Embed Size (px)
Citation preview

Facultad de Biología
Área de Fisiología
Departamento de Biología Funcional y Ciencias de la Salud
MODULACIÓN DE LOS CANALES DE POTASIO K2P POR PROTEÍNAS G Y
VÍAS DE SEGUNDOS MENSAJEROS EN NEURONAS SIMPÁTICAS DE
RATÓN EN CULTIVO
Tesis doctoral presentada por:
Paula Rivas Ramírez



Agradecimientos
V
AGRADECIMIENTOS
Parece que fue ayer cuando empecé a trabajar en el laboratorio, sin embargo ha pasado
mucho tiempo, durante el cual he aprendido y he cambiado tanto profesionalmente como
personalmente. Todo esto se lo debo a la gente que me rodea que me han enseñado cosas
nuevas día a día, al esfuerzo diario, a las ganas de aprender…pero también me han enseñado a
ser, me han servido como modelo a seguir, como profesionales a los que en ciertos aspectos
quiero semejarme. Aunque a veces a todos nos fallan las fuerzas, las ganas, la ilusión,…estos
son los valores que nos unen para trabajar y aprender cada día y que quiero agradecer a todas
las personas que me los han ofrecido, a veces sin darse cuenta.
En primer lugar quiero agradecer a mis directores de tesis: A José Antonio Lamas por
darme esta oportunidad y por confiar en mí, por su perfeccionismo y esmero diario y por
enseñarme a ser más ordenada. A Antonio Reboreda por explicármelo todo repetidas veces,
ayudarme siempre con todos los experimentos, las dudas… y tener una enorme paciencia.
Gracias a todos los compañeros que han pasado por el laboratorio. Aquellos que han
estado muchos años como Alba y Vanesa a las cuales le agradezco todo lo que me han
enseñado, tanto del trabajo del laboratorio como consejos vitales que te hacen más fuerte y
más independiente. A Diego, porque hemos empezado juntos el master y el doctorado y
estamos dando las últimas pinceladas casi a la par, porque ha sido un gran apoyo en los
momentos más difíciles y nos ha regalado muchos momentos divertidos.
Gracias a aquellas personas que han pasado un periodo de tiempo más corto en el
laboratorio realizando proyectos de máster y estancias. Especialmente a Natalia, porque a
pesar del poco tiempo que estuvo con nosotros, se ha convertido en una gran amiga que
transmite alegría, muchas ganas de trabajar y de aprender todos los días.
Gracias también a aquellas personas que aunque no pertenezcan ni hayan estado en el
laboratorio, me comprenden perfectamente cuando me fallan las fuerzas. A los chicos del
laboratorio “del fondo” y a todos los compañeros de congresos. Pero sobre todo a aquellos
verdaderos amigos que también han realizado tesis y que siempre han estado ahí: Nuria, Vero,
Dani, Carmen, Andrea, María, Lidia…por comprenderme y darme apoyo. Al grupo de
“comemos” porque todos los días las comida se convierten en una quedada con los amigos
donde desconectamos del trabajo y lo pasamos genial.

Agradecimientos
VI
Al resto de mis grupos de amigos por escucharme y entenderme cuando les hablo de mi
trabajo, que no es fácil, por animarme y comprenderme cuando no puedo quedar porque
“tengo que subir al Cuvi”.
Por último, y sobretodo quiero agradecer a toda mi familia. En especial a mis padres y a
mis hermanos, porque comprenden la importancia de mi trabajo, de las horas dedicadas,
siempre esperan, me comprenden y me apoyan incondicionalmente. Parte de esta tesis la
habéis “hecho” vosotros.
Gracias

VII
“La única forma de hacer un gran trabajo es amar lo que haces. Si todavía no lo has
encontrado sigue buscando, sabrás cuando lo hayas encontrado”. Steve Jobs.
A mi familia


Resumen
IX
RESUMEN
El potencial de reposo y la excitabilidad de las neuronas del ganglio cervical superior
(GCS) pueden ser modulados por agonistas muscarínicos y por bradiquinina. Clásicamente
este efecto era atribuido a la inhibición de la corriente M. El descubrimiento de la presencia
de canales TREK-2 en estas neuronas y su modulación a través de proteínas G, indican que el
canal TREK-2 también podría estar implicado en la modulación del potencial de membrana
en reposo y la excitabilidad.
En el presente trabajo se estudió mediante técnicas electrofisiológicas la modulación
muscarínica y por bradiquinina de la corriente de potasio a través de los canales TREK-2 en
neuronas del GCS de ratón en cultivo primario. Mediante la técnica de patch-clamp,
utilizando la modalidad de célula entera y la modalidad de canal individual, se determinó la
cascada de segundos mensajeros implicados en la modulación del canal TREK-2 por agentes
muscarínicos. Además se observó la implicación de este canal en el potencial de membrana en
reposo y la adaptación, en condiciones fisiológicas, utilizando protocolos de current-clamp y
voltage-clamp. Finalmente se comparó la contribución relativa de la corriente tipo M y de
TREK-2 a estos fenómenos.
Los resultados muestran que la corriente de potasio transportada a través de canales
TREK-2 es inhibida por el agonista muscarínico oxo-M y por la hormona bradiquinina,
siguiendo una vía de segundos mensajeros común, cuyo resultado final es el descenso de la
concentración de PIP2, cuya presencia es necesaria para mantener el canal activo. Por otro
lado hemos comprobado que los bloqueantes supuestamente selectivos para la corriente M,
linopirdina y XE991, también bloquean TREK-2 y ambos canales son sensibles a la
temperatura de forma similar. Por último este trabajo también nos indica que la contribución
relativa de ambos canales al valor del potencial de membrana en reposo a temperatura
fisiológica es similar.


Índice de Abreviaturas
XI
ÍNDICE DE ABREVIATURAS
2-APB: 2-aminoethoxydiphenylborano
4-AP: 4-amionopiridina
AC: Adenilato ciclasa
ACh: Acetilcolina
AMPc: Ácido adenosina-5fosfórico cíclico
B1: Solución de baño estándar
BK: Bradiquinina
CaM: Calmodulina
Canal BK: Big K+ channel
Canal SK: Small K+ channel
CHO: Chinese Hamster Ovary (Células derivadas de ovario de hamster chino)
CsCl: Cloruro de Cesio
DAG: Diacilglicerol
DMSO: Dimetil sulfóxido
EBSS: Earle´s Balanced Salt Solution
EGTA: Ethylene glycol tetraacetic acid
enº: Dominio extracelular
FCS: Suero bobino fetal (del inglés Fetal Calf Serum)
GCS: Ganglio Cervical Superior
GDP: Guanosin difosfato
GHK: Goldman-Hodgkin-Katz
GPCR: Receptores acoplados a proteínas G (del inglés G Protein Coupled Receptor)
GTP: Guanosin trifosfato
HBSS: Hank´s Balanced Salt Solution
HEPES: Ácido 4-(2-Hydroxyethyl)-1- piperazineethanesulfonico
I(-30): Corriente de las neuronas del GCS con el potencial de membrana fijado a -30 mV en presencia de cocktail
IC50: Dosis inhibitoria 50
IM: Corriente de potasio tipo M
inº: Dominio intracelular
IP3: Inositol 1,4,5-trifosfato
IRIL: Corriente activada por riluzol
ITEA: Corriente inhibida por TEA
IV: Intensidad-voltaje
IXE991: Corriente inhibida por XE991
K2P: Canales de potasio de doble dominio de poro (two pore domain potassium channels)
KOH: Hidróxido de potasio

Índice de Abreviaturas
XII
L-15: Medio Leibowitz
LHRH: Hormona liberadora de gonadotropina (del inglés Luteinizing-Hormone Realising Hormone)
NA: Noradrenalina
NGF: Factor de crecimiento nervioso (del inglés Nerve Growth Factor)
Oxo-M: Oxotremorina-M
PBS: Phosphate Buffered Saline
PCR: Reacción en cadena de la polimerasa
PIP2: Fosfatidilinositol 4,5-bifosfato
PI4K: Enzima fosfatidilinositol 4-quinasa
PKA: Proteinquinasa A
PKC: Proteinquinasa C
PLC: Fosfolipasa C
PTX: Toxina Pertussis
RE: Retículo endoplasmático
TALK: TWIK-related Alkaline pH activated K+ channel
TASK: TWIK-related Acid Sensitive K+ channel
TEA: Tetraetilamonio
THIK: Tandem-pore domain Halothane Inhibited K+ channel
TM: Segmento transmembrana
TREK: TWIK-Related K+ channel
TRESK: TWIK-Related Spinal cord K+ channel
TTX: Tetrodotoxina
TWIK: Tandem of P domain in Weak Inward rectifier K+ channel
XE991: 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone

Índice de Figuras
XIII
ÍNDICE DE FIGURAS
Figura 1: Ubicación del GCS
Figura 2: Inervación del GCS
Figura 3: Morfología de una neurona del GCS de ratón en cultivo primario
Figura 4: Sinapsis colinérgica
Figura 5: Esquema de la membrana neuronal
Figura 6: Estructura de una subunidad del canal de potasio tipo M
Figura 7: Estructura de tetraetilamonio (TEA), linopirdina y XE991
Figura 8: Efecto de linopirdina y XE991 sobre el potencial de membrana
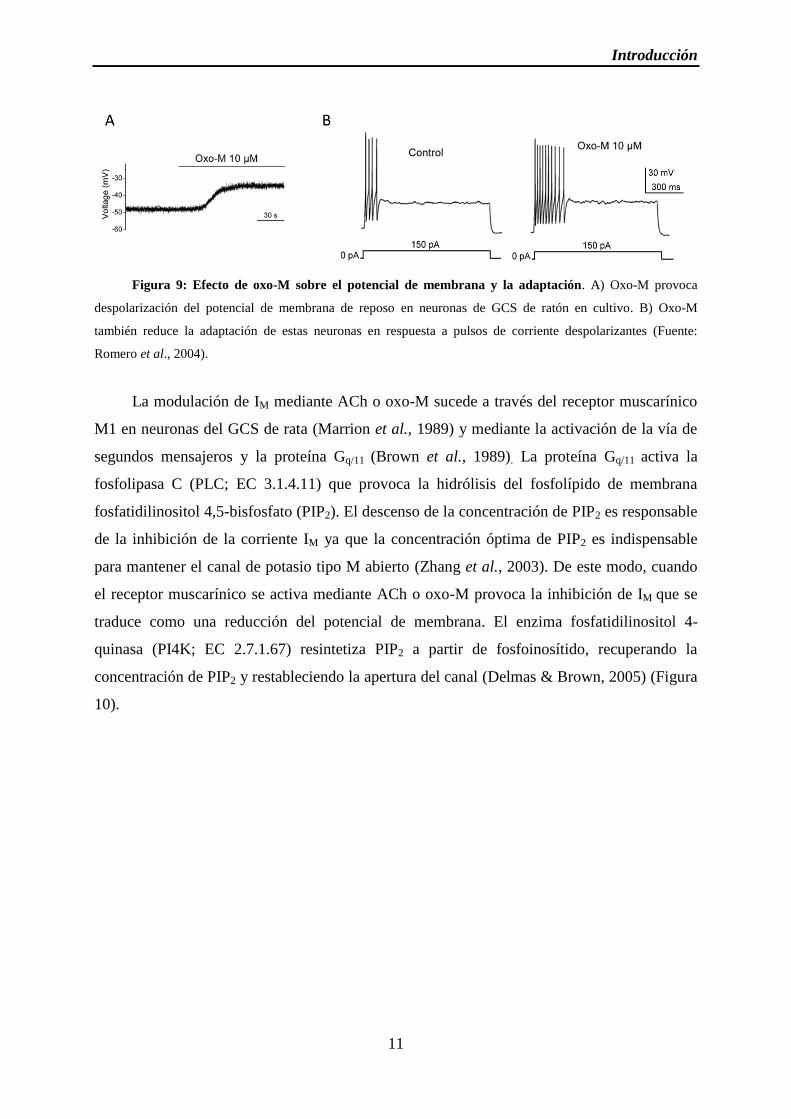
Figura 9: Efecto de oxo-M sobre el potencial de membrana y la adaptación
Figura 10: Posibles vías de regulación muscarínica y por BK del canal de potasio tipo M
Figura 11: Efecto de BK sobre la adaptación neuronal
Figura 12: Esquema de una subunidad y estructura tridimensional de un canal K2P
Figura 13: Estructura de riluzol
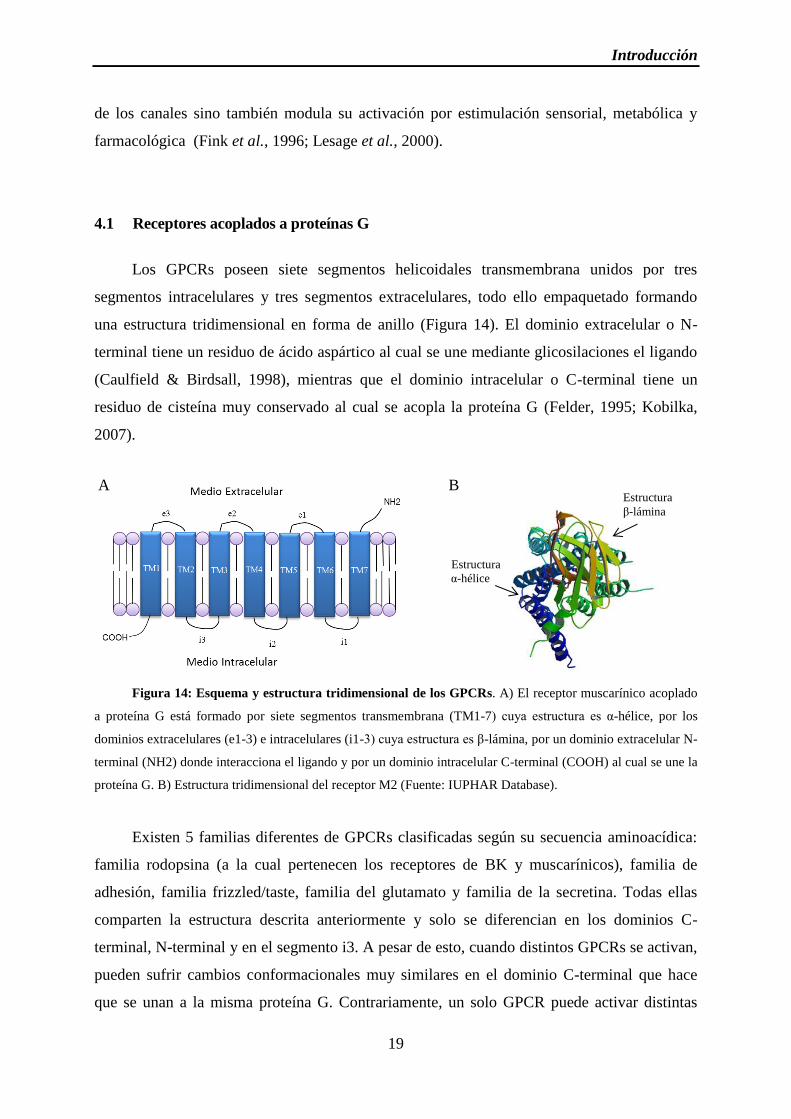
Figura 14: Esquema y estructura tridimensional de los GPCRs
Figura 15: Activación de la proteína G mediante GPCR
Figura 16: Estructura de acetilcolina, oxo-M y atropina
Figura 17: Posibles vías de regulación muscarínica de los canales de la subfamilia TREK
Figura 18: Estructura de BK
Figura 19: Posible vía de modulación por BK de la subfamilia TREK
Figura 20: Localización del GCS en el ratón
Figura 21: Esquema de los pasos de la extracción del GCS de ratón y el cultivo neuronal
Figura 22: Similitud de la membrana celular a un circuito eléctrico
Figura 23: Esquema de varias modalidades de la técnica patch-clamp
Figura 24: Esquema de las modalidades de patch-clamp utilizadas
Figura 25: Esquema del equipamiento utilizado para el registro electrofisiológico
Figura 26: Protocolo de current-clamp intensidad-voltaje (IV)
Figura 27: Protocolos de voltage-clamp de saltos y de rampas de voltaje
Figura 28: Protocolo de rampa de voltaje en la modalidad de canal individual
Figura 29: Esquema del protocolo utilizado para la aplicación del cocktail
Figura 30: Efecto del cocktail sobre la corriente basal registrada a -30 mV
Figura 31: Esquema del protocolo utilizado para la aplicación de riluzol
Figura 32: Efecto de riluzol sobre la neurona de GCS
Figura 33: Esquema del protocolo utilizado para la doble aplicación de riluzol
Figura 34: Efecto de la doble aplicación de riluzol
Figura 35: Esquema del protocolo utilizado para construir las curvas de activación
Figura 36: Curvas de activación de IRIL y de ITEA 3´
3´

Índice de Figuras
XIV
Figura 37: Esquema del protocolo utilizado para la aplicación de linopirdina y riluzol
Figura 38: Curva dosis respuesta de la inhibición de IRIL con linopirdina
Figura 39: Efecto de riluzol en presencia de linopirdina
Figura 40: Esquema del protocolo utilizado para la aplicación de XE991 y riluzol
Figura 41: Curva dosis respuesta de la inhibición de IRIL con XE991
Figura 42: Efecto de riluzol en presencia de XE991
Figura 43: Esquema del protocolo utilizado para la activación de las corrientes con la temperatura
Figura 44: Activación de las corrientes con la temperatura
Figura 45: La corriente ITEA e IXE991 son similares
Figura 46: Esquema del protocolo utilizado para la aplicación de oxo-M y riluzol
Figura 47: Efecto de riluzol en presencia de oxo-M
Figura 48: Esquema del protocolo utilizado para la aplicación de riluzol, atropina y oxo-M
Figura 49: Efecto de riluzol en presencia de oxo-M y atropina
Figura 50: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y pirenzepina
Figura 51: Efecto de riluzol en presencia de oxo-M y pirenzepina
Figura 52: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M e himbacina
Figura 53: Efecto de riluzol en presencia de himbacina y oxo-M
Figura 54: Porcentajes de inhibición de IRIL por oxo-M en presencia de antagonistas muscarínicos
Figura 55: Efecto de oxo-M y pirenzepina sobre la actividad de un canal TREK-2
Figura 56: Esquema del protocolo utilizado para la aplicación de riluzol y oxo-M en presencia de PTX
Figura 57: Efecto de riluzol en presencia de oxo-M y PTX
Figura 58: IRIL en presencia de oxo-M y PTX y porcentajes de inhibición de oxo-M
Figura 59: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y edelfosina
Figura 60: Efecto de riluzol en presencia de oxo-M y edelfosina
Figura 61: IRIL en presencia de edelfosina y oxo-M y porcentajes de inhibición de oxo-M
Figura 62: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y bisindolilmaleimida
Figura 63: Efecto de riluzol en presencia de oxo-M y bisindolilmaleimida
Figura 64: IRIL en presencia de oxo-M y bisindolilmaleimida y porcentajes de inhibición de oxo-M
Figura 65: Esquema del protocolo utilizado para la aplicación de riluzol, 2-APB y oxo-M
Figura 66: Efecto de riluzol en presencia de oxo-M y 2-APB
Figura 67: IRIL en presencia de oxo-M y 2-APB y porcentajes de inhibición de oxo-M
Figura 68: Esquema del protocolo utilizado para la aplicación de riluzol y wortmanina
Figura 69: Efecto de riluzol en presencia de wortmanina
Figura 70: Esquema del protocolo utilizado para la aplicación de riluzol y oxo-M en presencia de PIP2
Figura 71: Efecto de riluzol en presencia de oxo-M y PIP2
Figura 72: IRIL en presencia de PIP2 y oxo-M y porcentajes de inhibición de oxo-M
Figura 73: Vía de inhibición muscarínica de TREK-2
Figura 74: Esquema del protocolo utilizado para la aplicación de riluzol y BK
3´
3´
3´
3´
3´
3´
3´
3´
3´
3´

Índice de Figuras
XV
Figura 75: Efecto de riluzol en presencia de BK
Figura 76: Esquema del protocolo utilizado para la aplicación de riluzol, BK y oxo-M
Figura 77: Efecto de riluzol en presencia de oxo-M y BK
Figura 78: IRIL en presencia de oxo-M y BK y porcentajes de inhibición de oxo-M, de BK, y de oxo-M + BK
Figura 79: Esquema del protocolo utilizado para la aplicación de riluzol, bisindolilmaleimida y BK
Figura 80: Efecto de riluzol en presencia de bisindolilmaleimida y BK
Figura 81: IRIL en presencia de bisindolilmaleimida y porcentajes de inhibición de BK
Figura 82: Esquema del protocolo utilizado para la aplicación de riluzol, 2-APB y BK
Figura 83: Efecto de riluzol en presencia de BK y 2-APB
Figura 84: IRIL en presencia de BK y 2-APB y porcentajes de inhibición de BK
Figura 85: Esquema del protocolo utilizado para la aplicación de riluzol y BK en presencia de PIP2
Figura 86: Efecto de riluzol en presencia de BK y PIP2
Figura 87: IRIL en presencia de BK y PIP2 y porcentajes de inhibición de BK
Figura 88: Vía de inhibición por BK de TREK-2
Figura 89: Esquema del protocolo utilizado para la aplicación de oxo-M
Figura 90: Efecto de oxo-M sobre el potencial de membrana en reposo
Figura 91: Esquema del protocolo utilizado para la aplicación de TEA y XE991 a temperatura fisiológica
Figura 92: Efecto de TEA y de TEA + XE991 sobre el potencial de membrana en reposo
Figura 93: Esquema del protocolo utilizado para los experimentos de disparos de potenciales de acción
Figura 94: Número de potenciales de acción a temperatura ambiente, fisiológica y con XE991
Figura 95: Ejemplo de tren de disparos de potenciales de acción
Figura 96: Frecuencia y latencia a temperatura ambiente, a temperatura fisiológica y con XE991
Figura 97: Saltos de voltaje de corriente sensible a TEA a temperatura ambiente y fisiológica
Figura 98: Constante de tiempo de activación de IM a temperatura ambiente y fisiológica
Figura 99: Frecuencia y latencia a temperatura ambiente con XE99
3´
3´
3´
3´
3´
3´


ÍNDICE


Índice
XIX
INTRODUCCIÓN ..................................................................................................................... 1
1 Neuronas simpáticas del ganglio cervical superior ............................................................ 1
1.1 Sistema Nervioso Autónomo ....................................................................................... 1
1.2 Ganglio Cervical superior ............................................................................................ 1
1.3 Neuronas del GCS ....................................................................................................... 3
1.4 Potencial de membrana en reposo ............................................................................... 5
2 Corriente de potasio tipo M ................................................................................................ 8
3 Canales de doble dominio de poro ................................................................................... 13
3.1 Canales K2P en el GCS de ratón ............................................................................... 15
3.2 Subfamilia TREK ...................................................................................................... 16
4 Modulación mediante receptores acoplados a proteínas G .............................................. 18
4.1 Receptores acoplados a proteínas G .......................................................................... 19
4.2 Proteínas G ................................................................................................................ 20
4.3 Modulación muscarínica ............................................................................................ 21
4.4 Modulación mediante receptores de BK ................................................................... 25
4.5 Proteínas de anclaje y segundos mensajeros ............................................................. 27
4.6 Hipótesis de trabajo ................................................................................................... 28
OBJETIVOS ............................................................................................................................. 33
MATERIAL Y MÉTODOS ..................................................................................................... 37
1 Cultivo celular de neuronas de GCS ................................................................................ 37
1.1 Animales de experimentación ................................................................................... 37
1.2 Extracción del GCS de ratón ..................................................................................... 37
1.3 Cultivo primario de neuronas de GCS ....................................................................... 38
2 Registro electrofisiológico ............................................................................................... 39
2.1 Principios de electrofisiología ................................................................................... 39
2.2 Modalidades de patch-clamp ..................................................................................... 41
2.3 Electrodo de vidrio .................................................................................................... 44
2.4 Equipamiento ............................................................................................................. 45
2.5 Protocolos de registro ................................................................................................ 47
2.6 Soluciones de baño y pipeta ...................................................................................... 49
2.7 Análisis de datos y estadística ................................................................................... 50
RESULTADOS ........................................................................................................................ 55

