by-pass, tratamientos
secuenciales??
�Inmunoabsorción o plasmaféresis
� Previo a cirugías o precedimientos invasivos : profilaxis con agentes “by-pass”
*International recomendations on the diagnosis and treatment of patients with acquired Hemofilia A. Haematologica 2009

Hemofilia adquirida: Tto inmunosupresor
**Todos los pacientesTodos los pacientes diagnosticados de Hemofilia Adquirida diagnosticados de Hemofilia Adquirida deben deben recibir tratamiento inmunosupresorrecibir tratamiento inmunosupresor lo antes posible, con el fin de lo antes posible, con el fin de erradicar el inhibidorerradicar el inhibidor
�Green & Lechner 1991: Mortalidad 22%; Hemorragias mayores 87%
�Grenn 1991: En pacientes no tratados → Mortalidad 64%
�Delgado et al 2003 (meta-análisis, 249 pacientes):
�Mortalidad sin inmunosupresión: 41%�Mortalidad con imnunosupresión: 20%
� Relacionada directamente con inhibidor: 11%� Relacionada con complicaciones del Tto. Inmunosupresor o coomorbilidades: 89%
� Lottenberg et al 1987: (16 pacientes): remisión espontánea del inhibidor: 36%
*International recomendations on the diagnosis and treatment of patients with acquired Hemofilia A. Haematologica 2009
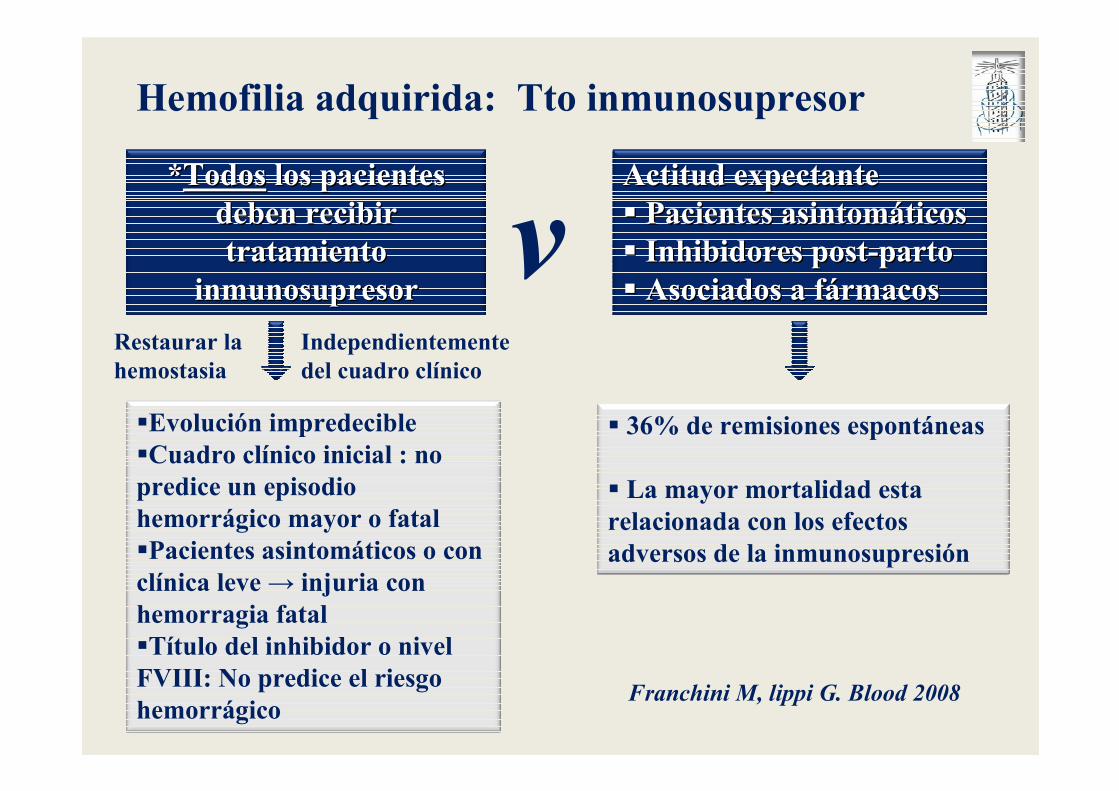
Hemofilia adquirida: Tto inmunosupresor
�Evolución impredecible�Cuadro clínico inicial : no predice un episodio hemorrágico mayor o fatal�Pacientes asintomáticos o con clínica leve → injuria con hemorragia fatal�Título del inhibidor o nivel FVIII: No predice el riesgo hemorrágico
**TodosTodos los pacientes los pacientes deben recibir deben recibir tratamiento tratamiento
inmunosupresorinmunosupresor
Actitud expectanteActitud expectante�� Pacientes asintomPacientes asintomááticosticos�� Inhibidores postInhibidores post--partoparto�� Asociados a fAsociados a fáármacosrmacos
vRestaurar la hemostasia
Independientemente del cuadro clínico
� 36% de remisiones espontáneas
� La mayor mortalidad esta relacionada con los efectos adversos de la inmunosupresión
Franchini M, lippi G. Blood 2008

Tto inmunosupresor: Estrategias terapéuticas
Primera lPrimera líínea de tratamientonea de tratamientoCorticosteroidesCorticosteroides + ciclofosfamida
Secunda lSecunda líínea de tratamientonea de tratamientoAnti-CD 20 ± corticosteroides
Tratamientos alternativosTratamientos alternativosAzatioprinaCiclosporinaMycofenolatoVincristina
No se recomienda No se recomienda Inmunoglobulinas intravenosas
Hemorragias que comprometan la vidaHemorragias que comprometan la vidaInmunoabsorciónInmunotolerancia
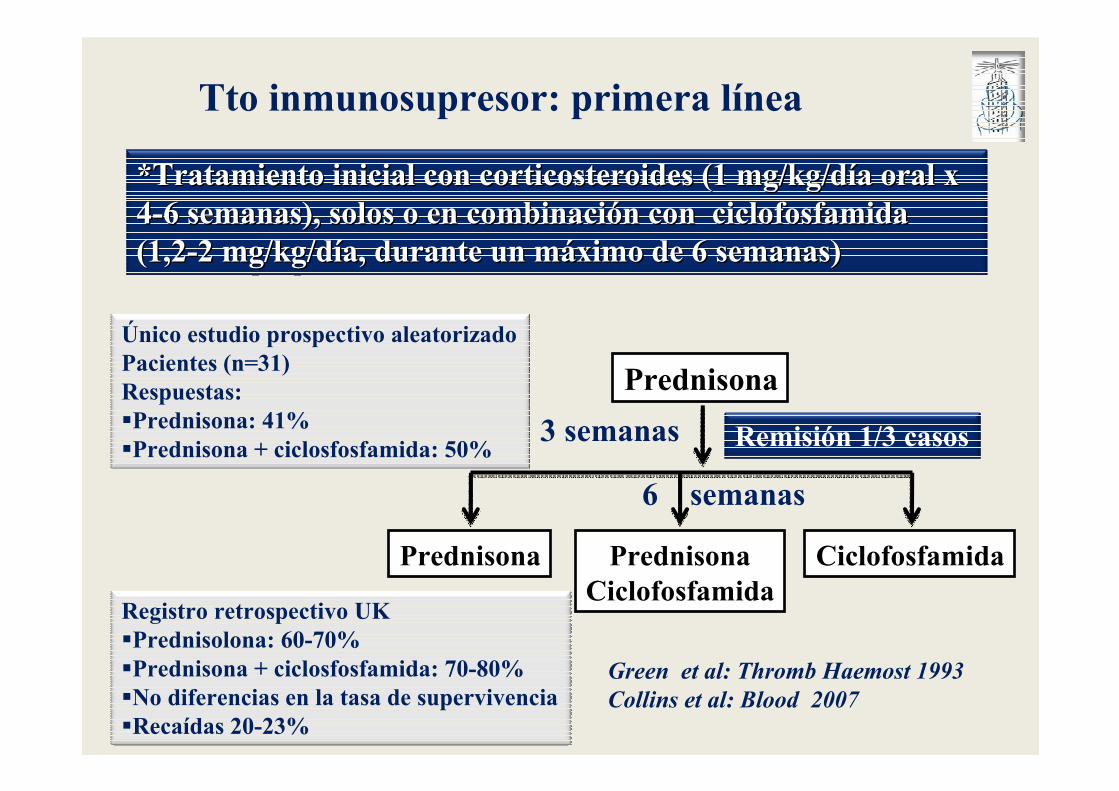
Tto inmunosupresor: primera línea
