Embed Size (px)
Citation preview

( 125 )
Capítulo 11 ... [Demencias]
1111.1.DEMENCIAS
DIAGNÓSTICO DIFERENCIAL
AGUDA SUBAGUDA CRÓNICA
SEMANAS MESES AÑOS
Enfermedad de Alzheimer
Hidrocefalia a presión normal
Enfermedad de Creutzfeldt-JakobEncefalitis
TiPoS De DeMenciaS> Demenciasdegenerativasprimarias:
• Corticales: Enfermedad de Alzheimer, de-mencia por cuerpos de Lewy, demencia frontotemporal, degeneración corticobasal
• Subcorticales:Parálisis supranuclear pro-gresiva, atrofia multisistémica, E. de Hun-tington, E. de Parkinson-demencia
> Demenciassecundarias:• Demencia vascular• Asociada a neoplasia: tumor primario o me-
tastásico, carcinomatosis meníngea, ence-falitis límbica paraneoplásica
• De origen infeccioso: neurosífilis, menin-goencefalitis por Borrelia o Brucella, me-ningoencefalitis tuberculosa, infecciones del SNC fúngicas o parasitarias, complejo demencia-SIDA, leucoencefalopatía mul-tifocal progresiva, encefalitis herpética, panencefalitis esclerosante subaguda (sa-rampión)
• Hidrocefalia a presión normal• De origen endocrino-metabólico: hipo e hi-
pertiroidismo/paratiroidismo/cortisolismo, hipoglucemia crónica, Wilson, degenera-ción hepatocerebral adquirida, encefalopa-tía hepática y urémica, porfirias, Whipple
• Carenciales: vit. B12, ácido fólico, niacina, tiamina
• De origen tóxico: alcohol, metales pesados, fármacos
• En enfermedades desmielinizantes• Postraumática• Por priones: Creutzfeldt-jakob, Gerstmann-
Straussler, insomnio familiar fatal, kuru• Errores innatos del metabolismo
enFerMeDaD De alZHeiMer
EPIDEMIOLOGÍA> Es la causa más frecuente de Demencia.> 45% en Mayores de 85 años. Más frecuente en
mujeres.> 10 - 20 % de Casos Familiares: Herencia AD.
Genes relacionados.
11DeMenciaS
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 126 )
07 neurología y neuroCirugía
11
ETIOPATOGENIA
» FACTORES GENÉTICOS• Cromosoma21:
~ Se codifica la proteína precursora de la amiloide (APP).
~ Esto explica la mayor frecuencia de Alzheimer precoz entre pacientes con Síndrome de Down.
• Cromosoma19:~ Gen APOE o gen para la apolipoproteína E. ~ El genotipo E4 se ha asociado con un
mayor riesgo de Alzheimer y con otras enfermedades (demencia con cuerpos de Lewy, enfermedades de Pick, Parkin-son y Creutzfeld jacob...
• Cromosoma14(presenilina1).• Cromosoma1(presenilina2).
~ Inicio precoz.~ En los casos ligados a presenilinas el
depósito de sustancia amiloide es del tipo beta 42 (43).
» OTROS FACTORES ASOCIADOS CON EL ALZHEIMER• Envejecimiento.• Traumatismo craneal (boxeadores): Asocia-
ción posible pero dudosa.• Bajo nivel educativo.• Aunque la prevalencia es mayor en mujeres
(seguramente por su mayor longevidad), los estrógenos son protectores.
• La ingesta crónica de antiinflamatorios pa-rece proteger.
• La hipertensión arterial crónica y otros fac-tores de riesgo vascular parecen aumentar también el riesgo de padecer Alzheimer.
ANATOMÍA PATOLÓGICA> Atrofia cerebral cortical difusa (Fronto-Tem-
poro-Parietal), con aumento de tamaño de los ventrículos.
> Reducción de Neuronas en el Nucleo Basal de Meynert, donde se produce la AcetilColin Transferasa con disminución de acetil colina (Alteración bioquímica más importante).
> Se ha visto asimismo reducción de los niveles de acetilcolintrasferasa y de receptores coli-nérgicos (sólo el receptor M1, presente en el hipocampo, está relativamente preservado).
> También hay reducción de Noradrenalina, Sero-tonina (5HT), Somatostatina, GABA...