Índice
XX
1 Distinción entre IM y la corriente de potasio de TREK-2................................................. 55
1.1 Riluzol aumenta la corriente de TREK-2 .................................................................. 55
1.1.1 La aplicación del cocktail inhibe la corriente de salida a -30 mV .................... 55
1.1.2 Riluzol incrementa la corriente de TREK-2 ....................................................... 56
1.1.3 IRIL no varía con la segunda aplicación ............................................................. 57
1.1.4 Las curvas de activación de IRIL y de ITEA son diferentes ................................... 58
1.2 Linopirdina y XE991 inhiben IRIL ............................................................................. 60
1.2.1 Linopirdina inhibe IRIL ....................................................................................... 60
1.2.2 XE991 inhibe IRIL ................................................................................................ 62
1.3 IM y la corriente de TREK-2 son sensibles a temperatura ......................................... 64
1.4 Resumen del apartado 1 ............................................................................................. 66
2 Modulación muscarínica de IRIL ....................................................................................... 66
2.1 Oxo-M inhibe IRIL ...................................................................................................... 67
2.2 La inhibición muscarínica sucede a través de M1 ..................................................... 68
2.2.1 La inhibición sucede a través de receptores muscarínicos ................................ 69
2.2.2 La inhibición muscarínica sucede a través de M1 ............................................. 70
2.2.3 La inhibición muscarínica no depende de M2/M4 ............................................. 72
2.2.4 Oxo-M inhibe la actividad individual de canales TREK-2 ................................ 75
2.3 La inhibición muscarínica sucede a través de proteínas G resistentes a PTX ........... 77
2.4 PLC está implicada en la inhibición muscarínica ...................................................... 80
2.5 PKC no participa en la inhibición muscarínica ......................................................... 82
2.6 El aumento de calcio no participa en la inhibición muscarínica ............................... 84
2.7 Implicación de PIP2 en la inhibición muscarínica ..................................................... 86
2.7.1 El descenso de la concentración de PIP2 inhibe IRIL ......................................... 86
2.7.2 La presencia de PIP2 bloquea la inhibición muscarínica .................................. 88
2.8 Resumen del apartado 2 ............................................................................................. 90
3 Modulación por bradiquinina de IRIL ................................................................................ 91
3.1 BK inhibe IRIL ............................................................................................................ 92
3.2 La inhibición muscarínica y por BK son oclusivas ................................................... 93
3.3 PKC no participa en la inhibición por BK ................................................................. 95
3.4 El aumento de calcio no participa en la inhibición por BK ....................................... 97
3.5 La presencia de PIP2 bloquea la inhibición por BK .................................................. 99
3.6 Resumen del apartado 3 ........................................................................................... 100
4 Implicación del canal TREK-2 en el potencial de membrana en reposo y la excitabilidad
neuronal .................................................................................................................................. 101

Índice
XXI
4.1 Implicación del canal TREK-2 en el potencial de membrana ................................. 102
4.1.1 La inhibición de TREK-2 no modifica el potencial de membrana a temperatura
ambiente ......................................................................................................................... 102
4.1.2 La inhibición de TREK-2 modifica el potencial de membrana a temperatura
fisiológica ....................................................................................................................... 103
4.2 Implicación del canal TREK-2 en la excitabilidad neuronal ................................... 104
4.2.1 TREK-2 e IM reducen la excitabilidad a temperatura fisiológica .................... 104
4.2.2 TREK-2 no afecta a la excitabilidad a temperatura ambiente......................... 108
4.3 Resumen del apartado 4 ........................................................................................... 110
DISCUSIÓN ........................................................................................................................... 115
CONCLUSIONES ................................................................................................................. 131
BIBLIOGRAFÍA .................................................................................................................... 135


INTRODUCCIÓN


Introducción
1
INTRODUCCIÓN
1 Neuronas simpáticas del ganglio cervical superior
1.1 Sistema Nervioso Autónomo
El sistema nervioso autónomo o vegetativo es el sistema encargado de controlar el
funcionamiento de los órganos de forma no voluntaria. Está distribuido entre el sistema
nervioso central y el sistema periférico. En este último se compone de las neuronas
preganglionares, los ganglios autónomos y las neuronas postganglionares.
El sistema nervioso autónomo se divide en sistema simpático y parasimpático. En el
sistema simpático los somas de las neuronas motoras preganglionares se encuentran en el asta
mediolateral de la médula espinal y hacen sinapsis en los ganglios paravertebrales y
prevertebrales, mientras que en la división parasimpática las neuronas motoras
preganglionares se encuentran en el tronco encefálico y en la médula sacra y realizan el relevo
sináptico en los ganglios torácicos o abdominales situados en las vísceras o próximo a ellas.
En ambas divisiones los axones de las neuronas preganglionares están mielinizados a
diferencia de las fibras postganglionares.
Las funciones del sistema nervioso autónomo simpático y parasimpático sobre los
órganos diana pueden ser antagónicas como ocurre sobre el músculo cardiaco, donde se
observa que el sistema simpático provoca un aumento de la frecuencia cardiaca mientras que
el parasimpático la disminuye; o sobre la pupila donde el sistema simpático activa la
contracción del musculo dilatador de la pupila mientras que el parasimpático activa el
músculo constrictor. Sin embargo, en otros órganos los efectos de estos sistemas son
sinérgicos, por ejemplo, la secreción de enzimas salivares se estimula con la activación del
sistema simpático, mientras que el sistema parasimpático controla el volumen de agua y de
electrolitos de la saliva. Otros órganos responden únicamente a aferencias simpáticas como
las glándulas sudoríparas, los vasos cutáneos y los músculos pilomotores, o únicamente a
aferencias parasimpáticas como las glándulas lacrimales (Aguilar et al., 1998).
1.2 Ganglio Cervical superior
El ganglio cervical superior (GCS) es el ganglio más rostral de un grupo de ganglios
llamados ganglios cervicales (superior, medio e inferior) que se encuentran en las cadenas

Introducción
2
paravertebrales a los lados de la columna vertebral. Este ganglio se localiza a nivel del cuello,
concretamente en la bifurcación de la arteria carótida (Figura 1).
El GCS pertenece a la división simpática del sistema nervioso autónomo. Inerva varios
órganos como el ojo donde provoca la vasoconstricción de las glándulas lacrimales
reduciendo la secreción lacrimal, también inerva el músculo dilatador de la pupila provocando
dilatación pupilar o midriasis, provoca la elevación del párpado superior, inerva los músculos
ciliares participando en mecanismos de acomodación en la visión lejana e inerva el lecho
vascular donde provoca vasoconstricción. Además, inerva la glándula parótida estimulando la
secreción de enzimas salivares y el músculo cardíaco estriado donde provoca el aumento de la
frecuencia cardíaca, la contractilidad y la vasoconstricción coronaria (Aguilar et al., 1998)
(Figura 2).
Figura 1: Ubicación del GCS. El GCS pertenece a la cadena de ganglios paravertebrales y se
encuentra en la bifurcación de la arteria carótida a nivel del cuello (Fuente: modificado de (Gray, 1918)).

Introducción
3
Figura 2: Inervación del GCS. El GCS inerva el músculo dilatador de la pupila, el párpado, las
glándulas lacrimales, la glándula parótida y el músculo cardíaco estriado (Fuente: modificado de (Aguilar et al.,
1998).
1.3 Neuronas del GCS
Las neuronas del GCS son bastante heterogéneas en tamaño, morfología de las dendritas
y perfil farmacológico. Tienen un tamaño medio de soma de aproximadamente 35 µm de
diámetro, pero la longitud de las dendritas puede alcanzar los 100 µm (Figura 3). Estas
neuronas se utilizan como modelo para el estudio del potencial de membrana de reposo y la
excitabilidad neuronal (Lamas et al., 2002) ya que han sido ampliamente estudiadas tanto en
mamíferos, sobre todo en rata, como en anfibios, por su tamaño, homogeneidad, resistencia y
fácil extracción. También son modelo para el estudio de la corriente de potasio tipo M (IM)
(Adams et al., 1982b; Brown & Selyanko, 1985; Brown et al., 1989; Romero et al., 2004) y
su modulación muscarínica (Brown et al., 1995; Romero et al., 2004). En nuestro laboratorio
se han utilizado estas neuronas como modelo para el estudio de los canales K2P (Cadaveira-
Mosquera et al., 2011, 2012). Además en estas neuronas también están presentes de forma
prominente los canales de potasio rectificadores tardíos, los de tipo A y los canales catiónicos
mixtos tipo H (Jobling & Gibbins, 1999).

Introducción
4
Figura 3: Morfología de una neurona del GCS de ratón en cultivo primario. Se observa el soma de la
célula y las proyecciones.
El valor del potencial de membrana de reposo en estas neuronas se encuentra en torno a
-60 mV y su capacidad en torno a 65 pF. Las neuronas del GCS se clasifican en fásicas
(disparan uno o pocos potenciales de acción y luego adaptan) y en tónicas (no adaptan).
Cuando se realiza un registro electrofisiológico del GCS intacto solamente el 29 % de las
neuronas adaptan, sin embargo cuando se registran estas neuronas en cultivo primario más del
80 % adaptan. Esto se debe a los cambios en la actividad y expresión de ciertos canales (como
por ejemplo el canal de potasio tipo M) durante la manipulación del ganglio y durante el
registro electrofisiológico (Springer et al., 2014).
Las neuronas preganglionares del sistema autónomo simpático liberan vesículas de ACh
al espacio sináptico donde se unirá a los receptores muscarínicos y nicotínicos de las neuronas
postsinápticas en el GCS (Figura 4). Las neuronas del GCS también presentan receptores para
bradiquinina (BK) que es un potente vasodilatador que induce y aumenta el dolor y la
inflamación. Ambas aferencias, colinérgicas y de BK, despolarizan ligeramente las neuronas
de este ganglio (Brown & Constanti, 1980; Babbedge et al., 1995; Jones et al., 1995).

Introducción
5
Figura 4: Sinapsis colinérgica. La sinapsis colinérgica en el GCS sucede entre los axones de una
neurona presináptica y las dendritas o el soma de una neurona postsináptica que forma parte del ganglio (Fuente:
(Koester et al., 2001)).
1.4 Potencial de membrana en reposo
Entre ambos lados de una membrana celular existe un gradiente electroquímico debido
a la distribución asimétrica de iones. Este gradiente electroquímico genera un potencial
eléctrico entre el exterior y el interior celular conocido como potencial de membrana. Cuando
las neuronas permanecen en reposo, es decir, cuando no están disparando potenciales de
acción, a esta diferencia de potencial se la llama potencial de membrana de reposo.
El valor del potencial de membrana de reposo, además de estar determinado por las
diferentes concentraciones de los iones a cada lado de la membrana celular y la distinta carga
de cada ión, también depende de la permeabilidad de la membrana a cada uno de ellos. El
valor del potencial de membrana en reposo suele tener un valor concreto y negativo para cada
neurona, si ese valor cambia hacia valores positivos y alcanza el umbral, normalmente la
neurona dispara un potencial de acción (Buño et al., 1998).
Los iones implicados en el mantenimiento del potencial de membrana de reposo son
principalmente el potasio, el sodio y el cloro. El ión potasio (K+) y el ión sodio (Na
+)
presentan carga positiva pero el primero se encuentra en mayor concentración en el interior
celular mientras que la concentración de sodio es mayor en el exterior. El ión cloro (Cl-)
presenta carga negativa y también suele encontrarse más concentrado en el exterior celular.

Introducción
6
Además de estos iones la presencia de aniones orgánicos impermeables y con carga negativa
en el citoplasma tienen gran influencia sobre el valor del potencial de membrana de reposo
(Buño et al., 1998) (Figura 5).
La permeabilidad de la membrana a un determinado ión depende del número y tipo de
canales abiertos por los cuales fluye dicho ión. En la mayoría de las neuronas de mamíferos
las membranas, en reposo, son más permeables al potasio que al sodio y al cloro, es decir, hay
más canales abiertos de potasio que de los otros iones y/o tienen una mayor conductancia
(Koester et al., 2001).
En este estado de reposo el ión potasio tiende a difundir hacia el exterior mientras que el
sodio tiende a entrar en la célula. Si esto ocurriera de forma constante, las concentraciones de
los iones tenderían a equilibrarse a ambos lados de la membrana reduciéndose la diferencia de
potencial. Para resolver esto, la membrana celular presenta una bomba electrogénica de
Na+/K
+ que constantemente introduce dos iones potasio por cada tres iones sodio que saca con
un gran consumo de energía en forma de ATP. Gracias a esta bomba las concentraciones
iónicas permanecen en equilibrio dinámico (Lamb & MacKinnon, 1971; Buño et al., 1998;
Lamas, 2005).
Figura 5: Esquema de la membrana neuronal. Se observa la diferente concentración de los iones K+,
Na+ y Cl
- y los aniones orgánicos a ambos lados de la membrana neuronal. También se representa la bomba de
Na+/K
+ y diferentes canales por los cuales los iones atraviesan la membrana.
El valor del potencial de equilibrio para un ión puede estimarse usando la ecuación de
Nernst que explica que cuando se alcanza el potencial de equilibrio el gradiente eléctrico de
cada ión (gradiente debido a la distinta proporción de cargas positivas y negativas a ambos
lados de la membrana) es igual pero de signo contrario que su gradiente químico (gradiente
debido a las distintas concentraciones del ión). Para calcular el valor del potencial de

Introducción
7
membrana, sabiendo que depende de los iones potasio, sodio y cloro en conjunto, se utiliza la
ecuación de Goldman-Hodgkin-Katz (GHK) que tiene en cuenta la concentración y la
permeabilidad relativa de la membrana a cada uno de ellos,
Vr=RT
Fln
PK [K ]e+PNa [Na]e+PCl [Cl]iPK [K ]i+PNa [Na ]i+PCl[Cl]e
donde Vr es el potencial de membrana de reposo (en voltios), R representa la constante
de los gases (8,314 JK-1
mol-1
), T es la temperatura absoluta en grados Kelvin , F es la
constante de Faraday (9,649 x 104
C mol-1
), P representa la permeabilidad de la membrana a
cada ión, []e y []i son las concentraciones extracelular e intracelular de cada ión
respectivamente (Goldman, 1943; Hodgkin & Katz, 1949).
Los canales implicados en el mantenimiento del potencial de membrana en reposo en
las neuronas del GCS pueden ser dependientes de voltaje como IM (Brown & Adams, 1980),
la corriente catiónica tipo H (Brown & Difrancesco, 1980) y la corriente de sodio persistente
(Wu et al., 2001; Rugiero et al., 2003) o corrientes independientes o ligeramente dependientes
de voltaje como los canales K2P (Cadaveira-Mosquera et al., 2011) y la bomba electrogénica
de Na+/K
+.
Canales dependientes de voltaje:
La corriente de potasio tipo M (IM) es una corriente de potasio que comienza a
abrirse a potenciales de membrana en reposo y carece de inactivación. Este canal tiene
una importante participación en el mantenimiento del potencial de membrana de
reposo, ya que cuando se bloquea provoca una ligera despolarización en el potencial
de membrana (Romero et al., 2004). Además se relaciona con la excitabilidad
neuronal ya que ACh, al igual que otros agonistas muscarínicos, y BK modulan el
potencial de membrana de reposo y la adaptación neuronal principalmente mediante la
inhibición de la corriente de potasio tipo M (Brown & Selyanko, 1985; Babbedge et
al., 1995; Jones et al., 1995).
La corriente H es una corriente catiónica mixta formada por iones de potasio y
de sodio. Estos canales permanecen abiertos a potenciales de reposo ya que se activan
con la hiperpolarización a potenciales de membrana de entre -30 y -40 mV, y se
cierran con la despolarización (Brown & Difrancesco, 1980). Se ha descubierto que

Introducción
8
esta corriente tiene un efecto despolarizante sobre el potencial de reposo de neuronas
del GCS de rata (Lamas, 1998)
La corriente de sodio persistente es una corriente dependiente de voltaje. Se
activa a potenciales cercanos al potencial de membrana de las neuronas del GCS (-60
mV) por lo que contribuye, con tendencia a la despolarización, al mantenimiento del
potencial de membrana de reposo (Wu et al., 2001; Rugiero et al., 2003; Lamas et al.,
2009).
Canales no dependientes o ligeramente dependientes de voltaje:
Los canales de doble dominio de poro o K2P son canales de potasio que están
abiertos a potenciales de membrana en reposo y son independientes de voltaje o
ligeramente dependientes. En apartados posteriores se desarrollan más profundamente
ya que son el objetivo de estudio de este trabajo.
Bomba electrogénica de Na+/K
+:
La bomba de Na+/K
+ es una proteína integrada en la membrana de las neuronas
que introduce dos iones K+ en el interior celular y extrae tres iones Na
+ en contra de
sus gradientes electroquímicos generando un efecto electrogénico. Por tanto la bomba
genera una corriente iónica neta hacia el exterior celular e independiente de voltaje,
que tiende a hiperpolarizar la membrana, modificando el valor del potencial de
membrana de reposo hacia valores más negativos (Koester et al., 2001; Lamas et al.,
2002).
2 Corriente de potasio tipo M
La corriente de potasio tipo M o IM en las neuronas del GCS es transportada
generalmente a través de un canal heteromérico compuesto por las subunidades KCNQ2 y
KCNQ3 que se ensamblan formando un canal funcional (Wang et al., 1998). El canal está
formado por dos subunidades KCNQ2 y dos subunidades KCNQ3. Cada subunidad está
compuesta por seis segmentos transmembrana y un dominio de poro (Figura 6). El cuarto
segmento transmembrana le ofrece la dependencia de voltaje ya que contiene aminoácidos
con carga positiva. Su dominio C-terminal, también llamado dominio A, se encuentra en el
interior celular y está implicado en la modulación del canal (Robbins, 2001).

Introducción
9
Figura 6: Estructura de una subunidad del canal de potasio tipo M. El canal está formado por cuatro
subunidades. Cada subunidad está formada por 6 segmentos transmembrana (M1-M6) y un dominio de poro (P).
Los dominios C-terminal (COOH) y N-terminal (NH2) son intracelulares.
Los canales de potasio tipo M comienzan a abrirse a partir de potenciales de membrana
alrededor de -60 mV, es decir, potenciales de membrana en reposo para las neuronas del GCS.
Estos canales alcanzan su máximo de activación a potenciales de -30 mV, se cierran con la
hiperpolarización, pero nunca inactivan (Brown & Selyanko, 1985). Su activación y
desactivación se producen de forma lenta lo que les proporciona una dependencia del tiempo
(Adams et al., 1982b). Estas características hacen que IM participe en la excitabilidad
neuronal y confiera adaptación ante estímulos despolarizantes a las neuronas del GCS
(Lamas, 2005). Por lo tanto la modulación del canal KCNQ2/KCNQ3 implica cambios en el
valor del potencial de membrana de reposo pero también en la excitabilidad y la adaptación
neuronal.
La IM es sensible al bloqueante clásico de canales de potasio tetraetilamonio (TEA)
(Figura 7), el cual provoca, en las neuronas del GCS de rata, una ligera despolarización del
potencial de membrana, sin afectar a la adaptación de las neuronas a un estímulo (Brown &
Constanti, 1980; Hadley et al., 2003). El bario también se considera un bloqueante clásico de
IM (Adams et al., 1982a). En nuestro laboratorio se ha observado que Ba++
provoca una
despolarización de aproximadamente 9 mV y reduce la adaptación en las neuronas del GCS
de ratón debido a la inhibición de esta corriente (Romero et al., 2004).
Linopirdina (DuP996) y su análogo XE991 (10,10-bis (4-pyridinylmethyl)-9(10H)-
anthracenone) (Figura 7) son considerados bloqueantes relativamente específicos de los
canales de potasio tipo M (Lamas et al., 1997; Wang et al., 1998). Ambas sustancias
provocan una ligera despolarización del potencial de membrana de reposo (Romero et al.,
2004) y altera el patrón de disparo (Aiken et al., 1995) (Figura 8). La inhibición de IM

Introducción
10
mediante linopirdina es independiente de la concentración de calcio intracelular y de las
proteínas G, lo que sugiere que linopirdina podría bloquear el canal desde la parte extracelular
y directamente o a través de una proteína asociada (Lamas et al., 1997; Costa & Brown,
1998).
Figura 7: Estructura de tetraetilamonio (TEA), linopirdina y XE991. Las tres sustancias son
bloqueantes de IM (Fuente: Sigmaaldrich.com).
Figura 8: Efecto de linopirdina y XE991 sobre el potencial de membrana. Linopirdina (A) y XE991
(B) provocan una ligera despolarización del potencial de membrana en reposo de las neuronas del GCS de ratón
(Fuente: Romero et al., 2004).
La IM, al igual que el potencial de membrana en reposo y la excitabilidad neuronal, se
modula mediante aferencias colinérgicas. La aplicación del agonista muscarínico
oxotremorina-M (oxo-M) despolariza el potencial de membrana y reduce la adaptación
provocando un incremento en la duración del tren de potenciales de acción en respuesta a
pulsos de corriente despolarizantes, como se observa en la figura 9 (Romero et al., 2004).
TEA Linopirdina XE991
A B
Introducción
11
Figura 9: Efecto de oxo-M sobre el potencial de membrana y la adaptación. A) Oxo-M provoca
despolarización del potencial de membrana de reposo en neuronas de GCS de ratón en cultivo. B) Oxo-M
también reduce la adaptación de estas neuronas en respuesta a pulsos de corriente despolarizantes (Fuente:
Romero et al., 2004).
La modulación de IM mediante ACh o oxo-M sucede a través del receptor muscarínico
M1 en neuronas del GCS de rata (Marrion et al., 1989) y mediante la activación de la vía de
segundos mensajeros y la proteína Gq/11 (Brown et al., 1989). La proteína Gq/11 activa la
fosfolipasa C (PLC; EC 3.1.4.11) que provoca la hidrólisis del fosfolípido de membrana
fosfatidilinositol 4,5-bisfosfato (PIP2). El descenso de la concentración de PIP2 es responsable
de la inhibición de la corriente IM ya que la concentración óptima de PIP2 es indispensable
para mantener el canal de potasio tipo M abierto (Zhang et al., 2003). De este modo, cuando
el receptor muscarínico se activa mediante ACh o oxo-M provoca la inhibición de IM que se
traduce como una reducción del potencial de membrana. El enzima fosfatidilinositol 4-
quinasa (PI4K; EC 2.7.1.67) resintetiza PIP2 a partir de fosfoinosítido, recuperando la
concentración de PIP2 y restableciendo la apertura del canal (Delmas & Brown, 2005) (Figura
10).

