Embed Size (px)
Citation preview

Determinação de ácidos haloacéticos em águas de
consumo humano por UPLC-MS/MS
Paula Alexandra de Sousa Rosa
Dissertação para obtenção do Grau de Mestre em
Engenharia Química
Orientadoras: Enga. Georgina Maria Sarmento Felisberto
Profa. Dra. Margarida Maria Portela Correia dos Santos Romão
Júri
Presidente: Profa. Dra. Benilde de Jesus Vieira Saramago
Vogais: Profa. Dra. Margarida Maria Portela Correia dos Santos Romão
Engª. Maria Paula Machado de Barros Viana
Dezembro de 2015


I
Agradecimentos
Em primeiro lugar gostaria de agradecer à minha orientadora, Engª Georgina Sarmento, por
toda a dedicação, pelo acompanhamento durante o trabalho realizado, pela paciência e orientação, e
pela disponibilidade.
À Professora Margarida Romão, pela oportunidade, pela orientação e pelas palavras de
incentivo ao longo de todo o trabalho, muito obrigada.
Aos meus colegas mentores, Ana Fernandes Cruz e Hélder Gageiro, o meu agradecimento por
todo o acompanhamento, por todo o conhecimento partilhado e amizade. Ao Jorge Lopes, muito
obrigada pelas palavras de incentivo durante o trabalho.
Por me terem recebido tão bem no LAIST e por toda a amizade, um muito obrigada a todos.
A todos os meus amigos, em especial à Ana Braz, Inês Ferreira, Joana Temido e Tiago Dias,
que sem o seu apoio o caminho teria sido muito mais difícil, um muito obrigada pela paciência e pelo
carinho.
Aos meus pais, Lina e Saúl, e à minha irmã Ângela, obrigada por nunca me deixarem desistir
dos meus sonhos e por me acompanharem sempre. À minha sobrinha Margarida, que foi uma força
constante em todo este processo, muito obrigada.
Um agradecimento especial ao Tiago Santos, pelo apoio incondicional, pela paciência e
amizade.

II

III
Resumo
O objetivo deste trabalho consistiu na validação de um método analítico para análise de ácidos
haloacéticos em águas para consumo humano.
Em termos de legislação, a USEPA impõe que o somatório das concentrações de HAAs5
(MCAA, DCAA, TCAA, MBAA e DBAA) não deverá exceder os 60 μg/L. A OMS dá como valores
referência para o MCAA, DCAA e TCAA os limites de 20 μg/L, 50 μg/L e 200 μg/L, respetivamente [1].
A ERSAR dita como valor de referência para o somatório de concentrações de HAAs3 (MCAA, DCAA,
TCAA), 100 μg/L [2].
Para a determinação destes compostos foi usado o método que combina a cromatografia
líquida de ultra eficiência com a espetroscopia de massa (UPLC-MS/MS).
Foi efetuada primeiramente, uma otimização das condições operatórias do espetrómetro de
massa, fazendo variar os parâmetros (voltagem de cone e energia de colisão) relacionados com a
formação dos iões precursores e iões produto de cada um dos HAAs.
De seguida foram otimizadas as condições cromatográficas de forma a obter uma maior
sensibilidade e resolução para cada um dos HAAs, no menor tempo possível.
Após a implementação do método analítico procedeu-se à sua validação.
Foram analisadas algumas amostras de água de consumo da zona de Lisboa e Algarve de
forma a averiguar a presença dos compostos halogenados em estudo, verificando-se uma presença
máxima de TCAA de 22 μg/L, para uma amostra. Em nenhuma das amostras analisadas os valores de
referência das entidades USEPA, OMS e ERSAR foram ultrapassados.
Palavras-Chave: ácidos haloacéticos, águas de consumo humano, UPLC-MS/MS

IV

V
Abstract
The objective of this work is the validation of an analytical method for analysis of haloacetic
acids in drinking water.
In terms of legislation, the US EPA requires that the limit for the concentration of HAAs5 (MCAA,
DCAA, TCAA, MBAA and DBAA) should not exceed 60 μg/L. WHO imposes to MCAA, DCAA and TCAA
the limits of 20 μg/L, 50 μg/L and 200 μg/L, respectively [1]. The ERSAR imposes that the limit for the
concentration of HAAs3 (MCAA, DCAA, TCAA) should not exceed 100 μg/L.
For the determination of these compounds it was used a method that combines the efficiency
of ultra-performance liquid chromatography with mass spectroscopy (UPLC-MS / MS).
First it was performed an optimization of the operating conditions of the mass spectrometer by
varying the parameters (cone voltage and collision energy) related to the formation of precursor ions
and product ions of each HAAs.
Then the chromatographic conditions were optimized so as to obtain a higher sensitivity and
resolution for each compound in the shortest possible time.
After the implementation of the analytical method it was carried out its validation.
Drinking water samples from the area of Lisbon and Algarve were analysed in order to examine
the presence of the halogenated compounds, verifying a maximum presence of TCAA of 22 μg/L in a
sample. In none of the samples analysed, the reference values of the entities USEPA, WHO and ERSAR
have been exceeded.
Keywords: haloacetic acids, drinking water, UPLC-MS/MS

VI

VII
Índice
Agradecimentos........................................................................................................................... I
Resumo...................................................................................................................................... III
Abstract ...................................................................................................................................... V
Índice ........................................................................................................................................ VII
Índice de Figuras ....................................................................................................................... XI
Índice de Tabelas ..................................................................................................................... XIII
Lista de Abreviaturas ................................................................................................................ XV
1. Âmbito do trabalho..................................................................................................................1
Enquadramento .........................................................................................................................1
Objetivo .....................................................................................................................................2
2. LAIST ......................................................................................................................................3
3. Águas ......................................................................................................................................5
3.1. Águas de consumo ...............................................................................................................5
3.2. Tratamento da Água .............................................................................................................5
3.2.1. Subprodutos da desinfeção ............................................................................................6
4. Ácidos Haloacéticos................................................................................................................9
4.1. Como se formam ..................................................................................................................9
4.2. Propriedades........................................................................................................................9
4.3. Toxicologia......................................................................................................................... 10
4.4. Legislação ......................................................................................................................... 10
4.5. Métodos de Análise ............................................................................................................ 11
4.6. Estado da Arte ................................................................................................................... 11
5. Cromatografia ....................................................................................................................... 13
5.1. Cromatografia Líquida ........................................................................................................ 13
5.1.1. Cromatografia Líquida de Ultra Eficiência...................................................................... 14
5.1.2. Deteção por Espetrometria de Massa ........................................................................... 16
5.2. Efeitos de Matriz................................................................................................................. 19
5.3. Cromatografia Gasosa ........................................................................................................ 20
5.3.1. Deteção de Captura de Eletrões ................................................................................... 20
6. Validação do Método Analítico – Qualidade .......................................................................... 21
6.1. Calibração ......................................................................................................................... 21
6.1.1. Método do Padrão Externo........................................................................................... 21
6.1.2. Método de Adição de Padrão ....................................................................................... 22
6.1.3. Método do Padrão Interno............................................................................................ 22
6.1.4. Método “Matrix-Matched” ............................................................................................. 23

VIII
6.2. Limiares Analíticos ............................................................................................................. 23
6.2.1. Limite de Deteção – LD................................................................................................ 23
6.2.2. Limite de Quanti ficação – LQ ....................................................................................... 23
6.3. Precisão ............................................................................................................................ 24
6.3.1. Repetibilidade ............................................................................................................. 24
6.3.2. Reprodutibilidade ........................................................................................................ 24
6.3.3. Precisão Intermédia..................................................................................................... 24
6.4. Exatidão ............................................................................................................................ 24
6.4.1. Ensaios de Recuperação ............................................................................................. 25
6.4.2. MRC ........................................................................................................................... 25
6.4.3. Ensaios Interlaboratoriais ............................................................................................. 25
6.4.4. Método de Referência.................................................................................................. 25
6.5. Robustez ........................................................................................................................... 25
6.6. Sensibilidade...................................................................................................................... 26
6.7. Seletividade e Especificidade .............................................................................................. 26
6.8. Incertezas .......................................................................................................................... 26
7. Técnica Experimental ............................................................................................................ 29
7.1. Equipamento...................................................................................................................... 29
7.2. Material .............................................................................................................................. 29
7.3. Reagentes ......................................................................................................................... 29
7.4. Preparação de Soluções ..................................................................................................... 30
7.5. Procedimento Experimental ................................................................................................ 31
7.5.1. Segurança .................................................................................................................. 31
7.5.2. Descontaminação de Material ...................................................................................... 31
7.5.3. Otimização dos parâmetros de espetrometria de massa ................................................ 31
7.5.4. Otimização dos parâmetros de cromatografia ................................................................ 32
7.5.5. Métodos de Calibração ................................................................................................ 33
7.5.6. Comparação de Técnicas ............................................................................................ 33
8. Apresentação e Discussão de Resultados ............................................................................ 35
8.1. Condições do Espetrómetro de Massa................................................................................. 35
8.2. Condições Cromatográficas ................................................................................................ 39
8.3. Validação do Método .......................................................................................................... 43
8.3.1. Linearidade ................................................................................................................. 43
8.3.2. Limiares Analíticos ...................................................................................................... 48
8.3.3. Precisão ..................................................................................................................... 48
8.3.4. Exatidão ..................................................................................................................... 49
8.4. Efeitos de Matriz................................................................................................................. 50

IX
8.5. Incertezas do Método ......................................................................................................... 53
8.6. Aplicação a Amostras Reais ................................................................................................ 54
8.7. Comparação de Métodos .................................................................................................... 55
8.8. Razão das Massas ............................................................................................................. 56
8.9. Procedimento Analítico ....................................................................................................... 57
9. Conclusões e Perspetivas Futuras........................................................................................ 59
10. Referências Bibliográficas .................................................................................................... 61
Anexo I ....................................................................................................................................... 67
Anexo II ...................................................................................................................................... 69
Anexo III ..................................................................................................................................... 71
Anexo IV..................................................................................................................................... 73
Anexo V...................................................................................................................................... 75

X

XI
Índice de Figuras
Figura 2.1 – Laboratório de Análises do Instituto superior Técnico (LAIST) [8]. ..................................3
Figura 3.1 – Sistema de tratamento de águas convencional. .............................................................5
Figura 3.2 – Relação do teor de cloro residual com a quantidade de cloro introduzida [11]. ................8
Figura 4.1 – Mecanismo de reação referente à formação de ácido monocloroacético em meio básico
[24]. ..............................................................................................................................................9
Figura 5.1 – (a) Diagrama que descreve a separação cromatográfica de dois compostos, A e B; (b)
Sinal do detetor ao longo do tempo de eluição [31]. ....................................................................... 14
Figura 5.2 – UPLC Acquity, Waters, semelhante ao usado no presente trabalho [33]. ...................... 14
Figura 5.3 – Representação das pontes de etileno com a sílica, características das colunas BEH [37].
................................................................................................................................................... 15
Figura 5.4 – Pré-coluna usual em UPLC [61]. ................................................................................ 16
Figura 5.5 – Espetrómetro de massa Xevo TQD, Waters, usado no presente trabalho. .................... 16
Figura 5.6 – Esquema simplificativo da sonda na posição típica em frente ortogonal em relação ao cone.
(adaptado de [41])........................................................................................................................ 17
Figura 5.7 – Esquema representativo do analisador de massas usado no trabalho a trabalhar em MRM
[44]. ............................................................................................................................................ 18
Figura 5.8 – Esquema do interior do equipamento de espetrometria usado no presente trabalho [46].
................................................................................................................................................... 19
Figura 6.1 – Representação gráfica exemplificativa do método da adição de padrão. ....................... 22
Figura 7.1 – Esquema representativo de uma sessão de trabalho na preparação de uma reta de
calibração. ................................................................................................................................... 31
Figura 8.1 – Espetro resultante da infusão do ácido monocloroacético para uma concentração de 2
mg/L............................................................................................................................................ 35
Figura 8.2 – Espetro resultante da infusão do ácido dicloroacético para uma concentração de 2 mg/L.
................................................................................................................................................... 36
Figura 8.3 – Espetro resultante da infusão do ácido tricloroacético para uma concentração de 2 mg/L.
................................................................................................................................................... 36
Figura 8.4 – Espetro resultante da infusão do ácido monobromoacético para uma concentração de 2
mg/L............................................................................................................................................ 37
Figura 8.5 – Espetro resultante da infusão do ácido dibromoacético para uma concentração de 2 mg/L.
................................................................................................................................................... 37
Figura 8.6 – Espetro resultante da infusão do ácido bromocloroacético para uma concentração de 2
mg/L............................................................................................................................................ 38
Figura 8.7 – Espetro resultante da infusão do ácido tribromoacético para uma concentração de 2 mg/L.
................................................................................................................................................... 38
Figura 8.8 – Janelas de tempo definidas para cada um dos HAAs, com 13 pontos por pico. ............. 39
Figura 8.9 – Comparação dos picos obtidos para o MCAA com uma concentração de 5 μg/L nas colunas
BEH C8 e C18. ............................................................................................................................ 40

XII
Figura 8.10 – Cromatograma resultante da injeção de uma solução conjunta dos 6 HAAs com uma
concentração de 10 μg/L. ............................................................................................................. 40
Figura 8.11 – Cromatograma com os TIC’s de cada HAA, para uma concentração de 50 μg/L, na coluna
dC18. .......................................................................................................................................... 41
Figura 8.12 – Programação da temperatura ótima da coluna cromatográfica. .................................. 41
Figura 8.13 – Programa de gradiente usado no presente trabalho em que A corresponde a Metanol e
C corresponde à Solução aquosa. ................................................................................................ 42
Figura 8.14 – a) Representação gráfica da curva de calibração para o DCAA pelo método de adição de
padrão. b) Representação gráfica da análise de res íduos respetiva. ............................................... 45
Figura 8.15 – a) Representação gráfica da curva de calibração para o DCAA pelo método de padrão
externo. b) Representação gráfica da análise de resíduos respetiva. .............................................. 45
Figura 8.16 – Representações gráficas dos ajustes a) linear e b) polinomial. ................................... 46
Figura 8.17 – Representação gráfica para os valores obtidos no teste de Rikilt para o MCAA. .......... 47
Figura 8.18 – Representação gráfica das retas obtidas para o MCAA, para diferentes matrizes. ....... 51
Figura 8.19 – Representação gráfica das retas obtidas para o DCAA, para diferentes matrizes. ....... 51
Figura 8.20 – Representação gráfica das retas obtidas para o TCAA, para diferentes matrizes. ........ 51
Figura 8.21 – Representação gráfica das retas obtidas para o MBAA, para diferentes matrizes. ....... 51
Figura 8.22 – Representação gráfica das retas obtidas para o DBAA, para diferentes matrizes. ....... 52
Figura 8.23 – Representação gráfica das retas obtidas para o BCAA, para diferentes matrizes. ....... 52
Figura 8.24 – Representação gráfica da variação das áreas em função da % de água da torneira para
o ácido monocloroacético. ............................................................................................................ 52
Figura 8.25 – Evolução das concentrações dos HAAs ao longo do tempo para a amostra X. ............ 54
Figura III.1 – Representação gráfica de um ensaio para o DCAA pelo método de padrão externo. .... 71
Figura III.2 – Representação gráfica de um ensaio para o MCAA pelo método de padrão externo. .... 71
Figura III.3 – Representação gráfica de um ensaio para o BCAA pelo método de padrão externo. .... 71
Figura III.4 – Representação gráfica de um ensaio para o MBAA pelo método de padrão externo. .... 71
Figura III.5 – Representação gráfica de um ensaio para o TCAA pelo método de padrão externo. .... 71
Figura III.6 – Representação gráfica de um ensaio para o DBAA pelo método de padrão externo. .... 71

XIII
Índice de Tabelas
Tabela 4-1 – Abreviatura, fórmula química, massa molecular (g/mol), constante de acidez e temperatura
de ebulição (ᵒC) dos ácidos haloacéticos estudados [23]. ............................................................... 10
Tabela 5-1 – Designação, estrutura, gama de pH e limite de temperatura (ᵒC) das colunas usadas no
trabalho [35]. ............................................................................................................................... 15
Tabela 7-1 – Parâmetros usados na otimização do espetrómetro de massa. ................................... 32
Tabela 8-1 – Valores otimizados dos parâmetros da espetrometria de massa para cada HAA. ......... 39
Tabela 8-2 – Resumo das condições ótimas para a cromatografia (1ª linha do gradiente, tempo total de
corrida, volume de injeção, fluxo e temperatura da coluna). ............................................................ 42
Tabela 8-3 – Tempos de retenção característicos de cada um dos HAAs, para as condições impostas.
................................................................................................................................................... 43
Tabela 8-4 – Valores dos declives (a) e coeficientes de correlação (R) das retas de calibração para
cada HAA pelo método de padrão externo. .................................................................................... 44
Tabela 8-5 – Valores dos declives (a) e coeficientes de correlação (R) nas retas de calibração para
cada HAA pelo método de adição de padrão. ................................................................................ 44
Tabela 8-6 – Valores obtidos para o teste de Rikilt relativamente a um ensaio para o MCAA. ........... 47
Tabela 8-7 – Valores obtidos nos testes (análise de resíduos, teste de Mandel e teste de Rikilt) para o
estudo da linearidade. .................................................................................................................. 48
Tabela 8-8 – Limiares analíticos obtidos pela reta de calibração e por repetibilidades (n=6), em μg/L.
................................................................................................................................................... 48
Tabela 8-9 – Repetibilidades para estudo da precisão com n=6. ..................................................... 49
Tabela 8-10 – Repetibilidades para estudo da precisão intermédia. ................................................ 49
Tabela 8-11 – Valores médios de recuperações obtidas com o respetivo desvio padrão, para cada HAA
em águas de consumo humano. ................................................................................................... 49
Tabela 8-12 – Condutividades (μs/cm) e pH dos diferentes tipos de água usados neste teste. .......... 50
Tabela 8-13 – Declives e coeficientes de correlação obtidos para cada tipo de água, para cada HAA.
................................................................................................................................................... 50
Tabela 8-14 – Resumo das incertezas estimadas para a determinação de cada ácido haloacético. .. 53
Tabela 8-15 – Valores de LQ do método com a incerteza associada para cada HAA. ...................... 53
Tabela 8-16 – Resultados das amostras provenientes do Algarve pelo método de adição de padrão.
................................................................................................................................................... 54
Tabela 8-17 – Valores obtidos da concentração de cada HAA e a sua comparação com o previsto na
matriz simulada. ........................................................................................................................... 55
Tabela 8-18 – Resultados obtidos na análise de HAAs por UPLC-MS/MS e GC-ECD. ..................... 55
Tabela 8-19 – Comparação das vantagens e desvantagens entre as duas técnicas. ........................ 56
Tabela 8-20 – Razão MRM1/MRM2 para cada um dos compostos em estudo. ................................ 57
Tabela II-1 – Volumes a retirar das soluções-mãe para se obter a solução intermédia em metanol. .. 69
Tabela II-2 – Volumes a retirar da solução intermédia para se obterem as soluções padrão para a reta
de calibração em balões de 5 mL em água. ................................................................................... 69

XIV
Tabela II-3 – Volumes a retirar das soluções-mãe para se obterem as soluções padrão individuais em
metanol em balões de 10 mL, para as infusões.............................................................................. 69
Tabela IV-1 – Áreas obtidas para uma concentração aproximada de 10 μg/L em diferentes matrizes.
................................................................................................................................................... 73
Tabela IV-2 – Variação da % de água ultrapura versus % de água da torneira e respetivas áreas para
o ácido monocloroacético. ............................................................................................................ 73

XV
Lista de Abreviaturas
BCAA – Ácido bromocloroacético
DCAA – Ácido dicloroacético
DBAA – Ácido dibromoacético
ECD – Detetor de Captura de Eletrões
ERSAR – Entidade Reguladora dos Serviços de Água e Resíduos
ESI – Ionização do tipo Electrospray
FE – Fase Estacionária
FM – Fase Móvel
GC – Cromatografia Gasosa
HAAs – Ácidos Haloacéticos
HPLC – Cromatografia Líquida de Alta Eficiência
IC – Cromatografia Iónica
LAIST – Laboratório de Análises do Instituto Superior Técnico
LD – Limite de Deteção
LQ – Limite de Quantificação
MBAA – Ácido monobromoacético
MCAA – Ácido monocloroacético
MRC – Material de Referência Certificado
MRM – Monitoramento de Reações Múltiplas
MTBE - Éter metil-terc-butílico
MS/MS – Espetrometria de Massa tandem
OMS – Organização Mundial de Saúde
RSD – Desvio Padrão Relativo
SIM – Monitorização Seletiva de Iões
TCAA – Ácido tricloroacético
THMs – Trihalometanos
TIC – Cromatograma do Ião Total
UPLC – Cromatografia Líquida de Ultra Eficiência
US EPA - Agência de Proteção Ambiental dos Estados Unidos da América

XVI

1
1. Âmbito do trabalho
Enquadramento
O abastecimento de água para consumo humano, assim como o controlo da sua qualidade, é
um serviço público essencial ao bem-estar dos cidadãos e à saúde pública. Pode-se afirmar que
Portugal se encontra avançado nesta obrigação, cobrindo praticamente a totalidade da população com
um serviço adequado de abastecimento. A nível nacional a água para consumo humano apresenta
excelente qualidade, com 98,4% de água segura (ERSAR – 2014), colocando Portugal ao nível dos
países mais avançados da União Europeia [3].
A qualidade exigida baseia-se não só na análise de parâmetros físico-químicos como no
cumprimento dos valores paramétricos definidos no decreto de Lei 306/2007, obrigando a água a ser
isenta de micro-organismos patogénicos [4]. Isto acontece uma vez que a água potável é uma das
principais fontes de patogénicos microbianos [5].
O tratamento a que a água destinada ao consumo humano é sujeita, é constituído por vários
processos, sendo um deles o da desinfeção. As substâncias químicas utilizadas como agentes
desinfetantes, tais como o cloro e o hipoclorito de sódio, reagem com a matéria orgânica natural
presente na água formando os chamados subprodutos da desinfeção. Existe uma grande variedade de
subprodutos de desinfeção que comportam sérios riscos à saúde pública, sendo os trihalometanos
(THMs) e os ácidos haloacéticos (HAAs), os principais [1].
Os compostos em estudo neste trabalho são os ácidos haloacéticos, representando o principal
grupo de subprodutos da desinfeção halogenados não-voláteis. Em termos de legislação atual, a
USEPA tem como valor de referência o limite de 60 μg/L para a soma de HAAs5 (ácido
monocloroacético (MCAA), ácido dicloroacético (DCAA), ácido tricloroacético (TCAA), ácido
monobromoacético (MBAA) e ácido dibromoacético (DBAA)) [6]. Já a OMS indica apenas para três
ácidos haloacéticos valores referência com os limites de 20 μg/L, 50 μg/L e 200 μg/L para o MCAA,
DCAA e TCAA, respetivamente [1]. A nível nacional, a ERSAR tem como valor de referência o limite
para a soma das concentrações de MCAA, DCAA e TCAA, o valor de 100 μg/L.
O interesse crescente nestes compostos rege-se pelo facto de apresentarem alguns efeitos
toxicológicos (segundo a USEPA, alguns são prováveis cancerígenos), razão pela qual o controlo
quantitativo dos mesmos deveria ser regulado [7].
Com a pesquisa bibliográfica realizada constatou-se que os métodos com maior aplicabilidade
na análise dos HAAs são a cromatografia gasosa com deteção de captura de eletrões (GC-ECD) e a
cromatografia iónica com deteção por espetrómetro de massa (IC-MS), sendo o primeiro método usado
no Laboratório de Análises do Instituto Superior Técnico (LAIST). Estes são métodos muito morosos,
sobretudo devido ao processo de preparação de amostras, e com gastos elevados de solventes, pelo
que faz todo o sentido implementar-se uma nova metodologia que permita a análise dos HAAs nas
águas de consumo humano, de forma mais eficiente [7].

2
Objetivo
Este trabalho permitiu a determinação de seis ácidos haloacéticos (MCAA, DCAA, TCAA,
MBAA, DBAA e BCAA) em águas de consumo humano por injeção direta da amostra, pelo método que
combina a cromatografia líquida de ultra eficiência com a deteção por espetrometria de massa com
ionização por electrospray (UPLC-ESI-MS/MS). Foram analisadas amostras reais de diferentes redes
públicas de abastecimento de água (Lisboa e Algarve), em que os resultados obtidos sugerem a
presença de alguns HAAs com valores inferiores aos valores de referência impostos pelas entidades
anteriormente referidas.

3
2. LAIST
No final do século XIX, ainda no Instituto Industrial e Comercial de
Lisboa, foi criado o Laboratório de Análises pelos Professores Charles LePierre
e Herculano de Carvalho.
O LAIST (Figura 2.1) encontra-se organizado por uma Área
Administrativa e por 5 diferentes núcleos analíticos (Análises Gerais aplicadas
a Águas; Gestão de Colheitas, Ambiente, Saúde e Segurança; Metais e Preparação de Amostras;
Microbiologia – Clássica e Novas Tecnologias; Análise de Compostos Orgânicos), definidos de acordo
com as suas principais áreas de atividade, sob a orientação de técnicos qualificados e dirigidos por um
coordenador técnico [8].
A atividade do LAIST é essencialmente de investigação aplicada e de prestação de serviços,
empregando variados métodos analíticos para doseamento de determinados compostos em diversos
produtos. Cerca de 80% de todo o trabalho realizado no Laboratório de Análises envolve a análise
química de águas, tanto minerais como as usadas para diferentes fins, tais como as de consumo
público, indústria e agricultura [9].
Figura 2.1 – Laboratório de Análises do Instituto superior Técnico (LAIST) [8].
O presente trabalho foi desenvolvido no núcleo de Análises de Compostos Orgânicos. Neste
núcleo estão implementados métodos analíticos para a caraterização de matrizes: águas de consumo
humano, naturais, residuais, lixiviados, eluatos, resíduos sólidos, lamas, sedimentos, solos, ar, entre
outro tipo de amostras utilizadas nas mais diversas finalidades. Os métodos respondem à legislação
(limites de deteção e quantificação, precisão e exatidão) em vigor para as diferentes origens de
amostras [8].

4

5
3. Águas
Existem cerca de 50 parâmetros distintos que são analisados regularmente a fim de controlar
a qualidade da água. Estas análises permitem avaliar as suas caraterísticas averiguando se os valore s
obtidos transpõem os limites legais a nível nacional. Entre estas caraterísticas encontram-se não só o
controlo de parâmetros físico-químicos mas também de indicadores de contaminação por parte de
microrganismos patogénicos, pesticidas e metais pesados entre outros [10].
3.1. Águas de consumo
A água potável pode ser obtida diretamente a partir de uma água subterrânea de elevada
qualidade ou indiretamente a partir de uma água não potável, posteriormente submetida a tratamentos
apropriados [11].
Em Portugal captam-se volumes de água a partir de captações superficiais superiores aos das
captações subterrâneas [12].
3.2. Tratamento da Água
O sistema de tratamento de águas mais convencional, segundo um artigo publicado pela EPA
em 2012, consiste resumidamente nos processos unitários que se apresentam na Figura 3.1. [12].
Pré-Oxidação
Nesta primeira etapa é adicionado um reagente (ozono, dióxido de cloro ou cloro) com o
objetivo de degradar a maior parte da matéria orgânica presente na água, através de uma reação de
oxidação e ainda inativar os microrganismos patogénicos que possam existir. Uma vez que a águ a
captada tem valores de pH ligeiramente ácidos dá-se a sua correção através da adição de dióxido de
carbono e hidróxido de cálcio. Este processo denomina-se por Remineralização [13] [14].
Coagulação/Floculação
A presença de partículas de natureza coloidal que levam a valores de turvação elevados,
caraterístico de águas de origem superficial, impõe a adição de um coagulante (sais de ferro e de
alumínio) numa câmara de mistura rápida com o intuito de destabilizar as partículas coloidais
(coagulação). Por consequência dá-se a agregação em flocos de grandes dimensões (floculação). Em
alguns casos, quando necessário, é utilizado carvão ativado em pó, que permite a remoção de cheiros,
sabores, toxinas e pesticidas presentes na água [13] [15].
Pré-Oxidação
Coagulação/ Floculação
Sedimentação Filtração Desinfeção
Figura 3.1 – Sistema de tratamento de águas convencional.

