Embed Size (px)
Citation preview

의료기기 GMP 적용 가이드라인
MDQMT-AG-2007-03
의료기기 소프트웨어
밸리데이션 가이드라인
(Guidelines for Software Validation
of Medical devices)
의 료 기 기 본 부
의료기기품질팀

- 1 -
Ⅰ. 소 트웨어 밸리데이션 근 방법
1. 개요
“검증(Verification)”, “밸리데이션(Validation)”이라는 용어는 다양한 산업과 다양한
표 에서 다양한 방식으로 정의하고 사용하기 때문에 혼동이 발생할 수 있다. 어떤
면에서 “의료기기 소 트웨어 개발”은 "의료기기 소 트웨어를 한 소 트웨어 밸리데
이션"이라고 부를 수도 있다.
우리의 품질 리기 (식약청 고시 제2005-69호)은 국제기 ISO 9000:2000과 조화를
이루어 검증과 밸리데이션을 분리된 별개의 용어로 사용한다. 그러나 다수의 소 트웨어
공학 련 문헌에서는 검증과 밸리데이션을 번갈아 사용하기도 하고, “소 트웨어 검증,
밸리데이션 시험(VV&T)”을 단일 개념으로 사용하기도 한다. 이는 소 트웨어 산업
에서 소 트웨어 VV&T의 목 은 소 트웨어가 의도한 환경에서 사용자가 사용하기에
합한지 확인하기 한 것으로 “의도한 랫폼에서 소 트웨어를 최종 으로 시험하는
것”으로 정의하는 경우도 많기 때문이다.
이러한 정의 는 다른 정의가 올바르다거나 잘못도니 것으로 간주하면 안 된다.
요한 것은 안 하고 효과 이며 신뢰성 있는 소 트웨어를 개발하고 유지하기에
필요한 모든 활동과 로세스 리를 이해하는 것이다.
소 트웨어 밸리데이션은 의료기기의 유용성과 신뢰도를 증가시킴으로 결함 발생률,
리콜 시정조치 감소, 환자 의료기기취 자에 한 험 감소, 의료기기 제조업체에
한 부담 경감을 한 것으로, 소 트웨어 밸리데이션은 의료기기에 소 트웨어를
결합시킨 후 실제 는 모의 사용환경에서 소 트웨어의 한 작동에 한 검을
포함하는 것이다. 용어 “소 트웨어 밸리데이션”을 사용하는 것은 안 하고 효과 인
소 트웨어를 보장하는 소 트웨어 개발의 모든 공식 인 측면을 정의하는 것일 수도
있다.
검증이란 “특정 요구사항이 충족되었음을 객 인 증거의 제공을 통하여 확인하는
것”을 의미하며, 설계 밸리데이션은 “의료기기가 사용자의 요구 사용 목 에 일치
함을 객 증거로 입증하는 것”을 의미한다. 소 트웨어의 밸리데이션은, 소 트웨어가
사용된 기능에 한 사용자 요구와 사용목 이 소 트웨어와 일치함을 객 증거로
입증하는 것이다.

- 2 -
넓은 범 에서 소 트웨어의 밸리데이션은 소 트웨어에 의해 자동화된 의료기기의
성능과 특성에 하여 모든 요구사항 사용자의 기 사항이 만족되었는지에 한
“신뢰의 수 ” 문제이다.
소 트웨어에 한 검증과 밸리데이션은 제조업체에서 계속 시험할 수도 없고 어느
정도의 정보로 충분한지 악하기가 곤란하기 때문에 어렵다. 일반 으로 모든 가능한
입력에 해 소 트웨어를 시험하는 것은 불가능하며 소 트웨어 실행 시 발생할 수
있는 모든 가능한 데이터 처리 경로를 시험하는 것도 불가능하다. 이는 가장 간단한
소 트웨어를 제외하고 소 트웨어는 철 하게 시험할 수 없음을 의미한다. 소 트웨어
제품을 완 하게 시험하 다고 확신할 수 있는 하나의 시험 형식이나 시험방법이 있는
것도 아니다. 소 트웨어의 모든 코드를 시험하는 것으로 그 소 트웨어에 모든 필요한
기능이 구축되어 있다는 것과 소 트웨어의 모든 기능 코드에 한 시험으로 소 트
웨어가 완 하게 정확함을 의미하는 것은 아니다. 소 트웨어 시험결과 오류가 발견되지
않은 것이 소 트웨어에 오류가 없는 것으로 명된 것은 아니다. 결과 으로 소 트웨어에
한 시험은 피상 이라고 볼 수도 있다.
의료기기에 포함되는 소 트웨어 자동화공정 소 트웨어의 밸리데이션은 조직의
책임과 무 하게 어떤 기본 원칙들이 소 트웨어 밸리데이션 활동에 용된다고 할 수
있다. 일반 으로 소 트웨어 밸리데이션 근법은 그림 1에서 규정한 일련의 순서에
따른다.
기본 으로 의료기기에 포함되거나, 제조업체에서의 생산 품질시스템 운 에
사용되는 소 트웨어는 품질경 시스템(의료기기 제조⋅품질 리기 )과 험 리시스템
(ISO 14971)과 조화를 이루는 범 에서 개발∙유지되고 사용되어야 한다.
설계에서의 소 트웨어 밸리데이션 활동은 소 트웨어에 한 포 인 시험과 소 트
웨어 개발 수명주기(Life-Cycle)1)의 각 단계에서 이미 완료한 다른 검증 작업에 상당히
의존한다. 기획, 검증, 추 성, 형상 리 소 트웨어 엔지니어링의 다양한 요소들은
소 트웨어가 유효하다는 결론 도출에 도움을 주는 요한 활동이다.
1) 요구사항의 정의에서부터 제조를 한 릴리즈에 이르기까지 소 트웨어의 수명 체를 포 하는 개념
구조로서,- 소 트웨어 제품 개발에 련된 로세스, 활동 업무를 확인(identify).- 활동과 업무 사이의 순서와 의존성을 설명.- 특정 산출물의 완벽성을 검증할 수 있는 일정을 확인(identify).
- 3 -
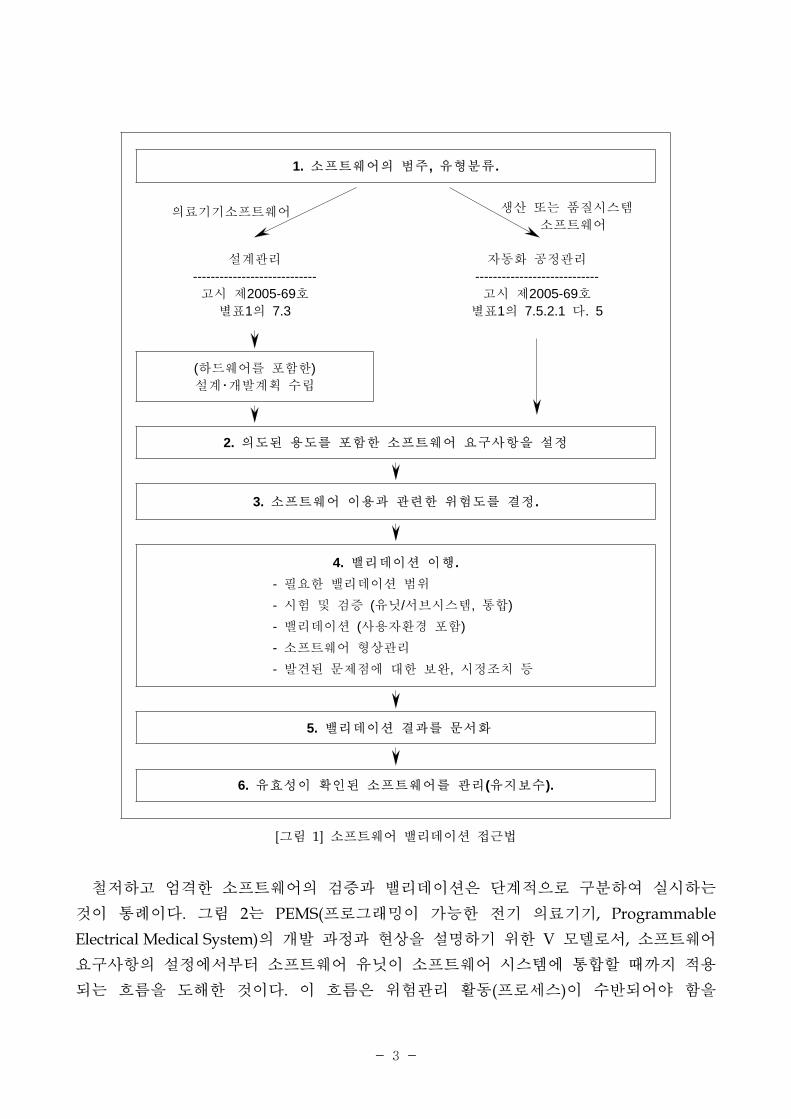
[그림 1] 소 트웨어 밸리데이션 근법
1. 소 트웨어의 범주, 유형분류.
의료기기소 트웨어 생산 는 품질시스템 소 트웨어
설계 리
----------------------------고시 제2005-69호별표1의 7.3
자동화 공정 리
----------------------------고시 제2005-69호별표1의 7.5.2.1 다. 5
(하드웨어를 포함한)설계・개발계획 수립
2. 의도된 용도를 포함한 소 트웨어 요구사항을 설정
3. 소 트웨어 이용과 련한 험도를 결정.
4. 밸리데이션 이행.- 필요한 밸리데이션 범- 시험 검증 (유닛/서 시스템, 통합)- 밸리데이션 (사용자환경 포함)- 소 트웨어 형상 리
- 발견된 문제 에 한 보완, 시정조치 등
5. 밸리데이션 결과를 문서화
6. 유효성이 확인된 소 트웨어를 리(유지보수).
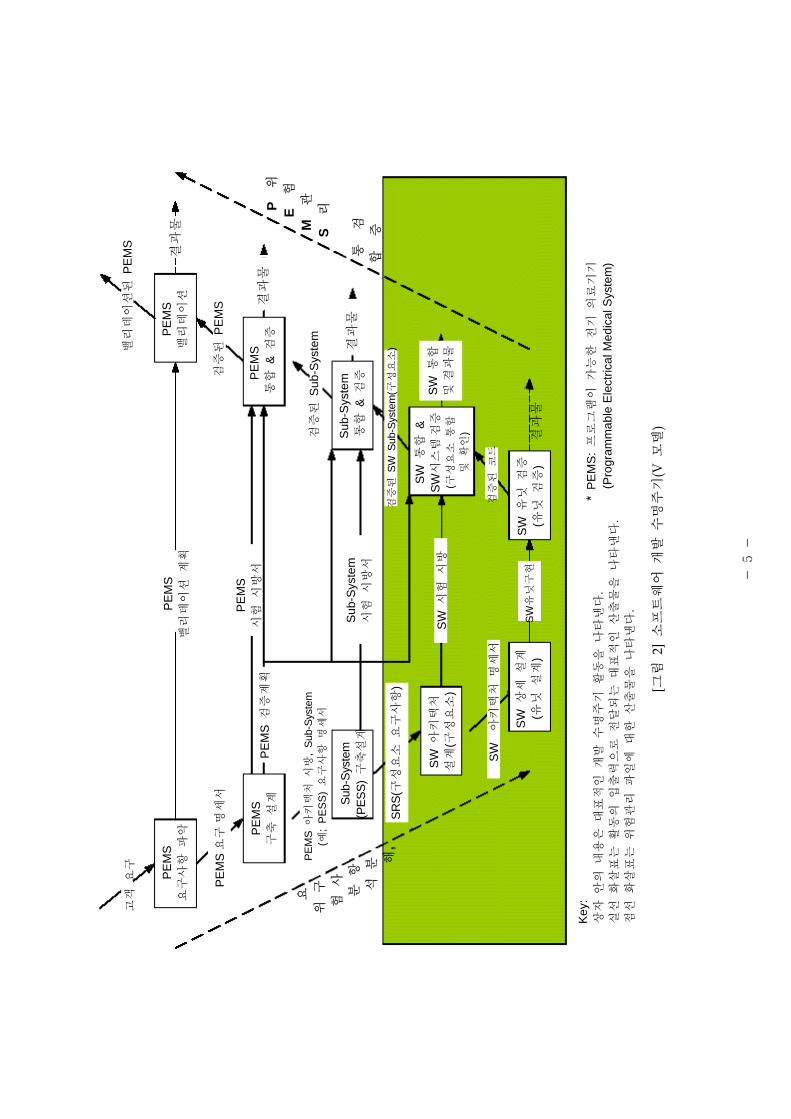
철 하고 엄격한 소 트웨어의 검증과 밸리데이션은 단계 으로 구분하여 실시하는
것이 통례이다. 그림 2는 PEMS( 로그래 이 가능한 기 의료기기, Programmable
Electrical Medical System)의 개발 과정과 상을 설명하기 한 V 모델로서, 소 트웨어
요구사항의 설정에서부터 소 트웨어 유닛이 소 트웨어 시스템에 통합할 때까지 용
되는 흐름을 도해한 것이다. 이 흐름은 험 리 활동( 로세스)이 수반되어야 함을

- 4 -
의미한다. 소 트웨어의 해요인(Hazard) 리는 체 인 의료기기 험 리의 일부,
특히 안 에 한 험으로, 따로 취 할 수는 없다. 해요인 리의 목 은 식별한
기능성에 있어 소 트웨어의 결함, 소 트웨어의 고장이나 상하지 못한 결과 등을
사 에 방지하기 함이다. 따라서 소 트웨어의 험 리와 련하여 그 험도의
등 에 따라 밸리데이션의 범 , 시험방법 그 수 (정도)는 달라질 것이다.
컴퓨터 자동화 설비 등 생산이나 품질시스템 운 에서 규정된 요구사항을 충족하기
해 제품성능에 향을 미치는 컴퓨터 소 트웨어를 사용하는 경우에는, 설계, 제조,
추 리 등 품질시스템의 모든 측면에서 사용하는 소 트웨어의 설치가 정확하며
그 의도한 용도⋅목 에 따라 유효한 결과를 만들어낸다는 것을 입증하기 하여 소 트
웨어의 밸리데이션에 한 문서화된 차를 수립하고 최 사용 에 밸리데이션을
실시하여 소 트웨어가 의도한 바와 같이 일 되게 기능을 발휘함을 밸리데이션을
통하여 보증할 필요가 있다.
자동화 설비나 운용되는 소 트웨어의 밸리데이션에 필요한 정도는 소 트웨어 이용과
련한 험과 비례한다. 즉, 자동화 운 에 따라 노출되는 험에 하며, 해당
소 트웨어 고유의 기능성뿐만 아니라 제조업체에서의 의도된 용도도 고려하여 밸리데
이션에 필요한 시험 방법과 범 가 설정될 것이다.
- 5 -
[그림
2]
소
트웨
어 개
발 수
명주
기(V
모델
)
* P
EM
S: 로그램이 가능한 기 의료기기
(P
rogr
amm
able
Ele
ctric
al M
edic
al S
yste
m)
Key
:상자 안의 내용은 표인 개발 수명주기 활동을 나타낸다
.실선 화살표는 활동의 입출력으로 달되는 표인 산출물을 나타낸다
.선 화살표는 험리 일에 한 산출물을 나타낸다
.
PEM
S 아키텍처 시방
, Su
b-Sy
stem
(
; P
ESS
) 요구사항 명세서
PE
MS
요구사항 악
PE
MS
밸리데이션 계획
PE
MS
구축 설계 Su
b-Sy
stem
(P
ESS)
구축설계
Sub
-Sys
tem
통합 &
검증P
EM
S 통합 &
검증P
EM
S밸리데이션
SW
통합 &
SW시스템검증
(구성요소 통합
확인
)
SW 아키텍처
설계
(구성요소
)
SW 유닛 검증
(유닛 검증
)SW
상세 설계
(유닛 설계
)
SRS(구성요소 요구사항
)검증된 S
W S
ub-S
yste
m(구성요소
)
SW 시험 시방
SW 아키텍처
명세서
SW유닛구
검증된 코드
SW 통합
결과물
고객 요구
PE
MS요구명세서
PE
MS
시험 시방서
밸리데이션된 P
EM
S
결과물
결과물
결과물
결과물
PE
MS
검증계획
검증된 P
EM
S
Sub
-Sys
tem
시험 시방서
검증된 S
ub-S
yste
m
요
구
험 사
분 항
석 분
해
,
P
E
험
M
S 리
통 검
합 증

- 6 -
2. 소 트웨어 분류 용기
본 가이드라인의 목 상, 밸리데이션이 필요한 소 트웨어는 의료기기의 사용목 에
따라 다음과 같이 분류한다.
a) 소 트웨어 자체가 의료기기이거나 는 의료기기의 구성품, 는 부속품으로
사용되는 소 트웨어
- 의료기기 목 으로 독립 (stand-alone) 사용되는 소 트웨어
- 의료기기의 기능에 향을 주거나 조 하는 소 트웨어
- 분석/모니터링이 목 인 의료기기에 의해 나온 환자의 데이터를 분석하는 소 트웨어
b) 자동화 설비(Automated Process Equipment) 품질시스템 운 에 사용되는
소 트웨어
- FA 제어용 소 트웨어
- PLC(Programmable Logic Controller)
- 생산 리, 환경 로세스 리, 포장, 라벨링, 추 성(traceability), 재고 리, SPC,
문서 리, 고객불만 리 등 품질시스템의 모든 측면에서 범 하게 사용되는
소 트웨어
의료기기 사용목 에 따른 소 트웨어의 분류별 밸리데이션은 다음과 같이 용한다.
․상기 2의 a)에 해당하는 소 트웨어는 “설계에서의 밸리데이션”을 용
․상기 2의 b)에 해당하는 소 트웨어는 “공정에서의 밸리데이션”을 용
3. 품질시스템과의 조화
의료기기 제조업체는 제조⋅수입 품질 리기 (식약청 고시)에 규정된 바와 같이,
수립된 품질시스템에서의 련 차에 따라 소 트웨어의 개발 유지를 한 활동을
수행하여야 한다. 본 가이드라인에서는 소 트웨어의 밸리데이션에 한 세부 활동을,
이미 품질시스템이 확립되어 있거나 수립 인 제조업체에서 그들의 차에 통합시켜
채택하거나 확장하여 실행될 수 있도록 제시할 뿐이다. 이는 소 트웨어의 밸리데이션이
필요한 정도(범 )에 따라 제조업체에서 한 방식으로 실행할 수 있음을 의미하는
것이다. 소 트웨어를 문 외주업체에 주문하여 개발할 경우, 소 트웨어 개발⋅유지
활동에서 설명한 차/활동이 외주업체에서 하게 용되는지에 한 보증의 책임은
제조업체에 있다.

- 7 -
표 1은 제조 품질 리기 별표 1의 요구사항과 본 가이드라인에서 언 된 소
트웨어 개발⋅유지활동과의 비교표이다.
[포 1] 품질시스템과 소 트웨어 개발⋅유지활동과의 계
제조 품질 리기 [별표 1] 련 조항 소 트웨어 개발⋅유지활동7.3.1 설계 개발 기획 소 트웨어 개발 기획
7.3.2 설계 개발 입력 소 트웨어 요구사항 분석
7.3.3 설계 개발 출력
소 트웨어 아키텍처 설계
소 트웨어 상세 설계/코딩
7.3.47.3.57.3.6
설계 개발 검토
설계 개발 검증
설계 개발 밸리데이션
소 트웨어 유닛 이행 검증
소 트웨어 통합 통합시험
소 트웨어 시스템 시험( 장시험)
소 트웨어 릴리즈
7.3.7(7.3.5)(7.3.6)
설계 개발 변경 리
(설계 개발 검증)(설계 개발 밸리데이션)
형상 리
문제 발견 수정 분석
수정 이행
험한 상황에 한 소 트웨어 향 분석
험 리 방법
험 리 방법의 검증
소 트웨어 변경의 험 리
7.5.3 식별 추 성
형상 리(형상식별, 형상상태)
변경 리
4. 험 리(Risk Management)와의 조화
이미 언 한 바와 같이 의료기기 소 트웨어는 기본 으로 품질시스템과 험 리
범 안에서 개발․유지되어야 한다.
소 트웨어에 한 해요인(Hazard)의 리는 의료기기의 체 인 험 리의 일부로서,

- 8 -
이를 따로 취 할 수는 없다. 소 트웨어가 포함된 의료기기의 안 성과 효과성의
확립에는 소 트웨어 사용 의도 소 트웨어 사용에 따른 허용할 수 없는 험이
발생하지 않고 그 의도가 충족된다는 사실의 증명이 필요하다. 소 트웨어로 인하여
해요인이 유발되거나 해요인의 감소 여부를 단하기 하여 제조업체는 의료기기
험 리를 이행할 수 있는 최소한의 차, 활동 업무를 수행하여야 한다.
험 리의 본질은 의료기기에서 발생할 수 있는 해요인을 식별하고, 그 해요인
으로 인한 험스러운 상황과 그에 응하는 험 리방법을 설정하는 것이다. 이 게
설정된 험 리 방법을 실행함으로 험스러운 상황의 발생가능성과 심각성을 허용
수 이하로 감소시킬 수 있을 것이다.
이러한 험 리활동은 이미 국제규격 ISO 14971에 규정되어 있다. ISO 14971에
정의된 험 리는 특히 안 에 한 험을 취 한다. FTA(Fault Tree Analysis, 결함수
분석), FMEA(Failure Mode and Effect Analysis, 고장형태 향 분석) 등 몇몇 기법들은
험 리의 체계 인 운 을 돕는데 사용된다. 소 트웨어의 밸리데이션 활동을 수행
하는 자는 담당 분야인 소 트웨어의 험 리를 한 최소한의 요구사항을 이해하여야
할 것이다. 표 2에 소 트웨어 개발⋅유지활동에 있어 ISO 14971과의 계를 표시한다.
의료기기 소 트웨어의 고장/결함은 성격상 체계 인 것이기 때문에, 이 고장 정도를
정 하게 추정하기는 어렵다. 체계 인 고장 추정에 합한 수 을 확실하게 설정하기가
곤란한 경우, 소 트웨어의 해요인 리의 엄격성, 련 문서의 상세한 수 경감의
성은 소 트웨어에 련된 해요인으로 발생할 수 있는 해(Harm)의 심각성에
따라 결정하여야 한다. 소 트웨어의 부분은 필수 인 안 기능을 직 실 하지
않으며, 경감시킨 경우에도 안 에 련된 소 트웨어에 부작용을 미칠 수 있으므로
소 트웨어에 의하여 발생할 수 있는 가장 심각한 해요인을 개발 로세스 련
해요인 경감의 정의에 있어 결정 인 요인으로 사용하여야 한다. 다음 3항에서 서술한
“소 트웨어의 안 성 분류”는 이러한 목 을 하여 설정된 것이다.
요한 것은 해요인을 분석할 때, 하드웨어와 소 트웨어 해요인을 포함하여
의료기기 사용목 과 련된 모든 의료기기 해요인을 고려하여야 한다. 식별된 각
해요인에 하여 해요인의 심각성, 해요인의 원인, 리방법( : 경고(alarm),
하드웨어 설계 등), 험스러운 상황을 제거하거나 감소하기 하여 취해지는 조치, 리
방법의 한 이행여부를 단하기 한 검증 활동 등은 문서화되어야 할 것이다.

- 9 -
[표 2] 험 리활동(ISO 14971)과의 계
ISO 14971:2000 조항 소 트웨어 개발⋅유지활동4.1 험 평가 차
4.2 의료기기 안 련 의도한 용도/목
특성의 식별
4.3 식별 는 측 가능한 해요인의
식별
- 험스러운 상황에 한 소 트웨어의 향
분석
4.4 각 해요인에 한 험 추정 - 소 트웨어 안 성 분류
5 험 평가
6.1 험 축소
6.2 옵션 분석 - 험 리 방법 정의
6.3 험 리 방법의 구- 소 트웨어에 구 된 험 리 방법
- 험 리 방법 검증
6.4 잔류 험 평가
6.5 험/장 분석
6.6 기타 발생 해요인 - 상황(Event)의 새로운 발생순서 문서화
6.7 험 평가 완료
7 체 잔류 험 평가
8 험 리 보고서 - 추 성 문서화
9 생산 후 정보 - 소 트웨어 변경에 한 험 리
소 트웨어의 유지⋅ 리과정에서 문제 해결이나 업그 이드 등을 하여 소 트
웨어, 하드웨어, 기성품(OTS) 소 트웨어 는 사용목 에 한 변경(change)이 이루어
지는 경우, 이러한 변경이 기존 시스템에 방해되는지 여부 다음의 단을 하여
분석하여야 한다.
․ 해요인에 한 새로운 경감이 필요한지 여부
․변경으로 새로운 해요인이 추가 발생하는지 여부
․기존 해요인에 한 새로운 소 트웨어 원인이 발생하는지 여부

- 10 -
5. 소 트웨어 안 성 분류
소 트웨어 밸리데이션 활동에 앞서 제조업체는 의료기기 소 트웨어의 고장이나
잠재 인 결함으로 환자, 작업자 는 기타 사람에게 미치는 해(Harm)의 향에 따른
소 트웨어의 안 성을 분류하고, 해당 소 트웨어의 가장 높은 안 성 등 을 결정하
여야 한다.
표 3의 안 성 분류는 1 4항에서 언 한 험 리 산출물이기도 하다. 다만, 이러한
안 성 분류는 설계⋅개발과정의 기 단계에서 결정되어야 할 것이다.
[표 3] 안 성 분류
등 설 명
하(Minor)고장, 잠재 설계 결함으로 환자나 조작자에게 어떤 부상이나 신체
피해가 발생할 가능성이 없는 경우
(Moderate)
고장이나 잠재 설계 결함이 직 원인이 되어 환자나 조작자에게
경상을 입힌 경우, 는 고장 는 잠재 결함이 간 인 원인으로
작용하여 부정확하거나 지연된 정보로 환자나 조작자에게 경상을 입힌
경우
상(Major)
고장이나 잠재 인 결함이 직 인 원인으로 환자나 조작자에게 사망
는 상을 입힌 경우, 는 고장이나 잠재 결함이 간 원인으로
작용하여 부정확하거나 지연된 정보로 인하여 환자나 조작자에게 사망
이나 상을 입힌 경우
용어 “안 성”은 “소 트웨어의 고장이나 잠재 인 결함으로 래될 수 있는 해의
심각성”으로 이해하여야 한다. 험 리시스템(ISO 14971)에서 험 평가는 식별된
각 해요인(hazard)으로 인한 해의 심각성과 발생확률(probability of occurrence)을
정성 는 정량 으로 산출하고 이 결과에 따라 험 리를 수행토록 규정되어 있으나,
소 트웨어의 경우 발생확률/고장률은 가용 정보나 데이터를 활용하여 쉽게 측할 수
없다는 특성을 갖고 있기 때문에 심각성에 근거한 소 트웨어 험을 리함이 보다
타당할 것이다.
본 가이드라인에서 제시하는 “안 성 분류”는 제조업체에서 수행하는 소 트웨어의
개발⋅유지활동 활동의 산출물( : 소 트웨어 요구사항 명세서(SRS), 소 트웨어

- 11 -
설계 규격(SDS) 등)에 어떤 제약을 두려 함이 아니다. 다만, 제조업체는 요구되는 소 트
웨어 안 성 분류, 소 트웨어의 규모 사용목 에 근거하여 그들이 수행하여야 할
개발⋅유지활동의 범 와 단계(세부 활동)를 설정하고, 이미 수립된 개발⋅유지 차에
이 범 와 단계(세부 활동)를 통합 는 확장하여 실행하도록 제안하는 것이다. 를
들어 소 트웨어의 안 성 등 이 낮으면서 작은 규모의 소 트웨어인 경우는 아주
은 밸리데이션 활동(시험⋅검증)으로도 충분히 입증될 수 있을 것이다.

- 12 -
Ⅱ. 설계에서의 소 트웨어 밸리데이션
본 가이드라인에서는 소 트웨어 개발에 한 품질보증활동을 “소 트웨어의 수명주기
로세스”와 조화될 수 있도록 “① 소 트웨어 개발 활동”과 “② 소 트웨어 유지보수
(변경 리) 활동”으로 구분하고, 각 활동별로 이행할 필요성이 있는 구체 인 세부
활동을 표 4와 같이 설정한다. 이 활동 분류의 목 은 제조업체에게 항상 이러한 세부
활동들을 요구하려 함이 아니다. 앞서 몇 차례 술한 바와 같이 제조업체가 스스로
설정한 소 트웨어 안 성 등 개발⋅유지하려는 소 트웨어의 규모, 복잡성에
따라 세부 활동의 통합이나 단축도 가능하다.
[표 4] 설계에서의 소 트웨어 밸리데이션 활동 개요
구 분 세 부 활 동
소 트웨어
개발 활동
1. 소 트웨어 기획(Planning)
2. 소 트웨어 요구사항 수립 평가
3. 소 트웨어 아키텍처(architecture) 설계 검증
4. 소 트웨어 상세 설계 유닛구
5. 소 트웨어 검증 밸리데이션 (V&V, Verification & Validation)- 유닛 시험(Unit Test)- 통합(Integration) 시험- 시스템 시험- 사용자 장 시험(User Site Testing)- 밸리데이션 결과 보고서
6. 소 트웨어 릴리즈(Release)
소 트웨어
유지보수
(변경 리) 활동
1. 변경 문제해결
2. 문서화
3. 형상 리

- 13 -
1. 소 트웨어 개발 활동
1.1 소 트웨어 기획(Planning) 활동
제조업체는 개발하려는 소 트웨어 시스템2)의 범 , 규모 소 트웨어 안 에
합한 개발 활동 진행을 하여 소 트웨어 개발 계획을 수립하여야 한다. 이 개발
계획의 목 은 소 트웨어에서 발생하는 험을 감소시키고 차와 목 을 개발
구성원에게 알리며 의료기기 소 트웨어에 한 품질 요구사항의 충족을 보장하는 개발
업무를 계획하는 것이다.
소 트웨어 개발 계획은 하드웨어를 포함한 의료기기 체 개발 계획에 통합할 수도
있고, 독자 으로 “○○소 트웨어 개발 계획서”와 같이 고유의 명칭을 부여하여 수립
하는 것도 무방하다. 수립된 개발 계획서는 자격이 있는 자가 검토 승인을 하도록 한다.
의료기기 소 트웨어 개발에 용되는 방침과 차를 이미 설정한 제조업체도 있을
수 있다. 이 경우 계획은 기존 방침과 차를 참조할 뿐이다. 이와 달리 개발하려는
소 트웨어 고유 계획을 각각의 특정 활동으로 자세히 규정한 개발 계획서를 작성하는
제조업체도 있을 수 있다. 한 의료기기 소 트웨어의 개발을 하게 조정한 계획도
있을 수 있다.
개발계획은 개발 수행에 필요한 상세 수 으로 지정하여야 하며, 험에 비례하여
수립하는 것이 요하다. 를 들어 험 수 이 높은 소 트웨어(시스템이나 항목)는
보다 엄격한 개발과정이 필요하며, 개발 계획서는 매우 상세하게 작성되어야 할 것이다.
일반 으로 소 트웨어 개발 계획서에는 소 트웨어 요구사항 분석, 아키텍처, 코딩,
통합, 시험, 설치 지원(훈련, 유지보수) 등 세부 활동을 구분하여 서술하며, 해당
내용을 포함한 독립 인 문서이거나, 다른 문서 일부 는 몇 개의 문서와 연결시킨
형태이어도 무방하다.
개발 계획서에 언 될 내용은 다음과 같다.
a) 소 트웨어 시스템 개발에 한 세부 활동
․개발 Life Cycle에서 수행되어야 할 구체 작업
․수행되어야 할 작업의 주요 일정 계획
․각 작업 방법, 차 지원 활동
2) Software System, 특정 기능이나 일련의 기능을 수행하도록 구성된 소 트웨어 항목(item)을 통합한 집합체

- 14 -
․작업 수용 기
․ 련 자원 책임
- 조직 내의 ( : Sub-Project 부서, 엔지니어링 부서, A/S 등), 서로 다른 개인
이나 그룹( : 외주 개발업체, 의료기기 취 자 등)
- 개발을 한 도구 기법, 그러한 도구 기법의 자격 부여 형상 리를 포함
b) 활동 (문서화를 포함한) 업무의 산출물
․ 로젝트 체에 한 입력 출력의 정의
- 입력 요구사항 확인을 평가할 수 있는 출력 정의 기
․각 작업을 한 입력
․각 작업에서 요구되는 출력
․각 단계에서 소 트웨어 산출물
c) 소 트웨어 시스템 요구사항, 시험(V&V)
․주요 품질요소( : 신뢰성, 유지성(maintainability) 유용성(usability) 등)의
문서화
․보안 사용자 요구사항의 문서화
․소 트웨어 검증 밸리데이션 계획
- 검증 밸리데이션 작업 수용 기
- 검증 밸리데이션 활동을 한 일정 자원
- 정규 설계 검토 요구사항
․기타 기술 요구사항
d) 소 트웨어 련 험 리
․소 트웨어 험 리 로세스의 활동과 업무 수행을 한 계획
- 개발과 련하여 발생 가능한 험, 가정(assumptions), 의존도 문제 의 분석
e) 소 트웨어 형상 변경 리
․형상 리 계획(Configuration Management Plan)
f) 개발 활동에서 확인(detected)되는 문제 취 을 한 해결
․문제 해결 차
․그 밖의 지원 활동
- 우발 인 사건, 바이러스 방지를 포함한 보 , 백업 보존을 한 차
범용 컴퓨터의 사용이 보다 활성화됨에 따라 기성품(OTS: Off-the-shelf) 소 트웨어가
의료기기와 혼합되는 경향도 있다. 의료기기에 OTS 소 트웨어를 용시키는 경우,
의료기기 사용 시에 지속되어야 하는 안 성과 유효성 문제 을 검토할 수 있도록
OTS 소 트웨어 리에 필요한 활동과 업무를 포함하여 개발계획을 수립하여야 한다.

- 15 -
OTS 소 트웨어를 사용할 때의 문제 은 용되는 의료기기마다 다르고, 해당 OTS
소 트웨어의 고장 발생시 환자, 조작자나 제3자에게 얼마나 향을 미치는가에 따라서
다르다. 그러므로 OTS 소 트웨어는 의료기기 설계 과정의 일부가 되는 험분석(risk
analysis)을 고려하는 것이 특히 요하다.
수립된 계발 계획은 개발의 진행에 따라 갱신하여야 한다. 개발계획이란 개발이
진행되는 동안에 계속 찰하고 갱신하는 회귀 인 활동이다. 따라서 악된 소 트웨어
시스템이나 험 리 활동 결과를 반 할 수 있도록 계획을 갱신하여 개발과정을 히
리하는 것이 요하다.
1.2 소 트웨어 요구사항 수립 평가
소 트웨어 요구사항의 수립
소 트웨어가 지나치게 복잡하거나 사용자의 직 인 상과 반 되는 설계로 인한
동작/사용상의 오류는 감독기 /규제기 에 보고되는 가장 지속 이고 한 문제
의 하나이다.
소 트웨어를 제조업체에서 자체 개발하거나 외주 개발업체(계약업체)에서 개발
하거나 는 기성품(OTS)로 구매하건 간에, 밸리데이션을 하여서는 소 트웨어 요구
사항을 분명히 문서화하는 것이 무엇보다 요하다. 사용자 요구와 사용의도가 명확하지
않을 경우 소 트웨어 시스템이 그 요구와 목 에 일 되게 충족하는지의 확인은 사실상
불가능하다는 것이 일반 인 통념이다.
소 트웨어 요구사항이란 소 트웨어가 어떤(what) 기능을, 어떻게(how) 수행할 것
인지에 하여 설명한 것으로, 소 트웨어가 수행할 수 있는 논리 이고 물리 인
표 으로 변환된 것이다.
로젝트의 복잡성 는 다양한 기술 책임의 수 을 지닌 사람들이 설계 정보를
명확하게 이해하도록 하기 하여 소 트웨어 요구사항은 설계 세부 설계 정보에
하여 높은 수 의 요약을 포함할 수 있다. 완성된 소 트웨어 요구사항은 개발 작업
자가 임기응변식으로 설계 결정을 하는 필요성을 경감시키고 설계 목 내에서 작업
하도록 한다.

- 16 -
제조업체는 의료기기에 사용되는 시스템 요구사항에서 소 트웨어 요구사항을 결정
하고 문서화하여야 한다. 소 트웨어 요구사항은 다음과 같은 사항들을 포함한다.
a) 물리 특성, 기능 성능 요구사항
․소 트웨어가 수행할 모든 기능, 신뢰성(reliability) 시간성(timing)
․요구되는 응답시간(response time)
․물리 특성( : 코드 언어, 기반(platform), 운 체제)
․소 트웨어의 사용 환경( : 사용되는 하드웨어, 메모리 크기, 처리장치, 네트워크)
․제어논리(control logic)를 포함한 논리 구조와 논리 처리단계( : 알고리즘)
․하드웨어, 소 트웨어 사용 환경과의 계를 포함하여 소 트웨어 시스템의 동작 의도를 설명한 소 트웨어 시스템의 개 (context)
․복수 SOUP3) 는 기타 장비( : 운 드라이버, 다른 응용 소 트웨어)와의
호환성
b) 소 트웨어 시스템 입력과 출력
․측정 는 기록되는 변수(Data) 특성( : 숫자, 숫자, 형식)
․소 트웨어가 수용할 수 있는 모든 범 , 한계 기본 값(value)
․데이터베이스 요구사항 데이터 흐름도
c) 소 트웨어 시스템 내⋅외부의 연계성
․모든 내부 소 트웨어와 시스템간의 인터페이스 모든 외부와 사용자 인터
페이스의 정의
․링크(communication links)( : 소 트웨어 내부 모듈간 링크, 보조 소 트웨어와
링크, 하드웨어와 링크 사용자와의 링크)
d) 소 트웨어 구동(driven) 경보, 경고 운 자 메시지
․오류 생성 요인과 처리방법
․오류, 경보(alarm) 경고 메시지
e) 물리 논리 인 보안 요구사항
․ 요 정보의 노출 험에 련되는 요구사항
․보안 수단
- 근 권한자(authentication)
- 인가
- 통신 무결성
f) 사람으로 인하여 발생하는 오류와 훈련에 민감한 인체공학 요구사항
․수동 운 지원
3) Software Of Unknown Provenance(기원이 확인되지 않은 소 트웨어)의 약어, 이미 개발되어 일반 으로
사용할 수 있거나, 의료기기에 채택할 목 으로 개발되지 않은 기성품(Off-the-shelf, OTS) 소 트웨어 는
이 에 개발되어 개발 로세스에 한 한 기록이 없는 소 트웨어

- 17 -
․인간-장치 상호작용(사용자가 시스템에 상호작용 할 수 있는 방법)
․담당자에 한 제약사항
․사람의 집 인 주의가 요구되는 역
h) 운 유지보수 장에 인도되는 의료기기 소 트웨어의 설치 인수 요구사항
i) 운 유지보수 방법과 련된 요구사항
․사용자 운 실행과 련하여 개발할 사용자 설명서
․사용자 유지보수 요구사항
j) 련 법규, 규제 요구사항
소 트웨어 요구사항에는 소 트웨어에서 수행되는 모든 안 요구사항뿐만 아니라
소 트웨어 결함으로 발생할 수 있는 잠재 인 해요인(hazard)도 명확히 규정하여야
한다. 한 험 리 활동에서 식별된 험 리 요구사항이 소 트웨어 요구사항과
련되는 경우 이들 요구사항은 험 리방법이 소 트웨어 요구사항까지 추 이 가능
하도록 소 트웨어 요구사항에서 구분할 수 있어야 한다.
소 트웨어 요구사항은 개발활동과 하게 연결된 기술 인 험 리활동에서
기인한다. 그러므로 제조업체는 하드웨어의 고장 잠재 인 소 트웨어 결함에 해
소 트웨어에 이행된 험 리방법, 특히 험분석(risk analysis)을 의료기기 소 트웨어
요구사항에 포함시켜야 한다. 이 사항은 소 트웨어 개발을 시작할 때는 용할 수 없을
수도 있으며, 소 트웨어가 설계되고 험 리 방법이 계속 정의되는 동안 변할 수도
있다.
소 트웨어 요구사항에서의 “기능 성능 요구사항”과 련하여 제조업체에서 OTS
소 트웨어를 사용하는 경우, 추가 으로 다음과 같은 소 트웨어 요구사항을 포함
시켜야 한다.
a) 사용될 OTS 소 트웨어 제목과 제조업체
b) OTS 소 트웨어의 한 운 지원에 필요한 시스템 하드웨어와 소 트웨어 ( ; 로세서 유형과 속도, 메모리 유형과 크기, 소 트웨어 시스템 유형, 디스
이 요구사항)
c) 버 수 , 릴리즈 날짜, 패치(patch) 번호 의료기기에 설치 시험할 업그 이드 명칭
d) OTS 소 트웨어 구성요소에 따른, 안 과 련된 요한 험 리 방법

- 18 -
수립된 요구사항 평가
소 트웨어 요구사항이 충분하게 명시되어 있고 하다는 것을 확인하기 하여,
소 트웨어 설계 작업을 시작하기 에 소 트웨어 요구사항을 평가하여야 한다. 이는
소 트웨어 요구사항에 의하여 소 트웨어에 이행되는 내용이 결정되기 때문에 “요구
사항 평가”는 매우 요하다.
이런 평가를 수행하려면 무엇보다도 의료기기 소 트웨어가 허용되는 작동상태를
나타내는지, 완성된 의료기기 소 트웨어를 즉시 사용할 수 있는지에 한 요구사항
확립이 필수 이다. 요구사항 로 이행될 수 있는지에 하여 입증하려면 요구사항이
객 으로 단할 수 있는 방식으로 표 하여야 한다.
이와 련하여 우리 품질 리기 (식약청 고시 제2005-69호, 7.3.2의 나)에서는 “요구
사항은 완 하고, 불명확하거나 다른 요구사항과 상충되지 않을 것”과 “ 정성을 검토,
승인”하도록 규정하고 있다. 제조업체에서 소 트웨어 요구사항을 결정한 이후 그 정
확성, 시험가능성(testability) 명확성(clarity) 등에 하여 다음과 같은 사항을 평가
하고 문서화한다.
a) 소 트웨어 요구사항은 험 리에 련된 해요인(hazard)에 하여 하며,
결함의 허용오차, 안 성 보안 요구사항은 완벽하고 정확하다;
b) 요구사항 내에서 내부 으로 서로 모순되거나, 상충하지 않는다.
c) 모든 수행 요구사항은 모호한 표 이 아닌 명확한 용어로 서술되어야 한다.
d) 기능 성능 기 에 충족여부를 단하기 한 시험 사항은 명확하며 하다.
․소 트웨어 설계 가능성 기능의 할당
․모든 요구사항은 측정 가능하거나 객 으로 검증할 수 있는 용어로 설명.
․검증 밸리데이션의 가능성(testability), 시기 허용 기 :
- 유닛4) 시험, 통합시험, 시스템시험
e) 시스템 요구사항이나 검증 밸리데이션을 한 시험, 이행된 험 리방법까지
추 할 수 있어야 한다.
․기기 요구조건에 한 추 성
․ 해요인 경감 요구사항의 합성
․ 해요인 분석과 검증/밸리데이션 시험간의 추 성
4) Software Unit, 다른 항목(item)으로 다시 분류할 수 없는 소 트웨어 항목, 소 트웨어 유닛은 소 트웨어
형상 리 는 시험 목 에 사용할 수 있음.

- 19 -
자주 혼동이 발생하는 것을 방지하려면, 고객 요구(Need), 설계 입력, 소 트웨어
요구사항(Requirement), 소 트웨어 기능 명세서(Functional Specification) 소 트웨어
설계 명세서(Design Specification)를 명확하게 구별(이해)하는 것이다.
- 설계 입력이란 고객 요구를 의료기기 요구사항으로 문서화한 것이며,
- 소 트웨어 요구사항이란 소 트웨어가 고객 요구와 설계 입력을 충족하도록 공식
으로 문서화된 것이다.
- 소 트웨어 기능 명세서는 소 트웨어가 요구사항을 충족하기 하여 수행해야 할
작업을 상세하게 정의한 것으로, 소 트웨어 요구사항에 포함시키는 경우가 많으며
다른 안도 가능하다.
- 소 트웨어 설계 명세서는 요구사항 기능 명세서의 구 을 하여 소 트웨어를
설계하고 분해하는 방법을 정의한 것이다.
일반 으로 소 트웨어 요구사항, 기능 명세서 설계 명세서는 하나 이상의 문서로
작성한다.
1.3 소 트웨어 아키텍처(architecture) 설계 검증
소 트웨어 아키텍처 설계
제조업체는 의료기기 소 트웨어 요구사항을 소 트웨어 구죠(structure)로 설명하며
소 트웨어 항목(item)5)을 식별하는 아키텍처로 변환하여야 한다. 소 트웨어 항목과
소 트웨어 항목 외부의 구성요소(소 트웨어와 하드웨어)간의 연계성과 소 트웨어
항목간 연계성을 한 아키텍처를 개발하고 문서화하여야 한다.
이 활동을 수행하려면 제조업체가 소 트웨어의 주요 구조 구성요소, 외부에서
확인할 수 있는 특성 특성간의 계를 정의하여야 한다. 어느 한 구성요소의 작동
상태가 다른 구성요소에 향을 경우 그 작동상태를 소 트웨어 아키텍처에 서술
하여야 한다. 이 서술은 소 트웨어 이외의 의료기기 구성요인에 향을 미칠 수 있는
작동상태에 특히 요하다. 다른 구성요인에 향을 수 있는 구성요인의 작동상태를
이해( 악)하지 못하면 시스템이 안 하다고 언 하는 것은 거의 불가능에 가깝다.
소 트웨어 아키텍처는 소 트웨어 요구사항의 정확한 이행에 필수 이다. 소 트웨어
아키텍처는 모든 소 트웨어 요구사항이 식별된 소 트웨어 항목으로 이행할 수 없는 한
완성되지 않는다. 소 트웨어 설계와 이행은 아키텍처에 따라 결정되기 때문에 아키텍처를
확인하여야 하며, 일반 으로 아키텍처의 확인은 기술 평가에 의해서 이루어진다.

- 20 -
소 트웨어 항목이 SOUP로 식별된 경우 제조업체는 의도한 사용에 필요한 SOUP
항목에 한 기능, 성능 요구사항 한 운 지원에 필요한 시스템 하드웨어와
소 트웨어( 로세서 유형과 속도, 메모리 유형과 크기, 시스템 소 트웨어 유형, 통신
디스 이 소 트웨어 요구사항이 포함하여)를 명시하여야 한다.
소 트웨어 아키텍처 검증
제조업체는 다음을 검증하고 문서화하여야 한다.
a) 소 트웨어 아키텍처가 험 리에 련된 요구사항을 포함해 시스템 소 트
웨어 요구사항을 이행한다.
b) 소 트웨어 아키텍처는 소 트웨어 항목 간의 연계성과 소 트웨어 항목
하드웨어 간의 연계성을 지원할 수 있다.
c) 의료기기 아키텍처는 모든 SOUP 항목의 한 운 을 지원한다.
1.4 소 트웨어 상세 설계 유닛구
소 트웨어 상세 설계
제조업체는 소 트웨어 아키텍처(architecture)를 소 트웨어 유닛(Unit)으로 표 될
때까지 개량하고, 각 소 트웨어 유닛에 한 상세 설계를 수행하고 검증하여야 한다.
한 제조업체는 소 트웨어 유닛 외부 구성요인(하드웨어나 소 트웨어) 사이의
연계성 소 트웨어 유닛 사이의 연계성을 한 상세 설계를 수행하고 문서화한다.
소 트웨어 유닛을 단일 기능, 로그램이나 모듈로 간주하는 경우가 많지만 이러한
견해가 항상 정확한 것은 아니다. 본 가이드라인에서는 소 트웨어 유닛을 더 작은 항
목으로 분해할 수 없는 소 트웨어 항목으로 정의한다. 소 트웨어 항목은 새로운
소 트웨어 항목 극히 일부만 원래 소 트웨어 항목의 안 성 련 요구사항을
이행하도록 분해할 수 있다.
이 활동을 수행하려면 아키텍처에서 정의한 소 트웨어 항목과 연계성을 다시 정의
하여 소 트웨어 유닛과 연계성을 작성하여야 한다. 제조업체는 소 트웨어 유닛의 상세
수 을 정의하고 알로기즘, 데이터 표 , 다양한 소 트웨어 유닛 사이의 연계성
소 트웨어와 데이터 구조사이의 연계성을 명시한다. 한 상세 설계는 소 트웨어
제품의 패키지구성에도 련한다. 소 트웨어 유닛을 정확히 실 할 수 있도록 각
소 트웨어 유닛의 연계성을 문서화하여야 한다.

- 21 -
구 은 상세 설계에 따라 결정되므로 활동을 완료하기 에 상세 설계를 검증하여야
한다. 상세 설계의 검증은 일반 으로 기술 검토에 의해 이루어진다. 제조업체는 소 트
웨어 상세 설계가 다음과 같은지 검증하고 문서화하여야 한다.
a) 소 트웨어 아키텍처를 이행한다.
b) 소 트웨어 아키텍처와 상충하지 않는다.
안 성에 요하다고 단될 수 있는 설계 특성 역시 검증하여야 한다. 그러한 특성
들의 몇 가지 는 다음과 같다.
․의도된 사상(Event), 입출력, 논리 흐름, CPU 할당, 메모리 자원의 할당
오류와 외 정의, 오류와 외 확인(isolation)과 오류 복구의 이행
․ 험한 상황을 유발할 수 있는 모든 결함이 사상과 환으로 취 되는 기본 상태의
정의변수, 메모리 리의 기화
․ 험 리 방법에 향을 수 있는 콜드 리셋과 웜 리셋, 기 기타 상태 변경
소 트웨어 유닛구
이 활동을 수행하려면 제조업체는 소 트웨어 유닛에 한 코드를 작성하고 검증해야
한다.
각 유닛에 한 코드가 정확히 구 되지 못할 경우 소 트웨어는 의도된 로 기능을
수행하지 않기 때문에 제조업체는 코드를 검증하여야 한다. 소스 코드 평가는 코드 실행
에 오류를 악할 수 있는 매우 효과 인 방법이다. 이는 격리된 상황에서 각 오류
시험을 가능하게 하고 추후 소 트웨어의 동 (dynamic) 시험에 집 할 수 있도록 해
다.
코딩은 세부 인 설계사양을 소스 코드(source code)로서 수행되는 소 트웨어 활동
으로, 코딩은 설계사양에 한 분해(decomposition)가 종료되고 실행 가능한 소 트웨
어의 구성(composition)이 시작되는 지 을 의미하며 소 트웨어 개발 로세스
가장 낮은 수 의 개념(abstraction)이다.
소 트웨어 코딩의 경우 다음과 같은 요구사항을 취 하여야 한다.
․코드가 정확하며 요구사항에 부합.
․코드가 안 요구사항을 정확히 실 .
․코드가 소 트웨어 유닛의 상세 설계에 문사화한 연계성과 일 성 유지.
․사용 코딩 방법과 표 이 .
․확립한 로그래 차나 코딩 표 과의 일치

- 22 -
․사용 검증 략의 합성
․시험 차의 정확성
․소 트웨어 통합 시험 가능성
․유지보수 가능성
1.5 소 트웨어 검증 밸리데이션(V&V, Verification & Validation)
개요
제 작업( : 코드 검사)이 성공 으로 완료되면 소 트웨어에 한 시험은 시작된다.
를 들어 소 트웨어 제품의 시험은 표 5에서와 같이 유닛, 통합 시스템 시험의
단계로 체계화될 수 있다. 이는 유닛시험에서 시작하고 소 트웨어 시스템시험에서 종
결됨을 의미한다. 통합시험과 시스템 시험을 단일 활동으로 결합할 수도 있다.
[표 5] 소 트웨어 시험 단계
유닛 시험 통합시험 시스템 시험 장시험(필요시)
시험
상
Black Box, White Box 경계조건
통합 유닛 인터페이
스 수행기능
시스템 요구사항
기능, 성능주요 기능, 문서
차
완료
시
완료된 유닛에 해
오류가 없다고 단
된 경우
통합 련 유닛에서
오류가 없다고 단
된 경우
모든 시스템 요구기
능이 만족된 경우사용자가 만족
시험
도구
시험사례, 커버리지 도구, 정 분석 등
빌드 검증 시험사례시험사례, Data생성기/비교기, 성능모의
인수시험, 시험사례, 시험 비교기
오류
보고
유닛시험 는 개발
담당자가 처리
발견된 오류/버그는 추 가능토록 기록
발견된 오류/버그는 추 가능토록 기록
발견된 오류/버그는 추 가능토록 기록
․유닛 시험은 보조 로그램(sub-program) 기능에 한 기 시험과 시스템에서
확인하기 곤란한 기능 검사가 확실히 이루어짐에 을 두고 있다.
․통합시험은 유닛 내․외부 인터페이스에서의 데이터 리의 이 에 을 두고
있다.
․시스템 시험은 소 트웨어 요구사항을 고려하여 완성된 기능 성능을 검증하는
소 트웨어의 신뢰성 보증에 을 두고 있다.
소 트웨어에 한 검증과 밸리데이션에 사용되는 시험은 화이트박스(White Box)
시험5), 블랙박스(Black Box) 시험6), 증 시험(Incremental Test), 연쇄식 시험(Thread
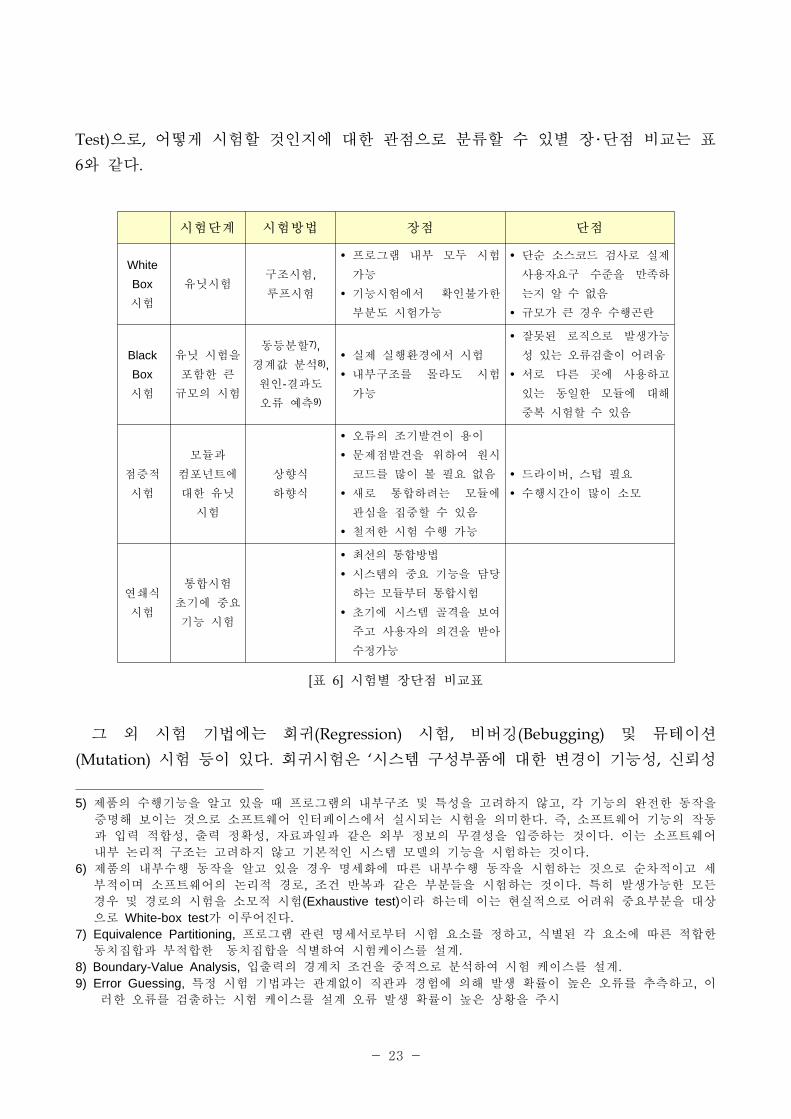
- 23 -
Test)으로, 어떻게 시험할 것인지에 한 으로 분류할 수 있별 장・단 비교는 표
6와 같다.
[표 6] 시험별 장단 비교표
시험단계 시험방법 장 단
WhiteBox시험
유닛시험구조시험, 루 시험
로그램 내부 모두 시험
가능
기능시험에서 확인불가한
부분도 시험가능
단순 소스코드 검사로 실제
사용자요구 수 을 만족하
는지 알 수 없음
규모가 큰 경우 수행곤란
BlackBox시험
유닛 시험을
포함한 큰
규모의 시험
동등분할7), 경계값 분석8), 원인-결과도오류 측9)
실제 실행환경에서 시험
내부구조를 몰라도 시험
가능
잘못된 로직으로 발생가능
성 있는 오류검출이 어려움
서로 다른 곳에 사용하고
있는 동일한 모듈에 해
복 시험할 수 있음
증
시험
모듈과
컴포 트에
한 유닛
시험
상향식
하향식
오류의 조기발견이 용이
문제 발견을 하여 원시
코드를 많이 볼 필요 없음
새로 통합하려는 모듈에
심을 집 할 수 있음
철 한 시험 수행 가능
드라이버, 스텁 필요수행시간이 많이 소모
연쇄식
시험
통합시험
기에 요
기능 시험
최선의 통합방법
시스템의 요 기능을 담당
하는 모듈부터 통합시험
기에 시스템 골격을 보여
주고 사용자의 의견을 받아
수정가능
그 외 시험 기법에는 회귀(Regression) 시험, 비버깅(Bebugging) 뮤테이션
(Mutation) 시험 등이 있다. 회귀시험은 ‘시스템 구성부품에 한 변경이 기능성, 신뢰성
5) 제품의 수행기능을 알고 있을 때 로그램의 내부구조 특성을 고려하지 않고, 각 기능의 완 한 동작을
증명해 보이는 것으로 소 트웨어 인터페이스에서 실시되는 시험을 의미한다. 즉, 소 트웨어 기능의 작동
과 입력 합성, 출력 정확성, 자료 일과 같은 외부 정보의 무결성을 입증하는 것이다. 이는 소 트웨어
내부 논리 구조는 고려하지 않고 기본 인 시스템 모델의 기능을 시험하는 것이다.6) 제품의 내부수행 동작을 알고 있을 경우 명세화에 따른 내부수행 동작을 시험하는 것으로 순차 이고 세
부 이며 소 트웨어의 논리 경로, 조건 반복과 같은 부분들을 시험하는 것이다. 특히 발생가능한 모든 경우 경로의 시험을 소모 시험(Exhaustive test)이라 하는데 이는 실 으로 어려워 요부분을 상
으로 White-box test가 이루어진다. 7) Equivalence Partitioning, 로그램 련 명세서로부터 시험 요소를 정하고, 식별된 각 요소에 따른 합한
동치집합과 부 합한 동치집합을 식별하여 시험 이스를 설계.8) Boundary-Value Analysis, 입출력의 경계치 조건을 으로 분석하여 시험 이스를 설계.9) Error Guessing, 특정 시험 기법과는 계없이 직 과 경험에 의해 발생 확률이 높은 오류를 추측하고, 이러한 오류를 검출하는 시험 이스를 설계 오류 발생 확률이 높은 상황을 주시

- 24 -
는 성능에 부정 향을 미치지 않으며 추가 결함이 발생하지 않음을 단하기에
필요한 시험’으로 오류 발견시 문제 수정을 확인하는 것으로 수정한 결과로 그 기 치를
만족하는지 는 수정한 부분으로 다른 부분에 향을 미치어 다른 오류를 발생
시키지 않는지 확인하는 것이고, 비버깅 시험이란 의도 인 오류를 포함하여 시험이
얼마나 효과 으로 오류를 감지하는지 확인하는 것이며, 뮤테이션 시험은 의도 으로
로그램에 약간의 수정을 가해 시험이 얼마나 효과 으로 수정한 것을 오류로 단하
는지 확인한다.
실제 작업환경에서는 이러한 모든 소 트웨어 시험 기법을 사용하는 것은 필요하지
않다. 보다 요한 것은 어떤 항목은 가 치를 높이고 어떤 것은 제외할 것인지를 정
하는 것 즉, 시험 리, 시험 계획, 시험 결과 분석 등의 시험 차를 수립하는 것이다.
유닛 시험(Unit Test)
각 유닛에 한 코드가 정확하지 못할 경우 의료기기 소 트웨어는 의도된 로
기능을 수행하지 않기 때문에 제조업체는 코드를 검증하여야 한다. 소스코드에 기 한
이러한 평가는 코드 실행 에 오류를 악할 수 있는 매우 효과 인 방법으로
white-box" 시험으로도 알려져 있다. 이는 소스코드, 구체 인 설계 규격과 다른 개발
문서로부터 획득된 지식에 기 하는 시험사례(Case)를 명확히 한다. 구조 인 시험은 로
그램이 실행될 경우 한 번도 수행되지 않는 dead 코드를 명확히 할 수 있다. 구조 인 시험은
유닛 시험으로 우선 수행되어지지만 소 트웨어 시험의 다른 수 까지 확 될 수는
없다.
이러한 코드평가는 격리된 상황에서 각 오류 시험을 가능하게 하고 추후 소 트웨어의
동 분석(dynamic Analysis)에 집 할 수 있도록 해 다. 동 분석도 밸리데이션의
요한 일부이지만 이것만으로 시스템 성능을 완벽하고 정확하게 입증하기는 사실상
불가능할 것이다. 어떤 시스템의 밸리데이션 결과는 시스템 개발과정의 체에 걸쳐
수행되는 다수의 검증 단계로도 입증된다. 이들 단계로는 문서 코드 검사, 웍스루
기술 검토와 같은 정 (static) 분석이 있다. 이 활동과 결과에 한 정보를 이용할
수 있을 경우 으로 다 야 할 시험들을 용이하게 구분할 수 있으며, 소 트웨어가
사용자의 요구와 사용목 을 충족하는지 검증하기 하여 사용자의 장에서 수행될
시스템 차원에서의 기능시험의 양을 일 수도 있다.
평가를 한 원시코드를 입수하지 못할 경우는 다음의 항목들을 이행함으로써 소 트
웨어 구조의 무결성에 한 유효성을 추론하여야 한다.

- 25 -
•유닛 사용 이력을 조사
1) 이미 알려진 로그램 한계의 악
2) 다른 사용자 경험들을 검토 평가
3) 알려진 소 트웨어 문제 과 해결방법의 악
•공 자의 소 트웨어 개발 활동을 평가하여 행 표 에 한 합성을 단;
이 평가는 최종사용자 조직 는 신뢰할 수 있으며 자격을 가진 제3자가 수행하는
평가 결과에 따르는 것이 바람직하다.
해당되는 경우, 제조업체는 규모가 큰 소 트웨어 항목으로 통합하기 소 트웨어
유닛에 한 허용기 을 확립하고, 소 트웨어 유닛이 허용기 을 충족하는지 검증
하여야 한다. 허용 기 의 는 아래와 같다.
․소 트웨어 코드가 험 리 방법을 포함하여 요구사항을 구 하는가?
․소 트웨어 코드가 소 트웨어 유닛의 상세 설계에 문서화한 연계성과 상충하지
않는가?
․소 트웨어 코드가 로그래 차나 코딩 표 과 일치하는가?
필요한 경우, 제조업체는 다음과 같은 추가 허용 기 을 포함시켜야 한다.
․ 한 사상(Event)의 순서
․데이터 리 흐름의 정확성
․계획한 자원 할당
․결함 취 (오류 정의, 확인 복구)
․변수의 기화
․자체 진단
․메모리 리 메모리 용량 과
․경계(boundary) 조건
제조업체는 소 트웨어 유닛 검증을 수행하고 그 결과를 문서화하여야 한다.
통합(Integration) 시험
이 활동을 수행하려면 소 트웨어 유닛을 집합 소 트웨어 항목으로 통합하기 한
계획을 설정하여야 한다. 이 통합계획에는 OTS 소 트웨어 구성요소를 모두 포함하여
근방식, 책임 순서가 포함되어야 하며, 체 으로 통합된 유닛을 통합 계획에 따라
시험하여, 소 트웨어가 의도한 바와 같이 작동하는지 확인하여야 한다.

- 26 -
제조업체는 통합계획에 따라 소 트웨어 통합에 한 다음 사항을 검증하고 기록한다.
이 검증은 항목(item)이 의도한 바와 같이 기능을 수행하는지에 한 검증이 아닌,
계획에 따라 항목(item)이 통합되었는지 검증하는 것이다.
․소 트웨어 유닛이 소 트웨어 항목과 소 트웨어 시스템으로 통합여부
․하드웨어 항목, 소 트웨어 항목 시스템의 수동 운 ( ; 사람 연계성,
On-Line 도움말 메뉴, 음성 식별, 음성 리)을 한 지원이 시스템에 통합여부
통합에 한 근방식은 비 증 (non-incremental) 통합에서부터 증 통합에
이르기까지 다양할 수 있다. 비 증 통합 방법을 사용하여 로그램을 구축한 경우
제조업체는 모든 유닛을 각각 시험을 실시하고 유닛시험 종료 후 모든 유닛들을 동시에
통합하여 시험하는 빅뱅(Big-bangt) 시험을 수행하며, 증 (incremental) 통합 방법을
사용한 경우 제조업체는 이 에 통합한 소 트웨어의 다른 곳에서 문제 이 발생하지
않음을 확실하게 입증하기에 합한 충분한 회귀분석10) 회귀시험을 수행하여야 한다.
소 트웨어 통합시험은 소 트웨어 항목의 내‧외부 연계성에서 데이터 달과 리에
집 한다. 외부 연계성은 운 체제 소 트웨어가 포함된 다른 소 트웨어와 의료기기
하드웨어와의 연계성을 의미한다.
소 트웨어 통합시험의 경우 제조업체는 통합된 소 트웨어 항목이 의도한 로 기능을
발휘하는지 확인한다. 통합시험 소 트웨어 시스템 시험은 단일 계획/활동으로
결합할 수도 있으며, 시험에 고려하여야 할 는 다음과 같다.
․소 트웨어에 요구되는 기능성
․ 험 리 방법의 이행
․지정한 시 기타 작동 상태
․내부 외부 연계성의 지정 기능
․ 측할 수 있는 오용을 포함해 비정상 조건에서 시험
소 트웨어 통합시험은 시뮬 이션 환경, 실제 상 하드웨어나 체 의료기기에서
수행할 수 있다. 통합시험 조건에는 정상 인 응답뿐 아니라 측되는 스트 스가 높은
최악 조건( : 많은 수의 사용자가 동시에 네트워크에 속 등)도 포함되어야 하다.
시험조건의 범 에는 경계값, 우발 데이터 입력, 오류 조건, 분기(branches), 데이터
흐름, 입력 조합 등을 포함한다.
10) 회귀분석(Regression analysis)은 변수들 사이의 계를 조사하여 모형화 시키는 통계 기법으로서 경제, 경 , 교육, 정치 등의 사회과학 그리고 물리, 화학, 생물, 공학, 농학, 의학 등 자연과학의 모든 분야에서 리 응용되고 있다. 즉, 변수들 간의 함수 인 련성을 규명하기 하여 수학 모형(통계모형)을 가정
하고, 측된 자료로부터 이 모형을 추정하는 통계분석방법으로 주로 측에 사용된다.

- 27 -
통합시험은 시뮬 이터를 사용해서 수행하기도 하며, 이 경우 개 실제의 사용자
산환경을 벗어난 오 라인으로 실행된다. 한 실제 운 조건으로 최종사용자의
산환경에서 수행11)되는 시험은 시스템이 범 한 조건과 사상을 겪기에 충분한
시간동안의 연속 조작들을 포함시켜 통상 인 활동 에는 명확하게 드러나지 않는
잠재 결함을 검출할 수 있어야 한다.
통합시험의 엄격성과 통합시험과 연 된 문서화의 상세 수 은 기기와 연 된 험,
잠재 으로 험한 기능에 한 기기의 의존성 험도가 높은 기기 기능에 있어
특정 소 트웨어의 역할에 합하여야 한다. 를 들어 모든 소 트웨어는 시험하여야
하지만 안 성에 향을 미치는 항목은 보다 직 이고 철 하며 상세한 시험의 상이
된다.
통합시험 계획에는 통합시험 일환으로 수행되는 화이트박스 시험 유형을 포함하도록
한다. 시스템이나 구성요소의 내부 메커니즘(구조)을 평가하는 화이트박스 시험은 glass
box, 구조 (structural) clear box 는 open box 시험이라고도 한다. 이 시험은 소 트
웨어 항목의 내부 작용에 한 명백한 지식을 시험데이터 선택에 사용하는 경우의
시험으로 소스코드, 구체 인 설계 규격 다른 개발 문서로부터 획득된 지식을 기 로
하는 시험사례들을 명확하게 한다. 화이트박스 시험은 출력의 찰을 하여 소 트웨
어의 특정 지식을 사용하며, 소 트웨어 항목(item)이 어떤 기능을 발휘하는지 시험자가
아는 경우에만 정 하게 시험을 수행할 수 있다. 이 경우 시험자는 소 트웨어 항목이
의도된 목표에서 벗어나는지 여부를 확인할 수 있다. 화이트박스 시험은 소 트웨어
항목 구 의 시험에 집 하기 때문에 체 시방서의 구 을 보장하지 못한다.
이와 달리 블랙박스 시험은 작동상태(behavioural), 기능, 불투명 박스(opaque-box)
closed- box 시험이라고 하며, 정의에 근거(definition-based)하거나 규격에 근거
(specific-based)하는 시험이다. 이 시험은 각 유닛에서부터 시스템 시험까지 소 트웨어
시험의 수 에 용될 수 있다. 블랙박스 시험은 기능 시방서에 집 하므로, 이행의
부분이 시험되었다고 보증하지 못한다. 이처럼 화이트박스 시험은 이행과 비교한
시험을 수행하여 임무의 결함을 확인함으로써 이행의 그 부분이 결함임을 나타내고,
블랙박스 시험은 이미 알려진 조건에서 규정 입력으로 로그램을 실행하고 결과를
문서화하여 설정된 기 값과 비교하는 과정을 수행하여 생략의 결함을 확인함으로써
시방서의 그 부분이 충족되지 않았음을 나타낸다.
11) Real time field test

- 28 -
소 트웨어에 한 완 한 시험을 하여 Black box와 White box 시험이 모두 필요
할 수 있다.
제조업체는 통합 시험결과를 문서화하여야 한다. 문서화된 통합시험 결과에는 시험
결과, 발견된 변종(anomalies), 시험한 소 트웨어의 버 12), 련 하드웨어와 소 트웨어
시험 형상, 련 시험 도구 시험자 신원을 포함하여야 한다.
시험결과 기록에는 단순한 정성 표 ( : 합격/불합격)보다는 정량 표 을 사용
하도록 한다. 이러한 정량 표 은 시험결과의 후속 검토, 독립 평가 시험을
반복할 수 있도록 해 다. 시험을 반복할 수 있는 충분한 기록은 를 들어 다음을
유지함으로써 가능할 수 있다.
․요구되는 조치와 상되는 결과를 나타내는 시험 사례 시방서
․장치 기록
․시험에 사용하는 시험 환경(소 트웨어 도구 포함)의 기록
시스템 시험
제조업체는 모든 소 트웨어 시스템이 요구사항을 충족하는지 확인하기 한 소 트
웨어 시스템 시험 수행을 한 계획( 근방식, 책임 순서 등) 일련의 시험(입력,
상 결과, 시험 기 ) 차를 수립하고 수행하여야 한다. 계획된 시스템 시험이 제조업체가
직 으로 리하지 않는 장(site)에서 수행될 때 시험계획은 사용범 가 설정되고
상된 시험결과의 정의 모든 시험 출력의 기록을 확실히 하기 하여 충분한 리를
필요로 한다. 한 이 의 통합시험 소 트웨어 시스템 시험은 단일 계획 활동
으로 결합할 수도 있다.
소 트웨어 시스템 시험은 다양한 지 에서 발생하고 다양한 조직에서 수행하기
때문에 시험에 한 책임이 분산될 수 있다. 그러나 업무 분산, 계약에 따른 계,
구성요인 발생원 는 개발 환경에도 불구하고 제조업체는 소 트웨어가 의도한 사용에
합한 기능을 발휘하는지 확인하는 궁극 인 책임을 져야 한다.
시스템 시험은 규격화된 모든 기능과 소 트웨어가 신뢰할 수 있음을 입증하는 것이다.
이 활동을 수행하려면 제조업체는 소 트웨어에 한 기능, 성능 사용목 등과
12) 형상 항목(item)의 식별된 인스턴스(instance)로 라벨은 디 토리 구조에서 산출물 경로 특정시 에서
어떤 의미를 갖는 버 임을 악하는데 유용한 형상식별 수단이다. 각종 소스의 버 을 리할 수 있도록
도와주는 도구인 CVS(공동 버 시스템, Concurrent Version System)이나 Visual Source Safe와 같은 리 도구의 이용을 권장한다.

- 29 -
련된 의료기기 소 트웨어의 다음과 같은 요소들이 성공 으로 이행되었는지 검증한다.
․성능; 시스템 실행시간, 응답시간, 처리능력 등
․스트 스 조건에서의 반응; 최 부하(load), 지속 인 사용에서의 반응(behavior)
․내‧외부 안 성(Security); 시스템 내‧외부로부터의 침입에 한 보안
․고장 복구 책(disaster recovery)을 포함한 회복(Recovering)의 유효성
․유용성(usability)
․다른 소 트웨어와의 호환성
․정의된 각 하드웨어 형상(configuration)에서의 반응
․문서/기록들의 정확성
소 트웨어 시스템시험은 통합 소 트웨어를 시험하며 의도된 운 환경에서 모의실험
(simulation), 실제 상 하드웨어 는 체 의료기기에서 수행하여 시스템이 범 한
조건과 사상을 겪기에 충분한 시간동안 연속 조작들을 포함시켜 통상 인 활동 에는
명확하게 드러나지 않는 잠재 결함을 검출할 수 있어야 한다.
시스템 시험조건은 보다 효율 으로 특정 시험들을 수행하고 스트 스 조건이나 결
함을 유발하며 격성(qualification) 시험의 코드 용범 확 를 하여, White box
시험이 바람직하더라도 Black box 시험에 집 하는 것을 권한다. 유형 단계별 시험
조직에 융통성을 부여할 수 있지만 요구사항, 해요인 경감, 활용성 결함, 설치, 스
트 스와 같은 시험유형에 한 용범 를 입증하고 문서화하여야 한다.
소 트웨어 시스템 시험 사소한 변경이라도, 변경이 발생할 경우
․문제해결에 있어서 변경의 유효성을 확인하기 하여 시험을 반복하거나, 수정한
시험을 수행하는 등의 추가 시험을 수행한다.
․의도치 않은 부작용이 발생하지 않음을 입증하기에 충분하도록 소 트웨어 시스템
시험을 완 히 반복하지 않는 이론 근거를 포함하여 회귀 시험을 수행한다.
․ 련 험 리 활동을 수행하여야 한다.
소 트웨어 시스템 시험과정에서 발견된 오류는 소 트웨어의 릴리즈(release) 에
등재(log), 분류(classify), 검토 해결하여야 한다. 시험 확인된 변종(anomalies)을
해결하기 한 조치를 취하지 않을 경우, 해요인 분석과 련하여 그러한 변종들을
검토하고, 의료기기의 안 과 효율성에 향을 미치지 않음을 입증하여야 한다. 한
변종의 근본 원인, 증상 해결/조치를 하지 않은 이론 근거를 문서화하여야 한다.