Introducción
12
Figura 10: Posibles vías de regulación muscarínica y por BK del canal de potasio tipo M. Ambas
sustancias, BK y ACh, activan la proteína Gq/11 que a su vez activa la PLC y ésta hidroliza PIP2 en DAG e IP3.
La inhibición muscarínica de IM se debe a la reducción de los niveles de PIP2, mientras que la inhibición por BK
se debe al incremento en la concentración de calcio citosólico provocado por IP3 en el retículo endoplasmático
(Fuente: Delmas & Brown, 2005).
Se ha visto en neuronas del GCS de rata que BK inhibe IM y provoca una ligera
despolarización del potencial de membrana en reposo y reduce la adaptación en estas células
(Figura 11), suponiendo un aumento en la excitabilidad del GCS de rata (Jones et al., 1995).
Figura 11: Efecto de BK sobre la adaptación neuronal. Se ha observado que la adaptación se reduce
en neuronas de GCS de rata debido a la inhibición de IM por BK (Fuente: Jones et al., 1995).
Esta inhibición por BK del canal de potasio tipo M no depende de la reducción de la
concentración de PIP2, como sucede durante la inhibición muscarínica de dicho canal, sino
que se ha visto que esta inhibición es causa del aumento en los niveles de calcio intracelular.
Control BK 10nM

Introducción
13
En este caso, tras la hidrolisis de PIP2 en DAG e IP3, el IP3 provoca la salida de calcio del
retículo endoplasmático y por consiguiente el aumento de la concentración de calcio. A
diferencia de la inhibición muscarínica, en la inhibición por BK el aumento del calcio
intracelular estimula la síntesis de PIP2, a través de PI4K, impidiendo la reducción de la
concentración de PIP2. Así la inhibición de la IM por BK se debe al aumento en la
concentración de calcio y no a la reducción de la concentración de PIP2 (Cruzblanca et al.,
1998; Delmas & Brown, 2005; Hernandez et al., 2008) (Figura 10).
Clásicamente se considera a IM como la corriente de potasio más importante en el
control de la excitabilidad neuronal en el GCS, ya que estos canales están abiertos a
potenciales de membrana en reposo, se modulan mediante agonistas muscarínicos y por BK y
su inhibición implica cambios en el potencial de membrana en reposo y en la excitabilidad.
Sin embargo, el descubrimiento de los canales de doble dominio de poro en 1996 (Lesage et
al., 1996) y su presencia en las neuronas del GCS (Cadaveira-Mosquera et al., 2012) pone en
duda la exclusividad de IM como la única corriente de potasio capaz de modular, mediante
aferencias colinérgicas y de BK, el potencial de membrana en reposo y la excitabilidad de las
neuronas del GCS.
3 Canales de doble dominio de poro
Si bloqueamos por completo todas las corrientes dependientes de voltaje mencionadas
anteriormente quedaría una corriente que se ha denominado corriente de fuga (Lamas et al.,
2002). Previo al descubrimiento de los canales K2P, la corriente de fuga era considerada
como una corriente independiente de voltaje o muy poco dependiente, que no se bloquea con
los bloqueantes clásicos que afectan a las corrientes de potasio y cuyo potencial de inversión
se acerca más al potencial de equilibrio del potasio que a los de los iones cloro y sodio
(Lamas, 2005). Desde el descubrimiento de los canales K2P o de doble dominio de poro
(Lesage et al., 1996; Lesage & Lazdunski, 2000) se planteó que pueden ser los responsables
de esta corriente de fuga ya que sus características se corresponden con las anteriormente
mencionadas.
La familia K2P de mamíferos está compuesta por 6 subfamilias codificadas por quince
genes. Estas subfamilias se clasifican según sus características físicas y químicas en TWIK
(Tandem of P domain in Weak Inward rectifier K+ channel), TREK (TWIK-Related K
+
channel), TASK (TWIK-related Acid Sensitive K+ channel), TALK (TWIK-related Alkaline

Introducción
14
pH activated K+ channel), THIK (Tandem-pore domain Halothane Inhibited K
+ channel) y
TRESK (TWIK-Related Spinal cord K+ channel).
Los canales K2P tienen características distintas al resto de los canales de potasio. Una
de esas características es que, por lo general, son insensibles a bloqueantes clásicos de canales
de potasio como TEA y 4-AP (4-amionopiridina), excepto TASK-2 y TRESK que son
sensibles a TEA (Lesage, 2003; Liu et al., 2004).
Se ha visto que algunos miembros de la familia K2P son bloqueados de forma voltaje-
dependiente con altas concentraciones de Ba+2
extracelular (a partir de 1 mM (Fink et al.,
1998; Cadaveira-Mosquera et al., 2011)), por ciertas sustancias como quinidina y quinina y
algunos canales K2P se inhiben con mibefradil (Czirják & Enyedi, 2006).
Ciertos canales K2P también son modulados por anestésicos generales volátiles como
cloroformo, halotano, isoflurano y dietil éter. Se ha visto que estos anestésicos utilizados a
concentraciones efectivas para el ser humano provocan la activación de los canales TREK-1 y
TREK-2 de forma directa e independiente de segundos mensajeros (Lesage et al., 2000). Sin
embargo el cloroformo y el dietil éter inhiben ligeramente el canal TASK, mientras que el
halotano provoca la apertura de este canal de forma rápida y reversible.
En general los canales K2P permanecen activos a todos los potenciales de membrana y
no inactivan con el voltaje, a excepción de TWIK-2 y TRESK (Lotshaw, 2007). La corriente
macroscópica de los canales K2P muestra rectificación de canal abierto, es decir la
conductancia del canal no sigue una línea recta como predice la ecuación de GHK en
condiciones fisiológicas debido a la diferencia de concentraciones de potasio entre el interior
y el exterior celular, sin embargo en condiciones simétricas de concentración de potasio la
conductancia seguiría una línea recta ya que estos canales apenas presentan rectificación por
voltaje (Goldstein et al., 2001; Lamas, 2012).
La estructura de los canales K2P es singular, ya que a diferencia de la mayoría de los
canales de potasio, éstos están formados por dos subunidades con 4 segmentos
transmembrana y dos dominios de poro cada una, denominados M1, M2, M3, M4, P1 y P2
respectivamente (Figura 12). El primer dominio de poro se encuentra entre M1 y M2 y
contiene de una a varias asparaginas implicadas en la glicosilación de este dominio y de una a
varias cisteínas que forman puentes entre las subunidades. El segundo dominio de poro se
encuentra entre M3 y M4 (Lesage et al., 1996). Los dominios C-terminal y N-terminal son
citosólicos. El C-terminal juega un importante papel en la modulación de la apertura del canal
mediante fosforilación por parte de proteínas citosólicas como por ejemplo el enzima

Introducción
15
proteinquinasa A (PKA; EC 2.7.11.11) y fosfoquinasa C (PKC; EC 2.7.11.13) (Fink et al.,
1996; Lesage & Lazdunski, 2000).
Figura 12: Esquema de una subunidad y estructura tridimensional de un canal K2P. A) Cada
subunidad está formada por 4 segmentos transmembrana (M1-M4) y dos dominios de poro (P1 y P2). Los
dominios C-terminal y N-terminal son intracelulares. Estas subunidades se dimerizan para formar el canal
funcional. B) Estructura tridimensional del canal TRAAK (Lolicato et al., 2014).
En la mayoría de las subfamilias de los canales K2P la dimerización de las dos
subunidades del canal se produce entre los residuos de cisteína de los segmentos M1-P1
mediante puente disulfuro (Lesage et al., 1996), sin embargo en la subfamilia TASK, el
miembro TASK-1 se cree que dimeriza entre los dominios M1 de cada subunidad (Hsu et al.,
2004). Algunas subunidades distintas se combinan entre ellas para formar heterodímeros
como por ejemplo TASK-1/TASK-3 (Czirják & Enyedi, 2002).
3.1 Canales K2P en el GCS de ratón
Estudios realizados en neuronas del GCS de ratón han demostrado, mediante técnicas de
inmunocitoquímica, la presencia de las subfamilias TREK (TREK-1, TREK-2 y TRAAK),
TRESK, TALK (TASK-2) y TASK (TASK-1 y TASK-3). La expresión de estas subfamilias
se ha podido determinar mediante PCR cuantitativa y estudio de las células que presentan
actividad de dichos canales mediante la técnica de registro de canal individual. Se ha
observado que la subfamilia TRESK es la más expresada seguido de TREK. Dentro de la
subfamilia TREK se expresa en mayor proporción TREK-2 (70%), seguido de TREK-1 (6%)
y en menor proporción TRAAK (2%), estos porcentajes de expresión son relativos al canal
B A

Introducción
16
TRESK cuya expresión se considera el 100%, ya que es el canal K2P más expresado en este
tipo neuronal (Cadaveira-Mosquera et al., 2012).
3.2 Subfamilia TREK
La subfamilia TREK está formada por los miembros TREK-1, TREK-2 y TRAAK. Esta
subfamilia presenta una apertura, ligeramente dependiente de voltaje, que es modulada por
estímulos físicos y químicos lo que les permite que sean además canales importantes en
procesos de transducción de estímulos térmicos, mecánicos, químicos y dolorosos. Además
son dianas de anestésicos, neuroprotectores y fármacos para la depresión (Franks & Honoré,
2004; Lamas, 2012).
La subfamilia TREK se distingue del resto de los canales K2P por su
mecanosensibilidad. De hecho se ha visto que estos canales se activan cuando se ejerce una
presión negativa sobre la membrana celular. Esta estimulación del canal no es permanente y el
canal puede sufrir desensibilización durante el mantenimiento del estímulo mecánico (Patel et
al., 1998; Maingret et al., 1999). Esta sensibilidad a estímulos mecánicos permanece aun
cuando se eliminan moléculas y segundos mensajeros citosólicos, sin embargo está
relacionado con el cuarto dominio transmembrana y el dominio C-terminal (Noël et al.,
2011).
El pH citosólico también modula la actividad espontánea de estos canales, pero la
respuesta de TREK-1 y TREK-2 es diferente a la de TRAAK. Los dos primeros son activados
mediante la acidificación del medio intracelular (pH 6,0 y pH<7,0 respectivamente), mientras
que TRAAK es estimulado por la alcalinización y no se ve afectada por la acidificación
teniendo su máxima actividad a pH>7,3 (Patel et al., 1998; Maingret et al., 1999). La
acidificación citosólica se puede conseguir mediante la adición de bicarbonato o de
dinitrofenol (DNP) en el medio externo y esta acidificación hace que sean más susceptibles a
activarse por estiramiento y por presión sobre la membrana celular (Fakler et al., 1996;
Honoré et al., 2002). La sensibilidad de TREK-1 a cambios de pH intracelular se ha asociado
al aminoácido glutamina 306 del dominio C-terminal, cuya mutación conlleva a la
permanente activación del canal TREK-1 (Noël et al., 2011).
Al igual que la mecanosensibilidad, la termosensibilidad también hace exclusiva a la
subfamilia TREK con respecto al resto de las subfamilias de K2P. Tanto en sistemas
heterólogos como en distintos tipos celulares, los cambios de temperatura modulan la
actividad de los canales TREK-1, TREK-2 y TRAAK (De la Peña et al., 2012; Maingret et

Introducción
17
al., 2000; Kang et al., 2005), de tal modo que a temperatura fisiológica estos canales están
muy activados y su control sobre el potencial de membrana es notable, respecto a la
temperatura ambiente a la cual se realizan habitualmente los experimentos de patch de
cultivos celulares (Noël et al., 2011). Este aumento de la actividad con la temperatura se debe
al aumento de la probabilidad de apertura, sin cambios en la conductancia de los canales
(Kang et al., 2005). La termosensibilidad también se relaciona con el dominio C-terminal y en
este caso se requiere la integridad celular, esto sugiere que la termosensibilidad podría
requerir proteínas auxiliares para poder manifestarse (Noël et al., 2011).
Los canales de la subfamilia TREK son estimulados mediante ciertos tipos de lípidos
como los ácidos grasos poliinsaturados y los fosfolípidos. Por ejemplo el ácido araquidónico,
que actúa como un neuroprotector reduciendo la muerte neuronal durante procesos
isquémicos (Lauritzen et al., 2000) activa los canales TREK-1, TREK-2 (Lesage et al., 2000)
y TRAAK (Fink et al., 1998) reversiblemente. Otros ácidos grasos insaturados como oleato,
linoleato, eicosapentaenoato y docosahexanoato también activan estos canales, sin embargo
los ácidos grasos saturados como palmitato, estearato y araquidato no tienen ningún efecto
sobre ellos (Lesage & Lazdunski, 2000). Lo mismo sucede con el PIP2 que, gracias a su
proximidad al canal TREK-1, aumenta la probabilidad de apertura del canal y por tanto la
corriente de potasio a través de éste (Chemin et al., 2005).
Otro estímulo químico que afecta a los canales TREK son ciertos anestésicos generales
como hemos mencionado antes. Los anestésicos volátiles (halotano, isoflurano y cloroformo)
aumentan la corriente de potasio a través de los canales TREK-1 y TREK-2 pero no afectan a
TRAAK (Patel et al., 1999). Esta similitud de respuesta a los anestésicos volátiles entre los
canales TREK-1 y TREK-2 y la diferencia con el canal TRAAK puede deberse a la estructura
de los canales ya que se observó en el canal TREK-1 que el extremo C-terminal es crucial
para la activación por anestésicos volátiles (Patel & Honoré, 2001).
Desde un punto de vista funcional los canales TREK están relacionados con la
neuroprotección, se ha visto que pueden contribuir a la protección de las neuronas durante
isquemias y convulsiones ya que estos canales contribuyen a mantener el potencial de
membrana en reposo y reducen la excitabilidad neuronal. Además se ha observado que el
agente neuroprotector riluzol (2-amino-6-trifluoromethoxy benzothiazole) (Figura 13),
utilizado para el tratamiento de la esclerosis lateral amiotrófica (Bensimon et al., 1994) y con
propiedades anticonvulsionante, sedante y antihisquémica (Malgouris et al., 1989), activa
exclusivamente, dentro de los canales K2P, a la subfamilia TREK. En sistemas hererólogos

Introducción
18
(Duprat et al., 2000; Lesage et al., 2000) y en neuronas del GCS de ratón (Cadaveira-
Mosquera et al., 2011) se ha demostrado que dicha activación, en el caso de los canales
TREK-1 y TREK-2, se muestra en dos fases, una activación seguida por una ligera inhibición.
La fase inhibitoria se atribuye al hecho de que el riluzol inhibe la actividad de la AMPc
fosfodiesterasa (EC 3.1.4.53) resultando un incremento en la concentración de AMPc y un
aumento en la fosforilación mediada por proteinquinasas lo que provoca la inhibición de
TREK-1 y TREK-2. TRAAK sin embargo, no muestra inhibición por AMPc y la estimulación
por riluzol es sostenida (Duprat et al., 2000).
Figura 13: Estructura de riluzol. Esta sustancia activa exclusivamente a la subfamilia TREK de las seis
subfamilias de K2P (Fuente: Sigmaaldrich.com).
En el GCS de ratón la corriente activada por riluzol es transportada a través de TREK-2
ya que riluzol provoca la activación de una corriente de potasio de forma no sostenida, cuyo
comportamiento en experimentos de canal individual es similar a la descrita para el canal
TREK-2 (Cadaveira-Mosquera et al., 2011). Esta corriente de potasio activada por riluzol
(IRIL) presenta una importante rectificación de salida (Cadaveira-Mosquera et al., 2011) como
se ha descrito para el canal TREK-1 y TRAAK expresados en oocitos de Xenopus laevis
(Fink et al., 1996, 1998) y para el canal TREK-2 expresado en células COS (Lesage et al.,
2000).
4 Modulación mediante receptores acoplados a proteínas G
La subfamilia TREK y algunos otros miembros de la familia K2P, al igual que el canal
de potasio tipo M, se modulan a través de vías activadas por receptores acoplados a proteínas
G (GPCRs). Estos receptores son proteínas diana de muchas sustancias químicas que a través
de segundos mensajeros activados por proteínas G modulan los canales iónicos. Los GPCRs
median una respuesta más lenta que los receptores ionotrópicos debido a que activan varias
proteínas citosólicas. Esta modulación no solo aumenta o disminuye la actividad espontánea
Introducción
19
de los canales sino también modula su activación por estimulación sensorial, metabólica y
farmacológica (Fink et al., 1996; Lesage et al., 2000).
4.1 Receptores acoplados a proteínas G
Los GPCRs poseen siete segmentos helicoidales transmembrana unidos por tres
segmentos intracelulares y tres segmentos extracelulares, todo ello empaquetado formando
una estructura tridimensional en forma de anillo (Figura 14). El dominio extracelular o N-
terminal tiene un residuo de ácido aspártico al cual se une mediante glicosilaciones el ligando
(Caulfield & Birdsall, 1998), mientras que el dominio intracelular o C-terminal tiene un
residuo de cisteína muy conservado al cual se acopla la proteína G (Felder, 1995; Kobilka,
2007).
Figura 14: Esquema y estructura tridimensional de los GPCRs. A) El receptor muscarínico acoplado
a proteína G está formado por siete segmentos transmembrana (TM1-7) cuya estructura es α-hélice, por los
dominios extracelulares (e1-3) e intracelulares (i1-3) cuya estructura es β-lámina, por un dominio extracelular N-
terminal (NH2) donde interacciona el ligando y por un dominio intracelular C-terminal (COOH) al cual se une la
proteína G. B) Estructura tridimensional del receptor M2 (Fuente: IUPHAR Database).
Existen 5 familias diferentes de GPCRs clasificadas según su secuencia aminoacídica:
familia rodopsina (a la cual pertenecen los receptores de BK y muscarínicos), familia de
adhesión, familia frizzled/taste, familia del glutamato y familia de la secretina. Todas ellas
comparten la estructura descrita anteriormente y solo se diferencian en los dominios C-
terminal, N-terminal y en el segmento i3. A pesar de esto, cuando distintos GPCRs se activan,
pueden sufrir cambios conformacionales muy similares en el dominio C-terminal que hace
que se unan a la misma proteína G. Contrariamente, un solo GPCR puede activar distintas
TM1 TM2
Estructura
β-lámina
Estructura
α-hélice
A B

Introducción
20
proteínas G a través de vías diferentes. Esto hace que la cascada posterior a la activación de
un GPCR sea muy compleja, ya que puede provocar distintas respuestas en la célula, o por lo
contrario, que la activación de varios GPCRs confluyan en una sola respuesta celular
(Kobilka, 2007).
La unión del agonista a un GPCR provoca otros cambios conformacionales en los
segmentos TM3, TM5 y TM6. Estos cambios son ligeras inclinaciones y giros de los
segmentos sobre sí mismos, que dependen del tipo de GPCR y del estado de activación que se
genere (Latek et al., 2012). Diferentes ligandos pueden producir respuestas diferentes en un
mismo GPCR dependiendo de su eficacia, de este modo podemos distinguir distintos tipos de
ligandos: los agonistas completos son aquellos que pueden generar una máxima respuesta a
través del GPCR, a diferencia de los agonistas parciales que no alcanzan nunca la máxima
respuesta, y los agonistas inversos que reducen la actividad basal del receptor. Esta
clasificación entre ligandos no depende de su estructura o de la afinidad por el GPCR sino del
estado de conformación al cual se convierte el GPCR, lo cual se traduce en diferente
magnitud y cinética en la vía de señalización a través de las proteínas G (Vilardaga, 2010).
4.2 Proteínas G
Los cambios conformacionales en los GPCRs comentados anteriormente provocan la
activación de las proteínas G acopladas al dominio C-terminal. Las proteínas G
heterotriméricas están compuestas por las subunidades α, β y γ (Miras et al., 1998). Cuando la
proteína G está inactivada las tres subunidades de la proteína G están unidas entre ellas y
además la subunidad α está unida a GDP (guanosín difosfato). Cuando el agonista se une al
receptor, se forma un complejo ternario formado por el agonista, el receptor y la proteína G.
Esta formación del complejo ternario aumenta la afinidad del receptor por el agonista y la
proteína G cambia su afinidad a favor de GTP (guanosín trifosfato) en lugar de GDP como
sucede en el estado de inactivación. Este cambio de afinidad también provoca la disociación
de la proteína G en la subunidad Gβγ y la subunidad Gα unida a GTP. La subunidad Gα-GTP
activa cascadas de transducción de la señal. Posteriormente el enzima GTPasa (EC3.6.5)
acelera la hidrolisis de GTP en GDP provocando la re-asociación de las subunidades de la
proteína G (Gαβγ) y finalizando la señalización (Latek et al., 2012) (Figura 15).

Introducción
21
Figura 15: Activación de la proteína G mediante GPCR. El GPCR inactivo permanece unido a la
proteína G heterotrimérica con sus tres subunidades ensambladas y la subunidad α unida a GDP (1), cuando un
ligando se une al GPCR (2), además de suceder un cambio conformacional en su estructura, provoca un cambio
en la proteína G. Ahora la proteína G se vuelve más afín por GTP (3) y la subunidad α se une a GTP y libera
GDP (4). Este cambio provoca que se disocien las subunidades de la proteína G (5) que interaccionarán por
separado con las proteínas efectoras. Posteriormente GTP se hidroliza a GDP (6) que se vuelve a unir a la
subunidad Gα y se ensambla de nuevo con la subunidad Gβγ finalizando la transducción de la señal (1) (Fuente:
Medical-institution.com).
La subunidad α se divide en cuatro familias según su secuencia genética: la familia Gαs,
la familia Gαi/o/t, la familia Gαq/11, y por último la familia Gα12/13 (Bastiani & Mendel, 2006).
La familia Gαs media su acción a través de la activación del enzima adenilato ciclasa (AC; EC
4.6.1.1) provocando un aumento en la concentración intracelular de AMPC, mientras que la
familia Gαi/o/t inhibe el enzima AC provocando una reducción de los niveles de AMPc. La
familia Gαq/11 activa la PLC. Sin embargo la transducción de la familia Gα12/13 no se conoce
bien por la falta de inhibidores específicos (Latek et al., 2012).
4.3 Modulación muscarínica
Los receptores muscarínicos son GPCRs estimulados por el ligando endógeno
acetilcolina (ACh, Figura 16). La ACh es un neurotransmisor que se sintetiza a partir de
colina y Acetil Co-A mediante el enzima Colina Acetiltransferasa (EC 2.3.1.6) (Fonnum,

Introducción
22
1969). La ACh se distribuye por todo el sistema nervioso central y periférico mediando la
transmisión del impulso nervioso (Macintosh, 1941) y por tanto se relaciona con la
excitabilidad neuronal. La ACh, tras la unión a los receptores de las neuronas postsinápticas,
se elimina mediante el enzima acetilcolinesterasa (EC 3.1.1.7) (Fonnum, 1969). Este enzima
tiene dos isoformas, una a nivel de la hendidura sináptica y otra a nivel sérica que degrada
rápidamente la ACh cuando se administra de forma intravenosa, lo que dificulta su uso
terapéutico y experimental (Taylor et al., 1981). Por ello se utilizan a menudo agonistas
muscarínicos como la oxotremorina metiodada (Oxo-M) (Figura 16) que, al igual que la
muscarina, actúan activando los receptores muscarínicos, mientras que se utilizan los
antagonistas muscarínicos, como la atropina (Figura 16) o la escopolamina para bloquear
dichos receptores.
Figura 16: Estructura de acetilcolina, oxo-M y atropina. A) Estructura de acetilcolina, activa de forma
nativa los receptores muscarínicos y nicotínicos. B) Estructura de oxo-M que es un agonista muscarínico. C)
Estructura de la atropina, antagonista del receptor muscarínico (Fuente: Sigmaaldrich.com).
Los receptores muscarínicos están presentes en neuronas del sistema nervioso central y
del sistema nervioso periférico, al igual que los receptores nicotínicos ionotrópicos. En el
sistema nervioso central, los receptores muscarínicos están implicados en funciones tan
importantes como el control motor, la regulación de la temperatura, la regulación
cardiovascular y la memoria, mientras que en el sistema nervioso periférico median funciones
como la contracción del músculo liso, la secreción glandular y la fuerza y frecuencia del
latido cardiaco (Caulfield & Birdsall, 1998).
Los receptores muscarínicos están clasificados en dos grupos según su secuencia
aminoacídica y sus propiedades farmacológicas. Los receptores muscarínicos pertenecientes
al primer grupo son los receptores M1, M3 y M5 y al segundo grupo pertenecen los
receptores M2 y M4. Estos dos grupos se diferencian entre ellos en la secuencia del segmento
i3 al cual están unidas las distintas proteínas G.
Acetilcolina Atropina Oxo-M
A B C

Introducción
23
La transducción de la señal desde el receptor hasta el canal sucede a través de proteínas
G y de segundos mensajeros intracelulares. Cuando el ligando se une al receptor provoca un
cambio conformacional en el dominio intracelular del receptor que está interaccionando con
la proteína G (Kobilka, 2006) (Figura 17). Los receptores muscarínicos M1, M3 y M5
típicamente están acoplados a la proteína Gq/11, mientras que los receptores M2 y M4 se
acoplan a Gi/o/t. Esto se debe a que las proteínas G se unen a regiones próximas a i2 e i3 de los
receptores, las cuales son diferentes para estos dos grupos de receptores muscarínicos. Sin
embargo la proteína Gs solamente puede unirse al receptor M4 (Caulfield & Birdsall, 1998).
Siguiendo la cascada de segundos mensajeros, la proteína Gq/11 activa la vía de la PLC,
mientras que las proteínas Gi/o/t inhiben la vía de la AC (Hamilton et al., 1997; Caulfield &
Birdsall, 1998) como se observa en la figura 17.
La PLC es un enzima presente en la membrana plasmática que hidroliza
específicamente al fosfolípido de membrana PIP2 (fosfatidilinositol 4,5-bifosfato)
convirtiéndolo en los segundos mensajeros IP3 (inositol 1, 4, 5-trifosfato) y DAG
(diacilglicerol) (Mathie, 2007). El IP3 difunde desde la membrana plasmática al retículo
endoplasmático (RE) donde se une al receptor de IP3 permitiendo la salida de calcio hacia el
citoplasma a favor de gradiente. Uno de los efectos del aumento de concentración de calcio en
el citoplasma es la activación de la PKC. El DAG coopera con el calcio en la activación de
PKC. La PKC fosforila proteínas que contienen la secuencia de residuos de serina y treonina
como pueden ser las proteínas que forman los canales iónicos (Lesage et al., 2000; Nelson &
Cox, 2009) (Figura 17).
Cuando el ligando se une a un receptor muscarínico M2 o M4, que está acoplada a una
proteína Gi/o/t, o Gs sucede un cambio conformacional en dichos receptores, esto provoca,
como en el caso de la proteína Gq/11, que las proteínas Gi/o/t o Gs reemplacen GDP por GTP
activándose. La proteína Gi/o/t inhibe la AC provocando la disminución del AMP cíclico
mientras que la proteína Gs activa la AC (Mathews et al., 1999). Altos niveles de AMP cíclico
provoca la activación de la PKA y también provoca un aumento en la concentración de calcio
intracelular a través de la activación de canales de calcio voltaje dependientes (Figura 17).
Se ha visto en ciertas ocasiones, mediante experimentos in vitro, una actividad
constitutiva del receptor muscarínico en el que se observa que estos receptores están activados
parcialmente en ausencia de agonistas (Jakubík et al., 1995).