> AlMicroscopioElectrónicoseobservan:• Placas Neuríticas (antes llamadas placas
seniles). ~ situadas entre axón y dendrita; com-
puestas por colecciones esféricas de Amiloide beta o proteína A4, rodeada por dendritas, microglia, etc.
~ Estas proteínas se codifican en el Cro-mosoma 21 (Angiopatía Congófila).
• Ovillos Neurofibrilares:~ Acúmulos de fibrillas intraneuronales,
sobre todo en el hipocampo, con dos proteínas anómalas: Tau hiperfosforila-da y Ubiquitina.
• Angiopatía amiloide o congófila.• Puede ser causa de hemorragias.• Inclusiones eosinófilas en las dendritas
(cuerpos de Hirano).
CLÍNICA> Evolución Crónica: Deterioro intelectual progre-
sivo (meses-años).> Diagnóstico Diferencial con Creutzfeld-jacob.
• Que también lleva Demencia + Mioclonía, pero la demencia es rápida.
> Lo primero es la pérdida de la Memoria reciente.> Fases:
• Primerafase~ Pérdida de Memoria reciente: olvida
nombres, citas, pierde objetos (olvida dónde los dejó), repite preguntas (olvi-da las respuestas)
~ Empobrecimiento del lenguaje: disno-mia, lenguaje estereotipado y repetitivo
~ Desorientación visoespacial: se pierde, dificultad para aparcar, para vestirse, para poner la mesa…
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 127 )
11
Capítulo 11 ... [Demencias]
~ Alteración de la conducta: pérdida de interés en el trabajo y las relaciones sociales, negligencia en la higiene y el vestido, trastornos del sueño,
• Segundafase:~ Mayor deterioro del lenguaje~ Pérdida de la memoria remota y discal-
culia~ Deja de reconocer a los familiares,
ideas delirantes de robo y celos. Pue-den desarrollar un Síndrome de Cap-gras.
~ Es caracteristico del Alzheimer el ma-yor deterioro por las noches.
• Tercerafase:~ Afasias, Apraxias (sobre todo ideatoria e
ideomotora: incapacidad en actividades instrumentales cotidianas como afeitar-se, comer con cubiertos, abrir con las llaves…) y Agnosias
~ Al final el paciente termina postrado sin comunicarse en absoluto.
~ Posible aparición de mioclonías, con-vulsiones...
> También puede haber:• Depresión: 50% pacientes.• Mioclonías, ansiedad, delirio, alucinacio-
nes en algunos.
DIAGNÓSTICO> Es eminentemente clínico.> TAC y RMN:
• Atrofia extensa fronto-parieto-temporal con aumento del tamaño ventricular.
> TEP (Tomografía de Emisión Positrónica): • Prueba que estudia el metabolismo cere-
bral.• Detecta menor consumo de glucosa a nivel
fronto-temporo-parietal.> Los marcadores moleculares genéticos son úti-
les en muy pocos pacientes.
TRATAMIENTO> Sintomas cognitivos:
• INHIBIDORES DE LA ACETILCOLINESTERASA ~ Donepezilo, Galantamina y RivastigminaCuidado en EPOC, arritmias, ulcus, epi-lepsia y síndrome miccional. Interaccionan con otros colinérgicos y con AINEs
• MEMANTINA~ Antagonista de receptores NMDA
> Síntomas conductuales: • Depresión: Antidepresivos tipo ISRS (ser-
tralina, citalopram)• Ansiedad: Ansiolíticos no benzodiacepíni-
cos (buspirona)• Insomnio: Hipnóticos no benzodiacepínicos
(zolpidem) o neurolépticos• Agitación, alucinaciones y delirio: Haloperi-
dol y risperidona
360_los fármacos anticolinesterásicos (donepezi-
lo y rivastigmina) en la enfermedad de alzheimer:1) Son eficaces sólo en los pacientes en estadio
leve de demencia2) Mejoran el rendimiento cognitivo, los defectos
funcionales y los trastornos de conducta3) Producen mejorías escasas en todos los pacientes4) Deben utilizarse en la dosis menor posible5) Tienen efectos secundarios cardiovasculares
potencialmente graves.