*Tratamiento inicial con corticosteroides (1 mg/kg/d*Tratamiento inicial con corticosteroides (1 mg/kg/díía oral x a oral x 44--6 semanas), solos o en combinaci6 semanas), solos o en combinacióón con ciclofosfamida n con ciclofosfamida (1,2(1,2--2 mg/kg/d2 mg/kg/díía, durante un ma, durante un mááximo de 6 semanas) ximo de 6 semanas)
Único estudio prospectivo aleatorizadoPacientes (n=31)Respuestas: �Prednisona: 41%�Prednisona + ciclosfosfamida: 50%
Prednisona
Prednisona PrednisonaCiclofosfamida
Ciclofosfamida
3 semanas Remisión 1/3 casos
6 semanas
Registro retrospectivo UK�Prednisolona: 60-70%�Prednisona + ciclosfosfamida: 70-80%�No diferencias en la tasa de supervivencia�Recaídas 20-23%
Green et al: Thromb Haemost 1993
Collins et al: Blood 2007

Agentes alquilantes: Efectos adversos
��Tasa de respuesta corticoides + ciclofosfamida superior a Tasa de respuesta corticoides + ciclofosfamida superior a corticosteroides solos corticosteroides solos ��Igual tasa de supervivenciaIgual tasa de supervivencia
Efectos adversos: 53%CitopeniasInfecciones relación neutropeniasHepatitis tóxicaCistitis hemorrágicasAlopecia Infertilidad
EACH 2008: infeccionesEACH 2008: infecciones11ªª causa mortalidad (12,2%)causa mortalidad (12,2%)
Tto individualizado� Pacientes ancianos→ Infecciones
� Inhibidores post-parto
Contraindicación� Fiebre alta de origen desconocido
� Sepsis o infección grave
� Enf. comprometan la vida
*Haematologica 2009

Tto inmunosupresor: segunda línea
*Anti CD 20: Rituximab*Anti CD 20: Rituximab®® (375 mg/m2/semana x 4 semanas) (375 mg/m2/semana x 4 semanas) ±± esteroides, si falla la primera lesteroides, si falla la primera líínea de tratamiento o esta nea de tratamiento o esta contraindicadacontraindicada
� Evidencia: casos clínicos o series cortas�No estudios prospectivos�Evidencia limitada pero prometedora�Pocos efectos adversos�Efectividad del Rituximab solo: No establecida�Mayoría de los casos asociado a esteroides u otros inmunosupresores
Sperr et al 2007
Meta-análisis (n=43)Remisiones:79%
¿¿Primera lPrimera líínea de tratamiento?nea de tratamiento?Futuro

Otros inmunosupresores
Ciclosporina (200Ciclosporina (200--300 mg/d300 mg/díía x 4 semanas) a x 4 semanas) ±± esteroidesesteroidesNiveles terapNiveles terapééuticos con monitorizaciuticos con monitorizacióón: 200n: 200--400 400 µµg/dLg/dL
�Muy eficaz en pacientes con Enf. Autoinmunes (LES)�Tasa de respuesta: ?? (casos clínicos o series muy limitadas)�Respuesta en 7-90 días�Toxicidad renal o hepática

Inmunoglobulinas
*No se recomienda el uso de Ig a altas dosis para la *No se recomienda el uso de Ig a altas dosis para la erradicacierradicacióón del inhibidor. n del inhibidor.
