Embed Size (px)
Citation preview

Corso di laurea in Scienze Biotecnologiche e Veterinarie
Tecnologia di coltura cellulare A.A. 2007/2008
A cura di: Federica Cheli – Eleonora Fusi Dipartimento di Scienze e Tecnologie Veterinarie per la
Sicurezza Alimentare Università degli Studi di Milano

PREMESSA
Le colture cellulari hanno acquisito negli ultimi anni un ruolo sempre più
preponderante all’interno della ricerca scientifica.
Sebbene le colture cellulari di origine animale fossero state impiegate
da Ross Harrison nel 1907, non è che nell’ultima metà degli anni 40 e
dei primi anni 50 che si attuarono forti sviluppi, che fecero in modo che
le colture cellulari fossero ampiamente disponibili come strumento per i
ricercatori. Primo ci fu uno sviluppo degli antibiotici, che consentì di
evitare facilmente molti dei problemi di contaminazione, che
affliggevano i primi tentativi di tenere le colture cellulari. Secondo ci fu
uno sviluppo di tecniche, come l’uso della tripsina per staccare le cellule
dai supporti di coltivazione, necessario per ottenere linee cellulari in
crescita continua. Terzo, usando queste linee cellulari i ricercatori
furono in grado di sviluppare terreni (media) di coltivazione
standardizzati, che rese molto più facile far crescere le cellule.
Queste sviluppi combinati consentirono a molti più ricercatori di usare
colture cellulari, tessutali o di organo nelle loro ricerche. Nel 1952 è
stata messa in coltura la prima linea cellulare stabilizzata umana,
proveniente da un tumore alla cervice: la linea HeLa.
Duranti gli anni 1960 e 1970, la commercializzazione di questa
tecnologia ha avuto un ulteriore impatto sulle colture cellulari che
continua sino ad oggi. Diverse compagnie hanno iniziato a sviluppare e
continuano sino ad ora a migliorare i materiali di plastica e di vetro dove
far crescere le cellule, i terreni di coltura in polvere o liquidi, la qualità
dei sieri, i fattori di crescita necessari alle diverse esigenze delle
tipologie cellulari.

TIPOLOGIA DELLE COLTURE CELLULARI
Che cosa è una coltura cellulare e una tessuto coltura o coltura
d’organo?
Tessuto coltura è il termine generico che viene impiegato per
identificare il prelievo di cellule, tessuti, o organi da piante o animali e il
loro conseguente posizionamento in un ambiente artificiale favorevole
alla crescita. Questo ambiente normalmente è costituito da materiale di
plastica, di vetro contenente un medium liquido o semisolido che
fornisce i nutrienti essenziali per la sopravvivenza e la crescita delle
cellule.
La coltura di un organo intatto o di parti di esso con lo scopo di studiare
il loro funzionamento o sviluppo viene denominata organo coltura.
Quando le cellule vengono rimosse dall’organo prima o durante la
coltivazione, distruggendo le normali relazioni esistenti con le cellule
vicine, danno vita a colture cellulari.
Come si ottengono le colture cellulari
COLTURE PRIMARIE
Quando le cellule vengono chirurgicamente rimosse dall’organismo e
poste in ambiente appropriato di coltura, esse si attaccheranno, si
divideranno e cresceranno. Questa viene definita coltura primaria.
Esistono fondamentalmente due sistemi per ottenere le colture
primarie.
Gli espianti di coltura sono piccoli pezzi di tessuti posti in idonei
contenitori di vetro o di plastica trattata a cui aderiscono e sono
mantenuti in terreni adatti. Dopo pochi giorni, le singole cellule si
muoveranno dall’espianto tissutale sulla superficie o sul substrato
presente nei contenitori, dove inizieranno a crescere e poi a dividersi.

Il secondo metodo molto più utilizzato, accelera questo processo
attraverso l’addizione di enzimi di digestione (proteolitici), come la
tripsina e la collagenasi, che consentono il dissolvimento del “cemento”
che unisce le cellule all’interno dei frammenti di tessuto. Questo
processo crea una sospensione di singole cellule che poste nei
contenitori idonei contenenti il medium di coltura possono aderire,
crescere e dividersi. Questo metodo è chiamato dissociazione
enzimatica.
Tali colture sono inizialmente eterogenee, ma più tardi saranno
dominate dalla tipologia fibroblastica, se non si provvede ad allontanare
questo tipo di cellula durante la preparazione.
La preparazione di colture primarie richiede un lavoro intenso e
laborioso. Queste possono essere mantenute in vitro solo per un
periodo limitato di tempo. Durante la loro vita limitata, queste cellule
primarie mantengono molte delle caratteristiche delle cellule presenti in
vivo.
SUBCOLTURA
Quando le cellule della coltura primaria nella flask sono cresciute e
hanno riempito tutto lo spazio disponibile, esse devono essere
sottoposte a subcoltura, per dar loro modo di poter continuare a
crescere e a replicarsi. Questo viene di solito fatto rimuovendo le cellule
molto delicatamente dal substrato, al quale sono adese, con gli enzimi.
Questi ultimi sono simili a quelli utilizzati nel corso dell’isolamento delle
cellule primarie e vengono utilizzati per rompere i legami proteici che
consentono l’adesione al substrato. Alcune linee cellulari possono essere
raccolte attraverso un delicato raschiamento delle stesse dal
contenitore. Una volta staccate, la sospensione cellulare può essere
suddivisa e posta in nuovi contenitori (piastre di Petri o flasks). Se fosse
presente un surplus di cellule, queste possono essere trattate con
agenti crioprotettivi adeguati, come il dimetilsulfossido (DMSO) o il

glicerolo, e congelate e mantenute a temperature criogeniche (sotto -
130°C) fino al loro successivo utilizzo.
ISOLAMENTO ORGANO
FRAMMENTAZIONE MECCANICA
FRAMMENTAZIONE
ENZIMATICA
ISOLAMENTO CELLULE

COLTURE CONTINUE
Le colture continue sono costituite da un singolo tipo di cellule che può
essere replicato in coltura sia per un numero limitato di divisioni
cellulari (approssimativamente 30) sia indefinitamente. Le linee cellulari
a vita determinata sono normalmente diploidi e mantengono un certo
grado di differenziazione. Il fatto che certe linee cellulari invecchia dopo
circa 30 cicli di divisione significa che è essenziale stabilire un sistema di
banche, al fine di mantenere queste linee per lunghi periodi.
Le linee cellulari continue che possono propagarsi indefinitamente
generalmente possiedono questa capacità poiché sono state trasformate
in cellule tumorali. Le linee cellulari tumorali spesso derivano da
isolamenti provenienti da neoplasie cliniche, ma la trasformazione può
essere tuttavia indotta utilizzando oncogeni virali o attraverso
trattamenti chimici.
Le linee cellulari trasformate presenta alcuni vantaggi quali la
disponibilità illimitata, ma di contro mantengono molto poco delle
caratteristiche presenti in vivo.
MORFOLOGIA DELLE COLTURE CELLULARI
Morfologicamente le colture cellulari possono crescere in sospensione
(come singole cellule o come piccoli clumps fluttuanti) o come
monostrato adeso al substrato costitutivo dei contenitori per la crescita.
La forma presa da una linea cellulare riflette il tessuto da cui ha preso
origine.
Es. linee cellulari derivate dal sangue (leucemia, linfoma) tendono a
crescere in sospensione, mentre le cellule isolate da tessuti solidi
(polmoni, reni) tendono a crescere come monostrato.
Ci sono casi in cui le colture cellulari possono crescere come semi-
aderenti (es. B95-8) dove appaiono costituiti da una popolazione mista
di cellule adese ed in sospensione. Per queste linee cellulari è essenziale

che entrambi i tipi cellulari vengano subcoltivati per mantenere la
natura eterogenea della coltura.
Tabella 1: Linee cellulari comunemente utilizzate
Attached Cell Lines
Name Species and tissue of origin Morphology
MRC-5 Human lung Fibroblast
HELA Human cervix Epithelial
VERO African Green Monkey Kidney Epithelial
NIH 3T3 Mouse embryo Fibroblast
L929 Mouse connective tissue Fibroblast
CHO Chinese Hamster Ovary Fibroblast
BHK-21 Syrian Hamster Kidney Fibroblast
LLC-PK1 Porcine Kidney Epithelial
HEPG2 Human Liver Epithelial
BAE-1 Bovine aorta Endothelial
Suspension Cell Lines
Name Species and tissue of origin Morphology
NSO Mouse myeloma Lymphoblastoid-like
U937 Human Hystiocytic Lymphoma Lymphoblastoid
Namalwa Human Lymphoma Lymphoblastoid
HL60 Human Leukaemia Lymphoblastoid-like
WEHI 231 Mouse B-cell Lymphoma Lymphoblastoid
YAC 1 Mouse Lymphoma Lymphoblastoid
U 266B1 Human Myeloma Lymphoblastoid
SH-SY5Y Human neuroblastoma Neuroblast

Banche cellulari (website e indirizzo):
http://www.nccc.com/
National Cell Culture Center
Mark Hirschel, Ph.D., Director
National Cell Culture Center
Scott Waniger, Manager
National Cell Culture Center
Phone: 800-325-1112 or 763-786-0302
FAX: 763-786-0915
Email: [email protected]
http://www.atcc.org/
American Type Culture Collection
12301 Parklawn Drive
Rockville Maryland
USA 20852
Sezione Italy
LGC Promochem
Via Venezia, 23
20099 Sesto San Giovanni, MI
Italy
E-mail: [email protected]
Tel: 39 02 24126830
Fax: 39 02 24126831
http://www.lgcpromochem-atcc.com/
http://www.ecacc.org.uk/
European Collection of Animal Cell Culture
Division of Biologics
PHLS Centre for Applied Microbiology & Research
Porton Down

Salisbury SP4 OJG
UK
http://www.bs.izs.it/servizi_db_cellule.htm#Centro%20Substr
ati%20Cellulari
Centro Substrati Cellulari
Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia
Romagna
Via Bianchi 9
25124 Brescia
Italia

TIPI DI COLTURE CELLULARI
COLTURE D’ORGANO
COLTURE CELLULARI:
COLTURE PRIMARIE
LINEE CELLULARI
La coltura primaria è una coltura originata da cellule o tessuti isolati
direttamente dall’organismo e mantenuta almeno per 24 ore.
La linea cellulare si ottiene dalla coltura primaria dopo la prima
subcoltura e può essere finita o continua.
Coltura finita: alcuni tipi di cellule normali (es. linfociti, cheratinociti,
fibroblasti, cellule endoteliali, …) possono essere coltivati in vitro per
moltiplicazione cellulare per un periodo di tempo limitato e per un
numero totale di divisioni cellulari limitato. Tali colture, in genere,
richiedono l’apporto di opportuni fattori di crescita per stimolare la
moltiplicazione cellulare.
Coltura continua: colture primarie di cellule trasformate in vivo (es.
tumori) o in vitro (es. per infezione con virus come EBV) possono dare
origine a linee cellulari caratterizzate dalla capacità di crescita
autonoma e illimitata in coltura. Tali cellule in genere non necessitano di
fattori di crescita.
Clone: è una popolazione cellulare derivata per mitosi da una singola
cellula.
Colture in sospensione (es. linfociti e linee trasformate da esse
derivanti).

Colture in monostrato (es. fibroblasti, macrofagi, epatociti, endotelio o
linee di tumori solidi).

SENESCENZA O SVILUPPO DI LINEE CONTINUE
Le linee cellulari possono essere mantenute inalterate per un limitato
numero di generazioni, superato il quale possono imboccare la strada
della senescenza e morte o divenire una linea continua.
La vita finita di una linea cellulare non è in relazione al tempo
cronologico di coltura, ma con il numero di duplicazioni cellulari. La
capacità di originare una linea continua da una coltura primaria dipende
da diversi fattori:
instabilità genetica del tipo cellulare utilizzato;
capacità di divenire indipendente da fattori di crescita estranei;
capacità di superare l’inibizione da contatto;
tendenza a sviluppare mutazione genetiche che condiziona i geni
coinvolti nella regolazione della proliferazione cellulare;
infezione con virus immortalizzanti (es. EBV).
Trasformazione neoplastica in vitro
Le linee cellulari possono essere mantenute inalterate per un limitato
numero di generazioni, superato il quale possono andare incontro a
morte

EVOLUZIONE DI UNA LINEA CELLULARE
a. EVOLUZIONE DI UNA LINEA NORMALE – Tipica delle cellule
umane. La fase I corrisponde al progressivo adattamento alle
condizioni in vitro della coltura primaria, seguita dalla fase II di
proliferazione a una velocità costante e dalla fase III di
senescenza cellulare, che porta alla morte di tutte le cellule.
b. IMMORTALIZZAZIONE SPONTANEA – Evento frequente nelle
cellule di roditore. Una coltura primaria che non si adatta alle
condizioni di coltura dando origine ad una linea cellulare, va
solitamente incontro ad una iniziale perdita della capacità
proliferativa ed entra in una fase di crisi in cui muoiono la maggior
parte delle cellule. Da questa fase, possono emergere delle cellule
selezionate per la capacità proliferativa, che daranno origine ad
una linea continua immortalizzata.