- 30 -
시험 차, 시험자료 시험결과는 객 인 검토 정을 하기에 하여야 하고
추후 발생할 수 있는 모든 회귀시험에서 사용하기에 하여야 한다. 제조업체는 시험
결과의 후속 검토, 독립 평가 시험을 반복할 수 있도록 시험 결과를 문서화하여야
한다. 문서화된 시험 결과에는 모든 시스템 구성요소가 시험과정에서 실행되었음과
시험한 소 트웨어 버 , 하드웨어, 시험 도구/장치, 시험에 사용되는 환경, 요구되는
조치와 상 결과를 나타내는 시험 사례, 비정상 상태/오류 목록, 개정 수 이나 개정일,
시험일 시험자 신원을 포함하여야 한다.
소 트웨어 시스템 시험의 결과를 평가해 상한 결과가 도출되는지 확인(ensure)
하여야 한다.
사용자 장 시험(User Site Testing)
이에 한 문용어는 다소 혼란스럽다. 베타(beta) 테스트, 장(site) 밸리데이션,
사용자 수용(user acceptance) 시험, 설치 검증 는 설치 시험과 같은 용어가 사용되어
왔다.
상황에 따라 사용자의 장에서 해당 소 트웨어의 시험이 이루어질 수도 있는데,
이 경우 시험은 설치되는 시스템 형상(configuration)의 일부가 될 수 있는 실제 하드웨어
소 트웨어를 갖춘 환경에서 수행되어야 한다. 시험계획은 안 을 확보하기 한
리기능을 명시하고, 의도한 용범 와 충족 여부를 확인하여야 한다. 소 트웨어
제조업체가 직 으로 리하지 않는 장에서 시험이 진행될 경우의 시험계획은
사용범 가 설정되고 상 시험결과의 정의 모든 시험출력의 기록을 확실히 하기
하여 충분한 리를 필요로 한다.
밸리데이션 결과의 문서화
소 트웨어의 모든 요구사항이 충족되었음을 보장하는 밸리데이션 활동은 소 트웨어
개발 마지막 단계뿐만 아니라 그 과정에서 실시될 수 있다. 일반 으로 소 트웨어는
하드웨어 시스템의 일부분이기 때문에 개발 활동의 각 단계에서 실시되는 시험, 검사,
분석 다른 검증 활동에 크게 향을 받는다. 소 트웨어의 모든 요구사항이 하고
완벽하게 되었음을 입증하는 밸리데이션 활동이 완료되면 그 결과를 문서화하여야 한다.

- 31 -
[그림 3] 소 트웨어 밸리데이션 문서화
소 트웨어
요구명세서
소 트웨어
험분석
소 트웨어
설계
소 트웨어
시험기
소 트웨어
시험기록
소 트웨어
밸리데이션 문서화
(패키지)
문서화된 보고서는 시험결과 등, 밸리데이션 결과를 자세히 기술하거나 요약하여
기술하고 문서화된 다른 상세한 증빙자료와 추 성이 가능하도록 연결될 수 있도록 한다.
보고서에는 단순하게 ‘합격/불합격’으로 결과를 표시하기보다 가능한 정량 으로 표
하고, 밸리데이션 과정에서 발견된 소 트웨어 결함 이를 해결하기 하여 취해진
조치에 해서도 언 되어야 한다.
문서화된 보고서에 포함되는 내용은 다음과 같다.
․밸리데이션 일자 활동 수행자
․ 상 소 트웨어의 버
․밸리데이션에 사용된 시험 방법 유형
․시험에 사용된 도구, 련 하드웨어
․밸리데이션/시험 결과
- 결함(Fault), 경고(Alarm), 확인된 비정상 상태(변종, 버그 등)
- 취해진 조치
․ 험 리
- 소 트웨어 안 성 등 , 해요인 분석
․해당되는 경우 OTS 소 트웨어의 식별
문서화된 보고서는 지정된 자가 검토하고 승인한 후 유지⋅ 리되어야 한다.
1.6 소 트웨어 릴리즈(Release)
소 트웨어를 릴리즈하기 에 모든 밸리데이션 활동을 만족스럽게 완료하 으며,
지정된 권한자의 승인여부를 확인하여야 한다. 제조업체는 개발한 소 트웨어가 재
릴리즈되는 소 트웨어인지 나타낼 수 있도록 릴리즈되는 소 트웨어의 버 과 설명서의

- 32 -
버 을 문서화하도록 한다. 한 제조업체는 릴리즈한 소 트웨어 제품이 개악이나 무단
변경 없이 사용지 까지 인도될 수 있는 차를 확립하여야 한다.
소 트웨어, 소스 코드 련 문서의 정본(master copies)을 장하고 유지하여야 한다.
릴리즈한 소 트웨어의 유지보수는 의료기기 소 트웨어의 생산 후 경험에 용된다.
제조업체는 품질시스템에서 수립한 차에 따라 소 트웨어에 수록된 매체의 손상과
오용이 없도록 생산과 취 (복제, 매체 라벨링, 포장, 보호, 보 , 인도)을 처리하여야
한다.
2. 소 트웨어 유지보수(변경 리) 활동
2.1 변경 문제해결
소 트웨어의 유지보수는 하드웨어에서와 같은 의미는 아니다. 하드웨어 소 트
웨어의 많은 차이 때문에 유지보수 활동방식도 다르다. 소 트웨어 유지보수 활동은
문제 보고서에 한 상 차원의 결정(문제의 존재 여부, 문제가 의료기기 안 성에
향을 미치는지 여부, 필요한 변경13) 변경의 이행 시 )을 취 하며, 소 트웨어
문제해결을 통하여 모든 암시를 악하고, 변경 필요가 있는 모든 형상 항목(item)과
필요한 모든 밸리데이션 활동을 식별하는 것이다.
소 트웨어의 변경은 다음과 같은 이유에서 발생할 수 있다.
․오류 발견 결 을 수정하기 한 디버깅(debugging)
․새로운 요구사항 발생 는 요구사항의 변경
․성능 는 다른 소 트웨어 시스템 속성(attribute)을 향상시키기 한 개선
소 트웨어 유지보수 활동의 은 소 트웨어의 릴리즈 후에 발생하는 피드백에
히 응하는 것이다. 이를 하여 다음과 같은 사항을 종합 으로 리 운 하여야
한다.
․발견된 문제 을 분석하고 문제의 모든 측면을 식별
․안 과 련된 문제 들을 해당 감독기 ( : 식품의약품안 청 등) 해당 사용자
에게 보고
․ 험 리를 포함하여 소 트웨어 항목의 일 성을 유지하면서 변경을 이행
․문제 해결 후속 문제 방지를 보장할 수 있도록 소 트웨어 변경 후 유효성을
13) 소 트웨어의 수정(modification), 향상(enhancement) 는 추가(addition)를 통칭한다.

- 33 -
재확인.
․ 향을 받을 수 있는 다른 소 트웨어 제품에 한 한 조치
소 트웨어 문제해결14)은 소 트웨어 유지활동의 요한 부분이다. 릴리즈된 소 트
웨어에 문제가 발생하거나 개선의 필요성 때문에 소 트웨어의 코드 련 문서가
수정되는 경우 유지보수 활동이 시작된다. 목 은 릴리즈된 소 트웨어 완 성
(integrity)을 보존하면서 소 트웨어를 수정하는 것이다.
제조업체는 보고받은 문제 과 그 향 등을 분석하여, 변경 사항을 이행하여야 한다.
제조업체는 릴리즈된 소 트웨어 제품의 문제 응에 있어 문제 해결은 물론 련 법
규를 수하여야 한다. 경우에 따라 릴리즈된 소 트웨어에서 발견된 문제 기타
변경 필요사항은 의료기기의 안 성이나 허가받은 내용에 향을 미칠 수 있다. 해당
되는 경우 련 감독기 등으로의 통보도 고려하여야 한다.
제조업체는 소 트웨어 개발 활동보다 은 규모의 자원으로 긴 한 문제를 신속하게
응할 수 있다. 제조업체에서의 문제해결을 한 세부 차는 다음과 같이 진행될
것이다.
․소 트웨어와 련된 문제 수신 문서화
․제조업체 내부 사용자에게 릴리즈된 소 트웨어에 한 피드백(추 성)을
모니터링
- 피드백이 문제로 간주되는지 여부를 결정
- 문제 발생시 모두 기록하고 그 향을 평가
․소 트웨어에 한 험 리를 실시
․소 트웨어 릴리즈 후 발생하는 문제 분석과 해결을 한 변경 활동 이행
- 갱신, 버그 해결, 패치, 폐기 등
․기존 소 트웨어의 변경 리를 하여 형상 리활동을 이행
․변경된 소 트웨어에 한 밸리데이션을 이행하고 승인
․변경된 소 트웨어를 다시 릴리즈.
소 트웨어 유지보수과정에서 발견된 모든 문제는 반드시 문서화되어야 한다. 각
문제는 그 문제가 확실하게 시정되었음을 입증하기 하여 기록하여야 하며, 소 트웨어
변경에 따라 개발활동에서 생성된 문서들의 변경여부를 주의 깊게 고려하여야 한다.
14) 소 트웨어 엔지니어링 문헌에서는 이를 “결함 추 (defect tracking)”이라 하는 경우가 많고, 국제규격 ISO/IEC 12207에서는 "문제해결(problem resolution)"으로, IEC 60601-1-4 규격에서는 "문제 보고(problem reporting)"라는 용어를 사용한다.

- 34 -
승인된 기존 문서( : SRS, 사용자 설명서 등)의 변경이 필요하다고 단되는 경우,
품질시스템에서의 문서 리 형상 리(Configuration Management) 차에 따라 변경
승인되어야 할 것이다.
소 트웨어의 다양한 특징 하나는 변화 속도 용이성이다. 이 때문에 많은
사람들은 소 트웨어 문제는 쉽게 수정될 수 있다고 믿으며, 하드웨어에서와 같은
엄격한 변경 리는 필요 없다고 생각하여 소 트웨어의 변경 리는 철 하게 이루어지지
않는 경우가 많다.
일부 산업에서는 제품을 구성하는 수많은 부품(하드웨어)을 리하기 해 부품을
구분하고 각 부품간의 계를 BOM(Bill of Material)에 정의하고, 완성된 제품의 설계
변경을 엄격하게 리하기 하여 ECC(Engineering Change Order)를 발행하고, 그
변경에 따른 향 분석을 실시한다. 그러나 소 트웨어의 경우, 변경이 용이하기 때문에
빈번하게 변경이 발생되며 한 이를 자연스럽게 생각한다. 요한 것은 하나의 변경
으로 인해 다른 부분의 안정성도 보장 받지 못할 수 있다. 따라서 변경으로 인한 향은
악해야 하고 변경을 받아들이기 해 어느 정도의 노력이 필요한지 알아야 한다.
사소한 변경이든 한 변경이든 변경은 통제 상에 놓여야 한다.
문제해결에서 매우 요한 것은 소 트웨어가 변경됨에 따라 소 트웨어/하드웨어의
다른 부분/ 역에 부작용이 발생하지 않음을 증명하는 것이다. 소 트웨어의 복잡성으로
인하여 소 트웨어에서 요하지 않은 것처럼 보이는 자그마한 변경이 시스템에 범
하게 향을 미치거나, 다른 부분에 상하지 못한 심각한 문제를 발생시킬 수 있다.
변경된 소 트웨어를 신규 개발한 소 트웨어로 취 하지 않는 한, 체 의료기기에
한 변경의 향을 분석하여야 하는 것이다.
소 트웨어의 각 변경이 의료기기 체 는 소 트웨어 시스템에 미치는 변경의
정도와 향을 평가하는데 필요한 검증 / 는 밸리데이션 활동의 범 는 변경 형태,
향을 받는 상에 따라 결정될 것이다. 최 개발활동에서 철 하고 완벽하게 이행
하고 그 결과물을 문서화하 다면, 변경의 향 평가에 필요한 밸리데이션은 작은
활동으로도 가능할 것이다.
2.2 문서화
소 트웨어의 개발 유지보수 과정에서 의료기기 소 트웨어가 어떻게 설계되었는지,
설계의도에 따라 어떻게 시험되었는지, 한 해요인을 식별하고 험 리가 효과
으로 이행되었는지 등을 입증하는 다양한 문서( 자매체 포함), 기록들이 산출될 것이다.

- 35 -
제조업체는 소 트웨어 개발 유지보수 활동에서 생성된 문서, 기록을 식별하여야
한다. 생성되는 문서, 기록의 범 종류는 개발⋅유지 리되는 소 트웨어 규모,
복잡성 소 트웨어 안 성 분류에 따라 다르게 된다. 소 트웨어 개발환경에서 활동
결과로 산출된 문서 기록들에 한 체계 인 리는 개발⋅유지보수 활동을 원활하게
해 뿐만 아니라 품질 리에도 큰 향을 미치게 된다. 한 이 문서 기록의 일부는
의료기기 허가/승인 등을 하여 계기 에 제출되기도 한다.
다양한 문서, 기록들은 검색이 용이하고 효율 인 코드, 날짜 등을 부여하여 개정⋅
갱신에 따른 혼란이 일어나지 않도록 품질시스템의 문서 리, 형상 리 기록 리
차에 따라 리하여야 한다.
제조업체에서 리가 필요할 주요 문서, 기록들은 다음과 같다.
․의료기기/소 트웨어 개발계획서
․소 트웨어 요구사항 명세서(SRS, Software Requirements Specification)
․소 트웨어 아키텍처 설계도(Software Architecture Design Chart)
․소 트웨어 설계 기술서(SDD, Software Design Description)
․소 트웨어 설계 명세서(SDS, Software Design Specification)
․소 트웨어 검증 밸리데이션
- 검증 밸리데이션 활동 기록, 활동 결과에 한 조치, 미해결된 변종(버그 는
결함) 등을 포함.
- 소 트웨어 형상 리(SCM, Software Configuration Management)
․기타 문서화
- 사용자를 한 문서화: 소 트웨어 설명서, 매뉴얼, 지침서 등
- 제조업체에서의 문서화: 소 트웨어 유지보수 매뉴얼 등
본 가이드라인에서 이러한 문서화를 언 하는 것은 이 범 를 한정하기 함이 아니다.
제조업체는 그들이 개발⋅유지하려는 소 트웨어의 사용목 , 규모/복잡성 안 성
분류에 따라 산출되는 문서화의 범 가 다를 수 있음을 인지하여야 한다.
소 트웨어 요구사항 명세서(SRS)
SRS는 소 트웨어가 무엇을 하는지에 하여 기술한 것이다. 형 으로 SRS에는
소 트웨어의 기능, 수행능력, 설계 제약, 속성 인터페이스 등에 하여 각각 명확히
기술하고, 이 요구사항들을 시험‧검사, 분석 등으로 객 인 검증 밸리데이션이
가능하도록 설정한다. 여기에는 다음과 같은 내용 등이 서술될 것이다.

- 36 -
․하드웨어 요구사항
- 마이크로 로세서(microprocessors), 메모리, 센서 등
․ 로그램 언어
- 메모리 (memory leaks) 리에 한 정보와 제한 는 로그램 크기(size)등을
포함
․인터페이스
- 시스템 구성요소들 간의 커뮤니 이션 린터, 모니터, 키보드, 마우스와 같은
유 (user)와의 커뮤니 이션 모두를 포함
․기능
- 소 트웨어로 인한 의료기기의 제한 조건(limitations), 오류와 인터럽트(interrupt)
처리, 소 트웨어 시험 확인, 결함 감지, 허용차(tolerance) 복구(recovery)
특성, 타이 (timing), 안 요구사항
- 치료, 진단, 감시, 경고, 분석을 한 알고리즘이나 제어 특성 필요한 경우 체
텍스트 참고문헌(references)이나 보조(supporting) 임상자료의 해석이 포함.
- 해당되는 경우 OTS 소 트웨어의 식별
소 트웨어 아키텍처 설계도(Software Architecture Design Chart)
네트워크 연결(networking)과 같이 하드웨어와 데이터 흐름과의 계를 포함하여 소
트웨어에서의 주요 기능별 유닛간 계에 한 흐름도(flowchart) 는 이와 유사한
설명을 기술한 것이다. 일반 으로 여기에 모든 기능 모듈을 포함시킬 필요는 없다.
다만, 소 트웨어의 기능 사용 목 에 련된 소 트웨어 아키텍처를 검토하기에
충분한 정보가 포함되어야 한다. 소 트웨어에 한 안 성 분류가 ‘ ’이거나 ‘상’으로
평가된 경우는 도해(diagrams)와 같이 소 트웨어 기능별 유닛간의 계를 명확히 묘
사한 상세한 정보가 유용할 수 있다.
소 트웨어 설계 기술서(SDD, Software Design Description)
SDD는 SRS 요구사항을 만족시키기 하여 어떻게 구성되어야 하는지 설명하는
문서이다. SDD는 데이터베이스와 내부 인터페이스를 포함하여 소 트웨어 설계의
구성요소와 하 구성요소들에 해서도 기술한다.
소 트웨어 설계 명세서(Software Design Specification)
SDS는 소 트웨어에 한 요구사항 실 (implementation)을 설명한 문서이다. SRS와
SDS 계에서, SRS는 ‘소 트웨어 기기가 무엇을 하는가?’를 설정한 것인 반면, SDS는
‘SRS에 설정한 요구사항을 어떻게 실 하는가?’로 정의된다. SDS에 기술된 내용은

- 37 -
소 트웨어를 만드는 설계 ⋅엔지니어가 설계 수행과정에서 즉흥 이거나 임시 인
설계 결정이 없도록 분명하고 명확하여야 한다.
소 트웨어 형상 리(SCM, Software Configuration Management)
형상 리는 소 트웨어 개발⋅유지보수 활동에서 매우 요한 요소이다. 형상 리는
2.3항에서 후술한다.
사용자를 한 문서화
사용자 문서화는 소 트웨어 설명서, 매뉴얼, 지침서 등과 같이 소 트웨어의 성공
실행을 해 요구되는 자료 제어입력, 입력 순서, 선택사항, 로그램 한계 다른
조치나 항목을 명시한 것들이다. 내장형(embedded) 소 트웨어이며 사용자와 직 인
상호작용이 없는 경우는 사용자 문서화가 필요하지 않다.
2.3 형상 리
소 트웨어 형상 리(SCM, Software Configuration Management)는 최근 몇 년 사이에
소 트웨어의 품질보증활동에서 아주 요하게 다 지고 있는 분야이다. 특히 소 트
웨어 규모가 커짐에 따라, 혹은 소 트웨어에서 품질 요소가 요한 치를 차지함에
따라 소 트웨어 형상 리의 요성이 더욱 부각되고 있다.
소 트웨어의 가장 큰 특징은 변경이고, 이 변경으로 인하여 개발과 유지보수 활동을
아주 어렵게 만들어 버린다. 변경이라는 소 트웨어의 큰 특징을 무시할 수 없기에 이를
효과 으로 해결할 수 있는 수많은 연구결과의 하나가 “소 트웨어 형상 리”이다.
소 트웨어에서 변경은 언제나 일어날 수 있기 때문에, 형상 리 활동은 ①변경을
알아내기 하여, ②변경을 리하기 하여, ③변경이 히 수행되고 있음을 확인하기
하여, ④변경과 련된 사람들에게 이것을 통보하기 한 것”이다.
소 트웨어 형상 리는 “형상 항목(Configuration Item)의 완벽성과 정확성 확보를
하여 소 트웨어 수명주기 반에 걸쳐 형상을 용하는 로세스15)”로 정의하고 있다.
형상 리에서 “형상” 용어의 의미를 의 으로 해석하면 “소 트웨어 그 자체”이다.
소 트웨어는 PC에서 실행되는 작은 규모도 있고 네트워크와 연결되어 복수의 서버에서
15) “형상항목을 식별하여 기능 물리 특성을 문서화하고, 그 특성에 한 변경을 제어하고, 변경 처리 상태를 기록 보고하고, 명시된 요구사항에 부합하는지 확인하는 일련의 사항에 하여 기술 행정 인
지침과 사후 리를 용하는 원칙”으로 정의하기도 한다.

- 38 -
운 되는 크고 복잡한 규모도 있다. 따라서 엄 하게 말하여 소 트웨어를 구성하는
모든 것으로서 소 트웨어를 개발하는 조직, 개발자, 소 트웨어가 포함되는 하드웨어,
소 트웨어 자체, 소 트웨어 개발활동 형상 리에 필요한 형상 리 도구와 개발
도구 모두가 해당된다고 할 수 있다.
형상 리 사용은 소 트웨어의 규모, 복잡성 험에 상응하는 정도(안 성 등 )에
의한 것으로, 형상 리의 가장 큰 목 은 변경에 의해 차 으로 변해가는 소 트웨
어의 형상을 리하는 것이다. 형상항목 변경은 특정 상태를 거쳐 이 지는데, 그 흐름은
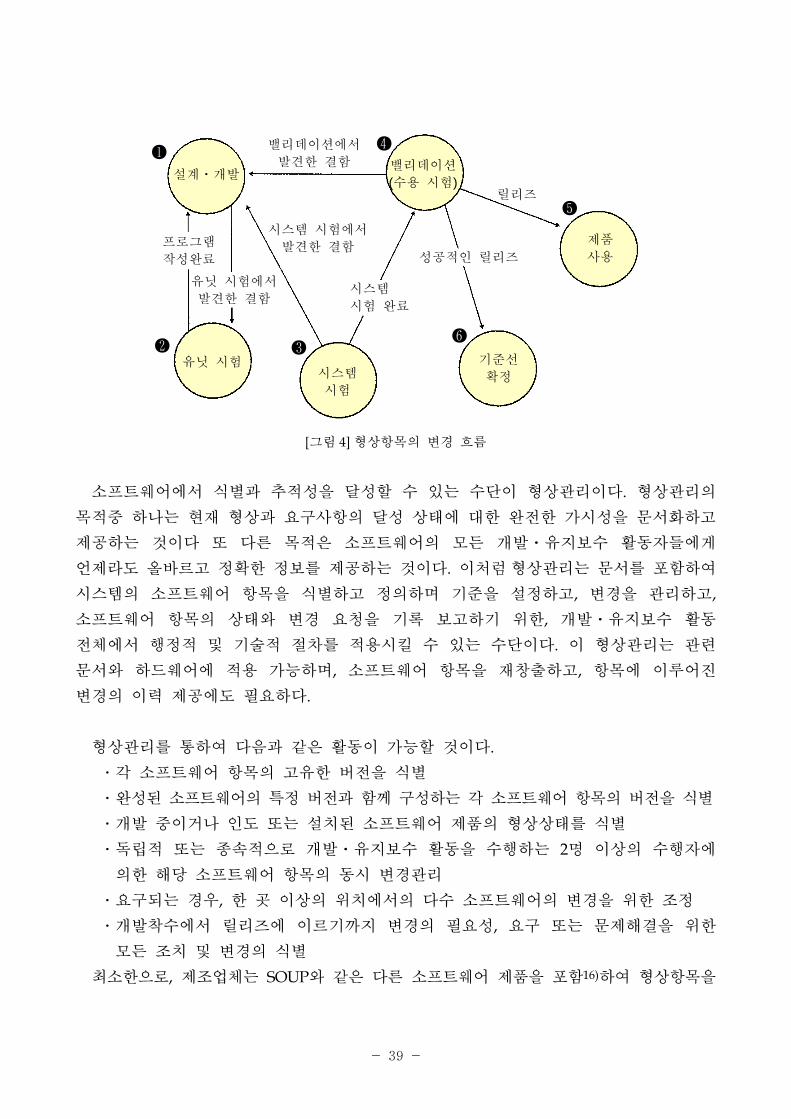
그림 4와 같다.
형상항목의 수명주기는 크게 6단계로 나뉠 수 있는데 개발 기에 설계⋅개발(①)
상태에 치하게 된다. 개발이 진행됨에 따라 유닛시험(②, Unit test)을 하고 이를
통과하면 시스템 시험(③) 비상태로 변하게 된다. 시스템 시험(③, System test)도
통과하면 밸리데이션(④)에서 소 트웨어의 수용(acceptance)여부를 결정하게 되고 이후
모든 결함, 문제 을 제거하면 기 선(⑥, baseline)을 확정하고 제품을 릴리즈(⑤)하게
된다. 이때 각각의 시험에서 문제 을 발견하면 그 의 상태로 돌아가고 발견된 모든
문제 이 제거될 때까지 과정을 반복하게 된다. 그림 4에서와 같이 우선 식별된 형상
항목을 최 로 작성하고 각각의 시험을 거치면서 형상 항목은 변경된다. 유닛시험에서
발견된 문제 은 수정되어 버 통제를 받게 될 것이고, 유닛시험을 통과한 유닛들은
의미 있는 집합(소 트웨어 항목)으로 묶여 시스템시험을 받게 되며 이때 결함이 발견
되면 해당 유닛들과의 추 (trace)을 포함한 변경 차가 진행된다. 이후 밸리데이션
에서는 소 트웨어의 요구사항과 직 련되므로 변경 차가 강화되고 요구사항과
변경과의 추 을 리하여야 하며, 소 트웨어의 수용이 결정되면 기 선이 확정되고
이 기 선은 변경할 수 없는 매우 요한 지표가 된다.
- 39 -
[그림 4] 형상항목의 변경 흐름
밸리데이션에서
발견한 결함
유닛 시험에서
발견한 결함
로그램
작성완료
시스템 시험에서
발견한 결함
시스템
시험 완료
설계⋅개발
시스템
시험
유닛 시험
밸리데이션
(수용 시험)릴리즈
제품
사용
기 선
확정
성공 인 릴리즈
❶
❷ ❸
❺
❻
❹
소 트웨어에서 식별과 추 성을 달성할 수 있는 수단이 형상 리이다. 형상 리의
목 하나는 재 형상과 요구사항의 달성 상태에 한 완 한 가시성을 문서화하고
제공하는 것이다 다른 목 은 소 트웨어의 모든 개발⋅유지보수 활동자들에게
언제라도 올바르고 정확한 정보를 제공하는 것이다. 이처럼 형상 리는 문서를 포함하여
시스템의 소 트웨어 항목을 식별하고 정의하며 기 을 설정하고, 변경을 리하고,
소 트웨어 항목의 상태와 변경 요청을 기록 보고하기 한, 개발⋅유지보수 활동
체에서 행정 기술 차를 용시킬 수 있는 수단이다. 이 형상 리는 련
문서와 하드웨어에 용 가능하며, 소 트웨어 항목을 재창출하고, 항목에 이루어진
변경의 이력 제공에도 필요하다.
형상 리를 통하여 다음과 같은 활동이 가능할 것이다.
․각 소 트웨어 항목의 고유한 버 을 식별
․완성된 소 트웨어의 특정 버 과 함께 구성하는 각 소 트웨어 항목의 버 을 식별
․개발 이거나 인도 는 설치된 소 트웨어 제품의 형상상태를 식별
․독립 는 종속 으로 개발⋅유지보수 활동을 수행하는 2명 이상의 수행자에
의한 해당 소 트웨어 항목의 동시 변경 리
․요구되는 경우, 한 곳 이상의 치에서의 다수 소 트웨어의 변경을 한 조정
․개발착수에서 릴리즈에 이르기까지 변경의 필요성, 요구 는 문제해결을 한
모든 조치 변경의 식별
최소한으로, 제조업체는 SOUP와 같은 다른 소 트웨어 제품을 포함16)하여 형상항목을

- 40 -
식별하기 한 방법을 수립하고, 이에 따라 리할 형상항목과 그 버 을 문서화하며,
형상항목에 한 이력(형상 상태)을 검색할 수 있는 기록을 유지하여야 할 것이다.
16) 를 들어 버 , 릴리즈 날짜, 패치 번호 는 업그 이드 명칭일 수도 있다.

- 41 -
Ⅲ. 공정에서의 소 트웨어 밸리데이션
의료기기 산업계에서 컴퓨터 자동화 설비는 범 하게 사용되고 있다. 소규모의
제조업체에서도 소 트웨어를 이용하여 제조공정을 자동화하고 아웃풋을 확 함으로서
생산성을 개선하며 인력 비용을 감소시키고 있다. 비록 이러한 부분의 소 트웨어
시스템들은 외부 외주업체에 주문 개발되지만 궁극 으로 의료기기 제품의 안 성과
합성을 보증할 일차 책임은 의료기기 제조업체에게 있다.
이러한 공정 소 트웨어의 는 다음과 같다.
a) 생산공정 는 품질시스템 활동에 한 정보를 작업자가 개입하지 않고 수집,
분석하여 활동을 리하고 결과를 검증하는 경우 그리고 그런 결과에 해서
조치를 취하는 programmable logic controller, Digital function controller 등과
같은 임베디드 시스템(embedded system) 설비
b) PCB 자부품 같은 반제품(Sub-Assembly)의 시험과 완제품의 최종 합격 정
시험에 사용되는 시험용 소 트웨어
c) 시험 검사 장치에 의한 물질 는 제품의 평가, 교정, DMR의 리, 불만처리,
설계 경향분석 같은 품질시스템 차를 실행하고 지원하는 데 사용되는 소
트웨어
d) 웨이 솔더 머신, 로 (Robotics), 컴퓨터수치제어(CNC) 기계, 환경 리 설비,
멸균장치, 연속조립공정 등의 각종 생산공정과 장치를 리하는 데 사용되는
생산 리 소 트웨어
우리의 의료기기 제조 품질 리기 별표 1의 7.5.2.1에서 “제조업자는 규정된
요구사항을 충족하기 하여 제품 성능에 향을 미치는 컴퓨터 소 트웨어 용(소
트웨어 / 는 용의 변경을 포함)의 밸리데이션을 한 문서화된 차를 수립하고,
이러한 소 트웨어 용에 있어 최 사용 에 유효성을 확인”하도록 요구하고 있다.
이 요구사항 목 은 설계, 제조, 유통, 추 리 등 품질시스템의 모든 측면에서 사용
되는 소 트웨어는 설치가 정확하며 의도한 용도⋅목 에 따라 유효한 결과가 산출됨을
입증하는 것이다. 따라서 제조업체에서는 설정된 밸리데이션 차(Protocol)에 따라
소 트웨어의 유효성을 확인하여 의도한 바와 같이 일 되게 기능을 발휘함을 보증할
수 있어야 한다.
의료기기 제조업체에서 사용하는 자동화 설비 소 트웨어 제품 품질의 통계,
경향분석에 사용되는 스 드시트, 그래픽처리, 의료기기 이력 는 고객 불만 리에

- 42 -
사용되는 다수의 소 트웨어는 자체 으로 는 계약에 따라 개발되거나 제3자로부터
구매한 기성품(OTS; off-the-shelf) 소 트웨어를 사용한다. 소 트웨어를 제조업체가
자체 으로, 는 주문계약에 의해 개발하거나 는 OTS 제품에 상 없이 제조업체는
생산 품질시스템에서 시용하는 소 트웨어에 한 밸리데이션을 어느 정도까지,
어떻게 수행할 것인지를 결정하여야 한다.
제조업체에서 곤란스러워 하는 것은 소 트웨어 밸리데이션 수 (정도)의 설정일 것이다.
고품질 소 트웨어에 한한 다양한 문헌과 국제표 의 유용성에도 불구하고, 많은
소규모 제조업체에서는 “소 트웨어 밸리데이션이 필요한 컴퓨터 자동화 설비가 체
무엇인가?”와 “소 트웨어 밸리데이션에 한 기록은 무엇인가?”라는 질문에 을
둘 것이다.
의료기기의 안 , 성능 신뢰성과 련된 자동화 설비 품질시스템에서의 소
트웨어 밸리데이션이 요구되는 수 은 소 트웨어 이용과 련한 험도와 일치한다고
할 수 있다. 부연하자면, 자동화 운 에서 제품 성능에 요하거나 는 안 성에 향을
미치는 요인은 노출되는 험에 하며, 안 하고 효과 인 의료기기 생산을 한
밸리데이션의 일부분으로서 확인하여야 할 것이다. 한 이러한 범 와 확인방법에는
해당 소 트웨어 고유의 완 한 기능성뿐만 아니라 해요인(hazard) 분석, 제조업체에서
의도한 용도도 고려되어야 한다.
제조업체에서 수행하는 소 트웨어의 밸리데이션은 표 7과 같이 몇 가지로 분류하여
고려할 수 있다.
‘범주 I’에 해당하는 소 트웨어 종류는 제품 품질의 통계, 경향분석에 사용되는 스
드시트, 그래픽처리, 의료기기 이력이나 고객 불만 리 등 품질 리를 한 소 트웨어
로서 개 PC, PC네트워크, 는 형 컴퓨터에 상주한다. 컴퓨터 수치제어설비
(CNC), 자동 선별기(시험⋅검사용 소 트웨어), PCB 시험도구 등과 같은 소 트웨어도
여기에 해당된다.
이 범주에 속하는 소 트웨어는 설비/장비에 내장되어 사용자가 메뉴에서 여러 가지
옵션을 선택하고 여러 라미터를 입력하여 보고서를 인쇄하며 제한된 데이터를 이용
하여 공정을 리할 수 있게 하여 다. 이러한 소 트웨어는 의료기기 제조업체에서
아주 은 노력과 시험으로도 가능하다. 경우에 따라 품질 리를 한 기성품(OTS)
소 트웨어는 제조업체의 의도된 용도로 출력여부를 확인(기능상의 확인)하는 것으로
충분할 수도 있다.

- 43 -
[표 7] 소 트웨어의 밸리데이션 유형
유 형 설명 밸리데이션 정도
범주 I- 공정 는 의료기기가 제품의 안 , 일 성, 성능에 향을 주지 않거
나, 경미하다.
- 방차원의 리
- 간단한 시험⋅검사 는 교정
범주 II
- 공정 는 의료기기가 제품의 안 , 일 성, 성능에 미치는 향을 무시
할 수 없다.- 소 트웨어 자체에 한 밸리데이션
이 곤란하다.
- 시험⋅검사- 체 는 부분 인 공정 밸리데
이션(설치, 운 , 성능 격성평가)
범주 III
- 공정 는 의료기기가 제품의 안 , 일 성, 성능에 주는 향이 지 하
거나, 향을 악할 수 없다.(소트웨어가 주문설계되거나, 신뢰성에 한 의심이 크다)
- 완 한 소 트웨어 밸리데이션
를 들어 CNC 로그램은 통상 기계도면 형식으로 기술되며 출력물은 일반 으로
기구류(부품, 구조물)이다. CNC 로그램은 다수의 부품들을 가공한 후 로그램에
사용된 도면의 모든 치수가 규정된 치수 허용한계 이내인지를 측정함으로서 밸리데이션이
가능하다. CNC 로그램을 네트워크에서 다운로드(download)를 받거나, 단독 인
자매체(CD, Disk) 형태로 CNC에 설치하는 경우도 있다. 이런 로그램은 최 개발
시 업그 이드의 변경 시 마다 밸리데이션이 필요하다. CNC 부품⋅공구는 마모
되는 경향이 있으므로 정기 인 조정(Adjust)이 필요할 수 있다. 통상 으로 이런 조정은
밸리데이션 활동이 아닌 설비의 유지보수 활동의 일환으로 이행된다.
‘범주 II’에 속하는 소 트웨어는 컴퓨터로 제어되는 공정이나 설비에 조작자(운 자)가
개입하지 않고 데이터를 수집, 분석하여 자동으로 조치를 수행하는 programmable
logic controller, Digital function controller 소 트웨어 등이다. 를 들어, 조작자가
미리 정해진 범 한계 내에서 시간, 온습도 등을 설정하고, 이를 컴퓨터로 조정⋅제어
하는 환경 리용 소 트웨어는 제조업체에서 소 트웨어 자체만의 밸리데이션이 불가능
하다. 이와 같이 하드웨어/설비와 같이 운용이 되는, “특별공정”으로 호칭되는 공정에서
사용되는 소 트웨어에 해서는 소 트웨어 밸리데이션이 아닌 공정 밸리데이션
(Process Validation)으로 평가될 필요가 있다. 공정 밸리데이션의 설치(Installation), 운용
격성평가(Operating Qualification) 성능 격성평가(Performance Qualification)를

- 44 -
통하여 밸리데이션이 보증되는 경우 내부 소 트웨어의 밸리데이션은 공정 밸리데이션
활동에 포함된다.
‘범주 III’에 속하는 소 트웨어는 “2 개발에서의 밸리데이션”에서 술한 바와 같이
소 트웨어의 의도된 용도(사용목 ), 의료기기 공정에 미치는 안 성 등을 평가하
여야 한다. 제조업체에서 소 트웨어를 문업체에 주문하여 개발하는 경우 제조업체는
제조품질 리기 기타 요건에 합한 소 트웨어를 요구하여야 한다. 이 경우
제조업체는 소 트웨어의 요건을 개발하고 검증과 합격 정 기 을 결정하는 것이
최상이다. 궁극 으로, 주문형 소 트웨어가 요구사항에 충족함을 보증하는 일은 제조
업체의 책임이다.
제조업체는 기성품(OTS) 소 트웨어를 구매하여 수정⋅변형하여 사용할 수도 있다.
기성품 소 트웨어를 수정⋅변형하는 경우 요구사항은 특정 응용 로그램과 계된
소 트웨어의 의도된 용도에 기 하여 결정되므로 소 트웨어와 함께 제공되는 사용
설명서에 근거를 둘 필요는 없다. 제조업체는 수정⋅변형된 소 트웨어가 밸리데이션을
통하여 그 수정사항 수정사항으로 인한 다른 부분/시스템에 미치는 향을 구체
으로 검토하여야 한다.
밸리데이션에 필요한 정보가 내⋅외부로부터 가능하지 않은 SOUP17)의 경우 소 트
웨어의 코드는 알 수 없거나 액세스될 수 없다. 이런 경우 소 트웨어의 기능성만
다 지며 코드 수 의 밸리데이션은 안되므로, 통상 으로 제조업체는 소 트웨어가
“사용자 필요 사용 의도”를 확립하기 한 충분한 “블랙박스(black box)” 시험을
실시하여 소 트웨어가 사용자 요구를 충족시키며 의도된 용도에 합한지 유효성을
확인하여야 한다. 즉, 제조자는 소 트웨어 환경에서 시험을 시행하기 해서 일련의
시험조건과 상 입력을 고안하여 설치와 기능의 함에 해서 소 트웨어를 평가
하여야 하며, 이 경우 상 입력은 공정오류의 가능성 등이다. 다양한 응용 로그램에
서는 블랙박스 검사 하나로는 충분하지 않을 수도 있다.
밸리데이션은 반드시 문서화된 계획(Protocol)에 따라 수행되어야 한다. 한 소 트
웨어의 밸리데이션 결과는 객 인 입증이 가능하도록 반드시 품질기록으로 유지⋅
리하여야 한다. 밸리데이션 결과기록에는 상 소 트웨어 버 (Version)의 식별,
밸리데이션 실시날짜, 합격/불합격 기 , 밸리데이션 방법 결과(시험 는 측정값)
등이 포함되어야 한다.
17) Software Of Unknown Provenance(기원이 확인되지 않은 소 트웨어)의 약어로, 이미 개발되어 일반 으
로 사용할 수 있거나, 의료기기에 채택할 목 으로 개발되지 않은 기성품(Off-the-shelf, OTS) 소 트웨어
는 이 에 개발되어 개발 로세스에 한 한 기록이 없는 소 트웨어

- 45 -
소 트웨어는 시간의 경과에 따라 문제 을 해결하거나 기능의 업그 이드 필요성
때문에 변경될 수 있으므로 소 트웨어와 소 트웨어변경은 최 사용 에 공식 으로
검토되고 승인되어야 한다. 제조업체가 어떤 이유로, 를 들어 용량을 늘리거나 문제 을
해결하기 하여 밸리데이션된 소 트웨어를 변경하는 경우는 변경 사용 에 변경된
소 트웨어의 유효성을 다시 확인하여 시스템 내에서 다른 소 트웨어 유닛에 한 향
제품 품질에 미치는 향 등을 평가하고 단하여야 한다. 변경의 특성은 문서화
해두며 밸리데이션 기록에는 변경사항과 그것이 미치는 향을 구체 으로 언 되어야
한다.
변경되지 않은 소 트웨어일지라도 정기 인 유효성 재확인(Revalidation)이 필요하다.
이는 “생산 서비스 제공 로세스의 유효성 재확인(Validation)18)”과 운용의 궤를
같이 한다. 이 주기가 규정된 바는 없으나 통상 으로 1년을 용한다.
컴퓨터 소 트웨어 련 문서의 정본(master copies)은 품질시스템의 문서 리
차에 따라 장하고, 개정이력을 리하여야 한다. 한 변경 소 트웨어(old
version)도 보존해 두는 것이 필요하다.
18) 의료기기 제조․수입 품질 리기 (식약청 고시 제2005-69호) 별표 1의 7.5.2.1 다. 5

- 46 -
Ⅳ. 참고 문헌
1. ISO 14971:2000 의료기기에 한 험 리의 용
2. ISO/IEC TR 15846:1998 소 트웨어 수명주기 로세스 - 형상 리
3. ISO 10007:1995 품질경 - 형상 리에 한 지침
4. ISO 90003:2004 소 트웨어 시스템 엔지니어링 - 컴퓨터 소 트웨어에 한 ISO
9001:2000 용을 한 지침
5. IEC 62304/FDiS(2006) 의료기기 소 트웨어 수명주기 로세스
6. IEC 60601-1-4:2000 의료용 기기기-안 일반 요구사항-부가규격: 로그램 가능한
기 의료시스템
7. IEC 60601-1-6:2004 의료용 기기기 - 안 일반 요구사항 - 보충 규격: 사용 합성
8. IEEE Std 828-1998 소 트웨어 형상 리 계획 표
9. IEEE Std 830-1993 소 트웨어 요구사양(SRS)에 한 권고기
10. IEEE STD 1012:1998 소 트웨어 검증 밸리데이션에 한 IEEE 표
11. IEEE STD 1012A:1998 소 트웨어 검증 밸리데이션에 한 IEEE 표 추가사항 :
IEEE/EIA 12207.1-1997에 한 내용
12. IEEE STD 1028:1998 소 트웨어 검토 심사(Audits)에 한 IEEE 표
13. FDA 지침 S/W 시 신고(2005) Guidance for the Content of Premarket
Submissions for Software Contained in
Medical Devices
14. FDA 지침 SW Validation 원칙(2002) General Principles of Software Validation
15. 21 CFR PART 11 자 기록; 자 서명
16. 21 CFR Part 820 Quality System Regulatory
17. ANSI/AAMI SW68:2001 의료기기 소 트웨어 수명주기 로세스
18. 93/42/EEC 유럽 의료기기 지침
19. MB-MED/2.2/Rec4 소 트웨어와 의료기기

- 47 -
[별첨 1]
국제규격화 동향
1. ISO/IEC 국제규격 분석
1990년도부터 시작된 소 트웨어 분야의 국제규격 활동은 국제규격화기구(ISO)
국제 기기술 원회(IEC)의 통합 기술 원회인 JTC1/SC7 소 트웨어 엔지니어링에서
주도하고 있다. 재 이 분야 WG 6에서의 국제규격화 작업은 소 트웨어 제품의 품질
특성 측정방법을 정의하는 ISO/IEC 9126 계열 14598 계열규격 제정을 완성하
으며, WG 10에서는 소 트웨어 개발업체의 능력과 성숙도를 평가하는 ISO/IEC TR
15504 계열규격(SPICE, Software Process Improvement and Capability dEtermination)를
제정하여 운 으로서, 이는 다음과 같이 소 트웨어 제품 평가, 로세스 평가,
품질시스템 분야로 분리하여 고려할 수 있다.
•소 트웨어 수명주기 련 규격
- ISO/IEC 12207:1995 (소 트웨어 수명주기 로세스)
- IEC 62304:200619) (의료기기 소 트웨어 수명주기20) 로세스)
•소 트웨어 제품의 품질특성 매트릭스(Software Quality Characteristics and
Metrics) 련 ISO/IEC 9126 계열규격
- ISO/IEC 9126-1 (소 트웨어 제품 품질-Part 1: 품질모델)
- ISO/IEC 9126-2 (소 트웨어 제품 품질-Part 2: 외부 메트릭)
- ISO/IEC 9126-3 (소 트웨어 제품 품질-Part 3: 내부 메트릭)
- ISO/IEC 9126-4 (소 트웨어 제품 품질-Part 4: 메트릭 사용에서의 품질)
•소 트웨어 제품의 평가(Software Product Evaluation) 련 ISO/IEC 14598 계열규격
- ISO/IEC 14598-1 (소 트웨어 제품 평가-Part 1: 개요)
- ISO/IEC 14598-2 (소 트웨어 제품 평가-Part 2: 기획 리)
19) IEC JTC(Joint Technical Committee) 1의 SC(Subcommittee) 62A Joint Working Group에서 2006년 5월 9일 발간되어 재 FDIS 상태인 규격으로서, ISO/TC 210, ISO/IEC JTC 1/SC 7, CENELEC TC 62외에도 TC 62, SC 62B/C/D, TC 66, TC 76외 다수 TC SC가 참여하여 합의한 규격
20) life cycle, 소 트웨어 제품의 개발, 운용 유지보수에 내포된 로세스, 활동 업무를 포함하고, 시스템 수명을 시스템 요구사항 정의에서 시스템 사용의 종료까지 보는 구조 (framework)로, 소 트웨어 개발
수명주기는 요구사항의 정의에서부터 제조를 한 릴리즈에 이르기까지 소 트웨어의 수명 체를 포 하
는 개념 구조.

- 48 -
- ISO/IEC 14598-3 (소 트웨어 제품 평가-Part 3: 개발자를 한 로세스)
- ISO/IEC 14598-4 (소 트웨어 제품 평가-Part 4: 취 자를 한 로세스)
- ISO/IEC 14598-5 (소 트웨어 제품 평가-Part 5: 평가자를 한 로세스)
- ISO/IEC 14598-6 (소 트웨어 제품 평가-Part 6: 평가모듈의 문서화)
•소 트웨어 개발업체 능력과 성숙성 등의 로세스 평가(Software Process
Assessment) 련 ISO/IEC TR15504 계열규격
- ISO/IEC TR15504-1 (소 트웨어 로세스 평가-Part 1: 개요)
- ISO/IEC TR15504-2 (소 트웨어 로세스 평가-Part 2: 로세스 능력에 한 모델)
- ISO/IEC TR15504-3 (소 트웨어 로세스 평가-Part 3: 평가 수행)
- ISO/IEC TR15504-4 (소 트웨어 로세스 평가-Part 4: 평가 지침)
- ISO/IEC TR15504-5 (소 트웨어 로세스 평가-Part 5: 평가 모델 지표 지침)
- ISO/IEC TR15504-6 (소 트웨어 로세스 평가-Part 6: 심사자의 능력 평가 지침)
- ISO/IEC TR15504-7 (소 트웨어 로세스 평가-Part 7: 로세스 개선 지침)
- ISO/IEC TR15504-8 (소 트웨어 로세스 평가-Part 8: 공 자 로세스 능력 측정 지침)
•기타 련 규격
- ISO/IEC 90003:2004 (컴퓨터 소 트웨어에 ISO 9001:2000 품질경 시스템(Quality
Management System) 용을 한 지침)
- ISO/IEC TR 15846:1998 (소 트웨어 수명주기 로세스에서의 형상 리
(Configuration Management))
1995년 8월에 발간된 ISO/IEC 12207은 항공, 자동차, 의학 등 모든 사업 분야에서
사용할 수 있는 소 트웨어의 수명주기 로세스, 활동 업무에 하여 종합 으로
규정하고 있으며, ISO/IEC 12207에 확립된 소 트웨어 수명주기 로세스를 한
체제를 사용하여 그 의도를 달하고 실 을 증명하기에 필요한 일련의 로세스,
활동 업무를 설명하고 있다. 이 규격은 소 트웨어 수명주기 체를 단계별로 구분
하고 해당 단계별로 이행하여야 할 활동을 언 하여, 소 트웨어 수명주기 체에서의
품질 리 활동을 수행하는 것이 가장 최선이며 효율 인 방법임을 규정하고 있다.
규모화・복잡화되고 있는 소 트웨어의 개발 유지보수 활동에서 소 트웨어
품질은 수명주기의 각 단계에서 모두 향을 미치고 있으며 특히 수명주기 기단계의
오류일수록 최종산출물에 치명 인 향을 미칠 수 있으므로 수명주기 기단계부터
각 단계별로 품질활동을 실시하고 그 결과물을 다음 단계로 피드백하여 필요한 리를

- 49 -
병행할 필요가 있다. 소 트웨어 개발에서의 품질 리 문제 은 개발이 완료된 후에
품질평가를 실시한다는 이었다. 이는 소 트웨어의 성능과 품질을 하시키는 요인
이 되기 때문에, 고품질의 소 트웨어 확보를 해서는 품질활동이 수명주기 단계
에 걸쳐 실시되어야 하며 각 단계에서 소 트웨어의 활동 결과가 다음 단계에서의
입력이 되는 피드백과 수정과정이 반복 으로 이루어져야 함을 의미한다.
소 트웨어 수명주기 로세스, 활동 업무에 하여 ISO/IEC 12207은 모든 사업
분야에서 용할 수 있는 규격임에 비하여 2006년 5월 9일 JTC 1 SC 62A JWG에서
발간한 IEC 62304규격은 재 FDIS 이지만, 의료기기 분야에서 용할 수 있는 소
트웨어 수명주기 로세스21) 규격으로 작성된 것이다. 규격 ISO/IEC 12207에서 소 트
웨어 제품(software product)의 정의는 ‘컴퓨터 로그램, 차서, 그리고 가능한 련
문서와 자료의 집합’으로 규정하고 있는데 비하여 IEC 62304에서는 ‘의료기기 소 트
웨어(Medical Device Software)’로 한정하고 있으며 이에 한 정의를 ‘개발 인 의료
기기에 채택할 목 으로 개발된 소 트웨어 시스템 는 자체 권한으로 의료기기로
사용하기 한 소 트웨어 시스템컴퓨터 로그램’으로 규정하고 있다.
IEC 62304 규격은 의료기기 소 트웨어의 개발, 운용 유지보수에 내포된 로세스,
활동 업무를 포함하고, 시스템의 수명을 요구사항의 정의에서부터 제조를 한
릴리즈에 이르기까지 소 트웨어의 수명 체를 포 하는 개념 구조(life cycle
model)에서 의료기기 소 트웨어의 안 한 설계와 유지보수에 필요한 활동과 업무에
련된 수명주기 로세스의 체제를 제공한다.
IEC 62304 규격은 의료기기 소 트웨어 개발과 유지보수에 용하며, 소 트웨어는
의료기기의 서 시스템(Sub-system) 는 그 자체가 의료기기로 간주한다. 수명주기
로세스는 소 트웨어 개발 로세스와 유지 리 로세스로 크게 분류하며, 수명주기
로세스에 한 요구사항을 제시하며, 각각의 수명주기 로세스를 일련의 활동으로
다시 분류하고, 각 활동을 일련의 업무로 분류하여, 품질경 시스템과 험경 시스템의
범 안에서 개발∙유지하도록 규정하고 있는데, 이는 장에서 발생하는 부분의
사고는 부 합한 소 트웨어 갱신과 업그 이드를 포함해 의료기기 시스템의 정비나
유지보수에 련되므로 소 트웨어 유지 리 로세스는 소 트웨어 개발 로세스만큼
21) 규격에서의 수명주기 모델은 V모델로, 폭포수형 모델은 Pure Waterfall, Winston W. Royce가 처음 개발한 소 트웨어의 개발 수명주기 략으로 로젝트를 리 가능한 단계(요구사항, 설계, 구 , 테스트 등)로 분류하고 마일스톤(Milestone)과 문서화 작업을 수립하며 각 단계의 종료마다 검토(review)하는 방법임. 즉, 폭포수형 모델은 문서 심의 모델로, 이는 단계에서 다음단계로 이동되는 주요 산출물이 문서임을
의미.

- 50 -
요하게 취 해야 한다. 소 트웨어 유지보수 로세스는 소 트웨어 개발 로세스와
매우 유사하다. 개발 로세스 활동에 하여 그림 2-1에 표시되어 있다.
림 2-1] 소 트웨어 개발 로세스 활동의 개요
8 소 트웨어 형상 리
9 소 트웨어 문제해결
이 규격의 범 에 포함되지 않는 활동
7 소 트웨어 험경
5.1소 트웨어
개발 기획
5.2소 트웨어 요구사항 분석
5.3소 트웨어
구축 설계
5.4소 트웨어
상세 설계
5.5소 트웨어 장비구
검증
5.6소 트웨어
통합 통합시험
5.7소 트웨어
시스템시험
5.8소 트웨어
릴리즈
시스템 개발 활동 ( 험경 포함)
고객 요구 충족고객 요구
이 규격과 련하여 ISO 13485(품질경 시스템) ISO 14971( 험경 시스템)과 같은
의료기기 국제규격은 제품을 개발하는 조직을 한 기 를 형성하는 리 환경을 제공
한다. IEC 60601-1(의료용 기기기의 기본 안 성 필수 성능에 한 공통 규격)
IEC 61010-1(측정, 제어 시험실용 기기기의 안 에 한 일반 요구사항)과
같은 안 련규격은 안 한 의료기기의 개발을 하여 특별히 용되는 지침을 제공
한다. 소 트웨어가 이러한 의료기기의 일부일 경우, 이 규격은 안 한 의료기기 소
트웨어 개발과 유지 리에 필요한 보다 상세한 지시를 제공한다. ISO/IEC 12207(소
트웨어 수명주기 로세스), IEC 61508-3(시스템 련 기‧ 자/ 로그램이 가능한
기 안 의 기능 안 성) ISO/IEC 90003(컴퓨터 소 트웨어에 ISO 9001:2000
용을 한 지침)과 같은 많은 다른 국제규격은 이 규격의 요구사항 구 에 사용할
수 있는 방법, 도구 기술로서 간주할 수 있다. 그림 2-2에는 이러한 다른 해당 규격
과의 계를 표시한다.
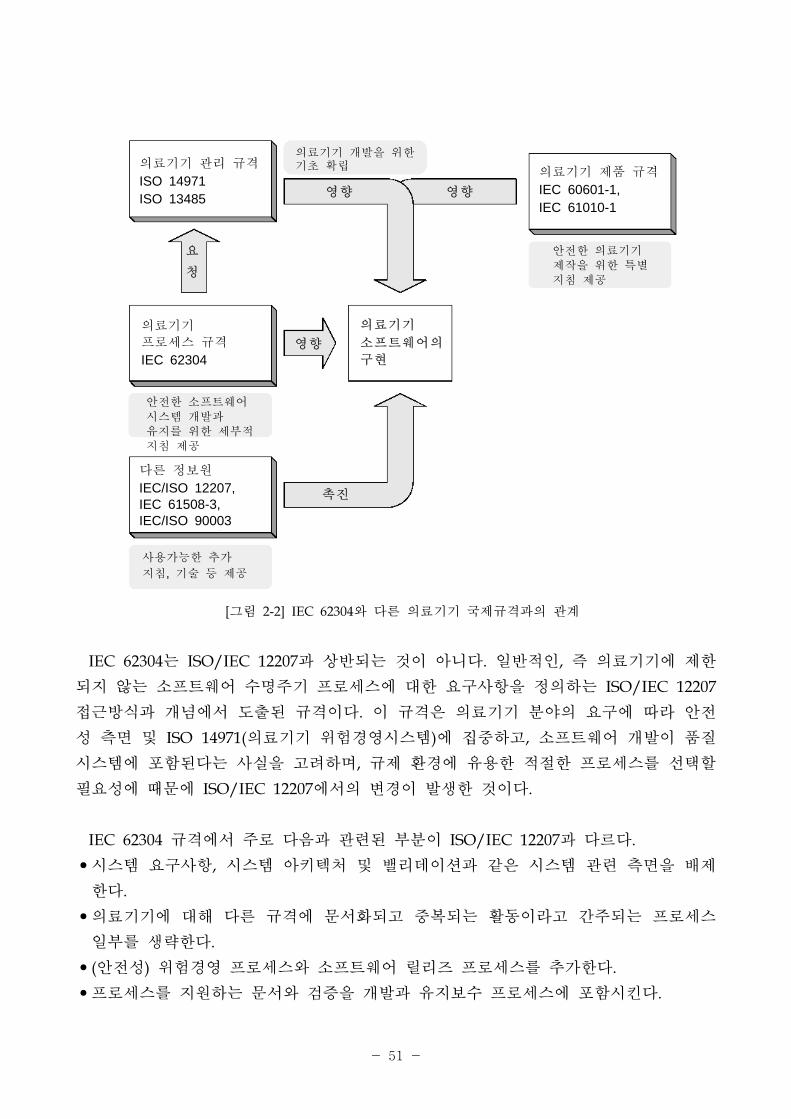
- 51 -
[그림 2-2] IEC 62304와 다른 의료기기 국제규격과의 계
향
요
청
진
향
향
의료기기 리 규격
ISO 14971ISO 13485
의료기기 제품 규격
IEC 60601-1,IEC 61010-1
의료기기
로세스 규격
IEC 62304
다른 정보원
IEC/ISO 12207,IEC 61508-3,IEC/ISO 90003
의료기기
소 트웨어의
구
의료기기 개발을 한 기 확립
안 한 소 트웨어 시스템 개발과 유지를 한 세부 지침 제공
사용가능한 추가
지침, 기술 등 제공
안 한 의료기기 제작을 한 특별 지침 제공
IEC 62304는 ISO/IEC 12207과 상반되는 것이 아니다. 일반 인, 즉 의료기기에 제한
되지 않는 소 트웨어 수명주기 로세스에 한 요구사항을 정의하는 ISO/IEC 12207
근방식과 개념에서 도출된 규격이다. 이 규격은 의료기기 분야의 요구에 따라 안
성 측면 ISO 14971(의료기기 험경 시스템)에 집 하고, 소 트웨어 개발이 품질
시스템에 포함된다는 사실을 고려하며, 규제 환경에 유용한 한 로세스를 선택할
필요성에 때문에 ISO/IEC 12207에서의 변경이 발생한 것이다.
IEC 62304 규격에서 주로 다음과 련된 부분이 ISO/IEC 12207과 다르다.
•시스템 요구사항, 시스템 아키텍처 밸리데이션과 같은 시스템 련 측면을 배제
한다.
•의료기기에 해 다른 규격에 문서화되고 복되는 활동이라고 간주되는 로세스
일부를 생략한다.
•(안 성) 험경 로세스와 소 트웨어 릴리즈 로세스를 추가한다.
• 로세스를 지원하는 문서와 검증을 개발과 유지보수 로세스에 포함시킨다.

- 52 -
ISO/IEC 62304 로세스 ISO/IEC 12207 로세스
활동 업무 활동 업무
5 소 트웨어 개발 로세스
5.3 개발 로세스6.1 문서화 로세스6.2 형상 리 로세스
6.4 검증 로세스6.5 밸리데이션 로세스6.8 문제해결 로7.1 경 로세스
5.1 소 트웨어
개발 기획
5.3.1 로세스 구
5.3.3 소 트웨어 구축 설계
5.3.7 소 트웨어 코딩 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
5.3.10 시스템 통합6.1.1 로세스 구
6.2.1 로세스 구
6.2.2 형상 식별6.4.1 로세스 구
6.5.1 로세스 구
6.8.1 로세스 구
7.1.2 기획7.1.3 실행 리
7.2.2 기반구조의 확립7.2.3 기반구조의 유지보수
5.1.1 소 트웨어 개발 계획5.3.1 로세스 구
7.1.2 기획
5.3.1.15.3.1.35.3.1.47.1.2.1
5.1.2 소 트웨어 개발 계획
갱신 유지7.1.3 실행 리 7.1.3.3
5.1.3 시스템 설계와 개발에 한 소 트웨어 개발
계획 참조
5.3.3 시스템 구축 설계5.3.10 시스템 통합6.5.1 로세스 구
5.3.3.15.3.10.16.5.1.4
•각 로세스의 로세스 구 과 기획을 개발과 유지보수 로세스의 단일 활동으로
통합한다.
•안 성 필요와 련한 요구사항을 분류한다.
• 로세스를 일차 는 지원 로세스로 명백히 분류하지 않으며, 로세스를 그룹으로
구성하지 않는다.
표 2-1에 IEC 62304와 ISO/IEC 12207과의 계를 표시한다.
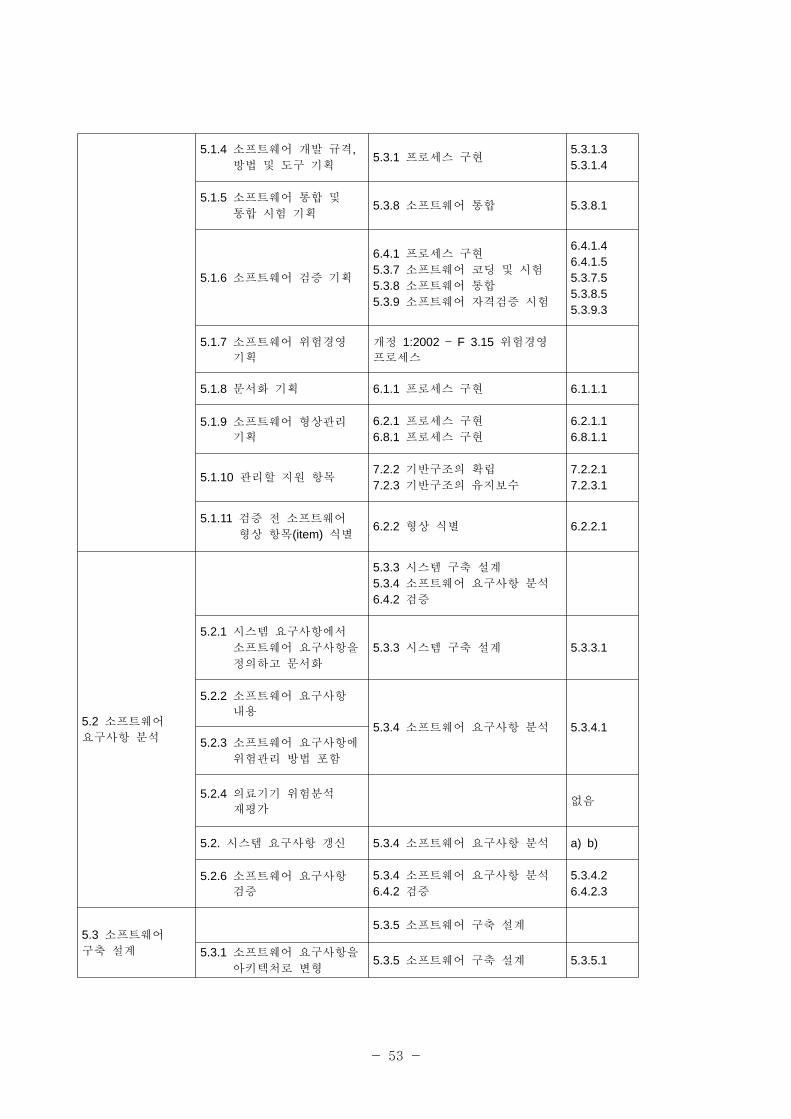
- 53 -
5.1.4 소 트웨어 개발 규격, 방법 도구 기획
5.3.1 로세스 구5.3.1.35.3.1.4
5.1.5 소 트웨어 통합
통합 시험 기획5.3.8 소 트웨어 통합 5.3.8.1
5.1.6 소 트웨어 검증 기획
6.4.1 로세스 구
5.3.7 소 트웨어 코딩 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
6.4.1.46.4.1.55.3.7.55.3.8.55.3.9.3
5.1.7 소 트웨어 험경
기획
개정 1:2002 – F 3.15 험경
로세스
5.1.8 문서화 기획 6.1.1 로세스 구 6.1.1.1
5.1.9 소 트웨어 형상 리
기획
6.2.1 로세스 구
6.8.1 로세스 구
6.2.1.16.8.1.1
5.1.10 리할 지원 항목7.2.2 기반구조의 확립7.2.3 기반구조의 유지보수
7.2.2.17.2.3.1
5.1.11 검증 소 트웨어
형상 항목(item) 식별6.2.2 형상 식별 6.2.2.1
5.2 소 트웨어
요구사항 분석
5.3.3 시스템 구축 설계5.3.4 소 트웨어 요구사항 분석
6.4.2 검증
5.2.1 시스템 요구사항에서 소 트웨어 요구사항을
정의하고 문서화
5.3.3 시스템 구축 설계 5.3.3.1
5.2.2 소 트웨어 요구사항
내용
5.3.4 소 트웨어 요구사항 분석 5.3.4.15.2.3 소 트웨어 요구사항에
험 리 방법 포함
5.2.4 의료기기 험분석 재평가
없음
5.2. 시스템 요구사항 갱신 5.3.4 소 트웨어 요구사항 분석 a) b)
5.2.6 소 트웨어 요구사항
검증
5.3.4 소 트웨어 요구사항 분석
6.4.2 검증5.3.4.26.4.2.3
5.3 소 트웨어
구축 설계
5.3.5 소 트웨어 구축 설계
5.3.1 소 트웨어 요구사항을
아키텍처로 변형5.3.5 소 트웨어 구축 설계 5.3.5.1
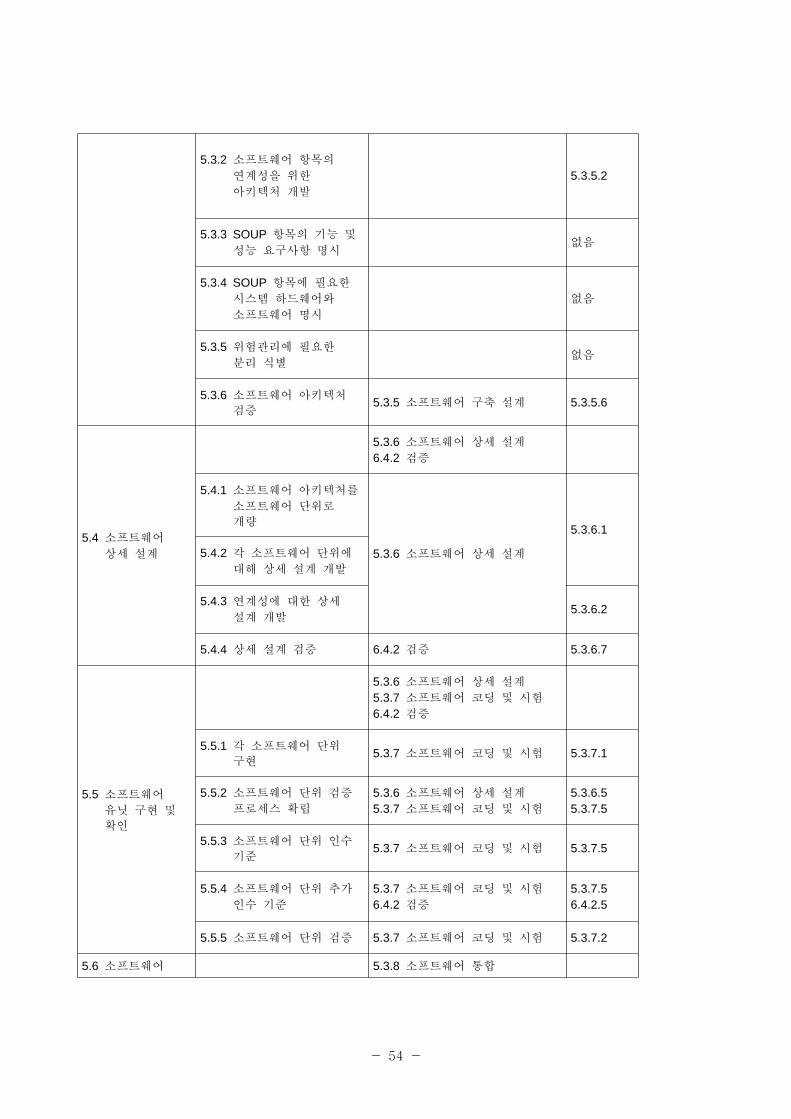
- 54 -
5.3.2 소 트웨어 항목의
연계성을 한
아키텍처 개발
5.3.5.2
5.3.3 SOUP 항목의 기능
성능 요구사항 명시없음
5.3.4 SOUP 항목에 필요한 시스템 하드웨어와
소 트웨어 명시
없음
5.3.5 험 리에 필요한
분리 식별없음
5.3.6 소 트웨어 아키텍처
검증5.3.5 소 트웨어 구축 설계 5.3.5.6
5.4 소 트웨어
상세 설계
5.3.6 소 트웨어 상세 설계
6.4.2 검증
5.4.1 소 트웨어 아키텍처를
소 트웨어 단 로
개량
5.3.6 소 트웨어 상세 설계
5.3.6.1
5.4.2 각 소 트웨어 단 에
해 상세 설계 개발
5.4.3 연계성에 한 상세 설계 개발
5.3.6.2
5.4.4 상세 설계 검증 6.4.2 검증 5.3.6.7
5.5 소 트웨어
유닛 구
확인
5.3.6 소 트웨어 상세 설계
5.3.7 소 트웨어 코딩 시험
6.4.2 검증
5.5.1 각 소 트웨어 단
구5.3.7 소 트웨어 코딩 시험 5.3.7.1
5.5.2 소 트웨어 단 검증
로세스 확립
5.3.6 소 트웨어 상세 설계
5.3.7 소 트웨어 코딩 시험
5.3.6.55.3.7.5
5.5.3 소 트웨어 단 인수
기5.3.7 소 트웨어 코딩 시험 5.3.7.5
5.5.4 소 트웨어 단 추가
인수 기
5.3.7 소 트웨어 코딩 시험
6.4.2 검증5.3.7.56.4.2.5
5.5.5 소 트웨어 단 검증 5.3.7 소 트웨어 코딩 시험 5.3.7.2
5.6 소 트웨어 5.3.8 소 트웨어 통합

- 55 -
통합 통합
시험
5.3.9 소 트웨어 자격검증 시험
5.3.10 시스템 통합6.4.1 로세스 구
6.4.2 검증
5.6.1 소 트웨어 단 통합 5.3.8 소 트웨어 통합 5.3.8.2
5.6.2 소 트웨어 통합 검증5.3.8 소 트웨어 통합
5.3.10 시스템 통합5.3.8.25.3.10.1
5.6.3 통합 소 트웨어 시험 5.3.9 소 트웨어 자격검증 시험 5.3.9.1
5.6.4 통합 시험 내용 5.3.9.3.
5.6.5 통합 시험 차 검증 6.4.2 검증 6.4.2.2
5.6.6 회귀시험 수행 5.3.8 소 트웨어 통합 5.3.8.2
5.6.7 통합시험 기록 내용 5.3.8 소 트웨어 통합 5.3.8.2
5.6.8 소 트웨어 문제해결
로세스의 사용6.4.1 로세스 구 6.4.1.6
5.7 소 트웨어
시스템 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
6.4.1 로세스 구
6.4.2 검증6.8.1 로세스 구
5.7.1 각 소 트웨어
요구사항에 한 시험
확립
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
5.3.8.45.3.9.1
5.7.2 소 트웨어 문제해결
로세스의 사용6.4.1 로세스 구 6.4.1.6
5.7.3 변경 후 재시험 6.8.1 로세스 구 6.8.1.1
5.7.4 소 트웨어 시스템
시험 검증
6.4.2 검증5.3.9 소 트웨어 자격검증 시험
6.4.2.25.3.9.3
5.7.5 각 시험 소 트웨어
시스템 시험 기록
내용에 한 데이터
문서화
5.3.9 소 트웨어 자격검증 시험 5.3.9.1
5.8 소 트웨어
릴리즈
5.3.9 소 트웨어 자격검증 시험
5.4.2 작동 시험6.2.5 형상 평가6.2.6 릴리즈 리 인도
5.8.1 소 트웨어 검증의 5.4.2 작동 시험 5.4.2.1

- 56 -
완벽성 확인(ensure) 6.2.6 릴리즈 리 인도5.4.2.26.2.6.1
5.8.2 확인(known)된 잔류 비정상 상태 문서화
6.2.5 형상 평가5.3.9 소 트웨어 자격검증 시험
6.2.5.15.3.9.35.8.3 확인(known)된 잔류
비정상 상태 평가
5.8.4 릴리즈 버 의 문서화
6.2.6 릴리즈 리 인도 6.2.6.1
5.8.5 릴리즈한 소 트웨어를
작성한 방법 문서화
5.8.6 활동과 업무의 완결 확인(ensure)
5.8.7 소 트웨어 장
5.8.8 소 트웨어 릴리즈의
반복 정 도 보장
6 소 트웨어 유지보수 로세스5.5 유지보수 로세스6.2 형상 리 로세스
6.1 소 트웨어
유지보수 계획
확립
5.5.1 로세스 구 5.5.1.1
6.2 문제 수정 분석
5.5.1 로세스 구
5.5.2 문제 코드화 분석5.5.3 수정 구5.5.5 감소(마이그 이션)
6.2.1 피드백 기록 평가
6.2.1.1 피드백 모니터링
5.5.1 로세스 구5.5.1.15.5.1.26.2.1.2 피드백 문서화
평가
6.2.1.3 안 에 한 문제
보고서 향의 평가5.5.2 문제 수정 분석
5.5.2.15.5.2.25.5.2.35.5.2.4
6.2.2 소 트웨어 문제해결
로세스의 사용5.5.1 로세스 구 5.5.1.2
6.2.3 변경 요청 분석 5.5.2 문제 수정 분석 5.5.2.1
6.2.4 변경 요청 승인 5.5.2 문제 수정 분석 5.5.2.5
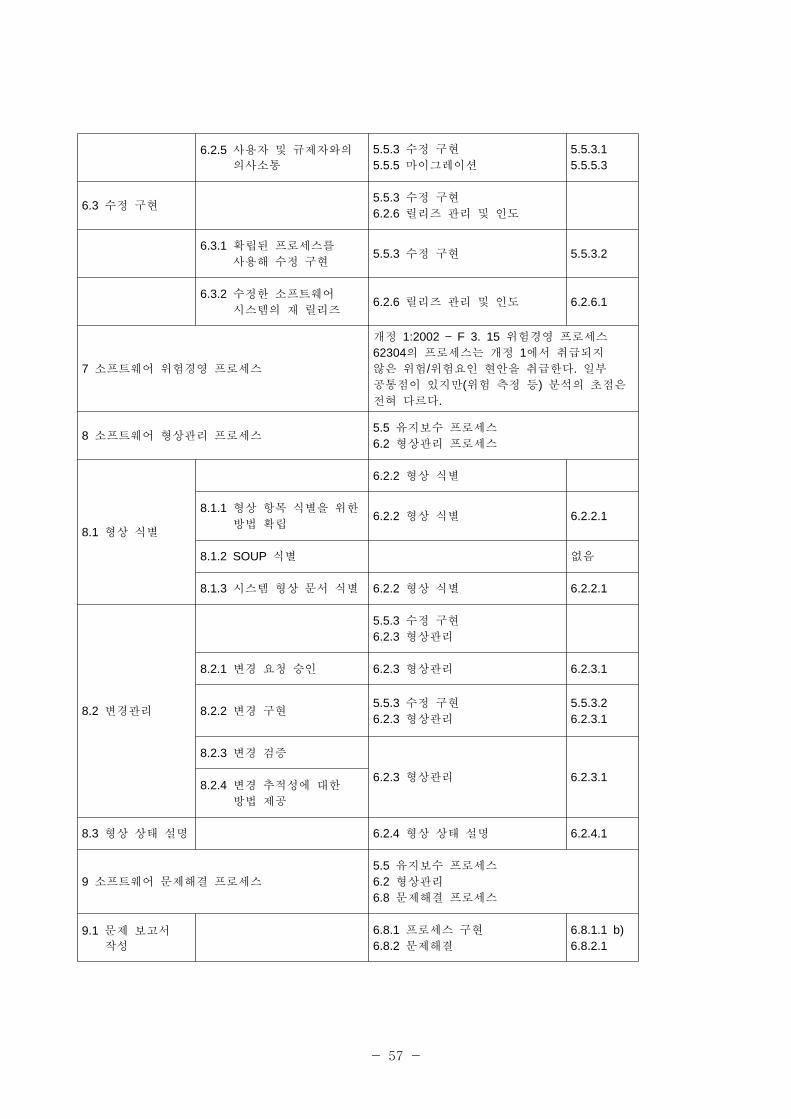
- 57 -
6.2.5 사용자 규제자와의 의사소통
5.5.3 수정 구5.5.5 마이그 이션
5.5.3.15.5.5.3
6.3 수정 구5.5.3 수정 구6.2.6 릴리즈 리 인도
6.3.1 확립된 로세스를 사용해 수정 구
5.5.3 수정 구 5.5.3.2
6.3.2 수정한 소 트웨어
시스템의 재 릴리즈6.2.6 릴리즈 리 인도 6.2.6.1
7 소 트웨어 험경 로세스
개정 1:2002 – F 3. 15 험경 로세스
62304의 로세스는 개정 1에서 취 되지
않은 험/ 험요인 안을 취 한다. 일부 공통 이 있지만( 험 측정 등) 분석의 은
다르다.
8 소 트웨어 형상 리 로세스5.5 유지보수 로세스6.2 형상 리 로세스
8.1 형상 식별
6.2.2 형상 식별
8.1.1 형상 항목 식별을 한
방법 확립6.2.2 형상 식별 6.2.2.1
8.1.2 SOUP 식별 없음
8.1.3 시스템 형상 문서 식별 6.2.2 형상 식별 6.2.2.1
8.2 변경 리
5.5.3 수정 구6.2.3 형상 리
8.2.1 변경 요청 승인 6.2.3 형상 리 6.2.3.1
8.2.2 변경 구5.5.3 수정 구6.2.3 형상 리
5.5.3.26.2.3.1
8.2.3 변경 검증
6.2.3 형상 리 6.2.3.18.2.4 변경 추 성에 한
방법 제공
8.3 형상 상태 설명 6.2.4 형상 상태 설명 6.2.4.1
9 소 트웨어 문제해결 로세스
5.5 유지보수 로세스6.2 형상 리
6.8 문제해결 로세스
9.1 문제 보고서 작성
6.8.1 로세스 구
6.8.2 문제해결6.8.1.1 b)6.8.2.1

- 58 -
[표 2-1] IEC 62304와 ISO/IEC 12207과의 계
9.2 문제 조사6.8.2 문제해결6.8.1 로세스 구
6.8.2.16.8.1.1 b)
9.3 련당사자에게 통보
6.8.1 로세스 구 6.8.1.1 a)
9.4 변경 리
로세스 사용
6.2.3 형상 리
5.5.3 수정 구
9.5 기록 유지 6.8.1 로세스 구 6.8.1.1 a)
9.6 문제 경향 분석6.8.1 로세스 구
6.8.2 문제해결6.8.1.1 b)6.8.2.1
9.7 소 트웨어
문제해결 검증6.8.1 로세스 구 6.8.1.1 d)
9.8 시험 문서의 내용
12207의 모든 시험
업무
문서화
1991년에 제정된 ISO/IEC 9126은 품질특성 메트릭을 정의하고 있는 국제규격으
로서 소 트웨어 품질을 기능성, 신뢰성, 사용성, 효율성, 유지보수성, 이식성의 6가지
특성으로 표 하며, 각 품질특성별 세부 메트릭을 제시하고 있다. 메트릭은 소 트웨어
개발과정에서 개발자들이 용할 수 있는 내부 메트릭과 소 트웨어 사용자들이 개발
기나 개발 완료 후에 용할 수 있는 외부 메트릭이 있다. ISO/IEC 9126은 소 트
웨어 제품에 한 품질 요구사항을 기술하는데 사용할 수 있으며 개발 에 있거나 는
개발 완료된 소 트웨어의 품질을 측정하는데 척도로 사용될 수 있다.
소 트웨어 품질특성은 공업제품의 품질특성보다 더욱 다양하고 복잡한 특성을 갖고
있기 때문에 여러 가지 품질특성을 고려하여 평가하여야 한다. 이러한 소 트웨어의
품질특성은 크게 외부특성과 내부특성으로 분류되고, 각 특성들은 다시 인자(요소)들로
구성되어 소 트웨어의 품질평가를 가능하게 한다. 어떤 제품을 비교 평가하는 경우,
동일 품질특성을 의미하는 어휘를 다른 의미로 이용하거나 동일특성에 한 의미를
다른 어휘로 사용한다면 명확한 평가가 이루어질 수 없다. 소 트웨어 제품 품질평가
에 한 국제규격인 ISO/IEC 9126에서 정의하고 있는 품질특성은 기능성, 신뢰성,
사용성, 효율성, 유지보수성, 이식성으로 각 품질특성의 개념은 다음과 같다.