6
Sedimentação
A remoção dos flocos formados na etapa anterior dá-se por ação gravítica na sua
sedimentação. Os flocos acumulam-se no fundo dos tanques formando deste modo as lamas do
processo de tratamento [13] [15].
Filtração
A etapa seguinte consiste na filtração da água proveniente da sedimentação, que permite a
remoção de partículas mais pequenas promovendo a clarificação final da água. De forma a garantir
uma remoção eficiente, os filtros são constituídos por camadas de cascalho e areia [15].
Desinfeção
“ A desinfeção da água assegura a proteção contra o risco de contrair doenças infeciosas de
origem hídrica, sendo este um objetivo prioritário e indispensável.” – (5/2007, IRAR [16])
A eficiência do tratamento de água na remoção de microrganismos patogénicos varia de mês
para mês, e mesmo quando a estação de tratamento está a atingir a remoção de 99,9%, haverá sempre
uma certa quantidade de patogénicos que permanecem na água. Isto significa que a desinfeção é
absolutamente vital para assegurar que quaisquer microrganismos decorrentes de contaminação fecal
da água sejam destruídos [14] [15].
A desinfeção da água pode efetuar-se através de diferentes métodos físicos ou químicos, tais
como a radiação ultravioleta e os processos químicos. Relativamente aos processos químicos, os
reagentes empregues são o cloro e seus derivados, o ozono e o dióxido de cloro. A generalização da
cloragem das águas no continente Europeu proporcionou a erradicação de epidemias , tais como a
febre tifoide e a cólera [11].
Em Portugal, o cloro é um dos principais desinfetantes utilizados no tratamento de água para
consumo humano devido ao seu baixo custo e pelo facto de a inativação de microrganismos
patogénicos ser relativamente rápida [17].
3.2.1. Subprodutos da desinfeção
A desinfeção é um passo essencial do processo de tratamento de águas que no entanto traz
um grande inconveniente, a formação de subprodutos. Os subprodutos da desinfeção ocorrem como
resultado de reações entre o desinfetante e outros compostos que se encontram na água.
De todos os subprodutos, os que requerem maior atenção são as substâncias
halogenadas: os trihalometanos e os ácidos haloacéticos. Estes são formados devido à adição
de cloro e hipoclorito na etapa de desinfeção [18].
Estas reações secundárias têm sido identificadas desde 1974, particularmente as que
envolvem alguns tipos de matéria orgânica naturalmente presentes na água, tais como ácidos húmicos
e fúlvicos [11].

7
Fatores que influenciam a sua formação
Existem diversos fatores que afetam a formação dos ácidos haloacéticos, tais como o pH, a
temperatura, o tipo e dose de desinfetante usado, assim como alguns parâmetros de qualidade da água
como a concentração e o caráter da matéria orgânica e a presença de bromo [19].
Características e concentração da matéria orgânica natural
Um dos principais precursores dos subprodutos da desinfeção é a matéria orgânica dissolvida
na água. Uma vez que a sua presença proporciona um maior crescimento bacteriano, acaba por impedir
a ação dos desinfetantes protegendo os microrganismos. Deste modo a quantidade de desinfetante a
adicionar no tratamento tem de ser superior, para ser eficaz [19].
pH e temperatura do meio
Existem estudos que demostram claramente a relação entre o pH e a formação dos ácidos
haloacéticos [20]. Já a temperatura tem um efeito diretamente proporcional na formação dos
subprodutos da desinfeção, isto é, no inverno os níveis de HAAs são inferiores aos níveis registados
no verão [20].
Tempo de contato
O aumento do tempo de contato com o agente oxidante implica um aumento na formação da
maioria dos subprodutos da desinfeção. No entanto há que ter em atenção, que contrariamente aos
trihalometanos (THMs), os HAAs degradam-se com o tempo [21].
Presença de Bromo
Com a presença de brometos na água, o cloro facilmente oxida estes a ácido hipobromoso que
quando reage com a matéria orgânica, dá origem a espécies organobromadas. É de notar que esta
reação dá-se 25 vezes mais rápida que a reação com o ácido hipocloroso [22].
Processo de Cloragem
Quando o cloro (Cl2) entra em contato com a água forma-se ácido hipocloroso (HOCl) e ácido
clorídrico (HCl) como se observa na Equação 3-1,
𝐶𝑙2(𝑔) + 𝐻2𝑂 (𝑙) ↔ 𝐻𝐶𝑙𝑂(𝑎𝑞) + 𝐻𝐶𝑙(𝑎𝑞) Equação 3-1
Em soluções diluídas a reação dá-se rapidamente. O ácido hipocloroso é um ácido fraco que
se dissocia originando o ião hipoclorito (ClO-). Tanto o ácido hipocloroso como o ião hipoclorito atuam
como desinfetantes, embora HOCl seja 80 vezes mais eficaz que ClO-. Deste modo e uma vez que a
pH 5 o cloro na água encontra-se sob a forma de ácido hipocloroso, faz todo o sentido que a cloragem
se dê a pH ácido de forma a tornar o processo de desinfeção mais eficiente [14].
O cloro também pode ser adicionado sob a forma de hipoclorito de sódio (NaClO) ou sob a
forma de hipoclorito de cálcio (Ca(ClO)2). Estes compostos são usados em detrimento do cloro gasoso
quando a água a tratar já possui alguma qualidade razoável. A Equação 3-2 e a Equação 3-3 traduzem
a interação destes compostos com a água.
8
𝑁𝑎𝐶𝑙𝑂(𝑎𝑞) + 𝐻2𝑂 (𝑙) ↔ 𝐻𝐶𝑙𝑂(𝑎𝑞) + 𝑁𝑎𝑂𝐻 (𝑎𝑞) Equação 3-2
𝐶𝑎(𝐶𝑙𝑂)2(𝑠) + 𝐻2𝑂 (𝑙) ↔ 2𝐻𝐶𝑙𝑂 (𝑎𝑞) + 𝐶𝑎(𝑂𝐻)2 Equação 3-3
Comportamento do cloro na água
Quando o cloro é adicionado à água dão-se diferentes reações químicas, pelo que é de extrema
importância que os seus mecanismos sejam bem conhecidos.
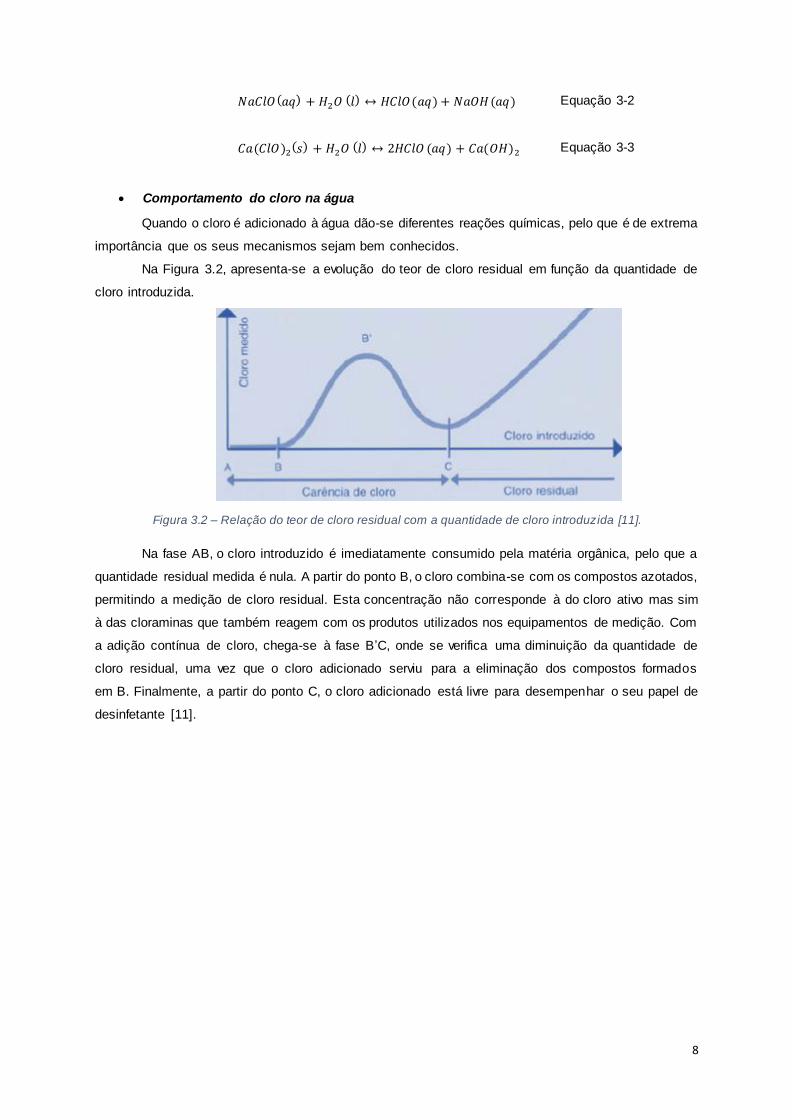
Na Figura 3.2, apresenta-se a evolução do teor de cloro residual em função da quantidade de
cloro introduzida.
Figura 3.2 – Relação do teor de cloro residual com a quantidade de cloro introduzida [11].
Na fase AB, o cloro introduzido é imediatamente consumido pela matéria orgânica, pelo que a
quantidade residual medida é nula. A partir do ponto B, o cloro combina-se com os compostos azotados,
permitindo a medição de cloro residual. Esta concentração não corresponde à do cloro ativo mas sim
à das cloraminas que também reagem com os produtos utilizados nos equipamentos de medição. Com
a adição contínua de cloro, chega-se à fase B’C, onde se verifica uma diminuição da quantidade de
cloro residual, uma vez que o cloro adicionado serviu para a eliminação dos compostos formados
em B. Finalmente, a partir do ponto C, o cloro adicionado está livre para desempenhar o seu papel de
desinfetante [11].

9
4. Ácidos Haloacéticos
Como já referido anteriormente, os ácidos haloacéticos são compostos não voláteis resultantes
da desinfeção realizada no tratamento de águas para consumo humano, constituindo o principal grupo
de subprodutos não-voláteis da desinfeção. No total existem 9 diferentes ácidos haloacéticos: ácido
monocloroacético, ácido dicloroacético, ácido tricloroacético, ácido monobromoacético, ácido
dibromoacético, ácido tribromoacético, ácido diclorobromoacético, ácido dibromocloroacét ico e o ácido
bromocloroacético [23]. No presente trabalho foram estudados os cinco ácidos haloacéticos legislados
pela US EPA (MCAA, DCAA, TCAA, MBAA e DBAA) e ainda dois outros HAAs (TBAA e BCAA).
4.1. Como se formam
A reação de formação dos ácidos haloacéticos pode ser catalisada tanto em meio ácido como
em meio básico. Os mecanismos envolvidos encontram-se na Figura 4.1.
Figura 4.1 – Mecanismo de reação referente à formação de ácido monocloroacético em meio básico [24].
Como se verifica pela Figura 4.1, a halogenação do ácido haloacético dá-se pela substituição
no carbono α. Em primeiro lugar forma-se o enolato em meio alcalino seguindo-se o ataque nucleófilo
ao halogénio. A substituição vai-se dando conforme a quantidade de grupos halogénios se encontra,
ou não, em excesso.
Uma vez que à medida que ocorre a substituição, a acidez vai aumentando devido à
deslocalização da carga implicando que a próxima halogenação dar-se-á mais rapidamente que a
anterior [24].
4.2. Propriedades
Os compostos em estudo são ácidos carboxílicos substituídos por grupos halogénios
(eletronegativos) conferindo desta forma aos HAAs maior acidez. Assim se justifica o valor de pKa de
cada ácido haloacético relativamente ao valor de pKa do ácido acético (4,75 a 25ᵒC), como se verifica
na Tabela 4-1.
Tratam-se de compostos polares com pontos de ebulição relativamente altos , de elevada
solubilidade em água.
1)
2)
H2O

10
Tabela 4-1 – Abreviatura, fórmula química, massa molecular (g/mol), constante de acidez e temperatura de ebulição
(ᵒC) dos ácidos haloacéticos estudados [23].
Composto Abreviatura Fórmula Química
Massa
Molecular (g/mol)
pKa teb (ᵒC)
Ácido Monocloroacético MCAA C2H3ClO2 94,50 2,86 189
Ácido Dicloroacético DCAA C2H2Cl2O2 128,9 1,25 194
Ácido Tricloroacético TCAA C2HCl3O2 163,4 0,630 197 Ácido Monobromoacético MBAA C2H3BrO2 138,9 2,87 208
Ácido Dibromoacético DBAA C2H2Br2O2 217,8 1,47 195
Ácido Tribromoacético TBAA C2HBr3O2 296,7 0,660 245 Ácido Bromocloroacético BCAA C2H2BrClO2 207,8 1,39 215
4.3. Toxicologia
O contato com água potável por parte dos humanos não se estabelece apenas pelo consumo
direto da água. As utilizações diárias quer para cozinhar como para higiene pessoal também são tidas
em conta [25] .
Com os estudos realizados sobre as consequências da exposição de compostos halogenados
nos seres humanos, o tema sobre os subprodutos da desinfeção de águas tem ganho maior
preocupação. Estes estudos alertam sobretudo para o facto deste tipo de compostos serem potenciais
carcinogénicos [1].
No caso especifico dos ácidos haloacéticos, os estudos realizados evidenciam diferentes
problemas para cada um deles. Começando pelo ácido monocloroacético, embora tenham sido
observadas alterações em alguns órgãos de ratos, é considerado como improvável
cancerígeno [1] [26]. Já o ácido dicloroacético é um provável cancerígeno uma vez que ratos expostos
a ele, demostraram diversos problemas tais como tumores hepáticos e toxicidade neuromuscular [27].
No caso do ácido tricloroacetico foram também evidenciados o aparecimento de tumores no fígado de
ratos, mas ainda é incerto que no caso dos seres humanos tal também aconteça. Para os ácidos
monobroacético, tribromoacético e bromocloroacético não existem dados suficientes que confirmem,
ou não, o perigo de ser cancerígeno. À semelhança do ácido dicloroacético, o ácido dibromoacetico
também é considerado provável cancerígeno devido ao aparecimento de tumores em diferentes órgãos
em animais após a sua exposição [26].
4.4. Legislação
Relativamente ao controlo da qualidade de água destinada ao consumo humano em Portugal,
encontra-se em vigor o Decreto-Lei nº 306/2007.
A nível nacional a ERSAR apresenta como valor de referência para o somatório de
concentrações para MCAA, DCAA e TCAA, de 100 μg/L [2].
Já a OMS apresenta valores específicos para alguns HAAs, nomeadamente 20 μg/L para o
MCAA, 50 μg/L para o DCAA e 200 μg/L para o TCAA [1].
A USEPA impõe um limite de 60 μg/L para o somatório das concentrações de cinco ácidos
haloacéticos (MCAA, MBAA, DCAA,DBAA e TCAA) [7].

11
4.5. Métodos de Análise
Existem algumas metodologias para a análise dos ácidos haloacéticos, tais como o método da
EPA – 557 (2009 - cromatografia iónica com deteção por espetrometria de massa tandem – IC-MS/MS)
e o método da EPA – 552 (1990 - cromatografia gasosa com deteção por captura de eletrões – GC-
ECD) que se encontra amplamente implementado nos laboratórios. Este último método implica que se
faça uma derivatização dos ácidos haloacéticos, sendo estes posteriormente extraídos.
No sentido de abreviar o tempo de análise e diminuir o consumo de reagentes, recentemente
têm sido publicados artigos para a análise dos HAAs usando a cromatografia líquida de ultra resolução
associada à espectroscopia de massa [7] [28].
4.6. Estado da Arte
Apresentam-se de seguida alguns estudos publicados referentes à análise de ácidos
haloacéticos em águas de consumo humano e em águas de piscinas:
Foram realizadas análises por UPLC-MS/MS em águas de torneira em 5 diferentes locais da
província de Shandong, China. Neste estudo foram detetados alguns ácidos haloacéticos,
sendo os mais abundantes o DCAA, TCAA, DBAA, e BCAA cuja soma representa mais de 90%
da concentração total dos HAAs em estudo. As concentrações de DCAA e TCAA foram de 22,5
e 9,71 μg/L, respetivamente, mantendo-se assim abaixo dos valores referência da OMS (50 e
200 μg/L, respetivamente). Do mesmo modo, a soma dos HAAs5 variou entre 5,36 e 32,2 μg/L,
respeitando o valor imposto pela US EPA (60 μg/L) (2010) [7].
Num outro estudo que incidiu sob a cidade de Bizerte, Tunísia, onde se verificaram
concentrações médias de 3,9 a 26,25 μg/L, de 2,76 a 23,19 μg/L e de 6,65 a 34,07 μg/L para
o TCAA, DCAA e MCAA, respetivamente. Neste estudo o método usado para a determinação
dos ácidos haloacéticos foi o de GC-ECD. Os valores obtidos para o caso do ácido
monocloroacético supera o valor de referência imposto pela OMS (MCAA – 20 μg/L). Os
restantes ácidos encontram-se em quantidades insignificantes até porque as quantidades de
bromo na água eram muito baixas [29].
Foi investigada a ocorrência de ácidos haloacéticos em diferentes amostras provenientes de 6
piscinas exteriores, 6 piscinas interiores e de 3 spas da Pensilvânia, EUA, e em 5 piscinas
exteriores e 9 piscinas interiores de Pequim, China. O método usado foi o de GC-ECD. Os
resultados indicam que os níveis de HAAs5 nas piscinas e spas da Pensilvânia variam entre
70 a 3980 μg/L sendo que nas piscinas testadas de Pequim os valores variam entre 13 e
332 μg/L. Estes valores superam os limites impostos de 60 μg/L para a soma dos cinco
principais HAAs [30].
Foi aplicado o método de GC-MS para a determinação de ácidos haloacéticos na urina de
nadadores para comparar com a urina de não nadadores em diferentes piscinas localizadas
em Espanha em 2010. Como seria previsível foram detetados níveis de MCAA, DCAA e TCAA
em todos os nadadores em contraste com os não nadadores que apresentaram valores
nulos [25].

12
Num estudo intenso realizado em 2011 pela Universidade de Cranfield, foram analisadas
diferentes amostras provenientes das saídas de estações de tratamento de águas para
consumo humano, pelo método de IC-MS/MS. Os resultados obtidos foram de 12,5 μg/L na
soma de HAA9 apresentando deste modo valores inferiores aos detetados em estudos
anteriores no mesmo local. Neste estudo comprovou-se ainda a influência que as diferentes
épocas do ano impõem na formação de ácidos haloacéticos [20].

13
5. Cromatografia
O objetivo de qualquer processo cromatográfico consiste na separação de analitos de uma
mistura.
Foi no início do século XX que o botânico russo Mickhail Tswett descobriu a cromatografia.
Utilizou a técnica para a separação de pigmentos de plantas, como as clorofilas, fazendo passar
soluções dessas espécies através de colunas de vidro com enchimento de carbonato de cálcio.
As espécies separadas apareceram como bandas coloridas na coluna, explicando assim o nome dado
à técnica: Chroma (palavra grega para cor) e Graphein (palavra grega para escrever) [31].
5.1. Cromatografia Líquida
Princípios Históricos
Em 1950 foi introduzido o conceito de cromatografia líquida com o uso de colunas de
enchimento com partículas irregulares de 100-200 μm com eficiências de somente 13 pratos/cm.
Nos anos 60 foi introduzido o conceito de Cromatografia Líquida de Alta Eficiência (HPLC)
tendo sido apresentadas colunas com enchimento de partículas rígidas de 40-50 μm, permitindo
eficiências na ordem dos66 pratos/cm. Já nos anos 70, as partículas porosas de diâmetro à volta de
10 μm vieram apresentar um ganho de eficiência de cerca de 400 pratos/cm.
Foi a partir dos anos 80, com a introdução de partículas esféricas que se começou a obter
eficiências mais elevadas, sendo que nos anos 90 com partículas esféricas porosas de 3-3,5 μm as
eficiências obtidas foram de 1500 pratos/cm. Deste modo as análises tornaram-se mais rápidas e
eficientes, uma vez que este tipo de partículas confere uma fase estacionária compacta de maior
homogeneidade. As partículas esféricas não porosas foram introduzidas no ano de 1996 aumentando
a eficiência para 2000 pratos/cm, oferecendo menor resistência à transferência de massa e maior
resistência a altas pressões e temperaturas, mas com o aspeto negativo de possuírem baixa área
superficial, relativamente às porosas, que se traduz em tempos de retenção menores.
Mais recentemente surgem então as partículas esféricas porosas de diâmetro inferior a 2 μm
que permitem melhores resoluções e altas eficiências. Nasce assim a Cromatografia Líquida de Ultra
Eficiência (UPLC). Esta é uma técnica já amplamente utilizada a nível laboratorial cujo equipamento
permite o uso de pressões elevadas (até 1000 bar) podendo deste modo dar resposta à necessidade
imposta pelas partículas de diâmetro inferior a 2 μm. Deste modo obtém-se uma análise mais rápida,
com menor consumo de solventes alcançando eficiências mais elevadas [32].
Princípios Básicos
A cromatografia é uma técnica que permite a separação de diferentes componentes de uma
mistura com base nas diferentes velocidades de migração caraterísticas de cada um dos compostos
quando estes são transportados por uma fase móvel (FM) através de uma fase estacionária (FE).

14
Na configuração mais usual a fase
estacionária está contida numa coluna como
se ilustra na Figura 5.1 e a fase móvel vai
preenchendo os espaços vazios entre as
partículas do enchimento da fase estacionária,
permitindo que os solutos fiquem mais ou
menos retidos, dependendo das afinidades
entre estes e as duas fases (a).
O cromatograma obtém-se com o
auxílio de um detetor situado após a coluna,
que permite relacionar o sinal emitido em
função do tempo (b).
5.1.1. Cromatografia Líquida de Ultra Eficiência
Como já referido anteriormente a cromatografia líquida de ultra eficiência devido às dimensões
das partículas de enchimento de diâmetro inferior a 2 μm, permite separações de compostos de forma
mais eficiente, uma vez que permite menores tempos de retenção com maior sensibilidade.
Instrumentação
Muito resumidamente o aparelho de cromatografia usado no presente trabalho (ver Figura 5.2)
é composto por três diferentes módulos: o primeiro onde se encontram as colunas cromatográficas, o
segundo que se denomina por injetor automático e o terceiro onde se encontram os sistemas que
bombeiam os eluentes.
Figura 5.2 – UPLC Acquity, Waters, semelhante ao usado no presente trabalho [33].
Figura 5.1 – (a) Diagrama que descreve a separação cromatográfica de dois compostos, A e B; (b) Sinal do detetor ao longo do tempo de
eluição [31].
15
Colunas Cromatográficas
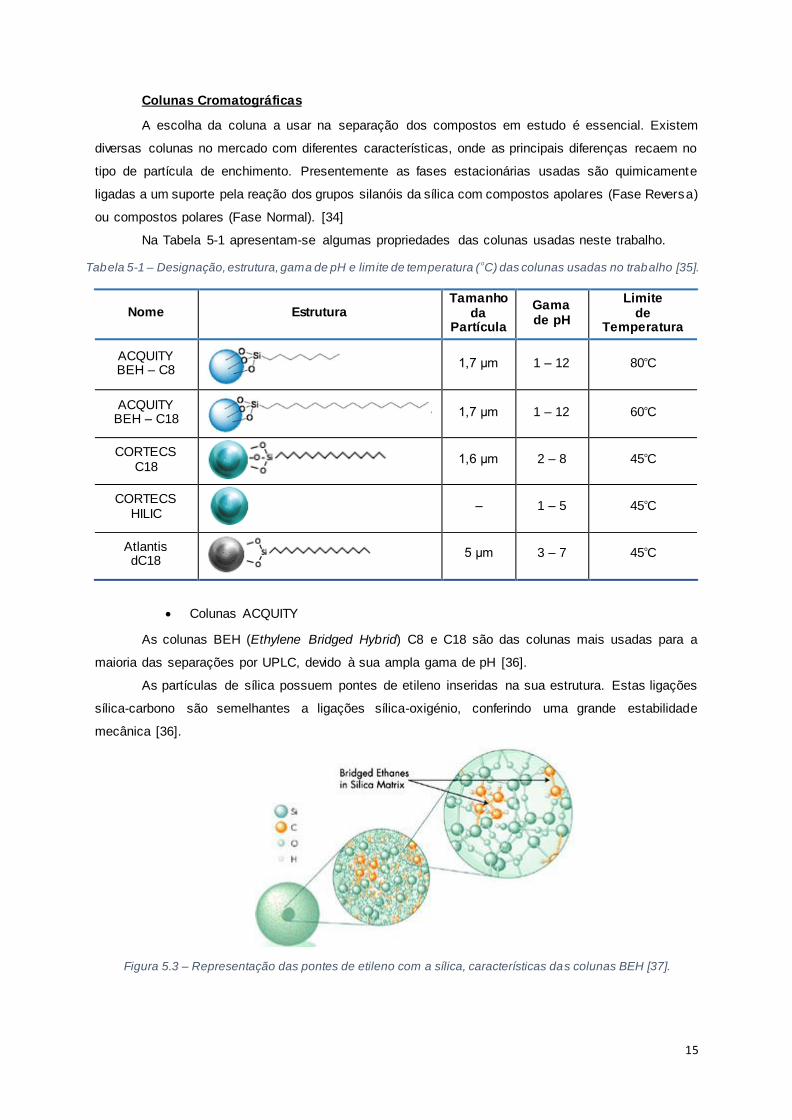
A escolha da coluna a usar na separação dos compostos em estudo é essencial. Existem
diversas colunas no mercado com diferentes características, onde as principais diferenças recaem no
tipo de partícula de enchimento. Presentemente as fases estacionárias usadas são quimicamente
ligadas a um suporte pela reação dos grupos silanóis da sílica com compostos apolares (Fase Reversa)
ou compostos polares (Fase Normal). [34]
Na Tabela 5-1 apresentam-se algumas propriedades das colunas usadas neste trabalho.
Tabela 5-1 – Designação, estrutura, gama de pH e limite de temperatura (ᵒC) das colunas usadas no trabalho [35].
Nome Estrutura Tamanho
da Partícula
Gama de pH
Limite de
Temperatura
ACQUITY BEH – C8
1,7 μm 1 – 12 80ᵒC
ACQUITY BEH – C18
1,7 μm 1 – 12 60ᵒC
CORTECS C18
1,6 μm 2 – 8 45ᵒC
CORTECS HILIC
– 1 – 5 45ᵒC
Atlantis dC18
5 μm 3 – 7 45ᵒC
Colunas ACQUITY
As colunas BEH (Ethylene Bridged Hybrid) C8 e C18 são das colunas mais usadas para a
maioria das separações por UPLC, devido à sua ampla gama de pH [36].
As partículas de sílica possuem pontes de etileno inseridas na sua estrutura. Estas ligações
sílica-carbono são semelhantes a ligações sílica-oxigénio, conferindo uma grande estabilidade
mecânica [36].
Figura 5.3 – Representação das pontes de etileno com a sílica, características das colunas BEH [37].

