Embed Size (px)
Citation preview

DFT Study of the ExBox·Aromatic Hydrocarbon Host−Guest ComplexSteven M. Bachrach*
Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States
*S Supporting Information
ABSTRACT: The structures of ExBox4+ 1 and its host−guest complexeswith the linear acenes benzene, naphthalene, anthracene, and tetracenewere optimized using DFT (ωB97X-D/6-311G(d,p)) in both the gas andsolution phases. The structure of 1 systematically varies as it moves fromthe gas to solution to the solid phase: the outward bending of the triarylfragment diminishes in this series. The structures of the complexes withanthracene and tetracene are in very good agreement with their X-raystructures. The gas phase binding energy is linearly related to the size of theacene, with the binding free energy of all complexes predicted to beexoergonic in both gas and solution phases.
■ INTRODUCTIONHost−guest chemistry involves the noncovalent interactionbetween a typically large host molecule with a smaller guestmolecule.1,2 The host molecule typically presents an interiorregion by assuming a concave shape. The guest binds bymatching the size and shape of the host, along with appropriatematching of features that permit favorable interaction, such asalignment of charges or dipoles.Host−guest chemistry has been utilized for a wide variety of
purposes, perhaps most famously by nature in designingenzymes to catalyze reactions, often with high selectivity andstereospecificity. Host−guest chemistry has been applied to theselective sequestration of molecules for a variety of purposes.This can include using hosts to remove compounds fromsolutions, hosts that catalyze reactions, and hosts for sensingpurposes.Isolation of polycyclic aromatic hydrocarbons (PAHs) is
important for environmental reasons, as these molecules can betoxic and carcinogenic. Extracting PAHs from crude mixtures iscritical toward removing these species from water supplies andother liquids. Cyclodextrins3−5 and calix[n]arene6,7 have beenused in this endeavor. Stoddart recently reported thedevelopment of a new compound, the so-called ExBox4+ 1,which was shown to exhibit strong and selective binding to avariety of PAHs.8 Subsequently, the Stoddart group reportedthe synthesis and binding capabilities of a larger variant calledEx2Box4+, which has two phenyl rings between the pyridiniumpairs,9 instead of the one phenyl ring in ExBox4+.The binding of a PAH to the interior of a host like ExBox4+
is likely to involve London dispersion and perhaps electrostaticinteractions. The weak, noncovalent dispersion interactionpresents a real challenge to computational chemistry.10
Dispersion is absent from the Hartree−Fock treatment, andso generally large configuration interaction computations orhigh-order perturbation methods are needed to properly
account for dispersion. Many widely utilized density functionalslack any accounting of dispersion, such as B3LYP. Instead,functionals that are corrected for dispersion are required,10 suchas with Grimme’s dispersion correction.11,12
We present here a study of the binding of ExBox4+ 1 withbenzene 2, naphthalene 3, anthracene 4, or tetracene 5evaluated using the ωB97X-D functional,13 a modern functionalthat includes a dispersion correction. The structures andbinding energies in both the gas and solution phases areexamined and compared with experiment. We demonstrate thatthe ωB97X-D functional performs well in predicting thebinding in these host−guest complexes and offers anappropriate method for further exploration of this new hostclass.
■ COMPUTATIONAL METHODSThe geometries of 1−5 along with the complexes formedbetween 1 and either 2, 3, 4, or 5 were completely optimized atωB97X-D/6-31G(d) and at ωB97X-D/6-311G(d,p).13 Geo-
Received: July 10, 2013Revised: August 6, 2013
Article
pubs.acs.org/JPCA
© XXXX American Chemical Society A dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXX

metries were constrained to appropriate point groups.Analytical frequencies were computed with both basis sets todetermine the nature of the structure: ground state having noimaginary frequencies, transition state having one imaginaryfrequency, or a higher-order saddle point. The unscaledfrequencies were used to compute the zero-point vibrationalenergy, along with obtaining the enthalpy and free energy at298.15 K. Some complexes were also optimized at B3LYP/6-31G(d) for comparison purposes. Solution phase computationswere performed using the conductor-like polarizable continuummodel (CPCM)14 with acetonitrile as the solvent, in order tocompare with the experiments. The complexes were reopti-mized at both CPCM/ωB97X-D/6-31G(d) and CPCM/ωB97X-D/6-311G(d,p), and frequencies were recomputed aswell. All computations were performed with the Gaussian09suite.15
Stoddart and Goddard et al. reported computations of someacene complexes with Ex2Box4+ at M06-2x/6-311G.9 Thesecomputations provided geometries in reasonable agreementwith experimental crystal structures. However, these computa-tions suffer from a basis set that lacks polarization functions.