Ig como único tratamiento�Dosis convencionales (0,4 mg/kg/día x 5 días ó 1g/kg/día x 2 días�Tasa de respuesta: 10%
Ig asociada a inmunosupresores�Delgado et al: Br J Hamatol 2003: Meta-análisis�Collins et al: Blood 2007: registro UK�No beneficios� Asociado a complicacciones tromboembólicas� Sí: en programas de inmunotolerancia

*Sugieren el uso de inmunotolerancia + inmunoabsorci*Sugieren el uso de inmunotolerancia + inmunoabsorcióón n en pacientes con hemorragias que comprometan la vida o en pacientes con hemorragias que comprometan la vida o en el en el contexto de estudios clcontexto de estudios clíínicosnicos
Protocolos de Inmunotolerancia
� Evidencia muy limitada. Estudios preliminares�Protocolo Budapest: �FVIII (bajas dosis 30 UI/kg/día x 7 días, 20 UI/kg/d x7d, 15 UI/kg x 7 d) + prednisona + ciclofosfamida
�Tasa de respuesta: 90%
�Protocolo Bonn-Malmo modificado: �FVIII (100 UI/kg/d)+ prednisona + Ig + inmunoabsorción
�Tasa de respuesta: 88% en 14 días
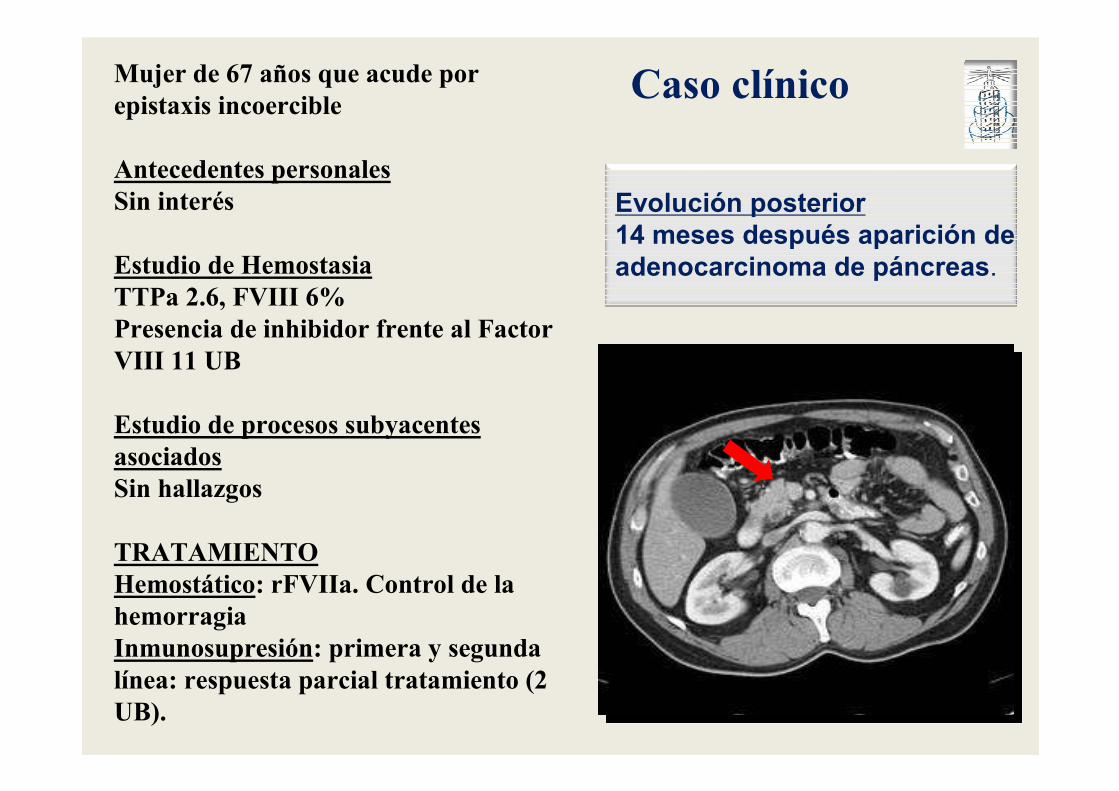
Mujer de 67 años que acude por epistaxis incoercible
Antecedentes personalesSin interés
Estudio de HemostasiaTTPa 2.6, FVIII 6%Presencia de inhibidor frente al Factor VIII 11 UB
Estudio de procesos subyacentes asociadosSin hallazgos