DeMencia FronToTeMPoral
> Atrofia cerebral circunscrita: muy localizada en Lóbulo Frontal (y en temporal).
> Puede ser esporádica o familiar, con herencia AD y mutaciones del gen tau en el cromosoma 17.
ANATOMÍA PATOLÓGICA> La atrofia cerebral afecta a la porción anterior de los
lóbulos frontales (más frecuente) y temporales.> Algunos pacientes tienen células globosas de
Pick: células neuronales balonizadas con cuer-pos de Pick. • Son argentófilas.• Tienen agregados esféricos densos en neu-
ronas.
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 128 )
07 neurología y neuroCirugía
11
> Para estos pacientes se reserva actualmente la denominación de Enfermedad de Pick.
> El tratamiento es menos eficaz que en el Alzheimer.
» DIFERENCIAS CON ENFERMEDAD DE ALZHEIMER
• Atrofia lobar muy localizada frontal y, me-nos, temporal.
• Pacientes más jóvenes.• Más Antecedentes Familiares.• Más frecuente en Mujeres (Alzheimer 1/1).• Evolución más rápida. Superviviencia me-
dia 7 años.Se clasifican como Tauopatías:1. Enfermedad de Pick.2. Degeneración corticobasal.3. Parálisis Supranuclear Progresiva.4. Demencia frontotemporal con parkinsonis-
mo y alteraciones en el cromosoma 17.
CLÍNICALa afasia y apraxia son más frecuentes. Dos patro-nes de alteración de la conducta> Síntomas frontales:
i. Pérdida de la iniciativaii. Apatía, abuliaiii. Reflejos de prensión y succión liberados
> Síntomas temporales:i. Verborreaii. Ansiedad en constante movimiento
enFerMeDaD De HunTingTon
ETIOPATOGENIA> Herencia AD. 100% de penetrancia.> Alteración del Cromosoma 4p.> El gen alterado es el IT 15, que codifica la hun-
tingtina.> Este trastorno genético motiva la repetición del
trinucleótido CAG, cuyo número es factor pro-nóstico.
> Suele manifestarse entre los 30 y los 40 años.
ANATOMÍA PATOLÓGICA> Atrofia de la cabeza del Nucleo Caudado g
típico.• También hay lesiones de Globo Pálido, Pu-
tamen, Tálamo y Corteza.
> Neurotransmisor déficitario:• GABA, encefalina y sustancia P.• También es déficitaria la Acetilcolina, por
déficit de AcetilColin Transferasa.> La consecuencia es una Hiperfunción Dopami-
nérgica.> También se alteran: Somatostatina y TRH están
elevados.
CLÍNICA> Corea + Demencia subcortical + historia fami-
liar con tipico comienzo entre 3ª y 5ª década.> La corea (movimientos involuntarios espasmó-
dicos arrítmicos y rápidos) es el principal sínto-ma, puede coexistir con movimientos atetósicos (movimientos involuntarios sinuosos arrítmicos
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 129 )
11
Capítulo 11 ... [Demencias]
y lentos), lo que se denomina coreoatetosis. Al progresar la enfermedad aparece un síndrome parkinsoniano y distonía (incapacitantes). La disfagia y la disartria también son síntomas co-munes.
> El deterioro cognitivo y los trastornos psiquiátri-cos pueden preceder al los síntomas motores: inestabilidad emocional, irritabilidad, conducta inapropiada, depresión (frecuentes ideas de suicidio) y
> Trastornos de memoria que evolucionan a una demencia subcortical (por alteración de los ganglios basales = no hay afasia ni agnosia, y sí hay movimientos involuntarios anormales).
> Variante de WESTPHAL:• Más grave en los más jovenes (niños)• Corea poco frecuente (acinético). Bradici-
nesia, rigidez y distonías son frecuentes.
DIAGNÓSTICO> Clínico, TC y RMN.> Confirmación y diagnóstico presintomático con
análisis de genética molecular (repetición del triplete CAG en el gen IT15).
> Puede hacerse Diagnóstico Prenatal: DNA en sangre fetal o Líquido Amniótico.