■ RESULTSStructure of ExBox4+ 1. The top and bottom of the
ExBox4+ molecule is a pyridinium−phenyl−pyridinium chain.This is analogous to the triphenylene molecule, which weexamined as part of a study of cycloparaphenylenes.16 Thethree phenyl rings of cycloparaphenylene can be oriented in analternating pattern or a helix, and the former is computed to beslightly more stable. The same conformational options wereexamined for 6, which can possess the alternating (6a) orhelical (6b) conformation. The alternating form 6a is slightlylower in enthalpy than 6b, by 0.06 kcal mol−1 at both ωB97X-D/6-31G(d) and ωB97X-D/6-311G(d,p).
This suggests that there are four symmetrical conformationsof 1 worth examining, where the top and bottom are eitherboth of the alternating type (leading to structures of pointgroups C2v or C2h) or both of the helical type (leading tostructures of point groups C2h or D2). Another possibility is forthe triaryl chains to be of different type, giving a C1 symmetryExBox4+. These five conformers are shown in Figure 1. Theirrelative energies are listed in Table 1. As expected theconformers having the alternating triaryl fragments are lowerin energy than those that contain the helical fragments.However, all five conformers are close in energy. Both basis setsgive quite similar relative energies of these conformers.The B3LYP functional remains quite popular despite
numerous studies that have detailed its deficiencies. Aparticularly notable deficiency is its inability to account fordispersion.17−19 The error arises even in structures such as 1awhere the rings are far apart. B3LYP/6-31G(d) predicts anExBox4+ structure with too large a cavity. The distance acrossthe ExBox4+, measured between the centers of the centralphenyl ring of each triaryl chain, is 8.955 Å at B3LYP, butshrinks to 8.641 Å when computed at ωB97X-D/6-31G(d),and decreases even further (to 8.603 Å) when the basis set isincreased to 6-311G(d,p).Visual inspection of the structures of 1 shown in Figure 1
indicate a curvature of the triaryl fragments, implying some
strain in this macrocycle. We have effectively employed thegroup equivalent reaction20 to assess the ring strain energy insmall and medium size molecules.16,21−24 Reaction 1 is one ofmany group equivalent reactions that might be employed toascertain the ring strain energy of 1. This reaction utilizes 6 tomimic the acyclic reference for the triaryl top and bottom ofExBox4+, 7 as the reference for the unstrained methylene group,and 8 as the reference for the phenyl groups on the sides. Theenthalpy of this reaction is computed to be 151.2 kcal mol−1 atωB97X-D/6-311G(d,p) (152.3 at ωB97X-D/6-31G(d)). Thisimplies an awfully large ring strain energy in 1.
Inspection of Reaction 1 reveals that while charge is properlyconserved in this reaction, the relative distances between thesecharges is not conserved. The positive charges of the fourmolecules of 7 map onto the positive charges on methylpyr-idinium, and all of these are infinitely far apart. However, thefour charges on the two molecules of 6 are brought togetherinto near proximity in forming 1. Therefore, some fraction ofthe energy of reaction 1 is due to ring strain and some is due toelectrostatic repulsion brought about in placing the fourpositive charges near each other.In order to (approximately) correct for this electrostatic
contribution to the energy of reaction 1, we employ the modelshown in reaction 2. Structure 9 is made up of two molecules of6 constrained to have the distances between the nitrogen atoms
Figure 1. Optimized structures of the conformers of 1.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXB
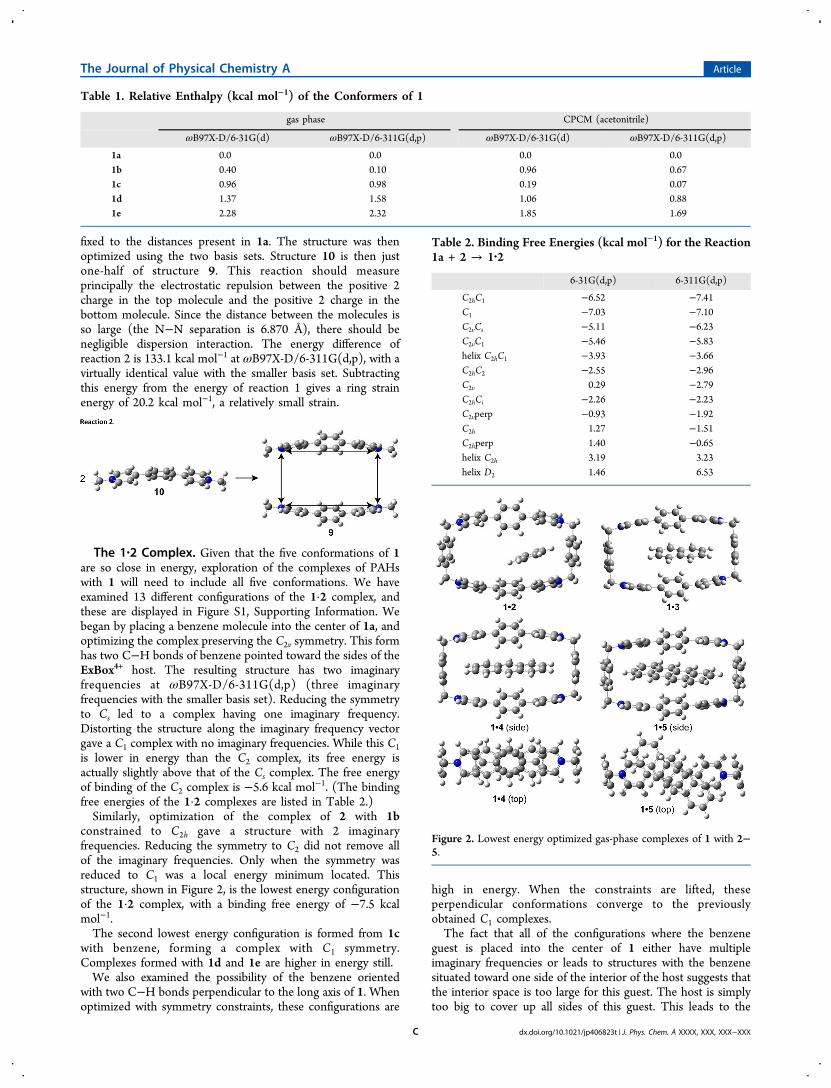