- 59 -
•기능성
소 트웨어가 특정 조건에서 사용될 때, 명시된 요구와 내재된 요구를 만족하는 기능을
제공하는 소 트웨어 제품의 능력으로, 다른 특성들은 주로 소 트웨어가 언제, 어떻게
하는가에 심을 두는 비해 이 특성은 요구를 충족하기 해서 소 트웨어가 무엇을
하는가에 심을 둔다. 이 특성의 명시된 요구와 내재된 요구에 해서는 품질의
정의부분에 기술된 참고를 용한다.
[그림 2-3기능성의 품질부특성
↳‘지정된 작업과 사용자 목 을 한 한 기능들을 제공하는
소 트웨어 제품의 능력’으로 운 성에 향을 미침.; 구성요소가 되는 부기능, 테이블 용량 등으로부터 나오는 작업 주의 복합 기능
↳ 올바른 혹의 동의된 효능 결과를 제공할 수 있는 소 트웨어
제품의 능력
↳ 하나 이상의 명세된 시스템과 상호작용할 수 있는 소 트웨어
제품의 능력으로, 용어 ‘ 치성(Replaceability)’과 혼동을 피하기 하여 호환성(Compatibility) 신에 사용.
↳ 권한이 없는 자, 시스템은 정보에 근하지 못하게 하여 정보를
보호하는 소 트웨어 제품의 능력으로 송 인 Data에도 용.
↳ 응용과 련된 규격, 례 는 법 규제 유사 규정을 고수
하는 소 트웨어 제품의 능력
•신뢰성
명세된 조건에서 소 트웨어를 사용할 경우, 성능을 유지할 수 있는 소 트웨어 제품의
능력으로 신뢰성의 한계는 요구사항, 설계 구 상의 결함에 기인한다. 결함으로
인한 고장은 사용 경과 시간보다는 소 트웨어 제품의 사용방법과 선정된 로그램
선택사항에 따라 달라질 수 있다. (그림 2-4 참조)
•사용성
소 트웨어를 규정된 조건에서 사용할 경우, 향을 받거나 의존하는 운 자, 최종
사용자 간 사용자가 이해, 학습, 사용하며 선호할 수 있는 소 트웨어 제품의
능력으로서 기능성, 신뢰성, 효율성 등의 몇몇 특징들은 사용성에 향을 수 있지만
ISO/IEC 9126의 목 상 사용성으로는 분류하지 않는다. (그림 2-5 참조)
합성
(Suitability)
기능성
Functionality
정확성
(Accuracy)
상호운용성
(Interoperability)
보안성
(Security)
수성
(Compliance)

- 60 -
[그림 2-4신뢰성의 품질부특성
↳ 로그램이나 데이터 결함으로 인한 기능장애를 피할 수 있는
능력
↳ 소 트웨어 결함이나 인테페이스 문제발생 시에도 지정된 수
의 성능을 유지하는 능력
↳ 장애발생시 지정된 수 의 성능을 회복하고 데이터를 복귀하는
능력
↳ 소 트웨어 신뢰성에 한 규격, 규정 례 등을 수하는
능력
[그림 2-5] 사용성의 품질부특성
↳ 사용자가 소 트웨어를 활용하기 한 방법이나, 조건, 성
등을 악할 수 있게 하는 소 트웨어의 능력
↳ 사용자가 소 트웨어의 응용을 배울 수 있게 하는 능력
↳ 사용자가 소 트웨어를 운 하고 제어할 수 있도록 하는 능력
↳ 소 트웨어의 색상, 그래픽 등 사용자에게 호감을 갖도록 하는 능력
↳ 사용성에 한 규격, 규정, 례 스타일 등을 수하는 능력
•효율성
명시된 조건에서 사용되는 자원(다른 소 트웨어 제품, 하드웨어 장비, 재료-인쇄용지나
디스켓 등을 포함)의 양에 따라 요구된 성능을 제공하는 소 트웨어 제품의 능력으
로서, 효율성의 품질부특성은 그림 2-6와 같다.
신뢰성
(Reliability)
결함허용성
(Fault tolerance)
회복성
(Recoverability)
성숙성
(Maturity)
수성
(Compliance)
사용성
(Usability)
이해성
(Understandability)
친 성
(Attractiveness)
학습성
(Learnability)
운용성
(Operability)
수성
(Compliance)

- 61 -
[그림 2-6] 효율성의 품질부특성
↳ 소 트웨어가 규정된 조건에서 기능을 수행할 때 한 응답
시간, 처리시간, 처리율을 제공하는 능력
↳ 소 트웨어가 규정된 조건에서 기능을 수행할 때 한 양과
종류의 자원을 사용하는 능력
↳ 효율성에 한 규격, 규정 등을 수하는 능력
•유지보수성
소 트웨어 제품이 변경되는 능력으로, 변경에는 환경과 요구사항 기능 명세에
따른 소 트웨어의 수정, 개선 등이 포함된다.
[그림 2-7] 유지보수성의 품질부특성
↳ 소 트웨어의 약 이나 장애 원인을 진단하고 변경될 부분을
식별할 수 있도록 하는 능력
↳ 지정된 변경사항이 구 될 수 있도록 하는 소 트웨어의 능력
↳ 소 트웨어 변경에 따른 상치 못한 결과를 최소화하는 능력
↳ 소 트웨어가 변경되었을 경우 검증받을 수 있는 능력
↳ 유지보수성에 한 규격, 규정 례 등을 수하는 능력
•이식성
조직, 하드웨어 혹은 소 트웨어를 포함하는 하나의 환경에서 다른 환경으로 이될
수 있는 소 트웨어 제품의 능력
수성
(Compliance)
자원효율성
(Resource Utilization)
시간반응성
(Time Behavior)
효율성
(Efficiency)
유지보수성
(Maintainability)
분석성
(Analyzability)
변경성
(Changeability)
안정성
(Stability)
시험성
(Testability)
수성
(Compliance)

- 62 -
[그림 2-8] 이식성의 품질부특성
↳ 소 트웨어가 상이한 환경에 응하는데 필요한 최소한의 조치
(화면 필드, 테이블 크기, 처리볼륨, 출력양식 등 조 )로 이식될 수 있는 능력
↳ 지정된 환경에서 설치될 수 있는 소 트웨어의 능력
↳ 공통 자원을 사용하여야 하는 환경에서 다른 독립 인 소 트
웨어와 해당 소 트웨어가 공존할 수 있는 능력
↳ 동일환경에서 같은 목 으로 사용되는 다른 소 트웨어를 신
하여 사용될 수 있는 능력(Up-Grade)
↳ 이식성에 한 규격, 규정 등을 수하는 능력
소 트웨어 제품의 품질을 측정하거나 평가하는데 필요한 방법과 차를 정의하고
있는 ISO/IEC 14598은 최 ISO/IEC 9126:1991에 품질 측정방법과 차를 개략 으로
포함하여 규정되었으나 1994년부터 별도 규격으로 제정작업을 추진하여 재는 6개의
계열규격에서 품질 평가주체를 소 트웨어 개발자, 구매자, 평가자로 구분하여 규격화
되었다.
평가를 시행하는 차로는 평가 요구사항 도출, 평가명세서 작성, 평가계획수립 평가
수행 결과도출 등의 단계를 제시하고 있고 평가명세서를 작성할 때는 ISO/IEC
9126에 따른 내‧외부 메트릭을 활용하도록 하고 있다.
메트릭을 용하여 평가를 수행하는 과정에서 객 성 로세스 확보를 하여
각 메트릭 용 차 기 등을 명시한 평가모듈 라이 러리를 이 규격에서 제공하
고 있기도 하다.
재 ISO/IEC 14598과 ISO/IEC 9126 계열규격에 한 면 인 개정을 검토 으로서,
이는 ISO 12119:1994(패키지 소 트웨어 품질요구사항 테스 지침) 규격의 제정 이후,
세계 으로 사용해 오면서 소 트웨어의 품질은 품질 모델에 기 하여 품질 요구
사항에 비되는 메트릭을 사용 평가해야 하므로 ISO/IEC 9126(소 트웨어 제품 품질
특성)과 이를 평가하는 ISO/IEC 14598을 보강․통합한 새로운 체계(Architecture)가
이식성
(Portability)
체성
(Replaceability)
응성
(Adaptability)
설치성
(Installability)
공존성
(Coexistence)
수성
(Compliance)

- 63 -
요구되었기 때문이다. 이러한 요구에 따라 ISO/IEC JTC1/SC7/WG622)은 새로운 소
트웨어 평가 모델인 SQuaRE (Software Quality Requirements and Evaluation) 로젝
트를 진행 이며, SQuaRE의 구축 후에는 ISO/IEC 9126과 ISO/IEC 14598 계열규격을
ISO/IEC 25000 계열규격으로 체하는 것을 목표로 추진 이다.
SQuaRE는 그림 2-9과 같이 4 + 1 구조를 갖는다.
• 품질 모델(25010 Quality Model Division)
• 품질 메트릭(25020 Quality Metrics Division)
• 품질 요구사항(25030 Quality Requirement Division)
• 품질 평가(25040 Quality Evaluation Division)
• +(plus) 체를 반 하는 부분(25000 Quality Management Division)
[그림 2-9] SQuaRE 체계(Architecture)
SQuaRE는 기존의 ISO/IEC 9126과 14598을 통합하는 과정에서 문서의 일 성 유지를
하여 약간 는 다수의 문서 내용을 변경(Minor/Major Editorial Revision)하고, 기술
으로 다수의 부분도 개정(Major Technical Revision)하 으며, 필요에 따라 새로운
하부 로젝트를 도입(New Subproject Proposal)하 다. 기본 으로 ISO/IEC 9126
ISO/IEC 14598의 통합과정에서 개발된 SQuaRE 모델과 세부 으로 측정 개념
(Concept)의 명확화, 측정 리미티 (Measurement Primitives)의 정의, 기능
(Functional) 요구사항을 포함하는 품질 요구사항(Quality requirement) 등에 한 내용이
변화의 근간을 이루고 있다.
22) ISO/IEC JTC1/SC7/WG6은 범용 소 트웨어 제품의 품질특성 평가, COTS 품질요구사항 제3자 평가에 한 규격을 수립함에 그 목 을 두고 있음.

- 64 -
새로이 도입된 부분은 SQuaRE의 체 인 개 (Overview)과 체 SQuaRE 체계
는 구조모델(Architecture Model)에 한 소개 ‘SQuaRE 개 소개(General
Overview and Guide to the SQuaRE)’, 메트릭 참조모델에 한 소개 ‘메트릭 참조
모델과 소개(Metrics Reference Model and Guide)’, 소 트웨어 개발 수명주기에서
공통 사용되는 메트릭에 한 설명 ‘기본 메트릭(Base Metrics)’, 소 트웨어 품질 요구
사항과 추 , 타당성 련성을 다루는 ‘품질 요구사항(Quality Requirement)’ 등이다.
기술 으로 다수 개정된 내용은 ‘외부 메트릭(External Metrics)’, ‘사용자 의 품질
메트릭(Quality In Use Metrics)’ 등이며 기타 부분은 규격 문서의 일 성 유지를 해
약간 는 상당부분 편집한 것이다.
SQuaRE 모델과 기술 수 의 계는 그림 2-10와 같이 소 트웨어 제품 련자
(Stakeholders)가 품질 요구사항을 제공하고 이러한 요구사항들이 품질평가(Product
Quality Evaluation)를 어떤 범 에서 어떻게 수행할지 결정한다. 품질 측정(Product
Quality Measurement)은 품질 요구사항과 품질 평가를 지원하는 역할을 하게 된다.
즉, 품질 모델과 품질 메트릭을 사용하여 품질 요구사항을 도출하고 도출된 요구사항은
품질평가를 한 평가항목이나 기 으로 작용한다. 여기서 품질 모델과 품질 메트릭은
품질 평가를 해서도 사용된다.
[그림 2-10] SQuaRE 모델을 기술 수 에서 본 계 모델
Quality Model
Quality Evaluation
Quality Requirement
Quality MetricsIs used byIs used by
Supplies Criteria for
Is used by Is used by
StakeholderStakeholderDeveloperEvaluator
측정 리미티 (Measurement Primitives) 정의와 련하여 SQuaRE에서의 메트릭
개념은 체 으로 상호 연 계를 갖는다. 품질 속성을 나타내는 측정 리미티 가
측정과 데이터 수집을 통해 이루어지고, 품질 측정을 하여 내‧외부/QIU 품질 측정
치가 필요하며 이는 품질부특성(Quality Sub-characteristic)을 나타낸다. 품질 측정치는
종합하여 품질 특성을 형성하게 되고 품질특성은 품질 평가에 필요하다. 이를 기반으로
- 65 -
품질평가보고서를 작성하여 소 트웨어 제품 품질을 평가하게 된다.
품질 요구사항(Quality Requirement)에서 요구명세서(Requirement Specification)는
기능 요구사항, 비기능 요구사항, 품질 요구사항을 포함하며, 품질 요구사항은 기능
성 요구사항, 신뢰성 요구사항, 사용성 요구사항 등을 포함한다. 이러한 요구사항은
소 트웨어 련자 (Stakeholders)의 요구(Needs)를 반 한 것으로 품질의 종류, 련자의
종류, 개발 생명주기 등 여러 가지 측면에서 품질 요구사항을 볼 수 있다.
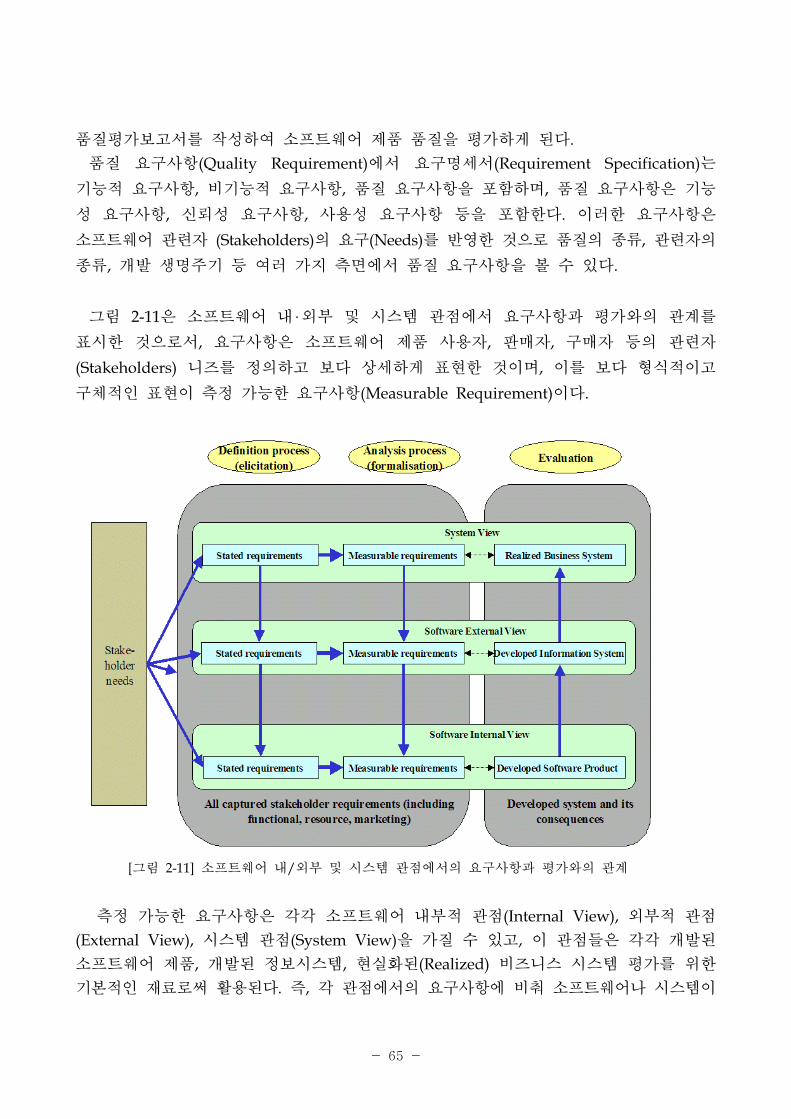
그림 2-11은 소 트웨어 내‧외부 시스템 에서 요구사항과 평가와의 계를
표시한 것으로서, 요구사항은 소 트웨어 제품 사용자, 매자, 구매자 등의 련자
(Stakeholders) 니즈를 정의하고 보다 상세하게 표 한 것이며, 이를 보다 형식 이고
구체 인 표 이 측정 가능한 요구사항(Measurable Requirement)이다.
[그림 2-11] 소 트웨어 내/외부 시스템 에서의 요구사항과 평가와의 계
측정 가능한 요구사항은 각각 소 트웨어 내부 (Internal View), 외부
(External View), 시스템 (System View)을 가질 수 있고, 이 들은 각각 개발된
소 트웨어 제품, 개발된 정보시스템, 실화된(Realized) 비즈니스 시스템 평가를 한
기본 인 재료로써 활용된다. 즉, 각 에서의 요구사항에 비춰 소 트웨어나 시스템이
- 66 -
규격 계열 규격 주요 내용
ISO/IEC 2500XISO/IEC 25000 SQuaRE에 한 일반 개요 안내
ISO/IEC 25001 계획과 리(ISO/IEC 14598-2에 해당)
ISO/IEC 2501X ISO/IEC 25010품질 모델(ISO/IEC 9126-1에 해당)- 품질모델 품질모델 사용에 한 안내
ISO/IEC 2502X
ISO/IEC 25020 메트릭 참조 모델과 안내
ISO/IEC 25021 기본 메트릭(SQuaRE에서 새로 제정됨)
ISO/IEC 25022 내부 메트릭(ISO/IEC 9126-3의 내부 메트릭)
ISO/IEC 25023 외부 메트릭(ISO/IEC 9126-2의 외부 메트릭)
ISO/IEC 25024 사용품질 메트릭(ISO/IEC 9126-4의 사용품질 메트릭)
ISO/IEC 25025 평가모듈의 문서화(ISO/IEC 14598-6에 해당)
ISO/IEC 2503X ISO/IEC 25030품질요구
- 품질요구에 한 일반 인 안내
- 품질요구를 한 요구사항
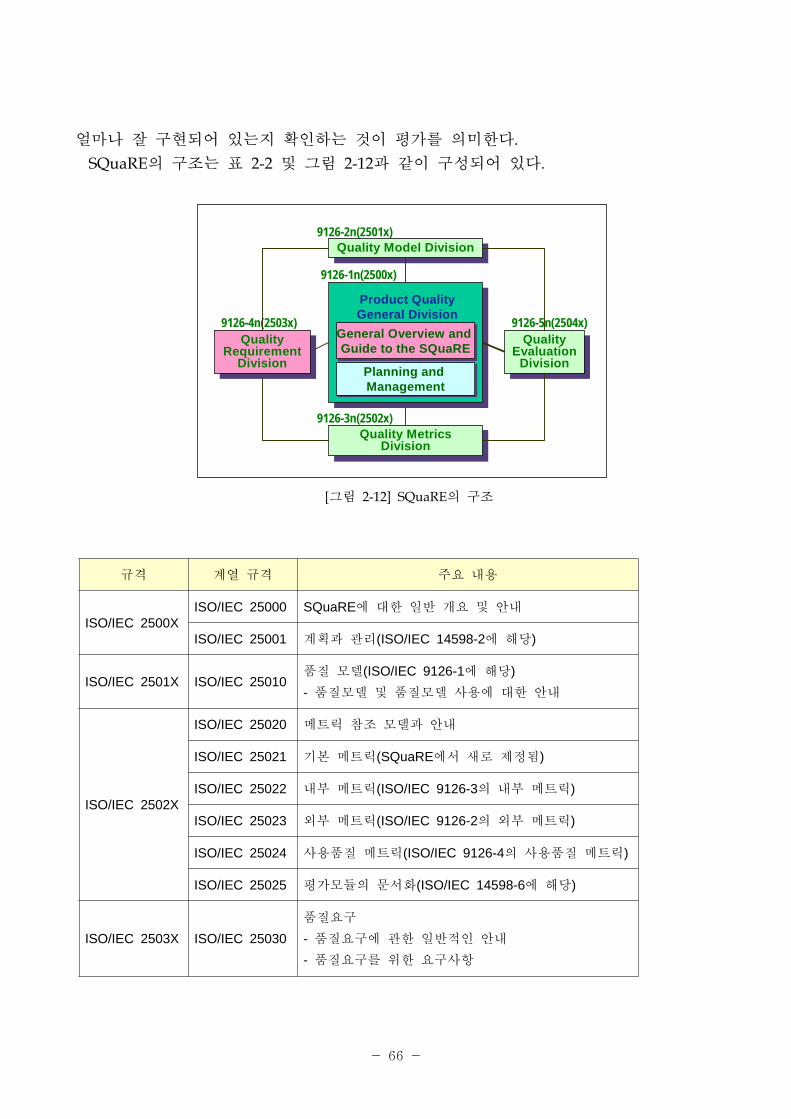
얼마나 잘 구 되어 있는지 확인하는 것이 평가를 의미한다.
SQuaRE의 구조는 표 2-2 그림 2-12과 같이 구성되어 있다.
[그림 2-12] SQuaRE의 구조
9126-2n(2501x)Quality Model DivisionQuality Model Division
Quality Evaluation
Division
Quality Evaluation
Division
Quality Metrics Division
Quality Metrics Division
9126-5n(2504x)9126-4n(2503x)
9126-3n(2502x)
9126-1n(2500x)
Planning and Management
Planning and Management
General Overview and Guide to the SQuaRE
General Overview and Guide to the SQuaRE
Product Quality General Division
Quality Requirement
Division
Quality Requirement
Division
- 67 -
[표 2-2] SQuaRE의 구조
- 사용품질 요구사항- 외부품질 요구사항- 내부품질 요구사항
ISO/IEC 2504X
ISO/IEC 25040 평가 로세스에 한 개요
ISO/IEC 25041 개발자의 로세스(ISO/IEC 14598-3을 일부 변경)
ISO/IEC 25042 구매자의 로세스(ISO/IEC 14598-4를 일부 변경)
ISO/IEC 25043 평가자의 로세스(ISO/IEC 14598-5를 일부 변경)
ISO/IEC 15504 규격은 소 트웨어의 로세서를 평가하고 개선함으로써 품질
생산성 향상을 한 규격으로, 로세스 평가 결과에 따른 부 정보다는 로 일
형태의 능력 수 을 결정함으로써 진 인 개선을 유도하고 있다는 에서 소 트웨어
제품을 평가하는데 용되는 ISO/IEC 9126, 14598과 차별화되며 보다 발 된 개념의
규격이라 할 수 있다.
ISO/IEC 15504에서는 소 트웨어 로세서 역을 ISO/IEC 12207(소 트웨어 수명
주기 로세스)에 거하여 일차(Primary) 로세스, 지원(Supporting) 로세스, 조직
(Organizational) 로세스로 구분하여 각 로세스 역별로 로세스 카테고리와 기본
로세스를 정의하고, 이들 로세스를 정립하여 수행하고 있는 수 에 따라 개발기
의 능력수 을 Incomplete, Performed, Managed Established, Predictable, Optimizing
level 등 6단계로 구분하고 있다. 한 이 규격에서는 소 트웨어 로세스 능력수
에 한 참조 모델을 제시함과 함께 참조모델을 토 로 실제로 소 트웨어 로세스를
평가하기 한 지침 심사원 자격 등에 한 사항을 명시하고 있다.
로세스 품질평가 SPICE(Software Process Improvement and Capability
dEtermination)는 소 트웨어 개발업체의 로세스 능력 평가를 하여 제정된
ISO/IEC TR 15504계열 규격의 평가 모델을 실증 으로 검증하기 한 목 으로
운 하고 있다. 재 미국, 유럽, 캐나다, 멕시코, 북아시아(한국, 일본), 남아시아 등 5개
지역별 센터(LNC)에서 소 트웨어 로세스 평가 심사자 양성업무를 수행하고
있으며 한국에는 KSPICE가 조직되어 운 에 있다. SPICE는 소 트웨어 수명주기
동안 조직의 능력 개발의 성숙성을 평가하는 내용으로 구성되어 있으며 표 2-3과
같이 6단계 수 으로 평가된다.
- 68 -
[표 2-3] SPICE 성숙도
Level 로세스 수 평가 내용
0 불완 로세스가 구 되지 않거나 그 목 을 달성하지 못함
1 실행 정의된 목 은 달성하나 활동계획의 추 불가능
2 리 로세스가 리되고 있으며 산출물을 생산함
3 확립 조직 반에 한 규격화된 로세스가 정의되어 있음
4 측가능 로세스별 정량 데이터가 리되고 일 성 있게 수행됨
5 최 화 로세스가 최 하게 수행되며 지속 인 개선 차가 확보됨
소 트웨어에 한 품질경 시스템 련 규격인 ISO 9000-3:1991은 컴퓨터 소 트웨어
제조업체에서 소 트웨어의 개발, 공 , 설치 유지보수에 한 ISO 9001:1994 (품질
경 시스템) 용을 한 계열 지침(Guideline)이었으나 ISO 9001:1994 규격이 2000년
에 면 개정됨에 따라, ISO/TC 176/SC 2에서는 이 규격을 폐지하 다. 이후
ISO/IEC JTC 1의 SC 7(소 트웨어 시스템 엔지니어링)에서 ISO 90003: 2004 규격
으로 새로이 제정하 다.
ISO 90003 규격은 조직에서 컴퓨터 소 트웨어의 획득, 지원, 개발, 작동 유지에
한 ISO 9001(품질경 시스템)의 용이한 용을 한 지침으로, 이 규격에서는 ISO
9001 요구사항 내용과 순서를 일치시켜 기술 사항, 수명주기 모델, 개발 차, 활동
순서 조직 구조 등을 언 하고 있다. 한 JTC 1/SC 7에서 발행된 특성 때문에
소 트웨어 련하여 ISO/IEC 12207, ISO/IEC 9126, ISO/IEC 12119, ISO/IEC 14598,
ISO/IEC TR 15271, ISO/IEC 15504, ISO/IEC TR 15846, ISO/IEC 15939, ISO/IEC TR
16326, ISO 10007, ISO/IEC 6592, ISO/IEC 19761, ISO/IEC 20926, ISO/IEC 20968,
ISO/IEC 14764, ISO/IEC 15026, ISO/IEC 15910, ISO/IEC 14102 규격들과 상호 연
되도록 구성하 다.
ISO 9001:2004 요구사항별 련 소 트웨어 규격간의 유용성 계를 표 2-4에 표시한다.
소 트웨어 개발 유지보수에 있어 형상 리(Configuration Management)가 많은
주목을 받고 있다. 형상 리는 1960년 에 등장하여 1970년 에 미 국방규격(Military
Standard)의 일부로 규격화되었으며 이후 지속 으로 발 하다가 1990년 들어 컴퓨
환경이 복잡해지고 품질기 이 엄격해지면서 본격 으로 용되어 1998년 11월에
ISO/IEC JTC1 SC7에서 ISO/IEC TR 15846:1998(소 트웨어 로세스에서의 형상 리)이 제
정되었다.

- 69
-
[표
2-4
] IS
O/
IEC
JT
C1/
SC
7,
ISO
/T
C 1
76
련 규
격과
IS
O 9
001
:200
0간의
이
행
련 유
용성
ISO
900
1:20
00
4 품질경시스템
4.1 일반 요구사항
XA
nnex
C
4.2 문서화 요구사항
6.1,
F.2
.1
5 경책임
5.1 경의지
5.2 고객심
5.3 품질방침
5.4 기 획
5.4.
1 품질목표
X
5.4.
2 품질경시스템기획
5.5 책임
,권한
의사소통
5.6 경검토
6 자원리
6.1 자원의 확보
6.2 인자원
6.2.
1 일반사항
F3.3
.1,
F3.3
2
- 70
-
ISO
900
1:20
00
6.2.
2 능력
,인식 교육
훈련
6.3 기반구조
7.2,
F.3
.2P
t2,
Pt3
X
6.4 업무환경
7 제품 실
7.1 제품 실
의 기획
5.2.
4, 5
.3.1
,6.
1to
6.8,
F.2
Pt
1P
t 2
6.2
6.2.
2
7.2 고객
련로세스
7.2.
1 제품
련요구사항
의 결정
5.3.
2to
5.3.
4,F.
1.3.
1,2,
4P
t 1
XX
7.2.
2 제품
련요구사항
의 검토
5.2.
1, 5
.2.6
, 6.
4.2.
1,6.
6, F
.3.1
.5
7.2.
3 고객과의사소통
5.2.
5.6,
5.2
.6,
5.2.
7,6.
6, F
.1.4
.26.
6.1,
7.3
.3,
8.2,
8.2
.3
7.3 설계
개발
F.1.
3.4,
F.1
.3.5
XX
XX
7.3.
1 설계
개발 기획
5.2.
4, 5
.3.1
6.2.
2
7.3.
2 설계
개발 입력
Pt
1
7.3.
3 설계
개발 출력
5.3.
5 to
5.3
.7
7.3.
4 설계
개발 검토
5.3.
4.2,
5.3
.5.6
,5.
3.6.
7, 6
.6.3
, F.
2.6
Anne
xA
- 71
-
ISO
900
1:20
00
7.3.
5 설계
개발 검증
5.3,
6.5
, F.
1.3,
F.2.
4
7.3.
6 설계
개발유효성
확인
5.3,
6.5
, F.
1.3,
F.2.
5P
t3,
Pt5
7.3.
7 설계
개발 변경
리
5.5.
2, 5
.5.3
, 6.
1,6.
2, F
.2.1
, F.
2.2
7.4 구매
Pt
1P
t 4
X
7.4.
1 구매 로세스
5.1,
F.1
.1P
t 3
7.4.
2 구매정보
5.1.
2, F
.1.1
.1
7.4.
3 구매제품의검증
5.1.
5, F
.1.1
.4
7.5 생산 서비스 확보
Pt
1X
XX
7.5.
1생산
서비스확보
리
5.3.
12,
5.4.
4, 5
.5,
6.3.
3, 6
.8,
F.1.
3.11
,F.
1.4.
2, F
.1.5
, F.
2.8
7.5.
2생산
서비스확보
로세스의 타당성 확인
7.5.
3 식별 추
성6,
F.2
.27
to12
X
7.5.
4 고객자산
7.5.
5 제품의 보존
7.6 모니터링
측정장치
리
- 72
-
ISO
900
1:20
00
8 측정
, 분석 개선
8.1 일반사항
7, F
.3.3
Pt2
,P
t3P
t 2
Pt
15
8.2 모니터링 측정
8.2.
1 고객만족
Pt
4
8.2.
2 내부감사
6.3,
6.7
, F.
2.3,
F.2.
7
8.2.
3 로세스 모니터링
측정
7.3.
2, 7
.3.3
, F.
3.3.
2P
t1,
Pt2
5
8.2.
4 제품 모니터링
측정
5.3,
F.1
.3P
t 1
Pt3,
Pt5
8.3 부합제품의
리6.
2, 6
.8,
F.2.
2,F.
2.8
XX
8.4 데이터의 분석
5.4
X
8.5 개선
8.5.
1 지속 개선
7.3,
F.3
.3X
8.5.
2 시정조치
6.8,
F.2
.8
8.5.
3 방조치
7.3.
2, F
.3.3
.2P
t 2

- 73 -
형상 리란 불안정한 구성요소를 가진 시스템 즉, 변경이 일어나는 형태에서의 리
기술을 의미하며, 형상 리에 해서 다수의 정의가 존재한다. IEEE Standard에서는
“형상 항목을 식별하여 기능 물리 특성을 문서화하고, 그 특성에 한 변경을 제어하고,
변경 처리 상태를 기록‧보고하고, 명시된 요구사항에 부합 여부를 확인하는 일련 사항에
하여 기술 행정 지침과 사후 리를 용하는 원칙(IEEE Standard Glossary of Software
Engineering Terminology 1991)”로 정의하고 있다. ISO/IEC 12207에서는 "형상 항목
(Configuration Item)은 특정의 기 에서 고유하게 식별할 수 있는 실체“로 ”버
(Version)은 형상 항목의 식별된 인스턴스(instance)로 규정하고 있다. 보편 으로 용되는
다른 정의는 “ 체 소 트웨어 공학 과정에 용되는 '보호(Umbrella)' 활동이다. 변경은
언제라도 일어날 수 있기 때문에, 소 트웨어 형상 리(SCM, Software Configuration
Management) 활동은 ①변경을 알아내기 해 ②변경을 제어하기 해 ③변경이 히
수행되고 있는 것을 확인하기 해 ④변경에 심을 가지고 있는 사람들에게 이것을 통보하는
것23)”이다.
이와 같은 정의에서 악할 수 있듯이 형상 리는 크게 5가지로 구분할 수 있다.
•형상 식별(configuration Identification): 형상 리의 상이 무엇인지 식별; 형상 리의
상이 되는 형상 항목의 개체를 기록하고, 기 선(Base line)으로 설정하는 활동
•형상통제(Configuration Control): 형상항목에 한 변경사항을 체계 으로 리하는
활동
•형상 감사(configuration Audit): 형상 리 활동이 제 로 수행되는지를 조사하는 활동
•상태 보고(status Reporting): 변경된 형상 항목을 기록하고, 보고하는 활동
•형상배포(Configuration Release): 형상항목을 련자 는 고객에게 통보하는 활동
형상식별 형상통제
형상상태 보고 형상배포
형상감사
형상항목
[그림 2-13] 형상 리 활동
ISO/IEC TR 15846 규격에서의 소 트웨어 형상 리(SCM)는 ISO/IEC 12207(소 트
웨어 수명주기 로세스)와 연계하여 소 트웨어 제품 수명주기에서의 CM 로세스를
지원한다. 이 규격에서 요구하는 SCM는 소 트 제품들을 포함하여 컴퓨터에 장될
수 있는 어떠한 정보도 제어가 가능하고, 한 다른 장소에 장되는 요 소 트웨어
23) Roger's S Pressman, Software Engineering; :A Practitioner's Approach, 3rd Edition(1995)

- 74 -
항목에 한 목록과 기록들을 리할 수도 있으며, 인도 가능한 소 트 제품들을 만들
거나, 유지하거나, 장(archive)하거나, 복원하는 소 트웨어 환경에서 도구(tools)로서
사용되는 소 트웨어 제품은 어떠한 소 트웨어 타입일지라도 SCM에 의한 리가
가능하도록 규정되어 있다. 다만 이 규격에서 SCM 로세스에서의 활동과 작업들의
실행 방법을 지정하고 있는 것은 아니며, 운 , 유지보수 개발 로세스의 반에
걸쳐 지속성을 제공하기 한 의도로 제정된 것이다.
ISO/IEC TR 15846의 SCM 요구사항은 운 , 유지보수 개발 로세스 내에서
가시성(visibility)과 가측성(accountability)의 개선에 을 두고 있다. SCM 요구사항
은 소 트웨어 제품 공 자에 작업을 주문하는 획득자(acquirer), 소 트웨어 제품의
인도에 책임을 지는 공 자 작업을 수행하는 외주업체 는 소 트웨어 문가로
구분될 수 있는 최소 3개의 연결고리에서 기인한 것으로, 에스크로(escrow)로 제3자
보 소 사용에 하여 획득자와 공 자가 동의하는 경우에는 4개의 연결고리 계가
존재할 수도 있다. 이러한 연결고리 하에서 SCM의 운 에 따른 이익은 다음과 같다.
•수명주기 로세스의 지원을 한, 한 문서화 자문서, 코드(Code),인터페
이스, 데이터베이스 등을 식별하고 수집하도록 하는 반복 인 체계(scheme)를 지원,
•요구사항, 기 , 방침과 지침, 조직 경 철학에 부합되는 개발, 유지보수 는 운
가동 방법론을 지원,
•기 선(baselines)의 상태, 변경, 릴리즈, 버 , 일보 (archives) 등과 련하여 리
제품 정보를 생산,
• 리될 요 개개의 항목들(items) 수 에 따라 SCIs를 순차 (recursively)으로 정의,
•그 상태 련 정보와 함께 SCIs에 장되는 라이 러리(libraries)를 제어,
•형상의 완 성(integrity)을 보증하기 한 ISO/IEC 12207 로세스를 인용,
•형상의 완 성 보증( ; 트래커(tracker),SCM 라이 러리 가디언, 릴리즈 빌더)과 그
도구들이 용( ; 운 체계)되도록, 소 트웨어 제품의 개발 검증에 사용되는
소 트웨어 도구를 포함하여, 유용한 수명 반에 걸쳐 소 트웨어 제품의 구성
(configure) 재구성이 가능하도록 소 트웨어 환경을 제어,
•소 트웨어 제품의 형상 개개의 SCIs에서의 변종/버그에 한 정보를 장 검색,
•면허(licences)나 작권과 같은 지 재산권 고려를 하여 소유권(ownership)을 기록
ISO/IEC TR 15846은 ISO/IEC 12207(소 트웨어 수명주기 로세스) ISO 10007
(형상 리를 한 품질경 지침)과 일 성을 유지한다. 표 2-5에 이 규격과 ISO/IEC
12207 ISO 10007과의 계를 표시한다.

- 75 -
ISO/IEC TR 15846 ISO/IEC 12207:1995 ISO 10007:1995
6 SCM 로세스
구
6.2.1 로세스 구 5 형상 리 로세스
6 형상 리 조직
6.1 개요
6.1 기 범 설정 5.1.1 기화 6.2 형상 리 구조
6.2 기획
5.1.2 제안에 한 요청 비 7.7 형상 리 계획
Annex A(권고) 형상 리 계획
구조 내용
5.1.3 계약 비 업데이트
5.1.4 공 자 모니터링
5.2.46.2.1
기획
로세스 구
7.1.2.1 e)
7.2 기반구조 로세스
7.3 개선 로세스
6.3 집행 통제
6.1.3.2, 6.1.4.1, 6.4.2.7 c)
6.8 문제해결 로세스
7.1.3
7.4 훈련 로세스
6.4SCM 로세스
검토 평가7.3 개선 로세스
6.5 종결 7.1.5 종결
7소 트웨어
형상식별(SCIs)
6.2.2 형상식별 5.2 형상식별
7.2 형상식별 차
7.1 SCIs의 식별 5.3.3.1
7.2 형상기 선 식별
7.3소 트웨어
라이 러리 식별
7.4 진상태
8 소 트웨어
형상 리
5.3.1.2 b) 5.3 형상 리
6.2.3 형상 리 7.4 형상 리 차
8.1 제안 변경

- 76 -
ISO/IEC TR 15846 ISO/IEC 12207:1995 ISO 10007:1995
8.2 제안된 변경 향
평가
8.3 변경의 이행
8.4 처리 의사소통 7.3
8.5 변경종결
9소 트웨어
형상상태 설명
6.2.4 형상상태 설명 5.4 형상상태 설명
7.5 형상상태 설명 차
9.1 식별 기록
9.2 추 성 변경
9.3 보고상태 설명
기록
10 소 트웨어
형상평가
5.2.6 검토 평가 5.5 형상감사
5.3.4.3 7.6 형상감사 차
5.3.9.5 b)
5.3.11.4b) 8 형상 리시스템 감사
6.2.5 형상 평가
6.4 검증 로세스
6.5 밸리데이션 로세스
6.6.1.1
6.6.3.1 c)
6.7.1.1
7.1.5 종결
7.3 개선 로세스
11소 트웨어
릴리즈 리
인도
5.2.7 승인 완료
5.4.2.1
6.2.6 릴리즈 리 인도
11.1 취 5.3.4.1 e)
11.2 장 5.5.6.1 b)
11.3 복제
11.4 포장5.4.4.35.5.5.3
11.5 인도
[표 2-5] ISO/IEC TR 15846, ISO/IEC 12207, ISO 10007과의 계

- 77 -
2. IEEE 규격 분석
컴퓨터 자분야 문가들의 참여로 구성된 IEEE의 규격들은 지나칠 정도로 상세
하게 규정되어 있다. IEEE24)(Institute of Electrical and Electronics Engineers, Inc.)기 의
성격 많은 IEEE 규격들이 이미 미국의 ANSI 규격으로 제정되어 있어 이들 규격의
분석은 미국 동향으로 분류할 수도 있으나 본 연구에서는 국제기 에 하는 것으로
가늠한다.
본 연구와 련된 IEEE의 주요 규격들은 표 2-6와 같다.
IEEE Std 730은 소 트웨어의 품질보증계획서(Software Quality Assurance Plans)에
한 규격으로, 소 트웨어의 개발, 검증 확인, 유지보수와 련된 품질보증계획서
(SQAP)의 비와 내용에 한 일 되고 수용 가능한 요구사항을 언 하고 있다.
이 규격에서 SQAP의 최소 문서화는 소 트웨어 요구사항 명세서(SRS), 소 트웨어
설계기술서(SDD), 소 트웨어검증 밸리데이션 계획서(SVVP), 소 트웨어 검증
밸리데이션 보고서(SVVR), 소 트웨어 형상 리 계획서(SCMP) 사용자 문서화 등을
요구하고 있다. SRS는 소 트웨어가 무엇을 할 것 인지에 한 서술로 외부 인터페이
스와 소 트웨어의 기본 요구사항들(기능, 수행능력, 설계 제약, 속성) 각각에 하여
명확하게 기술하고, 이 요구사항들을 시험‧검사, 분석 등으로 객 인 검증 밸리데
이션이 가능하도록 정의하고 있다. SDD는 소 트웨어가 SRS의 요구사항을 만족시키기
하여 어떻게 구성되어야 하는지 설명하는 문서이다. SDD는 데이터베이스와 내부
인터페이스를 포함하여 소 트웨어 설계의 구성요소와 하 구성요소들에 해서도
기술하여야 한다. 사용자 문서화에는 매뉴얼, 지침서와 같이 소 트웨어의 성공 인 실
행을 해 요구되는 자료와 제어입력, 입력 순서, 선택사항, 로그램 한계 다른 조
치나 항목을 명시하며, 모든 에러 메시지는 식별되고 시정조치를 설명하도록 규정되어
있다. 사용자와 직 인 상호작용이 없는 내장형(embedded) 소 트웨어의 경우는 사
용자 문서가 필요하지 않다. 이외에도 제조업체에서는 소 트웨어 개발계획서, 소 트
웨어 유지보수 매뉴얼 등을 문서화할 필요가 있다.
24) 국제 기 자기술자 회, 1884년 기 자 분야의 엔지니어 훈련을 해 설립되었으며, 1963년에 AIEE(American Institute of Electrical Engineers, formed in 1884) IRE(Institute of Radio Engineers, formed in 1912)가 통합되었다. 기‧ 자, 컴퓨터, 산 분야 등의 150개국 약 36만명의 엔지니어, 과학자 학생으로 구성된 단체이며 컴퓨터 자분야에 한 규격 제정, 세미나 회의 개최, 교육 등의 활
동을 수행.

- 78 -
IEEE std 규 격 명
730-1998 Standard for Software Quality Assurance Plans.
730.1-1995 Guide for Software Quality Assurance Planning.
828-1998 Standard for Software Configuration Management Plans.
829-1983 Standard for Software Test Documentation
830-1993 Recommended Practice for Software Requirements Specifications
982.1-1988 Standard Dictionary of Measures to Produce Reliable Software.
982.2-1988Guide for the Use of IEEE Standard Dictionary of Measures to Produce Reliable Software
983-1986 Guide for Software Quality Assurance Planning.
1008-1987 Standard for Software Unit Testing.
1012-1998 Standard for Software Verification and Validation.
1012a-1998Standard for Software Verification and Validation: Content Map to IEEE/EIA12207.1-1997
1016-1998 Recommended Practice for Software Design Descriptions.
1028-1988 Standard for Software Reviews and Audits.
1042-1987 Guide to Software Configuration Management.
1058a-1998Standard for Software Project Management Plans: Content Map to IEEE/EIA 12207.1-1997
1063-1987 Standard for Software User Documentation.
1074-1997 Standard for Developing Software life cycle Processes.
1228:1994 Standard for Software Safety Plans
1233-1998 Guide for Developing System Requirements Specifications.
[표 2-6] 주요 IEEE 규격 목록
IEEE Std 828은 소 트웨어 형상 리 계획에 한 규격으로 IEEE Std 730에서의 소
트웨어 형상 리 계획서(SCMP)와 일 된 사용을 가능하게 한다. 이와 련하여
IEEE Std 1042(소 트웨어의 형상 리)는 미국 국가규격(ANSI)으로 이미 채택되었다.
IEEE Std 828에서는 ‘형상(Configuration)’에 하여 다음과 같이 정의25)하고 있다.
(1) 컴퓨터 시스템의 배열 는 본질, 수 그리고 기능 단 들의 주요한 특성들에 의해서 정의
된 네트워크, 더 상세히 말하면 형상에는 하드웨어 형상과 소 트웨어 형상이 있다.
25) IEEE Std 828-1998의 3.1.4 참조

- 79 -
(2) 시스템 는 시스템 성분의 특별한 설명을 정의하는 구 , 설계, 그리고 요구사항.
(3) 기술 인 문서화작업에 나타나고 생산에 수행되는 하드웨어와 소 트웨어의 기능 , 물
리 특성.
표 2-7은 IEEE Std 828(소 트웨어 형상 리 계획)과 IEEE Std 1042(소 트웨어의 형
상 리)와의 상호 계를 표시한 것이다.
IEEE std 828-1998 해당 IEEE std 1042-1987 해당
1. 서론 1. 서론
― 2. 소 트웨어 리에서 SCM원칙
4. SCM계획 3. 소 트웨어 형상 리 계획
4.1 서론 3.1 서론
4.2 SCM조직 책임 3.2 리
4.3 SCM활동 3.3 SCM활동
4.3.1 형상 식별 3.3.1 형상 식별
4.3.2 형상 통제 3.3.2 형상 리
4.3.3 형상 상태의 기록 3.3.3 형상 상태의 기록
4.3.4 형상 감사 검토 3.3.4 감사 검토
4.3.5 인터페이스 제어 3.2.3 인터페이스 제어
4.3.6 외주업체/ 매자 리 3.5 공 자 리
4.4 SCM일정 3.2.4 SCM계획의 구
4.5 SCM자원 3.4 도구, 기법 방법론
4.6 SCM계획의 유지보수 2.5 SCM의 계획
5. SCM 계획 용 2.5 SCM의 계획
6. 규격에 한 합성 2.5 SCM의 계획
[표 2-7] IEEE Std 828과 IEEE Std 1042의 상호 계
IEEE 1012는 소 트웨어 검증 밸리데이션(V&V, Verification and Validation) 규격
으로 ISO/IEC 12207(소 트웨어 수명주기 로세스), IEEE 1074(수명주기 로세스
개발), IEEE/ EIA 12207.0(수명주기 로세스) 등의 규격과 일 성을 갖도록 V&V
로세스에 한 규격이다. 이러한 규격들은 모두 소 트웨어 검증 소 트웨어 밸리
데이션 계획에 한 요구사항을 규정하고, 이 요구사항들 간의 계를 설명함으로써
모든 규격을 만족하는 소 트웨어를 생산하는 제조업체들이 사용할 수 있도록 하는 것이다.

- 80 -
IEEE STD 1012는 소 트웨어 검증 밸리데이션 계획(SVVP)에 한 개념을 정의하
고 특정 활동과 련 작업에서의 검증 밸리데이션 활동을 규정하고 있다.
이 규격에서는 획득, 공 , 개발, 작동, 리 로세스 등을 포함하는 모든 소 트웨어
수명주기 로세스를 뒷받침하는 V&V 로세스 활동과 작업의 일반 골격(frames)을
구축하고, V&V 작업, 필요한 인풋과 아웃풋에 한 개념을 정의하며, 소 트웨어 완
성 수 에 상응하는 V&V 최소 작업을 표 2-8에서와 같이 4단계로 구분하여 소 트웨
어 V&V 계획(SVVP)의 개념을 정의한다.
요도 설명 수
상(High) 선택된 기능은 련 시스템의 주요 성능에 향을 미친다. 4
상(Major) 선택된 기능은 주요 시스템 성능에 향을 미친다. 3
(Moderate) 선택 기능은 시스템 성능에 향을 미치지만 효과 인 략을
이용하여 성능 상실을 보완한다. 2
하(Low) 선택된 기능은 시스템 성능에 한 향을 미치지만 그 기능
이 요구사항에 따라 이행되지 않으면 사용자에게 불편할 뿐이다. 1
[표 2-8] 소 트웨어 완 성 수 (Software integrity levels) 구분
이 소 트웨어 완 성 수 구분은 의료기기 험경 시스템에서의 심각도 분류와 유
사한 개념으로 다양한 소 트웨어 특성을 설명하기 하여 소 트웨어 완 성
(Software integrity)을 4단계의 수 으로 나 어 정의하고, 각 수 별로 필요한 V&V
최소한의 활동에 하여 언 한 것이다.
소 트웨어는 의도한 목 (용도)와 주요하거나 는 주요하지 않은 용도에 한 시스
템의 용에 기 한 다른 요도(criticality)를 갖고 있다. 일부 소 트웨어 시스템은 생
명 유지용 시스템에 한 향을 미치는 반면 다른 소 트웨어 시스템은 범용 , 독
립 인 연구용 툴이다. 소 트웨어 요도는 시스템 본래의 용도와 용을 말해 다.
이 규격에서는 소 트웨어 완 성 수 을 이용하여 소 트웨어 요도를 제한하고, 완
성 수 의 허용 한계 내에서 험(Risk)을 리하는데 필요한 소 트웨어 요도 범
를 설정하 다. 소 트웨어 속성으로는 안 성, 복잡성, 성능, 신뢰도 는 다른 특성
이 포함된다. 요도가 상(High)인 소 트웨어는 일반 으로 더 많고 더 정확한 V&V
작업의 용을 필요로 한다. 한 용된 소 트웨어 완 성 수 리스트 련
V&V 최소 작업의 지도(Map)를 SVVP에 첨부하여 어떤 활동들을 수행할 것인지 나타
내도록 한다. 다만, 규격에서 규정한 소 트웨어 완 성 수 리스트의 이용을 의무화
하지 않고 있다.

- 81 -
그림 2-14는 이러한 소 트웨어 V&V 활동에 한 개요를 표시한 것이다.
[그림 2-14] 소 트웨어 V&V 개요
소 트웨어 V&V 목 은 소 트웨어 수명주기 동안 제조업체에서 소 트웨어 품질을
구축하는데 있다. 소 트웨어 V&V 로세스에서 특정 범주의 개발 제품이 련 요구
사항을 따르고 있는지, 소 트웨어가 본래의 용도와 사용자 요구를 만족시키는지를
평가한다. 평가과정에서 소 트웨어 제품과 로세스의 심사, 분석, 평가, 검토, 조사
시험 내용을 고려한다. 소 트웨어 V&V는 소 트웨어 개발 결정 시 이 아닌, 소
트웨어 개발 시 에서 동시에 수행한다.

- 82 -
V&V 작업에 용되는 강도 정확도는 소 트웨어 완 성 수 에 따라 다르다. 여
기서 강도란 모든 정상 비정상 시스템 작동 조건에서 보다 넓은 분석 범주를 포함
한 것이며, 정확도란 더 형식 인 기술과 리코딩 차를 포함한다. 정확성, 일 성, 완
성, 읽기 쉬움 시험 가능성에 한 최소한의 기 을 포함하여 V&V 각 작업에
한 기 에는 해요인 분석(Hazard Analysis) 등의 평가를 포함한다.
V&V 로세스를 통하여 수명주기의 소 트웨어 제품과 모든 로세스를 객 으로
평가하고 이를 통하여 소 트웨어 요구사항과 시스템 요구사항의 정확성, 완 성, 일
성, 시험가능 여부를 입증하여 다음과 같은 목 을 달성한다.
• 소 트웨어 결함을 기에 검출하고 교정할 수 있도록 한다.
• 로세스와 제품 리스크에 한 경 식견을 높이도록 한다.
• 로그램 성능, 스 , 산 요구사항을 따르도록 수명주기 로세스를 뒷받침한다.
검증 로세스를 통해 소 트웨어와 련 제품이 다음과 같음을 증명한다.
• 각 수명주기 로세스(획득, 공 , 개발, 작동, 리)에서 모든 수명주기 활동에 한
요구사항( : 정확성, 완 성, 일 성 등)을 수한다.
• 수명주기 로세스 규격, 사례, 정을 만족시킨다.
• 각 수명주기 활동의 완료를 평가하고 다른 수명주기 활동을 개시하는 근거를 구축
한다.
밸리데이션 로세스를 통해 련 소 트웨어가 소 트웨어에 할당된 시스템 요구사
항을 만족시키고 문제를 바르게 해결하는지의 증거에 해서는 소 트웨어 검증 밸
리데이션 보고서(SVVR)에 기록한다.
표 2-9에 소 트웨어 검증 밸리데이션 계획의 제를 명시한다.

- 83 -
소 트웨어 V&V 랜 개요 (견본)1. 목
2. 참고 문헌
3. 개념 정의
4. V&V 개요4.1 조직4.2 마스터 스4.3 소 트웨어 무결성 수 일람표
4.4 자원 요약4.5 의무4.6 툴, 기술, 그리고 방법
5. V&V 로세스
5.1 로세스: 경5.1.1 활동: V&V의 경
5.2 로세스: 취득5.2.1 활동: 취득 지원 V&V
5.3 로세스: 공5.3.1 활동: V&V 계획
5.4 로세스: 개발5.4.1 활동: 개념 V&V5.4.2 활동: 요구사항 V&V5.4.3 활동: 설계 V&V5.4.4 활동: 이행 V&V5.4.5 활동: 테스트 V&V5.4.6 활동: 설치 체크아웃 V&V
5.5 로세스: 작동5.5.1 활동: 작동 V&V
5.6 로세스: 리
5.6.1 활동: 리 V&V6. V&V 보고에 한 요구사항7. V&V 리에 한 요구사항
7.1 외 해결과 보고
7.2 작업 반복 정책7.3 일탈 정책7.4 리 차
7.5 규격, 사례, 그리고 정8. V&V 기록에 한 요구사항
[표 2-9] SVVP 개요의 견본
1995년에 IEEE의 SESC26)(Software Engineering Standards Committee)는 ISO/IEC
12207(소 트웨어 수명주기 로세스) 국제규격 평가하고 그 규격이 IEEE 내의 수명
주기 과정에 한 근간으로 채택되도록 결정하 다. ISO/IEC 12207을 IEEE에서 채택
한 것이 IEEE/EIA 12207.0 규격이다. 이 규격은 ISO/IEC 12207 규격 요구사항을 포함
하여 개선시킨 합성 근법, 수명 주기 로세스의 구 성의 고려, 수명주기 데이터
26) 컴퓨터 학회 소트 웨어 공학 기 규격 원회

- 84 -
등과 같은 사항을 추가하여 IEEE/EIA 12207.1로 개정되었다.
IEEE/EIA 12207.1은 지침 규격이지만 구체 인 합성 요구사항을 갖는 기 규격으
로서 용에 필요한 내용도 포함하고 있다. 표 2-10은 이 규격과 ISO/IEC 12207의
계를 나타낸 것이다.
IEEE STD 1228 규격은 소 트웨어 안 성 계획서에 한 규격으로, 안 이 요한
소 트웨어의 안 성 개선을 한 과정 활동을 처리하기 하여 소 트웨어 안 성
계획서의 내용에 한 최소 허용 가능한 요구사항을 규정하고 있다. 이 규격의 취지는
소 트웨어는 시스템의 일부분이며, 시스템의 다른 부분으로 컴퓨터 하드웨어, 기타 장
치(기계 , 기 , 화학 는 방사선 련 장치를 포함) 사람들이 포함되는데, 소
트웨어 자체만으로는 안 성 문제가 있을 수 없고, 보다 더 큰 범 의 시스템이란
맥락 하에서만 문제가 된다. 따라서 소 트웨어 안 성은 시스템이란 큰 범 에서 시
작하여야 하며 소 트웨어 안 성은 반드시 련된 하드웨어, 환경 조작자라는 맥
락에서 고려할 것이다.
이 기 규격은 를 들면, 고장이 나게 되면 생명 손실, 상을 일으키거나 는 부
정 사회 충격을 확산시킬 수 있는 소 트웨어 제품과 같이 안 성이 요시되는 소
트웨어의 개발, 구매, 보 폐기에 사용되는 계획서에 용한다. 이 기 규격은
계획서가 시스템 안 성 로그램의 맥락 이내에서 작성될 것을 요구한다.
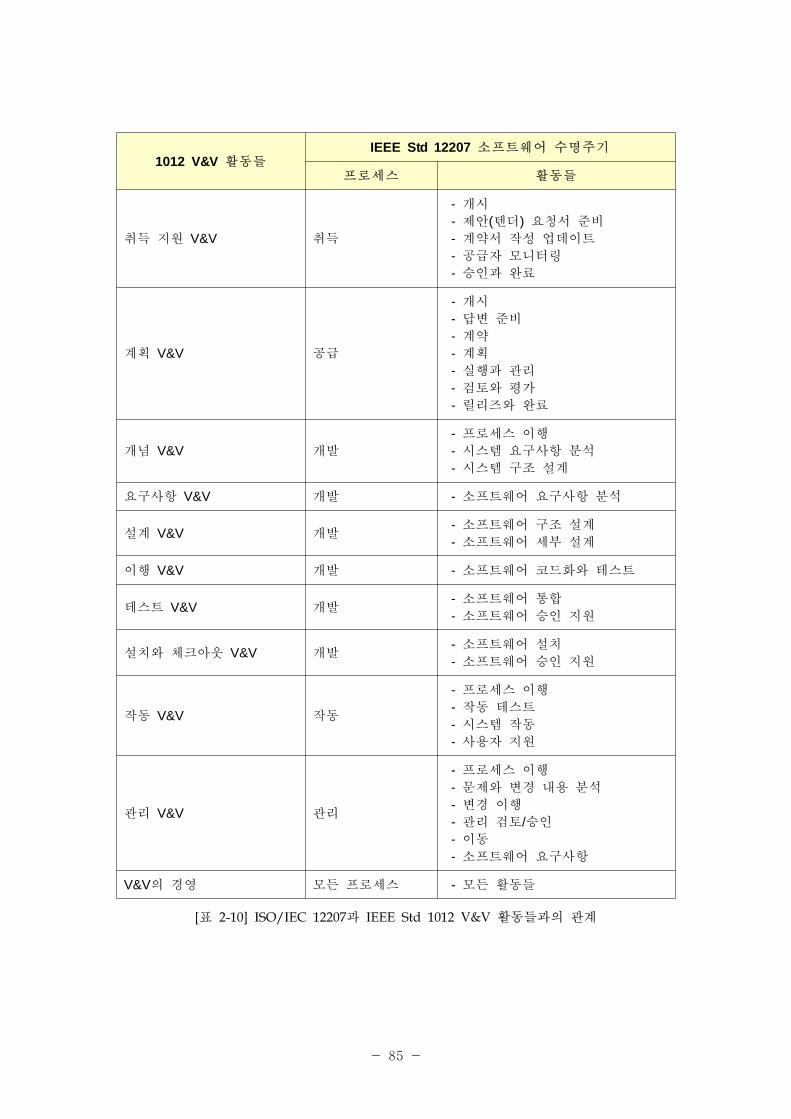
- 85 -
1012 V&V 활동들IEEE Std 12207 소 트웨어 수명주기
로세스 활동들
취득 지원 V&V 취득
- 개시 - 제안(텐더) 요청서 비 - 계약서 작성 업데이트 - 공 자 모니터링
- 승인과 완료
계획 V&V 공
- 개시 - 답변 비 - 계약 - 계획 - 실행과 리 - 검토와 평가 - 릴리즈와 완료
개념 V&V 개발
- 로세스 이행
- 시스템 요구사항 분석 - 시스템 구조 설계
요구사항 V&V 개발 - 소 트웨어 요구사항 분석
설계 V&V 개발 - 소 트웨어 구조 설계
- 소 트웨어 세부 설계
이행 V&V 개발 - 소 트웨어 코드화와 테스트
테스트 V&V 개발 - 소 트웨어 통합
- 소 트웨어 승인 지원
설치와 체크아웃 V&V 개발 - 소 트웨어 설치
- 소 트웨어 승인 지원
작동 V&V 작동
- 로세스 이행
- 작동 테스트 - 시스템 작동 - 사용자 지원
리 V&V 리
- 로세스 이행
- 문제와 변경 내용 분석 - 변경 이행 - 리 검토/승인 - 이동 - 소 트웨어 요구사항
V&V의 경 모든 로세스 - 모든 활동들
[표 2-10] ISO/IEC 12207과 IEEE Std 1012 V&V 활동들과의 계

- 86 -
[별첨 2]
ISO/IEC TR 15846:1998
정보기술 - 소 트웨어 수명주기 로세스 - 형상 리
(Medical electrical equipment - Part 1-6: General requirement for
safety-Collateral Standard: Usability)
1. 용범
이 기술 보고서에는 개발, 유지보수 운 을 한 컴퓨터소 트웨어 형상 리의
성능에 한 요구조건이 규정되어 있다. 이 기술 보고서는 ISO/IEC 12207의 형상 리
(CM) 로세스(이하 소 트웨어 형상 리(SCM) 로세스라 칭함)를 기 로 작성되었다.
이 기술 보고서는 다음에 용된다.
․모든 형태의 소 트웨어
․소 트웨어 제품 수명주기 개별 개발의 경우 그러한 수명 주기 유지보수와
운 로젝트, 하청업체나 벤더가 공 하는 소 트웨어
․소 트웨어 제품의 공 업체 구매자
이 기술 보고서는 당사자가 둘인 상황에 용되며, 두 당사자가 동일한 조직에 속할
경우 균등하게 용된다. 상황은 비공식 합의에서 공식 계약에 이르기까지 다양할 수
있다. 이 기술 보고서는 자체 부과 임무로서 단일 당사자가 사용하거나 기성품에 용
할 수 있다.
1.1 이 기술 보고서의 조정
일부 소 트웨어나 소 트웨어 수명주기에는 다른 해당 표 이나 계약에 규정한
요구조건이 용될 수 있으며, 지 실천요강이 채택될 수도 있다. SCM 로세스는
요구조건을 추가로 용함으로써 조정할 수 있다.
한 SCM 로세스는 특정 요구조건을 용할 필요가 없다고 단할 경우 이 기술
보고서의 요구조건을 무시하도록 조정할 수도 있다. ISO/IEC 12207에 따른 이 기술
보고서는 이 기술 보고서에 제공된 매핑을사용하면 쉽게 조정할 수 있다.(부속문서 A 참조)

- 87 -
1.2 로세스 역할
이 기술 보고서의 사용자는 구매자와 공 업체의 역할을 수행한다(그림 1 참조).
ISO/IEC 12207에 규정한 유지보수와 개발 로세스를 수행하는 소 트웨어 제품의
공 업체는 SCM의 구매자이다.
운 로세스의 경우 소 트웨어 제품의 인수 후 구매자는 최종 고객이나 소비자에
한 SCM 공 업체의 역할을 수행한다.
SCM 로세스(이하 "SCM 로세스"라 칭함)의 공 업체는 하청계약 는 벤더 제품
구매자의 역할을 수행한다.
2. 수성
해당 없음
3. 규격상 참고문헌
아래의 규격상 참고문헌에는 이 문서내용을 통해 이 기술 보고서의 규정을 구성하는
규정이 수록된다. 일자가 명기된 참고문헌의 경우 그러한 발간물의 후속 수정이나
개정은 용되지 않는다.
단, 이 기술 보고서에 기 한 합의에 한 당사자는 아래에 표시한규격상 참고문헌의
최신 버 을 용할 수 있는 가능성을 모색해야 한다. ISO와 IEC 구성원은 재 유효한
기술 보고서의 등록부를 유지한다.
․ISO/IEC 12207:1995, 정보기술 – 소 트웨어 수명주기 로세스
․ISO/IEC 2382-20:1990, 정보기술 – 용어 – 제20부: 시스템 개발
․ISO/IEC 2382-1:1993, 정보기술 – 용어, 제1부: 기본 용어
참조용 참고문헌은 부속문서 B에 수록되어 있다.
4. 정의
이 기술 보고서의 목 을 해 ISO/IEC 12207에 규정한 정의와 다음과 같은 정의가
용된다.

- 88 -
4.1 승인 받은 수정
SCI에 한 변경을 인가하는, 하나 이상으로 제안된 변경의 처리
참고) ‘제안된 변경’과 ‘승인된 수정’ 사이에는 다양한 계가 있을 수 있다. 제안된
변경에 의해 몇몇 SCI에 수정이 발생할 수 있다(코드와 시험 사례에만 해당되는
경우에도). 수정이 진행 인 동안 동시에 는 시간이 경과하면서 승인된 몇 가지
제안 변경에서 수정이 발생할 수 있다.
4.2 변경 권한
ISO 10007의 "형상 보드"
참고) 처리는 "변경/형상 리 보드"라고 그 동안 불렸던 지정 변경 권한에 의해 수행
된다. 이 권한에 따라 제안된 변경을 승인해 승인된 수정으로 변환하거나 제안된
변경을 승인하지 않거나 결정을 유보할 수 있다.
4.3 제안된 변경
아이디어를 기록한 시 부터 지정 변경 권한에 의한 처리까지 요구되거나 권고되는
강화가 포함되는 변경 보고서
참고 1) 처리는 거부, 후속 분석을 한 유 는 승인된다. 승인된 제안 변경은 허가
수정이 된다.
2) 제안된 변경과 허가된 수정 사이에는 일 일, 일 다자, 는 다자간 계가
있을 수 있다.
4.4 소 트웨어 형상 리(SCM)
SCI의 완벽성과 정확성 확보를 해 소 트웨어 수명주기 반에 걸쳐 형상 리를
용하는 로세스(ISO 10007 참조)
4.5 소 트웨어 라이 러리
개발, 운 유지보수 지원을 해 제어된 방식으로 수집한SCI 집합체
4.6 소 트웨어 툴
소 트웨어 수명주기 임무를 한 자동 지원을 제공하는 소 트웨어 제품
참고) 소 트웨어 툴에는 작성자의 지원 여부에 계없이 벤더 소 트웨어 자체
개발 툴이 포함된다. 툴에는 운 체제가 실행하는 소 트웨어와 운 체제 자체가
포함된다. 한 툴에는 매크로, 시험 스크립트 는 빌드 설명서와 같은 해석한
로그램이 포함된다.

- 89 -
5. 기호(약어 포함)
5.1 약어와 두문자
아래의 약어와 두문자가 이 기술보고서에 사용된다.
․CI : 형상 항목
․CM : 형상 리
․SCI : 소 트웨어 형상 항목
․SCM : 소 트웨어 형상 리
6. SCM 로세스 구
ISO/IEC 12207: 1995
6.2.1 로세스 구 . 이 활동은 다음과 같은 업무로 구성된다.
6.2.1.1 형상 리 계획을 개발해야 한다. 계획에는 다음에 한 설명을 포함해야 하다.
형상 리 활동, 그러한 활동의 수행을 한 차와 일정, 활동의 수행을 담당하는 조직,
그러한 조직과 소 트웨어 개발이나 유지보수를 담당하는 다른 조직과의 계. 계획을
문서화하고 구 해야 한다.
참고) 계획은 시스템 형상 리 계획의 일부일 수도 있다.
SCM 로세스는 운 , 유지보수 는 개발 로세스를 한 소 트웨어 수명주기
체 는 일부에 용해 구 된다.
6.1 개시 범 의 정의
6.1.1 SCM 로세스에 한 입력의 정의
SCM 로세스는 SCM 요구조건을 입력으로서 확보하고, SCM 요구조건이 완벽하며
쉽게 이해할 수 있는 것인지 확인해야 한다. 이러한 SCM 요구조건은 다음과 같다.
a) SCM 로세스의 일부가 되는 소 트웨어 제품
SCM 로세스가 SCM 계획에 규정한 것처럼 수행되는지 여부의 입증이나 보증
b) SCM 로세스의 소 트웨어 환경
소 트웨어 제품에 구매, 고객 공 , 하청계약에 따른 개발 는 벤더 SCI가 있을 경우
SCM 로세스는 외부 개발 SCI의 식별, 변경 제어, 상태 설명 형상평가("형상 심사"
라고도 함)를 수행해야 한다.

- 90 -
6.1.2 SCM 로세스에 하 자원과 제약사항의 정의
SCM 로세스는 SCM 활동이 다음의 정의에 의해 구 되는 기술 리 측면의
조직 상황을 구축해야 한다.
a) 기 으로 지정되는 SCI의 향을 받으며, SCM 활동에 참여하거나 그러한 활동을
담당하는 조직 단
b) 그러한 조직 단 에 한 SCM 역할과 책임
c) 조직 단 , 구매자 공 업체 사이의 계
SCM 로세스는 조직, 활동, 업무, 차, 정보와 보고서 계획입안을 한 형식
자원을규정하는 문서를 확립해 유지해야 한다.
SCM 로세스는 SCM 차, 표 , 용어 련 문서에 한 참조를 식별해야 한다.
6.1.3 책임과 권한의 할당
SCM 로세스는 SCM에 필요한 자원을 계획하고 획득하며 채택해야 한다.
SCM 로세스는 다음을 수행할 수 있는 권한과 능력이 있는 조직 단 에 SCM
활동을 할당해야 한다.
a) 기 의 확립
b) 기 의 변경에 한 승인/비승인
c) 소 트웨어 제품의 릴리즈
d) SCM 요구조건과의 편차에 한 승인/비승인
SCM 로세스는 통합 연락처를 식별하고 지명해야 한다.
SCM 로세스는 기 변경의 승인을 취득하기 한 기 을 결정해야 한다.
SCM 로세스는 권한의 변경을 식별하고 권한의 범 를 할당해야 한다.
6.1.4 SCI 선정을 한 기
SCM 로세스는 소 트웨어 제품에 기 해 SCI를 선정하기 한 다음과 같은 기 을
확립해야 한다.
a) 필수
b) 소 트웨어 환경에 의한 사용
c) 유도를 한 툴의 설명과 매개변수를 포함해 릴리즈의 유도에 사용

- 91 -
SCM 로세스는 SCI의 성능 매개변수와 물리 특성의 리에 충분한 SCI 선정을
한 기 을 정의해야 한다.
참고) SCM 로세스는 무 많은 SCI를 선정해 리의 가시성을 훼손하고 비용을
증가시키지 않도록 해야 한다.
6.1.5 SCM 로세스 결과의 정의
필요한 경우 SCM 로세스는 다음의 결과의 달을 계획해야 한다.
a) 소 트웨어 환경의 운 을 한 정보
b) SCI 식별 계획
c) SCI의 재작성을 한 툴과 소 트웨어 환경
d) SCI 버 제어를 한 계획
e) SCI 구조를 입증하는 문서
f) SCI 상태의 의미
g) SCI의 상태
h) SCI 상태의 완 성
I) SCI
6.2 계획입안
특정 소 트웨어 제품의 경우 SCM 로세스는 SCM 구 에 향을 주는 소 트웨어
수명주기 주요일정이나 안( 를 들어 인터페이스 제어의 도입)에 한 SCM 활동의
의존성을 계획해야 한다.
SCM 활동을 수행하거나 상호작용하는 조직의 표는 SCM 계획입안 정보를 검토하고
승인해야 한다.
필요한 경우 SCM 로세스는 SCM 로세스를 달하고 SCM 소 트웨어 제품을
개선해야 한다.
SCM 로세스는 필요한 경우 SCM 활동의 단을 계획해야 한다.
SCM 로세스는 변경의 반 을 해 SCM 계획입안 정보를 갱신해야 한다. SCM
로세스는 변경된 SCM 업무를 수행하기 그러한 업무를 검토하고 해당 계자의
승인을 받아야 한다.