16
Colunas CORTECS
Estas colunas estão disponíveis em duas opções: fase reversa e cromatografia de interação
hidrofílica (HILIC). São caraterizadas pela sua flexibilidade e capacidade de desenvolver métodos para
separações difíceis com rapidez [38].
Colunas Atlantis
Este tipo de colunas é líder na indústria para a retenção e separação de compostos polares em
HPLC [39]. A coluna da Atlantis, dC18, tem como principal diferença relativamente às colunas C18,
a dupla ligação que a sílica faz com o núcleo da partícula.
No Anexo I encontram-se outras propriedades das colunas cromatográficas usadas neste
trabalho.
É importante referir a existência da pré-coluna (Figura 5.4) que se
encontra no topo das colunas cromatográficas. Tem o papel semelhante dos
filtros (muito comum em HPLC) em proteger a coluna de eventuais
entupimentos. O uso das pré-colunas é vital para a vida útil da coluna
cromatográfica, uma vez que a filtração da fase móvel e das amostras pode
não ser suficiente para eliminar impurezas. Pode-se dar o caso de existir
“retenção irreversível” que consiste na afinidade do contaminante pela própria
coluna que por ser tão intensa, não permite a sua eluição, ficando retido. Isto
traz problemas de perda de eficiência e aumento de pressão. A pré-coluna
faz com que os contaminantes fiquem retidos num pequeno filtro que possui.
5.1.2. Deteção por Espetrometria de Massa
A espetrometria de massa é uma técnica analítica usada para identificar compostos e
quantificá-los. Os princípios pelos quais esta técnica se rege envolvem a geração de iões para a
posterior deteção de acordo com a razão massa/carga (m/z). O que torna esta técnica sofisticada
quando associada à cromatografia líquida é o método usado para a formação desses iões que ocorre
à pressão atmosférica.
Instrumentação
O espetrómetro de massa (Figura 5.5) é constituído
por 3 diferentes componentes principais: a fonte de ionização,
o analisador e o detetor. No caso do presente trabalho
utilizou-se como fonte de iões o mecanismo de ionização por
electrospray (ESI) e um analisador do tipo triplo quadrupolo.
Figura 5.4 – Pré-coluna
usual em UPLC [61].
Figura 5.5 – Espetrómetro de massa Xevo TQD, Waters, usado no presente
trabalho.

17
Fonte de ionização
A ionização da amostra torna-se crucial, uma vez que a base da espetroscopia de massa passa
pela leitura de iões. Na ionização à pressão atmosférica (API) inclui-se a ionização por electrospray
(ESI), capaz de gerar iões à pressão atmosférica, em vez de vácuo. Esta técnica tem como
características distintas: a capacidade de produzir iões multiplamente carregados, com elevado número
de cargas o que permite a análise de compostos de elevada massa molecular, visto que a razão m/z é
reduzida. [40]
A amostra dissolvida no solvente proveniente da coluna cromatográfica é bombeada através
de um capilar de aço inoxidável, formando-se deste modo o cone de Taylor. O líquido passa a aerossol
à medida que vai saindo do capilar à pressão atmosférica, permitindo às gotas de dessolvatação
libertarem os iões cujo fluxo segue para o espectrómetro de massa, induzido pelos efeitos combinados
de atração electroestática e vácuo (ver Figura 5.6) [41].
A solução proveniente do equipamento de
separação (neste trabalho, UPLC) é pulverizada
electrostaticamente com a formação de pequenas gotas
altamente carregadas, sendo este processo auxiliado por
um gás nebulizador, N2. A carga dessas mesmas gotas tanto
pode ser positiva como negativa, consoante o potencial
aplicado no cone.
A geração de iões por electrospray dá-se por dois
diferentes passos: dispersão das gotas carregadas quase à
pressão atmosférica seguida da evaporação dessas
mesmas gotas.
O mecanismo que leva à formação de iões é descrita por dois diferentes modelos, uma vez que
ainda não existe consenso relativamente a este tópico. O primeiro modelo foi proposto por Dole em
1968, denominado por Modelo de Resíduo de Carga. Este modelo considera que à medida que o
solvente se evapora, a densidade de carga à superfície aumenta até que as forças repulsivas de
Coulomb entre as cargas superficiais excedem a tensão superficial, levando deste modo à divisão da
primeira gota. A divisão é contínua, até que cada gota formada conterá apenas uma molécula que
retém parte da carga inicial, ou seja, tem-se a formação de macro iões. Já o Modelo da Evaporação
Iónica, introduzido por Iribarne e Thomson em 1976, sugere que a evaporação do solvente leva a uma
instabilidade das gotas com razões elevadas de densidade de carga/raio da gota. A energia
electroestática associada à gota carregada torna-se suficientemente grande para desadsorver iões do
analito para a fase gasosa [40] [42].
Analisador de Massa
O analisador de massas separa os iões de acordo com a relação massa/carga (m/z). Um dos
tipos de analisadores mais usuais, e usado neste trabalho, é o Quadrupolo que funciona como um filtro
de massas.
Figura 5.6 – Esquema simplificativo da sonda na posição típica em frente ortogonal em relação ao cone. (adaptado de [41])

18
Após a entrada dos iões no cone, estes passam pelo Túnel de Iões (Hexapolo) que tanto
redireciona os iões, como os acelera. Os iões seguem para o chamado primeiro quadrupolo (Q1) onde
se dá a seleção do ião precursor. De seguida, os iões dão entrada no segundo quadrupolo (Q2) também
designado por célula de colisão. Na célula de colisão, é importante a energia de colisão que se adota
para cada composto, uma vez que com o auxílio de um gás pesado (árgon), o ião precursor é
fragmentado dando origem a iões produto. Estes iões produto passam então pelo terceiro quadrupolo
(Q3) onde são separados por variação da voltagem de radiofrequência, tal como sucede no primeiro
quadrupolo, seguindo depois para o detetor de massas como se pode observar na Figura 5.7 [43].
Figura 5.7 – Esquema representativo do analisador de massas usado no trabalho a trabalhar em MRM [44].
Dos diferentes modos de operação, foi escolhido o que impõe um máximo de seletividade e
sensibilidade, MRM, Multiple Reaction Monitoring. Neste modo, os dois quadupolos trabalham no modo
SIM (Selected Ion Monitoring) selecionando e focando iões de razão m/z específicas.
Este modo de operação permite focar nos iões precursor e produto por maiores períodos de tempo,
conseguindo-se desta forma um aumento na sensibilidade e seletividade.
Neste sentido são escolhidas pelo menos duas transições ião precursor-ião produto (MRM1 e
MRM2), a primeira usada para quantificação e a segunda para confirmação qualitativa do composto. A
razão MRM1/MRM2 é um importante parâmetro na confirmação da identidade dos compostos em
estudo. Esta razão (Equação 5-1) pode ser calculada através da área ou altura do pico que resulta de
cada uma das transições.
𝑅 =𝐴𝑀𝑅𝑀1
𝐴𝑀𝑅𝑀2 Equação 5-1
Em que R corresponde à razão das transições para um determinado composto, 𝐴𝑀𝑅𝑀1 corresponde à
área ou altura de pico do composto para a transição MRM1 e 𝐴𝑀𝑅𝑀2 corresponde à área ou altura de
pico do composto para a transição MRM2.
Detetor
Ao detetor cabem as funções de detetar e amplificar o sinal da corrente de iões que vem do
analisador e transferir o sinal respetivo para o sistema de processamento de dados. A conversão de
iões em eletrões dá-se num dínodo de conversão, sendo estes redirecionados para o multiplicador de
eletrões, onde se consegue aumentar o número de eletrões de forma exponencial, obtendo-se assim
uma carga suficientemente alta para posterior análise [45].

19
Na Figura 5.8 encontra-se representado o interior do espetrómetro de massa.
Figura 5.8 – Esquema do interior do equipamento de espetrometria usado no presente trabalho [46].
Acoplamento UPLC-MS/MS
Como benefícios da utilização de cromatografia líquida de ultra eficiência acoplada à ionização
por electrospray com deteção por massa tandem, destacam-se a diminuição do tempo de análise sem
comprometer a resolução cromatográfica e a diminuição da largura do pico cromatográfico,
“concentrando” mais o analito de interesse ao entrar na interface com o MS, aumentando assim a
sensibilidade do método [41].
5.2. Efeitos de Matriz
Evitar os efeitos de matriz “é quase como uma arte”.– (1999 – Constantopoulos [47])
Uma das questões mais proeminentes num trabalho que envolva análise quantitativa e
qualificativa de compostos é o Efeito de Matriz. Os efeitos de matriz podem advir de diferentes fontes,
tais como a própria amostra e a sua preparação [48].
O efeito que a matriz pode impor numa análise afeta a exatidão do método podendo conduzir
a valores por excesso ou por defeito da concentração do analito.
No caso do método usado (UPLC-ESI-MS/MS) os efeitos de matriz refletem-se podendo
proporcionar supressão ou enriquecimento iónico.
A supressão iónica pode ocorrer por diferentes mecanismos. Um dos mecanismos surge
porque existe uma competição pelas cargas disponíveis e pelo acesso à superfície da carga entre o
analito e os restantes componentes da matriz. Este processo é intrinsecamente competitivo uma vez
que serão ionizados preferencialmente aqueles compostos que tenham maior facilidade de ionização
e com maior concentração. Uma outra explicação para a supressão iónica baseia-se no facto de poder
existir na matriz, interferentes que devido à sua elevada concentração, aumentem a viscosidade e
tensão superficial das gotas, alterando deste modo a eficiência da sua formação, o que interfere na
quantidade de iões do analito de interesse que chegam efetivamente ao analisador de massas [45].
No caso de existir enriquecimento iónico, este acontece devido à existência de interferentes
cujos iões fragmentados com determinado valor de m/z são muito semelhantes aos dos analitos em
estudo. Isto leva a uma sobreposição de picos, ou seja, está-se perante um caso de aumento de sinal,
conferindo deste modo um erro na análise efetuada [48].

20
Não existe uma solução universal para o problema, mas existem diferentes estratégias para
lidar com os efeitos de matriz.
5.3. Cromatografia Gasosa
Como já referido anteriormente, a análise de ácidos haloacéticos encontra-se implementada
pela técnica EPA502 no LAIST. Por esta razão é descrita muito sucintamente a cromatografia gasosa
com deteção por captura de eletrões.
Resumidamente numa separação por cromatografia gasosa, a amostra é introduzida pelo
injetor que se encontra aquecido a uma temperatura superior à do ponto de ebulição dos componentes
da amostra. Deste modo, a amostra é volatizada completa e rapidamente sendo então transportada
pela fase móvel (gás de arraste) através da coluna cromatográfica que contém a fase estacionária.
A separação dos componentes dá-se então na coluna onde a afinidade entre os analitos e a fase
estacionária ditam o tempo de eluição. Já com os compostos separados, o gás de arraste encarrega -
se de os transportar até ao detetor onde é gerado um sinal elétrico [31].
5.3.1. Deteção de Captura de Eletrões
O detetor de captura de eletrões (ECD) responde seletivamente a compostos orgânicos
contendo halogéneos como é o caso dos ácidos haloacéticos. Neste detetor, a amostra eluída da
coluna passa sobre uma fonte radioativa emissora. Um eletrão do emissor causa a ionização do gás,
produzindo uma rajada de eletrões, que gera um sinal elétrico. O sinal emitido por cada um dos analitos
em função do tempo revela-se no cromatograma, onde as áreas de cada pico são proporcionais à sua
concentração [31].

21
6. Validação do Método Analítico – Qualidade
Em análises químicas um dos principais requisitos é a qualidade dos resultados obtidos. Deste
modo a acreditação dos Laboratórios é essencial. Esta acreditação é efetuada através de uma
avaliação por um organismo de acreditação. Em Portugal, esse organismo é o Instituto Português de
Acreditação (IPAC) que impõe o cumprimento da norma ISO/IEC 17025. Esta norma estabelece
requisitos e procedimentos que permitem avaliar a competência técnica na realização de ensaios e
calibrações [49].
“É fundamental que os Laboratórios disponham de meios e critérios objetivos, para
demonstrarem, através da Validação, que os métodos internos de ensaio que executam conduzem a
resultados credíveis e adequados à qualidade pretendida.” – (2000, RELACRE [50])
O objetivo da validação de um método analítico passa por demonstrar que o método é
adequado para a quantificação de analitos numa determinada matriz de forma exata e precisa.
De seguida encontram-se breves definições dos principais critérios de validação que são
utilizados com a finalidade de validar um método analítico:
Âmbito do Método
O tipo de amostras a que o método se aplica, os compostos químicos que se determinam e as
possíveis interferências ao qual se está sujeito devem-se encontrar bem definidos [51].
Gama de Trabalho
A gama de trabalho pode ser obtida a partir de estudos de linearidade verificando-se a exatidão
e precisão nos extremos (Mínimo e Máximo). A concentração mais baixa deverá corresponder a
concentração expetável das amostras, e sempre que o valor obtido da amostra supere o valor máximo
imposto na gama, deverão ser feitas diluições [51].
6.1. Calibração
A calibração define-se como a relação estabelecida entre os valores obtidos pelo instrumento
de medição (sinal) e os valores correspondentes das grandezas estabelecidas por padrões
(concentrações). A regressão linear pelo método dos mínimos quadrados é o modelo habitualmente
utilizado para a calibração.
Para que as calibrações obtidas sejam aceites existem critérios que têm de ser estabelecidos,
nomeadamente a linearidade. Para este critério podem ser aplicados diferentes testes tais como a
análises de resíduos, os testes de Mandel e Rikilt e o coeficiente de correlação (R) [51] [52].
6.1.1. Método do Padrão Externo
Neste método são adicionadas diferentes quantidades de analito a uma quantidade de matriz
isenta do composto em análise. Após o traçar da curva, é analisada a amostra e o valor obtido da
concentração do analito na amostra é dado por interpolação da ,
𝑌 = 𝑎𝐶𝐴 + 𝑏 <=> 𝐶𝐴 =𝑌−𝑏
𝑎 Equação 6-1
Onde 𝐶𝐴 corresponde à concentração do analito da amostra, 𝑌 corresponde ao sinal do analito na
amostra, 𝑎 corresponde ao declive da curva e 𝑏 corresponde à ordenada na origem [52].

22
6.1.2. Método de Adição de Padrão
De modo a minimizar os efeitos de matriz usa-se o método de adição de padrão quando se
trabalha com matrizes muito complexas que provocam fortes interações com o analito.
Neste método fazem-se adições de diferentes quantidades de analito a quantidades iguais de
matriz que contem uma quantidade desconhecida do analito em questão. Constrói-se a curva de
calibração com os padrões referidos e é na interseção da curva com a linha das abcissas que se
encontra o valor da concentração (em módulo) da amostra sem a adição do analito
(ver Figura 6.1) [52].
Figura 6.1 – Representação gráfica exemplificativa do método da adição de padrão.
Para se determinar a concentração do analito na amostra basta igualar a zero a Equação 6-2
da curva de calibração,
𝑌 = 0 = 𝑎𝐶𝐴 + 𝑏 <=> 𝐶𝐴 = |0−𝑏
𝑎| Equação 6-2
Onde 𝐶𝐴 corresponde à concentração do analito da amostra, 𝑎 corresponde ao declive da curva e 𝑏
corresponde à ordenada na origem.
6.1.3. Método do Padrão Interno
Este método consiste na adição de um padrão interno tanto aos padrões da reta de calibração
como às amostras imediatamente antes destas serem analisadas. O padrão interno é um composto
similar ao analito que não pode estar presente na amostra antes da sua adição, não pode coeluir com
os analitos ou com impurezas. A quantidade adicionada aos padrões da reta é habitualmente a mesma
adicionada às amostras a serem analisadas de forma a verificar-se circunstância de igualdade.
Neste caso a Equação 6-3 corresponde à curva de calibração obtida,
𝑆𝐴
𝑆𝑃.𝐼.= 𝑎𝐶𝐴 + 𝑏 <=> 𝐶𝐴 =
𝑆𝐴𝑆𝑃.𝐼.
−𝑏
𝑎 Equação 6-3
Onde 𝑆𝐴 corresponde ao sinal do analito, 𝑆𝑃.𝐼. corresponde ao sinal do padrão interno, 𝐶𝐴 corresponde
à concentração do analito, 𝑎 corresponde ao declive da curva e 𝑏 corresponde à ordenada na
origem [52].
Sin
al
Concentração do analito adicionado à amostra
Concentração do
analito na amostra

23
6.1.4. Método “Matrix-Matched”
Este método de calibração recorre a padrões com sobreposição de matriz, isto é, padrões com
uma composição igual ou similar da amostra a analisar. A disponibilidade deste tipo de matriz é difícil
de encontrar, uma vez que a matriz em causa não poderá conter o analito que se quer determinar.
O método da sobreposição da matriz tem como objetivo minimizar os efeitos de matriz que
influenciam a análise dos analitos [53].
6.2. Limiares Analíticos
6.2.1. Limite de Deteção – LD
Este limite corresponde ao início da gama de trabalho em que é possível distinguir com uma
dada confiança estatística, o sinal do branco do sinal da amostra. A determinação deste limite pode ser
efetuada de diferentes formas [51] [52], sendo que em cromatografia se faz como descrito nos pontos
que se seguem,
- Quando se usa uma calibração linear tem-se que,
𝐿𝐷 =3.3×𝑆𝑦/𝑥
𝑎 Equação 6-4
Em que 𝑎 é o declive da curva de calibração, 𝑆𝑦/𝑥 o respetivo desvio-padrão e a corresponde ao declive
da curva.
- Por fim, pode ser baseado na razão sinal/ruído (S/N)> 3, nos métodos cujo ruído da linha de
base é possível de ser medido.
6.2.2. Limite de Quantificação – LQ
No caso do limite de quantificação, este corresponde à menor concentração medida a partir da
qual é possível a quantificação do analito com determinada exatidão e precisão. Em termos práticos
corresponde ao primeiro padrão de calibração (primeiro ponto da reta). De forma a ser validado, o
coeficiente de variação para os padrões repetidos de concentração correspondente ao LQ, não deverá
exceder os 10% [51].
Este pode ser calculado pela Equação 6-5,
𝐿𝑄 = 3,3 × 𝐿𝐷 Equação 6-5
- Quando se usa uma calibração linear tem-se o LQ pela Equação 6-6,
𝐿𝑄 = |10×𝑆𝑦/𝑥
𝑎| Equação 6-6
Em que a é o declive da curva de calibração, 𝑆𝑦/𝑥 o respetivo desvio-padrão, a corresponde ao
declive da curva [52].
- Através das repetibilidades verifica-se que o LQ pode ser calculado pela Equação 6-7,
𝐿𝑄 = |10 × 𝑆𝑦/𝑥 𝑟𝑒𝑝.| Equação 6-7
Em que 𝑆𝑦/𝑥𝑟𝑒𝑝.é o desvio-padrão para n repetibilidades.

24
- Pela análise do sinal de ensaios consecutivos de um branco (ver Equação 6-8),
𝐿𝑄 = 𝑆𝑏𝑟𝑎𝑛𝑐𝑜̅̅ ̅̅ ̅̅ ̅̅ ̅ + 10 × 𝑆𝑦/𝑥𝑏𝑟𝑎𝑛𝑐𝑜
Equação 6-8
Em que 𝑆𝑏𝑟𝑎𝑛𝑐𝑜̅̅ ̅̅ ̅̅ ̅̅ ̅ é a média do sinal obtido para 10 brancos e 𝑆𝑦/𝑥𝑏𝑟𝑎𝑛𝑐𝑜
é o desvio-padrão
respetivo.
6.3. Precisão
A precisão permite avaliar a dispersão de resultados entre ensaios independentes , repetidos
sobre uma mesma amostra. Esta dispersão é avaliada pela repetibilidade e pela reprodutibilidade.
Existe ainda uma precisão intermédia ou variabilidade intralaboratorial.
A precisão pode ser expressa em termos de desvio padrão ou pelo coeficiente de variação [51].
6.3.1. Repetibilidade
Esta medida exprime a precisão de um método de ensaio em condições idênticas, ou seja,
refere-se a ensaios efetuados sobre uma mesma amostra, nas condições tão estáveis quanto possível,
tais como: mesmo laboratório, mesmo analista, mesmo equipamento, mesmo tipo de reagentes entre
outros [51].
Assumindo uma distribuição normal e uma probabilidade de 95% tem-se a Equação 6-9,
𝑅𝑒𝑝 = 𝑟 = 1,96 × √2 × 𝑠𝑟 Equação 6-9
Em que 𝑠𝑟 corresponde ao desvio padrão da repetibilidade.
O coeficiente de variação para a repetibilidade é dado pela Equação 6-10
𝐶𝑉𝑟 =𝑠𝑟
𝑥̅× 100 Equação 6-10
6.3.2. Reprodutibilidade
A reprodutibilidade refere-se à precisão obtida quando se variam as condições de medição
(diferentes laboratórios, diferentes analistas, equipamentos) para a mesma amostra. Neste caso a
precisão é obtida através de ensaios interlaboratoriais.
O cálculo da reprodutibilidade é realizada do mesmo modo que a repetibilidade (Equação 6-9,
Equação 6-10) [51].
6.3.3. Precisão Intermédia
A precisão intermédia acaba por ser a ponte entre a repetibilidade e a reprodutibilidade, pois
permite identificar o efeito das variações dentro do mesmo laboratório fazendo variar o analista, o
equipamento ou diferentes dias de análise.
Esta precisão pode ser calculada através do coeficiente de variação, CV (Equação 6-10) [51].
6.4. Exatidão
Para um método ser exato, é necessário que não contenha erros sistemáticos, ou seja, os
resultados obtidos refletem o valor real do analito em estudo. Este parâmetro pode ser avaliado através
de diferentes processos, tais como, ensaios de recuperação, materiais de referência certificados,
ensaios interlaboratoriais e método de referência [51].

25
6.4.1. Ensaios de Recuperação
Nestes ensaios são adicionadas quantidades conhecidas do analito em estudo a amostras, que
tanto podem conter ou não o analito. Devem ser adicionadas quantidades correspondentes a
concentrações variadas: LQ, meio da gama de trabalho, máximo da gama de trabalho.
No fim de se analisarem estes ensaios, analisa-se o branco, que neste caso é a amostra sem
ser fortificada. Obtém-se deste modo a taxa de recuperação do analito na amostra, como se verifica
pela Equação 6-11,
%𝑅𝑒𝑐 =𝐶𝑠 −𝐶𝑟
𝐶𝑎× 100 Equação 6-11
Em que 𝐶𝑠 corresponde à concentração da amostra fortificada com o analito, 𝐶𝑟 corresponde à
concentração da amostra tal e qual e 𝐶𝑎 corresponde à concentração do analito adicionado à amostra.
São realizados n ensaios permitindo assim existir uma comparação estatística da taxa de
recuperação observada com a taxa de recuperação teórica (100%). Deve-se salientar que ao longo da
gama de trabalho a taxa de recuperação pode variar [51].
6.4.2. MRC
Os Materiais de Referência Certificados são uma ferramenta importante para a avaliação da
exatidão de um método.
Um MRC possui um valor de concentração do analito com a incerteza associada. Com esta
análise consegue-se avaliar o desempenho do laboratório, pois ao existir a comparação do valor obtido
na análise com o valor conhecido do MRC, consegue-se verificar a exatidão do método [51].
6.4.3. Ensaios Interlaboratoriais
Uma outra ferramenta importante na avaliação da exatidão de um método são os ensaios
interlaboratoriais. Estes ensaios permitem avaliar o resultado obtido apos a análise de uma amostra
cedida pela entidade certificada que coordena o ensaio, em comparação com o valor que amostra
realmente tem. A participação nestes ensaios tem duas grandes condicionantes, que passam pela
existência dos mesmos e os elevados custos associados. Para avaliar o desempenho na participação
num ensaio interlaboratorial, é calculado o fator de desempenho Z-score e o erro normalizado. [51]
6.4.4. Método de Referência
A comparação de resultados obtidos entre o método em estudo e um outro método de
referência permite também avaliar a exatidão do método.
Em termos práticos a mesma amostra é analisada por dois diferentes métodos, e os resultados
são comparados tendo em atenção a média e o desvio padrão de cada método. O nível de confiança
exigido é usualmente de 5% [51].
6.5. Robustez
A robustez de um método avalia a sensibilidade que este apresenta quando sujeito a pequenas
variações. Quanto mais robusto o método, maior a precisão visto que existe uma diminuição dos erros
aleatórios. Em análises cromatográficas, as variáveis que podem afetar a robustez estão diretamente
associadas ao tipo de colunas, às temperaturas, fluxos, entre outras. Neste teste podem ser usados,

26
por exemplo, MRC’s uma vez que é conhecida a concentração do analito, e fazendo-se variar uma
variável de cada vez verifica-se a influência de cada uma das variáveis em estudo [51].
6.6. Sensibilidade
Traduz-se como a capacidade de determinar e distinguir pequenas diferenças de
concentrações do analito na matriz. A sensibilidade pode ser definida como o quociente entre o
acréscimo do valor lido e a variação da concentração respetiva (Equação 6-12).
𝑆𝑒𝑛𝑠𝑖𝑏𝑖𝑙𝑖𝑑𝑎𝑑𝑒 =∆𝑆𝑖𝑛𝑎𝑙
∆𝐶𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎çã𝑜 Equação 6-12
Mais concretamente, a sensibilidade será igual ao declive da reta de calibração (polinómio de
1ºgrau) sendo constante ao longo de toda a gama de trabalho. Deste modo, um método será mais
sensível do que o outro se para a mesma variação de concentração a variação de sinal for
superior [51].
6.7. Seletividade e Especificidade
Define-se como a capacidade de identificar e distinguir um analito em particular numa matriz
complexa sem a interferência dos restantes compostos que a constituem. Quando se oferecem
garantias de que o resultado obtido provém apenas do analito em estudo e não de outras possíveis
substâncias presentes na amostra, diz-se que o método é específico. O estudo destes dois conceitos
é muito importante em análises laboratoriais devido à presença de interferentes que podem conduzir a
erros aleatórios e sistemáticos. Para se identificar a presença destes interferentes é usual recorrer-se
a testes de recuperação. Se as percentagens destes testes forem próximos de 100% e constantes, o
método analítico é considerado aplicável, isto é, específico e seletivo [51].
6.8. Incertezas
"Parâmetro associado ao resultado de uma medição que caracteriza a dispersão de valores
que se pode razoavelmente atribuir à grandeza medida" [50].
A incerteza global para cada composto foi estimada com base nas incertezas da precisão e da
exatidão como se verifica pela Equação 6-13 (ISO-11352).
𝑢𝑐𝑜𝑚𝑏𝑖𝑛𝑎𝑑𝑎 = √𝑢𝑝𝑎𝑑𝑟õ𝑒𝑠 𝑑𝑒 𝑐𝑜𝑛𝑡𝑟𝑜𝑙𝑜2 + 𝑢𝑑𝑢𝑝𝑙𝑖𝑐𝑎𝑑𝑜𝑠
2 + 𝑢𝑒𝑥𝑎𝑡𝑖𝑑ã𝑜2
Equação 6-13
De forma a majorar cada uma das incertezas, estima-se a incerteza expandida (Equação 6-14)
que consiste em duplicar o valor da incerteza combinada.
𝑢𝑒𝑥𝑝𝑎𝑛𝑑𝑖𝑑𝑎 = 2 × 𝑢𝑐𝑜𝑚𝑏𝑖𝑛𝑎𝑑𝑎
Equação 6-14
A incerteza associada à precisão, foi estimada com base nos dados de precisão intermédia
usando a Equação 6-15,
𝑢𝑝𝑟𝑒𝑐𝑖𝑠ã𝑜 =%𝑅𝑆𝐷
√3 Equação 6-15
Em que %RSD corresponde ao desvio padrão relativo para a concentração de limite de quantificação,
seja dos padrões de controlo, seja dos padrões em duplicado.