fixed to the distances present in 1a. The structure was thenoptimized using the two basis sets. Structure 10 is then justone-half of structure 9. This reaction should measureprincipally the electrostatic repulsion between the positive 2charge in the top molecule and the positive 2 charge in thebottom molecule. Since the distance between the molecules isso large (the N−N separation is 6.870 Å), there should benegligible dispersion interaction. The energy difference ofreaction 2 is 133.1 kcal mol−1 at ωB97X-D/6-311G(d,p), with avirtually identical value with the smaller basis set. Subtractingthis energy from the energy of reaction 1 gives a ring strainenergy of 20.2 kcal mol−1, a relatively small strain.
The 1·2 Complex. Given that the five conformations of 1are so close in energy, exploration of the complexes of PAHswith 1 will need to include all five conformations. We haveexamined 13 different configurations of the 1·2 complex, andthese are displayed in Figure S1, Supporting Information. Webegan by placing a benzene molecule into the center of 1a, andoptimizing the complex preserving the C2v symmetry. This formhas two C−H bonds of benzene pointed toward the sides of theExBox4+ host. The resulting structure has two imaginaryfrequencies at ωB97X-D/6-311G(d,p) (three imaginaryfrequencies with the smaller basis set). Reducing the symmetryto Cs led to a complex having one imaginary frequency.Distorting the structure along the imaginary frequency vectorgave a C1 complex with no imaginary frequencies. While this C1is lower in energy than the C2 complex, its free energy isactually slightly above that of the Cs complex. The free energyof binding of the C2 complex is −5.6 kcal mol−1. (The bindingfree energies of the 1·2 complexes are listed in Table 2.)Similarly, optimization of the complex of 2 with 1b
constrained to C2h gave a structure with 2 imaginaryfrequencies. Reducing the symmetry to C2 did not remove allof the imaginary frequencies. Only when the symmetry wasreduced to C1 was a local energy minimum located. Thisstructure, shown in Figure 2, is the lowest energy configurationof the 1·2 complex, with a binding free energy of −7.5 kcalmol−1.The second lowest energy configuration is formed from 1c
with benzene, forming a complex with C1 symmetry.Complexes formed with 1d and 1e are higher in energy still.We also examined the possibility of the benzene oriented
with two C−H bonds perpendicular to the long axis of 1. Whenoptimized with symmetry constraints, these configurations are
high in energy. When the constraints are lifted, theseperpendicular conformations converge to the previouslyobtained C1 complexes.The fact that all of the configurations where the benzene
guest is placed into the center of 1 either have multipleimaginary frequencies or leads to structures with the benzenesituated toward one side of the interior of the host suggests thatthe interior space is too large for this guest. The host is simplytoo big to cover up all sides of this guest. This leads to the
Table 1. Relative Enthalpy (kcal mol−1) of the Conformers of 1
gas phase CPCM (acetonitrile)
ωB97X-D/6-31G(d) ωB97X-D/6-311G(d,p) ωB97X-D/6-31G(d) ωB97X-D/6-311G(d,p)
1a 0.0 0.0 0.0 0.01b 0.40 0.10 0.96 0.671c 0.96 0.98 0.19 0.071d 1.37 1.58 1.06 0.881e 2.28 2.32 1.85 1.69
Table 2. Binding Free Energies (kcal mol−1) for the Reaction1a + 2 → 1·2
6-31G(d,p) 6-311G(d,p)
C2hC1 −6.52 −7.41C1 −7.03 −7.10C2vCs −5.11 −6.23C2vC1 −5.46 −5.83helix C2hC1 −3.93 −3.66C2hC2 −2.55 −2.96C2v 0.29 −2.79C2hCi −2.26 −2.23C2vperp −0.93 −1.92C2h 1.27 −1.51C2hperp 1.40 −0.65helix C2h 3.19 3.23helix D2 1.46 6.53
Figure 2. Lowest energy optimized gas-phase complexes of 1 with 2−5.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXC

relatively small free energy of binding of benzene in the gasphase. Further, it suggests that larger PAHs should bind moretightly.The 1·3 Complex. Optimization of the complex of
naphthalene 3 with either 1a or 1b at C2v and C2h, respectively,led to structures possessing one or more imaginary frequencies.Reducing the symmetry of the C2h structure to C2 by distortingalong the imaginary frequency gives the lowest energy 1·3complex, shown in Figure 2. Distorting the C2v structure to Csgives the second lowest energy configuration (shown in FigureS2, Supporting Information); it is 0.32 higher in free energythan the lowest energy structure.In these two local minima, the naphthalene guest lies along
