Embed Size (px)
Citation preview

H. Bartosz-BechowskiP. DavisJ. SlaninovaE. MalatynskaD. StropovaF. PorrecaH.I. YamamuraV.J. Hruby
Authors' af®liations:
H. Bartosz-Bechowski*, and V.J. Hruby,
Department of Chemistry, University of Arizona,
Tucson, AZ 85721 USA. *Present address:
Institute of Chemistry, University of Wroclaw, 14
F. Joliot Curie St., 50383 Wroclaw, Poland.
P. Davis, J. Slaninova{, E. Malatynska, D.
Stropova, F. Porreca and H.I. Yamamura,
Department of Pharmacology, University of
Arizona, Tucson, AZ 85721 USA. {Present
address: Institute of Organic Chemistry &
Biochemistry, Czech Academy of Sciences,
Flemingova 2, 11610 Prague 6, Czech Republic.
Correspondence to:
Dr Victor J. Hruby
Department of Chemistry,
University of Arizona,
Tucson, AZ 85721
USA
Dates:
Received 7 July 1998
Revised 6 August 1998
Accepted 6 October 1998
To cite this article:
Bartosz-Bechowski, H., Davis, P., Slaninova, J.,
Malatynska, E., Strpova, D., Porreca, F., Yamamura, H. I.
& Hruby, V. J. Cyclic enkephalin analogs that are hybrids
of DPDPE-related peptides and Met-enkephalin-Arg-Gly-
Leu: prohormone analogs that retain good potency and
selectivity for d opioid receptors.
J. Peptide Res., 1999, 53, 329±336.
Copyright Munksgaard International Publishers Ltd, 1999
ISSN 1397±002X
Cyclic enkephalin analogsthat are hybrids of DPDPE-
related peptides and Met-enkephalin-Arg-Gly-Leu:prohormone analogs thatretain good potency andselectivity for d opioid
receptors
Key words: DPDPE analogs; enzymatically cleavable peptides;
opioid peptides: prodrugs; structure±activity relationships
Abstract: We report here on the binding af®nity and bioassay
results of cyclic enkephalin analogs comprising a cyclic moiety
and C-terminal fragment of MERGL, where ME denotes
methionine enkephalin. MERGL (YGGFMRGL) has been suggested
to be cleaved enzymatically by membrane-bound enkephalinase
24.11 to leave ME and the tripeptide RGL. In our study we have
synthesized hybrids of DPDPE or DPLCE and the C-terminal
tripeptide RGL in order to mimic a prohormone able to cross the
blood±brain barrier. The study has shown that of the homologs
presented here, analogs of DPLCE often are more potent at delta
opioid receptors both in binding af®nity and in bioactivity at the
MVD, than DPDPE. Our hypothesis that hybrids (consisting of the
drug and the spacer for the carrier) could be designed which
would either have no opioid activity or, alternatively, be by
themselves very active, has been veri®ed.
Abbreviations: Symbols and abbreviations are in accord with the
recommendation of the IUPAC-IUB Commission on Nomenclature
(J. Biol. Chem. 1972, 247, 977±989). The optically active amino
acids are of L-chirality unless otherwise noted. Other
abbreviations included: DPLCE, c[D-Pen2,L-Cys5]enkephalin; DPDPE,
c[D-Pen2,D-Pen5]enkephalin; DPLPE, c[D-Pen2,L-Pen5]enkephalin; Na-
Boc, Na-tert-butyloxycarbonyl; BOP,
benzotriazolyltris(dimethylamino)phosphonium
hexa¯uorophosphate; DICI, diisopropylcarbodiimide; DCM,
dichloromethane; CTOP, c[D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-
NH2]; Pen, penicillamine; HOBT, N-hydroxybenzotriazole; TFA,
tri¯uoroacetic acid.
329

In our search for an enzymatically cleavable spacer for
enkephalin conjugates that might cross the blood±brain
barrier (BBB), we have focussed our attention on naturally
occurring extended enkephalins which undergo enzymatic
proteolysis to release the enkephalins from their precur-
sors. One such peptide is the octapeptide Tyr-Gly-Gly-
Phe-Met-Arg-Gly-Leu (methionine enkephalin-RGL,
MERGL). This native endogenous enkephalin homolog
was found in mammalian brains (1, 2) including humans
(3), and has opioid activity (4, 5). MERGL has some
preferences for the m-opioid receptor with a Ki of 23.7 nm
at the d-site and Ki 9.0 nm at the m-site (5±7). The MERGL
sequence is encoded in the DNA of preproenkephalin (7,
8). The release of MERGL from its precursor(s) is well
documented (9, 10). It was suggested some time ago that
methionine enkephalin (ME) is released from MERGL
upon the action of the membrane-bound form of endo-
peptidase 24.15 which is found in relatively high con-
centration in the brain (11). The release of methionine
enkephalin (ME) from MERGL must proceed via cleavage
of the C-terminal tripeptide Arg-Gly-Leu (RGL). For our
purposes the tripeptide RGL in conjunction with a
lipophilic peptide might play the role of a `Trojan horse'
enabling the conjugate to cross the BBB. Once the
conjugate reaches the brain it can enzymatically be
cleaved to release the enkephalin analog.
It is generally accepted that there are at least three types of
opioid receptors denoted as mu (m), delta (d) and kappa (k),
and much has been done to develop peptide ligands speci®c
for these receptors (12±15).