27
A incerteza associada à exatidão foi estimada com base nos dados das recuperações, como
se verifica na Equação 6-16,
𝑢𝑒𝑥𝑎𝑡𝑖𝑑ã𝑜 =
𝑆𝑟𝑒𝑐
𝑅𝑒𝑐̅̅ ̅̅ ̅
√𝑛
Equação 6-16
Em que 𝑅𝑒𝑐̅̅ ̅̅ ̅ corresponde à média da recuperação do analito, 𝑆𝑟𝑒𝑐 corresponde ao desvio-
padrão respetivo e 𝑛 corresponde ao número de ensaios.
Com o valor da incerteza expandida estimado, a concentração obtida numa amostra para
determinado analito deverá ser apresentada como se demonstra na Equação 6-17,
𝐶 = 𝐶𝑜𝑏𝑡𝑖𝑑𝑜 ± 𝐶𝑜𝑏𝑡𝑖𝑑𝑜 × 𝑢𝑒𝑥𝑝𝑎𝑛𝑑𝑖𝑑𝑎
Equação 6-17
“Quando a incerteza for corretamente estimada, ela pode ser considerada como uma medida
da qualidade do resultado, isto é, quanto menor a incerteza, maior a qualidade.” [50]

28

29
7. Técnica Experimental
7.1. Equipamento Cromatógrafo líquido de ultra eficiência – Modelo Acquity da Waters
- Coluna ACQUITY UPLC BEH C18 1.7μm
- Coluna ACQUITY UPLC BEH C8 1.7μm
- Coluna CORTECS™ UPLC C18 1.6 μm
- Coluna CORTECS™ UPLC HILIC 1.6 μm
- Coluna Atlantis dC18 5µm
- Desgaseificador
- Injetor
- Bomba quaternária
- Pré-coluna
Espetrómetro de massa – Modelo Xevo TQD, API (ESI)
- Fonte de Ionização do tipo electrospray
- Bombas de vácuo
- Analisador do tipo triplo quadrupolo
Software MassLynx V4.1
Sistema de obtenção de água ultra pura – Modelo Milli-Q
Balança analítica – Modelo AX205 Delta Range
7.2. Material
Seringas de vidro de 1000, 500, 250, 100, 50, 25 μL
Balões volumétricos de 5, 10, 20, 25, 50, 100, 500 mL
Vials de 2mL de vidro com tampa pré-furada para injeção automática (WATERS)
Micropipetas de 100, 1000 μL
7.3. Reagentes
Padrões puros no estado sólido (pureza>99%), de cada um dos HAAs (Riedel de H�̈�en)
[MCAA, DCAA, TCAA, MBAA, DBAA, TBAA, BCAA]
MTBE (Acros Organics)
Metanol (for HPLC-GOLD-Ultragradient – CARLOERBA reagentes)
Água ultra pura – Modelo Milli Q
Acetonitrilo (for HPLC-GOLD-Ultragradient – CARLOERBA reagentes)
Ácido Acético (ABSOLVE)
Formato de Amónio (ABSOLVE)
Ácido Fórmico (ABSOLVE)
Tiossulfato (Panreac)
Ácido ascórbico (ABSOLVE)
Árgon como gás de colisão e azoto produzido por gerador

30
7.4. Preparação de Soluções
Eluente
Foram usados diferentes eluentes consoante a coluna cromatográfica que se testava.
Na coluna HILIC, o eluente usado correspondeu a acetonitrilo e uma solução aquosa com 5 mM de
ácido fórmico com 10 mM de formato de amónio. Já nas colunas C18 (ACQUITY e BEH) o eluente
correspondeu a acetonitrilo e uma solução aquosa com 0,1% de ácido acético. Nas colunas C8
(ACQUITY) e dC18 foi testado o eluente anteriormente referido, verificando-se significativas melhorias
quando se usou o metanol, em vez de acetonitrilo. Esta fase móvel segue um programa de eluição com
gradiente entre uma solução aquosa contendo 0,2% de ácido acético (A), e metanol (B).
Apesar do equipamento proporcionar uma desgaseificação aos eluentes usados, de modo a
salvaguardar a não entrada de bolhas de ar no sistema, o eluente no fim de preparado é colocado no
banho do Ultrassons onde sofre desgaseificação por três minutos.
Soluções Padrão
As soluções-mãe foram preparadas a partir de padrões sólidos de cada HAA, em balões de
10mL em éter metil-terc-butílico (MTBE) com uma concentração aproximada de 1 g/L. Estas soluções
são guardadas no congelador em vials de 10 mL.
A partir das soluções-mãe prepararam-se em cada sessão de trabalho, 25 mL de um padrão
misto em metanol com uma concentração aproximada de 1 mg/L.
Por fim, as soluções padrão para traçar as curvas de calibração são feitas a partir da solução
intermédia, em balões de 5 mL. As concentrações para os 6 padrões da reta de calibração variam entre
5 a 50 μg/L nos dois métodos de calibração.
Todos os padrões são ainda acidificados com uma solução ácida, de modo a que cada padrão
contenha uma concentração de 0,1% de ácido acético e 30 mg/L de ácido ascórbico. Os cálculos para
as soluções intermédias e para as soluções padrão da reta de calibração encontram-se no Anexo II.
Na Figura 7.1 apresenta-se um esquema que resume as soluções necessárias para traçar a
reta de calibração, tanto pelo método de padrão externo como para o método de adição de padrão,

31
7.5. Procedimento Experimental
7.5.1. Segurança Durante todo o trabalho desenvolvido no LAIST foram respeitadas as regras pelo qual o
laboratório de rege, nomeadamente o manuseio de solventes ser efetuado nas hotes e a proteção
pessoal (bata, luvas e óculos).
7.5.2. Descontaminação de Material
Todo o material de vidro graduado utilizado no decorrer deste trabalho foi devidamente
descontaminado com os solventes usados.
7.5.3. Otimização dos parâmetros de espetrometria de massa
No sentido de se obter o ião precursor de cada HAA que resulta da ionização do analito, e os
iões produto que resultam da fragmentação do ião precursor, é realizada primeiramente a otimização
das condições de trabalho no espetrómetro de massa.
Figura 7.1 – Esquema representativo de uma sessão de trabalho na preparação de uma reta de calibração.
MC
AA
DC
AA
TC
AA
MB
AA
DB
AA
BC
AA
1 2 3 4 5 6
Solução – Intermédia ≈ 1 mg/L (em MeOH)
Soluções – Mãe ≈ 1 g/L (em MTBE)
Soluções – Curva de Calibração
≈ 5, 10, 20, 30, 40 e 50 μg/L
(em Água)
Solução Ácida (1:100) ≈ 10% Ácido Acético ≈ 3 g/L Ácido Ascórbico

32
Na página do Tune Page (Tabela 7-1) encontram-se os valores standard do equipamento
referentes à fonte de ionização em Electrospray no modo negativo, e referentes ao analisador de
massa.
Tabela 7-1 – Parâmetros usados na otimização do espetrómetro de massa.
Temperatura da fonte 150˚C
Gás de dessolvatação Azoto
Temperatura de dessolvatação 500˚C
Caudal de gás de dessolvatação 1000 L/h
Caudal de gás de cone 50 L/h
Gás de colisão Árgon
Modo de Ionização ESI negativo
Voltagem do Capilar 0,5 kV
As condições ótimas para a formação dos iões precursores de cada HAA foram obtidas com a
infusão de soluções individuais de concentração 2 mg/L. O modo de aquisição utilizado é em full scan
no primeiro quadrupolo para verificar quais os iões precursores mais intensos para cada compos to,
fazendo variar a voltagem do cone. Com os iões precursores bem definidos, procedeu-se à escolha
dos iões produto, impondo o primeiro quadrupolo a funcionar em modo SIM e o terceiro quadrupolo em
modo full scan. Para tal fez-se variar a energia de colisão no segundo quadrupolo (célula de colisão)
de modo a fragmentar o ião precursor. O objetivo de se obterem dois iões produto para cada composto,
resulta da necessidade de calcular a razão das massas (MRM1/MRM2), uma vez que é um parâmetro
importante na identificação do composto.
De seguida otimizou-se o Dwell de cada um dos ácidos haloacéticos (Tabela 8-1), isto é, o
tempo que o equipamento despende a procurar o ião em causa, para que cada pico tenha pontos
suficientes (13 pontos) aumentando deste modo a sensibilidade e seletividade ao analito.
A intensidade foi o fator determinante na escolha dos iões precursores tal como na escolha dos
iões produto. Os resultados obtidos encontram-se descriminados na Tabela 8-1.
É importante referir que é realizada mensalmente uma calibração do espetrómetro de massa
com uma solução da Waters XevoTQD – Sample Kit Calibration Solution, permitindo que o
espetrómetro de massa opere com o melhor desempenho possível.
7.5.4. Otimização dos parâmetros de cromatografia
Com o intuito de se otimizar a separação cromatográfica foram realizados diferentes testes
onde se impuseram diferentes condições, em diferentes colunas.
Uma vez que existiam diferentes colunas disponíveis no LAIST, foram feitas experiências em
cada uma delas de forma a verificar qual a que apresentava maior sensibilidade para com os ácidos
haloacéticos. As colunas testadas foram:
ACQUITY UPLC BEH C18 1.7μm;
CORTECS™ UPLC C18 1.6 μm;
CORTECS™ UPLC HILIC 1.6 μm;
Atlantis dC18 5 μm;
CORTECS™ UPLC BEH C8 1.7 μm

33
No Anexo I encontram-se as características com maior detalhe de cada uma das colunas
testadas.
Para um conjunto de condições experimentais fixas (parâmetros da espetrometria de massa)
selecionou-se a coluna cromatográfica que permitiu obter picos com maior altura e menor largura de
base. Com a coluna escolhida, seguiu-se então para um diferente conjunto de testes que passaram por
variar as composições do eluente, modificar o programa de eluição, variar a temperatura da coluna e o
volume de injeção, e ainda verificar a influência do solvente usado na preparação dos padrões.
7.5.5. Métodos de Calibração
Primeiramente o método de calibração usado foi o método do padrão externo. Quando se
procedeu à análise das recuperações verificou-se que os picos correspondentes aos ácidos
monohalogenados (MCAA e MBAA) apresentavam-se de forma irregular, com recuperações pequenas
(Rec ≤ 80%). Por esta razão foi testado o método de adição de padrão. Como já referido anteriormente
no capítulo 5.2, este método minimiza os problemas de matriz. Deste modo conseguem-se picos com
melhor forma apesar de as áreas permanecerem baixas, no caso do MCAA. Uma vez que se obtiveram
recuperações ≥ 80% para os restantes ácidos, o método de padrão externo continuou a ser usado de
forma a permitir uma comparação entre métodos de calibração.
A calibração é necessária em cada sessão de trabalho, uma vez que existem sempre flutuações
nos valores obtidos das retas de calibração, nomeadamente nos declives como se verifica pela Tabela
8-4 e Tabela 8-5.
7.5.6. Comparação de Técnicas
Uma vez que, como já referido, se encontra implementada no LAIST a técnica de análise de
ácidos haloacéticos por GC-ECD, faz todo o sentido existir uma comparação de resultados para uma
mesma amostra.
Este método encontra-se validado para a determinação de quatro ácidos haloacéticos: os
clorados e o dibromado. Devido à polaridade e tamanho dos HAAs, é necessária a derivatização e
posterior extração dos analitos em compostos que possam ser determinados por GC-ECD. O método
de calibração usado nesta técnica é o padrão interno e os limites de quantificação são 10 μg/L para o
MCAA, e 1 μg/L para o DCAA, TCAA e DBAA.
Foram realizados ensaios em diferentes amostras, permitindo a comparação dos resultados.

34
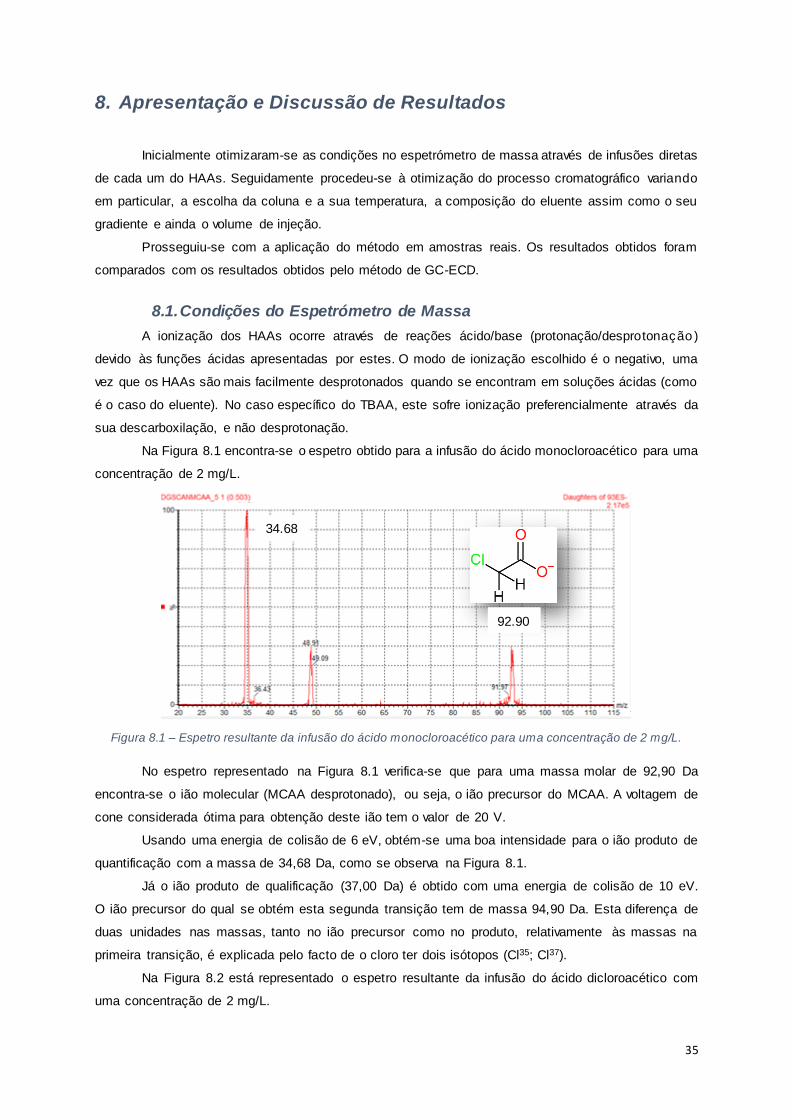
35
8. Apresentação e Discussão de Resultados
Inicialmente otimizaram-se as condições no espetrómetro de massa através de infusões diretas
de cada um do HAAs. Seguidamente procedeu-se à otimização do processo cromatográfico variando
em particular, a escolha da coluna e a sua temperatura, a composição do eluente assim como o seu
gradiente e ainda o volume de injeção.
Prosseguiu-se com a aplicação do método em amostras reais. Os resultados obtidos foram
comparados com os resultados obtidos pelo método de GC-ECD.
8.1. Condições do Espetrómetro de Massa
A ionização dos HAAs ocorre através de reações ácido/base (protonação/desprotonação )
devido às funções ácidas apresentadas por estes. O modo de ionização escolhido é o negativo, uma
vez que os HAAs são mais facilmente desprotonados quando se encontram em soluções ácidas (como
é o caso do eluente). No caso específico do TBAA, este sofre ionização preferencialmente através da
sua descarboxilação, e não desprotonação.
Na Figura 8.1 encontra-se o espetro obtido para a infusão do ácido monocloroacético para uma
concentração de 2 mg/L.
Figura 8.1 – Espetro resultante da infusão do ácido monocloroacético para uma concentração de 2 mg/L.
No espetro representado na Figura 8.1 verifica-se que para uma massa molar de 92,90 Da
encontra-se o ião molecular (MCAA desprotonado), ou seja, o ião precursor do MCAA. A voltagem de
cone considerada ótima para obtenção deste ião tem o valor de 20 V.
Usando uma energia de colisão de 6 eV, obtém-se uma boa intensidade para o ião produto de
quantificação com a massa de 34,68 Da, como se observa na Figura 8.1.
Já o ião produto de qualificação (37,00 Da) é obtido com uma energia de colisão de 10 eV.
O ião precursor do qual se obtém esta segunda transição tem de massa 94,90 Da. Esta diferença de
duas unidades nas massas, tanto no ião precursor como no produto, relativamente às massas na
primeira transição, é explicada pelo facto de o cloro ter dois isótopos (Cl35; Cl37).
Na Figura 8.2 está representado o espetro resultante da infusão do ácido dicloroacético com
uma concentração de 2 mg/L.
92.90
34.68
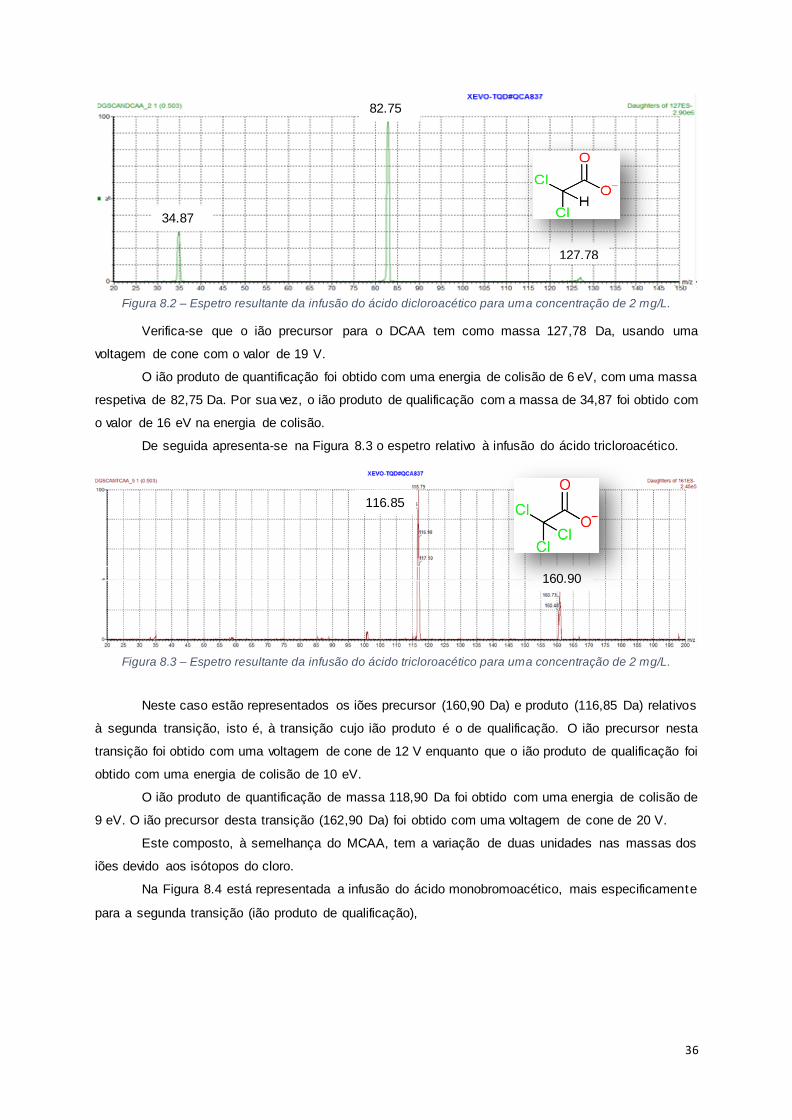
36
Figura 8.2 – Espetro resultante da infusão do ácido dicloroacético para uma concentração de 2 mg/L.
Verifica-se que o ião precursor para o DCAA tem como massa 127,78 Da, usando uma
voltagem de cone com o valor de 19 V.
O ião produto de quantificação foi obtido com uma energia de colisão de 6 eV, com uma massa
respetiva de 82,75 Da. Por sua vez, o ião produto de qualificação com a massa de 34,87 foi obtido com
o valor de 16 eV na energia de colisão.
De seguida apresenta-se na Figura 8.3 o espetro relativo à infusão do ácido tricloroacético.
Figura 8.3 – Espetro resultante da infusão do ácido tricloroacético para uma concentração de 2 mg/L.
Neste caso estão representados os iões precursor (160,90 Da) e produto (116,85 Da) relativos
à segunda transição, isto é, à transição cujo ião produto é o de qualificação. O ião precursor nesta
transição foi obtido com uma voltagem de cone de 12 V enquanto que o ião produto de qualificação foi
obtido com uma energia de colisão de 10 eV.
O ião produto de quantificação de massa 118,90 Da foi obtido com uma energia de colisão de
9 eV. O ião precursor desta transição (162,90 Da) foi obtido com uma voltagem de cone de 20 V.
Este composto, à semelhança do MCAA, tem a variação de duas unidades nas massas dos
iões devido aos isótopos do cloro.
Na Figura 8.4 está representada a infusão do ácido monobromoacético, mais especificamente
para a segunda transição (ião produto de qualificação),
127.78
160.90
82.75
34.87
116.85
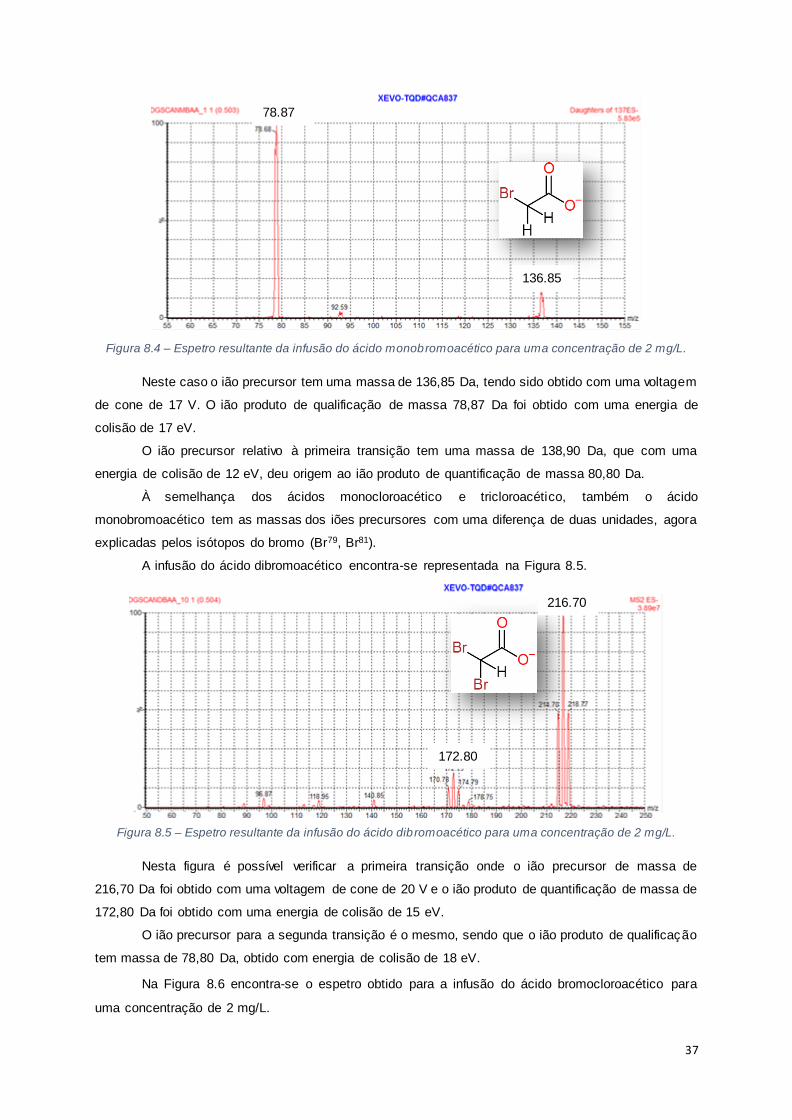
37
Figura 8.4 – Espetro resultante da infusão do ácido monobromoacético para uma concentração de 2 mg/L.
Neste caso o ião precursor tem uma massa de 136,85 Da, tendo sido obtido com uma voltagem
de cone de 17 V. O ião produto de qualificação de massa 78,87 Da foi obtido com uma energia de
colisão de 17 eV.
O ião precursor relativo à primeira transição tem uma massa de 138,90 Da, que com uma
energia de colisão de 12 eV, deu origem ao ião produto de quantificação de massa 80,80 Da.
À semelhança dos ácidos monocloroacético e tricloroacético, também o ácido
monobromoacético tem as massas dos iões precursores com uma diferença de duas unidades, agora
explicadas pelos isótopos do bromo (Br79, Br81).
A infusão do ácido dibromoacético encontra-se representada na Figura 8.5.
Figura 8.5 – Espetro resultante da infusão do ácido dibromoacético para uma concentração de 2 mg/L.
Nesta figura é possível verificar a primeira transição onde o ião precursor de massa de
216,70 Da foi obtido com uma voltagem de cone de 20 V e o ião produto de quantificação de massa de
172,80 Da foi obtido com uma energia de colisão de 15 eV.
O ião precursor para a segunda transição é o mesmo, sendo que o ião produto de qualificação
tem massa de 78,80 Da, obtido com energia de colisão de 18 eV.
Na Figura 8.6 encontra-se o espetro obtido para a infusão do ácido bromocloroacético para
uma concentração de 2 mg/L.
216.70
136.85
78.87
172.80

