Embed Size (px)
Citation preview

Aus dem Institut für
Pharmakologie, Toxikologie und Pharmazie
der Tierärztlichen Hochschule Hannover
und der
Experimentellen Unfallchirurgie
der Unfallchirurgischen Kinik
der medizinischen Hochschule Hannover
Der Einfluss von Fcγ-RIII und TNF-RI auf den Respiratory Burst
von neutrophilen Granulozyten in Abhängigkeit vom
Migrationsverhalten in einem
Ischämie-Reperfusions-Modell der Maus
INAUGURAL-DISSERTATION
Zur Erlangung des Grades einer
Doktorin der Veterinärmedizin
(Dr. med. vet.)
durch die Tierärztliche Hochschule Hannover
Vorgelegt von
Nina Zündel
aus Northeim
Hannover 2004

Wissenschaftliche Betreuung:
Univ.-Prof. Dr. med. vet. M. Kietzmann, Institut für Pharmakologie, Toxikologie und
Pharmazie der Tierärztlichen Hochschule Hannover
und
Prof. Dr. rer. biol. hum. Martijn van Griensven, Experimentelle Unfallchirurgie,
Unfallchirurgische Klinik der Medizinischen Hochschule Hannover
1. Gutachter: Univ.-Prof. Dr.med.vet. M. Kietzmann
2. Gutachter: Univ.-Prof. Dr.med.vet. H.-J. Hedrich
Tag der mündlichen Prüfung: den 4. Juni 2004

Meinen Eltern
&
Daggi und Werner
gewidmet


Inhaltsverzeichnis
1 EINLEITUNG.........................................................................................................1
2 LITERATURÜBERSICHT .....................................................................................3
2.1 Rolle des Immunsystems bei der Pathogenese des SIRS und MODS ................................................... 3
2.2 Rolle der neutrophilen Granulozyten bei der Pathogenese des SIRS und MODS............................... 8
2.3 Rolle der Adhäsionsmoleküle bei der Pathogenese des SIRS und MODS.......................................... 11
2.4 Rolle der Zytokine bei der Pathogenese des SIRS und MODS............................................................ 12 2.4.1 Tumor Nekrose Faktor α (TNF-α) ..................................................................................................... 13 2.4.2 Interleukin-6 ...................................................................................................................................... 16 2.4.3 Interleukin-10 .................................................................................................................................... 17
2.5 TNF-α-Rezeptoren................................................................................................................................... 18
2.6 Fcγ-Rezeptoren ........................................................................................................................................ 21 2.6.1 Fcγ-Rezeptorfunktionen auf neutrophilen Granulozyten................................................................... 23
1.7 Möglichkeiten der Prävention und der Therapie des SIRS und MODS............................................. 24
1.8 Grund für die Verwendung der Knockout-Mäuse ............................................................................... 27
3 FRAGESTELLUNG ............................................................................................29
4 MATERIAL UND METHODEN............................................................................30
4.1 Material .................................................................................................................................................... 30 4.1.1 Chemikalien und Medikamente......................................................................................................... 30 4.1.2 Geräte ................................................................................................................................................ 31 4.1.3 Verbrauchsmaterialien....................................................................................................................... 32
4.2 Methoden.................................................................................................................................................. 33 4.2.1 Versuchstiere ..................................................................................................................................... 33
4.2.1.1 Versuchstiergruppen...................................................................................................................... 34 4.2.2 Versuchsprotokoll.............................................................................................................................. 35 4.2.3 Blut und Organentnahme................................................................................................................... 36 4.2.4 Bronchoalveoläre Lavage (BAL) ...................................................................................................... 37 4.2.5 Kapilläre Permeabilität der Lunge..................................................................................................... 37 4.2.6 Proteinbestimmung nach Lowry........................................................................................................ 38 4.2.7 Harnstoffbestimmung ........................................................................................................................ 39
4.3 Myeloperoxidaseaktivität (MPO)........................................................................................................... 41
4.4 Histologische Untersuchung ................................................................................................................... 43 4.4.1 Durchflusszytometrie ( FACS-Analyse)............................................................................................ 45
4.4.1.1 Funktionsprinzip des Durchflusszytometers.................................................................................. 45 4.4.1.2 Durchführung der Messung ........................................................................................................... 46

4.5 Statistik..................................................................................................................................................... 48
5 ERGEBNISSE.....................................................................................................49
5.1 Lungenpermeabilität ............................................................................................................................... 49
5.2 Myeloperoxidaseaktivität........................................................................................................................ 51
5.3 Histologie.................................................................................................................................................. 53 5.3.1 Lungenödem...................................................................................................................................... 53 5.3.2 Granulozyteninfiltration ins Lungengewebe...................................................................................... 56 5.3.3 Leberödem......................................................................................................................................... 58 5.3.4 Granulozyteninfiltration in die Leber ................................................................................................ 61 5.3.5 Hydropische Degeneration in der Leber............................................................................................ 63 5.3.6 Nekrotische Veränderungen in der Milz............................................................................................ 65 5.3.7 Abgrenzbarkeit der weißen von der roten Milzpulpa ........................................................................ 67 5.3.8 Lymphozytendichte in der weißen Milzpulpa ................................................................................... 69 5.3.9 Dichte der Mesangiumzellen in den Glomeruli ................................................................................. 71 5.3.10 Granulozyteninfiltration in die Nieren............................................................................................... 73
5.4 Zytokine.................................................................................................................................................... 75 5.4.1 INF-γ-Konzentration ......................................................................................................................... 75 5.4.2 IL-12-Konzentration.......................................................................................................................... 77 5.4.3 TNF-α-Konzentration ........................................................................................................................ 79 5.4.4 IL-6-Konzentration............................................................................................................................ 81 5.4.5 IL-10-Konzentration.......................................................................................................................... 83 5.4.6 MCP-1-Konzentration ....................................................................................................................... 85
5.5 Mortalität ................................................................................................................................................. 87
6 DISKUSSION......................................................................................................89
6.1 Lungenpermeabilität ............................................................................................................................... 91
6.2 Myeloperoxidaseaktivität und Granulozyteninfiltration des Lungengewebes................................... 92
6.3 Lungenödem............................................................................................................................................. 95
6.4 Leberödem ............................................................................................................................................... 96
6.5 Granulozyteninfiltration in die Leber ................................................................................................... 98
6.6 Hydropische Degeneration der Leber.................................................................................................... 99
6.7 Nekrosen, Lymphozytendichte und Abgrenzbarkeit der weißen von der roten Pulpa in der Milz100
6.8 Dichte der Mesangiumzellen in den Glomeruli und Granulozyteninfiltration in die Nieren ......... 101
6.9 Zytokine.................................................................................................................................................. 102
7 ZUSAMMENFASSUNG ....................................................................................109

8 SUMMARY........................................................................................................111
9 LITERATURVERZEICHNIS ..............................................................................113
10 DANKSAGUNG .............................................................................................139

Abkürzungen
ARDS Adult Respiratory Distress Syndrome
BAL Bronchoalveoläre Lavage
BSA Bovine Serum Albumin
CARS Compensatory Antiinflammatory Response Syndrome
CD Cluster of Differentiation
CHAOS Cardiovascular shock, Homeostasis, Apoptosis, Organ
dysfunction, Immune suppression
DAG Diazylglyzerol
DHEA Dehydroepiandrosteron
DNA Desoxyribonucleicacid
ELAM Endothelial Leukocyte Adhesion Molecule
ELISA Enzyme Linked Immuno Sorbent Assay
F- Mäuse denen sowohl das Gen für den TNF-RI als auch für den
Fcγ-RIII fehlt
Fcγ-R Fcγ-Rezeptor
FMLP Formylmethionine Leucyl-Phenylalanine
GM-CSF Granulocyte-Makrophage-Colony-stimulating factor
HE Hämatoxylin-Eosin
ICAM Intercellular Adhesion Molecule
IFN Interferon
IgG Immunglobulin G
IL Interleukin
I/R Ischämie-Reperfusion
Isch. Ischämie
LPS Lipopolysaccharide
NO Stickstoffmonoxid
MARS Mixed Antagonist Response Syndrome
min Minuten

MODS Multiorgan Dysfunktions Syndrom
MOV Multiorganversagen
MPO Myeloperoxidase
NK-Zellen Natürliche Killerzellen
OD Optische Dichte
p55 Tumornekrosefaktorrezeptor I
p75 Tumornekrosefaktorrezeptor II
PMN Polymorphonuclear Granulocytes
SIRS Systemic Inflammatory Response Syndrome
sTNF löslicher Tumornekrosefaktor
T- Mäuse, denen das Gen für den TNF-Rezeptor I fehlt
TH T-Helferzellen
TNF Tumornekrosefaktor
TNF-R Tumornekrosefaktorrezeptor
WT Wildtyp-Mäuse (C57BL/6-Mäuse)
Ferner gelten die allgemeinen SI-Einheiten und die chemischen Elementsymbole.


Einleitung 1
1 Einleitung
Noch vor 50 Jahren war das Problem des Multiorganversagens (MOV) gänzlich unbekannt.
Aufgrund der noch wenig entwickelten rettungsmedizinischen Maßnahmen verstarben
Patienten, die einen hämorrhagischen Schock erlitten, wegen des großen Blutverlustes, bevor
sich ein derartiges Krankheitsbild entwickeln konnte. Auch schwere bakterielle Infektionen
stellten ein zusätzliches gravierendes Problem dar, weil die antibiotischen
Therapiemöglichkeiten noch sehr begrenzt waren.
Mit der konstanten Verbesserung rettungsmedizinischer Maßnahmen durch großzügige
Volumenersatztherapie war es möglich, polytraumatisierte Schockpatienten zu stabilisieren.
In diesem Zusammenhang trat eine Folge von Komplikationen auf, die bisher unbekannt
waren. Viele der Verletzten starben nun nicht mehr aufgrund des starken Blutverlustes,
sondern an schweren sekundären Beeinträchtigungen der Lungen- und Nierenfunktion.
Besonders häufig konnte diese Beobachtung während der 60 er Jahre gemacht werden, da mit
Hilfe von Hubschraubern eine schnelle Erstversorgung von verletzten Personen gewährleitset
werden konnte. Sie konnten somit zwar vor einem tödlichen Blutverlust bewahrt werden, es
entwickelte sich jedoch sekundär ein Krankheitsbild, was aus einem Komplex von Luftnot,
erhöhter Temperatur und zentraler Zyanose gekennzeichnet ist. Während dieser Zeit wurde
der Begriff der „Schocklunge“ geprägt. Zwar wurden die gleichen Symptome bereits 1932
durch MOON und 1950 durch JENKINS et al. beschrieben, den bis heute noch gültigen
Begriff des „Adult Respiratory Distress Syndrom“ (ARDS) führten ASHBAUGH et al.
jedoch erst 1967 ein. Verschiedene Autoren beschrieben zu jener Zeit eine Beeinträchtigung
der Lungenfunktion nach Bauchhöhlenoperationen (SKILLMAN 1969), Peritonitis (BURKE
1963) oder anderen Infektionen (CLOWES, Jr. 1968). Somit wurde klar, dass die pulmonale
Beeinträchtigung wahrscheinlich die Folge entfernter Prozesse im Körper ist. Das ARDS war
zu dieser Zeit die häufigste Todesursache bei schwerverletzten Patienten (ALBERTS 1999;
ASHBAUGH 1967; BAUE 1975; BURKE 1963; GORIS 2003). Die Überlebensrate an
ARDS erkrankter Patienten konnte durch weitere intensivmedizinische
Behandlungsfortschritte erheblich verbessert werden, so dass neben den zunächst

Einleitung 2
lebenslimitierenden Lungenveränderungen auch die Beeinträchtigungen anderer
Organsysteme in den Vordergrund rückten. TINLEY et al. beschrieben 1973 erstmals dieses
neu entstandene Syndrom, als drei Patienten nach rupturierten Aortenaneurysmen an den
Folgen von „distal organ failure“ verstarben. Von BAUE wurde dieses Syndrom 1975 als
Multiorganversagen (MOV) bezeichnet, „das Syndrom der Siebziger Jahre“. Trotz maximaler
Intensivtherapie konnten viele der Patienten auf Intensivstationen nicht mehr stabilisiert
werden, da sie ein multiples, progressives MOV entwickelten (CARRICO 1986; MANSHIP
1984; SCHUSTER 1989), was mit hoher Letalität einherging. Im Jahre 1991 wurden bei der
Konsensuskonferenz der Society of Critical Care Medicine und des American College of
Chest Physicians die Begriffe „SIRS“ (Systemic Inflammatory Response Syndrom) und
„Sepsis“ neu definiert. In diesem Rahmen wurde auch die Bezeichnung „MOV“ durch
„MODS“ (Multiorgan Dysfunktions Syndrom) ersetzt. Hierdurch soll das Kontinuum der
Organdysfunktion besser zum Ausdruck kommen und nicht nur der Endpunkt in Form des
Organversagens beschrieben werden.
Auch heute noch stellt das MODS die schwerwiegendeste Komplikation bei schwerverletzten
Patienten dar. Etwa 40% der Intensivpatienten entwickeln ein MODS, was mit
Mortalitätsraten von 50-80% einhergeht und somit die führende Todesursache auf
Intensivstationen ist (DEITCH 1992; MANSHIP 1984; MARSHALL 1995; RANGEL-
FRAUSTO 1995; SALVO 1995; TRAN 1990).

Literaturübersicht 3
2 Literaturübersicht
2.1 Rolle des Immunsystems bei der Pathogenese des SIRS und MODS
Das Immunsystem spielt nach neuen Erkenntnissen bei der Entstehung des MODS eine
besonders wichtige Rolle. Durch die Immunmediatoren wird das Immunsystem zu einer
Reaktion angeregt, welche das physiologische Ausmaß deutlich überschreitet. Es können
sowohl zu starke als auch zu geringe Immunantworten auf das auslösende Agens auftreten.
Die Folge ist ein sequentielles Versagen multipler Organsysteme wie der Lunge, der Leber,
des Intestinums und der Niere. Exogene Faktoren scheinen hierbei eine untergeordnete Rolle
zu spielen, sind es doch die endogenen Mediatoren, die auf das Immunsystem einwirken
(DEITCH 1992).
Eine Aufgabe des Immunsystems besteht darin, den Organismus vor der Invasion pathogener
Agentien zu schützen. Es besteht eine Proportionalität zwischen der Intensität der
Immunabwehr und der Stärke einer Infektion. Ziel dieser Immunabwehr ist es, das
schädigende Agens zu eliminieren.
Man unterscheidet funktionell zwischen dem angeborenen (unspezifischen) und dem
erworbenen (spezifischen) Immunsystem. Die angeborene Abwehr umfaßt unspezifische
physikalische, zelluläre und chemische Mechanismen, während das erworbene Abwehrsystem
aus einer zellulären (T-Zell-Antwort) und einer humoralen (Antikörper) Komponente besteht
(siehe Abbildung 1).

Literaturübersicht 4
angeborene, physikalische zelluläre chemische unspezifische Barrieren Abwehr Barrieren Abwehr Haut Granulozyten pH-Wert Mukosa Makrophagen Lipide NK-Zellen Enzyme Komplement Interleukine Akutphasenproteine erworbene, zelluläre humorale spezifische Abwehr Abwehr Immunität Lymphokine, Interleukine Antikörper Zytotoxische T-Zellen T-Helferzellen B-Zellen
Abbildung 1: Komponenten des Immunsystems (KAYSER 1998)
Bei einer physiologischen Immunantwort regulieren sich die funktionellen Einheiten des
Immunsystems gegenseitig, so dass es zu einer angemessenen Reaktion kommt, die innerhalb
bestimmter Grenzen bleibt, und rechtzeitig beendet wird. Kann das pathogene Agens jedoch
nicht eliminiert werden, wie zum Beispiel Bakterien oder Endotoxine bei einer Sepsis, ist es
möglich, dass eine derartig intensive Aktivierung des Immunsystems resultiert, welche zu
einem septischen Schock führt und schließlich in einem MODS endet (BUDELMANN 1969).

Literaturübersicht 5
Von einem Systemic Inflammatory Response Syndrom (SIRS) spricht man, wenn laut
Definition zwei oder mehr der folgenden Parameter diagnostiziert werden können:
1. Rektaltemperatur entweder > 38° C oder < 36° C
2. Herzfrequenz > 90 Schläge pro Minute
3. Atemfrequenz > 20 Atemzüge pro Minute oder
arterieller Kohlenstoffdioxidpartialdruck < 4,3 kPa
4. Leukozytenzahl> 12000 pro mm3 oder < 4000 pro mm3
oder 10% der Leukozyten bestehen aus unreifen Formen
Tritt das SIRS im Zusammenhang mit einer generalisierten Infektion auf, so spricht man in
diesem Falle von einer „Sepsis“. Ist zusätzlich der hämodynamische Status beeinträchtigt, so
liegt eine „schwere Sepsis“ vor. Das „MODS“ beschreibt den Zustand eines physiologischen
Ungleichgewichts, bei dem die Organe nicht in der Lage sind, die Homöostase aufrecht zu
halten (BONE 1992).
Die Entwicklung des SIRS kann in drei Stadien unterteilt werden:
• die lokale Immunantwort,
• die initiale systemische Immunantwort und
• die massive systemische Inflammation (BONE 1996a; DAVIES 1997)
Während der ersten Phase reagiert der Körper mit einer physiologischen lokalen
Immunantwort auf einen Infektionsreiz oder ein Trauma. Hierbei werden humorale und
zelluläre Immunmediatioren lokal aktiviert, um bereits aufgetretene Schäden zu beseitigen
und eine Ausbreitung zu verhindern (REGEL 1989). Um einem Überschießen der
proinflammatorischen Immunantwort vorzubeugen, werden regulativ auch
antiinflammatorische Mediatoren freigesetzt (DINARELLO 1993; PLATZER 1995). Ist die
lokale Immunantwort nicht in der Lage den initiierenden Reiz zu kontrollieren, gelangen
während der zweiten Phase Mediatoren und proteolytische Enzyme in die systemische
Zirkulation, um eine erhöhte Einwanderung peripherer immunkompetenter Zellen (z.B.:
Makrophagen, neutrophile Granulozyten, Thrombozyten und Koagulationsfaktoren)

Literaturübersicht 6
herbeizuführen. Dieser Prozeß endet erst mit dem Abklingen der Infektion oder der
Wiederherstellung der Homöostase (BONE 1996a; FUKUSHIMA 1994). Gelingt diese
Wiederherstellung jedoch nicht, kommt es während einer dritten Phase zu einer destruktiv
wirkenden systemischen Immunantwort und zur klinischen Manifestation des SIRS (BONE
1996b; REGEL 1991).
Aufgrund einer fortschreitenden endothelialen Dysfunktion kommt es zu einer erhöhten
mikrovaskulären Permeabilität mit Transsudation in die Organe (BARIE 1988; IBBOTSON
1989; KREUZFELDER 1988; LUCAS 1991; PAPE 1994; PETRAK 1989; SEEKAMP
1993b; STEPHENS 1988; VAN GRIENSVEN 1999b; WELBOURN 1991b). Des weiteren
bilden sich Mikrothrombi, welche die Mikrozirkulation negativ beeinflussen und so eine
lokale Ischämie hervorrufen können (GANDO 1996; SIGURDSSON 1992). Bei der
Wiederherstellung der Durchblutung kann es schließlich zu einem Reperfusionsschaden
aufgrund von Sauerstoffradikalen und Hitze-Schock-Proteinen kommen, welche im
Ischämiegebiet gebildet werden (CIPOLLE 1993; RINALDO 1990). Die Dysregulation der
vasodilatatorischen und vasokonstriktorischen Mechanismen ruft eine ausgeprägte Dilatation
der Gefäße hervor, so dass es zu einer zusätzlichen Verschlechterung der Blutzufuhr und einer
Steigerung der Transsudation kommt (GOMEZ-JIMENEZ 1995; MIYAUCHI 1990). Die
Folge kann eine Organdysfunktion oder bei Beteiligung mehrerer Organe eine
Multiorgandysfunktion sein. Hierbei sind außerdem klinisch die fünf klassischen Symptome
der Entzündung Rubor, Calor, Dolor, Tumor und Functio laesa festzustellen. Kann der Körper
die überschießende Inflammation nicht mit Hilfe von antiinflammatorischen Mediatoren unter
Kontrolle bringen, endet das MODS letal (REGEL 1991).
Kommt es in diesem Rahmen zu einer antiinflammatorischen Reaktion, die das
physiologische Maß überschreitet, tritt eine „Immunparalyse“ ein (KREMER 1996; MILLS
1989; RANDOW 1995; SYRBE 1994). In diesem Falle spricht man von einem Compensatory
Antiinflammatory Response Syndrom (CARS) (BONE 1996a; BONE 1996c; BONE 1996d;
DAVIES 1997). Während dieser Phase besteht eine besonders große Gefahr für eine Infektion
des Organismus mit Mikroorganismen, woraus leicht eine Sepsis und in dieser Folge ein
septischer Schock entstehen können (MOORE 1999; VOLK 1990)

Literaturübersicht 7
Es besteht außerdem die Möglichkeit, dass die Inflammation neben der einsetzenden
Aniinflammationsreaktion erhalten bleibt und somit ein Mixed Antagonist Response Syndrom
(MARS) hervorruft (BONE 1996b; BONE 1996d; DAVIES 1997). Auch diese beiden
klinischen Zustände können unter anderem zum MODS führen. Patienten, bei denen ein SIRS
oder MODS festgestellt wurde, erliegen demnach nicht zwangsläufig unkontrollierten
Infektionen, sondern unter Umständen der dysregulierten Entzündungsantwort (VAN
GRIENSVEN 1999a).
All diese möglichen Reaktionen (SIRS, CARS, MARS, MODS) werden unter dem Begriff
CHAOS (cardiovascular shock, homeostasis, apoptosis, organ dysfunction und immune
suppression) zusammengefaßt (BONE 1996b; DAVIES 1997).
Es wurde beobachtet, dass nicht alle Patienten bei gleicher Verletzungsschwere ein MODS
entwickeln. Daraus kann geschlossen werden, dass neben dem initialen Trauma verschiedene
andere Faktoren für die Entstehung dieses Syndroms verantwortlich sind (BERNARD 1990;
REGEL 1989). So besteht ein Zusammenhang zwischen sekundären pulmonalen
Veränderungen und Frakturen, weil hierbei aufgrund von Gefäßverletzungen Ischämien
hervorgerufen werden können. Besonders oft kann diese Beobachtung bei offenen Frakturen
gemacht werden, die in der Regel mit einer partiellen oder kompletten Ischämie einhergehen
(SUDKAMP 1989) und somit ein erhöhtes Risiko für posttraumatische Komplikationen
darstellen (AUFMKOLK 1996). Erst bei einer Reperfusion führt die entstandene Ischämie
anhand von Mediatoren, Proteasen, Sauerstoffradikalen und aktivierten Granulozyten zu
sekundären, distalen Organschäden (WELBOURN 1991b), deren pathologisches Muster in
allen Organen einheitlich ist (BERNARD 1994; BURCHARDI 1987).
Eine Ischämie mit anschließender Reperfusion führt zu einer sekundären Organschädigung,
unabhängig davon, in welche Gebiet die Ischämie vorlag. So konnten pulmonale Schäden
nach Ischämie/Reperfusion der Extremitäten (BELKIN 1989; GOLDMAN 1992; SEEKAMP
1993c; SEEKAMP 1993a; SEEKAMP 1994; SEIBERT 1993; WELBOURN 1991a), der
Leber (DUBAYBO 1988) und nach genereller Ischämie im Zusammenhang mit einem
hypovolämischen Schock festgestellt werden. Dabei ist jedoch zu beachten, dass die Schäden
erst dann auftreten, wenn das Gebiet reperfundiert wird (SEEKAMP 1993b). Verantwortlich

Literaturübersicht 8
sind hierfür neutrophile Granulozyten und reaktive Sauerstoffradikale (FLAHERTY 1991;
GOLDMAN 1992; GRECH 1994; KAZUI 1994; SEEKAMP 1993b; SEIBERT 1993;
WELBOURN 1991a; WELBOURN 1991b; WELBOURN 1990; YOKOTA 1989;
ZIMMERMAN 1990), die zum Teil von den neutrophilen Granulozyten gebildet werden und
teilweise Produkte von chemischen Reaktionen im reperfundierten Gebiet sind (KORTHUIS
1989; PARKS 1986). Die freien Radikale sind in der Lage durch Interaktionen mit
Desoxyribonucleinsäure DNA-Strangbrüche hervorzurufen, welche die Zelle irreparabel
schädigen können (BIELSKI 1979; BURGER 1980). Bei Reaktionen zwischen Radikalen und
ungesättigten Fettsäuren durch Lipidperoxidation kommt es zu Zellmembranschäden, die den
Untergang der Zelle verursachen können (AUST 1982; YOUNES 1987).
PAPE et al. konnten (1994) anhand von obduzierten Patienten, die aufgrund schwerer
Verletzungen verstorben waren, das typische Muster eines posttraumatischen
Multiorganversagens zeigen. Patienten, die innerhalb der ersten 24 Stunden nach dem Trauma
verstarben, erlagen grundsätzlich ihren Hirnverletzungen oder unstillbaren Blutungen. Starben
die Patienten nach zwei bis sieben Tagen, war die Beeinträchtigung der Lungenfunktion die
häufigste Ursache; ab dem siebten Tag post Trauma erlagen die meisten Patienten einem
MOV. Diese Beobachtungen konnten von REGEL et al. (1996) bestätigt werden. Sie
beschreiben, dass Patienten, die ein Multiorganversagen entwickeln, zu knapp 75% eine
starke Beeinträchtigung der Lungenfunktion zeigen. Etwa 65% dieser Patienten erlagen dieser
Dysfunktion. Die Lunge ist grundsätzlich das erste Organ, welches nach einer schweren
traumatischen Einwirkung auf den Organismus beeinträchtigt wird. Im Mittel tritt diese
Veränderung nach 3,7 ± 2,8 Tagen auf. Nur bei weniger als 5% der Patienten wurde eine
Beeinträchtigung der Nierenfunktion festgestellt.
2.2 Rolle der neutrophilen Granulozyten bei der Pathogenese des SIRS und
MODS
Neutrophile Granulozyten sind Zellen der myeloiden Reihe, welche sich im Knochenmark
innerhalb von etwa 14 Tagen aus Myeloblasten bis zu den reifen PMN mit einem lobulierten

Literaturübersicht 9
Zellkern differenzieren. Diese reifen Zellen verlassen das Knochenmark und treten
vorübergehend in die Blutbahn ein. Ihre Halbwertszeit beträgt hier etwa sechs Stunden.
Besteht jedoch der Bedarf einer unspezifischen Immunreaktion im Gewebe, so treten diese
Zellen mittels Diapedese ins Gewebe ein, wo ihre Funktionsfähigkeit nach etwa ein bis zwei
Tagen erlischt (BAINTON 1971). Bereits 1887 erkannte EHRLICH die sezernierende
Eigenschaft der neutrophilen Granulozyten, ca. 10 Jahre später, um 1900, wurde entdeckt,
dass diese Zellen außerdem zur Phagozytose befähigt sind. Ermöglicht werden ihnen diese
Eigenschaften durch den Gehalt von zwei verschiedenen Arten von Granula. Es handelt sich
hierbei einerseits um peroxidase-positive Granula, welche unter anderem Myeloperoxidase,
Elastase, Lysozym und β-Glycerophosphatase enthalten. Die peroxidase-negative Granula
hingegen beinhalten Lactoferrin, Prokollagenase, alkalische Phosphatase und Lysozym
(ACKERMAN 1971; BAINTON 1966; BAINTON 1971; SCOTT 1970). Die Möglichkeit der
Sekretion und der Phagozytose macht die neutrophilen Granulozyten zu einer der wichtigsten
Zellen des unspezifischen Immunsystems (GRISWOLD 1988). Sie werden durch Chemotaxis
auf die mikrobiellen Organismen hingewiesen. Die nun folgende Adhäsion an und Migration
der Zellen durch das Gefäßendothel kann in verschiedene Phasen unterteilt werden. Man
bezeichnet sie als Rolling, Attachment und Diapedese, welche jeweils von unterschiedlichen
Adhäsionsmolekülen abhängig sind (siehe Sektion 2.3). Über einen chemotaktischen
Gradienten verläuft die Migration an den Ort, wo die mikrobiellen Organismen mit Hilfe der
bereits oben erwähnten Granula phagozytiert und lysiert werden. Besonders in der frühen
Phase (bis zum 3. Tag) nach einem Trauma spielen die neutrophilen Granulozyten eine große
Rolle, im weiteren Verlauf (ab Tag 4) treten andere Zellen, wie Lymphozyten und Monozyten
in den Vordergrund (BOTHA 1995b; BOTHA 1995a).
Auch bei der Entstehung eines Gewebeschadens nach Ischämie-Reperfusion sind es gerade
die Produkte dieser Granula, die eine schädigende Funktion einnehmen, so dass den
neutrophilen Granulozyten hierbei eine Schlüsselposition zugesprochen werden kann
(FUJISHIMA 1995; KORTHUIS 1988; VEDDER 1989).
Welche entscheidende Rolle die neutrophilen Granulozyten bei der Entstehung des
Gewebeschadens nach Ischämie/Reperfusion spielen, zeigt außerdem die Tatsache, dass in
unterschiedlichen Tiermodellen eine Neutropenie dem Ischämie-Reperfusionsschaden
signifikant vorbeugte (CARDEN 1990; HERNANDEZ 1987). Neben den neutrophilen

Literaturübersicht 10
Granulozyten wird bestimmten Mediatoren bei der Entstehung des Ischämie-
Reperfusionsschadens eine große Bedeutung zugeschrieben. So konnten KLAUSNER et al.
(1989) bei einem derartigen Gewebeschaden erhöhte Thromboxanwerte feststellen. Die
Bedeutung dieses Mediators scheint jedoch gering zu sein, da die Inhibition der Bildung
dieser Moleküle durch Indomethacin keine Verringerung der Neutrophilenakkumulation im
pulmonalen Gewebe nachgewiesen werden konnte (PUNCH 1991). Auch bei Pferden, die
aufgrund eines Ileus einen Schock entwickelten, wurden unter anderem erhöhte
Thromboxanwerte im Plasma festgestellt werden. Sie ließen sich durch den Einsatz des
Medikamentes Flunixin-Meglumin signifikant absenken (GERDEMANN 1995). Ein weiterer
Mediator, dessen Plasmakonzentration bei einem Ischämie-Reperfusionsschaden erhöht ist, ist
das Leukotrien B4 (KLAUSNER 1988) Einerseits bewirkt dieses eine chemotaktische
Ausrichtung der neutrophilen Granulozyten (PALMBLAD 1981), andererseits aber auch die
direkte Freisetzung von Wasserstoffperoxidradikalen aus diesen Zellen, was ihre zentrale
Rolle bei der Pathogenese des Gewebeschadens unterstreicht.
Der initiale Schritt dieses Pathomechanismus stellt also die Chemotaxis dar. Dabei handelt es
sich um einen chemischer Reiz, welcher die Wanderungsrichtung phagozytierender Zellen in
Abhängigkeit vom Konzentrationsgradienten der reizauslösenden Substanz bestimmt. Diese
Substanz muß nicht unbedingt ein mikrobieller Organismus oder körperfremdes Antigen sein
(BAUER 1994; BILLING 1993; WARD 1991), auch IL-1, IL-6, TNF-α, Endotoxine,
Sauerstoffradikale, Komplementfaktoren C3a und C5a, Leukotriene (z.B. LTB4), Fragment D
der Fibrinolyse und Plättchenaktivierender Faktor stellen sehr potente Chemotaxine dar
(DEMLING 1985; KINDT 1991; WARREN 1989; WEIGELT 1988). Ein großer Teil dieser
Substanzen wird nach der Ischämie während der Reperfusionsphase direkt im Gewebe von
Endothelzellen und Gewebsmakrophagen gebildet.
Auch Immunkomplexe, Gerinnungsfaktoren, das Kallikrein-Kininsystem, Granulozyt-
Monozyt-Kolonieformender Faktor, Prostaglandine und Fragmente des geschädigten
Gewebes (wie zum Beispiel Kollagenfragmente) können die neutrophilen Granulozyten
aktivieren (CERASOLI, Jr. 1990; JONAS 1991; MALIK 1985; NEUHOF 1991; WARREN
1989; WARREN 1991; WEIGELT 1988). Bei dieser Aktivierung kommt es zu einer
vermehrten Expression von Adhäsionsmolekülen (siehe 2.3), wodurch die neutrophilen

Literaturübersicht 11
Granulozyten am kapillären Endothel adhärieren. Durch die Freisetzung der bereits oben
erwähnten Sauerstoffradikale und lysosomalen Produkte wird das sogenannte Zell-spreading
ausgelöst. Dabei nehmen die vorher runden Endothelzellen eine sternförmige Gestalt an, so
dass ihre Verformbarkeit deutlich vermindert wird(BAUER 1994; HOOVER 1987). Die
Folge ist eine erhöhte endotheliale Permeabilität, weshalb schädigende Mediatoren
einschließlich Zellen die Endothelbarriere einfacher passieren können. Auch
inflammatorische Mediatoren können eine Erhöhung der Endothelpermeabilität herbeiführen
(MARUO 1992; VAN GRIENSVEN 1999a).
2.3 Rolle der Adhäsionsmoleküle bei der Pathogenese des SIRS und MODS
Der entscheidende Schritt zur Migration der neutrophilen Granulozyten an den Ort der
Inflammation ist die Adhäsion dieser Zellen an das kapilläre Endothel (FURIE 1987; FURIE
1989; HARLAN 1987). Diese Adhäsion wird über bestimmte Adhäsionsmoleküle vermittelt,
welche in drei Gruppen unterteilt werden können: die Selektine, die Integrine und die
Immunglobulin-ähnlichen. Sie werden sowohl von den neutrophilen Granulozyten als auch
von den Endothelzellen auf der Zelloberfläche exprimiert (KUROSE 1994), wodurch es zu
einer hochaffinen spezifischen Ligand-Rezeptor-Bindung kommen kann, so dass eine
selektive Akkumulation der neutrophilen Granulozyten in dem inflammatorischen oder
reperfundierten Gewebe erfolgt.
Der Migrationsprozeß kann in die verschiedenen Stadien Rolling, Attachment (Aggregation)
und Diapedesis (Transmigration) unterteilt werden, welche jeweils von unterschiedlichen
Adhäsionsmolekülen abhängig sind. Besonders das von den neutrophilen Granulozyten
exprimierte L-Selektin spielt bei der initialen Adhäsion zwischen dem Gefäßendothel und den
PMN (Polymorphkernige Granulozyten) eine bedeutende Rolle(RICHTER 1994; SIMON
1995; SPERTINI 1991; WADDELL 1994; WADDELL 1995), somit kann insbesondere die
Gruppe der Selektine für das sogenannte Tethering und Rolling verantwortlich gemacht
werden (BARGATZE 1994; LEY 1991). Zunächst kommt es zu einer rein physikalischen
Adhäsion, da bei erniedrigter Flußgeschwindigkeit Scherkräfte zwischen dem Endothel und

Literaturübersicht 12
den neutrophilen Granulozyten auftreten. Später wird diese Anheftung mit Hilfe des
neutrophilen L-Selektins und des endothelialen P- und E-Selektins in eine transiente
Adhäsion umgewandelt (ANDERSON 1987; BUTCHER 1991; SPRINGER 1990).
Der nächste Schritt des Migrationsprozesses ist das sogenannte Attachment oder Abstoppen
der neutrophilen Granulozyten auf den Endothelzellen, welches durch die Gruppe der
Integrine ausgelöst wird, die einen stabilen Zell-Zellkontakt herstellen. Integrinmoleküle
bestehen aus mehreren Untereinheiten, von denen die β2 Untereinheit für die Adhäsion der
neutrophilen Granulozyten besonders wichtig ist. Das bekannteste Adhäsionsmolekül der
Immunglobulin-ähnlichen ist das interzelluläre Adhäsionsmolekül 1 (ICAM-1), welches
überwiegend von Endothelzellen exprimiert wird. SMITH et al. (1988, 1989) konnten das
ICAM-1 als endothelialen Liganden für die β2-Integrine identifizieren. Bei Patienten, die an
den Folgen einer Sepsis verstarben, wurde sowohl eine erhöhte Expression von ICAM am
Gefäßendothel der Lunge festgestellt, als auch eine vermehrte Expression von β2-Integrin auf
den neutrophilen Granulozyten. Traumapatienten hingegen, die eine Sepsis überlebten,
zeigten basale ICAM-Expressionswerte am Lungengefäßendothel (TSOKOS 2001).
2.4 Rolle der Zytokine bei der Pathogenese des SIRS und MODS
Nach bisherigen Untersuchungen steht die Expression der Adhäsionsmoleküle und die
Aktivität der neutrophilen Granulozyten in einem engen Zusammenhang mit der Freisetzung
von Zytokinen (GAMBLE 1985; KUIJPERS 1992; NATHAN 1989; POHLMAN 1986).
Bei einer physiologischen Reaktion des Körpers auf ein infektiöses oder traumatisches
Ereignis erfolgt zunächst die Ausschüttung der proinflammatorischen Zytokine TNF-α (siehe
2.4.1), IL-1 und IL-6 (siehe 2.4.2). Im weiteren Verlauf der Abwehrreaktion kommt es
aufgrund negativer Rückkopplungsmechanismen zu einer vermehrten Freisetzung
antiinflammatorischer Zytokine (DINARELLO 1996). Besonders wichtig sind hierbei das IL-
10 (siehe 2.4.3), IL-4 und das IL-13. Neben den erwähnten Zytokinen gibt es eine Vielzahl
anderer Mediatoren, welche ein hochdifferenziertes pro- oder antiinflammatorisches
Wirkungsprofil haben.