SVILUPPO DI UNA LINEA CONTINUA DA UNA COLTURA
PRIMARIA
ACQUISIZIONE DI NUOVE PROPRIETA’
Alterata morfologia cellulare
Perdita dell’inibizione da contatto
Perdita dell’inibizione densità-dipendente
Aumento della densità da saturazione
Perdita della dipendenza da ancoraggio
Modificazione del fenotipo
Capacità di andare incontro a numero illimitato di divisioni
Alterazioni del cariotipo
Acquisizione di capacità tumorigeniche
Capacità di crescita in terreni semi-solidi
Modificazione dello stato di differenziamento

COLTURE IN MONOSTRATO O IN SOSPENSIONE
MONOSTRATO
VANTAGGI
Facilità di studio di modificazioni morfologiche in
seguito a trattamenti con fattori biologicamente attivi
Utilizzazione di metodiche citochimiche e
immunoistochimiche direttamente sul supporto di
crescita
Studio di agenti con attività trasformanti
SVANTAGGI
Superficie di crescita correlata alla quantità di cellule
necessarie (possibilità di utilizzo di microcarriers)
SOSPENSIONE
VANTAGGI
Volume di crescita correlato con la quantità necessaria
Ridotte manipolazioni per l’utilizzazione sperimentale
SVANTAGGI
Vedi sopra monostrato

UTILIZZAZIONE DELLE COLTURE CELLULARI
APPLICAZIONI PRINCIPALI
Identificazione e quantizzazione di infezioni virali
Studio degli effetti tossici sia di composti farmaceutici che di
potenziali inquinanti
Produzione a scopi commerciali o scientifici di quantitativi di
fattori biologicamente attivi
Studio della risposta immunitaria verso antigeni ambientali o
antigeni espressi da cellule trasformate
Supporto alla crescita di altri tipi cellulari, studio del
differenziamento
Espressione di geni di cellule di eucariote
Studio dell’attività biologica di oncogeni
Studio di alterazioni metaboliche e cromosomiche e di malattie
genetiche
Campi d’uso
Controllo della crescita e del metabolismo, analisi
del differenziamento
Biologia cellulare
Ingegneria tissutale (coltura e produzione in vitro
di porzioni di tessuto: es. epidermide)
Biologia cellulare
Studio dei promoter e degli enhancer, di
espressione genica e di interi genomi
Biologia molecolare
Analisi dei mutanti e test di complementazione.
Mappaggio, trasfezione con DNA esogeno
Genetica
Diagnosi cliniche ( anche prenatali) cariologiche Citogenetica
Caratterizzazione di tumori Oncologia
Test di citotossicità Farmacologia
Produzione di proteine ricombinanti e di anticorpi
monoclonali
Immunologia

Le colture cellulari rappresentano dei metodi biologici alternativi,
caratterizzati da vantaggi, quali ad esempio gli studi tossicologici per la
specie umana, e svantaggi legati alle stesse caratteristiche delle colture
(estrema semplificazione sperimentale).
VANTAGGI SVANTAGGI
Sistemi semplificati e altamente
riproducibili
(costituiscono un materiale di
studio abbondante e omogeneo)
Sistemi semplificati rispetto ad un
organismo integrato
(perdita dei rapporti tridimensionali
all’interno di un tessuto, perdita
delle interazioni tra cellule diverse
nell’ambito dello stesso organo)
Analisi dei meccanismi cellulari e
molecolari del fenomeno studiato
Condizione di esposizione alle
sostanze diversa da quella in vivo
Controllo ambientale (consentono
di esercitare un preciso controllo
delle condizioni di utilizzo: es.
numero di cellule, caratteristiche di
crescita e di fenotipo, pH,
temperatura, pressione osmotica,
composizione del terreno di
coltura, tensione di CO2….)
Difficoltà nella correlazione delle
concentrazioni in vitro con quelle in
vivo
Economicità e rapidità di risposta
(esposizioni a concentrazioni
“ridotte” rispetto alle prove in vivo,
risposte nell’arco di ore e giorni)
Possibili contaminazioni con altri
tipi cellulari o con virus o
micoplasmi
Disponibilità: banche pubbliche di
linee cellulari
Natura delle linee continue rispetto
alle cellule in vivo
(trasformazione neoplastica con
alterazione delle caratteristiche
tipiche del tessuto di origine,
instabilità del corredo

cromosomico)
Si possono ottenere cloni e varianti
della cellula originale
Esperienza degli operatori.
Tecniche di coltura e di
sperimentazione con linee cellulari,
attrezzatura di laboratorio,
prevenzione delle contaminazioni
Si possono congelare e recuperare
a distanza di anni
Si evita il sacrificio di animali

Tabella 2. Proprietà delle colture cellulari.
TIPO DI
PREPARAZIONE
SISTEMI
INTATTI
COLTURE
D’ORGANO ESPIANTI AGGREGATI
COLTURE
PRIMARIE
LINEE
CELLULARI
Rapporto con la
situazione in vivo
Nessuna
alterazione
Rimozione delle influenze
dell’organismo
Distribuzione delle relazioni
intercellulari
Perdita
controllo della
crescita
Livello di
organizzazione del
tessuto
++++ ++++ +++ ++ + 0
Riproducibilità ++++ ++++ +++ ++ + ++++
Accessibilità + ++ ++ +++ ++++ ++++
Manipolazioni
genetiche + + + + + +++
Alterazioni
genetiche per
mutazione o
selezione
+ + + Da + a ++++
Controllo
ambientale + ++ +++ ++++

AREA DI LAVORO ED EQUIPAGGIAMENTO PER IL LABORATORIO
DI COLTURE CELLULARI
Uno degli aspetti, che spesso vengono presi in considerazione solo
marginalmente durante gli studi con le colture cellulari, è la necessità di
progettare strutture che assicurino che la buona qualità degli stessi
studi (sicurezza, efficienza e riproducibilità). La maggior parte delle
colture cellulari vengono trattate in laboratori, che sono stati adattati a
questo scopo e in condizioni quindi non ideali. Ci sono diversi aspetti
che devono essere considerati per allestire un buon laboratorio di
colture cellulari. Idealmente, il lavoro dovrebbe essere condotto in una
singola struttura che, se possibile, dovrebbe essere suddivisa in un’area
riservata per il trattamento del materiale appena ricevuto (area di
quarantena) e un’area per il materiale riconosciuto libero da possibili
contaminanti (il vero e proprio laboratorio di colture cellulari). Se
questo non fosse possibile, il lavoro dovrebbe essere suddiviso durante
l’arco di tempo lavorativo, con la manipolazione del “materiale pulito”
completata prima di quella coinvolgente il materiale “sporco”.
Dovrebbero essere presenti diversi incubatori. Inoltre, le superfici di
lavoro dovrebbero essere pulite e deterse nel corso delle diverse
attività. Tutti i materiali nuovi (linee cellulari, terreni, ecc.) dovrebbero
essere considerati “sporchi, da quarantena” fintanto che non si
dimostrino privi di batteri, funghi e soprattutto micoplasmi. Lavorare
con le colture cellulari in un laboratorio condiviso richiede una precisa
programmazione ed è essenziale che le buone pratiche di laboratorio
siano applicate al fine di ridurre al minimo il rischio di contaminazione.
Per la maggior parte delle linee cellulari, il laboratorio dovrebbe
appartenere almeno alla Categoria 2 [in relazione alle linee guida
fornite da Advisory Committee on Dangerous Pathogens (ACDP), 1985]
[Advisory Committee on Dangerous Pathogens (1985) Categorization of
Biological Agents According to Hazard and Categories of Containment,
4th edition, HSE books, Sudbury, UK]. Ciò nonostante, la categoria

appropriata è in relazione alla linea cellulare e alla natura del lavoro
programmato.
Tabella 3: Attrezzatura minima di laboratorio
1. Cappe sterili a flusso laminare, a flusso verticale, tipo biohazard
2. Incubatori a temperatura, umidità e atmosfera controllati
3. Microscopi (diretto e invertito a contrasto di fase)
4. Centrifughe refrigerate e non
5. Bagno termostatico ad acqua
6 Frigoriferi a 4°C (per la conservazione dei terreni)
7. Congelatori a -80°C (per la conservazione dei sieri e degli altri
reagenti deperibili)
8. Contenitori criogenici a -196°C per la conservazione delle colture
cellulari
9. Sistema di pipettamento automatico per il trasferimento di
liquidi
10. Materiale plastico sterile monouso per colture cellulari
11. Vetreria autoclavabile
12 Disponibilità di gas metano, linee o pompe a vuoto, sistema di
filtrazione e sterilizzazione dell’aria ambiente
A. Cappa a flusso laminare. La cappa biologica rappresenta forse
la più importante attrezzatura in un laboratorio di colture cellulari
al fine di operare correttamente. Questa infatti fornirà una
superficie idonea per il lavoro e nello stesso tempo proteggerà
l’operatore dagli aerosols. Ci sono due tipi di cappa a flusso
laminare: verticale e orizzontale. La cappa a flusso verticale,
chiamata anche cappa biologica, è la migliore per lavorare con
microrganismi o sostanze pericolose, poiché gli aerosols che si
possono generare nella cappa vengono filtrati prima di essere

rilasciati nell’ambiente. Le cappe orizzontali sono allestite in modo
tale che l’aria fluisce direttamente all’operatore, dunque non sono
le migliori per il lavoro con gli organismi pericolosi ma sono la
migliori protezione per le colture. Entrambi i tipi di cappa hanno
un continuo flusso di aria, che passa attraverso i filtri HEPA (high
efficiency particle), che rimuovono il particolato dall’aria. In una
cappa verticale, l’aria filtrata fluisce dalla parte superiore della
cappa, in una cappa orizzontale l’aria filtrata fluisce in modo
orizzontale verso l’operatore. Il livello di contenimento fornito
varia in relazione alla classe di cappa utilizzata. Queste non sono
cappe chimiche e non dovrebbero essere utilizzate per chimici
volatili o esplosivi, e non dovrebbero essere impiegate per lavori
con batteri o funghi. Le cappe sono equipaggiate con lampade UV
(onda corta), che possono essere accese per alcuni minuti prima
del lavoro, al fine di sterilizzare la superficie della cappa. Si
dovrebbe però considerare che solamente le superficie esposte
sono accessibili ai raggi UV. Non introdurre le mani o esporre la
faccia ai raggi UV quando sono accesi, poiché le onde corte
possono causare lesioni agli occhi e alla pelle. La cappa dovrebbe
essere accesa almeno 20 minuti prima dell’uso. Pulire tutte le
superfici con etanolo prima e dopo l’uso. Mantenere la cappa
libera da ingombri, perché questo può interferire con il flusso di
aria laminare. Il monitoraggio ambientale con piastre contenenti
Tryptose Soya Broth Agar posizionate nella cappa per un minimo
di 4 ore dovrebbe essere un buon indicatore del grado di pulizia
della cappa. Non si dovrebbero registrare crescita di funghi o
batteri su queste piastre. Nella maggior parte dei casi una cappa
di II classe è adeguata per le colture cellulari di origine animale.
Nonostante ciò, ciascun studio dovrebbe essere valutato per i suoi
pericoli e rischi e tutti possibili fattori addizionali, come infezioni
virali o di provenienza sconosciuta, richiedendo un più alto livello
di contenimento.

FiguraXX: Cappa a flusso laminare verticale (Danish Institute of
Agricultural Sciences, Department of Animal Health, Welfare and
Nutrition, Lab. Cell culture – Dr. Stig Purup)
B. Incubatori a CO2. Le colture cellulari richiedono un ambiente
strettamente controllato nel quale crescere. Speciali incubatori
sono usati di routine al fine di fornire le corrette condizioni per la
crescita, quali la temperatura, il grado di umidità e i livelli di CO2.
Generalmente, gli incubatori possono essere settati a
temperature comprese in un range compreso fra 28°C (per le
linee isolate da insetti) e 37°C (per le linee isolate da
mammiferi), a fornire la CO2 al livello richiesto (5-10%). Infatti le
cellule crescono di solito in una atmosfera con 5-10% di CO2
poiché il terreno di coltura usato è tamponato con sodio
carbonato/acido carbonico e il pH deve essere strettamente
mantenuto. Le flasks per le colture cellulari dovrebbero avere il
tappo aperto (dotato di filtro, se chiuso) per consentire gli scambi

gassosi. Le cellule non dovrebbero essere lasciate fuori
dall’incubatore a lungo, e la porta dell’incubatore non dovrebbe
essere lasciata aperta per lunghi periodi. L’umidità dovrebbe
essere conservata per quelle cellule in crescita nelle piastre grazie
ad una vaschetta che deve essere controllata e riempita di acqua
costantemente. In questa vaschetta possono essere aggiunte
anche soluzioni germicide, ma questo non deve precludere una
regolare pulizia. Alcuni incubatori hanno la possibilità di
controllare anche il livello di O2. Altri poi possono essere rivestiti
da rame, infatti sembra che questo rivestimento possa ridurre la
contaminazione microbica per l’attività inibitoria sostenuta dallo
stesso.