- 92 -
아래의 정보를 SCM 계획에 포함하거나 참조해야 한다.
a) 계약의 식별
b) 지정한 소 트웨어 수명주기 로세스에 한 SCM 지원의 범
c) 소 트웨어 제품이 인도되었는지 확인
d) 후속 유지보수에 필요하거나 c)에 식별한 항목의 완 성에 향을 주는 다른
소 트웨어 제품의식별
e) 조직 정의 상호 계
f) 역할과 책임
g) 필요한 자원의 목록 자원이 필요한 시
h) 하청업체 SCM 제어를 포함한활동의 인터페이스를 한 하드웨어나 시스템 CM
차와 SCM과의 계
i) 형식, 일정 배포를 포함해 상태 보고의 차
j) 변경 제안 SCI 변경과 개선을 해 할당한 권한을 포함해 변경을제어하는 차
k) 유지해야 할 버 의 번호를 포함해 이 버 지원에 한 정책
l) 다 버 으로 개발 고객 지원
m) 기 확인의 검토
n) 용할 SCM 로세스의 완 성 확인을 한 심사
o) SCM 로세스가 필요한 업무를 제공할 수 없어 SCM 업무의 원가, 일정 는
성능에 향을 주는 험
p) 릴리즈 리와 인도 차
q) 인터페이스 제어
6.3 실행 제어
SCM 로세스는 담당자가 SCM 계획에 규정한 SCM 업무를 수행하기에 충분한
시간으로 한 툴, 장치 훈련을 소 트웨어 환경에 제공해야 한다. SCM 로세스는
SCM 계획에 기록된 SCM 업무를 수행해야 한다.
6.4 SCM 로세스의 검토와 평가
SCM 로세스는 SCM 업무가 SCM 계획에 부합하는지 확인해야 한다. SCM 로세스는
문제해결과 로세스 개선 로세스와 같은 로세스를 수행해 SCM 계획과의 차이를
해결해야 한다.
6.5 마감
SCM 로세스는 필요한 경우 SCM 활동을 종료해야 한다.

- 93 -
7. 소 트웨어 형상식별
ISO/IEC 12207: 1995
6.2.2 형상 식별 이 활동은 다음과 같은 업무로 구성된다.
6.2.2.1 로젝트를 해 제어해야 할 소 트웨어 항목 버 의 식별을 한 계획을
확립해야 한다. 각 소 트웨어 항목과 버 의 경우 다음의 내용을 식별해야 한다. 기 을
확립하는 문서, 버 참조번호 기타 식별 내용이 그것이다.
SCM 로세스는 SCI와 기 으로서 제어되는 소 트웨어 제품에 한 식별 계획을
확립해야 한다.
7.1 SCI 식별
SCM 로세스는 각 SCI에 고유 식별번호를 부여해야 한다. SCM 로세스는 SCI
사이의 계를 문서화해야 한다.
SCM 로세스는 SCI 개발, 제어, 빌드, 확인, 부하 재작성에 사용하는 도구에 고유
식별번호를 부여해야 한다.
7.2 소 트웨어 형상 기 의 식별
SCM 로세스는 다음과 같은 에서 각 기 을 식별해야 한다.
a) 모든 기성제품 공 업자에게 사용권은 있지만 소유권은 없는 독 항목을
포함해 각 기 에서 제어되는 SCI
b) SCI를 기 에 입력하기 해 사용하는 차
c) 기 을 완벽하게 구성하고 확립하기 한 차
d) 기 정의에 필요한 소 트웨어 제품과 기록
e) 기 승인에 필요한 차
f) 기 승인에 필요한 권한
g) 기 구축을 한 툴
참고) 의 b)와 c) 차에는 확인 로세스를 사용해야 한다.
7.3 소 트웨어 라이 러리의 식별
SCM 로세스는 다음을 포함해, 고유의 이름이 부여되고 제어되는 소 트웨어 라이
러리를 식별해야 한다.
a) 치

- 94 -
b) 각 라이 러리에 한 매체
c) 동등한 내용 유지를 한 동일한 라이 러리와 메커니즘의 수
d) SCI 에서의 내용
e) SCI 상태 에서의 내용
f) 소 트웨어 라이 러리 내용과 호환되는 최소한의 상태를 포함해 SCI를 입력할
수 있는 조건
g) 효과 인 복구 차와 함께 악의 우발 해와 기능 하를 방지하기 한 규정
h) SCI의 검색, 복사나 제거를 하지 않은 상태의 사용과 복사를 한 상태의 사용
사이의 구별 SCI 제거를 한 조건
i) 각 라이 러리에 SCI 입력, 라이 러리에 수록된 목록과 내용의 확인, 각 라이
러리의 SCI 평가 복사 삭제라는 각 기능을 발휘하도록 허가된 담당자 그룹이나
유형에 한 근에 한 규정
7.4 개선 상태
SCM 로세스는 각 SCI 기 에 한 상태를 확립해야 한다.
SCM 로세스는 형상이 통제되는 각 SCI 기 의 개선을 명시해야 한다.
참고) SCM 로세스는 특히 "개방"과 "폐쇄"라는 의미에 있어 제안된 변경의 상태를
식별해야 한다.
8. 소 트웨어 형상통제
ISO/IEC 12207: 1995
6.2.3 형상통제: 이 활동은 다음과 같은 업무로 구성된다.
6.2.3.1 다음을 수행해야 한다. 변경 요청의 식별과기록, 변경 분석과 평가, 요청의
승인이나 비승인 수정된 소 트웨어 항목의 구 , 확인과 릴리즈가 그것이다. 각 수정,
수정의 이유 수정의 승인을 추 할수 있도록 심사 결과가있어야 한다. 안 과 보안에
요한기능을 취 하는 제어된 소 트웨어 항목에 한 모든 근을 통제하고 심사해야
한다.
SCM 로세스는 SCI 완 성, 기 SCI와 기 을 정의하는 소 트웨어 제품의
유지를 한 차를 확립하고 유지하며 운 해야 한다.

- 95 -
8.1 변경 제안
SCM 로세스는 변경 제안을 수하고 기 SCI에 한 변경을 처리해야 한다.
형상 통제에 따라 SCI 기 에 해 제안한 변경을 식별하고 기록하며, 승인/비승인
하고 추 해야 한다.
8.2 제안된 변경 향의 평가
SCM 로세스는 를 들어 유지보수 로세스를 사용해, 제안된 변경의 향 평가를
지원해야 한다.
SCM 로세스는 다음을 식별해야 한다.
a) 제안된 변경의 향을 받는 SCI 련 기
b) 식별한 SCI나 기 에 향을 주는 승인된 수정
SCM 로세스는 문제해결 조치를 한 로그램의 복제나 확인을 해 SCI를 구성
해야 한다.
SCM 로세스는 문제해결이나 로세스 개선 로세스를 사용해 강화에 한 편차,
오해 는 제안의 발생 원인을 추 하고 기록해야 한다.
8.3 변경의 구
SCM 로세스는 승인된 각 수정에 한 활동과업무의 순서를 기록해야 한다.
SCM 로세스는 승인된 수정만 기 에 포함되는지 확인해야 한다.
8.4 처리의 통보
SCM 로세스는 처리에 향을 받는 모든 계자에게 통보함으로써 제안된 각
변경의 승인, 비승인 는 유 에 해 지정된 변경권한 의사결정을 지원해야 한다.
주 1. 결정이 유 될 경우 SCM 로세스는 발의자에게 다시 검토를 진행하는 방법을
알려주어야 한다. 변경이 승인된 경우 SCM 로세스는 승인된 변경과 유 된
변경을 해당 SCI를 사용하는 계자에게 통보해야 한다.
주 2. 승인되지 않는 경우 SCM 로세스는 계자에게 제안된 변경을 무시할 것을
통보해야 한다.

- 96 -
8.5 변경의 마감
SCM 로세스는 승인된 수정에 따라 새로운 기 이 발생하는지 확인해야 한다.
9. 소 트웨어 형상 상태 설명
ISO/IEC 12207: 1995
6.2.4 형상상태 설명. 이 활동은 다음과 같은 업무로 구성된다.
6.2.4.1 기 을 포함해 제어된 소 트웨어 항목의 상태와 이력을 나타내는 리 기록과
상태 보고서를 작성해야 한다. 상태 보고서에는 로젝트에 한 변경의 숫자, 최신
소 트웨어 항목 버 , 릴리즈 식별자, 릴리즈의 숫자 릴리즈의 비교를 포함해야 한다.
구성 상태 설명 활동은 다른 SCM 로세스 활동의 기록을 수행한다.
9.1 식별번호의 기록
SCM 로세스는 새 SCI 수정한 SCI의 식별번호와 상태를 기록해야 한다.
SCI가 형상 통제될 경우 SCM 로세스는 각 후속 개선에 있어 버 과 상태를 유지
해야 한다.
9.2 변경 추
SCM 로세스는 제안된 변경의 상태와 승인된 수정의 구 상태를 추 , 기록
보고해야 한다.
참고 1. 제안된 변경의 추 은 비승인의 공식 통보나 하나 이상의 SCI 수정을 한
요구조건의 발표 는 발의자에 의한 철회를 통해 아이디어를 처음 통보한
시 부터 시작해야 한다.
2. 승인된 수정의 추 은 기 에 포함시키도록 하나 이상의 SCI를 수정해야 하는
요구조건 발표 후 시작해야 한다.
9.3 상태 설명 기록의 보고
SCM 로세스는 다음을 보고해야 한다.
a) 소 트웨어 제품의 구조
b) 수취인에 한 요도 수 에서 각 SCI의 상태
c) 제안된 변경의 상태
d) 승인된 수정 기 버
e) 릴리즈의 식별번호

- 97 -
SCM 로세스는 기 의 재 과거 버 에 한 상태를 보고해야 한다.
SCI에 확인된 편차가 있을 경우 SCM 로세스는 다음을 수행해야 한다.
a) 그러한 조건을 보고한다.
b) SCI를 식별한다.
c) 사후 향을 설명한다.
d) 일시 인 해결책을 제공한다.
필요한 경우 SCM 로세스는 해결되지 않은 수정, 편차 는 포기를 보고해야 한다.
참고. 소 트웨어 제품이 문제해결 로세스를 사용해 구매자에게 양보, 편차 는
포기의허용을 요청할 경우 SCM 로세스는 그 어떤 양보가 허용되는지
보고해야 한다.
소 트웨어 제품에 구매하거나 구매자가 공 한 제품이 포함될 경우 SCM 로
세스는 소유권의 추 성( 를 들어 라이선스 매와 작권)을 보고해야 한다.
10. 소 트웨어 형상평가
ISO/IEC 12207: 1995
6.2.5 형상평가 이 활동은 다음과 같은 업무로 구성된다.
6.2.5.1 다음을 단하고 확인해야 한다. 소 트웨어 시스템의 요구조건과 물리
완벽성과 비교한 소 트웨어 항목의 기능 완벽성(설계와 코드가 최신 기술을 반
하는지의 여부)
SCM 형상평가는 다음을 결정한다.
a) SCM 기록에 해당되는 제어된 라이 러리에 SCI가 장되어 있는지의 여부
b) SCI의 상태 SCI가 구축되는 승인된수정과 련해 소 트웨어 제품이
완벽하고 사용할 수 있다.
c) 기 SCI가 련 SCI 승인된 각 수정으로 구성된다.
SCM 로세스는 확인 심사 로세스를 지원해평가 인 SCI, 기 소 트웨어
제품의 완벽성을 확인해야 한다.
SCM 로세스는 형상평가를 수행해 기 을 구성하는 SCI가 안 하게 장되었는지
단해야 한다.

- 98 -
SCM 로세스는 형상평가 결과를 보고해야 한다.
편차가 악될 경우 SCM 로세스는 문제해결 는 로세스 개선 로세스를
수행해야 한다.
11. 소 트웨어 릴리즈 리 인도
ISO/IEC 12207: 1995
6.2.6 소 트웨어 릴리즈 리 인도. 이 활동은 다음과 같은 업무로 구성된다.
6.2.6.1 소 트웨어 제품과 문서의 릴리즈와 인도는 공식 으로 통제해야 한다. 코드와
설명서의 마스터복사본은 소 트웨어 제품 수명 동안 유지해야 한다. 안 보안에
요한기능이 수록된 코드와 설명서는 련 조직의 정책에 따라 취 , 보 , 포장
인도해야 한다.
SCM 로세스는 승인된 복수의 수정을 조정하고 완벽성과 정확성을 확립하며, SCI를
다시 구성하고 소 트웨어 제품을 달하기 한 차를 확립, 유지 수행해야 한다.
11.1 취
SCM 로세스는 릴리즈 리 인도에 한 모든 입력과 출력을 제어해야 한다.
SCM 로세스는 기 라이 러리에서 발표된 SCI를 이 버 이 필요한수 으로
유지되는 범 까지 미래에 다시 구성할 수 있는지확인해야 한다.
SCM 로세스는 소 트웨어 환경을 다시 구축할 수 있어야한다.
참고. 기 을 도출하는 차는 기 의 일부로 간주해야 한다. 소 트웨어 툴의 운 이나
소 트웨어 툴에 한 수정을 한 설명과 매개변수는 소 트웨어 툴의 일부로
다시 확립해야 한다.
SCM 로세스는 기 소 트웨어 라이 러리 소 트웨어 환경을 유지해야 한다.
11.2 보
SCM 로세스는 사용 매체나 라이 러리에 계 없이 보 된 SCI의 완 성을 다음을
수행함으로써 확인해야 한다.

- 99 -
a) 재발 오류나 성능 하의 최소화를 한 장 매체를 선택한다.
b) 매체의 장 수명에 합한 주기로 장된 SCI를 활용하고 새로 고친다.
c) 손실 험의 최소화를 해 제어된 치에 복사본을 이 으로 장한다.
재사용 SCI의 경우 SCM 로세스는 다음을 수행해야 한다.
a) 공공 이름을 사용한다.
b) 장 방법, 치 시 을 지정한다.
c) SCI의 사용을 담당하는 조직을 식별한다.
11.3 복제
복제는 복사본을 만들어 소 트웨어를 제조하는 단계이다.
SCM 로세스는 일 성 있고 완벽한 복제의 보증을 한 차를 확립해야 한다.
SCM 로세스는 릴리즈를 한 매체에 이질 항목( 를 들어 소 트웨어 바이러스나
데모에 부 합한 시험 데이터)이 없는지 확인해야 한다.
SCM 로세스는 합한 매체를 사용해 소 트웨어 제품이 제 로 복제되는지 확인
해야 한다. 매체는 공 품의 상 수명 동안 내용의 완 성을 보존할 수 있도록 선정
해야 한다.
11.4 포장
SCM 로세스는 인도된 매체가 승인된 차에 의해 비된 것인지 확인해야 한다.
SCM 로세스는 구매자가 확인할 수 있는 릴리즈의 식별번호를 명확히 표시해야 한다.
참고 1. 물리 매체( 를 들어 CD-ROM이나 자기 테이 )를 사용해 릴리즈할 경우
마크는 릴리즈가 포함된 매체에 표시해야 한다( 구 용기 포함). 자 릴리즈의
경우( 를 들어 운 라이 러리에 다운로드) 마크는 릴리스 안에 표시해야
한다.
2. SCM 로세스는 소 트웨어 제품과 함께 포장하는 다른 자료( 를 들어 라이
선스 계약서와 작권 명세서)를 포함해야 하며 복사본을 SCI에 장해야 한다.

- 100 -
11.5 인도
SCM 로세스는 인도 차를 따라야 한다.
12. 인터페이스 제어
SCM 로세스는 인터페이스( 를 들어 하드웨어, 시스템 소 트웨어, 지원 소 트웨어,
기성제품 병렬/동시 개발로 제작한 소 트웨어) 문서를 식별하고 제어해야 한다.
인터페이스는 상호 합의로 조정할 수 있으며( 를 들어 소 트웨어 통합자와 하청업체
소 트웨어 개발자) 어느 한 당사자가 지정할 수 있다( 를 들어 라이선스 사용자가
제품을 복제할 수 있는 기성 소 트웨어의 공 업체).
SCM 로세스는 다음을 식별해야 한다.
a) 인터페이스의 목
b) 인터페이스에서의 요구조건
c) 해당 조직
d) 인터페이스 되어 제어해야 할 문서
e) 인터페이스에 향을 주는 제안된 변경을 다른 계자에게 통보하고, 합동으로
는 별도로 인터페이스 체의 향 평가를 수행하는 차
f) 인터페이스 변경 권한을 포함해 승인, 변경 릴리즈할 인터페이스 문서의 차
g) 인터페이스 문서의 변경을다른 SCI에 한 변경으로 변환하기 한 차
h) 역할과 책임
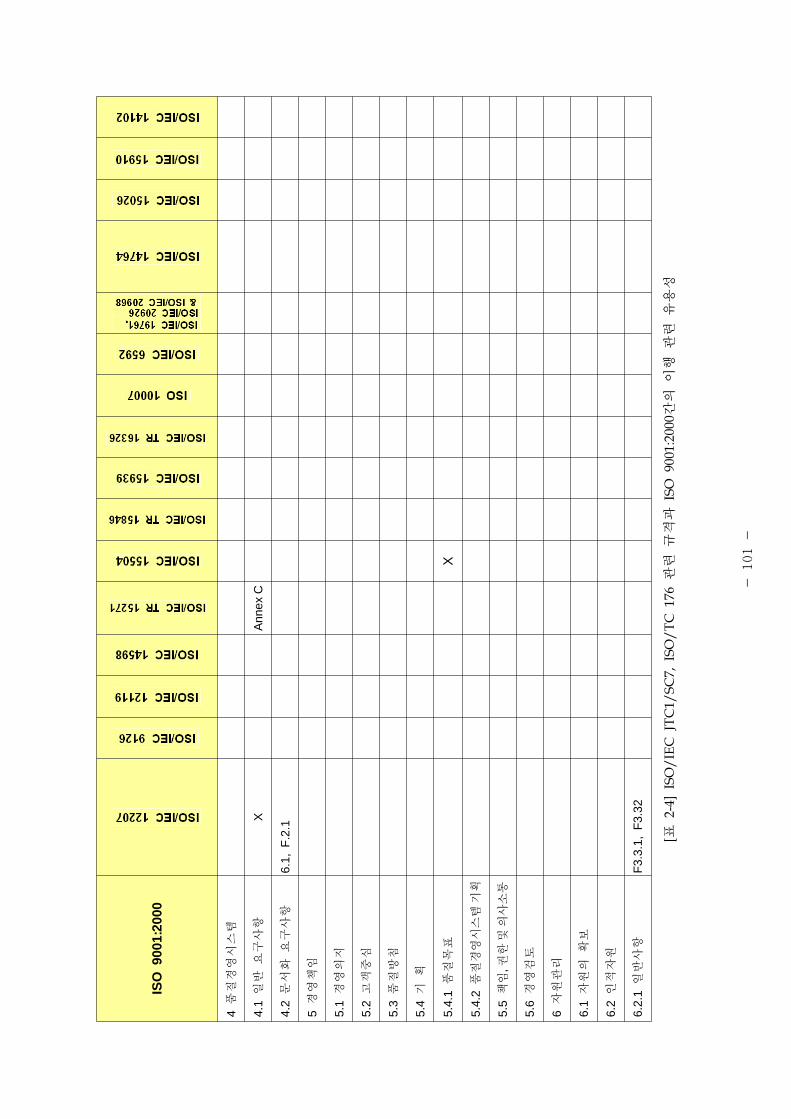
- 101
-
[표
2-4
] IS
O/
IEC
JT
C1/
SC
7,
ISO
/T
C 1
76
련 규
격과
IS
O 9
001
:200
0간의
이
행
련 유
용성
ISO
900
1:20
00
4 품질경시스템
4.1 일반 요구사항
XA
nnex
C
4.2 문서화 요구사항
6.1,
F.2
.1
5 경책임
5.1 경의지
5.2 고객심
5.3 품질방침
5.4 기 획
5.4.
1 품질목표
X
5.4.
2 품질경시스템기획
5.5 책임
,권한
의사소통
5.6 경검토
6 자원리
6.1 자원의 확보
6.2 인자원
6.2.
1 일반사항
F3.3
.1,
F3.3
2
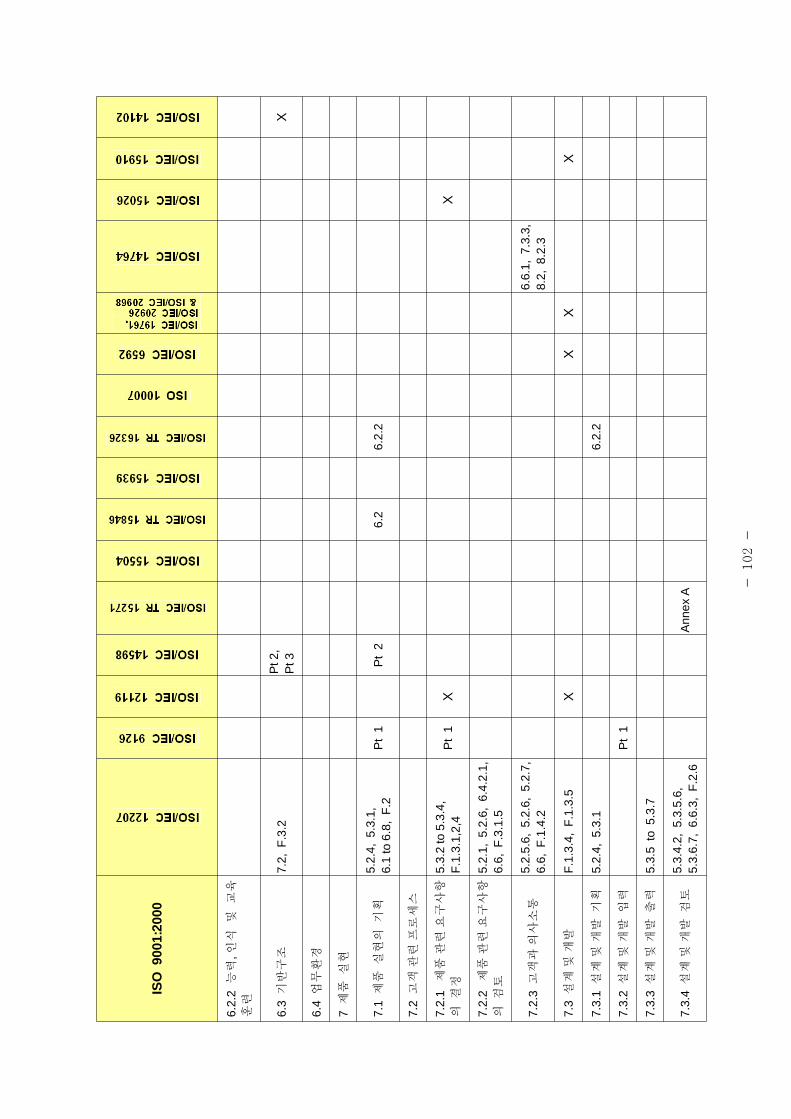
- 102
-
ISO
900
1:20
00
6.2.
2 능력
,인식 교육
훈련
6.3 기반구조
7.2,
F.3
.2P
t2,
Pt3
X
6.4 업무환경
7 제품 실
7.1 제품 실
의 기획
5.2.
4, 5
.3.1
,6.
1to
6.8,
F.2
Pt
1P
t 2
6.2
6.2.
2
7.2 고객
련로세스
7.2.
1 제품
련요구사항
의 결정
5.3.
2to
5.3.
4,F.
1.3.
1,2,
4P
t 1
XX
7.2.
2 제품
련요구사항
의 검토
5.2.
1, 5
.2.6
, 6.
4.2.
1,6.
6, F
.3.1
.5
7.2.
3 고객과의사소통
5.2.
5.6,
5.2
.6,
5.2.
7,6.
6, F
.1.4
.26.
6.1,
7.3
.3,
8.2,
8.2
.3
7.3 설계
개발
F.1.
3.4,
F.1
.3.5
XX
XX
7.3.
1 설계
개발 기획
5.2.
4, 5
.3.1
6.2.
2
7.3.
2 설계
개발 입력
Pt
1
7.3.
3 설계
개발 출력
5.3.
5 to
5.3
.7
7.3.
4 설계
개발 검토
5.3.
4.2,
5.3
.5.6
,5.
3.6.
7, 6
.6.3
, F.
2.6
Anne
xA
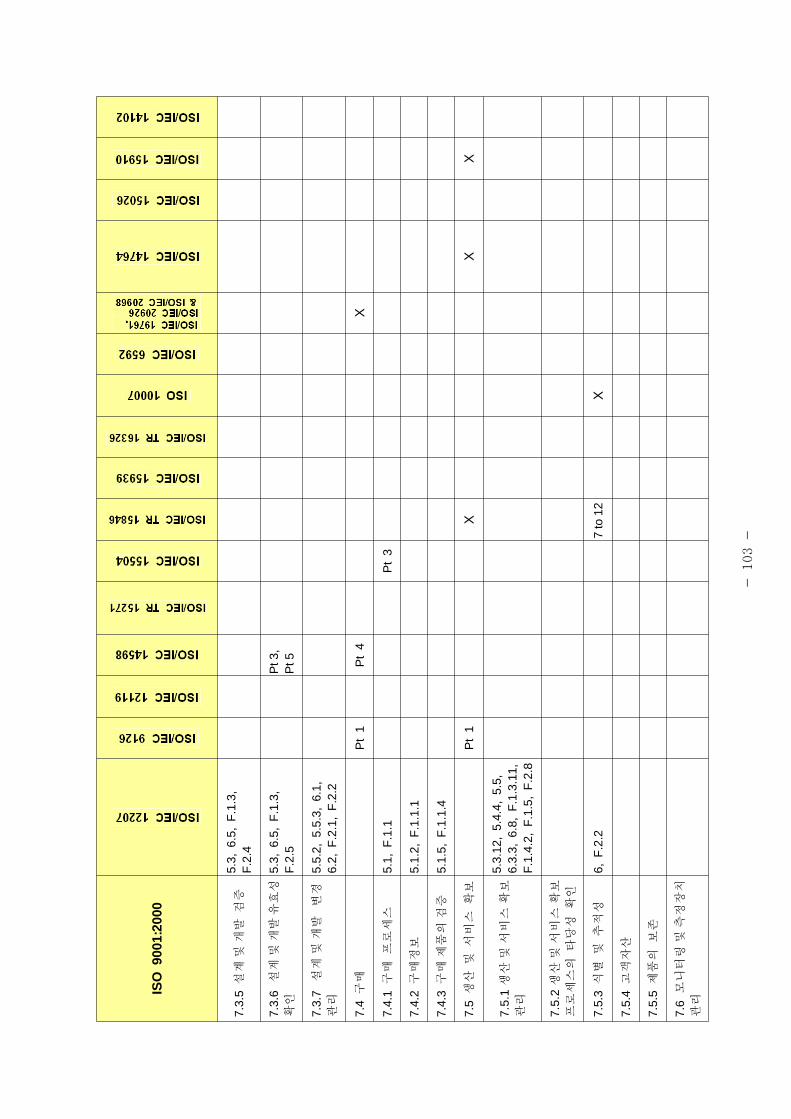
- 103
-
ISO
900
1:20
00
7.3.
5 설계
개발 검증
5.3,
6.5
, F.
1.3,
F.2.
4
7.3.
6 설계
개발유효성
확인
5.3,
6.5
, F.
1.3,
F.2.
5P
t3,
Pt5
7.3.
7 설계
개발 변경
리
5.5.
2, 5
.5.3
, 6.
1,6.
2, F
.2.1
, F.
2.2
7.4 구매
Pt
1P
t 4
X
7.4.
1 구매 로세스
5.1,
F.1
.1P
t 3
7.4.
2 구매정보
5.1.
2, F
.1.1
.1
7.4.
3 구매제품의검증
5.1.
5, F
.1.1
.4
7.5 생산 서비스 확보
Pt
1X
XX
7.5.
1생산
서비스확보
리
5.3.
12,
5.4.
4, 5
.5,
6.3.
3, 6
.8,
F.1.
3.11
,F.
1.4.
2, F
.1.5
, F.
2.8
7.5.
2생산
서비스확보
로세스의 타당성 확인
7.5.
3 식별 추
성6,
F.2
.27
to12
X
7.5.
4 고객자산
7.5.
5 제품의 보존
7.6 모니터링
측정장치
리
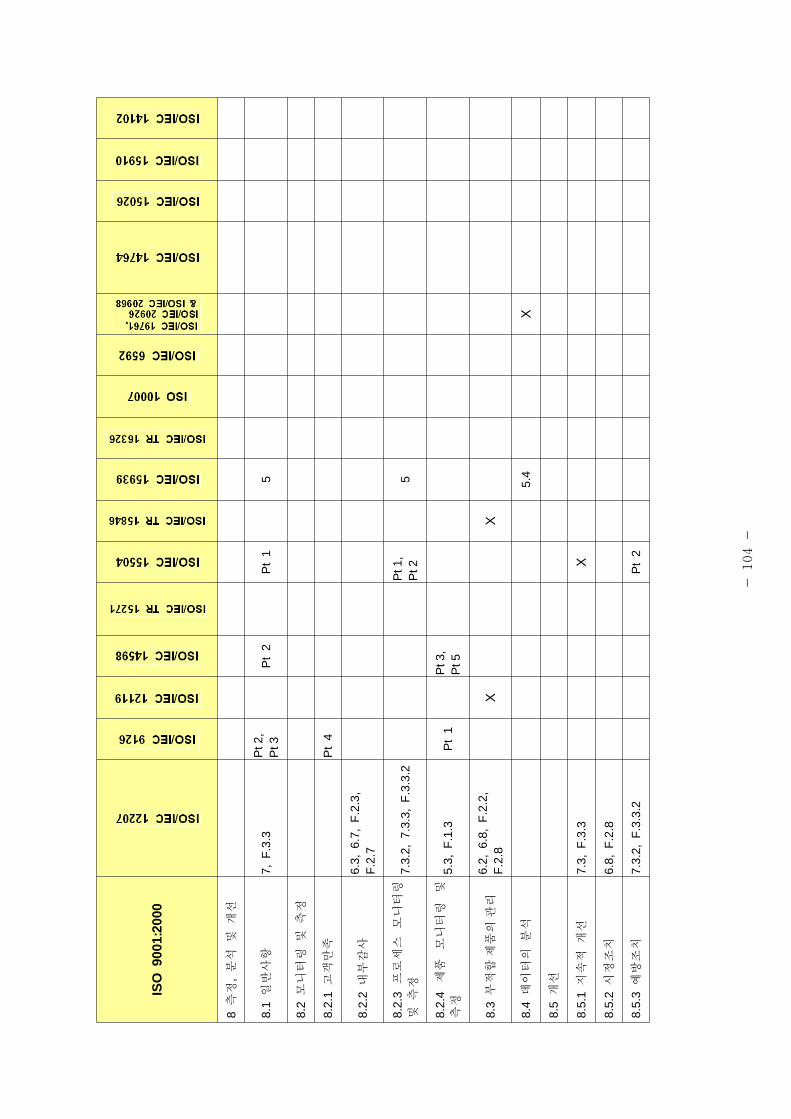
- 104
-
ISO
900
1:20
00
8 측정
, 분석 개선
8.1 일반사항
7, F
.3.3
Pt2
,P
t3P
t 2
Pt
15
8.2 모니터링 측정
8.2.
1 고객만족
Pt
4
8.2.
2 내부감사
6.3,
6.7
, F.
2.3,
F.2.
7
8.2.
3 로세스 모니터링
측정
7.3.
2, 7
.3.3
, F.
3.3.
2P
t1,
Pt2
5
8.2.
4 제품 모니터링
측정
5.3,
F.1
.3P
t 1
Pt3,
Pt5
8.3 부합제품의
리6.
2, 6
.8,
F.2.
2,F.
2.8
XX
8.4 데이터의 분석
5.4
X
8.5 개선
8.5.
1 지속 개선
7.3,
F.3
.3X
8.5.
2 시정조치
6.8,
F.2
.8
8.5.
3 방조치
7.3.
2, F
.3.3
.2P
t 2

- 105 -
[별첨 3]
의료기기에 포함된 소 트웨어의 시 신고 지침
(Guidance for the Content of Premarket Submissions for Software
Contained in Medical Devices)
FDA CDRH
2005. 5. 11 발행
이 문서는 의료기기에 포함된 소 트웨어의 시 신고에 한 지침(1998.5.29 발행)을
체하며, 액처리(Blood Establishment) 컴퓨터 소 트웨어의 시 신고에 한
지침(1997.1.13 발행)의 재검토이다.
CDRH에서 규제하는 의료기기와 련하여 본 문서에 의문 이 있으면 (301) 443-
8517의 David S. Buckles에게, CBER에서 규제하는 의료기기에 한 의문 이 있으면
(301) 827-6136의 Linda Weir에게 연락하기 바란다.
서 문
이 지침은 독립 으로 사용(stand-alone)되는 소 트웨어 하드웨어에 용 사용되는
소 트웨어를 포함한 소 트웨어 기기에 하여 시 신고에 제출 권고되는 문서와
련하여 산업계에 정보를 제공하는데 목 이 있다.
이 문서는 FDA의 권고사항을 더욱 명확히 하고 기술 발 에 따른 재의 시류를 보증
하기 한 노력의 결과이다. 한, 이 문서는 이 2종 지침에서의 권고사항을 단일
지침으로 통합한다.
최소 부담 근방법
이 지침에 명기된 사항(issues)은 의료기기의 시 에 수행되어야 함을 나타낸다.
지침 제정과정 동안 FDA는 감독기 (Agency)의 의사결정 련 법규들을 신 하게
검토했다. 한 이 지침 지침 명기사항에 따라 제조업체에서 발생될 수 있는 부담도
고려했다. FDA는 이 지침 명기사항의 해결을 하여 가장 효율 인 근방법(the least
burdensome approach)으로 고려하 다. 그러나 명기 사항들에 보다 효율 인 근법이
있다면, 다음 지침에 기술된 차에 따라야 한다.
A Suggested Approach to Resolving Least Burdensome Issues(지침),
http://www.fda.gov/cdrh/modact/leastburdensome.html.

- 106 -
이 지침을 포함한 FDA의 지침들은 법률 으로 시행 가능한 책임을 확정하지 않는다.
다만 지침은 주제에 한 재 감독기 의 생각을 기술하고 오직 권고사항으로 검토하
여야 한다. 그러나 특정 규제사항 는 법 요구사항은 소환된다. 감독기 지침에서
단어 사용은 제안 는 권고를 의미하지만, 강제사항은 아니다.
용범
이 문서의 목 상, 다음 사항과 같이 하나 이상의 소 트웨어 부품, 구성품 는 부속품을
포함하거나 는 소 트웨어로만으로 구성된 것을 “소 트웨어 기기(software
devices)”로 간주한다.
펌웨어 소 트웨어에 근거한 의료기기의 제어를 한 다른 수단
독립 사용(stand-alone) 소 트웨어의 용
일반 컴퓨터에 설치하도록 의도된 소 트웨어
의료기기 목 (dedicated)의 하드웨어/소 트웨어
소 트웨어를 구성하거나 포함되는 의료기기 부속품(accessories)
이 지침은 소 트웨어가 제3자 매자(vendor)에 의한 설치, 제조소 설치, 장 설치
는 업그 이드 간에 최종 사용자에게 인도되는 방법에 계없이 소 트웨어 기기에
용한다.
이 지침에서 제외하는 소 트웨어는 의료기기로서의 목 이 아닌 소 트웨어 제조
는 공정 리용으로 디자인된 소 트웨어를 포함한다. 보다 세부 인 정보 는 자사의
의료기기에 용 해당여부를 명확히 알기를 원하는 경우 FDA 련 검토부서에 문의하기
바란다.
이 지침은 다음과 같은 소 트웨어 기기에 한 시 신고의 모든 종류에 용한다.
통 (Traditional), 특별(Special) 약식(Abbreviated) 시 신고(510(k))
시 승인(PMA)
임상시험 의료기기의 용면제(IDE)
보상을 포함한 인도 의료기기의 용면제(HDE)
다른 문서와의 계
FDA 지침들
본 문서는 소 트웨어 련 권고사항을 제공하는 기존의 다른 지침을 보충하려는
의도이다. 를 들어, 본 문서에서는 소 트웨어 기기(단독 사용기기 는 기기의 부품,
구성품, 부속품을 포함) 련 권고사항으로 “소 트웨어 밸리데이션의 일반 원칙 지침”을

- 107 -
참조하도록 권한다. 의료기기가 OTS 소 트웨어를 사용하는 경우 “의료기기의 OTS
소 트웨어 사용에 한 지침”을 참조하도록 권한다.
소 트웨어 기기 제조업자들은 QSR(21 CFR part 820)의 요구사항에 일치하는 소
트웨어 련 문서를 수립 유지하여야 한다. 다른 FDA의 지침들 본 지침의 권고사항에
따르는 것은 QSR을 수하는 것으로 체할 수 없다는 것에 주의 하여야 한다.
소 트웨어 련 합의규격(Consensus Standards)
소 트웨어 련 합의규격의 출 은 험 리 험평가와 같은 정 한 활동 측면
에서 소 트웨어의 개발품질, 문서화 일 성 향상에 기여하 다. 본 문서에서 용어와
권고사항은 ISO 14971 AAMI SW68과 같은 소 트웨어 련 합의규격과 조화되도록
하 다.
문용어
검증 밸리데이션
이 문서에서는 QSR에서의 정의와 같이 “검증 밸리데이션”(V&V) 용어를 사용한다.
검증이란 “특정 요구사항에 충족된다는 객 증거 제시 검사에 의한 확인”을
의미한다. 소 트웨어 개발환경에서 소 트웨어 검증은 개발의 특정단계에서 출력이
그 단계의 모든 입력요건을 충족한다는 확인이다. 소 트웨어 시험은 소 트웨어 개발
출력이 그 입력요건을 충족함을 확인하기 한 여러 검증활동의 하나이다. 다른 검증
활동으로 다음 사항들을 포함한다.
워크스루(walk-through)
다양한 정 (static), 동 (dynamic) 분석
코드와 문서 조사
모듈 벨 시험
통합 시험
설계 밸리데이션은 “의료기기 규격이 사용자 요구 사용목 에 일치함을 객 인
증거로 입증하는 것”을 의미한다. 본 문서에서 밸리데이션은 “설계 밸리데이션”으로
제한하며 21 CFR 820.3(z)(1)에서 정의한 “공정 밸리데이션”은 포함하지 않는다.
설계 밸리데이션의 한 요소가 소 트웨어 밸리데이션이다. 소 트웨어 밸리데이션은
소 트웨어가 사용된 기능에 한 사용자 요구와 사용목 이 소 트웨어와 일치함을
객 증거로 입증하는 것이다. 소 트웨어의 밸리데이션은 완성된 의료기기의 설계

- 108 -
밸리데이션의 일부분이다. 이것은 의료기기에 소 트웨어를 결합시킨 후 실제 는 모의
사용 환경에서 소 트웨어의 한 작동에 하여 검하는 것을 포함한다. 소 트웨
어의 밸리데이션은 포 인 소 트웨어 시험과 소 트웨어 개발 수명주기의 각 단계
에서 이미 완료한 다른 검증 작업에 상당히 의존한다. 기획, 검증, 추 성, 형상 리
양호한 소 트웨어 엔지니어링의 다양한 요소들은 소 트웨어가 유효하다는 결론도출에
도움을 주는 요한 활동이다.
경미하고 심각한 상해
본 문서에서의 용어 “경미한 상해”는 21 CFR 803.3(bb)(1)의 정의와 같이 심각한 상해
(serious injury)에 해당되지 않는 것을 의미한다. 규정에서 심각한 상해 는 질병은
다음과 같이 정의한다.
생명에 이다.
인체기능의 구 장애 는 인체구조의 손상을 래한다.
인체기능의 구 장애 는 인체구조의 손상을 배제하기 한 의학 는 수
술이 필요하다.
본 문서에서 용어 “ 구(permanent)”는 “사소한 약화나 손상을 제외한 인체구조 는
기능에 한 회복가능성이 없는 장애 는 손상”으로 정의한다. 21 CFR 803.3(bb)(2).
심각도
서문
시 신고에 포함될 문서는 일반 으로 의료기기의 심각도에 의존한다. 심각도는
의료기기의 고장, 설계 결함 는 의료기기 사용목 에 따른 단순 사용의 결과로 환자나
조작자에게 직 는 간 으로 가해질 수 있는 상해의 심각성 평가를 의미한다.
환자 는 조작자에게 상해를 입힐 수 있는 험요인의 발생, 리 / 는 감소에 한
소 트웨어 역할을 명확히 기술하도록 권고한다. 한 이것들은 자사 의료기기의 해당
심각도를 결정하는 요인이다.
소 트웨어 기기에 한 시 제출 문서의 범 는 이러한 심각도에 비례한다.
심각도는 이러한 배경에서 정의하며, 의료기기의 등 분류(1,2,3등 ) 는 본질 인
험요인이나 험분석에 계하지 않는다.
심각도의 상, , 하
소 트웨어 기기에 해당되는 심각도 수 과 각 수 별 심각도에 따라 신고하는 문서에

- 109 -
한 권장사항을 설명한다. 련 험 요인을 감소시키기 에 심각도를 결정하도록
권한다. 즉, 심각도는 개별 험요인에 한 경감 향에 계없이, 경감 작업없이
험요인 분석으로 다루어져야 한다. FDA는 소 트웨어 기기에 하여 결정된 심각도를
제출문서에 명기하도록 권한다. 심각도는 다음과 같이 “상, , 하”가 있다. 한 어떻게
심각도를 결정하 는지에 하여 설명하도록 권한다. 심각도는 의료기기 기능에 연 된
소 트웨어 운 이 환자나 조작자에 미치는 향에 기 하여 결정한다. 향은 직
는 간 일 수도 있다.
상 (Major)
고장이나 잠재 인 결함이 직 인 원인으로 환자나 조작자에게 사망 는 상을
입힌 경우 심각도는 ‘상’에 해당된다. 고장이나 잠재 결함이 간 인 원인으로
작용하여 부정확하거나 지연된 정보로 환자나 조작자에게 사망 는 상을 입힌
경우에도 심각도는 ‘상’에 해당한다.
(Moderate)
고장이나 잠재 설계 결함이 직 원인이 되어 환자나 조작자에게 경상을 입힌
경우 심각도는 ‘ ’이며, 고장이나 잠재 결함이 간 원인으로 작용하여 부정확
하거나 지연된 정보로 환자나 조작자에게 경상을 입힌 경우 심각도는 ‘ ’에 해당한다.
하(Minor)
고장이나 잠재 인 설계, 결함으로 환자나 조작자에게 어떤 부상도 발생할 가능성
이 없는 경우 심각도는 ‘하’에 해당한다.
심각도 수 의 결정
심각도 결정을 하여 다음과 같은 핵심 질문을 제시한다. FDA는 험요인 경감을
시도하기 에 심각도를 평가하도록 권장한다. 즉, 험이 감소되지 않은 상태에서 이
질문들에 응답하여 소 트웨어 기기를 평가하여야 한다.
질문에 한 응답이 부정일 경우, 다음 질문으로 넘어간다. 나 에 상세히 설명하겠
지만, 시 제출 문서에 심각도를 결정한 근거를 포함하도록 권한다. 어떤 경우에도
소 트웨어 기기의 고장으로 발생하는, 최악의 가능성, 합리 상, 임상 결과의
에서 심각도를 평가하도록 권한다.

- 110 -
사용하는 소 트웨어 기기의 심각도에 의문이 있는 경우 언제라도 FDA의 검토부서에
문의하기 바란다. 심각도가 ‘상’으로 단되는 소 트웨어 기기에 한 시 신고를
아직 제출하지 않은 경우, 제출 에 FDA의 련 부서에서 해당 소 트웨어 기기에
하여 의할 것을 권한다.
소 트웨어 련 문서화
시 신고에서 제출할 소 트웨어 련 문서는 소 트웨어 기기의 사용목 ,
심각도 제출 형태에 일치하여야 한다. 본 장에서는 심각도에 따라 시 신고에
포함할 문서들을 언 한다.(표 3) 그러나 의료기기에 한 특정 지침이 있는 경우 그
권고사항에 따른다. 일반 으로 제출할 문서들은 다음과 같다.
[표 1] 상(Major) 심각도
질문의 답이 하나라도 yes인 경우 소 트웨어 기기의 심각도는 ‘상‘일 가능성이 높다.
소 트웨어 기기가 액처리(Blood Establishment) 컴퓨터 소 트웨어처럼 요한가?
( 액처리 컴퓨터 소 트웨어는 액과 액 성분의 제조에 사용하는 소 트웨어 제품 는
액처리 담당자가 헌 자의 합성을 단하거나 수 는 후속 제조를 한 액이나
액 성분 공 의 단에 사용하는 소 트웨어 제품으로 정의된다.)
소 트웨어 기기가 의약품이나 생물제재와 조합하여 사용하는가?
소 트웨어 기기는 심각도가 ‘상’인 의료기기의 부속품인가?
험요인을 경감시키기 , 소 트웨어 기기의 고장으로 환자나 의료기기 사용자의 사망이나
상이 발생될 수 있는가? 이러한 는 다음과 같다.
소 트웨어 기기가 생명보조(life supporting) 는 생명유지 기능을 제어하는가?
소 트웨어 기기가 방사선 치료 시스템, 제세동기 제 발생기(ablation generators)처럼 사망이나 상을 유발할 수 있는 잠재 으로 유해한 에 지의 달을 제어하는가?
소 트웨어 기기가 오류나 오작동(malfunction)이 발생할 경우 사망이나 상을 유발할 수
있는 치료나 요법(therapy)을 제어하는가?
잘못 용할 경우 상이나 사망을 유발할 수 있는 치료나 요법의 결정에 직 인 향을
주는 진단(diagnostic) 정보를 소 트웨어 기기가 제공하는가?
소 트웨어 기기는 의학 시술(intervention)이 필요한 잠재 인 생명 상황에서 생명
유지 신호(vital signs)의 모니터링 경고(alarms) 기능을 제공하는가?

- 111 -
[표 2] (Moderate) 심각도
소 트웨어 기기의 심각도가 ‘상’이 아니면서, 다음 질문에 하나라도 yes인 경우 심각도는 ‘ ’에 해당된다.
소 트웨어 기기는 심각도가 ‘ ’인 의료기기의 부속품인가?
험요인을 경감시키기 , 소 트웨어 기기의 고장으로 환자나 의료기기 사용자의 경상이
발생될 수 있는가?
소 트웨어 기기의 오작동 는 잠재 설계 결함으로 진단에 오류가 발생하거나 한 의
학 진료(care)가 지연되어 경상을 유발할 가능성이 있는가?
표 1과 2의 모든 질문에 한 답이 ‘No’인 경우, 그 심각도는 ‘하’에 해당된다.
의료기기 설계(design) 설명
설계가 어떻게 이행되었는지에 한 문서
설계에 따라 생산된 기기가 어떻게 시험되었는지의 입증
한 험요인 식별 효과 인 험 리의 증명
설계, 이행, 시험 험 리와 추 성의 연결
기기의 심각도를 핵심으로 하는, 제출하는 문서의 종류와 범 는 표 3에 요약한다.
이 권고사항은 설계 리를 포함한 효과 인 이행 QSR의 리에 근거한다.
제출 권장 문서들은 소 트웨어 기기의 개발과정에서 일반 으로 생성되는 문서이다.
그러므로 소 트웨어 개발환경에서 히 리되고 문서화된 제출문서는 자사 제품
개발 문서의 복사본일 수도 있다.
FDA가 제출을 권장하는 문서는 표 3의 뒤에서 설명하기로 한다. 심각도에 해
권장되는 문서는 신고서의 일부를 구성하는 경우도 있다. 소 트웨어 요구사항 규격
(SRS)과 같은 문서는 신청서에 복사하여 제출할 수도 있다.
심각도 수
소 트웨어 기기의 심각도는 어떠한 경감의 향이 있기 에 결정하도록 권한다.
심각도의 3수 자사 의료기기의 해당 수 을 명시하고 그 수 을 결정한 이론
근거를 련문서에 포함시킬 것을 권한다. 한 자사의 의사결정 과정(decision-making
process)을 문서화하도록 권장한다.

- 112 -
소 트웨어 설명서
소 트웨어가 제어하는 의료기기 특성(features)의 포 인 개요와 운 환경을 설명
하는 문서를 제출하도록 권한다. 일반 으로 정보는 단락 형식(paragraph format)으로
작성하고, 요하거나 운 에서 의미가 큰 소 트웨어의 특성을 언 하도록 권한다.
소 트웨어 설명서에는 다음의 정보를 포함하여야 한다.
로그래 언어
하드웨어 랫폼(platform)
해당되는 경우, 운 시스템(operating system)
해당되는 경우, OTS 소 트웨어 사용
의료기기에 기성품 소 트웨어를 사용할 경우 FDA의 “Guidance for Off-the-Shelf
Software Use in Medical Devices." 지침을 참조하기 바란다.
이 정보가 소 트웨어 요구사항 규격(SRS)과 같이 다른 문서에 포함되는 경우, 제출
문서에는 이 정보의 치를 나타내는 주석(annotation)과 참고사항(reference)을 명시하
여야 한다.
의료기기 험요인 분석
모든 소 트웨어 기기에 하여 의료기기 험요인 분석을 제출하도록 권한다. 의료
기기 험요인 분석은 하드웨어와 소 트웨어 험요인을 포함하여 의료기기 사용목 과
련된 모든 의료기기 험요인을 고려하여야 한다. 식별된 각 험요인에 한 정보를
표 형식(tabular form)으로 제출하도록 권한다. 이 문서는 ISO 14971에 명기된 험 리
요약서(Risk Management Summary)와 같이 포 인 험 리 문서에서 소 트웨어
련 항목의 발췌 형식일 수도 있다. 이러한 형식인 경우, 다음과 같은 정보를 포함하
여야 한다.
험스러운 사상(事象, event)의 식별
험요인의 심각도

- 113
-
[표
3] 심
각도
에 근
거한
문서
화
소트웨어 문서
하(M
inor
)(M
oder
ate)
상(M
ajor
)
심각도
심각도의 표시 해당 심각도에 한 이유를 기술
소트웨어 설명서
개략인 특징
(feat
ures
) 소
트웨어 운환경
(ope
ratin
g en
viro
nmen
t.) 요약
의료기기 험요인 분석
심각성 평가 감소를 포함시킨 하드웨어 소트웨어의 험요인 식별표
(Tab
ular
)
소트웨어 요구사항
규격 (
SR
S)
SR
S에서의 기능별
(func
tiona
l) 요구
사항 요약
완한 S
RS
문서
구조 설계도
제출 필요 문서 없음
기능별 유닛 소트웨어 모듈에 한 상세한 설명
.흐름도와 같은 d
iagr
ams 포함 가능
소트웨어 설계 규격
(SD
S)
제출 필요 문서 없음
완한 S
RS
(Sof
twar
e de
sign
spe
cific
atio
n) 문서
추성 분석
요구사항
, 설계 규격
, 식별된 험요인
, 험감소
, 검증 밸리데이션 시험간의 추
성.
소트웨어 개발 환경
설명서
제출 필요 문서 없음
형상
리 유지활동 요약서를 포
함한 소트웨어 개발 수명주기 계
획 요약서
.
소트웨어 개발 수명주기 계획 요약서
.형상
리 유지계획 문서를 포함하여
개발과정에서 작성된 문서들
검증 밸리데이션
문서
소트웨어 기능별 시험 계획
합부정기 결과
유닡
, 통합
시스템
벨에서
VV
&T 활동에 한 설명서
.합부
정기
, 시험결과를 포함한
시스템 벨의 시험계획
(pro
toco
ls)
유닡
, 통합 시스템 벨에서 V
V&
T 활
동에 한 설명서
.합부
정기
, 시험보고서
, 요약 시험
결과를 포함한 유니트
, 통합 시스템
벨의 시험계획
(pro
toco
ls) 로토콜
개정
이력
릴리즈
(rel
ease
) 버번호와 일자를 포함한 개정이력 일지
(log)
미해결된 변종
(버그 결함
)제출 필요 문서 없음
잔존하는 소트웨어 변종 목록
, 조작자 사용 인체요소
(hum
an f
acto
rs)를 포
함하여 안
는 효율성에 한 향을 설명한 주석
(ann
otat
e).

- 114 -
험요인의 원인
리방법( : 경고(alarm), 하드웨어 설계)
의료기기 설계/요구사항 측면에서의 설명을 포함하여, 험스러운 사상의 제거,
감소 는 경고(warn)와 같이 취해진 시정조치
리 방법의 올바른 이행여부에 한 검증
험요인 분석을 수행하는 경우, 의료기기의 의도 이거나 부주의한 오용(misuse)으
로 발생하는 험요인을 포함하여 상 가능한 모든 험요인을 다루도록 권한다.
소 트웨어 요구사항 규격
소 트웨어 요구사항 규격(SRS)에는 소 트웨어 요구사항이 규정되어 있다. 이 규
격에는 형 으로 소 트웨어의 기능, 성능, 인터페이스, 설계 개발요구사항들이
포함된다. 실제 이 문서로 소 트웨어 기기가 무엇을 하는지에 하여 기술한다. SRS
에 포함될 형 인 요구사항의 는 다음과 같다. 심각도가 ‘하’인 소 트웨어 기기는
OTS 소 트웨어의 식별을 포함하여 SRS에 기능 요구사항만을 제시하여도 충분하다.
‘ ’과 ‘상’의 심각도인 소 트웨어 기기는 완벽한 SRS를 제시하도록 권한다.
하드웨어 요구사항
일반 으로 포함된 하드웨어의 요구사항:
마이크로 로세서(microprocessors)
메모리 장치
센서
에 지 원(sources)
안 성
커뮤니 이션
로그램 언어 요구사항
메모리 (memory leaks) 리에 한 정보와 제한 는 로그램 크기(size) 요구사
항을 포함한 로그래 언어 요구사항
인터페이스 요구사항
시스템 구성요소들 간의 커뮤니 이션 린터, 모니터, 키보드, 마우스와 같은 유
(user)와의 커뮤니 이션 모두를 포함한 인터페이스 요구사항

- 115 -
소 트웨어 성능과 기능 요구사항
소 트웨어 성능 기능 요구사항에는 치료, 진단, 감시, 경고, 분석을 한 알고리
즘이나 제어 특성 필요한 경우 체 텍스트 참고문헌(references)이나 보조(supporting)
임상자료의 해석이 포함된다. 소 트웨어 성능과 기능 요구사항에는 다음과 같은 내용
이 포함될 수 있다.
소 트웨어로 인한 기기의 제한 조건(limitations)
내부 소 트웨어 시험 확인
오류와 인터럽트(interrupt) 처리
결함 감지, 허용차(tolerance) 복구(recovery)특성
안 요구사항
타이 (timing)과 메모리요구사항
해당되는 경우 OTS 소 트웨어의 식별
구조 설계도(Architecture Design Chart)
이 문서는 통상 네트워크 연결(networking)과 같이 하드웨어와 데이터 흐름과의 계
를 포함하여 소 트웨어 기기내의 주요 기능별 유닛(units)간 계에 한 흐름도
(flowchart) 는 이와 유사한 설명이다. 일반 으로 이 문서에 모든 기능 모듈을 포
함시킬 필요는 없다. 단, 소 트웨어 기기의 기능 사용 목 에 련된 소 트웨어
의 구조를 검토하기에 충분한 정보가 포함되어야 한다. 심각도가 ‘ ’과 ‘상’인 의료기기
의 경우, 도해(diagrams)와 같이 소 트웨어 기능별 유닛간의 계를 명확히 묘사한 상
세한 정보가 유용할 수 있다. 구조 설계도가 SRS처럼 다른 문서에 포함되어 있다면, 구
조 설계도의 치에 한 참고(reference)와 결과(effect)에 한 설명(statement)을 제출
문서에 포함하여야 한다.
소 트웨어 설계 규격(Software Design Specification)
소 트웨어 설계 규격(SDS)은 소 트웨어 기기에 한 요구사항의 실
(implementation)을 설명한 것이다. SRS SDS간의 계에서, SRS는 ‘소 트웨어 기
기가 무엇을 하는가?’인 반면 SDS는 ‘SRS에 기술된 요구사항을 어떻게 실 하는가?’로
정의된다. SDS에 기술된 정보는 소 트웨어 기기를 만든 소 트웨어 엔지니어가 수
행하는 작업이 임시 인 설계 결정이 없이 분명하고 명확함을 보증하기에 충분하여야
한다. SDS에는 상세한 소 트웨어 규격처럼 다른 문서와의 참조도 포함될 수 있다.
단, 제출문서에는 사용 목 , 기능성, 안 성 활용성과 같은 소 트웨어 요구사항에
한 실 계획을 검토하기에 합한 정보가 포함되어야 한다.

- 116 -
추 성(Traceability) 분석
추 성 분석은 제품 설계 요구사항, 설계 규격 시험 요구사항을 상호 연계한다. 한
식별된 험요인의 감소에 한 이행 시험을 연결하는 방법을 제공한다. 효과 인
제품 개발에 필수 임과 동시에 제품 설계, 개발, 시험 험요인 감소여부를 악
하는데 필수 인 이러한 활동 련 문서는 명확한 추 성 검토를 해 제출하도
록 권한다. 일반 으로 추 성 분석은 요구사항, 규격 시험에 한 항목과 험요
인 감소를 암시(pointers)하는 매트릭스로 구성된다. 공통번호 체계(common
numbering scheme)를 가진 분할된 조직구조(shared organizational structure)로서 문서
추 성을 용이하게 할 수 있다. 그러나 제출된 정보로 FDA 검토자에게 길잡이가 될 수
있는 매트릭스와 같은 구성(mechanism)을 포함하도록 권한다.
소 트웨어 개발환경 설명서
심각도가 ‘ ’과 ‘상’인 소 트웨어 기기의 경우, 제출문서에는 소 트웨어 개발 life
cycle 계획에 한 요약서를 포함하여야 한다. 요약서에는 소 트웨어 개발 life cycle
다양한 life cycle 활동들을 리하기 한 과정을 설명하여야 한다. 한 심각도가
‘상’인 소 트웨어 기기의 경우에는 소 트웨어 개발 과정동안 발생된 리/기
(control/baseline) 문서의 주석 (annotated) 목록 소 트웨어 코딩기 (coding
standards)의 목록이나 설명을 포함하여야 한다.
형상 는 변경 리는 소 트웨어 개발에 있어 매우 요한 요소이다. 최 시 후 소
트웨어 기기의 변경은, 필요한 경우 명확한 규격 잘 정의된(well-defined) 회귀시
험(regression testing)을 포함한 시험 계획으로 확실하게 리하여야 한다. 개발환경 설
명서에는 형상 리(configuration management) 소 트웨어 개발 life cycle 측면에서
의 유지 계획에 한 정보가 포함되어야 한다. 심각도가 ‘상’인 의료기기는 형상 리와
유지 계획을 완 히 악할 수 있는 충분한 상세 설명을 제공하여야 한다. 심각도가
‘ ’인 의료기기는 형상 리와 유지 계획에 한 요약서(summary)를 제시하면 된다.
검증 밸리데이션 문서
이 지침에서 “검증”과 “밸리데이션”에 해 소 트웨어 기기 시험의 두 단계와 련
하여 언 한 바 있다. 이 장에서는 소 트웨어 기기의 시 신고에 포함되는 시험 문
서(documentation)의 종류(type)에 하여 언 한다.
심각도가 ‘하’인 기기
심각도가 ‘하’인 기기의 경우, 시스템이나 기기의 벨 시험 해당되는 경우 통합

- 117 -
(integration) 시험에 한 문서를 제출하여야 한다. 제출문서에는 시스템이나 기기의
벨시험 합부 정기 (pass/fail criteria) 시험결과 요약서를 포함하여야 한다.
심각도가 ‘ ’인 기기
심각도가 ‘ ’인 기기의 경우, 검증 밸리데이션 활동의 요약 목록, 이러한 활동
의 결과 합부 정기 을 제출하여야 한다. 추 성 분석은 이러한 활동과 결과가
설계 요구사항 규격에 효과 으로 연계됨을 보장하여야 한다.
심각도가 ‘상’인 기기
심각도가 ‘상’인 기기의 경우 심각도가 ‘ ’인 의료기기에서 요구되는 정보와 불합
격된 시험에 한 설명을 제출하여야 한다. 한 불합격된 시험에 응하기 한 변
경(modification)과 그러한 변경이 효과 이었음을 입증하는 결과 문서를 포함시키도
록 권한다. 제출될 문서에는 유닛 통합 시험(unit integration testing)의 시(examples)
결과 요약서를 포함하여야 한다.
개정 (Revision Level) 이력
제출문서에는 제품 개발과정에서 발생한 소 트웨어의 개정(revisions) 이력을 포함하
여야 한다. 일반 으로 이력은 날짜, 버 이 버 과 개정 버 의 주요 변경 비교
설명을 포함하여 개발 주기 소 트웨어의 주요 변경항목을 표 형식(tabulation)으로
작성한다. 목록의 최종 기재내용은 출하된 기기에 채택된 최종 버 이어야 한다. 한,
기재내용에는 시험한 소 트웨어 버 과 출하된 버 간의 차이 기기의 안 성과
유효성 차이로 인한 잠재 향에 하여 평가한 것을 포함하여야 한다.
미해결된 변종(버그 는 결함)
심각도가 ‘ ’과 ‘상’인 소 트웨어 기기의 제출문서에는 미해결된 모든 소 트웨어 변종
(anomalies) 목록을 포함하여야 한다. 각 변종에 하여 다음 사항을 언 하도록 권한다.
문제
의료기기 성능에 미치는 향
해당되는 경우, 문제 해결을 한 계획이나 일정(time frames)
조작자 사용과 인체공학 문제를 포함한 의료기기 안 성이나 유효성에 한 변종의
향 설명을 각 항목별로 주석(annotate)을 달도록 권한다. 통상 이 목록은 미해결된
소 트웨어 변종의 평가와 처리를 하여 변경 리 원회(change control board) 는
유사한 조직(mechanism)에서 도출된 결과로 생성된다. 해당하는 경우 의료기기의

- 118 -
한 작동에 지원하도록, 이 목록을 최종 사용자(end user)에게 알릴 것을 권한다. 실행
이 가능한 모든 사례에서, 미해결된 변종을 감소시키거나 가능한 회피방법
(work-arounds)을 포함하여야 한다: 특히 이 권고사항은 액처리 컴퓨터 소 트웨어
에 용된다.
특별(Special) 510(k) 로그램
특별 510(k) 로그램에서 격성을 확인(qualify)하는 시 신고 상 의료기기는
이 에 받은 510(k) 의료기기의 수정(modification)한 의료기기가 되어야 한다. 단, 이러
한 수정으로 의료기기의 사용목 이나 근본 인 과학기술이 변경되어서는 안된다. 특
별 510(k)에서, 제출문서는 이 지침에 따라야 하지만 신고의 원인이 된 수정과 련된
문서만을 제출하면 된다. 를 들어 특별 510(k)에서의 요구사항 규격 제출문서는
수정된 부분에 을 맞추며, 의료기기의 모든 요구사항과 규격을 포함시킬 필요는
없다.
수정을 검증, 유효성 확인하기 하여 수행하는 회귀시험(regression testing)을 제출
하도록 권한다. 시험 자료보다도 오히려 시험 계획, 합부 정기 결과 요약서를
제출하도록 권한다. 어떠한 경우라도 제시할 소 트웨어 련 문서의 형태 세부 수
은 수정과 련된 의료기기 심각도 에 하여야 한다. 특별 510(k) 신고는 설계
리에 한 자사의 합성 선언(declaration of conformance)에 좌우되므로, 시험 는
선언에 따른 기타 활동을 완료하기 까지는 특별 510(k) 신고를 히 제출할 수 없
을 것이다. (연방 FD&C법 514(c)(1)(B)항(21 U.S.C. 360d(c)(1)(B) 참조).
약식(Abbreviated) 510(k) 로그램
약식 510(k) 신고는 21 CFR 807.87에 명시된 요구 요소(required elements)를 포함
하여야 한다. 약식 510(k)의 경우 이 지침에서 권고한 문서 내용은 21 CFR 807.87(f)나
(g)의 의미를 하게 입증하는 자료일 것으로 간주한다. 그러므로 이 지침에서 설명
한 문서를 제출하도록 권한다.
의료기기 설계나 시험에서 FDA가 인정한 표 (standard)에 따른 경우, 다음 하나
를 포함하면 된다.
제품 시 에 시험을 실시하고 시험이 규정된 허용기 (acceptance criteria)을
충족할 것이라는 진술서(statement)
기 에 한 합성 선언

- 119 -
합성 선언은 시험 결과에 근거하기 때문에, 표 에서 규정한 시험이 완료되기
에 합성 선언을 제출할 수 없다. 세부 인 사항은 법 514(c)(1)(B)항 FDA “Use of
Standards in Substantial Equivalence Determinations" 지침을 참조하기 바란다.
소 트웨어 기기에 해 권고되는 특정 시험이나 시험 방법으로 합성을 선언하는
경우, 합성 선언과 함께 합부 정기 련 시험 결과를 같이 제출하도록 권한다.
한 표 에서 규정된 시험과 시험 방법과의 차이(deviations)를 기록하고, 소 트웨어
기기의 안 성과 유효성에 미치는 향의 에서 이러한 차이를 설명하도록 권한
다. FDA가 인정한 합의표 (consensus standards) 목록은 CDRH 웹사이트에 있다.
추가 인 주제(Additional Topics)
험평가 리
배경
부 하거나 부 합한 소 트웨어 개발 life cycle 험 리 활동, 소 트웨어
기기의 부 합한 사용 는 작동상 오류는 다양한 잠재 인 고장이나 설계 결함
(flaws)의 결과일 수 있다. 이러한 고장이나 결함에는 안 하지 않거나 유효하지
않은 에 지, 약품 생명 보조나 생명 유지 기능의 달이 포함된다. 한 오진
는 잘못된 치료나 요법(therapy)의 선택을 야기하는 부정확, 불완 한 정보의 달은
특정 소 트웨어 기기와 련된 잠재 인 고장이다. 따라서 잠재 인 고장이나 설계
결함과 련된 험은 소 트웨어 기기의 검토과정에서 심의 상이다.
험 평가 심각도
이미 언 한 바와 같이,소 트웨어 기기와 련된 험 평가는 해당 심각도의 결정
에 도움이 된다. 한 동일한 유형(type)이나 사용 목 을 가진 다른 의료기기들의
심각도를 고려하도록 권한다. 자사의 의료기기에 해당하는 심각도 수 이 다르다고
단한 경우, 그 이론 근거의 상세 설명을 제출하도록 권한다.
험 리(Risk Management)
소 트웨어 기기와 련된 험은 ‘무시’에서부터 ‘매우 심각’까지 다양하다. 일반
으로 험은 상해의 심각성과 발생 가능성의 산물로 간주한다. 그러나 소 트웨어
고장은 특성상 시스템 인 사항이며, 이에 따라 발생 가능성은 형 인 통계
방법을 사용하여 결정할 수는 없다. 따라서 소 트웨어 기기에 한 험 추정
(estimation)은 고장 발생으로 야기되는 험요인의 심각성에 근거하도록 권한다. 한
ISO 14971과 같은 합의표 에 명기된 험식별 리기법을 사용하도록 권한다.

- 120 -
소 트웨어 변경 리
소 트웨어 수정 (revisions)의 설계, 개발, 시험 버 리는 시 신고에서 검
토되었던 소 트웨어의 개발 시험만큼이나 요하다. 소 트웨어 련 의료기기
의 리콜을 포함하여 장에서 발생하는 소 트웨어 련 의료기기의 다수의 문제
는 시 검토 이후 수정된 소 트웨어를 구 하는 의료기기에서 발생한다는 것이
FDA의 단이다. FDA의 검토가 요구되지 않는 수정 도 부작용(adverse events)과 리
콜을 래하 다. 이는 소 트웨어의 수정에 세심한 리가 필요함을 나타내는 것이
다.
액처리 컴퓨터 소 트웨어(Blood Establishment Computer Software)
액처리 컴퓨터 소 트웨어에 한 시 신고의 경우, 모든 제한사항의 설명을 포함
해 사용자에게 제공되는 사용자 설명서의 완 한 사본을 제출하여야 한다. 한 사
용자 설명서에 언 하지 않은 모든 요한 변종(anomalies) 는 소 트웨어 결함을
한 해결책(workarounds)과 함께 사용자게 제공할 경우 그러한 문서도 제출하여야 한
다.
근원(Pedigree)을 알 수 없는 소 트웨어(SOUP)
소 트웨어 기기에 포함되는 소 트웨어의 일부 는 부는 제3자가 개발한 것일 수
있다. 이러한 소 트웨어에 수반되는 문서의 형태와 품질은 매우 다양할 수 있다. 한
문서의 확보(obtain)가 어려운 소 트웨어를 ‘근원을 알 수 없는 소 트웨어’ 는 SOUP
라고 한다.
SOUP에 한 해당 설계 문서를 확보하거나 만들거나 재구성(reconstruct)하기는 어
려울 수 있다. 따라서 소 트웨어의 출처(origin)와 소 트웨어 문서의 주변 상황
(circumstances surrounding)을 설명하도록 권한다. 추가 으로 험요인 분석에는 잘
못되거나 불완 한 문서 는 시험 (prior testing) 문서의 락과 련하여 SOUP
연 험을 포함하여야 한다. 다만, 의료기기의 한 시험 한 소 트웨어
시험계획과 결론 문서의 제공에 한 책임은 신고인에게 있다.
바이러스 방지 소 트웨어
소 트웨어 기기를 포함한 정보 시스템을 유해하거나 악의 인 코드( ; 바이러스, 웜
등)로부터 보호하도록 설계된 소 트웨어 응용 로그램은, 의료기기의 상호연결 이
에 따른 외부정보환경으로의 노출 가능성 증가에 따라, 보다 상용화되고 있다. 바이러
스 방지 소 트웨어의 설치 시험 련 문제는 이 지침의 범 를 벗어난다. 이 주제
에 한 세부 사항은 CDRH OC(Office of Compliance)에 문의하기 바란다.

- 121 -
인터페이스, 네트워크 연결(Networking) 네트워크 기반구조
이미 언 한 바와 같이 소 트웨어 기기는 특정 의료기기와 특정 데이터의 교환을
하여 지 간(point-to-point) 인터페이스, 근거리(local)통신망과 역통신망(wide area
networks) 인터넷과의 속으로 상호 연결이 계속 증가하고 있다. 화, 근거리통
신망 역 연결과 같은 데이터 교환 통신 기반구조는 의료기기처럼 규제받지
는 않으나 이러한 송매체(carriers)로 인하여 소 트웨어 기기의 운 에 향을 미친
다. 를 들면 근거리통신망에 연결된 소 트웨어 기기는 네트워크 인터페이스에 문제
가 발생할 경우 원활한 운 이 단된다. 소 트웨어의 설계에서 의료기기에 제공되는
인터페이스의 용량(capabilities) 부하(liabilities) 모두를 고려하도록 권한다. 특히
험요인 분석과 감소 활동에는 이러한 문제(issues)를 포함하여야 한다.
조합제품(Combination Products)
일반 으로 이 지침의 권고사항은 의료기기 구성부품이 소 트웨어 기기의 정의에 부
합할 경우, 조합 제품( ; 의약품-의료기기 생물제재-의료기기 조합)의 구성부품에
용될 것이다. 자세한 사항은 OCP(Office of Combination Products) 는 조합 제
품에 한 검토를 안내하는 FDA 검토 부서에 문의하기 바란다.

- 122 -
참고문헌
ⅰ. 이 문서는 1998년 5월 29일에 발행된 “의료기기에 포함된 소 트웨어의 시 신
고 지침”의 체 1997년 1월 13일 발행된 액처리(Blood Establishment) 컴퓨
터 소 트웨어의 시 신고 지침“에 하여 재검토한 것을 조합한 것이다.
ii. “General Principles of Software Validation,” http://www.fda.gov/cdrh/comp/guidance/938.html
iii. “Guidance for Off-the-Shelf Software Use in Medical Devices” http://www.fda.gov/cdrh/ode/guidance/ 585.pdf
iv. 21 CFR 820.30 Subpart C – Design Controls of the Quality System Regulation
v. ISO 14971-1; Medical devices - Risk management - Part 1: Application of risk
analysis
vi. AAMI SW68:2001; Medical device software - Software life cycle processes.
vii. See “The New 510(k) Paradigm – Alternate Approaches to Demonstrating
Substantial Equivalence in Premarket Notifications – Final Guidance,” available on
the FDA Web site at http://www.fda.gov/ cdrh/ode/parad510.html
viii. For more information see Device Advice, “How to Prepare an Abbreviated 510(k),”
http://www. fda.gov/cdrh/devadvice/3145.html, in particular the section titled
“Information Required in an Abbreviated 510(k).”
ix. See “Required Elements for a Declaration of Conformity to a Recognized Standard
(Screening Checklist for All Premarket Notification [510(K)] Submissions),”
http://www.fda.gov/cdrh/ode/ reqrecstand.html
x. See “Use of Standards in Substantial Equivalence Determinations,”
http://www.fda.gov/cdrh/ ode/guidance/1131.html
xi. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm.
xii. For information on determining when revisions to software should result in a
new premarket submission, you should consult the relevant FDA guidances
such as “Deciding When to Submit a 510(k) for a Change to an Existing
Device,” http://www.fda.gov/cdrh/ode/510kmod. html. See also 21 CFR
807.81(a)(3)

- 123 -
[별첨 4]
소 트웨어 수명주기 로세스(IEC 62304/FDiS(2006))
(Medical device software–Software life cycle processes)
서 문
의료기기 기술에 소 트웨어가 요한 부분이 되는 경우가 많다. 소 트웨어가 포함
된 의료기기의 안 성과 효과성 확립에는 소 트웨어 사용 의도 소 트웨어 사용에
의해 허용할 수 없는 험의 발생 없이 그러한 의도가 충족된다는 사실의 증명이 필요
하다.
이 규격은 의료기기 소 트웨어의 안 한 설계와 유지보수에 필요한 활동과 업무에
련된 수명주기 로세스의 체제를 제공한다. 이 규격은 각 수명주기 로세스에
한 요구사항을 제공한다. 각 수명주기 로세스는 일련의 활동으로 다시 분류되며,
각 활동은 일련의 업무로 분류된다.
기본 으로 의료기기 소 트웨어는 품질경 시스템(4.1 참조)과 험 리시스템(4.2
참조) 범 안에서 개발∙유지된다. 험 리 로세스는 이미 국제규격 ISO 14971에
규정되어 있다. 따라서 IEC 62304는 단순히 ISO 14971 규정을 참조함으로써 이 장 을
활용한다. 특히 험요인에 련해 향을 주는 소 트웨어 요인의 식별 역의 경우
일부 사소한 기경 요구사항이 소 트웨어에 추가로 필요하다. 이러한 요구사항은 제
7장에 IEC 험 리 로세스에 요약 설명되어 있다.
소 트웨어가 험요인에 한 기여 요소인지의 여부는 험 리 로세스의 험요
인 식별 활동 단된다. 소 트웨어에 의해 간 으로 발생할 수 있는 험요인
( ; 잘못된 정보를 제공해 치료가 부 하게 수행되는 경우)은 소 트웨어가 발생 요
인인지 여부를 결정할 때 고려하여야 한다. 험 제어에 소 트웨어를 사용할 것인지
의 여부는 험 리 로세스의 험제어 활동 결정된다. 이 표 에 필요한 소 트
웨어 험 리 로세스는 ISO 14971에 따라 의료기기 험 리 로세스에 내재되어
야 한다.