Literaturübersicht 13
Durch das beschriebene Zusammenspiel der pro- und antiinflammatorischen Zytokine ist es
dem Organismus möglich, eine zeitlich und qualitativ begrenzte Entzündungsreaktion
hervorzurufen. So ist bei einer limitierten Immunreaktion nach wenigen Tagen am
Plasmazytokinspiegel der Übergang von der inflammatorischen Primärphase zur
kompensatorischen hypoinflammatorischen Sekundärphase zu erkennen. Gerät dieser
Regelmechanismus aus dem Gleichgewicht, kann es zur Manifestation folgeschwerer
Syndrome kommen. Bei einer überschießenden systemischen proinflammatorischen
Immunreaktion spricht man vom SIRS, welches schließlich in einem MODS enden kann
(siehe 2.1). Überwiegen hingegen die Rückkopplungsmechanismen mit Freisetzung von
antiinflammatorischen Zytokinen, kann eine Hyporeaktivität des Immunsystems resultieren,
welche man als „Immunparalyse“ (CARS, siehe 2.1) bezeichnet. Liegt hingegen eine
persistierende proinflammatorische Aktivität trotz aktiver antiinflammatorischer
Immunantwort vor, bezeichnet man diesen Zustand als MARS (siehe 2.1) (BONE 1996a;
BONE 1996b; BONE 1996c; DAVIES 1997; KREMER 1996; MILLS 1989; RANDOW
1995; SEEKAMP 1998; SYRBE 1994).
2.4.1 Tumor Nekrose Faktor α (TNF-α)
Derzeit werden der TNF-α und TNF-β voneinander unterschieden. Beide Formen haben
nahezu identische Eigenschaften, jedoch konnten REMICK et al. (1987) zeigen, dass der
TNF-α bei der inflammatorischen Reaktion die bedeutendere Rolle spielt. So wird der TNF-α
von Monozyten, Makrophagen und T-Lymphozyten sezerniert (siehe Abbildung 2), der
TNF-β hingegen nur von den Lymphozyten (BELLOMO 1992; CERASOLI, Jr. 1990;
MULLIGAN 1993; WARD 1991; WARREN 1989). Geringe Mengen von TNF-α sind für
eine adäquate Immunantwort von Nutzen. So verschlechterte die Gabe von Anti-TNF-
Antikörpern vor Induktion einer experimentellen Peritonitis in Mäusen die Überlebensraten
(ECHTENACHER 1990), wohingegen eine vorangehende Injektion geringer TNF-α- oder
Endotoxindosen wahrscheinlich durch Toleranzinduktion die Überlebensraten verbesserte
(ECHTENACHER 1995). TNF-α löst in den Endothelzellen verschiedene Mechanismen aus,

Literaturübersicht 14
die bei der Entstehung des MODS eine bedeutende Rolle spielen (siehe Abbildung 2). Es
induziert zum Beispiel die Freisetzung von IL-1, IL-6 und IL-8 aus den Endothelzellen
(LOPPNOW 1989; NAWROTH 1986a), zusätzlich wird die Endothelpermeabilität im
Zusammenspiel mit IL-1 gesteigert (BRETT 1989; MARCUS 1996; ROYALL 1989; VAN
GRIENSVEN 1999b). Ferner konnte gezeigt werden, dass TNF-α die Expression zellulärer
Adhäsionsmoleküle der Endothelzellen wie ICAM-1, ELAM-1 (Endothelial Leukocyte
Adhesion Molecule) und E-Selektin anregt (MULLIGAN 1993; WONG 1992), so dass die
Gewebeschäden, besonders von den neutrophilen Granulozyten verursacht, verstärkt werden
(CHEN 1998; VARANI 1994). Zusätzlich muß die gesteigerte prokoagulierende Aktivität des
Gefäßendothels erwähnt werden (NAWROTH 1986b; SAWDEY 1991).
Eine kausale Rolle des TNF-α konnte auch bei der Entstehung eines septischen Schocks
nachgewiesen werden(BEUTLER 1985a; BEUTLER 1985b), da in humanem Serum in
diesen Fällen erhöhte TNF-α-Werte festgestellt werden konnten. Mit Hilfe dieser Werte kann
eine prognostische Aussage über die Überlebenswahrscheinlichkeit der Patienten gemacht
werden (CALANDRA 1991; DAMAS 1989; DEBETS 1989). So korrelieren hohe TNF-α
Werte mit einer schlechten Prognose(CALANDRA 1991; CASEY 1993). Bei Tieren konnte
ebenfalls durch Verabreichung von TNF-α ein dem septischen Schock ähnliches
Krankheitsbild hervorgerufen werden, was ein weiterer Beweis für die kausale Rolle dieses
Zytokins bei einem Schockgeschehen ist (JOHNSON 1989; REDL 1990; TRACEY
1986).Auch im Hinblick auf das SIRS und MODS wurde die Rolle des TNF-α untersucht
(BROUCKAERT 1996), wobei gezeigt werden konnte, dass für Patienten, die nach Trauma
ständig erhöhte TNF-α Plasmaspiegel aufwiesen, eine schlechte Prognose besteht (CASEY
1993; THIJS 1995). Bei an ARDS verstorbenen Patienten konnten sowohl in der
bronchoalveolären Lavage als auch im Plasma stark erhöhte TNF-α-Plasmakonzentrationen
festgestellt werden (MEDURI 1995b; MEDURI 1995a). Eine verläßliche prognostische
Aussage über den Ausgang der Situation konnte in diesem Falle jedoch nicht gemacht werden
(MEDURI 1995a).
Die Sekretion von TNF-α kann ebenfalls durch Ischämie hervorgerufen werden. So erzeugten
eine Ischämie der Leber (COLLETTI 1990; WANNER 1996), des Darmes (DEITCH 1994)

Literaturübersicht 15
oder der hinteren Extremitäten (MA 1993; WELBOURN 1991c) eine erhöhte TNF-α
Plasmakonzentration.
Interessanterweise sezernieren Monozyten von Traumapatienten, die einen erhöhten TNF-α
Serumwert haben, nach Stimulation mit Endotoxin weniger TNF-α als üblich. Diese Tatsache
geht mit einer verminderten mRNA-Expression des TNF-α einher (ERTEL 1995). Es besteht
somit die Gefahr der Entwicklung eines CARS.
Zytokine, Adhäsionsmoleküle, Koagulationsfaktoren, TNF, NO-Synthese IL-1, IL-6 Antikörper Endothelzellen LPS Monozyten B-Lymophozyten Monozyten / Makrophagen TNF-α T-Lymphozyten IL-2, IFN-γ Fibroblasten
Gehirn Fieber Fettzellen Proliferation Kachexie Abbildung 2: Biologische Wirkungen des TNF-α (VAN GRIENSVEN 2000)
IL= Interleukin; IFN= Interferon; TNF= Tumor Nekrose Faktor; LPS= Lipopolysaccharid; NO=
Stickstoffmonoxid

Literaturübersicht 16
2.4.2 Interleukin-6
Dem Zytokin IL-6 kann eine bedeutende Stellung sowohl bei der Immunabwehr als auch bei
der Hämatopoese zugeschrieben werden (AKIRA 1993; HIRANO 1994; KISHIMOTO 1992;
VAN SNICK 1990). Das Molekül ist ein Polypeptid mit einer Länge von 212 Aminosäuren.
HIRANO et al. (1986) gelang es, die genetische Sequenz, die das Protein kodiert, zu
entschlüsseln. Die bioaktive Form des IL-6, welche aus 184 Aminosäuren besteht, entsteht
erst nach Abspaltung einer 28 Aminosäuren langen hydrophoben Signalsequenz. Das
Molekulargewicht des IL-6 kann zwischen 21,5 und 28 kD liegen, da an den
Aminosäurepositionen 73 und 172 unterschiedliche Glykosylierungen und
Phosphorylierungen möglich sind.
IL-6 wird von einer Vielzahl von Zellen wie T- und B-Lymphozyten,
Monozyten/Makrophagen, Fibroblasten, Hepatozyten, Astrozyten, Endothelzellen,
Mesangiumzellen, Osteoblasten, Sertolizellen sowie in Karzinomen, Sarkomen, Myelomen
und Melanomen exprimiert. Durch Mitogene, Antigene, Lipopolysaccharide, Zytokine (IL-1
und TNF) und Viren wird die Bildung von IL-6 angeregt (AARDEN 1985; NAVARRO 1989;
WAAGE 1990). Auch durch Ischämie, Hämorrhagie und das Ischämie-Reperfusionsmodell
der hinteren Extremitäten kann eine gesteigerte IL-6 Produktion induziert werden (O'NEILL
1994; WANG 1997; YASSIN 1996). Eine Inhibition der IL-6 Produktion wird hingegen von
den Zytokinen IL-4, IL-10 und IL-13 hervorgerufen. Bei einer Vielzahl von pathologischen
Prozessen kann im Serum ein erhöhter IL-6 Spiegel festgestellt werden. Dazu gehören
Traumen, Sepsis, baterielle und virale Infektionen, Autoimmunerkrankungen und Malignome
(AKIRA 1993; HIRANO 1994; KISHIMOTO 1992; VAN SNICK 1990). Anhand
unmittelbar nach einem traumatischen Ereignis gemessener IL-6-Werte kann eine Aussage
über die Schwere eines Traumas gemacht werden, welche zum Beispiel mit dem Injury
Severity Score (ISS) beschrieben werden kann. Mit Hilfe dieses Scores werden die Patienten
in verschieden Risikogruppen unterteilt, um so eine optimale Versorgung der Gruppe
entsprechend gewährleisten zu können (GEBHARD 2000; PAPE 1999). Das IL-6 wird
deswegen heute an vielen intensivmedizinischen Kliniken als Routineparameter bestimmt
(CALANDRA 1991; HACK 1989; HELFGOTT 1989; WAAGE 1989; ZABEL 1989).
Hierdurch kann eine Aussage über das voraussichtliche Outcome bei septischem Schock oder

Literaturübersicht 17
SIRS gemacht werden. Ein kontinuierlich hoher Plasmaspiegel des IL-6 ab dem Tag der
Aufnahme in der Klinik weist auf eine schlechte Prognose hin (HACK 1989; PAPE 1999;
THIJS 1995; WAAGE 1989). Für die Bewertung der Überlebenswahrscheinlichkeit der
Patienten mit Hilfe der IL-6 Konzentration in Abhängigkeit vom Untersuchungszeitpunkt
nach dem Trauma, wurden spezielle Score-Systeme entwickelt (LEMESHOW 1993).
TERREGINO et al. (1997) zeigten, dass Patienten, die ein SIRS überlebten, ein niedrigeres
IL-6 Niveau aufweisen. Außerdem konnte beobachtet werden, dass Kinder mit einer Sepsis
und einem hohen IL-6 Plasmaspiegel häufiger ein MODS entwickeln als Kinder ohne
Steigerung des IL-6 Wertes (DOUGHTY 1996). Auch das Auftreten von ARDS in
Traumapatienten korreliert mit erhöhten IL-6 Plasmawerten(CLERICI 1993; MEDURI
1995b; MEDURI 1995a).
In vitro konnte die Endothelpermeabilität mit Hilfe von IL-6 dosis- und zeitabhängig
gesteigert werden, nach Zugabe eines spezifischen Antikörpers konnte diese Reaktion jedoch
wieder rückgängig gemacht werden (MARCUS 1996; MARUO 1992; VAN GRIENSVEN
1999b).
2.4.3 Interleukin-10
Besonders für die Regulation inflammatorischer Prozesse ist das antiinflammatorisch
wirkende Interleukin 10 ein sehr wichtiger Immunmediator. IL-10 wird von einer Vielzahl
unterschiedlicher Zellen exprimiert, wie zum Beispiel aktivierten Th2-Zellen,
Monozyten/Makrophagen, B-Lymphozyten, Keratinozyten, fetalen Thymozyten und
Gliazellen (MOORE 1993; MOSMANN 1994). Besonders TNF-α regt die Sekretion von IL-
10 an (VAN DER 1994). Das IL-10 ist ein Polypeptid mit einer Länge von 178 Aminosäuren.
Die genetische Sequenz des Proteins konnte entschlüsselt werden. Dabei wurde eine
Homologie der Aminosäuzesequenzen zwischen humanem und murinem IL-10 von 73%
festgestellt. Die bioaktive Form des Proteins, welche aus 160 Aminosäuren besteht, bildet
sich erst nach Abspaltung einer hydrophoben Precursorsignalsequenz. Besonders nach

Literaturübersicht 18
Traumen beziehungsweise operativen Eingriffen können erhöhte IL-10 Werte im Plasma
festgestellt werden.
Das Zytokin ist befähigt, die Produktion bestimmter anderer Mediatoren zu inhibieren. Hierzu
zählen IL-1, IL-6, IL-8, IL-12, TNF, Granulozyten-Makrophagen-Kolonie-stimulierender
Faktor (GM-CSF) und IFN-γ, welche von aktivierten Th1-Zellen, NK-Zellen und
Monozyten/Makrophagen gebildet werden.
Durch IL-10 wird besonders stark das IFN-γ gehemmt, wobei sich die Inhibition nicht direkt
auf die Zytokinsekretion durch Th1-Zellen auswirkt, sondern indirekt durch eine
Beeinträchtigung der Antigenpräsentation und Sekretion proinflammatorischer Zytokine
durch die Makrophagen (ENK 1993; FIORENTINO 1991) vermittelt wird. Neben der
Einschränkung dieser Makrophagenfunktion ist das IL-10 in der Lage die zytotoxische
Aktivität der Zellen herabzusetzen. Zusätzlich ist es dem Zytokin möglich, die Proliferation
und Differenzierung von Mastzellen und T- und B-Lymphozyten zu induzieren
(FIORENTINO 1991; MOORE 1993; MOSMANN 1994). Im Falle einer Überproduktion des
IL-10 kann es zur Entwicklung eines CARS kommen (BONE 1996b).
2.5 TNF-α-Rezeptoren
TNF-α bindet an zwei verschiedene membrangebundene Rezeptoren, welche als TNF-RI
(55 kD = p55) und TNF-RII (75 kD = p75) Rezeptor bezeichnet werden (siehe Abbildung 3).
Sie sind auf allen Zelltypen außer Erythrozyten zu finden. Beide Rezeptoren sind ebenfalls in
löslicher Form (sTNFR) im Plasma nachweisbar, wo sie bioaktives TNF-α binden und somit
antagonistisch wirken können (MOLDAWER 1994; PESCHON 1998). Drei Stunden nach
Trauma können erhöhte Werte für die lösliche Form beider Rezeptoren im Plasma festgestellt
werden, welche sich bei ungestörtem Verlauf nach 12 Stunden wieder normalisieren
(HENSLER 2002). Bei Patienten, die kritisch an einer Sepsis erkrankt waren und daran
verstarben, konnte ein erhöhter sTNF-RI-Spiegel festgestellt werden. Überlebende Patienten
hingegen zeigten verminderte Werte (ROGY 1994).

Literaturübersicht 19
Die biologische Funktion der beiden Rezeptoren ist unterschiedlich und verläuft über zwei
verschiedene Signaltransduktionswege. Viele der Effekte, die durch Endotoxine ausgelöst
werden, werden über den TNF-RI vermittelt, welcher außerdem für die Entwicklung von
Lymphfollikeln in den peripheren Lymphknoten verantwortlich (MATSUMOTO 1996).
Fibroblasten werden über den TNF-RI zur Proliferation und zur Produktion von IL-6 und
GM-CSF angeregt (MACKAY 1994). Die Apoptose der zytotoxischen T-Zellen wird
ebenfalls über diesen Rezeptor vermittelt (SPEISER 1996), bei anderen Zellen scheinen für
die Apoptoseinduktion jedoch beide TNF-Rezeptoren verantwortlich zu sein
(VANDENABEELE 1995). Auch bei einer akuten Graft-versus-host Reaktion spielt der
TNF-RI eine wichtige Rolle (SPEISER 1996) (siehe Abbildung 3).
Der TNF-RII vermittelt die durch TNF- α induzierte T-Lymphozytenproliferation (GRELL
1998; TARTAGLIA 1993). Bei SIRS-Patienten konnte eine verminderte Expression dieses
TNF-Rezeptors festgestellt werden (HUBL 1999).
MODZELEWSKI (1997, 2003) stellte fest, dass bei Sepsispatienten eine erhöhte
Konzentration von TNF-α -einhergehend mit verminderten sTNF-RI-Werten- auf eine
schlechte Prognose hindeuten. Entscheidend sind hierbei jedoch nicht die absoluten
Blutwerte, sondern das Verhältnis von TNF-α zu seinen löslichen Rezeptoren. Es konnte
außerdem gezeigt werden, dass der sTNFRI einen prediktiven Wert für die Entwicklung eines
SIRS/MODS nach schwerwiegenden operativen Eingriffen wie beispielsweise einem
kardiopulmonalen Bypass hat. Vergleicht man post- und preoperative Werte miteinander, so
ist das Verhältnis bei SIRS/MODS-Patienten deutlich höher als bei Patienten mit einem
unkomplizierten Verlauf (EL BARBARY 2002).
Mit Hilfe von Knockout-Mäusen, denen entweder das Gen für den TNF-RI oder den TNF-RII
fehlt, konnten in dieser Hinsicht neue Erkenntnisse gewonnen werden. Bei TNFRp55-/-
Mäusen, welche defekte Peyer`sche Platten besitzen, wurde eine Resistenz gegenüber
niedrigen Endotoxindosen festgestellt. Die Verabreichung hoher Dosen hingegen war für die
Tiere letal (ROTHE 1993). Bei Infektionsversuchen dieser Knockout-Mäuse mit dem Erreger
Listeria monocytogenes wurde eine schlechte Abwehr der Mikroorganismen und ein Erliegen
der Tiere aufgrund der Infektion beobachtet (PFEFFER 1993). Die Suszeptibilität für Listeria
monocytogenes wird polygenetisch determiniert und ist stark stammesabhängig. Während
BALB/c-Tiere bei Infektionsversuchen erkranken, stellen sich C57BL/6-Mäuse als resistent

Literaturübersicht 20
heraus. Versuche, bei denen der Erreger Streptococcus pneumoniae eingesetzt wurde,
erbrachten ähnliche Ergebnisse (O`BRIAN et al. 1999). ACTON et al. (1996) stellten zwar
eine Resistenz der TNFRp55-/- Mäuse gegenüber E. coli-Endotoxinen fest, eine Peritonitis
konnte jedoch mit Hilfe dieser Erreger ausgelöst werden. Außerdem wurde bei diesen Tieren
nach Verabreichung von TNF-α eine verminderte Expression von VCAM-1 und E-Selektin
auf Endothelzellen beobachtet, was eine verminderte Leukozyteninfiltration in Leber, Lunge
und Niere zur Folge hat (MACKAY 1993; NEUMANN 1996). VAN GRIENSVEN et al.
(2001) zeigten in einem Sepsismodell mit TNFRp55-/- Mäusen, dass eine Behandlung mit
dem Hormon Dehydroepiandrostenon (DHEA) die Mortalität signifikant senkte. Wobei sich
der protektive Effekt bei Mäusen, denen das Gen für den TNF-RI fehlt, deutlicher zeigte als
bei den Wildtypen. Aufgrund der Tatsache, dass in beiden Gruppen eine positive Wirkung des
Hormons zu bemerken war, wird davon ausgegangen, dass es sowohl einen zytokinabhängige
als auch eine zytokinunabhängige Wirkungsweise des DHEA geben muß (VAN
GRIENSVEN 2001). Der TNF-RI hat demnach ebenfalls eine wichtige Funktion im Rahmen
einer Sepsis, die aufgrund eines traumatischen Ereignisses entsteht.
Tieren, denen das Gen für den TNF-RII fehlt, zeigen eine weniger ausgeprägte
Gewebsnekrose nach subkutanen TNF-Injektionen als Vergleichstiere (ERICKSON et al.
1994).

Literaturübersicht 21
Monozyten/Makrophagen TNF-α p55-Rezeptor p75-Rezeptor Entwicklung von Lymphfollikeln Apoptose Gewebsnekrosen und Peyer`schen Platten Fieber T-Zellproliferation Endotoxinschock Adhäsionsmolekülexpression auf Endothelzellen Fibroblastenproliferation Apoptose von peripheren Zytotoxischen T-Zellen
Lyse von Tumorzellen
Abbildung 3: Wirkungen des TNF-α in Bezug auf die verschiedenen Rezeptoren (VAN GRIENSVEN 2000)
TNF= Tumor Nekrose Faktor; p55= Tumor Nekrose Faktor-Rezeptor I; p75= Tumor Nekrose Faktor-Rezeptor II
2.6 Fcγ-Rezeptoren
Im humanen System sind drei Klassen von Fcγ-Rezeptoren (FcγR) bekannt: Fcγ-RI, Fcγ-RII
und der Fcγ-RIII. Sie bilden eine Familie von Zelloberflächenrezeptoren, die genetisch,
strukturell und funktionell eng miteinander verwandt sind (RAVETCH 1991). Beim 4th
International Workshop on Human Leukocyte Differentiation Antigens 1989 in Wien wurden
diese Rezeptoren mit den Bezeichnungen CD64 für den Fcγ-RI, CD32 für den Fcγ-RII und

Literaturübersicht 22
CD16 für den Fcγ-RIII belegt(KNAPP 1989). Die Rezeptoren besitzen die Eigenschaft, den
Fc-Teil des Immunglobulins G (IgG) zu binden und stellt somit ein wesentliches
Verbindungsglied zwischen der humoralen Antigenerkennung durch Immunglobuline und der
Aktivierung von Effektorzellen dar. Auf diese Weise wird eine Vielzahl immunologischer
Funktionen wie die antikörperabhängige zelluläre Zytotoxizität, Phagozytose, Degranulation
und Immunkomplex-Clearance vermittelt. Die Rezeptoren besitzen ebenfalls eine
immunregulatorische Funktion indem sie die Lymphozytenproliferation beeinflussen und die
Antikörperproduktion steuern (VAN DE WINKEL 1991a; WITTE 1992).
Alle Gene des Fcγ-R sind auf dem Chromosom 1 lokalisiert. Bisher sind zwei
unterschiedliche Gene für den Fcγ-RI und drei verschiedene Gene mit insgesamt sechs
Transkripten für den Fcγ-RII bekannt (BROOKS 1989; VAN DE WINKEL 1991b). Die
beiden Gene des Fcγ-RIII werden zellspezifisch exprimiert (RAVETCH 1989). Auf
natürlichen Killerzellen liegt das Gen Fcγ-RIIIA transkribiert als transmembranöse Form des
Fcγ-RIII (Fcγ-RIIIa) vor. Auf der Membran der neutrophilen Granulozyten hingegen findet
man den Fcγ-RIIIB. Es enthält die Informationen für eine glykosylphosphatidylinositol-
verankerte Form des Fcγ-RIII, den Fcγ-RIIIb. Strukturell handelt es sich bei den Fcγ-R mit
Ausnahme des Fcγ-RIIIb auf neutrophilen Granulozyten um integrale Membranproteine. Sie
besitzen einen extrazellulären Anteil mit IgG-ähnlichen Domänen, die die Fcγ-R der Ig-
Superfamilie zuordnen, einen transmembranen und einen intrazellulären Anteil. Alle Fcγ-R
sind mehrfach glykosyliert.
Die Rezeptoren binden das IgG mit unterschiedlicher Affinität. Nur der Fcγ-RI ist in der Lage
monomeres IgG mit hoher Affinität zu binden(ANDERSON 1980). Fcγ-RII und Fcγ-RIII
hingegen interagieren aufgrund ihrer niedrigen Affinität zu monomeren IgG vorwiegend mit
IgG-Immunkomplexen. Sie werden deshalb als niedrig-affine Fcγ-R bezeichnet (HUIZINGA
1989; VAN DE WINKEL 1989). Fcγ-R sind besonders im peripheren Blut auf einer Vielzahl
von Zellen lokalisiert. Fcγ-RI auf Monozyten und aktivierten neutrophilen Granulozyten nach
IFN-γ-Produktion bei Streptokokkeninfektionen (GUYRE 1990) oder bei Patienten mit
schwerer kongenitaler Neutropenie, welche mit rekombinantem humanen Granulozyten
Kolonie-stimulierendem Faktor behandelt wurden (ELSNER 1992). Fcγ-RII sind vorwiegend
auf Monozyten, B-Zellen, Thrombozyten und Granulozyten zu finden. Die Fcγ-RIII befinden
sich hauptsächlich auf natürlichen Killerzellen, neutrophilen Granulozyten und

Literaturübersicht 23
Teilpopulationen von Monozyten und T-Zellen (UCIECHOWSKI 1992; VAN DE WINKEL
1991a).
2.6.1 Fcγ-Rezeptorfunktionen auf neutrophilen Granulozyten
Auf den neutrophilen Granulozyten befinden sich demnach die niedrig-affinen Fcγ-R: Fcγ-RII
und Fcγ-RIIIb. Zwar binden sie beide IgG-Immunkomplexe, unterscheiden sich jedoch in
ihrer Membranverankerung. Während der Fcγ-RII ein Transmenbranprotein ist (BROOKS
1989), ist der Fcγ-RIII auf den Granulozyten über eine Glykosylphosphatidylinositolstruktur
mit der äußeren Zellmembran verbunden (RAVETCH 1989). Diese Form der
Membranverankerung erhöht die Lateralbeweglichkeit der Rezeptoren, da sie nicht über einen
intrazytoplasmatischen Anteil an interne Proteine des Zytoskelettes gebunden sind (BROOKS
1989). Die Expression der Rezeptoren auf der Zelloberfläche wird durch hydrolytische
Enzyme wie Phospholipase C, welche den Glykolipidanker spaltet, reguliert. Auf ruhenden
neutrophilen Granulozyten werden zwischen 10.000 und 40.000 Moleküle des Fcγ-RII und
zwischen 80.000 und 300.000 Moleküle des Fcγ-RIIIb exprimiert. Eine Stimulation der
neutrophilen Granulozyten mit dem Tripeptid FMLP (N-Formylmethionin Leucyl-
Phenylalanin) oder dem Phorbolester Phorbolmyristatazetat setzt Fcγ-RIII von der
Zelloberfläche frei (HUIZINGA 1988). Die lösliche Form des Fcγ-RIII kann auch im Plasma
von gesunden Spendern nachgewiesen werden (HUIZINGA 1990).
Beide Fcγ-R induzieren Degranulation und Phagozytose (HUIZINGA 1989; HUIZINGA
1990) der neutrophilen Granulozyten, wodurch ihre wichtige Rolle bei der Entstehung des
SIRS und MODS unterstrichen wird. Die wichtigsten bei der Degranulation freigesetzten
Produkte sind Wasserstoffperoxid, Superoxidanion und das Stickstoffoxid. Der Vorgang, bei
dem diese Substanzen produziert werden, heißt respiratory burst und wird hauptsächlich vom
Fcγ-RIII vermittelt. Findet der respiratory burst jedoch in gesteigerter Form im Gewebe statt,
so wird die Organfunktion dadurch nachhaltig gestört. Ein Beispiel hierfür ist die
Beeinträchtigung der Lungenfunktion nach einem Ischämie-Reperfusionsschaden.
Der Fcγ-RIIIb auf den neutrophilen Granulozyten zeigt einen Polymorphismus, welcher als
Neutrophilen-Antigensystem (NA) bezeichnet wird (ORY 1989). Es konnten funktionelle

Literaturübersicht 24
Unterschiede zwischen NA1+ und NA2+ neutrophilen Granulozyten festgestellt werden. NA1
homozygote Zellen phagozytieren IgG-opsonierte Erythrozyten besser als NA2 homozygote
(SALMON 1990). Die über Fcγ-R induzierten zellulären Funktionen werden durch
intrazelluläre Botenstoffe vermittelt. Die Signaltransduktion der FMLP-Rezeptoren auf den
neutrophilen Granulozyten ist am genauesten untersucht worden. Auch diese Rezeptorart
bewirkt eine Degranulation und Sauerstoffradikalproduktion, womit sie ein ähnliches
funktionelles Spektrum aufweist wie die Fcγ-R. Der FMLP-Rezeptor wirkt über einen
intrazellulären Kalziumanstieg, welcher über folgende Kaskade vermittelt wird: Der Ligand-
Rezeptor-Komplex aktiviert G-Proteine, die Phospholipasen C stimulieren. Diese spalten
Phosphatidylinositolbiphosphat in Inositoltriphosphat und Diazylglyzerol (DAG).
Inositoltriphosphat setzt daraufhin Kalzium aus intrazellulären Speichern im
endoplasmatischen Retikulum frei (DILLON 1988). DAG und Kalzium initiieren dann über
die Proteinkinase C die Sauerstoffradikalproduktion (HEYWORTH 1990). Die bei diesem
Vorgang involvierten G-Proteine können durch Pertussistoxin in ihrer Funktion gehemmt
werden. Auch für den Fcγ-RII ist eine Pertussistoxin-Sensitivität in Bezug auf die
Sauerstoffradikalproduktion beschrieben worden, so dass auf eine Beteiligung der G-Proteine
an der Signaltransduktion der Fcγ-RII geschlossen werden kann (FEISTER 1988). Beim Fcγ-
RIII wurde ein intrazellulärer Kalziumanstieg nach Kreuzvernetzung des Rezeptors
nachgewiesen (KIMBERLY 1990). Es existieren somit Hinweise für Parallelen in der
Signaltransduktion von FMLP und Fcγ-Rezeptoren.
1.7 Möglichkeiten der Prävention und der Therapie des SIRS und MODS
Eine ausreichend wirksame Therapie des SIRS und MODS konnte bis heute leider nicht
entwickelt werden. Zur Verbesserung der Prognose und zur Verringerung der Mortalität beim
Auftreten dieser Syndrome gibt es jedoch unterschiedliche Präventions- und Therapieansätze.
Eine Möglichkeit besteht darin, bereits der Entwicklung des SIRS und MODS vorzubeugen,
indem Adhäsionsmoleküle selektiv inhibiert werden, um so das Rolling der neutrophilen

Literaturübersicht 25
Granulozyten und die Akkumulation dieser Zellen im Gewebe zu verhindern. Den daraus
resultierenden Organschäden könnte damit eventuell vorgebeugt werden.
Im experimentellen Modell erschien eine Therapie mit monoklonalen Antikörpern gegen
Integrin-Adhäsionsmoleküle der neutrophilen Granulozyten (Anti-CD18, Anti-CD11b)
durchaus erfolgversprechend (ARFORS 1987; GASIC 1991; TALBOTT 1994; WINN 1993).
Eine selektive Inhibition des L-Selektins, welches für das Rolling der Granulozyten an den
Gefäßendothelzellen verantwortlich ist, konnte bereits mit Hilfe monoklonalen Antikörper
EL-246 (anti-Schaf-L-Selektin) (VAN GRIENSVEN 1999b), DREG-200 (humanisiertes anti-
human-L-Selektin) (MA 1993) und CY-1747 (synthetisches sialyl Lewisx Analog) (RUBIO-
AVILLA 1997) erreicht werden. Anhand dieser Methode konnten zwar gute Erfolge erzielt
werden, jedoch muß beachtet werden, dass es bei mehrfacher Applikation einer solchen
Substanz zu einer idiopathischen Anti-Antikörperreaktion kommen kann, wodurch die
Wirksamkeit der monoklonalen Antikörper verloren geht. Zusätzlich besteht bei der
Behandlung des Patientens über mehrere Tage die Gefahr einer allergischen Reaktion auf das
Fremdeiweiß.
Aufgrund dieser Nebenwirkungen ist es vorteilhaft, Substanzen einzusetzen, die zwar das L-
Selektin inhibieren, jedoch nur eine geringe allergene Wirkung besitzen und keine Anti-
Antikörperreaktion hervorrufen. Möglich ist hier der Einsatz niedermolekularer Substanzen
auf der Basis der Oligosaccharide, wie zum Beispiel das Fucoidin. SEEKAMP et al. (2001)
konnten zwar eine gewisse protektive Wirkung des Fucoidins auf die Adhärenz der
neutrophilen Granulozyten feststellen, da der Wirkstoff jedoch ein Antagonist für alle drei
Selektine (P, E und L) ist, besteht die Gefahr der unspezifischen Bindung mit durchaus sehr
unterschiedlichen Ergebnissen. Außerdem hat das Fucoidin Heparin-ähnliche Eigenschaften,
weshalb es nach der Applikation zu Blutungen kommen kann. Von einem Einsatz bei
klinischen Studien wird aus diesen Gründen abgeraten.
Spezifische Therapieversuche mit monoklonalen Antikörpen gegen IL-1, TNF-α oder den
Thrombozyten-aggregierenden Faktor erbrachten kein zufriedenstellendes Ergebnis, es war
sogar möglich, durch die Beeeinträchtigung der Immunantwort einen negativen Effekt
hervorzurufen (DINARELLO 1993).

Literaturübersicht 26
Ein anderer Therapieansatz ist die topische und parenterale Applikation von Antibiotika.
Hiermit soll eine selektive digestive Dekontamination des oberen Verdauungstraktes von
gramnegativen und anderen potentiell pathogenen Erregern erreicht werden. Zwar konnte die
nosokomiale Infektion damit verringert werden, die Inzidenz des MODS und die Mortalität
blieben jedoch unverändert(CERRA 1992; GASTINNE 1992). Der Versuch einer Anti-
Endotoxin-Therapie mit Antiseren, hyperimmunem polyklonalem Immunglobulin,
monoklonalen IgM-Antikörpern, welche entweder gegen den Kern des Lipopolysaccharids
oder gegen die Lipid-A-Region des Endotoxinmoleküls gerichtet waren, blieben ohne
nennenswerten Erfolg (MANTHOUS 1993; ZANETTI 1993). Bei Pferden, die in Folge einer
Kolikerkrankung eine Endotoxämie entwickelten, wurde die Wirkung eines polyklonalen,
anti-endotoxin-spezifischen Immunglobulinpräparates (Stegantox 60®) untersucht. Es konnte
kein signifikanter Unterschied der Prostaglandin E2-Konzentration im Plasma der
Kontrollgruppe und der mit dem Präparat behandelten Gruppe festgestellt werden. Die
Thromboxan B2-Plasmakonzentrationen hingegen waren, auf den gesamten
Untersuchungszeitraum bezogen, geringer als der Wert der Kontrollgruppe (SPILLE 1997).
Auch die Therapie mit verschiedenen Antioxidantien wie Eisenchelatbildner, Katalase,
Superoxiddismutase, Allopurinol und N-Acetylcystein, welche die Entstehung freier Radikale
vermindern sollen, erbrachten keine deutlichen Erfolge (SCHILLER 1993). Positive
Ergebnisse wurden mit der Anwendung von Glucan als Immunmodulator und Antithrombin
III erzielt (BABINEAU 1994; YU 1993).
Über die kontinuierliche Hämofiltration als Therapiemöglichkeit bei SIRS und MODS-
Patienten herrschen sehr unterschiedliche Meinungen. HIRASAWA et al. (1996) und
KODAMA et al. (1995) sehen darin eine effektive Möglichkeit im Plasma enthaltene
Endotoxine und Immunmediatoren, besonders TNF-α, IL-1 und IL-6, zu verringern und den
Sauerstoffpartialdruck zu verbessern. SCHETZ et al. (1995) hingegen konnten keine
Verringerung der Immunmediatoren nach dieser Methode feststellen und befürchten sogar
eine zusätzliche Aktivierung von Leukozyten.
Da bis heute keine gezielte Therapie entwickelt werden konnte, bleibt die symptomatische
Prävention und Therapie des MODS in Form adäquater Notfalltherapie das Mittel der ersten
Wahl Dazu gehören die Sicherung des Gasaustausches, ausreichende Volumen- und

Literaturübersicht 27
Erythrozytenkonzentratsubstitution und die Verabreichung vasopressiver Substanzen zur
Aufrechterhaltung der Gewebeperfusion. Die Morbidität und die Mortalität können auf diese
Art und Weise jedoch nicht verringert werden (VINCENT 1996).
Es ist wichtig, anhand von Zytokinen (IL-6) im Plasma den immunologischen Status der
Patienten zu bestimmen. Je nachdem in welchem Status (hyper- oder hypoinflammatorisch)
sich der Patient befindet, kann eine Behandlung in Form von antiinflammatorischen oder
immunfördernden Maßnahmen eingeleitet werden, denn entscheidend für das Überleben
dieser Patienten ist ein zeitlich adäquates Management der Therapie. Der Status muß deshalb
für jeden Patienten mehrfach individuell bestimmt werden (VAN GRIENSVEN 2003).
1.8 Grund für die Verwendung der Knockout-Mäuse
Wie bereits erwähnt, induzieren sowohl der Fcγ-RII als auch der Fcγ-RIII auf den
neutrophilen Granulozyten die Phagozytose von Partikeln und die Degranulation
schädigender Agentien zur Abtötung des phagozytierten Objektes. Die wichtigsten dabei
freigesetzten Produkte sind Wasserstoffperoxid, Superoxidanion und das Stickstoffoxid. Der
Vorgang, bei dem diese Substanzen produziert werden, heißt respiratory burst und wird
hauptsächlich vom Fcγ-RIII vermittelt. Findet der respiratory burst jedoch in gesteigerter
Form im Gewebe statt, so wird die Organfunktion dadurch nachhaltig gestört. Ein Beispiel
hierfür ist die Beeinträchtigung der Lungenfunktion nach einem Ischämie-
Reperfusionsschaden.
Auch an der Aktivierung der neutrophilen Granulozyten über das Komplementsystem ist der
Fcγ-R beteiligt. Bakterien werden durch die Anlagerung des Komplementfaktors C3b
opsoniert, wodurch sie für phagozytierende Strukturen leichter erkennbar werden. C3b allein
kann jedoch über seinen speziellen Rezeptor die Phagozytose nicht anregen. Hierzu ist eine
zusätzliche Bindung von Immunglobulin G an den Fcγ-R des neutrophilen Granulozyten
notwendig.
TNF-α ist ein bedeutendes Zytokin bei der Inflammationsreaktion eines Organismus. Die
inflammatorische Wirkung wird hauptsächlich über den TNF-RI vermittelt (siehe 2.5). Um

Literaturübersicht 28
den Einfluß des TNF-α bei der Entwicklung eines SIRS/MODS nach einem Ischämie-
Reperfusionsschaden abschätzen zu können, wurden in der vorliegenden Arbeit sowohl an
Einfach- (Tnfrsf1atm1lmx) als auch an Doppel-Knockout-Mäusen (Fcgr3tmiSjv Tnfrsf1atm1lmx) ein
Ischämie-Reperfusionsversuch durchgeführt.

Fragestellung 29
3 Fragestellung
Es ist bekannt, dass eine Ischämie, die von einer Reperfusion gefolgt wird, eine Schädigung
verschiedener Organsysteme bewirkt. Zuerst ist hiervon die Lunge betroffen, im weiteren
zeitlichen Verlauf treten jedoch auch pathologische Veränderungen an der Leber auf.
Besonders neutrophile Granulozyten sind für die Entstehung dieses Schadens verantwortlich,
da sie ins Parenchym migrieren und dort unter anderem Sauerstoffradikale freisetzen, welche
das Gewebe beeinträchtigen. Chemotaktische Zytokine spielen somit eine wichtige Rolle bei
der Pathogenese des Ischämie-Reperfusions-Schadens. Im Rahmen dieser Arbeit soll geklärt
werden, welchen Einfluß der TNF-RI und der Fcγ-RIII auf die Entwicklung der
Organbeeinträchtigung haben.
1. Welche Rolle spielen die Rezeptoren TNF-RI und Fcγ-RIII bei der Infiltration der
neutrophilen Granulozyten ins Lungen- und Lebergewebe?
2. Ist dabei ein zeitabhängiger Effekt zu beobachten?
3. Kann ein Unterschied in der Mortalität zwischen genetisch unveränderten Tieren und
Tieren ohne TNF-RI bzw. ohne TNF-RI und Fcγ-RIII festgestellt werden?
4. Ist ein synergistischer oder addditiver Effekt der TNF-RI und Fcγ-RIII zu bemerken?
5. Welche Rolle spielen die Zytokine IL-12, TNF-α, IFN-γ, MCP-1, IL-10 und IL-6 bei der
Entstehung des I/R-Schadens?
6. Ist eine zeitabhängige Ausschüttung der Zytokine zu beobachten?
7. Welchen Effekt haben der TNF-RI und Fcγ-RIII auf die Zytokinsekretion und –wirkung?
8. Welche Rolle spielen der TNF-RI und Fcγ-RIII beim respiratory burst nach Ischämie-
Reperfusion?

Material und Methoden 30
4 Material und Methoden
4.1 Material
4.1.1 Chemikalien und Medikamente
Carprofen (Rimadyl®, Pfizer GmbH, Karlsruhe)
Desinfektionsmittel Softasept® (Braun, Melsungen)
Diethylether (Rüsch Hospital, Böblingen)
Formalin (Merck, Darmstadt)
Heparinlösung (Liquemin® N 25000) (Hoffmann-La Roche AG, Grenzach-Wyhlen)
Ketamin-Lösung (Ketanest®) (Parke-Davis GmbH, Karlsruhe)
Natriumchloridlösung 0,9% (Braun, Melsungen)
Ringer-Lactat-Lösung (Braun, Melsungen)
Stickstoff (Linde, Hannover)
Waschlotion Bactolin® basic (Bode Chemie, Hamburg)
Xylazin-Lösung (Rompun® 2%) (Bayer, Leverkusen)
Zytokin-Assay (Zytokin-CBA) (BD Biosciences, Heidelberg)
Allgemeine Laborchemikalien (u.a. verschiedene Salze für Pufferlösungen) wurden in
handelsüblicher p.A.-Qualität (Sigma-Aldrich Chemie GmbH, Taufkirchen) bezogen.