38
Figura 8.6 – Espetro resultante da infusão do ácido bromocloroacético para uma concentração de 2 mg/L.
Da figura anterior verifica-se que para uma massa molar de 170,82 Da encontra-se o ião
molecular (BCAA desprotonado) obtido com uma voltagem de cone de 18 V. As transições 126,82 Da
e 78,88 Da, respetivamente 1ª e 2ª transições encontram-se também evidentes neste espetro tendo
sido obtidas com uma energia de colisão de 10 eV.
No caso particular do ácido tribromoacético, apresenta-se igualmente o espetro obtido na sua
infusão. É visível na Figura 8.7 a razão pela qual se tornou impossível trabalhar com este composto.
A massa para o ião precursor do TBAA é de 250,68 Da (TBAA descarboxilado), e apesar de se ter
imposto uma concentração elevada de 2 mg/L, o pico correspondente a esta massa possui uma
intensidade muito baixa. Intensidade esta que iria baixar consideravelmente quando se passasse para
separação cromatográfica pelo que não se prosseguiu com a sua determinação.
Figura 8.7 – Espetro resultante da infusão do ácido tribromoacético para uma concentração de 2 mg/L.
Os resultados correspondentes às infusões realizadas para os 6 ácidos haloacéticos
encontram-se descritos na Tabela 8-1.
170.82
126.82
78.88
250.68
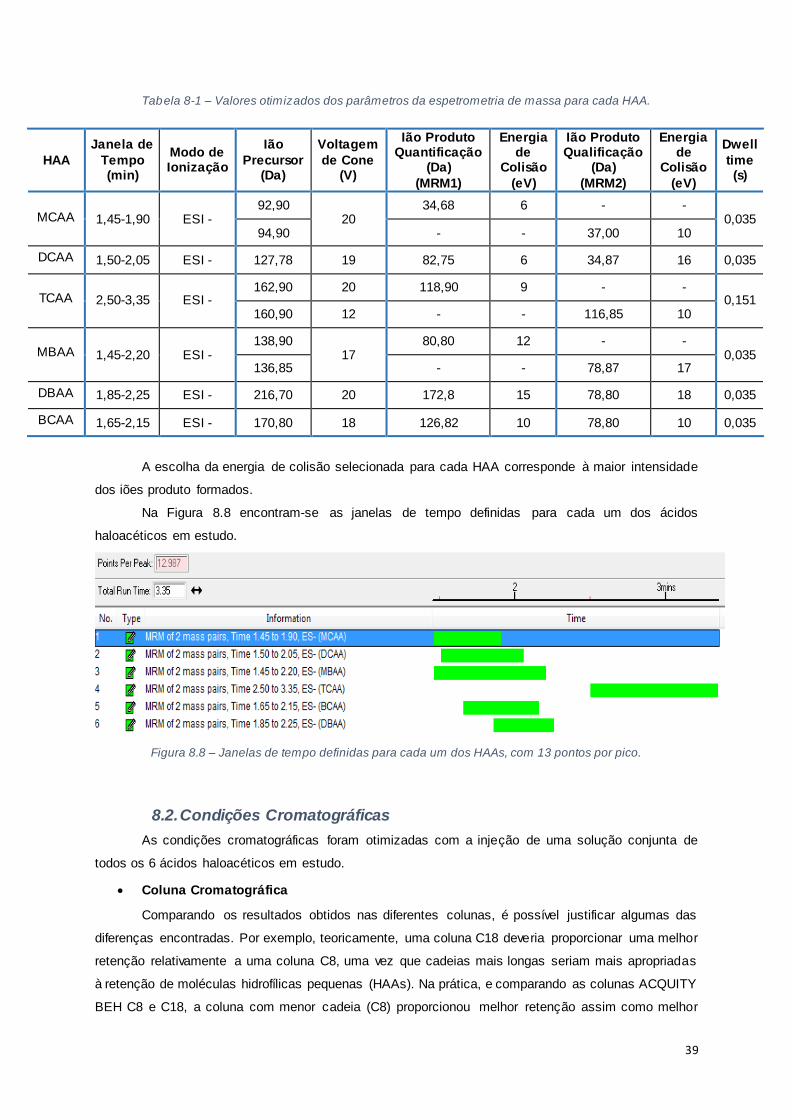
39
Tabela 8-1 – Valores otimizados dos parâmetros da espetrometria de massa para cada HAA.
HAA
Janela de
Tempo (min)
Modo de Ionização
Ião
Precursor (Da)
Voltagem
de Cone (V)
Ião Produto Quantificação
(Da)
(MRM1)
Energia de
Colisão
(eV)
Ião Produto Qualificação
(Da)
(MRM2)
Energia de
Colisão
(eV)
Dwell
time (s)
MCAA 1,45-1,90 ESI - 92,90
20 34,68 6 - -
0,035 94,90 - - 37,00 10
DCAA 1,50-2,05 ESI - 127,78 19 82,75 6 34,87 16 0,035
TCAA 2,50-3,35 ESI - 162,90 20 118,90 9 - -
0,151 160,90 12 - - 116,85 10
MBAA 1,45-2,20 ESI - 138,90
17 80,80 12 - -
0,035 136,85 - - 78,87 17
DBAA 1,85-2,25 ESI - 216,70 20 172,8 15 78,80 18 0,035
BCAA 1,65-2,15 ESI - 170,80 18 126,82 10 78,80 10 0,035
A escolha da energia de colisão selecionada para cada HAA corresponde à maior intensidade
dos iões produto formados.
Na Figura 8.8 encontram-se as janelas de tempo definidas para cada um dos ácidos
haloacéticos em estudo.
Figura 8.8 – Janelas de tempo definidas para cada um dos HAAs, com 13 pontos por pico.
8.2. Condições Cromatográficas
As condições cromatográficas foram otimizadas com a injeção de uma solução conjunta de
todos os 6 ácidos haloacéticos em estudo.
Coluna Cromatográfica
Comparando os resultados obtidos nas diferentes colunas, é possível justificar algumas das
diferenças encontradas. Por exemplo, teoricamente, uma coluna C18 deveria proporcionar uma melhor
retenção relativamente a uma coluna C8, uma vez que cadeias mais longas seriam mais apropriadas
à retenção de moléculas hidrofílicas pequenas (HAAs). Na prática, e comparando as colunas ACQUITY
BEH C8 e C18, a coluna com menor cadeia (C8) proporcionou melhor retenção assim como melhor
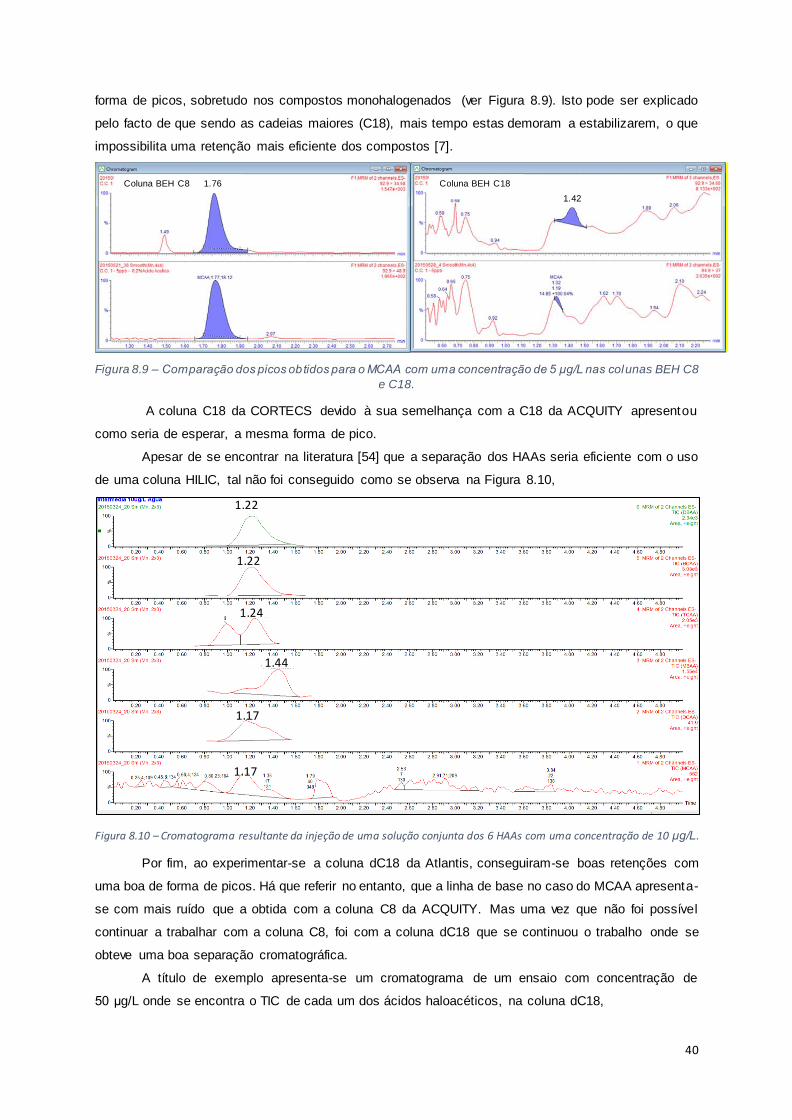
40
forma de picos, sobretudo nos compostos monohalogenados (ver Figura 8.9). Isto pode ser explicado
pelo facto de que sendo as cadeias maiores (C18), mais tempo estas demoram a estabilizarem, o que
impossibilita uma retenção mais eficiente dos compostos [7].
Figura 8.9 – Comparação dos picos obtidos para o MCAA com uma concentração de 5 μg/L nas colunas BEH C8
e C18.
A coluna C18 da CORTECS devido à sua semelhança com a C18 da ACQUITY apresentou
como seria de esperar, a mesma forma de pico.
Apesar de se encontrar na literatura [54] que a separação dos HAAs seria eficiente com o uso
de uma coluna HILIC, tal não foi conseguido como se observa na Figura 8.10,
Figura 8.10 – Cromatograma resultante da injeção de uma solução conjunta dos 6 HAAs com uma concentração de 10 μg/L.
Por fim, ao experimentar-se a coluna dC18 da Atlantis, conseguiram-se boas retenções com
uma boa de forma de picos. Há que referir no entanto, que a linha de base no caso do MCAA apresenta-
se com mais ruído que a obtida com a coluna C8 da ACQUITY. Mas uma vez que não foi possível
continuar a trabalhar com a coluna C8, foi com a coluna dC18 que se continuou o trabalho onde se
obteve uma boa separação cromatográfica.
A título de exemplo apresenta-se um cromatograma de um ensaio com concentração de
50 μg/L onde se encontra o TIC de cada um dos ácidos haloacéticos, na coluna dC18,
1.22
1.22
1.24
1.44
1.17
1.17
1.76
1.42
Coluna BEH C8 Coluna BEH C18

41
Temperatura da Coluna Cromatográfica
A temperatura da coluna é um parâmetro importante na separação cromatográfica pois tem
direta influência na transferência de massa. Uma vez que quanto maior a temperatura, menor a
viscosidade da fase móvel, o processo de transferência de massa aumenta conferindo maior
sensibilidade na separação cromatográfica. Por outro lado, a temperaturas mais baixas os compostos
ficam mais retidos na coluna devido ao aumento da viscosidade da fase móvel. Neste trabalho
estudaram-se 3 diferentes temperaturas de operação da coluna: 30, 40 e 45ᵒC. Uma vez que a
temperatura máxima suportada pela coluna dC18 é de 45ᵒC, esta temperatura foi desde logo excluída.
Foi necessário chegar a um compromisso entre as duas restantes temperaturas . Tendo em conta que
não se notou grande diferença entre os tempos de retenção obtidos para 30 e 40ᵒC, foi estabelecida
como temperatura ótima da coluna, os 40ᵒC. Deste modo consegue-se uma boa separação de picos
(tempos de retenção diferentes para cada analito) num tempo de corrida total de 9 minutos
(ver Figura 8.12).
Figura 8.12 – Programação da temperatura ótima da coluna cromatográfica.
2.03;8844;103901
1.84;8343;99866
2.80;1631;15902
1.83;1702;19449
1.70;103;1488
1.60;597;6924
Figura 8.11 – Cromatograma com os TIC’s de cada HAA, para uma concentração de 50 μg/L, na coluna dC18 .
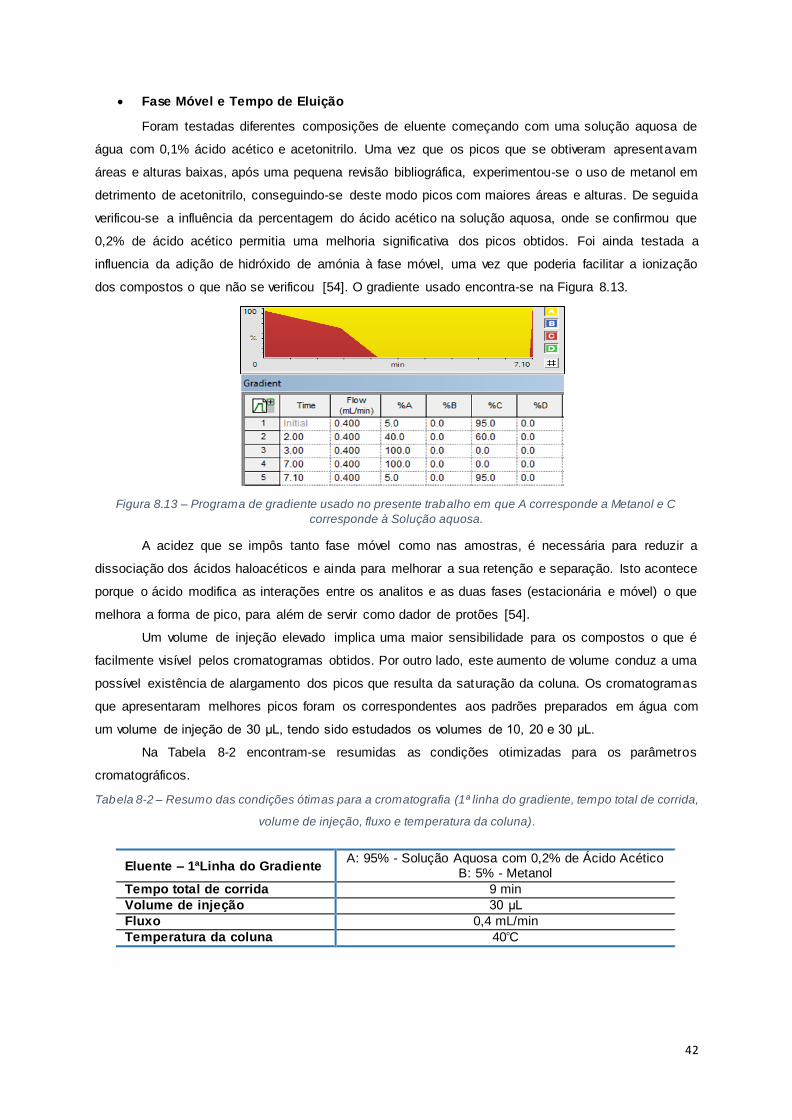
42
Fase Móvel e Tempo de Eluição
Foram testadas diferentes composições de eluente começando com uma solução aquosa de
água com 0,1% ácido acético e acetonitrilo. Uma vez que os picos que se obtiveram apresentavam
áreas e alturas baixas, após uma pequena revisão bibliográfica, experimentou-se o uso de metanol em
detrimento de acetonitrilo, conseguindo-se deste modo picos com maiores áreas e alturas. De seguida
verificou-se a influência da percentagem do ácido acético na solução aquosa, onde se confirmou que
0,2% de ácido acético permitia uma melhoria significativa dos picos obtidos. Foi ainda testada a
influencia da adição de hidróxido de amónia à fase móvel, uma vez que poderia facilitar a ionização
dos compostos o que não se verificou [54]. O gradiente usado encontra-se na Figura 8.13.
Figura 8.13 – Programa de gradiente usado no presente trabalho em que A corresponde a Metanol e C
corresponde à Solução aquosa.
A acidez que se impôs tanto fase móvel como nas amostras, é necessária para reduzir a
dissociação dos ácidos haloacéticos e ainda para melhorar a sua retenção e separação. Isto acontece
porque o ácido modifica as interações entre os analitos e as duas fases (estacionária e móvel) o que
melhora a forma de pico, para além de servir como dador de protões [54].
Um volume de injeção elevado implica uma maior sensibilidade para os compostos o que é
facilmente visível pelos cromatogramas obtidos. Por outro lado, este aumento de volume conduz a uma
possível existência de alargamento dos picos que resulta da saturação da coluna. Os cromatogramas
que apresentaram melhores picos foram os correspondentes aos padrões preparados em água com
um volume de injeção de 30 μL, tendo sido estudados os volumes de 10, 20 e 30 μL.
Na Tabela 8-2 encontram-se resumidas as condições otimizadas para os parâmetros
cromatográficos.
Tabela 8-2 – Resumo das condições ótimas para a cromatografia (1ª linha do gradiente, tempo total de corrida,
volume de injeção, fluxo e temperatura da coluna).
Eluente – 1ªLinha do Gradiente A: 95% - Solução Aquosa com 0,2% de Ácido Acético
B: 5% - Metanol
Tempo total de corrida 9 min
Volume de injeção 30 μL
Fluxo 0,4 mL/min
Temperatura da coluna 40ᵒC

43
Dependendo sobretudo da coluna usada, os tempos de retenção podem variar. Os tempos de
retenção obtidos para a coluna eleita neste trabalho (dC18) para as condições definidas, encontram-se
na Tabela 8-3,
Tabela 8-3 – Tempos de retenção característicos de cada um dos HAAs, para as condições impostas.
Ácido Haloacético Tempo de retenção (min)
MCAA 1,61
DCAA 1,70
TCAA 2,80
MBAA 1,83
DBAA 2,03
BCAA 1,84
Do ponto de vista da separação cromatográfica pode-se concluir que as condições utilizadas
permitem obter picos bem definidos e com boa resolução.
8.3. Validação do Método
8.3.1. Linearidade
A validação do método começou por ser feita através de alguns testes para verificar a
linearidade. Para tal foram usados 4 diferentes testes: o coeficiente de correlação das retas de
calibração, a análise de resíduos, o teste de Mandel e o teste de Rikilt.
Usaram-se como métodos de calibração o do padrão externo e o da adição de padrão.
A título de exemplo encontram-se no Anexo III, as curvas de calibração pelo método de padrão
externo obtidas num ensaio.
Coeficiente de Correlação
Como já referido anteriormente, devido às variações que os declives das retas de calibração,
pelos dois métodos apresentam, a calibração foi feita em cada sessão de trabalho. Na Tabela 8-4 e na
Tabela 8-5 apresentam-se os valores obtidos para os declives e coeficientes de correlação para
diferentes ensaios pelo método do padrão externo e da adição de padrão, respetivamente.
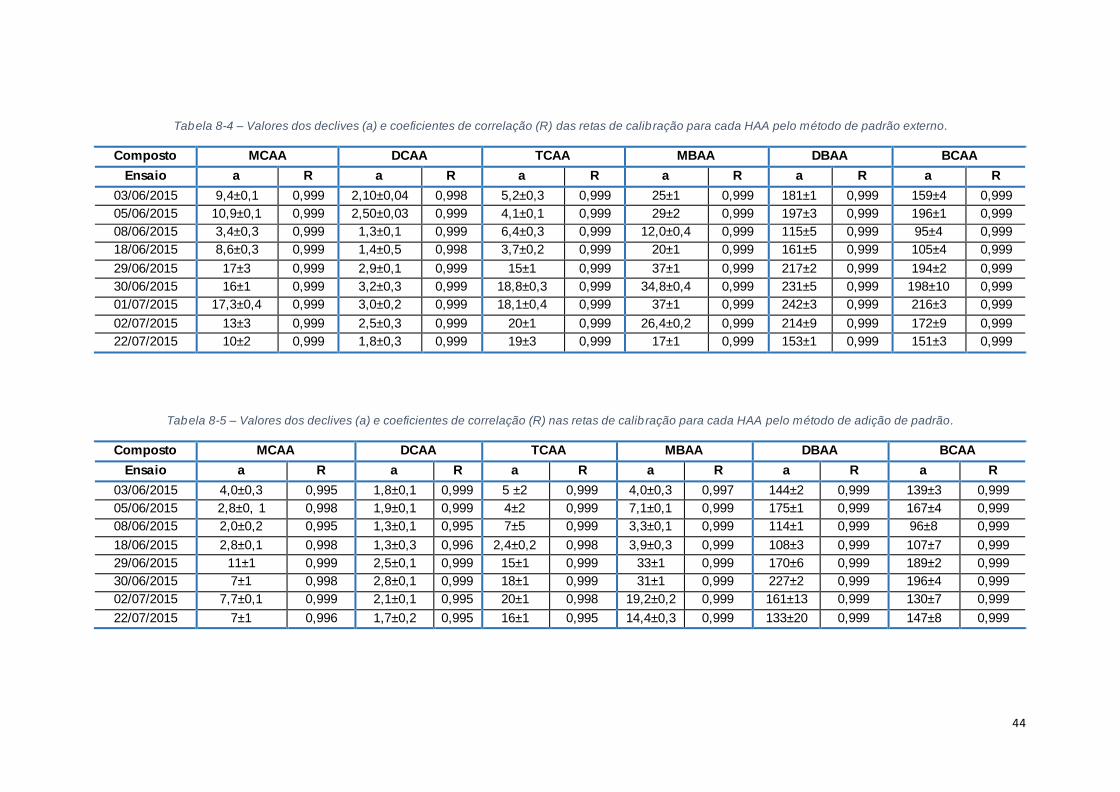
44
Tabela 8-4 – Valores dos declives (a) e coeficientes de correlação (R) das retas de calibração para cada HAA pelo método de padrão externo.
Composto MCAA DCAA TCAA MBAA DBAA BCAA
Ensaio a R a R a R a R a R a R
03/06/2015 9,4±0,1 0,999 2,10±0,04 0,998 5,2±0,3 0,999 25±1 0,999 181±1 0,999 159±4 0,999
05/06/2015 10,9±0,1 0,999 2,50±0,03 0,999 4,1±0,1 0,999 29±2 0,999 197±3 0,999 196±1 0,999
08/06/2015 3,4±0,3 0,999 1,3±0,1 0,999 6,4±0,3 0,999 12,0±0,4 0,999 115±5 0,999 95±4 0,999
18/06/2015 8,6±0,3 0,999 1,4±0,5 0,998 3,7±0,2 0,999 20±1 0,999 161±5 0,999 105±4 0,999
29/06/2015 17±3 0,999 2,9±0,1 0,999 15±1 0,999 37±1 0,999 217±2 0,999 194±2 0,999
30/06/2015 16±1 0,999 3,2±0,3 0,999 18,8±0,3 0,999 34,8±0,4 0,999 231±5 0,999 198±10 0,999
01/07/2015 17,3±0,4 0,999 3,0±0,2 0,999 18,1±0,4 0,999 37±1 0,999 242±3 0,999 216±3 0,999
02/07/2015 13±3 0,999 2,5±0,3 0,999 20±1 0,999 26,4±0,2 0,999 214±9 0,999 172±9 0,999
22/07/2015 10±2 0,999 1,8±0,3 0,999 19±3 0,999 17±1 0,999 153±1 0,999 151±3 0,999
Tabela 8-5 – Valores dos declives (a) e coeficientes de correlação (R) nas retas de calibração para cada HAA pelo método de adição de padrão.
Composto MCAA DCAA TCAA MBAA DBAA BCAA
Ensaio a R a R a R a R a R a R
03/06/2015 4,0±0,3 0,995 1,8±0,1 0,999 5 ±2 0,999 4,0±0,3 0,997 144±2 0,999 139±3 0,999
05/06/2015 2,8±0, 1 0,998 1,9±0,1 0,999 4±2 0,999 7,1±0,1 0,999 175±1 0,999 167±4 0,999
08/06/2015 2,0±0,2 0,995 1,3±0,1 0,995 7±5 0,999 3,3±0,1 0,999 114±1 0,999 96±8 0,999
18/06/2015 2,8±0,1 0,998 1,3±0,3 0,996 2,4±0,2 0,998 3,9±0,3 0,999 108±3 0,999 107±7 0,999
29/06/2015 11±1 0,999 2,5±0,1 0,999 15±1 0,999 33±1 0,999 170±6 0,999 189±2 0,999
30/06/2015 7±1 0,998 2,8±0,1 0,999 18±1 0,999 31±1 0,999 227±2 0,999 196±4 0,999
02/07/2015 7,7±0,1 0,999 2,1±0,1 0,995 20±1 0,998 19,2±0,2 0,999 161±13 0,999 130±7 0,999
22/07/2015 7±1 0,996 1,7±0,2 0,995 16±1 0,995 14,4±0,3 0,999 133±20 0,999 147±8 0,999

45
De acordo com os valores obtidos para os coeficientes de correlação, R ≥ 0,995, (valor este
também imposto no LAIST) pode concluir-se que as curvas de calibração são lineares.
Análise de Resíduos
O teste da análise de resíduos consiste em calcular a diferença entre o valor observado e o
valor estimado pela equação de regressão linear obtida. Os resultados obtidos deverão apresentar-se
de forma aleatória em torno da linha de ordenada zero.
A título de exemplo encontram-se na Figura 8.14 (a, b) as representações gráficas de uma reta
de calibração pelo método da adição de padrão com a respetiva representação gráfica dos resíduos
(ensaio do dia 22 de Julho).
Figura 8.14 – a) Representação gráfica da curva de calibração para o DCAA pelo método de adição de padrão. b) Representação gráfica da análise de resíduos respetiva.
Verifica-se que os resíduos da reta de calibração representada na Figura 8.14 a), apresentam
valores inferiores a 20 %, pelo que se conclui que esta apresenta linearidade.
Também pelo método de calibração de padrão externo se verifica que a análise de resíduos
não ultrapassa os 20 %, como se verifica na Figura 8.15 (a, b) (ensaio do dia 22 de Julho).
Figura 8.15 – a) Representação gráfica da curva de calibração para o DCAA pelo método de padrão externo.
b) Representação gráfica da análise de resíduos respetiva.
O teste estatístico da análise de resíduos permitiu também concluir que existe de facto
linearidade, uma vez que os valores obtidos se encontram inferiores a 20%, valor este imposto pelos
laboratórios.
0
30
60
90
0 20 40 60
Áre
a
Concentração (μg/L)
Adição de Padrão - DCAA
-20
-10
0
10
20
0 20 40%
De
svio
Concentração (μg/L)
Resíduos - DCAAa) b)
0
50
100
0 20 40 60
Áre
a
Concentração (μg/L)
Padrão externo - DCAA
-20
-10
0
10
20
0 20 40 60
De
svio
(%
)
Concentração (μg/L)
Resíduos - DCAAb) a)