Introducción
24
Figura 17: Posibles vías de regulación muscarínica de los canales de la subfamilia TREK. El ligando
(L) puede interaccionar con los receptores muscarínicos M1, M3 o M5 que están acoplados a la proteína Gq/11
(Gq), o bien a los receptores M2 o M4 acoplados a la proteína Gi/o/t (Gi) o a la proteína Gs cuyos efectos son
contrarios. La activación de la proteína Gq/11 a su vez activa la PLC, que fosforila e hidroliza PIP2 dando lugar a
IP3 y DAG. Estos segundos mensajeros activan directa o indirectamente a PKC que fosforila el canal (C). La
activación de la proteína Gi/o/t inhibe la AC, reduciendo los niveles de AMPc y provocando la inactivación de
PKA mientras que Gs provoca la activación de PKA.
La modulación muscarínica de la subfamilia TREK es muy controvertida y no se ha
aclarado completamente a lo largo de los últimos años. En primer lugar no se conoce con
exactitud qué receptor muscarínico (M1-M5) y qué proteína o proteínas G (Gq/11, Gi/o/t o Gs)
participan en la modulación muscarínica de la subfamilia TREK (Figura 17). En segundo
lugar, algunos autores describen que los canales de esta subfamilia se inhiben mediante la
fosforilación directa por PKC (Gu et al., 2002; Murbartián et al., 2005), excepto el canal
TRAAK que no se ve afectado (Fink et al., 1998). Sin embargo otros investigadores afirman
que estos canales se pueden modular directamente mediante DAG e IP3 resultados de la
hidrólisis de PIP2 (Chemin et al., 2003) o incluso se ha observado que el canal TREK-1 se
inhibe cuando se reduce la concentración de PIP2 que estaría activando el canal
permanentemente (Lopes et al., 2005). Por último, se ha observado que el canal TREK-1 se
inhibe cuando aumenta la concentración de calcio intracelular (Enyeart et al., 2005).
En el caso de que el receptor muscarínico implicado esté asociado a la proteína Gs, sería
la PKA la que modularía los canales TREK-1 y TREK-2, sin embargo no modula al canal

Introducción
25
TRAAK. Si seguimos la vía de modulación de la proteína Gi/o/t conllevaría a un resultado
inverso ya que la activación de esta proteína G provoca la reducción de la concentración de
AMP cíclico y esto conllevaría a una reducción de la fosforilación llevada a cabo por PKA
(Fink et al., 1998).
4.4 Modulación mediante receptores de BK
La BK (Figura 18) es una hormona peptídica nativa que actúa como un potente
vasodilatador y genera inflamación y dolor (Greaves & Shuster, 1967), incrementa la
permeabilidad de la barrera hemato-encefálica y la extravasación del plasma que suceden
durante el periodo inflamatorio, y provoca hipotensión lo que confiere a la BK una función
cardioprotectora. Además se relacionan con el cáncer ya que ciertas células cancerígenas
expresan, sin previa estimulación, receptores de BK y estimula el crecimiento de dichas
células. La participación de los receptores de BK en dichas enfermedades los ha señalado
como dianas para nuevos fármacos. La administración de BK también induce síntomas tales
como bronco-constricción en enfermos asmáticos, rinitis y dolor de garganta (Sharma & Al-
Sherif, 2006).
Figura 18: Estructura de BK. (Fuente: sigmaaldrich.com).
La hormona peptídica BK se genera a parir del precursor calicreína que circula por el
plasma sanguíneo, hasta que un daño en el tejido desencadena la síntesis de BK (Sharma &
Al-Sherif, 2006). La BK ya sintetizada se une a GPCRs específicos. En los mamíferos existen
dos tipos de GPCRs para la BK, el receptor B1 y el receptor B2. La afinidad de la BK es
diferente para cada uno, siendo mayor la afinidad por el receptor B2, mientras que los

Introducción
26
quininógenos (péptidos que contienen en su estructura la secuencia de la BK) son más afines
al receptor B1 (Leeb-Lundberg et al., 2005).
Ambos receptores de BK se expresan en numerosos tipos celulares como neuronas,
células endoteliales, células del músculo liso, fibroblastos, células epiteliales y tumorales. Sin
embargo, el receptor B2 se expresa en mayor proporción que el receptor B1 (Dray et al., 1988).
En general, el receptor B1 se expresa raramente de forma constitutiva, ya que su expresión es
inducida por la activación del receptor B2 (Babbedge et al., 1995). La BK se une al receptor
B2 en las neuronas sensoriales contribuyendo a la activación del dolor agudo, enrojecimiento
e hinchazón, y a través del aumento en la expresión local del receptor B1 propaga la respuesta
inflamatoria (Leeb-Lundberg et al., 2005). De este modo el receptor B2 está implicado en la
fase inicial del proceso inflamatorio, rápidamente se fosforila e internaliza, mientras que el
receptor B1 aumenta y prolonga esta fase aumentando el dolor (Perkins & Kelly, 1993). Por
ello los antagonistas de estos receptores han sido estudiados como tratamiento antinflamatorio
y para el estudio del dolor crónico. En este último caso se han estudiado los antagonistas del
receptor B1 que realizan su acción en los receptores de los terminales nerviosos del asta dorsal
o de las neuronas de la médula espinal e interrumpen la señalización del dolor (Leeb-
Lundberg et al., 2005). Además la BK excita las neuronas del GCS de rata, sobre las cuales
además de provocar una ligera despolarización y modular la excitabilidad de éstas (Figura
10), provoca en los terminales simpáticos la liberación de prostanoides, los cuales inducen
sensibilización de los nociceptores (Jones et al., 1995).
La unión de BK a los receptores B1 y B2 desata una cascada de señalización que
comienza con la activación de la proteína G (Figura 19). En neuronas del GCS de rata la BK
se une principalmente al receptor B2 que activa la proteína Gq/11 (Jones et al., 1995), a pesar
de que pueda interaccionar con otras proteínas G como Gi/o/t, Gs o G12/13 (Gutowski et al.,
1991), mientras que el receptor B1 puede interaccionar con las proteínas Gq/11 y Gi/o/t.
Posteriormente esta proteína Gq/11, ahora activada, provoca la activación de la PLC, y ésta a su
vez provoca la hidrólisis de PIP2 generando los segundos mensajeros IP3 y DAG. Finalmente
el IP3 provoca un aumento de la concentración de calcio intracelular (Yano et al., 1985)
mientras que el DAG, en cooperación con el calcio, activa la PKC (Tippmer et al., 1994). No
se ha determinado con exactitud la implicación de estos segundos mensajeros en la vía de
modulación por BK de los canales de la subfamilia TREK en sistemas heterólogos ni en
sistemas autólogos. Sin embargo podemos sugerir que la inhibición por BK podría ser el
resultado de la fosforilación de los canales TREK por PKC activada por el incremento del ión

Introducción
27
calcio, como sucede en la inhibición de la corriente M en neuronas simpáticas (Delmas &
Brown, 2005), o bien dicha inhibición puede deberse al descenso de la concentración de PIP2
durante la hidrólisis de éste como ocurre durante la inhibición por BK de canales de sodio
(Zaika et al., 2011) o en la inhibición muscarínica de los canales TREK-1 expresados en
oocitos de Xenopus (Lopes et al., 2005).
Figura 19: Posible vía de modulación por BK de la subfamilia TREK. La modulación por BK sucede
a través del receptor B2 que está ampliamente expresado en mamíferos. Este receptor está acoplado a la proteína
Gq/11 (Gq), cuya activación provoca la activación del enzima PLC, que fosforila PIP2 dando lugar a IP3 y DAG.
Estos segundos mensajeros provocan la liberación de calcio al citosol y activan PKC que podría fosforilar el
canal (C).
4.5 Proteínas de anclaje y segundos mensajeros
La modulación de los canales puede ser directa o indirecta a través de proteínas y
segundos mensajeros que fosforilan los dominios del canal. El dominio C-terminal de los
canales de la subfamilia TREK juega un papel muy importante en su regulación por
temperatura, por cambios en el pH intracelular, estímulos mecánicos y por anestésicos
volátiles, como ya se mencionó anteriormente, pero también en la modulación muscarínica y
por BK. En el dominio C-terminal de los canales TREK-1 y TREK-2, pero no de TRAAK, se

Introducción
28
une la proteína de anclaje AKAP (Sandoz et al., 2006). La función de esta proteína es unir
varias proteínas que fosforilan el dominio C-terminal de los canales, entre las que se
encuentran PKC y PKA (Sandoz et al., 2006) y el complejo Ca++
-calmodulina (Faux & Scott,
1997). Sin embargo, el complejo Ca++-calmodulina podría estar unido directamente al
dominio C-terminal del canal TREK-2, como ocurre con el canal de potasio tipo M (Yus-
Najera et al., 2002).
La proteína AKAP además se une al fosfolípido de membrana PIP2. La unión AKAP-
PIP2 es modulada por las quinasas PKC y PKA que fosforilan esta proteína de anclaje
(Dell’Acqua et al., 1998). La asociación de AKAP al canal permite que éste permanezca
completamente abierto e imposibilita que pueda ser activado por ácido araquidónico, por
cambios en el pH intracelular o por estímulos mecánicos (Sandoz et al., 2006).
La presencia de la proteína AKAP es fundamental para la inhibición muscarínica y por
BK de la corriente de potasio tipo M en sistemas heterólogos (Bal et al., 2010) y en neuronas
simpáticas (Hoshi et al., 2003). La activación de los receptores muscarínicos y de BK provoca
la activación de las proteínas G y posteriormente de diversos segundos mensajeros entre los
que se encuentras las quinasas PKA y PKC y se incrementa la concentración de calcio
intracelular. Las quinasas PKA y PKC fosforilan la proteína AKAP reduciendo la unión
AKAP-PIP2 y provocando así la inhibición del canal (Dell’Acqua et al., 1998). Sin embargo
el complejo Ca++
-calmodulina compite con la proteína AKAP por el sitio de unión al canal,
inhibiendo la unión de AKAP al canal y por lo tanto se bloquea el canal. Además la unión del
complejo Ca++
-calmodulina a la proteína AKAP imposibilita la fosforilación por parte de
PKC (Faux & Scott, 1997).
La proteína Mtap2 (también conocida como MAP2) es una proteína que está asociada a
los microtúbulos y se une también al dominio C-terminal de los canales TREK. La proteína
Mtap2 y la proteína AKAP pueden encontrarse simultáneamente unidas al canal. A diferencia
de la proteína AKAP, la proteína Mtap2 modula la densidad de los canales TREK en la
membrana celular y por consiguiente la corriente de TREK en la célula (Sandoz et al., 2008).
4.6 Hipótesis de trabajo
El potencial de membrana en reposo y la excitabilidad neuronal están determinados por
los canales iónicos que permanecen abiertos a dicho potencial de membrana. Algunos de estos
son los canales de sodio persistente (Wu et al., 2001), la corriente catiónica tipo H (Brown &

Introducción
29
Difrancesco, 1980), la corriente de potasio tipo M (Brown & Selyanko, 1985) y los canales
K2P (Patel et al., 2000). Además la bomba de sodio/potasio también ejerce un importante
efecto sobre su valor (Koester et al., 2001; Lamas et al., 2002). El potencial de membrana en
reposo de las neuronas del GCS de ratón se modula mediante agonistas muscarínicos y BK, al
igual que la corriente de potasio tipo M, por lo que se responsabilizaba exclusivamente a esta
corriente de la modulación muscarínica y por BK del potencial de membrana (Babbedge et
al., 1995; Jones et al., 1995; Delmas & Brown, 2005). Sin embargo, experimentos previos
realizados en nuestro laboratorio demuestran que el agente neuroprotector riluzol activa una
corriente de potasio transportada principalmente por el canal TREK-2 (IRIL) en las neuronas
del GCS de ratón (Cadaveira-Mosquera et al., 2011, 2012), y se ha determinado en sistemas
heterólogos que este canal se modula mediante agonistas muscarínicos (Kang et al., 2006).
Por esta razón consideramos que el canal TREK-2 es un excelente candidato, junto con la
corriente M, para modular el potencial de membrana mediante agonistas muscarínicos, y
posiblemente por BK, en neuronas del GCS de ratón.
Tanto los canales TREK-2 como los canales tipo M, son selectivos para el potasio,
observándose una corriente de salida cuando se estudian mediante la técnica de fijación de
voltaje o voltage-clamp (principal técnica utilizada en este trabajo). Diferenciar la corriente de
potasio transportada por cada uno de ellos es imprescindible (aunque muy complejo) para
poder estudiar los canales de forma independiente.


OBJETIVOS


Objetivos
33
OBJETIVOS
El objetivo general de este trabajo fué estudiar la modulación muscarínica y por BK del
canal TREK-2 en neuronas de GCS de ratón en cultivo primario. Para ello los objetivos
específicos fueron:
1. Diferenciar la corriente de potasio a través del canal TREK-2 de la corriente de
potasio a través del canal tipo M.
2. Observar y cuantificar la modulación de IRIL en presencia del agonista muscarínico
oxo-M y caracterizar la vía de segundos mensajeros implicados en la modulación
muscarínica.
3. Observar y cuantificar la modulación de IRIL en presencia de bradiquinina y
caracterizar la vía de segundos mensajeros implicados en dicha modulación.
4. Determinar la implicación fisiológica del canal TREK-2 en el potencial de membrana.


MATERIAL Y MÉTODOS


Material y Métodos
37
MATERIAL Y MÉTODOS
1 Cultivo celular de neuronas de GCS
Para el estudio de los canales iónicos nativos se han utilizado neuronas de GCS
extraídas de ratones y mantenidas en cultivo primario.
1.1 Animales de experimentación
Se utilizaron ratones Swiss CD1 de una edad comprendida entre 20 y 60 días y de
ambos sexos. Se criaron y mantuvieron en un estabulario donde fueron alimentados con
pienso comercial y agua ad libitum. El estabulario presenta un ciclo de luz/oscuridad de 12/12
horas y una temperatura ambiente constante (22-23ºC).
Todos los procedimientos utilizados con los ratones cumplen la directiva española y
europea de protección de animales de experimentación (RD53/2013; 2010/63/EU). Estos
procedimientos también fueron aprobados por el Comité Ético de Bienestar Animal (CEBA)
de la Universidad de Vigo.
1.2 Extracción del GCS de ratón
La extracción de los ganglios se realiza tras sacrificar al ratón con CO2 en una cámara
de eutanasia. Se decapita a nivel de la parte inferior del cuello con material de cirugía. En la
cara ventral de la cabeza del ratón se separa la piel para poder observar la tráquea. La tráquea
se retira hacia la parte más rostral poniendo así de manifiesto las arterias carótidas que están a
ambos lados de la misma (Figura 20). Bajo una lupa se localiza la bifurcación de la arteria
carótida sobre la que se sitúa el ganglio, se extrae todo junto y se coloca en un pocillo con
medio Leibowitz (L-15).
En la bifurcación de la arteria carótida se encuentra el GCS. Éste se separa de la arteria
y se introduce en otro pocillo con L-15 donde se retiran los restos de sangre y de tejido
adiposo que pueda tener adosado. Ya en un tercer pocillo, se realiza un corte profundo pero
sin llegar a cortarlo del todo con el fin de facilitar la disgregación mecánica posterior.

Material y Métodos
38
Figura 20: Localización del GCS en el ratón. El GCS se encuentra en la bifurcación de la arteria
carótida tras separar la tráquea. (Fuente: modificado de (Fedoroff & Richardson, 2001)).
1.3 Cultivo primario de neuronas de GCS
En condiciones de esterilidad dentro de una campana de flujo laminar se lavan los
ganglios con la solución HBSS+HEPES (2 lavados) y luego se incuban con una solución
enzimática de colagenasa (purificada de Clostridium histolyticum) en HBSS+HEPES durante
15 minutos en un incubador a 37 ºC y 5 % de CO2. A continuación y después de dos lavados
con HBSS+HEPES se incuban de nuevo en una solución enzimática de tripsina (purificada de
páncreas de bovino) en HBSS y HEPES durante 30 minutos en las mismas condiciones.
Después de las incubaciones enzimáticas, los ganglios se disocian mecánicamente en un
tubo con medio de cultivo haciendo pasar los ganglios a través de una pipeta Pasteur de vidrio
hasta que se disgreguen los ganglios completamente. Se pasa el medio de cultivo a otro tubo
con L-15 que se somete a centrifugación durante tres minutos a 500 rcf. El “pellet” se
resuspende en medio de cultivo que se siembra en placas “petri” de 35 mm previamente
tratadas con el substrato laminina. La laminina, cuya concentración es de 10 µg/ml, se incuba
previamente a 37ºC y 5% de CO2 durante dos horas y luego se lava el pocillo con L-15 para
eliminar su exceso (Figura 21).
Las neuronas se mantienen en cultivo durante un día a 37ºC y 5% de CO2 en un
incubador hasta realizar los registros electrofisiológicos al día siguiente.
El medio de cultivo se compone de medio L-15 suplementado con: NaHCO3 24 mM,
FCS 10%, glucosa 38 mM, L-glutamina 2 mM, penicilina 100 UI/ml, estreptomicina 100
μg/ml y NGF 50 ng/ml. La solución de HBSS+HEPES se compone de la solución “Hank´s
balanced salt solution” y la solución de tampón “4-(2-Hydroxyethyl)-1-
Ganglio Cervical Superior Arteria Carótida

Material y Métodos
39
piperazineethanesulfonic acid” 10 mM. La soluciones enzimáticas de colagenasa (2,5 mg/ml)
y de tripsina (1 mg/ml) contienen albúmina (6mg/ml) y HBSS+HEPES.
Todos los productos y reactivos utilizados para el cultivo celular fueron comprados a
Sigma-Aldrich (Madrid, España).
Figura 21: Esquema de los pasos de la extracción del GCS de ratón y el cultivo neuronal.
Posteriormente a la extracción del ganglio, éste se somete a disgregación enzimática, seguida de disgregación
mecánica. Posteriormente las neuronas individualizadas se centrifugan y se recogen en el pellet. A continuación
se disgregan y se siembran en pocillos con medio de cultivo.
2 Registro electrofisiológico
El registro electrofisiológico de las neuronas de GCS se realizó mediante la técnica de
“Patch-Clamp” desarrollada por Neher y Sakmann en 1976 (Hamilton et al., 1981; Neher &
Sakmann, 1976). Esta técnica permite medir y caracterizar la corriente a través de los canales
iónicos de las membranas celulares.
2.1 Principios de electrofisiología
La técnica se basa en asumir que las células se comportan como un circuito eléctrico,
similar al que podemos observar en la figura 22, de tal modo que:
La membrana celular se comporta como un condensador ya que se trata de una bicapa
lipídica aislante que separa dos soluciones salinas (solución de baño o extracelular y la
solución citosólica o intracelular) con diferentes concentraciones iónicas que se

Material y Métodos
40
comportan como dos conductores y que generan una diferencia de potencial
electroquímico.
La fuerza electromotriz determinada por la diferencia electroquímica de los iones
supone la pila, es decir, la diferencia de voltaje del circuito.
Los canales iónicos se comportan como resistencias ya que solamente a través de ellos
pueden pasar los iones, de forma selectiva (muchos canales son selectivos a uno o
varios iones) y en un determinado momento (ya que los canales iónicos pueden estar
abiertos, cerrados o inactivados). Los canales iónicos nos determinan la conductancia
de la membrana respecto a los iones, que no es más que la inversa de la resistencia.
Figura 22: Similitud de la membrana celular a un circuito eléctrico. En experimentos
electrofisiológicos la membrana neuronal se comporta como un circuito eléctrico. La membrana en sí misma
(lípidos) funciona como un condensador, la fuerza electromotriz determinada en este caso por la diferencia
electroquímica del ión K+ actúa como una pila, mientras que los canales nos determinan la conductancia (inversa
de la resistencia) de la membrana respecto al ión de K+ (Fuente: (Hille, 1940)).
Por tanto una neurona, al igual que el circuito eléctrico, sigue la ley de Ohm, pero en
este caso cambiando la diferencia de voltaje de un circuito eléctrico por la fuerza
electroquímica,
gE=I sería equivalente a )( KKK EEg=I
Esto no es más que una representación de la ley de Ohm para un circuito eléctrico,
donde I es la intensidad de corriente (en amperios, A), g es la conductancia (en siemens, s) y
E es la diferencia de potencial (en voltios, V). En una neurona la ley de Ohm se modifica de
tal manera que la intensidad de corriente para un ión (por ejemplo el ión potasio, representado
por IK) viene determinada por la conductancia de la membrana para ese ión gK (que mide la
facilidad con que dicho ión atraviesa la membrana y es inversa a la resistencia, por tanto está
determinado por el número y tipo de canales iónicos y del estado en que se encuentran, es

Material y Métodos
41
decir, abiertos o cerrados) y por la fuerza electroquímica, E-EK, que representa la diferencia
entre el potencial de membrana y el potencial de equilibrio para ese ión (Hille, 1940).
Utilizando la técnica de Pacth-clamp, y basándonos en la ley de Ohm modificada,
podemos medir y determinar la corriente a través de un canal o de todos los canales de la
célula, así como el potencial de membrana de la célula en cada momento del experimento.
Para ello debemos realizar un sello entre la membrana celular y una pipeta de vidrio en cuyo
interior se encuentra el electrodo de plata clorada (electrodo de patch). Además del electrodo
de patch que contacta con el interior celular, el electrodo de referencia (también de plata
clorada) se sumerge en el baño donde se encuentran las células. De este modo podemos medir
la diferencia de potencial entre los dos lados de la membrana celular (Sakmann & Neher,
1984).
2.2 Modalidades de patch-clamp
A lo largo del tiempo se han desarrollado distintas modalidades de la técnica de patch-
clamp con el fin de mejorar y facilitar el estudio dependiendo de lo que queramos medir y
analizar en la célula. Por un lado podemos utilizar la célula entera si lo que nos interesa es
analizar la corriente a través de los canales de toda la membrana celular, o bien podemos
utilizar la modalidad de canal individual si queremos trabajar con la corriente que pasa a
través de un solo canal. Por otra parte podemos manipular la célula (Figura 23) de tal modo
que la parte interna de la membrana celular nos quede en contacto con la solución de baño
mediante la modalidad de inside-out y cuyo fin es estudiar la modulación intracelular de los
canales iónicos, o bien reducir el tamaño de la célula mediante la modalidad outside-out de tal
forma que la parte externa de la membrana siga en contacto con la solución de baño con el fin
de estudiar más fácilmente la modulación de los canales iónicos desde la parte externa de la
membrana (Shakmann & Neher, 1984). En este trabajo hemos utilizado la modalidad de
célula entera en su variante de sello perforado y la modalidad de canal individual (cell-
attached).