Material und Methoden 31
4.1.2 Geräte
Durchflusszytometer FACSCalibur ( BD Biosciences, Heidelberg)
Feinwaage A 120 S (Sartorius, Göttingen)
Kühlzentrifuge (Heraeus, Hanau)
Mikroskop (Zeiss, Jena)
pH-Meter 537 (Wissenschaftlich-Technische Werkstätten, Weilheim)
Photometer Biophotometer (Eppendorf, Hamburg)
Photometer Ultrospec 2000 (Pharmacia Biotech, Cambridge, England)
Rüttler Typ VF 2 (Janke & Kunkel, IKA Labortechnik, Staufen i. Br.)
Schermaschine (Aesculap Typ H 585)
Tischzentrifuge (Heraeus, Hanau)
Ultraschallhomogenisator Sonoplus GM 2070 (Bandelin-Electronic, Berlin)
Waage (Mettler-Waagen GmbH, Gießen)
Zentrifuge 3200 (Eppendorf, Hamburg)

Material und Methoden 32
4.1.3 Verbrauchsmaterialien
Combitips 50 ml (Eppendorf, Hamburg)
Einmalhandschuhe, unsteril (Kimberly Clark, Roswell, USA)
FACS-Röhrchen, 5 ml (Sarstedt, Nümbrecht)
Faden Prolene®, 1,5 metric (Ethicon, Norderstedt)
Faden Mersilene®, Polyester, geflochten, steril, 4 metric (Braun, Melsungen)
Kanülen (27 G und 24 G) (Braun, Melsungen)
Kompressen, 10 x 20 cm (Lohmann, Neuwied)
Kryoröhrchen mit Schraubdeckel (Nunc, Wiesbaden)
Pipettenspitzen (Sarstedt, Nümbrecht)
Reaktionsgefäße, 1,5 ml (Sarstedt, Nümbrecht)
Skalpellklingen No.11 (Feather, Industries Limited, Tokyo, Japan)
Spritzen (1 ml und 2 ml) (Braun, Melsungen)
Wattestäbchen (NOBA Verbandsmittel Danz GmbH, Wetter)
Zentrifugenröhrchen, 15 ml (Greiner GmbH, Nürtingen)

Material und Methoden 33
4.2 Methoden
4.2.1 Versuchstiere
Versuchstierstämme:
• C57BL/6-Mäuse, Zentrales Tierlaboratorium der MHH (werden im folgenden Teil der
Arbeit als Wildtypen (WT)bezeichnet)
• B6;129P2-Fcgr3tmiSjv TNFrsf1atm1Imx, Zentrales Tierlaboratorium der MHH (Knockout-
Mäuse für den TNF-Rezeptor I und den Fc-γ-Rezeptor III, sie werden im folgenden Teil
der Arbeit als
F--Mäuse bezeichnet)
• B6;129S-TNFrsf1atm1Imx , Zentrales Tierlaboratorium der MHH (Knockout-Mäuse für den
TNF-Rezeptor I, werden im folgenden Teil der Arbeit als T--Mäuse bezeichnet)
Die durchgeführten Versuche wurden von der Bezirksregierung Hannover unter dem
Aktenzeichen 03/663 genehmigt.
Es wurden Untersuchungen an insgesamt 120 männlichen Mäusen vorgenommen. Bei 57
Tieren handelte es sich um Mäuse des Wildtypstammes. 33 der verwendeten Versuchstiere
fehlten die Gene für den TNF-Rezeptor I und den Fc-γ-Rezeptor III. Weiteren 30 Tieren
fehlte nur das Gen für den TNF-Rezeptor I.
Alle Mäuse wurden im Zentralen Tierlaboratorium der Medizinischen Hochschule Hannover
gezüchtet, ihr mittleres Körpergewicht betrug 23,8 ± 2,1 g bei einem durchschnittlichen Alter
von 6-8 Wochen.
Während der Versuchsdurchführung wurden die Mäuse bei einer Gruppengröße von 5 bis 10
Tieren in Typ-III-Käfigen (810 cm2 Grundfläche) gehalten. Futter in pelletierter Form und
Wasser stand ihnen ad libitum zur Verfügung. Die Raumtemperatur betrug 22 ± 2 °C, es war
ein Zyklus mit 12 Stunden Licht und 12 Stunden Dunkelheit gegeben.
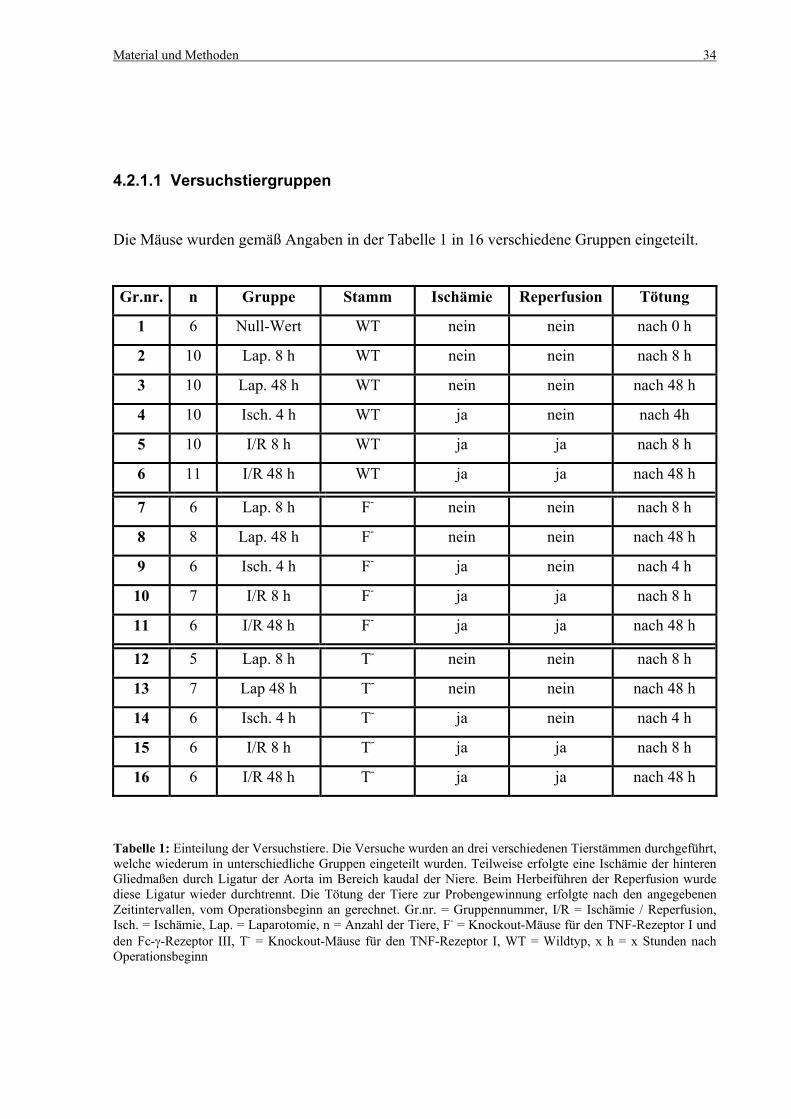
Material und Methoden 34
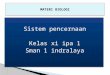
4.2.1.1 Versuchstiergruppen
Die Mäuse wurden gemäß Angaben in der Tabelle 1 in 16 verschiedene Gruppen eingeteilt.
Gr.nr. n Gruppe Stamm Ischämie Reperfusion Tötung
1 6 Null-Wert WT nein nein nach 0 h
2 10 Lap. 8 h WT nein nein nach 8 h
3 10 Lap. 48 h WT nein nein nach 48 h
4 10 Isch. 4 h WT ja nein nach 4h
5 10 I/R 8 h WT ja ja nach 8 h
6 11 I/R 48 h WT ja ja nach 48 h
7 6 Lap. 8 h F- nein nein nach 8 h
8 8 Lap. 48 h F- nein nein nach 48 h
9 6 Isch. 4 h F- ja nein nach 4 h
10 7 I/R 8 h F- ja ja nach 8 h
11 6 I/R 48 h F- ja ja nach 48 h
12 5 Lap. 8 h T- nein nein nach 8 h
13 7 Lap 48 h T- nein nein nach 48 h
14 6 Isch. 4 h T- ja nein nach 4 h
15 6 I/R 8 h T- ja ja nach 8 h
16 6 I/R 48 h T- ja ja nach 48 h
Tabelle 1: Einteilung der Versuchstiere. Die Versuche wurden an drei verschiedenen Tierstämmen durchgeführt, welche wiederum in unterschiedliche Gruppen eingeteilt wurden. Teilweise erfolgte eine Ischämie der hinteren Gliedmaßen durch Ligatur der Aorta im Bereich kaudal der Niere. Beim Herbeiführen der Reperfusion wurde diese Ligatur wieder durchtrennt. Die Tötung der Tiere zur Probengewinnung erfolgte nach den angegebenen Zeitintervallen, vom Operationsbeginn an gerechnet. Gr.nr. = Gruppennummer, I/R = Ischämie / Reperfusion, Isch. = Ischämie, Lap. = Laparotomie, n = Anzahl der Tiere, F- = Knockout-Mäuse für den TNF-Rezeptor I und den Fc-γ-Rezeptor III, T- = Knockout-Mäuse für den TNF-Rezeptor I, WT = Wildtyp, x h = x Stunden nach Operationsbeginn

Material und Methoden 35
4.2.2 Versuchsprotokoll
Zur Durchführung der Versuche wurden die Tiere mit Ketamin (100 mg/kg Körpergewicht)
und Xylazin (16 mg/kg Körpergewicht) narkotisiert und bei erhaltener Spontanatmung
operiert. Die Verabreichung der Medikamente erfolgte in einer Mischspritze durch subcutane
Injektion.
Nach Eintritt der Narkose wurde das Abdomen der Mäuse geschoren, gereinigt (Bactolin®
basic Waschlotion) und desinfiziert (Softasept®). Alle Tiere erhielten subcutan Heparin
(Liquemin®, 12,5 IE/Tier), um eine Thrombosierung der distalen Gefäße während der
Ischämiephase zu verhindern. Zusätzlich wurde allen Tieren subcutan Carprofen (Rimadyl®)
als Analgetikum appliziert. Die initiale Dosis betrug 5 mg/kg, an den folgenden Tagen wurde
den Mäusen alle 24 Stunden 2,5 mg/kg des Medikamentes verabreicht. Die Versuchstiere
wurden auf einer Korkplatte ausgebunden und ein ca. 2 cm langer Schnitt in der Medianlinie
des Abdomens (Linea alba) vorgenommen. Das Darmpaket wurde aus der Bauchhöhle
vorgelagert und in eine mit Ringer-Lactatlösung befeuchtete Kompresse gehüllt, um einer
Austrocknung vorzubeugen. Zur Erzeugung der Ischämie der unteren Extremitäten erfolgte
eine infrarenale Ligatur der Aorta abdominalis und der Vena cava caudalis mit einem
Polyesterfaden (Mersilene®) (siehe Abbildung 4). Während der nachfolgenden vierstündigen
Ischämiephase blieben die Mäuse narkotisiert. Die Laparotomiewunde wurde mit
Diagonalheften verschlossen (Prolene® Faden). Zur Verringerung des Wärmeverlustes
während der Ischämiephase wurden die Tiere auf dem Bauch liegend unter einer
Wärmelampe platziert.
Nach vier Stunden wurde die Bauchhöhle erneut eröffnet, um die Ligatur mit Hilfe einer
Skalpellklinge zu entfernen. Nach dem Verschließen der Wunde wurden die Tiere während
des gesamten Beobachtungszeitraumes (je nach Gruppe 4 oder 48 h) unter der Wärmelampe
gehalten. Futter und Wasser stand ihnen während dieser Zeit ad libitum zur Verfügung.
Die Kontrolltiere wurden narkotisiert (siehe oben) und sofort für die Organentnahme durch
Blutentzug aus dem Herzen getötet (Exsanguation, siehe 4.2.3).
Material und Methoden 36
Abbildung 4: Infrarenale Ligatur der Aorta abdominalis und Vena cava caudalis.
4.2.3 Blut und Organentnahme
Zur Probengewinnung wurden die narkotisierten Tiere (siehe Kapitel 4.2.2) durch
Exsanguation getötet. Nachdem sowohl die Bauch- als auch die Brusthöhle eröffnet wurden,
erfolgte die Punktion des Herzens mit einer Spritze, deren Konus mit Heparin gefüllt war, um
eine Gerinnung des gewonnenen Blutes zu verhindern. Das Blut wurde daraufhin für 2
Minuten bei 13000 rpm zentrifugiert und das so gewonnene Serum in einem Eppendorfgefäß
bis zu den weiteren Untersuchungen bei –80° C aufbewahrt.
Weiterhin wurde das Herz-Lungen-Paket entnommen und eine bronchoalveoläre Lavage
(BAL) durchgeführt (siehe Kapitel 4.2.4). Danach wurde ein Lungenflügel für eine
Myeloperoxidasebestimmung kryokonserviert, der andere wurde hingegen mit 5%-iger
phosphatgepufferter Formalinlösung perfundiert und schließlich in einem Glasbehältnis mit
Formalin aufbewahrt. Des weiteren wurden die Milz, eine Niere und ein Teil der Leber für
histologische Untersuchungen entnommen und ebenfalls in Formalin fixiert.

Material und Methoden 37
4.2.4 Bronchoalveoläre Lavage (BAL)
Für die bronchoalveoläre Lavage wurde eine stumpfe Kanüle in die Trachea eingeführt (siehe
Abbildung 5) und fixiert. Danach erfolgte die Spülung beider Lungenflügel mit ca. 1 ml
steriler isotonischer Kochsalzlösung. Die bei der BAL erhaltene Spülflüssigkeit wurde bis zu
den weiteren Untersuchungen in einem Eppendorfgefäß bei –80°C gelagert.
Abbildung 5: Die Kanüle wurde für die BAL in die Trachea eingeführt.
4.2.5 Kapilläre Permeabilität der Lunge
Mit Hilfe der in der BAL-Flüssigkeit gemessenen Protein- und Harnstoffkonzentration läßt
sich eine Aussage über die Schädigung der Endothelzellen und damit der kapillären
Permeabilität der Lunge in vivo machen (VAN GRIENSVEN 1999). Da jedoch
unterschiedliche Mengen an Lavageflüssigkeit zurückgewonnen wurden, muss der absolute
Proteingehalt über die gleichzeitig gemessene Harnstoffkonzentration im Serum und in der
BAL korrigiert werden, damit eine volumenunabhängige Aussage der Proteinkonzentration in
der BAL gemacht werden kann. Hierzu wird folgende Formel verwendet:
SerumBAL
SerumBAL
]Protein[]Harnstoff[]Harnstoff[]Protein[
altProteingehrelativer×
×=

Material und Methoden 38
4.2.6 Proteinbestimmung nach Lowry
Der Proteingehalt wurde sowohl im Mäuseserum als auch in der BAL nach Lowry bestimmt
(FOLIN u. CIOCALTEU, 1927). Grundprinzip dieser Eiweißbestimmung ist eine
Farbänderung der Reagenzien, die mit Hilfe eines Photometers bei 595 nm bestimmt werden
kann. Proteine reduzieren Cu(II)-Ionen zu Cu(I)-Ionen, die wiederum mit Phosphomolybdat-
Phosphowolframat einen blaugrünen Komplex bilden und somit photometrisch bestimmt
werden können. Die gemessene Extinktion ist proportional zur Proteinkonzentration.
Lösungen:
Lösung A: 2% Na₂CO₃ 0,02% Na+-K+-Tatrat
0,4% NaOH
Lösung B: 0,5% CuSO₄
Lösung I: 49 Teile Lösung A + 1 Teil Lösung B
Lösung II: Folin-Ciocalteu-Phenolreagenz
(Phosphomolybdat-Phosphowolframat 1:1 mit H2O verdünnt)
BSA-Stammlösung: 1 mg BSA/ml in Lösung I
Durchführung:
Zur Erstellung der Eichkurve wurde mit Hilfe des BSA eine Standardreihe mit verschiedenen
Proteingehalten angelegt. Für die Messung der BAL wurde ein Bereich zwischen 5 µg/ml und
500 µg/ml gewählt, die Standardreihe für die Serummessung lag zwischen 200 µg/ml und
2000 µg/ml. Aus den gemessenen Extinktionen der bekannten Konzentrationen der Proben
wurden die Standardkurven und die dazugehörigen beschreibenden Funktionen mittels
linearer Regression erstellt.

Material und Methoden 39
Die Probenmessungen wurden analog zur Standardmessung wie folgt durchgeführt: 200 µl
BAL-Flüssigkeit wurden mit 2300 µl Lösung I versetzt und danach 15 Minuten bei
Raumtemperatur inkubiert. Anschließend wurden 200 µl Lösung II hinzugegeben, eine
Inkubation für 30 Minuten, ebenfalls bei Raumtemperatur, schloss sich an. Direkt im
Anschluss erfolgte die Auswertung bei 595 nm.
Die Proteinbestimmung im Serum folgte dem gleichen Prinzip; jedoch wurden die Proben
1:50 mit Lösung I vorverdünnt. Es wurden 50 µl verdünntes Serum eingesetzt, die mit
2450 µl Lösung I versetzt wurden. Die folgenden Schritte waren identisch mit der
Proteinbestimmung in der BAL-Flüssigkeit.
Mit Hilfe der erstellten Standardkurven und deren zugehörigen Funktionen konnten die
Proteinkonzentrationen für die Serum- und BAL-Proben errechnet werden:
baOD +×= ]Protein[
abOD −
=]Protein[
OD = optische Dichte [Protein] = Protein-Konzentration a , b = Konstanten
4.2.7 Harnstoffbestimmung
Der quantitativen Bestimmung des Harnstoffgehaltes in der BAL und im Serum liegt als
Prinzip ebenfalls eine Farbreaktion zugrunde: Urease katalysiert die Hydrolyse von Harnstoff
zu Ammoniak und Kohlendioxid. Das entstehende Ammoniak reagiert mit Hypochlorit und
Phenol zu Indolphenol, welches photometrisch erfasst wird. Die gemessene
Extinktionsänderung ist proportional zum Harnstoffgehalt.

Material und Methoden 40
Lösungen:
Urease-Lösung : 10 U in PBS/Glycerin
(1,25 ml Urease und 8,75 ml PBS)
Urea-Standardlösung: 30 mg ad 10 ml H₂O
Phenol/Natriumprussidlösung: 25 mg Na-Prussid
5 ml Phenol (pH 6-8)
ad 500 ml H₂O
Hypochloritlösung: 2,5 g NaOH
2,5 ml Na- Hypochlorit
ad 500 ml H₂O
Durchführung:
Zur Erstellung der Eichkurve wurde eine Standardreihe mit Harnstoffkonzentrationen
zwischen 150 µg/ml und 3000 µg/ml verwendet. Sie galt sowohl für die BAL- als auch für die
Serumbestimmungen. Aus den bei 595 nm gemessenen Extinktionen der bekannten
Standardkonzentrationen wurde eine Standardkurve mit einer dazugehörigen Funktion erstellt.
200 µl BAL-Flüssigkeit wurden mit 100 µl Ureaselösung versetzt, gut gemischt und 15
Minuten bei 37°C inkubiert. Nach der Zugabe von jeweils 5 ml Phenol/Natriumprussid und
Hypochloritlösung wurden die Ansätze erneut gut durchmischt und für 45 Minuten bei 37°C
inkubiert. Im Anschluß erfolgte die photometrische Auswertung bei 595 nm. Die
Durchführung der Serummessung erfolgte nach dem oben beschriebenen Prinzip. Jedoch
mussten die Proben 1:5 mit PBS-Lösung vorverdünnt werden. Im Unterschied zur BAL-
Messung wurden nur 50 µl eingesetzt, die außer mit 100µl Ureaselösung noch mit 150 µl PBS
versetzt wurden, bevor der erste Inkubationsschritt begann. Alle nun folgenden Schritte waren
identisch.

Material und Methoden 41
Mit Hilfe der erstellten Standardkurve und ihrer zugehörigen Funktion konnten nun die
Harnstoffkonzentrationen in den Serum- und BAL-Proben errechnet werden:
baOD +×= ]Harnstoff[
abOD −
=]Harnstoff[
OD = optische Dichte [Harnstoff] = Harnstoff-Konzentration a, b = Konstanten
4.3 Myeloperoxidaseaktivität (MPO)
Nach ALLAN et al. (1985) besteht ein direkter Zusammenhang zwischen der MPO-Aktivität
eines Gewebes und der darin enthaltenen Anzahl von neutrophilen Granulozyten. Um indirekt
den Gehalt an neutrophilen Granulozyten bestimmen zu können, wurde daher die MPO-
Aktivität nach folgendem Prinzip ermittelt:
Lösungen:
0,02 M Kalium-Phosphatpuffer pH 7,4: 0,696 g K₂HPO₄
2,18 g KH₂PO₄ ad 1 l aqua dest.
HTAB-Puffer pH 6,0: 7,84 g K₂HPO₄
0,68 g KH₂PO₄ 5,0 g HTAB
ad 1 l aqua dest.
TMB: 700 µl 3,3´,5,5´-Tetramethylbenzidin-Substrat

Material und Methoden 42
Durchführung:
Die kryokonservierten Gewebeproben wurde in einer Reibschale in flüssigem Stickstoff fein
zermahlen und in Eppendorfgefäße verbracht. Um die eingefüllte Menge zu ermitteln, wurden
die Gefäße vor und nach dem Befüllen gewogen. Anschließend wurden 700 µl Kalium-
Phosphatpuffer (4°C) hinzugefügt und die Proben gut durchmischt. Es folgte eine
Zentrifugation für 15 Minuten bei 13 000 rpm. Der Überstand wurde verworfen und die
verbleibenden Pellets für 2 Stunden bei 60°C inkubiert. Daraufhin erfolgte eine Resuspension
der Pellets in 1 ml HTAB-Puffer (Raumtemperatur) und eine Ultraschallhomogenisation für
10 Sekunden. Nachdem die Proben in flüssigem Stickstoff dreimal eingefroren und wieder
aufgetaut wurden, wurde eine erneute Ultraschallhomogenisation für 10 Sekunden
durchgeführt. Es folgte eine abschließende Zentrifugation für 15 Minuten bei 13 000 rpm.
70 µl des Überstandes wurden mit 700 µl TMB versetzt, gut durchmischt und bei 655 nm
photometrisch gemessen.
Die Eichkurve für das Photometer wurde mit Hilfe von Meerrettichperoxidase erstellt, indem
Konzentrationsstufen von 1, 3, 10, 30 und 100 mU Peroxidase pro ml Testflüssigkeit
hergestellt wurden. Die jeweiligen Extinktionsänderungen pro Minute (∆E) konnten daraufhin
bei einer Wellenlänge von 655 nm gemessen werden.
Der Gehalt an Peroxidase pro g Lungengewebe wurde anhand der im Versuch ermittelten
Extinktionsänderung pro Minute und der vorliegenden Eichkurve nach folgender Formel
berechnet:
∆E x 100 = mU Peroxidaseaktivität
mU = milliUnits

Material und Methoden 43
4.4 Histologische Untersuchung
Für die histologische Untersuchung wurden den Versuchstieren post mortem jeweils ein
Lungenflügel (siehe Kapitel 4.2.3.), die Milz, die linke Niere und ein Teil der Leber
entnommen und für mindestens 7 Tage in phosphatgepuffertem 5%igen Formalin aufbewahrt.
Die entnommenen Gewebeproben wurden für die lichtmikroskopische Untersuchung in
Paraffin eingebettet und daraus 3 µm dicke Präparate angefertigt. Die Anfärbung dieser
Schnitte erfolgte mit Hämatoxylin und Eosin. Die mikroskopische Beurteilung der Präparate
wurde mit 63-, 100- und 400-facher Vergrößerung durchgeführt.
Bewertung der Organproben:
In den Präparaten der Lunge wurden das interstitielle Ödem und die Infiltration mit
neutrophilen Granulozyten beurteilt. Dabei wurden Präparate ohne besonderen Befund mit 0,
geringgradige Verdickungen der Septen bzw. geringgradige Infiltrationen mit neutrophilen
Granulozyten mit 1 und mittel- bis hochgradige Verdickungen bzw. Infiltrationen mit 2
bewertet. Die Beurteilung der Leberschnitte erfolgte nach dem gleichen Scoresystem, wobei
neben dem interstitiellen Ödem und der Granulozyteninfiltration zusätzlich die hydropische
Degeneration der Leberzellen betrachtet wurde. Bei der histologischen Beurteilung der
Milzpräparate wurde die Schwere der Nekrosen nach oben beschriebenem System mit Zahlen
von 0 bis 2 bewertet. Ließ sich des weiteren die rote von der weißen Pulpa abgrenzen, so
wurde das Organ mit 1 bewertet, war dies jedoch nicht der Fall, so erfolgte die Bewertung mit
einer 2. Zusätzlich wurde die Fülle der weißen Pulpa betrachtet: wenig gefüllte Follikel
wurden mit 1, stark gefüllte mit 2 beurteilt. Die Präparate der Niere wurden ebenfalls auf
Infiltrationen mit neutrophilen Granulozyten nach oben beschriebenem Prinzip (0-2)
untersucht, außerdem erfolgte die Beurteilung der Fülle der Glomeruli (Anzahl der Zellkerne)
mit 1 für weniger stark gefüllte und 2 für stark gefüllte Glomeruli (siehe Tabelle 2).
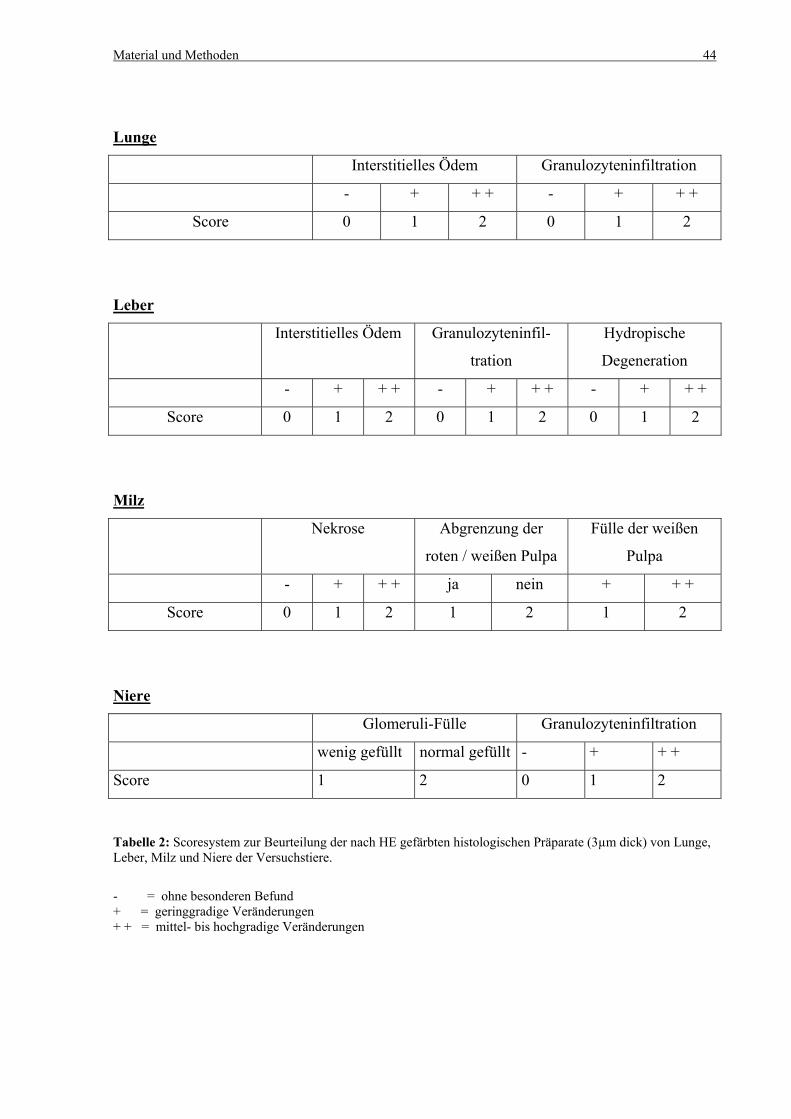
Material und Methoden 44
Lunge
Interstitielles Ödem Granulozyteninfiltration
- + + + - + + +
Score 0 1 2 0 1 2
Leber
Interstitielles Ödem Granulozyteninfil-
tration
Hydropische
Degeneration
- + + + - + + + - + + +
Score 0 1 2 0 1 2 0 1 2
Milz
Nekrose Abgrenzung der
roten / weißen Pulpa
Fülle der weißen
Pulpa
- + + + ja nein + + +
Score 0 1 2 1 2 1 2
Niere
Glomeruli-Fülle Granulozyteninfiltration
wenig gefüllt normal gefüllt - + + +
Score 1 2 0 1 2
Tabelle 2: Scoresystem zur Beurteilung der nach HE gefärbten histologischen Präparate (3µm dick) von Lunge, Leber, Milz und Niere der Versuchstiere.
- = ohne besonderen Befund + = geringgradige Veränderungen + + = mittel- bis hochgradige Veränderungen

Material und Methoden 45
4.4.1 Durchflusszytometrie ( FACS-Analyse)
Die Durchflusszytometrie bietet durch die Kombination der Detektion von
Lichtstreuungseigenschaften und Fluoreszenzemission eine effektive Möglichkeit der
qualitativen und quantitativen Messung einzelner Zellen. In der vorliegenden Arbeit wurden
keine Zellen, sondern Beads (engl.: Perle, Tropfen) gemessen. Die FACS-Analyse
(Fluorescent Activated Cell Scanner) hat gegenüber einem herkömmlichen ELISA (Enzyme
Linked Immuno Sorbent Essay) mehrere Vorteile: Es wird nur 1/6 des ursprünglichen
Probenvolumens für die gleiche Anzahl Ergebnisse benötigt, welche außerdem in nur einem
Durchlauf gemessen werden können. Zusätzlich ist die Genauigkeit der Ergebnisse durch die
Einzelbeadmessung größer als bei einem ELISA.
4.4.1.1 Funktionsprinzip des Durchflusszytometers
Ein Durchflusszytometer ist ein Messsystem, das optische Signale, Streulicht und Fluoreszenz
einzelner fokussierter Partikel analysiert. Die zu untersuchende Flüssigkeit wird durch eine
bestimmte Analysestrecke geleitet und von den Beads einzeln durchlaufen. Hierbei werden
die Partikel von einem Argonionenlaser mit einer Wellenlänge von 488 nm angeregt und mit
Hilfe eines optischen Detektionssystems sowohl die Fluoreszenz als auch die
Streulichtemissionen der einzelnen Partikel ermittelt. Anhand des Vorwärtsstreulichtes
(Forwardscatter = FSC) kann eine Aussage über die Partikelgröße gemacht werden, während
das Seitwärtsstreulicht (Sidescatter = SSC) ein Maß für die Granularität des Partikels darstellt.
In der vorliegenden Arbeit wurde eine Auswahl von Zytokinen (IL-6, IL-10, IL-12p70, TNF-
α, IFN-γ und MCP-1) im Serum der Versuchstiere gemessen. Das Prinzip der Analyse besteht
darin, dass die verschiedenen im Serum enthaltenen Zytokine mit Hilfe von spezifischen
Antikörpern, denen am Fc-Teil ein Bead angehängt ist, für den Laserstrahl sichtbar gemacht
werden. Je nach Zytokinzugehörigkeit besitzen diese Beads verschiedene
Fluoreszenzintensitäten, um eine Unterscheidung möglich zu machen. Ein weiterer gegen das
Zytokin gerichteter Detektionsantikörper ist an seinem Fc-Teil mit einem PE-Farbstoff

Material und Methoden 46
gekoppelt. Die Menge dieses Farbstoffes wird vom FACS-Gerät ermittelt, sie ist proportional
zur vorhandenen Menge des Zytokins.
Die Darstellung der Messwerte erfolgte über einen angeschlossenen Computer mit dem
Programm Cell Quest Pro (BD Biosciences). Die mit Hilfe von rekombinanten Zytokinen
erstellten Standardkurven wurden als Regressionskurven mit dazugehörigen beschreibenden
Funktionen dargestellt. Mit Hilfe dieser Funktionen wurden die Serumkonzentrationen der
Zytokine in den Proben berechnet und anhand eines Punktdiagrammes (dot plot) und in
tabellarischer Form dargestellt.
4.4.1.2 Durchführung der Messung
Reagenzien:
Mouse Inflammation Capture Beads: Jedes Gefäß enthält Antikörper, die gegen ein
bestimmtes Zytokin gerichtet sind. Um eine Unterscheidung möglich zu machen, sind die
enthaltenen Antikörper an einen Bead gekoppelt, welcher mit einer für das Zytokin
spezifischen Intensität fluoresziert. Die Zytokine sind in abnehmender Fluoreszenzstärke
geordnet:
IL-6 (stärkste Fluoreszenz)
IL-10
MCP-1
IFN-γ
TNF-α
IL-12p70 (schwächste Fluoreszenz)

Material und Methoden 47
Cytometer Setup Beads: Beads enthaltende Flüssigkeit zur Kalibrierung des Zytometers
Mouse Inflammation PE Detection Reagent: mit PE-Farbstoff kombinerte Antikörper, die
gegen die verschiedenen Zytokine gerichtet sind
PE Positive Control Detector: mit PE-Farbstoff konjugierte Antikörper zur anfänglichen
Einstellung des Zytometers
FITC Positive Control Detector: mit FITC-Farbstoff konjugierte Antikörper zur anfänglichen
Einstellung des Zytometers
Mouse Inflammation Standards: murine Zytokine enthaltende Flüssigkeit zur Erstellung einer
Standardreihe. Mit Hilfe dieser Standardreihe können Fluoreszenzintensitäten bei bestimmten
Zytokinkonzentrationen am Zytometer eingestellt werden.
Verdünnungsflüssigkeit: gepufferte Proteinlösung
Waschpuffer: PBS-Lösung
Durchführung:
Erstellung der Standardreihe: Der Mouse Inflammation Standard wurde mit 0,2 ml
Verdünnungsflüssigkeit versetzt und nach dem Mischen für mindestens 15 Minuten stehen
gelassen. Aus dieser Lösung erfolgte die Erstellung einer Standardreihe mit folgenden
Verdünnungsstufen: Standard mit höchster Konzentration, 1:2, 1:4, 1:8, 1:16, 1:32, 1:64,
1:128 und 1:256. Die Konzentrationen der Standardreihe reichten von 5000 pg/ml (Standard
mit höchster Konzentration) bis zu 20 pg/ml (1:256). Als Negativkontrolle diente die Assay-
Flüssigkeit.
Erstellung der Mixed Capture Beads: Da die verschiedenen Antikörper in unterschiedlichen
Fläschchen aufbewahrt wurden, mussten sie vor der Messung gemischt werden. Es wurde aus
allen Behältnissen jeweils die gleiche Menge Antikörperflüssigkeit entnommen (10µl pro
Serumprobe, Standardlösung und Negativkontrolle) und in ein Röhrchen gegeben.

Material und Methoden 48
Serumproben: 50µl der Mixed Capture Beads und 50µl Serum wurden in die verschiedenen
Probenröhrchen gegeben. Danach erfolgte eine Zugabe von 50µl Mouse Inflammation PE
Detection Reagent, worauf sich eine Inkubation über zwei Stunden bei Raumtemperatur an
einer abgedunkelten Stelle anschloß. Nach diesem Inkubationsschritt wurde jeweils 1ml
Waschpuffer in die Röhrchen gegeben und sie bei 200 x g für 5 Minuten zentrifugiert. Der
Überstand wurde vorsichtig abgesaugt und verworfen. Erneut wurden 300µl Waschpuffer
hinzugefügt und das Bead-Pellet resuspendiert. Die Proben konnten mit Hilfe des
Durchflusszytometers ausgewertet werden.
4.5 Statistik
Für die einzelnen Gruppen wurden Mittelwerte und der Standardfehler (SEM) berechnet. Alle
statistischen Berechnungen erfolgten mit dem Programm SigmaStat 2.03 (SPSS Inc., San
Rafael, USA).
Signifikante Unterschiede zwischen den Gruppenmittelwerten wurden mit dem ungepaarten
Student`s t-test ermittelt. Bei ungleicher Varianz oder nicht normal verteilter Population
wurde ein Mann-Whitney Rank Sum Test durchgeführt. Die Überlebenskurven wurden mit
dem sogenannten Fisher exakter Test geprüft (p < 0,05 = signifikanter Unterschied).
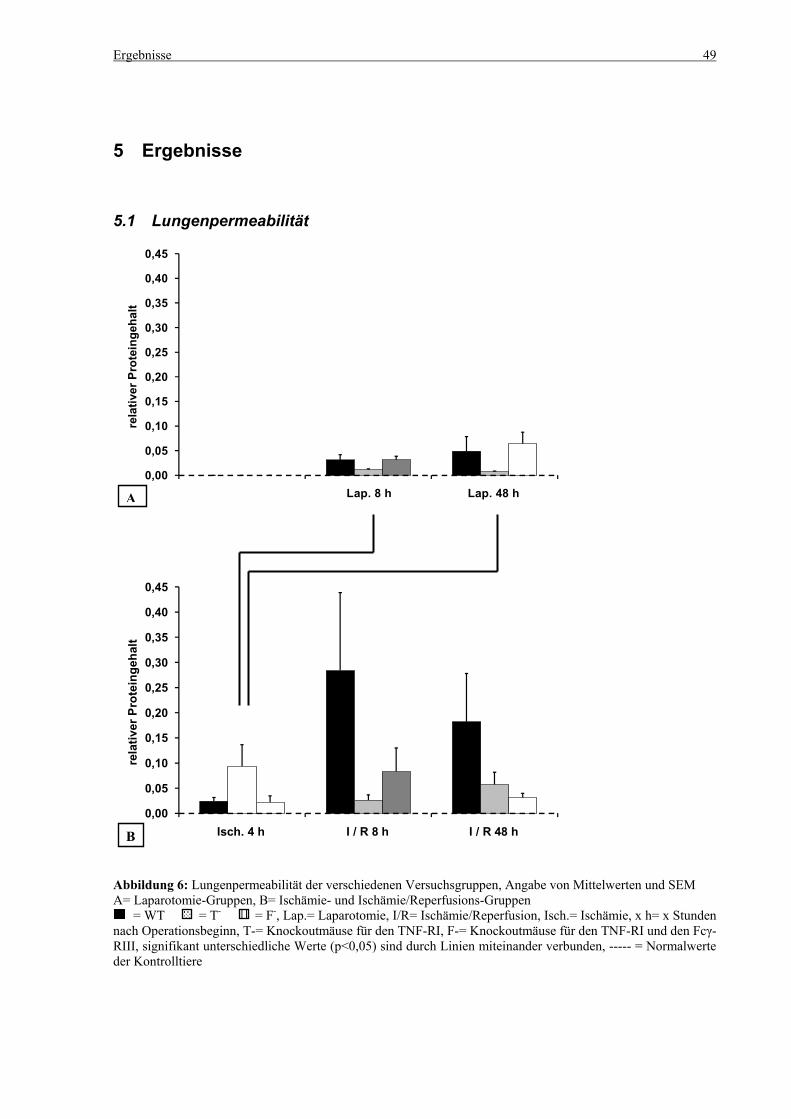
Ergebnisse 49
5 Ergebnisse
5.1 Lungenpermeabilität
Abbildung 6: Lungenpermeabilität der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
Lap. 8 h Lap. 48 h
rela
tiver
Pro
tein
geha
lt
A
B
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
Isch. 4 h I / R 8 h I / R 48 h
rela
tiver
Pro
tein
geha
lt

Ergebnisse 50
Abbildung 6 zeigt die kapilläre Lungenpermeabilität der verschiedenen Versuchsgruppen. Es
fällt auf, dass fast alle Gruppen der T--und F--Knockout-Tiere geringere Werte aufweisen als
die entsprechenden Gruppen der WT-Tiere. Ausnahmen bilden hierbei die Gruppe T--
Ischämie 4 h und F--Laparotomie 48 h. Die Lungenpermeabilität in der Gruppe T--
Laparotomie 8 h (0,01 ± 0,01) ist gegenüber der Gruppe T--Ischämie 4 h (0,09 ± 0,04)
signifikant niedriger (p<0,05). Auch der Wert der Gruppe T--Laparotomie 48 h ist signifikant
niedriger als der der Gruppe Ischämie 4 h (0,09 ± 0,04) (p<0,05).
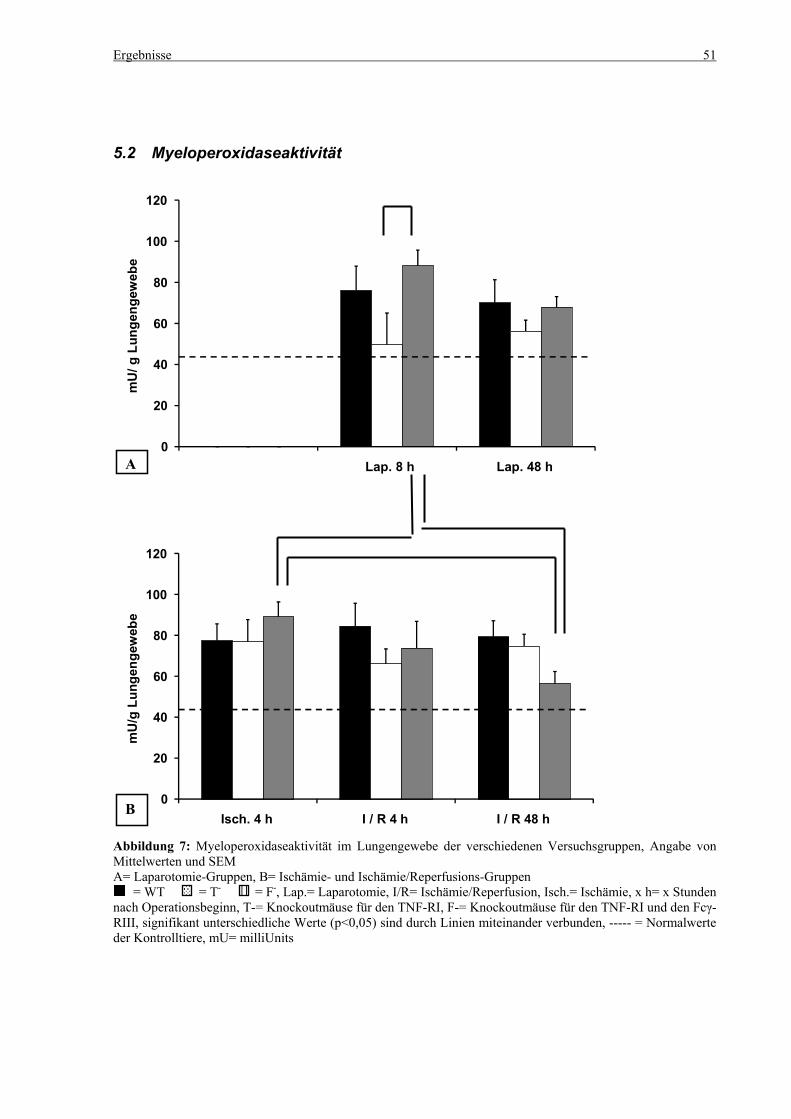
Ergebnisse 51
5.2 Myeloperoxidaseaktivität
Abbildung 7: Myeloperoxidaseaktivität im Lungengewebe der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, mU= milliUnits
0
20
40
60
80
100
120
Lap. 8 h Lap. 48 h
mU
/ g L
unge
ngew
ebe
A
B0
20
40
60
80
100
120
Isch. 4 h I / R 4 h I / R 48 h
mU
/g L
unge
ngew
ebe