TRATAMIENTOHemostático: rFVIIa. Control de la hemorragiaInmunosupresión: primera y segunda línea: respuesta parcial tratamiento (2 UB).
Evolución posterior
14 meses después aparición de
adenocarcinoma de páncreas.
Caso clínico
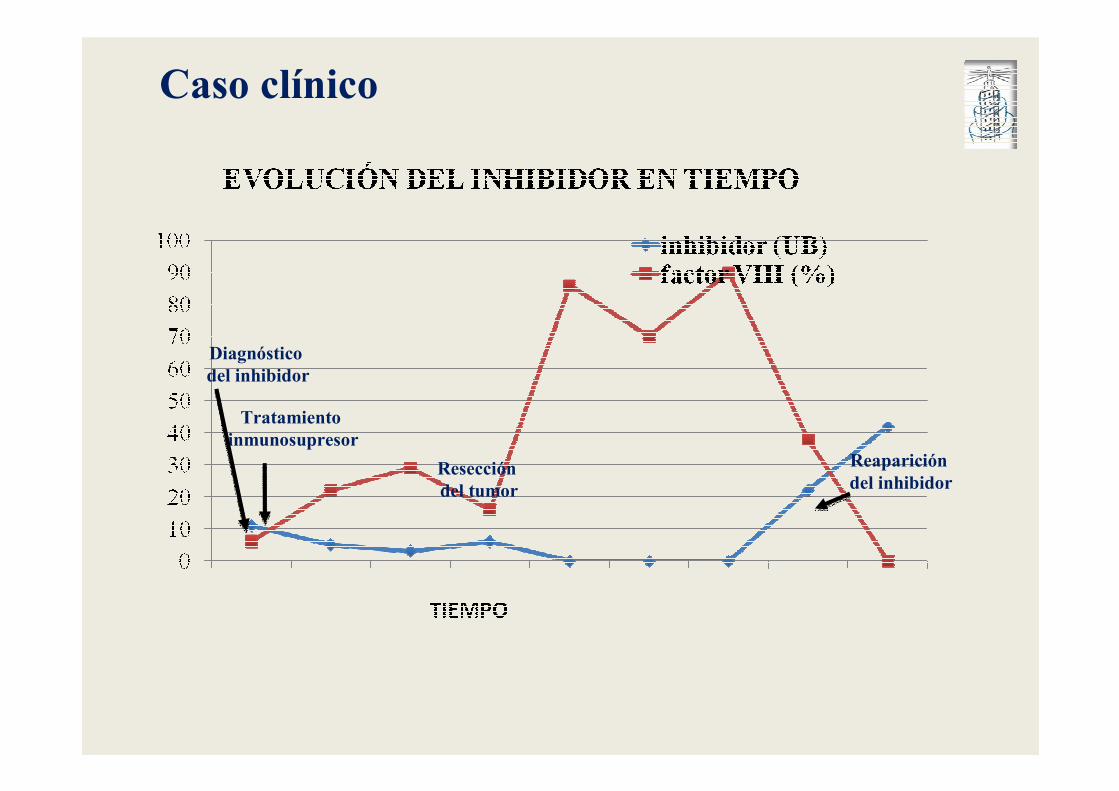
Diagnóstico del inhibidor
Tratamiento inmunosupresor
Resección del tumor
Reaparición del inhibidor
Caso clínico
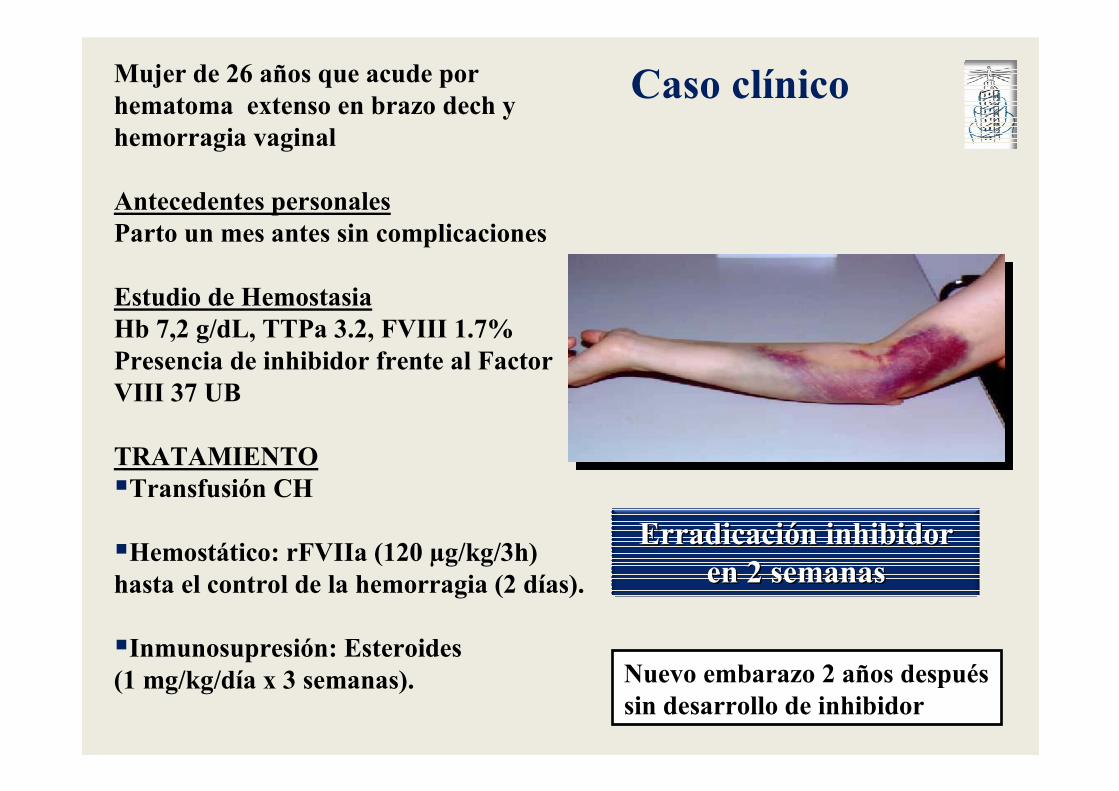
Mujer de 26 años que acude por hematoma extenso en brazo dech y hemorragia vaginal
Antecedentes personalesParto un mes antes sin complicaciones
Estudio de HemostasiaHb 7,2 g/dL, TTPa 3.2, FVIII 1.7%Presencia de inhibidor frente al Factor VIII 37 UB
TRATAMIENTO�Transfusión CH
�Hemostático: rFVIIa (120 µg/kg/3h) hasta el control de la hemorragia (2 días).
�Inmunosupresión: Esteroides (1 mg/kg/día x 3 semanas).
Caso clínico
ErradicaciErradicacióón inhibidor n inhibidor en 2 semanasen 2 semanas
Nuevo embarazo 2 años después sin desarrollo de inhibidor

Síndrome de von Willebrand adquirido
¿Desorden extremadamente raro o desconocido?

SVWaSVWa
� Síndrome raro
� Probablemente infradignosticado
� Similar a la EVW hereditaria
�Cuadro clínico y severidad
�Pruebas de laboratorio
�Pacientes adultos sin historia previa personal o familiar de