FiguraXX: Incubatore a CO2 (Danish Institute of Agricultural Sciences,
Department of Animal Health, Welfare and Nutrition, Lab. Cell culture –
Dr. Stig Purup)
C. Microscopi. I microscopi ottici rovesciati sono utilizzati per
visualizzare le cellule viventi. I microscopi dovrebbero essere
coperti e la luce spenta quando non in uso. Prima di utilizzare il

microscopio o tutte le volte che viene cambiato l’obiettivo,
controllare che gli anelli di fase siano allineati.
D. Centrifughe. Le centrifughe sono usate di routine all’interno di
laboratori di colture cellulari, durante i processi di subcoltura e di
preparazione delle cellule per la crioconservazione. Per la loro
stessa natura, le centrifughe producono aerosols ed è perciò
necessario ridurre al minimo i rischi. Questo potrebbe essere
raggiunto acquistando modelli, che hanno i canestri sigillati.
Idealmente, la centrifuga dovrebbe avere un coperchio
trasparente così che le condizioni del contenuto possano essere
osservate senza aprire il coperchio. Questo riduce i rischi per
l’operatore di essere esposto a materiali pericolosi, se un
contenitore si rompe durante la centrifugazione. Bisognerebbe far
attenzione a non riempire troppo i contenitori e a bilanciarli
correttamente. Queste precauzioni dovrebbero ridurre i rischi per
la generazione di aerosols. La centrifuga dovrebbe essere situata
in un luogo di facile accesso e pulizia, dovrebbe essere controllata
spesso per eventuali segni di corrosione.
E. Contenitori. Le cellule che crescono adese in monostrato
richiedono una superficie non tossica, biologicamente inerte e
otticamente trasparente, che consenta loro di attaccarsi e
consenta loro movimenti necessari per la crescita. I contenitori
più convenienti sono quelli in plastica, polistirene trattato in modo
speciale, che risultano forniture sterili e sempre disponibili. Questi
includono diversi modelli le piastre di Petri, le piastre
multipozzetto, le micropiastre, le bottiglie, le flasks T-25, T-75, T-
150 (cm2 di area di superficie). Le cellule in sospensione vengono
mantenute in agitazione e possono crescere in contenitori idonei
o identici a quelli delle cellule che crescono adese.

Tabella: Alcuni dati utili per l’uso di flask nelle colture cellulari
FLASK DENSITA’ CELLULARE MEDIA VOLUME TERRENO
25 cm2 2*106 5-7.5 ml
75 cm2 7.5*106 15-22.5 ml
150 cm2 1.5*107 30-45 ml
175 cm2 1.75*107 35-52.5 ml
225 cm2 2.25*107 45-67.5 ml
DENSITA’ CELLULARE MEDIA = si considera come 100% di coltura confluente 1*105
cells/cm2
Tabella: Alcuni dati utili per l’uso di piastre di Petri nelle colture cellulari.
PETRI AREA DI
CRESCITA
DENSITA’
CELLULARE MEDIA
VOLUME
TERRENO
35 mm 8 cm2 8*105 1.6-2.4 ml
60 mm 21 cm2 2.1*106 4.2-6.3 ml
100 mm 55 cm2 5.5*106 10-15 ml
150 mm 148 cm2 1.48*107 30-45 ml
245 mm 500 cm2 5.0*107 100-150 ml
DENSITA’ CELLULARE MEDIA = si considera come 100% di coltura confluente 1*105
cells/cm2
F. Conservazione. Le cellule vengono conservate in appositi
contenitori con azoto liquido.
G. Superfici di lavoro e pavimenti. Al fine di mantenere un
ambiente di lavoro pulito, le superfici del laboratorio, compresi i
banconi, le pareti e i pavimenti dovrebbero essere lisci e facili da
pulire. Essi dovrebbero essere resistenti all’acqua e ad una
varietà di chimici (come acidi, alcali, solventi e disinfettanti).

Nell’area di stoccaggio del materiale con bidoni con azoto liquido,
il pavimento dovrebbe essere resistente a tale composto (se
dell’azoto liquido dovesse rovesciarsi e cadere a terra). Le
superfici di lavoro dovrebbero essere posizionate in modo tale da
rendere confortevole il lavoro.
FiguraXX: Termostato agitato (Danish Institute of Agricultural Science,
Animal Welfare, Lab. Cell culture – Dr. Stig Purup)

MANUTENZIONE E CURA DEI LABORATORI
Al fine di mantenere un ambiente di lavoro pulito e sicuro, la cura e la
pulizia sono i principali elementi da considerare, tutte le attenzioni
dovrebbero essere prese. Le pulizie di routine dovrebbero comprendere
tutte le superfici di lavoro (compresi interno/esterno della cappa), i
pavimenti e tutti gli strumenti presenti. Gli incubatori rappresentano
una potenziale area di sviluppo di batteri e funghi, essi rappresentano
un potenziale rischio, che potrà essere ridotto con la regolare pulizia
dell’incubatore. Le cappe biologiche dovrebbero essere controllate ogni
6 mesi per garantire la loro efficienza e sicurezza sia per i prodotti che
per gli operatori. I tests che vengono effettuati valutano i flussi dell’aria
e il corretto funzionamento dei filtri HEPA. La temperatura
dell’incubatori dovrebbe essere regolarmente controllata con un
campione NAMAS (National Accreditation of Measurement and
Sampling, UK), o con un termometro calibrato. Anche i livelli di CO2 e
O2 dovrebbero essere controllati periodicamente.

ASPETTI DI SICUREZZA ALL’INTERNO DI LABORATORI DI
COLTURE CELLULARI
VALUTAZIONE DEL RISCHIO
Lo scopo principale della valutazione del rischio è quello di prevenire
danni a persone e cose. La valutazione dei rischi deve essere effettuata
prima di iniziare qualsiasi attività. La valutazione consiste in due
elementi:
• Identificazione e valutazione dei rischi;
• Definizione dei modi per minimizzare o evitare questi rischi.
Le direttive inerenti alla salute e sicurezza sul lavoro sono presenti sul
sito dell’Agenzia Europea per la sicurezza e la salute sul lavoro
www.europe.osha.eu.int
Per le colture cellulari di origine animale il livello di rischio dipende dalla
linea che deve essere utilizzata e se questa è in grado di provocare
danni all’uomo.
RISCHIO BASSO
Linee cellulari continue non umane o derivate da primati; linee di
origine umana ben caratterizzate
RISCHIO MEDIO
Linee cellulari poco caratterizzate
RISCHIO ELEVATO
Linee cellulari di origine umana o derivate da primati o ematiche o
con patogeni endogeni
Fonte consultabile: Advisory Committee on Dangerous Pathogens
(1985) Categorization of Biological Agents According to Hazard and
Categories of Containment, 4th edition, HSE books, Sudbury, UK.

Una banca cellulare come l’ ECACC, raccomanda come minimo livello di
contenimento una volta data una linea cellulare la Categoria 2 (questo
per la maggior parte delle cellule). Questo livello potrebbe essere
accresciuto a Categoria 3 nel caso in cui si maneggiassero linee
particolari o grandi quantitativi. Per le linee cellulari isolate da pazienti
con HIV or HTLV il livello sale a 3. Il contenimento rappresenta il mezzo
più ovvio di contenimento del rischio. Altri mezzi meno ovvi possono
includere una restrizione dei movimenti dello staff e la movimentazione
fuori/dentro della strumentazione dai laboratori. La buona pratica di
laboratorio e le buone tecniche di base come delimitare le aree di lavoro
sono scontate, i reagenti devo essere contrassegnati, correttamente
etichettati e stoccati sempre per garantire un ambiente con rischi ridotti
al minimo e sicuro per gli operatori. La preparazione degli operatori e
l’uso di procedure e regole standard scritte potranno garantire una
riduzione dei rischi.

DISINFEZIONE
I metodi deputati alla disinfezione/decontaminazione dei rifiuti prodotti
dal laboratorio di colture cellulari, delle superfici di lavoro e delle
strumentazioni rappresenta un mezzo importante per abbattere i
pericoli.
Le principali categorie di disinfettanti:
1. Ipocloriti (es. cloro)
In generale sono dei buoni disinfettanti, attivi contro i virus,
ma corrosivi per i metalli e perciò non dovrebbero essere
utilizzati su tali superfici (es. centrifughe). Sono
rapidamente inattivati dal materiale organico e per questo
dovrebbero essere preparati quotidianamente. Dovrebbero
essere utilizzati a concentrazioni pari 1000 ppm (1000
mg/kg) per la disinfezione generale delle superfici, 2500
ppm per i rifiuti (es. pipette, puntali), 10000 ppm per i rifiuti
proventi dalle colture cellulari (es. medium e altri liquidi).
Attenzione ai processi di fumigature delle cappe: quando
vengono effettuati usando la formaldeide, tutti gli ipocloriti
dovrebbero essere rimossi dal locale poiché i due prodotti
chimici assieme possono dare origine a prodotti carcinogeni.
2. Fenoli (es. Sudol, Hycolin)
Non sono reattivi nei confronti dei virus, ma rimangono
attivi in presenza di materiale organico.
3. Alcool (es. etanolo, isopropanolo)
Le concentrazioni più efficaci sono date dal 70% per
l’etanolo, 60-70% per l’isopropanolo. Questi agiscono
mediante disidratazione e fissazione. Sono entrambi efficaci
nei confronti dei batteri: l’etanolo risulta attivo anche nei
confronti della maggior parte dei virus, ma non di quelli privi

di envelope; l’isopropanolo non è efficace nei confronti dei
virus.
4. Aldeidi (es. gluteraldeide, formaldeide)
Le aldeidi sono composti irritanti e il loro uso dovrebbe
essere limitato per i problemi di sensibilizzazione. La
gluteraldeide potrebbe essere impiegata in situazioni dove
l’uso dell’ipoclorito sconsigliato (es. pulizia della centrifuga o
di materiali costruiti in acciaio inossidabile che può essere
danneggiato e corrotto dall’uso di soluzioni di ipoclorito).
Gestione dei rifiuti
Ciascun frequentatore del laboratorio deve provvedere allo smaltimento
dei rifiuti in modo appropriato. Tutto il materiale di consumo (puntali,
piastre, pipette, ecc.) deve andare in bidone dei rifiuti speciali, nel
contenitore adeguato ci vanno aghi, lame per bisturi e materiale in
vetro (es. Pasteur). I liquidi come le saline possono essere smaltiti
seguendo le vie comuni (lavandino-scarico), mentre i liquidi chimici
devono essere smaltiti seguendo le regole generali (Bidoni di
smaltimento).
Es. Medium di coltura → inattivazione overnight con soluzione di
ipoclorito di sodio o forti agenti ossidanti secondo le indicazioni
presenti su MSDS.
Pipette contaminate → in attivazione con ipoclorito di sodio
overnight.
Rifiuti solidi → es. flasks, provette, ecc. meglio incenerire che
autoclavare.

ISOLAMENTO DI CELLULE DA FRAMMENTI DI TESSUTO
a. PRELIEVO
BIOPSIA O EXERESI CHIRURGICA
Il reperto viene prelevato sterilmente e posto in un apposito recipiente
contenente il terreno pre-riscaldato ed addizionato con siero fetale
bovino inattivato al calore (HI-FBS), glutammina 200 mM 1% e
antibiotici (Penicillina 100 IU/ml + Streptomicina 100 µg/ml o
Gentamicina 40 mg/ml).
Il frammento sarà stato preventivamente ripulito da grasso, parti
necrotiche o non tumorali, a seconda delle necessità.
LIQUIDO ASCITICO EFFUSIONE PLEURICA
I liquidi vengono prelevati sterilmente mediante siringa in apposite
bottiglie eparinizzate alla dose di 100 IU/ml. I fluidi vengono trattati tali
e quali o sottoposti a gradienti di Ficoll-Hypaque per eliminare le cellule
non neoplastiche e/o morte.
SANGUE EPARINATO
b. PROCEDIMENTO
DISSOCIAZIONE MECCANICA
DISSOCIAZIONE ENZIMATICA
DISSOCIAZIONE MECCANICA
Porre il pezzo di tessuto in una capsula (piastra) di Petri sterile (∅
pari a 10 cm) e si aggiungono 5-10 ml di soluzione di trattamento

costituita da RPMI 1640 (soluzione salina) addizionata di
antibiotici (Penicillina, Streptomicina o Gentamicina).
Tagliare delicatamente il reperto con bisturi e pinze, fino ad
ottenere frammenti delle dimensioni pari a 2-3 mm3, spremendo i
pezzettini con le pinzette.
Filtrare il tessuto così ottenuto raccogliendo il filtrato in un
protettone da 50 ml a fondo conico (Falcon da 50 ml).
Addizionare al tessuto sminuzzato rimasto sul filtro 4-5 ml di
soluzione di trattamento e filtrare per raccogliere ulteriori cellule.
Centrifugare per 10 minuti a 700 rpmi, freno 9.
Risospendere il pellet ed eseguire la conta delle cellule in presenza
di un colorante vitale (Trypan Blu 0.05%)
In presenza di emazie
Trattamento del pellet per 10 min con 10-15 ml di soluzione ACK
(NH4Cl), un lavaggio con RPMI 1640 + FBS 10% (per inattivare l’attività
di ACK).
In presenza di cellule morte (se superiore al 20% della popolazione
totale)
Trattamento per circa 2 min del pellet con tripsina EDTA + DNAsi (250
U/ml) a 37°C, pipettare gentilmente durante il trattamento. Due lavaggi
con RPMI 1640 + HI-FBS 10% (per inattivare l’attività degli enzimi).
DISSOCIAZIONE ENZIMATICA
Preparare l’enzima o il pool di enzimi proteolitici secondo il
protocollo prescelto, sciogliendoli in soluzione salina o nel terreno
di coltura specifico e completo.
Filtrare e conservare a 4°C.

Aggiungere ai frammenti di tessuto (tumorale o non) in piastra di
Petri la soluzione contente l’enzima nella quantità (mg/ml)
prevista dal protocollo di dissociazione adottato (il più classico è
quello di Slocum-Pavelic che prevede 2ml/g).
Incubare a 37°C per almeno 3 ore, sotto agitazione. Dopo
l’incubazione filtrare senza ulteriore manipolazione dei residui di
tessuto non disgregato.
Raccogliere le cellule e passarle attraverso una siringa con un ago
da 18G o 25G per eliminare i possibili aggregati cellulari.
Lavare due volte con soluzione salina o con RPMI 1640.
Centrifugare per 10 minuti a 700 rpmi, freno 9.
Risospendere il pellet ed eseguire la conta delle cellule.