Ergebnisse 52
In Abbildung 7 ist die Myeloperoxidaseaktivität im Lungengewebe der verschiedenen
Versuchsgruppen dargestellt. Signifikante Unterschiede konnten besonders innerhalb der F--
Gruppen festgestellt werden. Die MPO-Aktivität der Gruppe F--Ischämie 4 h (87,99 ± 7,74
mU/g Lungengewebe) war signifikant höher als die der Gruppe F--Ischämie-Reperfusion 48 h
(56,45 ± 5,79 mU/g Lungengewebe) (p<0,05). Auch zwischen der MPO-Aktivität der Gruppe
F--Ischämie 4 h (89,02 ± 7,03 mU/g Lungengewebe) und der Gruppe F--Ischämie-Reperfusion
48 h (56,45 ± 5,79 mU/g Lungengewebe) war ein signifikanter Unterschied zu beobachten
(p<0,05). Die MPO-Aktivität der Gruppe F--Laparotomie 48 h (67,93 ± 5,12 mU/g
Lungengewebe) ist signifikant niedriger als die der Gruppe F--Ischämie 4 h (89,02 ± 7,74
mU/g Lungengewebe) (p<0,05).
Weiterhin konnte ein signifikanter Unterschied zwischen den Knockout-Gruppen T--
Laparotomie 8 h (49,68 ± 15,33) und F--Laparotomie 8 h (87,99 ± 7,74) festgestellt werden
(p<0,05).
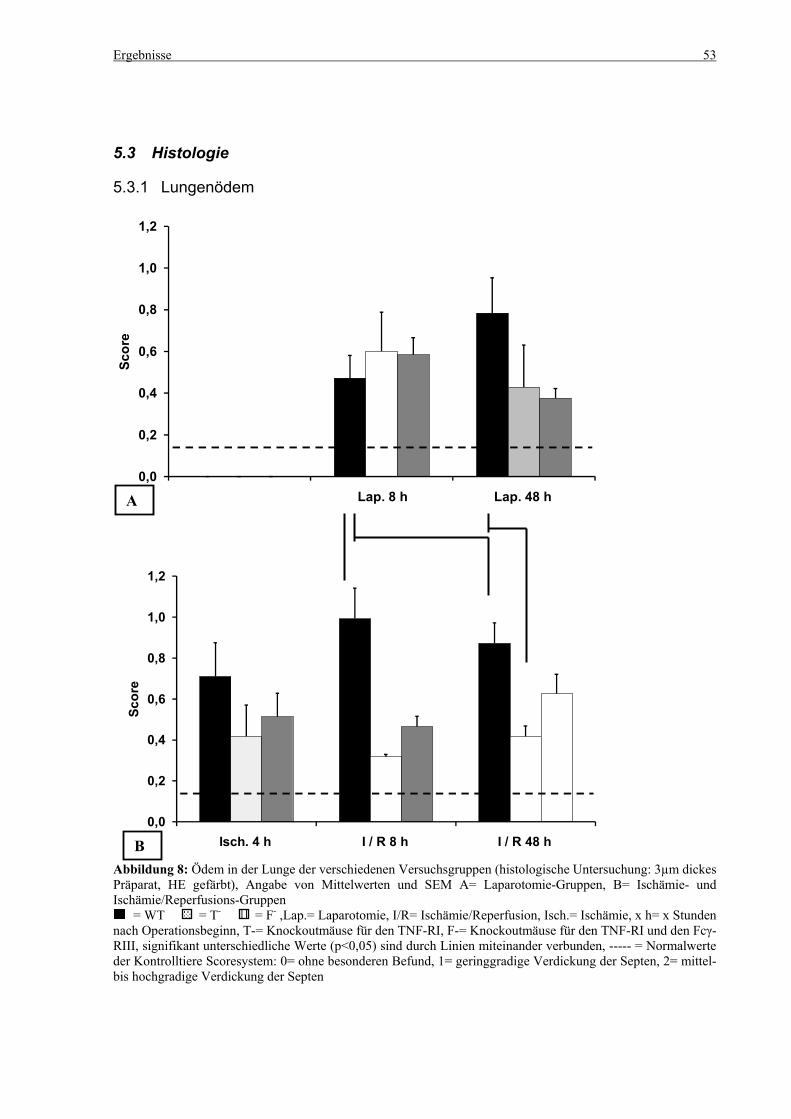
Ergebnisse 53
5.3 Histologie
5.3.1 Lungenödem
Abbildung 8: Ödem in der Lunge der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F- ,Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere Scoresystem: 0= ohne besonderen Befund, 1= geringgradige Verdickung der Septen, 2= mittel- bis hochgradige Verdickung der Septen
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Lap. 8 h Lap. 48 h
Scor
e
A
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 54
Abbildung 8 zeigt das Lungenödem der verschiedenen Versuchsgruppen. Bis auf in der
Gruppe Laparotomie 8 h wurden für die Knockout-Tiere geringere Lungenödeme gegenüber
den Wildtyp-Tieren festgestellt (siehe Abbildung 9 und 10). Sowohl die Werte in der F--
Gruppe Ischämie-Reperfusion 8 h (0,46 ± 0,05) als auch die Werte in der Gruppe T--
Ischämie-Reperfusion 8 h (0,32 ± 0,01) waren gegenüber den Werten für die Wildtyp-Tiere
der entsprechenden Gruppe (0,99 ± 0,15) signifikant niedriger (p<0,05). Das Lungenödem der
Gruppe WT-Ischämie-Reperfusion 48 h (0,87 ± 0,10) waren im Vergleich zur Gruppe T--
Ischämie-Reperfusion 48 h (0,42 ± 0,05) signifikant höher (p<0,05).
Auch die Vergleiche der Gruppe WT-Laparotomie 8 h (0,47 ± 0,11) mit den Gruppen WT-
Ischämie-Reperfusion 8 h (0,99 ± 0,15) und WT-Ischämie-Reperfusion 48 h (0,87 ± 0,10)
ergaben jeweils ein signifikant stärkeres Lungenödem bei den I/R-Gruppen (p<0,05).
Abbildung 9: Histologisches Präparat der Lunge ohne besonderen Befund (Gruppe WT Lap. 8 h), HE-Färbung,
100x
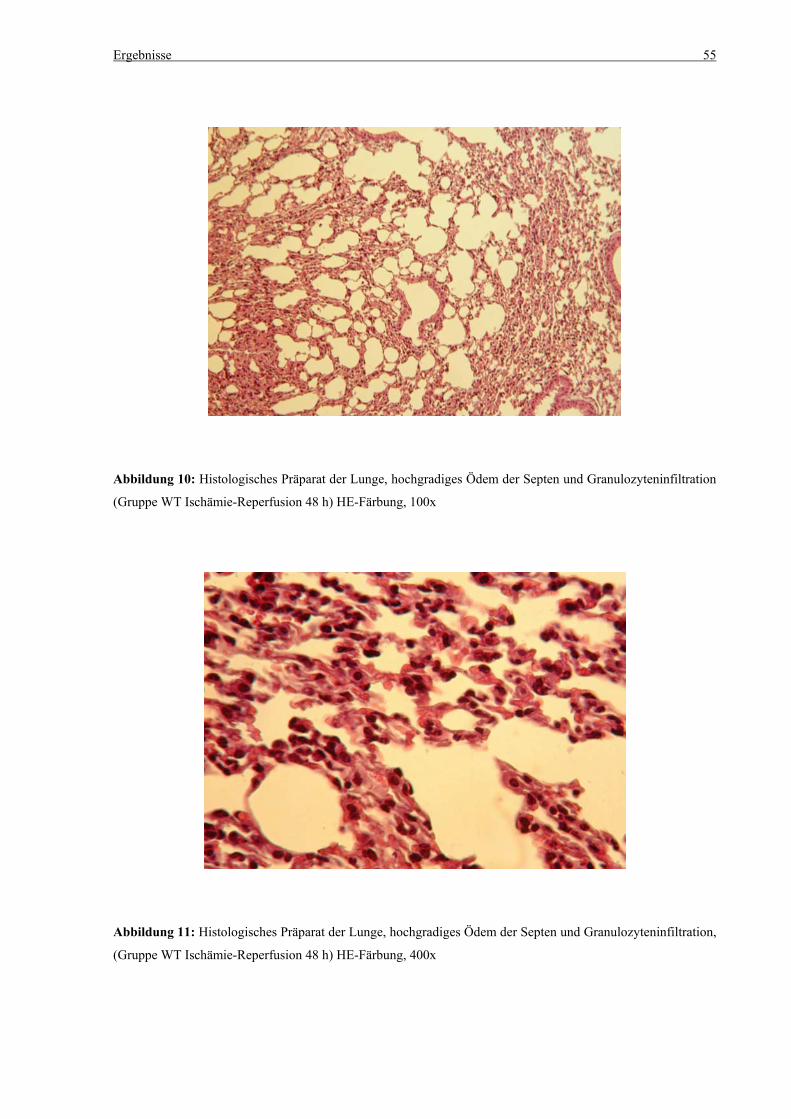
Ergebnisse 55
Abbildung 10: Histologisches Präparat der Lunge, hochgradiges Ödem der Septen und Granulozyteninfiltration
(Gruppe WT Ischämie-Reperfusion 48 h) HE-Färbung, 100x
Abbildung 11: Histologisches Präparat der Lunge, hochgradiges Ödem der Septen und Granulozyteninfiltration,
(Gruppe WT Ischämie-Reperfusion 48 h) HE-Färbung, 400x
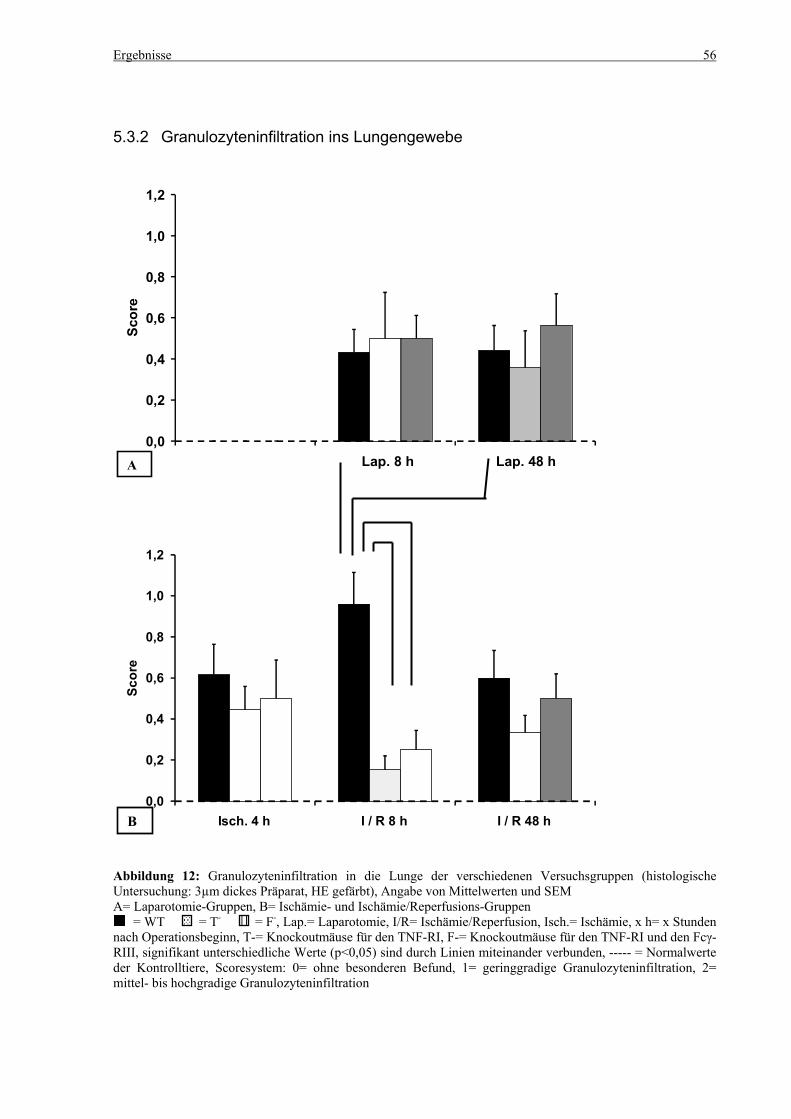
Ergebnisse 56
5.3.2 Granulozyteninfiltration ins Lungengewebe
Abbildung 12: Granulozyteninfiltration in die Lunge der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= geringgradige Granulozyteninfiltration, 2= mittel- bis hochgradige Granulozyteninfiltration
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Lap. 8 h Lap. 48 h
Scor
e
A
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 57
Abbildung 12 zeigt die Granulozyteninfiltration in die Lunge der verschiedenen
Versuchsgruppen (siehe Abbildung 11 und 13). In der Ischämie 4 h Gruppe und in den
Ischämie-Reperfusions-Gruppen sind die Werte der Knockout-Tiere geringer als bei den
Wildtyp-Tieren. Dabei ist sowohl die Granulozyteninfiltration der F--Tiere der Gruppe
Ischämie-Reperfusion 8 h (0,25 ± 0,09) als auch die Granulozyteninfiltration der T--Tiere der
gleichen Gruppe (0,15 ± 0,07) signifikant niedriger im Vergleich zu den Wildtyp-Tieren
dieser Gruppe (0,96 ± 0,15) (p<0,05).
Unter den Wildtyp-Tieren konnte ein signifikanter Unterschied bei der
Granulozyteninfiltration der Gruppen Laparotomie 8 h (0,43 ± 0,11) und Laparotomie 48 h
(0,44 ± 0,12) gegenüber der Gruppe Ischämie-Reperfusion 8 h (0,96 ± 0,15) beobachtet
werden (p<0,05).
Abbildung 13: Histologisches Präparat der Lunge, hochgradige Granulozyteninfiltration (Gruppe WT Ischämie-
Reperfusion 48 h), HE-Färbung, 400x
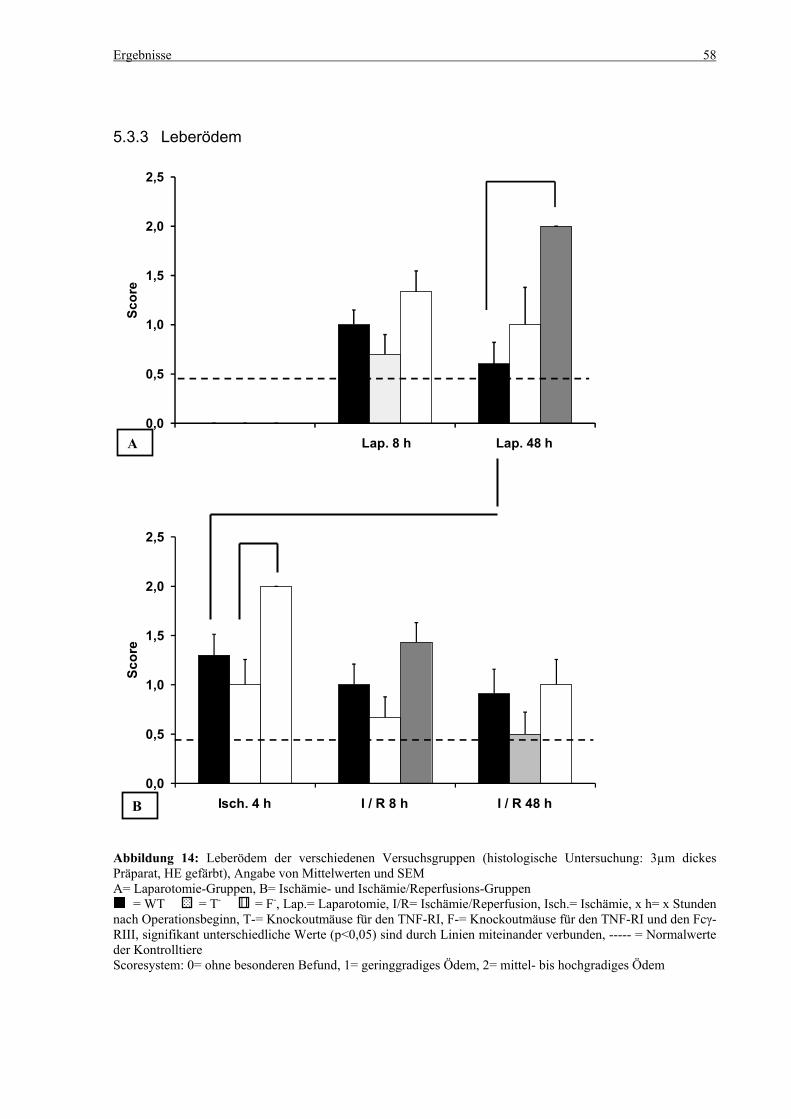
Ergebnisse 58
5.3.3 Leberödem
Abbildung 14: Leberödem der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere Scoresystem: 0= ohne besonderen Befund, 1= geringgradiges Ödem, 2= mittel- bis hochgradiges Ödem
0,0
0,5
1,0
1,5
2,0
2,5
Lap. 8 h Lap. 48 h
Scor
e
A
0,0
0,5
1,0
1,5
2,0
2,5
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B
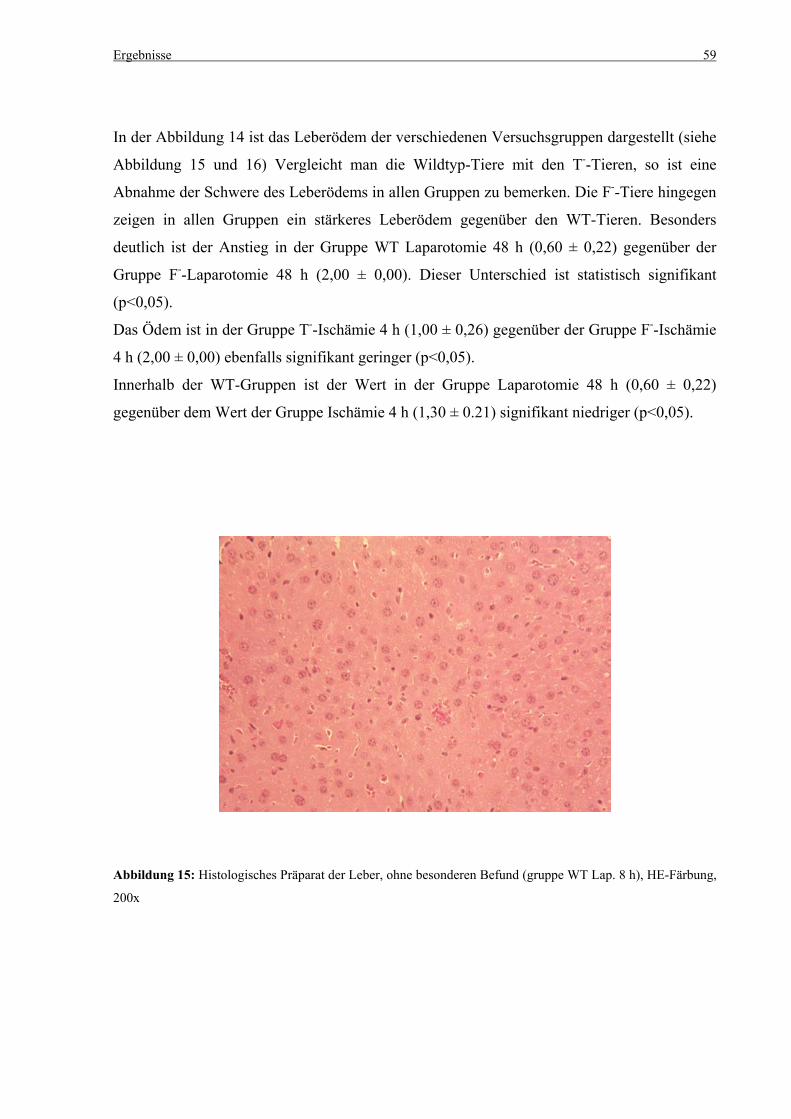
Ergebnisse 59
In der Abbildung 14 ist das Leberödem der verschiedenen Versuchsgruppen dargestellt (siehe
Abbildung 15 und 16) Vergleicht man die Wildtyp-Tiere mit den T--Tieren, so ist eine
Abnahme der Schwere des Leberödems in allen Gruppen zu bemerken. Die F--Tiere hingegen
zeigen in allen Gruppen ein stärkeres Leberödem gegenüber den WT-Tieren. Besonders
deutlich ist der Anstieg in der Gruppe WT Laparotomie 48 h (0,60 ± 0,22) gegenüber der
Gruppe F--Laparotomie 48 h (2,00 ± 0,00). Dieser Unterschied ist statistisch signifikant
(p<0,05).
Das Ödem ist in der Gruppe T--Ischämie 4 h (1,00 ± 0,26) gegenüber der Gruppe F--Ischämie
4 h (2,00 ± 0,00) ebenfalls signifikant geringer (p<0,05).
Innerhalb der WT-Gruppen ist der Wert in der Gruppe Laparotomie 48 h (0,60 ± 0,22)
gegenüber dem Wert der Gruppe Ischämie 4 h (1,30 ± 0.21) signifikant niedriger (p<0,05).
Abbildung 15: Histologisches Präparat der Leber, ohne besonderen Befund (gruppe WT Lap. 8 h), HE-Färbung,
200x
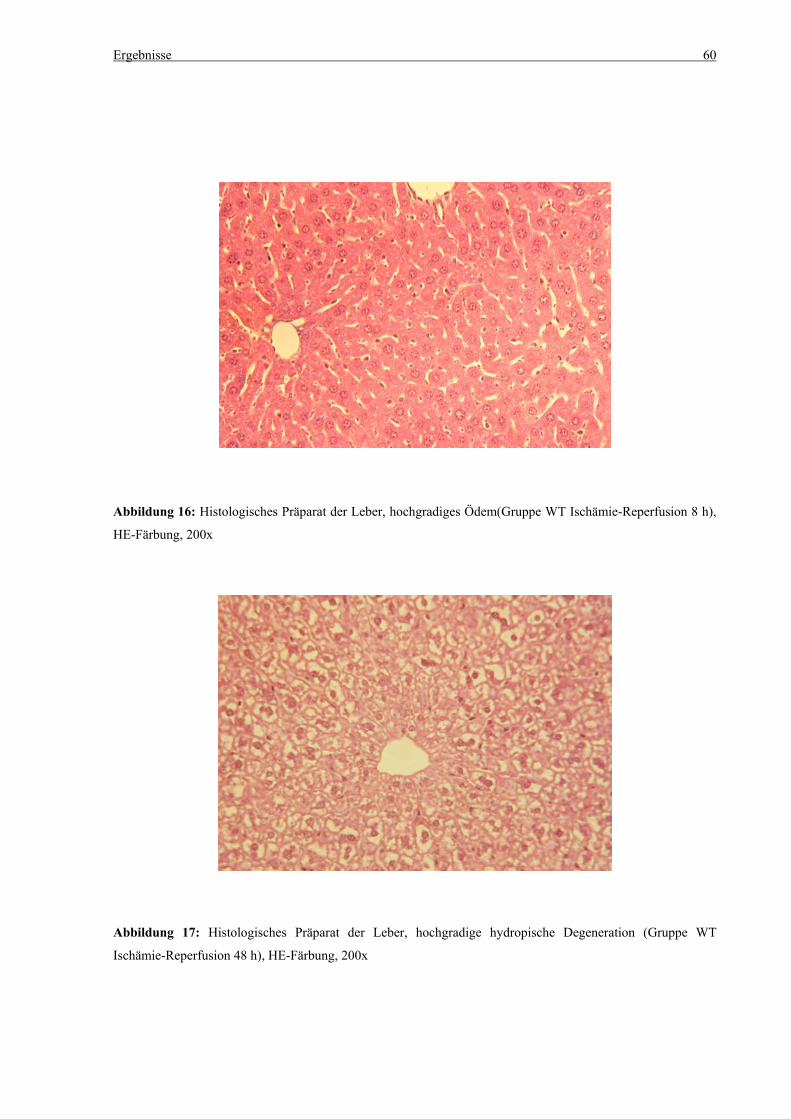
Ergebnisse 60
Abbildung 16: Histologisches Präparat der Leber, hochgradiges Ödem(Gruppe WT Ischämie-Reperfusion 8 h),
HE-Färbung, 200x
Abbildung 17: Histologisches Präparat der Leber, hochgradige hydropische Degeneration (Gruppe WT
Ischämie-Reperfusion 48 h), HE-Färbung, 200x
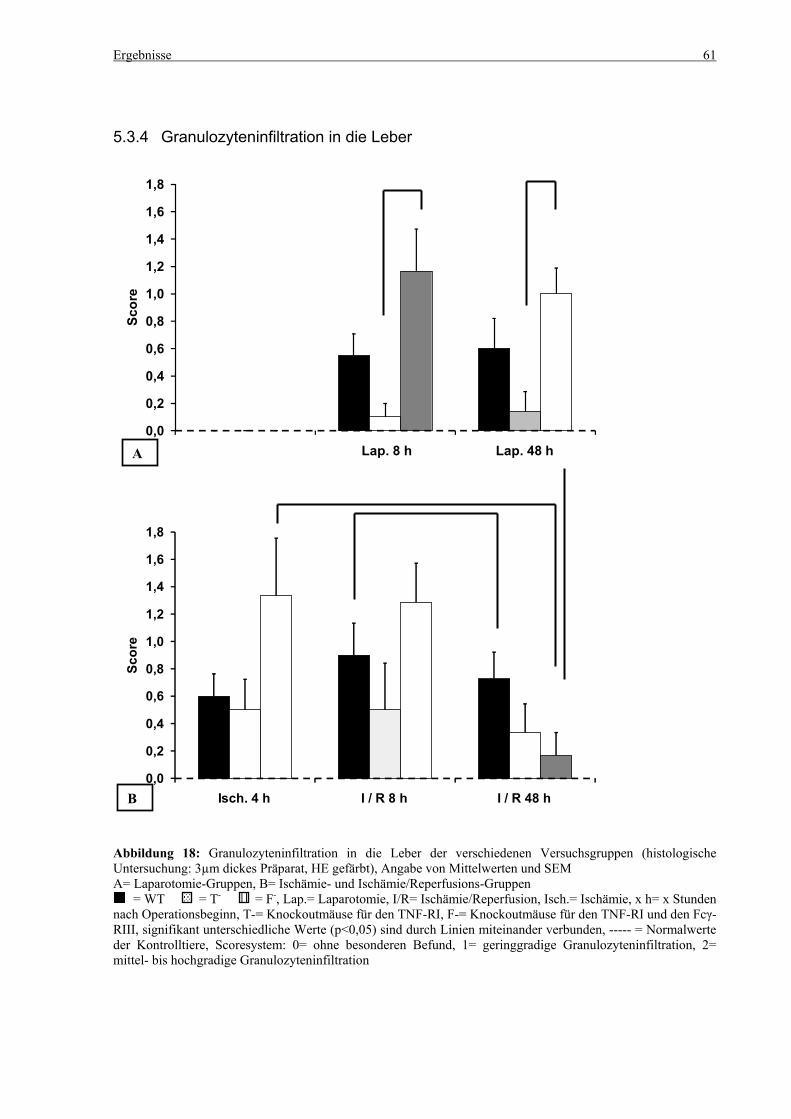
Ergebnisse 61
5.3.4 Granulozyteninfiltration in die Leber
Abbildung 18: Granulozyteninfiltration in die Leber der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= geringgradige Granulozyteninfiltration, 2= mittel- bis hochgradige Granulozyteninfiltration
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
Lap. 8 h Lap. 48 h
Scor
e
A
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 62
In Abbildung 18 ist die Granulozyteninfiltration der verschiedenen Versuchsgruppen
dargestellt. In allen Gruppen der T--Mäuse kann eine geringere Granulozyteninfiltration in das
Lebergewebe gegenüber der WT-Mäuse beobachtet werden. Hingegen ist bei den F--Mäusen,
außer in der Gruppe Ischämie-Reperfusion 48 h, gegenüber der WT-Tiere eine verstärkte
Zellinfiltration zu bemerken. Die Granulozyteninfiltration der Gruppe Ischämie-Reperfusion 8
h (1,29 ± 0,29) gegenüber der Gruppe Ischämie-Reperfusion 48 h (0,17 ± 0,17) der F--Mäuse
ist signifikant (p<0,05). Weiterhin besteht ein signifikanter Unterschied in der Stärke der
Zellinfiltration zwischen den Gruppen F--Ischämie 4 h (1,33 ± 0,22) und F--Ischämie-
Reperfusion 48 h (0,17 ± 0,17) (p<0,05). Auch gegenüber der Gruppe F--Laparptomie 48 h
(1,00 ± 0,19) ist die Anzahl der Granulozyten im Lebergewebe bei der Gruppe F--Ischämie-
Reperfusion 48 h signifikant geringer (0,17 ± 0,17) (p<0,05).
Vergleicht man die Laparotomie 8 h-Gruppe der T--Tiere (0,10 ± 0,10) mit den F--Tieren
(1,17 ± 0,31), so fällt eine signifikant höhere Zellzahl in der Leber der F--Tieren auf (p<0,05).
Auch 48 h nach der Laparotomie bleibt dieser signifikante Unterschied bestehen (T-: 0,14 ±
0,14; F-: 1,00 ± 0,19) (p<0,05).
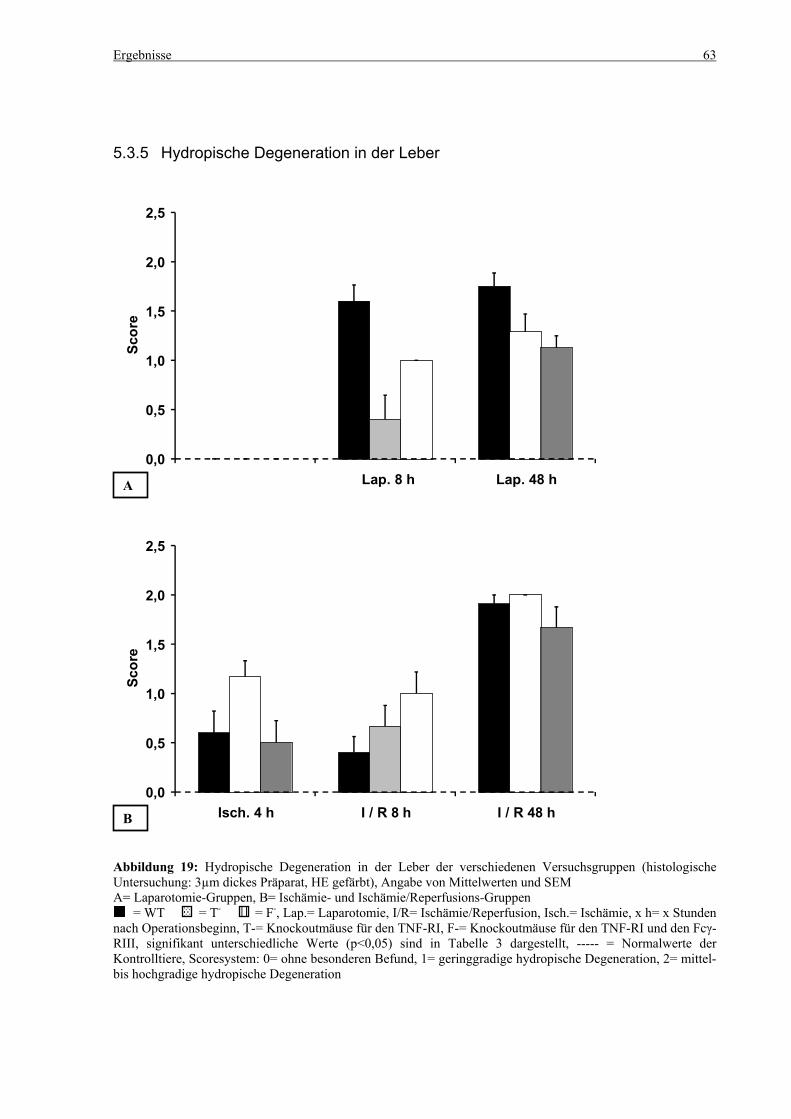
Ergebnisse 63
5.3.5 Hydropische Degeneration in der Leber
Abbildung 19: Hydropische Degeneration in der Leber der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind in Tabelle 3 dargestellt, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= geringgradige hydropische Degeneration, 2= mittel- bis hochgradige hydropische Degeneration
0,0
0,5
1,0
1,5
2,0
2,5
Lap. 8 h Lap. 48 h
Scor
e
A
0,0
0,5
1,0
1,5
2,0
2,5
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B
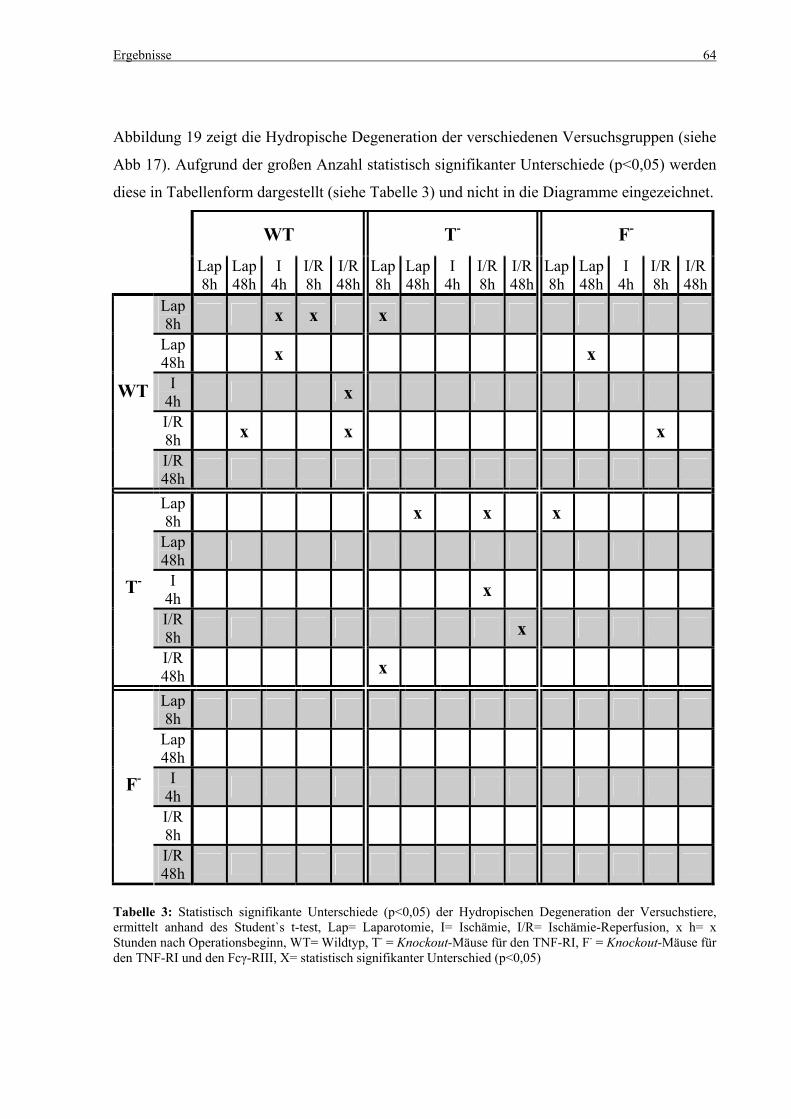
Ergebnisse 64
Abbildung 19 zeigt die Hydropische Degeneration der verschiedenen Versuchsgruppen (siehe
Abb 17). Aufgrund der großen Anzahl statistisch signifikanter Unterschiede (p<0,05) werden
diese in Tabellenform dargestellt (siehe Tabelle 3) und nicht in die Diagramme eingezeichnet.
WT T- F-
Lap 8h
Lap 48h
I 4h
I/R 8h
I/R48h
Lap8h
Lap48h
I 4h
I/R8h
I/R48h
Lap 8h
Lap 48h
I 4h
I/R8h
I/R48h
Lap 8h x x x
Lap 48h x x
I 4h x I/R 8h x x x
WT
I/R 48h
Lap 8h x x x
Lap 48h
I 4h x I/R 8h x
T-
I/R 48h x
Lap 8h
Lap 48h
I 4h I/R 8h
F-
I/R 48h
Tabelle 3: Statistisch signifikante Unterschiede (p<0,05) der Hydropischen Degeneration der Versuchstiere, ermittelt anhand des Student`s t-test, Lap= Laparotomie, I= Ischämie, I/R= Ischämie-Reperfusion, x h= x Stunden nach Operationsbeginn, WT= Wildtyp, T- = Knockout-Mäuse für den TNF-RI, F- = Knockout-Mäuse für den TNF-RI und den Fcγ-RIII, X= statistisch signifikanter Unterschied (p<0,05)
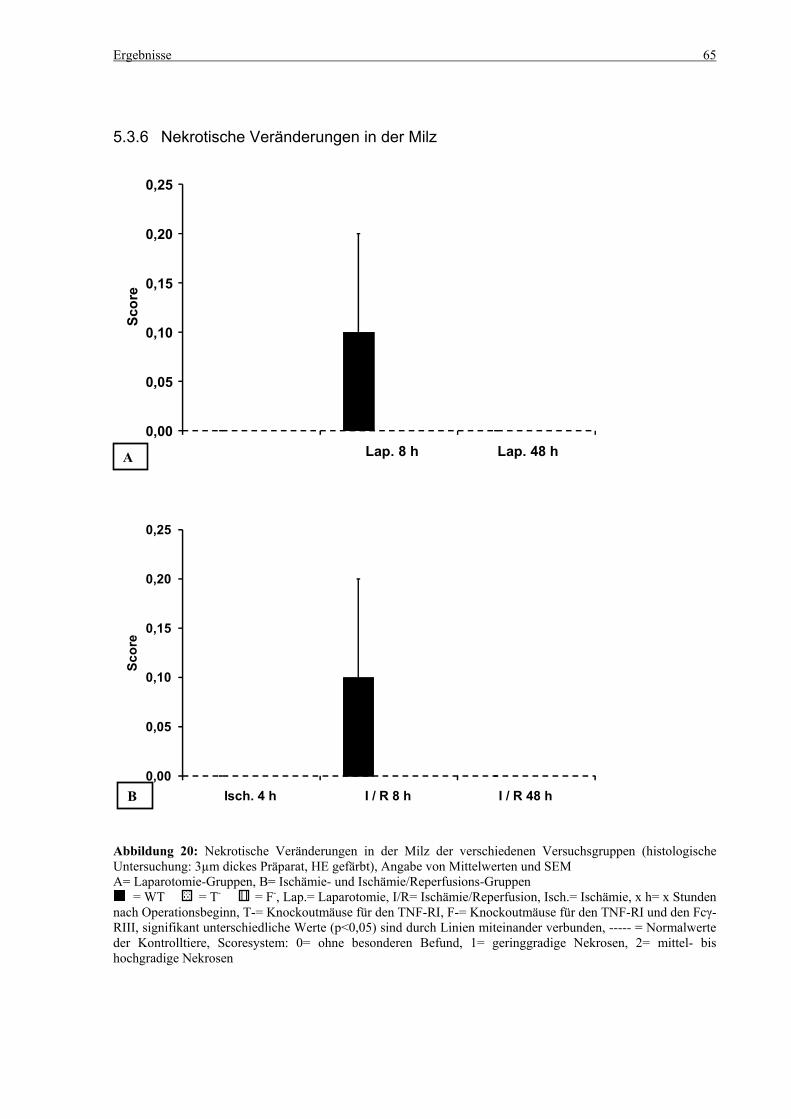
Ergebnisse 65
5.3.6 Nekrotische Veränderungen in der Milz
Abbildung 20: Nekrotische Veränderungen in der Milz der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= geringgradige Nekrosen, 2= mittel- bis hochgradige Nekrosen
0,00
0,05
0,10
0,15
0,20
0,25
Lap. 8 h Lap. 48 h
Scor
e
A
0,00
0,05
0,10
0,15
0,20
0,25
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 66
Abbildung 20 zeigt die nekrotischen Veränderungen in der Milz der verschiedenen
Versuchsgruppen. Bei der Beurteilung dieser Veränderungen konnten keine signifikanten
Unterschiede festgestellt werden. Lediglich bei den WT-Tieren in den Gruppen Laparotomie
4 h und Ischämie-Reperfusion 8 h konnten Nekrosen festgestellt werden.
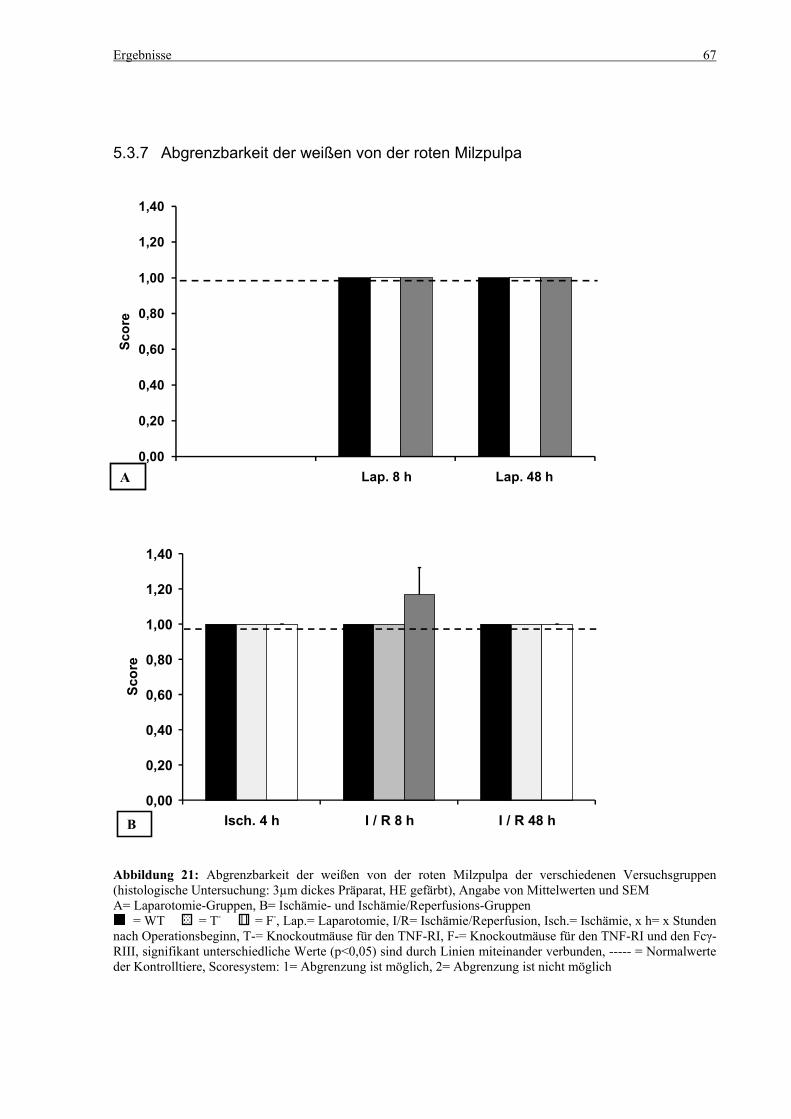
Ergebnisse 67
5.3.7 Abgrenzbarkeit der weißen von der roten Milzpulpa
Abbildung 21: Abgrenzbarkeit der weißen von der roten Milzpulpa der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 1= Abgrenzung ist möglich, 2= Abgrenzung ist nicht möglich
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
Lap. 8 h Lap. 48 h
Scor
e
A
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 68
In der Abbildung 21 ist die Abgrenzbarkeit der roten von der weißen Milzpulpa der
verschiedenen Versuchsgruppen dargestellt. Hierbei wurden keine signifikanten Unterschiede
festgestellt. Die Abgrenzbarkeit war bei allen außer einem Tier gegeben.
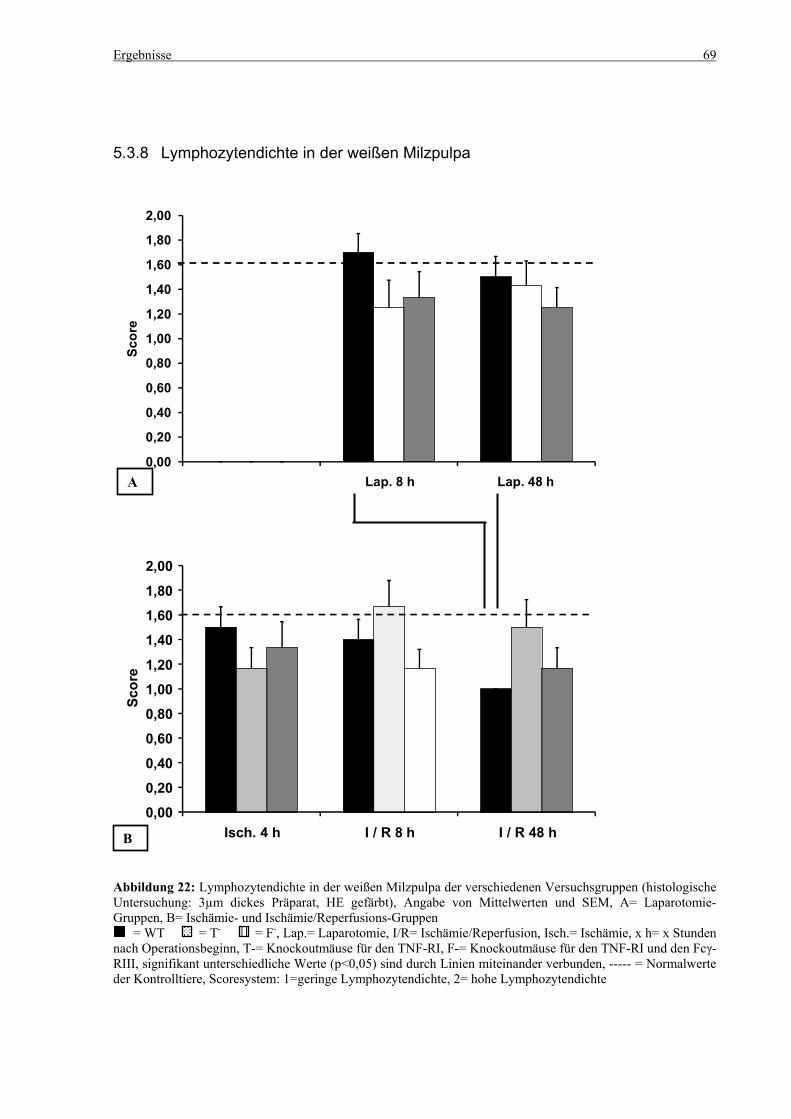
Ergebnisse 69
5.3.8 Lymphozytendichte in der weißen Milzpulpa
Abbildung 22: Lymphozytendichte in der weißen Milzpulpa der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM, A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 1=geringe Lymphozytendichte, 2= hohe Lymphozytendichte
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
2,00
Lap. 8 h Lap. 48 h
Scor
e
A
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
2,00
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 70
In Abbildung 22 ist die Dichte der Lymphozyten in der weißen Milzpulpa der verschiedenen
Versuchsgruppen dargestellt. Das Diagramm zeigt einen signifikanten Unterschied der
Pulpafülle zwischen der Gruppe WT Laparotomie 8 h (1,70 ± 0,15) und der Gruppe WT
Ischämie-Reperfusion 48 h (1,00 ± 0,00) (p<0,05). Die Lymphozytendichte der Gruppe WT
Laparotomie 48 h (1,50 ± 0,17) ist signifikant höher als die der Gruppe WT Ischämie-
Reperfusion 48 h (1,00 ± 0,00) (p<0,05).
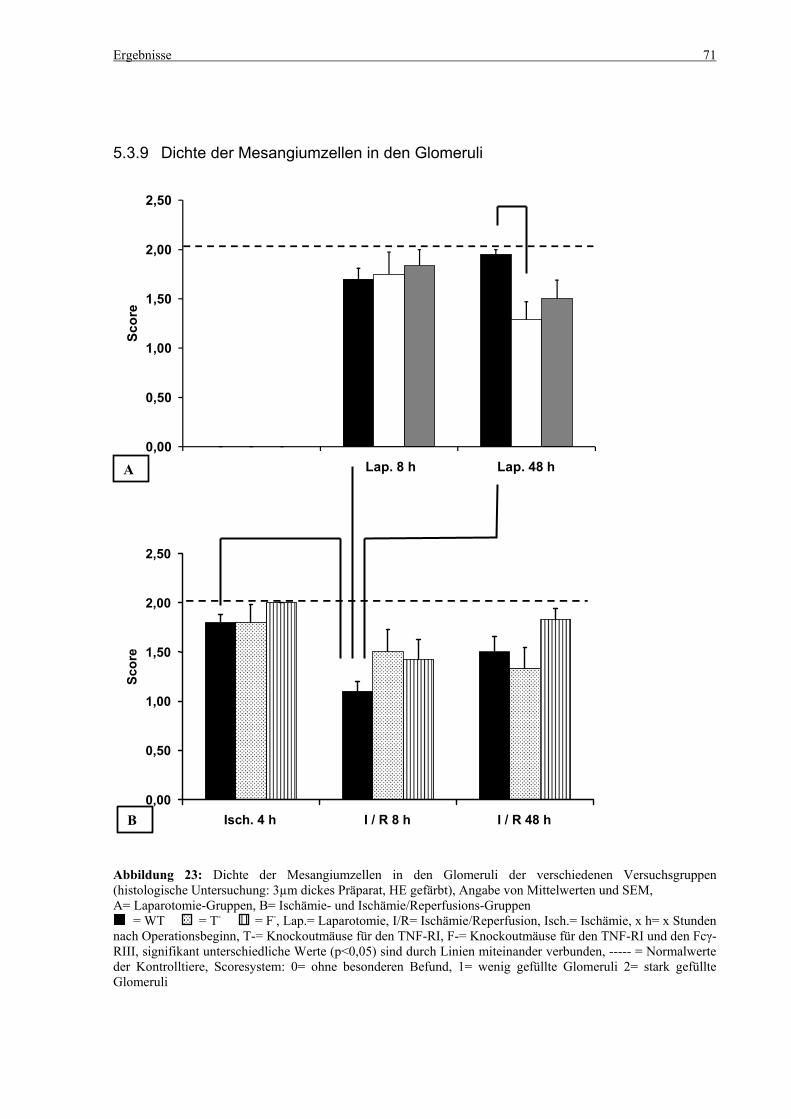
Ergebnisse 71
5.3.9 Dichte der Mesangiumzellen in den Glomeruli
Abbildung 23: Dichte der Mesangiumzellen in den Glomeruli der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM, A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= wenig gefüllte Glomeruli 2= stark gefüllte Glomeruli
0,00
0,50
1,00
1,50
2,00
2,50
Lap. 8 h Lap. 48 h
Scor
e
A
0,00
0,50
1,00
1,50
2,00
2,50
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 72
Abbildung 23 zeigt die Dichte der Mesangiumzellen in den Glomeruli der verschiedenen
Versuchstiergruppen. Sowohl in der Gruppe WT Laparotomie 8 h (1,70 ± 0,11) als auch in
der Gruppe WT Laparotomie 48 h (1,95 ± 0,05) ist eine signifikant stärkere Dichte der Zellen
im Vergleich zur Gruppe WT Ischämie-Reperfusion 8 h (1,10 ± 0,10) festzustellen (p<0,05).
Auch zwischen den Gruppen WT Ischämie 4 h (1,80 ± 0,08) und WT Ischämie-Reperfusion 8
h (1,10 ± 0,10) ist einen signifikanter Unterschied zu beobachten (p<0,05). Die
Mesangiumzelldichte der Tiere der Gruppe WT Laparotomie 48 h (1,95 ± 0,05) ist signifikant
höher als die der Gruppe T--Laparotomie 48 h (1,29 ± 0,19) (p<0,05).
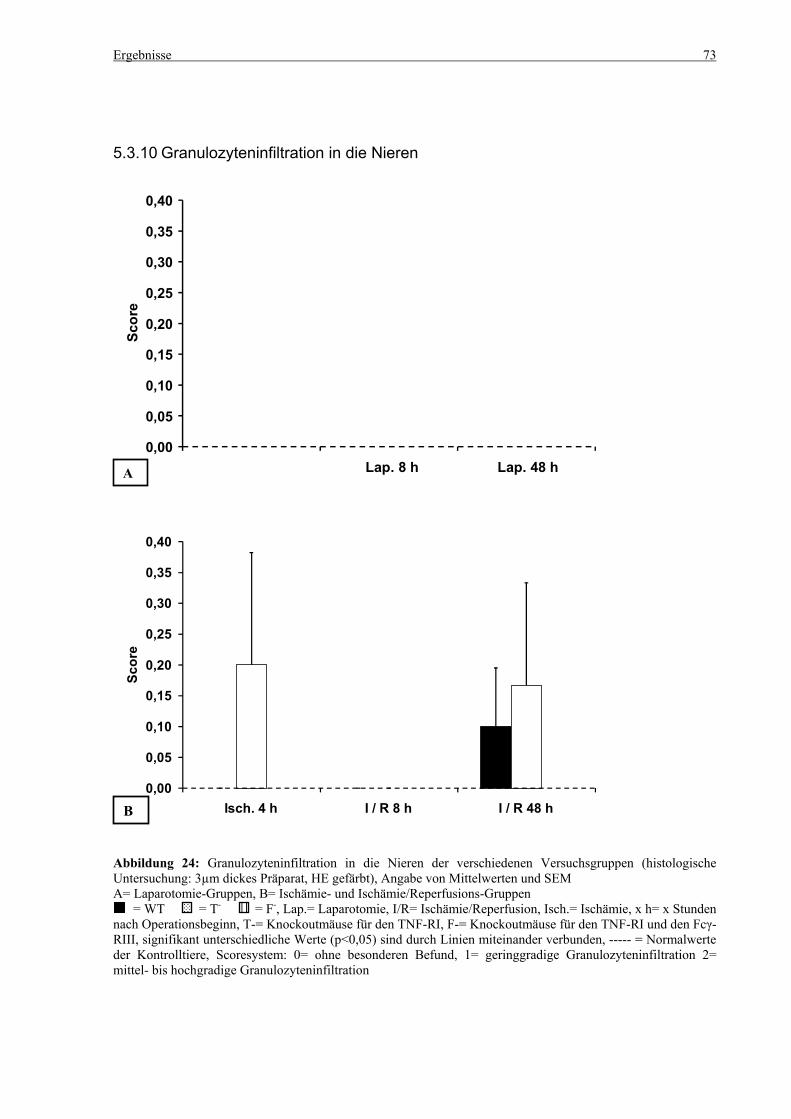
Ergebnisse 73
5.3.10 Granulozyteninfiltration in die Nieren
Abbildung 24: Granulozyteninfiltration in die Nieren der verschiedenen Versuchsgruppen (histologische Untersuchung: 3µm dickes Präparat, HE gefärbt), Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere, Scoresystem: 0= ohne besonderen Befund, 1= geringgradige Granulozyteninfiltration 2= mittel- bis hochgradige Granulozyteninfiltration
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
Lap. 8 h Lap. 48 h
Scor
e
A
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
Isch. 4 h I / R 8 h I / R 48 h
Scor
e
B