diátesis hemorrágica
� Asociada a múltiples procesos
� Clínica expresión de alteración en hemostasia primaria:
�Hemorragias mucocutáneas o gastrointestinales (o ambas)
� La mayoría asintomáticos: con hemorragias en relación
con traumatismos, cirugías, procedimientos invasivos etc
Federici et al: J Thromb Haemost 2008
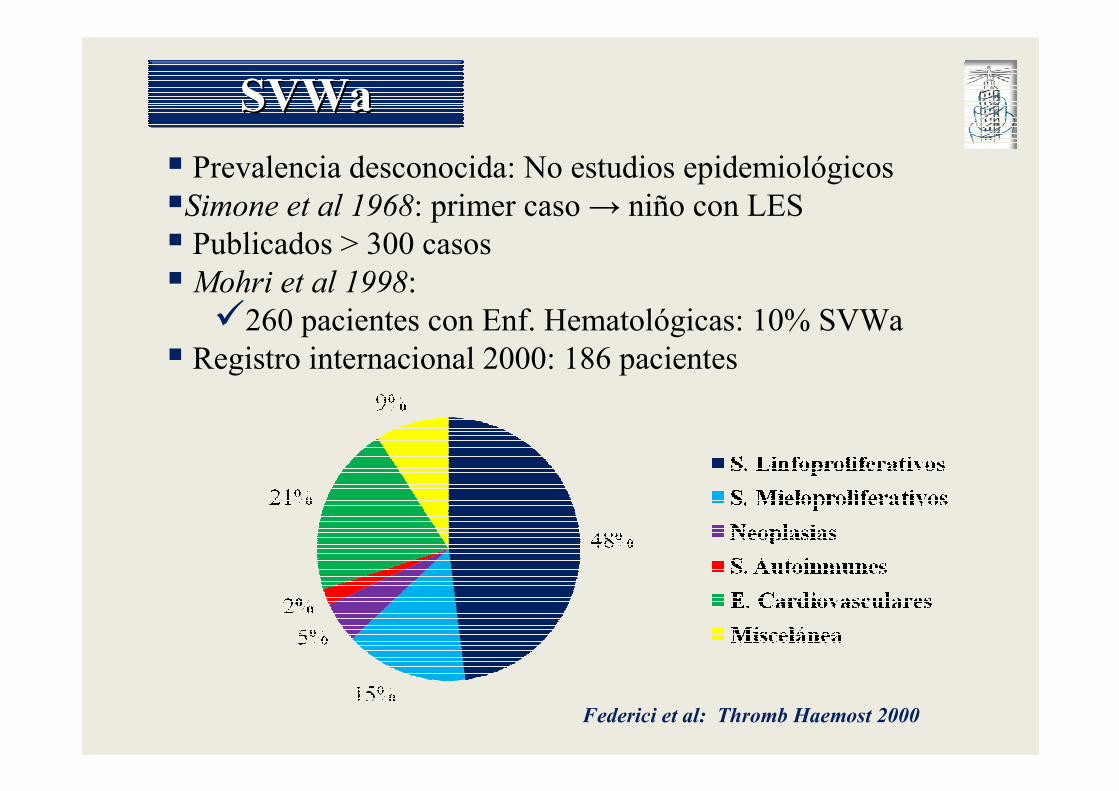
SVWaSVWa
� Prevalencia desconocida: No estudios epidemiológicos
�Simone et al 1968: primer caso → niño con LES
� Publicados > 300 casos
� Mohri et al 1998:�260 pacientes con Enf. Hematológicas: 10% SVWa
� Registro internacional 2000: 186 pacientes
Federici et al: Thromb Haemost 2000
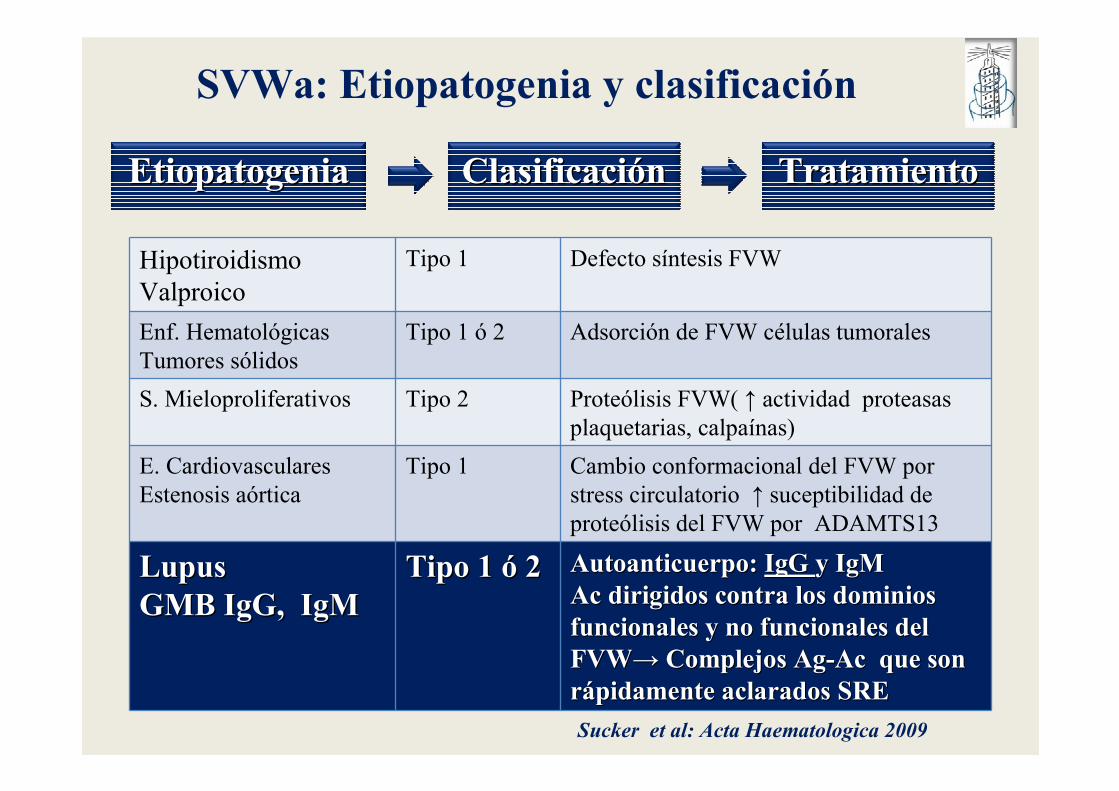
SVWa: Etiopatogenia y clasificación
EtiopatogeniaEtiopatogenia ClasificaciClasificacióónn TratamientoTratamiento
Hipotiroidismo
Valproico
Tipo 1 Defecto síntesis FVW
Enf. Hematológicas
Tumores sólidos
Tipo 1 ó 2 Adsorción de FVW células tumorales
S. Mieloproliferativos Tipo 2 Proteólisis FVW( ↑ actividad proteasas
plaquetarias, calpaínas)
E. Cardiovasculares
Estenosis aórtica
Tipo 1 Cambio conformacional del FVW por
stress circulatorio ↑ suceptibilidad de
proteólisis del FVW por ADAMTS13
LupusLupusGMB IgG, IgMGMB IgG, IgM