PROTOCOLLO PER LA DISGREGAZIONE CELLULARE SECONDO IL METODO MECCANICO ENZIMATICO SLOCUM-PAVELIC TESSUTO IN GHIACCIO Porre in piastra di Petri RPMI1640 + HI-FBS 10% (freddo) Triturare con il bisturi Filtrare su retino calibrato (100 µm) lasciando drenare spontaneamente le cellule
Cellule rilasciate nel mezzo SOSPENSIONE CELLULARE DELLA DISSOCIAZIONE MECCANICA
Riporre delicatamente il tessuto residuo in Petri, aggiungere terreno fresco, triturare ulteriormente e filtrare Cellule rilasciate Il tessuto rimanente viene posto in RPMI 1640 + HI-FBS 10% Collagenasi II (146 U/mg) 0.8 g % 5 ml/2.5 g tessuto DNasi I (3085 KU/mg solido) 0.002 g %
CONTA VITALE
Incubare a 37°C per 2 ore in atmosfera umidificata CO2 5% Filtrare su retino (100 µm) il tessuto digerito
Cellule rilasciate SOSPENSIONE CELL. DELLA DISSOC. ENZIMAT
Aggiungere RPMI 1640 + HI-FBS 10% al tessuto rimasto nella Petri, tagliare brevemente e riversare sul filtro Cellule rilasciate Lavare con terreno fino a che non si ottengono più cellule

Dissociazione enzimatica dei tessuti
Lo scopo principale delle procedure di dissociazione enzimatica dei
tessuti è quello di disgregare la matrice extra-cellulare che trattiene le
cellule, e di rompere i legami inter-cellulari (desmosomi, tight junctions
e gap junctions).
Tale dissociazione è necessaria sia per il rilascio di cellule da tessuti che
per il distacco di cellule dal substrato di coltura.
La matrice è una miscela complessa di proteine, glicoproteine, lipidi,
glicolipidi e mucopolisaccaridi. Contiene tre principali proteine capaci di
formare fibre: collagene, elastina e fibronectina. Tali fibre sono
interconnesse in un gel idratato formato da catene di glicosaminoglicani.
La dissociazione dei tessuti deve avvenire tramite dissoluzione della
matrice extracellulare e tramite rottura dei contatti fra cellule, ma senza
rompere la membrana cellulare e senza danneggiare la superficie
cellulare o strutture intracellulari.
Figura: Tipologie di legami fra cellule.

ENZIMI UTILIZZATI NELLA DISSOCIAZIONE DEI TESSUTI
COLLAGENASI
Proteasi con specificità per il legame X-GLY nella sequenza PRO-X-GLY-
PRO. Tale sequenza è presente con alta frequenza nel collagene. Capace
di attaccare e degradare le fibrille native a tripla elica del collagene.
Inibitori: EDTA, EGTA, 2-Mercaptoetanolo.
TRIPSINA
Proteasi con specificità per i legami peptidici che coinvolgono il gruppo
carbossilico degli aminoacidi arginina e lisina. Utilizzata da sola non è
efficace per la dissociazione dei tessuti (manca di selettività per le
proteine extra-cellulari), da utilizzarsi in combinazione con altri enzimi,
come elastasi e ialuronidasi. Efficace nel distacco di colture cellulari
(monostrati) dal substrato di crescita. L’esposizione prolungata alla sua
azione danneggia le cellule.
Inibitori: siero.
DISPASI
E’ una amino-endo peptidasi. Idrolizza i legami peptidici dal lato N-
terminale di aminoacidi non polari. E’ indicata per la sua attività di
dissociazione dei tessuti. Utile ad esempio per separare l’epidermide dal
derma o i foglietti epiteliali intatti dal substrato. Utile anche per il
distacco di colture primarie e linee dal substrato. Agisce tagliando i
legami a livello di membrana basale.
Inibitori: EDTA, EGTA.
DNasi I
Endonucleasi specifica per DNA a doppia elica. Utile per la dissociazione
di tessuti e per la realizzazione di sospensioni cellulari.

La dissociazione dei tessuti è sempre associata a lisi cellulare con
rilascio di DNA, che si svolge a formare materiale vischioso capace di
aggregare le cellule.
Si utilizza in genere in associazione con altri enzimi (collagenasi o
tripsina).
Inibitori: EDTA, EGTA, SDS.
ELASTASI
E’ una proteasi che idrolizza i legami peptidici dal lato C-terminale di
aminoacidi neutri con catene laterali alifatiche. Digerisce molte proteine,
ma è l’unica attiva sull’elastina (proteina fibrinosa del tessuto
connettivo). Si utilizza in associazione con altri enzimi come collagenasi
e tripsina, per dissociare tessuti ricchi di fibre intercellulari.
Possibili applicazioni: isolamento di cellule polmonari e digestione di
fibre elastiche delle arterie.
Inibitori: α-antitripsina, α2-macroglobulina.
IALURONIDASI
E’ una glicosidasi con specificità per acido ialuronico e condroitina.
Entrambe queste molecole sono glicosaminoglicani, che costituiscono la
matrice extra-cellulare e si ritrovano in alte concentrazioni in tutti i
tessuti connettivi.
Utile per la dissociazione dei tessuti in associazione con altri enzimi
come collagenasi e tripsina.
Inibitori: eparina, siero.
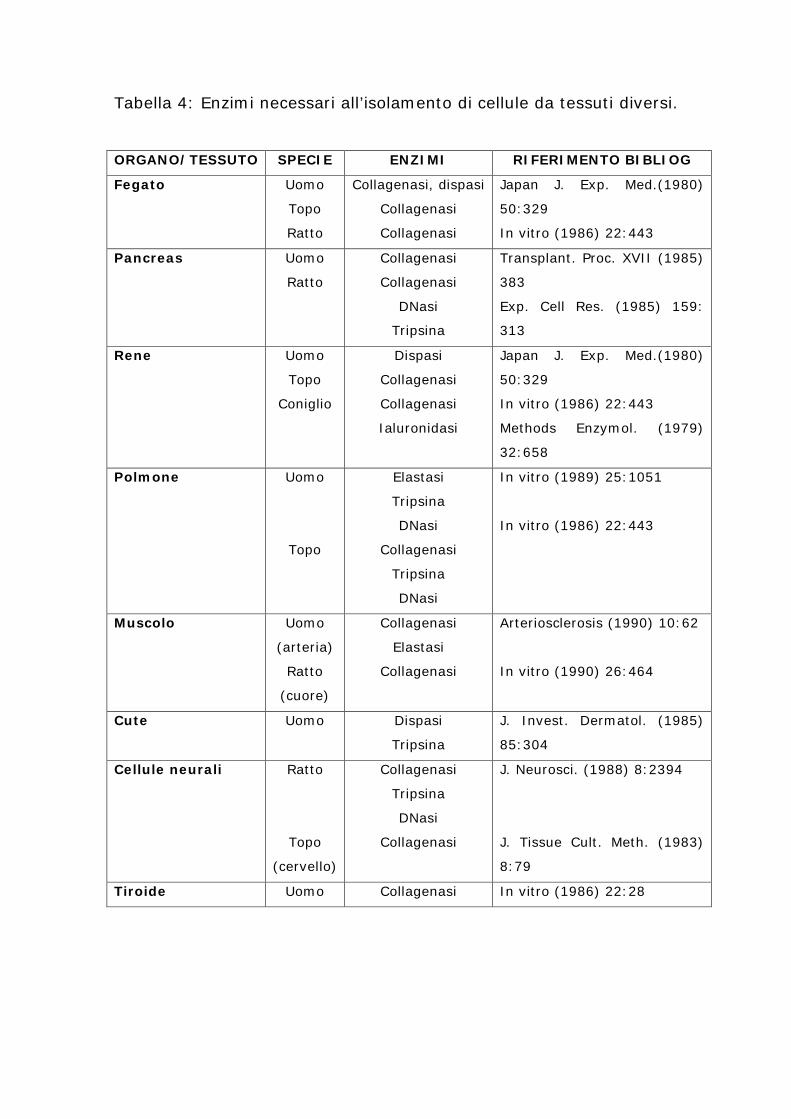
Tabella 4: Enzimi necessari all’isolamento di cellule da tessuti diversi.
ORGANO/TESSUTO SPECIE ENZIMI RIFERIMENTO BIBLIOG
Fegato Uomo
Topo
Ratto
Collagenasi, dispasi
Collagenasi
Collagenasi
Japan J. Exp. Med.(1980)
50:329
In vitro (1986) 22:443
Pancreas Uomo
Ratto
Collagenasi
Collagenasi
DNasi
Tripsina
Transplant. Proc. XVII (1985)
383
Exp. Cell Res. (1985) 159:
313
Rene Uomo
Topo
Coniglio
Dispasi
Collagenasi
Collagenasi
Ialuronidasi
Japan J. Exp. Med.(1980)
50:329
In vitro (1986) 22:443
Methods Enzymol. (1979)
32:658
Polmone Uomo
Topo
Elastasi
Tripsina
DNasi
Collagenasi
Tripsina
DNasi
In vitro (1989) 25:1051
In vitro (1986) 22:443
Muscolo Uomo
(arteria)
Ratto
(cuore)
Collagenasi
Elastasi
Collagenasi
Arteriosclerosis (1990) 10:62
In vitro (1990) 26:464
Cute Uomo
Dispasi
Tripsina
J. Invest. Dermatol. (1985)
85:304
Cellule neurali Ratto
Topo
(cervello)
Collagenasi
Tripsina
DNasi
Collagenasi
J. Neurosci. (1988) 8:2394
J. Tissue Cult. Meth. (1983)
8:79
Tiroide Uomo Collagenasi In vitro (1986) 22:28

FATTORI DI ADESIONE
La crescita normale e lo sviluppo di molti tipi cellulari sono dipendenti
da fattori di adesione. Alcune cellule li sintetizzano, ma altri tipi cellulari
li richiedono come fattori esogeni. Terreni di coltura serum free sono
privi di fattori di adesione normalmente presenti nel siero e tali fattori
devono spesso essere aggiunti a colture mantenute con questi terreni.
FATTORE DI ADESIONE TIPO CELLULARE
COLLAGENE TIPO I Muscolo, epatociti, cellule del ganglio spinale
COLLAGENE TIPO II Condrociti
COLLAGENE TIPO IV Cellule endoteliali, epiteliali, muscolari, nervose
FIBRONECTINA Cellule epiteliali, neuronali, endoteliali, fibroblasti
GELATINA Adesione di vari tipi cellulari
LAMININA Cellule endoteliali, epiteliali, muscolari, epatociti,
tumori
POLI-LISINA Adesioni di vari tipi cellulari
VITRONECTINA Cellule endoteliali, melanoma,osteosarcoma
Preparazione del collagene (tipo I) da code di ratto
Nelle preparazione dei diversi tipi di collagene, prima fra tutti emerge la
scelta di una fonte appropriata come una adeguata procedura per la
costituzione di tale preparato. Sebbene il collagene di tipo I possa
essere virtualmente preparato a partire qualsiasi tessuto o organo, le
fonti più convenienti sono date dal derma, dai tendini e dalle ossa di
organismi relativamente giovani. Ciascuno di questi tessuti contiene in
abbondanza il collagene. Le fibre dei tendini e le ossa sono quasi
esclusivamente costituite da collagene di tipo I, ed è considerevolmente
semplice ottenere il collagene da queste fonti.

Procedura
Parte sporca
1. le code di ratto sono poste in etanolo 70% per circa 30 minuti.
2. Passato questo lasso di tempo, le fibre di collagene vengono
isolate: si incide la cute della coda con un bisturi e si cerca di
mettere a nudo i fasci tendinei.
3. Con bisturi e pinza si isolano solo i fasci bianchi, che vengono
deposti in una piastra di Petri contenente etanolo al 70%.
4. Si pesano circa 5 g di fibre di collagene in una piastra pulita
(questo quantitativo serve a costituire 1 litro di collagene).
5. Le fibre vengono quindi ricoperte da etanolo al 70% e messe ad
incubare per 30 minuti.
Parte pulita
6. Sotto Cappa: si depongono le fibre recuperate con una pinzetta
sterile in una Erlenmeyer flask e si aggiunge un litro di acqua
sterile.
7. A questa soluzione viene aggiunto 1 ml di acido acetico glaciale e
un magnete sterile.
8. La soluzione rimane per 48-72 ore a 4°C in agitazione (tutto il
week end).
9. Centrifugare la soluzione (di solito al termine del week end) a
11000 rpm a 4°C per 1 ora e trasferire il surnatante in una
bottiglia sterile.
10. Questa nuova soluzione viene sottoposta ad un secondo
ciclo di centrifugazione a 17000 rpm a 4°C per 45 minuti.
11. Il collagene così isolato in una nuova bottiglia sterile viene
stoccato a 4°C (termine max per uso: 30 gg).