Ergebnisse 74
Abbildung 24 zeigt die Granulozyteninfiltration in die Nieren der verschiedenen
Versuchsgruppen. Größtenteils konnten keine Zellinfiltrationen beobachtet werden, bei
einigen Tieren konnten geringgradige Veränderungen festgestellt werden. Signifikante
Unterschiede waren hierbei nicht zu ermitteln.
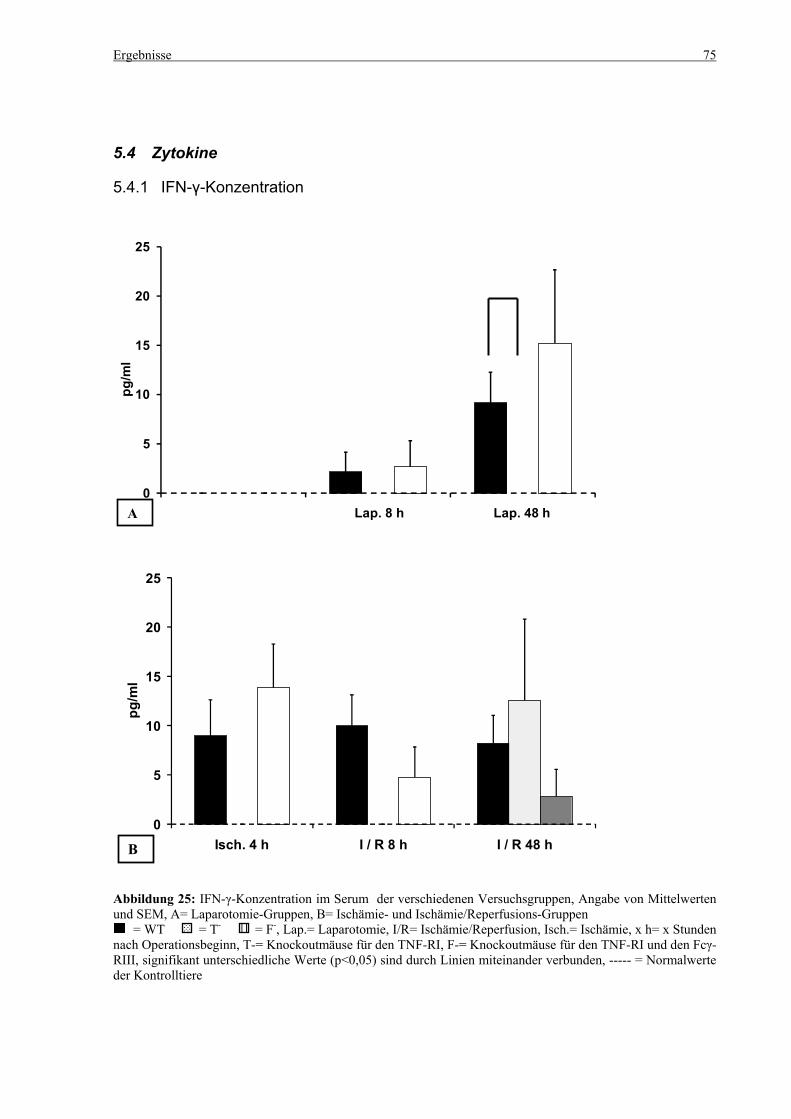
Ergebnisse 75
5.4 Zytokine
5.4.1 IFN-γ-Konzentration
Abbildung 25: IFN-γ-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM, A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere
0
5
10
15
20
25
Lap. 8 h Lap. 48 h
pg/m
l
A
0
5
10
15
20
25
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B

Ergebnisse 76
Abbildung 25 zeigt die INF-γ-Konzentration der verschiedenen Versuchsgruppen. Es konnte
nur zwischen der Gruppe Laparotomie 48 h (9,14 ± 3,14) und der Gruppe T-Laparotomie 48 h
(0,00 ± 0,00) ein statistisch signifikanter Unterschied (p<0,05) festgestellt werden.
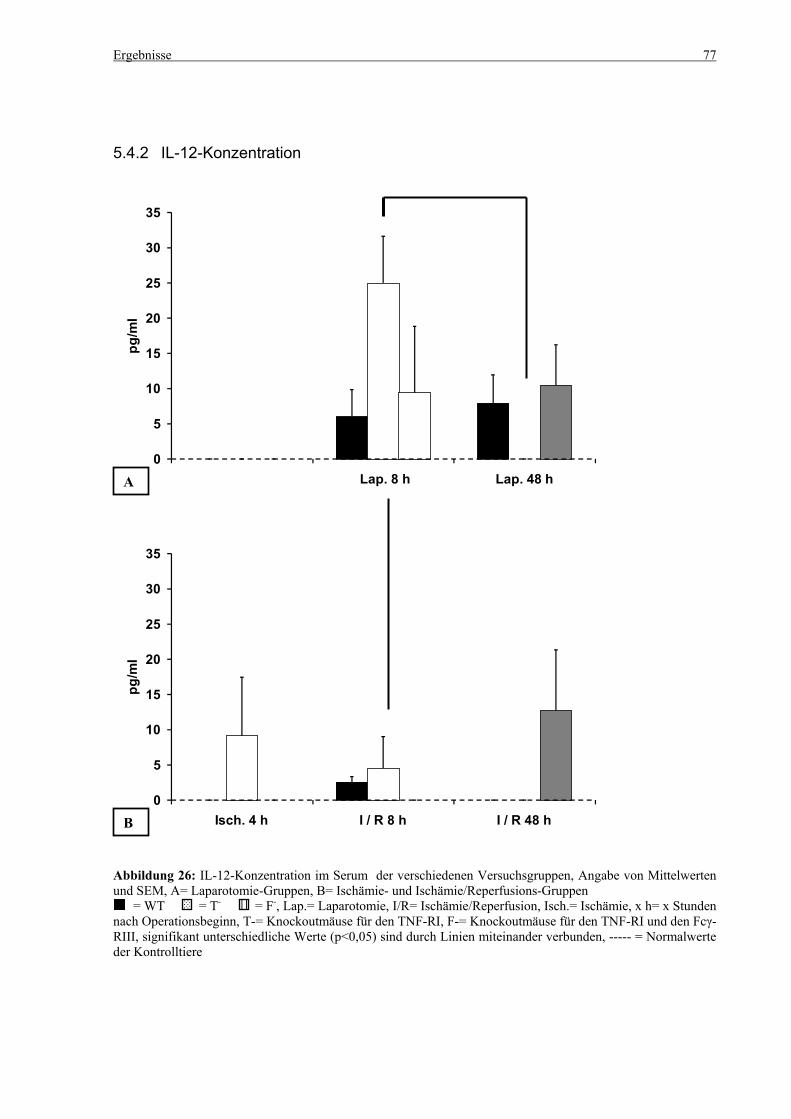
Ergebnisse 77
5.4.2 IL-12-Konzentration
Abbildung 26: IL-12-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM, A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere
0
5
10
15
20
25
30
35
Lap. 8 h Lap. 48 h
pg/m
l
A
0
5
10
15
20
25
30
35
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B

Ergebnisse 78
Abbildung 26 zeigt die IL-12-Konzentration im Serum der verschiedenen Versuchsgruppen.
Nur innerhalb der T--Knockout-Mäuse sind signifikante Unterschiede festzustellen. Die IL-
12-Konzentration der Gruppe T--Laparotomie 8 h (24,98 ± 6,65) ist signigfikant höher als die
der Gruppen T--Laparotomie 48 h (0,00 ± 0,00) und T--Ischämie/Reperfusion 8 h (4,52 ±
4,52) (p<0,05).
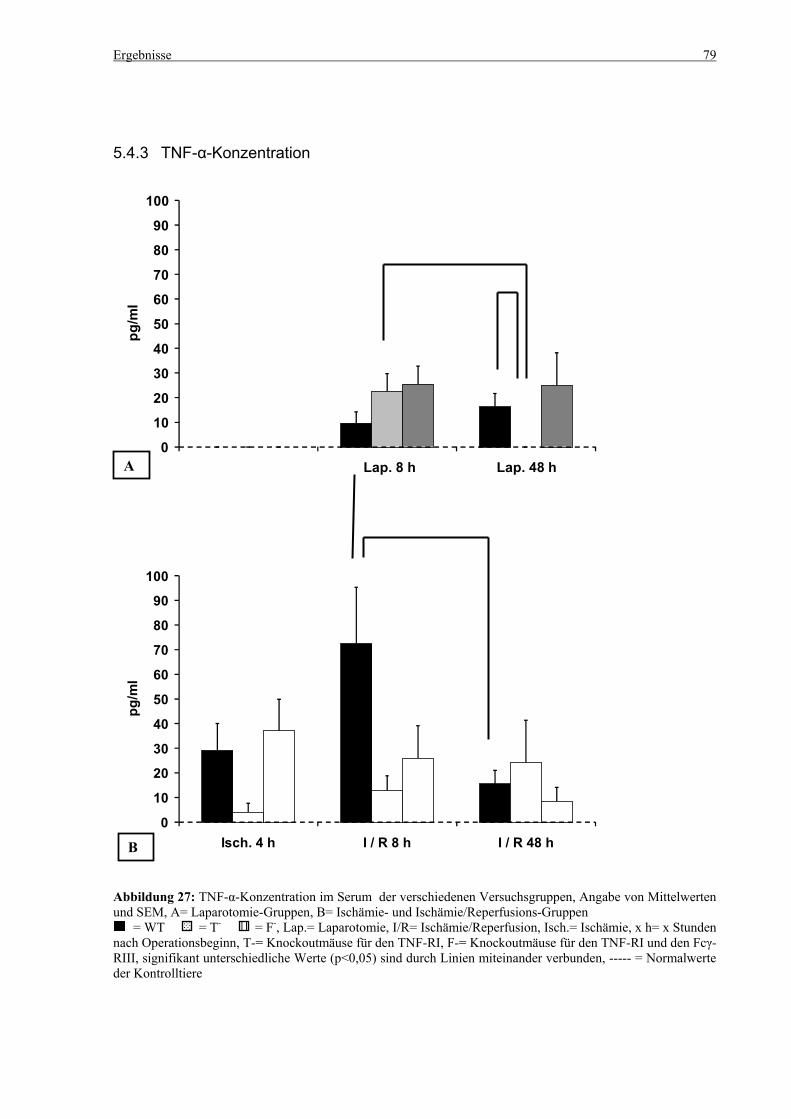
Ergebnisse 79
5.4.3 TNF-α-Konzentration
Abbildung 27: TNF-α-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM, A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere
0
10
20
30
40
50
60
70
80
90
100
Lap. 8 h Lap. 48 h
pg/m
l
A
0
10
20
30
40
50
60
70
80
90
100
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B

Ergebnisse 80
Abbildung 27 zeigt die TNF-α-Konzentration im Serum der verschiedenen Versuchsgruppen.
Hierbei fällt ein statistisch signifikanter Unterschied zwischen den Gruppen Laparotomie 48 h
(16,22 ± 5,49) und T--Laparotomie 48 h auf (0,00 ± 0,00) (p<0,05). Die Zytokinkonzentration
im Serum der Mäuse der Gruppe Ischämie/Reperfusion 8 h (72,41 ± 22,90) ist signifikant
höher (p<0,05) als die Konzentration der Gruppe Laparotomie 8 h (9,54 ± 4,97). Auch
zwischen den Gruppen Ischämie/Reperfusion 8 h (72,41 ± 22,90) und Ischämie/Reperfusion
48 h (15,48 ± 5,51) besteht ein statistisch signifikanter Unterschied(p<0,05). Weiterhin
konnte festgestellt werden, dass die TNF-α-Konzentration der Gruppe T--Laparotomie 8 h
(22,58 ± 7,17) signifikant höher war (p<0,05) als die der Gruppe T--Laparotomie 48 h (0,00 ±
0,00).
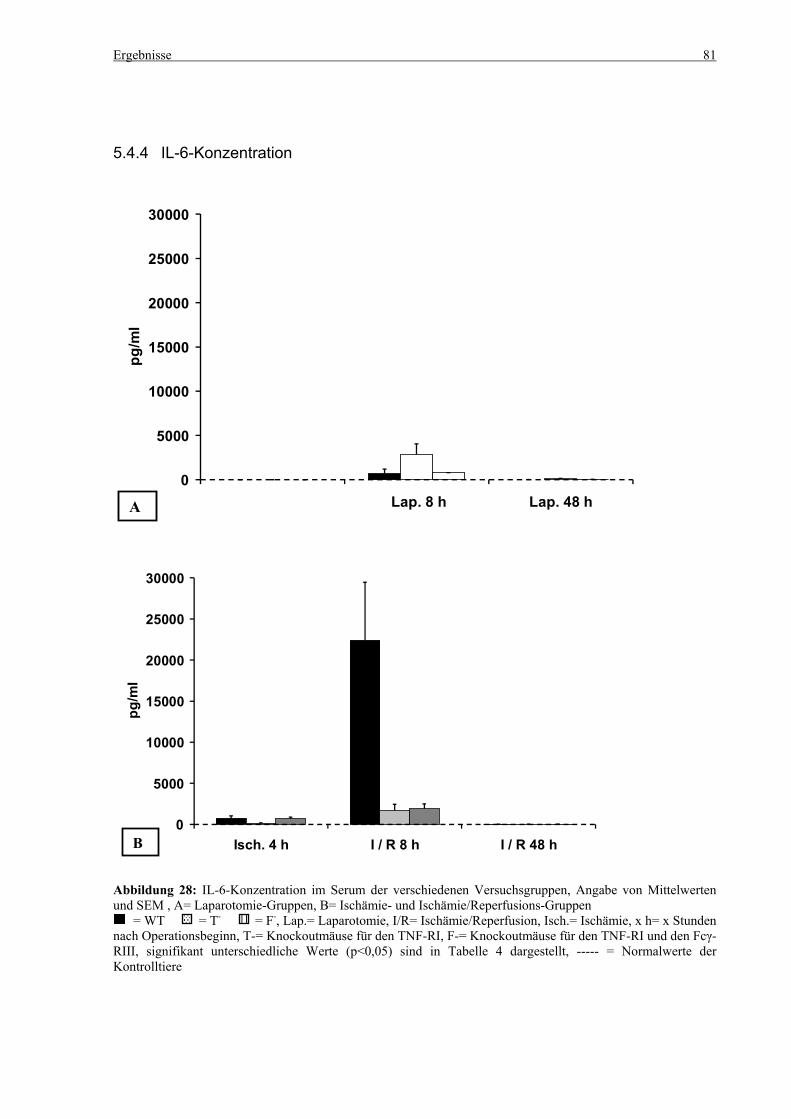
Ergebnisse 81
5.4.4 IL-6-Konzentration
Abbildung 28: IL-6-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM , A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind in Tabelle 4 dargestellt, ----- = Normalwerte der Kontrolltiere
0
5000
10000
15000
20000
25000
30000
Lap. 8 h Lap. 48 h
pg/m
l
A
0
5000
10000
15000
20000
25000
30000
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B
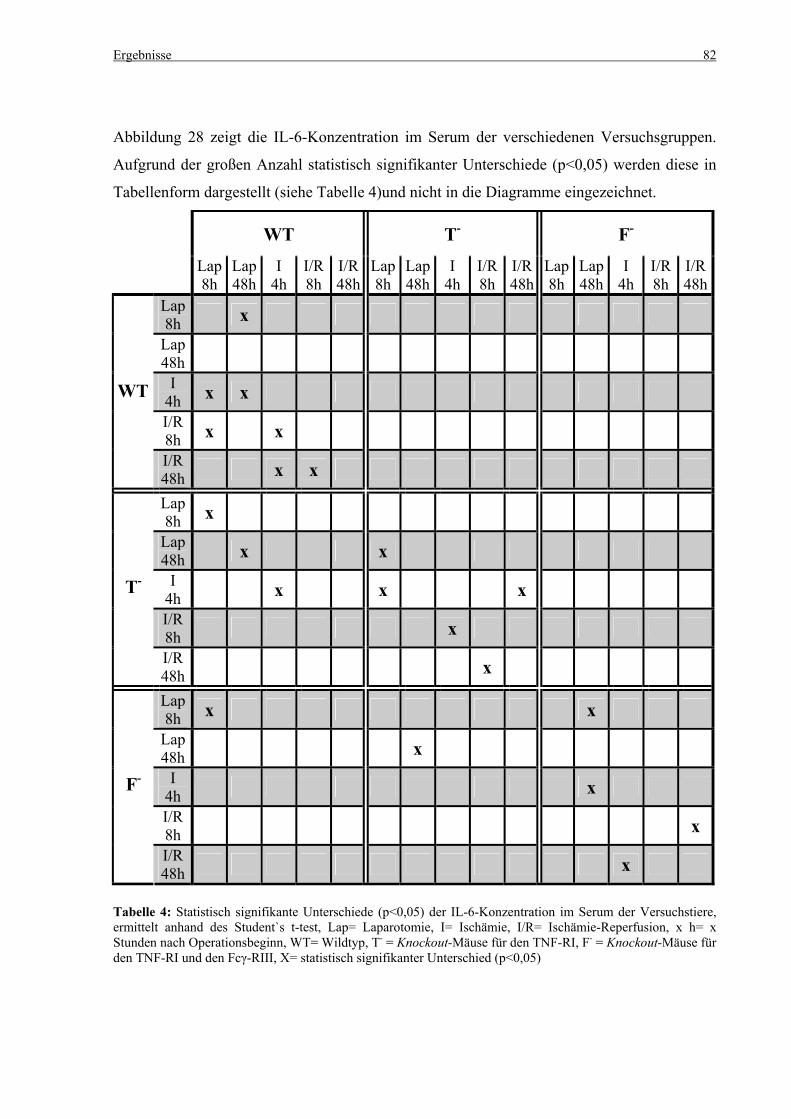
Ergebnisse 82
Abbildung 28 zeigt die IL-6-Konzentration im Serum der verschiedenen Versuchsgruppen.
Aufgrund der großen Anzahl statistisch signifikanter Unterschiede (p<0,05) werden diese in
Tabellenform dargestellt (siehe Tabelle 4)und nicht in die Diagramme eingezeichnet.
WT T- F-
Lap 8h
Lap 48h
I 4h
I/R 8h
I/R48h
Lap8h
Lap48h
I 4h
I/R8h
I/R48h
Lap 8h
Lap 48h
I 4h
I/R8h
I/R48h
Lap 8h x
Lap 48h
I 4h x x I/R 8h x x
WT
I/R 48h x x
Lap 8h x
Lap 48h x x
I 4h x x x I/R 8h x
T-
I/R 48h x
Lap 8h x x
Lap 48h x
I 4h x I/R 8h x
F-
I/R 48h x
Tabelle 4: Statistisch signifikante Unterschiede (p<0,05) der IL-6-Konzentration im Serum der Versuchstiere, ermittelt anhand des Student`s t-test, Lap= Laparotomie, I= Ischämie, I/R= Ischämie-Reperfusion, x h= x Stunden nach Operationsbeginn, WT= Wildtyp, T- = Knockout-Mäuse für den TNF-RI, F- = Knockout-Mäuse für den TNF-RI und den Fcγ-RIII, X= statistisch signifikanter Unterschied (p<0,05)
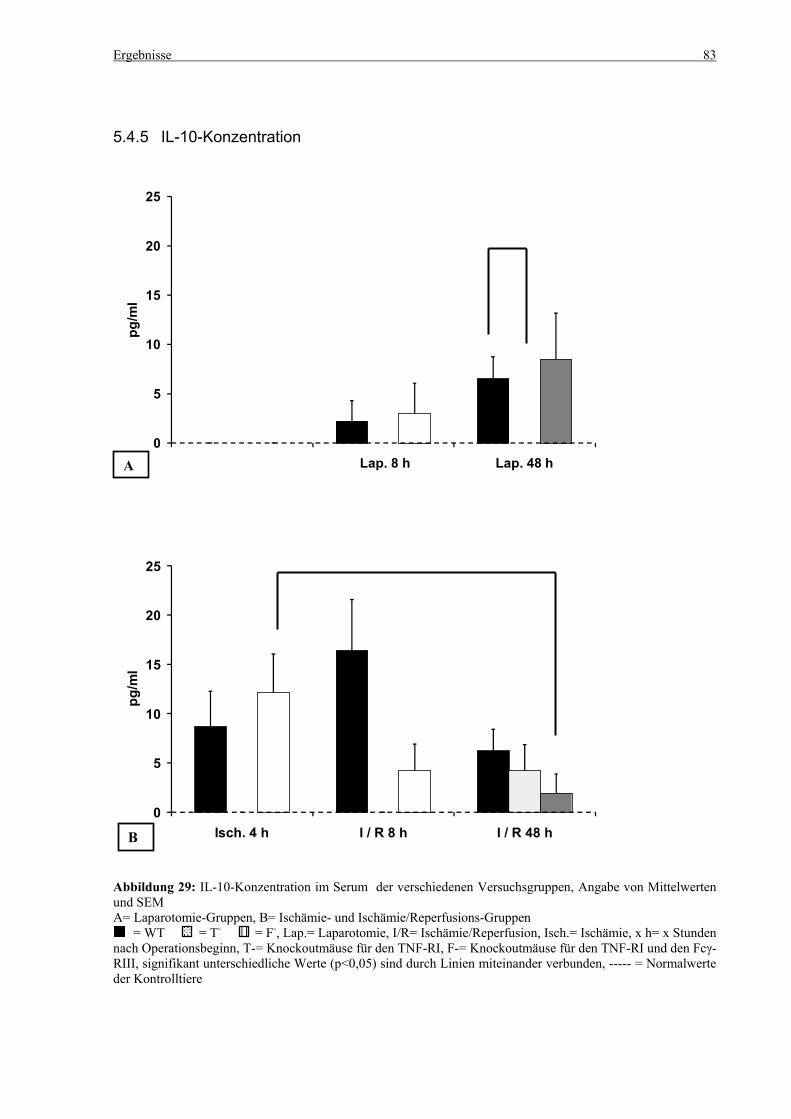
Ergebnisse 83
5.4.5 IL-10-Konzentration
Abbildung 29: IL-10-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind durch Linien miteinander verbunden, ----- = Normalwerte der Kontrolltiere
0
5
10
15
20
25
Lap. 8 h Lap. 48 h
pg/m
l
A
0
5
10
15
20
25
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B

Ergebnisse 84
Abbildung 29 zeigt die IL-10-Konzentration im Serum der verschiedenen Versuchsgruppen.
Zwischen den Gruppen F--Ischämie 4 h (12,10 ± 3,93) und F--Ischämie/Reperfusion 48 h
(1,93 ± 1,93) liegt ein statistisch signifikanter Unterschied vor (p<0,05). Die IL-10-
Konzentration der Gruppe Laparotomie 48 h (6,52 ± 2,23) ist signifikant (p<0,05) höher als
die der Gruppe T--Laparotomie 48 h (0,00 ± 0,00).
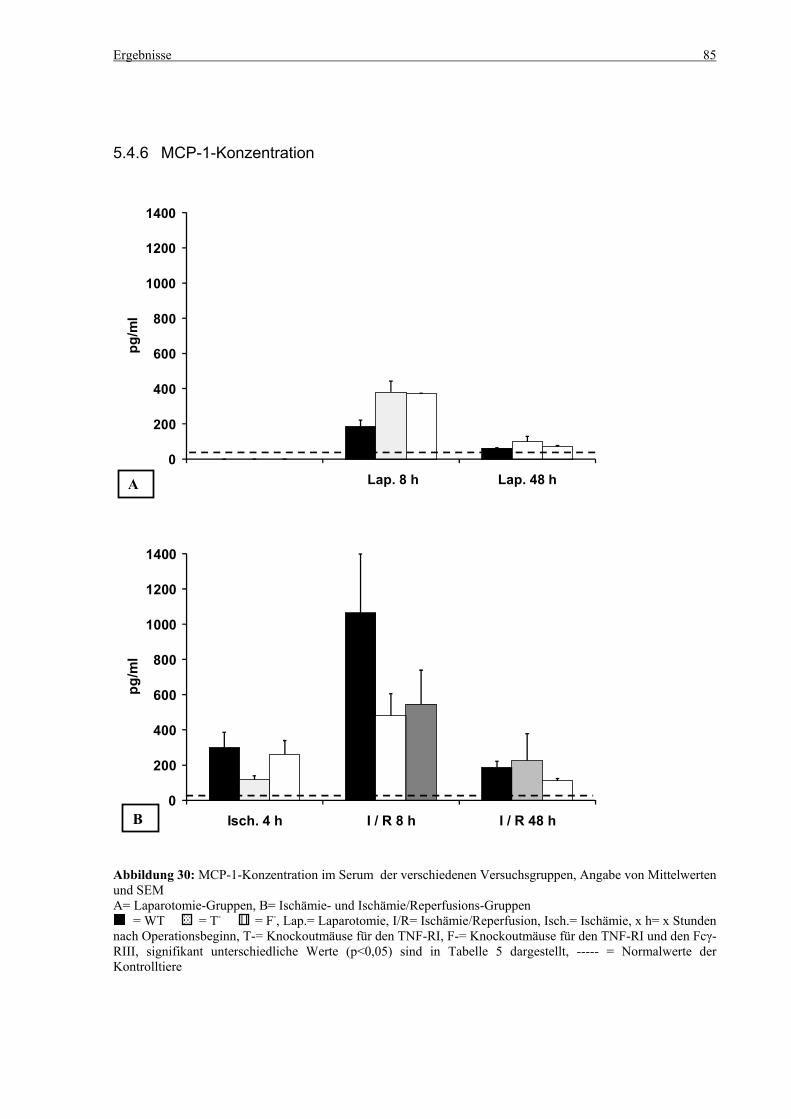
Ergebnisse 85
5.4.6 MCP-1-Konzentration
Abbildung 30: MCP-1-Konzentration im Serum der verschiedenen Versuchsgruppen, Angabe von Mittelwerten und SEM A= Laparotomie-Gruppen, B= Ischämie- und Ischämie/Reperfusions-Gruppen = WT = T- = F-, Lap.= Laparotomie, I/R= Ischämie/Reperfusion, Isch.= Ischämie, x h= x Stunden nach Operationsbeginn, T-= Knockoutmäuse für den TNF-RI, F-= Knockoutmäuse für den TNF-RI und den Fcγ-RIII, signifikant unterschiedliche Werte (p<0,05) sind in Tabelle 5 dargestellt, ----- = Normalwerte der Kontrolltiere
0
200
400
600
800
1000
1200
1400
Lap. 8 h Lap. 48 h
pg/m
l
A
0
200
400
600
800
1000
1200
1400
Isch. 4 h I / R 8 h I / R 48 h
pg/m
l
B
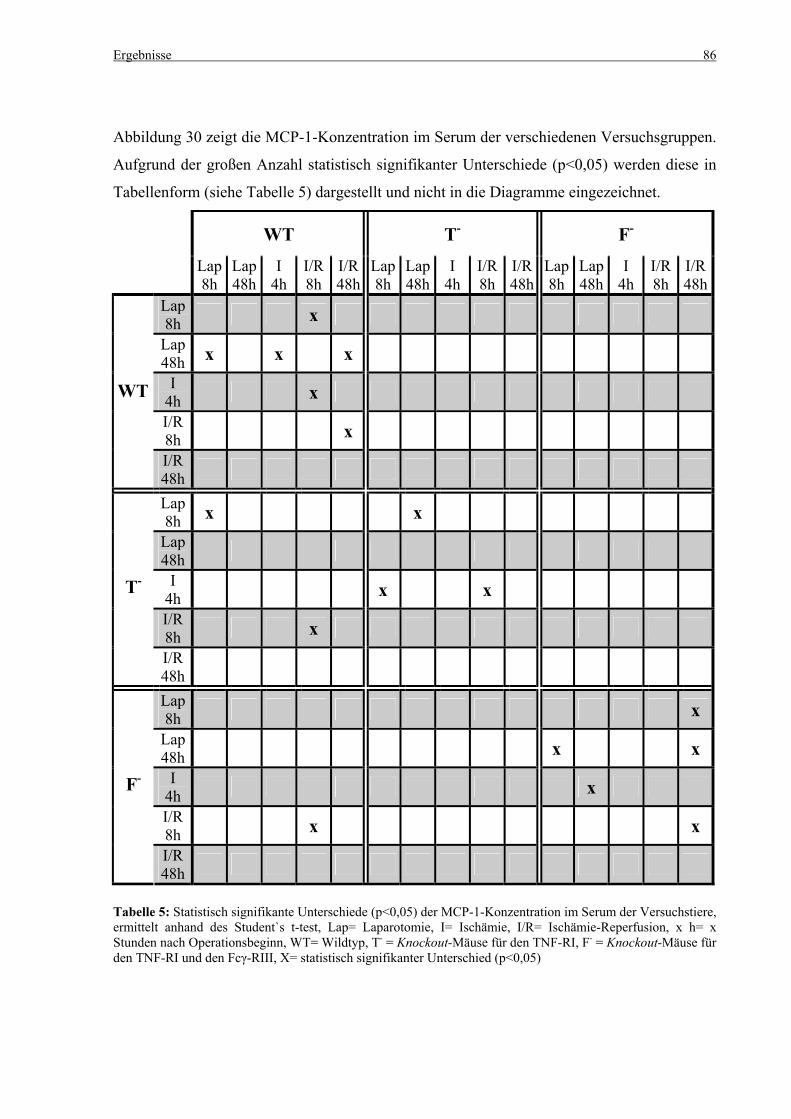
Ergebnisse 86
Abbildung 30 zeigt die MCP-1-Konzentration im Serum der verschiedenen Versuchsgruppen.
Aufgrund der großen Anzahl statistisch signifikanter Unterschiede (p<0,05) werden diese in
Tabellenform (siehe Tabelle 5) dargestellt und nicht in die Diagramme eingezeichnet.
WT T- F-
Lap 8h
Lap 48h
I 4h
I/R 8h
I/R48h
Lap8h
Lap48h
I 4h
I/R8h
I/R48h
Lap 8h
Lap 48h
I 4h
I/R8h
I/R48h
Lap 8h x
Lap 48h x x x
I 4h x I/R 8h x
WT
I/R 48h
Lap 8h x x
Lap 48h
I 4h x x I/R 8h x
T-
I/R 48h
Lap 8h x
Lap 48h x x
I 4h x I/R 8h x x
F-
I/R 48h
Tabelle 5: Statistisch signifikante Unterschiede (p<0,05) der MCP-1-Konzentration im Serum der Versuchstiere, ermittelt anhand des Student`s t-test, Lap= Laparotomie, I= Ischämie, I/R= Ischämie-Reperfusion, x h= x Stunden nach Operationsbeginn, WT= Wildtyp, T- = Knockout-Mäuse für den TNF-RI, F- = Knockout-Mäuse für den TNF-RI und den Fcγ-RIII, X= statistisch signifikanter Unterschied (p<0,05)
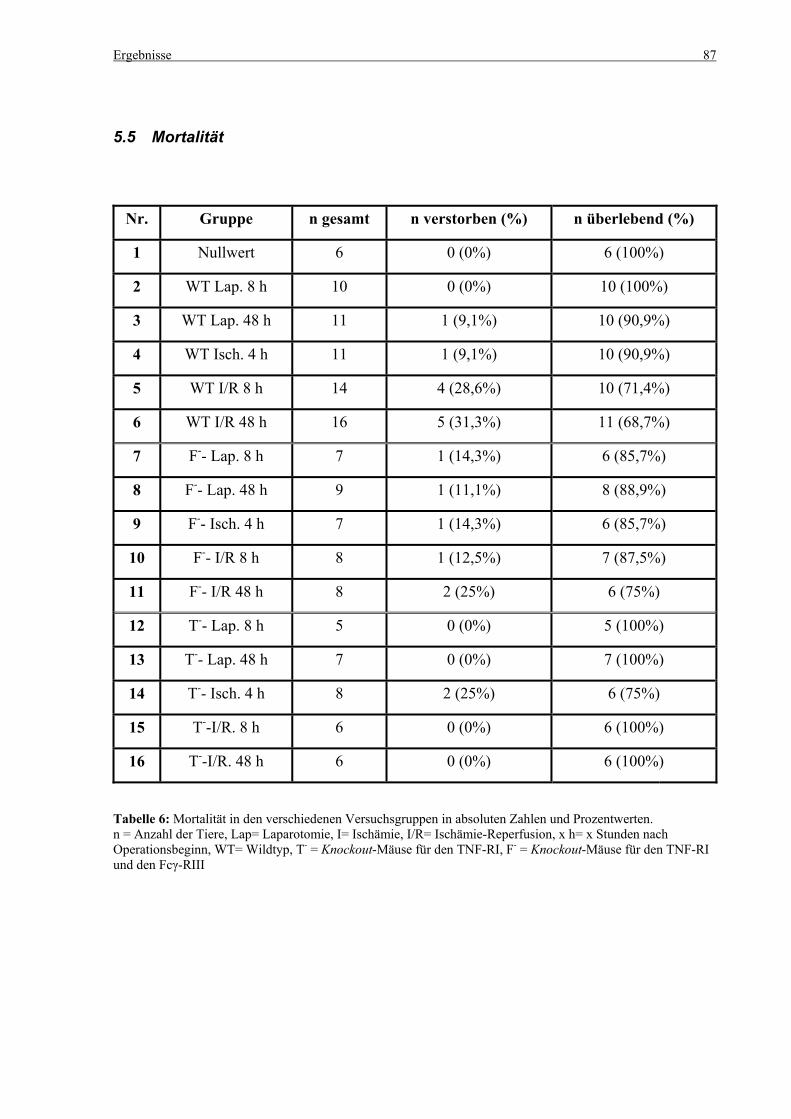
Ergebnisse 87
5.5 Mortalität
Nr. Gruppe n gesamt n verstorben (%) n überlebend (%)
1 Nullwert 6 0 (0%) 6 (100%)
2 WT Lap. 8 h 10 0 (0%) 10 (100%)
3 WT Lap. 48 h 11 1 (9,1%) 10 (90,9%)
4 WT Isch. 4 h 11 1 (9,1%) 10 (90,9%)
5 WT I/R 8 h 14 4 (28,6%) 10 (71,4%)
6 WT I/R 48 h 16 5 (31,3%) 11 (68,7%)
7 F-- Lap. 8 h 7 1 (14,3%) 6 (85,7%)
8 F-- Lap. 48 h 9 1 (11,1%) 8 (88,9%)
9 F-- Isch. 4 h 7 1 (14,3%) 6 (85,7%)
10 F-- I/R 8 h 8 1 (12,5%) 7 (87,5%)
11 F-- I/R 48 h 8 2 (25%) 6 (75%)
12 T-- Lap. 8 h 5 0 (0%) 5 (100%)
13 T-- Lap. 48 h 7 0 (0%) 7 (100%)
14 T-- Isch. 4 h 8 2 (25%) 6 (75%)
15 T--I/R. 8 h 6 0 (0%) 6 (100%)
16 T--I/R. 48 h 6 0 (0%) 6 (100%)
Tabelle 6: Mortalität in den verschiedenen Versuchsgruppen in absoluten Zahlen und Prozentwerten. n = Anzahl der Tiere, Lap= Laparotomie, I= Ischämie, I/R= Ischämie-Reperfusion, x h= x Stunden nach Operationsbeginn, WT= Wildtyp, T- = Knockout-Mäuse für den TNF-RI, F- = Knockout-Mäuse für den TNF-RI und den Fcγ-RIII

Ergebnisse 88
Es konnten keine signifikanten Unterschiede im Hinblick auf die Mortalitätsraten (siehe
Tabelle 6) festgestellt werden. Auffällig ist jedoch eine hohe Sterblichkeit in den Gruppen
WT I/R 8 h und 48 h (28,6% und 31,3%) im Vergleich zu den entsprechenden Gruppen der
T--Tiere (0%).