376_un hombre de 45 años presenta un trastorno
del comportamiento, con irritabilidad y desinhibición intermitente, al que se han añadido recientemente sacudidas irregulares, frecuentes y bruscas de las extremidades y del tronco, así como disartria. tiene antecedentes de una enfermedad similar en su padre. ¿cuál sería su sospecha diagnóstica?:
1) Una enfermedad por expansión de la repetición de tripletes de ADN.
2) Una enfermedad por cuerpos de Lewy.3) Una enfermedad desmielinizante.4) Una enfermedad con placas amiloides neuríticas
y ovillos neurofibrilares.5) Una enfermedad con inclusiones neuronales con
cuerpos de Pick.
TRATAMIENTO> No tiene tratamiento eficaz.> Sólo Sintomático:
• AntiDopaminérgicos:~ Reserpina.~ Neurolépticos.
- FENOTIACINAS.• Clorpromacina.
- BUTIROFENONAS.• Haloperidol.
• Antidepresivos.
DeMencia MulTiinFarTo> Segunda causa de Demencia tras Enfermedad
de Alzheimer.> Antecedentes de Arteriosclerosis HTA y ACV.
CLÍNICA> Inicio agudo, curso fluctuante y deterioro esca-
lonado.> Múltiples pequeños infartos que dejan atrofia
definitiva.> Confusión nocturna y Depresión.> Otra demencia de origen vascular es la enfer-
medad difusa de la sustancia blanca, denomi-nada Encefalopatía de Binswanger.
TRATAMIENTO> Prevención con Nimodipino.
• Es antagonista del Calcio. Vasodilatador ce-rebral.
HiDroceFalia norMoTenSiVa
ETIOPATOGENIA> Es una hidrocefalia comunicante con acueducto
de Silvio permeable.> Parece provocado por obliteración del Espacio
Sub-Aracnoideo (ESA).> Puede aparecer como secuela de meningitis,
traumatismos y hemorragia subaracnoidea.
CLÍNICA> Triada de Hakim-Adams:
• Demencia.
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 130 )
07 neurología y neuroCirugía
11
• Apraxia o ataxia de la Marcha.• Incontinencia Urinaria.
> En ocasiones puede presentarse como un par-kinsonismo.
DIAGNÓSTICO> ESTUDIO DEL LCR:
• Estudio de la dinámica del LCR: ~ cálculo del Rout por test de katzman y
Marmarou• Monitorización de la presión de LCR
~ No elevada de forma basal.~ Elevaciones intermitentes (ondas A y
B): registros de PIC incluyendo la fase de sueño.
> TAC. • Dilatación de ventrículos laterales que pue-
da cursar sin atrofia (a diferencia de otras hidrocefalias).
• A veces se observa edema periventricular.• Tampoco hay cefalea ni papiledema.
TRATAMIENTO> Muchos pacientes tienen mejoría transitoria tras
la extracción de LCR.> El tratamiento consiste en la derivación de LCR
para pacientes seleccionados, pues la mejoría no es constante ni definitiva y el tratamiento no está exento de complicaciones (hematoma sub-dural, infecciones).
420_hombre de 70 años que consulta por un tras-
torno de marcha y un deterioro cognitivo subagudo. nos indican que el diagnóstico de presunción del pa-ciente es hidrocefalia a presión normal. en este caso, ¿cuál de los siguientes datos no esperaría encontrar?:
1) Una hidrocefalia comunicante con acueducto de Silvio permeable en la resonancia cerebral.
2) Un trastorno de la marcha tipo apráxico.3) Un LCR con leve elevación de la presión de aper-
tura y con un aumento de células y proteínas.4) La realización de una punción lumbar evacuadora
(30 ml de LCR) puede mejorar la marcha del pa-ciente.
5) Ausencia de signos de atrofia cortical cerebral.
DegeneraciÓn corTico BaSal
> Actualmente clasificada como síndrome parkin-soniano (tauopatía).
> Demencia de evolución lenta, en cuyo inicio se observa rigidez unilateral, distonía y apraxia de brazo y mano.• Es típico el signo de la mano no propia o
alienígena.> En fases finales presenta disartria, marcha lenta,
temblor y demencia.> A veces se asocia con signos de demencia fron-
totemporal.
enFerMeDaD DiFuSa con cuerPoS De lewy
> Demencia fluctuante, pero de curso progresivo.> Parkinsonismo.