the long axis of 1. Placing 3 so that it is perpendicular to the 1means that some of each of the two phenyl rings of 3 extendspast the width of 1. One might expect that this orientationwould be less favorable as it diminishes the possible dispersioninteraction between the host and guest. In fact, not only do theC2v and C2h perpendicular structures lie significantly higher inenergy (more than 8 kcal mol−1 above the lowest energy 1·3complex), these perpendicular structures possess multipleimaginary frequencies.As with benzene, naphthalene is too small to fully fill the
interior of 1. Just as with benzene, naphthalene slides out of thecenter of 1 to maximize the dispersion interactions with thehost. Naphthalene does bind more tightly to 1 than doesbenzene: −14.5 for 3 vs −7.4 kcal mol−1 for 2 (see Table 3). Aneven larger PAH is needed to fill the interior of 1.
The 1·4 Complex. The lowest energy configuration of thecomplex formed between 1 and anthracene 4 has C2v symmetryand is shown in Figure 2 from both a side and top view. Theanthracene host sits in the center of 1 and is held within thewalls of the host.The crystal structure8 of the 1·4 complex has two different
host−guest complexes in the unit cell, both of C2 symmetry,but one has near helical triaryl fragments and the other hasalternating triaryl fragments. This latter experimental host−
guest structure is very similar to the computed C2v structure.The symmetry plane of the C2v structure that reflects the toptriaryl fragment into the bottom one is absent in the crystalstructure. This manifests in the anthracene lying not along themiddle of 1, as in the computed C2v structure, but rather lies ata diagonal. The other major difference is the size of the host;the host puckers out more in the computed structure that in thecrystal structure. This can be quantified by comparing thedistance between the center of the middle ring of anthracene tothe center of the phenyl ring of the triaryl fragment. In thecrystal structure this distance is 3.683 Å, while in the computedstructure the distance is 3.942 Å. This compression of theExBox4+ host in the crystal structure is likely a result of crystalpacking forces. This is supported in part by the optimizedstructure of the C2v configuration of 1·4 in acetonitrile solution;here the intraring distance is 3.769 Å, a value less than 0.1 Ålonger than that found in the experimental crystal structure.Optimization of anthracene inside 1b, a structure with C2h
symmetry, leads to a structure with one imaginary frequency.Reducing the symmetry to Ci gives a structure (shown in FigureS3, Supporting Information) that is 0.6 kcal mol−1 above theC2v structure discussed above. Complexes that have theanthracene perpendicular to the host, preserving either C2h orC2v symmetry, have two imaginary frequencies.Anthracene appears to fit nicely within the confines of the
ExBox4+ interior. This is reflected in its large binding freeenergy of −22.8 kcal mol−1, significantly larger than the bindingfree energy of 1 with either benzene or naphthalene.The inherent deficiencies of the B3LYP functional are clearly
evident in examination of the optimized C2v structure for 1·4 itpredicts. This structure is shown in Figure 3. In the top viewone can readily observe that the anthracene molecule hasmoved out of the host cavity and weakly interacts with thehydrogens of the central phenyl rings. While B3LYP/6-31G(d)does predict that the formation of this complex is exothermic(ΔH = −8.1 kcal mol−1), it is endergonic (ΔG = +1.7 kcalmol−1). Because of the importance of dispersion in theinteraction between 1 and 4 and the fact that B3LYP doesnot account for dispersion, B3LYP fails to properly predict thegeometry or binding energy of the 1·4 complex.