Diskussion 89
6 Diskussion
Bereits seit Mitte der 70er Jahre sind bei Traumapatienten das SIRS und das MODS als
Spätkomplikationen gefürchtet. Da trotz intensiver Forschung bis heute keine
zufriedenstellende Therapie dieser Syndrome entwickelt werden konnte, stellen sie weiterhin
die führende Todesursache auf chirurgischen Intensivstationen dar (RANGEL-FRAUSTO
1995). Die Behandlungsmöglichkeiten beschränken sich somit auf die symptomatische
Therapie und ein bestmögliches intensiv-medizinisches Management.
Im Rahmen eines Traumas kommt es häufig in Folge einer Hypovolämie aufgrund starker
Blutungen zu einem Ischämiezustand mit anschließender Reperfusion, die durch das
Auffüllen des Gefäßsystems mit Infusionslösung während der Rettung oder des
anschließenden Klinikaufenthaltes herbeigeführt wird. Außerdem kann eine
Gefäßkonstriktion oder -ruptur aufgrund von Frakturen eine Ischämie auslösen. Um eine
derartige Situation und deren Auswirkungen bei Mäusen zu imitieren, wurde in der
vorliegenden Arbeit ein Ischämie-Reperfusionsversuch der unteren Extremitäten durch
Abklemmen der Aorta abdominalis vorgenommen. Zum Schutze des Organismus bewirkt ein
traumatisches Ereignis die Aktivierung des Immunsystems (REGEL 1989). Primär werden
dabei lokal besonders die Zytokine TNF-α und IL-1β freigesetzt, welche unter anderem die
sekundäre Sekretion von IL-6 bewirken. Werden diese proinflammatorischen Zytokine jedoch
nicht nur lokal, sondern auch systemisch sezerniert, kommt es zu einer überschießenden
Inflammationsreaktion des gesamten Organismus (BONE 1996b;DAVIES 1997). Hieraus
entwickelt sich ein SIRS, welches schließlich durch die sekundäre Beeinträchtigung
verschiedener Organsysteme in einem MODS enden kann. Auch eine gegenregulierende
antiinflammatorische Immunantwort mit gesteigerter IL-10-Produktion kann die ausgelöste
Entzündungsreaktion häufig nicht mehr in ein physiologisches Maß eindämmen. In einigen
Fällen kann es jedoch zu einem Überschießen der antiinflammatorischen Reaktion kommen,
welche als CARS bezeichnet wird und mit erhöhter Gefahr der Entstehung einer Sepsis
einhergeht.
TNF-α ist ein sehr potentes proinflammatorisches Zytokin, welches von Monozyten,
Makrophagen und T-Lymphozyten sezerniert wird. Besonders über den TNF-Rezeptor I ist

Diskussion 90
dieses Zytokin an der Inflammationsreaktion beteiligt. Geringe TNF-α-Mengen sind für eine
adäquate Immunantwort unabdingbar, dauerhaft erhöhte Werte schädigen den Organismus
jedoch nachhaltig. Grund hierfür ist die Tatsache, dass TNF-α eine vermehrte Expression
zellulärer Adhäsionsmoleküle der Endothelzellen (ICAM, ELAM, E-Selektin) und eine
gesteigerte Permeabilität des Gefäßendothels bewirkt (BRETT 1989; MULLIGAN 1993).
Neutrophile Granulozyten können dadurch leicht aus dem Gefäßbett in das umliegende
Gewebe auswandern und schädigen dies durch die Reaktionsprodukte des respiratory burst.
Zusätzlich bewirkt TNF-α eine verstärkte Freisetzung von IL-1, IL-6 und IL-8 aus den
Endothelzellen, welche wiederum eine proinflammatorische Wirkung aufweisen (LOPPNOW
1989; NAWROTH 1986a). Auf den eingewanderten neutrophilen Granulozyten befinden sich
unter anderem Fcγ-Rezeptoren, die den Fc-Teil des Immunglobulins G binden und somit ein
wichtiges Element für die Verbindung der humoralen und der zellulären Abwehr darstellen.
Die Degranulation und Phagozytose der Granulozyten werden hauptsächlich über die Fcγ-RI
und Fcγ-RIII vermittelt. HELLER et al. (1999) zeigten anhand von F--Mäusen, bei denen
experimentell eine Immunkomplex-Peritonitis ausgelöst wurde, dass die Migration
neutrophiler Granulozyten in die Bauchhöhle bei diesen Tieren gegenüber Wildtypen
unbeeinträchtigt ist. Weiterhin konnte dabei aber eine geringere TNF-Sekretion beobachtet
werden, da diese besonders durch die Bindung von Immunglobulin an den Fcγ-RIII vermittelt
wird (HELLER 1999).
Die oben beschriebenen Tatsachen bestätigen die zentrale Rolle der neutrophilen
Granulozyten bei der Entstehung der sekundären Organschäden im Verlaufe eines SIRS
(VAN GRIENSVEN 1999a; VAN GRIENSVEN 2003). In der vorliegenden Arbeit wurde mit
Hilfe von Knockout-Tieren die Rolle der Rezeptoren TNF-RI und Fcγ-RIII und deren Einfluß
auf die posttraumatische Situation der Versuchstiere ermittelt.
Mit Hilfe eines Ischämie-Reperfusionsversuchs der unteren Extremitäten wurde in der
vorliegenden Arbeit an Mäusen ein Trauma imitiert und so ein reproduzierbarer
Lungenschaden hervorgerufen. SEEKAMP et al. (1993, 1994) nahmen ein entsprechendes
Versuchsmodell an Ratten vor.

Diskussion 91
Die im folgenden Teil der Arbeit diskutierten Ergebnisse sind jedoch nur unter Vorbehalt zu
interpretieren. Grund hierfür ist der Vergleich von rein-stämmigen Kontrolltieren mit
gemischt-stämmigen Knockout-Versuchstieren. Demnach ist es durchaus möglich, dass die
Ergebnisse nicht durch das Fehlen bestimmter Gene, sondern durch den Stammunterschied
hervorgerufen wurden. Diese Tatsache ist bei allen Parametern zu beachten.
6.1 Lungenpermeabilität
Um den Grad der Lungenschädigung quantifizieren zu können, sind verschiedene Methoden
bekannt. Heute wird besonders die Bestimmung des relativen Proteingehaltes der BAL in
Kombination mit der Quantifizierung der neutrophilen Granulozyten mittels Histologie oder
MPO-Messung genutzt. Nach PETERSON (1992) ist die Verwendung zweier
unterschiedlicher Methoden ausreichend, um eine pathophysiologisch signifikante
Veränderung der endothelialen Permeabilität in vivo nachzuweisen. Die pulmonale
Proteinextravasation und deren Quantifizierung durch eine BAL mit Berechnung des relativen
Proteingehaltes wurde sowohl in Klein- als auch in Großtiermodellen hinreichend validiert
(KLAUSNER 1989; SEEKAMP 1993b). Werte von 0,1 bis 0,2 gelten hierbei als
physiologisch, liegen sie jedoch über 0,7, so gilt dies als Nachweis für eine defekte
Kapillarpermeabilität in vivo.
Die in der vorliegenden Arbeit ermittelten Werte aller Gruppen lagen zwar unter dem
pathologischen Grenzwert von 0,7, jedoch war ein Unterschied zwischen den Wildtyp-Tieren
und den Knockout-Tieren festzustellen.
Innerhalb der Wildtyp-Gruppen ist der Proteingehalt der Gruppe Ischämie 4 h am niedrigsten.
Die I/R-Gruppen hingegen zeigen deutlich jedoch nicht signifikant erhöhte Werte. Diese
Beobachtung bestätigt, dass die Lungenfunktion nicht durch die Ischämie, sondern erst durch
eine nachfolgende Reperfusion geschädigt wird (WELBOURN 1991b).
Betrachtet man die Laparotomie- und I/R-Gruppen der WT, so stellt man fest, dass der
Proteingehalt in der BAL im zeitlichen Verlauf im Vergleich zu den 8 h Werten nach jeweils
48 h bereits wieder abgenommen hat. Diese Tatsache ist dadurch zu erklären, dass zwar eine
Schädigung des pulmonalen Endothels vorliegt, die aber durch Regenerationsmaßnahmen zu

Diskussion 92
beheben ist. Die Integrität und Funktionsfähigkeit des Gefäßbettes ist somit wieder
hergestellt.
Die Werte der I/R-Gruppen der T-- und F--Tiere sind deutlich niedriger als die der
entsprechenden WT-Gruppen. Diese Unterschiede sind statistisch jedoch nicht signifikant.
Das Fehlen des TNF-RI scheint einen protektiven Einfluß vor einer Erhöhung der
Lungenpermeabilität zu haben. Bestärkt wird diese Vermutung durch die Feststellung von
VAN GRIENSVEN (1999), wonach die pulmonale Endothelpermeabilität bei einem In-vitro-
Versuch durch TNF-α gesteigert werden kann. Es besteht allerdings die Möglichkeit, dass die
unterschiedlichen Werte durch die verschiedenen Stämme der Versuchs- und Kontrolltiere
verursacht wurden.
Da sich die Werte der F--Gruppen nur unwesentlich von denen der anderen Knockout-Tiere
unterscheiden, kann in diesem Falle kein synergistischer oder additiver Effekt beim
zusätzlichen Fehlen des Fcγ-Rezeptors beobachtet werden.
6.2 Myeloperoxidaseaktivität und Granulozyteninfiltration des Lungengewebes
Nach ALLAN et al. (1985) ist eine Quantifizierung der neutrophilen Granulozyten mittels
MPO-Messung sensitiver und bezogen auf das gesamte Lungengewebe repräsentativer als
eine Auszählung der PMN in einem histologischen Präparat. Der Nachteil einer
histologischen Beurteilung ist, dass nur jeweils ein kleiner Ausschnitt des Organs bewertet
werden kann.
Die Myeloperoxidaseaktivität im Lungengewebe der Versuchstiere zeigte in der vorliegenden
Arbeit größtenteils keine deutlichen Unterschiede. Bei den Wildtyp-Tieren sind
erwartungsgemäß die Werte der I/R-Gruppen geringgradig höher als die der
Laparotomiegruppen. Verursacht wird dies durch eine Aktivierung der neutrophilen
Granulozyten aufgrund des Reperfusionsreizes und einem Auswandern dieser Zellen in das
pulmonale Gewebe. Der Abfall der MPO-Aktivität im zeitlichen Verlauf der I/R-Gruppen
wird begründet durch die kurze Halbwertszeit der neutrophilen Granulozyten im Gewebe von

Diskussion 93
circa 12-24 Stunden. Somit ist ein Großteil der Zellen nach 48 h bereits nicht mehr
vorhanden.
Für die T--Tiere wurden ähnliche Werte festgestellt wie für die entsprechenden Gruppen der
WT-Tiere. Auch bei den F--Tieren ist ein Abfall der Werte im zeitlichen Verlauf der I/R-
Gruppen zu beobachten und kann wie bei den WT-Tieren mit einem Zerfall der neutrophilen
Granulozyten nach circa einem Tag begründet werden.
Bei einer Myeloperoxidasebestimmung nach dieser Methode muss jedoch immer bedacht
werden, dass es zu Abweichungen kommen kann, da die Gewebeprobe gesamthaft in die
Messung eingeht und somit nicht nur die Granulozyten gemessen werden, die sich im
Parenchym befinden, sondern auch jene, die noch im Gefäßbett enthalten sind. Um einer
Verfälschung der Ergebnisse aufgrund dieser Tatsache vorzubeugen, müsste das Gefäßsystem
der Lunge nach der Entnahme des Organs perfundiert werden. Die Bewertung der Ergebnisse
sollte deshalb immer unter Berücksichtigung der histologischen Auszählung der neutrophilen
Granulozyten erfolgen. Auch in der vorliegenden Arbeit können Abweichungen zwischen der
histologischen Auswertung und der MPO-Messung beobachtet werden. In diesem Falle sollte
der Histologie eine größere Beachtung zukommen, da es sich um die tatsächlich im Gewebe
vorkommenden neutrophilen Granulozyten handelt. Hierbei ist eine Abnahme der Zellen im
Lungengewebe beider Knockout-Stämme gegenüber der WT-Tiere zu bemerken.
Erwartungsgemäß zeigen die Wildtyp-Tiere der I/R-Gruppen gegenüber den laparotomierten
Tieren eine stärkere Granulozyteninfiltration. Diese Tatsache bestätigt die Schlüsselrolle der
neutrophilen Granulozyten bei der Entstehung eines Ischämie-Reperfusionsschadens
(FUJISHIMA 1995). Da die Halbwertszeit dieser Zellen im Gewebe allerdings nur circa 24 h
beträgt, ist 48 Sunden nach der Reperfusion die Granulozytenzahl bereits deutlich gesunken.
Vergleicht man nun die beiden Knockout-Gruppen I/R 8 h mit der Gruppe WT-I/R 8 h, so
fällt jeweils eine statistisch signifikant geringere Granulozyteninfiltration in das
Lungengewebe auf. Es ist wahrscheinlich, dass der TNF-α bei den T--Tieren seine biologische
Funktion nicht ausüben kann, da der TNF-Rezeptor I bei diesen Knockout-Mäusen nicht
existent ist. TNF-α bewirkt normalerweise in Kombination mit IL-1 eine gesteigerte
Endothelpermeabilität (BRETT 1989) und eine erhöhte Expression zellulärer
Adhäsionsmoleküle (ICAM-1, ELAM-1, E-Selektin) der Endothelzellen (MULLIGAN 1993).
Das L-Selektin ist ein wichtiges Molekül beim initialen Schritt des Migrationsprozesses der

Diskussion 94
neutrophilen Granulozyten (“Rolling“) in das Gewebe. Nach dem Verringern der
Fließgeschwindigkeit des Blutes bewirkt es eine transiente Adhäsion der Granulozyten, indem
es eine Bindung mit dem neutrophilen L-Selektin eingeht (ANDERSON 1987). Auch die
immunglobulinähnlichen Adhäsionsmoleküle (ICAM, ELAM) spielen bei den nun folgenden
Migrationsschritten eine wichtige Rolle, da sie die endothelialen Liganden für das neutrophile
β2-Integrin darstellen (SMITH 1989). Können die beschriebenen Prozesse bei den T--Tieren
aufgrund des Gendefektes jedoch nicht oder nur in verringerter Form ablaufen, so wird eine
Einwanderung der neutrophilen Granulozyten wirkungsvoll verhindert. BURNE et al. (2001)
zeigten allerdings mit Hilfe von IL-1-RI- und TNF-RI-Knockout-Tieren, dass es einen IL-1
und TNF unabhängigen Weg für die gesteigerte Expression von ICAM-1 und VCAM-1 nach
Ischämie-Reperfusion geben muß. In der vorliegenden Arbeit konnte jedoch ein positiver
Effekt auf die Verhinderung der Granulozyteninfiltration in die Lunge durch das Fehlen des
TNF-RI bemerkt werden. Erneut muss jedoch darauf hingewieden werden, dass es sich um
einen Effekt handeln kann, der durch das Vergleichen verschieden-stämmiger Tiere
hervorgerufen sein kann.
GRUTKOSKI et al. (2002) beschrieben einen hemmenden Effekt von neutrophilen
Granulozyten, die mit TNF-α stimuliert wurden, auf die Migration und Funktion anderer
neutrophiler Granulozyten, auch wenn die Zellen nicht in direktem Kontakt zueinander
standen. Es wird vermutet, dass TNF-α an einer negative feedback-Funktion beteiligt ist. Die
geringere Infiltration der PMN in der vorliegenden Arbeit könnte auf eine derartige
Hemmung zurückzuführen sein.
Auffallend ist jedoch, dass die Anzahl der Zellen 48 h nach der Reperfusion deutlich höher ist
als nach 8 h. Es ist bekannt, dass der TNF-α das primär freigesetzte Zytokin nach einem
traumatischen Einfluß auf den Organismus ist (VAN GRIENSVEN 2003). Der TNF bewirkt
daraufhin sehr schnell die Sekretion anderer proinflammatorischer Zytokine wie zum Beispiel
IL-1 und IL-6. Kann der TNF-α aufgrund des Genotyps der Knockout-Mäuse jedoch seine
proinflammatorische Funktion nicht ausüben, so muß die Aktivierung der Kaskade zur
Freisetzung anderer Zytokine über Kollateralwege erfolgen. Diese Umwege nehmen mehr
Zeit in Anspruch und auch die Infiltration der neutrophilen Granulozyten in das
Lungengewebe ist dadurch verlangsamt. Dies wird in der vorliegenden Arbeit durch die
vermehrte Anzahl von neutrophilen Granulozyten im Lungengewebe beider Knockout-Sämme

Diskussion 95
48 h nach Reperfusion verdeutlicht. Wie bereits in 2.2 beschrieben, bewirkt diese vermehrte
Infiltration die Ausbildung eines stärkeren Ödems.
Die Werte für die F--Tiere haben das gleiche Muster wie die der T--Tiere, sind jedoch
geringgradig höher. Demnach kann von einem minimal synergistischen Effekt beim
zusätzlichen Fehlen des Fcγ-RIII in Bezug auf die Granulozyteninfiltration gesprochen
werden.
6.3 Lungenödem
Bei den WT-Tieren ist innerhalb der I/R-Gruppen ein deutlich stärkeres Lungenödem im
Vergleich zu den Laparotomie-Gruppen festzustellen. Diese Beobachtung bestätigt die
Tatsache, dass eine Ischämie, gefolgt von einer Reperfusion, einen sekundären
Lungenschaden bedingt (SEEKAMP 1993b). Dabei spielt es keine Rolle, welcher Bereich des
Körpers von der Ischämie betroffen war (DUBAYBO 1988; GOLDMAN 1992). Im Verlauf
der Reperfusion werden Zytokine freigesetzt, die unter anderem auf das Endothel der Gefäße
einwirken und eine Permeabilitätserhöhung bewirken (VAN GRIENSVEN 1999a).
Infolgedessen kommt es zu einer verstärkten Extravasation von Zellen, Blutbestandteilen und
Flüssigkeit ins Gewebe.
Die Ausprägung des Lungenödems ist bei beiden Knockout-Stämmen deutlich geringer als bei
den Wildtypen. Einige dieser Unterschiede waren statistisch signifikant. Auch in diesem Falle
muss jedoch der unterschiedliche genetische Hintergrund der Versuchs- und Kontrolltiere
beachtet werden. Es ist bekannt, dass TNF-α im Zusammenspiel mit IL-1 die
Endothelpermeabilität steigert (VAN GRIENSVEN 1999a). Zusätzlich bewirkt es eine
vermehrte Expression der zellulären Adhäsionsmoleküle der Endothelzellen. Vermutlich ist
das weniger stark ausgeprägte Lungenödem der T--Tiere durch eine Verringerung der
Endothelpermeabilität und eine geringere Infiltration von neutrohpilen Granulozyten in das
umgebende Gewebe bedingt, da der TNF-α zwar im Körper vorhanden ist, er jedoch nicht an
den TNF-RI binden kann und somit keine Wirkung hat. Zusätzlich konnte anhand der
Zytokinmessung gezeigt werden, dass neben der Wirksamkeit auch die Produktion des TNF-α

Diskussion 96
bei den Knockout-Tieren deutlich vermindert ist. So ist der Zytokinwert der WT-Tiere 8 h
nach I/R fast 6 mal höher (T-) bzw. fast 3 mal höher (F-) als der der Knockout-Tiere.
Auffällig ist allerdings, dass die Gruppen I/R 48 h der Knockout-Tiere ein stärkeres
Lungenödem aufweisen als die Gruppen I/R 8 h. Ursache hierfür kann die stärkere
Granulozyteninfiltration bei diesen Gruppen sein, was mit Hilfe der histologischen Präparate
und teilweise der MPO-Messung gezeigt werden konnte. Die Infiltration dieser Zellen führt
aufgrund der bereits beschriebenen Steigerung der Endothelpermeabilität und der Schädigung
des Lungenparenchyms zu einer Ödembildung. Für die Knockout-Tiere kann somit eine
Verschlimmerung des Krankheitsbildes im zeitlichen Verlauf nach Ischämie-Reperfusion
beobachtet werden. Möglich ist eine langsamere Infiltration der neutrophilen Granulozyten
nach dem Reperfusionsreiz, da der TNF-RI bei beiden Knockout-Stämmen nicht vorhanden
ist. TNF-α wird üblicherweise nach einem Trauma primär freigesetzt und bewirkt die
Sekretion anderer proinflammatorischer Zytokine wie IL-1 und IL-6 (VAN GRIENSVEN
2003). Kann diese Kaskade jedoch aufgrund des Gendefektes nicht ablaufen, so muss eine
Aktivierung der sekundär sezernierten Zytokine auf einem Umweg, der mehr Zeit in
Anspruch nimmt, erfolgen. Die Granulozyteninfiltration ist daher verzögert. BURNE et al.
(2001) konnten mit Hilfe von B6;129S-Tnfrsf1atm1imx Il1r1tm1lmx-Mäusen zeigen, dass es für
die Infiltration von neutrophilen Granulozyten einen IL-1- und TNF-α-unabhängigen Weg
geben muß. Möglicherweise ist auch dieser Weg langsamer als eine Aktivierung der Zellen
über den TNF-α.
6.4 Leberödem
Bei der Betrachtung der Stärke des Leberödems ist auffällig, dass jeweils die Ischämie 4 h-
Gruppen die höchsten Werte innerhalb der verschiedenen Typen aufwiesen. Ein möglicher
Grund hierfür ist der durch die Ischämie herbeigeführte hypoxische Zustand, in dem sich die
Leber befindet. Das Setzen der Ligatur bewirkt eine Beeinträchtigung der Blutzirkulation.
Somit entsteht ein intrahepatischer Blutstau, der einen erhöhten hydrostatischen Druck in den
Kapillargefäßen bewirkt. Zusätzlich läuft aufgrund des Sauerstoffmangels ein anaerober
Stoffwechsel ab, der sowohl das Leberparenchym als auch das Kapillarendothel schädigt.

Diskussion 97
Eiweißreiche Flüssigkeit ist so in der Lage, die Gefäßwände zu passieren und sammelt sich in
Form von Ödemflüssigkeit im interstitiellen Raum. Des weiteren treten zelluläre Bestandteile
aus den Blutgefäßen aus, was den osmotischen Effekt im Gewebe und die Ödembildung
zusätzlich steigert.
Wird die Ischämie von einer Reperfusion gefolgt, so ist eine Abnahme des Leberödems bei
allen Maus-Stämmen zu beobachten. Die bereits nach 8 h verringerten Werte zeigen nach 48
h nochmals einen Abfall. Eine mögliche Erklärung für diese Tatsache ist, dass der Blutfluß
mit dem Durchtrennen der Ligatur wiederhergestellt ist und somit auch die Leber einer
physiologischen Durchblutung unterworfen wird. Während dieser Phase kommt es aufgrund
der aeroben Stoffwechselsituation zu Regenerationsmaßnahmen des Gefäßendothels. Die
interstitielle Ödemflüssigkeit wird nach und nach über das Lymphsystem abgeführt, wodurch
es im zeitlichen Verlauf zu einer kontinuierlichen Verbesserung kommt. Neben den
erwähnten hämodynamischen Kompensationsmechanismen sind auch zelluläre Faktoren
bekannt, die einen Abtransport der Ödemflüssigkeit bewirken (PILLER 1980). Hierbei
handelt es sich um Gewebsmakrophagen, welche die Fähigkeit besitzen interstitielle Proteine
zu phagozytieren, wodurch der osmotische Druck und damit auch die
Flüssigkeitsansammlung vermindert wird. Sekundärschäden, wie sie in der Lunge beobachtet
werden können, spielen in der Leber bei diesem Modell anscheinend eine untergeordnete
Rolle.
Auffällig ist, dass die T--Tiere ein geringeres Leberödem, die F--Tiere jedoch ein stärkeres
Ödem als die WT-Tiere aufweisen. Demnach bewirkt ein zusätzliches Fehlen des Fcγ-RIII
keine Verringerung, sondern eine Verstärkung des Ödems nach Ischämie-Reperfusion. Grund
hierfür ist bei diesen Gruppen wahrscheinlich die erhöhte Granulozyteninfiltration in die
Leber. Es ist bekannt, dass die Aktivierung der neutrophilen Granulozyten eine Schädigung
des Gefäßbettes und somit eine erhöhte Permeabilität hervorrufen. Zusätzlich wird das
Organparenchym durch die Stoffwechselprodukte (Sauerstoffradikale) migrierter Zellen
beeinträchtigt. Diese Faktoren führen zu einer Ödembildung in dem betroffenen Organ. Ein
weiterer Grund für das Auftreten eines stärkeren Leberödems bei den F--Tieren ist die
Tatsache, dass die oben beschriebene Phagozytose der interstitiellen Bestandteile aufgrund
des Fehlens des Fcγ-RIII auf Makrophagen und neutrophilen Granulozyten nicht stattfinden

Diskussion 98
kann. Ein wichtiger Kompensationsmechanismus für die Verringerung des Leberödems ist
dadurch ausgeschaltet.
6.5 Granulozyteninfiltration in die Leber
Innerhalb der WT-Gruppen ist eine ungefähr gleich starke Infiltration von nur wenigen
Granulozyten ins Lebergewebe zu beobachten. Lediglich der Wert der Gruppe I/R 8 h ist im
Vergleich zu den anderen WT-Gruppen geringgradig höher. Die Granulozyteninfiltration der
T--Tiere ist niedriger als die der entsprechenden WT-Gruppen. Auffallend ist, dass die Werte
der F--Tiere, ebenso wie bei der Beurteilung des Leberödems, deutlich über denen der WT
liegen. Nur die Gruppe F--I/R 48 h bildet hierbei eine Ausnahme. Vergleicht man die
Granulozyteninfiltration in das Lungengewebe mit der Infiltration ins Lebergewebe, so stellt
man ähnliche Werte für die WT- und T--Gruppen fest, bei den F--Gruppen fällt jedoch eine
größtenteils stärkere Zellmigration ins Lebergewebe auf. Wiederum muss beim Vergleich der
Knockout- mit den Kontrolltieren auf den unterschiedlichen genetischen Hintergrund
hingewiesen werden, wodurch es zu abweichenden Ergebnissen kommen kann.
Die Leber ist ein Organ, in dem besonders zahlreich Bestandteile des Reticulo-Endothelialen-
Systems vorhanden sind, welches bei den Wildtyp-Tieren mit dem Fcγ-RIII ausgestattet ist.
Fehlt dieser Rezeptor jedoch bei den Knockout-Mäusen, so kann die phagozytierende
Tätigkeit des Reticulo-Endothelialen-Systems nicht ausgeführt werden. Als
Ausweichmöglichkeit wird eine verstärkte Infiltration neutrophiler Granulozyten ins
Lebergewebe veranlasst. Die Stärke der Granulozyteninfiltration stimmt mit der Ausprägung
des Leberödems überein. Es ist bekannt, dass die neutrophilen Granulozyten durch die
Produkte des respiratory burst (Sauerstoffradikale) das Gefäßendothel schädigen und somit
eine gesteigerte Permeabilität hervorrufen (FUJISHIMA 1995). Demnach müssten jedoch die
F--Tiere geringere Werte für das Leberödem aufweisen, da der respiratory burst durch die
Abwesenheit des Fcγ-RIII stark gehemmt wird. Es liegt also nahe, dass für die Entstehung des
Ödems weitere Faktoren verantwortlich sind. Möglicherweise ist der Blutstau in Verbindung
mit der Hypoxie, welche durch die Ischämie herbeigeführt wird, ein Hauptgrund für die
Ödembildung.

Diskussion 99
Zu klären bleibt allerdings, warum die F--Tiere insgesamt eine deutlich höhere
Granulozyteninfiltration, die T--Tiere jedoch eine niedrigere Zellzahl als die WT-Tiere
aufweisen.
Der Abfall der Granulozytenanzahl nach jeweils 48 h kann dadurch erklärt werden, dass diese
Zellen im Gewebe eine geringere Halbwertszeit als im Blut haben. Sie beträgt circa 12 bis 24
Stunden. Nach 48 Stunden ist somit bereits ein großer Teil der Granulozyten nicht mehr im
Gewebe vorhanden.
6.6 Hydropische Degeneration der Leber
Die hydropische Degeneration ist eine Art der Zellschädigung, die besonders nach Hypoxie
auftritt. Es handelt sich dabei um einen gestörten Wasserstoffwechsel der Zelle, der sich
aufgrund eines mangelhaften Sauerstoffangebotes und einer energetischen Insuffizienz
entwickelt. Der sogenannte point of no return ist dabei überschritten und es entsteht eine
dauerhafte Beeinträchtigung der Zellfunktion, was zu einem programmierten Zelltod, der
Apoptose, führen kann. Die hydropische Degeneration ist ein langsamer Untergang von
Leberzellen, bei dem es durch einen Ausfall der Ionenpumpen zu einem Einstrom von Wasser
in die Zelle kommt. Durch das Ligieren der Aorta abdominalis wird die Leber in einen
Zustand der Minderversorgung sowohl mit sauerstoff- als auch mit energierreichem Blut
versetzt, wodurch die oben beschriebenen pathologischen Vorgänge ausgelöst werden.
Betrachtet man in der vorliegenden Arbeit die Stärke der hydropischen Degeneration in der
Leber, so fällt auf, dass die höchsten Werte bei allen Maus-Stämmen 48 Stunden sowohl nach
Laparotomie als auch nach I/R zu beobachten sind. Die stärkste Degeneration ist hierbei 48
Stunden nach Reperfusion zu beobachten ist. Diese Ergebnisse bestätigen die Tatsache, dass
die hydropische Degeneration der Leber als ein typischer Spätschaden nach Ischämie-
Reperfusion beschrieben werden kann. Der TNF-RI und der Fcγ-RIII scheinen auf die Stärke
des Schadens keinen Einfluss zu haben.
RUDIGER und CLAVIEN (2002) zeigten an T--Mäusen, dass TNF-α ein wichtiges Zytokin
bei der Apoptoseinduktion von Leberzellen nach einer Ischämie dieses Organs darstellt. Auch
TIETZE et al. (2000) beobachteten bei erhöhten TNF-α-Werten eine verstärkte Apoptose von

Diskussion 100
Leberzellen. Für die Entstehung der hydropischen Degeneration nach Ischämie-Reperfusion
der unteren Extremitäten konnte diese Beobachtung jedoch nicht gemacht werden. In der
vorliegenden Arbeit war der Schaden sowohl bei den WT- als auch bei den Knockout-Tieren
etwa gleich stark. Begründet werden kann diese Tatsache mit der Beobachtung, dass
besonders das P-Selektin eine Schlüsselfunktion bei der Entstehung eines Leberschadens nach
Ischämie-Reperfusion dieses Organs einnimmt. Untersuchungen an Patienten mit einem
chronischen Leberschaden zeigten, dass das P-Selektin im Blut signifikant erhöht gegenüber
Kontrollpatienten war (TACKE 2003). Der TNF-α bewirkt zwar eine erhöhte Expression von
ICAM-1, ELAM-1 und E-Selektin, auf die Expression von P-Selektin hat er jedoch keinen
Einfluß (MULLIGAN 1993). Demnach ist durch das Fehlen des TNF-RI kein protektiver
Einfluss auf die Ausbildung eines Leberschadens in Form einer hydropischen Degeneration
nach Ischämie-Reperfusion zu erwarten.
Warum allerdings die laparotomierten WT-Tiere in der vorliegenden Arbeit eine sehr starke
Schädigung der Leber aufweisen, kann nicht erklärt werden.
6.7 Nekrosen, Lymphozytendichte und Abgrenzbarkeit der weißen von der roten Pulpa in der Milz
Bei der histologischen Beurteilung der Milznekrosen konnten keine signifikanten
Unterschiede festgestellt werden. Lediglich bei jeweils einem Tier in den Gruppen WT-Lap. 8
h und WT-I/R 8 h konnten nekrotische Veränderungen beobachtet werden. Dabei handelt es
sich um Zufallsbefunde. Das Ischämie-Reperfusions-Versuchsmodell hat demnach keinen
Einfluss auf die Entstehung von Nekrosen in diesem Organ. Ähnlich verhält es sich mit der
Abgrenzbarkeit der weißen von der roten Milzpulpa. Auch hier war lediglich bei einem Tier
die Abgrenzung nicht möglich. Es handelt sich demnach ebenfalls um einen Zufallsbefund.
Die Dichte der Lymphozyten in der weißen Pulpa variiert deutlich zwischen den einzelnen
WT-Gruppen. Hierbei sind statistisch signifikante Unterschiede festzustellen. Die Werte der
laparotomierten Wildtypen sind signifikant höher als die der Gruppe WT-I/R 48 h. Aufgrund
der Aktivierung des Immunsystems nach einem I/R-Zustand kommt es zu einer Migration der

Diskussion 101
Lymphozyten aus der Milz in das Blutgefäßsystem und somit zu einer Abnahme der Zellen in
den Lymphfollikeln.
Die Lymphozytendichte in den F--Gruppen variiert kaum. Die Werte der I/R-Gruppen sind
jedoch geringgradig niedriger als die der übrigen F--Tiere. Begründet werden kann diese
Tatsache ebenfalls durch die Aktivierung des Immunsystems nach erfolgtem I/R-Reiz. Dass
die Dichte der Lymphozyten bei den F--Tieren insgesamt niedriger als bei den WT-Mäusen
ist, bestätigt die Beobachtung von PASPARAKIS et al. (1997), wonach der TNF-RI eine
wichtige Rolle bei der Entstehung von Lymphfollikeln und Keimzentren in der Milz spielt.
Die I/R-Gruppen der T--Tiere zeigen jedoch eine höhere Lymphozytendichte in der Pulpa als
die Tiere der entsprechenden WT-Gruppen. Möglicherweise wird durch das Fehlen des TNF-
RI die inflammatorische Reaktion auf den Ischämie-Reperfusionsreiz derartig abgeschwächt,
dass es zu einer verminderten Aktivierung der Lymphozyten und damit zu einer geringeren
Mobilisierung dieser Zellen kommt. WITTWER (2001) beschrieb ein Ausbleiben oder eine
Verzögerung der spezifischen humoralen Immunantwort durch eine fehlende B-Zell-
Aktivierung bei T--Mäusen. Eine verstärkte Präsenz der Lymphozyten in der Milz nach I/R ist
auch in der vorliegenden Arbeit zu bemerken. Da jedoch auch den F--Tieren der TNF-RI
fehlt, wären hier ähnliche Ergebnisse zu erwarten gewesen. Die Werte dieser Tiere sind
jedoch niedriger als die der T--Tiere, das heißt, es hat eine stärkere Aktivierung und Migration
der Lymphozyten stattgefunden.
6.8 Dichte der Mesangiumzellen in den Glomeruli und Granulozyteninfiltration in die Nieren
Es konnte nur bei wenigen Tieren eine geringgradige Infiltration von neutrophilen
Granulozyten in das Nierengewebe beobachtet werden, alle anderen Tiere zeigten in dieser
Hinsicht keine pathologischen Veränderungen. Signifikante Unterschiede konnten hierbei
nicht ermittelt werden.
Die Dichte der Mesangiumzellen in den Glomeruli zeigte hingegen deutlichere Unterschiede.
Innerhalb der WT-Tiere konnten für die I/R-Gruppen signifikant niedrigere Werte als für die
laparotomierten und ischämischen Tiere beobachtet werden. Vergleicht man die Werte der

Diskussion 102
Mesangiumzelldichte der T--Tiere, so fällt auf, dass auch hier die Werte der I/R-Gruppen am
geringsten sind. Bei den F--Mäusen hingegen konnte die geringste Zelldichte sowohl bei der
Gruppe I/R 8 h als auch bei der Gruppe Laparotomie 48 h festgestellt werden. Die Niere ist
ein Organ, das ebenfalls durch den vorausgegangenen Ischämie-Reperfusions-Reiz der
unteren Extremitäten sekundär geschädigt wird. Die pathologischen Veränderungen sind
allerdings nicht so stark wie zum Beispiel in der Lunge oder der Leber. Hauptsächlich die
Glomeruli werden bei diesem Versuchsmodell beeinträchtigt. Die Hypoxie aufgrund der
eingeschränkten Blutzirkulation bewirkt eine Schädigung der Mesangiumzellen, die in einer
Apoptoseinduktion enden kann. Diese Veränderungen sind jedoch in der vorliegenden Arbeit
nur geringgradig. Es konnte weiterhin keine deutliche Beeinflussung der Parameter durch den
Genotyp der Versuchstiere festgestellt werden.
6.9 Zytokine
Der TNF-α ist ein Zytokin, das von Makrophagen, NK-Zellen und stimulierten neutrophilen
Granulozyten gebildet wird. Er wirkt als endogenes Pyrogen, erzeugt lokale Entzündungen
und bewirkt eine Aktivierung des Endothels, wodurch es zur Thromboseneigung und einer
gestörten Mikrozirkulation kommt. Die Störung des Blutflusses ist in Verbindung mit anderen
Faktoren ein Grund für die Entstehung des ARDS. Der TNF-α wirkt zusätzlich chemotaktisch
auf PMN und verstärkt deren Adhärenz mit dem Endothel, wodurch eine Auswanderung in
das Gewebe erleichtert wird. Weiterhin hat das Zytokin Einfluß auf die PMN-Funktion: Es
steigert die Zytotoxizität und Phagozytosefähigkeit dieser Zellen (BONAVIDA 1998;
OPPENHEIM JJ. 2001). All diese Tatsachen machen den TNF-α zu einem zentralen Zytokin
bei der Entstehung des I/R-Schadens. Die Auswirkungen des Fehlens des TNF-RI oder des
TNF-RI und Fcγ-RIII auf die Immunreaktion nach einem Ischämie-/Reperfusions-Reiz
wurden im Laufe dieser Arbeit ermittelt.
Neben den oben beschriebenen Einflüssen bewirkt der TNF-α eine Freisetzung anderer
proinflammatorischer Zytokine wie IL-1 und IL-6. Ein hoher IL-6 Wert inhibiert allerdings
die TNF-α Produktion, so dass hierbei von einer negativen Rückkopplung gesprochen werden

Diskussion 103
kann. IFNγ hingegen verstärkt die Bildung von TNF-α. Für eine physiologische
Immunantwort ist das Zytokin TNF-α unabdingbar.
In der internationalen Literatur wird beschrieben, dass aufgrund der Höhe des TNF-α-Wertes
eine Aussage über das Outcome eines Traumapatienten gemacht werden kann (SEEKAMP
1998). GEBHARD et al. (2000) können derartige Beobachtungen aufgrund einer von ihnen
durchgeführten Studie nicht bestätigen. Ihrer Meinung nach läßt sich innerhalb der ersten 24
Stunden kein Zusammenhang zwischen den TNF-α-Werten im Serum der Traumapatienten
und dem Schweregrad oder Schwerpunkt des Traumas herstellen.
In der vorliegenden Arbeit betragen die TNF-α Serum-Konzentrationen bei allen Gruppen
zwischen 0 pg/ml und 37,03 pg/ml. Einzige Ausnahme bildet hierbei die Gruppe I/R 8 h der
WT-Tiere, welche einen TNF-α-Wert von 72,41 pg/ml aufweist. Dieser ist gegenüber den
Konzentrationen anderer Gruppen signifikant erhöht. Die TNF-α Konzentration der T--Tiere
(12,80 pg/ml) und F--Tiere (25,89 pg/ml), die den gleichen Versuchsbedingungen ausgesetzt
waren, sind deutlich niedriger. Da der Normalwert der Kontrolltiere 0 pg/ml ist, kann man
erkennen, dass der TNF-α nach einer Laparotomie, besonders jedoch nach der Durchführung
einer Ischämie mit anschließender Reperfusion, gebildet wird. Die WT-Tiere sind in der I/R 8
h Gruppe deutlich stärker betroffen als die beiden verschiedenen Knockout-Stämme der
gleichen Gruppen. Bei der Beurteilung dieser Ergebnisse muss jedoch bedacht werden, dass
rein-stämmige Kontrolltiere mit gemischt-stämmigen Knockout-Tieren verglichen wurden
und es allein dadurch zu Unterschieden kommen kann. Da bei den T- und F--Tieren der TNF-
RI defizient ist, kann das gebildete Zytokin hier nicht in vollem Umfang die oben
beschriebenen Effekte hervorrufen, so dass von einem Schutz vor schädlichen Auswirkungen
ausgegangen werden kann. Die geringere Beeinträchtigung der Tiere beider Knockout-
Stämme zeigte sich in der vorliegenden Arbeit besonders anhand der Lungenmesswerte.
Hierbei fielen sowohl eine geringere Lungenpermeabilität als auch eine geringere
Granulozyteninfiltration und Ödembildung auf.
Beim IL-6 handelt es sich ebenfalls um ein sehr wichtiges proinflammatorisches Zytokin. Wie
bereits erwähnt, wird die IL-6 Produktion von TNF-α und IL-1 angeregt. Das Zytokin wird
hauptsächlich von Monozyten, Makrophagen, B- und T-Zellen und Endothelzellen freigesetzt.