• Los cuerpos de Lewy son positivos para ubi-quitina y sinucleína alfa.
> Alucinaciones.• EL delirio puede ser más intenso o desen-
cadenarse por tratamiento con levodopa, al ser diagnosticado el enfermo de “Parkin-son”.
> Caídas frecuentes.
TRATAMIENTO> Pueden beneficiarse del tratamiento con antico-
linesterásicos.> oJo!: Si hay síntomas psicóticos, deben em-
plearse los fármacos con mucha precaución, pues pueden exacerbar los síntomas parkinso-nianos.
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana

( 131 )
11
Capítulo 11 ... [Demencias]
422_un hombre de 77 años es traído a la consulta
por su esposa para evaluación. ella refiere que duran-te los últimos seis meses su marido ha experimentado fuertes alucinaciones visuales y auditivas e ideas de-lirantes paranoides. asimismo, durante el último año los déficit cognitivos progresivos se han vuelto cada vez más evidentes para ella y para otros miembros de la familia. estos déficit, todavía en una fase leve, implican la memoria, las habilidades matemáticas, la orientación y la capacidad de aprender nuevas habili-dades. aunque se han observado importantes fluctua-ciones en la capacidad cognitiva día a día o semana a semana, es aparente un curso de declive definido. el paciente no ha estado tomando ningún medicamento. el examen físico revela un temblor en reposo de la ex-tremidad superior derecha y una leve rigidez en rueda dentada. la marcha del paciente se caracteriza por pasos cortos que los arrastra y una disminución del balanceo de los brazos. ¿Qué síndrome clínico es más compatible con los síntomas de este hombre?:
1) Enfermedad de Parkinson.2) Demencia de Creutzfeldt-Jacob.3) Demencia con cuerpos de Lewy.4) Demencia vascular.5) Demencia del lóbulo frontal.
ParÁliSiS SuPranuclear ProgreSiVa
> Incluida dentro de los síndromes parkinsonia-nos (ver más adelante).
> Cuadro de demencia de tipo frontotemporal con rigidez, caídas frecuentes y limitación de los movimientos en el plano vertical.
oTraS DeMenciaS(Estudiadasenotroscapítulos)
ENFERMEDAD DE CREUTZFELD JACOB
ENCEFALOPATÍA DE WERNICkE
ENFERMEDAD DE MARCHIAFAVA BIGNAMI
DÉFICIT DE VITAMINA B12
DÉFICIT DE ÁCIDO NICOTÍNICO (PELAGRA)
INFECCIONES
ENCEFALOPATÍA VIH
SÍFILIS
NEOPLASIAS
ENCEFALITIS LÍMBICA> Síndrome paraneoplásico por carcinomas ocul-
tos, generalmente pulmonares.
EXPOSICIÓN A METALES> Plomo, mercurio, arsénico, aluminio (diálisis).
HEMATOMA SUBDURAL CRÓNICO
COMPLEJO ELA-PARkINSON-DEMENCIA DE LA ISLA DE GUAM
OTROS TRASTORNOS SISTÉMICOS> Hipotiroidismo.> Encefalopatía de origen hepático, renal, pulmonar.> Vasculitis.
• Existe incluso una vasculitis aislada del SNC.
SeuDoDeMencia DePreSiVa
> Realmente es una importante Depresión Mayor en ancianos.
> Produce déficits cognitivos bruscos.> Sobrevaloración subjetiva de su incapacidad.> Humor deprimido continuado.> Su razonamiento es correcto.> Diagnóstico Diferencial: las Demencias sue-
len ser menos bruscas.> Tratamiento:
• Antidepresivos.