The 1·5 Complex. Optimization of the complex oftetracene placed in the interior of 1, with the long axis of themolecule coincident with the long axis of ExBox4+ led to a C2vcomplex. However, the tetracene molecule has moved out ofthe interior of the ring, and the phenyl sides of 1 have twistedopen (see Figure S4, Supporting Information). Tetracene isclearly too big to fit entirely within the interior of 1.This is not a surprise as the X-ray structure8 of the 1·5
complex shows the tetracene oriented at an angle to the host,
Table 3. Gas Phase Binding Enthalpies and Free Energies(kcal mol−1) for the Reaction 1a + X → 1·X
ΔH ΔG
1:X 6-31G(d,p) 6-311G(d,p) 6-31G(d,p) 6-311G(d,p)
1·2 −18.26 −18.48 −6.52 −7.411·3 −25.60 −26.28 −14.07 −14.501·4 −37.86 −38.50 −21.70 −22.811·5 −44.88 −45.58 −29.50 −30.57
Figure 3. B3LYP/6-31G(d) optimized structure of the 1·4 complex.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXD

with its two terminal phenyl rings protruding outside of theconfines of 1. Embedding tetracene in such a fashion into 1band optimizing gives a Ci complex that is the lowest energyconfiguration. As seen in the top view presented in Figure 2, theterminal phenyls of 5 stick outside of 1. The complex formed of1a and tetracene optimizes to a Cs structure that is 2.9 kcalmol−1 above the Ci complex. Other configurations were alsooptimized starting with 1d; these are 5−7 kcal mol−1 higher inenergy than the Ci complex.The lowest energy 1·5 complex, with Ci symmetry is similar
to the experimentally observed structure. Both have Ci
symmetry with the tetracene oriented at an angle to the longaxis of the ExBox4+, with its ends protruding outside theinterior. The computed 1·5 structure is puckered out more thanin the experiment, but not as much as in the anthracene case.The distance between the center of the phenyl ring of thetriaryl fragment to the midpoint of the C5a−C11a bond (thebond shared by the B and C rings) is 3.614 Å in theexperimental structure and 3.677 Å in the computed structure.Again, crystal packing is the likely cause of the more compactExBox4+ structure in the solid phase than in the gas phase.Despite the fact that tetracene extends outside of 1, its
binding free energy is quite large: −30.6 kcal mol−1. Thisbinding energy is larger than the values for the smaller acenes.Solution-Phase Complexes. While computed gas phase
structures allow one to probe the inherent nature of theinteractions that make up the complexes, ExBox4+ is of interestfor its behavior as a sequestering agent in solution. Thus, wereoptimized structures mimicking an acetonitrile solution usingthe CPCM procedure with the ωB97X-D functional and usingboth the 6-31G(d) and 6-311G(d,p) basis sets.All five conformers of 1 were reoptimized with CPCM. The
relative free energies of these solvated ExBox4+ conformationsare listed in Table 1. The lowest energy conformer in solutionis 1a, the same as in the gas phase. However, the second lowestenergy conformer in solution is 1c, and it is only slightly higherin energy than 1a.While the lowest energy conformation of 1 has the same
symmetry in both solution and gas phase, there are someimportant differences in its structure in the two phases. Thetriaryl fragments bow out more in the gas phase than in
solution. This is seen in the distance between the centers of thecentral ring of the triaryl fragment: 7.485 Å in solution and8.603 Å in the gas phase. The long axis of the box, measured bythe distance between the centers of the phenyl group on eachside, changes in concert with the short axis. So, the more bowedout structure, present in the gas phase, has the shorter long axis(13.961 Å) than in solution (14.43 Å).The Stoddardt group has reported the X-ray crystal structure
of 1; two different geometries are observed in the unit cell.8
Both have C2 symmetry, with alternating rings along the triarylfragment. Both structures are slightly distorted from C2v
symmetry. The distances for the short axis are 6.791 and7.106 Å, while the distances of their long axis are 14.805 and14.637 Å. The ExBox4+ structure becomes less bowed out as itmoves from the gas to solution to solid phase.The two lowest energy configurations of the ExBox4+·
aromatic complexes were reoptimized with CPCM. For the 1·2complex, the C1 complex with 1 in a C2v-like conformation isslightly lower in energy than the C1 complex with 1 in a C2h-likeconformation, opposite that found in the gas phase. Thegeometry of this lowest energy configuration found inacetonitrile solution is displayed in Figure 4.A similar reversal is seen in the energy ordering of the 1·3
and 1·4 complexes. The 1·3 complex with 1 in a C2h-likearrangement, which is the lowest energy gas phase config-uration, is 2.3 kcal mol−1 higher in free energy than theconfiguration with 1 in a C2v-like arrangement. The C2v
configuration of 1·4, the lowest energy configuration in thegas phase, is 0.3 kcal mol−1 above the complex with 1 in a C2h-like conformation. These lowest energy configurations are alsoshown in Figure 4.For the 1·5 complex, the relative energy ordering remains the
same in the gas and acetonitrile solution phases; the lowestenergy configuration has Ci symmetry, and the solution phasestructure is given in Figure 4. However, the energy differencebetween the Ci configuration and the Cs configurationdiminishes from 2.9 kcal mol−1 in the gas phase to 0.7 kcalmol−1 in solution.The solution phase binding energies of the ExBox4+
complexes are listed in Table 4. As with the gas phase results,both the enthalpies and free energies are quite similar when
Figure 4. Lowest energy optimized solution-phase (acetonitrile) complexes of 1 with 2−5.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXE

computed with either the 6-31G(d) or 6-311G(d,p) basis set.While the formation of all four complexes are predicted to beexorgonic, the binding is not as strong in solution as in the gasphase. As in the gas phase, binding energy increases with thesize of the acene.