The physiological roles for these receptors are not yet fully
understood and are a matter of intense investigation. The mreceptors prefer morphine-like drugs and are blocked by
naloxone. It has been suggested that morphine-induced
analgesia might be mediated by m1 receptors, whereas the
excitation of the m2 receptors might be involved in
respiratory depression, a serious side-effect in the clinical
use of morphine (16±18). The kappa receptors are recognized
by ketocyclazocine and related drugs, and presumably their
endogenous peptide counterparts are the Dynorphines (13),
but these potent endogenous peptides are not highly k
receptor selective.
In all cases, the stimulation of opioid receptors
produces analgesia (antinociception), generally by the
reduction of central autonomic and endocrine response to
a pain stimulus. However, the use of m opiates has several
disadvantages such as constipation, respiratory depres-
sion, tolerance, and addiction. Since the question of the
physiological roles of this receptor diversity is still to be
answered, the search for highly potent and selective
opioid ligands has continued to help address this issue.
Highly selective and potent opioid ligands, both agonists
and antagonists, should enable the precise description of
how these particular receptors act in the human body and
how they interact together. It has been suggested recently
that the binding sites for d opioid agonists and antago-
nists are different (19). The possibility of `switching on±
off' of particular receptor types or subtypes should lead to
a better understanding of the interactions that lead to
desired pharmacological effects as well as elimination of
undesirable side-effects. This approach will lead to
clinically useful drugs for pain relief in long-term therapy
and/or for the replacement of m opiates (such as
morphine) in high-dose treatments at terminal stages of
cancer.
During the last 15 years many efforts have led to the
development of potent and selective ligands for each type
of opioid receptor (12±14). One of the more d selective
ligands is cyclic-[dPen2,dPen5]enkephalin (DPDPE)
synthesized in our laboratory (20±22). The cyclization of
the peptide chain via a disul®de bridge has enabled these
peptides to adapt a biologically active conformation that
greatly prefers d opioid receptors rather than m or k
receptors. In binding studies, DPDPE showed favorable
binding properties towards d opioid receptors. In bioassays
based on electrically-induced smooth muscle contraction
of the mouse vas deferens (MVD assay) and the guinea pig
ileum longitudinal muscle-myenteric plexus tissue (GPI
assay), DPDPE was found to be over 2000 times more
potent at d receptors than at m receptors. Introduction of p-
halogen substituted Phe4 residues in place of Phe4 led to
further improvement in the biological potencies and
selectivities of DPDPE and its analogs (23±26). Moreover,
the chloro substituted analog had better antinociceptive
properties than DPDPE in the hot-plate test after i.c.v.
administration (24).
As for the use of possible precursor conjugates, it is
necessary to know what biological properties the conjugates
and their fragments have. In this report we have studied the
binding and bioassay properties of hybrids consisting of the
highly d-selective cyclic enkephalin analogs, and the
tripeptide ArgGlyLeu derived from MERGL. Several of these
analogs have very high potency at d opioid receptors, and
selectivity for d versus m opioid receptors. Binding and
bioassays at k receptors were not determined because
DPDPE analogs have very little interaction with kappa
opioid receptors (20).
Bartosz-Bechowski et al . Cyclic enkephalin analogs
330 | J. Peptide Res. 53, 1999 / 329±336

Experimental Procedures
General methods of peptide synthesis
The peptides were synthesized by solid-phase methods of
peptide synthesis. The protected amino acids and chlor-
omethylated polystyrene resin (cross-linked with 1% of
divinylbenzene, 1 meq/g) were purchased from Bachem
(Torrance, CA, USA). d-Pen(S-p-MeBzl) was obtained from
Peptides International (Louisville, KY, USA). The C-term-
inal amino acids were attached to the resin by the Gisin
method (Cs salt in DMF for 18 h at 508C) (27), and
substitution levels of 0.6±0.84 meq/g were achieved. The
Na-Boc protecting group was removed using TFA±DCM±
anisole mixture (48/50/2, v/v). The protected amino acids
were coupled using diisopropycarbodiimide (DICI) as cou-
pling reagent. The amino acids and DICI were used in
threefold excess. To diminish the cost, the para-substituted
phenylalanines were coupled to the growing peptide chain
by means of BOP in the presence of HOBT used in 1.2 excess
(see details below). The peptides and the protecting groups
were removed using liquid HF. The crude sulfhydryl
peptides were oxidized without puri®cation. The sulfhydryl
peptides were obtained in greater than 90% purity by HPLC.
Oxidation
The peptides were oxidized by methods described previously
(25, 28). Brie¯y, the peptide was dissolved in methanol (300±
400 mg in 50 mL) and added from a syringe pump to a well-
stirred solution of oxidant (1±1.5 L). The oxidant solution
was prepared by dissolving potassium ferricyanide
(K3Fe(CN)6) in fourfold excess in 0.05 m ammonium acetate
buffer pH 8.5. The use of buffer allowed easy maintenance of
slightly basic conditions, and control of pH was unneces-
sary. The addition rate was calculated to be < 10 mg of
peptide/h/L of oxidant. In this way the formation of peptide
oligomer was diminished greatly.
After addition of the peptide was completed, the reaction
mixture was stirred for an additional 5±6 h and acidi®ed
carefully with glacial acetic acid. The excess of ferro- and
ferricyanide ions were removed by ion-exchange resin
Amberlite IRA-45 (Cl± form). The resin was ®ltered off,
the solution concentrated under diminished pressure at
temperatures below 408C, and lyophilized. The lyophilized
powder was dissolved again in acetic acid, ®ltered to remove
inorganic salts formed, and re-lyophilized.