Material y Métodos
42
Figura 23: Esquema de varias modalidades de la técnica patch-clamp. La modalidad de cell-attached
(1), consiste en formar un sello aplicando presión negativa sobre la membrana celular. Mediante una segunda
aplicación de presión negativa rompemos la membrana en contacto con el electrodo (2). De esta forma podremos
obtener la modalidad de célula entera (3), o bien, retirando el electrodo (4), podemos obtener un pequeño
fragmento de membrana con la parte externa hacia fuera, lo que conocemos como modalidad outside-out (5). A
partir de la modalidad cell-attached también podemos retirar el electrodo sin romper la membrana previamente
(6), para obtener una esfera que en contacto con el aire se romperá (7) de tal forma que la parte interna de la
membrana queda en contacto con la solución de baño. Esta modalidad se conoce como inside-out (8) (Fuente:
modificado de bem.fi/book y (Hamill et al., 1981)).
Modalidad de célula entera
Esta modalidad permite medir la corriente de todos los canales de la membrana celular,
es decir medir la corriente de toda la célula. Para llevar a cabo esta modalidad se realiza
primero la modalidad de cell-attached, que consiste en formar un sello de alta tresistencia
(GΩ) entre el electrodo de vidrio y la membrana celular aplicando una ligera presión negativa
cuando tenemos el electrodo tocando la membrana, de esta forma parte de la membrana
celular se introducirá ligeramente en la punta del electrodo (Figura 23). A partir de la
modalidad de cell-attached podemos realizar las modalidades de canal individual, de inside-

Material y Métodos
43
out y outside-out antes mencionadas. Para realizar la modalidad de célula entera se vuelve a
aplicar presión negativa pero esta vez más intensa y corta para que se rompa la membrana
celular que queda dentro del electrodo de vidrio. Ahora tenemos acceso al interior celular por
lo que podemos medir la corriente como si tuviéramos el electrodo de plata en el interior
celular. Sin embargo en esta modalidad al romper la membrana celular ciertos componentes
celulares escapan hacia el electrodo de vidrio diluyéndose con la solución interna del
electrodo, por tanto para el estudio de la implicación de segundos mensajeros no nos sirve
esta modalidad y debemos utilizar una variante conocida como sello perforado.
Modalidad de célula entera en su variante de sello perforado
En este trabajo para estudiar las corrientes que pasan a través de toda la membrana
celular se ha utilizado la modalidad de célula entera en su variante de sello perforado. Para
llevar a cabo esta variante se realiza primero un sello mediante la modalidad de cell-attached,
pero a diferencia de la modalidad de célula entera, en esta variedad no se rompe la membrana
celular sino que se perfora usando el antibiótico anfotericina-B (100 μg/ml) que suplementa la
solución intracelular en el electrodo de patch. Al formar poros en la membrana de la neurona
que queda dentro del sello nos permite tener acceso eléctrico al interior celular sin romper la
membrana celular y por tanto sin perder componentes intracelulares, ya que a través de estos
poros que genera la anfotericina-B solo pueden pasar cationes monovalentes (Figura 24).
Modalidad de canal individual o Single-channel
Para estudiar la corriente iónica que pasa a través de un solo canal utilizamos la
modalidad de canal individual o single-channel. Para ello utilizamos la modalidad de cell-
attached pero esta vez con un electrodo de patch con la punta de menor diámetro, cuya
resistencia es de 10-12 MΩ de tal modo que nos permita registrar solamente uno o dos canales
circunscritos al sello. Para esta modalidad utilizamos la misma solución para el baño
(solución extracelular) y para introducir en el electrodo de patch (solución intracelular), es
decir, dos soluciones equimolares de potasio que hacen que el potencial de equilibrio del

Material y Métodos
44
potasio sea 0. Esta vez no utilizamos anfotericina-B ya que lo que queremos observar y medir
es la corriente transportada a través del canal que se encuentra dentro del sello (Figura 24).
Figura 24: Esquema de las modalidades de patch-clamp utilizadas. En ambas figuras se representa el
sello que se forma entre la pipeta de vidrio y la neurona durante la técnica de Patch-clamp. A) En la variante de
sello perforado se introduce anfotericina-B para perforar el fragmento de membrana que queda dentro del sello
para poder registrar los canales de la célula entera. B) En la variante de canal individual no perforamos la
membrana sino que registramos solamente el canal o canales del fragmento de membrana que queda dentro del
sello.
2.3 Electrodo de vidrio
El electrodo de patch es en realidad un capilar de vidrio acabado en punta que se sella
íntimamente con la membrana celular y con un hilo de plata clorado en su interior. Este sello
debe tener una resistencia muy alta (del orden de gigaohmios) para que podamos medir y
observar la corriente a través de los canales iónicos minimizando al máximo el ruido eléctrico.
El electrodo de vidrio se construye a partir de un capilar de vidrio de borosilicato (Harvard
Apparatus), con un diámetro interior de 1,17 mm y un diámetro exterior de 1,5 mm. En su
interior e incrustado en la pared contiene un hilo de fibra a lo largo del electrodo que ayuda a
rellenarlo de solución intracelular sin que queden atrapadas burbujas de aire. El capilar de
vidrio se estira y se parte por la mitad en dos gracias a un estirador de pipetas (“puller”)
horizontal (Sutter Instrument) mediante dos pasos: en el primer paso el calor hace que se
A B

Material y Métodos
45
estire el vidrio y en el segundo paso hace que se parta en dos. Ambos electrodos resultantes
presentan una punta muy fina que se pule con la ayuda de una microforja (MF 830, Narishige)
obteniéndose un diámetro de la punta tan pequeño que nos permita obtener una resistencia del
electrodo de 2 a 5 MΩ para los experimentos en la modalidad de célula entera en la variante
de sello perforado y de 10-12 MΩ para la modalidad de canal individual.
Durante los experimentos de canal individual recubrimos la punta del electrodo, sin
llegar al extremo final, con una fina capa de resina Silgar (se usa una mezcla de resina Silgar
con un secante en la proporción 10:1) para aumentar el grosor de la pared del electrodo y así
reducir la capacidad del electrodo y mejorar su compensación.
2.4 Equipamiento
Para realizar la técnica de patch-clamp hemos utilizado un equipamiento (Figura 25)
compuesto por:
Una mesa (Wentworth Laboratories) con una cámara de nitrógeno en su interior que
amortigua las vibraciones evitando que lleguen al sello.
Para mover el cabezal donde se encuentra el electrodo de vidrio de forma segura y
lenta utilizamos un micromanipulador (Sutter Instruments).
Sobre la mesa se encuentra un microscopio invertido de contraste de fases (Nikon
Eclipse TE300) donde se coloca el pocillo de 35 cm de diámetro que contiene el
cultivo celular.
Una caja de Faraday evita que interfieran en el registro los campos electromagnéticos.
Desde el pocillo sale un circuito de tubos por donde circula la solución de baño. Este
sistema pasa por una bomba peristáltica (Dinko) que mantiene el baño sobre las
células en perfusión constante a una velocidad de 7 ml/min. Este sistema también está
conectado a varias jeringas donde se añaden las soluciones de baño y las drogas. Para
seleccionar la jeringuilla donde se encuentra la solución de baño que queremos en
cada momento utilizamos una llave de paso de pasos múltiples. Por tanto este sistema
de tuberías forma un circuito cerrado que hace pasar la solución de baño desde las
jeringuillas hasta el pocillo y vuelve a las jeringuillas, o bien se puede abrir el circuito
desechando la solución de baño una vez que pasa por el pocillo.

Material y Métodos
46
Para los experimentos en los que tenemos que modificar la temperatura de la solución
de baño utilizamos un controlador de temperatura (Warner Instruments) que nos
permite calentar la temperatura de las camisas que rodean las jeringuillas y el cabezal
por donde entra la solución de baño en el pocillo. Una sonda dentro de la solución de
baño en el pocillo nos permite conocer y controlar la temperatura del baño en cada
momento.
Los electrodos, interno y externo, están conectados al cabezal o headstage que nos
permite amplificar la corriente de la célula para que llegue al amplificador y a la
tarjeta convertidora. El amplificador Axopatch 200B (Molecular Devices) además de
amplificar y filtrar la señal, también nos permite medir y aplicar pulsos de corriente a
la célula para así poder realizar los protocolos utilizados que se nombran en el
siguiente apartado.
La tarjeta convertidora Digidata 1440A (Molecular Devices) convierte las señales
analógicas en digitales y viceversa, para que podamos observarlas en el ordenador.
Mediante el software pClamp 10 (Molecular Devices) hemos aplicado los protocolos
electrofisiológicos para medir la corriente y el voltaje y estudiar el comportamiento de
las corrientes iónicas. Los datos se almacenaron gracias al programa Clampfit 10
(Molecular Devices) y se analizaron y se representaron utilizando los programas
Clampfit 10 y Origin 8 (OriginLab Corporation).

Material y Métodos
47
Figura 25: Esquema del equipamiento utilizado para el registro electrofisiológico. Sobre la mesa de
registro se encuentra el microscopio invertido con el cual observamos las células del pocillo. El pocillo está
conectado a un sistema de tuberías conectadas a una bomba peristáltica y a las jeringuillas donde añadimos las
soluciones de baño y las drogas. En el pocillo también se encuentran los electrodos de patch y de referencia que
están conectados a una tarjeta convertidora, a un amplificador y al ordenador donde observamos las corrientes.
La mesa utiliza un sistema antivibratorio para proteger el sello de vibraciones que lo puedan dañar y sobre ésta
se sostiene una caja de Faraday que lo protege del ruido eléctrico exterior.
2.5 Protocolos de registro
Los registros electrofisiológicos en la modalidad de célula entera en su variante de sello
perforado se realizaron utilizando varios protocolos de fijación de voltaje o voltage-clamp y
de fijación de corriente o current-clamp. En los experimentos de voltage-clamp la frecuencia
de muestreo fue de 2 KHz y la señal se filtró a 1 KHz, mientras que para los experimentos de
current-clamp la frecuencia de muestreo fue de 10 KHz y se filtró a 5 KHz. Los protocolos de
registro utilizados para la modalidad de célula entera en su variante de sello perforado fueron:
Protocolo de voltage-clamp con el potencial de membrana fijado en -30 mV, ya que a
este potencial de membrana la fuerza electroquímica para el potasio es mayor y por lo
Microscopio
Mesa
PC
Tarjeta
Amplificador
Caja de Faraday
Jeringas
Llave de pasos
Bomba

Material y Métodos
48
tanto las corrientes son más fáciles de visualizar. También realizamos experimentos
con el potencial de membrana fijado a -60 mV que es aproximadamente el potencial
de reposo de las neuronas del GCS de ratón.
Protocolos de current-clamp dejando el potencial de membrana sin fijar por lo que la
célula permanece en su potencial de membrana en reposo, y experimentos en los que
hemos fijado manualmente el potencial de membrana a -30 mV y a -60 mV.
Protocolos de current-clamp intensidad-voltaje (IV) en los que se aplicaron distintas
intensidades de corriente durante un segundo en intervalos de 25 pA o 50 pA como, se
observa en la figura 26, para observar el patrón de disparos de potenciales de acción.
Protocolos de voltage-clamp en los que hemos realizado rampas de voltaje desde -30
mV a -100 mV durante 7 segundos y fijando la célula de nuevo en -30 mV y protocolo
de voltage-clamp en los cuales se aplicó saltos de voltaje desde -30 mV a -50 mV
durante 2 segundos (Figura 27).
Figura 26: Protocolo de current-clamp intensidad-voltaje (IV). Este protocolo consiste en aplicar
pulsos de un segundo de duración desde -50 pA a 175 pA con incrementos de 25 pA para observar el patrón de
disparos de potenciales de acción.
Figura 27: Protocolos de voltage-clamp de saltos y de rampas de voltaje. En ambos casos fijamos el
potencial de membrana de la célula en -30 mV. A) En el protocolo de saltos de voltaje se aplican saltos desde -
30 mV a -50mV durante 2 segundos, tras los cuales el potencial de membrana vuelve a -30 mV. B) En el
protocolo de rampas de voltaje aplicamos rampas desde -30 mV a -100 mV de 7 segundos de duración tras los
cuales el potencial de membrana vuelve a -30 mV.

Material y Métodos
49
En los registros electrofisiológicos utilizados para la modalidad de canal individual la
señal fue filtrada utilizando un filtro de 2 KHz y con una frecuencia de muestreo de 20 KHz.
Se descartaron las aperturas menores del 50 % de la amplitud máxima, al igual que las
aperturas más rápidas de 50 µs. También se descartaron los sellos en los cuales se detectaron
las aperturas de más de un nivel. Los registros de canal individual se realizaron utilizando dos
protocolos:
Se realizaron experimentos de rampa de voltaje de -80 a +80 mV con un incremento
de 25 mV/segundo como se observa en la figura 28.
Se realizaron experimentos fijando el potencial de membrana en -60 mV.
Figura 28: Protocolo de rampa de voltaje en la modalidad de canal individual. El protocolo utilizado
consiste en una rampa de voltaje desde -80 a 80 mV con un incremento de 25 mV/s.
2.6 Soluciones de baño y pipeta
La solución de baño estándar (B1) extracelular para la modalidad de sello perforado
contiene NaCl 140 mM, KCl 3 mM, MgCl2 1 mM, CaCl2 2 mM, D-glucosa 10 mM, HEPES
10 mM con un pH de 7.2 que se consigue con la adición de Trizma-base. También se ha
utilizado una solución con baja concentración de potasio en la cual se ha cambiado la
concentración de KCl de 3 mM a 1.234 mM. La solución de pipeta o solución intracelular está
compuesta por KAc 90 mM, KCl 20 mM, MgCl2 3 mM, CaCl2 1 mM, EGTA 3 mM, HEPES
40 mM, pH 7.2 ajustado con NaOH. Además la pipeta también contiene anfotericina-B 75
μg/ml para los registros electrofisiológicos en la variante de sello perforado.
Para la modalidad de canal individual utilizamos la misma solución para el baño en
perfusión que para rellenar el electrodo de vidrio. Esta solución contiene HEPES 10 mM,
EGTA 5 mM, KCl 150 mM y MgCl2 1 mM, con un pH 7.2 ajustado con KOH.
Se ha utilizado un cocktail de varias drogas con el fin de inhibir las corrientes iónicas
que no nos interesan. Este cocktail está compuesto por tetrodotoxina (TTX, 0.5 µM), CsCl (1
mM) y tetraetilamonio (TEA, 15 µM) y es aplicado directamente en la solución de baño en
perfusión durante los experimentos de la modalidad de sello perforado. La TTX bloquea los

Material y Métodos
50
canales de sodio voltaje dependiente (Wen et al., 1994) y la corriente de sodio persistente
(Davis & Stuart, 1988), el CsCl bloquea la corriente catiónica h (Fu et al., 1997) y el TEA los
canales de potasio rectificadores tardío (Decoursey et al., 1984; Lipton & Tauck, 1987;
Cobbett et al., 1989), de potasio tipo A o transitorio (Lipton & Tauck, 1987; Cobbett et al.,
1989), canales de potasio dependientes de calcio (Lipton & Tauck, 1987) y la corriente de
potasio tipo M (Storm, 1989).
Las drogas, como riluzol, oxo-M, atropina, pirenzepina, himbacina, edelfosina,
bisindolilmaleimida, 2-APB (2-Aminoethyl diphenylborinato), wortmanina, BK, linopirdina y
XE991 también son aplicadas directamente a la solución de baño en perfusión. La toxina
Pertussis (PTX) fue pre-incubada con el cultivo neuronal de 17 a 30 horas previas al registro
electrofisiológico en un incubador a 37 ºC y 5 % de CO2.
El fosfolípido diC8-PIP2 (D-myo-Phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2))
se internaliza en la célula con la ayuda de un carrier (Shuttle PIPTM
Carrier 2). Para ello el día
del registro se mezclan a partes iguales el diC8-PIP2 y el carrier a partir de sus respectivos
stocks (1 µg/µl de PIP2 en agua milliQ y 0,5 mM de carrier en agua milliQ). Posteriormente
esta mezcla se diluye con más agua milliQ para obtener una concentración final durante el
registro de 0,5 µM de diC8-PIP2. La mezcla se añade a la solución de pipeta con anfotericina-
B en el momento de realizar el registro electrofisiológico y al medio de cultivo en gota para
pre-incubarlo durante una hora previa al registro electrofisiológico en un incubador a 37 ºC y
5 % de CO2 (Sohn et al., 2007).
Los reactivos y productos pertenecen a Sigma-Aldrich (Madrid, España), excepto la
PTX y TTX que se compraron a Tocris Bioscience (Bristol, UK) y diC8-PIP2 y el carrier que
pertenecen a Echelon biosciences (Salt Lake City, UT, USA).
2.7 Análisis de datos y estadística
La media de los resultados estadísticos se expresan en media ± s.e.m. (s.e.m., error
estándar de la media). Las medias fueron comparadas mediante la prueba estadística “t-
student” cuando se relacionan datos de muestras independientes y “t-student pareada”
cuando se relacionan datos de muestras dependientes. Las diferencias entre las medias fueron
consideradas significativas cuando p < 0,05.

Material y Métodos
51
Las curvas de activación de IRIL y de inhibición de IM por TEA se construyeron
mediante la conductancia hallada mediante la ley de Ohm a cada voltaje y posteriormente se
ajustaron a una curva sigmoidea mediante la ecuación de Boltzmann (Origin8, OriginLab),
2
21
01 A
e
AAy
)/dxx(x
donde A2 es el valor máximo o final, A1 es el valor mínimo o inicial, dx es la pendiente
o constante de tiempo y x0 indica el centro o punto de inflexión. Posteriormente se
compararon las curvas de activación mediante el test F.
Las curvas dosis-respuesta de linopirdina y XE991 se ajustaron a una curva sigmoidea
mediante la ecuación de Hill modificada (Hill1 en Origin8, OriginLab),
nn
n
xk
xSTARTENDSTART=y
)(
donde START es el valor inicial, END es el valor final, k es la constante de Michaelis, y
n es el coeficiente de Hill. La constante de Michaelis indica la mitad de la inhibición máxima
(IC50), mientras que el coeficiente de Hill indica la cooperación entre las moléculas del
ligando de tal modo que un coeficiente de 1 indica que la unión es un proceso independiente,
los coeficientes mayores a 1 indican una cooperación positiva que favorece a la unión,
mientras que aquéllos menores a 1 indican una cooperación negativa.
Las curvas de activación de temperatura de la corriente sensible a TEA y de la corriente
sensible a XE991 se ajustaron a una curva exponencial mediante la siguiente ecuación,
𝑦 = 𝑦0 ∗ 𝐴 ∗ exp(𝑅0 ∗ 𝑥)
donde y0 es el offset, A es el valor inicial y R0 es el ratio. Posteriormente se halló tau
(63.2 % de la corriente activada) de ambas corriente mediante la fórmula
tau = 1/R0
En los experimentos de canal individual o Single-channel, la media de la amplitud y la
media del tiempo de apertura del canal fueron calculados mediante el ajuste de los

Material y Métodos
52
histogramas de distribución a una curva de Gauss. La probabilidad de apertura (Po) del canal
se calculó mediante la siguiente ecuación,
Po=to/T
donde to es el tiempo que el canal permanece abierto mientras que T es tiempo total
observado.

RESULTADOS


Resultados
55
RESULTADOS
1 Distinción entre IM y la corriente de potasio de TREK-2
El objetivo de este grupo de experimentos es diferenciar la corriente de potasio que
fluye a través de los canales TREK-2 de la corriente de potasio tipo M, con el fin de poder
estudiar la corriente de potasio a través de TREK-2 en los apartados siguientes.
1.1 Riluzol aumenta la corriente de TREK-2
Mediante estos experimentos se pretende cuantificar la corriente de potasio que fluye a
través del canal TREK-2 que se activa con la aplicación de riluzol (IRIL), al mismo tiempo que
se bloquea la corriente de potasio tipo M con TEA. Estos experimentos se realizaron en
presencia de un cocktail de drogas, que además de contener TEA, presenta otros bloqueantes
para inhibir otras corrientes que podrían interferir en los resultados.
Los registros electrofisiológicos se realizaron mediante la modalidad de sello perforado
en neuronas de GCS de ratón en cultivo mediante protocolos de voltage-clamp con el
potencial de membrana celular fijado en -30 mV o mediante protocolos de rampas de voltaje.
1.1.1 La aplicación del cocktail inhibe la corriente de salida a -30 mV
La neuronas del GCS muestran una clara corriente persistente de salida cuando se fija su
membrana a -30 mV en la solución estándar (ver figura 30 a la izquierda del trazo). En varios
de los experimentos descritos a continuación, y previo a la aplicación de riluzol, se añadió a la
solución de baño estándar (B1) un cocktail compuesto por los bloqueantes TTX (0,5 µM),
CsCl (1 mM) y TEA (15 µM) del modo en que muestra la figura 29. El cocktail provocó una
reducción significativa de la corriente basal de salida a -30 mV (de -139.48 ± 10.66 pA; p =
1*10-5
n = 29), desde 339.06 ± 25.53 pA en ausencia de cocktail, hasta 199.58 ± 16.57 pA en
presencia de cocktail (Figura 30). A la corriente de membrana que queda a -30 mV después de
ser inhibida por el cocktail se le ha denominado I(-30) en los experimentos posteriores.