- 124 -
소 트웨어 개발 로세스는 몇 가지 활동으로 구성된다. 이들 활동은 그림 1에 표
시되어 있으며, 제5조에 설명된다. 장에서 발생하는 부분의 사고는 부 합한 소 트
웨어 갱신과 업그 이드를 포함해 의료기기 시스템의 정비나 유지보수에 련되므로
소 트웨어 유지 리 로세스는 소 트웨어 개발 로세스만큼 요하게 취 해야 한
다. 소 트웨어 유지보수 로세스는 소 트웨어 개발 로세스와 매우 유사하다. 이
로세스는 그림 2에 표시되어 있으며, 제6조에 설명된다.
이 표 은 안 한 의료기기 소 트웨어 개발에 필수 인 것으로 간주되는 두 가지
추가 로세스를 식별한다. 즉, 형상 리 로세스(제8조) 소 트웨어 문제해결 로
세스(제9조)가 그것이다.
이 표 은 제조업체에 한 조직 구조 는 조직의 어떤 부서가 어떤 로세스, 활
동 는 업무를 수행할 것인지 규정하지 않는다. 이 표 에는 이 표 의 수성을 확보할
수 있도록 로세스, 활동 는 업무가 완료되어야 한다는 사실만 요구된다.
이 표 은 제작할 문서의 명칭, 형식 는 명확한 내용을 규정하지 않는다. 이 표
에 따라 업무의 문서화가 필요하지만, 이 문서를 구성하는 방법은 표 의 사용자가 결
정한다.
이 표 은 특정 수명주기 모델을 규정하지 않는다. 이 표 사용자는 소 트웨어
로젝트에 한 수명주기 모델을 선택하고, 그 모델에 이 표 의 로세스, 활동 업
무를 매핑할 책임이 있다.
부록 A에는 이 표 의 조항에 한 이론 근거가 제공된다. 부록 B에는 이 표 의
규정에 한 지침이 제공된다.
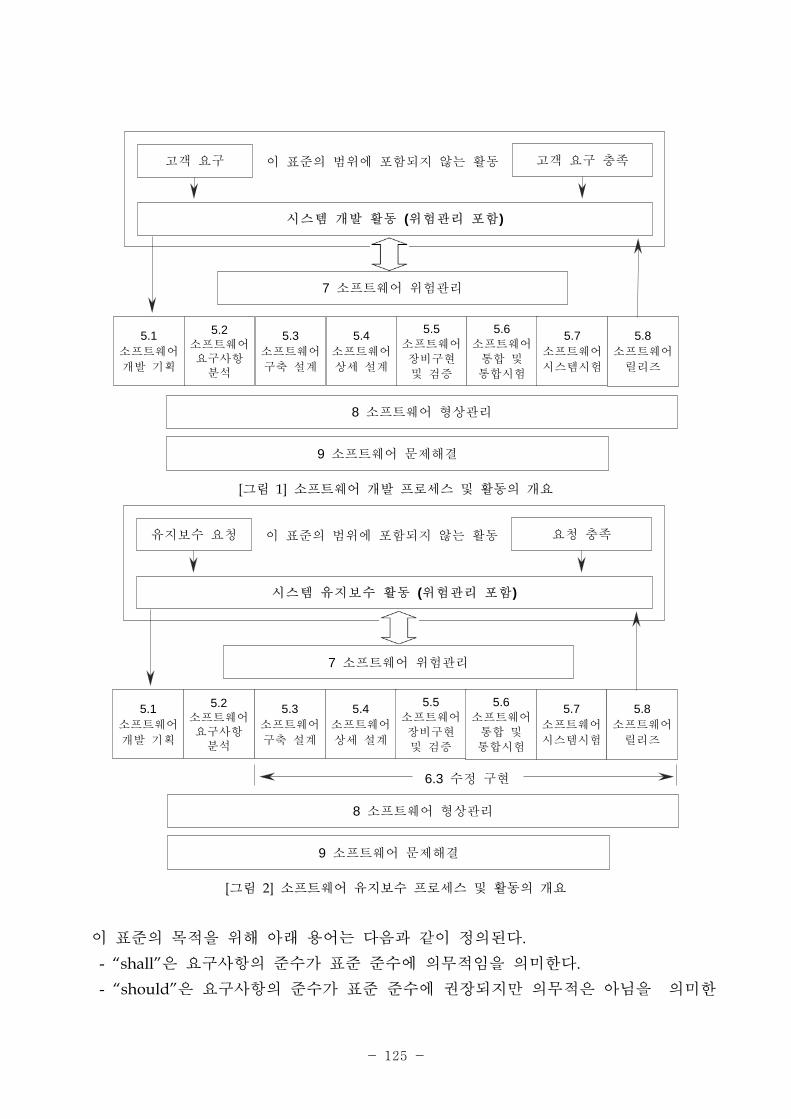
- 125 -
[그림 1] 소 트웨어 개발 로세스 활동의 개요
8 소 트웨어 형상 리
9 소 트웨어 문제해결
이 표 의 범 에 포함되지 않는 활동
7 소 트웨어 험 리
5.1소 트웨어
개발 기획
5.2소 트웨어 요구사항 분석
5.3소 트웨어
구축 설계
5.4소 트웨어
상세 설계
5.5소 트웨어 장비구
검증
5.6소 트웨어
통합 통합시험
5.7소 트웨어
시스템시험
5.8소 트웨어
릴리즈
시스템 개발 활동 ( 험 리 포함)
고객 요구 충족고객 요구
[그림 2] 소 트웨어 유지보수 로세스 활동의 개요
6.3 수정 구
8 소 트웨어 형상 리
9 소 트웨어 문제해결
시스템 유지보수 활동 ( 험 리 포함)
이 표 의 범 에 포함되지 않는 활동유지보수 요청 요청 충족
5.1소 트웨어
개발 기획
5.2소 트웨어 요구사항 분석
5.3소 트웨어
구축 설계
5.4소 트웨어
상세 설계
5.5소 트웨어
장비구 검증
5.6소 트웨어통합 통합시험
5.7소 트웨어
시스템시험
5.8소 트웨어
릴리즈
7 소 트웨어 험 리
이 표 의 목 을 해 아래 용어는 다음과 같이 정의된다.
- “shall”은 요구사항의 수가 표 수에 의무 임을 의미한다.
- “should”은 요구사항의 수가 표 수에 권장되지만 의무 은 아님을 의미한

- 126 -
다.
- “may”는 요구사항 수를 해 허용되는 방법의 설명에 사용된다.
- “establish”는 정의, 문서화 구 을 의미한다.
- 이 표 에서 “as appropriate”라는 용어를 요구되는 로세스, 활동, 임무 는 산
출과 연계해 사용할 경우 그 사용 목 은 제조업체가 그러한 로세스, 활동, 업
무 는 산출을 사용하지 않아도 되는 근거를 문서화할 수 없는한 제조업체가 그
러한 로세스, 활동, 업무 는 산출을 사용해야 한다는 것이다.
1. 범
1.1 목
이 표 에는 의료기기 소 트웨어에 한 수명주기 요구사항이 정의된다. 이 표 에
설명한 로세스, 활동 업무는 의료기기 소 트웨어 수명주기 로세스에 한 공통
인 체제를 확립한다.
1.2 용분야
이 표 은 의료기기 소 트웨어 개발과 유지보수에 용된다.
이 표 은 소 트웨어 자체가 의료기기이거나, 소 트웨어가 최종 의료기기에 내장
되거나 구성 부분인 경우 의료기기 소 트웨어 개발과 유지보수에 용된다.
이 표 은 의료기기가 소 트웨어로만 구성된 경우에도 의료기기의 밸리데이션과 최
종 릴리스는 취 하지 않는다.
1.3 다른 표 과의 계
이 의료기기 소 트웨어 수명주기 표 은 의료기기를 개발할 때 다른 해당 표 과
함께 사용된다. 부록 C에는 이 표 기타 련 표 과의 계가 표시되어 있다.
1.4 수성
이 표 의 수성은 소 트웨어 안 등 에 따라 이 표 에서 확인(identified)된 모
든 로세스, 활동 업무를 구 하는 것으로 정의된다.

- 127 -
주 : 각 요구사항에 할당된 소 트웨어 안 등 은 요구사항 뒤의 규정 텍스트에 설명
된다.
수성은 험 리 일 소 트웨어 안 등 에 필요한 로세스, 활동 업무
의 평가를 포함해, 이 표 이 요구하는 모든 문서화의 검사에 의해 결정된다. 부록 D
참조.
주 1 : 이 평가는 내부감사나 외부감사로 수행할 수 있다.
주 2 : 지정 로세스, 활동 업무를 수행하는 경우에도 그러한 로세스의 구 과
그러한 활동과 업무 수행의 방법에는 융통성이 있다.
주 3 : 요구사항에 “as appropriate”라는 용어가 포함되어 있지만 수행되지 않은 경우
이 평가에는 그 정당화에 한 문서화가 필요하다.
주 4 : 일치성이라는 용어는 ISO/IEC 12207에 사용되며, 이는 이 표 에 사용되는
수성이라는 용어에 해당된다.
2. 규격 참고문헌
아래에 인용한 문서는 이 문서의 용에 필수불가결이다. 날짜가 명시된 참고문헌의
경우 해당 만 용된다. 일자가 명기되지 않은 참고문헌의 경우 인용한 문서 최신
(수정 포함)이 용된다.
ISO 14971, 의료기기 – 의료기기 험 리의 용
3. 용어 정의
이 문서에는 다음과 같은 용어와 정의가 용된다.
3.1 활동(ACTIVITY)
하나 이상의 상호 련 는 상호작용 업무
3.2 비정상 상태(ANOMALY)
요구사항 시방서, 설계문서, 표 등에 기 한 상치 는 인식이나 경험에서 벗어
나는 조건. 비정상 상태는 소 트웨어 제품 는 해당 문서의 검토, 시험, 분석, 작성
는 사용 확인(be found)할 수 있다. [IEEE 1044:1993, 정의 3.1]
3.3 아키텍처(ARCHITECTURE) :
시스템이나 구성부품의 조직 구조. [IEEE 610.12:1990]

- 128 -
3.4 변경 요청(CHANGE REQUEST)
소 트웨어 제품에 이루어지는 변경을 문서화한 시방서
3.5 형상 항목(CONFIGURATION ITEM)
특정 기 에서 고유하게 확인(identified)할 수 있는 실체
주 : ISO/IEC 12207:1995, 정의 3.6에 기 .
3.6 산출물(DELIVERABLE)
활동이나 업무에서 요구되는 결과 는 산출(문서화 포함)
3.7 평가(EVALUATION)
실체가 지정 기 을 충족하는 범 에 한 체계 인 단. [ISO/IEC 12207:1995, 정
의 3.9]
3.8 해(HARM)
사람의 보건에 한 신체 상해나 피해 는 양쪽 모두. 는 재산이나 환경에
한 피해 [ISO/IEC Guide 51:1999, 정의 3.1]
3.9 험요인(HAZARD)
해의 잠재 발생원 [ISO/IEC Guide 51:1999, 정의 3.5]
3.10 제조업체(MANUFACTURER)
의료기기의 설계, 제조, 포장 는 라벨링, 시스템 조립 는 해당 계인이나 해당
계인을 리한 제3자가 운 을 수행하는지의 여부에 계 없이 시 사용 의
료기기의 채택을 담당하는 자연인이나 법인. [ISO 14971:2000, 정의 2.6]
3.11 의료기기(MEDICAL DEVICE)
다음과 같은 특정 목 하나 는 그 이상을 해 인간에 해 단독으로 는 조
합으로 사용되는, 제조업체의 기구, 장비, 도구, 기계, 기구, 이식 조직편, 체외 시약
는 측정기, 소 트웨어, 자재 는 기타 유사하거나 련된 품목
- 질병의 진단, 방, 모니터링, 치료 는 경감
- 상해의 진단, 모니터링, 치료, 경감 는 보상
- 비정상 상태 는 생리학 로세스의 조사, 체, 수정 는 지지
- 생명 지지 는 유지

- 129 -
- 임신 리
- 의료기기의 소독
- 인체에서 채취한 표본의 체외 검사를 사용해 의료 목 을 한 정보 제공 한
약리학, 면역학 는 신진 사 수단으로 인체에 의도된 주된 조치를 달성하는
것이 아니라 기능이 그러한 수단의 지원을 받을 수 있는 것
주 1 : 이 정의는 의료기기 국제조화기기(GHTF)가 개발한 것이다. 참고문헌[15] (ISO
13485:2003) 참조. [ISO 13485:2003, 정의 3.7]
주 2 : 각국의 법규에서 사용하는 정의에 어느 정도 차이가 발생할 수 있다.
3.12 의료기기 소 트웨어(MEDICAL DEVICE SOFTWARE)
개발 인 의료기기에 채택할 목 으로 개발된 소 트웨어 시스템 는 자체 권한으
로 의료기기로서 사용하기 한 소 트웨어 시스템
3.13 문제 보고서(PROBLEM REPORT)
사용자나 다른 이해 계 당사자가 의도된 사용에 불안 하고 하지 않거나 시방
서와 다르다고 생각하는 소 트웨어 제품의 실제 는 잠재 작동상태의 기록
주 1 : 이 표 에 다르면 모든 문제 보고서에 따라 소 트웨어 제품을 변경할 필요
는 없다. 제조업체는 문제 보고서를 오해, 오류 는 사소한 사상(事象)으로 간
주해 배척할 수 있다.
주 2 : 문제 보고서는 이미 릴리즈된 소 트웨어 제품 도는 재 개발 인 소
트웨어 제품에 련될 수 있다.
주 3 : 이 표 에 따라 규제 조치를 식별하고 구 할 수 있도록 이미 릴리즈된 제품
에 련된 문제 보고서에 해 추가 의사결정 단계(제6조 참조)를 수행해야
한다.
3.14 로세스(PROCESS)
입력을 출력으로 변환하는 일련의 상호 련 는 상호작용 활동 [ISO 9000:2000, 정
의 3.4.1]
주 : “활동”이란 용어에는 자원의 사용도 포함된다.
3.15 회귀시험(REGRESSION TESTING)
시스템 구성부품에 한 변경이 기능성, 신뢰성 는 성능에 부정 인 향을 미치
지 않으며 추가 결함이 발생하지 않았다고 단하기에 필요한 시험 [ISO/IEC
90003:2004, 정의 3.11]

- 130 -
3.16 험(RISK)
해 해 심각도 발생 가능성의 조합 [ISO/IEC Guide 51:1999 정의 3.2]
3.17 험 분석(RISK ANALYSIS)
해를 식별하고 험을 추정하기 한 가용 정보의 체계 인 사용 [ISO/IEC Guide
51:1999 정의 3.10]
3.18 험 리(RISK CONTROL)
의사결정이 이루어지며 험이 지정 수 으로 감소하거나 지정 수 안에 유지되는
로세스 [ISO 14971:2000 정의 2.16]
3.19 험 리(RISK MANAGEMENT)
험 분석, 평가 리 업무에 한 경 정책, 차 방법의 체계 인 용[ISO
14971:2000 정의 2.18]
3.20 험 리 일(RISK MANAGEMENT FILE)
험 리 로세스에서 생성되는 기록과 기타 문서. 단, 연속 일 필요는 없다. [ISO
14971:2000 정의 2.19]
3.21 안 성(SAFETY)
허용할 수 없는 험의 부재 [ISO/IEC Guide 51:1999 정의 3.1]
3.22 보안(SECURITY)
미승인 계자나 시스템이 정보와 데이터를 읽을 수 없게 만들며, 승인 계자나 시
스템이 정보와 데이터에 한 액세스가 거부되지 않도록 정보와 데이터를 보호하는 활
동. [ISO/IEC 12207:1995 정의 3.25]
3.23 상(SERIOUS INJURY)
직 는 간 으로 다음과 같은 부상이나 질병
a) 생명을 한다.
b) 신체 기능에 한 구 훼손이나 신체 구조에 한 구 피해가 발생한다.
c) 신체 기능에 한 구 훼손이나 신체 구조에 한 구 피해 배제에 의료
는 수술 개입이 필요하다.
주 : 구 훼손은 사소한 훼손이나 피해를 제외하고, 신체 구조나 기능에 한
회복 불가능한 훼손이나 피해를 의미한다.

- 131 -
3.24 소 트웨어 개발 수명주기 모델(SOFTWARE DEVELOPMENT LIFE CYCLE MODEL)
요구사항의 정의에서부터 제조를 한 릴리즈에 이르기까지 소 트웨어의 수명 체
를 포 하는 개념 구조로서,
- 소 트웨어 제품 개발에 련된 로세스, 활동 업무를 확인(identify)한다.
- 활동과 업무 사이의 순서와 의존성을 설명한다.
- 특정 산출물의 완벽성을 검증할 수 있는 일정을 확인(identify)한다.
주 : ISO/IEC 12207:1995, 정의 3.11에 기 .
3.25 소 트웨어 항목(SOFTWARE ITEM)
컴퓨터 로그램의 확인 가능한 부분(identifiable part)[ISO/IEC 90003:2004, 정의 3.14]
주 : 세 가지 용어가 소 트웨어 분해(decomposition)를 구분한다. 최상 수
(Level)은 소 트웨어 시스템이다. 더 이상 분해할 수 없는 최하 수 이 소
트웨어 유닛이다. 최상 와 최하 가 포함된 모든 수 의 구성을 소 트웨어 항
목(item)이라고 부를 수 있다. 즉, 소 트웨어 항목은 하나 이상의 소 트웨어
항목으로 구성되며, 각 소 트웨어 항목은 하나 이상의 소 트웨어 유닛이나 분
해 가능한 소 트웨어 항목으로 구성된다. 소 트웨어 항목과 소 트웨어 유닛
의 정의(definition )와 입도(granularity)를 제공하는 것은 제조업체의 책임이다.
3.26 소 트웨어 제품(SOFTWARE PRODUCT)
컴퓨터 로그램, 차 연 문서와 데이터 [SIO/IEC 12207:1995 정의 3.26]
3.27 소 트웨어 시스템(SOFTWARE SYSTEM)
특정 기능이나 일련의 기능을 수행하도록 구성된 소 트웨어 항목(item)을 통합한 집합체
3.28 소 트웨어 유닛(SOFTWARE UNIT)
다른 항목(item)으로 다시 분류할 수 없는 소 트웨어 항목(item)
주 : 소 트웨어 유닛은 소 트웨어 형상 리 는 시험 목 에 사용할 수 있다.
3.29 기원이 확인되지 않은 소 트웨어(SOUP; Software Of Unknown Provenance (acronym))
이미 개발되어 일반 으로 사용할 수 있으며, 의료기기에 채택할 목 으로 개발되지
않은 소 트웨어 항목(item)(“기성품 소 트웨어”라고도 함) 는 개발 로세스에
한 한 기록이 없는, 이 에 개발된 소 트웨어
3.30 시스템(SYSTEM)
하나 이상의 로세스, 하드웨어, 소 트웨어, 시설 명시한 요구나 목 을 충족시킬
수 있는 성능을 제공하는 사람으로 통합 구성된 합성체. [ISO/IEC 12207:1995, 정의 3.31]

- 132 -
3.31 업무(TASK)
완수해야 할 작업의 한 부분
3.32 추 성(TRACEABILITY)
개발 로세스의 두 개 이상 제품 사이에 확립할 수 있는 계의 정도 [IEEE
610.12:1990]
3.33 검증(VERIFICATION)
특정 요구사항이 충족되었다는 객 증거의 제공을 통해 확인(confirmation)하는
것
주 1 : ‘Verified(검증된)’이라는 용어는 해당 상태의 지정에 사용된다. [ISO
9000:2000, 정의 3.8.4]
주 2 : 설계와 개발의 경우 검증은 일반 으로 특정 활동에 해 명시한 요구사항과
의 합성을 단하기 해 그러한 활동의 결과를 조사하는 로세스와 연
된다.
3.3.4 버 (VERSION)
형상 항목(item)의 식별된 인스턴스(instance).
주 1 : 새 버 이 작성되며 형상 리 조치가 필요한, 소 트웨어 제품 버 의 수정
주 2 : ISO/IEC 12207:1995, 정의 3.37에 기 .
4. 일반 요구사항
4.1 품질경 시스템
의료기기 소 트웨어 제조업체는 일 성 있게 고객 요구사항 해당 규제 요구사
항을 충족하는 의료기기 소 트웨어를 제공할 수 있는 능력을 입증해야 한다.
주 1 : 이 능력은 다음을 수하는 품질경 시스템의 사용으로 입증할 수 있다.
- ISO 13485 [7] 는
- 국가 품질경 시스템 규격, 는
- 국가 법규에 따라 요구되는 품질경 시스템
주 2 : 소 트웨어에 한 품질경 시스템 요구사항 용에 한 지침은 ISO/IEC
90003 [11]에 수록되어 있다.

- 133 -
4.2 험 리
제조업체는 ISO 14971을 수하는 험 리 로세스를 용해야 한다.
4.3 소 트웨어 안 성 분류
a) 제조업체는 소 트웨어 시스템에서 발생하며, 환자, 작업자 는 기타 사람에
한 해의 향에 따라 각 소 트웨어 시스템에 소 트웨어 안 성 등 을 할당
해야 한다(A, B 는 C).
소 트웨어 안 성 등 은 기에는 다음과 같은 심각도에 기 해 할당된다.
▪등 A: 부상이나 건강 피해가 없다.
▪등 B: 상이 가능하지 않다.
▪등 C: 사망이나 상이 가능하다.
소 트웨어 시스템이 지정한 로 작동하지 않아 해가 발생할 경우 그러한
고장의 발생 가능성은 100퍼센트로 간주되어야 한다.
b) 소 트웨어 고장에서 발생하는 사망이나 상의 험을 고장의 결과를 감소시키거
나 고장에서 발생하는 사망이나 상의 발생 가능성을 감소함으로써 하드웨어 험
리 방법으로 허용할 수 있는 수 (ISO 14971에 정의)으로 축소할 수 있는 경우
소 트웨어 안 성 분류는 C에서 B로 축소할 수 있다. 한 소 트웨어 고장에서
발생하는 비 상의 험이 하드웨어 험 리 방법으로 유사하게 허용 가능한 수
으로 감소될 경우 소 트웨어 안 성 분류는 B에서 A로 축소할 수 있다. 제조업
체는 험 리 방법이 리 인 향에 기 한 소 트웨어 안 성 등 을 험
리 방법의 구 에 기여하는 각 소 트웨어 시스템에 할당해야 한다.
c) 제조업체는 각 소 트웨어 시스템에 할당된 소 트웨어 안 성 등 을 험 리 일
에 문서화해야 한다.
d) 소 트웨어 시스템을 소 트웨어 항목(item)으로 분해하고 소 트웨어 항목(item)을
다시 다른 소 트웨어 항목(item)으로 분해하면 제조업체가 다른 소 트웨어 안
성 등 으로의 분류에 한 이론 근거를 문서화하지 않는 한 그러한 소 트웨어
항목(item)은 원래 소 트웨어 항목(item)( 는 소 트웨어 시스템)의 소 트웨어 안
성 분류를 계승한다. 그러한 이론 근거에는 새로운 소 트웨어 항목(item)이 별
도로 분류될 수 있도록 분리되는 방법의 설명이 포함되어야 한다.

- 134 -
e) 제조업체는 소 트웨어 안 성 등 이 분해에 의해 생성된 소 트웨어 항목(item)
등 과 다를 경우 각 소 트웨어 항목(item)의 소 트웨어 안 성 등 을 문서화해야
한다.
f) 이 표 의 수성을 해 특정 분류의 소 트웨어 항목(item)에 로세스가 필요
하거나 소 트웨어 항목(item) 그룹에 로세스를 용해야 할 때마다 제조업체가
낮은 수 의 분류 사용에 한 이론 근거를 험 리 일에 문서화하지 않는
한 제조업체는 그룹 가장 상 로 분류된 로세스와 업무를 사용해야 한다.
g) 각 소 트웨어 시스템의 경우 소 트웨어 안 성 등 이 할당될 때까지 등 C 요
구사항이 용된다.
주 : 뒤따르는 요구사항에서 요구사항을 수행해야 하는 소 트웨어 안 성 등 은
[등 …]의 형식으로 요구사항 뒤에 식별된다.
5. 소 트웨어 개발 로세스
5.1 소 트웨어 개발 기획
5.1.1 소 트웨어 개발 계획
제조업체는 개발 상 소 트웨어 시스템의 범 , 규모 소 트웨어 안 성 분류
에 합한 소 트웨어 개발 로세스의 활동 진행을 한 소 트웨어 개발 계획을 확
립해야 한다. 소 트웨어 개발 수명주기 모델을 계획에 완벽히 정의하거나 참조해야 한
다. 계획은 다음의 내용을 취 해야 한다.
a) 소 트웨어 시스템의 개발에 사용되는 로세스(주 4 참조)
b) 활동 업무의 산출물(문서화 포함)
c) 시스템 요구사항, 소 트웨어 요구사항, 소 트웨어 시스템 시험 소 트웨어
에 구 되는 험 리 방법 사이의 추 성
d) SOUP 형상 항목(item) 개발 지원에 사용되는 소 트웨어를 포함하여 소
트웨어 형상 변경 리
e) 수명주기의 각 단계에서 소 트웨어 제품, 산출물 활동 확인(detected)되는
문제 취 을 한 소 트웨어 문제해결 [등 A, B, C]
주 1 : 소 트웨어 개발 수명주기 모델은 소 트웨어 시스템의 각 소 트웨어 항목
(item)에 한 소 트웨어 안 성 분류에 따라 서로 다른 소 트웨어 항목
(item)에 해 서로 다른 요소( 로세스, 활동, 업무 산출물)를 확인(identify)
할 수 있다.

- 135 -
주 2 : 이 활동과 업무는 복되거나 상호 작용할 수 있으며, 반복 으로 는 귀납
으로 수행할 수 있다. 단, 특정 수명주기 모델을 사용해야 한다고 암시하
는 것이 아니다.
주 3 : 다른 로세스는 개발 로세스와는 별도로 이 표 에 설명된다. 단, 다른
로세스가 별도 활동과 업무로서 구 되어야 한다고 암시하는 것은 아니
다. 다른 로세스의 활동과 업무는 개발 로세스에 통합할 수 있다.
주 4 : 소 트웨어 개발 계획은 기존 로세스를 참조하거나 새 로세스를 정의
할 수 있다.
주 5 : 소 트웨어 개발 계획은 체 시스템 개발 계획에 통합할 수 있다.
5.1.2 갱신된 소 트웨어 개발 계획
제조업체는 개발이 진행되면 그에 상응하게 계획을 갱신해야 한다. [등 A, B, C]
5.1.3 시스템 설계와 개발에 한 소 트웨어 개발 계획 참조
a) 소 트웨어 요구사항은 소 트웨어 개발을 한 입력으로서 제조업체가 소 트
웨어 개발계획에 참조해야 한다.
b) 제조업체는 4.1의 충족을 하여 소 트웨어 개발과 설계 개발 유효성 확인을
일원화하기 해 소 트웨어 개발 계획 차를 포함시키거나 참조해야 한다. [등
A, B, C]
주 : 소 트웨어 시스템이 자립형 시스템(소 트웨어로만 구성된 장비)인 경우 소
트웨어 시스템 요구사항과 시스템 요구사항에는 차이가 있을 수 있다.
5.1.4 소 트웨어 개발 표 , 방법 도구 기획
제조업체는 소 트웨어 개발 계획에 다음 사항을 포함시키거나 참조해야 한다.
a) 표
b) 방법
c) 도구
이들 사항은 등 C의 소 트웨어 항목(item) 개발에 련된다. [등 C]
5.1.5 소 트웨어 통합 통합 시험 기획
제조업체는 소 트웨어 항목(item)(SOUP 포함)을 통합하고 통합 시험을 수행하기
한 계획을 소 트웨어 개발 계획에 포함시키거나 참조해야 한다.[등 B, C]
주 : 통합 시험 소 트웨어 시스템 시험을 단일 계획 활동으로 결합할 수 있다.

- 136 -
5.1.6 소 트웨어 검증 기획
제조업체는 소 트웨어 개발 계획에 다음과 같은 검증 정보를 포함시키거나 참조해
야 한다. [등 A, B, C]
a) 검증이 필요한 산출물
b) 각 수명주기 활동에 요구되는 검증업무
c) 산출물을 검증하는 일정
d) 산출물 검증 인수 기
5.1.7 소 트웨어 험 리 기획
제조업체는 SOUP에 련된 험의 경 을 포함해 소 트웨어 험 리 로세스의
활동과 업무 수행을 한 계획을 소 트웨어 개발 계획에 포함시키거나 참조해야 한
다. [등 A, B, C]
주 : 제7조 참조
5.1.8 문서화 기획
제조업체는 소 트웨어 개발 수명주기 생산되는 문서에 한 정보를 소 트웨어
개발 계획에 포함시키거나 참조해야 한다. 식별된 각 문서나 문서 유형에 있어 다음과 같
은 정보를 포함시키거나 참조해야 한다. [등 A, B, C]
a) 제목, 명칭 명칭 표기법
b) 목
c) 문서의 사용 상자
d) 개발, 검토, 승인 수정을 한 차와 책임
5.1.9 소 트웨어 형상 리 기획
제조업체는 소 트웨어 개발 계획에 소 트웨어 형상 리 정보를 포함시키거나 참조
해야 한다. 소 트웨어 형상 리 정보에 다음 내용을 포함시키거나 참조해야 한다. [등
A, B, C]
a) 리 상 항목(item)의 등 , 유형, 범주 는 목록
b) 소 트웨어 형상 리 활동 업무
c) 소 트웨어 형상 리와 활동의 수행을 담당하는 조직
d) 소 트웨어 개발 는 유지보수와 같은 다른 조직과의 계
e) 항목(item)을 형상 리 상으로 정하는 시기
f) 문제해결 로세스를 사용할 시기

- 137 -
5.1.10 리할 지원 항목(item)
리할 항목(item)에는 의료기기 소 트웨어 개발에 사용되며 의료기기 소 트웨어에
향을 수 있는 도구, 항목(item) 설정이 포함된다. [등 B, C]
주 : 그러한 항목의 에는 컴 일러/어셈블러 버 , 제작 일, 배치 일 특정
환경 설정이 포함된다.
5.1.11 검증 소 트웨어 형상 항목(item) 리
제조업체는 형상 항목(item)을 검증하기 문서화된 형상 리 제어 아래에 형상 항
목(item)을 배치하도록 계획해야 한다. [등 B, C]
5.2 소 트웨어 요구사항 분석
5.2.1 시스템 요구사항에서 소 트웨어 요구사항을 정의하고 문서화
의료기기에 사용되는 각 소 트웨어 시스템의 경우 제조업체는 시스템 차원 요구사
항에서 소 트웨어 시스템 요구사항을 정의하고 문서화해야 한다.[등 A, B, C]
주 : 소 트웨어 시스템이 자립형 시스템(소 트웨어로만 구성된 장비)인 경우 소
트웨어 시스템 요구사항과 시스템 요구사항에는 차이가 있을 수 있다.
5.2.2 소 트웨어 요구사항 내용
의료기기 소 트웨어에 합하도록 제조업체는 소 트웨어 요구사항에 다음을 포함
시켜야 한다.[등 A, B, C]
a) 기능 성능 요구사항
주 1 : 에는 다음이 포함된다.
- 성능( 를 들어 소 트웨어 목 , 시 요구사항)
- 물리 특성( 를 들어 코드 언어, 랫폼, 운 체제)
- 소 트웨어가 수행되는 산환경( 를 들어 하드웨어, 메모리 크기, 처리
장치, 시간 , 네트워크 기반구조)
- 업그 이드나 복수 SOUP 는 기타 장비 버 과의 호환성 요구
b) 소 트웨어 시스템 입력과 출력
주 2 : 에는 다음이 포함된다.
- 데이터 특성( 를 들어 숫자, 숫자, 형식)
- 범
- 한계
- 기본값

- 138 -
c) 소 트웨어 시스템과 다른 소 트웨어 사이의 연계성
d) 소 트웨어 구동 경보, 경고 운 자 메시지
e) 보안 요구사항
주 3 : 에는 다음이 포함된다.
- 요한 정보의 노출 험에 련되는 요구사항
- 정체확인(authentication)
- 인가
- 심사추
- 통신 무결성
f) 사람에 의해 발생하는 오류와 훈련에 민감한 활용성 공학 요구사항
주 4 : 에는 다음에 련된 요구사항이 포함된다.
- 수동 운 지원
- 인간-장치 상호작용
- 담당자에 한 제약사항
- 사람의 주의 집 이 필요한 역
주 5 : 활용성 공학 요구사항에 한 정보는 IEC 60601-1-6에 수록되어 있다.
g) 데이터 정의 데이터베이스 요구사항
주 6 : 에는 다음이 포함된다.
- 형태
- 응성
- 기능
h) 운 유지보수 장에 인도된 의료기기 소 트웨어의 설치 인수 요구사항
i) 운 유지보수 방법에 련된 요구사항
j) 개발할 사용자 설명서
k) 사용자 유지보수 요구사항

- 139 -
l) 규제 요구사항
주 7 : 이러한 모든 요구사항은 소 트웨어 개발을 시작할 때에는 사용하지 못할 수
있다.
주 8 : ISO/IEC 9126-1[8]에는 소 트웨어 요구사항 정의에 유용할 수 있는 품질 특성
에 한 정보가 수록되어 있다.
5.2.3 소 트웨어 요구사항에 험 리 방법 포함
제조업체는 하드웨어 고장 잠재 소 트웨어 결함에 해 소 트웨어에 구 한
험 리 방법을 의료기기 소 트웨어에 합한 요구사항에 포함시켜야 한다. [등 B,
C]
주 : 이러한 요구사항은 소 트웨어 개발을 시작할 때 사용할 수 없을 수 있으며, 소
트웨어가 설계되고 험 리 방법이 계속 정의되는 동안 변할 수 있다.
5.2.4 의료기기 험분석의 재평가
제조업체는 소 트웨어 요구사항이 확립되고 갱신되면 의료기기 험평가를 재평가
해야 한다. [등 A, B, C]
5.2.5 시스템 요구사항 갱신
제조업체는 시스템 요구사항을 포함해 기존의 요구사항이 소 트웨어 요구사항 분석
활동의 결과에 합하게 재평가되고 갱신되는지 확인(ensure)해야 한다. [등 A, B, C]
5.2.6 소 트웨어 요구사항 검증
제조업체는 소 트웨어 요구사항이 다음과 같은지 검증하고 문서화해야 한다. [등
A, B, C]
a) 험 리에 련된 요구사항을 포함해 시스템 요구사항을 구 한다.
b) 서로 상충하지 않는다.
c) 모호한 표 을 방지하는 용어로 표 된다.
d) 시험 기 이 충족되는지 단하기 해 시험 기 과 시험 성능을 확립할 수 있
는 용어로 언 된다.
e) 서로 다르게 식별할 수 있다.
f) 시스템 요구사항이나 다른 발생원까지 추 할 수 있다.
주 : 이 표 에 따르면 공식 시방서 언어를 사용할 필요는 없다.

- 140 -
5.3 소 트웨어 구축 설계
5.3.1 소 트웨어 요구사항을 아키텍처로 변형
제조업체는 의료기기 소 트웨어에 한 요구사항을 소 트웨어 구조를 설명하고 소
트웨어 항목을 식별하는 문서화된 아키텍처로 변환해야 한다. [등 B, C]
5.3.2 소 트웨어 항목의 연계성을 이한 아키텍처 개발
제조업체는 소 트웨어 항목과 소 트웨어 항목 외부의 구성요인(소 트웨어와 하드
웨어) 사이의 연계성과 소 트웨어 항목 사이의 연계성을 한 아키텍처를 개발하고
문서화해야 한다. [등 B, C]
5.3.3 SOUP 항목의 기능 성능 요구사항 명시
소 트웨어 항목이 SOUP로 식별된 경우 제조업체는 의도한 사용에 필요한 SOUP
항목에 한 기능 성능 요구사항을 명시해야 한다. [등 B, C]
5.3.4 SOUP 항목에 필요한 시스템 하드웨어와 소 트웨어 명시
소 트웨어 항목이 SOUP로 식별된 경우 제조업체는 SOUP 항목의 한 운 지원
에 필요한 시스템 하드웨어와 소 트웨어를 명시해야 한다. [등 B, C]
주 : 에는 로세서 유형과 속도, 메모리 유형과 크기, 시스템 소 트웨어 유형, 통
신 디스 이 소 트웨어 요구사항이 포함된다.
5.3.5 험 리에 필요한 분리 식별
제조업체는 험 리에 필수 인 소 트웨어 항목 사이의 분리를 식별하고 분리가
유효한지 확인(ensure)하는 방법을 명시해야 한다. [등 C]
주 : 분리의 한 는 소 트웨어 항목이 다른 로세서에서 실행되도록 만드는 것이
다. 분리의 유효성은 로세서 사이에 자원을 공유하지 않으면 확인(ensure)할
수 있다.
5.3.6 소 트웨어 아키텍처 검증
제조업체는 다음을 검증하고 문서화해야 한다. [등 B, C]
a) 소 트웨어 아키텍처가 험 리에 련된 요구사항을 포함해 시스템 소
트웨어 요구사항을 구 한다.
b) 소 트웨어 아키텍처는 소 트웨어 항목 사이의 연계성과 소 트웨어 항목
하드웨어 사이의 연계성을 지원할 수 있다.
c) 의료기기 아키텍처는 모든 SOUP 항목의 한 운 을 지원한다.

- 141 -
5.4 소 트웨어 상세 설계
5.4.1 소 트웨어 아키텍처를 소 트웨어 유닛으로 개량
제조업체는 소 트웨어 유닛으로 표 될 때까지 소 트웨어 아키텍처를 개량해야 한
다. [등 B, C]
5.4.2 각 소 트웨어 유닛에 해 상세 설계 개발
제조업체는 소 트웨어 항목의 각 소 트웨어 유닛에 한 상세 설계를 개발하고 문
서화해야 한다. [등 C]
5.4.3 연계성에 한 상세 설계 개발
제조업체는 소 트웨어 유닛 외부 구성요인(하드웨어나 소 트웨어) 사이의 연계
성 소 트웨어 유닛 사이의 연계성을 한 상세 설계를 개발하고 문서화해야 한다.
[등 C]
5.4.4 상세 설계 검증
제조업체는 소 트웨어 상세 설계가 다음과 같은지 검증하고 문서화해야 한다. [등
C]
a) 소 트웨어 아키텍처를 구 한다.
b) 소 트웨어 아키텍처와 상충하지 않는다.
5.5 소 트웨어 유닛 구 검증
5.5.1 각 소 트웨어 유닛 구
제조업체는 각 소 트웨어 유닛을 구 해야 한다. [등 A, B, C]
5.5.2 소 트웨어 유닛 검증 로세스 확립
제조업체는 각 소 트웨어 유닛 검증을 한 략, 방법 차를 확립해야 한다. 시
험을 검증할 경우 시험 차의 정확성을 평가해야 한다. [등 B, C]
주 : 통합 시험 소 트웨어 시스템 시험을 단일 계획 활동으로 결합할 수 있
다.
5.5.3 소 트웨어 유닛 인수 기
제조업체는 해당되는 경우 규모가 큰 소 트웨어 항목으로 통합하기 소 트웨어
유닛에 한 인수 기 을 확립하고, 소 트웨어 유닛이 인수 기 을 충족하는지 확인
해야 한다. [등 B, C]
주 : 인수 기 의 는 아래와 같다.
- 소 트웨어 코드가 험 리 방법을 포함해 요구사항을 구 하는가?

- 142 -
- 소 트웨어 코드가 소 트웨어 유닛의 상세 설계에 문서화한 연계성과 상충하
지 않는가?
- 소 트웨어 코드가 로그래 차나 코딩 표 과 일치하는가?
5.5.4 소 트웨어 유닛 추가 인수 기
제조업체는 설계에 필요한 경우 다음과 같은 추가 인수 기 을 포함시켜야 한다. [등
C]
a) 한 사상의 순서
b) 데이터 리 흐름
c) 계획한 자원 할당
d) 결합 취 (오류 정의, 확인 복구)
e) 변수의 기화
f) 자체 진단
g) 메모리 리 메모리 용량 과
h) 경계 조건
5.5.5 소 트웨어 유닛 검증
제조업체는 소 트웨어 유닛 검증을 수행하고 결과를 문서화해야 한다. [등 B, C]
5.6 소 트웨어 통합 통합 시험
5.6.1 소 트웨어 유닛 통합
제조업체는 통합계획(5.1.5 참조)에 따라 소 트웨어 유닛을 통합해야 한다.[등 B, C]
5.6.2 소 트웨어 통합 검증
제조업체는 통합계획(5.1.5 참조)에 따라 소 트웨어 통합에 한 다음과 같은 사항을
검증하고 기록해야 한다. [등 B, C]
a) 소 트웨어 유닛이 소 트웨어 항목(item)과 소 트웨어 시스템으로 통합되었다.
b) 하드웨어 항목(item), 소 트웨어 항목(item) 시스템의 수동 운 ( ; 사람 연계
성, 온라인 도움말 메뉴, 음성 식별, 음성 리)을 한 지원이 시스템에 통합되었
다.
주 : 이 검증은 항목(item)이 의도된 로 기능을 수행하는지 검증이 아니라, 계획에
따라 항목(item)이 통합되었는지 검증하는 것이다. 이 검증은 몇 가지 형태의
검사에 의해 구 된다.

- 143 -
5.6.3 통합 소 트웨어 시험
제조업체는 통합계획(5.1.5 참조)에 따라 통합된 소 트웨어 유닛을 시험하고 결과를
문서화해야 한다. [등 B, C]
5.6.4 통합 시험 내용
소 트웨어 통합 시험의 경우 제조업체는 통합된 소 트웨어 항목이 의도한 로 기능
을 발휘하는지 확인(address)해야 한다. [등 B, C]
주 1 : 고려해야 할 는 다음과 같다.
- 소 트웨어에 요구되는 기능성
- 험 리 방법의 구
- 지정한 시 기타 작동상태
- 내부 외부 연계성의 지정 기능
- 측할 수 있는 오용을 포함해 비정상 조건에서 시험
주 2 : 통합 시험 소 트웨어 시스템 시험을 단일 계획 활동으로 결합할 수
있다.
5.6.5 통합 시험 차 검증
제조업체는 통합 시험 차의 정확성을 평가해야 한다. [등 B, C]
5.6.6 회귀시험 수행
소 트웨어 항목을 통합할 때 제조업체는 이 에 통합한 소 트웨어에 결함이 발생
하지 않았음을 입증하기에 합한 회귀시험을 수행해야 한다. [등 B, C]
5.6.7 통합시험 기록 내용
제조업체는 다음을 수행해야 한다. [등 B, C]
a) 시험 결과를 문서화한다(합격/불합격 비정상 상태 목록).
b) 시험을 반복할 수 있도록 충분한 기록을 유지한다.
c) 시험자를 식별한다.
주 : 요구사항 b)는 를 들어 다음을 유지함으로써 구 할 수 있다.
- 요구되는 조치와 상되는 결과를 나타내는 시험 사례 시방서
- 장치 기록
- 시험에 사용하는 시험 환경(소 트웨어 도구 포함)의 기록

- 144 -
5.6.8 소 트웨어 문제해결 로세스의 사용
제조업체는 소 트웨어 통합 통합 시험 확인(found)한 비정상 상태를 소 트
웨어 문제해결 로세스에 입력해야 한다. [등 B, C]
주 : 제9조 참조
5.7 소 트웨어 시스템 시험
5.7.1 소 트웨어 요구사항에 한 시험 확립
제조업체는 모든 소 트웨어 요구사항이 포함되도록, 입력 진, 상 출력, 합격/불
합격 기 과 차로 표 되는 시험을 확립하고 수행해야 한다. [등 B, C]
주 1 : 통합 시험 소 트웨어 시스템 시험을 단일 계획 활동으로 결합할 수
있다. 한 조기 단계에서 소 트웨어 요구사항을 시험할 수도 있다.
주 2 : 특히 요구사항 사이에 의존성이 있을 경우 각 요구사항에 한 별도의 시험
뿐 아니라 요구사항의 결합에 한 시험도 수행할 수 있다.
5.7.2 소 트웨어 문제해결 로세스의 사용
제조업체는 소 트웨어 시스템 시험 확인(found)한 비정상 상태를 소 트웨어 문제
해결 로세스에 입력해야 한다. [등 B, C]
5.7.3 변경 후 재시험
소 트웨어 시스템 시험 변경이 있을 경우 제조업체는 다음을 수행해야 한다. [등
B, C]
a) 문제해결에 있어 변경의 유효성 검증을 해 시험을 반복하건, 수정한 시험을 수행
하거나 추가 시험을 수행한다.
b) 의도하지 않은 부작용이 발생하지 않음을 입증하기에 합한 시험을 수행한다.
c) 7.4에 정의한 것처럼 련 험 리 활동을 수행한다.
5.7.4 소 트웨어 시스템 시험 검증
제조업체는 다음을 검증한다. [등 B, C]
a) 사용한 검증 략과 시험 차가 하다.
b) 소 트웨어 시스템 시험 차가 소 트웨어 요구사항까지 추 한다.
c) 모든 소 트웨어 요구사항을 시험하거나 검증했다.
d) 시험 결과가 요구되는 합격/불합격 기 을 충족한다.

- 145 -
5.7.5 소 트웨어 시스템 시험 기록 내용
제조업체는 다음을 수행해야 한다. [등 B, C]
a) 시험 결과를 문서화한다(합격/불합격 비정상 상태 목록).
b) 시험을 반복할 수 있도록 충분한 기록을 유지한다.
c) 시험자를 식별한다.
주 : 요구사항 b)는 를 들어 다음을 유지함으로써 구 할 수 있다.
- 요구되는 조치와 상되는 결과를 나타내는 시험 사례 시방서
- 장치 기록
- 시험에 사용하는 시험 환경(소 트웨어 도구 포함)의 기록
5.8 소 트웨어 릴리즈
5.8.1 소 트웨어 확인의 완벽성 확인(ensure)
제조업체는 소 트웨어 검증이 완료되었으며 소 트웨어를 릴리즈 하기 결과가
평가되었는지 확인(ensure)해야 한다. [등 B, C]
5.8.2 확인(known)된 잔류 비정상 상태 문서화
제조업체는 모든 확인(known)된 잔류 비정상 상태를 문서화해야 한다. [등 B, C]
5.8.3 확인(known)된 잔류 비정상 상태 평가
제조업체는 확인(known)한 모든 잔류 비정상 상태가 허용되지 않는 험을 발생시
키지 않는지 확인(ensure)하기 해 그러한 비정상 상태가 평가되었는지 확인(ensure)해
야 한다. [등 B, C]
5.8.4 릴리즈 버 의 문서화
제조업체는 릴리즈 인 소 트웨어 제품의 버 을 문서화해야 한다.[등 A, B, C]
5.8.5 릴리즈한 소 트웨어를 작성한 방법 문서화
제조업체는 릴리즈한 소 트웨어 작성에 사용한 차와 환경을 문서화해야 한다.[등
B, C]
5.8.6 활동과 업무의 완결 확인(ensure)
제조업체는 모든 련 문서화와 함께 모든 활동과 업무가 완결되었는지 확인(ensure)
해야 한다. [등 B, C]

- 146 -
5.8.7 소 트웨어 장
제조업체는 최소 제조업체가 정의한 장비의 수명시간과 련 규제 요구사항에 의해
명시된 시간 긴 시간으로 결정된 기간 동안 다음을 장해야 한다. [등 B, C]
a) 소 트웨어 제품 형상 항목
b) 문서
5.8.8 소 트웨어 릴리즈의 반복 정 도 보장
제조업체는 릴리즈한 소 트웨어 제품이 개악이나 무단 변경 없이 신뢰성 있게 사용
지 까지 인도되는지 확인(ensure)할 수 있는 차를 확립해야 한다. 이 차는 다음을
포함해 소 트웨어 제품이 수록된 매체의 생산과 취 을 처리해야 한다. [등 B, C]
- 복제
- 매체 라벨링
- 포장
- 보호
- 보
- 인도
6. 소 트웨어 유지보수 로세스
6.1 소 트웨어 유지보수 계획 확립
제조업체는 유지보수 로세스의 활동과 업무 진행을 한 소 트웨어 유지보수 계획
을 확립해야 한다. 계획은 다음의 내용을 취 해야 한다. [등 A, B, C]
a) 다음에 한 차
- 수신
- 문서화
- 평가
- 문제해결
- 추 ; 의료기기 소 트웨어의 릴리즈 후 발생하는 피드백
b) 피드백이 문제로 간주되는지 여부를 결정하기 한 기
c) 소 트웨어 험 리 로세스의 사용
d) 의료기기 소 트웨어 릴리즈 후 발생하는 문제 분석과 해결을 한 소 트웨어
문제해결 로세스의 사용
e) 기존 소 트웨어 수정 리를 해 형상 리 로세스(제8조) 사용
f) 평가 구 을 한 차

- 147 -
- 갱신
- 버그 해결
- 패치
- SOUP의 폐기
6.2 문제 수정 분석
6.2.1 피드백 문서화 평가
6.2.1.1 피드백 모니터링
제조업체는 자체 조직 내부와 사용자로부터 릴리즈된 소 트웨어 제품에 한 피드
백을 모니터링 해야 한다. [등 A, B, C]
6.2.1.2 피드백 문서화 평가
릴리즈한 소 트웨어 제품에 문제가 존재하는지 여부를 단하기 해 피드백을 문
서화하고 평가해야 한다. 그러한 문제는 모두 문제 보고서로서 기록해야 한다(제9조 참
조). 문제 보고서에는 실제 는 잠재 부정 사상 시방서와의 편차가 포함되어
야 한다. [등 A, B, C]
6.2.1.3 안 에 한 문제 보고서 향의 평가
문제가 릴리즈된 소 트웨어 제품에 어떤 향을 주는지, 릴리즈한 소 트웨어 제
품에 한 변경 때문에 문제의 처리가 필요한지 여부의 결정을 해 각 문제 보고서를
평가해야 한다. [등 A, B, C]
6.2.2 소 트웨어 문제해결 로세스의 사용
제조업체는 문제 보고서 처리를 해 소 트웨어 문제해결 로세스(제9조 참조)
를 사용해야 한다. [등 A, B, C]
주 : 이 활동이 완료되면 소 트웨어 시스템이나 소 트웨어 항목의 안 등 의 변경
을 악해야 한다.
6.2.3 변경 요청 분석
제9조에서 요구하는 분석 이외에도 제조업체는 조직, 릴리즈한 소 트웨어 제품
제품이 연계성을 갖는 시스템에 한 향의 악을 해 각 변경 요청을 분석해야
한다. [등 B, C]

- 148 -
6.2.4 변경 요청 승인
제조업체는 릴리스한 소 트웨어 제품을 수정하는 변경 요청을 평가하고 승인해야
한다. [등 A, B, C]
6.2.5 사용자 규제자와의 의사소통
제조업체는 승인된 변경 요청 릴리스한 소 트웨어 제품에 향을 주는 변경 요청
을 식별해야 한다.
제조업체는 지역 규정에 의해 요구되는 경우 다음의 내용을 사용자와 규제자에게 알
려야 한다. [등 A, B, C]
a) 릴리스한 소 트웨어 제품의 문제 변경하지 않고 계속 사용함으로써 발생
하는 결과
b) 릴리즈한 소 트웨어 제품에 사용할 수 있는 변경의 성격 변경 내용을 확보
하고 설치하는 방법
6.3 수정 구
6.3.1 확립된 로세스를 사용해 수정 구
제조업체는 소 트웨어 개발 로세스(제5조 참조)나 수정의 구 을 해 확립한 유
지보수 로세스를 사용해야 한다. [등 A, B, C]
주 : 소 트웨어 변경의 험 리에 련된 요구사항은 7.4를 참조하라.
6.3.2 수정한 소 트웨어 시스템의 재 릴리즈
제조업체는 5.8에 따라 수정한 소 트웨어 시스템을 릴리즈해야 한다. 수정은 소 트
웨어 시스템의 체 재 릴리즈 일환으로서, 는 변경된 소 트웨어 키트와 기존 소
트웨어 시스템에 변경을 수정으로서 설치하기에 필요한 도구로 구성된 수정 키트로서
릴리즈할 수 있다. [등 A, B, C]
7. 소 트웨어 험 리 로세스
7.1 험한 상황에 한 소 트웨어 향의 분석
7.1.1 험한 상황에 향을 수 있는 소 트웨어 항목의 식별
제조업체는 ISO 14971의 의료기기 분석활동에서 식별된 험한 상황에 향을 수
있는 소 트웨어 항목을 식별해야 한다(4.2 참조). [등 B, C]
주 : 험한 상황은 소 트웨어 고장의 직 인 결과, 는 소 트웨어에 구 되는
험 리 방법의 결함 결과에 의한 것일 수 있다.

- 149 -
7.1.2 험한 상황에 향을 수 있는 잠재 원인의 식별
제조업체는 험한 상황에 향을 주는 것으로 에서 식별한 소 트웨어 항목의 잠
재 원인을 식별해야 한다. [등 B, C]
제조업체가 고려해야 할 잠재 원인은 다음과 같다.
a) 기능성의 부정확하거나 불완 한 시방서
b) 식별한 소 트웨어 항목 기능성에 있어 소 트웨어의 결함
c) SOUP의 고장 는 상하지 못한 결과
d) 측할 수 없는 소 트웨어 운 을 야기할 수 있는 하드웨어 고장이나 기타 소
트웨어 결함
e) 타당한 수 으로 측할 수 있는 오용
7.1.3 발표된 SOUP 비정상 상태 목록의 평가
SOUP의 고장이나 상하지 못한 결과가 소 트웨어 항목이 험한 상황에 향을
주는 잠재 원인일 경우 제조업체는 최소한 의료기기에 사용되는 SOUP 항목의 버
과 련해 SOUP 항목의 공 업체가 발표한 비정상 상태 목록을 평가해 확인(known)된
비정상 상태에 의해 험한 상황을 야기할 수 있는 일련의 사상이 발생했는지 단해
야 한다. [등 B, C]
7.1.4 잠재 원인의 문서화
제조업체는 험한 상황에 향을 주는 소 트웨어 항목의 잠재 원인을 험 리
일에 문서화해야 한다(ISO 14971 참조). [등 B, C]
7.1.5 사상 발생순서 문서화
제조업체는 7.1.2에서 식별한 험한 상황을 야기할 수 있는 사상의 발생순서를 험
리 일에 문서화해야 한다. [등 B, C]
7.2 험 리 방법
7.2.1 험 리 방법 정의
험 리 일에 문서화한, 험한 상황에 향을 주는 소 트웨어 항목의 각 잠재
원인에 있어 제조업체는 험 리 방법을 정의하고 문서화해야 한다. [등 B, C]
주 : 험 리 방법은 하드웨어, 소 트웨어, 작업환경 는 사용자 설명서에 구 할
수 있다.

- 150 -
7.2.2 소 트웨어에 구 된 험 리 방법
험 리 방법이 소 트웨어 항목의 기능 일부로서 구 된 경우 제조업체는 다음과
같이 수행해야 한다. [등 B, C]
a) 소 트웨어 요구사항에 험 리 방법을 포함시킨다.
b) 험 리 방법이 리 인 해의 향에 기 해 소 트웨어 항목에 소 트웨
어 안 성 등 을 할당한다.
c) 제5조에 따라 소 트웨어 항목을 개발한다.
주 : 이 요구사항은 ISO 14971의 험 리 요구사항에 한 상세한 설명을 추가로
제공한다.
7.3 험 리 방법의 검증
7.3.1 험 리 방법 검증
7.2에 문서화한 각 험 리 방법의 구 은 검증되어야 하며, 검증 결과를 문서화해야
한다. [등 B, C]
7.3.2 사상의 새로운 발생순서 문서화
험 리 방법을 소 트웨어 항목으로서 구 한 경우 제조업체는 험 리 방법을
평가해 험한 상황을 야기할 수 있는 사상의 새로운 발생순서를 식별하고 험 리
일에 문서화해야 한다. [등 B, C]
7.3.3 추 성 문서화
제조업체는 소 트웨어 험요인의 추 성을 다음과 같이 문서화해야 한다. [등 B,
C]
a) 험한 상황부터 소 트웨어 항목까지
b) 소 트웨어 항목부터 특정 소 트웨어 원인까지
c) 소 트웨어 원인이나 험 리 방법까지
d) 험 리 방법부터 험 리 방법의 검증까지
주 : ISO 14971 – 험 리 보고서 참조
7.4 소 트웨어 변경의 험 리
7.4.1 안 성과 련해 의료기기 소 트웨어에 한 변경 분석
제조업체는 다음의 내용을 악하기 해 의료기기 소 트웨어(SOUP 포함)에 한
변경을 분석해야 한다. [등 A, B, C]
a) 험한 상황에 향을 수 있는 추가 잠재 원인
b) 추가로 필요한 소 트웨어 험 리 방법

- 151 -
7.4.2 기존 험 리 방법에 한 소 트웨어 변경의 향 분석
제조업체는 SOUP에 한 변경을 포함해 소 트웨어에 한 변경을 분석해 소 트
웨어 수정이 기존 험 리 방법을 방해할 수 있는지의 여부를 단해야 한다. [등
B, C]
7.4.3 분석에 기 해 험 리 활동 수행
제조업체는 이러한 분석에 기 해 7.1, 7.2 7.3에 정의한 련 험 리 활동을 수
행해야 한다. [등 B, C]
8. 소 트웨어 형상 리 로세스
8.1 형상 식별
8.1.1 형상항목(item)을 식별하기 한 방법 확립
제조업체는 로젝트를 해 리해야 하는 형상 항목 그 버 의 고유 식별을
한 계획을 수립해야 한다. 이 계획에는 SOUP와 문서와 같은 다른 소 트웨어 제품이
나 실체가 포함되어야 한다. [등 A, B, C]
8.1.2 SOUP 식별
표 라이 러리를 포함해 사용 인 각 SOUP 형상항목의 경우 제조업체는 각 형
상항목에 해 다음을 문서화해야 한다. [등 A, B, C]
a) 제목
b) 제조업체
c) 고유 SOUP 지정자
주 : 고유 SOUP 지정자는 를 들어 버 , 릴리즈 날짜, 패치 번호 는 업그 이드
명칭일 수 있다.
8.1.3 시스템 형상 문서 식별
제조업체는 소 트웨어 시스템 형상을 구성하는 형상항목과 그 버 을 문서화해야
한다. [등 A, B, C]
8.2 변경 리
8.2.1 변경 요청 승인
제조업체는 승인된 변경요청에 해서만 형상항목을 변경해야 한다. [등 A, B, C]
주 1 : 변경요청의 승인 결정은 변경 리 로세스의 일부이거나 다른 로세스
일부일 수 있다. 이 부 에 따르면 변경을 승인할 경우 구 을 신한다.

- 152 -
주 2 : 계획에 언 된 것처럼 수명주기의 다양한 단계에서 다양한 인수 로세스를
변경 요청에 사용할 수 있다. 5.1.1 e) 6.1 e) 참조.
8.2.2 변경 구
제조업체는 변경요청에 명시한 것처럼 변경을 구 해야 한다. 제조업체는 소 트웨
어 시스템과 소 트웨어 항목의 소 트웨어 안 성 분류에 한 변경을 포함해, 변경의
결과로서 반복해야 할 활동을 식별하고 수행해야 한다. [등 A, B, C]
주 : 이 부 에는 한 변경 리 달성을 해 변경을 구 하는 방법이 설명된다. 단,
구 이 변경 리 로세스의 필수 인 부분이라고 암시하는 것은 아니다. 구 은
계획한 로세스를 사용해야 한다. 5.1.1 e) 6.1 e) 참조.
8.2.3 변경 검증
제조업체는 변경에 의해 무효가 된 버 의 반복과 5.7.3 9.7의 고려를 포함해
변경을 검증해야 한다. [등 A, B, C]
주 : 이 부 에 따르면 변경만 검증하면 된다. 단, 검증이 변경 리 로세스의 필수
인 부분이라고 암시하는 것은 아니다. 검증은 계획한 로세스를 사용해야
한다. 5.1.1 e) 6.1 e) 참조.
8.2.4 변경 추 성에 한 방법 제공
제조업체는 다음을 추 할 수 있는 경우 그에 해 심사 추 을 작성해야 한다. [등
A, B, C]
a) 변경 요청
b) 련 문제 보고서
c) 변경요청의 승인
8.3 형상 상태 설명
제조업체는 시스템 형상이 포함된, 리된 형상 항목에 해 검색할 수 있는 이력 기록을
유지해야 한다. [등 A, B, C]
9. 소 트웨어 문제해결 로세스
9.1 문제 보고서 작성
제조업체는 소 트웨어 제품에서 검출된 각 문제에 해 문제 보고서를 작성해야 한다.
문제 보고서는 다음과 같이 분류해야 한다. [등 A, B, C]

- 153 -
a) 유형: 제 1 교정, 방 는 새 환경에 한 응성 유형
b) 범 : 제 2 변경 규모, 해당 장비 모델의 수, 지원되는 해당 부속품, 련 자원
변경할 시간
c) 심각도: 제 3 성능, 안 성 는 보안에 한 향
주 : 문제는 제조업체 조직 내부나 외부에서 릴리즈 는 이후 발견할 수 있다.
9.2 문제 조사
제조업체는 다음을 수행해야 한다. [등 A, B, C]
a) 문제를 조사하고, 가능한 경우 원인을 식별한다.
b) 소 트웨어 험 리 로세스를 사용해 안 성에 련된 문제를 평가한다(제7
조)
c) 조사와 평가의 결과를 문서화한다.
d) 문제 해결에 필요한 조치에 해 변경 요청을 작성하거나, 조치를 취하지 않을
경우 그 이론 근거를 문서화한다.
주 : 문제가 안 과 련되지 않은 경우 제조업체는 소 트웨어 문제해결 로세스
의 수를 해 문제를 해결할 필요는 없다.
9.3 련 당사자에게 통보
제조업체는 문제가 있는 경우 그를 련 당사자에게 통보해야 한다. [등 A, B, C]
주 : 문제는 제조업체 조직 내부나 외부에서 릴리즈 는 이후 발견할 수 있다. 제조
업체는 상황에 따라 련 당사자를 결정한다.
9.4 변경 리 로세스의 사용
제조업체는 변경 리 로세스의 요구사항을 수하면서 모든 변경요청을 승인하고
구 해야 한다(8.2 참조). [등 A, B, C]
9.5 기록 유지
제조업체는 문제 보고서와, 검증을 포함해 문제해결의 기록을 유지해야 한다.
제조업체는 필요한 경우 험 리 일을 갱신해야 한다(7.4 참조). [등 A, B, C]
9.6 문제 경향 분석
제조업체는 문제 보고서에서 경향을 확인(detect)하기 한 분석을 수행해야 한다. [등
A, B, C]

- 154 -
9.7 소 트웨어 문제해결 검증
제조업체는 다음을 단하기 해 해결을 검증해야 한다. [등 A, B, C]
a) 문제가 해결되었고 문제 보고서가 마감되었다.
b) 부정 인 경향이 반 되었다.
c) 변경요청이 해당 소 트웨어 제품 활동에 구 되었다.
d) 문제가 추가로 발생했다.
9.8 시험 문서의 내용
변경 후 소 트웨어 항목과 시스템을 시험, 재시험 는 회귀 시험할 때 제조업체는
시험 문서에 다음 내용을 포함시켜야 한다. [등 A, B, C]
a) 시험 결과
b) 확인(found)한 비정상 상태
c) 시험한 소 트웨어의 버
d) 련 하드웨어와 소 트웨어 시험 형상
e) 련 시험 도구
f) 시험 날짜
g) 시험자의 신원

- 155 -
부록 A (참조용) 이 표 의 요구사항에 한 이론 근거
이 시험의 조항에 한 이론 근거가 이 부록에 제공된다.
A.1 이론 근거
이 표 의 일차 인 요구사항은 로세스가 의료기기 소 트웨어의 개발과 유지보
수에 따라 진행되며, 로세스의 선택이 환지 다른 사람에 한 험에 해야
한다는 것이다. 이러한 요구사항은 소 트웨어의 시험으로는 안 하게 운 되는지
단하기 충분하지 않다는 단에 따른 것이다.
이 표 에 요구되는 로세스는 두 가지 범주로 분류할 수 있다.
- 소 트웨어의 각 소 트웨어 항목 운 에서 발생하는 험의 단에 필요한 로
세스
- 결정한 험을 기 으로 선택한, 각 소 트웨어 항목에 한 소 트웨어 고장의
하게 낮은 발생가능성의 달성에 필요한 로세스
이 표 에 따르면 첫 번째 범주는 모든 의료기기 소 트웨어에, 두 번째 범주는 선
택한 소 트웨어 항목에 해 수행해야 한다.
따라서 이 표 수성 주장에는 소 트웨어가 포함되며 험한 상황을 야기할 수
있는 측 가능한 발생순서가 식별되는 문서화된 험분석이 포함되어야 한다. 소 트
웨어에 의해 간 으로 발생할 수 있는 험요인( 를 들어 잘못된 정보를 제공해 치
료가 부 하게 수행되는 경우)은 이 험분석에 포함되어야 한다.
로세스의 첫 번째 범주의 일부로서 필요한 모든 활동은 규정 텍스트에 “[등 A,
B, C]”로 표시되어, 로세스가 용되는 소 트웨어의 분류와 련이 없음을 나타낸다.
다음과 같은 이유 때문에 등 A, B와 C 체에 활동이 필요하다.
- 활동은 험 리 는 소 트웨어 안 성 분류에 련된 계획을 생성한다.
- 활동은 험 리 는 소 트웨어 안 성 분류에 한 입력인 출력을 생성한다.
- 활동은 험 리 는 소 트웨어 안 성 분류의 일부이다.
- 활동은 험 리 는 소 트웨어 안 성 분류를 지원하는 행정 시스템, 문서화
기록유지 시스템을 확립한다.

- 156 -
- 일반 으로 활동은 련 소 트웨어의 분류를 확인(unknown)할 수 없을 경우 발생
한다.
- 활동에 따라서 연 소 트웨어의 재 소 트웨어 안 성 분류를 무효화할 수
있는(invalidate) 변경이 발생할 수 있다. 여기에는 릴리즈 후 안 성 련 문제의
확인(discovery)과 분석이 포함된다.
다른 로세스는 소 트웨어 안 성 등 B나 C로 분류된 소 트웨어 시스템이나 소
트웨어 항목에만 필요하다. 이러한 로세스의 일부로서 필요한 활동은 규정 텍스트
에 “[등 B, C]” 는 “[등 C]”로 표시되어, 로세스가 용되는 소 트웨어의 분
류에 선택 으로 의존해야 함을 나타낸다.
다음과 같은 이유 때문에 등 B와 C의 소 트웨어에 선택 활동이 필요하다.
- 활동은 설계, 시험 기타 검증에 있어 보다 상세한 내용이나 엄격성 강화가 필
요하기 때문에 소 트웨어의 신뢰성을 강화한다.
- 활동은 등 B나 C에 필요한 다른 활동을 지원하는 행정 활동이다.
- 활동은 안 련 문제 해결을 지원한다.
- 활동은 안 련 소 트웨어의 설계, 구 , 검증 릴리즈의 기록을 생성한다.
다음과 같은 이유 때문에 C의 소 트웨어에 선택 활동이 필요하다.
- 활동은 설계, 시험 기타 검증에 있어 보다 상세한 내용이나 강화된 엄격성
는 주의가 필요하기 때문에 소 트웨어의 신뢰성을 한층 강화한다.
이 표 에 정의한 모든 로세스와 활동은 고품질 소 트웨어 개발과 유지보수 확보
에 귀 한 것으로 간주된다. 험요인을 발생시킬 수 없는 등 A 소 트웨어에 한
요구사항으로서 이러한 로세스 활동을 다수 생략하는 경우에도 그러한 로세스
와 활동에 가치가 없거나 권장되지 않음을 의미하는 것은 아니다. 생략의 목 은 의료
기기 설계 일차 으로 종합 인 밸리데이션 활동(이는 이 표 의 범 에 포함되지
않음) 일부 단순한 소 트웨어 수명주기 리를 통하여 안 성과 효과성에 있어
험요인을 유발할 수 없는 소 트웨어를 쉽게 확인(assure)할 수 있는 소 트웨어를 확
인(recognize)하는 것이다.
A.2 등 별 요구사항의 요약
표 A.1에는 각 요구사항에 할당된 소 트웨어 안 성 등 이 요약 설명된다. 이 표
는 참고용이며, 사용상 편의 목 만을 해 제공된다. 규정 부분에 각 요구사항에 한
소 트웨어 안 성 등 이 설명된다.

- 157 -
[표 A.1] 소 트웨어 안 성 등 별 요구사항의 요약
조항 부 등 A 등 B 등 C제4조 체 요구사항 x x x제5.1조 5.1.1, 5.1.2, 5.1.3, 5.1.6, 5.1.7, 5.1.8, 5.1.9 x x x
5.1.5, 5.1.10, 5.1.11 x x5.1.4 x
제5.2조 5.2.1, 5.2.2, 5.2.4, 5.2.5, 5.2.6 x x x5.2.3 x x
제5.3조 5.3.1, 5.3.2, 5.3.3, 5.3.4, 5.3.6 x x5.3.5 x
제5.4조 5.4.1 x x5.4.2, 5.4.3, 5.4.4 x
제5.5조 5.5.1 x x x5.5.2, 5.5.3, 5.5.5 x x5.5.4 x
제5.6조 체 요구사항 x x제5.7조 체 요구사항 x x제5.8조 5.8.4, x x x
5.8.1, 5.8.2, 5.8.3, 5.8.5, 5.8.6, 5.8.7, 5.8.8 x x제6.1조 6.1 x x x제6.2조 6.2.1, 6.2.2, 6.2.4, 6.2.5 x x x
6.2.3 x x제6.3조 체 요구사항 x x x제7.1조 체 요구사항 x x제7.2조 체 요구사항 x x제7.3조 체 요구사항 x x제7.4조 7.4.1 x x x
7.4.2, 7.4.3 x x제8조 체 요구사항 x x x제9조 체 요구사항 x x x

- 158 -
부록 B (참조용) 이 표 의 규정에 한 지침
B.1 범
B.1.1 목
이 표 의 목 은 고품질의 안 한 의료기기 소 트웨어를 일 성 있게 생산하는 개
발 로세스를 제공하는 것이다. 이 목 의 달성을 해 이 표 은 소 트웨어가 신뢰
성 높고 안 한 소 트웨어 제품을 생산할 수 있는 방식으로 개발되었다는 확신을 제
공하기 해 달성해야 하는 최소한의 활동과 업무를 식별해야 한다.
이 부록에는 이 표 의 요구사항 용에 한 지침이 제공된다. 이 부록은 이 표
의 요구사항에 새로운 요구사항을 추가하거나 기존 요구사항을 변경하지 않는다. 이
부록은 이 표 의 요구사항을 보다 잘 이해하기 해 사용할 수 있다.
이 표 에서 활동은 로세스 안에서 필요한 부 이며, 업무는 활동 안에 정의된다.
를 들어, 소 트웨어 개발 로세스를 해 정의한 활동에는 소 트웨어 개발 기획,
소 트웨어 요구사항 분석, 소 트웨어 구축설계, 소 트웨어 상세설계, 소 트웨어 유
닛 구 과 검증, 소 트웨어 통합과 통합 시험, 소 트웨어 시스템 시험 소 트웨어
릴리즈가 포함된다. 이러한 활동 안의 업무는 개별 요구사항이다.
이 표 에는 특정 소 트웨어 개발 수명주기 모델이 필요 없다. 단, 로세스는 다른
로세스에 의해 생성되기 때문에 이 표 에 따라 로세스 사이의 의존성이 있다고 암
시하는 것은 아니다. 를 들어 험분석 로세스에 따라 소 트웨어 시스템의 고장
에서 발생할 수 있는 해를 확인(establish)한 후 소 트웨어 시스템의 소 트웨어 안
성 분류를 완료해야 한다.
로세스 사이의 이러한 논리 의존성 때문에 이 표 의 로세스를 발생순서 로,
즉 “일 수행”이나 “일회성” 수명주기 모델로 설명하는 것이 가장 쉽다. 단, 다른 수
명주기도 사용할 수 있다. ISO/IEC 12207[9]에 정의한 일부 개발(모델) 략에는 다음
이 포함된다(표 B.1 참조).
- 일 수행 Waterfall ; “일 수행”이라고도 하는 “일회성” 략은 개발 로세스를
한번 수행하는 것으로 구성된다. 단순화: 고객 요구를 결정하고, 요구사항을 정의하
며, 시스템을 설계하고, 시스템을 구 하고 시험, 수리 인도한다.
- 증분 Incremental ; “증분” 략은 고객이 요구를 결정하고 시스템 요구사항을 정
의한 다음 빌드 순서로 나머지 개발을 수행한다. 첫째 빌드는 계획한 능력의 일부
- 159 -
를 사용하며, 둘째 빌드는 능력을 더 추가하는 식으로 시스템이 완료될 때까지 지
속된다.
- 진화 Evolutionary ; “진화” 략 한 시스템을 빌드로 개발하지만, 사용자 요구가
완 히 이해되지 않으며 모든 요구사항을 처음부터 정의할 수 없다는 사실을 인정
한다는 에서 증분 략과는 차이를 보인다. 이 략에서 고객 요구와 시스템 요
구사항 일부가 처음에 정의되며, 후속 빌드에서 개량된다.
[표 B.1] ISO/IEC 12207에 정의한 개발(모델) 략
개발 략 모든 요구사항을 처음부터 정의하는가?
개발 주기가 복수인가?
임시 소 트웨어를 배포하는가?
일 수행
(일회성)아니오 아니오
증분
(계획한 제품 개선)가능
진화 아니오
어떤 수명주기를 선택하건 시방서, 설계 문서 소 트웨어와 같은 로세스 출력
사이의 논리 의존성을 유지해야 한다. 일 수행 수명주기 모델은 로세스의 입력이 완
료되어 승인될 때까지 로세스의 시작을 지연함으로써 이러한 의존성을 달성한다.
다른 수명주기, 특히 진화 수명주기를 사용하면 로세스에 한 입력을 모두 사용
할 수 있기 로세스 출력을 생성할 수 있다. 를 들어 체 소 트웨어 형상이
완성되기 새 소 트웨어 항목(item)을 지정, 분류, 구 검증할 수 있다. 그러한
수명주기는 한 로세스의 변경이나 개발 때문에 다른 로세스 출력을 무효화하는
(invalidate) 험을 발생한다. 따라서 모든 수명주기는 종합 인 형상 리 시스템을 사
용해 모든 로세스 출력을 일 성 있는 상태로 유지하며, 의존성을 유지한다.
아래에 설명하는 원칙은 소 트웨어 개발 수명주기 종류에 계 없이 요하다.
- 모든 로세스 출력은 일 성 있는 상태로 유지되어야 한다. 로세스 출력이 작
성되거나 변경될 때마다 련된 모든 로세스 출력은 서로 일 성을 유지하고,
이 표 이 명시 으로 는 묵시 으로 요구하는 모든 의존성 유지를 해 즉시
갱신해야 한다.
- 모든 로세스 출력은 소 트웨어에 한 후속 작업에 입력으로 필요한 경우 사
용할 수 있어야 한다.