The crude cyclic peptides were puri®ed by preparative
HPLC on an ODS column (Vydac 218TP152050), 5 3 25 cm,
using a Rainin HPLX instrument with detection at 220 and
254 nm. The pure fractions were pooled and lyophilized.
The purity of the peptides was checked by analytical HPLC
(ODS column 4.6 3 25 cm, Vydac 218TP104) using a
Hewlett-Packard 1090 instrument (detection at 220, 254,
280 nm) with 0.1% aqueous TFA and a 0±50% acetonitrile
gradient over 30 min. The chromatograms were analyzed by
a computer program provided by the manufacturer (Hewlett-
Packard) and the peptides were shown to be . 98% pure.
TLC was performed in four solvent systems on silica gel and
visualized by ninhydrin and iodine vapors; all peptides
showed single spots (Table 1). The amino acids analyses
were performed at the University of Arizona Biotechnology
Core Facility and gave satisfactory results (u 10% of
theoretical values). The system was a dedicated Applied
Biosystem Model 420A amino acid analyzer with automatic
hydrolysis (vapor phase at 1608C for 1 h 40 min using 6 n
HCl) and precolumn phenylthiocarbamoyl-amino acid
(PTA-AA) analysis (d-Pen could not be detected). FAB-MS
spectra were in agreement with the amino acid sequence and
the composition of each analog. The analytical data of the
compounds synthesized in this paper are given in Table 1.
H-Tyr-c[D-Pen2,Cys5]enkephalin-Arg6-Gly7-Leu8-OH (H-Tyr-c[D-Pen-
Gly-Phe-Cys]-Arg-Gly-Leu-OH, 1)
The peptide was obtained by stepwise elongation of the
peptide resin by the method outlined above starting from 3 g
of Na-Boc-Leu-resin, substitution level 0.6 meq/g. The
following amino acids were coupled to the resin: Boc-Gly,
Boc-Arg(Tos), Boc-Cys(p-MeBzl), Boc-Phe, Boc-Gly, Boc-d-
Table 1. Analytical properties of new peptides
TLCa Rf values HPLCb FAB-MS
Peptide I II III IV red.c ox.d calc. obs.
1 0.38 20.48 944 0.75 0.63 0.17 20.01 944
2 0.27 17.32 831 0.55 0.56 0.09 15.74 831
3 0.39 22.0 978 0.77 0.62 0.20 20.92 978
4 0.32 19.36 865 0.68 0.55 0.16 19.04 865
5 0.48 23.66 972 0.81 0.67 0.27 20.42 972
6 0.36 31.05e 860 0.72 0.66 0.23 27.05e 861
a. Silica gel plates (Analtech), 0.25 mm, solvent path 8 cm. Eluants used areas follows: I, n-butyl alcohol±acetic acid±water 4 : 1:1; II, n-butyl alcohol±acetic acid±pyridine±water 13 : 3:12 : 10; III, isopropyl alcohol±ammonia±water 3 : 1:1; IV, n-butyl alcohol±acetic acid±ethyl acetate±water 1 : 1:1 : 1.b. Retention times (min) for the following system: Hewlett-Packard 1090,column C-18 (Vydac 218TP104) 4.6 mm 3 25 cm, buffer A 0.1% TFA inacetonitrile, buffer B 0.1% TFA in water, gradient 0±50% A in 30 min, ¯owrate 1.0 mL/min, simultaneous detection at 225, 254 and 280 nm. c.Retention times (min) of nonoxidized linear peptides. d. Retention times(min) of pure oxidized (cyclic) peptides e. Gradient used: 0±50% A in 60 min.
Bartosz-Bechowski et al . Cyclic enkephalin analogs
J. Peptide Res. 53, 1999 / 329±336 | 331

Pen(p-MeBzl) and Boc-Tyr(2,6-Cl2Bzl). After the peptide was
assembled, the Na-Boc group was removed by TFA, the resin
was washed several times with DCM and dried overnight
under diminished pressure over KOH; yield was 4.65 g. The
peptide resin was mixed with 4.5 mL of a mixture 1 : 1
cresol and p-thiocresol and then < 45 mL of liquid HF was
added. The reaction mixture was stirred for 1 h at 08C, and
then the HF was distilled off in vacuo. The scavengers were
removed by washing the residue three times with dry ether
and the resin with precipitated peptide was dried in a
desiccator. The peptide was extracted three times with
acetic acid and the acetic solutions were pooled and
lyophilized; yield of crude peptide was 1.89 g. The crude
peptide was cyclized as described above. The peptide was
then puri®ed by preparative HPLC; gradient 15 min of 0.1%
TFA and then 0±60% acetonitrile in 120 min. The main
fractions were pooled, concentrated on a rotary evaporator
and lyophilized; yield was 178 mg. The purity was assessed
to be at least 98%. An additional 112 mg was obtained with
a purity of < 92±95%.
Amino acid analysis: Tyr 0.92 (1.0), Gly 2.0 (2.0), Phe 1.04
(1.0), Arg 0.98 (1.0), Leu 1.05 (1.0). Additional analytical data
are found in Table 1.
H-Tyr-c[D-Pen2,Cys5]enkephalin-Arg6-Gly7-OH (Tyr-c[D-Pen-Gly-
Phe-Cys]-Arg-Gly-OH, 2)
This peptide was synthesized by a similar method to that
described above (from 3 g of resin, 0.67 meq/g). The yields
were: 5.0 g of peptide resin, 1.61 g of crude peptide and
93 mg of pure peptide.