Resultados
56
Figura 29: Esquema del protocolo utilizado para la aplicación del cocktail. A los 0.5 minutos tras el
inicio del registro se aplicó el cocktail compuesto por TTX (0,5 µM), CsCl (1 mM) y TEA (15 µM) en la
solución de baño (B1) en perfusión constante. El tiempo de aplicación del cocktail previo a la aplicación de
riluzol en los siguientes experimentos fue de mínimo 5 minutos.
Figura 30: Efecto del cocktail sobre la corriente basal registrada a -30 mV. La aplicación del cocktail
a la solución de baño provocó una reducción significativa de la corriente de salida de -139.48 ± 10.66 pA (p =
1*10-5
, n = 29) en las neuronas del GCS de ratón fijadas a -30 mV.
1.1.2 Riluzol incrementa la corriente de TREK-2
Se aplicó el agente neuroprotector riluzol (300 µM), en solución de baño estándar con
cocktail de bloqueantes, del modo que se muestra en la figura 31. Se realizó este experimento
en 16 neuronas y en todas se observó el mismo efecto a partir del primer minuto
aproximadamente tras la aplicación de riluzol. Este efecto se muestra en la figura 32 como un
incremento en la corriente de salida seguida de una ligera inhibición. La media del incremento
de corriente fue de (249.43 ± 24.12 pA, p = 2.31*10-8
, n = 16). El efecto de riluzol sobre la
corriente de potasio se recupera con el lavado. En 9 de las 16 células el lavado de riluzol
provoca un ligero descenso de la línea de base inicial que se recupera tras varios minutos.
Esta corriente activada por riluzol (IRIL) ya se ha adscrito principalmente a la actividad
de los canales TREK-2 tanto en neuronas del GCS como en las neuronas del ganglio nodoso
(Cadaveira-Mosquera et al., 2011; 2012).
3´
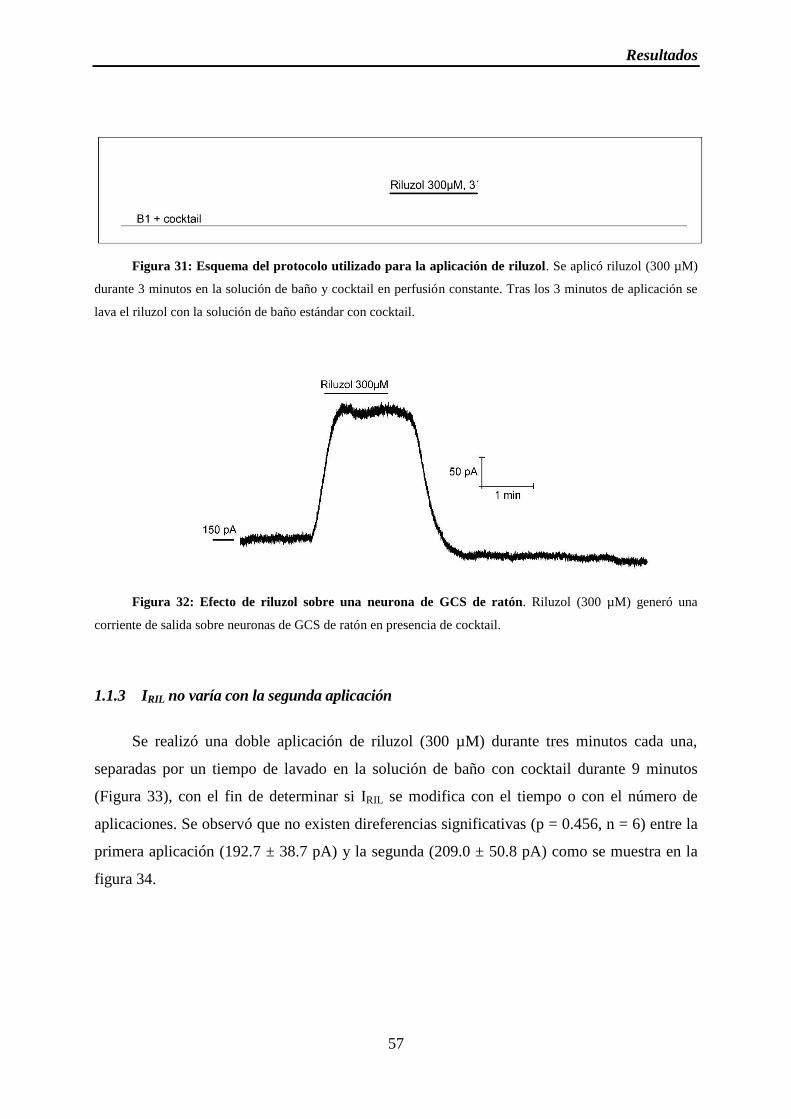
Resultados
57
Figura 31: Esquema del protocolo utilizado para la aplicación de riluzol. Se aplicó riluzol (300 µM)
durante 3 minutos en la solución de baño y cocktail en perfusión constante. Tras los 3 minutos de aplicación se
lava el riluzol con la solución de baño estándar con cocktail.
Figura 32: Efecto de riluzol sobre una neurona de GCS de ratón. Riluzol (300 µM) generó una
corriente de salida sobre neuronas de GCS de ratón en presencia de cocktail.
1.1.3 IRIL no varía con la segunda aplicación
Se realizó una doble aplicación de riluzol (300 µM) durante tres minutos cada una,
separadas por un tiempo de lavado en la solución de baño con cocktail durante 9 minutos
(Figura 33), con el fin de determinar si IRIL se modifica con el tiempo o con el número de
aplicaciones. Se observó que no existen direferencias significativas (p = 0.456, n = 6) entre la
primera aplicación (192.7 ± 38.7 pA) y la segunda (209.0 ± 50.8 pA) como se muestra en la
figura 34.
3´
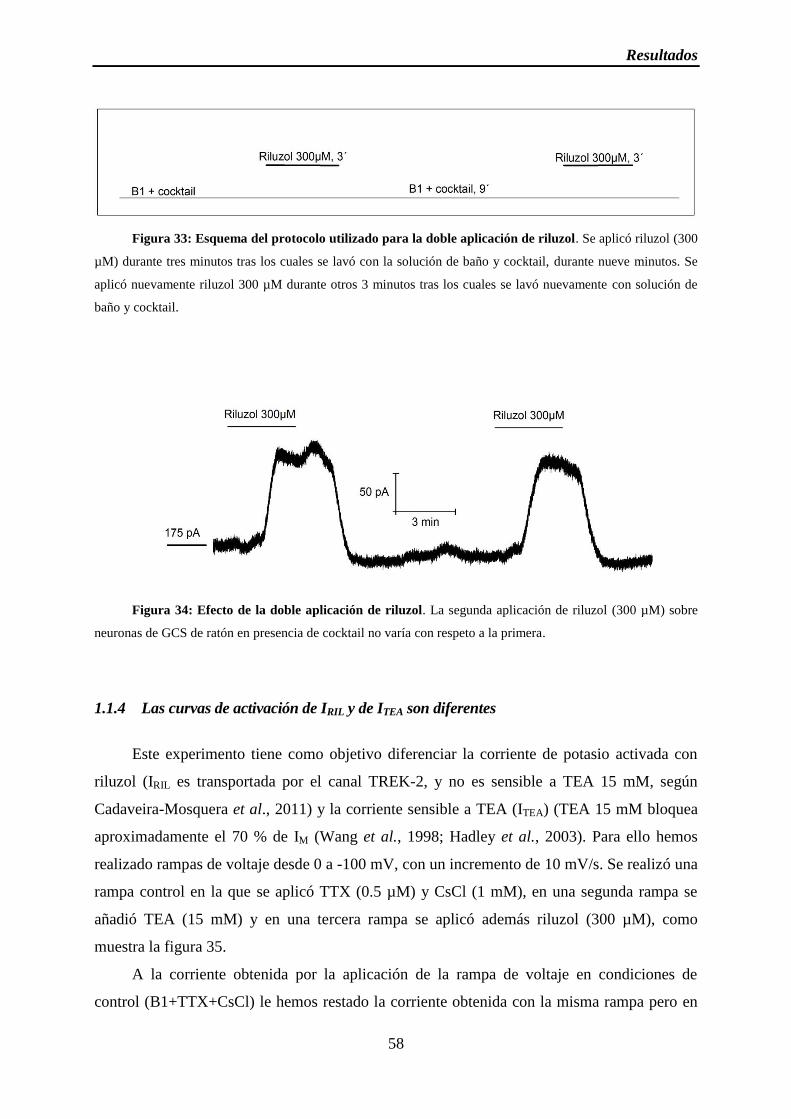
Resultados
58
Figura 33: Esquema del protocolo utilizado para la doble aplicación de riluzol. Se aplicó riluzol (300
µM) durante tres minutos tras los cuales se lavó con la solución de baño y cocktail, durante nueve minutos. Se
aplicó nuevamente riluzol 300 µM durante otros 3 minutos tras los cuales se lavó nuevamente con solución de
baño y cocktail.
Figura 34: Efecto de la doble aplicación de riluzol. La segunda aplicación de riluzol (300 µM) sobre
neuronas de GCS de ratón en presencia de cocktail no varía con respeto a la primera.
1.1.4 Las curvas de activación de IRIL y de ITEA son diferentes
Este experimento tiene como objetivo diferenciar la corriente de potasio activada con
riluzol (IRIL es transportada por el canal TREK-2, y no es sensible a TEA 15 mM, según
Cadaveira-Mosquera et al., 2011) y la corriente sensible a TEA (ITEA) (TEA 15 mM bloquea
aproximadamente el 70 % de IM (Wang et al., 1998; Hadley et al., 2003). Para ello hemos
realizado rampas de voltaje desde 0 a -100 mV, con un incremento de 10 mV/s. Se realizó una
rampa control en la que se aplicó TTX (0.5 µM) y CsCl (1 mM), en una segunda rampa se
añadió TEA (15 mM) y en una tercera rampa se aplicó además riluzol (300 µM), como
muestra la figura 35.
A la corriente obtenida por la aplicación de la rampa de voltaje en condiciones de
control (B1+TTX+CsCl) le hemos restado la corriente obtenida con la misma rampa pero en

Resultados
59
presencia de TEA para obtener la corriente sensible a TEA (ITEA). Del mismo modo
obtenemos la corriente sensible a riluzol (IRIL), restando la rampa de corriente en presencia de
TEA de aquella obtenida en presencia de TEA y Riluzol, como se muestra en la figura 36A. A
continuación se hallaron las conductancias a cada voltaje y se representaron las curvas de
activación para IRIL y para ITEA (Figura 36B). Las curvas de activación se ajustaron a una
curva sigmoidea y se compararon mediante el test F que nos indicó que son diferentes
significativamente (p = 3.43*10-7
, n = 6). El voltaje al cual la corriente está activada el 50 %
(V1/2) de IRIL es -52.21 ± 1.99 mV y de ITEA es -21.25 ± 2.59 mV, la pendiente de la curva de
activación de IRIL es 10.29 ± 1.89 y de ITEA es 17.22 ± 1.54, el potencial umbral de activación
de IRIL es de -80 mV, mientras que de ITEA es -60 mV y el potencial máximo de activación de
IRIL es -20 mV y de ITEA es 0 mV.
Este experimento demuestra que IM e IRIL son diferentes.
Figura 35: Esquema del protocolo utilizado para construir las curvas de activación. Las curvas de
activación se han construido utilizando tres rampas de voltaje con tratamientos diferentes. La solución de baño
utilizada en la rampa que utilizaremos como control contiene CsCl (1 mM) y TTX (0.5 µM). Posteriormente
añadimos TEA (15 mM) para realizar la segunda rampa de voltaje. La tercera rampa de voltaje además contiene
riluzol (300 µM).
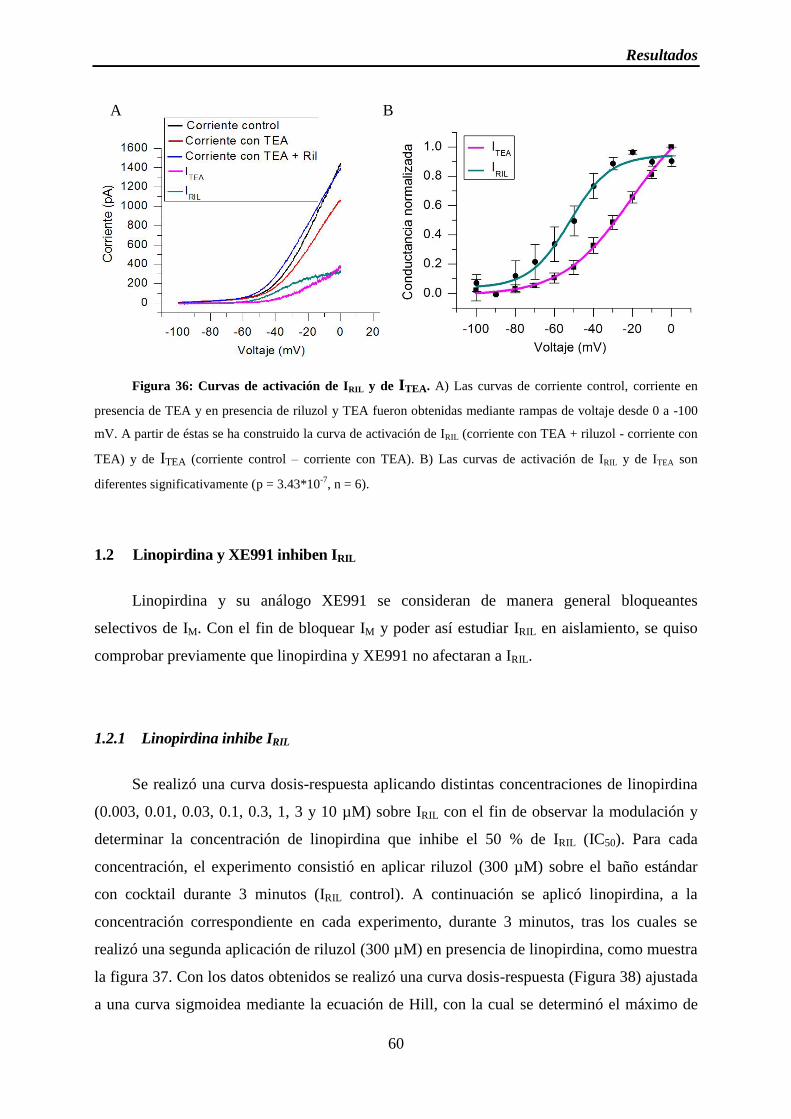
Resultados
60
Figura 36: Curvas de activación de IRIL y de ITEA. A) Las curvas de corriente control, corriente en
presencia de TEA y en presencia de riluzol y TEA fueron obtenidas mediante rampas de voltaje desde 0 a -100
mV. A partir de éstas se ha construido la curva de activación de IRIL (corriente con TEA + riluzol - corriente con
TEA) y de ITEA (corriente control – corriente con TEA). B) Las curvas de activación de IRIL y de ITEA son
diferentes significativamente (p = 3.43*10-7
, n = 6).
1.2 Linopirdina y XE991 inhiben IRIL
Linopirdina y su análogo XE991 se consideran de manera general bloqueantes
selectivos de IM. Con el fin de bloquear IM y poder así estudiar IRIL en aislamiento, se quiso
comprobar previamente que linopirdina y XE991 no afectaran a IRIL.
1.2.1 Linopirdina inhibe IRIL
Se realizó una curva dosis-respuesta aplicando distintas concentraciones de linopirdina
(0.003, 0.01, 0.03, 0.1, 0.3, 1, 3 y 10 µM) sobre IRIL con el fin de observar la modulación y
determinar la concentración de linopirdina que inhibe el 50 % de IRIL (IC50). Para cada
concentración, el experimento consistió en aplicar riluzol (300 µM) sobre el baño estándar
con cocktail durante 3 minutos (IRIL control). A continuación se aplicó linopirdina, a la
concentración correspondiente en cada experimento, durante 3 minutos, tras los cuales se
realizó una segunda aplicación de riluzol (300 µM) en presencia de linopirdina, como muestra
la figura 37. Con los datos obtenidos se realizó una curva dosis-respuesta (Figura 38) ajustada
a una curva sigmoidea mediante la ecuación de Hill, con la cual se determinó el máximo de
A B
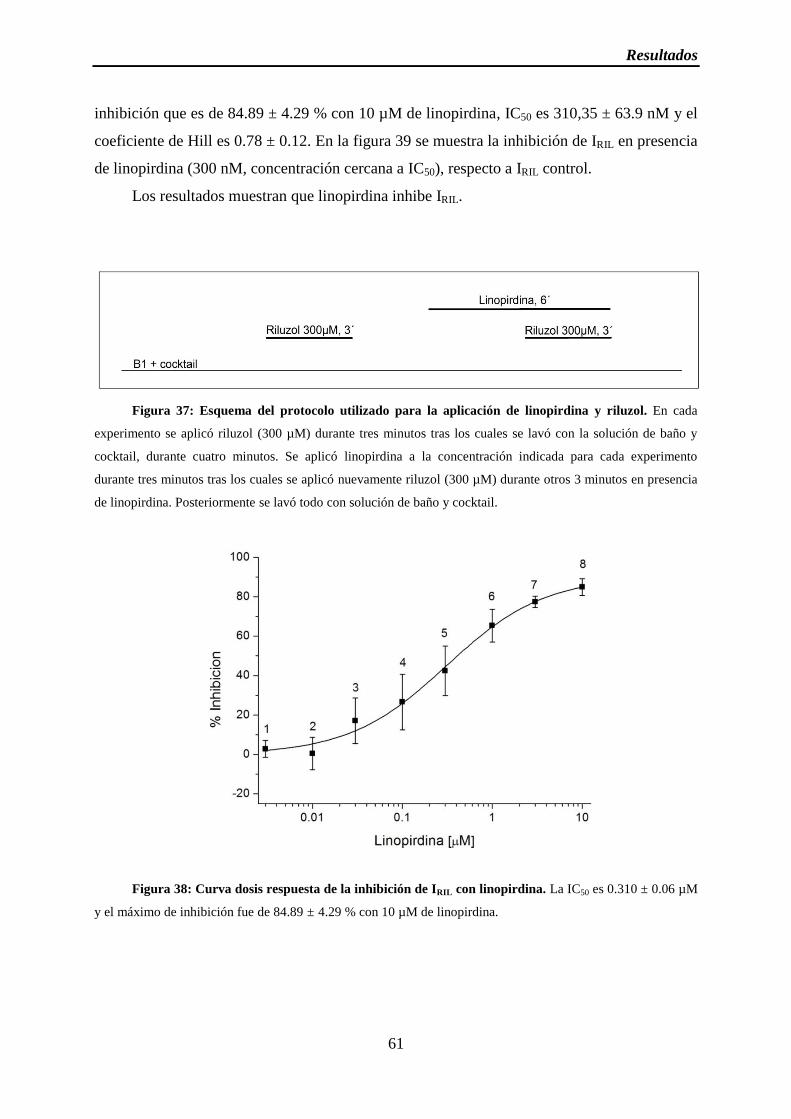
Resultados
61
inhibición que es de 84.89 ± 4.29 % con 10 µM de linopirdina, IC50 es 310,35 ± 63.9 nM y el
coeficiente de Hill es 0.78 ± 0.12. En la figura 39 se muestra la inhibición de IRIL en presencia
de linopirdina (300 nM, concentración cercana a IC50), respecto a IRIL control.
Los resultados muestran que linopirdina inhibe IRIL.
Figura 37: Esquema del protocolo utilizado para la aplicación de linopirdina y riluzol. En cada
experimento se aplicó riluzol (300 µM) durante tres minutos tras los cuales se lavó con la solución de baño y
cocktail, durante cuatro minutos. Se aplicó linopirdina a la concentración indicada para cada experimento
durante tres minutos tras los cuales se aplicó nuevamente riluzol (300 µM) durante otros 3 minutos en presencia
de linopirdina. Posteriormente se lavó todo con solución de baño y cocktail.
Figura 38: Curva dosis respuesta de la inhibición de IRIL con linopirdina. La IC50 es 0.310 ± 0.06 µM
y el máximo de inhibición fue de 84.89 ± 4.29 % con 10 µM de linopirdina.
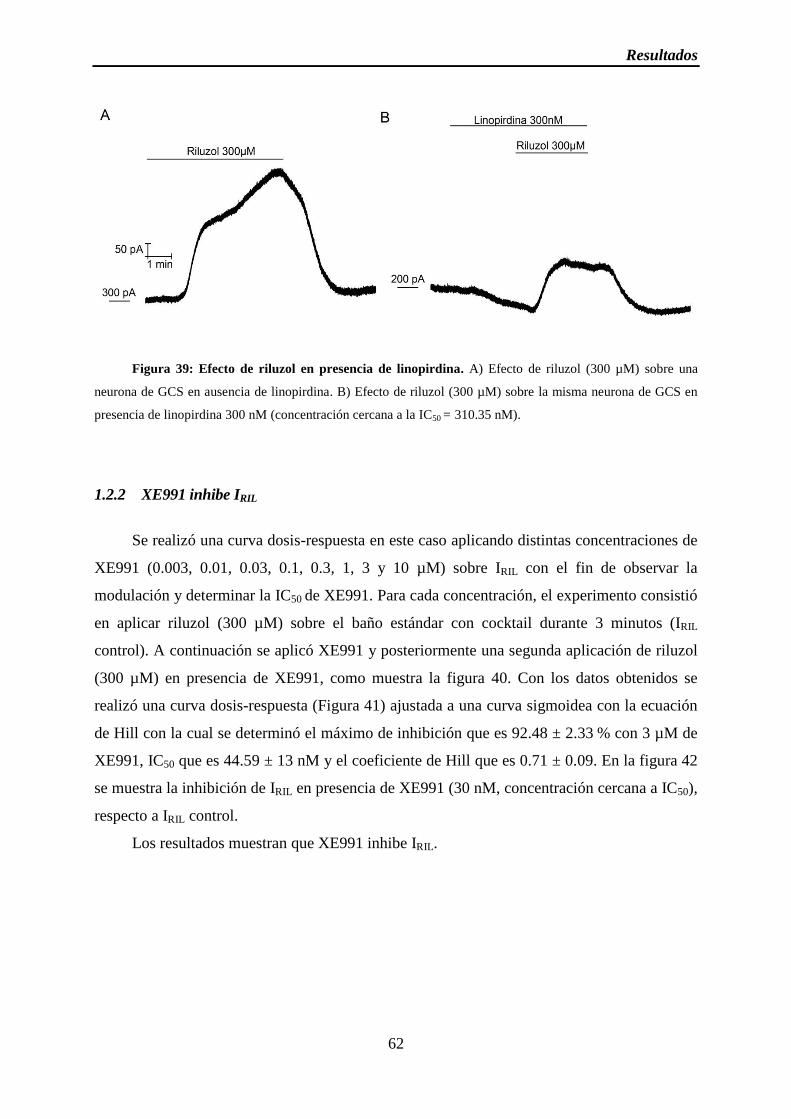
Resultados
62
Figura 39: Efecto de riluzol en presencia de linopirdina. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS en ausencia de linopirdina. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en
presencia de linopirdina 300 nM (concentración cercana a la IC50 = 310.35 nM).
1.2.2 XE991 inhibe IRIL
Se realizó una curva dosis-respuesta en este caso aplicando distintas concentraciones de
XE991 (0.003, 0.01, 0.03, 0.1, 0.3, 1, 3 y 10 µM) sobre IRIL con el fin de observar la
modulación y determinar la IC50 de XE991. Para cada concentración, el experimento consistió
en aplicar riluzol (300 µM) sobre el baño estándar con cocktail durante 3 minutos (IRIL
control). A continuación se aplicó XE991 y posteriormente una segunda aplicación de riluzol
(300 µM) en presencia de XE991, como muestra la figura 40. Con los datos obtenidos se
realizó una curva dosis-respuesta (Figura 41) ajustada a una curva sigmoidea con la ecuación
de Hill con la cual se determinó el máximo de inhibición que es 92.48 ± 2.33 % con 3 µM de
XE991, IC50 que es 44.59 ± 13 nM y el coeficiente de Hill que es 0.71 ± 0.09. En la figura 42
se muestra la inhibición de IRIL en presencia de XE991 (30 nM, concentración cercana a IC50),
respecto a IRIL control.
Los resultados muestran que XE991 inhibe IRIL.
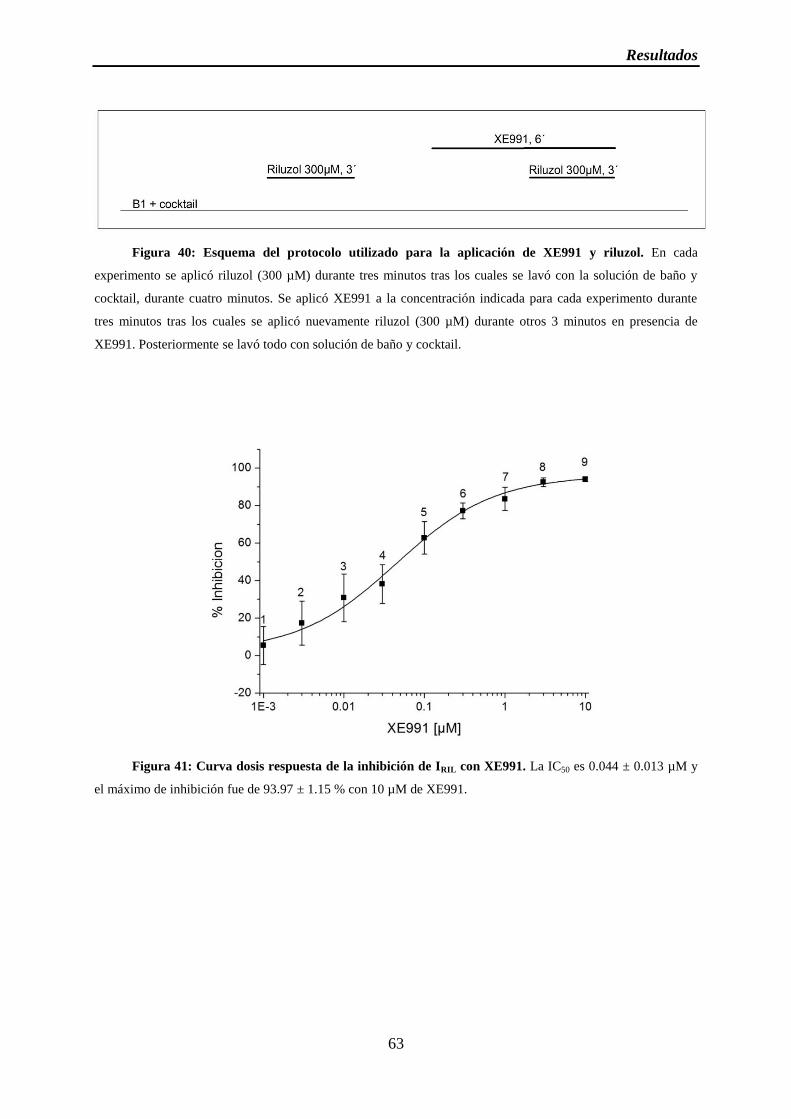
Resultados
63
Figura 40: Esquema del protocolo utilizado para la aplicación de XE991 y riluzol. En cada
experimento se aplicó riluzol (300 µM) durante tres minutos tras los cuales se lavó con la solución de baño y
cocktail, durante cuatro minutos. Se aplicó XE991 a la concentración indicada para cada experimento durante
tres minutos tras los cuales se aplicó nuevamente riluzol (300 µM) durante otros 3 minutos en presencia de
XE991. Posteriormente se lavó todo con solución de baño y cocktail.
Figura 41: Curva dosis respuesta de la inhibición de IRIL con XE991. La IC50 es 0.044 ± 0.013 µM y
el máximo de inhibición fue de 93.97 ± 1.15 % con 10 µM de XE991.
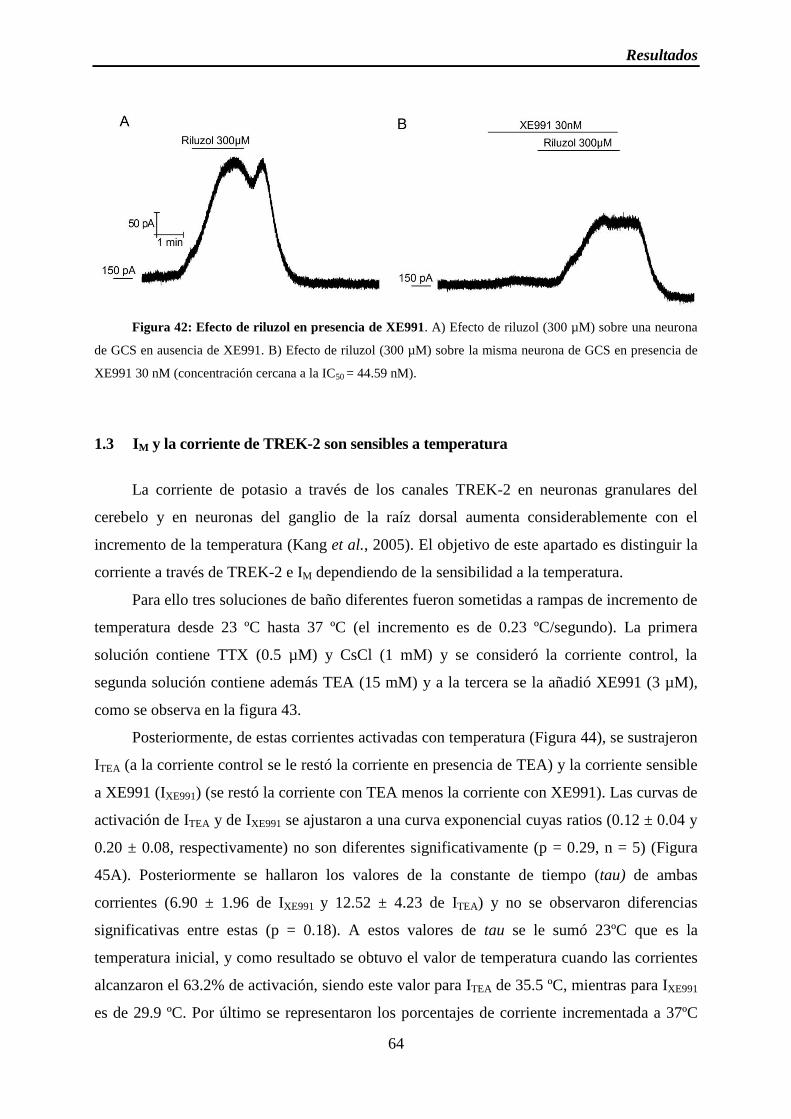
Resultados
64
Figura 42: Efecto de riluzol en presencia de XE991. A) Efecto de riluzol (300 µM) sobre una neurona
de GCS en ausencia de XE991. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en presencia de
XE991 30 nM (concentración cercana a la IC50 = 44.59 nM).
1.3 IM y la corriente de TREK-2 son sensibles a temperatura
La corriente de potasio a través de los canales TREK-2 en neuronas granulares del
cerebelo y en neuronas del ganglio de la raíz dorsal aumenta considerablemente con el
incremento de la temperatura (Kang et al., 2005). El objetivo de este apartado es distinguir la
corriente a través de TREK-2 e IM dependiendo de la sensibilidad a la temperatura.
Para ello tres soluciones de baño diferentes fueron sometidas a rampas de incremento de
temperatura desde 23 ºC hasta 37 ºC (el incremento es de 0.23 ºC/segundo). La primera
solución contiene TTX (0.5 µM) y CsCl (1 mM) y se consideró la corriente control, la
segunda solución contiene además TEA (15 mM) y a la tercera se la añadió XE991 (3 µM),
como se observa en la figura 43.
Posteriormente, de estas corrientes activadas con temperatura (Figura 44), se sustrajeron
ITEA (a la corriente control se le restó la corriente en presencia de TEA) y la corriente sensible
a XE991 (IXE991) (se restó la corriente con TEA menos la corriente con XE991). Las curvas de
activación de ITEA y de IXE991 se ajustaron a una curva exponencial cuyas ratios (0.12 ± 0.04 y
0.20 ± 0.08, respectivamente) no son diferentes significativamente (p = 0.29, n = 5) (Figura
45A). Posteriormente se hallaron los valores de la constante de tiempo (tau) de ambas
corrientes (6.90 ± 1.96 de IXE991 y 12.52 ± 4.23 de ITEA) y no se observaron diferencias
significativas entre estas (p = 0.18). A estos valores de tau se le sumó 23ºC que es la
temperatura inicial, y como resultado se obtuvo el valor de temperatura cuando las corrientes
alcanzaron el 63.2% de activación, siendo este valor para ITEA de 35.5 ºC, mientras para IXE991
es de 29.9 ºC. Por último se representaron los porcentajes de corriente incrementada a 37ºC
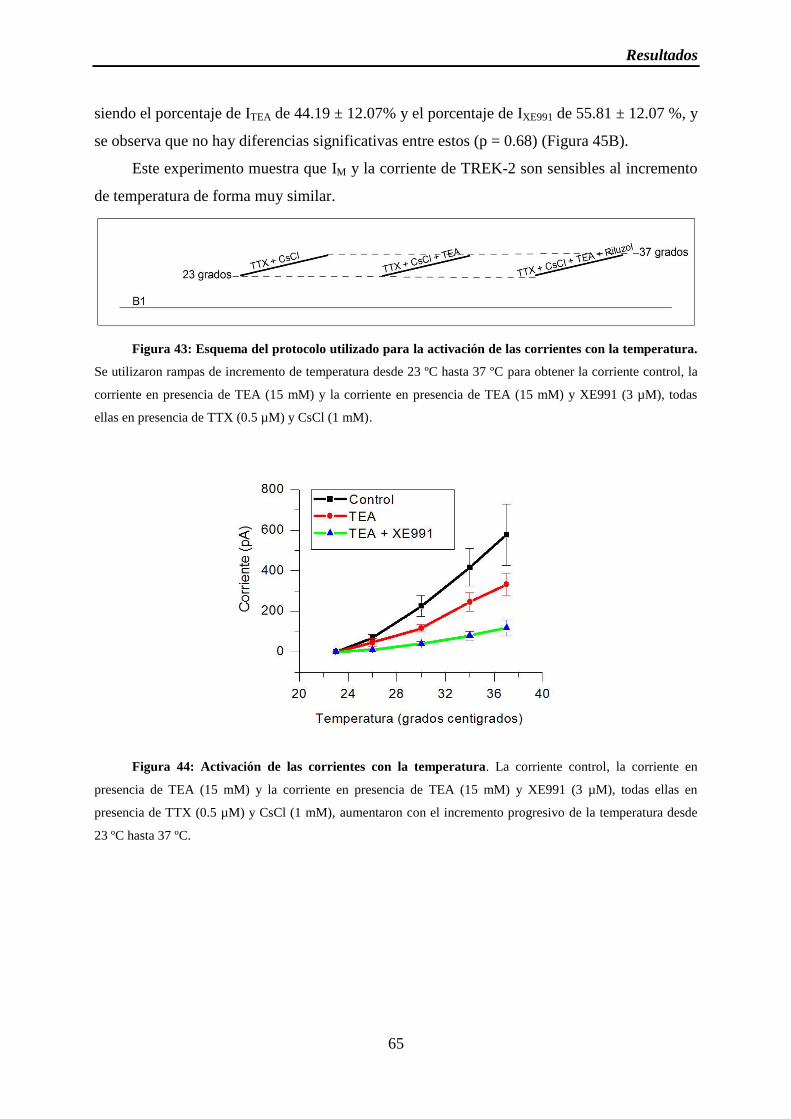
Resultados
65
siendo el porcentaje de ITEA de 44.19 ± 12.07% y el porcentaje de IXE991 de 55.81 ± 12.07 %, y
se observa que no hay diferencias significativas entre estos (p = 0.68) (Figura 45B).
Este experimento muestra que IM y la corriente de TREK-2 son sensibles al incremento
de temperatura de forma muy similar.
Figura 43: Esquema del protocolo utilizado para la activación de las corrientes con la temperatura.
Se utilizaron rampas de incremento de temperatura desde 23 ºC hasta 37 ºC para obtener la corriente control, la
corriente en presencia de TEA (15 mM) y la corriente en presencia de TEA (15 mM) y XE991 (3 µM), todas
ellas en presencia de TTX (0.5 µM) y CsCl (1 mM).
Figura 44: Activación de las corrientes con la temperatura. La corriente control, la corriente en
presencia de TEA (15 mM) y la corriente en presencia de TEA (15 mM) y XE991 (3 µM), todas ellas en
presencia de TTX (0.5 µM) y CsCl (1 mM), aumentaron con el incremento progresivo de la temperatura desde
23 ºC hasta 37 ºC.
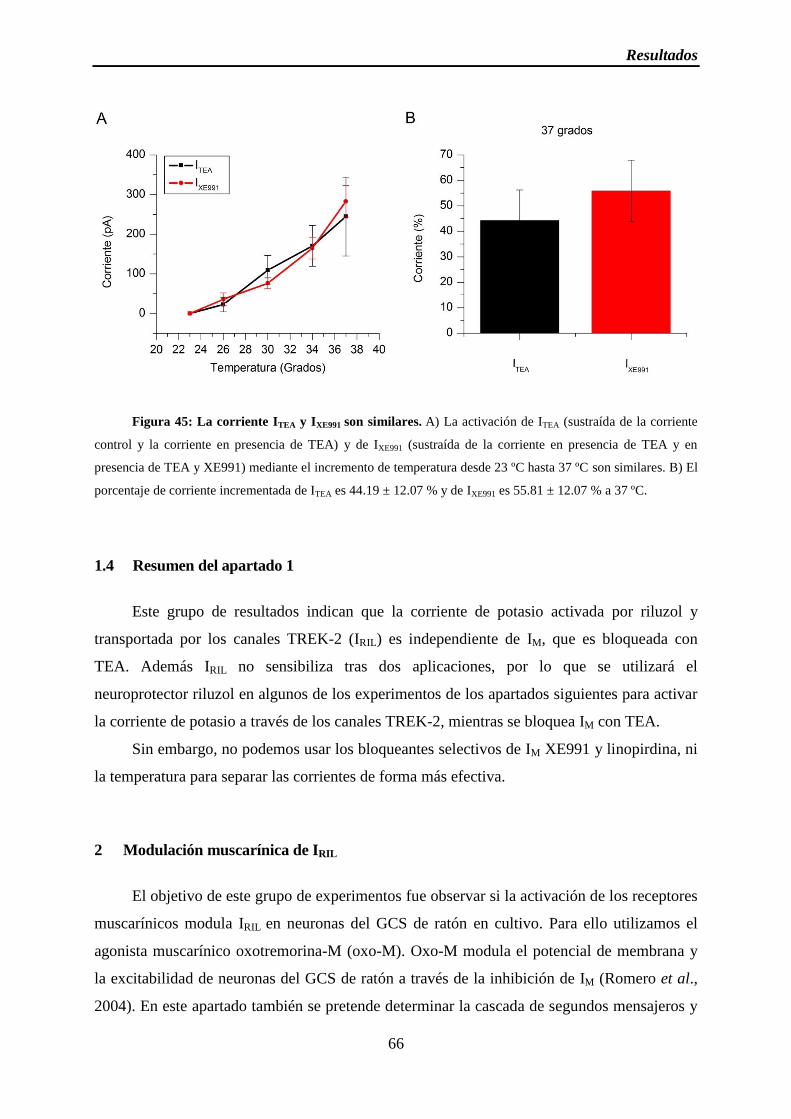
Resultados
66
Figura 45: La corriente ITEA y IXE991 son similares. A) La activación de ITEA (sustraída de la corriente
control y la corriente en presencia de TEA) y de IXE991 (sustraída de la corriente en presencia de TEA y en
presencia de TEA y XE991) mediante el incremento de temperatura desde 23 ºC hasta 37 ºC son similares. B) El
porcentaje de corriente incrementada de ITEA es 44.19 ± 12.07 % y de IXE991 es 55.81 ± 12.07 % a 37 ºC.
1.4 Resumen del apartado 1
Este grupo de resultados indican que la corriente de potasio activada por riluzol y
transportada por los canales TREK-2 (IRIL) es independiente de IM, que es bloqueada con
TEA. Además IRIL no sensibiliza tras dos aplicaciones, por lo que se utilizará el
neuroprotector riluzol en algunos de los experimentos de los apartados siguientes para activar
la corriente de potasio a través de los canales TREK-2, mientras se bloquea IM con TEA.
Sin embargo, no podemos usar los bloqueantes selectivos de IM XE991 y linopirdina, ni
la temperatura para separar las corrientes de forma más efectiva.
2 Modulación muscarínica de IRIL
El objetivo de este grupo de experimentos fue observar si la activación de los receptores
muscarínicos modula IRIL en neuronas del GCS de ratón en cultivo. Para ello utilizamos el
agonista muscarínico oxotremorina-M (oxo-M). Oxo-M modula el potencial de membrana y
la excitabilidad de neuronas del GCS de ratón a través de la inhibición de IM (Romero et al.,
2004). En este apartado también se pretende determinar la cascada de segundos mensajeros y