Diskussion 104
IL-6 ist das zentrale Zytokin der acute-phase-response nach Trauma oder Infektion, das unter
anderem die Bildung der Akuten-Phase-Proteine in den Hepatozyten anregt. Des weiteren
bewirkt es eine Differenzierung und Aktivierung von B- und T-Zellen mit einer gesteigerten
Ig-Sekretion. Zusätzlich fördert es die Ausreifung hämatopoetischer Vorläuferzellen zu
Makrophagen und Granulozyten (OPPENHEIM JJ. 2001). Die IL-6 Antwort ist abhängig
vom Traumaschweregrad und stellt somit einen guten und frühen Marker für die Intensität
eines Traumas dar (GEBHARD 2000). Sind bei einem Patienten lang anhaltende sehr hohe
zirkulierende IL-6 Werte festzustellen, so sind diese mit Komplikationen und/oder einer
erhöhten Mortalität verbunden. TANIGUCHI et al. (1999) beschrieben, dass nicht die
absoluten Werte, sondern das Verhältnis von IL-6- zu IL-10-Konzentration entscheidend für
das Outcome eines Traumapatienten sei. Ein Anstieg der Ratio korreliere demnach mit einer
ungünstigen Prognose.
In der vorliegenden Arbeit sind die IL-6 Serumwerte der Kontrolltiere 0 pg/ml. Bereits 8 h
nach Laparotomie sind bei allen Versuchstier-Stämmen erhöhte Zytokinkonzentrationen von
bis zu 2847,53 pg/ml (T-) zu beobachten, welche im zeitlichen Verlauf jedoch wieder
abnehmen, so dass in den Laparotomie 48 h-Gruppen die Werte auf maximal 115,37 pg/ml
abgesunken sind. Besonders deutlich gestiegen ist die IL-6-Konzentration in der Gruppe WT
I/R 8 h. Der Wert von 22375,60 pg/ml ist im Vergleich zu den Knockout-Tieren (T-: 1656,58
pg/ml, F-: 1890,23 pg/ml) etwa um das 12-fache höher. Bereits 48 h nach I/R sind die Werte
aller drei Versuchstier-Stämme nur noch im niedrigen zweistelligen Bereich (zwischen 15,82
pg/ml (WT) und 29,52 pg/ml (T-)). Deutlich wird die Verbesserung der posttraumatischen
Situation der Versuchstiere zusätzlich durch das Absinken der Mortalitätsrate. Verstarben in
der Gruppe I/R 8 h bei den WT noch 28,6 % der Tiere, so sank die Zahl bei den F--Tieren um
etwa die Hälfte auf 12,5 %. Bei den T--Tieren fiel die Mortalitätsrate sogar auf 0 % ab. Erneut
muss darauf hingewiesen werden, dass Versuchstiere mit einem unterschiedlichen
genetischen Hintergrund miteinander verglichen werden.
Es kann demnach bestätigt werden, dass es sich bei dem Zytokin IL-6 um ein Protein handelt,
dessen Antwortstärke sich nach dem Traumaschweregrad richtet und hauptsächlich in der
akuten Phase der Immunantwort ausgeschüttet wird. Außerdem konnte beim Fehlen des TNF-
RI oder des TNF-RI und Fcγ-RIII eine deutlich geringere IL-6-Ausschüttung beobachtet
werden. Zu erklären ist diese Tatsache damit, dass der TNF-α, der bei den WT-Tieren eine

Diskussion 105
erhöhte IL-6-Produktion auslöst, durch das Fehlen des TNF-RI bei den Knockout-Tieren
keinen Angriffspunkt hat. Die „Produktions-Kette“ TNF-α → IL-1→ IL-6 → IL-10 ist somit
unterbrochen. Es ist demnach zu erwarten, dass auch die IL-10 Konzentration bei den
Knockout-Tieren herabgesetzt ist, oder erst zu einem späteren Zeitpunkt ansteigt, da die
Produktion auf Umwegen aktiviert werden muß. In den I/R-Gruppen konnte bei beiden
Knockout-Stämmen tatsächlich eine wesentlich geringere IL-10-Konzentration im Serum
festgestellt werden. Die Werte der laparotomierten Knockout-Tiere sind allerdings höher als
die der Wildtyp-Tiere. Eine Möglichkeit zur Herstellung der Zytokine IL-1, IL-6 und IL-10
trotz des vorhandenen Gendefektes besteht in der Aktivierung der sogenannten toll-like-
receptors. Sie befinden sich unter anderem auf Makrophagen, Granulozyten und
Dendritischen Zellen. Ihre Aufgabe besteht darin, Pathogene zu erkennen und daraufhin die
Produktion proinflammatorischer Zytokine wie IL-6, IL-12, IL-1, TNF-α und IFN-γ zu
induzieren (OZATO 2002).
Das IL-10 ist ein wichtiges antiinflammatorisches Zytokin, da es durch seine inhibitorische
Wirkung auf die Makrophagenfunktion eine Herunter-Regulation der Immunantwort
hervorruft. Zusätzlich verursacht IL-10 in TH1-Zellen eine Inhibition der IFNγ-Produktion.
Hauptsächlich T-Zellen und Makrophagen setzen es frei. Läuft eine Immunreaktion in
physiologischem Ausmaß ab, so wird, wie bereits oben beschrieben, zuerst TNF-α freigesetzt,
welches die Produktion weiterer proinflammatorischer Zytokine wie IL-1 und IL-6 anregt und
schließlich gleichzeitig eine Gegenregulation der Immunantwort durch die IL-10 Sekretion
auslöst (OPPENHEIM JJ. 2001).
Der Normalwert der Kontrolltiere für IL-10 in der vorliegenden Arbeit ist 0 pg/ml. Die
weiteren Ergebnisse zeigen, dass die IL-10 Konzentration besonders bei den WT 8 h nach I/R
ansteigt (16,40 pg/ml). Bei den Knockout-Tieren liegen die entsprechenden Werte hingegen
bei 0 pg/ml (T-) und 4,20 pg/ml (F-). Die Zytokin-Konzentration fällt 48 h nach I/R bei den
WT- und den T--Tieren ab. Anders hingegen verhält es sich bei den F--Tieren: Hier steigt der
Wert von 0 pg/ml auf 4,20 pg/ml. Es ist zu vermuten, dass die IL-10 Produktion bei den F--
Tieren im Vergleich zu den WT-Tieren durch das Fehlen der Rezeptoren in verringertem
Maße abläuft. Warum allerdings bei den T--Tieren erst 48 h nach dem I/R-Reiz eine IL-10
Sekretion meßbar ist, bleibt zu klären. Möglicherweise wird die IL-10 Freisetzung auf einem

Diskussion 106
anderen als dem oben beschriebenen Weg bewirkt, welcher mehr Zeit in Anspruch nimmt.
Erstaunlich ist jedoch, dass die F--Tiere, denen zusätzlich der Fcγ-RIII fehlt, mit einer
schnelleren IL-10 Produktion reagieren als die T--Tiere.
Beim Monocyte Chemotactic Peptide (MCP-1) handelt es sich um ein Zytokin, welches dem
IL-8 in der Struktur und Wirkung sehr ähnlich ist. Deswegen wurde es in der vorliegenden
Arbeit als Messgröße verwandt.
Beim MCP-1 handelt es sich um ein Zytokin, das hauptsächlich von stimulierten Monozyten,
Makrophagen, Endothelzellen, aber auch von verschiedenen anderen Zellen produziert wird.
Es hat eine proinflammatorische Wirkung, da es chemotaktisch auf alle bisher identifizierten
migratorischen Immunzellen wirkt. Zeitlich ist es jedoch dem IL-6 nachgeordnet
(OPPENHEIM JJ. 2001; VAN GRIENSVEN 2003). Für die Entstehung des I/R-Schadens ist
das MCP-1 ein besonders wichtiges Zytokin, weil es chemotaktisch auf neutrophile
Granulozyten wirkt und eine verstärkte Expression von Adhäsionsmolekülen verursacht.
Zusätzlich wandelt es das Rolling der PMN auf der Endothelwand in eine stabile Bindung
zwischen Zelle und Gefäßwand, wodurch eine Migration der Granulozyten erst ermöglicht
wird. Nach einem Kontakt der Endothelwand mit aktivierten Thrombozyten wird MCP-1
unter anderem direkt von den Gefäßwandzellen produziert. Ebenfalls wichtig für die
Pathogenese des I/R-Schadens ist die Tatsache, dass IL-8 spezifisch neutrophile Granulozyten
aktiviert, indem es einen Anstieg von Calcium im Zytosol, eine erhöhte Freisetzung von
Enzymen aus den Granula bewirkt und zusätzlich den Metabolismus der Sauerstoffradikale
heraufsetzt. Die Anwesenheit von IL-1 und TNF-α stimulieren die Freisetzung von MCP-1.
Der in dieser Arbeit ermittelte Normalwert der Kontrolltiere ist 45,53 pg/ml. Die Werte der
Laparotomie 48 h Gruppen steigen auf die maximal doppelte Konzentration an (bis 96,41
pg/ml). Liegt die Laparotomie nur 8 h zurück, so sind die Werte deutlich höher und liegen
zwischen 184,18 pg/ml (WT) und 380,60 pg/ml (T-). Es ist erkennbar, dass allein die
Laparotomiewunde das Immunsystem der Tiere aktiviert, indem es vermehrt MCP-1
ausschüttet, um so einer Infektion vorzubeugen. Neutrophile Granulozyten als
phagozytierende Zellen werden auf diese Weise schnell auf einen möglichen Infektionsherd
aufmerksam gemacht. Bereits nach 48 h nimmt die Produktion dieses Zytokins deutlich ab.

Diskussion 107
Weiterhin konnte beobachtet werden, dass die MCP-1 Konzentration der Gruppe WT I/R 8 h
auf etwa das 23-fache des Normalwertes der Kontrolltiere ansteigt (1062,74 pg/ml). Die
Werte der Knockout-Tiere sind im Vergleich hierzu deutlich geringer und steigen auf das
maximal 11-fache (T-: 480,78 pg/ml; F-: 358,53 pg/ml). Es müssen allerdings wie bereits
erwähnt Einschränkungen bei der Beurteilung gemacht werden, da rein-stämmige und
gemischt-stämmige Tiere miteinander verglichen wurden. Bereits 48 h nach I/R ist die MCP-
1 Konzentration aller Versuchstier-Stämme auf ähnliche Werte im Bereich zwischen 109,12
(F-) und 223,98 (T-) abgesunken.
Der Gendefekt scheint sich demnach auf die Höhe der MCP-1 Konzentration der
Versuchstiere auszuwirken. Wie bereits oben erwähnt stimuliert die Anwesenheit des TNF-α
die Produktion von MCP-1. Da dieser jedoch bei den Knockout-Tieren nur in geringerem
Maße produziert wird und das Zytokin aufgrund des Fehlens des TNF-RI nicht wirken kann,
scheint auch der Einfluß auf die Sekretion des MCP-1 gehemmt.
Von mehreren Autoren wurde beschrieben, dass das MCP-1 ein wichtiges Zytokin bei der
Entstehung des ARDS bei Traumapatienten ist. MATSUBARA et al. (1996) identifizierten
das IL-8 als einen Hauptchemoattractor für die neutrophilen Granulozyten in das
Lungengewebe. Sie konnten nachweisen, dass die IL-8-Konzentration mit der Zahl der PMN
in der BAL korreliert. Auch AGGARWAL et al. (2000) sehen einen Zusammenhang
zwischen der Anzahl neutrophiler Granulozyten im Lungengewebe und der Höhe der IL-8-
Konzentration. Da das Zytokin zusätzlich die Phagozytose, Degranulation und
Sauerstoffradikalfreisetzung der PMN verstärkt, ist eine Schädigung des Lungengewebes
nach der Migration nicht mehr zu verhindern. Ist der IL-8-Wert (neben IL-1β, IL-6 und
TNF-α) bei Traumapatienten permanent erhöht, so ist mit einem schlechten Outcome zu
rechnen (IKUTA 1996; MEDURI 1995b). KURDOWSKA et al. (2001) sehen jedoch keinen
prediktiven Wert für das Outcome des ARDS bei alleiniger Bestimmung der IL-8-
Konzentration. Es müsse der Komplex aus Zytokin und Anti-IL-8-Antikörpern betrachtet
werden. Bei verstorbenen Patienten seien die Werte für diesen Komplex besonders hoch.
In der vorliegenden Arbeit konnte festgestellt werden, dass ein Fehlen des TNF-RI oder des
TNF-RI in Verbindung mit dem Fcγ-RIII eine reduzierte IL-8-Freisetzung zur Folge hat. Dies
wiederum bewirkt eine verringerte Migration von neutrophilen Granulozyten und somit die
Ödembildung in Lunge und Leber.

Diskussion 108
In der vorliegenden Arbeit wurde das IL-12p70 stellvertretend für das IL-12 gemessen, da
dieses Zytokin aus einer p70 und einer r35 Untereinheit besteht. Das IL-12 wird hauptsächlich
von B-Lymphozyten produziert und stellt einen growth-factor für T- und NK-Zellen dar.
Damit löst es unter anderem die zelluläre Immunantwort aus. Außerdem ist das IL-12 in der
Lage, die Bildung von IFN-γ in peripheren Lymphozyten zu induzieren (OPPENHEIM JJ.
2001). Da die Produktion von IFN-γ und IL-12 eng miteinander verknüpft sind, konnten wie
bei dem IFN-γ auch für das IL-12 nur schwach variierende Konzentrationen im Serum der
Versuchstiere festgestellt werden und schwanken zwischen 0 und 24,98 pg/ml. Der
Normalwert der Kontrolltiere beträgt 0 pg/ml. Auch das IL-12 wird demnach nur in geringem
Maße nach einem Ischämie-/Reperfusions-Reiz gebildet und spielt somit bei der Entstehung
des I/R-Schadens keine entscheidende Rolle.
IFN-γ ist ein Zytokin, was hauptsächlich von T- und NK-Zellen produziert wird. Es hat
sowohl eine antivirale als auch eine antiparasitäre Wirkung, außerdem werden Makrophagen
durch dieses Zytokin aktiviert. Im Serum von Traumapatienten kann man häufig erhöhte
IFN-γ-Werte feststellen.
In der vorliegenden Arbeit fallen für dieses Zytokin sehr homogene Messwerte auf: Sie
variieren bei allen Gruppen zwischen 0 und 15,21 pg/ml. Der Normalwert der Kontrolltiere ist
0 pg/ml. Demnach ist keine Relevanz für die Entstehung des Ischämie/Reperfusions-Schadens
zu beobachten. Grund hierfür ist die Tatsache, dass besonders neutrophile Granulozyten für
die Organschäden verantwortlich gemacht werden können. Da IFN-γ jedoch hauptsächlich
von T-Zellen produziert wird, sind die Zytokinwerte erwartungsgemäß sehr niedrig. Eine
Beeinflussung der Messdaten aufgrund des Genotypes der Versuchstiere war ebenfalls nicht
festzustellen.
Wie bereits am Anfang der Diskussion erwähnt, müssen alle Ergebnisse unter
Berücksichtigung der Tatsache betrachtet werden, dass rein-stämmige Kontrolltiere mit
gemischt-stämmigen Versuchstieren verglichen wurden. Hierdurch könnten Unterschiede
beobachtet worden sein, die nicht auf das Fehlen bestimmter Gene, sondern auf den
Stammunterschied der Mäuse zurück zuführen sind.

Zusammenfassung 109
7 Zusammenfassung
Nina Zündel:
Der Einfluss von Fcγ-RIII und TNF-RIII auf den Respiratory Burst von neutrophilen
Granulozyten in Abhängigkeit vom Migrationsprozess in einem Ischämie-Reperfusions-
Modell der Maus
Die neutrophilen Granulozyten spielen eine entscheidende Rolle bei der Pathogenese des
Ischämie/Reperfusions-Schadens. Sie gelangen, von Zytokinen chemotaktisch geleitet, über
das Endothel ins Organparenchym, wo sie eine schädliche Wirkung ausüben und schließlich
zu einem Multiorgan-Dysfunktion-Syndrom führen können. Die Schädigung wird maßgeblich
von den Produkten des respiratory burstes hervorgerufen.
In der vorliegenden Arbeit soll mit Hilfe von Knockout-Mäusen die Rolle verschiedener
Zytokine und der Rezeptoren TNF-RI und Fcγ-RIII bei der Organschädigung untersucht
werden. Die Ergebnisse dieser Arbeit sind jedoch nur unter Vorbehalt zu interpretieren, da bei
den Versuchen rein-stämmige Kontrolltiere mit gemischt-stämmigen Knockout-Tieren
verglichen wurden. Es besteht deshalb die Möglichkeit, dass die beobachteten Effekte nicht
durch das Fehlen einzelner Gene, sondern durch den Stammunterschied der Versuchstiere
hervorgerufen wurden.
Die Ergebnisse zeigen eine Schädigung der Organe aufgrund von PMN-Infiltrationen und
Ödembildung. In der Frühphase nach Ischämie-Reperfusion ist eine besonders starke
Affektion der Lunge (gemessen anhand der Lungenpermeabilität, des interstitiellen Ödems,
der Granulozyteninfiltration im histologischen Präparat und der MPO-Aktivität) zu
beobachten. Sie ist bereits nach acht Stunden festzustellen und bleibt auch nach 48 Stunden
bestehen. Eine Schädigung der Leberzellen, ausgedrückt durch die hydropische Degeneration,
konnte besonders in der Spätphase nach Ischämie-Reperfusion beobachtet werden, wobei die
stärkste Degeneration nach 48 h auftrat. Bei beiden Knockout-Stämmen ist eine geringere
Beeinträchtigung der Organe zu bemerken als bei den WT-Tieren. Somit wirkt sich das
Fehlen eines oder beider Rezeptoren positiv auf die Situation der Organe aus. Zusätzlich
konnte bei den Knockout-Tieren eine signifikant geringere Sekretion der für den I/R-Schaden
wichtigen Zytokine TNF-α, MCP-1 und IL-6 beobachtet werden. Bedeutende Unterschiede

Zusammenfassung 110
zwischen den T--und F--Tieren wurden bei den jeweiligen Untersuchungen nicht festgestellt,
so dass ein additiver oder synergistischer Effekt beim Fehlen beider Rezeptoren
ausgeschlossen werden kann. Insgesamt ist ein statistisch signifikanter Zusammenhang
zwischen der Art des Traumas und dem Outcome zu verzeichnen. Tiere, die einer I/R
ausgesetzt waren, verstarben häufiger als Tiere, die lediglich einer Laparotomie oder Ischämie
unterworfen wurden. Zwischen den verschiedenen Stämmen und dem Outcome war kein
signifikanter Unterschied zu bemerken, jedoch wurde in den Knockout-Gruppen eine deutlich
geringere Letalitätsrate als bei den WT beobachtet. Die Ergebnisse zeigen, dass ein Fehlen
des TNF-RI durchaus einen protektiven Effekt vor der Ausbildung von Organschäden hat.
Der Fcγ-RIII hat für die Pathogenese des I/R-Schadens keine Bedeutung, möglicherweise ist
er jedoch an der Vorbeugung einer Sepsis beteiligt.

Summary 111
8 Summary
Nina Zündel:
The influence of Fcγ-RIII and TNF-RI on the respiratory burst of neutrophilic
granulocytes dependent on the migration process in a murine ischemia-reperfusion-
model
Neutrophilic granulocytes are very important for the pathogenesis of a damage caused by
ischemia-reperfusion. They get into the parenchyma through the endothelium conducted
chemotactically by cytokines, where they have a damaging effect and finally lead to a MODS.
The damage is substantially caused by the products of the respiratory burst.
The influence of different cytokines and the receptors TNF-RI and Fcγ-RIII on the organic
damage is to be investigated in the paper on hand with the help of knockout-mice. Because
pure-background controll animals were compared to mixed-background knockout-mice there
is a limitation in the interpretation of the results. It might be possible that the shown
differences are reasoned by the different type of background and not by the knocked out
genes.
The results show a damage of the organs caused by PMN-infiltration and formation of
oedemas. In an early phase after ischemia-reperfusion a very strong affection of the lung can
be observed (studied by the permeability of the capillaries in the lungs, the interstitial oedema,
the infiltration of PMN into the histological preparation and the MPO-activity). It can be
diagnosed even after 8 hours and continues to exist after 48 hours. A damage of the livercells
showed by the hydropic degeneration could be observed especially in the late phase post
trauma, whereas the strongest degeneration was found after 48 h. Both knockout-lines show a
lower interference of the organs than the wildtype-animals. Obviously the lack of one or both
receptors has a positive effect on the situation of the organs. In addition the knockout-animals
showed a significantly lower secretion of the cytokines TNF-α, MCP-1 and IL-6 causing the
I/R-damage. Eminent differences between the T-- and F--animals weren’t found during the
tests so that the lack of both of the receptors excludes an additive or synergistic effect.

Summary 112
Altogether there really is a statistically significant correlation between the kind of trauma and
the outcome. Animals which were exposed to a I/R died more frequently than those which
only underwent a laparotomy or ischemia. Between the different lines and the outcome no
significant difference was found, but the groups of both knockout-lines showed a clearly
recognizable lower mortality-rate than the WT. The results show that lacking the TNF-RI
absolutely has a protective effect on the development of the organic harm. However, the Fcγ-
RIII is without meaning for the pathogenesis of the I/R-damage but possibly it is necessary
during the development of sepsis.

Literaturverzeichnis 113
9 Literaturverzeichnis
AARDEN,L.A., P.LANDSDORP, und E.DE GROOT (1985): A growth factor B cell hybridomas produced by human monocytes. Lymphokines 10:175-185
ACKERMAN,G.A. (1971): The human neutrophilic myelocyte. A correlated phase and electron microscopic study. Z.Zellforsch.Mikrosk.Anat. 121:153-170
AKIRA,S., T.TAGA, und T.KISHIMOTO (1993): Interleukin-6 in biology and medicine. Adv.Immunol. 54:1-78
ALBERTS,K.A., B.M.BELLANDER, und G.MODIN (1999): Improved trauma care after reorganisation: a retrospective analysis. Eur.J.Surg. 165:426-430
ANDERSON,C.L. und G.N.ABRAHAM (1980): Characterization of the Fc receptor for IgG on a human macrophage cell line, U937. J.Immunol. 125:2735-2741
ANDERSON,D.C. und T.A.SPRINGER (1987): Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu.Rev.Med. 38:175-194
ARFORS,K.E., C.LUNDBERG, L.LINDBOM, K.LUNDBERG, P.G.BEATTY, und J.M.HARLAN (1987): A monoclonal antibody to the membrane glycoprotein complex CD18 inhibits polymorphonuclear leukocyte accumulation and plasma leakage in vivo. Blood 69:338-340
ASHBAUGH,D.G., D.B.BIGELOW, T.L.PETTY, und B.E.LEVINE (1967): Acute respiratory distress in adults. Lancet 2:319-323
AUFMKOLK,M., E.DOMINGUEZ, R.LETSCH, F.NEUDECK, und W.NIEBEL (1996): [Results of peripheral arterial vascular injury in polytraumatized patients]. Unfallchirurg 99:555-560
AUST,S.D. und SVINGEN B.A. (1982): The role of iron in enzymatic lipid peroxidation. In: Free radicals in biology by W.A.Pryor (Hrsg.)

Literaturverzeichnis 114
Academic, New York, S. 1-28
BABINEAU,T.J., P.MARCELLO, W.SWAILS, A.KENLER, B.BISTRIAN, und R.A.FORSE (1994): Randomized phase I/II trial of a macrophage-specific immunomodulator (PGG-glucan) in high-risk surgical patients. Ann.Surg. 220:601-609
BAINTON,D.F. und M.G.FARQUHAR (1966): Origin of granules in polymorphonuclear leukocytes. Two types derived from opposite faces of the Golgi complex in developing granulocytes. J.Cell Biol. 28:277-301
BAINTON,D.F., J.L.ULLYOT, und M.G.FARQUHAR (1971): The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J.Exp.Med. 134:907-934
BARGATZE,R.F., S.KURK, G.WATTS, T.K.KISHIMOTO, C.A.SPEER, und M.A.JUTILA (1994): In vivo and in vitro functional examination of a conserved epitope of L- and E-selectin crucial for leukocyte-endothelial cell interactions. J.Immunol. 152:5814-5825
BARIE,P.S. und R.J.MULLINS (1988): Experimental methods in the pathogenesis of limb ischemia. J.Surg.Res. 44:284-307
BAUE,A.E. (1975): Multiple, progressive, or sequential systems failure. A syndrome of the 1970s. Arch.Surg. 110:779-781
BAUER,M. und I.MARZI (1994): [The leukocyte-endothelial interaction as an expression of a hepatic inflammation reaction during an experimental shock syndrome]. Anasthesiol.Intensivmed.Notfallmed.Schmerzther. 29:46-50
BELKIN,M., W.L.LAMORTE, J.G.WRIGHT, und R.W.HOBSON (1989): The role of leukocytes in the pathophysiology of skeletal muscle ischemic injury. J.Vasc.Surg. 10:14-18
BELLOMO,R. (1992): The cytokine network in the critically ill. Anaesth.Intensive Care 20:288-302
BERNARD,G.R. (1990): Potential of N-acetylcysteine as treatment for the adult respiratory distress syndrome. Eur.Respir.J.Suppl 11:496s-498s

Literaturverzeichnis 115
BERNARD,G.R., A.ARTIGAS, K.L.BRIGHAM, J.CARLET, K.FALKE, L.HUDSON, M.LAMY, J.R.LEGALL, A.MORRIS, und R.SPRAGG (1994): Report of the American-European Consensus conference on acute respiratory distress syndrome: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus Committee. J.Crit Care 9:72-81
BEUTLER,B., I.W.MILSARK, und A.C.CERAMI (1985a): Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229:869-871
BEUTLER,B.A., I.W.MILSARK, und A.CERAMI (1985b): Cachectin/tumor necrosis factor: production, distribution, and metabolic fate in vivo. J.Immunol. 135:3972-3977
BIELSKI,B.H. und G.G.SHIUE (1979): Reaction rates of superoxide radicals with the essential amino acids. In: Oxygen free radicals and tissue damage by Ciba Foundation New York (Hrsg.) Elsevier, Amsterdam, S. 43-56
BILLING,A. (1993): Immunologische Probleme der Peritonitis. In: Peritonitis by R.Härig (Hrsg.) Thieme, Stuttgart, S. 25-31
BONAVIDA,B. (1998): Tumor necrosis Factor- cachectin and related cytokines. In: Karger, Basel, S. 43-64
BONE,R.C. (1996a): Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann.Intern.Med. 125:680-687
BONE,R.C. (1996b): Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 24:1125-1128
BONE,R.C. (1996c): Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med. 24:163-172
BONE,R.C. (1996d): Why sepsis trials fail. JAMA 276:565-566

Literaturverzeichnis 116
BONE,R.C., R.A.BALK, F.B.CERRA, R.P.DELLINGER, A.M.FEIN, W.A.KNAUS, R.M.SCHEIN, und W.J.SIBBALD (1992): Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644-1655
BOTHA,A.J., F.A.MOORE, E.E.MOORE, F.J.KIM, A.BANERJEE, und V.M.PETERSON (1995a): Postinjury neutrophil priming and activation: an early vulnerable window. Surgery 118:358-364
BOTHA,A.J., F.A.MOORE, E.E.MOORE, A.SAUAIA, A.BANERJEE, und V.M.PETERSON (1995b): Early neutrophil sequestration after injury: a pathogenic mechanism for multiple organ failure. J.Trauma 39:411-417
BRETT,J., H.GERLACH, P.NAWROTH, S.STEINBERG, G.GODMAN, und D.STERN (1989): Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J.Exp.Med. 169:1977-1991
BROOKS,D.G., W.Q.QIU, A.D.LUSTER, und J.V.RAVETCH (1989): Structure and expression of human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J.Exp.Med. 170:1369-1385
BROUCKAERT,P. und W.FIERS (1996): Tumor necrosis factor and the systemic inflammatory response syndrome. Curr.Top.Microbiol.Immunol. 216:167-187
BUDELMANN,G. (1969): [Hugo Schottmuller, 1867-1936. The problem of sepsis]. Internist (Berl) 10:92-101
BURCHARDI,H. (1987): [Acute lung failure--a critical analysis]. Langenbecks Arch.Chir 372:53-58
BURGER,R.M., A.R.BERKOWITZ, J.PEISACH, und S.B.HORWITZ (1980): Origin of malondialdehyde from DNA degraded by Fe(II) x bleomycin. J.Biol.Chem. 255:11832-11838
BURKE,J.F., H.PONTOPPIDAN, und C.E.WELCH (1963): HIGH OUTPUT RESPIRATORY FAILURE: AN IMPORTANT CAUSE OF DEATH ASCRIBED TO PERITONITIS OR ILEUS. Ann.Surg. 158:581-595

Literaturverzeichnis 117
BUTCHER,E.C. (1991): Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67:1033-1036
CALANDRA,T., J.GERAIN, D.HEUMANN, J.D.BAUMGARTNER, und M.P.GLAUSER (1991): High circulating levels of interleukin-6 in patients with septic shock: evolution during sepsis, prognostic value, and interplay with other cytokines. The Swiss-Dutch J5 Immunoglobulin Study Group. Am.J.Med. 91:23-29
CARDEN,D.L., J.K.SMITH, und R.J.KORTHUIS (1990): Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ.Res. 66:1436-1444
CARRICO,C.J., J.L.MEAKINS, J.C.MARSHALL, D.FRY, und R.V.MAIER (1986): Multiple-organ-failure syndrome. Arch.Surg. 121:196-208
CASEY,L.C., R.A.BALK, und R.C.BONE (1993): Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann.Intern.Med. 119:771-778
CERASOLI,F., JR., P.J.MCKENNA, D.L.ROSOLIA, K.H.ALBERTINE, S.P.PETERS, und M.H.GEE (1990): Superoxide anion release from blood and bone marrow neutrophils is altered by endotoxemia. Circ.Res. 67:154-165
CERRA,F.B., M.A.MADDAUS, D.L.DUNN, C.L.WELLS, N.N.KONSTANTINIDES, S.L.LEHMANN, und H.J.MANN (1992): Selective gut decontamination reduces nosocomial infections and length of stay but not mortality or organ failure in surgical intensive care unit patients. Arch.Surg. 127:163-167
CHEN,X. und N.V.CHRISTOU (1998): Protective effect of plasma in polymorphonuclear neutrophil-mediated cytotoxicity of endothelial cells in the systemic inflammatory response syndrome. J.Leukoc.Biol. 63:68-74
CIPOLLE,M.D., M.D.PASQUALE, und F.B.CERRA (1993): Secondary organ dysfunction. From clinical perspectives to molecular mediators. Crit Care Clin. 9:261-298
CLERICI,M. und G.M.SHEARER (1993): A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol.Today 14:107-111

Literaturverzeichnis 118
CLOWES,G.H., JR., W.ZUSCHNEID, S.DRAGACEVIC, und M.TURNER (1968): The nonspecific pulmonary inflammatory reactions leading to respiratory failure after shock, gangrene, and sepsis. J.Trauma 8:899-914
COLLETTI,L.M., D.G.REMICK, G.D.BURTCH, S.L.KUNKEL, R.M.STRIETER, und D.A.CAMPBELL, JR. (1990): Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J.Clin.Invest 85:1936-1943
DAMAS,P., A.REUTER, P.GYSEN, J.DEMONTY, M.LAMY, und P.FRANCHIMONT (1989): Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit Care Med. 17:975-978
DAVIES,M.G. und P.O.HAGEN (1997): Systemic inflammatory response syndrome. Br.J.Surg. 84:920-935
DEBETS,J.M., R.KAMPMEIJER, M.P.VAN DER LINDEN, W.A.BUURMAN, und C.J.VAN DER LINDEN (1989): Plasma tumor necrosis factor and mortality in critically ill septic patients. Crit Care Med. 17:489-494
DEITCH,E.A. (1992): Multiple organ failure. Pathophysiology and potential future therapy. Ann.Surg. 216:117-134
DEITCH,E.A., D.XU, L.FRANKO, A.AYALA, und I.H.CHAUDRY (1994): Evidence favoring the role of the gut as a cytokine-generating organ in rats subjected to hemorrhagic shock. Shock 1:141-145
DEMLING,R.H. (1985): Mechanisms of pulmonary vascular injury in sepsis. In: The pulmonary circulation and acute lung injury by S.I.Said (Hrsg.) Futura Publishing Co.Inc., Mount Kisco, S. 403-427
DILLON,S.B., M.W.VERGHESE, und R.SNYDERMAN (1988): Signal transduction in cells following binding of chemoattractants to membrane receptors. Virchows Arch.B Cell Pathol.Incl.Mol.Pathol. 55:65-80
DINARELLO,C.A. (1996): Cytokines as mediators in the pathogenesis of septic shock. Curr.Top.Microbiol.Immunol. 216:133-165

Literaturverzeichnis 119
DINARELLO,C.A., J.A.GELFAND, und S.M.WOLFF (1993): Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. JAMA 269:1829-1835
DOUGHTY,L.A., S.S.KAPLAN, und J.A.CARCILLO (1996): Inflammatory cytokine and nitric oxide responses in pediatric sepsis and organ failure. Crit Care Med. 24:1137-1143
DUBAYBO,B.A. und R.W.CARLSON (1988): Post-infectious ARDS: mechanisms of lung injury and repair. Crit Care Clin. 4:229-243
ECHTENACHER,B., W.FALK, D.N.MANNEL, und P.H.KRAMMER (1990): Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J.Immunol. 145:3762-3766
ECHTENACHER,B., L.HULTNER, und D.N.MANNEL (1995): Cellular and molecular mechanisms of TNF protection in septic peritonitis. J.Inflamm. 47:85-89
EL BARBARY,M. und K.S.KHABAR (2002): Soluble tumor necrosis factor receptor p55 predicts cytokinemia and systemic inflammatory response after cardiopulmonary bypass. Crit Care Med. 30:1712-1716
ELSNER,J., J.ROESLER, A.EMMENDORFFER, C.ZEIDLER, M.L.LOHMANN-MATTHES, und K.WELTE (1992): Altered function and surface marker expression of neutrophils induced by rhG-CSF treatment in severe congenital neutropenia. Eur.J.Haematol. 48:10-19
ENK,A.H., V.L.ANGELONI, M.C.UDEY, und S.I.KATZ (1993): Inhibition of Langerhans cell antigen-presenting function by IL-10. A role for IL-10 in induction of tolerance. J.Immunol. 151:2390-2398
ERTEL,W., J.P.KREMER, J.KENNEY, U.STECKHOLZER, D.JARRAR, O.TRENTZ, und F.W.SCHILDBERG (1995): Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood 85:1341-1347
FEISTER,A.J., B.BROWDER, H.E.WILLIS, T.MOHANAKUMAR, und S.RUDDY (1988): Pertussis toxin inhibits human neutrophil responses mediated by the 42-kilodalton IgG Fc receptor. J.Immunol. 141:228-233

Literaturverzeichnis 120
FIORENTINO,D.F., A.ZLOTNIK, P.VIEIRA, T.R.MOSMANN, M.HOWARD, K.W.MOORE, und A.O'GARRA (1991): IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J.Immunol. 146:3444-3451
FLAHERTY,J.T. (1991): Myocardial injury mediated by oxygen free radicals. Am.J.Med. 91:79S-85S
FUJISHIMA,S. und N.AIKAWA (1995): Neutrophil-mediated tissue injury and its modulation. Intensive Care Med. 21:277-285
FUKUSHIMA,R., J.W.ALEXANDER, J.Z.WU, J.X.MAO, K.SZCZUR, A.M.STEPHENS, J.D.OGLE, und C.K.OGLE (1994): Time course of production of cytokines and prostaglandin E2 by macrophages isolated after thermal injury and bacterial translocation. Circ.Shock 42:154-162
FURIE,M.B. und D.D.MCHUGH (1989): Migration of neutrophils across endothelial monolayers is stimulated by treatment of the monolayers with interleukin-1 or tumor necrosis factor-alpha. J.Immunol. 143:3309-3317
FURIE,M.B., B.L.NAPRSTEK, und S.C.SILVERSTEIN (1987): Migration of neutrophils across monolayers of cultured microvascular endothelial cells. An in vitro model of leucocyte extravasation. J.Cell Sci. 88 ( Pt 2):161-175
GAMBLE,J.R., J.M.HARLAN, S.J.KLEBANOFF, und M.A.VADAS (1985): Stimulation of the adherence of neutrophils to umbilical vein endothelium by human recombinant tumor necrosis factor. Proc.Natl.Acad.Sci.U.S.A 82:8667-8671
GANDO,S., T.KAMEUE, S.NANZAKI, und Y.NAKANISHI (1996): Disseminated intravascular coagulation is a frequent complication of systemic inflammatory response syndrome. Thromb.Haemost. 75:224-228
GASIC,A.C., G.MCGUIRE, S.KRATER, A.I.FARHOOD, M.A.GOLDSTEIN, C.W.SMITH, M.L.ENTMAN, und A.A.TAYLOR (1991): Hydrogen peroxide pretreatment of perfused canine vessels induces ICAM-1 and CD18-dependent neutrophil adherence. Circulation 84:2154-2166