Amino acid analysis: Tyr 0.87 (1.0), Gly 2.02 (2.0), Phe
0.97 (1.0), Arg 0.98 (1.0). Additional analytical data are
included in Table 1.
H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-Leu-OH, 3
This compound was synthesized by the method described
above starting from 2.5 g of resin (substitution level 0.6
meq/g). The Na-Boc-Phe(p-Cl) was coupled to the resin using
BOP (1.2 equiv), HOBT (1.2 equiv) and DIPEA (3 equiv) in N-
methylpyrrolidinone. Yields were 4.5 g of peptide resin,
1.72 g of crude peptide, and about 100 mg pure peptide.
Amino acid analysis: Tyr 0.91 (1.0), Gly 2.12 (2.0), Phe(p-
Cl) 1.01 (1.0), Leu 1.03 (1.0), Arg 0.99 (1.0). Additional
analytical data are included in Table 1.
H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-OH, 4
The title compound was synthesized starting from 2 g of
resin (substitution level 0.67 meq/g) applying the above
method which yielded 3.50 g of peptide resin, 884 mg of
crude peptide, and 232 mg of pure peptide.
Amino acid analysis: Tyr 0.84 (1.0), Gly 2.01 (2.0), Arg 1.00
(1.0). Additional analytical data are included in Table 1.
H-Tyr-c[D-Pen2,D-Pen5]-Arg6-Gly7-Leu8-enkephalin-OH, 5
This analog was synthesized in the same manner from 2.5 g
resin (0.60 meq/g). Yields were: 5.6 g of peptide resin, 1.10 g
of crude peptide, and 53 mg of pure peptide.
Amino acid analysis: Tyr 0.90 (1.0), Gly 2.03 (2.0), Phe
0.96 (1.0), Arg 0.92 (1.0). Additional analytical data are
included in Table 1.
H-Tyr-c[D-Pen2,D-Pen5]-Arg6-Gly7-enkephalin, 6
This analog was synthesized in the same manner from 2.5 g
resin (0.80 meq/g). Yields were 3.41 g of peptide resin,
824 mg of crude peptide, and 205 mg of pure peptide.
Amino acid analysis: Tyr 1.06 (1.0), Gly 2.12 (2.0), Phe 1.0
(1.0), Arg 0.98 (1.0). Additional analytical data are included
in Table 1.
Radioligand binding methods
Membranes were prepared from whole (less cerebellum)
brain taken from adult male Sprague-Dawley rats (250±
300 g) obtained from Harlan Sprague-Dawley, Inc., IN, USA.
Following decapitation, the brain was removed, dissected
and homogenized at 08C in 20 volumes of 50 mm Tris-HCl
buffer adjusted to pH 7.4 using a Te¯on glass homogenizer.
The membrane fraction obtained by centrifugation at
48 000 g for 15 min at 48C was resuspended in 20 volumes
of fresh Tris buffer and incubated at 258C for 30 min to
dissociate any receptor-bound endogenous opioid peptides.
The incubated homogenate was centrifuged again and the
®nal pellet resuspended in 20 volumes of fresh Tris buffer.
Radioligand-binding inhibition assay samples were prepared
in an assay buffer consisting of 50 mm Tris-HCl, 1.0 mg/mL
bovine serum albumin, 30 mm bestatin, 50 mg/mL bacitra-
cin, 10 mm captopril, and 0.1 mm toluenosulfonyl ¯uoride,
pH 7.4. The radioligands used were [3H]c[d-Pen2,p-Cl-
Phe4,d-Pen5]enkephalin (31) at a concentration of 0.75 nm
and [3H]CTOP (32) (New England Nuclear, Boston, MA,
USA) at a concentration of 0.5 nm. Peptide analogs were
Bartosz-Bechowski et al . Cyclic enkephalin analogs
332 | J. Peptide Res. 53, 1999 / 329±336
dissolved in assay buffer prior to each experiment and added
to duplicate assay tubes at 10 concentrations over an 800-
fold range. Control (total) binding was measured in the
absence of any inhibitor while nonspeci®c binding was
measured in the presence of 10 mm naltrexone. The ®nal
volume of the assay samples was 1.0 mL of which 10%
consisted of the membrane preparation in 0.1 mL of Tris-
HCl buffer. Incubations were performed at 258C for 3 h after
which the samples were ®ltered through poly (ethylenei-
mine)-treated GF/B glass ®ber ®lter strips. The ®ltrates were
washed three times with 4.0 mL of ice-cold normal saline
before transfer to scintillation vials. The ®ltrate radio-
activity was measured after adding 10 mL of cocktail
consisting of 16 g of Crystal Fluor (West Chemical, San
Diego, CA) in 1.0 L of Triton X-100 and 2.0 L of toluene to
each vial and allowing the samples to equilibrate over 8 h at
48C. The data were analyzed by using nonlinear least-
squares regression analysis on the Apple II Plus computer.
Programs were generously written by Susan Yamamura.
GPI and MVD bioassays
Electrically-induced smooth muscle contractions of mouse
vas deferens and strips of guinea pig ileum longitudinal
muscle-myenteric plexus were used for the bioassays.
Tissues came from male ICR mice weighing 25±40 g, and
from male Hartley guinea pigs weighing 250±500 g. The
tissues were tied to a gold chain with suture silk, suspended
in 20-mL baths containing 378C oxygenated (95% O2, 5%
CO2) Krebs bicarbonate solution (magnesium free for the
MVD), and allowed to equilibrate for 15 min. The tissues
were then stretched to optimal length, previously deter-
mined to be 1 g tension (0.5 g for MVD), and allowed to
equilibrate for 15 min. The tissues were stimulated trans-
murally between platinum wire electrodes at 0.1 Hz, 0.4-ms
pulses (2.0-ms pulses for MVD), and supra maximal voltage.