46
Teste de Mandel
A linearidade pode ser avaliada estatisticamente pelo teste de Mandel [55].
Com o conjunto de resultados obtidos (y=sinal vs x=concentração), calcula-se a função de
calibração linear e a função de calibração não-linear, assim como os seus respetivos desvios padrão
residuais de acordo com as Equações 8-1 e 8-2:
𝑆𝑦/𝑥 = √∑ (𝑦𝑖−𝑦𝑖)2𝑁
𝑖=1
𝑁−2 Equação 8-1
𝑆𝑦2 = √∑ (𝑦𝑖 −𝑦𝑖2)2𝑁
𝑖 =1
𝑁−3 Equação 8-2
Onde 𝑁 é o número de padrões de calibração, 𝑦𝑖 é o sinal obtido para um padrão de
determinada concentração, 𝑦𝑖 é o sinal estimado pela função de calibração linear para um padrão da
mesma concentração e o 𝑦𝑖2 é o sinal estimado pela função de calibração polinomial do segundo grau
para um padrão da mesma concentração.
De seguida calcula-se a diferença de variâncias (𝐷𝑆2) através da Equação 8-3,
𝐷𝑆2 = (𝑁 − 2) × 𝑠2𝑦/𝑥 − (𝑁 − 3) × 𝑠2
𝑦2 Equação 8-3
O valor teste é então dado pela Equação 8-4,
𝑉𝑇 =𝐷𝑆2
𝑠2𝑦2
Equação 8-4
Este valor teste é comparado com o valor tabelado da distribuição F para um grau de confiança
de 95%.
Os critérios de decisão são:
Se 𝑉𝑇 ≤ 𝐹 , então a função de calibração é linear
Se 𝑉𝑇 > 𝐹 , então a função de calibração é não linear
A título de exemplo apresenta-se o teste de Mandel aplicado ao ensaio do dia 22 de Julho, para
o TCAA pelo método de padrão externo. Na Figura 8.16 encontram-se as representações gráficas das
calibrações por ajuste linear e polinomial.
Figura 8.16 – Representações gráficas dos ajustes a) linear e b) polinomial.
Uma vez que o valor teste deu um valor de 0,351 (inferior ao F95%=10,128) conclui-se que a
calibração apresenta linearidade.
0
200
400
600
800
1000
0 20 40 60
Áre
a
Concentração (μg/L)
Ajuste linear
y = 4,4E-03x2 + 18,8x - 41,5R² = 0,999
0
200
400
600
800
1000
0 20 40 60
Áre
a
Concentração (μg/L)
Ajuste polinomiala) b)
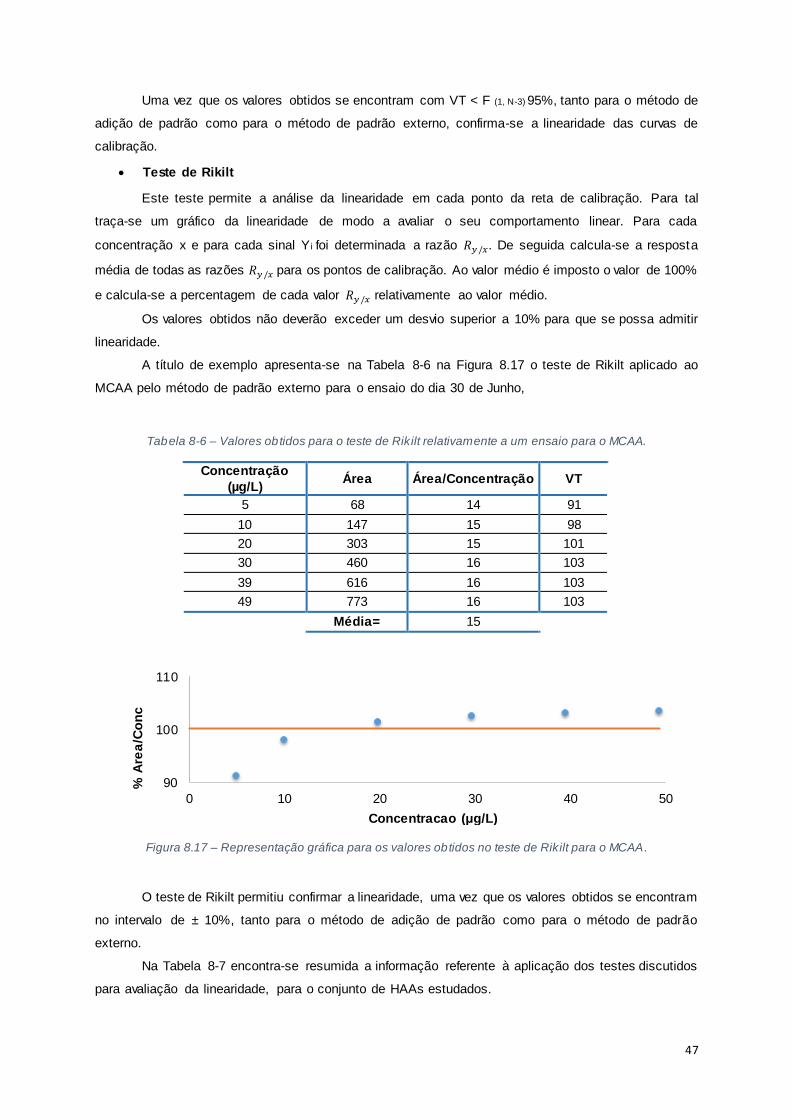
47
Uma vez que os valores obtidos se encontram com VT < F (1, N-3) 95%, tanto para o método de
adição de padrão como para o método de padrão externo, confirma-se a linearidade das curvas de
calibração.
Teste de Rikilt
Este teste permite a análise da linearidade em cada ponto da reta de calibração. Para tal
traça-se um gráfico da linearidade de modo a avaliar o seu comportamento linear. Para cada
concentração x e para cada sinal Y i foi determinada a razão 𝑅𝑦 /𝑥. De seguida calcula-se a resposta
média de todas as razões 𝑅𝑦 /𝑥 para os pontos de calibração. Ao valor médio é imposto o valor de 100%
e calcula-se a percentagem de cada valor 𝑅𝑦 /𝑥 relativamente ao valor médio.
Os valores obtidos não deverão exceder um desvio superior a 10% para que se possa admitir
linearidade.
A título de exemplo apresenta-se na Tabela 8-6 na Figura 8.17 o teste de Rikilt aplicado ao
MCAA pelo método de padrão externo para o ensaio do dia 30 de Junho,
Tabela 8-6 – Valores obtidos para o teste de Rikilt relativamente a um ensaio para o MCAA.
Concentração
(µg/L) Área Área/Concentração VT
5 68 14 91
10 147 15 98
20 303 15 101
30 460 16 103
39 616 16 103
49 773 16 103
Média= 15
Figura 8.17 – Representação gráfica para os valores obtidos no teste de Rikilt para o MCAA.
O teste de Rikilt permitiu confirmar a linearidade, uma vez que os valores obtidos se encontram
no intervalo de ± 10%, tanto para o método de adição de padrão como para o método de padrão
externo.
Na Tabela 8-7 encontra-se resumida a informação referente à aplicação dos testes discutidos
para avaliação da linearidade, para o conjunto de HAAs estudados.
90
100
110
0 10 20 30 40 50
% A
rea
/Co
nc
Concentracao (μg/L)
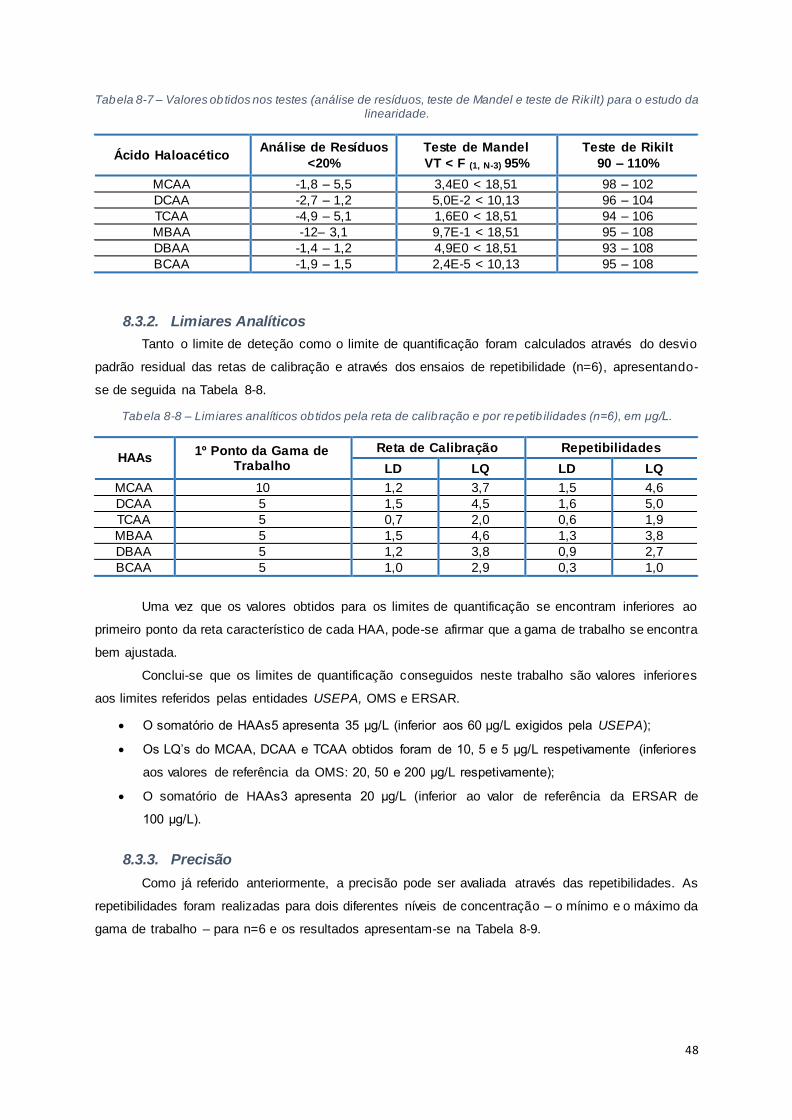
48
Tabela 8-7 – Valores obtidos nos testes (análise de resíduos, teste de Mandel e teste de Rikilt) para o estudo da linearidade.
Ácido Haloacético Análise de Resíduos
<20%
Teste de Mandel
VT < F (1, N-3) 95%
Teste de Rikilt
90 – 110%
MCAA -1,8 – 5,5 3,4E0 < 18,51 98 – 102
DCAA -2,7 – 1,2 5,0E-2 < 10,13 96 – 104
TCAA -4,9 – 5,1 1,6E0 < 18,51 94 – 106
MBAA -12– 3,1 9,7E-1 < 18,51 95 – 108
DBAA -1,4 – 1,2 4,9E0 < 18,51 93 – 108
BCAA -1,9 – 1,5 2,4E-5 < 10,13 95 – 108
8.3.2. Limiares Analíticos
Tanto o limite de deteção como o limite de quantificação foram calculados através do desvio
padrão residual das retas de calibração e através dos ensaios de repetibilidade (n=6), apresentando-
se de seguida na Tabela 8-8.
Tabela 8-8 – Limiares analíticos obtidos pela reta de calibração e por repetib ilidades (n=6), em μg/L.
HAAs 1º Ponto da Gama de
Trabalho
Reta de Calibração Repetibilidades
LD LQ LD LQ
MCAA 10 1,2 3,7 1,5 4,6
DCAA 5 1,5 4,5 1,6 5,0
TCAA 5 0,7 2,0 0,6 1,9
MBAA 5 1,5 4,6 1,3 3,8
DBAA 5 1,2 3,8 0,9 2,7
BCAA 5 1,0 2,9 0,3 1,0
Uma vez que os valores obtidos para os limites de quantificação se encontram inferiores ao
primeiro ponto da reta característico de cada HAA, pode-se afirmar que a gama de trabalho se encontra
bem ajustada.
Conclui-se que os limites de quantificação conseguidos neste trabalho são valores inferiores
aos limites referidos pelas entidades USEPA, OMS e ERSAR.
O somatório de HAAs5 apresenta 35 μg/L (inferior aos 60 μg/L exigidos pela USEPA);
Os LQ’s do MCAA, DCAA e TCAA obtidos foram de 10, 5 e 5 μg/L respetivamente (inferiores
aos valores de referência da OMS: 20, 50 e 200 μg/L respetivamente);
O somatório de HAAs3 apresenta 20 μg/L (inferior ao valor de referência da ERSAR de
100 μg/L).
8.3.3. Precisão
Como já referido anteriormente, a precisão pode ser avaliada através das repetibilidades. As
repetibilidades foram realizadas para dois diferentes níveis de concentração – o mínimo e o máximo da
gama de trabalho – para n=6 e os resultados apresentam-se na Tabela 8-9.
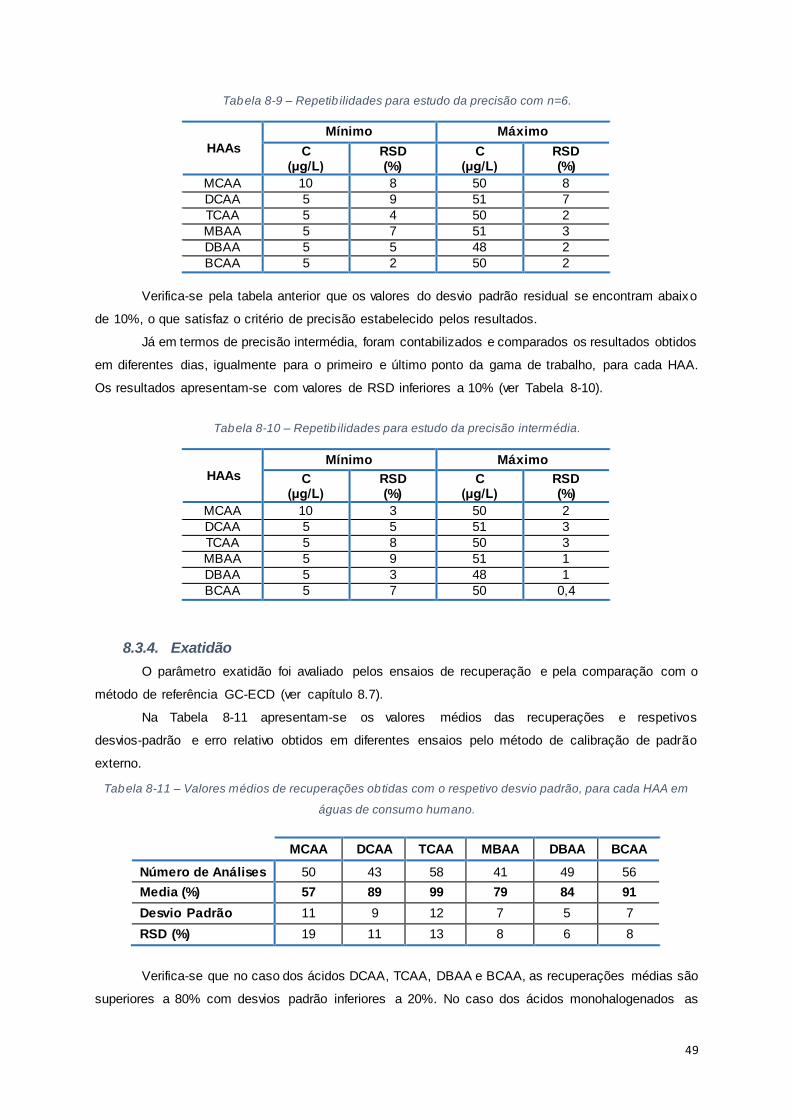
49
Tabela 8-9 – Repetib ilidades para estudo da precisão com n=6.
HAAs
Mínimo Máximo
C (μg/L)
RSD (%)
C (μg/L)
RSD (%)
MCAA 10 8 50 8
DCAA 5 9 51 7
TCAA 5 4 50 2
MBAA 5 7 51 3
DBAA 5 5 48 2
BCAA 5 2 50 2
Verifica-se pela tabela anterior que os valores do desvio padrão residual se encontram abaixo
de 10%, o que satisfaz o critério de precisão estabelecido pelos resultados.
Já em termos de precisão intermédia, foram contabilizados e comparados os resultados obtidos
em diferentes dias, igualmente para o primeiro e último ponto da gama de trabalho, para cada HAA.
Os resultados apresentam-se com valores de RSD inferiores a 10% (ver Tabela 8-10).
Tabela 8-10 – Repetib ilidades para estudo da precisão intermédia.
HAAs
Mínimo Máximo
C (μg/L)
RSD (%)
C (μg/L)
RSD (%)
MCAA 10 3 50 2
DCAA 5 5 51 3
TCAA 5 8 50 3
MBAA 5 9 51 1
DBAA 5 3 48 1
BCAA 5 7 50 0,4
8.3.4. Exatidão
O parâmetro exatidão foi avaliado pelos ensaios de recuperação e pela comparação com o
método de referência GC-ECD (ver capítulo 8.7).
Na Tabela 8-11 apresentam-se os valores médios das recuperações e respetivos
desvios-padrão e erro relativo obtidos em diferentes ensaios pelo método de calibração de padrão
externo.
Tabela 8-11 – Valores médios de recuperações obtidas com o respetivo desvio padrão, para cada HAA em
águas de consumo humano.
MCAA DCAA TCAA MBAA DBAA BCAA
Número de Análises 50 43 58 41 49 56
Media (%) 57 89 99 79 84 91
Desvio Padrão 11 9 12 7 5 7
RSD (%) 19 11 13 8 6 8
Verifica-se que no caso dos ácidos DCAA, TCAA, DBAA e BCAA, as recuperações médias são
superiores a 80% com desvios padrão inferiores a 20%. No caso dos ácidos monohalogenados as
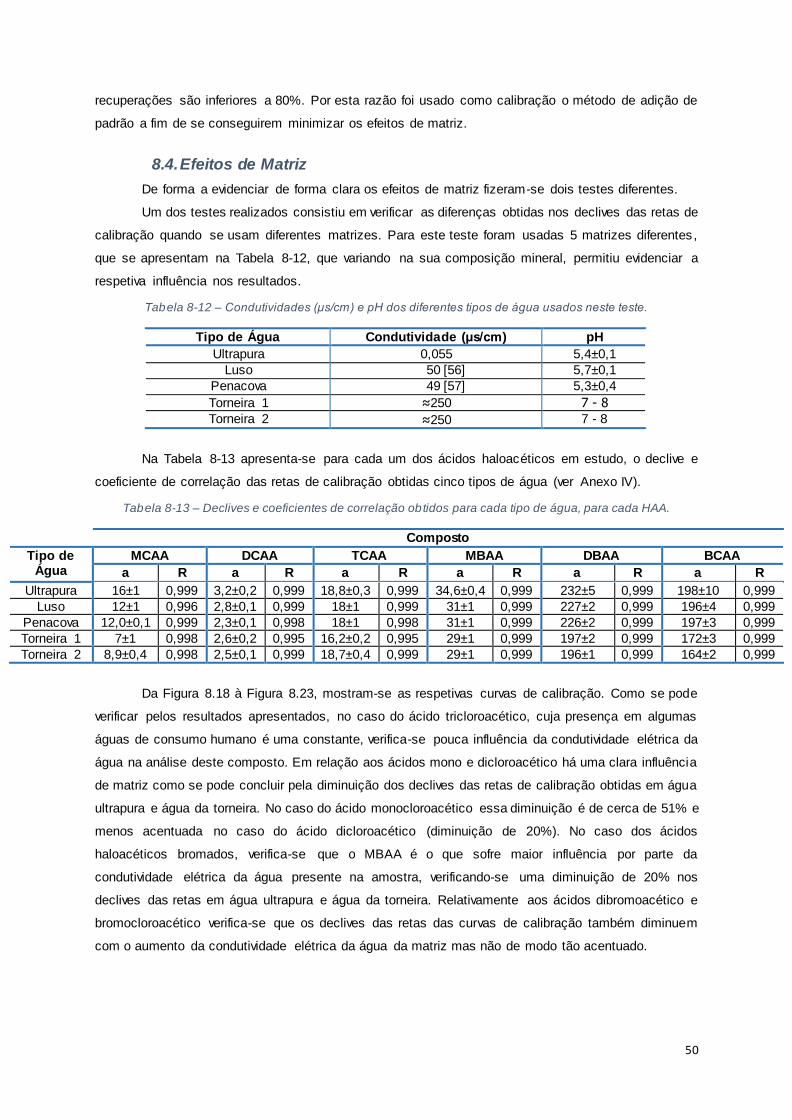
50
recuperações são inferiores a 80%. Por esta razão foi usado como calibração o método de adição de
padrão a fim de se conseguirem minimizar os efeitos de matriz.
8.4. Efeitos de Matriz
De forma a evidenciar de forma clara os efeitos de matriz fizeram-se dois testes diferentes.
Um dos testes realizados consistiu em verificar as diferenças obtidas nos declives das retas de
calibração quando se usam diferentes matrizes. Para este teste foram usadas 5 matrizes diferentes,
que se apresentam na Tabela 8-12, que variando na sua composição mineral, permitiu evidenciar a
respetiva influência nos resultados.
Tabela 8-12 – Condutividades (μs/cm) e pH dos diferentes tipos de água usados neste teste.
Tipo de Água Condutividade (μs/cm) pH
Ultrapura 0,055 5,4±0,1
Luso 50 [56] 5,7±0,1
Penacova 49 [57] 5,3±0,4
Torneira 1 ≈250 7 - 8
Torneira 2 ≈250 7 - 8
Na Tabela 8-13 apresenta-se para cada um dos ácidos haloacéticos em estudo, o declive e
coeficiente de correlação das retas de calibração obtidas cinco tipos de água (ver Anexo IV).
Tabela 8-13 – Declives e coeficientes de correlação obtidos para cada tipo de água, para cada HAA.
Composto
Tipo de Água
MCAA DCAA TCAA MBAA DBAA BCAA
a R a R a R a R a R a R
Ultrapura 16±1 0,999 3,2±0,2 0,999 18,8±0,3 0,999 34,6±0,4 0,999 232±5 0,999 198±10 0,999
Luso 12±1 0,996 2,8±0,1 0,999 18±1 0,999 31±1 0,999 227±2 0,999 196±4 0,999
Penacova 12,0±0,1 0,999 2,3±0,1 0,998 18±1 0,998 31±1 0,999 226±2 0,999 197±3 0,999
Torneira 1 7±1 0,998 2,6±0,2 0,995 16,2±0,2 0,995 29±1 0,999 197±2 0,999 172±3 0,999
Torneira 2 8,9±0,4 0,998 2,5±0,1 0,999 18,7±0,4 0,999 29±1 0,999 196±1 0,999 164±2 0,999
Da Figura 8.18 à Figura 8.23, mostram-se as respetivas curvas de calibração. Como se pode
verificar pelos resultados apresentados, no caso do ácido tricloroacético, cuja presença em algumas
águas de consumo humano é uma constante, verifica-se pouca influência da condutividade elétrica da
água na análise deste composto. Em relação aos ácidos mono e dicloroacético há uma clara influência
de matriz como se pode concluir pela diminuição dos declives das retas de calibração obtidas em água
ultrapura e água da torneira. No caso do ácido monocloroacético essa diminuição é de cerca de 51% e
menos acentuada no caso do ácido dicloroacético (diminuição de 20%). No caso dos ácidos
haloacéticos bromados, verifica-se que o MBAA é o que sofre maior influência por parte da
condutividade elétrica da água presente na amostra, verificando-se uma diminuição de 20% nos
declives das retas em água ultrapura e água da torneira. Relativamente aos ácidos dibromoacético e
bromocloroacético verifica-se que os declives das retas das curvas de calibração também diminuem
com o aumento da condutividade elétrica da água da matriz mas não de modo tão acentuado.