PREVENZIONE DELLE CONTAMINAZIONI
Gli agenti contaminanti che possono infestare una coltura sono
generalmente batteri, funghi, micoplasmi e cellule di tipo diverso da
quello coltivato (contaminazioni crociate).
Come appaiono al microscopio:
Batteri appaiono come dei granellini neri che si
agitano intensamente.
Funghi (muffe) appaiono come miceli filamentosi
ASPETTI DELLE CONTAMINAZIONI
INVISIBILE causa MICOPLASMI
VISIBILE causa BATTERI e FUNGHI
Alterazioni del terreno di coltura (intorbidimento, aspetto a
“sabbia” sul monostrato), acidificazione
Alterazione cellulare (aree di lisi, detriti cellulari)
NORME GENERALI
Laboratori riservati per le colture cellulari
Stanze chiuse e separate
Lampade UV
Cappe a flusso verticale
Pipettatrici elettriche
Sterilizzazione della vetreria
Utilizzazione di materiale plastico monouso
Uso di antimicrobici nei terreni di coltura
Minimizzare il numero di linee diverse nello stesso incubatore o
sotto la stessa cappa

METODI PER EVIDENZIARE LE CONTAMINAZIONI DI COLTURE
CELLULARI
Esame microscopico a fresco o dopo la colorazione di Gram
Esame colturale in terreni liquidi
Esame colturale in terreni solidi
Isolamento ed identificazione di micoplasmi
Immunofluorescenza con coloranti per il DNA
Microscopia elettronica
Contaminazione micoplasmica di colture cellulari
I micoplasmi sono parassiti di superficie delle cellule coltivate in vitro, in
quanto si ritrovano adesi alla membrana plasmatici cellulare. Questi
organismi, largamente diffusi in natura, possono essere definiti come le
più piccole entità dotate di una autonomia replicativa. Si caratterizzano
per l’assenza di parete cellulare e l’esclusiva presenza di una membrana
cellulare delimitante. Questo conferisce loro un notevole pleiomorfismo.
Le fonti di contaminazione comprendono
il personale (la maggior parte delle specie isolate risiedono nella
cavità orale o in altri distretti dell’organismo umano);
il siero bovino;
la tripsina di origine porcina.
Gli effetti della contaminazione da micoplasmi sono dati da alterazioni
cromosomiche, modificazione della sintesi delle proteine e degli acidi
nucleici ed inibizione della replicazione virale mediante sottrazione di
metabolici, competizione per i precursori degli acidi nucleici e
aumentata sintesi di interferone.

Tabella 5: Antibiotici utili per il controllo della contaminazione da
micoplasmi di colture cellulari.
ANTIBIOTICO STABILITA’
NEI TERRENI
CONC. CITOTOX
µg/ml
MIC
µg/ml
CONC. CONSIG#
µg/ml
Cloramfenicolo Alta 30 15 20
Tetraciclina Moderata 35 2 10
Clorotetraciclina Molto bassa 80 40 60
Idrossitetraciclina Moderata 35 5 10
Eritromicina Moderata 300 15 50
Acido fusidico Alta 40 20 20
Gentamicina Alta 3000 1 200
Kanamicina Molto alta 10000 25 200
Neomicina B Molto alta 3000 15 50
Novobiocina Bassa 200 10 50
Paromicina Alta 5000 20 50
Spiramicina Moderata 1000 1 50
Tilosina Moderata 300 1 10
Stabilità: Molto alta = semivita 8 giorni ; Alta = 6 gg; Moderata = 4 gg; Bassa = 2 gg;
Molto bassa = < 2 gg.
MIC = Minima Concentrazione Inibente i micoplasmi in terreni artificiali.
#Concentrazione consigliate per controllare i micoplasmi in colture cellulari con
cambiamenti del terreno di coltura ad intervalli di tre giorni.

ANTIMICROBICI
ANTIBATTERICI (ampia possibilità di scelta)
ANTIMICOTICI (disponibilità limitata)
ANTIMICOPLASMA (validi solo a livello preventivo)
Gli antimicrobici vanno aggiunti al terreno di coltura
contemporaneamente al siero ed ai fattori di crescita.
Tabella 6: Antibiotici e colture cellulari. ANTIBIOTICO SPETTRO D’AZIONE TOSSICITA’ CONC. CONS.
PENICILLINA G+ 10000 100
STREPTOMICINA G- 20000 100
DIIDROSTREPTOMICINA G- 30000 100
GENTAMICINA G+, G-, MYC 3000 200
NEOMICINA G+, G-, MYC 3000 50
KANAMICINA G+, G-, MYC 10000 100
PAROMOMICINA G+, G-, MYC 5000 100
VIOMICINA G+, G-, MYC 3000 50
POLIMIXINA B G- 3000 50
CLORAMFENICOLO G+, G-, MYC 30 5
TETRACICLINA G+, G-, MYC 30 10
CLOROTETRACICLINA G+, G-, MYC 80 10
IDROSSITETRACICLINA G+, G-, MYC 25 5
NISTATINA FUNGHI e LIEVITI 600 50
ANFOTERICINA B FUNGHI, LIEVITI, MYC 30 2.5
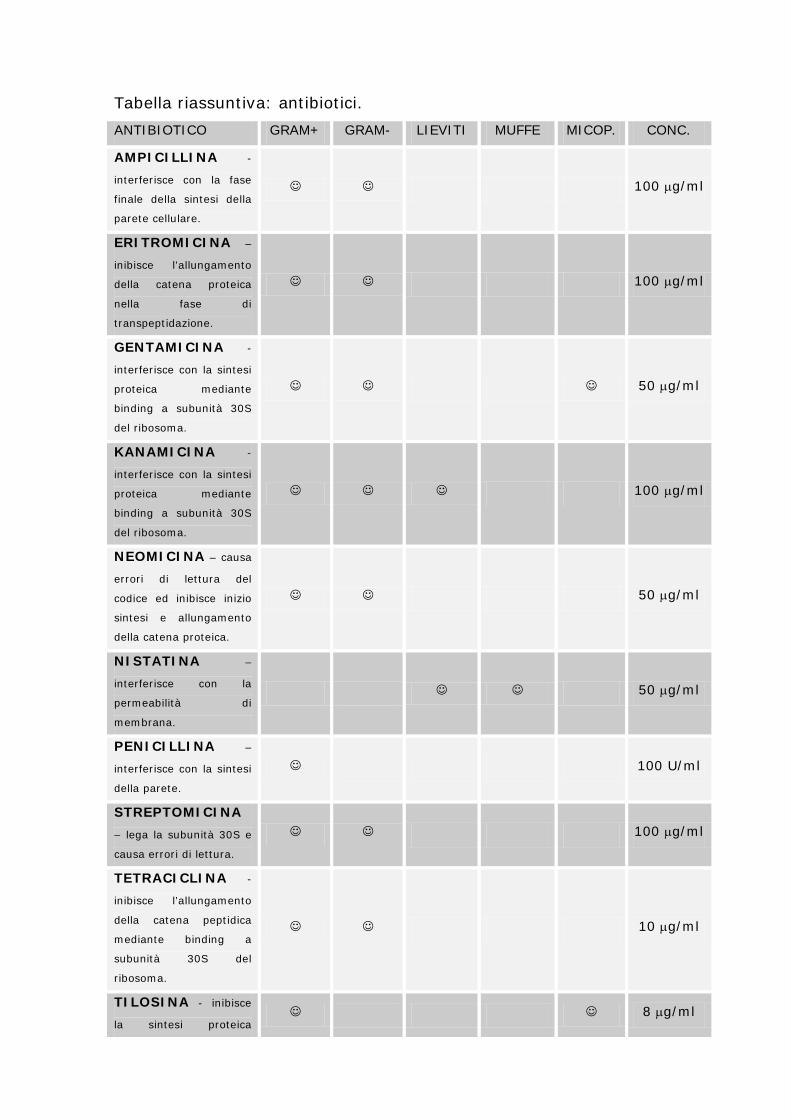
Tabella riassuntiva: antibiotici. ANTIBIOTICO GRAM+ GRAM- LIEVITI MUFFE MICOP. CONC.
AMPICILLINA -
interferisce con la fase
finale della sintesi della
parete cellulare.
100 µg/ml
ERITROMICINA –
inibisce l’allungamento
della catena proteica
nella fase di
transpeptidazione.
100 µg/ml
GENTAMICINA -
interferisce con la sintesi
proteica mediante
binding a subunità 30S
del ribosoma.
50 µg/ml
KANAMICINA -
interferisce con la sintesi
proteica mediante
binding a subunità 30S
del ribosoma.
100 µg/ml
NEOMICINA – causa
errori di lettura del
codice ed inibisce inizio
sintesi e allungamento
della catena proteica.
50 µg/ml
NISTATINA –
interferisce con la
permeabilità di
membrana.
50 µg/ml
PENICILLINA –
interferisce con la sintesi
della parete.
100 U/ml
STREPTOMICINA
– lega la subunità 30S e
causa errori di lettura.
100 µg/ml
TETRACICLINA -
inibisce l’allungamento
della catena peptidica
mediante binding a
subunità 30S del
ribosoma.
10 µg/ml
TILOSINA - inibisce
la sintesi proteica 8 µg/ml

mediante binding a
subunità 50S del
ribosoma.

TERRENI DI COLTURA
Sono state descritte numerose formulazioni di terreni per la coltivazione
in vitro di cellule di eucarioti. Solo alcuni di questi terreni sono in realtà
utilizzati in larga scala e si possono acquistare come terreni liquidi già
sterilizzati oppure come miscele di componenti in polvere, da sciogliere
e sterilizzare il laboratorio. Nella maggior parte dei casi i terreni
richiedono l’aggiunta di un quantitativo di siero di origine animale o
umana, come fonte di fattori di crescita non facilmente sostituibili
tramite terreni sintetici. Alcuni terreni di recente introduzione non
richiedono l’aggiunta di siero per la presenza nella formulazione di
fattori di crescita purificati.
Requisiti fondamentali del terreno di coltura
Deve fornire tutti i composti necessari alle cellule (sostanze
elementari per la costruzione delle macromolecole, substrati per il
metabolismo energetico, vitamine, ecc.);
Deve contenere un sistema tampone per mantenere il pH entro
limiti fisiologici (7.2-7.4);
Non deve contenere fattori tossici per le cellule.
GRUPPI DI SOSTANZE PRESENTI IN UN TERRENO A SECONDA DELLA
LORO FUNZIONE
1. SALI INORGANICI – Contribuiscono a mantenere il potenziale di
membrana e l’osmolarità del terreno.
a. La pressione osmotica ottimale varia nelle diverse linee
cellulari, ma risulta in genere compresa tra valori di 260-
320 mOsm/kg.

b. La pressione osmotica necessaria viene ottenuta con
un’adeguata concentrazione di una soluzione salina
bilanciata.
c. I sali di sodio, magnesio, potassio, e calcio sono
fondamentali per l’equilibrio osmotico, il calcio inoltre
influisce in modo determinante sulla capacità delle cellule di
aderire al substrato.
d. Il bicarbonato di sodio viene aggiunto al terreno come
fattore necessario a molte reazioni biosintetiche, inoltre in
molte formulazioni esso agisce come costituente del sistema
tampone bicarbonato/acido carbonico.
NaHCO3 + H2O
Na+ + HCO3- + H2O
Na+ + H2CO3 + OH-
Na+ + OH-+ H2O +CO2
Da ciò si deduce che la CO2 viene rilasciata nell’atmosfera
esterna al terreno di coltura con conseguente aumento dell’alcalinità
(OH-). L’aumento degli ioni idrossile è proporzionale alla quantità di
bicarbonato aggiunto alla soluzione. Per controllare questa reazione
si fornisce artificialmente CO2 all’atmosfera dell’incubatore (5%) in
modo da impedire al gas di formarsi ed impedire l’aumento degli ioni
idrossile. Un controllo più fine del pH viene attuato anche grazie
all’aggiunta di soluzioni tampone quali HANKS o HEPES.
2. ZUCCHERI – Il glucosio viene aggiunto come fonte di atomi di
carbonio per il metabolismo cellulare.
3. AMINOACIDI – Per le cellule in coltura a lungo termine vengono
considerati essenziali 13 aminoacidi. Spesso i terreni contengono
tutti i 20 aminoacidi per evitare un dispendio di energia
metabolica alle cellule.

4. VITAMINE – Costituiscono cofattori necessari al funzionamento di
molte vie enzimatiche.
5. ACQUA – Per la preparazione in laboratorio dei terreni acquistati
in polvere deve essere priva di pirogeni e tridistillata.
6. SIERI – Il più usato è quello di bovino. In alcuni casi, come ad
esempio la coltura a lungo termine di linfociti umani, si deve
utilizzare siero umano da donatori. I sieri vengono aggiunti in
percentuali variabili tra il 5 e il 20%.
7. MINERALI – Ferro, rame, zinco e selenio sono in genere già
presenti nel siero o come contaminanti nei terreni e sono
necessari come costituenti di enzimi.
8. SUPPLEMENTI ORGANICI – Nucleosidi, piruvati, lipidi si rendono
necessari quando viene ridotta la concentrazione di siero.
9. ORMONI – Si aggiungono nei mezzi privi di siero (es. insulina)
10. FATTORI DI CRESCITA – Sono in genere provvisti dal siero,
ma non sempre in concentrazione sufficiente. Contribuiscono
segnali specifici necessari per la proliferazione cellulare delle linee
continue.
Tutti i terreni pronti all’uso (dopo addizione di antibiotici, soluzioni
tampone, siero ed eventuali additivi) devono essere filtrati sterilmente
tramite filtri con pori di dimensioni pari o inferiori a 0.45 µm (qualora
uno dei componenti non sia sterile).

REALIZZAZIONE DI COLTURE CELLULARI
Scelta del terreno
1. Linee cellulari ottenute da banche pubbliche – il tipo di terreno, gli
eventuali additivi e la natura e quantità di siero vengono
specificati al momento dell’acquisto.
2. Linee cellulari umane prodotte in laboratorio da prelievi tumorali –
in genere è necessario utilizzare terreni come RPMI 1640
addizionati con alte concentrazioni di siero bovino (15-20%).
3. Linee cellulari di altre specie (comunemente murine) – in caso di
cellule a crescita aderente si utilizza MEM o DMEM addizionati con
10% di siero. Nel caso di linee in sospensione (es. leucemie o
ibridomi) si utilizza RPMI 1640 addizionato con HI-FBS.
4. Colture a tempo determinato di cellule normali (es. linfociti
umani) – RPMI 1640 addizionato con siero umano al 10-20%. Il
siero umano deve essere un pool da diversi donatori.
MANTENIMENTO DI UNA COLTURA CELLULARE
Cambiamento del mezzo o terreno di crescita
Una coltura cellulare, sia essa primaria o una linea cellulare, necessita
di periodiche sostituzioni del mezzo di crescita (terreno di crescita). La
frequenza dei cambi di terreno dipende dalle caratteristiche di crescita e
di attività metabolica delle cellule.
Parametri fondamentali per decidere i cambi di terreno
Il pH del terreno – Ogni tipo cellulare in crescita determina, per
effetto della propria attività metabolica, un abbassamento del pH
rilevabile come tendenza del terreno a cambiare colore (da rosso-
arancio a giallo) per effetto della presenza di un indicatore di pH
(in genere il rosso fenolo) che viene normalmente aggiunto al
terreno.