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana
( 132 )
07 neurología y neuroCirugía
11
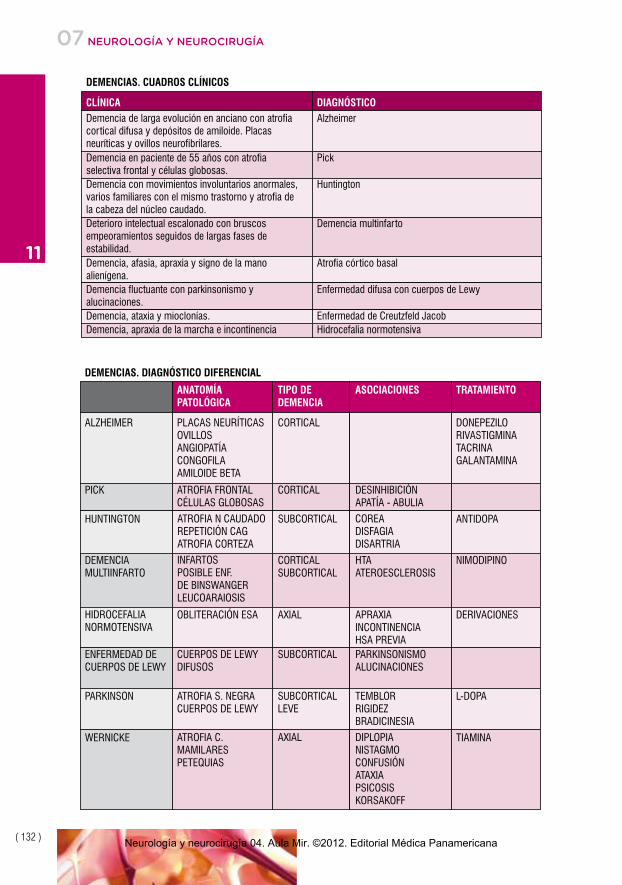
CLÍNICA DIAGNÓSTICO
Demencia de larga evolución en anciano con atrofiacortical difusa y depósitos de amiloide. Placasneuríticas y ovillos neurofibrilares.
Alzheimer
Demencia en paciente de 55 años con atrofiaselectiva frontal y células globosas.
Pick
Demencia con movimientos involuntarios anormales,varios familiares con el mismo trastorno y atrofia dela cabeza del núcleo caudado.
Huntington
Deterioro intelectual escalonado con bruscosempeoramientos seguidos de largas fases deestabilidad.
Demencia multinfarto
Demencia, afasia, apraxia y signo de la manoalienígena.
Atrofia córtico basal
Demencia fluctuante con parkinsonismo yalucinaciones.
Enfermedad difusa con cuerpos de Lewy
Demencia, ataxia y mioclonías. Enfermedad de Creutzfeld JacobDemencia, apraxia de la marcha e incontinencia Hidrocefalia normotensiva
DEMENCIAS. CUADROS CLÍNICOS
ANATOMÍAPATOLÓGICA
TIPO DEDEMENCIA
ASOCIACIONES TRATAMIENTO
ALZHEIMER PLACAS NEURÍTICASOVILLOSANGIOPATÍACONGOFILAAMILOIDE BETA
CORTICAL DONEPEZILORIVASTIGMINATACRINAGALANTAMINA
PICK ATROFIA FRONTALCÉLULAS GLOBOSAS
CORTICAL DESINHIBICIÓNAPATÍA - ABULIA
HUNTINGTON ATROFIA N CAUDADOREPETICIÓN CAGATROFIA CORTEZA
SUBCORTICAL COREADISFAGIADISARTRIA
ANTIDOPA
DEMENCIAS. DIAGNÓSTICO DIFERENCIAL
DEMENCIAMULTIINFARTO
INFARTOSPOSIBLE ENF.DE BINSWANGERLEUCOARAIOSIS
CORTICALSUBCORTICAL
HTAATEROESCLEROSIS
NIMODIPINO
HIDROCEFALIANORMOTENSIVA
OBLITERACIÓN ESA AXIAL APRAXIAINCONTINENCIAHSA PREVIA
DERIVACIONES
ENFERMEDAD DECUERPOS DE LEWY
CUERPOS DE LEWYDIFUSOS
SUBCORTICAL PARKINSONISMOALUCINACIONES
PARKINSON ATROFIA S. NEGRACUERPOS DE LEWY
SUBCORTICALLEVE
TEMBLORRIGIDEZBRADICINESIA
L-DOPA
WERNICKE ATROFIA C.MAMILARESPETEQUIAS
AXIAL DIPLOPIANISTAGMOCONFUSIÓNATAXIAPSICOSISKORSAKOFF
TIAMINA
Neurología y neurocirugía 04. Aula Mir. ©2012. Editorial Médica Panamericana



