Tipo 1 Tipo 1 óó 22 Autoanticuerpo: Autoanticuerpo: IgG IgG y IgMy IgMAc dirigidos contra los dominios Ac dirigidos contra los dominios funcionales y no funcionales del funcionales y no funcionales del FVWFVW→→ Complejos AgComplejos Ag--Ac que son Ac que son rráápidamente aclarados SREpidamente aclarados SRE
Sucker et al: Acta Haematologica 2009
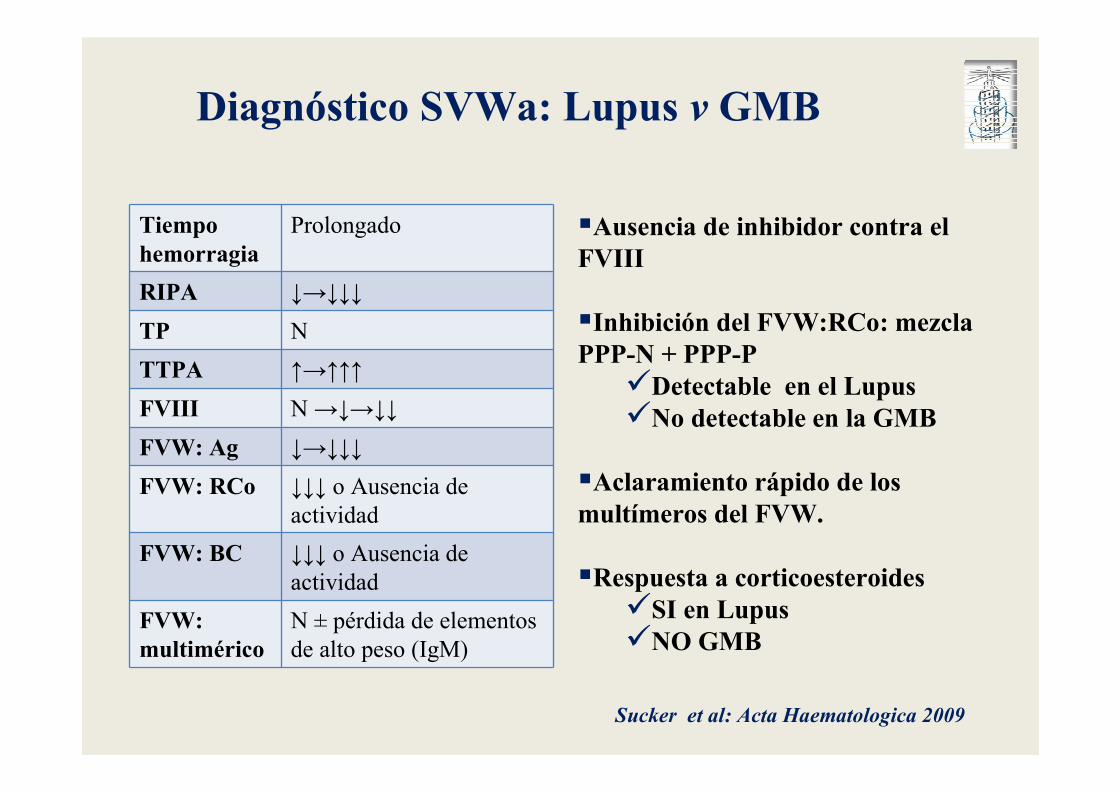
Tiempo hemorragia
Prolongado
RIPA ↓→↓↓↓
TP N
TTPA ↑→↑↑↑
FVIII N →↓→↓↓
FVW: Ag ↓→↓↓↓
FVW: RCo ↓↓↓ o Ausencia de
actividad
FVW: BC ↓↓↓ o Ausencia de
actividad
FVW: multimérico
N ± pérdida de elementos
de alto peso (IgM)
Diagnóstico SVWa: Lupus v GMB
�Ausencia de inhibidor contra el FVIII
�Inhibición del FVW:RCo: mezcla PPP-N + PPP-P
�Detectable en el Lupus�No detectable en la GMB
�Aclaramiento rápido de los multímeros del FVW.
�Respuesta a corticoesteroides�SI en Lupus�NO GMB
Sucker et al: Acta Haematologica 2009
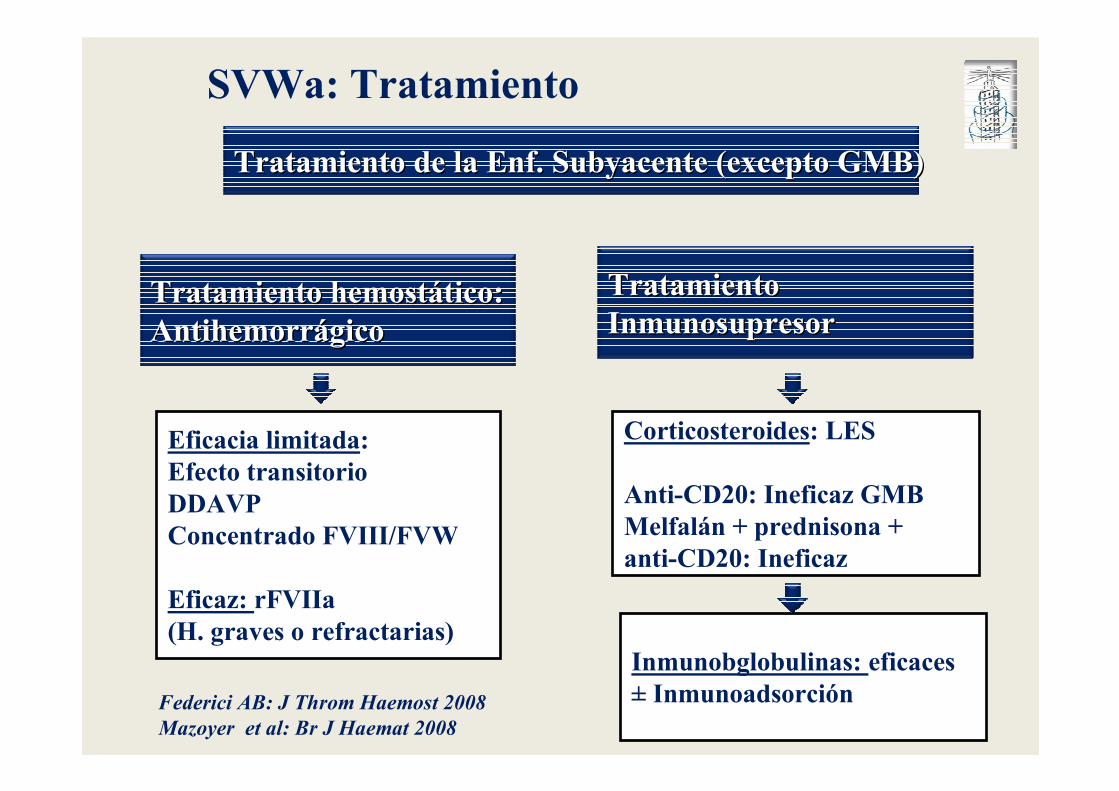
Tratamiento Tratamiento InmunosupresorInmunosupresor
SVWa: Tratamiento
Tratamiento hemostTratamiento hemostáático:tico:AntihemorrAntihemorráágicogico
Tratamiento de la Enf. Subyacente (excepto GMB)Tratamiento de la Enf. Subyacente (excepto GMB)
Eficacia limitada:Efecto transitorioDDAVPConcentrado FVIII/FVW
Eficaz: rFVIIa (H. graves o refractarias)
Corticosteroides: LES