La concentrazione cellulare – Occorre distinguere tra colture in
sospensione e in monostrato. Nel primo caso, la maggior parte
delle colture primarie o linee cellulari in fase di crescita
logaritmica non possono crescere a concentrazioni superiori a
1*106 cells/ml. Nel secondo caso, la crescita cellulare viene
ostacolata dopo il raggiungimento della confluenza.
Arresto della crescita per cellule normali o deterioramento per
cellule trasformate.
Presenza di granuli in prossimità del nucleo o di vacuoli nel
citoplasma.
SUBCOLTURA (O PASSAGGIO) DI CELLULE IN MONOSTRATO
La prima subcoltura rappresenta per la coltura un’importante fase di
transizione; il primo passaggio viene effettuato quando la coltura
primaria sia cresciuta in modo tale da occupare tutto il substrato
disponibile, quando cioè viene a costituire un “monostrato confluente”.
Poiché l’adesività cellulare dipende da glicoproteine e da calcio e
magnesio, si preferisce eseguire enzimaticamente il distacco del
substrato mediante breve incubazione con tripsina o con sostanze
chelanti come l’EDTA.
TRIPSINIZZAZIONE
Materiale necessario
Medium appropriato per la coltura cellulare considerata. Questo
dovrebbe contenere tutti i fattori (siero, glutamina, ecc.)
necessari alle cellule prese in esame.
Flasks con colture cellulari sane e prossime alla confluenza
Calcium- and Magnesium-Free Phosphate-Buffered Saline CMF-
PBS (10mL). Questa soluzione salina viene utilizzata per
mantenere il pH e il bilancio osmotico corretti, mentre le cellule

vengono lavate per rimuovere gli inibitori delle proteasi, presenti
nel siero.
Soluzione 0.05% Tripsina/EDTA (0.05%). La tripsina viene
normalmente utilizzata in concentrazioni comprese fra 0.05% e
0.25%. Le concentrazioni di lavoro sono generalmente
determinate utilizzando la più bassa concentrazione di tripsina che
sia in grado di staccare le cellule dal substrato e di formare una
sospensione cellulare in un lasso di tempo relativamente breve (5-
10 minuti). Le soluzioni di tripsina sono spesso integrate con altri
enzimi (collagenasi) o con agenti chelanti (EDTA) per migliorare la
sua attività.
Tubi da centrifuga da 15-50 ml
Emocitometro
Centrifuga
Microscopio a contrasto di fase
Pipette
Procedura
1. Aspirare (sterilmente) il medium dal fondo della flask (o
piastra di Petri).
2. Lavare il monostrato 2-3 volte con il PBS, al fine di rimuove
tutte le tracce di siero (e spt gli inibitori delle proteasi). Il
volume dei lavaggi è in relazione alle dimensioni dove è
presente il monostrato (es. flask da 75 cm2 = 10 ml).
3. Aggiungere la soluzione di Tripsina/EDTA (disponibile
commercialmente come soluzione salina bilanciata
contenente EDTA a 0.02% e tripsina a 0.05%) al fondo della
flask. Girare delicatamente il contenitore, affinché la
soluzione tocchi il monostrato. Incubare per 5 minuti in
incubatore a 37 °C con 5% CO2. Dopo alcuni minuti,
controllare il monostrato al microscopio per poter rilevare il
distaccamento. Generalmente, più bassa è la

concentrazione di tripsina nelle soluzioni utilizzate e meno
tempo le cellule ne stanno a contatto, meglio è. Una volta
che le cellule assumono l’aspetto rotondeggiante o il
monostrato si stacca dal substrato (di solito si può
osservare un’area opaca che con dei colpetti gentili al
contenitore si riduce progressivamente a favore di una
sospensione cellulare).
4. Recuperare tutte le cellule mediante fasi di pipettamento e
di lavaggio del fondo del contenitore e raccogliere il tutto in
tubi da centrifuga (15 o 50 ml).
5. Aggiungere 5 ml di terreno completo di siero per inattivare
l’attività della tripsina.
6. Centrifugare le cellule per 5-10 minuti a 700-1500 rpm
(freno 9).
7. Rimuovere il medium surnatante il pellet cellulare.
8. Aggiungere 10 ml di terreno di coltura e risospendere le
cellule (fasi di pipettaggio). Mettere in ghiaccio.
9. Recuperare un’aliquota per il conteggio con l’emocitometro.
SUBCOLTURA (O PASSAGGIO) DI CELLULE IN SOSPENSIONE
1. Mescolare la coltura.
2. Prelevare un’aliquota della coltura, lavare con eccesso di
terreno fresco e contare.
3. Risospendere in terreno fresco e seminare la sospensione in
nuovi contenitori per la coltura cellulare alla concentrazione
dipendente da
• Velocità di crescita del tipo cellulare.
• Intervallo di tempo previsto prima dell’utilizzo.
• Concentrazione minima dipendente dal tipo
cellulare necessaria per la propagazione della
linea.

COLTURE IN MEZZO SEMISOLIDO
Alcune colture cellulari possono essere realizzate in mezzo semisolido,
nel quale il normale terreno di coltura viene addizionato con piccole
quantità di agar o di metilcellulosa in modo da realizzare una forma di
“gelatina” di consistenza tale da consentire la crescita localizzata delle
cellule senza che si abbia rimescolamento come nei terreni liquidi.
Tali mezzi semisolidi sono in genere utilizzati per
Clonaggio di linee tumorali
Crescita, differenziamento e analisi morfologica di colonie
midollari da progenitori emopoietici.
La crescita in terreno semisolido consente di individuare ed
eventualmente isolare piccole colonie o veri cloni cellulari sulla base
della morfologia in modo altrimenti impossibile con i mezzi di coltura
liquidi.
Realizzazione di coltura cellulare in terreno semisolido
1. Preparare la soluzione madre di agar al 5% in soluzione salina
bilanciata mediante ebollizione.
2. Trasferire una quantità di soluzione madre di agar alla
temperatura di fusione (42°C) nel terreno di coltura in modo da
realizzare una concentrazione finale di agar pari a 0.7%.
3. Preparare una sospensione cellulare a temperatura di 37°C e
unire tale sospensione in rapporto 1:1 con la soluzione di agar allo
0.7% (concentrazione finale agar = 0.35%).
4. Aliquotare la sospensione di agar/terreno/cellule in piastre di Petri
e consolidare il terreno mediante incubazione a 4°C.
5. Incubare a 37°C.

EMOCITOMETRO
Conteggio cellulare
L’emocitometro è un camera di vetro incisa su di un vetrino, con parti
sollevate che sorreggono un vetrino coprioggetto di quarzo esattamente
0.1 mm sopra la camera. La camera di conteggio (o camera di Burker)
è incisa su di una superficie totale di 9 mm2 (Figura 1).
Figura 1. Dimensioni di un emocitometro.
La camera dell’emocitometro è divisa, da linee bianche triple in 9 aree
quadrate; ogni quadrato (dei 16 presenti) risulta delimitato da 3 righe
parallele ravvicinate ed ha una superficie di 1 mm2.
La valutazione della concentrazione è basata sul volume che sottostà il
vetrino coprioggetto.
Considerando che la camera, quando viene sovrapposto il vetrino
coprioggetto, è di 0.1 mm, il volume complessivo di un quadrato sarà
uguale a:
1*0.1 =0.1 mm3 = 10-4 ml (1 cm3 = 1 ml)
1. Fissare il vetrino coprioggetto sulla camera (controllarne sempre
l’aderenza, devono comparire i caratteristici “anelli di Newton”)
2. Caricare la sospensione cellulare toccando il margine del vetrino
coprioggetto con una pipetta Pasteur o con il puntale di una
micropipetta (l’emocitometro viene riempito attraverso processi di
capillarità): espellere molto lentamente in modo tale che la
sospensione cellulare vada a riempire progressivamente tutti gli
spazi.

3. Contare almeno 3 quadrati, se si osservano cellule sui bordi
conteggiare due lati su quattro
4. Il cerchio indica l’area approssimativa coperta all’ingrandimento
microscopico 100X (10X oculare e 10X obiettivo).
Figura 2.
Partendo dalla prima camera contare le cellule nel quadrato centrale da
1mm e nei 4 quadrati da 1 mm agli angoli (Figura 2).
Contare le cellule in alto e a sinistra che toccano la linea centrale del
perimetro di ciascun quadrato. Non contare le cellule che toccano la
linea centrale in basso e a destra (Figura 3)
Figura 3.
5. il numero delle cellule per ml è dato da
[n° cells contate * diluizione (eventuale)/ n° quadrati contati]
* 10-4
5. il numero delle cellule totali
cells/ml*volume originale campione

Di seguito, vengono riportati 2 metodi semplici per il conteggio cellulare
basati sul area di superficie dell’emocitometro. Si possono utilizzare
anche altri schemi di conteggio. La scelta dei metodi dipende dalla
concentrazione cellulare presente, l’accuratezza della procedura è in
relazione al numero di cellule contate. Quando la concentrazione è
bassa, si dovrebbero contare più griglie.
Figure 4. Procedure di conteggio secondo il metodo A (parte sinistra) e B (parte destra).
Metodo A
Contare il numero di cellule in 4 quadrati esterni (vedi parte sinistra
Figura 4)
La concentrazione cellulare viene calcolata come segue:
Cells/ml = Totale cellule contate in 4 quadrati x 2500 x fattore di
diluizione
Es. Se uno ha contato 450 cellule dopo la diluizione di una
aliquota di sospensione cellulare 1:10,
la concentrazione cellulare originale = 450 x 2500 x 10 =
11,250,000/ml
Metodo B
Stimare la concentrazione cellulare attraverso il conteggio di 5 quadrati
nel quadrato centrale più grande (vedi parte destra Figura 4)
La concentrazione cellulare viene calcolata come segue
Cells/ml = Totale cellule contate in 5 quadrati x 50,000 x fattore di
diluizione

Es. Se uno ha contato 45 cellule dopo la diluizione di una
aliquota di sospensione cellulare 1:10,
la concentrazione cellulare originale = 45 x 50,000 x 10 =
22,500,000/ml.
La colorazione delle cellule spesso facilita la visualizzazione e il
conteggio delle cellule stesse.
Miscelare le cellule con un eguale volume di Trypan Blue [0.4% (w/v)
Tyrypan Blue in PBS] [es. 20 μl di sospensione cellulare con 20 μl di
sospensione di Trypan Blue (0.4%)] e lasciare riposare per 5-15 minuti
al fine di determinare la conta delle cellule vive/morte (le cellule morte
sono blue).
Uccidere le cellule con formalina al 10% e poi colorarle con Trypan Blue
o altra colorazione per migliorare la visualizzazione di tutte le cellule.

TEST DI VITALITA’ CELLULARE
Materiale necessario
• Sospensione cellulare
• Pipette sterili
• Soluzione stock 0.2% (w/v) di Trypan Blue
• Emocitometro e microscopio ottico
Procedura
1. Agitare gentilmente la sospensione cellulare per distribuire
uniformemente le cellule. Prelevare asetticamente un piccolo
quantitativo di cellule (0.1 ml) dalla sospensione. Porre il
campione in un contenitore falcon (non è necessario che sia
sterile).
2. Diluire 4 parti di stock Trypan Blue con una parte di salina 5X.
3. Aggiungere 0.1 ml di colorante diluito al campione, posto nella
falcon. Mixare gentilmente.
4. Allestire l’emocitometro e il relativo vetrino coprioggetto. Porre
una goccia di colorante/sospensione cellulare a contatto con
l’emocitometro (ricordarsi di utilizzare entrambi i lati per il
conteggio) e aspettare 1 minuto.
5. Osservare le cellule al microscopio a basso ingrandimento.
Contare il numero totale di cellule e il numero di cellule colorate.
6. Calcolare la concentrazione delle cellule vitali per ml di coltura
cellulare.
Note
Il Trypan Blue è un colorante che viene mantenuto attivamente fuori
dalle cellule vitali, ma che rapidamente entra e colora le cellule morte.
Perciò, le cellule che risultano colorate in blu, sono morte. La differenza
fra il numero totale di cellule e il numero di cellule morte sarebbe il

numero di cellule vitali in una certa aliquota della coltura cellulare. Il
sistema Trypan Blue sovrastima significativamente il numero di cellule
vitali, ma è sufficiente agli scopi di laboratorio. Approssimativamente il
30% delle cellule misurate come vitali con il Trypan Blue non è in grado
di continuare a crescere oltre un periodo di 24 ore.
VITALITA’ CELLULARE (%) = TOTALE CELLULE VIVE (NON COLORATE)
TOTALE CELLULE (COLORATE+NON COLORATE)
[CELLS/ml] = n cells contate * fattore di diluizione * 104
N quadrati contati
Es.
Volume iniziale = 5ml
50µl di campione e 100µl di soluzione di Trypan blue = DILUIZIONE 1:3
Conta media per quadrato di 16 quadratini = 65 cells
Cells/ml = 65 *3 *104 = 1.95 * 106 / ml
Cellule totali = 1.95 * 106 * 5 = 9.75 * 106