Resultados
67
las proteínas G implicadas en la modulación muscarínica de IRIL. Los experimentos de este
apartado se realizaron mediante registros electrofisiológicos en la modalidad de sello
perforado en neuronas de GCS de ratón en cultivo mediante protocolos de voltage-clamp con
el potencial de membrana fijado a -30 mV y mediante protocolos de rampas y saltos de
voltaje en la modalidad de canal individual.
2.1 Oxo-M inhibe IRIL
En este experimento se aplicó riluzol (300 µM) antes y después de la aplicación de oxo-
M (10 µM), ambos registros se realizaron en presencia del cocktail de bloqueantes como
muestra la figura 46.
La primera aplicación de riluzol provocó una corriente de salida (260.7 ± 17.2 pA)
significativamente mayor (p = 0.00048) que en presencia de oxo-M (122.5 ± 22.1 pA) (n = 7,
figura 47). El porcentaje de inhibición de la corriente IRIL en presencia de oxo-M fue de 53.20
± 7.10 %. Se comprobó mediante la prueba estadística “t-student” que este porcentaje de
inhibición es significativo (p = 0.00013), figura 54.
La aplicación de oxo-M (10 µM) provocó un descenso significativo (p = 0.012) de -69.7
± 14.7 pA en I(-30) (Figura 47B).
Figura 46: Esquema del protocolo utilizado para la aplicación de oxo-M y riluzol. Se aplicó riluzol
(300 µM) en la solución de baño con cocktail. Tras el lavado durante 4 minutos, se aplicó oxo-M (10 µM)
durante 4 minutos. A continuación se añadió por segunda vez riluzol (300 µM), en presencia de oxo-M, durante
tres minutos tras los cuales se lavó todo con solución de baño y cocktail.
3´
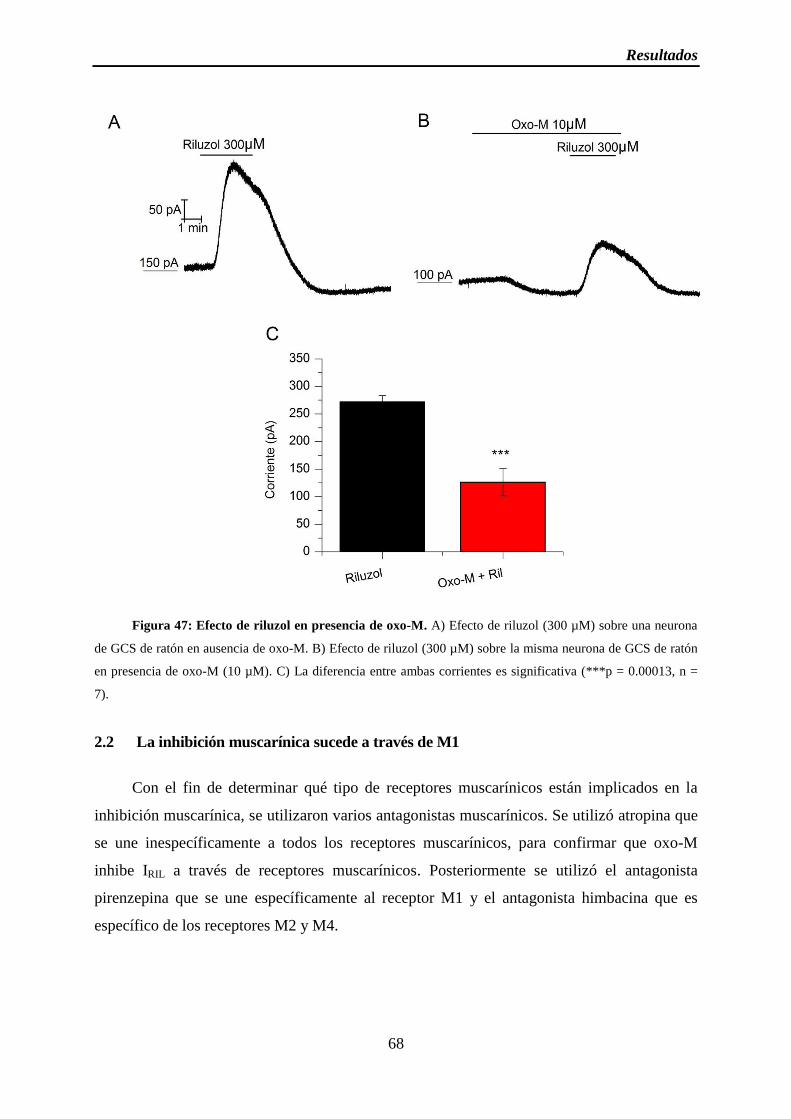
Resultados
68
Figura 47: Efecto de riluzol en presencia de oxo-M. A) Efecto de riluzol (300 µM) sobre una neurona
de GCS de ratón en ausencia de oxo-M. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS de ratón
en presencia de oxo-M (10 µM). C) La diferencia entre ambas corrientes es significativa (***p = 0.00013, n =
7).
2.2 La inhibición muscarínica sucede a través de M1
Con el fin de determinar qué tipo de receptores muscarínicos están implicados en la
inhibición muscarínica, se utilizaron varios antagonistas muscarínicos. Se utilizó atropina que
se une inespecíficamente a todos los receptores muscarínicos, para confirmar que oxo-M
inhibe IRIL a través de receptores muscarínicos. Posteriormente se utilizó el antagonista
pirenzepina que se une específicamente al receptor M1 y el antagonista himbacina que es
específico de los receptores M2 y M4.

Resultados
69
2.2.1 La inhibición sucede a través de receptores muscarínicos
Este experimento consiste en aplicar riluzol (300 µM) en presencia de oxo-M (10 µM) y
de atropina (3 µM), del modo que se muestra en la figura 48, con el fin de comprobar el
efecto de oxo-M cuando se bloquean los receptores muscarínicos.
En este caso IRIL sola (298.80 ± 45.60 pA) no difiere significativamente (p = 0.079, n =
6) de IRIL en presencia de atropina y oxo-M (247.30 ± 31.90 pA), figura 49. La reducción de
IRIL con oxo-M en presencia de atropina fue del 15.50 ± 6.10%. Este porcentaje de inhibición
no es significativo (p = 0.076, n = 6), figura 54.
La aplicación de atropina sola provocó un incremento significativo (p = 0.035, n = 5) de
I(-30) de 19.30 ± 4.10 pA. Posteriormente la aplicación de oxo-M, en presencia de atropina,
provocó en esta corriente una variación no significativa (p = 0.19, n = 5) de -7.90 ± 4.30 pA
(Figura 49B).
Se comprobó que la presencia de atropina, no influye sobre IRIL cuando se realizó el
mismo experimento en ausencia de oxo-M. En este grupo de células riluzol (300 µM) activó
una corriente de 266.10 ± 63.10 pA en ausencia de atropina y de 206.80 ± 52.70 pA en
presencia de atropina (p = 0.079, n=4).
Este experimento demuestra que la inhibición de IRIL por oxo-M sucede a través de
receptores muscarínicos ya que cuando se bloquean con atropina, la inhibición desaparece.
Figura 48: Esquema del protocolo utilizado para la aplicación de riluzol, atropina y oxo-M. Se
aplicó riluzol (300 µM) en la solución de baño en presencia de cocktail. Tras el lavado durante 7 minutos, se
aplicó atropina (3 µM). Trascurridos 4 minutos desde la aplicación de atropina, se añadió oxo-M (10 µM)
durante 4 minutos más. A continuación se añadió por segunda vez riluzol (300 µM), en presencia de oxo-M y
atropina, durante 3 minutos. Finalmente se lavó todo con solución de baño y cocktail.
3´
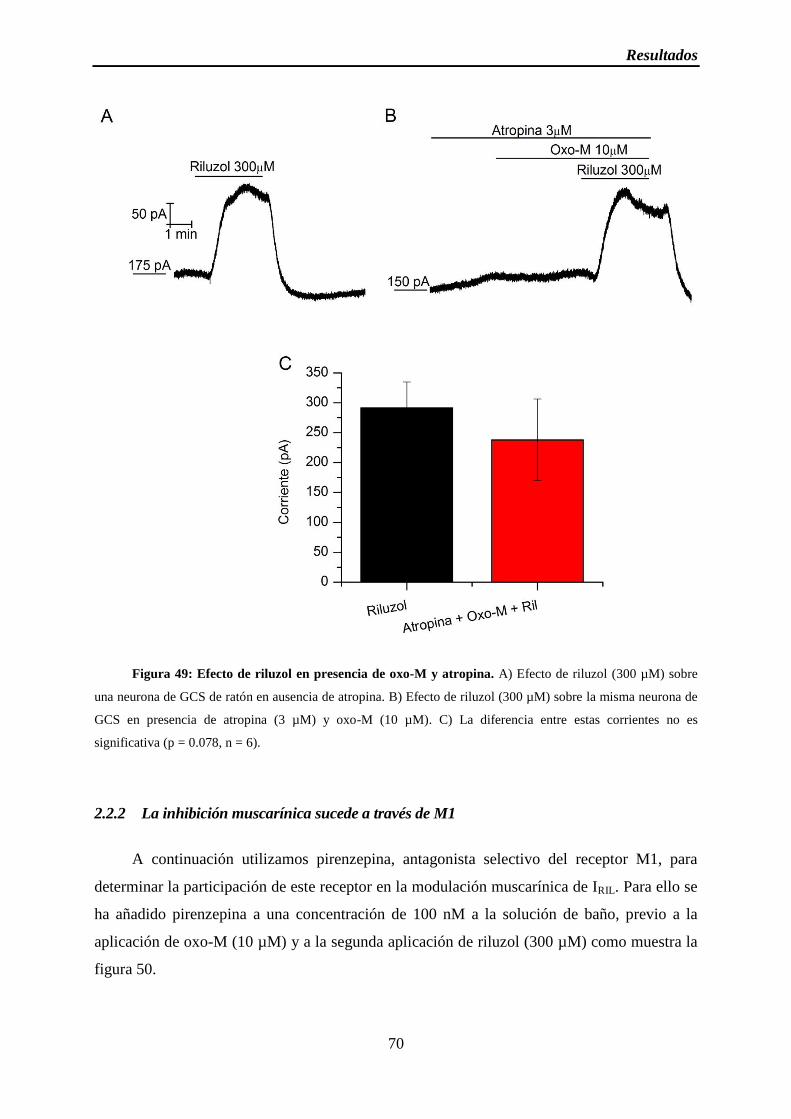
Resultados
70
Figura 49: Efecto de riluzol en presencia de oxo-M y atropina. A) Efecto de riluzol (300 µM) sobre
una neurona de GCS de ratón en ausencia de atropina. B) Efecto de riluzol (300 µM) sobre la misma neurona de
GCS en presencia de atropina (3 µM) y oxo-M (10 µM). C) La diferencia entre estas corrientes no es
significativa (p = 0.078, n = 6).
2.2.2 La inhibición muscarínica sucede a través de M1
A continuación utilizamos pirenzepina, antagonista selectivo del receptor M1, para
determinar la participación de este receptor en la modulación muscarínica de IRIL. Para ello se
ha añadido pirenzepina a una concentración de 100 nM a la solución de baño, previo a la
aplicación de oxo-M (10 µM) y a la segunda aplicación de riluzol (300 µM) como muestra la
figura 50.