Literaturverzeichnis 121
GASTINNE,H., M.WOLFF, F.DELATOUR, F.FAURISSON, und S.CHEVRET (1992): A controlled trial in intensive care units of selective decontamination of the digestive tract with nonabsorbable antibiotics. The French Study Group on Selective Decontamination of the Digestive Tract. N.Engl.J.Med. 326:594-599
GEBHARD,F., H.PFETSCH, G.STEINBACH, W.STRECKER, L.KINZL, und U.B.BRUCKNER (2000): Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch.Surg. 135:291-295
Gerdemann, R. Prostanoide bei Pferden mit Ileus - Nachweis und medikamentöse Beeinflussungmit Flunixin-Meglumin. 1995. Tierärztliche Hochschule Hannover, Klinik für Pferde und Veterinärmedizinische Fakultät der Universität Leipzig, Institut für Pharmakologie, Pharmazie und Toxikologie. Ref Type: Thesis/Dissertation
GOLDMAN,G., R.WELBOURN, J.M.KLAUSNER, L.KOBZIK, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1992): Mast cells and leukotrienes mediate neutrophil sequestration and lung edema after remote ischemia in rodents. Surgery 112:578-586
GOMEZ-JIMENEZ,J., A.SALGADO, M.MOURELLE, M.C.MARTIN, R.M.SEGURA, R.PERACAULA, und S.MONCADA (1995): L-arginine: nitric oxide pathway in endotoxemia and human septic shock. Crit Care Med. 23:253-258
GORIS,R.J. und J.DRAASIMA (2003): Causes of death after blunt trauma. Trauma 22:141-146
GRECH,E.D., C.M.BELLAMY, M.J.JACKSON, R.A.MUIRHEAD, E.B.FARAGHER, und D.R.RAMSDALE (1994): Free-radical activity after primary coronary angioplasty in acute myocardial infarction. Am.Heart J. 127:1443-1449
GRELL,M., F.M.BECKE, H.WAJANT, D.N.MANNEL, und P.SCHEURICH (1998): TNF receptor type 2 mediates thymocyte proliferation independently of TNF receptor type 1. Eur.J.Immunol. 28:257-263
GRISWOLD,J. und R.V.MAIER (1988): Neutrophil phagocytosis during endotoxin-induced lung injury. J.Surg.Res. 44:417-424
GUYRE,P.M., A.S.CAMPBELL, W.D.KNIFFIN, und M.W.FANGER (1990): Monocytes and polymorphonuclear neutrophils of patients with streptococcal pharyngitis express increased numbers of type I IgG Fc receptors. J.Clin.Invest 86:1892-1896

Literaturverzeichnis 122
HACK,C.E., E.R.DE GROOT, R.J.FELT-BERSMA, J.H.NUIJENS, R.J.STRACK VAN SCHIJNDEL, A.J.EERENBERG-BELMER, L.G.THIJS, und L.A.AARDEN (1989): Increased plasma levels of interleukin-6 in sepsis. Blood 74:1704-1710
HARLAN,J.M. (1987): Neutrophil-mediated vascular injury. Acta Med.Scand.Suppl 715:123-129
HELFGOTT,D.C., S.B.TATTER, U.SANTHANAM, R.H.CLARICK, N.BHARDWAJ, L.T.MAY, und P.B.SEHGAL (1989): Multiple forms of IFN-beta 2/IL-6 in serum and body fluids during acute bacterial infection. J.Immunol. 142:948-953
HELLER,T., J.E.GESSNER, R.E.SCHMIDT, A.KLOS, W.BAUTSCH, und J.KOHL (1999): Cutting edge: Fc receptor type I for IgG on macrophages and complement mediate the inflammatory response in immune complex peritonitis. J.Immunol. 162:5657-5661
HENSLER,T., S.SAUERLAND, B.BOUILLON, M.RAUM, D.RIXEN, H.J.HELLING, J.ANDERMAHR, und E.A.NEUGEBAUER (2002): Association between injury pattern of patients with multiple injuries and circulating levels of soluble tumor necrosis factor receptors, interleukin-6 and interleukin-10, and polymorphonuclear neutrophil elastase. J.Trauma 52:962-970
HERNANDEZ,L.A., M.B.GRISHAM, B.TWOHIG, K.E.ARFORS, J.M.HARLAN, und D.N.GRANGER (1987): Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am.J.Physiol 253:H699-H703
HEYWORTH,P.G. und J.A.BADWEY (1990): Protein phosphorylation associated with the stimulation of neutrophils. Modulation of superoxide production by protein kinase C and calcium. J.Bioenerg.Biomembr. 22:1-26
HIRANO,T. (1994): Interleukin 6. In: The cytokine Handbook 2nd ed. Academic Press, New York, S. 145-172
HOOVER,R.L., J.M.ROBINSON, und M.J.KARNOVSKY (1987): Adhesion of polymorphonuclear leukocytes to endothelium enhances the efficiency of detoxification of oxygen-free radicals. Am.J.Pathol. 126:258-268

Literaturverzeichnis 123
HUBL,W., G.WOLFBAUER, J.STREICHER, S.ANDERT, G.STANEK, S.FITZAL, und P.M.BAYER (1999): Differential expression of tumor necrosis factor receptor subtypes on leukocytes in systemic inflammatory response syndrome. Crit Care Med. 27:319-324
HUIZINGA,T.W., M.DE HAAS, M.KLEIJER, J.H.NUIJENS, D.ROOS, und A.E.DEM BORNE (1990): Soluble Fc gamma receptor III in human plasma originates from release by neutrophils. J.Clin.Invest 86:416-423
HUIZINGA,T.W., C.E.VAN DER SCHOOT, C.JOST, R.KLAASSEN, M.KLEIJER, A.E.DEM BORNE, D.ROOS, und P.A.TETTEROO (1988): The PI-linked receptor FcRIII is released on stimulation of neutrophils. Nature 333:667-669
HUIZINGA,T.W., F.VAN KEMENADE, L.KOENDERMAN, K.M.DOLMAN, A.E.DEM BORNE, P.A.TETTEROO, und D.ROOS (1989): The 40-kDa Fc gamma receptor (FcRII) on human neutrophils is essential for the IgG-induced respiratory burst and IgG-induced phagocytosis. J.Immunol. 142:2365-2369
IBBOTSON,G.C. und J.L.WALLACE (1989): Inhibitory effects of dexamethasone in endotoxic shock and its relation to PAF-acether synthesis in the gastrointestinal tract and lung. J.Lipid Mediat. 1:273-282
IKUTA,N., H.TANIGUCHI, Y.KONDOH, K.TAKAGI, und T.HAYAKAWA (1996): Sustained high levels of circulatory interleukin-8 are associated with a poor outcome in patients with adult respiratory distress syndrome. Intern.Med. 35:855-860
JOHNSON,J., B.MEYRICK, G.JESMOK, und K.L.BRIGHAM (1989): Human recombinant tumor necrosis factor alpha infusion mimics endotoxemia in awake sheep. J.Appl.Physiol 66:1448-1454
JONAS,E., A.DWENGER, B.LUEKEN, und U.BOEHME (1991): Simultaneous measurement of endothelial cell damage, elastase release and chemiluminescence response during interaction between polymorphonuclear leukocytes and endothelial cells. J.Biolumin.Chemilumin. 6:19-27
Kayser, F. H., Bienz, K. A., Eckert, J., and Zinkernagel, R. M. Medizinische Mikrobiologie. 9, 44. 1998. Stuttgart, Georg Thieme Verlag.

Literaturverzeichnis 124
KAZUI,M., K.A.ANDREONI, G.M.WILLIAMS, B.A.PERLER, G.B.BULKLEY, C.BEATTIE, R.T.DONHAM, S.S.SEHNERT, J.F.BURDICK, und T.H.RISBY (1994): Visceral lipid peroxidation occurs at reperfusion after supraceliac aortic cross-clamping. J.Vasc.Surg. 19:473-477
KIMBERLY,R.P., J.W.AHLSTROM, M.E.CLICK, und J.C.EDBERG (1990): The glycosyl phosphatidylinositol-linked Fc gamma RIIIPMN mediates transmembrane signaling events distinct from Fc gamma RII. J.Exp.Med. 171:1239-1255
KINDT,G.C., J.E.GADEK, und J.E.WEILAND (1991): Initial recruitment of neutrophils to alveolar structures in acute lung injury. J.Appl.Physiol 70:1575-1585
KISHIMOTO,T., M.HIBI, M.MURAKAMI, M.NARAZAKI, M.SAITO, und T.TAGA (1992): The molecular biology of interleukin 6 and its receptor. Ciba Found.Symp. 167:5-16
KLAUSNER,J.M., H.ANNER, I.S.PATERSON, L.KOBZIK, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1988): Lower torso ischemia-induced lung injury is leukocyte dependent. Ann.Surg. 208:761-767
KLAUSNER,J.M., I.S.PATERSON, G.GOLDMAN, L.KOBZIK, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1989): Thromboxane A2 mediates increased pulmonary microvascular permeability following limb ischemia. Circ.Res. 64:1178-1189
KNAPP,W., B.DORKEN, P.RIEBER, R.E.SCHMIDT, H.STEIN, und A.E.DEM BORNE (1989): CD antigens 1989. Blood 74:1448-1450
KORTHUIS,R.J., M.B.GRISHAM, und D.N.GRANGER (1988): Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am.J.Physiol 254:H823-H827
KORTHUIS,R.J., J.K.SMITH, und D.L.CARDEN (1989): Hypoxic reperfusion attenuates postischemic microvascular injury. Am.J.Physiol 256:H315-H319
KREMER,J.P., D.JARRAR, U.STECKHOLZER, und W.ERTEL (1996): Interleukin-1, -6 and tumor necrosis factor-alpha release is down-regulated in whole blood from septic patients. Acta Haematol. 95:268-273

Literaturverzeichnis 125
KREUZFELDER,E., T.JOKA, H.O.KEINECKE, U.OBERTACKE, K.P.SCHMIT-NEUERBURG, J.A.NAKHOSTEEN, D.PAAR, und N.SCHEIERMANN (1988): Adult respiratory distress syndrome as a specific manifestation of a general permeability defect in trauma patients. Am.Rev.Respir.Dis. 137:95-99
KUIJPERS,T.W., B.C.HAKKERT, M.H.HART, und D.ROOS (1992): Neutrophil migration across monolayers of cytokine-prestimulated endothelial cells: a role for platelet-activating factor and IL-8. J.Cell Biol. 117:565-572
KUROSE,I., D.C.ANDERSON, M.MIYASAKA, T.TAMATANI, J.C.PAULSON, R.F.TODD, J.R.RUSCHE, und D.N.GRANGER (1994): Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ.Res. 74:336-343
LEMESHOW,S., D.TERES, J.KLAR, J.S.AVRUNIN, S.H.GEHLBACH, und J.RAPOPORT (1993): Mortality Probability Models (MPM II) based on an international cohort of intensive care unit patients. JAMA 270:2478-2486
LEY,K., P.GAEHTGENS, C.FENNIE, M.S.SINGER, L.A.LASKY, und S.D.ROSEN (1991): Lectin-like cell adhesion molecule 1 mediates leukocyte rolling in mesenteric venules in vivo. Blood 77:2553-2555
LOPPNOW,H. und P.LIBBY (1989): Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol. 122:493-503
LUCAS,C.E., A.M.LEDGERWOOD, W.J.RACHWAL, D.GRABOW, und J.M.SAXE (1991): Colloid oncotic pressure and body water dynamics in septic and injured patients. J.Trauma 31:927-931
MA,X.L., A.S.WEYRICH, D.J.LEFER, M.BUERKE, K.H.ALBERTINE, T.K.KISHIMOTO, und A.M.LEFER (1993): Monoclonal antibody to L-selectin attenuates neutrophil accumulation and protects ischemic reperfused cat myocardium. Circulation 88:649-658
MACKAY,F., H.LOETSCHER, D.STUEBER, G.GEHR, und W.LESSLAUER (1993): Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J.Exp.Med. 177:1277-1286
MACKAY,F., J.ROTHE, H.BLUETHMANN, H.LOETSCHER, und W.LESSLAUER (1994): Differential responses of fibroblasts from wild-type and TNF-R55-deficient mice to mouse and human TNF-alpha activation.

Literaturverzeichnis 126
J.Immunol. 153:5274-5284
MALIK,A.B. (1985): Mediators of pulmonary vascular injury and edema after thrombin. In: The pulmonary circulation and acute lung injury by S.I.Said (Hrsg.) Futura Publishing Co. Inc., Mount Kisco, S. 429-454
MANSHIP,L., R.D.MCMILLIN, und J.J.BROWN (1984): The influence of sepsis and multisystem and organ failure on mortality in the surgical intensive care unit. Am.Surg. 50:94-101
MANTHOUS,C.A., J.B.HALL, und R.W.SAMSEL (1993): Endotoxin in human disease. Part 2: Biologic effects and clinical evaluations of anti-endotoxin therapies. Chest 104:1872-1881
MARCUS,B.C., C.W.WYBLE, K.L.HYNES, und B.L.GEWERTZ (1996): Cytokine-induced increases in endothelial permeability occur after adhesion molecule expression. Surgery 120:411-416
MARSHALL,J.C., D.J.COOK, N.V.CHRISTOU, G.R.BERNARD, C.L.SPRUNG, und W.J.SIBBALD (1995): Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 23:1638-1652
MARUO,N., I.MORITA, M.SHIRAO, und S.MUROTA (1992): IL-6 increases endothelial permeability in vitro. Endocrinology 131:710-714
MATSUMOTO,M., S.MARIATHASAN, M.H.NAHM, F.BARANYAY, J.J.PESCHON, und D.D.CHAPLIN (1996): Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science 271:1289-1291
MEDURI,G.U., S.HEADLEY, G.KOHLER, F.STENTZ, E.TOLLEY, R.UMBERGER, und K.LEEPER (1995a): Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107:1062-1073
MEDURI,G.U., G.KOHLER, S.HEADLEY, E.TOLLEY, F.STENTZ, und A.POSTLETHWAITE (1995b): Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest 108:1303-1314

Literaturverzeichnis 127
MILLS,C.D., M.D.CALDWELL, und D.S.GANN (1989): Evidence of a plasma-mediated "window" of immunodeficiency in rats following trauma. J.Clin.Immunol. 9:139-150
MIYAUCHI,T., Y.TOMOBE, R.SHIBA, T.ISHIKAWA, M.YANAGISAWA, S.KIMURA, Y.SUGISHITA, I.ITO, K.GOTO, und T.MASAKI (1990): Involvement of endothelin in the regulation of human vascular tonus. Potent vasoconstrictor effect and existence in endothelial cells. Circulation 81:1874-1880
MOLDAWER,L.L. (1994): Biology of proinflammatory cytokines and their antagonists. Crit Care Med. 22:S3-S7
MOORE,F.A. (1999): The role of the gastrointestinal tract in postinjury multiple organ failure. Am.J.Surg. 178:449-453
MOORE,K.W., A.O'GARRA, M.R.DE WAAL, P.VIEIRA, und T.R.MOSMANN (1993): Interleukin-10. Annu.Rev.Immunol. 11:165-190
MOSMANN,T.R. (1994): Properties and functions of interleukin-10. Adv.Immunol. 56:1-26
MULLIGAN,M.S., A.A.VAPORCIYAN, M.MIYASAKA, T.TAMATANI, und P.A.WARD (1993): Tumor necrosis factor alpha regulates in vivo intrapulmonary expression of ICAM-1. Am.J.Pathol. 142:1739-1749
NATHAN,C., S.SRIMAL, C.FARBER, E.SANCHEZ, L.KABBASH, A.ASCH, J.GAILIT, und S.D.WRIGHT (1989): Cytokine-induced respiratory burst of human neutrophils: dependence on extracellular matrix proteins and CD11/CD18 integrins. J.Cell Biol. 109:1341-1349
NAVARRO,S., N.DEBILI, J.F.BERNAUDIN, W.VAINCHENKER, und J.DOLY (1989): Regulation of the expression of IL-6 in human monocytes. J.Immunol. 142:4339-4345
NAWROTH,P.P., I.BANK, D.HANDLEY, J.CASSIMERIS, L.CHESS, und D.STERN (1986a): Tumor necrosis factor/cachectin interacts with endothelial cell receptors to induce release of interleukin 1. J.Exp.Med. 163:1363-1375

Literaturverzeichnis 128
NAWROTH,P.P. und D.M.STERN (1986b): Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J.Exp.Med. 163:740-745
NEUHOF,H. (1991): Actions and interactions of mediator systems and mediators in the pathogenesis of ARDS and multiorgan failure. Acta Anaesthesiol.Scand.Suppl 95:7-13
NEUMANN,B., A.LUZ, K.PFEFFER, und B.HOLZMANN (1996): Defective Peyer's patch organogenesis in mice lacking the 55-kD receptor for tumor necrosis factor. J.Exp.Med. 184:259-264
O'NEILL,P.J., L.M.COBB, A.AYALA, M.H.MORRISON, und I.H.CHAUDRY (1994): Aggressive fluid resuscitation following intestinal ischemia-reperfusion in immature rats prevents metabolic derangements and down regulates interleukin-6 release. Shock 1:381-387
OPPENHEIM,J.J. (2001): Cytokine reference: a compendium of cytokines and other mediators. San Diego. ORY,P.A., M.R.CLARK, E.E.KWOH, S.B.CLARKSON, und I.M.GOLDSTEIN (1989): Sequences of complementary DNAs that encode the NA1 and NA2 forms of Fc receptor III on human neutrophils. J.Clin.Invest 84:1688-1691
OZATO,K., H.TSUJIMURA, und T.TAMURA (2002): Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques Suppl:66-8, 70, 72
PALMBLAD,J., C.L.MALMSTEN, A.M.UDEN, O.RADMARK, L.ENGSTEDT, und B.SAMUELSSON (1981): Leukotriene B4 is a potent and stereospecific stimulator of neutrophil chemotaxis and adherence. Blood 58:658-661
PAPE,H., M.STALP, M.GRIENSVEN, A.WEINBERG, M.DAHLWEIT, und H.TSCHERNE (1999): [Optimal timing for secondary surgery in polytrauma patients: an evaluation of 4,314 serious-injury cases]. Chirurg 70:1287-1293
PAPE,H.C., A.DWENGER, M.GROTZ, V.KAEVER, R.NEGATSCH, W.KLEEMANN, G.REGEL, J.A.STURM, und H.TSCHERNE (1994): Does the reamer type influence the degree of lung dysfunction after femoral nailing following severe trauma? An animal study. J.Orthop.Trauma 8:300-309

Literaturverzeichnis 129
PARKS,D.A. und D.N.GRANGER (1986): Contributions of ischemia and reperfusion to mucosal lesion formation. Am.J.Physiol 250:G749-G753
PESCHON,J.J., D.S.TORRANCE, K.L.STOCKING, M.B.GLACCUM, C.OTTEN, C.R.WILLIS, K.CHARRIER, P.J.MORRISSEY, C.B.WARE, und K.M.MOHLER (1998): TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J.Immunol. 160:943-952
PETRAK,R.A., R.A.BALK, und R.C.BONE (1989): Prostaglandins, cyclo-oxygenase inhibitors, and thromboxane synthetase inhibitors in the pathogenesis of multiple systems organ failure. Crit Care Clin. 5:303-314
PFEFFER,K., T.MATSUYAMA, T.M.KUNDIG, A.WAKEHAM, K.KISHIHARA, A.SHAHINIAN, K.WIEGMANN, P.S.OHASHI, M.KRONKE, und T.W.MAK (1993): Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457-467
PILLER,N.B. (1980): Lymphoedema, macrophages and benzopyrones. Lymphology 13:109-119
PLATZER,C., C.MEISEL, K.VOGT, M.PLATZER, und H.D.VOLK (1995): Up-regulation of monocytic IL-10 by tumor necrosis factor-alpha and cAMP elevating drugs. Int.Immunol. 7:517-523
POHLMAN,T.H., K.A.STANNESS, P.G.BEATTY, H.D.OCHS, und J.M.HARLAN (1986): An endothelial cell surface factor(s) induced in vitro by lipopolysaccharide, interleukin 1, and tumor necrosis factor-alpha increases neutrophil adherence by a CDw18-dependent mechanism. J.Immunol. 136:4548-4553
PUNCH,J., R.REES, B.CASHMER, K.OLDHAM, E.WILKINS, und D.J.SMITH, JR. (1991): Acute lung injury following reperfusion after ischemia in the hind limbs of rats. J.Trauma 31:760-765
RANDOW,F., U.SYRBE, C.MEISEL, D.KRAUSCH, H.ZUCKERMANN, C.PLATZER, und H.D.VOLK (1995): Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J.Exp.Med. 181:1887-1892
RANGEL-FRAUSTO,M.S., D.PITTET, M.COSTIGAN, T.HWANG, C.S.DAVIS, und R.P.WENZEL (1995): The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA 273:117-123

Literaturverzeichnis 130
RAVETCH,J.V. und J.P.KINET (1991): Fc receptors. Annu.Rev.Immunol. 9:457-492
RAVETCH,J.V. und B.PERUSSIA (1989): Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J.Exp.Med. 170:481-497
REDL,H., G.SCHLAG, und H.LAMCHE (1990): TNF- and LPS-induced changes of lung vascular permeability: studies in unanesthetised sheep. Circ.Shock 31:183-192
REGEL,G., A.DWENGER, K.F.GRATZ, M.L.NERLICH, J.A.STURM, und H.TSCHERNE (1989): [Humoral and cellular changes of non-specific immune response following severe trauma]. Unfallchirurg 92:314-320
REGEL,G., J.A.STURM, H.C.PAPE, K.F.GRATZ, und H.TSCHERNE (1991): [Multiple organ failure. Reflection of generalized cell damage of all organs following severe trauma]. Unfallchirurg 94:487-497
RICHTER,J. und E.ZETTERBERG (1994): L-selectin mediates downregulation of neutrophil TNF receptors. J.Leukoc.Biol. 56:525-527
RINALDO,J.E., M.GORRY, R.STRIETER, H.COWAN, R.ABDOLRASULNIA, und V.SHEPHERD (1990): Effect of endotoxin-induced cell injury on 70-kD heat shock proteins in bovine lung endothelial cells. Am.J.Respir.Cell Mol.Biol. 3:207-216
ROGY,M.A., S.M.COYLE, H.S.OLDENBURG, C.S.ROCK, P.S.BARIE, K.J.VAN ZEE, C.G.SMITH, L.L.MOLDAWER, und S.F.LOWRY (1994): Persistently elevated soluble tumor necrosis factor receptor and interleukin-1 receptor antagonist levels in critically ill patients. J.Am.Coll.Surg. 178:132-138
ROTHE,J., W.LESSLAUER, H.LOTSCHER, Y.LANG, P.KOEBEL, F.KONTGEN, A.ALTHAGE, R.ZINKERNAGEL, M.STEINMETZ, und H.BLUETHMANN (1993): Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364:798-802
ROYALL,J.A., R.L.BERKOW, J.S.BECKMAN, M.K.CUNNINGHAM, S.MATALON, und B.A.FREEMAN (1989): Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am.J.Physiol 257:L399-L410

Literaturverzeichnis 131
RUBIO-AVILLA,J., J.M.PALMA-VARGAS, J.T.COLLINS, R.SMEJKAL, J.MCLAREN, L.M.PHILLIPS, und L.H.TOLEDO-PEREYRA (1997): Sialyl Lewis(x) analog improves liver function by decreasing neutrophil migration after hemorrhagic shock. J.Trauma 43:313-318
SALMON,J.E., J.C.EDBERG, und R.P.KIMBERLY (1990): Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J.Clin.Invest 85:1287-1295
SALVO,I., W.DE CIAN, M.MUSICCO, M.LANGER, R.PIADENA, A.WOLFLER, C.MONTANI, und E.MAGNI (1995): The Italian SEPSIS study: preliminary results on the incidence and evolution of SIRS, sepsis, severe sepsis and septic shock. Intensive Care Med. 21 Suppl 2:S244-S249
SAWDEY,M.S. und D.J.LOSKUTOFF (1991): Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J.Clin.Invest 88:1346-1353
SCHILLER,H.J., P.M.REILLY, und G.B.BULKLEY (1993): Tissue perfusion in critical illnesses. Antioxidant therapy. Crit Care Med. 21:S92-102
SCHUSTER,H.P. (1989): [Infection as a cause of multiple organ failure. Definition, pathophysiology and diagnostic parameters]. Anasth.Intensivther.Notfallmed. 24:206-211
SCOTT,R.E. und R.G.HORN (1970): Ultrastructural aspects of neutrophil granulocyte development in humans. Lab Invest 23:202-215
SEEKAMP,A., M.JOCHUM, M.ZIEGLER, M.VAN GRIENSVEN, M.MARTIN, und G.REGEL (1998): Cytokines and adhesion molecules in elective and accidental trauma-related ischemia/reperfusion. J.Trauma 44:874-882
SEEKAMP,A., M.S.MULLIGAN, G.O.TILL, C.W.SMITH, M.MIYASAKA, T.TAMATANI, R.F.TODD, III, und P.A.WARD (1993a): Role of beta 2 integrins and ICAM-1 in lung injury following ischemia-reperfusion of rat hind limbs. Am.J.Pathol. 143:464-472
SEEKAMP,A., G.O.TILL, M.S.MULLIGAN, J.C.PAULSON, D.C.ANDERSON, M.MIYASAKA, und P.A.WARD (1994): Role of selectins in local and remote tissue injury following ischemia and reperfusion. Am.J.Pathol. 144:592-598

Literaturverzeichnis 132
SEEKAMP,A. und P.A.WARD (1993b): Ischemia-reperfusion injury. Agents Actions Suppl 41:137-152
SEEKAMP,A., J.S.WARREN, D.G.REMICK, G.O.TILL, und P.A.WARD (1993c): Requirements for tumor necrosis factor-alpha and interleukin-1 in limb ischemia/reperfusion injury and associated lung injury. Am.J.Pathol. 143:453-463
SEIBERT,A.F., J.HAYNES, und A.TAYLOR (1993): Ischemia-reperfusion injury in the isolated rat lung. Role of flow and endogenous leukocytes. Am.Rev.Respir.Dis. 147:270-275
SIGURDSSON,G.H., H.A.YOUSSEF, und S.STEPHEN (1992): Multiple organ platelet sequestration, hemodynamics, and gas exchange in endotoxin shock and the effects of aspirin-terbutaline treatment. Am.J.Physiol Imaging 7:66-72
SIMON,S.I., A.R.BURNS, A.D.TAYLOR, P.K.GOPALAN, E.B.LYNAM, L.A.SKLAR, und C.W.SMITH (1995): L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J.Immunol. 155:1502-1514
SKILLMAN,J.J., L.S.BUSHNELL, und J.HEDLEY-WHYTE (1969): Peritonitis and respiratory failure after abdominal operations. Ann.Surg. 170:122-127
SMITH,C.W., S.D.MARLIN, R.ROTHLEIN, C.TOMAN, und D.C.ANDERSON (1989): Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J.Clin.Invest 83:2008-2017
SPEISER,D.E., E.SEBZDA, T.OHTEKI, M.F.BACHMANN, K.PFEFFER, T.W.MAK, und P.S.OHASHI (1996): Tumor necrosis factor receptor p55 mediates deletion of peripheral cytotoxic T lymphocytes in vivo. Eur.J.Immunol. 26:3055-3060
SPERTINI,O., F.W.LUSCINSKAS, G.S.KANSAS, J.M.MUNRO, J.D.GRIFFIN, M.A.GIMBRONE, JR., und T.F.TEDDER (1991): Leukocyte adhesion molecule-1 (LAM-1, L-selectin) interacts with an inducible endothelial cell ligand to support leukocyte adhesion. J.Immunol. 147:2565-2573

Literaturverzeichnis 133
Spille, C. Prostanoide nach präoperativer Applikation von Stegantox 60 bei Pferden mit Kolik. 1997. Tierärztliche Hochschule Hannover, Klinik für Pferde und Veterinärmedizinische Fakultät der Universität Leipzig, Institut für Pharmakologie, Pharmazie und Toxikologie. Ref Type: Thesis/Dissertation
SPRINGER,T.A. (1990): Adhesion receptors of the immune system. Nature 346:425-434
STEPHENS,K.E., A.ISHIZAKA, J.W.LARRICK, und T.A.RAFFIN (1988): Tumor necrosis factor causes increased pulmonary permeability and edema. Comparison to septic acute lung injury. Am.Rev.Respir.Dis. 137:1364-1370
SUDKAMP,N., N.HAAS, P.J.FLORY, H.TSCHERNE, und A.BERGER (1989): [Criteria for amputation, reconstruction and replantation of extremities in multiple trauma patients]. Chirurg 60:774-781
SYRBE,U., A.MEINECKE, C.PLATZER, A.MAKKI, K.ASADULLAH, und C.KLUG (1994): Improvement of monocyte function - a new therapeutic approach? In: Sepsis: Current perspectives in pathophysiology and therapie by K.Reinhart, K.Eyrich, und C.L.Sprung (Hrsg.) Springer Verlag, Berlin, S. 473-500
TACKE,F., P.SCHOFFSKI, C.TRAUTWEIN, T.LUEDDE, A.GANSER, M.P.MANNS, und M.VON DEPKA (2003): Plasma P-selectin levels are elevated in patients with chronic liver disease. Blood Coagul.Fibrinolysis 14:319-325
TALBOTT,G.A., S.R.SHARAR, J.M.HARLAN, und R.K.WINN (1994): Leukocyte-endothelial interactions and organ injury: the role of adhesion molecules. New Horiz. 2:545-554
TARTAGLIA,L.A., D.V.GOEDDEL, C.REYNOLDS, I.S.FIGARI, R.F.WEBER, B.M.FENDLY, und M.A.PALLADINO, JR. (1993): Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. J.Immunol. 151:4637-4641
THIJS,L.G. und C.E.HACK (1995): Time course of cytokine levels in sepsis. Intensive Care Med. 21 Suppl 2:S258-S263
TRACEY,K.J., B.BEUTLER, S.F.LOWRY, J.MERRYWEATHER, S.WOLPE, I.W.MILSARK, R.J.HARIRI, T.J.FAHEY, III, A.ZENTELLA, J.D.ALBERT, und . (1986): Shock and tissue injury induced by recombinant human cachectin. Science 234:470-474

Literaturverzeichnis 134
TRAN,D.D., A.B.GROENEVELD, M.J.VAN DER, J.J.NAUTA, R.J.STRACK VAN SCHIJNDEL, und L.G.THIJS (1990): Age, chronic disease, sepsis, organ system failure, and mortality in a medical intensive care unit. Crit Care Med. 18:474-479
TSOKOS,M. und F.FEHLAUER (2001): Post-mortem markers of sepsis: an immunohistochemical study using VLA-4 (CD49d/CD29) and ICAM-1 (CD54) for the detection of sepsis-induced lung injury. Int.J.Legal Med. 114:291-294
UCIECHOWSKI,P., J.E.GESSNER, R.SCHINDLER, und R.E.SCHMIDT (1992): Fc gamma RIII activation is different in CD16+ cytotoxic T lymphocytes and natural killer cells. Eur.J.Immunol. 22:1635-1638
VAN DE WINKEL,J.G. und C.L.ANDERSON (1991a): Biology of human immunoglobulin G Fc receptors. J.Leukoc.Biol. 49:511-524
VAN DE WINKEL,J.G., G.J.BOONEN, P.L.JANSSEN, A.VLUG, N.HOGG, und W.J.TAX (1989): Activity of two types of Fc receptors, Fc gamma RI and Fc gamma RII, in human monocyte cytotoxicity to sensitized erythrocytes. Scand.J.Immunol. 29:23-31
VAN DE WINKEL,J.G., L.K.ERNST, C.L.ANDERSON, und I.M.CHIU (1991b): Gene organization of the human high affinity receptor for IgG, Fc gamma RI (CD64). Characterization and evidence for a second gene. J.Biol.Chem. 266:13449-13455
VAN DER,P.T., J.JANSEN, M.LEVI, H.TEN CATE, J.W.TEN CATE, und S.J.VAN DEVENTER (1994): Regulation of interleukin 10 release by tumor necrosis factor in humans and chimpanzees. J.Exp.Med. 180:1985-1988
VAN GRIENSVEN, M. (1999a) Multi Organ Dysfunktion Syndrom: Ein Zusammenspiel von Granulozyten, Endothelzellen und Zytokinen. Dissertation, Med. Hochschule Hannover, Unfallchirurgische Klinik.
VAN GRIENSVEN,M., C.KRETTEK, und H.C.PAPE (2003): Immune reactions after trauma. European Journal of Trauma 4:344-387
VAN GRIENSVEN,M., M.STALP, und A.SEEKAMP (1999b): Ischemia-reperfusion directly increases pulmonary endothelial permeability in vitro. Shock 11:259-263

Literaturverzeichnis 135
VAN GRIENSVEN,M., T.WITTWER, N.BRAUER, und H.PAPE (2001): DHEA wirkt protektiv bei einer experimentellen polymikrobiellen Sepsis durch CLP- besteht eine pathogenetische Bedeutung des TNF-α. Chirurgisches Forum 30: 267-279
VAN SNICK,J. (1990): Interleukin-6: an overview. Annu.Rev.Immunol. 8:253-278
VANDENABEELE,P., W.DECLERCQ, B.VANHAESEBROECK, J.GROOTEN, und W.FIERS (1995): Both TNF receptors are required for TNF-mediated induction of apoptosis in PC60 cells. J.Immunol. 154:2904-2913
VARANI,J. und P.A.WARD (1994): Mechanisms of endothelial cell injury in acute inflammation. Shock 2:311-319
VEDDER,N.B., B.W.FOUTY, R.K.WINN, J.M.HARLAN, und C.L.RICE (1989): Role of neutrophils in generalized reperfusion injury associated with resuscitation from shock. Surgery 106:509-516
VINCENT,J.L. (1996): Prevention and therapy of multiple organ failure. World J.Surg. 20:465-470
VOLK,H.D., T.LOHMANN, und S.HEYM (1990): Decrease of the proportion of HLA-DR and monocytes as prognostic parameter for the clinical outcome of septic disease. In: Immunotherapeutic prospescts of infectious disease by L.W.Masihi K.N. (Hrsg.) Springer, Berlin, S. 298-301
WAAGE,A., P.BRANDTZAEG, A.HALSTENSEN, P.KIERULF, und T.ESPEVIK (1989): The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J.Exp.Med. 169:333-338
WAAGE,A., G.SLUPPHAUG, und R.SHALABY (1990): Glucocorticoids inhibit the production of IL6 from monocytes, endothelial cells and fibroblasts. Eur.J.Immunol. 20:2439-2443
WADDELL,T.K., L.FIALKOW, C.K.CHAN, T.K.KISHIMOTO, und G.P.DOWNEY (1994): Potentiation of the oxidative burst of human neutrophils. A signaling role for L-selectin. J.Biol.Chem. 269:18485-18491

Literaturverzeichnis 136
WADDELL,T.K., L.FIALKOW, C.K.CHAN, T.K.KISHIMOTO, und G.P.DOWNEY (1995): Signaling functions of L-selectin. Enhancement of tyrosine phosphorylation and activation of MAP kinase. J.Biol.Chem. 270:15403-15411
WANG,P., Z.F.BA, und I.H.CHAUDRY (1997): Severe hypoxemia in the absence of blood loss depresses hepatocellular function and up-regulates IL-6 and PGE2. Biochim.Biophys.Acta 1361:42-48
WANNER,G.A., W.ERTEL, P.MULLER, Y.HOFER, R.LEIDERER, M.D.MENGER, und K.MESSMER (1996): Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock 5:34-40
WARD,P.A. und M.S.MULLIGAN (1991): New insights into mechanisms of oxyradical and neutrphil mediated lung injury. Klin.Wochenschr. 69:1009-1011
WARREN,J.S. (1991): Intrapulmonary interleukin 1 mediates acute immune complex alveolitis in the rat. Biochem.Biophys.Res.Commun. 175:604-610
WARREN,J.S., K.R.YABROFF, D.G.REMICK, S.L.KUNKEL, S.W.CHENSUE, R.G.KUNKEL, K.J.JOHNSON, und P.A.WARD (1989): Tumor necrosis factor participates in the pathogenesis of acute immune complex alveolitis in the rat. J.Clin.Invest 84:1873-1882
WEIGELT,J.A., D.E.CHENOWETH, K.R.BORMAN, und J.F.NORCROSS (1988): Complement and the severity of pulmonary failure. J.Trauma 28:1013-1019
WELBOURN,C.R., G.GOLDMAN, I.S.PATERSON, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1991a): Neutrophil elastase and oxygen radicals: synergism in lung injury after hindlimb ischemia. Am.J.Physiol 260:H1852-H1856
WELBOURN,C.R., G.GOLDMAN, I.S.PATERSON, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1991b): Pathophysiology of ischaemia reperfusion injury: central role of the neutrophil. Br.J.Surg. 78:651-655
WELBOURN,R., G.GOLDMAN, L.KOBZIK, I.PATERSON, D.SHEPRO, und H.B.HECHTMAN (1991c): Interleukin-2 induces early multisystem organ edema mediated by neutrophils. Ann.Surg. 214:181-186

Literaturverzeichnis 137
WELBOURN,R., G.GOLDMAN, L.KOBZIK, I.PATERSON, C.R.VALERI, D.SHEPRO, und H.B.HECHTMAN (1990): Neutrophil adherence receptors (CD 18) in ischemia. Dissociation between quantitative cell surface expression and diapedesis mediated by leukotriene B4. J.Immunol. 145:1906-1911
WINN,R.K., D.LIGGITT, N.B.VEDDER, J.C.PAULSON, und J.M.HARLAN (1993): Anti-P-selectin monoclonal antibody attenuates reperfusion injury to the rabbit ear. J.Clin.Invest 92:2042-2047
WITTE,T. und R.E.SCHMIDT (1992): [Fc gamma receptors: structure, function, and clinical significance]. Immun.Infekt. 20:76-80
WONG,D. und K.DOROVINI-ZIS (1992): Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J.Neuroimmunol. 39:11-21
YASSIN,M.M., A.A.BARROS D'SA, G.PARKS, A.S.ABDULKADIR, I.HALLIDAY, und B.J.ROWLANDS (1996): Mortality following lower limb ischemia-reperfusion: a systemic inflammatory response? World J.Surg. 20:961-966
YOKOTA,J., J.P.MINEI, G.A.FANTINI, und G.T.SHIRES (1989): Role of leukocytes in reperfusion injury of skeletal muscle after partial ischemia. Am.J.Physiol 257:H1068-H1075
YOUNES,M., A.MOHR, M.H.SCHOENBERG, und F.W.SCHILDBERG (1987): Inhibition of lipid peroxidation by superoxide dismutase following regional intestinal ischemia and reperfusion. Res.Exp.Med.(Berl) 187:9-17
YU,M. und G.TOMASA (1993): A double-blind, prospective, randomized trial of ketoconazole, a thromboxane synthetase inhibitor, in the prophylaxis of the adult respiratory distress syndrome. Crit Care Med. 21:1635-1642
ZABEL,P., D.T.WOLTER, M.M.SCHONHARTING, und U.F.SCHADE (1989): Oxpentifylline in endotoxaemia. Lancet 2:1474-1477
ZANETTI,G., M.P.GLAUSER, und J.D.BAUMGARTNER (1993): Anti-endotoxin antibodies and other inhibitors of endotoxin. New Horiz. 1:110-119

Literaturverzeichnis 138
ZIMMERMAN,B.J., M.B.GRISHAM, und D.N.GRANGER (1990): Role of oxidants in ischemia/reperfusion-induced granulocyte infiltration. Am.J.Physiol 258:G185-G190

Danksagung 139
10 Danksagung
Herrn Prof. Dr. med. vet. M. Kietzmann danke ich für die bereitwillige Übernahme der Betreuung und die gute & humorvolle Zusammenarbeit. Außerdem möchte ich mich für die schnelle Korrektur der Arbeit bedanken. Leider konnte ich Sie bis heute nicht davon überzeugen, dass die Haare ROT waren und nicht blau... Prof. Dr. rer. biol. hum. M. van Griensven: Martijn, Dir gilt ein besonders großes „Dankeschön“. Nur durch Deine einzigartige Kooperation und die unkomplizierte Art war es möglich, die Arbeit so schnell erfolgreich abzuschließen. Du hast alle Fragen (sowohl fachlich als auch den Computer betreffend) geduldig beantwortet (...wenn es sein mußte, auch zweimal...) und hattest für alle kleinen und großen Problem während der Experimente ein offenes Ohr. Auch in der sehr hektischen „Endphase“ hast Du mich großartig unterstützt. VIELEN, VIELEN DANK! Frau Claudia Pütz, Frau Iris Albers, Frau Dr. Tanja Bakhausen und Frau Diana Dudacy möchte ich für die hervorragende Unterstützung im Labor und die schnelle Anfertigung der histologischen Präparate danken. In den Mittagspausen und Inkubationsphasen entstanden sehr häufig interessante und lustige Gespräche. Seither bin ich fest davon überzeugt, dass man Leute herbeireden kann. Meinen Eltern danke ich ganz besonders, da Ihr mir sowohl das Tiermedizinstudium als auch die Anfertigung der Doktorarbeit ermöglicht habt, obwohl beides mit großen Entbehrungen für Euch selbst verbunden war. Besonders die Fußmassagen von Dir, Mutti, haben zur Wortfindung in dieser Arbeit und zur Seelentröstung während der Prüfungszeiten beigetragen. Vati, Dir möchte ich für die einzigartige detektivische Leistung beim Aufspüren der Fehler in dieser Arbeit danken. Auf Deine zweifelnde Frage vom April 1980: „ Vater, was bist Du für ein Vater?“ antworte ich Dir: „Ein guter.“ Daggi, an Dich geht ebenfalls ein großer Dank für das Korrekturlesen und besonders für die finanzielle Unterstützung in der Anfangsphase der Dr.-Arbeit. Vielen Dank, Werner, dass Du das Kuckucksei in Deinem Nest geduldet und sogar unterstützt hast... Meiner Schwester Janka möchte ich danken, dass sie schon früh meinen Ehrgeiz maßgeblich beeinflußt hat (weil sie grundsätzlich besser war als ich...). Dir, Tanja, danke ich für eine außergewöhnliche Freundschaft seit dem ersten Semester! „Keiner ist so verrückt, dass er nicht einen noch Verrückteren findet, der ihn versteht.“ (F. Nietzsche) „Viele Menschen treten in Dein Leben, aber nur wenige hinterlassen Spuren.“ Michel, ich danke Dir für Deine realistischen Einschätzungen während dieser Arbeit, auch, wenn ich sie meist nicht hören wollte. Für alles, was Du für mich bist: Danke.














![RIII[1]ROCAS EXOGENAS](https://img.pdfslide.tips/doc/110x75/552494994a795934498b47ab/riii1rocas-exogenas.jpg)