■ DISCUSSIONThe host−guest complexes of ExBox4+ 1 with polycyclicaromatic compounds constitute a set of supermolecular systemsthat pose challenges to computational chemistry. Theinteraction between the host and guest is noncovalent andweak, including π−π stacking, that requires careful consid-eration of the proper methodology. The popular B3LYPfunction does not properly account for dispersion and thus failsto describe both the ExBox4+ molecule itself and the complexesit forms with PAHs. For example, in the complex withanthracene, B3LYP predicts that anthracene will be positionedoutside of the interior of 1 (see Figure 3), while in fact the X-ray crystal structure shows anthracene nicely held within theconfines of the box.Computations with the ωB97X-D functional eliminate these
problems. The structures of the computed gas and solutionphases of 1 are geometrically similar, having the samealternating triaryl motifs and symmetry. There is an interestingtrend in the size of the cavity: the ExBox4+ molecule is mostbowed out in the gas phase, less so in solution, and even less soin the solid phase. This can be attributed to an increase in theinteractions with neighboring molecules as it moves from thegas to solution to solid phase, causing 1 to collapse inward.These intermolecular interactions tend to make the cavity morecompact.Stoddart and Goddard’s DFT computations9 of Ex2Box4+
suggest a strong electrostatic component to the binding whenthe guest is electron rich. We will address the role ofelectrostatics in the binding of PAHs to 1 in a later study.The poor accounting of binding by the B3LYP functional,which omits any dispersion, strongly implicates dispersion inthe binding process of 1 with PAHs.More importantly, the ωB97X-D functional predicts that all
four of the linear acenes examined here will bind to the interiorof the ExBox4+ molecule. Benzene is much smaller than theinterior of 1 and so its preferred position is not right in themiddle of the ring, but rather it slides over to one side tomaximize its dispersion interaction with the much largerExBox4+ ring. Naphthalene too does not fill up the interior of 1and its favored position is also off to a side. The optimalposition of anthracene is right in the center of 1, suggesting thatit essentially fills the interior of the host. These gas-phase andsolution phase 1·4 structures (see Figures 2 and 4) are quitesimilar to that found in the X-ray crystal structure.Tetracene is too long to fit within the cavity of 1, and so the
molecule aligns at an angle with both ends protruding outside
of the host. Again, the computed gas and solution phasestructures are quite similar to that observed in the crystalstructure.Having part of the guest protrude out of the interior of the
host 1 is less favorable than the case where the host is entirelycontained within 1. For example, if we compare the freeenergies of the C2h structure of 1·3 to the structure wherenaphthalene is rotated by 90° so that the ends of naphthalenestick out of the host (Scheme 1), the former structure is lower
in energy by 5.4 kcal mol−1. Similarly, the C2v configuration of1·4 having anthracene entirely within 1 is 9.7 kcal mol−1 lowerin free energy that the rotated version (Scheme 1).The above result, along with our understanding of dispersion
as being related to surface contact, suggests that the binding ofthe acene to 1 should increase with the size of the acene(perhaps even linearly) up to anthracene. However, sincetetracene does not fit wholly within 1, one might expect thatthis linear relationship at least might break. Figure 5 presents aplot of the binding free energy relative to the number of ringsin the linear acene.In fact, in the gas phase, there is a linear relationship between
the binding free energy and the number of rings of the acene.So, even though some fraction of each terminal ring oftetracene extends outside the interior of 1, the binding freeenergy falls right on the line in Figure 5. This is not true insolution; the binding energy of tetracene is only 1.3 kcal mol−1
Table 4. Solution Phase (Acetonitrile) Binding Enthalpiesand Free Energies (kcal mol−1) for the Reaction 1a + X →1·X
ΔH ΔG
1:X 6-31G(d,p) 6-311G(d,p) 6-31G(d,p) 6-311G(d,p)
1·2 −16.01 −15.94 −5.54 −4.881·3 −25.47 −25.00 −9.97 −13.681·4 −33.80 −34.74 −17.29 −18.791·5 −37.78 −38.16 −19.89 −20.10
Scheme 1. Top View of the 1·3 and 1·4 Complexes with theGuest Aligned Inside and Perpendicular to the Host;Relative Free Energies in kcal mol−1
Figure 5. Computed binding free energy (kcal mol−1) vs. number ofrings in the guest acene. Filled circles are for the gas phase, and openboxes are for the solution phase.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXF

greater than that of anthracene. There is certainly some levelingoff with tetracene. Detailed analysis of the nature of the bindingenergy is currently underway.One final consideration is the comparison of the computed
solution-phase binding energy of 1·4. Its predicted binding freeenergy is −18.8 kcal mol−1, a very exoergic reaction. Theexperimental value for the binding free energy, obtained byITC, is −4.01 ± 0.073 kcal mol−1.8 There is a large discrepancybetween the computed and experimental value. A number ofassumptions can be at fault. Grimme has demonstrated thataccurate computation of frequencies, needed in determining theentropy and free energy, are very sensitive to the basis set, andwe are forced to use a rather small basis set here.25,26 Second,our solution phase computations neglect any local solvationeffects, which might be important given the large charge on 1.In addition, the solution computations neglect any role that thegegenion might have in the equilibrium. Given theseapproximations, it might be best to simply accept that thesolution phase computations are properly indicating that thereaction is exoergonic. Further study of the trends in thebinding energy are underway.