Drugs were added to the baths in 14±60 mL volumes. The
agonists remained in contact with the tissue for 3 min
before the addition of the next cumulative doses, until
maximum inhibition was reached. Percentage inhibition
was calculated by using the average contraction height for
1 min preceding the addition of the agonist divided by the
contraction height 3 min after exposure to the dose of the
agonist. IC50 values represent the mean of not less than four
tissues. IC50 estimates, relative potency estimates, and their
associated standard errors were determined by ®tting the
mean data to the Hill equation by using a computerized
nonlinear least-squares method. Each assay was repeated at
least three times in duplicate for an n = 6 or more.
Results and Discussion
The peptides were synthesized by the solid-phase method of
peptide synthesis. The protected peptides were cleaved from
the resin and the side chain-protecting groups removed by
published methods (see Experimental Procedures). The
linear peptides (with free ±SH groups) were oxidized by
K3[Fe(CN)6] in 0.05 m ammonium acetate buffer at pH = 8.5
as described previously (25, 28). Brie¯y, the linear peptide
was dissolved in methanol and was added dropwise by
means of syringe pump to the oxidant solution with the rate
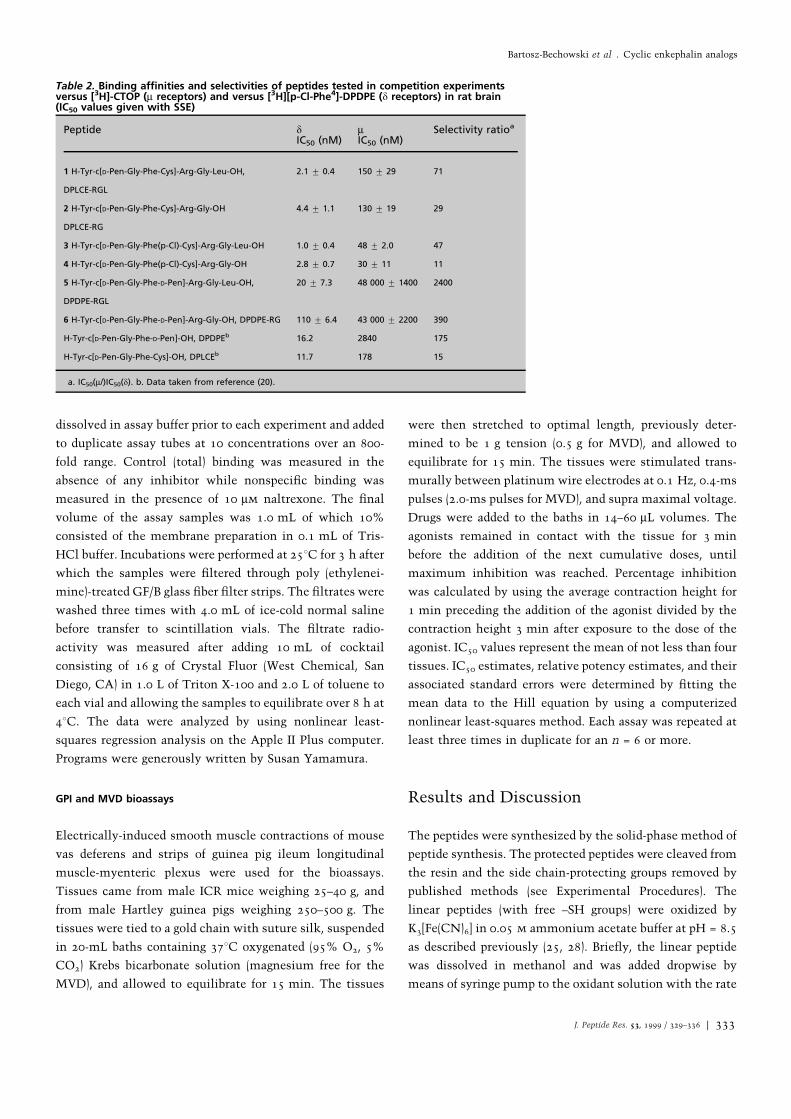
Table 2. Binding af®nities and selectivities of peptides tested in competition experimentsversus [3H]-CTOP (m receptors) and versus [3H][p-Cl-Phe4]-DPDPE (d receptors) in rat brain(IC50 values given with SSE)
Peptide dIC50 (nM)
mIC50 (nM)
Selectivity ratioa
1 H-Tyr-c[D-Pen-Gly-Phe-Cys]-Arg-Gly-Leu-OH,
DPLCE-RGL
2.1 u 0.4 150 u 29 71
2 H-Tyr-c[D-Pen-Gly-Phe-Cys]-Arg-Gly-OH
DPLCE-RG
4.4 u 1.1 130 u 19 29
3 H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-Leu-OH 1.0 u 0.4 48 u 2.0 47
4 H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-OH 2.8 u 0.7 30 u 11 11
5 H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-Arg-Gly-Leu-OH,
DPDPE-RGL
20 u 7.3 48 000 u 1400 2400
6 H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-Arg-Gly-OH, DPDPE-RG 110 u 6.4 43 000 u 2200 390
H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-OH, DPDPEb 16.2 2840 175
H-Tyr-c[D-Pen-Gly-Phe-Cys]-OH, DPLCEb 11.7 178 15
a. IC50(m/)IC50(d). b. Data taken from reference (20).