CRIOCONSERVAZIONE E STOCCAGGIO DELLE LINEE CELLULARI
Lo scopo principale della crio-conservazione delle colture cellulari è
quello di garantire quantitativi sufficienti di cellule stoccate, al fine di
evitare di mantenere le stesse linee cellulari in coltura tutto il tempo.
Questo è estremamente prezioso, quando si tratta di cellule con una
vita limitata nel tempo. Gli altri principali vantaggi di questo
procedimento sono:
riduzione dei rischi di contaminazione microbica;
riduzione di rischi di cross contaminazione con le altre linee
cellulari;
riduzione dei rischi di drift genetici e cambiamenti morfologici;
il lavoro può essere condotto utilizzando cellule ad un passaggio
costante;
riduzione dei costi (di materiale consumabile e di personale).
Sono stati messi a punto diversi protocolli per garantire validi processi
di crioconservazione e di ripresa di un’ampia varietà di linee cellulari, di
diversi tipi cellulari. La base per la riuscita di un processo di
crioconservazione è quello di un lento congelamento e di un rapido
scongelamento. Sebbene le richieste possano differire in relazione alle
diverse tipologie cellulari, in generale le cellule dovrebbero essere
raffreddate a 1 – 3°C al minuto e rapidamente scongelate mediante
una incubazione in una bagnetto termostatato a 37°C per 3-5 minuti.
Se questo e altri punti elencati successivamente vengono rispettati, il
processo di crioconservazione va a buon fine.
Le colture dovrebbero essere sane con un tasso di vitalità
cellulare superiore al 90% e non presentare segni di
contaminazione microbica.
Le colture dovrebbero essere in fase di crescita logaritmica
(questi può essere ottenuto utilizzando colture pre-confluenti;

es. colture che sono sotto la loro densità cellulare massima ed al
cambio del terreno di coltura 24 ore prima del congelamento).
Dovrebbe essere impiegato un alto rapporto siero/proteine
(>20%), in molti casi il siero viene usato al 90%.
L’uso di un crioprotettore come il dimetil sulfossido (DMSO) o del
glicerolo aiuta a proteggere le cellule dalla rottura per la
formazione dei cristalli di ghiaccio. Il crioprotettore più
comunemente utilizzato è il DMSO ad una concentrazione finale
pari al 10%. Questo non risulta appropriato per linee cellulari
come le HL60, dove il DMSO è utilizzato per indurre la
differenziazione. In tali casi una alternativa come il glicerolo
dovrebbe essere considerata.
LE TEMPERATURE DI STOCCAGGIO ULTRA-BASSE DELLE LINEE
CELLULARI
Mantenendo costante la discesa delle temperature per il congelamento,
le linee cellulari possono essere crioconservate in questo stato per
periodi di tempo indefiniti, garantendo una temperatura inferiore a
135°C. Queste temperature ultra-basse possono solamente essere
raggiunte da freezer elettrici particolari o, molto più frequentemente,
attraverso l’immersione in azoto liquido o nei suoi vapori. Tutti i sistemi
presentano sia vantaggi che svantaggi come descritto nella tabella
sottostante.
Tabella . Confronto fra i diversi sistemi per lo stoccaggio delle
line cellulari.
Metodi Vantaggi Svantaggi
Freezer
(-135ºC)
• Facile mantenimento
• Temperature costanti
• Bassi costi di gestione
• Necessita dell’azoto liquido
per back-up
• Meccanicamente complesso
• Temperature di stoccaggio

alte se confrontate con l’azoto
liquido
Azoto liquido
• Temperature costante
(-196ºC)
• Semplicità e affidabilità
meccanica
• Necessita di costanti
rabbocchi di azoto liquido
• Alti costi di gestione
• Rischi di cross
contaminazione via azoto
liquido
Vapori di azoto
• Nessun rischio di cross
contaminazione
dall’azoto liquido
• Acquisizione di basse
temperature
• Semplicità e sicurezza
• Necessita regolare fornitura
di azoto liquido
• Alti costi di gestione
• Fluttuazione delle
temperature fra - 135ºC e -
190ºC
Lo stoccaggio in azoto liquido permette di raggiungere le temperature
più basse per la conservazione, mantenerle richiede grossi quantitativi
di azoto liquido e contenitori ermetici. Infatti, sono documentati casi di
cross contaminazione di virus patogeni via azoto liquido. Per questo
motivo le temperature ultra-basse possono essere mantenute con i
vapori di questo composto.
Per la conservazione con i vapori di azoto liquido, le vials vengono
posizionate sopra una riserva poco profonda di azoto liquido, le cui
dimensioni devono essere controllate e mantenute costantemente.
All’interno delle fase gassosa esiste un gradiente verticale per le
temperature, le cui estremità dipendono dai livelli della fase liquida,
dalla struttura del contenitore e dalla frequenza con cui si apre. Le
variazioni della temperatura negli strati superiori della fase gassosa
possono essere drastiche se non viene eseguito un controllo costante.

I contenitori per la conservazione ad azoto liquido dovrebbero avere
degli allarmi per la segnalazione dei bassi livelli di azoto. Questo risulta
particolarmente importante quando si tratta di conservazione con
vapori. Il contenitore per la conservazione con l’azoto liquido non
dovrebbe scendere mai sotto la metà del possibile quantitativo di
liquido, prima di essere rabboccato. Questo garantirebbe che almeno
una spedizione potrebbe andare a vuoto senza conseguenze
catastrofiche sulle cellule conservate.
Controllo del materiale stoccato
Tutti i sistemi per lo stoccaggio delle cellule a basse temperature
dovrebbero avere un sistema di classificazione, strutturato in modo tale
da permettere l’organizzazione dei materiali contenuti per garantire
rapidità e sicurezza nelle sistemazione e nel recupero. Questo dovrebbe
essere supportato da un accurato registro ed controllo includente
Ciascuna vials dovrebbe essere etichettata singolarmente,
mostrando il tipo cellulare, il numero di lotto o passaggio e
la data di congelamento, sigla operatore (es. BME-UV1, P
52; 10/07/2005; EF);
La localizzazione di ciascuna vial dovrebbe essere registrata
su di un registro (cartaceo ed elettronico) (meglio tenere
sempre un doppio registro per evitare le perdite).
Considerazioni per la sicurezza
E’ importante che il personale venga istruito sull’uso dei bidoni per lo
stoccaggio con azoto liquido e sull’equipaggiamento da indossare e da
utilizzare per se stessi (es. guanti e occhiali di protezione) e per le
cellule (es. vials a tenuta e ricarica dei bidoni). Come descritto in tutte
le procedure di laboratorio, l’equipaggiamento personale dovrebbe
essere indossato tutte le volte che si deve maneggiare l’azoto liquido,
incluso visori facciali di protezione, guanti termicamente isolati, oltre al

camice di laboratorio. Una corretta educazione e l’uso di un
equipaggiamento di protezione minimizzano i possibili rischi di ustioni
da congelamento e altri incidenti minori.
Rischi di asfissia
La considerazione più importante concernente la sicurezza è il
potenziale rischio di asfissia dovuto ad elevati livelli di azoto, che
possono portare ad una deplezione dell’ossigeno. Questo risulta
particolarmente critico, poichè la mancanza di ossigeno può portare
molto rapidamente a perdita di coscienza, senza preavviso. Di
conseguenza, i contenitori con l’azoto liquido dovrebbero essere allocati
in aree ben ventilate, al fine di ridurre al minimo questo rischio.
Misure preventive
• Sensori per la determinazione del quantitativo di ossigeno settati
su 18% (v/v);
• Educazione del personale. Questo dovrebbe essere istruito in
modo da evacuare l’area immediatamente al suono dell’allarme e
farvi ritorno solo quando il livello di ossigeno sarà pressoché
normale (~ 20% v/v);
• Due persone devo essere presenti quando si maneggia l’azoto
liquido;
• E’ proibito utilizzare l’azoto liquido al di fuori delle ore di lavoro;
• Sistemi di ventilazione (dovrebbero essere presenti o altrimenti
installati).

BUONE PRATICHE PER LA CONSERVAZIONE DELLE COLTURE
CELLULARI (CREAZIONE DI UNA BANCA)
Non è una pratica corretta mantenere una linea cellulare per un periodo
di tempo prolungato per le seguenti ragioni:
rischi di contaminazione microbica;
perdita delle caratteristiche di interesse (es. Antigene di
superficie o espressione di un anticorpo monoclonale);
genetic drift, in particolare in cellule riconosciute avere un
cariotipo instabile (es. CHO, BHK 21);
perdita di linee cellulari per esaurimento del tempo vitale;
rischi di cross contaminazioni con altre cellule;
aumento dei costi per materiale e staff.
Tutti questi potenziali fattori di rischio possono essere ridotti al minimo
attraverso l’implementazione del sistema di stoccaggio delle cellule.
Questo tipo di sistema è conosciuto come Master Cell Banking System o
sistema stratificato.
Al primo arrivo in laboratorio una nuova coltura cellulare dovrebbe
essere trattata come potenziale fonte di contaminazione per batteri,
funghi e micoplasmi e dovrebbe essere maneggia come in condizioni di
quarantena, fino a prova negativa. Di seguito all’iniziale proliferazione,
3-5 ampolle dovrebbero essere congelate e stoccate come stock di
base, prima che la Master Bank sia preparata. Una di queste vials
dovrebbe essere poi scongelata e fatta moltiplicare al fine di produrre
almeno una Master Bank di 10-20 ampolle, in relazione alla tipologia di
lavoro che si vuole intraprendere.
2-3 ampolle di questa banca dovrebbero essere destinate al controllo
qualità comprendente la conferma che la conta cellulare e la vitalità del
lotto siano accettabili e che il lotto sia privo di batteri/funghi e
micoplasmi. Ulteriori tests (come quelli di screening virale e di
autenticità) possono essere richiesti. Una volta che questi tests siano

stati completati con successo, una vials dalla Master Bank dovrebbe
essere scongelata e mantenuta in coltura al fine di dar vita alla Working
Bank. Le dimensioni di questa banca dipenderanno dal livello previsto
della domanda (ovvero dal carico di lavoro). I tests per il controllo di
qualità (conta cellulare e vitalità, in assenza di contaminanti microbici)
sono di nuovo richiesti prima dell’utilizzo delle colture cellulari per le
sperimentazioni di routine o per la produzione. E’ importante che a
questo stadio ci sia la conferma che la Master sia geneticamente
identica alla Working Bank, mediante le tecniche per la determinazione
del profilo del DNA.
Implementare questo sistema significa e assicura che
• il materiale a disposizione è sufficiente e di qualità eccellente;
• gli esperimenti vengono eseguiti utilizzando colture allo stesso
numero di passaggio;
• le cellule sono in coltura solo quando necessario;
• le caratteristiche originali delle linee cellulari sono mantenute.
Il numero di vials preparato per la banca di stoccaggio e per quella di
lavoro dipende dalla domanda prevista per il loro uso. Il numero di
ampolle campionate per il controllo di qualità dipende dalle dimensione
della banca. Idealmente il 5-10% dello stoccato dovrebbe essere
testato prima dell’uso. Le vials appartenenti alla banca di lavoro
dovrebbero essere messe in coltura sequenzialmente per non oltre un
determinato numero di passaggi per il raddoppio. Questo numero
dovrebbe essere minimo, nel caso in cui le linee cellulari hanno una vita
finita o comunque limitata (es. linee diploidi). La banca di lavoro
dovrebbe essere rifornita con una vials della Master Bank. Questo
dovrebbe essere fatto in tempo sufficiente a consentire che il controllo
qualità venga effettuato. Una nuova Master Bank dovrebbe essere
preparata prima che il numero dello stock originale scenda sotto le 5
vials. Il pannello dei tests per il controllo della qualità dipende dall’uso
che si intende fare delle cellule.

CONGELAMENTO
L’azoto liquido viene utilizzato per preservare le colture tissutali, sia in
fase liquida (-196°C) che in fase gassosa (-156°C). Il congelamento può
essere letale per le cellule in relazione agli effetti dannosi causati dai
cristalli di ghiaccio, dalle alterazione nelle concentrazioni degli elettroliti,
dalla disidratazione e dai cambiamenti di pH. Per minimizzare questi
effetti, si possono prendere diverse precauzioni. Primo, utilizzare un
agente crioprotettivo in grado di abbassare il punto di congelamento,
come il glicerolo o il DMSO. Il medium di congelamento, che usualmente
viene utilizzato è costituito da 80-90% siero, 10% DMSO. Inoltre, è
meglio usare cellule in salute che stanno crescendo durante la fase
logaritmica e sostituire il medium 24 ore prima del congelamento.
Anche le cellule sono portate lentamente dalla temperatura ambiente a
-80°C, al fine di permettere all’acqua di uscire dalle cellule prima del
congelamento. Il tasso ottimale di raffreddamento è 1-3°C per minuto.
Alcuni laboratori hanno la possibilità di utilizzare camere di
congelamento per regolare questo processo al tasso ottimale attraverso
una periodica immissione di azoto liquido. Noi utilizziamo uno strumento
chiamato Mr. Frosty.
Il Mr. Frosty è un contenitore riempito con 200 ml di isopropanolo a
temperatura ambiente e le vials di congelamento contenenti le cellule
sono posizionate in un rack posto all’interno del contenitore stesso.