- 160 -
- 의료기기 소 트웨어를 릴리즈하기 모든 로세스 출력은 서로 일 성을 갖추
어야 하며, 이 표 이 명시 으로 는 묵시 으로 요구하는 로세스 사이의 모
든 의존성이 수되어야 한다.
B.1.2 용분야
이 표 은 의료기기 소 트웨어의 개발과 유지보수는 물론 SOUP가 포함된 의료
기기 개발과 유지보수에 용된다.
이 표 을 사용하려면 제조업체는 ISO 14971에 따라 의료기기 험 리을 수행해야
한다. 따라서 의료기기 소 트웨어 아키텍처에 SOUP가 포함된 린터/ 로터와 같은
구성요소가 포함될 경우(구입한 구성요소이거나 기원을 알 수 없는 구성요소일 수
있다) 그러한 구성요소는 제조업체가 책임을 담당해야 하며, 의료기기 험 리에 포함
되어야 한다. 의료기기 험 리을 히 수행할 경우에만 제조업체는 그 구성요소를
이해하고, 그 구성요소에 SOUP가 포함된 사실을 확인(recognize)할 수 있다. 이 표 을
사용하는 제조업체는 험 리 로세스를 의료기기 험 리 로세스의 일환으로 사
용할 수 있을 것이다.
릴리즈한 의료기기 소 트웨어의 유지보수는 의료기기 소 트웨어와의 생산 후 경험
에 용된다. 소 트웨어 유지보수에는 요구되는 성능과 릴리즈한 소 트웨어 제품에
련된 수정 요청을 수행할 수 있는 상태로 항목(item)을 유지하거나 그러한 상태로 복귀
시키기 해 문제 보고서에 따라 조치를 취하기 한 감독 조치가 포함된 모든 기술
행정 방법의 조합이 포함된다. 를 들어, 여기에는 문제 확인(rectification), 규제
보고, 타당성 재확인 방 조치가 포함된다. ISO/IEC 14764 [10] 참조.
B.2 규격상 참고문헌
ISO/IEC 90003[11]은 품질경 시스템을 소 트웨어 개발에 용하기 한 지침을 제
공한다. 이 지침은 이 표 에서는 요구되지 않지만 강력히 권장되는 지침이다.
B.3 용어 정의
가능한 한 용어는 국제 표 의 정의를 사용해 정의했다.
이 표 은 소 트웨어 시스템(최상 )의 분해 설명에 세 가지 용어를 사용하기로 선
택했다. 소 트웨어 시스템은 의료기기의 서 시스템(IEC 60601-1-4[2] 참조)이거나 의
료기기 그 자체일 수 있다. 시험이나 소 트웨어 형상 리의 목 을 해 더 이상 분

- 161 -
해할 수 없는 최 수 이 소 트웨어 유닛이다. 최상 와 최하 가 포함된 모든 수
의 형상을 소 트웨어 항목이라고 부를 수 있다. 즉, 소 트웨어 항목은 하나 이상의
소 트웨어 항목으로 구성되며, 각 소 트웨어 항목은 하나 이상의 소 트웨어 유닛
는 분해가능 소 트웨어 항목으로 구성된다. 소 트웨어 항목과 소 트웨어 유닛의 정
의와 입도를 제공하는 것은 제조업체의 책임이다. 이러한 용어를 애매한 상태로 유지
하면 다양한 의료기기에 사용되는 소 트웨어의 개발방법과 소 트웨어 유형에 이러한
용어를 용할 수 있다.
B.4 일반 요구사항
어떤 종류의 소 트웨어에도 안 성을 100% 보장할 수 있는 방법은 없다.
의료기기 소 트웨어의 안 성을 향상시키는 주요 원칙에는 세 가지가 있다.
- 험 리
- 품질경
- 소 트웨어 엔지니어링
안 한 의료기기 소 트웨어를 개발하고 유지 보수하려면 한 소 트웨어 엔지니
어링 방법 기술에 한 체 인 체제로서의 품질경 시스템의 일부로서 험 리을
확립해야 한다. 이러한 세 가지 개념을 조합하면 의료기기 제조업체가 잘 구성되고 일
성 있게 반복 정 도가 높은 의사결정 로세스를 사용해 의료기기 소 트웨어의 안
성을 향상시킬 수 있다.
B.4.1 품질경 시스템
잘 구성되고 효과 인 소 트웨어 로세스에는 경 , 기반구조, 개선 훈련과 같은
조직의 로세스가 포함된다. 복을 방지하고 이 표 을 소 트웨어 엔지니어링에 집
하기 해 이러한 로세스를 이 표 에서 생략했다. 이러한 로세스는 품질경 시스
템이 담당한다. ISO 13485[7]은 의료기기에 품질경 개념을 용하기 한 국제표 이
다. ISO 13485 품질경 시스템 요구사항을 수한다 하더라도 국가 는 지역 규제 요
구사항과의 수성이 자동으로 이루어지는 것은 아니다. 련 규제 요구사항과의 수
성을 식별하고 확립하는 책임은 제조업체의 몫이다.
B.4.2 험 리
의료기기 소 트웨어와 련해 타당한 수 으로 상할 수 있는 모든 험을 고려할
수 있도록 소 트웨어 개발은 험 리 활동에 충분히 참여한다.

- 162 -
이 소 트웨어 엔지니어링 표 에서 해당 험 리 로세스를 정의하기보다는 제조
업체가 의료기기에 한 험 리을 명백히 취 하며, ISO 14971을 수하는 험 리
로세스를 용하는 것이 바람직하다. 소 트웨어가 원인 하나가 되는 험요인에서
발생하는 특정 소 트웨어 험 리 활동은 제7조의 지원 로세스에 설명되어 있다.
B.4.3 소 트웨어 안 성 분류
의료기기 일부로서, 의료기기 부속품으로서, 는 의료기기 자체로서의 소 트웨어에
련된 험은 소 트웨어 안 성 분류 계획에 한 입력을 사용되며, 이 경우 소 트
웨어 개발과 유지보수 사용되는 로세스가 결정된다.
험은 부상 심각도 발생 가능성의 조합으로 간주된다. 단, 재래식 통계 방법을
사용해 소 트웨어 고장 발생 가능성을 결정하는 방법에는 어떤 합의도 없다. 따라서
이 표 에서 소 트웨어 시스템 분류는 고장이 발생한다고 가정해, 소 트웨어의 고정
에서 발생하는 험요인의 심각도에 기 해 결정된다. 험 리 방법의 구 에 기여하
는 소 트웨어 시스템은 시스템이 리하고 있는 험요인의 심각도에 기 해 분류된
다.
소 트웨어 시스템을 소 트웨어 항목을 분해할 경우 각 소 트웨어 항목에는 자체
소 트웨어 안 성 분류가 할당된다.
소 트웨어 항목의 고장에 련된 험은 다음과 같은 경우에만 결정할 수 있다.
- 시스템 아키텍처와 소 트웨어 아키텍처가 그 목 다른 소 트웨어와 하드
웨어 항목과의 연계성 에서 소 트웨어 항목의 역할을 정의할 경우
- 시스템에 한 변경이 리 일 경우
- 지정된 아키텍처와 험 리 방법에 험분석을 수행한 후
이 표 에는 모든 등 의 소 트웨어에 해 에 규정한 조건을 달성하는 최소한
의 활동이 필요하다.
소 트웨어 아키텍처 활동의 종료 시 은 체 소 트웨어 항목이 정의되고 험
리 활동에 따라 소 트웨어 항목이 안 성에 어떤 련이 있는지 식별되는 경우 개발
의 최 시 이 된다. 따라서 이 시 은 소 트웨어 항목을 안 성 역할에 따라 명확하
게 분류할 수 있는 최 의 시 이 된다.
이 시 은 험 리가 ISO 14971에서 시작하는 시 에 해당된다.

- 163 -
이 시 이 험 리 로세스는 를 들어 보호 서 시스템 추가 는 소 트웨
어 고장이 해를 발생시킬 수 있는 가능성 축소와 같은 구조 험 리 방법을 식
별한다. 이 시 이후 험 리 로세스는 소 트웨어 항목 고장 발생가능성 축소에
집 하는 로세스를 사용한다. 즉, 소 트웨어 항목의 분류에 따라 그 항목에 용되
는 로세스 기반 험 리 방법이 지정된다.
제조업체는 를 들어 조사 항 역에 주의를 집 하면 이 시 이 소 트웨어
를 쉽게 분류할 수 있지만, 그러한 분류는 비 인 것으로 간주되어 로세스 생략의
정당화에는 사용할 수 없다.
소 트웨어 안 성 분류 계획의 목 은 ISO 14971의 험 분류와 일치시키는 것이
아니다. ISO 14971 계획은 험을 심각도와 가능성에 따라 분류하지만, 소 트웨어 안
성 분류 계획은 개발 유지보수에 용되는 로세스에 따라 소 트웨어 시스템과
소 트웨어 항목을 분류한다.
설계가 진행되면 새로운 험이 발생할 수 있다. 따라서 험 리은 개발 로세스
의 일부로 용해야 한다. 이 경우 안 한 운 과 결함이 해를 야기하는 것을 방지하는
운 의 확보를 해 기능이 정확하게 발휘되어야 하는 소 트웨어 항목이 포함된 체
소 트웨어 항목을 식별할 수 있는 구축설계를 개발할 수 있다.
소 트웨어 아키텍처는 안 한 운 에 필요한 소 트웨어 항목의 분리를 진해야
하며, 그러한 소 트웨어 항목의 효과 인 분리 확보에 사용되는 방법을 설명해야
한다.
B.3에서 언 한 것처럼, 이 표 은 소 트웨어 시스템(최상 )의 분해 설명에 세 가
지 용어를 사용하기로 선택한다.
그림 B.1은 소 트웨어 시스템 내부 소 트웨어 항목을 분할하는 방법과 분해 소
트웨어 항목 그룹에 소 트웨어 안 성 등 을 용하는 방법이 설명된다.

- 164 -
[그림 B.1] 소 트웨어 항목 분할의
소 트웨어 시스템/소 트웨어 항목
(등 C)
소 트웨어 항목 W
(등 B)
소 트웨어 항목 Z
(등 C)
소 트웨어 항목 X
(등 A)
소 트웨어 항목 Y
(등 C)
이 의 경우 개발 인 의료기기 소 트웨어의 유형 때문에 제조업체는 소 트웨어
시스템에 한 비 소 트웨어 안 성 등 이 소 트웨어 안 성 등 C라는 사실을
안다. 소 트웨어 구축설계 제조업체는 그림처럼 세 가지 소 트웨어 항목, X, W
Z로 시스템을 분할할 것을 결정했다. 제조업체는 사망이나 상을 발생할 수 있는
험요인에 한 모든 소 트웨어 시스템 향을 소 트웨어 항목 Z로 분리하고, 비
상을 발생시킬 수 있는 험요인에 한 나머지 모든 소 트웨어 시스템을 소 트웨
어 항목 W로 분리할 수 있다. 소 트웨어 항목 W는 소 트웨어 안 성 등 B로 분류
되며, 소 트웨어 항목 Z는 소 트웨어 안 성 등 C로 분류된다. 따라서 소 트웨
어 항목 Y는 4.3d)에 따라 등 C로 분류되어야 한다. 한 이 요구사항에 따라 소 트
웨어 시스템은 소 트웨어 안 성 등 C로 분류된다. 소 트웨어 항목 X는 소 트웨어
안 성 등 A로 분류되었다. 제조업체는 분리의 무결성 확보를 해 소 트웨어 항목
X와 Y 소 트웨어 항목 W와 Z 사이의 분리에 한 이론 근거를 문서화할 수
있다. 분할이 불가능할 경우 소 트웨어 항목 X와 Y는 소 트웨어 안 성 등 C로 분
류되어야 한다.
B.5 소 트웨어 개발 로세스
B.5.1 소 트웨어 개발 기획
이 활동의 목표는 소 트웨어에 의해 발생하는 험을 이고 차와 목 을 개발
구성원에게 알리며 의료기기 소 트웨어에 한 시스템 품질 요구사항이 충족되는
지 확인(ensure)하기 해 소 트웨어 개발 업무를 계획하는 것이다.

- 165 -
소 트웨어 개발 기획 활동에 다라 업무를 단일 계획이나 복수 계획에 문서화할 수
있다. 모든 의료기기 소 트웨어의 개발에 용되는 정책과 차를 이미 확립한 제조
업체가 있을 수 있다. 이 경우 계획은 기존 정책과 차를 참조할뿐 이다. 특정 활동을
자세히 규정하고 일반 인 차를 참조하는 각 의료기기 소 트웨어 제품의 개발 고유
의 계획을 작성하는 제조업체도 있을 수 있다. 한 각 의료기기 소 트웨어 제품
의 개발에 합하게 조정한 계획도 있을 수 있다. 기획은 개발 로세스 수행에 필요
한 상세 수 으로 지정되어야 하며, 험에 비례해야 한다. 를 들어 험 수 이
높은 시스템이나 항목은 보다 엄격한 개발 로세스의 상이 되며, 매우 상세하게 작
성되어야 한다.
기획은 개발이 진행되는 동안 계속 찰하고 갱신해야 하는 회귀 활동이다. 계획
은 시스템을 더 잘 이해하고 시스템 개발에 어느 정도 노력이 요구됨에 따라 더 많은
양호한 정보를 채택하도록 진화할 수 있다. 를 들어 시스템의 기 소 트웨어 안
성 분류는 험 리 로세스를 활용한 결과와 소 트웨어 아키텍처의 개발에 따라
변화할 수 있다. 한 시스템에 SOUP를 채택하도록 결정할 수도 있다. 재 악된
시스템의 정보와 시스템이나 시스템 내부 항목에 필요한 엄격성 수 을 반 할 수 있
도록 계획을 갱신해 개발 로세스를 히 리하는 것이 요하다.
B.5.2 소 트웨어 요구사항 분석
이 활동을 수행하려면 제조업체는 의료기기 소 트웨어에 한 소 트웨어 요구사
항을 확립하고 검증해야 한다. 구축할 내용을 결정하고 의료기기 소 트웨어가 허용되
는 작동상태를 나타내는지 단하며 완성한 의료기기 소 트웨어를 즉시 사용할 수 있
다고 입증하려면 검증할 수 있는 요구사항의 확립이 필수 이다. 요구사항이 원하는
로 구 되었는지 입증하려면 요구사항이 정확히 구 되었는지 단하기 한 객 기
을 수립할 수 있는 방식으로 표 해야 한다. 기기 험 리 로세스가 식별한 험
제어를 한 요구사항을 소 트웨어에 부과한 경우 이러한 요구사항은 험 리 방법
을 소 트웨어 요구사항까지 추 할 수 있도록 소 트웨어 요구사항에서 식별할 수 있
어야 한다. 모든 소 트웨어 요구사항은 요구사항과 소 트웨어 시스템 시험 사이의
추 성을 입증할 수 있는 방식으로 식별해야 한다. 일부 국가에서 규정에 의거한 승인
을 받으려면 특정 규정이나 국제 표 을 수해야 할 경우 이 수성 요구사항은 소
트웨어 요구사항에 문서화해야 한다. 소 트웨어 요구사항에 의해 소 트웨어에 구 되
는 내용이 결정되기 때문에 요구사항 분석 활동을 완료하기 요구사항을 평가해야
한다.

- 166 -
혼동이 자주 발생하는 분야는 고객 요구, 설계 입력, 소 트웨어 요구사항, 소 트웨
어 기능 시방서 소 트웨어 설계 시방서를 구별하는 것이다. 설계 입력은 고객 요
구를 공식 으로 문서화한 의료기기 요구사항으로 해석한 것이다. 소 트웨어 요구사항
은 소 트웨어가 고객 요구와 설계 입력을 충족해야 하는, 공식 으로 문서화된 시방서
이다. 소 트웨어 기능 시방서는 소 트웨어 요구사항에 포함되는 경우가 많으며, 많은
다른 안도 요구사항을 충족할 수 있지만, 소 트웨어가 그 요구사항을 충족하기 해
수행해야 할 작업을 상세하게 정의한다. 소 트웨어 설계 시방서는 요구사항과 기능 시
방서 규 을 해 소 트웨어를 설계하고 분해하는 방법을 정의한다.
일반 으로 소 트웨어 요구사항, 기능 시방서 설계 시방서는 하나 이상의 문서
로 작성되었다. 이제는 이 정보를 공통 데이터베이스 안의 데이터 항목으로 간주할 수
있다. 각 항목에는 항목의 목 데이터베이스 내 다른 항목과의 연결성을 정의하는
속성이 하나 이상 있다. 이 근방식을 사용하면 상 사용자( 를 들어 마 담당자,
제조업체, 시험자 심사자)에 가장 합한 정보를 다양하게 표 하고 인쇄할 수 있으
며, 모든 요구사항의 한 구 을 입증하고 용범 를 시험할 수 있다. 이 근방식
을 지원하기 한 도구는 HTML 하이퍼링크를 사용하는 단순한 하이퍼텍스트 문서일
수도 있고 컴퓨터 지원 소 트웨어 엔지니어링(CASE) 도구와 요구사항 분석 도구처럼
복잡하지만 성능이 뛰어난 도구일 수도 있다.
시스템 요구사항 로세스는 이 표 에서는 다루지 않는다. 단, 의료기기 기능성을
소 트웨어에 구 하고자 하는 결정은 일반 으로 시스템 설계 이루어진다. 일부 는
체 시스템 요구사항은 소 트웨어에서 구 되도록 할당된다. 소 트웨어 요구사항
분석 활동은 시스템 요구사항 로세스에서 소 트웨어에 할당한 요구사항의 분석과
할당된 요구사항을 반 하는 종합 인 소 트웨어 요구사항을 도출하는 것으로 구성된
다.
시스템 무결성 확보를 해 제조업체는 상 시스템 요구사항이나 소 트웨어 요구
사항의 실성 결여, 일 성 결여 는 모호성 해결을 해 시스템 요구사항에 한 변
경과 해명의 교섭을 한 메커니즘을 제공해야 한다.
시스템 소 트웨어 요구사항의 포착과 분석 로세스는 회귀 일 수 있다. 이 표
에 따르면 로세스는 엄격하게 두 층으로 분리할 필요는 없다. 사실상, 시스템 아키
텍처와 소 트웨어 아키텍처는 동시에 설명되는 경우가 많으며, 시스템과 소 트웨어
요구사항은 층으로 구성된 양식으로 문서화된다.

- 167 -
B.5.3 소 트웨어 구축 설계
이 활동을 수행하려면 제조업체가 소 트웨어의 주요 구조 구성요인, 외부에서 확인
할 수 있는 특성 특성 사이의 계를 정의해야 한다. 어느 한 구성요인의 작동상태
가 다른 구성요인에 향을 경우 그 작동상태를 소 트웨어 아키텍처에 기술해야 한
다. 이 설명은 소 트웨어 이외의 의료기기 구성요인에 향을 수 있는 작동상태에
특히 요하다. 험 리 방법 구 에 있어 구축 결정은 매우 요하다. 다른 구성요
인에 향을 수 있는 구성요인의 작동상태를 이해(문서화 포함)하지 못하면 시스템
이 안 하다고 표시하기는 거의 불가능에 가깝다. 소 트웨어 아키텍처는 소 트웨어
요구사항의 정확한 구 에 필요하다. 소 트웨어 아키텍처는 모든 소 트웨어 요구
사항이 식별된 소 트웨어 항목으로 구 할 수 없는 한 완성되지 않는다. 소 트웨어
의 설계와 구 은 아키텍처에 따라 결정되기 때문에 아키텍처를 확인해 이 활동을 완성
해야 한다. 아키텍처의 확인은 일반 으로 기술 평가에 의해 이루어진다.
소 트웨어 아키텍처 활동 소 트웨어 항목을 분류하면 소 트웨어 로세스의
후속 선택을 한 기 가 마련된다. 분류의 기록은 험 리 일의 일부로 변경 리
의 상이 된다.
다양한 후속 사상 때문에 분류가 무효로 되는(invalidate) 경우가 있다. 를 들면 다음
과 같다.
- 시스템 시방서, 소 트웨어 시방서 는 아키텍처의 변경
- 특히 상할 수 없는 험요인과 같은 오류를 험분석에서 발견
- 특히 험제어 방법과 같은 요구사항의 실 불가능성 확인(discovery)
따라서 소 트웨어 아키텍처 설계 후 모든 활동 소 트웨어 시스템과 소 트웨어
항목(item)의 분류는 다시 평가해야 하며, 이 경우 개정이 필요할 수도 있다. 이 경우 높
은 수 의 등 으로 업그 이드되기 때문에 소 트웨어 항목(item)에 로세스를 추가
로 용하기 한 재작업이 발생할 수 있다. 소 트웨어 형상 리 로세스(제8조)는
필요한 모든 재작업이 식별되고 완료되는지 확인(ensure)할 경우 사용된다.
B.5.4 소 트웨어 상세 설계
이 활동을 수행하려면 제조업체가 아키텍처에 정의된 소 트웨어 항목 연계성을
다시 정의해 소 트웨어 유닛과 연계성을 작성해야 한다. 소 트웨어 유닛은 단일 기능
이나 모듈로 간주되는 경우가 많지만 이 견해가 항상 정확한 것은 아니다. 여기에서는
소 트웨어 유닛을 더 작은 항목으로는 분할할 수 없는 소 트웨어 항목으로 정의한

- 168 -
다. 소 트웨어 유닛은 별도로 시험할 수 있다. 제조업체는 소 트웨어 유닛의 상세 수
을 정의해야 한다. 상세 설계는 알고리즘, 데이터 표 , 다양한 소 트웨어 유닛 사
이의 연계성 소 트웨어와 데이터 구조 사이의 연계성을 명시한다. 한 상세 설
계는 소 트웨어 제품의 패키지 구성에도 련된다. 소 트웨어 유닛을 정확히 구 할
수 있도록 각 소 트웨어 유닛의 설계와 연계성을 문서화해야 한다. 상세 설계에는 소
트웨어 구성에 필요한 상세 내용이 수록된다. 상세 설계는 로그래머가 임시로
설계 결정을 하지 않아도 될 정도로 완벽해야 한다.
소 트웨어 항목은 새 소 트웨어 항목 극히 일부만 원래 소 트웨어 항목의 안
성 련 요구사항을 구 하도록 분해할 수 있다. 나머지 소 트웨어 항목은 안 성
련 기능을 구 하지 않으며, 낮은 수 의 소 트웨어 안 성 등 으로 다시 분류할 수
있다. 단, 이러한 작업에 한 결정은 험 리 로세스의 일부이며, 험 리 일에
문서화된다.
구 은 상세 설계에 따라 결정되기 때문에 활동을 완료하기 상세 설계를 검증해
야 한다. 상세 설계의 검증은 일반 으로 기술 검토에 의해 이루어진다. 부 5.4.4에 따
라 제조업체는 상세설계 활동의 산출을 검증해야 한다. 설계는 요구사항이 구 되는 방
식을 지정한다. 설계에 결함이 있을 경우 코드는 요구사항을 정확히 구 할 수 없다.
제조업체는 안 성에 요하다고 제조업체가 단하는 설계 특성을 검증해야 한다.
그러한 특성의 몇 가지는 아래와 같다.
- 의도된 사상, 입력, 출력, 논리 흐름, CPU 할당, 메모리 자원의 할당 오류와
외 정의, 오류와 외 확인(isolation)과 오류 복구의 구
- 험한 상황을 유발할 수 있는 모든 결함이 사상과 환으로 취 되는 기본 상태
의 정의
- 변수, 메모리 리의 기화
- 험 리 방법에 향을 수 있는 콜드 리셋과 웜 리셋, 기 기타 상태 변
경
B.5.5 소 트웨어 유닛 구 검증
이 활동을 수행하려면 제조업체는 소 트웨어 유닛에 한 코드를 작성하고 검증해
야 한다. 상세 설계는 원시 코드로 변환해야 한다. 코딩은 시방서 분해가 종료되고 실
행 가능한 소 트웨어의 구성이 시작되는 지 을 의미한다. 원하는 코드 특성을 일 성

- 169 -
있게 달성하려면 코딩 표 을 사용해 선호하는 코딩 스타일을 지정해야 한다. 코딩
표 에는 이해성, 언어 사용 규칙이나 제약조건 복잡성 리에 한 요구사항이 포
함된다. 각 유닛에 한 코드를 검증해 상세 설계에서 지정한 로 기능을 발휘하며
지정한 코딩 표 을 수하는지 확인(ensure)해야 한다.
부 5.5.5에 따라 제조업체는 코드를 검증해야 한다. 코드가 설계를 정확히 구
하지 못할 경우 의료기기 소 트웨어는 의도된 로 기능을 수행하지 않는다.
B.5.6 소 트웨어 통합 통합 시험
이 활동을 수행하려면 개발자는 소 트웨어 유닛을 집합 소 트웨어 항목으로 통합
하고 소 트웨어 항목을 높은 수 의 집합 소 트웨어 항목으로 통합하기 한 계획을
입안하고 그러한 통합을 실행해야 하며, 소 트웨어가 의도된 로 작동하는지 확인해
야 한다.
통합에 한 근방식은 비 증 통합에서 증 통합에 이르기까지 다양할 수 있다.
조립되는 소 트웨어의 특성에 따라 통합을 해 선택한 방법이 표시된다.
소 트웨어 통합 시험은 소 트웨어 항목의 내부와 외부 연계성에서의 데이터 달
과 리에 집 한다. 외부 연계성은 운 체제 소 트웨어가 포함된 다른 소 트웨어와
의료기기 하드웨어와의 연계성을 의미한다.
통합 시험의 엄격성과 통합 시험과 연 된 문서화의 상세 수 은 기기와 연 된
험, 잠재 으로 험한 기능에 한 기기의 의존성 험도가 높은 기기 기능에 있어
특정 소 트웨어의 역할에 합해야 한다. 를 들어 모든 소 트웨어는 시험해야 하
지만 안 성에 향을 미치는 항목은 보다 직 이고 철 하며 상세한 시험의 상이
된다.
해당되는 경우 통합 시험 결과는 로그램 작동상태가 입력과 출력 도메인의 경계에
있다는 사실을 나타내며, 로그램이 유효하지 않고(invalid) 상할 수 없는 특수 입력
에 응답한다고 확인(confirm)한다. 로그램의 조치는 입력 는 상하지 못한 입력 순
서의 조합이나 정의한 타이 요구사항이 반되는 경우 밝 진다. 계획의 시험 요구
사항에는 통합 시험의 일환으로 수행되는 화이트박스 시험의 유형이 포함되어야 한
다.

- 170 -
glass box, 구조 (structural) clear box open box 시험이라고도 하는 White box
시험은 시험 인 소 트웨어 항목의 내부 작용에 한 명백한 지식을 시험데이터 선
택에 사용하는 경우의 시험 기술이다. 화이트박스 시험은 출력의 찰을 해 소 트
웨어의 특정 지식을 사용한다. 시험은 소 트웨어 항목(item)이 어떤 기능을 발휘하는
지 시험자가 아는 경우에만 정 하게 수행할 수 있다. 이 경우 시험자는 소 트웨어
항목이 의도된 목표에서 벗어나는지 여부를 확인(see)할 수 있다. 화이트박스 시험은
소 트웨어 항목(item) 구 의 시험에 집 하기 때문에 체 시방서의 구 을 보장하지
못한다. 작동상태(behavioural), 기능 (functional), 불투명 박스(opaque-box)
closed-box 시험이라고도 하는 Black box 시험은 기능 시방서에 집 하며, 구 의 모
든 부분이 시험되었다고 보증하지 못한다. 따라서 블랙박스 시험은 시방서와 비교한
시험을 수행하며, 생략의 결함을 확인(discover)함으로써 시방서의 그 부분이 충족되지
않았음을 나타낸다. 화이트박스 시험은 구 과 비교한 시험을 수행하며, 임무의 결함
을 확인(discover)함으로써 구 의 그 부분이 결함임을 나타낸다. 소 트웨어 제품을
완 히 시험하려면 블랙박스와 화이트박스 시험이 모두 필요할 수 있다.
5.6과 5.7에 설명한 계획 시험 문서는 개발이나 진화 로토타입의 특정 단계에
련된 개발 문서일 수 있다. 한 단일 문서나 일련의 문서가 복수 소구분의 요구사
항을 취 할 수 있도록 조합할 수도 있다. 문서의 부 는 일부는 하드웨어와 소
트웨어의 모든 측면을 취 하는 소 트웨어나 로젝트 품질보증 계획 는 종합 인
시험 계획과 같은 상 로젝트 문서의 일부로 구성할 수 있다. 그러한 경우 다양한
로젝트 문서가 각 소 트웨어 통합 업무와 어떻게 련되는지 식별하기 한 교차
참조를 작성해야 한다.
소 트웨어 통합 시험은 시뮬 이션 환경, 실제 상 하드웨어 는 체 의료기기
에서 수행할 수 있다.
부 5.6.2에 따라 제조업체는 소 트웨어 통합 활동의 산출을 검증해야 한다. 소
트웨어 통합 활동의 산출이 통합된 소 트웨어 항목이다. 이러한 통합 소 트웨어 항
목은 정확하고 안 하게 기능을 수행하려면 체 의료기기 소 트웨어에서 제 로 기능
을 발휘해야 한다.
B.5.7 소 트웨어 시스템 시험
이 활동을 수행하려면 제조업체는 소 트웨어에 한 요구사항이 성공 으로 구 되
었는지 검증함으로써 소 트웨어의 기능성을 검증해야 한다.

- 171 -
소 트웨어 시스템 시험에 따라 지정 기능성 존재 여부가 밝 진다. 이 시험은 소
트웨어에 한 요구사항과 련해 구축된 로그램의 기능성과 성능을 검증한다.
소 트웨어 시스템 시험은 보다 효율 으로 특정 시험을 수행하고 스트 스 조건이
나 결함을 유발하며 자격검증 시험의 코드 용범 의 확 를 해 화이트박스 시험
(이 부 참조)을 사용하는 것이 바람직할 경우에도 기능(블랙박스) 시험에 집 한다. 유
형 시험 단계별 시험 조직에는 융통성이 부여되지만 요구사항, 험 리, 활용성
시험 유형( 를 들어 결함, 설치, 스트 스)에 한 용범 를 입증하고 문서화해야
한다.
소 트웨어 시스템시험은 통합 소 트웨어를 시험하며 시뮬 이션 환경, 실제 상
하드웨어 는 체 의료기기에서 수행할 수 있다.
소 트웨어 시스템이 변경될 경우(사소한 변경이라도) 회귀 시험의 정도(개별 변경의
시험에 국한되지 않음)를 단하여 의도되지 않은 부작용이 발생하지 않는지 확인
(ensure)해야 한다. 이 회귀 시험(소 트웨어 시스템 시험을 완 히 반복하지 않는 이
론 근거 포함)을 계획하고 문서화해야 한다.
소 트웨어 시스템 시험은 다양한 지 에서 발생하고 다양한 조직에서 수행하기 때문
에 시험에 한 책임이 분산될 수 있다. 그러나 업무의 분산, 계약에 따른 계, 구성요인
발생원 는 개발 환경에도 불구하고 기기 제조자는 소 트웨어가 의도한 사용에 합하
게 기능을 발휘하는지 확인(ensuring)하는 궁극 인 책임을 져야 한다.
시험 발견된 비정상 상태가 반복되지만 그러한 비정상 상태의 해결을 한 결정
을 하지 않은 경우 그러한 비정상 상태는 험요인 분석과 련해 평가함으로써 기기
의 안 성에 향을 주지 않는지 검증해야 한다. 그러한 비정상 상태의 근본 원인과
증상을 이해해야 하며, 해결하지 않기로 결정한 이론 근거를 문서화해야 한다.
부 5.7.4에 따라 소 트웨어 시스템 시험의 결과를 평가해 상한 결과가 도출되
는지 확인(ensure)해야 한다.

- 172 -
B.5.8 소 트웨어 릴리즈
이 활동을 수행하려면 제조업체는 릴리즈되는 의료기기 소 트웨어의 버 을 문서
화하고 소 트웨어를 작성한 방법을 명시하며 소 트웨어 릴리즈에 해 합한
차를 따라야 한다.
제조업체는 개발 로세스를 사용해 개발한 소 트웨어가 재 릴리즈되는 소 트
웨어인지 나타낼 수 있어야 한다. 한 제조업체는 향후 필요할 경우 소 트웨어
소 트웨어 생성에 사용한 도구를 검색할 수 있어야 하며, 소 트웨어의 손상과 오용을
최소화하도록 소 트웨어를 보 , 포장 공 해야 한다. 정의된 차를 확립해 이러한
업무가 히 수행되며 일 성 있는 결과를 도출하는지 확인(ensure)해야 한다.
B.6 소 트웨어 유지보수 로세스
B.6.1 소 트웨어 유지보수 계획 확립
소 트웨어 유지보수 로세스는 소 트웨어 개발 로세스와는 두 가지가 다르
다.
- 제조업체는 체 소 트웨어 개발 로세스보다 은 규모의 로세스를 사용
해 긴 한 문제의 응에 있어 신속한 변경을 구 할 수 있다.
- 릴리즈된 제품과 련해 소 트웨어 문제 보고서에 응함에 있어 제조업체는 문제
해결은 물론 지역 법규를 수해야 한다(주로 제출된 문제 데이터를 취합하고
문제에 해 사용자와 규제자와 의사소통하기 한 향 조사 계획을 실행한
다).
부 6.1에 따라 이들 로세스는 유지보수 계획에 확립되어야 한다.
이 활동을 수행하려면 제조업체는 유지보수 활동과 업무를 구 하는 차를 작성하
거나 식별해야 한다. 시정조치를 구 하고 유지보수 변경을 리하며 개정 소 트웨
어의 릴리즈를 리하려면 제조업체는 문제 보고서와 사용자의 요청을 기록하고 문제
를 해결해야 하며, 의료기기 소 트웨어에 한 수정을 리해야 한다. 문제가 발생하거
나 개선 는 용의 필요성 때문에 의료기기 소 트웨어의 코드 련 문서가 수정
되는 경우 이 로세스가 시작된다. 목 은 릴리즈된 의료기기 소 트웨어 무결성을 보
존하면서 소 트웨어를 수정하는 것이다. 이 로세스에는 원래는 릴리즈되지 않은 환
경이나 랫폼에 한 의료기기 소 트웨어의 마이그 션이 포함된다. 이 조항에 규정
한 활동은 유지보수 로세스에 한 것이다. 단, 유지보수 로세스에는 이 표 의
다른 로세스도 사용될 수 있다.
제조업체는 유지보수 로세스의 활동과 업무를 수행하는 방법을 계획해야 한다.

- 173 -
B.6.2 문제 수정 분석
이 활동을 수행하려면 유지보수자는 그 향에 한 피드백을 분석하고, 보고 받은
문제를 검증하며 수정 옵션의 구 을 고려하고 선택하며 승인을 확보해야 한다. 문제
기타 변경 요청은 의료기기의 성능 안 성이나 규제 허가에 향을 미칠 수 있다.
문제 보고서 때문에 향이 발생하는지의 여부 는 문제를 해결하고 요청을 구 하기
한 수정에 의해 향이 발생하는지 여부의 단에 분석이 필요하다. 소 트웨어
유지보수 활동의 일환으로 구 되는 소 트웨어 변경에 의해 기기에 내장된 험
리 수단이 변경되거나 수정되는지 여부를 추 이나 회귀 분석을 통해 검증하는 것이 특
히 요하다. 한 이 에는 험요인을 발생하거나 험을 최소화하지 않았던 소 트
웨어에 수정된 소 트웨어가 험요인을 발생하거나 험을 최소화하지 않는다는 사실
의 검증도 요하다. 소 트웨어 항목(item)의 소 트웨어 안 성 분류는 소 트웨어
수정으로 험요인이 발생하거나 험이 최소화할 경우 변경될 수 있다.
소 트웨어 유지보수(제6조)와 소 트웨어 문제해결(제9조)을 구별하는 것이 요
하다.
소 트웨어 유지보수 로세스의 은 소 트웨어 제품의 릴리즈 후 발생하는 피드
백에 히 응하는 것이다. 의료기기의 일부인 소 트웨어 유지보수 로세스는 다
음을 확인(ensure)해야 한다.
- 안 련 문제보고서가 처리되었으며, 해당 규제당국과 해당 사용자에게 보고되
었다.
- 문제의 해결과 후속 문제 방지를 보장하는 공식 리장치로 수정 후 소 트웨어
제품이 타당성 재확인되거나 다시 릴리즈되었다.
- 제조업체가 향을 받을 수 있는 다른 소 트웨어 제품을 고려하며, 한 조치
를 취한다.
소 트웨어 문제해결의 은 다음과 같은 종합 인 리 시스템을 운 하는 것이
다.
- 문제 보고서를 분석하고 문제의 모든 측면을 식별한다.
- 변경의 숫자를 결정하고 부작용을 모두 식별한다.
- 험 리 일을 포함해 소 트웨어 형상 항목의 일 성을 유지하는 동시에 변경
을 구 한다.
- 변경의 구 을 검증한다.

- 174 -
소 트웨어 유지보수 로세스는 소 트웨어 문제해결 로세스를 사용한다. 소
트웨어 유지보수 로세스는 문제 보고서에 한 상 차원의 결정을 취 하며(문제가
존재하는지의 여부, 문제가 안 성에 상당한 향을 미치는지의 여부, 필요한 변경
변경의 구 시 ), 소 트웨어 문제해결 로세스를 사용해 모든 암시를 악하고,
변경할 필요가 있는 모든 형상 항목(item)과 필요한 모든 검증 단계를 식별하는 변경
요청을 생성한다.
B.6.3 수정 구
이 활동을 수행하려면 제조업체는 수정을 해 확립한 로세스를 사용해야 한다. 유지
보수 로세스가 정의되어 있지 않은 경우 해당 개발 로세스 업무를 사용해 수정을
수행할 수도 있다. 한 제조업체는 수정에 의해 의료기기 소 트웨어의 다른 부분에
부작용이 발생하지 않는다고 확인(ensure)해야 한다. 의료기기 소 트웨어를 새로 개발
한 소 트웨어로 취 하지 않는 한 체 의료기기 소 트웨어에 한 수정의 향을 분
석해야 한다. 수정되지 않은 의료기기 소 트웨어 부분이 수정 과 같은 성능을 발휘
할 수 있도록 수행되는 회귀 시험의 정도를 정당화하는 이론 근거를 마련해야 한다.
B.7 소 트웨어 험 리 로세스
소 트웨어 험 리은 체 인 의료기기 험 리의 일부이며, 따로 취 할 수는
없다. 이 표 을 사용하려면 ISO 14971을 수하는 의료기기 험 리 로세스를 사
용해야 한다. ISO 14971에 정의된 험 리은 특히 의료기기 사용에 연 된 험의
효과 인 경 을 한 체제를 취 한다. 험분석 식별한 각 험요인에 련해
식별한 험의 리에 ISO 14971 일부가 련된다. 이 표 에 규정한 소 트웨어 험
리 로세스의 목 은 험 분석 험한 상황을 잠재 으로 발생할 것으로 식
별된 소 트웨어 는 의료기기 험 리에 사용되는 소 트웨어가 포함된 소 트웨어
를 한 험 리의 추가 요구사항을 제공하는 것이다. 소 트웨어 험 리 로세스
는 다음과 같은 두 가지 이유 때문에 이 표 에 포함되었다.
a) 이 표 의 상 사용자는 담당 분야인 소 트웨어의 험 리 수단을 한 최소
한의 요구사항을 이해해야 한다.
b) 이 표 에 규정 참조로 제공되는 일반 험 리 표 ISO 14971은 소 트웨어에
한 험 리 소 트웨어 개발 수명주기에서의 험 리를 제 로 취 하
지 못한다.
소 트웨어 험 리은 체 인 의료기기 험 리의 일부이다. 소 트웨어 험

- 175 -
리 활동에 필요한 계획, 차 문서는 일련의 독립된 문서이거나 단일 문서일 수
있으며, 이 표 의 모든 요구사항이 충족될 경우 의료기기 험 리 활동과 문서에
통합할 수도 있다.
B.7.1 험한 상황에 한 소 트웨어 향의 분석
의료기기 험요인 분석에 따라 험한 상황과 그에 응하는 험 리 방법이 악
되어 그러한 험한 상황의 발생가능성과 심각도를 허용되는 수 으로 일 수 있을
것으로 기 된다. 한 험 리 방법은 그러한 험 리 방법을 구 할 것으로 상
되는 소 트웨어 기능에 할당될 것으로 상된다.
단, 소 트웨어 아키텍처를 생성할 때까지 모든 의료기기의 험한 상황이 식별될 것으
로 상되지는 않는다. 그러한 경우 소 트웨어 기능이 소 트웨어 요소에서 구 되는
방법과, 소 트웨어 기능에 할당된 실질 인 험 리 방법을 평가할 수 있는 방법을
악할 수 있다. 이 경우 의료기기 험요인 분석은 다음을 포함하도록 개정되어야 한다.
- 개정된 험한 상황
- 개정된 험 리 방법 소 트웨어 요구사항
- 를 들어 인간에 의해 발생되는 요인에 련된 험한 상황과 같이, 소 트웨어
에서 발생하는 새로운 험한 상황
소 트웨어 아키텍처에는 소 트웨어 요소가 불안 한 방식으로는 상호 작용할 수
없도록 소 트웨어 요소를 분리하기 한 신뢰성 있는 략이 포함되어야 한다.
B.8 소 트웨어 형상 리 로세스
소 트웨어 구성 리 로세스는 문서를 포함해 시스템의 소 트웨어 항목을 식별하
고 정의하며 기 을 정하고, 수정을 리하고 수정을 리하고 항목을 릴리즈하며, 항목
의 상태와 변경 요청을 기록하고 보고하기 해 소 트웨어 수명주기 체에 행정
기술 차를 용하는 로세스이다. 소 트웨어 구성 리는 소 트웨어 항목을
재창출하고 구성부품을 식별하며 항목에 이루어진 변경의 이력 제공에 필요하다.
B.8.1 형상 식별
이 활동을 수행하려면 제조업체는 소 트웨어 항목과 버 을 별도로 식별해야 한다.
의료기기 소 트웨어에 포함되는 소 트웨어 형상 항목과 버 의 식별에 이 식별이 필
요하다.

- 176 -
B.8.2 변경 리
이 활동을 수행하려면 제조업체는 소 트웨어 형상 항목의 변경을 리하고, 변경
요청을 식별하며 성격에 한 문서를 제공하는 정보를 기록해야 한다. 소 트웨어 형
상 항목에 무단 는 의도되지 않은 변경이 발생하지 않으며 승인된 변경요청이 구
되고 완 히 검증되도록 보장하려면 이 활동이 필요하다.
변경 요청은 소 트웨어 형상 리 계획에 따라 변경 리 원회나 리자 는 선임
기술자가 승인할 수 있다. 승인된 변경요청은 소 트웨어의 실제 수정과 검증까지
추 할 수 있어야 한다. 요구사항은 실질 인 각 변경이 변경 요청에 연계되며, 변경
요청 승인을 나타내는 문서가 존재해야 한다는 것이다. 문서는 변경 리 원회의 의
사록, 승인 서명 는 데이터베이스의 기록일 수 있다.
B.8.3 형상 상태 설명
이 활동을 수행하려면 제조업체는 소 트웨어 형상 항목(item) 이력의 기록을 유지해
야 한다. 변경이 이루어진 시 과 이유 단에 이 활동이 필요하다. 소 트웨어 형상
항목에 승인된 수정 내용만 수록되는지 확인(ensure)하려면 이 정보에 액세스해야 한
다.
B.9 소 트웨어 문제해결 로세스
소 트웨어 문제해결 로세스는 개발, 유지보수 는 기타 로세스 실행 확인
(discovered)된 것을 포함해 성격이나 발생원에 계 없이 문제(비 수성 포함)를 분
석하고 해결하는 로세스이다. 이 로세스의 목 은 확인(discovered)된 문제를 분석하
고 해결하며 경향을 확인(recognized)할 수 있도록 시에 하고 문서화된 방법을
제공하는 것이다. 소 트웨어 엔지니어링 문서에서는 이 로세스를 ‘결함 추 ’이라고
하는 경우가 많다. 이를 ISO/IEC 12207[9]와 IEC 60601-1-4[2] 부록 1에서는 “문제해
결”이라고 한다. 이 표 에서는 “소 트웨어 문제해결”이라고 칭하기로 한다.
이 활동을 수행하려면 제조업체는 소 트웨어 문제나 비 수성이 식별된 경우 문제
해결 로세스를 사용해야 한다. 발견된 문제가 안 성(ISO 14971에 명시된)에 련되
었는지 분석하고 평가하려면 이 활동을 수행해야 한다.
5.1에서 요구되는 것과 같은 소 트웨어 개발 계획이나 차에는 문제나 비 수성을
취 할 방법이 설명되어야 한다. 여기에는 수명주기의 각 단계에서 공식 으로 문서화되
는 소 트웨어 문제해결 로세스의 측면은 물론 문제 비 수성을 소 트웨어 문
제해결 로세스에 입력해야 하는 시 의 지정이 포함된다.

- 177 -
부록 C (참조용) 다른 표 과의 계
C.1 일반
이 표 은 의료기기 소 트웨어 개발과 유지보수에 용된다. 소 트웨어는 의료
기기의 서 시스템 는 그 자체가 의료기기로 간주된다. 이 표 은 의료기기를 개발
할 때 다른 해당 표 과 함께 사용해야 한다.
ISO 13485[7](C.2 부록 D 참조)과 ISO 14971(부록 C.3 참조)과 같은 의료기기 리
표 은 제품을 개발하는 조직을 한 기 를 형성하는 리 환경을 제공한다. IEC
60601-1[1](부록 C.4참조)과 IEC 61010-1[4](부록 C.5 참조)와 같은 안 성 표 은 안
한 의료기기 생성에 특별히 용되는 지침을 제공한다. 소 트웨어가 이러한 의료기
기의 일부일 경우 IEC 62304가 안 한 의료기기 소 트웨어 개발과 유지에 필요한 보
다 상세한 지시를 제공한다. ISO/IEC 12207[9](부록 C.6 참조), IEC 61508-3[3](부록 C.7
참조) ISO/IEC 90003[11]과 같은 많은 다른 표 은 IEC 62304의 요구사항의 구 에
사용할 수 있는 방법, 톨 기술로서 간주할 수 있다. 그림 C.1에는 이러한 표 사이
의 계가 표시된다.
다른 표 의 조항이나 요구사항이 인용되는 경우 인용된 항목에 정의된 용어는 이
표 에 정의된 용어가 아니라 다른 표 에 정의된 용어이다.
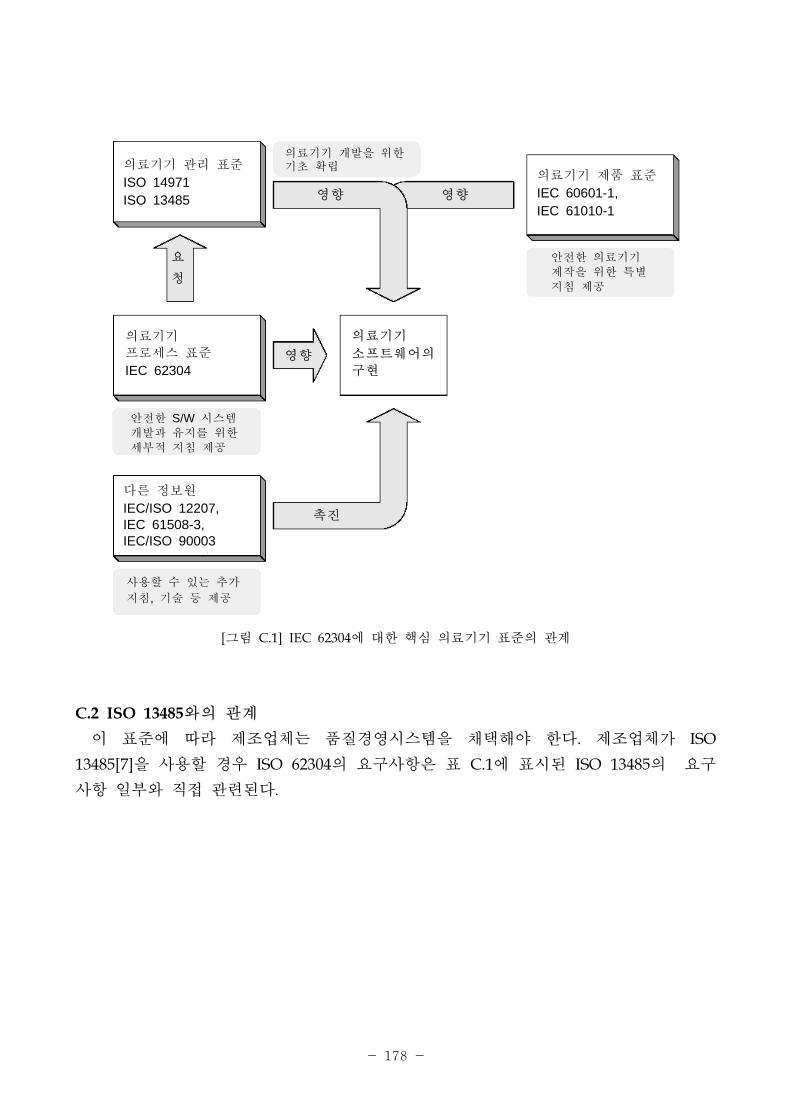
- 178 -
[그림 C.1] IEC 62304에 한 핵심 의료기기 표 의 계
향
요
청
진
향
향
의료기기 리 표
ISO 14971ISO 13485
의료기기 제품 표
IEC 60601-1,IEC 61010-1
의료기기
로세스 표
IEC 62304
다른 정보원
IEC/ISO 12207,IEC 61508-3,IEC/ISO 90003
의료기기
소 트웨어의
구
의료기기 개발을 한 기 확립
안 한 S/W 시스템 개발과 유지를 한 세부 지침 제공
사용할 수 있는 추가
지침, 기술 등 제공
안 한 의료기기 제작을 한 특별 지침 제공
C.2 ISO 13485와의 계
이 표 에 따라 제조업체는 품질경 시스템을 채택해야 한다. 제조업체가 ISO
13485[7]을 사용할 경우 ISO 62304의 요구사항은 표 C.1에 표시된 ISO 13485의 요구
사항 일부와 직 련된다.

- 179 -
[표 C.1] ISO 13485:2003과의 계
IEC 62304 조항 ISO 13485:2003의 련 조항
5.1 소 트웨어 개발 기획 7.3.1 설계 개발 기획
5.2 소 트웨어 요구사항 분석 7.3.2 설계 개발 입력
5.3 소 트웨어 아키텍처 설계
5.4 소 트웨어 상세 설계
5.5 소 트웨어 유닛 구 검증
5.6 소 트웨어 통합 통합 시험
5.7 소 트웨어 시스템 시험7.3.3 설계 개발 출력
7.3.4 설계 개발 검토
5.8 소 트웨어 릴리즈7.3.5 설계 개발 검증
7.3.6 설계 개발 밸리데이션
6.1 소 트웨어 유지보수 계획 확립 7.3.7 설계 개발 변경 리
6.2 문제 수정 분석
6.3 수정 구7.3.5 설계 개발 검증
7.3.6 설계 개발 밸리데이션
7.1 험한 상황에 한 소 트웨어 향의 분석
7.2 험 리 방법
7.3 험 리 방법의 검증
7.4 소 트웨어 변경의 험 리
8.1 형상 식별 7.5.3 식별 추 성
8.2 변경 리 7.5.3 식별 추 성
8.3 형상 상태 설명
9 소 트웨어 문제해결 로세스
C.3 ISO 14971와의 계
표 C.2에는 ISO 14971에 의해 요구되는 험 리 로세스에 한 요구사항을 IEC
62304가 강조하는 역이 표시된다.

- 180 -
[표 C.2] ISO 14971:2000과의 계
ISO 14971:2000 조항 ISO 62304의 련 조항
4.1 험 평가 차
4.2 의료기기 안 련 의도한 용도/ 목 특성의 식별
4.3 식별 는 측가능한 험요인의 식별 7.1 험한 상황에 한 소 트웨어 향 분석
4.4 각 험요인에 한 험 추정 4.3 소 트웨어 안 성 분류
5 험 평가
6.1 험 축소
6.2 옵션 분석 7.2.1 험 리 방법 정의
6.3 험 리 방법의 구7.2.2 소 트웨어에 구 된 험 리 방법
7.3.1 험 리 방법 검증
6.4 잔류 험 평가
6.5 험/장 분석
6.6 기타 발생 험요인 7.3.2 사상의 새로운 발생순서 문서화
6.7 험 평가 완료
7 체 잔류 험 평가
8 험 리 보고서 7.3.3 추 성 문서화
9 생산 후 정보 7.4 소 트웨어 변경의 험 리
C.4 IEC 60601-1:2004의 PEMS 요구사항에 한 계
C.4.1 일반
소 트웨어에 한 요구사항은 로그램이 가능한 기 의료기기(PEMS)에 한 요
구사항의 하 요구사항이다. 이 표 은 PEMS을 한 IEC 60601-1[1] 요구사항에 추가
되지만 그 요구사항들과 호환되지 않는 소 트웨어에 한 요구사항에 한 것이다.
PEMS에는 소 트웨어가 아닌 요소도 포함되므로 PEMS에 한 IEC 60601-1의 요구사
항이 모두 이 표 에서 취 되는 것은 아니다.
C.4.2 PEMS 개발과의 소 트웨어 계
PEMS 개발 발생하는 상의 설명을 해 그림 C.2에 표시된 V 모델을 사용하면
소 트웨어 요구사항의 지정에서부터 소 트웨어 항목을 소 트웨어 시스템에 통합할
때까지 PEMS 요소 수 에서 이 소 트웨어 수 의 요구사항이 용되는 것을 확인할
수 있다. 이 소 트웨어 시스템은 PEMS의 일부인 로그램이 가능한 기 서 시스템
(PESS)의 일부이다.

- 181 -
C.4.3 개발 로세스
이 표 의 소 트웨어 개발 로세스(제5조)와의 수성을 유지하려면 소 트웨어 개발
계획을 지정하고 그를 따라야 한다. 여기에는 특정 수명주기 모델의 사용은 필요하지
않지만 계획에는 특정 아키텍처와 속성이 포함되어야 한다.
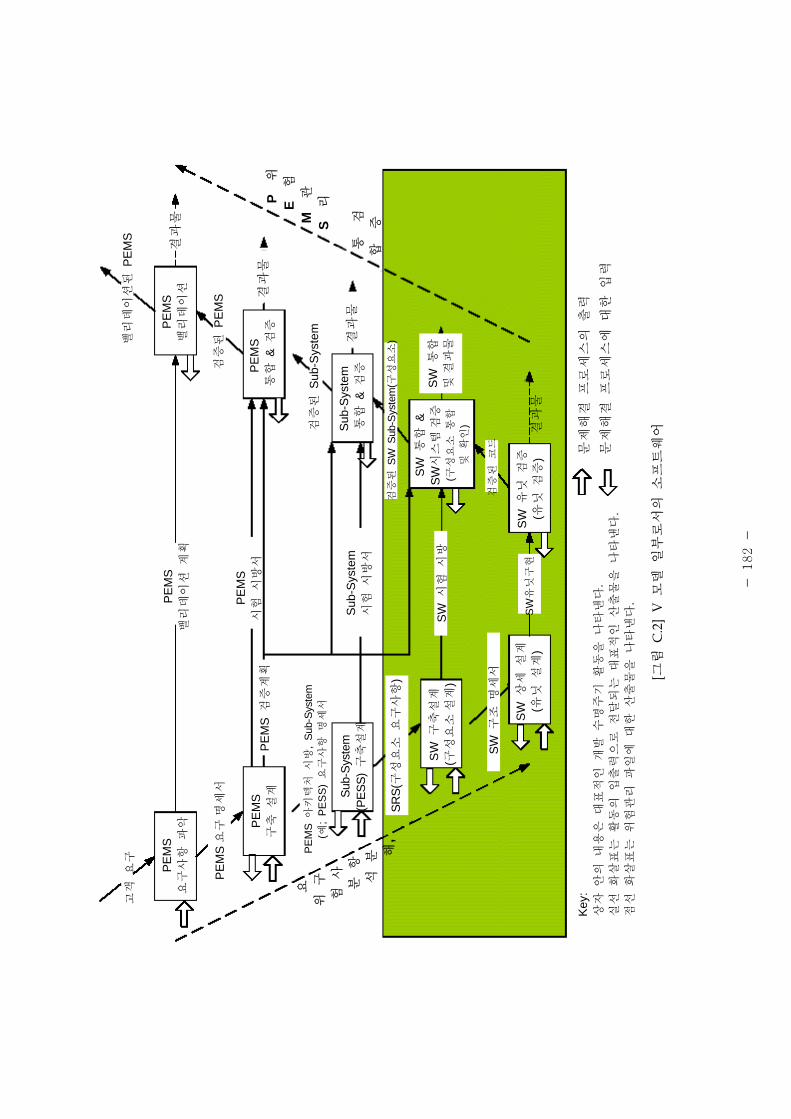
- 182
-
[그림
C.2
] V
모
델 일
부로
서의
소트
웨어
문제해결 로세스의 출력
문제해결 로세스에 한 입력
Key
:상자 안의 내용은 표인 개발 수명주기 활동을 나타낸다
.실선 화살표는 활동의 입출력으로 달되는 표인 산출물을 나타낸다
.선 화살표는 험리 일에 한 산출물을 나타낸다
.
PE
MS
아키텍처 시방
, Su
b-Sy
stem
(
; P
ESS
) 요구사항 명세서
PE
MS
요구사항 악
PE
MS
밸리데이션 계획
PE
MS
구축 설계 Su
b-Sy
stem
(P
ESS)
구축설계
Sub
-Sys
tem
통합 &
검증P
EM
S 통합 &
검증P
EM
S밸리데이션
SW 통합 &
SW시스템검증
(구성요소 통합
확인
)
SW
구축설계
(구성요소설계
)
SW
유닛 검증
(유닛 검증
)SW
상세 설계
(유닛 설계
)
SRS(구성요소 요구사항
)검증된 S
W S
ub-S
yste
m(구성요소
)
SW 시험 시방
SW 구조 명세서
SW유닛구
검증된 코드
SW
통합
결과물
고객 요구
PE
MS요구명세서
PE
MS
시험 시방서
밸리데이션된 P
EM
S
결과물
결과물
결과물
결과물
PE
MS
검증계획
검증된 P
EM
S
Sub
-Sys
tem
시험 시방서
검증된 S
ub-S
yste
m
요
구
험 사
분 항
석 분
해
,
P
E
험
M
S 리
통 검
합 증

- 183 -
IEC 60601-1:005의 PEMS 요구사항PEM의 소 트웨어 서 시스템에 련된
IEC 62304의 요구사항
14.1 일반
이 요구사항은 다음 경우를 제외하고 PEMS에 용
된다.
- PESS가 기본 안 성이나 필수 성능을 제공하지
않는 경우
- ISO 14971 용 결과 PESS 결함으로도 허용할 수
없는 험이 발생하지 않음을 확인(demonstrate)될
경우
4.3 소 트웨어 안 성 분류
IEC 60601-1의PEMS 요구사항은 소 트웨어 안 성
등 B와 C에만 용된다. 이 표 에는 소 트웨어
안 성 등 A에 한 요구사항이 일부 포함된다.
14.2 문서화
ISO 14971에서 요구되는 기록과 문서 이외 제14조 4.2 험 리
이러한 요구사항은 개발 수명주기, 요구사항 시방서, 아키텍처, 설계와 구 검증
에 한 IEC 60601-1의 PEMS 요구사항에 련된다. 이 표 의 요구사항은 IEC
60601-1보다 소 트웨어 개발에 해 더 많은 정보를 제공한다.
C.4.4 유지보수 로세스
이 표 의 소 트웨어 유지보수 로세스(제6조)와의 수성을 유지하려면 소 트웨
어를 변경할 때 차를 확립하고 그를 따라야 한다. 이러한 요구사항은 PEMS의 수정
에 한 IEC 60601-1의 요구사항에 해당된다. 소 트웨어 유지보수를 한 이 표 의
요구사항은 소 트웨어 유지보수를 해 수행하는 것과 련해 IEC 60601-1의 PEMS
수정에 한 요구사항보다 더 많은 정보를 제공한다.
C.4.5 기타 로세스
이 표 의 기타 로세스는 IEC 60601-1의 PEMS에 한 유사한 요구사항 이외의 소
트웨어 요구사항을 추가로 지정한다. 부분의 경우 이 표 의 로세스가 확장되는
IEC 60601-1의 PEMS에 한 일반 인 요구사항이 있다.
이 표 의 소 트웨어 험 리 로세스는 IEC 60601-1의 PEMS에 추가로 식별된
험 리 요구사항에 해당되며, 이 표 의 소 트웨어 문제해결 로세스는 IEC 60601-1
의 PEMS에 한 문제해결 험 리 요구사항에 해당된다.
이 표 의 소 트웨어 형상 리 로세스는 문서화를 제외하고 IEC 60601-1의 PEMS
에 해 제시되지 않는 요구사항을 추가로 지정한다.
C.4.6 IEC 60601-1의 PEMS 요구사항의 취 범
표 C.3에는IEC 60601-1의 PEMS 요구사항 이 표 의 해당 요구사항이 수록된다.

- 184 -
용에 따라 발생하는 문서를 유지해야 하며, 그러
한 문서는 험 리 일의 일부를 형성해야 한다.
제14조에 따라 요구되는 문서는 공식 문서 리
차에 따라 검토, 승인, 발 변경되어야 한다.
5.1 소 트웨어 개발 기획
소 트웨어 개발기획 활동의 특정 요구사항 이외에
ISO 14971에 따라 험 리 일의 일부인 문서가
유지되어야 한다. 한 품질 시스템이 필요로 하는
문서의 경우 ISO 13485[7]에 따라 문서를 리해야
한다.
14.3 험 리 계획
ISO 14971의3.5에 따라 요구되는 험 리 계획에는
PEMS 밸리데이션 계획에 한 기 도 포함되어야
한다(14.11 참조).
특별히 필요하지 않다.
특정 소 트웨어 밸리데이션 계획이 없다. PEMS 밸
리데이션 계획 시스템 차원에서 이루어지므로 이 소
트웨어 표 의 범 에 포함되지 않는다. 이 표 에
는 험 리 방법에 한 특정 소 트웨어 원인에
한 험요인부터 험 리 방법의 검증까지의 추
성이 필요하지 않다(7.3 참조).
14.4 PEMS 개발 수명주기
PEMS 개발 수명주기를 문서화해야 한다.
5.1 소 트웨어 개발 기획
5.1.1 소 트웨어 개발 계획
소 트웨어 개발 계획이 처리하는 항목(item)은 소
트웨어 개발 수명주기를 구성한다.
PEMS 개발 수명주기에는 정의된 일정이 포함되어
야 한다.
각 일정에서 완료해야 할 활동과 그러한 활동에
용되는 검증 방법을 정의해야 한다.
5.1.6 소 트웨어 검증 기획
검증 업무, 일정 인수 기 을 계획해야 한다.
각 활동은 입력과 출력을 포함해 정의해야 한다.
5.1.1 소 트웨어 개발 계획
활동은 이 표 에서 정의된다. 제작할 문서가 각 활
동에서 정의된다.
각 일정은 일정 완료해야 하는 험 리 활동을
식별해야 한다.
PEMS 개발 수명주기는 활동, 일정 스 이 자
세히 규정된 계획을 작성함으로써 특정 개발에
합하게 조정해야 한다.
5.1.1 소 트웨어 개발 계획
이 표 에 따라 개발 수명주기를 개발 계획에 문서
화할 수 있다. 즉, 개발 계획에는 조정된 개발 수명
주기가 포함된다.
PEMS 개발 수명주기에는 문서화 요구사항이 포함
되어야 한다.
5.1.1 소 트웨어 개발 계획
5.1.8 문서화 기획
14.5 문제해결
필요한 경우 PEMS 개발 수명주기의 모든 단계와
활동 사이의 문제해결에 해 문서화한 시스템을
개발하고 유지해야 한다.
9 소 트웨어 문제해결 로세스
제품 유형에 따라 문제해결 시스템은 다음과 같을
수 있다.
- PEMS 개발 수명주기 일환으로 문서화된다.
- 기본 안 성이나 필수 성능에 향을 주는 잠재
는 기존 문제를 보고한다.
- 연 험에 한 각 문제의 평가가 포함된다.
- 마감해야 할 안에 하여 충족해야 할 기 을
식별한다.
- 각 문제해결을 해 취해야 할 조치를 식별한다.
5.1.1 소 트웨어 개발 계획
9.1 문제 보고서 작성
14.6 험 리 로세스 7 소 트웨어 험 리 로세스
14.6.1 확인(known)되거나 측할 수 있는 험요인 7.1 험한 상황에 한 소 트웨어 향의 분석
- 185 -
의 식별
확인(known)되거나 측 가능한 험요인의 목록을
작성할 때 제조업체는 네트워크/데이터 결합, 제3자
원본 구성요소와 거시 서 시스템에 련된 것
을 포함해 PEMS의 소 트웨어와 하드웨어 측면에
련된 험요인을 고려해야 한다.
이 표 에는 네트워크/데이터 결합은 언 되지 않는
다.
14.6.2 험 리
각 험 리 방법을 구 하려면 하게 밸리데이
션된 도구와 차를 선택하고 식별해야 한다. 이러
한 도구와 차는 각 험 리 방법이 식별된 험
을 만족스러운 수 으로 축소하는지 확인(assure)할
수 있도록 해야 한다.
5.1.4 소 트웨어 개발 표 , 방법 도구 기획
이 표 은 각 험 리 방법이 아닌, 개발에 일반
으로 사용되는 특정 도구와 방법을 식별해야 한다.
14.7 소 트웨어 시방서
PEMS와 그 서 시스템( 를 들어 PESS에 한)의
경우 문서화한 요구사항 시방서가 있어야 한다.
5.2 소 트웨어 요구사항 분석
이 표 은 PEMS의 소 트웨어 서 시스템만 취
한다.
시스템이나 서 시스템에 한 요구사항 시방서에
는 그 시스템이나 서 시스템이 구 한 핵심 성능
험 리 방법이 포함되어야 한다.
5.2.1 시스템 요구사항에서 소 트웨어 요구사항을
정의하고 문서화
5.2.2 소 트웨어 요구사항 내용
5.2.3 소 트웨어 요구사항에 험 리 방법 포함
이 표 에 따르면 필수 성능과 험 리 방법을 다
른 요구사항과 구별할 필요는 없지만 모든 요구사항
은 각각 식별해야 한다.
14.8 아키텍처
PEMS과 각 서 시스템의 경우 아키텍처는 요구사
항 시방서를 충족하도록 지정되어야 한다.
5.3 소 트웨어 구축 설계
필요한 경우 험을 허용 수 으로 축소하려면 아
키텍처 시방서는 아래의 내용을 사용해야 한다.
a) 무결성이 높은 특성의 구성요소
b) 결함이 발생하지 않는 기능
c) 복성
d) 다양성
e) 기능성의 분할
f) 방어 설계. 일례로 가용 출력 워를 제한하거나
작동기 왕복운동을 제한하는 방법을 채택함으로
써 잠재 으로 험한 향을 제한.
5.3.5 험 리에 필요한 분리 식별
분할은 식별된 유일한 기술이며, 분할의 무결성을 보
장하는 방법을 나타내야 한다는 요구사항이 있기 때
문에 유일하게 식별된다.
아키텍처 시방서는 다음을 고려해야 한다.
g) 험 리 방법을 PEMS의 서 시스템과 구성요
소에 할당
h) 구성요소의 결함 모드 그 향
i) 결함의 공통 원인
j) 시스템 결함
k) 시험 간격과 진단 용범
l) 유지보수 가능성
m) 타당한 수 으로 측할 수 있는 오용의 방지
n) 해당되는 경우 네트워크/데이터 결합 시방서
이 표 에는 포함되지 않는다.
14.9 설계 구
필요한 경우 설계는 각각 설계와 시험 시방서가 있
는 서 시스템으로 분해해야 한다.
5.4 소 트웨어 상세 설계
5.4.2 각 소 트웨어 유닛에 해 상세 설계 개발
이 표 에 따르면 상세 설계에는 시험 시방서가 필
요하지 않다.
설계환경에 련된 설명 데이터를 험 리 일이
포함해야 한다.5.4.2 각 소 트웨어 유닛에 해 상세 설계 개발
14.10 검증 5.1.6 소 트웨어 검증 기획

- 186 -
기본 안 성, 필수 성능 는 험 리 방법을 구
하는 모든 기능을 검증해야 한다. 각 활동에 검증이 필요하다.
이러한 기능을 검증해야 하는 방법을 나타내기
한 검증 계획을 작성해야 한다. 계획에는 다음과 같
은 내용이 포함되어야 한다.
- 각 기능에 해 검증을 수행해야 하는 시
- 검증 략, 활동, 기술 검증을 수행하는 담당자
의 한 독립성 수 의 선택과 문서화
- 검증 도구의 선택과 활용
- 검증을 한 용 범 기
5.1.6 소 트웨어 검증 기획
이 표 에는 담당자 독립성은 포함되지 않는다. 이
독립성은 ISO 13485에서 취 한다.
검증 계획에 따라 검증을 수행해야 한다. 검증 활동
의 결과는 문서화해야 한다.부분의 활동에는 검증 요구사항이 있다.
14.11 PEMS 밸리데이션
PEMS 밸리데이션 계획에는 기본 안 성과 필수 성
능의 밸리데이션이 포함되어야 하며, PEMS의 의도
되지 않은 기능 여부를 검해야 한다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
PEMS 밸리데이션은 PEMS 밸리데이션 계획에 따라
수행해야 한다. PEMS 밸리데이션 활동의 결과는
문서화해야 한다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
PEMS 밸리데이션에 해 반 인 책임을 지는 담
당자는 설계 과 독립되어야 한다. 제조업체는 독
립성 수 에 한 이론 근거를 문서화해야 한다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
설계 의 구성원은 자신의 설계에 한 PEMS 밸
리데이션을 담당할 수 없다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
PEMS 밸리데이션 구성원과 설계 구성원의
모든 문 계를 험 리 일에 문서화해야
한다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
PEMS 밸리데이션의 방법과 결과에 한 참고문서
가 험 리 일에 포함되어야 한다.
이 표 은 소 트웨어 밸리데이션을 처리하지 않는
다. PEMS 밸리데이션은 시스템 차원 활동이므로 이
표 의 범 에 포함되지 않는다.
14.12 수정
설계 일부나 부가 이 설계의 수정에 의한 것일
경우 수정에 의한 설계가 새로운 설계인 것으로 간
주해 이 조항을 부 용하거나, 이 설계 문서의
연속 유효성을 문서화된 수정/변경 차에 따라 평
가해야 한다.
6 소 트웨어 유지보수 로세스
이 표 은 소 트웨어 유지보수를 기획해야 하고 수
정의 구 이 소 트웨어 개발 로세스나 확립된 소
트웨어 유지보수 로세스를 사용해야 한다는
근방식을 선택한다.
14.13 다른 장치에 한 Network/Data 연계에 의한
PEMS 연결
PEMS 제조업체의 리 역을 벗어나는 다른 장치
와의 네트워크/데이터 연계를 사용해 PEMS를 연결
할 경우 기술 설명은 다음과 같아야 한다.
a) 의도된 사용의 달성을 해 PEMS에 필요한 네
트워크/데이터 연계의 특성을 명시한다.
b) 명시한 특성 제공을 해 네트워크/데이터 연계
의 실패에서 발생하는 험한 상황을 나열한다.
c) 해당 조직에 다음을 알린다.
- 다른 장치가 포함된 네트워크/Data 연계에
한 PEMS 연결에 따라 이 에 확인되지 않은
(unidentified) 험이 환자, 운 자 는 제3자
에게 발생할 수 있다.
이 표 에는 네트워크/데이터 연계 요구사항은 포함
되지 않는다.

- 187 -
[표 C.3] IEC 60601-1과의 계
- 해당 조직은 이러한 험을 식별, 분석, 평가
리해야 한다.
- 네트워크/Data 연계에 한 후속 변경 때문에
새로운 험이 발생할 수 있으므로 분석을 추
가로 수행해야 한다.
- 네트워크/데이터 연계의 변경에는 다음이 포
함된다.
- 네트워크/Data 형상의 변경
- 네트워크/Data 연계에 추가 항목 연결
- 네트워크/Data 연계에서 항목 분리
- 네트워크/Data 연계에 연결된 장치의 갱신
- 네트워크/Data 연계에 연결된 장치의 업그
이드
IEC 60601-1-4:1996 개정 1:1999의 PEMS 요구사항 IEC 62304의 련 요구사항
6.8 첨부 문서6.8.210 4.2와 4.3 c)52.201 문서화52.201.1 4.152.201.2 4.1와 4.252.201.3 4.252.202 험 리 계획
52.202.1 4.252.202.2 5.1.1, 5.1.552.202.3 4.1, 5.1.1 g52.203 개발 수명주기52.203.1 5.1.152.203.2 5.1.152.203.352.203.4 5.1.752.203.5 752.204 험 리 로세스
52.204.1 4.2
C.4.7 IEC 60601-1-4의 요구사항에 한 계
IEC 60601-1-4는 IEC 60601-1:2005에 한 환 기간이 완료될 때까지 계속 사용된
다.
표 C.4에는IEC 60601-4[2]의 요구사항 이 표 의 해당 요구사항이 수록된다. 이
표는 이 표 의 련 요구사항이 IEC 60601-1-4의 요구사항을 모두 처리한다고 명시하
지 않는다. 60601-1-4 요구사항의 부분은 ISO 14971과의 수성에 따라 처리된다.
IEC 60601-1-4의 일부 요구사항은 IEC 62304가 처리하지 않는다.