51
Figura 8.18 – Representação gráfica das retas obtidas para o MCAA, para diferentes matrizes.
Figura 8.19 – Representação gráfica das retas obtidas para o DCAA, para diferentes matrizes.
Figura 8.20 – Representação gráfica das retas obtidas para o TCAA, para diferentes matrizes.
Figura 8.21 – Representação gráfica das retas obtidas para o MBAA, para diferentes matrizes.
0
200
400
600
800
1000
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
MCAAÁgua Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
0
40
80
120
160
200
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
DCAAÁgua Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
0
400
800
1200
1600
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
TCAA
Água Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
0
500
1000
1500
2000
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
MBAA
Água Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
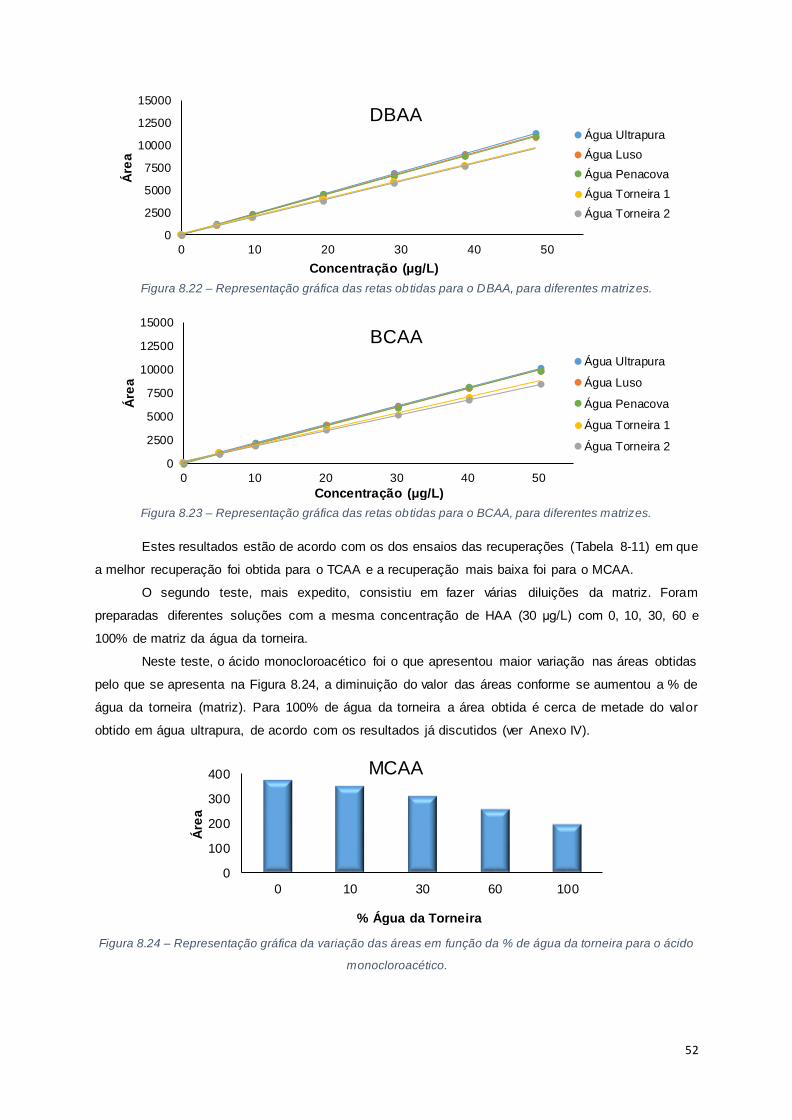
52
Figura 8.22 – Representação gráfica das retas obtidas para o DBAA, para diferentes matrizes.
Figura 8.23 – Representação gráfica das retas obtidas para o BCAA, para diferentes matrizes.
Estes resultados estão de acordo com os dos ensaios das recuperações (Tabela 8-11) em que
a melhor recuperação foi obtida para o TCAA e a recuperação mais baixa foi para o MCAA.
O segundo teste, mais expedito, consistiu em fazer várias diluições da matriz. Foram
preparadas diferentes soluções com a mesma concentração de HAA (30 μg/L) com 0, 10, 30, 60 e
100% de matriz da água da torneira.
Neste teste, o ácido monocloroacético foi o que apresentou maior variação nas áreas obtidas
pelo que se apresenta na Figura 8.24, a diminuição do valor das áreas conforme se aumentou a % de
água da torneira (matriz). Para 100% de água da torneira a área obtida é cerca de metade do valor
obtido em água ultrapura, de acordo com os resultados já discutidos (ver Anexo IV).
Figura 8.24 – Representação gráfica da variação das áreas em função da % de água da torneira para o ácido
monocloroacético.
0
2500
5000
7500
10000
12500
15000
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
DBAAÁgua Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
0
2500
5000
7500
10000
12500
15000
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
BCAA
Água Ultrapura
Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
0
100
200
300
400
0 10 30 60 100
Áre
a
% Água da Torneira
MCAA
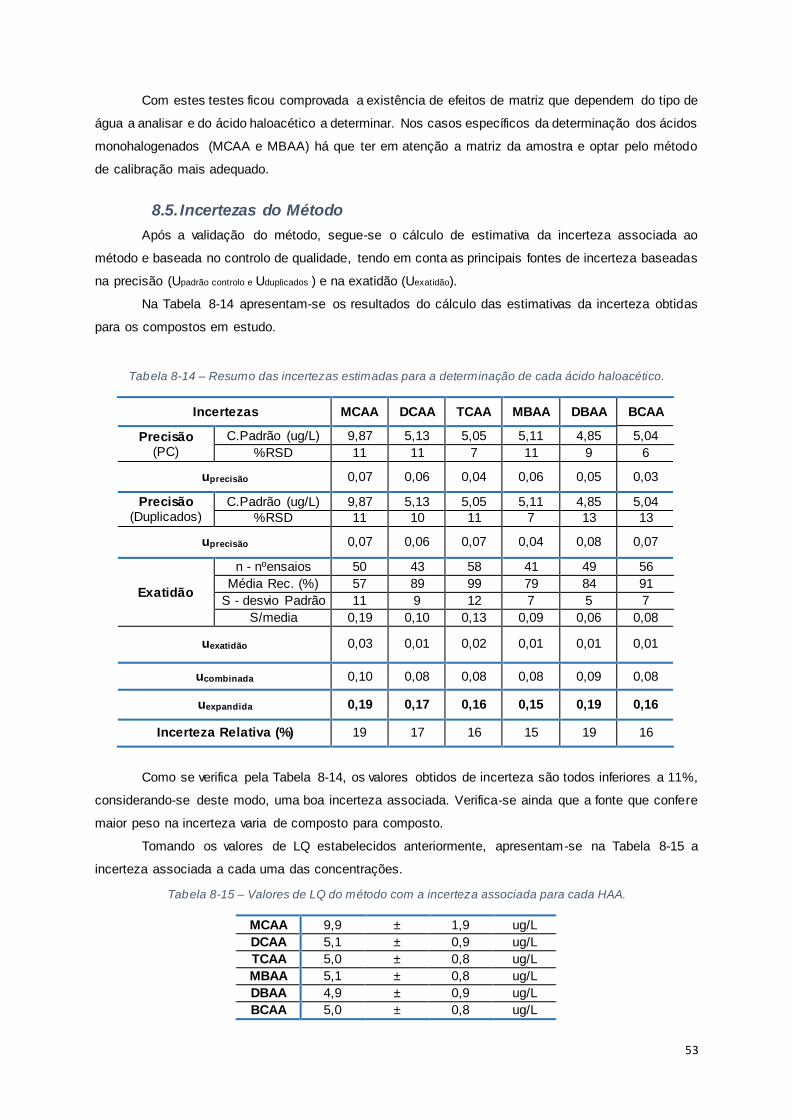
53
Com estes testes ficou comprovada a existência de efeitos de matriz que dependem do tipo de
água a analisar e do ácido haloacético a determinar. Nos casos específicos da determinação dos ácidos
monohalogenados (MCAA e MBAA) há que ter em atenção a matriz da amostra e optar pelo método
de calibração mais adequado.
8.5. Incertezas do Método
Após a validação do método, segue-se o cálculo de estimativa da incerteza associada ao
método e baseada no controlo de qualidade, tendo em conta as principais fontes de incerteza baseadas
na precisão (Upadrão controlo e Uduplicados ) e na exatidão (Uexatidão).
Na Tabela 8-14 apresentam-se os resultados do cálculo das estimativas da incerteza obtidas
para os compostos em estudo.
Tabela 8-14 – Resumo das incertezas estimadas para a determinação de cada ácido haloacético.
Incertezas MCAA DCAA TCAA MBAA DBAA BCAA
Precisão (PC)
C.Padrão (ug/L) 9,87 5,13 5,05 5,11 4,85 5,04
%RSD 11 11 7 11 9 6
uprecisão 0,07 0,06 0,04 0,06 0,05 0,03
Precisão (Duplicados)
C.Padrão (ug/L) 9,87 5,13 5,05 5,11 4,85 5,04
%RSD 11 10 11 7 13 13
uprecisão 0,07 0,06 0,07 0,04 0,08 0,07
Exatidão
n - nºensaios 50 43 58 41 49 56
Média Rec. (%) 57 89 99 79 84 91
S - desvio Padrão 11 9 12 7 5 7
S/media 0,19 0,10 0,13 0,09 0,06 0,08
uexatidão 0,03 0,01 0,02 0,01 0,01 0,01
ucombinada 0,10 0,08 0,08 0,08 0,09 0,08
uexpandida 0,19 0,17 0,16 0,15 0,19 0,16
Incerteza Relativa (%) 19 17 16 15 19 16
Como se verifica pela Tabela 8-14, os valores obtidos de incerteza são todos inferiores a 11%,
considerando-se deste modo, uma boa incerteza associada. Verifica-se ainda que a fonte que confere
maior peso na incerteza varia de composto para composto.
Tomando os valores de LQ estabelecidos anteriormente, apresentam-se na Tabela 8-15 a
incerteza associada a cada uma das concentrações.
Tabela 8-15 – Valores de LQ do método com a incerteza associada para cada HAA.
MCAA 9,9 ± 1,9 ug/L
DCAA 5,1 ± 0,9 ug/L
TCAA 5,0 ± 0,8 ug/L
MBAA 5,1 ± 0,8 ug/L
DBAA 4,9 ± 0,9 ug/L
BCAA 5,0 ± 0,8 ug/L
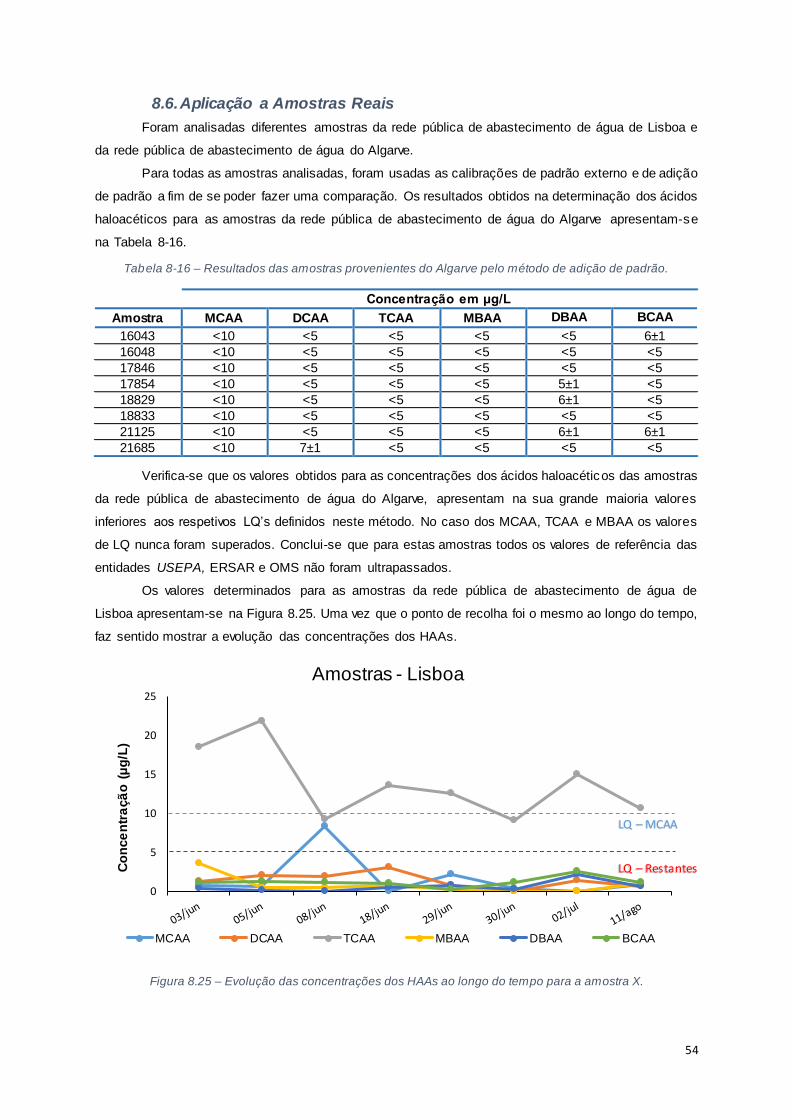
54
8.6. Aplicação a Amostras Reais
Foram analisadas diferentes amostras da rede pública de abastecimento de água de Lisboa e
da rede pública de abastecimento de água do Algarve.
Para todas as amostras analisadas, foram usadas as calibrações de padrão externo e de adição
de padrão a fim de se poder fazer uma comparação. Os resultados obtidos na determinação dos ácidos
haloacéticos para as amostras da rede pública de abastecimento de água do Algarve apresentam-se
na Tabela 8-16.
Tabela 8-16 – Resultados das amostras provenientes do Algarve pelo método de adição de padrão.
Concentração em μg/L
Amostra MCAA DCAA TCAA MBAA DBAA BCAA
16043 <10 <5 <5 <5 <5 6±1
16048 <10 <5 <5 <5 <5 <5
17846 <10 <5 <5 <5 <5 <5
17854 <10 <5 <5 <5 5±1 <5
18829 <10 <5 <5 <5 6±1 <5
18833 <10 <5 <5 <5 <5 <5
21125 <10 <5 <5 <5 6±1 6±1
21685 <10 7±1 <5 <5 <5 <5
Verifica-se que os valores obtidos para as concentrações dos ácidos haloacéticos das amostras
da rede pública de abastecimento de água do Algarve, apresentam na sua grande maioria valores
inferiores aos respetivos LQ’s definidos neste método. No caso dos MCAA, TCAA e MBAA os valores
de LQ nunca foram superados. Conclui-se que para estas amostras todos os valores de referência das
entidades USEPA, ERSAR e OMS não foram ultrapassados.
Os valores determinados para as amostras da rede pública de abastecimento de água de
Lisboa apresentam-se na Figura 8.25. Uma vez que o ponto de recolha foi o mesmo ao longo do tempo,
faz sentido mostrar a evolução das concentrações dos HAAs.
Figura 8.25 – Evolução das concentrações dos HAAs ao longo do tempo para a amostra X.
0
5
10
15
20
25
Co
nce
ntr
açã
o (
μg
/L)
Amostras - Lisboa
MCAA DCAA TCAA MBAA DBAA BCAA
LQ – MCAA
LQ – Restantes
55
Nas amostras da rede pública de abastecimento de água de Lisboa o ácido tricloroacético
apresenta sempre valores superiores ao LQ definido neste método, nunca superando o valor de
referência da OMS (200 μg/L). Os restantes valores dos HAAs estão abaixo do LQ e como tal nunca
excedem o valor de referência da USEPA (HAAs5 < 60 μg/L).
Amostra Cega
Uma vez que as amostras reais analisadas apresentavam valores, para a maioria dos ácidos
haloacéticos abaixo dos limites de quantificação definidos neste método, foi realizado um ensaio com
uma amostra cega. A matriz utilizada foi uma água engarrafada e por isso mesmo, isenta de ácidos
haloacéticos. A esta matriz foi adicionada uma determinada concentração de cada HAA, e foi analisada
pelo método de adição de padrão a fim de se verificar se as concentrações obtidas iam de encontro ao
que tinha sido adicionado previamente.
Após a análise da matriz, compararam-se os resultados com as concentrações espetáveis
como se verifica na Tabela 8-17.
Tabela 8-17 – Valores obtidos da concentração de cada HAA e a sua comparação com o previsto na matriz
simulada.
Composto Concentração
Esperada (μg/L) Concentração Obtida (μg/L)
|Δ| (%)
MCAA 20 21±4 7
DCAA 21 24±4 18
TCAA 20 20±3 3
MBAA 20 24±4 18
DBAA 19 23±4 19
BCAA 20 24±4 18
Verifica-se que os resultados obtidos, com a incerteza associada, vão ao encontro dos valores
das concentrações esperadas, podendo assim concluir que o método se encontra bem implementado
na análise de amostras reais, para os seis ácidos haloacéticos analisados.
8.7. Comparação de Métodos
Resultados das Amostras
Foram realizadas análises em simultâneo com a técnica por GC-ECD em amostras reais. Os
resultados obtidos encontram-se na Tabela 8-18.
Tabela 8-18 – Resultados obtidos na análise de HAAs por UPLC-MS/MS e GC-ECD.
Amostra 16043 16048 18833 21685 X
Método UPLC-
MS/MS
GC-
ECD
UPLC-
MS/MS
GC-
ECD
UPLC-
MS/MS
GC-
ECD
UPLC-
MS/MS
GC-
ECD
UPLC-
MS/MS
GC-
ECD
MCAA < 10 < 10 < 10 < 10 < 10 < 10 < 10 < 10 < 10 < 10
DCAA < 5 5±1 < 5 4±1 < 5 3±1 7±1 5±1 < 5 6±1
TCAA < 5 1,0±0,2 < 5 2,0±0,4 < 5 < 1 < 5 1,0±0,2 11±2 12±2
MBAA < 5 - < 5 - < 5 - < 5 - < 5 -
DBAA < 5 5±1 < 5 4±1 < 5 4±1 < 5 5±1 < 5 1,0±0,2
BCAA 6±1 - < 5 - < 5 - < 5 - < 5 -
Nota: a incerteza associada a cada HAA pelo método de GC-ECD apresenta valor de 20%.

56
Verifica-se que os valores obtidos na técnica de análise por UPLC-MS/MS apresentam
coerência face aos valores obtidos pela técnica de análise por GC-ECD. Este resultado valida a
exatidão do método desenvolvido.
Tempo de Preparação de Amostra
O tempo necessário para um ensaio por UPLC-MS/MS é consideravelmente menor que por
GC-ECD, tanto na preparação de amostra como na análise.
Por exemplo, numa sessão de trabalho onde existem 4 amostras a serem analisadas, o tempo
despendido pelo método por GC-ECD necessita de 6h, já descontando as 2h necessárias para a
derivatização, enquanto que pelo método de UPLC-MS/MS não existe preparação de amostra.
Tempo de Análise
Uma vez que a injeção no método de UPLC-MS/MS é automática e a injeção no método de
GC-ECD é manual, este último necessita de mais mão-de-obra durante a análise.
Quantidades de Solventes e Reagentes
As quantidades de solventes e reagentes que se usa na técnica por GC-ECD são superiores
às da técnica por UPLC-MS/MS, nomeadamente o uso de derivatizante. Por UPLC-MS/MS a
quantidade solvente utilizado recai sobretudo na fase móvel.
Segurança
Pelas razões já referidas, tais como, a quantidade de solventes utilizada em cada análise, a
injeção no método por UPLC-MS/MS ser automática e tempo de contato entre o analista e os solventes
e reagentes na preparação de amostra, conclui-se que o método UPLC-MS/MS oferece grandes
vantagens em termos de segurança.
Tempo de Resposta ao Cliente
Relativamente ao tempo de resposta ao cliente, o método por UPLC-MS/MS permite que este
seja mais curto, uma vez que, como já referido, não há preparação de amostra.
Na Tabela 8-19 apresentam-se as vantagens e desvantagens para as duas técnicas,
podendo-se concluir que a técnica por UPLC-MS/MS oferece significativas vantagens em relativamente
à técnica por GC-ECD.
Tabela 8-19 – Comparação das vantagens e desvantagens entre as duas técnicas.
Parâmetros UPLC – MS/MS GC – ECD
Tempo de Preparação de Amostra X
Tempo de Análise X
Quantidade de Solventes e Reagentes X
Segurança X
Tempo de Resposta ao Cliente X
8.8. Razão das Massas
Como já referido anteriormente no subcapítulo 5.1.2, a razão entre as duas transições de um
composto (MRM1 e MRM2) pode ser utilizada para confirmar a sua identidade. Ao longo da gama de
trabalho esta razão foi estudada para cada composto, para se confirmar a sua estabilidade.

57
Na Tabela 8-20 encontram-se as razões mássicas para os 6 ácidos haloacéticos com os
respetivos desvios padrão relativos,
Tabela 8-20 – Razão MRM1/MRM2 para cada um dos compostos em estudo.
HAAs Número de Ensaios Média %RSD Intervalo de aceitação
MCAA 102 3,7 10 ±25%
DCAA 81 4,4 13 ±25%
TCAA 98 1,4 16 ±20%
MBAA 140 1,5 7 ±20%
DBAA 145 9,1 6 ±30%
BCAA 146 4,4 6 ±25%
Todos os HAAs possuem valores de desvio padrão residual concordantes com o intervalo de
aceitação.
8.9. Procedimento Analítico
A partir do estudo feito e seguindo as diretivas utilizadas no Laboratório de Análises do Instituto
Superior Técnico, elaborou-se um procedimento analítico que resume as condições de trabalho para
posteriores execuções de ensaios que se apresenta no Anexo V.

58

59
9. Conclusões e Perspetivas Futuras
O objetivo do trabalho foi conseguido, uma vez que foi possível a determinação dos ácidos
haloacéticos (MCAA, DCAA, TCAA, MBAA, DBAA e BCAA) em águas de consumo humano, cujo
somatório de limites de quantificação (HAAs5 = 35 μg/L) não excede os valores de referência das
entidades: USEPA (HAAs5 < 60 μg/L), OMS (MCAA < 20 μg/L; DCAA < 50 μg/L; TCAA < 200 μg/L) e
ERSAR (HAAs3 < 100 μg/L). O método desenvolvido consiste na separação cromatográfica por
cromatografia líquida de ultra eficiência, com ionização por electrospray e com deteção por
espetrometria de massa (UPLC-ESI-MS/MS).
As otimizações realizadas, tanto a nível cromatográfico como a nível de espetrometria de
massa, permitiram desenvolver um método eficiente na separação e quantificação dos 6 ácidos
haloacéticos. A razão das massas, assim como os tempos de retenção permitiram a identificação de
cada um dos ácidos haloacéticos.
Em termos de validação do método os testes realizados apresentam respostas positivas, pelo
que se pode afirmar que o método se encontra validado. Na linearidade, os quatro testes usados para
a sua verificação, todos apresentam valores que obedecem aos critérios de cada um dos deles. Sobre
os limiares analíticos, seria interessante baixar os valores dos limites de quantificação nos ácidos
haloacéticos, que o permitissem, diminuindo assim a soma atual de 35 μg/L. O interesse de se
conseguir baixar os valores dos limites de quantificação, passa pela possibilidade de diretivas futuras
que exijam valores inferiores aos que se encontram atualmente em vigor. Em particular, uma vez que
a forma e altura de pico para o MCAA (ácido haloacético com LQ de 10 μg/L) no teste realizado na
coluna BEH C8 deram melhor, seria interessante voltar a trabalhar nesta coluna, até porque o valor de
referência da OMS para este ácido é de 20 μg/L.
Conclui-se que se trata de um método preciso, uma vez que as repetibilidades e a precisão
intermédia apresentam desvios inferiores a 10%.
Relativamente à exatidão, as médias das recuperações para os ácidos DCAA, TCAA, DBAA e
BCAA apresentam valores superiores a 80% enquanto as médias dos ácidos monohalogenados
(MCAA e MBAA) apresentam valores inferiores a 80%. Relativamente aos efeitos de matriz, verifica-se
que o ácido monocloroacético é o mais afetado de todos os ácidos haloacéticos em estudo. Esta
dificuldade é ultrapassada usando o método de adição de padrão, pois os efeitos de matriz são deste
modo minimizados.
O método desenvolvido neste trabalho (UPLC-MS/MS) apresenta diversas vantagens face ao
método por GC-ECD, tais como menor tempo de preparação de amostra, análise e de resposta ao
cliente, assim como menor exposição a solventes para o analista, sendo portanto um método mais
seguro.
As incertezas expandidas estimadas para cada um dos ácidos haloacéticos apresentam
valores aceitáveis abaixo de 20%.
A metodologia desenvolvida foi aplicada na determinação dos HAAs em amostras reais de
água para consumo humano de diferentes pontos de recolha (redes públicas de abastecimento de água
de Lisboa e do Algarve). Os resultados obtidos demonstram que a presença de HAAs é vestigial para

60
a maioria dos analitos em estudo, onde as concentrações apresentam valores inferiores aos exigidos
pelas entidades USEPA, OMS e ERSAR. A água proveniente de Lisboa, que foi testada em variadas
sessões, apresentou uma concentração de TCAA sempre superior a 9 μg/L, que no entanto nunca
superou os valores de referência das entidades já referidas.
Uma vez que as águas de piscinas sofrem um processo de cloragem de maior dimensão, seria
interessante a análise destas no futuro, até porque segundo a literatura já existem estudos com este
tipo de matriz onde se verificam concentrações que ultrapassam os limites impostos pela USEPA.
Como perspetivas futuras sugere-se a tentativa de implementação e validação do método na
análise do TBAA e ainda um estudo aprofundado dos efeitos de matriz que, como ficou comprovado
pelos testes realizados, influenciam a determinação dos ácidos haloacéticos em águas de consumo
humano.

61
10. Referências Bibliográficas
[1] WHO - World Health Organization, “Guidelines for Drinking-water Quality,” 2011. [Online].
Available: http://whqlibdoc.who.int/publications/2011/9789241548151_eng.pdf. [Acedido em
Março 2015].
[2] ERSAR, Recomendação ERSAR n.º 02/2011 - ESPECIFICAÇÃO TÉCNICA PARA A
CERTIFICAÇÃO DO PRODUTO ÁGUA PARA CONSUMO HUMANO, 2011.
[3] Entidade Reguladora dos Serviços de Águas e Resíduos, “A qualidade da água - O consumidor
e os serviços de águas e resíduos,” ERSAR, Portugal, Abril, 2013.
[4] I. Zilberman, Introdução à Engenharia Ambiental, Ulbra, 1997.
[5] N. J. Ashbolt, “Microbial contamination of drinking water and disease outcomes in developing
regions,” Toxicology, vol. 198, p. 360, 2004.
[6] EPA United States Environmental Protection Agency, “Water: Basic Information about Regulated
Drinking Water Contaminants,” Dezembro 2013. [Online]. Available: http://www.clsud.com/wp-
content/uploads/2008/06/EPA-Basic-Fluoride-Information.pdf. [Acedido em Março 2015].
[7] L. Meng, S. Wu, F. Ma, A. Jia e J. Hu, “Trace determination of nine haloacetic acids in drinking
water by liquid chromatography-electrospray tandem mass spectrometry,” Journal of
Chromatography A, vol. 1426, pp. 1-252, 2010.
[8] LAIST, “LAIST - Laboratório de Análises do Instituto Superior Técnico,” [Online]. Available:
http://la.tecnico.ulisboa.pt/index.html. [Acedido em Junho 2015].
[9] “Qualidade em Química -51,” O Laboratório de Análises Químicas do I.S.T., pp. 48-49, 1993.
[10] Entidade Reguladora dos Serviços de Águas e Resíduos, “A qualidade da água na torneira, ”
ERSAR, Abril, 2013.
[11] Organização Mundial de Saúde, Desinfeção da Água, Gabinete Regional para a Europa: OMS,
1996.
[12] T. Martins, Sistemas de Abastecimento de Água para Consumo Humano, Bragança: IPB, 2014.
[13] Grupo Águas de Portugal, “Águas do Centro,” [Online]. Available:
http://www.aguasdocentro.pt/index.asp. [Acedido em Março 2015].
[14] N. F. Gray, Drinking Water Quality, Dublin: Cambridge University Press, 2008.
[15] R. C. Gaur, Basic Environmental Engineering, New Age International Publishers, 2008.
[16] IRAR, “Recomendação IRAR n.º 05/2007,” 2007. [Online]. Available:
http://www.ersar.pt/website/ViewContent.aspx?Section=News&FolderPath=&SubFolderPath=& F
inalPath=Not%C3%ADcias&Name=Recomenda%C3%A7%C3%A3oIRARn.%C2%BA052007.
[Acedido em Agosto 2015].

62
[17] Naturlink, “Naturlink - A ligação à natureza,” [Online]. Available: http://naturlink.sapo.pt/Natureza-
e-Ambiente/Gestao-Ambiental/content/Tecnologias-de-Tratamento-de-aguas-Residuais-
Urbanas/section/3?bl=1. [Acedido em Maio 2015].
[18] C. Schuck, Ocorrência de Trihalometanos e Ácidos Haloacéticos na Desinfeção de Efluentes
Tratados Biológicamente, Porto Alegre: UFRGS, 2004.
[19] D. F. Pereira, DETECÇÃO DE SUBPRODUTOS DA DESINFECÇÃO, Porto, Portugal, 2007.
[20] P. R. R. K. Mark Harman, “Evaluation of Haloacetic Acid Concentrations in Treated Drinking
Water,” 2011. [Online]. Available: http://dwi.defra.gov.uk/research/completed -
research/reports/DWI70_2_242.pdf.
[21] S. J. L. P. Rodriguez M, “Behavior of trihalomethanes and haloacetic acids in a drinking water
distribution system,” Water Research, vol. 38, nº 20, p. 4367–4382, 2004.
[22] H. Baribeau, Formation and Decay of Disinfection By-products in the Distribution System, USA:
AWWA Research Foundation, 2006.
[23] K. Srinivasan, R. Lin e C. Pohl, Recent Advances in Analysis of Haloacetic Acids by Two-
Dimensional Matrix Elimination Ion Chromatography, USA: Dionex Corporation.
[24] B. C. Fryhle e T. G. Solomons, Organic Chemistry, USA: JOHN WILEY & SONS, INC, 2011.
[25] G. M. Cardador MJ, “Determination of haloacetic acids in human urine by headspace gas
chromatography–mass spectrometry,” J Chromatogr B, vol. 878, p. 21, 2010.
[26] Health Canada, “Guideline Technical Document - Haloacetic Acids,” Julho 2008. [Online].
Available: http://www.hc-sc.gc.ca/ewh-semt/pubs/water-eau/haloaceti/index-eng.php. [Acedido
em Maio 2015].
[27] V. C. M. e. all, “Neurotoxicity Produced by Dibromoacetic Acid in Drinking Water of Rats,”
Toxicological Sciences, vol. 122, p. 22, 2004.
[28] Q. Luo, D. Wang, Z. Wei e Z. Wang, “Optimized chromatographic conditions for separation oh
halogenated acetic acids by ultra-performance liquid chromatography-electrospray ionization-
mass spectrometry,” Journal of Chromatography A, vol. 1426, pp. 1-252, 2012.
[29] B. H. e. all, “Assessment of Haloacetic Acids in Drinking Water in Bizerte, Tunisia,” CLEAN – Soil,
Air, Water, vol. 42, nº 8, p. 1052–1059, 2013.
[30] X. W. e. all, “Haloacetic acids in swimming pool and spa water in the United States and China,”
Frontiers of Environmental Science & Engineering, vol. 8, nº 6, pp. 820-824, 2014.
[31] D. A. Skoog, D. M. West, F. J. Holler e S. R. Crouch, Fundamentals of Analytical Chemistry,
Belmont: Thomson-Brooks/Cole, 2004.
[32] I. Jardim e L. Maldaner, “O Estado da Arte da Cromatografia Líquida de Ultra Eficiência,” Química
Nova, vol. 32, p. 1, 2008.

63
[33] WATERS, “ACQUITY UPLC H-Class Bio,” [Online]. Available:
http://www.waters.com/waters/pt_BR/ACQUITY-UPLC-H-Class-
Bio/nav.htm?locale=pt_BR&cid=10166246. [Acedido em Abril 2015].
[34] Química Suprema, “O que é a Cromatografia?,” 2013. [Online]. Available:
http://www.quimicasuprema.com/2013/12/o-que-e-cromatografia.html. [Acedido em Julho 2015].
[35] WATERS, “Chromatographic Columns,” 2015. [Online]. Available:
http://www.waters.com/waters/en_US/HPLC-Columns/nav.htm?cid=511505&locale=en_US.
[Acedido em Maio 2015].
[36] WATERS, “Colunas ACQUITY UPLC,” [Online]. Available:
http://www.waters.com/waters/pt_PT/ACQUITY-UPLC-
Columns/nav.htm?locale=pt_PT&cid=513206. [Acedido em 2015].
[37] Michael E. Swartz e Brian Murphy, “New Frontiers in Chromatography,” American Laboratory ,
2005. [Online]. Available: http://www.americanlaboratory.com/914-Application-Notes/19157-New-
Frontiers-in-Chromatography/. [Acedido em Setembro 2015].
[38] WATERS, “Colunas CORTECS,” [Online]. Available: http: //www.waters.com/waters/pt_PT/Solid -
Core-Particle-Columns/nav.htm?cid=134740934&alias=Alias_cortecs_chem&locale=pt_PT.
[Acedido em 2015].
[39] WATERS, “Atlantis dc18 Column,” [Online]. Available:
http://www.waters.com/waters/partDetail.htm?partNumber=186001329. [Acedido em 2015].
[40] SOQUÍMICA, “Espectrometria de Massa de EIectrospray,” Boletim da Sociedade Portuguesa de
Química, vol. Nº80, nº Química, pp. 36-42, 2001.
[41] “WATERS - The Science of What's Possible,” WATERS, 2015. [Online]. Available:
http://www.waters.com/waters/en_US/Common-Ionization/nav.htm?cid=10073251. [Acedido em
Março 2015].
[42] R. Vessecchi, J. Lopes e N. Lopes, “ESPECTROMETRIA DE MASSAS COM IONIZAÇÃO POR
“ELECTROSPRAY”: PROCESSOS QUÍMICOS ENVOLVIDOS NA FORMAÇÃO DE IÕES DE
SUBSTÂNCIAS ORGÂNICAS DE BAIXO PESO MOLECULAR,” Química Nova, vol. 29, nº 2,
2006.
[43] WATERS, “New Mass Spectrometer Packs Impressive Performance into a Compact Design,”
Junho 2014. [Online]. Available:
http://www.waters.com/waters/newsDetail.htm?locale=en_US&id=134803229. [Acedido em Abril
2015].
[44] R. H. Boja ES, “The Path to Clinical Proteomics Research: Integration of Proteomics, Genomics,
Clinical Laboratory and Regulatory Science,” Korean J Lab Med, vol. 31, nº 2, pp. 61-71, 2011.
[45] A. H. Iglesias, Introdução ao Acoplamento Cromatografia Líquida - Espetrometria de Massas,
Waters Technologies do Brasil, Barueri, SP.