Anti-CD20: Ineficaz GMBMelfalán + prednisona + anti-CD20: Ineficaz
Inmunobglobulinas: eficaces± InmunoadsorciónFederici AB: J Throm Haemost 2008
Mazoyer et al: Br J Haemat 2008
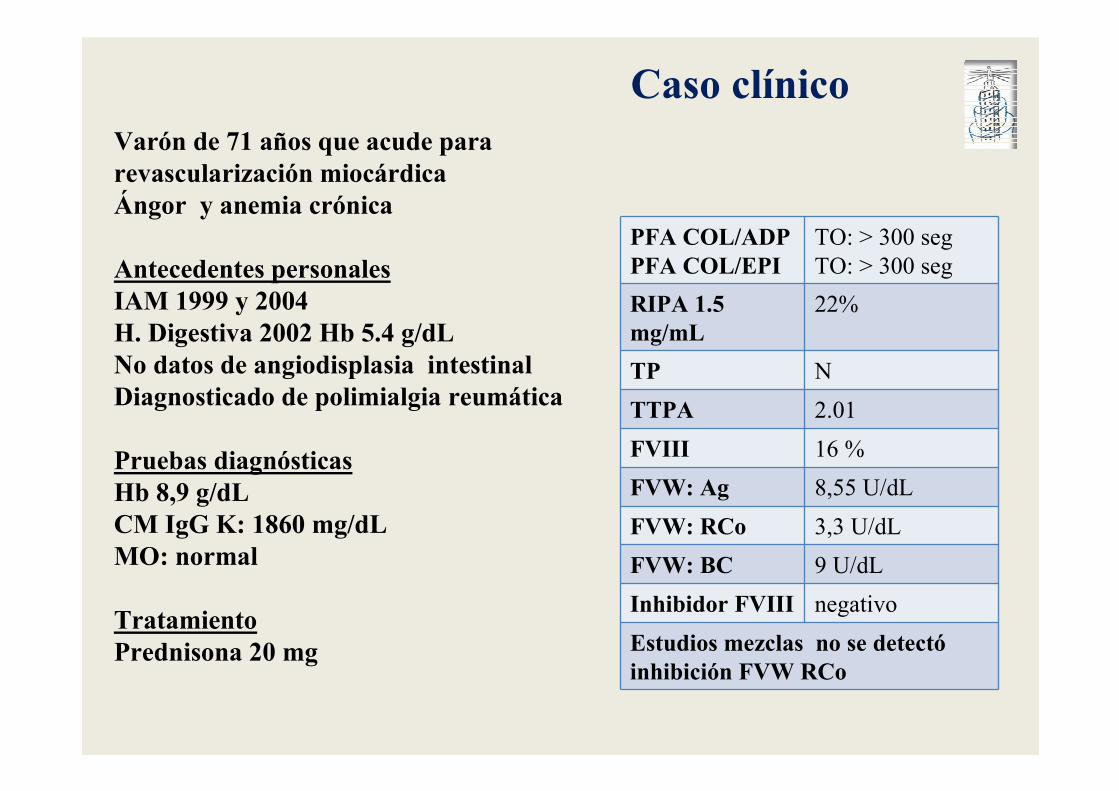
Varón de 71 años que acude para revascularización miocárdicaÁngor y anemia crónica
Antecedentes personalesIAM 1999 y 2004H. Digestiva 2002 Hb 5.4 g/dLNo datos de angiodisplasia intestinalDiagnosticado de polimialgia reumática
Pruebas diagnósticasHb 8,9 g/dLCM IgG K: 1860 mg/dLMO: normal
TratamientoPrednisona 20 mg
Caso clínico
PFA COL/ADPPFA COL/EPI
TO: > 300 seg
TO: > 300 seg
RIPA 1.5 mg/mL
22%
TP N
TTPA 2.01
FVIII 16 %
FVW: Ag 8,55 U/dL
FVW: RCo 3,3 U/dL
FVW: BC 9 U/dL
Inhibidor FVIII negativo
Estudios mezclas no se detectóinhibición FVW RCo
DAKO A0082
1/16 1/4 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/1 1/4
PP 1
PP 1: Plasma paciente: 10060037/04
PN: ELF (11/05)
HP: Haemate P 1U/mL
EVW 3+aloAc
PN
DAKO A0082
Pool 1.06 HP 1/1 1/2 HP 1/1 1/2 HP 1/1 1/2 HP 1/1 1/2 HP HP 1 U Pool 1.06 1 U Pool 1.06 1 U Pool 1.06 1 U Pool 3.05 1 U 1 U
24.03.06
PN 11.05 PN 10.05 PN 11.05
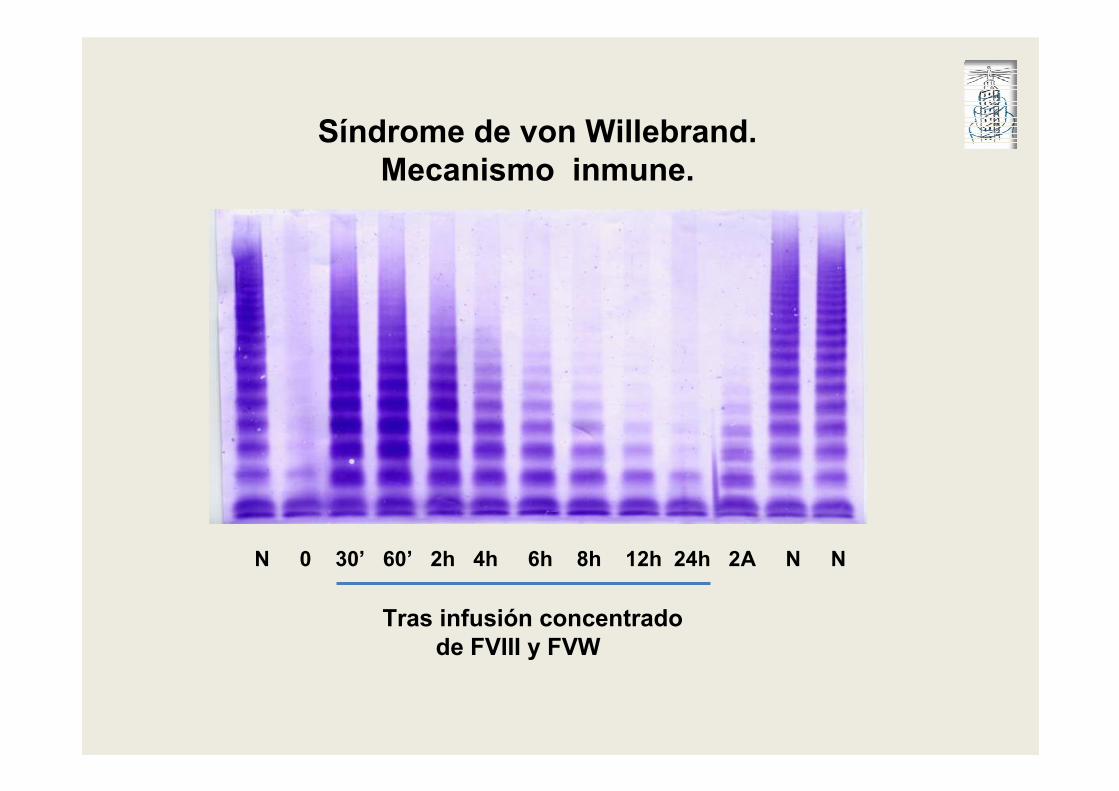
N 0 30’ 60’ 2h 4h 6h 8h 12h 24h 2A N N
Tras infusión concentrado
de FVIII y FVW
Síndrome de von Willebrand.