- 188 -
52.204.2 4.2, 752.204.352.204.3.152.204.3.1.1 4.2, 7.152.204.3.1.2 4.2, 7.1.252.204.3.1.3 4.252.204.3.1.4 4.2, 7.1.2 e)52.204.3.1.5 4.2, 7.1.252.204.3.1.6 4.2, 7.152.204.3.1.7 4.252.204.3.1.8 4.252.204.3.1.9 4.252.204.3.1.10 4.252.204.3.252.204.3.2.1 4.252.204.3.2.2 4.2, 4.352.204.3.2.352.204.3.2.452.204.3.2.5 4.252.204.452.204.4.1 4.252.204.4.2 4.252.204.4.3 4.252.204.4.4 4.252.204.4.552.204.4.6 4.252.205 담당자의 자격검증 4.152.206 요구사항 시방서52.206.1 5.252.206.2 7.2.252.206.352.207 아키텍처52.207.1 5.3.152.207.2 5.352.207.352.207.452.207.552.208 설계 구
52.208.1 552.208.252.209 검증52.209.1 5.7.152.209.2 5.1.5, 5.1.652.209.3 5.2.6, 5.3.6, 5.4.4, 5.5.5, 5.6, 5.752.209.4

- 189 -
[표 C.4] IEC 60601-4와의 계
52.210 밸리데이션52.210.1 4.152.210.2 4.152.210.3 4.152.210.452.210.552.210.652.210.752.211 수정52.211.1 652.211.2 4.1, 652.212 평가52.212.1 4.1
C0.5 IEC 61010-1에 한 계
IEC 61010-1[4]의 범 에는 기 시험과 측정장치, 기 리 장치와 기 실험실 장치
가 포함된다. 의료 환경이나 체외 진단장치(IVD)에는 실험실 장치만 사용된다.
법규 는 규제 참고문헌 때문에 IVD 장치는 의료기기로 할당되지만, IEC
60601-1[1]의 범 안에 든다. 그 이유는 엄격히 말해 IVD 계측기는 환자와 직
하는 의료기기가 아니라는 사실과 그러한 제품은 다양한 실험실에서 다양한 용도를
해 제조된다는 사실에서 비롯된다. 따라서 IVD 계측기나 IVD 계측기의 부속품으로서
사용하는 경우는 드물다.
실험실 장치를 IVD 장치로 사용할 경우 특정 결과는 의료 기 에 따라 평가해야 한
다. 험 리에는 ISO 14971의 용이 필요하다. 한 그러한 제품에 를 들어 소 트
웨어 결함에 의한 의료 데이터(측정결과)의 불필요한 변경과 같은 험요인을 발생시킬
수 있는 소 트웨어가 포함된 경우 IEC 62304를 고려해야 한다.
그림 C.3의 흐름도를 보면 험 리 로세스와 IEC 62304의 용에 한 일차 인
방법을 쉽게 이해할 수 있다.
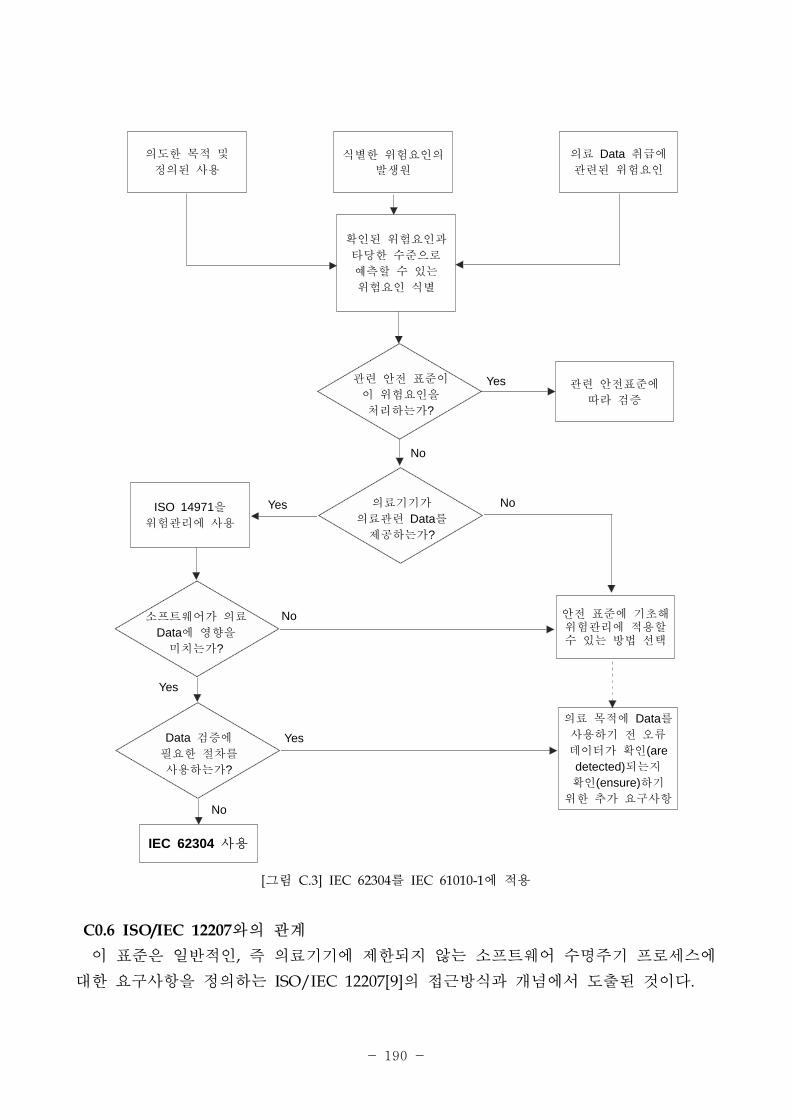
- 190 -
[그림 C.3] IEC 62304를 IEC 61010-1에 용
의도한 목
정의된 사용
식별한 험요인의
발생원
의료 Data 취 에
련된 험요인
확인된 험요인과
타당한 수 으로
측할 수 있는
험요인 식별
련 안 표 에
따라 검증
ISO 14971을 험 리에 사용
IEC 62304 사용
련 안 표 이
이 험요인을
처리하는가?
안 표 에 기 해 험 리에 용할 수 있는 방법 선택
의료 목 에 Data를 사용하기 오류
데이터가 확인(are detected)되는지 확인(ensure)하기 한 추가 요구사항
Yes
No
Yes
No
Yes
No
Yes
No
Data 검증에 필요한 차를
사용하는가?
의료기기가
의료 련 Data를 제공하는가?
소 트웨어가 의료
Data에 향을 미치는가?
C0.6 ISO/IEC 12207와의 계
이 표 은 일반 인, 즉 의료기기에 제한되지 않는 소 트웨어 수명주기 로세스에
한 요구사항을 정의하는 ISO/IEC 12207[9]의 근방식과 개념에서 도출된 것이다.

- 191 -
ISO/IEC 62304 로세스 ISO/IEC 12207 로세스
활동 업무 활동 업무
5 소 트웨어 개발 로세스
5.3 개발 로세스6.1 문서화 로세스6.2 형상 리 로세스
6.4 검증 로세스6.5 밸리데이션 로세스6.8 문제해결 로세스7.1 경 로세스
이 표 은 주로 다음과 련해 ISO/IEC 12207과는 다르다. 이 표 의 성격은 다음과
같다.
- 시스템 요구사항, 시스템 아키텍처 밸리데이션과 같은 시스템 련 측면을 배
제한다.
- 의료기기에 해 다른 표 에 문서화되고 복되는 활동이라고 간주되는 로세
스 일부를 생략한다.
- (안 성) 험 리 로세스와 소 트웨어 릴리즈 로세스를 추가한다.
- 로세스를 지원하는 문서와 검증을 개발과 유지보수 로세스에 포함시킨다.
- 각 로세스의 로세스 구 과 기획을 개발과 유지보수 로세스의 단일 활동으
로 통합한다.
- 안 성 필요와 련한 요구사항을 분류한다.
- ISO/IEC 12207과는 달리 로세스를 일차 는 지원 로세스로 명백히 분류
하지 않으며, 로세스를 그룹으로 구성하지 않는다.
이러한 변경의 부분은 표 을 의료기기 분야의 요구에 따라 다음과 같은 방식으
로 조정하고자 하는 필요성에 의해 발생한다.
- 안 성 측면 의료기기 험 리 표 ISO 14971에 집 한다.
- 규제된 환경에 유용한 한 로세스를 선택한다.
- 소 트웨어 개발이 품질시스템에 포함되어 있다는 사실을 고려한다(ISO/IEC
12207의 로세스와 요구사항 일부 취 ).
- 사용 용이성의 진을 해 추상 인 수 을 내린다.
이 표 은 ISO/IEC 12207과 상반되는 것이 아니다. ISO/IEC 12207은 이 표 의 요구
사항이 포함되는, 잘 구성된 소 트웨어 개발 수명주기 모델의 설정에 유용하다.
표 C.5에 IEC 62304와 ISO/IEC 12207과의 계를 표시한다.

- 192 -
5.1 소 트웨어
개발 기획
5.3.1 로세스 구
5.3.3 소 트웨어 구축 설계
5.3.7 소 트웨어 코딩 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
5.3.10 시스템 통합6.1.1 로세스 구
6.2.1 로세스 구
6.2.2 형상 식별6.4.1 로세스 구
6.5.1 로세스 구
6.8.1 로세스 구
7.1.2 기획7.1.3 실행 리7.2.2 기반구조의 확립7.2.3 기반구조의 유지보수
5.1.1 소 트웨어 개발 계획5.3.1 로세스 구
7.1.2 기획
5.3.1.15.3.1.35.3.1.47.1.2.1
5.1.2 소 트웨어 개발 계획
갱신 유지7.1.3 실행 리 7.1.3.3
5.1.3 시스템 설계와 개발에 한 소 트웨어 개발
계획 참조
5.3.3 시스템 구축 설계5.3.10 시스템 통합6.5.1 로세스 구
5.3.3.15.3.10.16.5.1.4
5.1.4 소 트웨어 개발 표 , 방법 도구 기획
5.3.1 로세스 구5.3.1.35.3.1.4
5.1.5 소 트웨어 통합
통합 시험 기획5.3.8 소 트웨어 통합 5.3.8.1
5.1.6 소 트웨어 검증 기획
6.4.1 로세스 구
5.3.7 소 트웨어 코딩 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
6.4.1.46.4.1.55.3.7.55.3.8.55.3.9.3
5.1.7 소 트웨어 험 리
기획
개정 1:2002 – F 3.15 험 리
로세스
5.1.8 문서화 기획 6.1.1 로세스 구 6.1.1.1
5.1.9 소 트웨어 형상 리
기획
6.2.1 로세스 구
6.8.1 로세스 구
6.2.1.16.8.1.1
5.1.10 리할 지원 항목7.2.2 기반구조의 확립7.2.3 기반구조의 유지보수
7.2.2.17.2.3.1
5.1.11 검증 소 트웨어 6.2.2 형상 식별 6.2.2.1
- 193 -
형상 항목(item) 식별
5.2 소 트웨어
요구사항 분석
5.3.3 시스템 구축 설계5.3.4 소 트웨어 요구사항 분석
6.4.2 검증
5.2.1 시스템 요구사항에서 소 트웨어 요구사항을
정의하고 문서화
5.3.3 시스템 구축 설계 5.3.3.1
5.2.2 소 트웨어 요구사항
내용
5.3.4 소 트웨어 요구사항 분석 5.3.4.15.2.3 소 트웨어 요구사항에
험 리 방법 포함
5.2.4 의료기기 험분석 재평가
없음
5.2. 시스템 요구사항 갱신 5.3.4 소 트웨어 요구사항 분석 a) b)
5.2.6 소 트웨어 요구사항
검증
5.3.4 소 트웨어 요구사항 분석
6.4.2 검증5.3.4.26.4.2.3
5.3 소 트웨어
구축 설계
5.3.5 소 트웨어 구축 설계
5.3.1 소 트웨어 요구사항을
아키텍처로 변형
5.3.5 소 트웨어 구축 설계
5.3.5.1
5.3.2 소 트웨어 항목의
연계성을 한
아키텍처 개발
5.3.5.2
5.3.3 SOUP 항목의 기능 성능 요구사항 명시
없음
5.3.4 SOUP 항목에 필요한 시스템 하드웨어와
소 트웨어 명시
없음
5.3.5 험 리에 필요한
분리 식별없음
5.3.6 소 트웨어 아키텍처
검증5.3.5 소 트웨어 구축 설계 5.3.5.6
5.4 소 트웨어
상세 설계
5.3.6 소 트웨어 상세 설계
6.4.2 검증
5.4.1 소 트웨어 아키텍처를
소 트웨어 유닛으로
개량5.3.6 소 트웨어 상세 설계 5.3.6.1
5.4.2 각 소 트웨어 유닛에
- 194 -
해 상세 설계 개발
5.4.3 연계성에 한 상세 설계 개발
5.3.6.2
5.4.4 상세 설계 검증 6.4.2 검증 5.3.6.7
5.5 소 트웨어
유닛 구
확인
5.3.6 소 트웨어 상세 설계
5.3.7 소 트웨어 코딩 시험
6.4.2 검증
5.5.1 각 소 트웨어 유닛
구5.3.7 소 트웨어 코딩 시험 5.3.7.1
5.5.2 소 트웨어 유닛 검증
로세스 확립
5.3.6 소 트웨어 상세 설계
5.3.7 소 트웨어 코딩 시험
5.3.6.55.3.7.5
5.5.3 소 트웨어 유닛 인수
기5.3.7 소 트웨어 코딩 시험 5.3.7.5
5.5.4 소 트웨어 유닛 추가
인수 기
5.3.7 소 트웨어 코딩 시험
6.4.2 검증5.3.7.56.4.2.5
5.5.5 소 트웨어 유닛 검증 5.3.7 소 트웨어 코딩 시험 5.3.7.2
5.6 소 트웨어
통합 통합
시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
5.3.10 시스템 통합6.4.1 로세스 구
6.4.2 검증
5.6.1 소 트웨어 유닛 통합 5.3.8 소 트웨어 통합 5.3.8.2
5.6.2 소 트웨어 통합 검증5.3.8 소 트웨어 통합
5.3.10 시스템 통합5.3.8.25.3.10.1
5.6.3 통합 소 트웨어 시험 5.3.9 소 트웨어 자격검증 시험 5.3.9.1
5.6.4 통합 시험 내용 5.3.9.3.
5.6.5 통합 시험 차 검증 6.4.2 검증 6.4.2.2
5.6.6 회귀시험 수행 5.3.8 소 트웨어 통합 5.3.8.2
5.6.7 통합시험 기록 내용 5.3.8 소 트웨어 통합 5.3.8.2
5.6.8 소 트웨어 문제해결
로세스의 사용6.4.1 로세스 구 6.4.1.6
5.7 소 트웨어
시스템 시험
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
6.4.1 로세스 구
6.4.2 검증
- 195 -
6.8.1 로세스 구
5.7.1 각 소 트웨어
요구사항에 한 시험
확립
5.3.8 소 트웨어 통합
5.3.9 소 트웨어 자격검증 시험
5.3.8.45.3.9.1
5.7.2 소 트웨어 문제해결
로세스의 사용6.4.1 로세스 구 6.4.1.6
5.7.3 변경 후 재시험 6.8.1 로세스 구 6.8.1.1
5.7.4 소 트웨어 시스템
시험 검증
6.4.2 검증5.3.9 소 트웨어 자격검증 시험
6.4.2.25.3.9.3
5.7.5 각 시험 소 트웨어
시스템 시험 기록
내용에 한 데이터
문서화
5.3.9 소 트웨어 자격검증 시험 5.3.9.1
5.8 소 트웨어
릴리즈
5.3.9 소 트웨어 자격검증 시험
5.4.2 작동 시험6.2.5 형상 평가6.2.6 릴리즈 리 인도
5.8.1 소 트웨어 검증의
완벽성 확인(ensure)5.4.2 작동 시험6.2.6 릴리즈 리 인도
5.4.2.15.4.2.26.2.6.1
5.8.2 확인(known)된 잔류 비정상 상태 문서화
6.2.5 형상 평가5.3.9 소 트웨어 자격검증 시험
6.2.5.15.3.9.35.8.3 확인(known)된 잔류
비정상 상태 평가
5.8.4 릴리즈 버 의 문서화
6.2.6 릴리즈 리 인도 6.2.6.1
5.8.5 릴리즈한 소 트웨어를
작성한 방법 문서화
5.8.6 활동과 업무의 완결 확인(ensure)
5.8.7 소 트웨어 장
5.8.8 소 트웨어 릴리즈의
반복 정 도 보장
6 소 트웨어 유지보수 로세스5.5 유지보수 로세스6.2 형상 리 로세스
6.1 소 트웨어
유지보수 계획
확립
5.5.1 로세스 구 5.5.1.1
6.2 문제 수정 5.5.1 로세스 구
- 196 -
분석
5.5.2 문제 코드화 분석5.5.3 수정 구5.5.5 감소(마이그 이션)
6.2.1 피드백 기록 평가
6.2.1.1 피드백 모니터링
5.5.1 로세스 구5.5.1.15.5.1.26.2.1.2 피드백 문서화
평가
6.2.1.3 안 에 한 문제
보고서 향의 평가5.5.2 문제 수정 분석
5.5.2.15.5.2.25.5.2.35.5.2.4
6.2.2 소 트웨어 문제해결
로세스의 사용5.5.1 로세스 구 5.5.1.2
6.2.3 변경 요청 분석 5.5.2 문제 수정 분석 5.5.2.1
6.2.4 변경 요청 승인 5.5.2 문제 수정 분석 5.5.2.5
6.2.5 사용자 규제자와의 의사소통
5.5.3 수정 구5.5.5 마이그 이션
5.5.3.15.5.5.3
6.3 수정 구5.5.3 수정 구6.2.6 릴리즈 리 인도
6.3.1 확립된 로세스를 사용해 수정 구
5.5.3 수정 구 5.5.3.2
6.3.2 수정한 소 트웨어
시스템의 재 릴리즈6.2.6 릴리즈 리 인도 6.2.6.1
7 소 트웨어 험 리 로세스
개정 1:2002 – F 3. 15 험 리 로세스
62304의 로세스는 개정 1에서 취 되지 않은
험/ 험요인 안을 취 한다. 일부 공통 이
있지만( 험 측정 등) 분석의 은 다르다.
8 소 트웨어 형상 리 로세스5.5 유지보수 로세스6.2 형상 리 로세스
8.1 형상 식별
6.2.2 형상 식별
8.1.1 형상 항목 식별을 한
방법 확립6.2.2 형상 식별 6.2.2.1
8.1.2 SOUP 식별 없음
8.1.3 시스템 형상 문서 식별 6.2.2 형상 식별 6.2.2.1
8.2 변경 리5.5.3 수정 구6.2.3 형상 리
- 197 -
8.2.1 변경 요청 승인 6.2.3 형상 리 6.2.3.1
8.2.2 변경 구5.5.3 수정 구6.2.3 형상 리
5.5.3.26.2.3.1
8.2.3 변경 검증
6.2.3 형상 리 6.2.3.18.2.4 변경 추 성에 한
방법 제공
8.3 형상 상태 설명
6.2.4 형상 상태 설명 6.2.4.1
9 소 트웨어 문제해결 로세스
5.5 유지보수 로세스6.2 형상 리
6.8 문제해결 로세스
9.1 문제 보고서 작성
6.8.1 로세스 구
6.8.2 문제해결6.8.1.1 b)6.8.2.1
9.2 문제 조사6.8.2 문제해결6.8.1 로세스 구
6.8.2.16.8.1.1 b)
9.3 련당사자에게 통보
6.8.1 로세스 구 6.8.1.1 a)
9.4 변경 리
로세스 사용
6.2.3 형상 리
5.5.3 수정 구
9.5 기록 유지 6.8.1 로세스 구 6.8.1.1 a)
9.6 문제 경향 분석
6.8.1 로세스 구
6.8.2 문제해결6.8.1.1 b)6.8.2.1
9.7 소 트웨어
문제해결 검증6.8.1 로세스 구 6.8.1.1 d)
9.8 시험 문서의 내용
12207의 모든 시험
업무 문서화
C.7 IEC 61508과의 계
안 성이 요한 소 트웨어의 설계와 련된 이 표 이 IEC 61508의 원칙을 수해
야 하는지에 한 의문이 제기되었다. 아래에는 이 표 의 범 가 설명된다.
IEC 61508은 세 가지 주요 안을 취 한다.
1) 험 리 수명주기와 수명주기 로세스

- 198 -
2) 안 무결성 수 의 정의
3) 소 트웨어 개발을 한 기술, 도구 방법의 권장사항 다양한 업무 수행
담당자의 독립성 수
안 1)은 이 표 에서 ISO 14971( 험 리에 한 의료기기 부문 표 )에 한 규정
참조에 의해 취 된다. 이 참조의 향은 험 리에 한 ISO 14971의 근방식을 의
료기기 소 트웨어에 한 소 트웨어 로세스 일부로 채택한다는 것이다.
안 2)의 경우 이 표 은 IEC 61508보다 단순한 근방식을 선택한다. IEC 61508은
신뢰성 목표의 에서 정의한 네 개 “소 트웨어 무결성 수 ”으로 소 트웨어를
분류한다. 신뢰성 목표는 소 트웨어 결함에 의해 발생하는 해의 심각도와 가능성을
모두 정량화하는 험 분석 후 식별된다.
이 표 은 분류 소 트웨어 결함의 발생가능성의 고려를 허용하지 않음으로써
안 2)을 단순화한다. 세 개 소 트웨어 안 성 등 으로 분류하는 것은 결함에 의해 발
생되는 해의 심각도에만 기 해 결정된다. 분류 후 다양한 소 트웨어 안 성 등
에 다양한 로세스가 필요하다. 그 의도는 소 트웨어 결함 발생가능성을 더욱 축소
하는 것이다.
안 3)은 이 표 에서는 처리되지 않는다. 이 표 의 사용자는 IEC 61508을 양호
한 소 트웨어 방법, 기술 도구를 제공하는 표 으로 사용하는 것이 바람직하다. 단,
재 사용되고 미래에 개발될 다른 근방식도 동일하게 양호한 결과를 제공할 수 있다.
이 표 은 소 트웨어 활동( 를 들어 확인) 하나를 담당하는 담당자와 다른 활동(
를 들어 검증) 담당자 사이의 독립성에 련해 어떤 권고도 하지 않는다. 특히, 이 표
은 독립 인 안 성 평가자에 해 어떤 요구사항도 제시하지 않는다. 이는 ISO
14971의 범 이기 때문이다.

- 199 -
부록 D (참조용) 구
D.1 서문
이 부록에는 이 표 을 제조업체 로세스에 구 할 수 있는 방법이 개 으로 설명
된다. 한 이 부록에서는 ISO 13485[7]과 같은 다른 표 이 하고 유사한 로세스
를 필요로 한다고 간주한다.
D.2 품질경 시스템
표 에서 의료기기 소 트웨어가 포함되는 의료기기 제조업체의 경우 품질경
시스템(QMS)이 4.1에서 요구된다. 이 표 에 의하면 QMS를 꼭 인증할 필요는 없다.
D.3 품질경 로세스 평가
확립되고 문서화된 QMS 로세스가 얼마나 잘 소 트웨어 수명주기의 로세스를
담당하는지 제조업체의 책임으로 심사, 검사 는 분석을 통해 평가할 것이 권장된다.
식별한 간극은 QM 로세스를 확 해 처리할 수도 있고 별도로 설명할 수도 있다. 소
트웨어의 개발, 검증 밸리데이션을 규제하는 로세스 설명을 제조업체가 이미
악한 경우 이러한 설명도 평가해 이 표 에 얼마나 잘 부합하는지 단해야 한다.
D.4 이 표 의 요구사항을 제조업체의 품질경 로세스에 통합
이 표 은 이미 QMS 시스템에 확립되어 있거나 새 로세스에 통합된 로세스를
채택하거나 확장하면 구 할 수 있다. 이 표 은 그러한 구 방법은 명시하지 않는다.
제조업체가 한 방식으로 구 할 수 있다.
자체에 문서화된 QMS가 없는 자기제품 생산자(OEM)나 하청업체가 의료기기 소
트웨어를 개발할 때 제조업체는 이 표 에 설명한 로세스가 히 용되는지 확인
(ensuring)할 책임이 있다.
D.5 인증된 QMS가 없는 소규모 제조업체를 한 검목록
제조업체는 소 트웨어의 가장 높은 안 성 분류(A, B 는 C)를 결정해야 한다. 표
D.1에는 이 표 에 설명한 활동이 모두 수록되어 있다. ISO 13485를 참조하면 QMS에
해당되는 수 을 쉽게 정의할 수 있을 것이다. 제조업체는 요구되는 소 트웨어 안
성 등 에 기 해 요구되는 각 로세스를 기존 로세스와 비교해 평가해야 한다. 요
구사항이 이미 취 된 경우 련 로세스 설명을 참고해야 한다.차이가 있을 경우 로
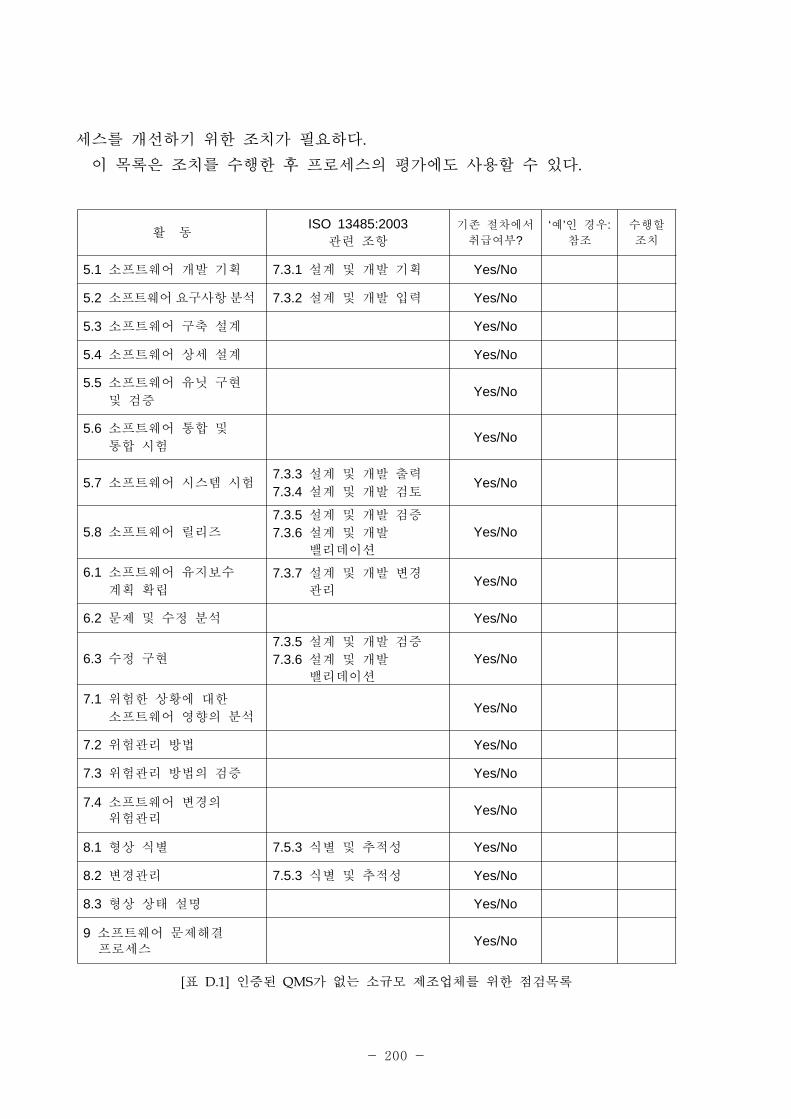
- 200 -
세스를 개선하기 한 조치가 필요하다.
이 목록은 조치를 수행한 후 로세스의 평가에도 사용할 수 있다.
[표 D.1] 인증된 QMS가 없는 소규모 제조업체를 한 검목록
활 동ISO 13485:2003
련 조항
기존 차에서
취 여부?‘ ’인 경우: 참조
수행할
조치
5.1 소 트웨어 개발 기획 7.3.1 설계 개발 기획 Yes/No
5.2 소 트웨어요구사항분석 7.3.2 설계 개발 입력 Yes/No
5.3 소 트웨어 구축 설계 Yes/No
5.4 소 트웨어 상세 설계 Yes/No
5.5 소 트웨어 유닛 구
검증Yes/No
5.6 소 트웨어 통합
통합 시험Yes/No
5.7 소 트웨어 시스템 시험7.3.3 설계 개발 출력7.3.4 설계 개발 검토 Yes/No
5.8 소 트웨어 릴리즈
7.3.5 설계 개발 검증7.3.6 설계 개발
밸리데이션
Yes/No
6.1 소 트웨어 유지보수
계획 확립
7.3.7 설계 개발 변경 리
Yes/No
6.2 문제 수정 분석 Yes/No
6.3 수정 구7.3.5 설계 개발 검증7.3.6 설계 개발
밸리데이션
Yes/No
7.1 험한 상황에 한
소 트웨어 향의 분석Yes/No
7.2 험 리 방법 Yes/No
7.3 험 리 방법의 검증 Yes/No
7.4 소 트웨어 변경의 험 리
Yes/No
8.1 형상 식별 7.5.3 식별 추 성 Yes/No
8.2 변경 리 7.5.3 식별 추 성 Yes/No
8.3 형상 상태 설명 Yes/No
9 소 트웨어 문제해결 로세스
Yes/No

- 201 -
참고문헌
[1] IEC 60601-1, Ed. 3: Medical electrical equipment - Part 1: General requirements
for basic safety and essential performance
[2] IEC 60601-1-4:1996, Medical electrical equipment - Part 1-4: General requirements
for safety - Collateral standard: Programmable electrical medical systems
Amendment 1 (1999)
[3] IEC 61508:1998, Functional safety of electrical/electronic/programmable electronic
safety related systems
[4] IEC 61010-1:2001, Safety requirements for electrical equipment for measurement,
control, and laboratory use - Part 1: General requirements
[5] ISO 9000:2000, Quality management systems - Fundamentals and vocabulary
[6] ISO 9001:2000, Quality management systems - Requirements
[7] ISO 13485:2003, Medical devices - Quality management systems - Requirements
for regulatory purposes
[8] ISO/IEC 9126-1:2001, Software engineering - Product quality - Part 1: Quality
model
[9] ISO/IEC 12207:1995, Information technology - Software life cycle processes
Amendment 1 (2002), Amendment 2 (2004)
[10] ISO/IEC 14764:1999, Information technology - Software maintenance
[11] ISO/IEC 90003:2004, Software and system engineering - Guidelines for the
application of ISO 9001:2000 to computer software
[12] ISO/IEC Guide 51:1999, Safety aspects - Guidelines for their inclusion in
standards
[13] IEEE 610.12:1990, IEEE standard glossary of software engineering terminology

- 202 -
[별첨 5]
소 트웨어 밸리데이션의 일반 원칙
(Guidance for Industry, GENERAL PRINCIPLES OF
SOFTWARE VALIDATION)
발행일: 2002. 1. 11
Section 1. 목
이 지침은 소 트웨어 의료기기 는 의료기기의 설계⋅개발 제조에 사용되는 소
트웨어의 밸리데이션에 용되는 FDA의 일반 원칙을 제시한다. 이 최종 지침
(Version 2.0)는 1997.7.9 소 트웨어 밸리데이션의 일반원칙(Version 1.1)을 체하는
것이다.
Section 2. 용 범
이 지침은 의료기기 품질 리규정(Quality System regulation)이 소 트웨어에 어떻
게 용이 되며 FDA(agency)에서 밸리데이션 시스템을 평가하는 근법을 보여주고
있다. 를 들면 이 문서는 소 트웨어 밸리데이션에 있어 FDA에서의 수용가능한 요소
들 목록을 제시하 다. 그러나 법령을 수함에 있어 용되는 모든 활동과 작업을 제시
한 것은 아니다.
이 지침의 용 범 는 엄 한 의미의 밸리데이션 용 범 보다 넓다. 계획
(planning), 검증, 시험, 추 성, 형상 리(configuration management) 기타 이 지침
내에서 언 되는 우수 소 트웨어 공학(good software engineering)의 많은 측면은 소
트웨어가 유효하다는 최종 결론을 뒷받침할 수 있는 요한 활동이다.
이 지침은 소 트웨어 life cycle 리와 험 리 활동의 통합을 권장한다. 개발되
는 소 트웨어와 련된 사용목 안 험(safety risk)에 기 하여 소 트웨어
개발자는 구체 인 근방법, 사용되는 기술의 조합, 용되는 노력(effort)의 수 을 결
정하여야 한다. 이 지침은 특정 life cycle 모델, 기술 는 방법을 권장하지는 않지만,
소 트웨어 밸리데이션 검증 활동이 소 트웨어 체 life cycle동안 행해져야 한다는
것은 권장하고 있다.

- 203 -
소 트웨어가 기기 제조업자 이외의 다른 개발자에 의하여 개발( : OTS 소 트웨어
)되는 경우, 소 트웨어 개발자는 FDA 규정에 합해야 하는 직 인 책임이 있는
것은 아니다.
이런 이유 때문에 법 책임이 있는 자( : 기기 제조업자)는 OTS 소 트웨어 개발자
의 활동이 한지 평가할 필요가 있고 기기 제조업자의 사용목 을 하여 소 트웨
어가 유효한지를 보장하기 하여 어떤 추가 인 노력이 필요한지 결정하여야 한다.
2.1. 용
이 지침의 용 범 는 다음과 같다.
•의료기기의 구성 요소, 부분, 는 부속품으로서 사용되는 소 트웨어
•소 트웨어 자체가 의료기기인 것( : 액기기(blood establishment) 소 트웨어)
•의료기기 생산에 사용되는 소 트웨어( : 제조 장비내의 programmable logic
controller)
•의료기기 제조업자의 품질시스템 운 에 사용되는 소 트웨어( : 기기 이력 기
록 리하는 소 트웨어)
이 문서는 일반 인 소 트웨어 밸리데이션 원칙에 기 하므로 어떠한 소 트웨어에
도 용될 수 있다. FDA의 목 상, 식품의약품화장품법의 201(h)조항 FDA 소 트
웨어와 규제 정책에 정의된 바와 같이 이 지침은 규제 상이 되는 의료기기와 련된
어떠한 소 트웨어에도 용된다. 이 문서는 어떠한 소 트웨어가 규제 상인지 특별
히 규정하고 있지는 않다.
2.2. 상(AUDIENCE)
이 지침은 다음과 같은 상에게 유용한 정보와 권고(recommendation)를 제공한다.
•의료기기 품질시스템 규정에 용을 받는 사람
•의료기기 소 트웨어의 설계, 개발, 제작의 책임이 있는 사람
•의료기기의 설계, 개발 는 제조를 하여 사용되는 자동화 장비의 설계, 개발,
제작 는 획득(procurement)이나 품질시스템 이행에 사용되는 소 트웨어 툴
(Tool)에 한 책임이 있는 사람
•FDA 감시원(Investigators)
•FDA 합성 심사원(Compliance office)
•FDA 기술문서 검토자(Scientific reviewer)

- 204 -
2.3. 가장 부담이 은 근방법
의료기기 규정의 모든 범 내에서 가장 부담이 은 근법을 용하여야 하는 것이
바람직하다. 이 지침은 련된 과학 , 법 요구사항에 한 철 한(careful) 검토를
반 하고, 그 요구사항에 부합하기 하여 제조업자가 수하여야 할 가장 부담이
은 방법을 제시한 것이다. 그러나 보다 효율 인 근법이 있다면, 이를 고려해볼 수
있다.
이 지침의 서문에 있는 사람이나 CDRH 옴부즈맨에게 서면으로 당신의 견해를
보낼 수 있다. CDRH의 옴부즈맨과의 연락방법 등은 다음의 인터넷 사이트에서 알 수
있다.
http://www.fda.gov/cdrh/resolvingdisputes/ombudsman.html
2.4. 소 트웨어 밸리데이션을 한 법 요구사항
1992~1998년 동안 실시된 3,140건의 의료기기 리콜(recall) 242건(7.7%)은 소 트
웨어 불량이 원인이라고 FDA 분석결과 나타났다. 소 트웨어와 련된 리콜 192건
(79%)은 소 트웨어가 최 생산 시 후 변경되었을 때 발생된 소 트웨어 결함에
의한 것이었다. 이 지침에서 다루는 소 트웨어 밸리데이션 기타 련된 우수 소
트웨어공학 기 은 이러한 결함 리콜을 피할 수 있는 원칙 인 방법을 제시한 것
이다.
소 트웨어 밸리데이션은 품질시스템 규정(1996.9.7 공포, 1997.6.1 시행)의 요구사항
이다. 밸리데이션은 의료기기 구성 요소로 사용되는 소 트웨어, 그 자체가 의료기기인
소 트웨어, 의료기기 생산에 사용되거나 제조업자의 품질시스템 운 에 사용되는 소
트웨어에 용되는 요구사항이다.
특별히 등 분류규정에서 제외된 것이 아니라면, 1997.6.1 이후 개발된 의료기기 소
트웨어 제품은 기기등 에 계없이 설계 리(21 CFR 820.30 참조)를 용할 수 있
다. 이 요구사항은 재 개발 인 로젝트, 신규 개발 로젝트 기존의 의료기기
소 트웨어의 모든 변경사항에 용된다. 의료기기 소 트웨어의 밸리데이션을 한
특정 요구사항은 21 CFR 820.30(g) 조항을 참조바란다. 한 의료기기 소 트웨어에
해서 계획, 입력, 검증, 검토와 같은 설계 리가 요구되어진다.(21 CFR 820.30 참조) 이
러한 활동에 따른 문서화 결과는 의료기기 소 트웨어가 유효한지 결정하는데 도움
이 될 것이다.

- 205 -
품질시스템의 구성요소이거나 의료기기 생산 공정의 자동화에 사용되는 소 트웨어
는 21 CFR 820.70(i)에서 요구되는 사용목 에 하여 밸리데이션을 하여야 한다. 이는
기기 설계, 시험, 구성요소 수락(acceptance), 제조, 라벨링, 포장, 배포, 불만 처리를 자
동화하거나 품질시스템의 다른 측면을 자동화하는 모든 소 트웨어에 용되는 요구사
항이다.
한 자기록의 작성, 변경, 유지 자서명 리에 사용되는 컴퓨터 시스템도 밸
리데이션이 요구된다(21 CFR 11.10(a) 참조). 이런 컴퓨터 시스템은 정확성, 신뢰성,
의도된 성능과의 일치성 효력이 상실되고 변경된 기록의 식별능력을 확실히 하기
하여 유효성이 확인되어야 한다.
상기와 같은 용을 한 소 트웨어는 조직 내 는 계약에 의해 개발된다. 그러
나 소 트웨어는 간혹 특별한 사용목 을 하여 기성품(off-the-shelf)으로 구입하기도
한다. OTS로 구매된 소 트웨어일지라도 본래의 사용 목 에 맞게 유효하다는 것을
입증하기 하여 모든 생산 / 는 품질시스템 소 트웨어는 본래 의도된 사용 목 ,
비교가능한 시험결과 증거에 한 정보를 규정한 문서화된 요구사항을 수립하여야
한다.
자동화 의료기기, 자동화 생산 품질시스템에서 OTS 소 트웨어의 사용이 증가
하고 있다. OTS 소 트웨어가 다양한 기능을 가지고 있더라도 의료기기 제조업자는 그
일부 기능만 필요로 할 수 있다. 의료기기 제조업자는 해당 의료기기 는 그 의료기
기의 생산에 사용되는 소 트웨어가 한지에 한 책임이 있다. 의료기기 제조업자
는 OTS 소 트웨어의 구매시 해당 소 트웨어가 의도된 목 로 작동할지를 확실히
하여야 한다. 이 문서 Section 6.3에 제조 는 품질시스템에 사용되는 OTS 소 트웨어
에 한 추가 지침이 있다. 의료기기 소 트웨어에 유용한 추가 정보는 FDA의
'Guidance for Industry, FDA Reviewers, and Compliance on Off-The-Shelf Software
Use in Medical Devices'를 참조하기 바란다.
2.5. 품질시스템 규정과 시 제출자료
이 문서는 소 트웨어 밸리데이션의 이행을 포함한 품질시스템규정(QSR) 련 사항
을 언 하고 있다. 소 트웨어 밸리데이션 과정의 리를 한 지침을 제시한다. 소 트
웨어 밸리데이션 과정의 리와 자동화 공정에서의 공정 밸리데이션과 같은 다른 밸리
데이션 요구사항을 혼동해서는 아니 된다.

- 206 -
의료기기 제조업자는 FDA에 제출하는 시 자료처럼 품질시스템 설계 리 요
구사항에 합함을 증명하는 동일한 차 기록을 사용할 수 있다. 이 문서는 소
트웨어 밸리데이션과 련하여 특정한 안 성 는 유효성 사항을 언 하지 않으며,
규제되는 소 트웨어의 시 제출 자료를 한 설계사항 문서화 요구사항에
하여 다루고 있지 않다. 시 제출 자료에서 요구되는 안 성, 유효성 문서화 요
구사항과 련된 특정 사항은 Office of Device Evaluation (ODE), Center for Devices
and Radiological Health(CDRH) 는 Office of Blood Research and Review, Center
for Biologics Evaluation and Research(CBER)에서 다루어진다. 시 제출 자료를
한 FDA 지침은 Appendix A를 참조하기 바란다.
Section 3. 소 트웨어 밸리데이션의 개
많은 사람들이 소 트웨어 밸리데이션과 련하여 품질시스템 규정에 합함을 증명
하기 하여 하여야 할 특별한 지침을 문의한다. 이 문서에 제시된 소 트웨어 밸리데이
션 련 정보는 새로운 것이 아니다. Section 4, 5에 제시된 원칙과 작업(tasks)을 이용
한 소 트웨어 밸리데이션은 소 트웨어 산업계에서는 20여년 동안 실시되어 온 것이
다.
의료기기의 종류, 공정 제조시설의 다양성 때문에 용 가능한 모든 밸리데이션
요소들을 이 문서 하나에서 다룰 수는 없다. 그러나 포 다수 개념의 일반 인
용이 밸리데이션을 한 지침에서는 유용하게 용될 수 있다. 이러한 포 개념은
밸리데이션을 한 반 근법을 설계하는 체계를 제공할 것이다. 추가 인 특정정보
는 부속서 A에 수록된 참고문헌에서 참고할 수 있다.
3.1. 용어의 정의
품질시스템 규정 는 기타 하 규정에서 정의되지 않은 이 지침에서 사용된 모든
다른 용어들은 “FDA Glossary of Computerized System and Software Development
Terminology” 최신 에 정의되어 있다.
의료기기 품질시스템규정(21 CFR 820.3(k))에서 “establish”는 “정의하다(define), 문서화
하다(document), 수행하다(implement)”로 정의하고 있다. 이 지침에서 “establish”
“established”는 와 같은 의미로 해석되어야 한다.
의료기기 품질시스템규정에서 일부 정의는 소 트웨어 산업계에서 사용되는 일반
용어와 비교했을 때 혼동될 수 있다. 일례로 요구사항(requirements), 규격
(specification), 검증(verification) 밸리데이션(validation)을 들 수 있다.

- 207 -
3.1.1 요구사항과 규격
품질시스템 규정은 설계 입력 요구사항을 반드시 문서화하고 특정 요구사항은 반드
시 검증하도록 하고 있으나 ‘요구사항’과 ‘규격’의 차이 을 명확히 설명하고 있지 않
다. ‘요구사항(requirement)’은 시스템 는 소 트웨어에 필요하거나 상될 수 있
는 것을 말한다. 요구사항은 명시되거나 함축된 고객의 요구를 반 하며 조직의 내부,
시장의 요구(market-based), 계약상 는 법 요구사항일 수도 있다. 다양한 종류의
요구사항이 있을 수 있다( : 설계, 기능(functional), 실행, 인터페이스, 성능 는 물
리 요구사항). 소 트웨어 요구사항은 형 으로 소 트웨어에 배정된 시스템의
기능 인 측면에 한 요구사항으로부터 기인한다. 소 트웨어 요구사항은 일반 으로
개발과정에서 기능 인 용어로 규정, 정의, 수정 갱신된다. 소 트웨어 요구사항을
정확하고 완벽하게 문서화하는데 성공하는 것은 결과물인 소 트웨어의 성공 인 밸리
데이션을 하여 결정 요소이다.
규격(specification)’은 “요구사항을 설명한 문서”로 정의되어 있다(21 CFR 820.3(y)).
이는 도면(drawing), 도안(pattern) 는 그 밖의 련 문서를 참조하거나 포함할 수 있
고 요구사항에 합함을 확인할 수 있는 방법 기 을 가리킨다. 이 규격은 시스템
요구사항 규격, 소 트웨어 설계 규격, 소 트웨어 시험 규격서, 소 트웨어 통합 규격
등과 같이 여러 종류가 있다. 이러한 문서들은 “특정 요구사항”을 설정하고 있으며 검
증이 필요한 다양한 형태의 설계 출력이다.
3.1.2 검증과 밸리데이션
품질시스템 규정은(QSR) ISO 8402:1994와의 조화를 이루어 “검증”과 “밸리데이션”을
분리된 별개의 용어로서 사용한다. 반면, 많은 소 트웨어 공학 의 논설 교과서
는 “검증”과 ”밸리데이션"을 번갈아 사용하거나 어떤 경우에는 소 트웨어 “검증, 밸리데
이션 시험(VV&T)"을 단일 개념으로 사용하고 있다.
소 트웨어 검증은 소 트웨어 개발 life cycle의 특정 단계의 설계 출력이 그 단계
를 한 특정 요구사항을 모두 만족하는 것을 객 으로 증명하는 것이다. 소 트웨
어 검증은 소 트웨어 련 문서의 일 성(consistency), 완 성(completeness), 정확
성(correctness)을 평가하고 소 트웨어가 유효하다는 결론을 뒷받침할 수 있게 한다.
소 트웨어 시험은 소 트웨어 개발 출력이 그 입력 요구사항을 만족시킨다는 것을 확
인하기 하여 의도된 많은 검증 활동 의 하나이다. 다른 검증활동에는 다양한 정
․동 분석, 코드 문서검사, walkthroughs, 그리고 그 밖의 다른 기술사항들을
포함한다.

- 208 -
소 트웨어 밸리데이션은 완성된 의료기기를 한 설계 밸리데이션의 한 부분이지
만 품질시스템 규정 내에서 독립 으로 정의되어 있지 않다. 이 지침의 목 을 하여
FDA는 소 트웨어 밸리데이션을 “시험에 의한 확인과 사용자의 요구사항․의도된 사용
목 을 한 소 트웨어 규격확인 소 트웨어에 의해 실시된 특정 요구사항이
일 되게 실시될 수 있음을 보여주는 객 증거의 제공”으로 간주한다.
모든 요구사항이 충족되었음을 보장하기 하여 소 트웨어 밸리데이션은 소 트웨
어 개발 life cycle의 마지막 단계뿐만 아니라 그 과정에서 실시될 수 있다. 일반 으로
소 트웨어는 종합 인 하드웨어 시스템의 일부분이기 때문에 소 트웨어 밸리데이션
은 모든 소 트웨어 요구사항이 하고 완벽하게 실시되었고 시스템 요구사항에 추
성이 가능하도록 증빙자료를 포함하고 있다. 소 트웨어가 유효하다는 결론은 소 트
웨어 개발 life cycle의 각 단계에서 실시되는 종합 소 트웨어 시험, 검사, 분석 다
른 검증 활동에 크게 향을 받는다. 모의사용 환경(simulated use environment)에서 의
료기기 소 트웨어 성능시험과 사용자 장 시험은 자동화된 소 트웨어 의료기기의
체 인 설계 밸리데이션 로그램의 요소로써 포함된다.
소 트웨어 검증과 밸리데이션은 개발자가 계속 시험할 수도 없고 어느 정도의 정보
로 충분한지 알기 어렵기 때문에 어렵다. 넓은 범 에서 소 트웨어 밸리데이션은 소
트웨어에 의하여 자동화된 의료기기의 성능과 특성에 하여 모든 요구사항 사용
자 기 사항이 만족되었는지에 한 “신뢰의 수 (level of confidence)” 문제이다. 규
격 문서, 잠재 인 결함의 평가, 시험범 다른 기법에서 나타나는 결함에 한 측
정들은 제품을 인도하기 에 수용 가능한 신뢰의 수 을 개발함에 있어 모두 활용된
다.
신뢰의 수 , 필요한 소 트웨어 밸리데이션․검증․시험의 수 은 기기의 자동화 기능
에 의해 제기되는 안 험(safety risk) 는 해요소(hazard)에 따라 달라질 수 있다.
소 트웨어의 안 한 험 리를 한 추가 지침은 FDA의 Guidance for the Content
of Pre-market Submissions for Software Contained in Medical devices의 Section 4,
Appendix A에 있는 국제기 인 ISO/IEC 14971-1 IEC 60601-1-4를 참조하기 바란
다.
3.1.3 IQ/OQ/PQ
오랫동안 FDA와 규제 상 업체들은 소 트웨어 밸리데이션을 공정 밸리데이션의
범 에서 이해하고 정의하려 하 다. 를 들어 업계의 문서 다른 FDA의 밸리데이
션 지침에서 사용자 환경 소 트웨어 밸리데이션을 설치 격성확인(installation

- 209 -
qualification, IQ), 운 격성확인(operational qualification, OQ), 성능 격성확인
(performance qualification, PQ)의 용어로 정의하고 있다. IQ/OQ/PQ의 용어 정의와
련 추가 정보는 FDA의 Guideline on General Principles of Process
Validation(1987.5.11) Glossary of Computerized System and Software
Development Terminology(1995.8)에서 나타나 있다.
IQ/OQ/PQ 용어가 그 목 에 하게 사용되고 사용자 환경에서 소 트웨어 밸리
데이션 작업을 체계화하는 많은 합법 인 방법 하나이지만, 이 용어는 많은 소 트웨
어 문가들 사이에서 충분히 이해되지 못하고 이 문서의 다른 부분에서는 사용되고
있지 않다. 그러나 FDA 직원 의료기기 제조업자는 소 트웨어 밸리데이션과 련
하여 질문하고 정보를 제공함으로써 이 용어의 차이 을 인식할 필요가 있다.
3.2. 시스템 설계의 일부로서 소 트웨어 개발
소 트웨어 사용에 의한 시스템 기능 실행에 한 결정은 일반 으로 시스템 설계과
정에서 이루어진다. 소 트웨어 요구사항은 반 인 시스템 요구사항과 소 트웨어
를 이용하여 실행되는 시스템 측면의 설계에서 기인한다. 이는 완성품에 한 사용자
요구사항 의도된 사용목 이지만, 사용자는 이러한 요구사항이 하드웨어, 소 트웨
어 는 이 두 가지의 복합에 의해 만족되는지의 여부를 일일이 언 하지 않는다. 따라
서 소 트웨어 밸리데이션은 시스템을 한 체 인 설계 밸리데이션의 맥락에서 고
려되어야 한다.
개발 제품의 문서화된 요구사항 규격은 사용자 요구사항 사용목 을 나타낸다. 소
트웨어 밸리데이션의 요한 목표는 모든 완성된 소 트웨어 제품이 모든 문서화된
소 트웨어 시스템 요구사항에 합함을 증명하는 것이다. 시스템 요구사항과 소
트웨어 요구사항에 있어 정확성, 완 성은 설계 밸리데이션의 일부로서 반드시 검
토하여야 한다. 밸리데이션은 모든 소 트웨어 규격에 합함과 모든 소 트웨어 요구
사항이 시스템 규격에 소 가능함을 확인(confirmation)하는 것을 포함한다. 확인
(confirmation)은 의료기기의 모든 측면이 사용자 요구사항과 사용목 에 합함을 보
증하기 한 반 인 설계 밸리데이션의 요한 부분이다.
3.3. 하드웨어와 소 트웨어의 차이
소 트웨어는 하드웨어처럼 유사한 기술을 많이 공유하고 있으나 요한 차이 이 있다.
를 들면,
• 다수 소 트웨어 문제는 설계 개발과정동안 발생한 오류(error)로 추 가능
하다. 하드웨어 제품 품질은 설계 개발, 제조에서 크게 향을 받는 반면, 소
트웨어 제품 품질은 소 트웨어 제조를 한 최소한의 고려사항과 함께 주로

- 210 -
설계 개발에서 향을 받는다. 소 트웨어 제조는 쉽게 검증될 수 있는 재생
산으로 이루어진다. 원본과 동일한 기능을 가지는 수천 개의 로그램 복사본을
생산하는 것은 어렵지 않다; 어려운 것은 원본 로그램이 모든 규격을 만족하도
록 하는 것이다.
•소 트웨어의 요한 특징 하나는 랜칭(branching, 다른 입력 기반의 명령어
들을 실행시키는 능력)이다. 이러한 특징은 소 트웨어의 복잡성(complexity)과 같
은 다른 특징에도 요하게 작용한다. 간단한 로그램이라 할지라도 매우 복잡하
고 충분히 이해하는데 어려울 수 있다.
•소 트웨어가 완벽하고 정확한지, 시험만으로 완벽히 검증할 수 없다. 종합 인 밸리
데이션 근을 확실히 하기 하여 다른 검증 기술 체계화되고 문서화된 개발
공정이 병행되어야 한다.
•하드웨어와는 달리 소 트웨어는 실체가 있는 것이 아니고 없어지는 것도 아니
다. 소 트웨어는 잠재 인 결함이 발견되고 삭제되는 것처럼 시간이 지나면서
진화할 수 있다. 그러나 소 트웨어가 업데이트 변경되는 동안 새로운 결함이
소 트웨어에 발생함으로 그러한 개선 활동이 무효화되는 경우도 있다.
•하드웨어 결함과는 달리 소 트웨어 결함은 사 경고 없이 발생한다. 실행되는
동안 경로들을 다르게 따라가도록 하는 랜칭은 소 트웨어 제품이 시장에 출시
된 후에도 약간의 잠재 인 결함을 내재하고 있을 수 있다.
•소 트웨어와 련한 다른 특징은 변화 속도 용이성이다. 이로 인하여 소
트웨어 문가와 비(非) 문가들은 소 트웨어 문제가 쉽게 수정될 수 있다는 믿
음을 야기시킨다. 소 트웨어에 한 이해부족은 리자들이 하드웨어를 한 엄
격한 리기술이 소 트웨어에는 필요 없다고 생각하도록 할 수 있다. 사실은 이
와 반 이다. 그 복잡성 때문에 개발 로세스에서 추후에 쉽게 발견될 수
없는 문제의 발생을 방하기 하여 소 트웨어는 하드웨어보다 더 엄격하게
리되어야 한다.
•소 트웨어에서 요하지 않은 것처럼 보이는 변경이 소 트웨어 로그램과 다른
곳에서 상하지 못한 심각한 문제를 발생시킬 수 있다. 소 트웨어 개발 로세스
는 소 트웨어 변경으로 인한 상치 않은 결과를 발견하고 수정하기 하여 충분
히 계획, 리 문서화 되어야 한다.
•소 트웨어 문가 첨단 모바일 종사자에게는 높은 요구사항이 주어지는 반면
소 트웨어 변경을 유지․보수하는 소 트웨어 종사자는 원본 소 트웨어 개발과정
에 참여하지 않을 수 있다. 그래서 정확하고 체 인 문서화가 필요하다.
•소 트웨어 구성요소는 하드웨어 구성요소처럼 자주 규격화, 호환
(interchangeable) 되지 않았다. 그러나 의료기기 소 트웨어 개발자들은 구성요

- 211 -
소 기반(component-based)의 개발도구와 기술을 사용하기 시작했다. 목 -지향 방
법론(Object-oriented methodologies)과 OTS 소 트웨어 구성 요소의 사용으로 소
트웨어 개발은 빨라지고 비용이 감되었다. 그러나 구성요소 기반의 근은
통합(integration)할 때 매우 주의하여야 한다. 통합 에 재사용 소 트웨어 코드
를 충분히 정의, 개발하고 규격 구성요소의 기능을 충분히 이해할 수 있는 시
간이 필요하다.
이러한 이유로 소 트웨어 기술은 하드웨어 기술보다 더 높은 단계의 리․감독
이 필요하다.
3.4. 소 트웨어 밸리데이션의 장 (BENEFITS)
소 트웨어 밸리데이션은 의료기기 소 트웨어 소 트웨어 자동화공정의 품질을
보증하는데 사용되는 요한 수단이다. 소 트웨어 밸리데이션은 의료기기의 유용성과
신뢰도를 증가시킴으로 결함 발생율, 리콜 시정조치 감소, 환자 사용자에 한 험
감소, 의료기기 제조업자에 한 부담 경감을 가져올 것이다. 소 트웨어 밸리데이션
은 소 트웨어의 변경 변경의 유효성 재확인을 쉽고 은 비용으로 가능하게 하여
장기 으로는 비용을 감소시킬 수 있다. 소 트웨어 life cycle에서의 체 비용에 있어
소 트웨어 유지․보수는 많은 부분을 차지할 수 있다. 수립된 체 소 트웨어 밸리
데이션 로세스는 추후 소 트웨어의 출고(release)를 한 밸리데이션 비용을 감함
으로써 소 트웨어의 장기 비용을 이는데 도움이 된다.
3.5 설계 검토
설계 검토는 설계요구사항의 성(adequacy)을 평가하고 이런 요구사항에 부응하
는 설계 능력평가와 문제 식별을 한 문서화된 반 ․체계 인 설계의 시험이다.
소 트웨어 개발과정에서 개발 내부의 많은 약식(informal) 기술검토가 있는 반면 정
식 인 설계 검토는 좀 더 체계화되고 개발 외부인원이 참여한다. 정식 인 설계 검
토는 다른 정식․약식 검토에서 나온 결과를 참조 는 포함할 수 있다. 소 트웨어가
하드웨어와 함께 시스템에 통합된 후에 소 트웨어를 해서 별도로 설계 검토를 실시할
수 있다. 설계 검토는 개발 계획의 시험, 요구사항 규격, 설계 규격, 시험 계획․공정,
로젝트 련 문서․활동, 규정된 life cycle의 각 단계의 검증결과 의료기기를 한
반 밸리데이션 결과를 포함해야 한다.
설계 검토는 개발 로젝트를 리하고 평가하기 한 주요도구이다. 를 들어 정
식 설계 검토는 경 자가 소 트웨어 밸리데이션 계획에서 정의된 모든 목표가 수행되
었음을 확인하도록 한다.

- 212 -
품질시스템규정은 최소 1회의 정식 설계 검토가 의료기기 설계과정에서 수행 되도록
요구하고 있다. 그러나 각 소 트웨어 life cycle 활동의 마지막 단계 는 다음 활동
진행 과 같이 다양한 설계 검토를 실시하도록 권장한다. 정식 설계 검토는 주요 자
원(resource)이 특정 설계 해결(solutions)로 되기 인 요구사항 충족활동의 마지막 단
계에서 매우 요하다. 이 시 에서 발견된 문제 들은 보다 쉽게 해결되고 시간과 비
용의 감, 요한 문제 을 놓치는 것과 같은 실수를 감소시킬 것이다.
다음과 같이 요한 질문사항에 한 답을 정식 설계 검토과정에서 문서화하여야
한다.
•각 소 트웨어 life cycle 활동별 한 작업, 상 결과, 출력물 는 제품을 설
정하 는가?
•각 소 트웨어 life cycle 활동의 작업, 상 결과, 출력물 는 제품은 다음을 만
족 하는가;
√ 정확성, 완 성, 일 성 정확성의 에서 다른 소 트웨어 life cycle 활동
의 요구사항에 합한가?
√ 그 활동의 표 , 규정 약정을 만족하는가?
√ 추후 소 트웨어 life cycle 활동에 한 기 작업을 하여 한 근거를 설
정하 는가?
Section 4. 소 트웨어 밸리데이션의 원칙
이번 장에서는 소 트웨어 밸리데이션에 고려되어야 할 일반 인 원칙을 제시한다.
4.1. 요구사항
문서화된 소 트웨어 요구사항의 규격은 밸리데이션 검증을 한 기 을 제공한
다. 소 트웨어 밸리데이션 로세스는 설정된 소 트웨어 요구사항의 규격없이 완료
될 수는 없다(21 CFR 820.3(z), (aa) 820.20(f), (g)).
4.2. 결함 방
소 트웨어 품질보증은 소 트웨어 개발 로세스에 있어 결 발생을 방지하고 코딩
후 소 트웨어 코드에 시험 품질을 시도하지 않는데 을 둘 필요가 있다. 소 트웨
어 시험은 소 트웨어 코드 내에 잠재하는 모든 결 을 확인함에 있어 매우 제한 이
다. 를 들어 부분 소 트웨어가 복잡하여 시험검사를 통하여 소 트웨어가 철
하게 테스트되지 못하게 한다. 소 트웨어 시험은 꼭 필요한 작업이다. 그러나 부
분의 경우 자체 인 소 트웨어 시험은 소 트웨어가 그 사용목 에 합한지 나

- 213 -
타내기 해서는 충분하지 않다. 이 신뢰성을 수립하기 하여 소 트웨어 개발자는
소 트웨어 오류를 방지하고 발생하는 소 트웨어 오류를 발견하기 하여 방법 기
법을 병행하여야 한다. 이 방법들의 “최상의 병행(best mix)”은 개발환경, 응용, 로젝
트 규모, 언어 험을 포함한 많은 요소들에 달려있다.
4.3. 시간과 노력(EFFORT)
소 트웨어가 유효한지를 단하기 해서는 시간과 노력이 요구된다. 소 트웨어 밸리
데이션을 한 비는 설계․개발 계획과 설계 입력과 같이 기에 실시해야 한다. 소
트웨어 밸리데이션에 한 최종 결정은 소 트웨어 life cycle 동안 수행된 계획
인 노력으로부터 수집한 증빙자료를 기 로 한다.
4.4. 소 트웨어 life cycle(LIFE CYCLE)
소 트웨어 밸리데이션은 설정된 소 트웨어 life cycle의 환경 내에서 실시한다. 소
트웨어 life cycle는 소 트웨어 기술 작업과 소 트웨어 밸리데이션 노력을 제공하기
하여 필요한 문서화를 포함한다. 소 트웨어 life cycle는 소 트웨어 사용 목 에
합한 특별한 검증 밸리데이션 작업을 포함한다. 이 지침은 소 트웨어 개발 로젝
트를 해서 선택되고 사용돼야만 하는 특정 life cycle 모델을 제시하지는 않는다.
4.5. 계획
소 트웨어 밸리데이션 로세스는 계획의 활용을 통해서 정의되고 리된다. 소
트웨어 밸리데이션 계획은 소 트웨어 밸리데이션 노력(effort)을 통해 “무엇(what)”을
수행할 것인지를 정의한다. 소 트웨어 밸리데이션 계획은 요한 품질시스템 수단이
다. 소 트웨어 밸리데이션 계획은 용범 , 근(approach), 자원, 일정, 활동의 형
태․범 (extent), 작업 업무요소(items)를 구체화 한다.
4.6. 공정
소 트웨어 밸리데이션 로세스는 차의 활용을 통해서 실행된다. 이러한 차
는 “어떻게(how)” 소 트웨어 밸리데이션 노력을 실시할 것인지를 설정한다. 이 차
는 개별 인 밸리데이션 활동, 작업 업무 사항을 완성하기 하여 수행되어야 하는
특정 활동 는 활동의 연속(sequence)을 명백하게 해 다.
4.7. 변경후 소 트웨어 밸리데이션
소 트웨어의 복잡성으로 인하여 좁은 범 의(local) 변경으로 인해 시스템에 범
하게 향(impact)을 미칠 수 있다. 소 트웨어에 어떠한 변경(매우 작은 변화라

- 214 -
할지라도)이 발생한 경우 소 트웨어의 밸리데이션 상태가 재설정되어야할 필요가 있다.
소 트웨어가 변경될 때마다 개별 인 변경에 한 밸리데이션뿐만 아니라 체 소
트웨어 시스템에 미치는 변경의 정도 향을 결정하기 하여 밸리데이션 분
석이 실시되어야 한다. 이 분석에 기 하여 소 트웨어 개발자는 변경되지 않는
(unchanged) 시스템의 취약한 부분이 악 향을 받지 않는다는 것을 입증하기 하여
한 소 트웨어 회귀시험(regression testing)의 수 을 결정하여야 한다. 설계
리 한 회귀시험은 소 트웨어 변경 후에 소 트웨어가 유효하다는데 한 신뢰
성을 제공한다.
4.8. 밸리데이션 용범 (COVERAGE)
밸리데이션 용범 는 소 트웨어의 복잡성과 안 험(safety risk)에 기 하여야
한다. - 업소 규모 는 자원제약에 기 하지 않음. 밸리데이션 활동, 작업 업무사항
의 선택은 소 트웨어 설계의 복잡성과 특정한 사용목 을 한 소 트웨어의 사용에
련 있는 험에 상응하여 이루어져야 한다. 험도 의료기기에 해서는 기본 인
밸리데이션 활동이 실시될 수 있다. 험증가에 따라 추가 인 밸리데이션 활동이 추
가 인 험처리를 하여 확 되어야(added) 한다. 밸리데이션 문서화는 모든 소 트
웨어 밸리데이션 계획과 차가 하게 실시되었음을 증명하기에 충분해야 한다.
4.9. 검토의 독립성
밸리데이션 활동은 “검토의 독립성(independence of review)”이라는 기본 인 품질
보증규칙(precept)에 의하여 실시되어야 한다. 자체 밸리데이션(self-validation)은 매우 어
렵다. 가능한 독립 인 평가가 바람직하며, 고도의 험성이 있는 경우 더욱 그러하
다.
제3자에 의한 독립 인 검증 밸리데이션을 하여 탁을 하는 업체들도 있으나
이것이 항상 가능한 것은 아니다. 다른 방법은 특정 계발 는 실행부서 소속되지 않
은 로젝트 평가와 검증․밸리데이션 실시에 하여 역량이 있는 내부 직원을 선정
하는 것이다. 소규모 업체일수록 어떻게 작업을 구성하고 내부 인 검토의 독립성을 유
지할 것인가에 하여 독창 일 필요가 있다.
4.10. 유연성 신뢰도
이러한 소 트웨어 밸리데이션 원칙은 구체 으로 실행함에 있어 매우 다양하게 용
된다. 의료기기 제조업자는 이러한 밸리데이션 원칙을 어떻게 용할 것인지를 선택하는
데 유연성을 가지되 소 트웨어기 유효하다는 것을 입증함에 있어 최종 인 책임이 있다.

- 215 -
소 트웨어는 범 한 환경조건 다양한 험수 을 가진 의료기기를 고려하여
설계, 개발, 밸리데이션 규제되어야 한다. FDA는 다음과 같은 소 트웨어를 포함하
는 의료기기 응용 로그램을 규제하고 있다.
•의료기기의 구성 요소, 부분품, 는 부속품(accessory)인 소 트웨어
•그 자체가 의료기기인 소 트웨어
•제조, 설계, 개발, 는 품질시스템의 다른 부분에 사용되는 소 트웨어
각 환경에서 여러 가지 원인(source)으로 인하여 소 트웨어 구성요소는 응용 로그
램을 만드는데 사용될 수 있다( : 사내 개발된 소 트웨어(in-house developed
software), OTS 소 트웨어(off-shelf-software), 계약 소 트웨어, 셰어웨어). 한 소
트웨어 구성요소는 다양한 형태로 나타난다( : 응용 로그램 소 트웨어, 운 시스
템, 컴 일러(compiler), 디버거(debugger), 구성 리도구(configuration management
tool) 등). 이런 조건에서 소 트웨어 밸리데이션은 복합해질 수 있다. 따라서 이러한
모든 소 트웨어 밸리데이션 원칙은 소 트웨어 밸리데이션 로세스를 설계할 때 고
려되는 것이 하다. 결과 으로 소 트웨어 밸리데이션 로세스는 시스템, 의료기기
는 로세스와 련된 안정 험에 상응하여 이루어져야 한다.
소 트웨어 밸리데이션 활동 작업은 다른 장소에서 발생하고 다른 조직으로
되어 분산될 수 있다. 그러나 작업분배․계약 계․구성요소 원인 는 개발환경에
계없이 의료기기 제조업자 는 특정 개발자는 소 트웨어가 유효하다는 것을 입증함에
있어 최종 인 책임이 있다.
Section 5. 활동과 작업(TASKS)
소 트웨어 밸리데이션은 소 트웨어 개발 life cycle의 다양한 단계에서 계획․실행
되는 연속 인 활동과 작업을 통하여 실시되어야 한다. 이런 작업은 소 트웨어 로젝트
로세스로서 사용되는 life cycle 모델 발생되는 변경 범 에 향을 받아서 단발
는 반복 으로 실시될 수 있다.
5.1. 소 트웨어 life cycle 활동
이 지침은 특정 소 트웨어 life cycle 모델을 권장하지는 않는다. 소 트웨어 개발자
는 해당 제품 조직에 한 소 트웨어 life cycle 모델을 설정하여야 한다. 선택된
소 트웨어 life cycle 모델은 제품 기부터 폐기 때까지 용되어야 한다. 일반 인 소
트웨어 life cycle 모델의 활동은 다음과 같다.

- 216 -
•품질 계획
•시스템 요구사항 정의
•구체 인 소 트웨어 요구사항에 한 규격(specification)
•소 트웨어 디자인 규격
•구성 는 코딩(coding)
•시험
•설치
•운 유지(support)
•유지․보수(maintenance)
•폐기(retirement)
소 트웨어 밸리데이션을 증명하는 검증, 시험 다른 작업들이 이런 활동의 각 단
계에서 발생한다. life cycle모델은 다양한 방식으로 소 트웨어 개발 활동을 구성하고
소 트웨어 개발 로젝트를 모니터링하고 리하기 한 체계를 제공한다. 몇 개의
소 트웨어 life cycle 모델(아주 빠른(waterfall), spiral, 빠른(rapid prototyping), 단계
인 개발(incremental development) 등)은 FDA의 Glossary of Computerized System
and Software Development Terminology(1995.8)에 정의 되어있다. 이런 life cycle 모델
은 Appendix A의 참고문헌에 설명되어 있다.
5.2. 밸리데이션을 한 작업
각 소 트웨어 life cycle 활동은 소 트웨어가 유효하다는 결론을 뒷받침하는 “일반
인” 작업이다. 그러나 구체 인 수행 작업, 수행 작업순서 수행 작업의 반복․시간
성(timing)이 선택된 특정 소 트웨어 life cycle 모델과 소 트웨어 용과 련한
안 험에 의하여 서술(dictated)되어야할 것이다. 험도의 응용 로그램은 특정
작업이 필요하지 않을 수 있다. 그러나 소 트웨어 개발자는 최소한 이러한 각
작업들을 고려하고 해당되는 특정한 용에 한지의 여부를 정의하고 문서화하여야
한다.
다음의 논의는 특정 소 트웨어 life cycle 모델 는 수행되는 특정 작업을 설명한
것이 아니라 일반 인 것이다.
5.2.1. 품질 기획
설계 개발 계획은 필요한 작업, 외사항(anomaly) 보고 해결 차, 필요한 자원
그리고 정식 설계 검토를 포함하여 경 검토 요구사항을 규정한 계획을 통하여 완성
된다. 각 소 트웨어 life cycle 활동을 하여 필요한 작업뿐만 아니라 소 트웨어 life
cycle 모델 련 활동도 정의되어야만 한다. 이러한 계획은 다음 사항을 포함한다.