64
[46] Waters Corporation Food Safety, “SlideShare,” 2015. [Online]. Available: Analysis of Mycotoxins
by UPLC and Tandem Quadrupole Mass Spectrometry - Waters Corporation Food Safety.
[Acedido em Junho 2015].
[47] G. J. C. E. T.L. Constantopoulos, Journal of the American Society for Mass Spectrometry, vol. 10,
pp. 625-634, 1999.
[48] L. L. Jessome e D. A. Volmer, “Ion Suppression: A Major Concern in Mass Spectrometry,” LCGC
North America, vol. 24, nº 5, Maio, 2006.
[49] “A Acreditação de Laboratórios - ISO/IEC 17025,” PortalQAS®, [Online]. Available:
http://www.portalqas.pt/acreditacao-de-laboratorios-np-en-isoiec-17025.html. [Acedido em
Outubro 2015].
[50] Relacre, “Guia Relacre 13 - Validação de métodos internos de ensaio em análise química,”
Fevereiro 2000. [Online]. Available:
http://www.relacre.pt/assets/relacreassets/files/commissionsandpublications/Guia%20RELACRE
%2013.pdf. [Acedido em Setembro 2015].
[51] C. T. R. CTR03, “Guia Relacre 3 - Validação de Resultados em Laboratórios Químicos,”
RELACRE, 1996.
[52] M. S. Romão, Análise Química, Lisboa: Secção de Folhas, 2013/2014.
[53] J. Z. e. all, “Alternative calibration approaches to compensate the effect of co-extracted matrix
components in liquid chromatography–electrospray ionisation tandem mass spectrometry analysis
of pesticide residues in plant materials,” Journal of Chromatography A, vol. 973, nº 1-2, pp. 13-26,
2002.
[54] C. Y. Chen, S. N. Chang e G. S. Wang, “Determination of Ten Haloacetic Acids in Drinking Water
Using High Performance and Ultra Performance Liquid Chromatography - tandem Mass
Spectrometry,” J Chromatogr Sci., vol. 47, nº 1, pp. 67-74, 2009.
[55] “Teste de Mandel ou Teste de Fisher/Snedecor,” [Online]. Available:
http://repositorio.ul.pt/bitstream/10451/252/27/20027_27_Anexos_10_Mandel.pdf. [Acedido em 8
Setembro 2015].
[56] CESAB, “Cronograma relativo à Zona de Abastecimento: Luso,” 2012. [Online]. Available:
http://www.cm-mealhada.pt/ficheiros/ambiente/aguassaneamento/za_luso4tri11.pdf. [Acedido
em 2015].
[57] CALDAS DE PENACOVA, “SOP Business Centers,” 2015. [Online]. Available: http://sop-
business-centers.webnode.pt/products/agua-caldas-de-penacova-0-33-lt/. [Acedido em 2015].
[58] HT-LABS, “ESI/MS - Atmosphere Pressure Ionization,” [Online]. Available: http://www.ht-
labs.com/Analytical_Lab/esi_ms.html. [Acedido em Março 2015].
[59] S. Lindsay, High Performance Liquid Chromatography, Londres: John Wiley & Sons, 1992.

65
[60] WATERS, “Partícula característica das colunas HILIC - Care and Use Manual,” 2013. [Online].
Available:
https://webmail.tecnico.ulisboa.pt/rc/?_task=mail&_framed=1&_action=get&_mbox=Sent& _uid=2
26&_part=4&_frame=1. [Acedido em Junho 2015].
[61] D. Snehal V.Chopade, “Introduction to new chromatography technique - UPLC,” Pharmatutor,
[Online]. Available: http://www.pharmatutor.org/articles/new-chromatographic-technique-uplc -
ultra-performance-liquid-chromatography.
[62] WATERS, “VanGuard Pre-column,” [Online]. Available:
http://www.waters.com/waters/partDetail.htm?partNumber=186004629. [Acedido em 2015].

66

67
Anexo I
Colunas Cromatográficas
Benefícios de desempenho: Esta coluna possui elevada estabilidade a pH’s extremos e tem
elevada aplicabilidade para a maioria das classes de compostos. [35]
Ligação: C18 com 3 grupos funcionais com pontes de
etileno, BEH
Área Superficial: 185 m2/g
Carga de Carbono: 18%
Densidade do Ligando: 3.1 μmol/m2
Benefícios de desempenho: Esta coluna possui elevada estabilidade a pH’s extremos e tem
elevada aplicabilidade para a maioria das classes de compostos. [35]
Ligação: C18 com 3 grupos funcionais com pontes de
etileno, BEH
Área Superficial: 185 m2/g
Carga de Carbono: 13%
Densidade do Ligando: 3.2 μmol/m2
Benefícios de desempenho: Esta coluna possui elevada eficiência na retenção de analitos
solúveis em água, básicos e extremamente polares. [35]
Ligação: N/A
Área Superficial: 100 m2/g
Carga de Carbono: 0%
Densidade do Ligando: N/A
Benefícios de desempenho: Esta coluna é ideal para analises em fase-reversa em particular
para compostos ácidos, básicos e neutros a pH baixo e médio. [35]
Ligação: C18 com 3 grupos funcionais
Área Superficial: 100 m2/g
Carga de Carbono: 6.6%
Densidade do Ligando: 2.7 μmol/m2
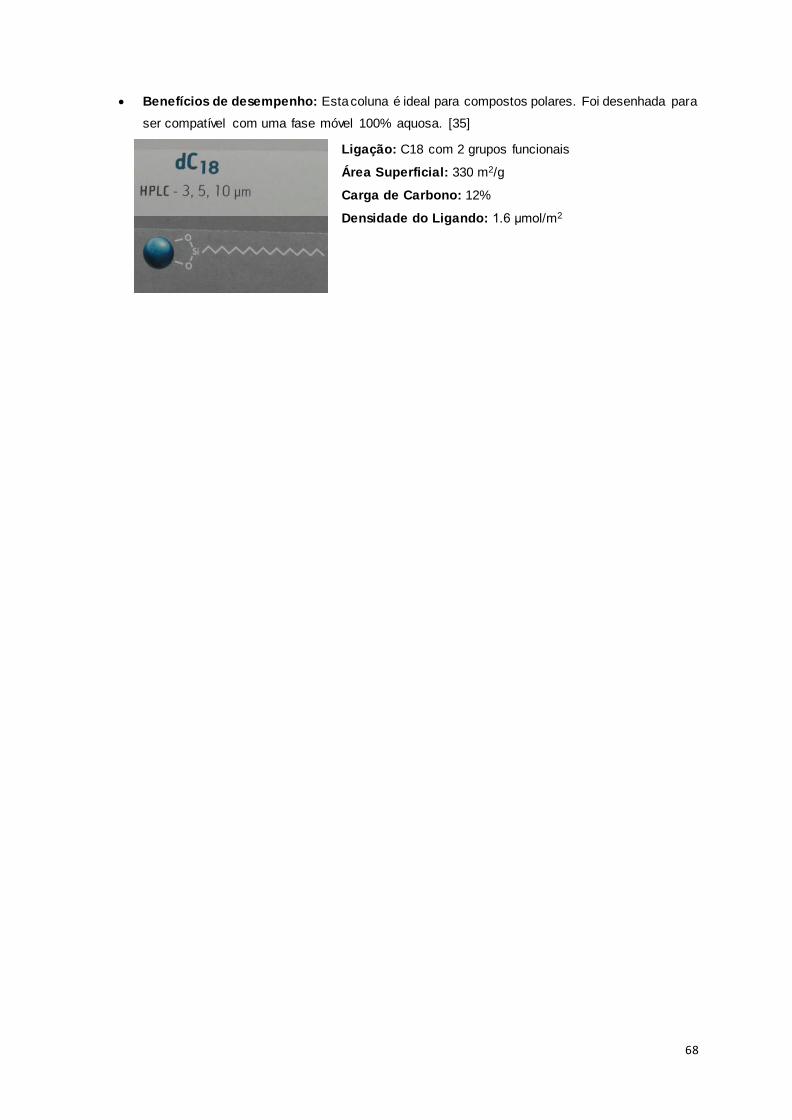
68
Benefícios de desempenho: Esta coluna é ideal para compostos polares. Foi desenhada para
ser compatível com uma fase móvel 100% aquosa. [35]
Ligação: C18 com 2 grupos funcionais
Área Superficial: 330 m2/g
Carga de Carbono: 12%
Densidade do Ligando: 1.6 μmol/m2
69
Anexo II
Cálculos – Soluções
Tabela II-1 – Volumes a retirar das soluções-mãe para se obter a solução intermédia em metanol.
HAAs
Soluções-Mãe
Volume Transferido
Solução-Intermédia
Balão de 10 mL Balão de 25 mL
Massa (g) g/L em MTBE (μL) mg/L em Metanol
MCAA 0,00987 0,987 25 0,987
DCAA 0,0151 1,51 17 1,03
TCAA 0,0105 1,05 24 1,01
MBAA 0,0106 1,06 24 1,02
DBAA 0,00970 0,970 25 0,970
BCAA 0,0229 2,29 11 1,01
Tabela II-2 – Volumes a retirar da solução intermédia para se obterem as soluções padrão para a reta de
calibração em balões de 5 mL em água.
(mg/L) (μg/L)
HAAs Solução
Intermédia
Volume a Retirar da Solução Intermédia (μL)
25 50 100 150 200 250
MCAA 0,987 4,94 9,87 19,7 29,6 39,5 49,3
DCAA 1,03 5,13 10,3 20,5 30,8 41,0 51,3
TCAA 1,01 5,05 10,1 20,2 30,3 40,4 50,5
MBAA 1,02 5,11 10,2 20,4 30,6 40,9 51,1
DBAA 0,970 4,85 9,69 19,4 29,1 38,8 48,5
BCAA 1,01 5,04 10,1 20,1 30,2 40,3 50,4
Tabela II-3 – Volumes a retirar das soluções-mãe para se obterem as soluções padrão individuais em metanol
em balões de 10 mL, para as infusões.
HAAs Ca Va Cb
mg/L μL mg/L
MCAA 987 20 1,97
DCAA 1509 15 2,26
TCAA 1052 20 2,10
MBAA 1064 20 2,13
DBAA 969 20 1,94
TBAA 1203 20 2,41
BCAA 2289 10 2,29

70

71
Anexo III
Calibrações Seguem as representações gráficas da sessão de 3 de Junho, com os respetivos valores para
o coeficiente de correlação, R, pelo método de calibração do padrão externo,
0
40
80
120
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - DCAA
0
150
300
450
600
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - MCAA
0
400
800
1200
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - MBAA
0
75
150
225
300
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - TCAA
Figura III.2 – Representação gráfica de um ensaio para o MCAA pelo método de padrão externo.
Figura III.1 – Representação gráfica de um ensaio
para o DCAA pelo método de padrão externo.
Figura III.4 – Representação gráfica de um ensaio
para o MBAA pelo método de padrão externo.
Figura III.5 – Representação gráfica de um ensaio
para o TCAA pelo método de padrão externo.
Figura III.6 – Representação gráfica de um ensaio para
o DBAA pelo método de padrão externo.
Figura III.3 – Representação gráfica de um ensaio
para o BCAA pelo método de padrão externo.
0
2500
5000
7500
10000
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - DBAA
0
2500
5000
7500
0 10 20 30 40 50
Áre
a
Concentração (μg/L)
Reta de calibração - BCAA

72
73
Anexo IV
Efeitos de Matriz
Tabela IV-1 – Áreas obtidas para uma concentração aproximada de 10 μg/L em diferentes matrizes.
C≈10 μg/L
Ácidos Haloacéticos Água
Ultrapura Água Luso
Água Penacova
Água Torneira 1
Água Torneira 2
MCAA 147 109 117 82 79
DCAA 23 31 22 25 8
TCAA 191 197 198 404 340
MBAA 360 345 325 311 288
DBAA 2357 2193 2260 1970 1966
BCAA 2150 2046 2063 1958 1913
Tabela IV-2 – Variação da % de água ultrapura versus % de água da torneira e respetivas áreas para o ácido
monocloroacético.
%Água Ultrapura %Água Torneira Área
100 0 370
90 10 348
70 30 307
40 60 253
0 100 192

74

75
Anexo V
Procedimento Analítico
Determinação de Compostos Orgânicos por Injeção Direta
em UPLC-MS-MS
Introdução
A determinação de ácidos haloacéticos (HAAs) em amostras de água de consumo humano é
necessária para determinar o grau de poluição destas.
Um método simples, rápido e sensível foi implementado em Cromatografia Liquida de Ultra
Eficiência com detetor de espectrometria de massa (UPLC-MS-MS), com o objetivo de analisar
baixas concentrações de ácidos haloacéticos em amostras de águas de consumo humano, sem
que estas necessitem de sofrer previamente um processo de concentração, sendo as mesmas
injetadas diretamente no equipamento.
Por esta metodologia podem ser analisados diversos ácidos haloacéticos.
1.1. Tipo de Amostras
Águas para consumo humano e outras águas limpas cloradas.
1.2. Princípio do Método
O equipamento de Cromatografia Liquida de Ultra Eficiência foi desenhado para resistir a
pressões muito elevadas que surgem devido ao uso de colunas de tamanho de partícula infer ior
a 2 µm.
A vantagem deste tipo de cromatografia sobre a cromatografia convencional de HPLC deve-se
ao melhoramento da resolução, menor tempo de retenção, uma maior sensibilidade e menor
consumo de solventes.
A amostra é injetada diretamente no equipamento UPLC-MS-MS onde os HAAs são separados
na coluna selecionada, são detetados através do tempo de retenção e da razão dos sinais das
transições MRM1 e MRM2 e de seguida quantificados através de curva de calibração por
método de adição de padrão.
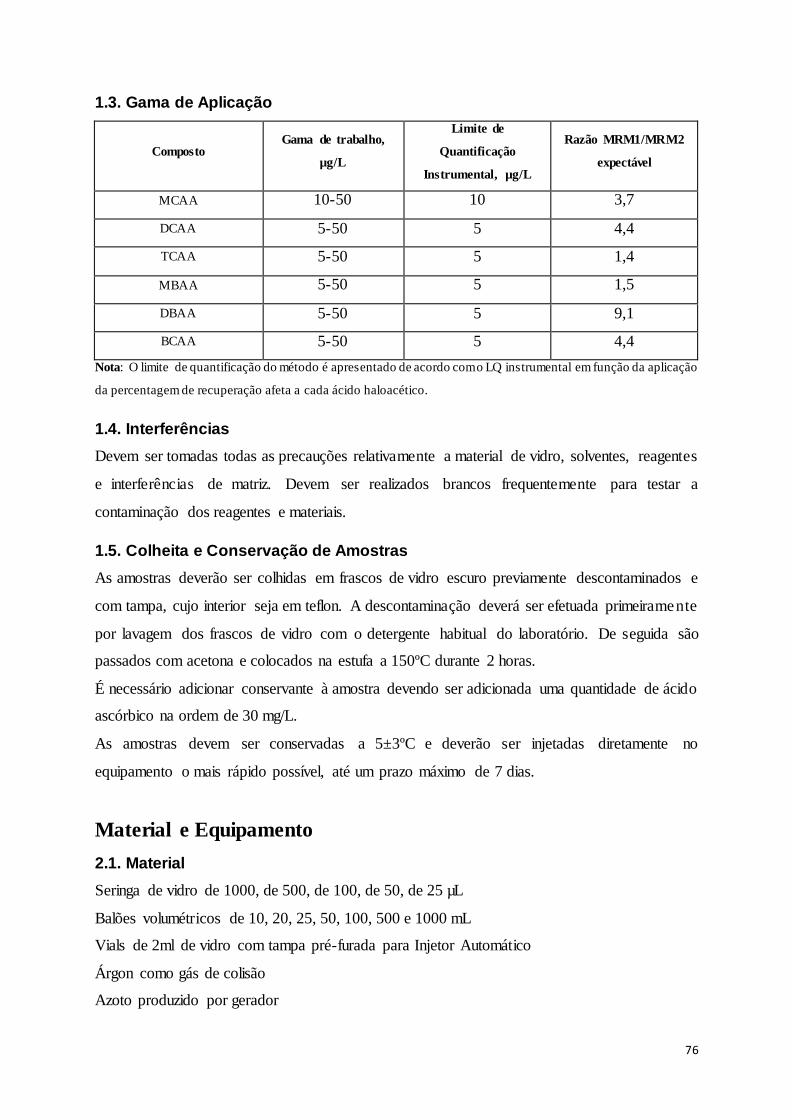
76
1.3. Gama de Aplicação
Composto Gama de trabalho,
µg/L
Limite de
Quantificação
Instrumental, µg/L
Razão MRM1/MRM2
expectável
MCAA 10-50 10 3,7
DCAA 5-50 5 4,4
TCAA 5-50 5 1,4
MBAA 5-50 5 1,5
DBAA 5-50 5 9,1
BCAA 5-50 5 4,4
Nota: O limite de quantificação do método é apresentado de acordo com o LQ instrumental em função da aplicação
da percentagem de recuperação afeta a cada ácido haloacético.
1.4. Interferências
Devem ser tomadas todas as precauções relativamente a material de vidro, solventes, reagentes
e interferências de matriz. Devem ser realizados brancos frequentemente para testar a
contaminação dos reagentes e materiais.
1.5. Colheita e Conservação de Amostras
As amostras deverão ser colhidas em frascos de vidro escuro previamente descontaminados e
com tampa, cujo interior seja em teflon. A descontaminação deverá ser efetuada primeiramente
por lavagem dos frascos de vidro com o detergente habitual do laboratório. De seguida são
passados com acetona e colocados na estufa a 150ºC durante 2 horas.
É necessário adicionar conservante à amostra devendo ser adicionada uma quantidade de ácido
ascórbico na ordem de 30 mg/L.
As amostras devem ser conservadas a 5±3ºC e deverão ser injetadas diretamente no
equipamento o mais rápido possível, até um prazo máximo de 7 dias.
Material e Equipamento
2.1. Material
Seringa de vidro de 1000, de 500, de 100, de 50, de 25 µL
Balões volumétricos de 10, 20, 25, 50, 100, 500 e 1000 mL
Vials de 2ml de vidro com tampa pré-furada para Injetor Automático
Árgon como gás de colisão
Azoto produzido por gerador

77
Micropipeta de 100-1000 µL
Micropipeta de 10 – 100 µL
Balança
Estufa
2.2. Reagentes
Água millipore (ou outra isenta de contaminantes)
Acetona p.a.
Metanol p.a.
Metanol para HPLC ou UPLC
Acetonitrilo para HPLC ou UPLC
Ácido Acético
Ácido Ascórbico
MTBE
Padrão puro no estado sólido (pureza >99%), de cada um dos HAAs
2.3. Equipamento UPLC-MS-MS
2.3.1. Condições do UPLC
ACQUITY UPLC
Coluna: Waters Atlantis dC18, 2.1x150 mm de 5µm (part number 186001301) ou equivalente
Pré-Coluna
Temperatura da coluna: 40ºC
Volume de injeção: 30µl
Fluxo: 0,4 ml/min em sistema gradiente:
Fase móvel A – Água com 0,2% de ácido acético
Fase móvel B - Metanol
Gradiente:
Tempo
(min)
Solvente
A
Solvente
B
Fluxo
(ml/min) Curva
0 95 5 0,4 6
2 60 40 0,4 6
3 0 100 0,4 6
7 0 100 0,4 6
7,1 95 5 0,4 6
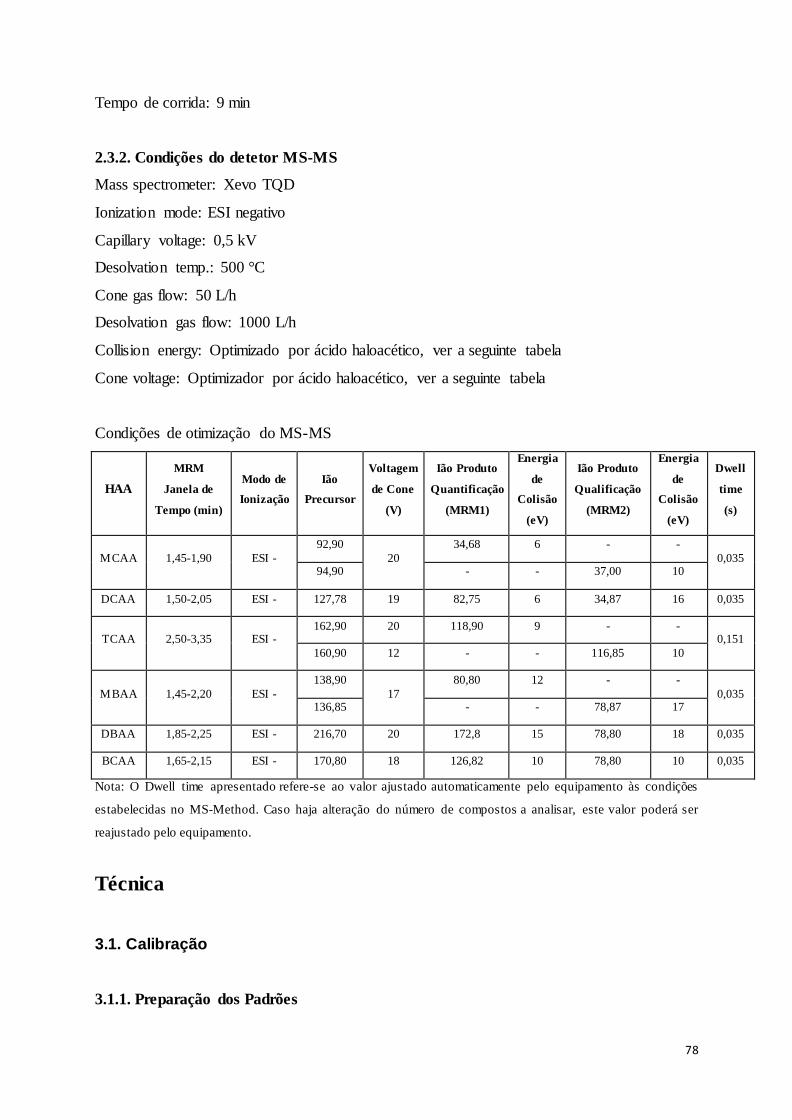
78
Tempo de corrida: 9 min
2.3.2. Condições do detetor MS-MS
Mass spectrometer: Xevo TQD
Ionization mode: ESI negativo
Capillary voltage: 0,5 kV
Desolvation temp.: 500 °C
Cone gas flow: 50 L/h
Desolvation gas flow: 1000 L/h
Collision energy: Optimizado por ácido haloacético, ver a seguinte tabela
Cone voltage: Optimizador por ácido haloacético, ver a seguinte tabela
Condições de otimização do MS-MS
HAA
MRM
Janela de
Tempo (min)
Modo de
Ionização
Ião
Precursor
Voltagem
de Cone
(V)
Ião Produto
Quantificação
(MRM1)
Energia
de
Colisão
(eV)
Ião Produto
Qualificação
(MRM2)
Energia
de
Colisão
(eV)
Dwell
time
(s)
MCAA 1,45-1,90 ESI -
92,90
20
34,68 6 - -
0,035 94,90 - - 37,00 10
DCAA 1,50-2,05 ESI - 127,78 19 82,75 6 34,87 16 0,035
TCAA 2,50-3,35 ESI - 162,90 20 118,90 9 - -
0,151
160,90 12 - - 116,85 10
MBAA 1,45-2,20 ESI -
138,90
17
80,80 12 - -
0,035 136,85 - - 78,87 17
DBAA 1,85-2,25 ESI - 216,70 20 172,8 15 78,80 18 0,035
BCAA 1,65-2,15 ESI - 170,80 18 126,82 10 78,80 10 0,035
Nota: O Dwell time apresentado refere-se ao valor ajustado automaticamente pelo equipamento às condições
estabelecidas no MS-Method. Caso haja alteração do número de compostos a analisar, este valor poderá ser
reajustado pelo equipamento.
Técnica
3.1. Calibração
3.1.1. Preparação dos Padrões

79
Os padrões são pesados individualmente em balões de 10ml, de forma a obter uma concentração
próxima de 1 g/L e dissolvidos em MTBE.
Após a utilização de cada um destes padrões tomar nota do respetivo peso.
Guardar os padrões no frigorífico. Estes padrões conservam-se durante um período máximo de 12
meses.
Antes de cada utilização, aguardar que o balão atinja a temperatura ambiente e depois acertar o peso
com o solvente usado, caso aplicável.
Preparar uma solução intermédia A em metanol dos vários ácidos haloacéticos com uma concentração
na ordem de 1 mg/L, retirando o volume conveniente de cada balão mãe. Esta solução deverá ser
preparada em todos os ensaios.
3.1.2. Traçado das Curvas de Calibração – Método da Adição de Padrão
A partir da solução intermédia A, retirar os volumes necessários para preparar as soluções padrão a
usar na calibração pelo método da adição de padrão.
Devem ser preparados diariamente, no mínimo, quatro padrões na matriz da amostra com uma
concentração na ordem de 5 µg/L a 50 µg/L.
A cada padrão preparado será adicionada uma solução ácida de forma a que no final apresente
uma concentração de 30 mg/L de ácido ascórbico e 0,1% de ácido acético.
Colocar cada padrão em vial de 2ml com tampa pré-furada e de seguida no amostrador do
equipamento. Injetar cada padrão segundo as condições definidas no ponto 2.3.
Traçar as curvas de calibração representando área do padrão de cada composto versus concentração
do padrão injetado, utilizando o método dos mínimos quadrados.
3.1.3. Modo de Operação do equipamento
Qualquer dúvida que surja em relação ao manuseamento do aparelho UPLC-MS-MS, recorrer
às instruções do fabricante que se encontram descritas no "Manual de Instruções" que o
acompanha.
3.2. Preparação da Amostra
Deixar a amostra atingir a temperatura ambiente.
Proceder ao método de adição de padrão acidificando no fim cada solução.
Colocar os padrões da reta da amostra em vials de 2mL e proceder à sua análise no UPLC-MS-MS.

80
Resultados
No cromatograma obtido o equipamento identifica o tempo de retenção de cada pico e integra
a área e altura correspondente à primeira transição MRM1 usada para quantificação e a área e
altura correspondente à segunda transição MRM2 usada para qualificação. Os parâmetros
tempo de retenção e razão das áreas ou alturas MRM1/MRM2 são usados para identificar
inequivocamente o composto correspondente a um determinado pico por comparação com o do
padrão puro.
A concentração de cada composto existente na amostra é determinada pela interseção da reta
obtida com o eixo da concentração.
Curva de calibração:
Conc. de cada ácido haloacético na amostra (µg/L) =m
b
b – ordenada na origem
m – declive
A razão das transições MRM1 e MRM2 é calculada através da área ou altura de pico resultante
da transição MRM1 e a área ou altura de pico resultante da transição MRM2, para o respetivo
composto de acordo com a seguinte expressão:
2
1
MRM
MRM
A
AR
em que:
R - Razão das transições para um determinado composto
AMRM1 - Área ou altura de pico do composto para a transição MRM1
AMRM2 - Área ou altura de pico do composto para a transição MRM2
Critério de Aceitação de Resultados
Os critérios de aceitação de resultados encontram-se definidos no CAR NII/ØØ.
Bibliografia
Consulta de artigos publicados em revistas da especialidade e na Internet.

81





