Bartosz-Bechowski et al . Cyclic enkephalin analogs
J. Peptide Res. 53, 1999 / 329±336 | 333

of the addition calculated to be < 10 mg of the crude peptide
per 1 L of oxidant per h to get the best yield of monocyclic
peptide. The structures of the new analogs are given in
Table 2.
All of the peptides prepared (Table 2) showed some
preference in binding to the d opioid receptor. In the binding
studies all DPLCE hybrids (hepta- or octapeptides 1±4)
showed slightly improved af®nity for the d receptor as
compared to the parent pentapeptide DPLCE (Tyr-c[d-Pen-
Gly-Phe-Cys]). The compounds 1±4 are 2.5±10 times more
potent than DPLCE, but in terms of absolute IC50 values the
differences are relatively small. The IC50 value of DPLCE is
11.7 nm, and for the most potent analog, [p-Cl-Phe4]DPLCE-
RGL, the IC50 value is 1.0 nm. The attachment of a fragment
at the C-terminus has only a modest in¯uence on the
binding properties of the hybrids at the central d opioid
receptor. The IC50 changes from 11.7 nm to 4.4 nm in
heptapeptide 2, and to 2.1 nm for the octapeptide 1. In other
words, the attachment of an additional hydrophobic Leu
residue has caused only a small effect on binding af®nity.
We have shown previously that the introduction of a para
halogen substituted phenylalanine at position 4 of the cyclic
peptide resulted in a substantial improvement in binding
af®nity and d opioid receptor selectivity (25, 26). In the
present study the introduction of halogen has lowered the
IC50 value by approximately a factor of 2. For example, for
octapeptide 3, the chlorine atom lowers the IC50 value from
2.1 nm to 1.0 nm.
In the case of DPDPE, both hepta- and octapeptide hybrids
are less potent than the parent DPDPE. The heptapeptide 6
(Table 2) showed weak agonist properties at central d
receptors. Probably, in the case of DPDPE, the constraints
induced by two additional methyl groups do not allow the
longer peptides to adapt the conformation optimal for
interacting with central d receptors.
At the central m receptor, the DPLCE-derived hybrids are
slightly more potent than the parent DPLCE. However, the
hepta- and octapeptides 1 and 2 differ insigni®cantly from
DPLCE. The chlorine atom has only a small in¯uence on the
af®nity. As a result all the DPLCE hybrids are not very
selective in binding assays (Table 2). The most selective and
most potent is the octapeptide DPLCE-RGL with a
selectivity ratio of 71.
In the case of DPDPE hybrids the addition of a peptide
fragment at the C-terminus led to analogs with somewhat
higher IC50 values at the central m opioid receptor.
Surprisingly, the octapeptide 5 with an additional Leu has
d binding activity similar to that of DPDPE, but it binds very
weakly to the m receptor and as a result in the binding study
the octapeptide DPDPE-RGL shows very good selectivity,
although this peptide is not highly potent.
In the bioassay studies (Table 3) the potency of all DPLCE-
derived peptides at the peripheral d receptors are similar to
that of the DPLCE. The length of the peptide chain added at
the C-terminus seems not to be signi®cant. Some improve-
ment resulted from the introduction of a p-chloro atom,
which led to the IC50 value of 0.2 nm in the case of the
heptapeptide 4 [p-Cl-Phe4]DPLCE-RG). Thus potencies for
compounds 1±4 in the MVD (d) assay (Table 3) closely
parallel those seen in the binding af®nity assays (Table 2).
Any addition to DPDPE at the C-terminus showed weaker
potencies in the peripheral d receptor assays (Table 3). Both
extended hepta and octapeptides 5 and 6 are much less
potent than the parent DPDPE (Table 3). This is quite
interesting since the binding af®nities (Table 2) are not
affected so dramatically. Perhaps a conformational interac-
Table 3. Potencies and selectivities of Tyr-c[D-Pen-Gly-Phe-Cys(dPen)] hybrids inbioassays (MVD for d receptors and GPI for m receptors) IC50 values in SSE
Peptide MVD (d)(IC50 nm)
GPI (m)(IC50) (nm)
Ratioa
1 H-Tyr-c[D-Pen-Gly-Phe-Cys]-Arg-Gly-Leu-OH DPLCE-RGL 2.8 u 0.41 230 u 30 83
2 H-Tyr-c[D-Pen-Gly-Phe-Cys]-Arg-Gly-OH DPLCE-RG 1.3 u 0.10 140 u 18 110
3 H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-Leu-OH 0.79 u 0.08 63 u 6.9 80
4 H-Tyr-c[D-Pen-Gly-Phe(p-Cl)-Cys]-Arg-Gly-OH 0.20 u 0.01 91 u 10 450
5 H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-Arg-Gly-Leu-OH 360 u 69 17 000 u 3200 46
6 H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-Arg-Gly-OH 2800 u 270 21 000 u 4000 8
H-Tyr-c[D-Pen-Gly-Phe-D-Pen]-OH DPDPEb 2.19 6900 3160
H-Tyr-c[D-Pen-Gly-Phe-Cys]-OH DPLCEb 0.32 213 670
a. IC50(m)/IC50(d). b. Data taken from reference (20).