Questo posto in un freezer -80°C. Il ruolo dell’isopropanolo è quello di
consentire alle vials di raggiungere la temperatura di congelamento
lentamente, circa 1°C al minuto. Una volta che il contenitore ha
raggiunto -80°C (in 4 ore o più, overnight), le vials devono essere
rimosse dal Mr. Frosty e immediatamente poste nel bidone dell’azoto
liquido. Le cellule sono stoccate alle temperature dell’azoto liquido,
perché lo sviluppo dei cristalli di ghiaccio è ritardata al di sotto dei -
130°C. Per massimizzare il recupero delle cellule dopo lo
scongelamento, le cellule devono essere riscaldate molto rapidamente,
posizionando le vials direttamente dall’azoto liquido nel bagnetto
riscaldato (leggermente agitato) a 37°C. Non appena l’ultimo cristallo di
ghiaccio si è sciolto, le cellule devo essere diluite nel medium di coltura
pre-riscaldato.
Materiale necessario
NON STERILE
Cappa a flusso laminare
Pipette e pipettatrice automatica
Mister Frosty
Microscopio ottico a contrasto di fase
Contenitore con del ghiaccio
Emocitometro
STERILE
Medium appropriato per le cellule che devono essere congelate
Flasks con colture cellulari sane e prossime alla confluenza
Pipette e puntali
Tubi per la centrifuga
PBS (senza calcio e magnesio) – a differenza HBSS e EBSS, il
PBS è stato approntato proprio per mantenere un pH fisiologico in
un sistema aperto. Il calcio e il magnesio non sono stati inclusi

nella soluzione, perché questi cationi giocano un ruolo
fondamentale nei rapporti di adesione cellula-cellula.
Tripsina o agenti di disgregazione diluiti in PBS privo di calcio e
magnesio. La concentrazione della tripsina dovrebbe essere
ottimizzata affinché possa rimuovere le cellule il più velocemente
possibile, provocando il minimo stress o danno possibile.
Criovials da 2 ml.
Medium per il congelamento - questo è dato dal medium di
coltura delle cellule completo contenente il 10% di DMSO (sterile).
Se non fosse a disposizione del DMSO sterile, si può rendere tale
attraverso la filtrazione del composto con filtri dotati di membrane
di nylon 0.2 µm (es Corning Catalog # 431224) in contenitori di
polipropilene o acciaio inossidabile (5 ml).
Trypan Blue per il test di vitalità cellulare.
Procedura
Prima di eseguire il congelamento, le cellule dovrebbero essere coltivate
e presentarsi alla sub-confluenza, ciò in uno stato di crescita attiva per
assicurare il massimo livello di salute e di buona raccolta. Idealmente ,
il medium dovrebbe essere cambiato il giorno precedente. Usando il
microscopio a contrasto di fase, bisogna controllare l’aspetto generale
della coltura, e controllare se ci sono segni di una eventuale
contaminazione microbica. E’ molto importante esaminare la coltura
cellulare direttamente nella flask, per evidenziare l’eventuale presenza
di piccole colonie fungine che possono flottare a livello dell’interfaccia
medium-aria, tuttavia invisibili con il microscopio. Sarebbe meglio che le
colture vengano mantenute con un medium privo di antibiotici per
almeno una settimana prima del congelamento, per aiutare a svelare le
possibili contaminazioni.
Le cellule andrebbero trattate gentilmente durante la raccolta, poiché è
molto difficile per le cellule danneggiate durante questa fase riuscire a
sopravvivere agli stress, che vengono applicati durante i processi di

congelamento e scongelamento. In generale, bisognerebbe essere in
grado di ottenere delle flasks da 75 cm2 un quantitativo di cellule pari a
1.5*107 prossime alla confluenza (attenzione dipende sempre dal tipo
cellulare e dal grado di confluenza). Questo dovrebbe essere sufficiente
per poter allestire diverse vials contenenti 2*106 cellule.
1. Utilizzando una pipetta sterile rimuovere e buttare tutto il
vecchio medium di coltura.
2. Per una flask da 75 cm2, ricoprire il monostrato cellulare con 5
ml di PBS (senza calcio e magnesio), agitare gentilmente per
allontanare tutte le tracce del siero fetale, quindi allontanare il
prodotto del lavaggio.
3. Aggiungere 4-5 ml di tripsina alla flask e mettere le cellule in
incubatore a 37°C per almeno un minuto(un prelavaggio con
l’enzima riduce i tempi di esposizione; prelevare circa 3 ml
della soluzione di tripsina e lasciare che le cellule si stacchino).
4. Controllare lo stato della coltura cellulare: le cellule si devono
staccare e devono avere un aspetto rotondeggiante. Dare dei
colpetti alla flask in modo da ottenere il distaccamento
completo delle cellule dalla superficie di plastica. Aggiungere
quindi 5 ml di medium di coltura alla sospensione cellulare e
utilizzando la stessa pipetta, sciacquare vigorosamente le
cellule rimaste ancora adese alla flask.
5. Raccogliere la sospensione cellulare in una falcon da 15 ml e
metterla in ghiaccio. Prendere una aliquota per la conta
cellulare e poi centrifugare a 1000 g per 5 minuti al fine di
ottenere un pellet di cellule. Mentre le cellule sono in centrifuga
è bene eseguire una conta vitale (con il Trypan blu) e calcolare
il numero di cellule/ml e quindi il numero totale.
6. Allontanare il surnatante ottenuto dopo la centrifugazione e
risospendere il pellet cellulare in un quantitativo sufficiente di
medium di congelamento contenente il 10% di DMSO per

ottenere una concentrazione finale di cellule pari a 1-2*106
cellule/ml. Sebbene non sia direttamente tossico, il DMSO è un
potente solvente ed è in grado di penetrare attraverso la pelle
intatta (lasciando un sapore di pesce o di aglio in bocca). Tutte
le precauzioni di dovere devono essere messe in atto quando si
usa questo composto.
7. Contrassegnare le vials con il numero o la sigla della linea
cellulare, il passaggio e la data di congelamento. Quindi
aggiungere 1.5-1.8 ml della sospensione cellulare con DMSO
per ciascuna vials e sigillare.
8. Posizionare le vials in un contenitore (Mr. Frosty) e mettere il
tutto in un congelatore -80°C. Dopo 24 ore trasferire il
materiale in azoto liquido per lo stoccaggio definitivo.
9. Registrare tutte le informazioni inerenti le cellule su adeguato
supporto: tipo di coltura, nome linea, passaggio, data di
congelamento, medium di congelamento e metodo utilizzato,
numero di cellule per vials, numero di vials prodotte e
congelate inizialmente e numero rimante, loro localizzazione e
risultati dei tests eseguiti (sterilità, micoplasmi, specie,
cariotipo).

SCONGELAMENTO
1. Utilizzando un appropriato equipaggiamento di sicurezza (guanti –
occhiali), prendere la vial dalla sede di stoccaggio (bidone azoto
liquido), facendo attenzione che sia proprio quella che si vuole
prendere (CTR etichetta)!!!
2. Posizionare la vial nel bagnetto agitato preventivamente riscaldato
a 37°C, controllando che il contenuto si scongeli rapidamente (60-
90 secondi). Questo passaggio è estremamente importante per
poter ottenere il massimo recupero di tutte le cellule congelate.
Infatti il rapido scongelamento riduce o previene i danni legati alla
formazione di cristalli di ghiaccio all’interno delle cellule durante la
reidratazione.
3. Poiché alcuni agenti crioprotettivi possono danneggiare le cellule
in seguito ad una prolungata esposizione, rimuovere al più presto
questi composti. Diversi sono gli approcci in relazione al
crioprotettore utilizzato e alle caratteristiche delle cellule:
a. La maggior parte delle cellule viene raccolta in una
contenitore con 10 ml terreno di coltura riscaldato (37°C) e
viene eseguita una centrifugazione (1200 rpm per 5 min). Il
pellet di cellule recuperato dopo l’allontanamento del
surnatante viene risospeso (10 ml di terreno) e seminato in
piastre o flask.
b. Alcune linee cellulari possono essere direttamente seminate,
dopo lo scongelamento, in piastre o flasks utilizzando il
terreno di coltura appropriato. Si attuerà un cambio di
medium entro 6-8 ore dallo scongelamento, quando le
cellule saranno adese al substrato di crescita.
c. Altre ancora più resistenti possono aspettare il giorno
successivo (si verifica sempre e comunque una diluizione
del crioprotettore nel terreno di coltura).

MTT ASSAY
L’MTT test misura l’attività metabolica delle cellule vitali, è utile per la
valutazione della proliferazione cellulare, la vitalità cellulare e la
citotossicità. Questo è un saggio che non prevede l’uso di molecole
radioattive, è necessario utilizzare un microplate reader.
Il principio base su cui si regge questo saggio è la riduzione dei sali di
tetrazolio da parte della succinato deidrogenasi mitocondriale delle
cellule vitali a sali di formazano. Questi sono sali insolubili in acqua e
possono essere solubilizzati impiegando DMSO.

L’assorbanza letta è direttamente correlate al numero di cellule vitali.

INCORPORAZIONE DI TIMIDINA TRIZIATA
Durante la proliferazione cellulare il DNA deve essere replicato prima
che la cellula si divida in due cellule figlie. Questa stretta associazione
fra sintesi del DNA e duplicazione cellulare fa si che la misurazione della
sintesi del DNA sia molto interessante per la valutazione della
proliferazione cellulare.
Se i precursori del DNA marcati fossero aggiunti alla coltura cellulare, le
cellule che stanno per duplicarsi andrebbero ad incorporare i nucleotidi
marcati nel loro DNA.
Tradizionalmente questi saggi prevedono
l’impiego di nucleosidi radiomarcati come la timidina triziata ([3H]-
TdR). Il quantitativo di timidina triziata incorporata nel DNA delle cellule
viene ad essere misurata con la radioattività in soluzione attraverso
l’aggiunta del liquido di scintillazione (LSC).

Il liquido di scintillazione o liquido scintillatore è un solvente organico
contenente molecole di fluor, che quando viene colpito da un elettrone
o da un fotone emette un fotone. Un fotorilevatore converte il segnale
luminoso in segnale elettrico. In questo modo si ottiene un dato
espresso in [cpm] (colpi per minuti), ma dato che una parte delle
disintegrazioni non viene rilevata è una sottostima delle disintegrazioni
per minuto. Approssimativamente cpm= 30% dpm (Andrea Telatin,
htpp//www.neofox.it/bioblog).

COSA FARE E COSA NON FARE
COSA FARE
1. Uso da parte del personale di tutto l’equipaggiamento protettivo
(camice, guanti, occhiali di protezione) durante il lavoro. Inoltre
guanti isolati termicamente, visore facciale e grembiule
antispruzzo dovrebbero essere indossati quando si maneggia
l’azoto liquido.
2. Le superfici di lavoro dovrebbero essere pulite e prive di
ingombri.
3. Marcare correttamente i reagenti, le falsks e le ampolle con il
nome del contenuto e la data di preparazione.
4. Manipolare una linea cellulare per volta (se possibile). Questo
consente di ridurre al minimo i fenomeni di cross contaminazione
o di scambio di identificativo. Inoltre in questo modo si riduce la
diffusione di batteri e micoplasmi attraverso la produzione di
aerosols fra le diverse bottiglie di terreni e flasks aperte sotto
cappa.
5. Pulire le superfici di lavoro con i prodotti disinfettanti appropriati
(es. etanolo al 70%) negli intervalli fra le diverse operazioni e
aspettare un minimo di 15 minuti quando si cambia linea cellulare
di lavoro.
6. Dove possibile mantenere separate le bottiglie dei terreni per
ciascuna linea cellulare in coltivazione.
7. Esaminare le colture cellulari e i terreni giornalmente per
mettere in evidenza le possibili contaminazioni da batteri o
funghi. Questo include i terreni acquistati.
8. Controllare i certificati di qualità di tutti i reagenti e terreni prima
dell’uso.
9. Ridurre al minimo i contenitori di cartone e di altro genere in tutte
le aree dove si lavora con le colture cellulari.

COSA NON FARE
1. Non utilizzare in modo spropositato gli antibiotici, perché questo
comporterebbe inevitabilmente la selezione di ceppi resistenti
compromettendo la validità della linea cellulare utilizzata.
2. Non consentire ad un accumulo dei rifiuti nei locali dove sono
presenti l’incubatore e la cappa biologica.
3. In laboratorio di colture cellulari non devono essere presenti
troppe persone (soprattutto contemporaneamente).
4. Non maneggiare senza precauzioni cellule la cui fonte non sia
stabilita e prima di tutti i test di controllo (utilizzare la
quarantena).
5. Evitare di mantenere in continuazione cellule in coltura (meglio
congelare).
6. Evitare le che cellule siano alla piena confluenza. Sempre meglio
eseguire una subcoltura quando queste hanno raggiunto il 70-
80% della confluenza o dal dato suggerito da chi ha fornito il
materiale cellulare.
7. Non utilizzare materiale scaduto. L’emivita è di sole 6 settimane a
+4°C, una volta addizionati glutamina e siero.
8. Evitare di utilizzare bagni termostatati privi di soluzioni per il
controllo della carica batterica.
9. Non utilizzare strumenti privi di controllo e di taratura. Le cappe
biologiche come tutti gli altri strumenti devono essere sottoposti
a periodici controlli di sicurezza e taratura.

Riferimenti
Sigma-Aldrich and ECACC would like to thank Dr. Alison Stacey,
Cambridge, UK and Labcaire, Clevedon, UK for their help in compiling
this laboratory handbook.
Corning Incorporated
Life Sciences
http://catalog.corning.com/Lifesciences/greater_europe/en/techInfoInd
ex.asp?catalog_name=TechDocs&category_name=Technical_Informati
on(root)®ion=ge&language=en&Page=1
web site at www.sivb.org/edu_terminology.asp





