■ CONCLUSIONSExBox4+ 1 is slightly strained, by about 20 kcal mol−1, whenproperly corrected for the high charge density on thecompound. The geometry of 1 is well-predicted by ωB97X-D/6-311G(d,p), but B3LYP predicts a geometry that is toobent. The lack of accounting for dispersion by the B3LYPfunctional leads to this defect. The dimensions of 1 aredependent on phase; the compound is quite bowed out in thegas phase, less so in solution, and even less bowed in the solidstate.The interior of 1 provides an excellent environment for a
linear acene. Both benzene and naphthalene are smaller thanthe interior space of 1; each slides to one side of the interior tomaximize interactions with the host. For both the gas andsolution phases, anthracene sits right in the middle of 1, in ageometry very similar to that found in the X-ray crystalstructure of the 1·4 complex. Tetracene is too large of theinterior of 1 and it sits at an angle to the larger ring, with bothends protruding outside of the interior of ExBox4+. Thesecomputed gas and solution phase structures are quite similar tothe X-ray structure of the 1·5 complex. Common functionalsthat omit dispersion are unsuited for study of these complexes;B3LYP does not predict a geometry with the acene in theinterior of 1.The binding free energy of 1 with the acenes increases as the
size of the linear acene increases. This increase is linear (in thegas phase) with respect to the number of rings of the acene,even though tetracene does not fully lie within 1. The bindingenergy in solution is not linear and appears to be leveling offwith tetracene.
■ ASSOCIATED CONTENT*S Supporting InformationFull citation for ref 15, figures and relative energies of allExBox4+·acene complexes, and coordinates and absoluteenergies of all computed structures. This material is availablefree of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
The author thanks Dr. Charlotte Stern for providing CIF andres files of the X-ray structure of 1·5, a reviewer for pointing outref 9, and Trinity University for the computational resourcesutilized in this project.
■ REFERENCES(1) Steed, J. W.; Atwood, J. L. Supramolecular Chemistry; Wiley-VCH:Weinheim, Germany, 2009.(2) Cragg, P. A Practical Guide to Supramolecular Chemistry; JohnWiley & Sons: Chichester, U.K., 2005.(3) Blyshak, L. A.; Dodson, K. Y.; Patonay, G.; Warner, I. M.; May,W. E. Determination of Cyclodextrin Formation Constants UsingDynamic Coupled-Column Liquid Chromatography. Anal. Chem.1989, 61, 955−960.(4) Ravelet, C.; Ravel, A.; Grosset, C.; Villet, A.; Geze, A.;Wouessidjewe, D.; Peyrin, E. Stoichiometry and Formation Constantsof Six PAHs with γ-Cyclodextrin, Determined by HPLC Using aCyano Stationary Phase. J. Liq. Chromatogr. Relat. Technol. 2002, 25,421−432.(5) Butterfield, M. T.; Agbaria, R. A.; Warner, I. M. Extraction ofVolatile PAHs from Air by Use of Solid Cyclodextrin. Anal. Chem.1996, 68, 1187−1190.(6) Bandela, A.; Chinta, J. P.; Hinge, V. K.; Dikundwar, A. G.; Row,T. N. G.; Rao, C. P. Recognition of Polycyclic Aromatic Hydrocarbonsand Their Derivatives by the 1,3-Dinaphthalimide Conjugate ofCalix[4]arene: Emission, Absorption, Crystal Structures, and Compu-tational Studies. J. Org. Chem. 2011, 76, 1742−1750.(7) Li, H.; Qu, F. Selective Inclusion of Polycyclic AromaticHydrocarbons (PAHs) on Calixarene Coated Silica NanospheresEnglobed with CdTe Nanocrystals. J. Mater. Chem. 2007, 17, 3536−3544.