Mecanismo inmune.
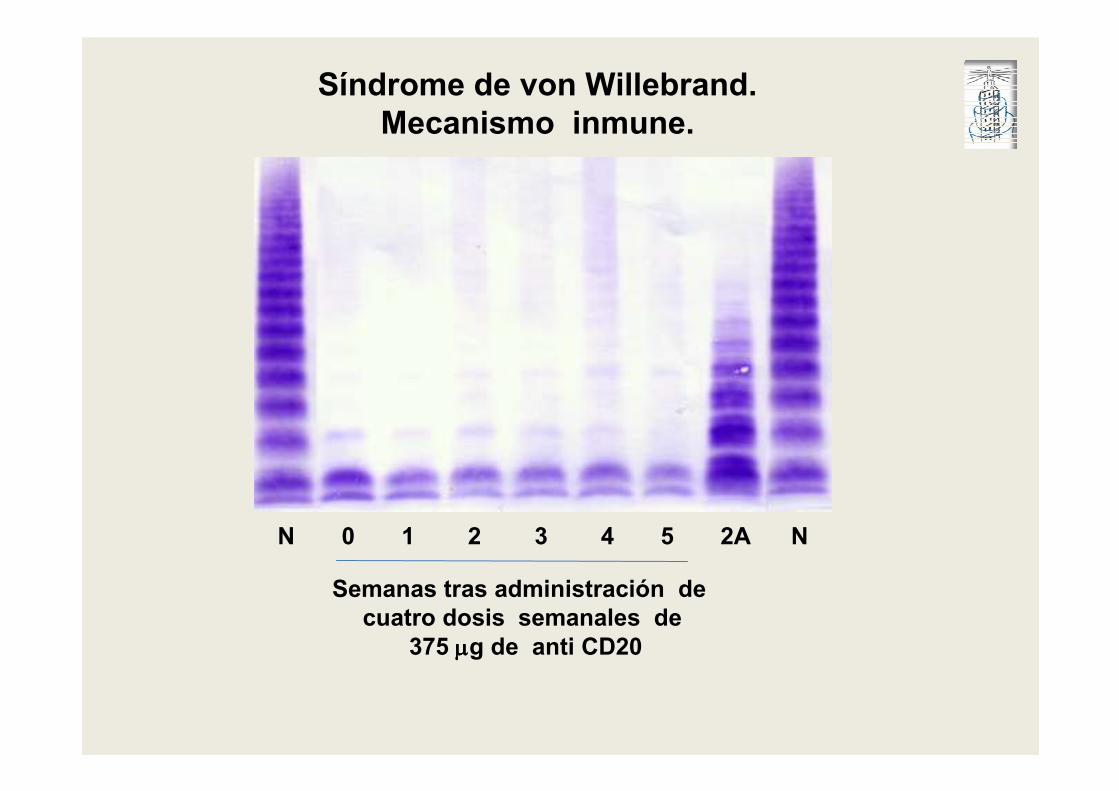
N 0 1 2 3 4 5 2A N
Semanas tras administración de
cuatro dosis semanales de
375 µµµµg de anti CD20
Síndrome de von Willebrand.
Mecanismo inmune.
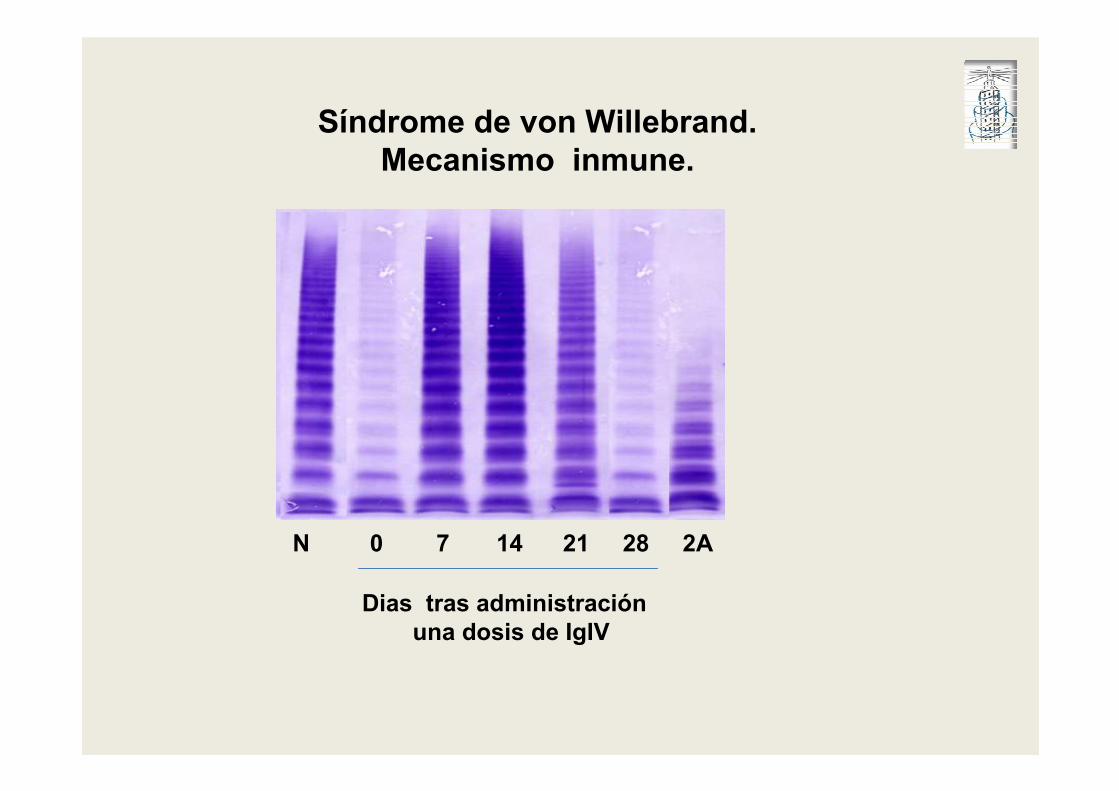
N 0 7 14 21 28 2A
Dias tras administración
una dosis de IgIV
Síndrome de von Willebrand.
Mecanismo inmune.

Agradecimientos
Dr Batlle
Pilar EcheverríaTeresa IglesiasPilar GestalJulia Carnero
Almudena PérezEster LourésÁngela Rodríguez Trillo
Joana Costa Pinto





