- 217 -
•각 life cycle 활동을 한 구체 작업
• 요한 품질요소의 열거( : 신뢰성, 유지성(maintainability) 유용성(usability)
•각 작업을 한 방법 차
•작업 수용 기
•입력 요구사항 확인을 평가할 수 있는 출력 정의 문서화에 한 기
•각 작업을 한 입력
•각 작업의 출력
•각 작업을 한 규칙(role), 자원 책임
• 험과 가정(assumptions)
•사용자 요구사항의 문서화
리부서는 한 소 트웨어 개발환경 자원을 악하고 제공하여야 한다(21
CFR 820.20(b)(1) and (2)). 일반 으로 각 작업은 물 자원뿐만 아니라 인 자원
(personnel)을 필요로 한다. 이런 계획에는 인력, 각 작업을 한 시설․장비자원
험 리를 한 역할(role)을 명시하여야 한다. 형상 리 계획(configuration
management plan)은 병렬 으로 수행되는 다양한 개발 활동을 리하고 한 의사
소통 문서화를 확실히 하도록 개발되어야 한다. 리는 구체 인 문서, 소스 코드
(source code), 오 젝트 코드(object code) 소 트웨어 시스템을 포함하는 시험원
(test suites)이 승인된 모든 version 내에서 정확하게 일치됨을 확실히 하는 것이 필요
하다. 한 근 가능한 승인된 version을 정확하게 식별하여야 한다.
차는 밸리데이션 는 다른 활동을 통하여 발생하는 소 트웨어 외사항을 보
고․해결하기 하여 만들어야 한다. 리는 기록을 식별하고 내용(contents), 형식
각 보고를 한 책임 조직의 구성요소를 명시하여야 한다. 한 검토 승인을 하
여 책임있는 조직의 구성원을 포함한 소 트웨어 개발 결과의 검토․승인을 하여
차가 필요하다.
형 작업 - 품질 계획
• 험 리 계획
•형상 리 계획(Configuration Management Plan)
•소 트웨어 품질 승인 계획(Software Quality Assurance Plan)
- 소 트웨어 검증 밸리데이션 계획
․검증 밸리데이션 작업 수용 기
․소 트웨어 검증 밸리데이션 활동을 한 일정 자원 배분

- 218 -
․보고 시 요구사항
- 정규 설계 검토 요구사항
- 그 밖의 기술 검토 요구사항
•문제 보고 해결 차
•그 밖의 지원 활동
5.2.2. 요구사항
요구사항 개발은 의료기기 사용목 에 한 정보의 식별, 분석 문서화를 포함
한다. 특별히 요한 범 는 하드웨어/소 트웨어, 운 조건, 사용자 특성, 잠재
해요소(hazard) 상 작업에 한 시스템 기능의 배분(allocation)을 포함한다. 한
요구사항은 소 트웨어의 사용목 을 정확하게 명시하여야 한다.
소 트웨어 요구사항을 규정한 문서는 소 트웨어 기능에 한 문서화된 규정을 포함해
야 한다. 이는 미리 확정되고 문서화된 소 트웨어 요구사항 없이는 소 트웨어의 밸리데이션
을 수행하는 것은 불가능하다. 일반 으로 소 트웨어 요구사항은 다음을 명확히 한다.
•모든 소 트웨어 시스템 입력
•모든 소 트웨어 시스템 출력
•소 트웨어가 수행할 모든 기능
•소 트웨어가 만족시켜야 하는 모든 수행 요구사항(데이터 처리량(throughput), 신
뢰성(reliability) 시간성(timing))
•모든 내부의 소 트웨어와 시스템간의 인터페이스뿐만 아니라 모든 외부와 사용자
인터페이스의 정의
•사용자가 시스템에 상호작용 할 수 있는 방법
•오류 생성 요인과 처리방법
•요구되는 응답시간(response time)
•설계 제약이 있을시 소 트웨어를 한 의도된 사용 환경( : 하드웨어 기반
(platform), 운 시스템)
•소 트웨어가 수용할 수 있는 모든 범 , 한계, 결함 특정 값(value)
•소 트웨어에서 수행될 요구사항, 규격, 특징(feature) 는 기능과 련한 모든 안 성
소 트웨어 안정성 요구사항은 시스템 요구사항 개발 로세스와 하게 결합된
기술 인 험 리 로세스에서 기인한다. 소 트웨어 요구사항 규격은 소 트웨어에
서 수행되는 모든 안정성 요구사항뿐만 아니라 시스템에서 소 트웨어 결함으로 발생할
수 있는 잠재 인 해요소도 명확히 규정하여야 한다. 소 트웨어 결함의 향은 결함을

- 219 -
완화시키는 방법과 함께 평가되어야 한다( : 하드웨어 완화, 방어 로그래 등). 이런
분석으로부터 험(harm)을 방하는데 필요한 가장 한 방법을 명시하는 것이 가
능하다.
품질시스템 규정은 불완 하고 불확실(ambiguous) 는 일치하지 않는(conflicting)
요구사항을 검토하기 한 메커니즘을 요구한다(21 CFR 820.30(c)). 소 트웨어 요구사
항 규격에 명시된 각 요구사항( : 하드웨어, 소 트웨어, 사용자 운 인터페이스
안 성)은 정확성(accuracy), 완료성(completeness), 일 성(consistency), 시험가능성
(testability), 정확성(correctness) 명확성(clarity))을 하여 평가되어야 한다. 를
들어 소 트웨어 요구사항은 다음 사항을 검증하기 해 평가되어야 한다.
•요구사항 내에서 내부 으로 모순된 (inconsistency)이 없어야 한다;
•시스템을 한 모든 수행 요구사항은 명쾌하게 설명되어야 한다;
•결함의 허용오차, 안 성 보안 요구사항은 완벽하고 정확하다;
•소 트웨어 기능의 할당은 정확하고 완 하다;
•소 트웨어 요구사항은 시스템 해요소(hazard)에 하여 하다;
•모든 요구사항은 측정 가능하거나 객 으로 검증할 수 있는 용어로 설명 되어야
한다.
소 트웨어 요구사항에 한 추 성 분석은 시스템 요구사항 험 분석결과에
하여 소 트웨어 요구사항을 추 하기 하여 실시되어야 한다. 소 트웨어 요구사
항을 검증하기 하여 사용되는 모든 분석 문서화 외에, 요구사항이 충분히 명시
화되어 있고 하다는 것을 확인하기 하여 체 인 소 트웨어 설계 작업이 시작
되기 에 정식 설계 검토과정이 권장된다. 요구사항은 계속 으로 승인되고 배포될 수
있으나 소 트웨어( 하드웨어) 요구사항간의 상호 향 인터페이스가 하게 검
토, 분석 리하는 것에 주의를 기울여야 한다.
형 작업 - 요구사항
•사 험 분석
•추 성 분석
- 시스템 요구사항에 한 소 트웨어 요구사항(반 의 경우도 포함)
- 험 분석에 한 소 트웨어 요구사항
•사용자 특성에 한 서술
•1차 2차 메모리의 특성과 제한에 한 목록

- 220 -
•소 트웨어 요구사항 평가
•소 트웨어 사용자 인터페이스 요구사항 분석
•시스템 시험 계획의 생성(generation)
•승인 시험 계획의 생성
•모호성(ambiguity)에 한 검토 는 분석
5.2.3. 설계
설계 과정에서 소 트웨어 요구사항 규격은 소 트웨어가 수행할 수 있는 논리 이
고 물리 인 표 으로 변환된다. 소 트웨어 설계 규격은 소 트웨어가 어떤(what) 기
능을, 어떻게(how) 수행할 것인지에 하여 설명한 것이다.
로젝트의 복잡성 는 다양한 기술 책임의 수 을 지닌 사람들이 설계 정보를
명확하게 이해하도록 하기 하여 설계 규격은 설계 세부 설계 정보에 하여 높은
수 의 요약을 포함할 수 있다. 완성된 소 트웨어 설계 규격은 동의된 요구사항 설
계 목 내에서 로그래머/코더(coder)가 작업하도록 한다. 완성된 소 트웨어 설계
규격은 로그래머가 임기응변(ad hoc)으로 설계 결정을 내려야 하는 필요성을 경감시
킨다.
소 트웨어 설계는 인 요소(human factor)를 명확히 하는 것이 필요하다. 지나치게
복잡하거나 동작(operation)에 한 사용자의 직 인 상에 반 되는 설계로 인한
사용상의 오류는 FDA에 보고되는 가장 지속 이고 한 문제 의 하나이다. 소
트웨어 설계는 이러한 사용상 오류에 있어서 한 가지 요소이다. 인 요소공학(human
factors engineering)은 의료기기 설계 요구사항, 분석 시험을 포함한 체 인 설
계 개발 로세스에 포함되어야 한다. 의료기기 안 성 유용성(usability) 문제
는 설계 흐름도(flowchart), 상태도, 표 (prototyping) 도구 시험계획을 할 때 고려되
어야 한다. 한 작업과 기능에 한 분석, 험 분석, 표 시험, 검토 충분한(full)
유용성 시험이 수행되어야 한다. 사용자 집단(population)으로부터의 참가자는 이러한
방법론을 용할 때 포함되어져야 한다.
소 트웨어 설계 규격에는 아래의 사항들을 포함해야만 한다.:
•소 트웨어 수용에 있어 결정된 기 을 포함한 소 트웨어 요구사항 규격
•소 트웨어 험 분석
•개발 차 코딩 지침( 는 다른 로그래 차)

- 221 -
•하드웨어, 소 트웨어 물리 환경과의 계를 포함하여 로그램이 동작하기
로 의도한 시스템의 개 (context)을 설명한 시스템 문서( : 서술 는 개 도)
•사용되는 하드웨어
•측정 는 기록되는 변수
•논리 구조(제어논리(control logic) 포함)와 논리 처리단계( : 알고리즘)
•데이터 구조와 데이터 흐름도
•변수(제어와 데이터)의 정의 사용되는 곳에 한 기술
•오류, 경보(alarm) 경고 메시지
•보조 로그램( : 운 시스템, 드라이버, 다른 응용 소 트웨어)
• 달 연결(communication links)(소 트웨어 내부 모듈 사이의 연결, 보조 소
트웨어와의 연결, 하드웨어와의 연결 사용자와의 연결)
•보안 수단(물리 논리 인 보안)
•상기 요소들에서 언 되지 않은 모든 추가 인 constraints
의 요소 처음의 4개는 소 트웨어 설계규격에 참고문헌으로 포함되는 것으로
별도의 미리 존재하는 문서이다. 소 트웨어 요구사항 규격은 소 트웨어 험 분석으로
서 이 Section에서 언 하 다. 문서화된 개발 차는 조직의 지침으로 제공되고 문
서화된 로그래 차는 각 로그래머에 한 지침으로서 제공된다. 소 트웨어가 기
능하는 시스템의 맥락에 한 지식이 없이는 소 트웨어의 밸리데이션이 될 수 없기
때문에 문서가 참고 되어진다.
의 요소 몇몇이 소 트웨어에 포함되어 있지 않을 경우 명확히 언 될 때 소
트웨어에 한 향후 검토자 유지․보수하는 사람에게 도움이 될 것이다.( : 이
로그램에 오류 메시지가 없음)
소 트웨어 설계과정에서 발생하는 활동은 여러 가지 목 이 있다. 소 트웨어 설계
평가는 설계의 완 성, 정확성, 일 성, 불확실성, 실행가능성 지속성 여부를 결정
하기 하여 수행된다. 설계과정에서 소 트웨어 구성(architecture, : 모듈구조)에
한 한 고려는 소 트웨어 변경이 필요할 때 향후 밸리데이션 노력의 양을 일
수 있다. 소 트웨어 설계 평가는 제어 흐름, 데이터 흐름, 복잡성, 시간성, 크기, 메모
리 할당, 임계(criticality) 분석 다른 설계측면에 한 분석을 포함할 수 있다. 추
성 분석(traceability analysis)은 소 트웨어 설계가 모든 소 트웨어 요구사항을
수행하는지를 검증하기 하여 수행되어야 한다. 요구사항에 한 식별을 하기 한 기
술이 불충분할 경우 추 성 분석은 모든 설계측면이 소 트웨어 요구사항에 하여 추

- 222 -
가능하다는 것을 검증할 수 있어야 한다. 상호의사소통 연결의 분석은 하드웨어, 사
용자 련 소 트웨어 요구사항을 고려하여 제안된 설계인지를 평가하기 하여 수
행되어진다. 소 트웨어 험분석은 모든 추가 인 해요인의 식별 여부 설계에
의하여 새로운 해요인이 유도되었는지의 여부를 결정하기 하여 재시험되어야 한
다.
소 트웨어 설계 활동의 최종 단계에서 정규 설계 검토(Formal Design Review)는 설계
를 수행하기 에 설계의 옳음, 일 성, 완 성, 정확성 시험가능성을 검증하기
하여 수행되어야 한다. 설계의 부분들은 수행을 하여 단계 으로 승인 배포될 수
있다. 그러나 다양한 요소사이의 상호작용(interaction) 의사소통연결을 히 검토,
분석 제어하여야 한다.
부분의 소 트웨어 개발 모델은 반복될 것이다. 이는 소 트웨어 요구사항 규격
과 소 트웨어 설계 규격이 여러 가지 version으로 나오는 것과 같다. 모든 승인된 버
은 수립된 형상 리 차에 따라서 구성 제어되어야 한다.
형 작업 - 설계
•업데이트 된 소 트웨어 험분석
•추 성 분석 - 소 트웨어 요구사항에 한 설계 규격(설계 요구사항에 한 소
트웨어 규격)
•소 트웨어 설계 평가
•설계 상호의사소통 연결 분석
•모듈 시험 계획 시기(generation)
•통합 시험 계획 시기
•설계 시험 시기(모듈, 통합, 시스템 수용)
5.2.4 구조Construction) 는 코딩(Coding)
소 트웨어는 새로운 응용 로그램 사용을 하여 코딩( 로그래 )하거나 미리 코
딩된 소 트웨어 구성요소들( : 코드 라이 러리, OTS 소 트웨어 등)을 같이 조합하
여 구성될 수 있다. 코딩은 세부 인 설계 규격이 소스 코드(source code)로서 수행되
는 소 트웨어 활동이다. 코딩은 소 트웨어 개발 로세스 가장 낮은 수 의 작용
(abstraction)이다. 모듈 규격이 로그래 언어로 변환(translation)되는 것은 소 트웨
어 요구사항의 분해(decomposition)에 있어서는 최종 단계 이다.

- 223 -
코딩은 보통 높은 수 의 로그래 언어의 사용을 필요로 하지만 임계시간
(time-critical) 동작을 하여 어셈블리 언어(assembly language)( 는 마이크로코드)의
사용을 수반할 수도 있다. 소스 코드는 상 하드웨어 기반에서 사용을 하여 컴 일
되거나 변환(interpreted)될 수 있다. 로그래 언어와 소 트웨어 빌드 도구(어셈블
러(assembler), 링커(linker), 컴 일러(compiler))의 선택에 한 결정은 결과 인 품질
평가 작업( : 선택된 언어를 한 디버깅 시험도구의 가능성)에 미치는 향을 고
려하여야 한다. 몇몇 컴 일러는 코드 디버깅시 보조 으로 오류확인을 한 선택 수
(optional level) 명령어를 제공한다. 오류 확인의 여러 가지 수 은 코딩과정을
통하여 이용될 수 있고 컴 일러에서 오는 경고 는 다른 메시지는 기록되거나 그
지 아니할 수 있다. 그러나 코딩 디버깅 로세스의 최종 단계에는 무슨 컴 일
이션(compilation) 오류가 소 트웨어에 여 히 남아있는지를 문서화하기 하여 가장
엄격한 오류 확인 수 이 이용되어진다. 가장 엄격한 오류 확인 수 이 최종 소스
코드 환시 사용되지 않을 경우 덜 엄격한 환 오류 확인의 사용을 한 조정
(justification)이 문서화되어야 한다. 한 최종 컴 일 이션을 하여 모든 경고 는
컴 일에서 오는 메시지를 포함한 컴 일 이션 과정 그 결과 는 미결된 문제
을 남기는 결정을 한 조정에 하여 문서화 하여야 한다.
업소는 소 트웨어 코딩 로세스와 련하여 품질방침 차를 설정하는 규격 코
딩 지침을 채택한다. 소스 코드는 규격화된 코딩 지침에 합함을 검증하기 하여
평가되어야 한다. 이러한 지침은 명확성(clarity), 형식, 복잡성, 리 해설과 련하
여 코딩 합의사항을 포함하여야 한다. 코드 해설은 상 입․출력, 참조 변수, 상 데이
터 형식 수행 동작을 포함한 모듈을 해 유용하고 기술 인 정보를 제공하여야 한
다. 소스 코드는 세부 인 설계 규격에 합함을 검증하기 하여 평가되어야 한다. 통
합 시험을 한 모듈은 코딩 지침 다른 용되는 품질방침․ 차에 합하도록
문서화 하여야 한다.
소스 코드 평가는 코드 검사 코드 walkthrough로서 수행될 때도 있다. 이런 통계
분석은 코드의 실행 에 오류를 악할 수 있는 매우 효과 인 방법이다. 이는 격
리된 상황에서 각 오류의 시험을 가능하게 하고 추후 소 트웨어의 동 인 시험에 집
할 수 있도록 해 다. 업소는 일 성과 독립성을 확실히 하기 한 한 리 기능
을 가진 수동 확인을 사용할 수 있다. 소스 코드 평가는 모듈과 이어(수평 , 수직
인터페이스) 그 설계 규격에 합성 사이의 내부 인 연계에 한 검증까지 확 되
어야 한다. 사용되는 차의 문서화 소스코드 평가의 결과는 설계 검증의 일부분
으로써 리되어야 한다.

- 224 -
소스 코드의 추 성 분석은 모든 코드가 설정된 규격 시험 차와 연결이 되어있
는지를 검증하기 한 요한 도구이다. 소스 코드 추 성 분석은 다음과 같은 사항을
검증하기 하여 수행 문서화 되어야 한다.
•소 트웨어 설계 규격의 각 요소들은 코드로 구 되었다
•코드로 구 된 모듈 기능은 소 트웨어 설계 규격의 요소와 험분석까지 추
될 수 있다.
•모듈 기능에 한 시험은 소 트웨어 설계 규격의 각 요소와 험분석까지 추
될 수 있다.
•모듈 기능에 한 시험은 동일한 모델과 기능을 한 소스코드까지 추 될 수
있다.
형 작업 - 구조 는 코딩
•추 성 분석
- 설계 규격을 한 소스코드( 소스코드를 한 설계 규격)
- 소스코드 설계 규격을 한 시험 사례(cases)
•소스코드 소스코드 문서화 평가
•소스코드 인터페이스 분석
•시험 차 시험 제 시기(모듈, 통합, 시스템 수용)
5.2.5 소 트웨어 개발자에 의한 시험
소 트웨어 시험은 미리 규정된 상과 비교될 수 있는 규정된 입력 문서화된 출
력의 조건하에서 소 트웨어 제품을 실행을 하는 것이다. 이는 시간 소모 이며 어렵
고 불완 한 활동이다. 한 효과 이고 효율성 있기 해서는 기에 계획되는 것이 요
구된다.
시험 계획과 시험 사례는 가능한 소 트웨어 개발 로세스 기에 수행되어야 한
다. 이것들은 일정, 환경, 자원(인력, 도구 등), 방법론, 사례(입력, 차, 출력 상 결과),
문서화 보고 기 을 명확히 하여야 한다. 시험 로세스를 통하여 용되는 노력의
크기는 복잡성, 임계(criticality), 신뢰성 / 는 안 성의 문제( : 요구 기능 는 그
결함 허용(tolerance) 형상의 요한 시험에 용되는 특별한 결과를 생성하는 모듈)와
연결될 수 있다. 소 트웨어 소 트웨어 시험 노력의 범주에 한 설명은 여러 문
헌에 나타나 있다. 를 들면 다음과 같다.
•NIST Special Publication 500-235, Structured Testing: A Testing Methodology
Using the Cyclomatic Complexity Metric;
•NUREG/CR-6293, Verification and Validation Guidelines for High Integrity

- 225 -
Systems; and
•IEEE Computer Society Press, Handbook of Software Reliability Engineering.
소 트웨어 시험 계획은 각 개발 단계에서 수행되는 특별한 작업을 명확히 하고 련
완 성 기 에 의하여 나타내어지는 노력 수 의 정당화를 포함하여야 한다.
소 트웨어 시험은 특정 소 트웨어 제품의 시험을 계획할 때에 인지하고 고려
해야 하는 한계를 가지고 있다. 가장 간단한 로그램을 제외하고 소 트웨어는 철 하게
시험될 수는 없다. 일반 으로 모든 가능한 입력을 가지고 소 트웨어 제품을 시험하는
것은 불가능하고 로그램 실행시 발생할 수 있는 모든 가능한 데이터 로세싱 경로
를 시험하는 것도 불가능하다. 특정 소 트웨어 제품이 완 하게 시험되었다고 확신할 수
있는 단 한 가지 시험 형식 는 시험방법론이 있는 것은 아니다. 한 로그램의 모든 코드
를 시험하는 것이 로그램에 모든 필요한 기능이 있다는 것을 의미하는 것은 아니다.
모든 로그램 기능 코드에 한 시험이 로그램이 100% 정확하다는 것을 의미하
는 것은 아니다. 오류가 없음을 나타내는 소 트웨어 시험결과가 소 트웨어 제품에 오류
가 없다는 것으로 해석되어서는 아니 된다. 이는 시험이 피상 이라는 것을 의미할 수
있다.
소 트웨어 시험사례의 필수 요소는 결과 이다. 실제 인 시험 결과의 객 인 평가를 허용
하는 것이 요 사항이다. 필수 시험 정보는 련성이 있고 미리 정의된 정의 는 규격
으로부터 획득된다. 소 트웨어 규격 문서는 무엇을(what), 언제(when), 어떻게(how), 왜(why) 등
을 명확히 하여야 하고 그것이 시험을 통하여 확인되기 하여 구체 인 공학( : 측정가
능하거나 객 으로 검증이 가능한)수 으로 수행되어야 한다. 효과 인 소 트웨어
시험의 실제 인 노력은 시험의 결과보다는 무엇을 시험해야 하는지 규정하는 것이
다.
소 트웨어 시험 로세스는 소 트웨어 제품의 효과 인 시험을 진하려는 원칙을
기반으로 하고 있다. 응용 소 트웨어 시험은 다음의 사항을 포함한다.
• 상되는 시험 결과는 미리 정의한다.
•우수 시험사례는 오류 노출(exposing an error)의 가능성이 높다.
•성공 인 시험은 오류를 발견하는 것이다
•코딩으로부터 독립 이다.
•사용자 소 트웨어( 로그래 ) 문가가 고용되어진다.

- 226 -
•시험자는 코더와는 다른 도구를 사용한다.
•단지 일반 인 사례를 시험하는 것은 불충분하다
•시험 문서화는 재사용이 가능하고 추후 검토과정에서 시험결과의 합(pass)/부
합(fail) 상태에 한 독립 인 확인이 가능하다
제가 되는 작업( : 코드 검사)이 성공 으로 완료되면 소 트웨어 시험은 시작된
다. 이는 단 수 시험에서 시작하고 시스템 수 의 시험에서 종결된다. 여기에는 시
험수 의 통합이 별도로 있을 수 있다. 소 트웨어 제품은 그 내부 인 구조에 기 한
시험사례 외부의 규격에 기 한 시험사례를 가지고 수행되어야 한다. 이 시험은 소
트웨어의 기능성, 실행 인터페이스 규정 요구사항의 합성에 한 체 이
고 엄격한 시험을 제공하여야 한다.
코드 기반(code-based) 시험은 구조 시험 는 “white-box" 시험27)으로도 알려져 있다.
이는 소스코드, 구체 인 설계 규격 다른 개발 문서로부터 획득된 지식을 기 로 하는
시험 사례들을 명확히 한다. 이 시험사례들은 로그램에서 만들어진 리결정(control
decision) 형상(configuration) 표를 포함하는 로그램의 데이터 구조를 수행한다. 구
조 인 시험은 로그램이 실행될 때 한번도 수행되지 않는 dead 코드를 명확히 할 수 있다.
구조 인 시험은 단 (모듈) 수 시험으로 우선 으로 수행되어지지만 소 트웨어
시험의 다른 수 까지 확 될 수는 없다.
구조 인 시험의 수 은 구조 인 시험과정에서 평가되는 소 트웨어 구조의 비율이
얼마인지를 보여주기 한 측정기 (metrics)을 사용하여 평가될 수 있다. 이런 측정기 은
일반 으로 “범 (coverage)"로 참고 되고 시험 선택기 을 고려한 완 성의 측정이
다. 구조 인 범 의 크기는 소 트웨어에 의하여 제기된 험의 수 에 동일하여야
한다. ”범 “라는 용어의 사용은 일반 으로 100% 커버리지(coverage)를 의미한다. 를
들면 시험 로그램이 ”명령문 커버리지(statement coverage)"를 수행하 다면 이는 소
트웨어의 명령문 100%가 어도 한번은 실행되었다는 것을 의미한다. 일반 인 구조
인 측정기 은 다음의 사항을 포함한다.
•명령문 범 (Statement Coverage) - 이 기 은 최소 한번은 각 로그램 명령
27) 제품의 내부수행동작을 알고 있을 때 명세화에 따른 내부수행 동작을 시험하는 것으로 순차 이고 세부
이며 소 트웨어의 논리 인 경로, 조건 반복과 같은 부분들을 시험하는 것이다. 발생가능한 모든 경우
경로를 시험하는 것을 특히 소모 시험(Exhaustive test)이라 하는데 이는 실 으로 어려워 요부
분을 상으로 White-box test가 이루어진다.

- 227 -
어가 수행되기 하여 충분한 시험사례들을 요구한다. 그러나 이들의 수행은 소
트웨어 로그램의 반응에서 신뢰성을 제공하는 데는 불충분하다.
•결정(Decision)(분기, Branch) 범 - 이 기 은 각 가능한 결과가 최소 한번은 발생
할 수 있도록 실행되어지는 각 로그램 결정 는 분기를 한 충분한 시험사례
를 요구한다. 이는 부분의 소 트웨어 제품을 한 범 의 최소 수 으로 고려
되어지나 결정 범 단독으로는 높은 수 의 통합 용(high-integrity application)
을 해서는 불충분하다.
•조건 범 – 이 기 은 최소 한번은 모든 가능한 결과에서 수행하여야 하는
로그램 결정에서 각 조건을 한 충분한 시험사례를 요구한다. 이는 단지 다양한
조건이 결정에 이르기 하여 평가되어져야 할 때의 분기 범 와는 다르다.
•다양한 조건 범 – 이 기 은 로그램 결정에서 모든 가능한 조건 조합을 수행
하기 한 충분한 시험사례를 요구한다.
•루 (loop) 범 – 이 기 은 기화(initialization), 일반 인 실행 종료(한계,
boundary) 조건을 포함하는 0, 1, 2 많은 반복을 하여 실행되는 모든 로그
램 루 를 한 충분한 시험사례를 요구한다.
•경로 범 – 이 기 은 정의된 로그램 구획의 시작부터 종료까지 최소 한번
은 실행되는 각 실행 가능한 경로, 기본 경로 등을 하여 충분한 시험사례를 요
구한다. 소 트웨어 로그램을 통틀어 가능한 경로의 수가 매우 많기 때문에 일
반 으로 경로 범 는 수될 수는 없다. 보통 경로 범 의 크기(amount)는 시험
하에서 소 트웨어의 험 는 임계(criticality)에 기 하여 설정된다.
•데이터 흐름 범 (Data Flow Coverage) – 이 기 은 최소 한번은 수행되는 각 실
행 가능한 데이터 흐름을 하여 충분한 시험사례를 요구한다. 많은 데이터 흐
름 시험 략이 가능하다.
정의기반(definition-based) 는 규격기반(specific-based) 시험은 한 기능 시험 는
“black-box”시험28)으로 알려져 있다. 이는 소 트웨어 제품(한 개의 단 (unit)((모듈)
28) 제품의 수행기능을 알고 있을 때 각 기능의 완 한 동작을 증명해 보이는 것으로 소 트웨어 인터페이스
에서 실시되는 시험을 의미한다. 즉, 소 트웨어 기능의 작동과 입력의 합성, 출력의 정확성, 자료 일과
같은 외부 정보의 무결성을 입증하는 것이다. 이는 소 트웨어 내부 논리 구조는 고려하지 않고 기본
인 시스템 모델의 기능을 시험하는 것이다.

- 228 -
는 완 한 로그램)이 목 으로 한 동작에 하여 정의내린 것에 기 한 시험사례
에 하여 명확히 하여야 한다. 이 시험사례는 사용목 는 로그램의 기능성
로그램의 내․외 인터페이스를 수행한다. 기능 시험은 각 단 에서부터 시스템 수
의 시험까지 소 트웨어 시험의 수 에 용될 수 있다.
다음의 기능 시험의 형식은 일반 으로 노력의 수 향상을 포함한다.
일반 사례 – 일반 인 입력에 한 검사가 필요하다. 그러나 상되는 확실한
입력만을 가지는 소 트웨어 제품에 한 시험은 그 소 트웨어 제품에 한 완벽한
시험은 아니다. 그것만으로는 일반 인 사례 시험은 소 트웨어 제품의 확실성
(dependability)에 있어 충분한 신뢰를 제공할 수 없다.
출력 forcing – 선택된( 는 모든) 소 트웨어 출력을 확실히 하기 한 시험 입력
을 선택하는 것은 시험에 의하여 이루어진다.
Robustness – 소 트웨어 시험은 상하지 못한 무효한(invalid) 입력이 주어진 경우
소 트웨어 제품이 정확하게 동작한다는 것을 증명하여야 한다. 이런 충분한 일
련의 시험 사례를 명확히 하기 한 방법으로 동일 등 구분(Equivalence Class
Partitioning), 경계값 분석(Boundary Value Analysis) 특별 사례 확인(Special
Case Identification)(Error Guessing)이 있다.
입력의 조합 – 에서 명시된 모든 기능 시험 방법이 개별 는 단일 시험 입력
을 강조하고 있다. 부분의 소 트웨어 제품은 그 사용 환경에서 다양한 입력을
가지고 동작한다. 소 트웨어 제품 시험은 소 트웨어 단 는 시스템이 동작과
정에서 일으키는 입력의 조합을 고려하여야 한다.
기능 구조 인 소 트웨어 시험사례 확인(identification) 기술은 임의의(random)
시험 입력보다 시험을 한 특정 입력을 제공한다. 이 기술에서의 결 (weakness)은
소 트웨어 제품의 신뢰도(reliability)를 한 구조․기능 시험 완료 기 연결(linking)
에 있어 어려움이 있다. 통계 시험과 같이 향상된 소 트웨어 시험방법은 소 트웨
어의 신뢰성을 좀 더 확실히 하기 하여 이용될 수 있다. 통계 시험은 수행도
(operational profile)( : 소 트웨어의 상된 사용, 험한 사용 는 부당한 사용)에
기 하여 정의된 분포(distribution)로부터 생성된 시험 데이터를 임의로 사용한다. 시험
데이터의 많은 부분은 소 트웨어 제품의 개발자 는 시험검사자가 상하지 못한 개별
이고 다양한 외 인 동작 환경을 확인하기 한 증가된 가능성을 제공하는 특정한
역 는 문제 을 방어하기 하여 수행되고 목표로 한다. 통계 시험은 한 높은
구조 용(coverage)을 제공한다. 이는 안정된 소 트웨어 제품을 요구하는 것은 아
니다. 그러므로 구조․기능 시험은 소 트웨어 제품의 통계 시험을 하여 필수 이다.

- 229 -
소 트웨어 시험의 다른 측면은 소 트웨어 변경의 시험이다. 변경은 소 트웨어 개발
과정에서 자주 발생한다. 이러한 변경은 1) 오류 발견 수정하는 디버깅(debugging),
2) 신규 는 변경된 요구사항(“requirements creep") 3) 더 효과 이고 효율 인 실
행이 악될 경우 변경된 설계의 결과이다. 한 소 트웨어 제품이 승인되어지면 그
제품을 한 모든 변경은 시험을 포함한 자체의 ”최소 life cycle(mini life cycle)"를 가
져야 한다. 변경된 소 트웨어 제품의 시험은 추가 인 노력을 요구한다. 이는 그 변경
이 정확하게 수행되어진다는 것뿐만 아니라 시험은 한 변경사항이 소 트웨어 제품
의 다른 역에 부작용을 일으키지 않는다는 것을 증명하여야 한다. 회귀분석
(Regression analysis)29) 시험은 변경이 소 트웨어 제품의 다른 곳에서 문제를 발생
하지 않는다는 것을 확실히 하기 하여 이용되어진다. 회귀분석은 실행되는 필요한
회귀 시험을 확실히 하기 하여 련 문서( : 소 트웨어 요구사항 규격, 소 트웨어
설계 규격, 소스 코드, 시험 계획, 시험 사례, 시험설명서 등)의 검토에 기 한 변경
의 향에 한 문서화이다. 회귀검사는 로그램이 본래 정확하게 수행해온 시험사례
의 재실행(rerunning)이고 의도하지 않은 소 트웨어 변경의 효과를 악하기 하여
재의 결과를 이 의 결과와 비교하는 것이다. 한 회귀분석 회귀검사는 신규로
통합된 모듈이 이 에 통합된 모듈의 동작에 반 향을 주지 않는다는 것을 확실
히 하기 한 소 트웨어 제품을 설계하기 하여 통합방법을 사용하여 소 트웨어 제
품을 설계할 때 도입된다.
철 하고 엄격한 소 트웨어 제품의 검사(examination)를 하여 개발 시험은 일반
으로 단계 (level)으로 설정되어 있다. 를 들어 소 트웨어 제품의 시험은 단 , 통합
시스템 시험의 단계로 체계화될 수 있다.
1) 단 (모듈 는 구성요소) 수 의 검사는 보조 로그램(sub-program) 기능에
한 기 시험과 시스템 단계에서 보이지 않는 기능이 시험에 의해 검사된다는 것
을 확실히 하는데 을 두고 있다. 단 시험(unit testing)은 품질 소 트웨어
단 가 최종 소 트웨어 제품으로의 통합을 하여 제공된다는 것을 확실히 한다.
2) 통합 단계 시험(integration level testing)은 로그램의 내․외부 인터페이스에 걸친
데이터 제어(control)의 이 에 을 두고 있다. 외부 인터페이스는 소 트웨
어(운 시스템 소 트웨어 포함), 시스템 하드웨어 사용자가 있고 달 연결
(communication link)처럼 묘사될 수 있다.
29) 회귀분석(Regression analysis) 이란 변수들 사이의 계를 조사하여 모형화 시키는 통계 기법으로서 경
제, 경 , 교육, 정치 등의 사회과학 그리고 물리, 화학, 생물, 공학, 농학, 의학 등 자연과학의 거의 모든 분야에서 리 응용되고 있다. 즉, 변수들 간의 함수 인 련성을 규명하기 하여 수학 모형(통계모형)을 가정하고, 측된 자료로부터 이 모형을 추정하는 통계분석방법으로 주로 측에 사용된다.

- 230 -
3) 시스템 단계 검사(system level testing)는 모든 규격화된 기능이 있고 소 트웨어
제품이 신뢰할 만하다는 것을 증명한다. 이 시험은 규격화된 운 기반(operating
platform)에 표시된 것처럼 소 트웨어 제품을 한 요구사항을 고려한 완성된
(as-built) 로그램의 기능 성능을 검증한다. 시스템 단계 소 트웨어 시험은
기능 문제(concern) 사용목 과 련 의료기기 소 트웨어의 다음과 같은 요
소들을 다룬다.
•성능 문제(performance issues)( : 반응시간, 신뢰도 측정)
•가혹조건(stress conditions)에 한 반응. : 최 하 (load), 지속 인 사용하
의 반응(behavior)
•내․외부 보안 특성(feature)의 운
•손실 회복(disaster recovery)을 포함한 회복 차의 유효성
•유용성(usability)
•다른 소 트웨어 제품에 한 호환서(compatibility)
•정의된 각 하드웨어 형상(configuration)에서의 반응
•문서화의 정확성(accuracy)
제어 방법(control measures)( : 추 성 분석)은 의도한 범 가 수행되었는지를 확실
히 하기 하여 사용되어야 한다.
한 시스템 단계 시험은 의도한 운 환경에서 소 트웨어 제품의 반응을 나타낸
다. 이러한 시험의 치는 목표(target) 운 환경을 한 소 트웨어 개발자의 능력에
좌우된다. 환경에 따라 잠재 인 사용자 치에서의 모의실험(simulation) / 는 시
험은 이용될 수 있다.
계획된 시스템 단계 시험이 소 트웨어 개발자에 의하여 직 으로 리되지 않는
장(site)에서 수행될 때 시험계획은 사용범 가 설정되고 한 문서화가 비되었
는지를 확실히 하기 하여 필요한 리(control)을 명확히 하여야 한다. 한 FDA 승
인(clearance) 인체에 사용되는 의료기기 는 그 구성요소인 소 트웨어 제품에
하여 인체 피실험자(human subject)를 포함하는 시험은 IDE(Investigational Device
Exemption) 는 IRB(Institutional Review Board) 승인을 요구할 수 있다.
시험 차, 시험 데이터 시험결과는 이루어진 객 인 합/부 합 결정을 수락
하는 방식(manner)에서 문서화되어야 한다. 한 검토 시험 실시를 수반하는 객
인 결정을 내리는 것에 하여야 하고 추후에 발생하는 모든 회귀시험에서의 사용
에 하여야 한다. 시험과정에서 발견되는 오류는 소 트웨어 배포(release) 에 기록

- 231 -
(log), 분류(classify), 검토 해결(resolve)되어야 한다. 개발life cycle(development life
cycle)동안 수집 분석된 소 트웨어 오류 데이터는 상업 매를 한 배포를 하
여 소 트웨어의 합성을 결정하는데 사용될 수 있다. 시험 보고서는 련 시험계획
의 요구사항에 합하여야 한다.
의료기기 는 해당 기기의 생산에서 유용한 기능을 수행하는 소 트웨어 제품은 복
잡한 것도 있다. 소 트웨어 시험도구는 이러한 소 트웨어 제품의 시험에서 일 성
(consistency), 완벽성(thoroughness) 효율성(efficiency)을 확실히 하고 계획된 시험
활동 요구사항을 만족하기 하여 자주 사용된다. 이러한 도구는 상업용 소 트웨어
시험도구 뿐만 아니라 단 (모듈) 시험 수반되는 통합시험( : 드라이버 stub)을
도와주기 하여 자체 개발된(built-in-house) 보조 소 트웨어를 포함할 수 있다. 이러
한 도구는 개발에 사용되는 소 트웨어 제품과 같은 품질의 등 (degree)을 가져야
한다. 그 사용목 을 한 소 트웨어 도구의 밸리데이션 증거를 제공하는 한 문서
화가 유지되어야 한다(동 지침의 Section 6 참조)
형 인 작업 - 소 트웨어 개발자에 의한 시험
•검사 계획
•구조 시험사례 확인(Structural Test Case Identification)
•기능 시험사례 확인(Functional Test Case Identification)
•추 성 분석(Traceability Analysis) - 시험
- 세부 인 설계를 한 단 (모듈) 시험
- 고수 (high level) 설계를 한 통합 시험
- 소 트웨어 요구사항을 한 시스템 시험
•단 (모듈) 시험 실행
•통합시험 실행
•기능 시험 실행
•시스템 시험 실행
•수용 시험 실행(Acceptance Test Execution)
•시험결과 평가
•오류 평가/해결
•최종 시험 보고
5.2.6. 사용자 장 시험(User Site Testing)
사용자 환경에서의 시험은 소 트웨어 밸리데이션에서 필수 인 부분이다. 품질 리

- 232 -
규정은 한 설치를 증명하기 한 심사(inspection) 시험의 문서화뿐만 아니라
설치 심사 차( 한 장소에서의 시험 포함)를 요구한다(21 CFR 820.170 참고).
한 제조장비는 규격화된 요구사항을 반드시 만족하여야 하고 자동화된 시스템은 그
사용목 을 하여 반드시 밸리데이션되어야 한다(21 CFR 820.70(g) 820.70(i) 참고).
사용자 환경시험에 한 문용어는 혼란스러울 수 있다. 베타 시험(beta test), 환경
밸리데이션(site validation), 사용자 수용 시험(user acceptance test), 설치 검증
(installation verification) 설치 시험(installation testing)과 같은 용어는 사용자 환경
시험을 설명하기 하여 사용되어 왔다. 이 지침의 목 에 따라 “사용자 환경시험”이
라는 용어는 이러한 모든 것을 포함하고 개발자 조정 환경 범 의 외부에서 일어나는
다른 모든 시험도 포함한다. 이 시험은 설치되는 시스템 형상(configuration)의 일부가
될 수 있는 실제 하드웨어 소 트웨어를 갖춘 사용자 환경에서 수행되어야 한다.
이 시험은 목 하는 기능 범 내에서 시험되는 소 트웨어의 실제 는 가상사용을
통하여 수행되어진다.
동 지침은 일반 이고 모든 사용자 환경시험에 용될 수 있는 것을 포함하고 있다.
그러나 일부 범 ( : 액 설정 시스템 등)에서는 사용자 환경시험 계획에서 고려되어
져야할 특별한 환경 밸리데이션 문제가 있을 수 있다. 시험 계획자는 사용자 환경시
험을 하여 추가 인 법 요구사항이 있는지의 여부를 결정하기 한 유사한 제품
권한(product jurisdiction)에 하여 FDA 센터에 확인하여야 한다.
사용자 환경시험은 정식 시험 요약 정식 수용(formal acceptance) 기록을 가진 미리
정의된 문서화된 계획을 따라야 한다. 모든 시험 차, 시험 입력 데이터 시험 결과
의 문서화된 증거는 유지되어야 한다.
하드웨어 소 트웨어가 규격 로 설치 구성되었다는 증거가 있어야 한다. 방
법(measures)은 모든 시스템 구성요소가 시험과정에서 실행되고 이 구성요소의 버젼
(version)이 규격화되어 있다는 것을 명확히 하여야 한다. 시험 계획은 운 조건의 모
든 범 에 걸친 시험을 규격화하고, 일반 인 활동에서 나타나지 않은 어떤 잠재 인
결 을 악하기 한 노력의 과정에서 시스템이 직면하는 다양한 사건과 조건에 하
여 충분한 시간동안 지속됨을 명확히 하여야 한다.
개발자 환경에서 소 트웨어 개발자에 의하여 기에 운 된 일부 평가는 실제 사용
자 환경에서 반복되어야 한다. 이는 데이터의 고용량(high volume), 부담(load or

- 233 -
stress), 보완, 결 시험(fault testing)(무효(avoidance), 발견, 허용(tolerance) 해결),
오류 메시지 안 성 요구사항의 수행을 한 시험을 포함할 수 있다. 개발자는 이러한
목 을 하여 사용되는 시험 데이터세트(test data set)의 일부를 사용자에게 제공할 수
있다.
목 한 기능을 히 수행하기 한 시스템의 능력 평가에 더하여 정확한 인터페
이스 시스템 사용자 능력의 평가도 이루어져야 한다. 운 자(operator)는 목 한 기
능을 수행하고 모든 경보, 경고 오류 메시지에 하여 한 방법으로 처리할 수 있
어야 한다. 사용자 환경 시험과정에서 기록은 한 시스템 성능 발생된 모든 시스
템 결 을 유지하여야 한다. 이 사용자 환경 시험과정에서 발견된 결 을 보정하기 한
시스템의 개정(revision)은 다른 소 트웨어 변경을 하여 같은 차 리를 따라
야 한다.
소 트웨어 개발자는 사용자 환경시험에 참여 는 불참할 수 있다. 개발자 참여
시 설계수 (design-level) 시스템 시험의 최종 부분을 사용자 환경을 하여 수행할 수
있다. 그 지 않은 경우 사용자는 철 한(careful) 시험계획의 요성, 상 시험결과의
정의 모든 시험 출력의 기록을 이해하는 사람을 필요로 한다.
형 작업 - 사용자 환경시험
•수용 시험 실시
•시험결과 평가
•오류 평가/해결
•최종 시험 보고
5.2.7. 유지보수와 소 트웨어 변경
소 트웨어에 용시 유지보수(maintenance)라는 용어는 하드에어에 용한 것과 같은
의미는 아니다. 하드웨어 소 트웨어의 부 합/오류 메커니즘(mechanism)이 다르기
때문에 유지보수의 운 방식도 다르다. 하드웨어 유지보수는 일반 으로 방차원의 하
드웨어 유지보수 활동, 구성요소 교체(replacement) 시정 변경(corrective change)을
포함한다. 소 트웨어 유지보수는 시정, 향상(perfective) 개조(adaptive)하는 유지보
수를 포함하지만 방차원의 유지보수 활동 는 소 트웨어 구성요소 교체를 포함하
지는 않는다.
소 트웨어의 오류 결 을 교정하기 하여 수행된 변경은 시정(corrective) 유지
보수이다. 성능, 지속성(maintainability) 는 다른 소 트웨어 시스템의 속성(attribute)을

- 234 -
향상시키기 하여 소 트웨어에 일어난 변경은 완성 (perfective) 유지보수이다. 변경
된 환경에 소 트웨어 시스템이 사용가능하도록 하기 한 소 트웨어 변경은 응
(adaptive) 유지보수 이다.
소 트웨어 시스템에 변경이 발생하는 경우 기 개발 는 배포 후 유지보수 과정
에서 변경되지 않은 소 트웨어의 부분이 반 향을 받지 않는다는 것을 증명하기
하여 충분한 회귀분석 시험이 수행되어야 한다. 이는 수행된 변경의 정확성
(correctness)을 평가하는 시험에 추가 인 것이다.
각 소 트웨어 변경을 하여 필요한 특정 밸리데이션 노력은 변경의 형태, 향 받는
개발 제품 소 트웨어 동작시 그 제품의 향에 따라 결정된다. 설계 구조의 철
하고 완벽한 문서화와 다양한 모듈, 인터페이스 등의 상호 계는 변경이 발생했을 때 필
요한 밸리데이션 노력을 한정할 수 있다.
변경에 한 충분한 밸리데이션을 하여 필요한 노력의 수 은 원본 소 트웨어의
밸리데이션이 문서화되고 수행된 정도에 따라 좌우된다. 를 들어 추후 회귀분석을
수행하기 하여 필요할 경우에는 시험 문서화, 시험사례 이 검증․밸리데이션 시험
의 결과는 수행될 필요가 있다. 추후 사용을 하여 이 정보를 보 하는데 실패하는
것은 노력의 수 변경 수행 후에 소 트웨어 유효성 재확인(revalidation) 하는 비
용을 크게 증가시킬 수 있다.
표 소 트웨어 개발 로세스의 일부분인 소 트웨어 검증 밸리데이션 작업에
추가하여 다음과 같은 유지보수 작업이 개정된 소 트웨어의 밸리데이션을 지원하기
하여 설정되어야 한다.
•소 트웨어 밸리데이션 계획 변경(Software Validation Plan Revision) - 이 에
밸리데이션된 소 트웨어에 하여 재 소 트웨어 밸리데이션 계획은 개정된 소
트웨어의 밸리데이션을 제공하기 하여 개정되어야 한다.
• 외사항 평가(Anomaly Evaluation) - 소 트웨어 조직은 발견된 소 트웨어 외
사항 각 외사항을 해결하기 하여 취해진 특정 시정조치를 설명하는 소 트
웨어 문제 보고서와 같은 문서화를 지속한다. 그러나 소 트웨어 개발자는 문제의
근본 원인을 결정하고 문제의 재발(recurrence) 방지를 하여 필요한 로세스
차상의 변경을 수행하기 한 그 다음 단계를 취하지 않아서 실수가 자주 반

- 235 -
복된다. 소 트웨어 외사항은 시스템 운 안 성에서 심각성(severity)과 효
과의 에서 평가되어야 한다. 그러나 이러한 외사항은 품질시스템에서 부
합(deficiency) 로세스의 증상으로 취 되어서는 아니 된다. 외사항의 근본 인
원인 분석은 특정 품질시스템 부 합을 명확히 할 수 있다. 경향이 식별되는( :
유사 소 트웨어 외사항의 재발) 곳에서 한 시정 방조치가 반드시 수
행되고 유사한 품질 문제의 재발을 방지하기 하여 문서화되어야 한다(21 CFR
820.100 참조)
•문제 식별 해결책 기록(Problem Identification and Resolution Tracking) -
소 트웨어 유지보수과정에서 발견된 모든 문제는 반드시 문서화되어야 한다. 각
문제 의 해결책은 참고이력 경향에 하여 그 문제가 수정되었음을 확실히
하기 하여 기록되어야 한다.
•제시된 변경 평가(Proposed Change Assessment) - 모든 제시된 수정
(modification), 향상(enhancement) 는 추가사항(addition)은 시스템에 하여 각
변경이 가져올 향을 결정하기 하여 평가되어야 한다. 이런 정보는 반복되기
하여 필요한 검증 / 는 밸리데이션 작업의 범 를 결정하여야 한다.
•작업 반복(Task Iteration) - 승인된 소 트웨어 변경에 하여 모든 필요한 검증
밸리데이션 작업은 계획된 변경이 정확하게 수행되고, 모든 문서화가 완료
최신으로 갱신(up to date)되고, 받아들이기 어려운 변경이 소 트웨어 성능에 일어
나지 않았음을 확실히 하기 하여 실시되어야 한다.
•문서화 갱신(Documentation Updating) - 문서화는 문서가 변경에 따라 여향을 받았
음을 결정하기 하여 주의 깊게 검토되어야 한다. 향 받게 되는 모든 승인된 문
서( : 규격, 시험 차, 사용자 설명서 등)는 형상 리 유지 로세스(configuration
management process)에 따라 갱신되어야 한다. 규격은 모든 유지보수 소 트웨
어 변경이 수행되기 에 갱신되어야 한다.
Section 6. 자동화 공정설비의 밸리데이션과 품질시스템 소 트웨어
품질 시스템 규정은 “컴퓨터 는 자동화 데이터 로세싱 시스템이 생산 는 품질
시스템의 일부로서 사용될 시 의료기기 제조업자는 설정된 로토콜에 따라 사용목
을 하여 컴퓨터 소 트웨어를 밸리데이션해야 한다”고 요구하고 있다(21 CFR
820.20.70(i) 참조). 이는 1978년 이후 FDA의 의료기기 GMP 규정의 법 요구사항이다.
의 밸리데이션 요구사항에 추가하여 의료기기 제조업자의 생산 로세스 는 품질

- 236 -
시스템( 는 모든 다른 FDA 규정에 의하여 요구되는 기록을 생성 유지하기 하여
필요한)의 일부분으로서 수행되는 컴퓨터 시스템은 자기록(Electronic Record); 자
서명(Electronic Signature) 규정을 필요로 한다(21 CFR Part 11 참조). 이 규정은 기록이
자 으로 생성 는 리될 때에 추가 인 보안, 데이터 통합 밸리데이션 요구사항
을 설정한다. 이 추가 인 Part 11 요구사항은 시스템 요구사항 시스템을 유지하는
모든 자동화된 기록에 한 소 트웨어 요구사항은 신 하게 고려되고 포함되어야 한
다. 시스템 밸리데이션 소 트웨어 밸리데이션은 모든 Part 11요구사항이 충족됨을
증명하여야 한다.
컴퓨터 자동화 장비는 의료기기 설계, 실험실 시험 분석, 제품 검사 수용,
생산 로세스 리, 환경 리, 포장, 라벨링, 추 성(traceability), 문서 리, 고객
불만 리의 모든 측면 품질시스템의 많은 다른 측면을 통하여 범 하게 사용
되어진다. 자동화 공장설비 운 (automated plant floor operation)은 다음과 같은 사항
에서 임베디드 시스템(embedded system)의 범 한 사용을 포함한다.
• 로그래머블 로직 컨트롤러(PLC, programmable logic controller)
•디지털 기능 컨트롤러(digital function controller)
•통계 로세스 제어(statistical process control)
•슈퍼바이 (supervisory, 운 체제의 심 부분에서 하드웨어의 능력을 최 한 활용
할 수 있도록 체계를 감시․제어하는 로그램) 제어 데이터 획득
•로보틱스(robotics)
•인간-기계 인터페이스(human-machine interface)
•입/출력기기
•컴퓨터 운 시스템
소 트웨어 도구는 자동화된 의료기기에 들어가는 소 트웨어를 설계, 제작(build)
시험하기 하여 자주 사용된다. 워드 로세서, 스 드시트, 데이터베이스
로 차트 소 트웨어와 같이 많은 다른 상업용 소 트웨어 용은 품질시스템을 수행
하기 하여 사용된다. 이 모든 용은 소 트웨어 밸리데이션을 한 요구사항을 필요
로 하지만 각 용을 하여 필요한 밸리데이션 근은 매우 범 할 수 있다.
제품 는 품질시스템 소 트웨어가 의료기기 제조업자에 의하여 자체 으로
(in-house) 개발, 계약자에 의한 개발 는 규격품(off-the-shelf) 구입일 경우 이 지침의
다른 부분에서 개략 으로 서술된 기본 인 원칙을 이용하여 개발되어야 한다. 의료기

- 237 -
기 제조업자는 그 소 트웨어의 밸리데이션이 어떻게 수행되었는지를 정의하는데 있어
범 융통성(flexibility)을 가지지만 밸리데이션은 어떻게, 구에 의하여 소 트웨
어가 개발될 것인지 는 구로부터 구매할 것인지를 결정하는데 요한 고려사항이
어야 한다. 소 트웨어 개발자는 life cycle 모델을 정의하여야 한다. 밸리데이션은 일반
으로 다음과 같은 사항에 의하여 지원된다.
•소 트웨어 개발 life cycle의 각 단계로부터 출력의 검증
•의료기기 제조업자의 사용 목 에서 완성된 소 트웨어의 한 운 에 한 확인
6.1.밸리데이션을 입증하는 자료는 얼마나 필요한가?(HOW MUCH VALIDATION
EVIDENCE IS NEEDED?)
밸리데이션 노력의 수 은 자동화 운 에 의하여 제시된 험에 하여야 한다. 험
성에 추가하여 소 트웨어 로세스의 복잡성과 의료기기 제조업자가 안 하고 효과
인 의료기기를 생산하기 한 자동화된 로세스에 의존하고 있는지의 정도와 같은
다른 요인들은 밸리데이션 노력의 일부분으로서 필요한 시험의 특징 범 를 결정한
다. 자동화 로세스의 문서화된 요구사항 험 분석은 소 트웨어가 사용목 을
하여 유효하다는 것을 증명하기 하여 필요한 증거의 범 를 결정한다. 를 들어
의료기기 제조업자가 운 의 출력이 배포 의 규격에 반하여 충분히 검증됨을 보여
수 있다면 자동화 링(milling) 기계는 아주 은 시험을 요구할 수 있다. 반면에 범
한 시험은 다음의 사항을 하여 필요할 수 있다.
•plant-wide 자기록 자서명 시스템
•멸균 주기를 한 자동화 컨트롤러
•생명유지/보조 의료기기의 완성된 회로보드의 검사 수용을 하여 사용되는 자
동화 시험장비
수많은 상업용 소 트웨어 용은 품질시스템( : 품질시스템 추측(calculation)을 하
여 사용되는 스 드시트 는 통계 패키지, 경향 분석을 하여 사용되는 그래픽 패
키지, 의료기기 이력 는 고객 불만 리 기록을 하여 사용되는 상업용 데이터베이스)
의 일부로서 사용될 수 있다. 이러한 소 트웨어를 하여 필요한 밸리데이션 증거의
범 는 제조업자의 문서화된 소 트웨어의 사용목 에 따른다. 를 들어 매업체가
제공한 소 트웨어의 모든 기능을 사용하지 않는 의료기기 제조업자는 사용될 소 트
웨어 기능과 생산 는 품질시스템의 일부로서 소 트웨어 결과에 의존하는 의료기기

- 238 -
제조업자를 해서만 밸리데이션이 필요하다. 그러나 고 험도의 응용 로그램은 비
록 그 소 트웨어 기능이 사용되지 않더라도 밸리데이션되지 않은 소 트웨어 기능을
가지고 동일한 운 환경에서 실행되어서는 아니 된다. 메모리 분할(memory
partitioning) 는 자원(resource) 보호 근과 같은 험 감소 기술(risk mitigation
technique)은 고 험도 응용 로그램 험도 응용 로그램이 동일 운 환경에
서 사용될 때 고려될 필요가 있을 수 있다. 소 트웨어가 갱신 는 어떤 변경이 소 트
웨어에 발생한 경우 의료기기 제조업자는 이 변경을 소 트웨어의 “사용되던 부분
(used portion)"에 어떤 향을 미치는지 고려하고 사용되는 소 트웨어의 그 부분의
유효성을 반드시 재확인 하여야 한다(21 CFR 820.70(i) 참조)
6.2. 규정된 사용자 요구사항
소 트웨어 밸리데이션을 하여 아주 요한 건은 다음과 같은 사항을 정의하는
규격화된 사용자 요구사항 규격이다.
•소 트웨어 는 자동화 장비의 “사용 목 ”
•우수한(quality) 의료기기의 생산을 하여 의료기기 제조업자가 소 트웨어 는
장비에 의존하는 범
의료기기 제조업자(사용자)는 모든 요구된 하드웨어 소 트웨어 형상
(configuration), 소 트웨어 버젼, 유틸리티(utility) 등을 포함한 상되는 운 환경을
정의하는 것이 필요하다. 한 사용자는 다음과 같은 것이 필요하다.
•시스템 성능, 품질, 오류 처리(handling), 시작(startup), 종료(shutdown), 보안 등을
한 문서 요구사항
•센서, 경보, 연동장치(인터락(interlock) : 진행 인 동작이 끝날 때까지 다음 동작
이 개시되지 않도록 하는 일(장치)), 논리 로세싱 단계 는 명령어 배열
(command sequence)과 같은 기능 는 특징과 련된 모든 안 성의 확인
•수용 가능한 성능을 결정하기 한 객 인 기 의 정의
밸리데이션은 반드시 문서화된 로토콜에 따라 수행되어야 하고 밸리데이션 결과
는 반드시 문서화 되어야 한다(21 CFR 820.70(i) 참조). 시험사례는 미리 정의된 기 (그
부분의 기(critical) 변수들을 하여)에 하여 그 성능을 조사하기 한 시스템을
수행할 것을 문서화하여야 한다. 시험사례는 오류 경보 조건, 시작, 종료, 모든 용
가능한 사용자 기능 운 자 제어, 잠재 인 운 자 오류, 인정되는 값의 최
최소 범 장비의 사용목 을 하여 용될 수 있는 가혹(stress) 조건을 확인하여

- 239 -
야 한다. 시험사례는 실행되어야 하고 소 트웨어가 그 사용목 을 하여 유효하다는
결론에 하여 그 결과가 지지하는지의 여부를 결정하기 하여 기록 평가되어야
한다.
의료기기 제조업자는 자체 인 인력을 이용하여 밸리데이션을 실시할 수 있고 장비
/소 트웨어 매업자 는 컨설턴트와 같은 제3자(소 트웨어나 주변장치 등의 제조
자)에 의지할 수도 있다. 어떤 경우든 의료기기 제조업자는 생산 품질시스템 소
트웨어가 다음과 같음을 확실히 하기 한 최종 인 책임을 유지하여야 한다.
•특정 사용목 을 한 문서화된 차에 따라 밸리데이션되는가;
•선택된 응용 로그램에서 의도한 로 운 할 것인가
의료기기 제조업자는 그 사용목 을 하여 소 트웨어가 밸리데이션되었음을 객
으로 확인할 수 있도록 다음 사항을 포함하여 문서화 하여야 한다.
•정의된 사용자 요구사항
•사용되는 밸리데이션 로토콜
•수용 기
•시험사례 결과
•밸리데이션 요약
6.3. OTS 소 트웨어 자동화 설비의 밸리데이션
의료기기 제조업자들에 의해 사용된 부분의 자동화 설비와 시스템은 제 3의 매
업체에 의해 제공되는 기성품(OTS; off-the-shelf)으로 구입되어 진다. 기기 제조업자는
OTS소 트웨어 개발자에 의해 사용되는 제품개발방법론(product development
methodologies)이 기기제조업자의 의도된 사용에 합하고 충분한지 확인을 한 책임
이 있다. OTS소 트웨어와 장비를 해 기기제조업자는 매업체의 소 트웨어 확인
문서에 근할 수도 있다. 만약 매업체가 그들의 시스템 요구사항, 확인 로세스와
확인결과에 한 정보를 제공할 수 있다면, 의료기기 제조업자는 밸리데이션을 한
문서화에 있어 출발 이 되는 정보를 활용할 수 있다. 검사 로토콜과 검사결과, 소스
코드, 설계명세서, 요구사항정의서와 같은 매업체의 life cycle 문서는 확인된 소 트
웨어 확립에 있어 사용이 가능하다.
기기제조업자는 매업자의 OTS소 트웨어의 구조에 있어 설계와 개발방법론 감사
를 반드시 고려해야 하고 OTS소 트웨어를 해 산출된 개발 확인문서를 평가하여

- 240 -
야 한다. 그러한 감사(audit)는 기기제조업자나 자격을 가진 제3의 기 에 의해 수행되
어질 수 있다. 이 감사는 반드시 매업체의 공정을 증명하여야 한다. 규정된 환경 내
에서 운 하는 것에 익숙하지 않은 몇몇 제조업체는 기기제조업자의 확인요구사항을 제
공할 수 있는 문서화된 life cycle 로세스를 가지고 있지 않을 수도 있다. 다른 매
업체는 감사를 수락하지 않을 수도 있다.
필요한 확인정보가 매업체로부터 가능하지 않을 경우, 기기제조업자는 소 트웨어
가 “사용자 필요성과 사용의도”를 확립하기 한 충분한 시스템단계 “black box" 검사
수행을 필요로 할 것이다. 많은 응용 로그램에 있어서의 black box 검사 하나로는 충
분하지 않다. 기기생산의 험요소여하로 만약 합한 다른 가능한 방법이 있다면, 로
세스 내 OTS 소 트웨어의 역할, 매업체를 감시할 수 있는 능력, 충분한 매업체
제공 정보, 소 트웨어의 사용 는 장비는 합할 수도 있고 그 지 않을 수도 있다.
기기제조업자는 한 지속 인 유지보수와 OTS소 트웨어 제품의 제공을 한 련사
항들을 고려해야 한다.
이를테면 소 트웨어 컴 일러, 링커, 에디터, 운 시스템 같은 몇몇의 OTS 소
트웨어 개발도구를 하여 기기제조업자에 의한 철 한 블랙박스 검사는 실용 이
아닐 수 도 있다. 그러한 검사(확인작업의 핵심요소)가 없다면 그것은 아마도 소 트웨
어 도구를 확인하기에는 불가능할 지도 모른다. 그러나 그것의 당한 운 은 다른
의미에 의해 만족하게 추론될 수도 있다. OTS 운 시스템은 독립된 로그램과 같이
확인되지 않을 필요가 있다.
그러나 응용소 트웨어의 시스템단계 확인검사는 최 로딩 조건, 일 운 , 시스템
에러조건의 핸들링, 그리고 메모리 리를 포함하여 반드시 사용된 모든 운 시스템
서비스를 제공하여야 한다.
제조 공정 소 트웨어 Validation에 한 더 자세한 사항은 부속서 A를 참고 하기
바란다.

- 241 -
부록 A 참고문헌(REFERENCES)
- Design Control Guidance for Medical Device Manufacturers, Center for Devices and
Radiological Health, Food and Drug Administration, March 1997.
- Do It by Design, An Introduction to Human Factors in Medical Devices, Center
for Devices and Radiological Health, Food and Drug Administration, March 1997.
- Electronic Records; Electronic Signatures Final Rule, 62 Federal Register 13430
(March 20, 1997).
- Glossary of Computerized System and Software Development Terminology,
Division of Field Investigations, Office of Regional Operations, Office of
Regulatory Affairs, Food and Drug Administration, August 1995.
- Guidance for the Content of Pre-market Submissions for Software Contained in
Medical Devices, Office of Device Evaluation, Center for Devices and Radiological
Health, Food and Drug Administration, May 1998.
- Guidance for Industry, FDA Reviewers and Compliance on Off-the-Shelf Software
Use in Medical Devices, Office of Device Evaluation, Center for Devices and
Radiological Health, Food and Drug Administration, September 1999.
- Guideline on General Principles of Process Validation, Center for Drugs and
Biologics, & Center For Devices and Radiological Health, Food and Drug
Administration, May 1987.
- Medical Devices; Current Good Manufacturing Practice (CGMP) Final Rule;
Quality System Regulation, 61 Federal Register 52602 (October 7, 1996).
- Reviewer Guidance for a Pre-Market Notification Submission for Blood
Establishment Computer Software, Center for Biologics Evaluation and Research,
Food and Drug Administration, January 1997
- Student Manual 1, Course INV545, Computer System Validation, Division of
Human Resource Development, Office of Regulatory Affairs, Food and Drug
Administration, 1997.
- Technical Report, Software Development Activities, Division of Field Investigations,
Office of Regional Operations, Office of Regulatory Affairs, Food and Drug
Administration, July 1987.
▪Other Government References
- W. Richards Adrion, Martha A. Branstad, John C. Cherniavsky. NBS Special

- 242 -
Publication 500-75, Validation, Verification, and Testing of Computer Software,
Center for Programming Science and Technology, Institute for Computer Sciences
and Technology, National Bureau of Standards, U.S. Department of Commerce,
February 1981.
- Martha A. Branstad, John C Cherniavsky, W. Richards Adrion, NBS Special
Publication 500-56, Validation, Verification, and Testing for the Individual
Programmer, Center for Programming Science and Technology, Institute for
Computer Sciences and Technology, National Bureau of Standards, U.S.
Department of Commerce, February 1980.
- J.L. Bryant, N.P. Wilburn, Handbook of Software Quality Assurance Techniques
Applicable to the Nuclear Industry, NUREG/CR-4640, U.S. Nuclear Regulatory
Commission, 1987.
- H. Hecht, et.al., Verification and Validation Guidelines for High Integrity Systems.
NUREG/CR-6293. Prepared for U.S. Nuclear Regulatory Commission, 1995.
- H. Hecht, et.al., Review Guidelines on Software Languages for Use in Nuclear
Power Plant Safety Systems, Final Report. NUREG/CR-6463. Prepared for U.S.
Nuclear Regulatory Commission, 1996.
- J.D. Lawrence, W.L. Persons, Survey of Industry Methods for Producing Highly
Reliable Software, NUREG/CR-6278, U.S. Nuclear Regulatory Commission, 1994.
- J.D. Lawrence, G.G. Preckshot, Design Factors for Safety-Critical Software,
NUREG/CR-6294, U.S. Nuclear Regulatory Commission, 1994.
- Patricia B. Powell, Editor. NBS Special Publication 500-98, Planning for Software
Validation, Verification, and Testing, Center for Programming Science and Technology,
Institute for Computer Sciences and Technology, National Bureau of Standards, U.S.
Department of Commerce, November 1982.
- Patricia B. Powell, Editor. NBS Special Publication 500-93, Software Validation,
Verification, and Testing Technique and Tool Reference Guide, Center for
Programming Science and Technology, Institute for Computer Sciences and
Technology, National Bureau of Standards, U.S. Department of Commerce,
September 1982.
- Delores R. Wallace, Roger U. Fujii, NIST Special Publication 500-165, Software
Verification and Validation: Its Role in Computer Assurance and Its Relationship
with Software Project Management Standards, National Computer Systems
Laboratory, National Institute of Standards and Technology, U.S. Department of

- 243 -
Commerce, September 1995.
- Delores R. Wallace, Laura M. Ippolito, D. Richard Kuhn, NIST Special Publication
500-204, High Integrity Software, Standards and Guidelines, Computer Systems
Laboratory, National Institute of Standards and Technology, U.S. Department of
Commerce, September 1992.
- Delores R. Wallace, et.al. NIST Special Publication 500-234, Reference Information
for the Software Verification and Validation Process. Computer Systems
Laboratory, National Institute of Standards and Technology, U.S. Department of
Commerce, March 1996.
- Delores R. Wallace, Editor. NIST Special Publication 500-235, Structured Testing: A
Testing Methodology Using the Cyclomatic Complexity Metric. Computer Systems
Laboratory, National Institute of Standards and Technology, U.S. Department of
Commerce, August 1996.
▪International and National Consensus Standards
- ANSI / ANS-10.4-1987, Guidelines for the Verification and Validation of Scientific
and Engineering Computer Programs for the Nuclear Industry, American National
Standards Institute, 1987.
- ANSI / ASQC Standard D1160-1995, Formal Design Reviews, American Society for
Quality Control, 1995.
- ANSI / UL 1998:1998, Standard for Safety for Software in Programmable
Components, Underwriters Laboratories, Inc., 1998.
- AS 3563.1-1991, Software Quality Management System, Part 1: Requirements.
Published by Standards Australia [Standards Association of Australia], 1 The
Crescent, Homebush, NSW 2140.
- AS 3563.2-1991, Software Quality Management System, Part 2: Implementation Guide.
Published by Standards Australia [Standards Association of Australia], 1 The
Crescent, Homebush, NSW 2140.
- IEC 60601-1-4:1996, Medical electrical equipment, Part 1: General requirements for
safety, 4. Collateral Standard: Programmable electrical medical systems.
International Electrotechnical Commission, 1996.
- IEC 61506:1997, Industrial process measurement and control – Documentation of
application software. International Electrotechnical Commission, 1997.
- IEC 61508:1998, Functional safety of electrical/electronic/programmable electronic

- 244 -
safety-related systems. International Electrotechnical Commission, 1998.
- IEEE Std 1012-1986, Software Verification and Validation Plans, Institute for
Electrical and Electronics Engineers, 1986.
- IEEE Standards Collection, Software Engineering, Institute of Electrical and Electronics
Engineers, Inc., 1994. ISBN 1-55937-442-X.
- ISO 8402:1994, Quality management and quality assurance – Vocabulary.
International Organization for Standardization, 1994.
- ISO 9000-3:1997, Quality management and quality assurance standards - Part 3:
Guidelines for the application of ISO 9001:1994 to the development, supply,
installation and maintenance of computer software. International Organization for
Standardization, 1997.
- ISO 9001:1994, Quality systems – Model for quality assurance in design,
development, production, installation, and servicing. International Organization for
Standardization, 1994.
- ISO 13485:1996, Quality systems – Medical devices – Particular requirements for
the application of ISO 9001. International Organization for Standardization, 1996.
- ISO/IEC 12119:1994, Information technology – Software packages – Quality
requirements and testing, Joint Technical Committee ISO/IEC JTC 1, International
Organization for Standardization and International Electrotechnical Commission,
1994.
- ISO/IEC 12207:1995, Information technology – Software life cycle processes, Joint
Technical Committee ISO/IEC JTC 1, Subcommittee SC 7, International
Organization for Standardization and International Electrotechnical Commission,
1995.
- ISO/IEC 14598:1999, Information technology – Software product evaluation, Joint
Technical Committee ISO/IEC JTC 1, Subcommittee SC 7, International
Organization for Standardization and International Electrotechnical Commission,
1999.
- ISO 14971-1:1998, Medical Devices – Risk Management – Part 1: Application of
Risk Analysis. International Organization for Standardization, 1998.
- Software Considerations in Airborne Systems and Equipment Certification. Special
Committee 167 of RTCA. RTCA Inc., Washington, D.C. Tel: 202-833-9339.
Document No. RTCA/DO-178B, December 1992.

- 245 -
▪Production Process Software References
- The Application of the Principles of GLP to Computerized Systems, Environmental
Monograph #116, Organization for Economic Cooperation and Development
(OECD), 1995.
- George J. Grigonis, Jr., Edward J. Subak, Jr., and Michael Wyrick, "Validation Key
Practices for Computer Systems Used in Regulated Operations," Pharmaceutical
Technology, June 1997.
- Guide to Inspection of Computerized Systems in Drug Processing, Reference
Materials and Training Aids for Investigators, Division of Drug Quality
Compliance, Associate Director for Compliance, Office of Drugs, National Center
for Drugs and Biologics, &Division of Field Investigations, Associate Director for
Field Support, Executive Director of Regional Operations, Food and Drug
Administration, February 1983.
- Daniel P. Olivier, "Validating Process Software", FDA Investigator Course: Medical
Device Process Validation, Food and Drug Administration.
- GAMP Guide For Validation of Automated Systems in Pharmaceutical Manufacture,
Version V3.0, Volume 1, Part 1: User Guide & Part 2: Supplier Guide & Volume
2: Best Practice for User and Suppliers
- Good Automated Manufacturing Practice (GAMP) Forum, March 1998: Technical
Report No. 18, Validation of Computer-Related Systems. PDA Committee on
Validation of Computer-Related Systems. PDA Journal of Pharmaceutical Science
and Technology, Volume 49, Number 1, January-February 1995 Supplement.
- Validation Compliance Annual 1995, International Validation Forum, Inc.
▪General Software Quality References
- Boris Beizer, Black Box Testing, Techniques for Functional Testing of Software and
Systems, John Wiley &Sons, 1995. ISBN 0-471-12094-4.
- Boris Beizer, Software System Testing and Quality Assurance, International
Thomson Computer Press, 1996. ISBN 1-85032-821-8.
- Boris Beizer, Software Testing Techniques, Second Edition, Van Nostrand Reinhold,
1990. ISBN 0-442-20672-0.
- Richard Bender, Writing Testable Requirements, Version 1.0, Bender &Associates,
Inc., Larkspur, CA 94777, 1996.
- Frederick P. Brooks, Jr., The Mythical Man-Month, Essays on Software

- 246 -
Engineering, Addison-Wesley Longman, Anniversary Edition, 1995. ISBN
0-201-83595-9.
- Silvana Castano, et.al., Database Security, ACM Press, Addison-Wesley Publishing
Company, 1995. ISBN 0-201-59375-0.
- Computerized Data Systems for Nonclinical Safety Assessment, Current Concepts
and Quality Assurance, Drug Information Association, Maple Glen, PA, September
1988.
- M. S. Deutsch, Software Verification and Validation, Realistic Project Approaches,
Prentice Hall, 1982.
- Robert H. Dunn and Richard S. Ullman, TQM for Computer Software, Second
Edition, McGraw-Hill, Inc., 1994. ISBN 0-07-018314-7.
- Elfriede Dustin, Jeff Rashka, and John Paul, Automated Software Testing –
Introduction, Management and Performance, Addison Wesley Longman, Inc., 1999.
ISBN 0-201-43287-0.
- Robert G. Ebenau and Susan H. Strauss, Software Inspection Process,
McGraw-Hill, 1994. ISBN 0-07-062166-7.
- Richard E. Fairley, Software Engineering Concepts, McGraw-Hill Publishing
Company, 1985. ISBN 0-07-019902-7.
- Michael A. Friedman and Jeffrey M. Voas, Software Assessment - Reliability,
Safety, Testability, Wiley-Interscience, John Wiley &Sons Inc., 1995. ISBN
0-471-01009-X.
- Tom Gilb, Dorothy Graham, Software Inspection, Addison-Wesley Publishing
Company, 1993. ISBN 0-201-63181-4.
- Robert B. Grady, Practical Software Metrics for Project Management and Process
Improvement, PTR Prentice-Hall Inc., 1992. ISBN 0-13-720384-5.
- Les Hatton, Safer C: Developing Software for High-integrity and Safety-critical
Systems, McGraw-Hill Book Company, 1994. ISBN 0-07-707640-0.
- Janis V. Halvorsen, A Software Requirements Specification Document Model for
the Medical Device Industry, Proceedings IEEE SOUTHEASTCON '93, Banking on
Technology, April 4th -7th, 1993, Charlotte, North Carolina.
- Debra S. Herrmann, Software Safety and Reliability: Techniques, Approaches and
Standards of Key Industrial Sectors, IEEE Computer Society, 1999. ISBN
0-7695-0299-7.
- Bill Hetzel, The Complete Guide to Software Testing, Second Edition, A

- 247 -
Wiley-QED Publication, John Wiley &Sons, Inc., 1988. ISBN 0-471-56567-9.
- Watts S. Humphrey, A Discipline for Software Engineering. Addison-Wesley
Longman, 1995. ISBN 0-201-54610-8.
- Watts S. Humphrey, Managing the Software Process, Addison-Wesley Publishing
Company, 1989. ISBN 0-201-18095-2.
- Capers Jones, Software Quality, Analysis and Guidelines for Success, International
Thomson Computer Press, 1997. ISBN 1-85032-867-6.
- J.M. Juran, Frank M. Gryna, Quality Planning and Analysis, Third Edition, ,
McGraw-Hill, 1993. ISBN 0-07-033183-9.
- Stephen H. Kan, Metrics and Models in Software Quality Engineering,
Addison-Wesley Publishing Company, 1995. ISBN 0-201-63339-6.
- Cem Kaner, Jack Falk, Hung Quoc Nguyen, Testing Computer Software, Second
Edition, Vsn Nostrand Reinhold, 1993. ISBN 0-442-01361-2.
- Craig Kaplan, Ralph Clark, Victor Tang, Secrets of Software Quality, 40
Innovations from IBM, McGraw-Hill, 1995. ISBN 0-07-911795-3.
- Edward Kit, Software Testing in the Real World, Addison-Wesley Longman, 1995.
ISBN 0-201-87756-2.
- Alan Kusinitz, "Software Validation", Current Issues in Medical Device Quality
Systems, Association for the Advancement of Medical Instrumentation, 1997. ISBN
1-57020-075-0.
- Nancy G. Leveson, Safeware, System Safety and Computers, Addison-Wesley
Publishing Company, 1995. ISBN 0-201-11972-2.
- Michael R. Lyu, Editor, Handbook of Software Reliability Engineering, IEEE
Computer Society Press, McGraw-Hill, 1996. ISBN 0-07-039400-8.
- Steven R. Mallory, Software Development and Quality Assurance for the
Healthcare Manufacturing Industries, Interpharm Press,Inc., 1994. ISBN
0-935184-58-9.
- Brian Marick, The Craft of Software Testing, Prentice Hall PTR, 1995. ISBN
0-13-177411-5.
- Steve McConnell, Rapid Development, Microsoft Press, 1996. ISBN 1-55615-900-5.
- Glenford J. Myers, The Art of Software Testing, John Wiley &Sons, 1979. ISBN
0-471-04328-1.
- Peter G. Neumann, Computer Related Risks, ACM Press/Addison-Wesley
Publishing Co., 1995. ISBN 0-201-55805-X.

- 248 -
- Daniel Olivier, Conducting Software Audits, Auditing Software for Conformance to
FDA Requirements, Computer Application Specialists, San Diego, CA, 1994.
- William Perry, Effective Methods for Software Testing, John Wiley &Sons, Inc.
1995. ISBN 0-471-06097-6.
- William E. Perry, Randall W. Rice, Surviving the Top Ten Challenges of Software
Testing, Dorset House Publishing, 1997. ISBN 0-932633-38-2.
- Roger S. Pressman, Software Engineering, A Practitioner's Approach, Third
Edition, McGraw-Hill Inc., 1992. ISBN 0-07-050814-3.
- Roger S. Pressman, A Manager’s Guide to Software Engineering, McGraw-Hill
Inc., 1993 ISBN 0-07-050820-8.
- A. P. Sage, J. D. Palmer, Software Systems Engineering, John Wiley &Sons, 1990.
- Joc Sanders, Eugene Curran, Software Quality, Addison-Wesley Publishing Co.,
1994. ISBN 0-201-63198-9.
- Ken Shumate, Marilyn Keller, Software Specification and Design, A Disciplined
Approach for Real-Time Systems, John Wiley &Sons, 1992. ISBN 0-471-53296-7.
- Dennis D. Smith, Designing Maintainable Software, Springer-Verlag, 1999. ISBN
0-387-98783-5.
- Ian Sommerville, Software Engineering, Third Edition, Addison Wesley Publishing
Co., 1989. ISBN 0-201-17568-1.
- Karl E. Wiegers, Creating a Software Engineering Culture, Dorset House
Publishing, 1996. ISBN 0-932633-33-1.
- Karl E. Wiegers, Software Inspection, Improving Quality with Software Inspections,
Software Development, April 1995, pages 55-64.
- Karl E. Wiegers, Software Requirements, Microsoft Press, 1999. ISBN 0-7356-0631-5.





