(8) Barnes, J. C.; Jurícek, M.; Strutt, N. L.; Frasconi, M.; Sampath, S.;Giesener, M. A.; McGrier, P. L.; Bruns, C. J.; Stern, C. L.; Sarjeant, A.A.; Stoddart, J. F. ExBox: A Polycyclic Aromatic HydrocarbonScavenger. J. Am. Chem. Soc. 2012, 135, 183−192.(9) Juricek, M.; Barnes, J. C.; Dale, E. J.; Liu, W.-G.; Strutt, N. L.;Bruns, C. J.; Vermeulen, N. A.; Ghooray, K.; Sarjeant, A. A.; Stern, C.L.; Botros, Y. Y.; Goddard, W. A.; Stoddart, J. F. Ex2Box:Interdependent Modes of Binding in a Two-Nanometer-LongSynthetic Receptor. J. Am. Chem. Soc. 2013, DOI: 10.1021/ja4052763.(10) Grimme, S. Density Functional Theory with London DispersionCorrections. WIREs Comput. Mol. Sci. 2011, 1, 211−228.(11) Grimme, S. Accurate Description of van der Waals Complexesby Density Functional Theory Including Empirical Corrections. J.Comput. Chem. 2004, 25, 1463−1473.(12) Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent andAccurate ab Initio Parametrization of Density Functional DispersionCorrection (DFT-D) for the 94 Elements H−Pu. J. Chem. Phys. 2010,132, 154104−154119.(13) Chai, J.-D.; Head-Gordon, M. Long-Range Corrected HybridDensity Functionals with Damped Atom−Atom Dispersion Correc-tions. Phys. Chem. Chem. Phys. 2008, 10, 6615−6620.(14) Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies,Structures, and Electronic Properties of Molecules in Solution with theC-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669−681.(15) Frisch, M. J.; et al. Gaussian 09, revision A.2; Gaussian, Inc.:Wallingford, CT, 2009.(16) Bachrach, S. M.; Stuck, D. DFT Study of Cycloparaphenylenesand Heteroatom-Substituted Nanohoops. J. Org. Chem. 2010, 75,6595−6604.(17) Bachrach, S. M. Computational Organic Chemistry. Ann. Rep.Prog. Chem., Sect. B: Org. Chem. 2008, 104, 394−426.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXG

(18) Grimme, S.; Schreiner, P. R. Steric Crowding Can Stabilize aLabile Molecule: Solving the Hexaphenylethane Riddle. Angew. Chem.,Int. Ed. 2011, 50, 12639−12642.(19) Schreiner, P. R.; Chernish, L. V.; Gunchenko, P. A.;Tikhonchuk, E. Y.; Hausmann, H.; Serafin, M.; Schlecht, S.; Dahl, J.E. P.; Carlson, R. M. K.; Fokin, A. A. Overcoming Lability ofExtremely Long Alkane Carbon−Carbon Bonds through DispersionForces. Nature 2011, 477, 308−311.(20) Bachrach, S. M. The Group Equivalent Reaction: An ImprovedMethod for Determining Ring Strain Energy. J. Chem. Educ. 1990, 67,907−908.(21) Bachrach, S. M. DFT Study of [2.2]-, [3.3]-, and [4.4]-Paracyclophanes: Strain Energy, Conformations, and RotationalBarriers. J. Phys. Chem. A 2011, 115, 2396−2401.(22) Bachrach, S. M. On the Destabilization (Strain) Energy ofBiphenylene. J. Phys. Chem. A 2008, 112, 7750−7754.(23) Bachrach, S. M.; Demoin, D. W. Computational Studies ofEthynyl- and Diethynyl-Expanded Tetrahedranes, Prismanes, Cubanes,and Adamantanes. J. Org. Chem. 2006, 71, 5105−5116.(24) Bachrach, S. M. Structure, Deprotonation Energy, and CationAffinity of an Ethynyl-Expanded Cubane. J. Phys. Chem. A 2003, 107,4957−4961.(25) Kruse, H.; Goerigk, L.; Grimme, S. Why the Standard B3LYP/6-31G* Model Chemistry Should Not Be Used in DFT Calculations ofMolecular Thermochemistry: Understanding and Correcting theProblem. J. Org. Chem. 2012, 77, 10824−10834.(26) Grimme, S. Supramolecular Binding Thermodynamics byDispersion-Corrected Density Functional Theory. Chem.Eur. J.2012, 18, 9955−9964.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp406823t | J. Phys. Chem. A XXXX, XXX, XXX−XXXH





