Bartosz-Bechowski et al . Cyclic enkephalin analogs
334 | J. Peptide Res. 53, 1999 / 329±336

tion between the d-Pen5 residue and the C-terminal
extension of 5 and 6, or of the C-terminal extension directly
with the receptor, may modify ligand±receptor interactions
in such a way as to reduce the transduction agonist effects in
a highly signi®cant way. On the other hand, the peripheral mreceptor the peptides derived from DPLCE have similar
potency as DPLCE itself. However, in combination with the
high delta activity, the most potent and most selective
compound in this series is the heptapeptide 4, having a
substituted phenylalanine and a dipeptide addition at the C-
terminus with a selectivity ratio of 450.
In conclusion, in all cases the DPLCE-extended peptides
showed higher potency and better selectivity than those
derived from DPDPE. Probably, the peptides derived from
DPLCE are better candidates for a prodrug approach to
introduce enkephalin analogs into the brain. It is important
to emphasize the potential of DPLCE hybrids as prodrugs.
Both the parent pentapeptide DPLCE as well as its longer
analogs are very potent. The di- or tripeptides attached at
the C-terminus of DPLCE did not greatly affect the activity
of the hybrids. Examination of enzyme cleavage and blood±
brain permeability of some of these analogs both in vitro
and in vivo (29, 30) are generally consistent with this
hypothesis. For example, the extended DPDPE analog 6 is
completely stable in both serum and 15% brain homo-
genates, whereas the DPLCE analog 2 is readily converted
to DPLCE in 15% brain homogenates and the analog
DPLCE-RPA is readily cleaved by brain homogenates but
not by serum (30).
Acknowledgment: This work was supported by grant from NIDA,
P01 DA06284. The views expressed are those of the authors and
do not necessarily re¯ect the views of the US Public Health
Service.
References
1. Lindberg, I. & Yang, H.Y.T. (1984)
Distribution of Met-enkephalin-Arg-Gly-Leu
immunoreactive peptides in rat brain. Brain
Res. 299, 73±78.
2. Soinila, S., Bach, N. & Mpitsos, G.J. (1991)
Distribution of Met-enkephalin-Arg-Gly-Leu
immunoreactivity in the rat and mouse
pituitary gland. Reg. Peptides 36, 271±281.
3. Yoshio, J., Kazawa, N., Takadi, Y., Makoto,
S., Mitsuoki, S., Noborn, Y. & Hiroo, Y. (1983)
Parallel distribution of MERGL with ME, LE
and MERF in human and bovine brains. Life
Sci. 33, Suppl. 1, 65±68.
4. Morgan, J., Paterson, S.J. & Kosterlitz, H.W.
(1982) Extended enkephalin sequences. Life
Sci. 31, 1360±1367.
5. Garzon, J., Sanchez-Blazqueez, P., Hollt, V.,
Lee, N.M. & Loh, H.H. (1983) Endogenous
opioid peptides. Comparative evolution of
their receptor af®nities in the mouse brain.
Life Sci. 33, Suppl. 1, 291±294.
6. Zajac, J.M., Ling, N., Rossier, J. & Roques,
B.P. (1983) Receptor speci®city of Met-
enkephalin-Arg-Gly-Leu. Eur. J. Pharmacol.
90, 147±148.
7. Gubler, U., Seeberg, P., Hoffman, B.J., Gage,
L.P. & Udenfriend, S. (1982) Molecular
cloning establishes proenkephalin as
precursor of enkephalin-containing peptides.
Nature 295, 206±208.
8. Noda, M., Furutari, Y., Takahashi, H.,
Toyosoto, M., Hirose, T., Inayama, S.,
Nakanishi, S. & Numa, S. (1982) Cloning and
sequence analysis of DNA for bovine adrenal
preproenkephalin. Nature 295, 202±206.
9. Udenfriend, S. (1984) Preproenkephalin and
the products derived from its processing. In
Opioid Modulation of Endocrine Function
(Deitalia, G., ed.) Raven Press, NY, pp. 1±10.
10. Patey, G., Cupo, A., Chaminade, M., Morget,
J.L. & Rossier, J. (1983) Release of the
heptapeptide Met-enkephalin-Arg-Phe and
octapeptide Met-enkephalinarg-Gly-Leu
from striatum in vitro and their rapid
degradation. Life Sci. 33, Suppl. 1, 117±120.
11. Acker, G.R., Molineux, Ch. & Orlowski, M.
(1987) Membrane-bound form of
endopeptidase 24.15 generates Leu-
enkephalin from dynorphin 1±8, a and b-
neoendorphin and Met-enkephalin from Met-
enkephalin-Arg-Gly-Leu. J. Neurochem. 48,
284.
12. Hruby, V.J. & Gehring, C.A. (1989) Recent
development in the design of receptor speci®c
opioid peptides. J. Med. Res. Rev. 9, 343±401.
13. Holt, V. (1986) Opioid peptides processing
and receptor selectivity. Annu. Rev.
Pharmacol. Toxicol. 26, 59±79.
14. Schiller, P. (1991) Development of receptor-
speci®c opioid peptide analogues. In Progress
in Medicinal Chemistry, vol. 28. (Ellis, G.P.,
West, G.B., eds). Elsevier Science,
Amsterdam, pp. 304±340.
15. Wolozin, B.L. & Pasternak, G.D. (1981)
Classi®cation of multiple morphine and
enkephalin binding sites in the central
nervous systems. Proc. Natl. Acad. Sci. USA
78, 6181±6185.
16. Ling, G.S.F., Spiegel, K., Lockhard, S.H. &
Pasternak, G. (1985) Separation of opioid
analgesia from respiratory depression:
evidence for different receptor mechanisms. J.
Pharmacol. Exp. Ther. 232, 149±153.
17. Paakkari, P., Paakkari, I., Vonhof, S.,
Fuerstein, G. & Siren, A.L. (1993)
Dermorphin analogue Tyr-d-Arg-Phe-
sarcosine induces opioid analgesia and
respiratory stimulation: The role of m1-
receptors. J. Pharmacol. Exp. Ther. 266, 544±
548.
18. Kosterlitz, H.W. & Paterson, S.J. (1985) Types
of opioid receptors. Relation to
antinociception. Phil. Trans. R. Soc. London
B. 308, 291±297.
19. Kong, H., Raynor, K., Yasuda, K., Moe, S.T.,
Portoghese, P.S., Bell, G.I. & Reisine, T.
(1993) A single residue aspartic acid 95 in the
d opioid receptor speci®es selective high
af®nity agonist binding. J. Biol. Chem. 268,
23055±23058.
20. Mosberg, H.I., Hurst, R., Hruby, V.J., Gee, K.,
Yamamura, H.I., Galligan, J.J. & Burks, T.F.
(1983) Bis-penicillamine enkephalins show
pronounced delta opioid receptor selectivity.
Proc. Natl. Acad. Sci. USA 80, 5871±5874.
21. Hruby, V.J. (1992) Strategies in the
development of peptide antagonists. In
Progress in Brain Research, vol.92, 18.
(Joosee, J., Buijs, R.M., Tilders, F.J.H., eds).
Elsevier Science, Amsterdam, pp. 215±224.
Bartosz-Bechowski et al . Cyclic enkephalin analogs
J. Peptide Res. 53, 1999 / 329±336 | 335

22. Akiyama, K., Gee, K.W., Mosberg, H.I.,
Hruby, V.J. & Yamamura, H.I. (1985)
Characterization of [3H][2-d-penicillamine,5-
d-penicillamine]-enkephalin binding to delta
opiate receptors in the rat brain and
neuroblastoma-glioma hybrid cell line (NG
108-15). Proc. Natl. Acad. Sci. USA 82, 2543±
2547.
23. Toth, G., Kramer, T.H., Knapp, R., Lui, G.,
Davis, P., Burks, T.F., Yamamura, H.I. &
Hruby, V.J. (1990) [d-Pen2,d-Pen5]enkephalin
analogues with increased Af®nity and
selectivity for d opioid receptors. J. Med.
Chem. 33, 249±253.
24. Weber, S.J., Greene, D.L., Sharma, S.D.,
Yamamura, H.I., Kramer, T.H., Burks, T.F.,
Hruby, V.J., Hersh, L.B. & Davis, T.P. (1994)
Distribution and analgesia of [3H][d-Pen2,d-
Pen5]-enkephalin and two halogenated
analogues after intravenous administration. J.
Pharmacol. Exp. Ther. 259, 1109±1117.
25. Bartosz-Bechowski, H., Davis, P., Zalewska,
T., Slaninova, J., Porreca, F., Yamamura, H.I.
& Hruby, V.J. (1994) Cyclic enkephalin
analogs with exceptional potency at
peripheral d opioid receptors. J. Med. Chem.
37, 146±150.
26. Hruby, V.J., Bartosz-Bechowski, H., Davis, P.,
Slaninova, J., Zalewska, T., Stropova, D.,
Porreca, F. & Yamamura, H.I. (1997) Cyclic
enkephalin analogues with exceptional
potency and selectivity for delta opioid
receptors. J. Med. Chem. 40, 3957±3962.
27. Gisin, B.F. (1973) The preparation of
Merri®eld-resin through esteri®cation with
cesium salts. Helv. Chem. Acta. 56, 1476±
1482.
28. Misicka, A. & Hruby, V.J. (1994)
Optimization of disul®de bond formation.
Polish J. Chem. 68, 893±899.
29. Weber, S.J., Abbruscato, T.J., Brownson, E.A.,
Lipkowski, A.W., Polt, R., Misicka, A.,
Haaseth, R.C., Bartosz-Bechowski, H.,
Hruby, V.J. & Davis, T.P. (1993) Assessment
of an in vitro blood±brain barrier model using
several [Met5]enkephalin opioid analogues. J.
Pharmacol. Exp. Ther. 266, 1649±1655.
30. Greene, D.L., Hau, V.S., Abbruscato, T.J.,
Bartosz, H., Misicka, A., Lipkowski, A.W.,
Hom, S., Gillespie, T.J., Hruby, V.J. & Davis,
T.P. (1996) Enkephalin analog prodrugs:
assessment of in Vitro conversion, enzyme
cleavage characterization and blood±brain
permeability. J. Pharmacol. Exp. Ther. 277,
1366±1375.
31. Vaughn, L.K., Knapp, R.J., To th, G., Wan,
Y.-P., Hruby, V.J. & Yamamura, H.I. (1989)
A high af®nity, highly selective ligand for
he delta opioid receptor: [3H]-[d-Pen2,pCl-
Phe4,d-Pen5]enkephalin. Life Sci. 45, 1001±
1008.
32. Hawkins, K.N., Knapp, R.J., Lui, G.K., Gulya,
K., Kazmierski, W.M., Wan, Y.-P., Pelton,
J.T., Hruby, V.J. & Yamamura, H.I. (1989)
[3H]-c[H-d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-
Thr-NH2], a potent and highly selective
peptide for mu opioid receptors in rat brain. J.
Pharmacol. Exp. Ther. 248, 73±81.
Bartosz-Bechowski et al . Cyclic enkephalin analogs
336 | J. Peptide Res. 53, 1999 / 329±336





