Resultados
71
En este experimento se ha visto que IRIL control (240.50 ± 27.20 pA) no es diferente
significativamente (p = 0.078, n = 7) a IRIL en presencia de oxo-M y de pirenzepina (215.70 ±
32.10 pA) (Figura 51). El porcentaje de inhibición en este caso tampoco es significativo (p =
0.49) siendo del 12.20 ± 8.80 %, figura 54.
Se aplicó pirenzepina en ausencia de oxo-M para observar si pirenzepina afecta
directamente a IRIL. La aplicación de pirenzepina, en ausencia de oxo-M, no afectó
significativamente (p = 0.47) a IRIL, siendo IRIL control de 162.10 ± 20.60 pA y en presencia
de pirenzepina de 142.40 ± 17.50 pA (n = 3).
La aplicación de pirenzepina incrementó I(-30) de forma significativa (p = 0.036, n = 6,
figura 51B) en 21.4 ± 4.40 pA, mientras que la aplicación de oxo-M, en presencia de
pirenzepina, provocó un descenso no significativo (p = 0.068, n = 6) sobre esta corriente de -
19.87 ± 5.72 pA, de manera muy similar a lo que ocurre al utilizar atropina (ver apartado
anterior).
Los resultados de este experimento sugieren que la inhibición de IRIL por oxo-M es
mediada por receptores muscarínicos de tipo M1.
Figura 50: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y pirenzepina. Se
aplicó riluzol (300 µM) durante 3 minutos en la solución de baño con cocktail. Tras el lavado durante 7 minutos,
se aplicó pirenzepina (100 nM). Trascurridos 4 minutos desde la aplicación de pirenzepina, se añadió oxo-M (10
µM) durante 4 minutos, a continuación se añadió por segunda vez riluzol (300 µM), en presencia de oxo-M y
pirenzepina, durante tres minutos. Finalmente se lavó todo con solución de baño y cocktail.
3´
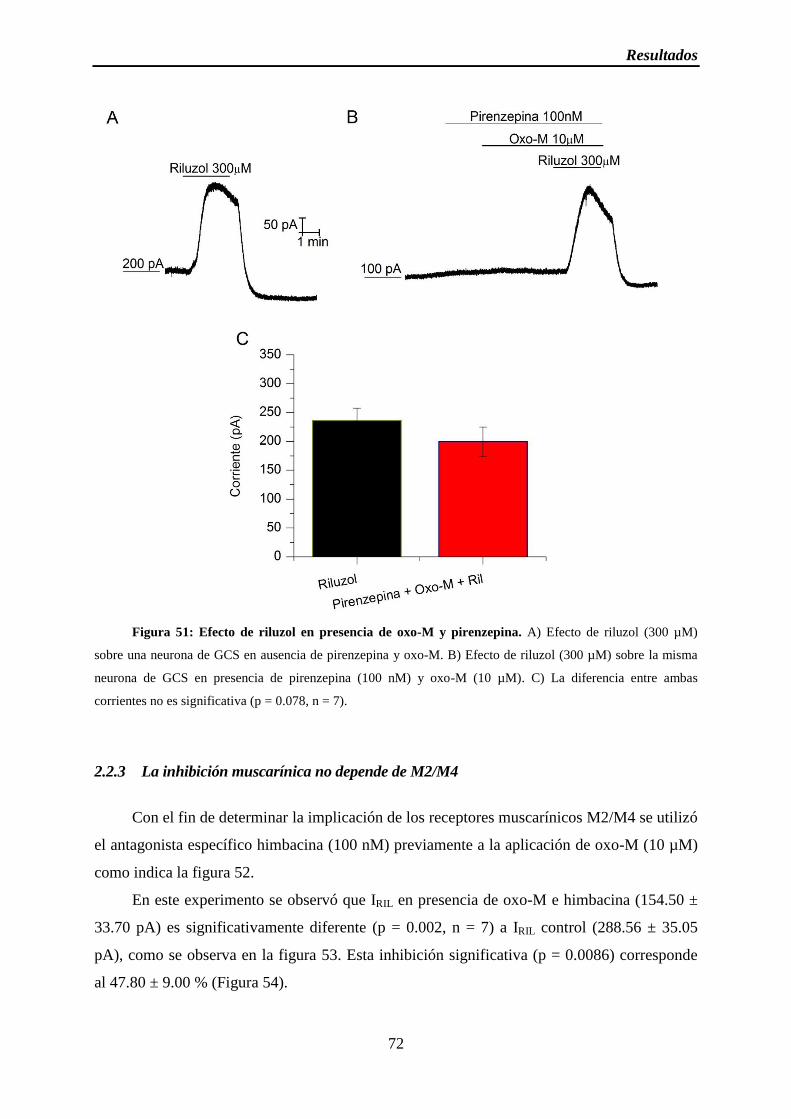
Resultados
72
Figura 51: Efecto de riluzol en presencia de oxo-M y pirenzepina. A) Efecto de riluzol (300 µM)
sobre una neurona de GCS en ausencia de pirenzepina y oxo-M. B) Efecto de riluzol (300 µM) sobre la misma
neurona de GCS en presencia de pirenzepina (100 nM) y oxo-M (10 µM). C) La diferencia entre ambas
corrientes no es significativa (p = 0.078, n = 7).
2.2.3 La inhibición muscarínica no depende de M2/M4
Con el fin de determinar la implicación de los receptores muscarínicos M2/M4 se utilizó
el antagonista específico himbacina (100 nM) previamente a la aplicación de oxo-M (10 µM)
como indica la figura 52.
En este experimento se observó que IRIL en presencia de oxo-M e himbacina (154.50 ±
33.70 pA) es significativamente diferente (p = 0.002, n = 7) a IRIL control (288.56 ± 35.05
pA), como se observa en la figura 53. Esta inhibición significativa (p = 0.0086) corresponde
al 47.80 ± 9.00 % (Figura 54).

Resultados
73
Se realizó otro experimento similar en el cual se aplicó himbacina sin oxo-M para
observar si este antagonista afecta a IRIL. Se observó que IRIL control (205.70 ± 17.90 pA) no
difiere significativamente (p = 0.382, n = 10) de IRIL en presencia de himbacina (180.90 ±
26.40 pA). La aplicación de himbacina provocó un incremento no significativo (p = 0.12, n =
7) de I(-30) de 9.30 ± 5.20 pA. Sin embargo, la aplicación de oxo-M provocó una disminución
significativa (p = 0.023, n = 7) de I(-30) de -79.90 ± 20.50 pA, como se observa en la figura
53B.
Estos resultados demuestran que la inhibición de IRIL por oxo-M no sucede a través de
los receptores muscarínicos M2/M4.
Figura 52: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M e himbacina. Se
aplicó riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se
aplicó himbacina (100 nM). Trascurridos 4 minutos desde la aplicación de himbacina, se añadió oxo-M (10 µM)
durante 4 minutos, a continuación se añadió por segunda vez riluzol (300 µM), en presencia de oxo-M e
himbacina, durante tres minutos. Posteriormente se lavó todo con solución de baño y cocktail.
3´
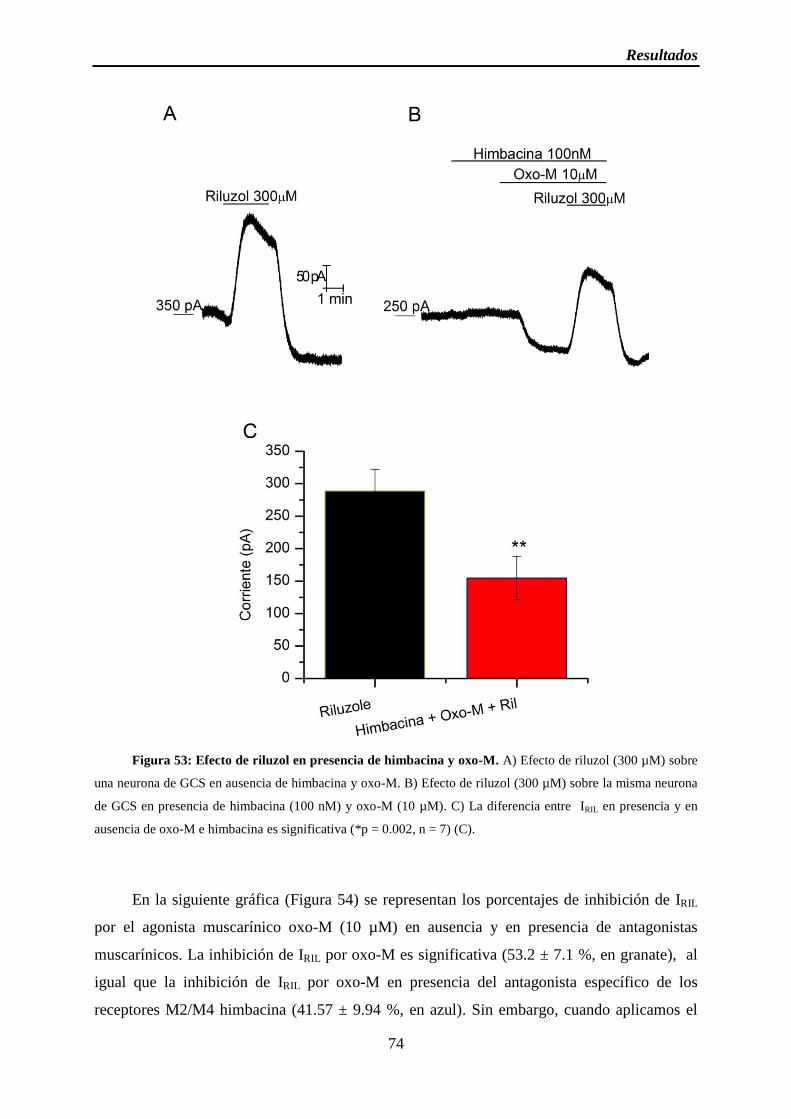
Resultados
74
Figura 53: Efecto de riluzol en presencia de himbacina y oxo-M. A) Efecto de riluzol (300 µM) sobre
una neurona de GCS en ausencia de himbacina y oxo-M. B) Efecto de riluzol (300 µM) sobre la misma neurona
de GCS en presencia de himbacina (100 nM) y oxo-M (10 µM). C) La diferencia entre IRIL en presencia y en
ausencia de oxo-M e himbacina es significativa (*p = 0.002, n = 7) (C).
En la siguiente gráfica (Figura 54) se representan los porcentajes de inhibición de IRIL
por el agonista muscarínico oxo-M (10 µM) en ausencia y en presencia de antagonistas
muscarínicos. La inhibición de IRIL por oxo-M es significativa (53.2 ± 7.1 %, en granate), al
igual que la inhibición de IRIL por oxo-M en presencia del antagonista específico de los
receptores M2/M4 himbacina (41.57 ± 9.94 %, en azul). Sin embargo, cuando aplicamos el
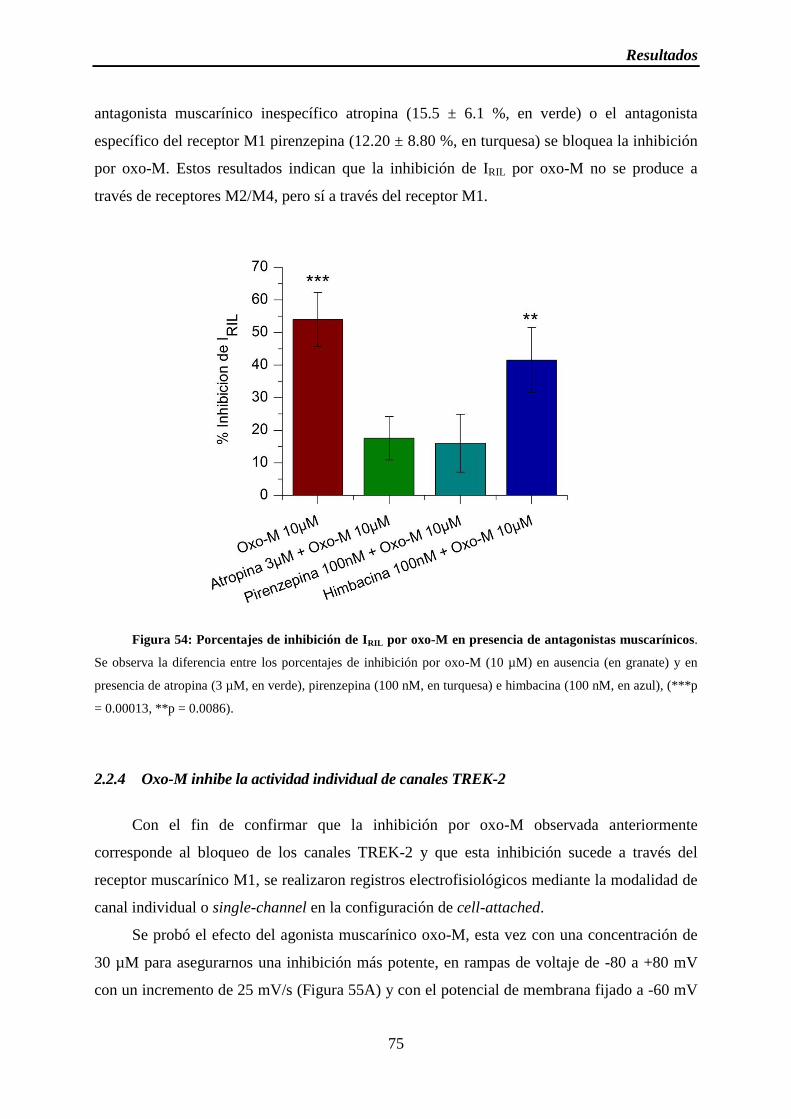
Resultados
75
antagonista muscarínico inespecífico atropina (15.5 ± 6.1 %, en verde) o el antagonista
específico del receptor M1 pirenzepina (12.20 ± 8.80 %, en turquesa) se bloquea la inhibición
por oxo-M. Estos resultados indican que la inhibición de IRIL por oxo-M no se produce a
través de receptores M2/M4, pero sí a través del receptor M1.
Figura 54: Porcentajes de inhibición de IRIL por oxo-M en presencia de antagonistas muscarínicos.
Se observa la diferencia entre los porcentajes de inhibición por oxo-M (10 µM) en ausencia (en granate) y en
presencia de atropina (3 µM, en verde), pirenzepina (100 nM, en turquesa) e himbacina (100 nM, en azul), (***p
= 0.00013, **p = 0.0086).
2.2.4 Oxo-M inhibe la actividad individual de canales TREK-2
Con el fin de confirmar que la inhibición por oxo-M observada anteriormente
corresponde al bloqueo de los canales TREK-2 y que esta inhibición sucede a través del
receptor muscarínico M1, se realizaron registros electrofisiológicos mediante la modalidad de
canal individual o single-channel en la configuración de cell-attached.
Se probó el efecto del agonista muscarínico oxo-M, esta vez con una concentración de
30 µM para asegurarnos una inhibición más potente, en rampas de voltaje de -80 a +80 mV
con un incremento de 25 mV/s (Figura 55A) y con el potencial de membrana fijado a -60 mV

Resultados
76
(valor cercano al potencial de membrana de reposo en las neuronas del GCS de ratón (Romero
et al., 2004)) (Figura 55B). Mediante el protocolo de rampas de voltaje y en ausencia de oxo-
M podemos observar que el canal presenta rectificación de entrada, típica de los canales
TREK-2, observándose una conductancia de 127.5 ± 6.9 pS a -60 mV y de 48.4 ± 6.5 pS a
+60 mV (n = 5). En presencia de oxo-M (30 µM) el canal se bloqueó completamente,
recuperándose la conductancia del canal con el lavado de oxo-M. Sin embargo oxo-M en
presencia de pirenzepina 100 nM no tuvo ningún efecto sobre el canal, al igual que la
aplicación de pirenzepina sola.
Se calculó la probabilidad de apertura (NPo) cuando la célula fue fijada a -60 mV. La
aplicación de oxo-M bloqueó significativamente la probabilidad de apertura del canal desde
0.027 ± 0.010 a 0.008 ± 0.004 (p = 0.037, n = 10). Este efecto desaparece con el lavado de
oxo-M y con la aplicación de pirenzepina (100 nM) en presencia de oxo-M. Sin embargo, la
amplitud de la corriente no disminuyó significativamente con la aplicación de oxo-M, siendo
de 8.21 ± 0.36 pA en ausencia de oxo-M y de 7.94 ± 0.45 pA en presencia de éste (p = 0.105,
n = 10), ni tampoco afectó al tiempo de apertura del canal TREK-2 siendo de 2.21 ± 0.34 ms
en ausencia de oxo-M y de 1.95 ± 0.27 ms en presencia de este agonista muscarínico (p =
0.183, n = 10).

Resultados
77
Figura 55: Efecto de oxo-M y pirenzepina sobre la actividad de un canal TREK-2. A) Reducción de
la actividad del canal en presencia de oxo-M (30 µM) en rampas de voltaje desde -80 a 80 mV. La presencia de
pirenzepina (100 nM) bloquea el efecto de oxo-M. B) Reducción de la actividad del canal en presencia de oxo-M
(30 µM) y bloqueo de este efecto con pirenzepina (100 nM) con el potencial de membrana fijado a -60 mV.
2.3 La inhibición muscarínica sucede a través de proteínas G resistentes a PTX
Una vez determinado el tipo de receptor muscarínico implicado en la inhibición de IRIL,
se investigó el siguiente paso de la vía. Se utilizó la toxina Pertussis (PTX) para determinar
qué tipo de proteína G está implicada. La PTX bloquea las proteínas Gi, Go y Gt pero no
afecta a Gq/11.
La adición de riluzol (300 µM), en presencia de PTX (1 µg/ml) pre-incubada durante
17-30 horas (Figura 56), generó una corriente de salida de 162.45 ± 14.83 pA (n = 11) como
representa la figura 57A. La segunda aplicación de riluzol, en presencia de oxo-M (10 µM) y

Resultados
78
PTX (1 µg/ml), generó una corriente de salida de 98.83 ± 16.35 pA como se observa en la
figura 57B. La diferencia entre IRIL en presencia y en ausencia de oxo-M fue significativa (p =
0.0011, n = 11) como se muestra en la figura 58A. El porcentaje de inhibición por oxo-M en
presencia de PTX fue significativo (p = 2.17*10-4
) de 39.71 ± 7.05 %. Este porcentaje de
inhibición no fue significativamente diferente (p = 0.19) al porcentaje de inhibición por oxo-
M control (53.20 ± 7.10 %, ver apartado 2.1), como se observa en la figura 58B.
La aplicación de oxo-M, en presencia de PTX, provocó un descenso significativo (p =
0.00104, n = 11) de I(-30) de 69.44 ± 15.22 pA, figura 57B.
Se realizó un control positivo para determinar la efectividad de PTX que consistió en
cuantificar y comparar la inhibición de IM por oxo-M (10 µM) en presencia y en ausencia de
PTX (1 µg/ml) pre-incubada, ya que la modulación muscarínica de IM sucede a través de
proteínas G resistentes a PTX. Para ello se utilizaron rampas de voltaje desde -30 a -100 mV y
de 7 segundos de duración en presencia de CsCl (1 mM). Los resultados muestran que la
inhibición de IM por oxo-M en ausencia (60.05 ±8.72 %, n = 6) y en presencia de PTX (34.73
± 5.87 %, n = 5) son diferentes significativamente (p = 0.046), por lo que se afirma que la pre-
incubación de PTX en las neuronas del GCS en cultivo primario fue efectiva.
Estos resultados descartan la implicación de las proteínas Gi, Go y Gt en la inhibición de
IRIL por oxo-M sugiriendo que la proteína G implicada es la Gq/11.
Figura 56: Esquema del protocolo utilizado para la aplicación de riluzol y oxo-M en presencia de
PTX. Se aplicó riluzol (300 µM) en la solución de baño estándar en presencia del cocktail sobre el cultivo
celular pre-incubado con PTX (1 µg/ml) durante 17-30 horas. Tras el lavado de riluzol durante 7 minutos se
aplicó oxo-M (10 µM) durante 4 minutos previos a la aplicación de riluzol. Luego se añadió por segunda vez
riluzol (300 µM) en presencia de oxo-M durante 3 minutos, tras los cuales, se lavó todo con solución de baño y
cocktail durante 7 minutos. Hay que tener en cuenta que PTX siempre está presente en el interior celular pues no
se pierde con el lavado.
3´
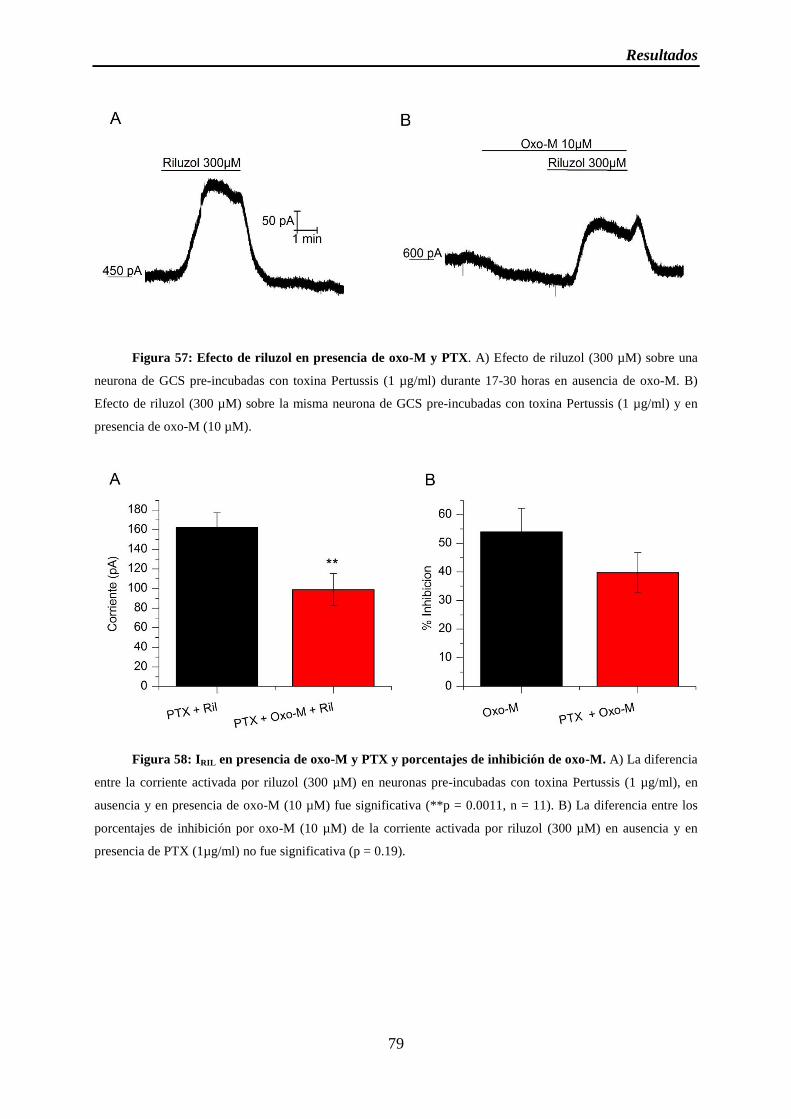
Resultados
79
Figura 57: Efecto de riluzol en presencia de oxo-M y PTX. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS pre-incubadas con toxina Pertussis (1 µg/ml) durante 17-30 horas en ausencia de oxo-M. B)
Efecto de riluzol (300 µM) sobre la misma neurona de GCS pre-incubadas con toxina Pertussis (1 µg/ml) y en
presencia de oxo-M (10 µM).
Figura 58: IRIL en presencia de oxo-M y PTX y porcentajes de inhibición de oxo-M. A) La diferencia
entre la corriente activada por riluzol (300 µM) en neuronas pre-incubadas con toxina Pertussis (1 µg/ml), en
ausencia y en presencia de oxo-M (10 µM) fue significativa (**p = 0.0011, n = 11). B) La diferencia entre los
porcentajes de inhibición por oxo-M (10 µM) de la corriente activada por riluzol (300 µM) en ausencia y en
presencia de PTX (1µg/ml) no fue significativa (p = 0.19).

Resultados
80
2.4 PLC está implicada en la inhibición muscarínica
Las proteínas Gq/11 están principalmente acopladas a la activación de la PLC por lo que
el siguiente paso fue determinar la implicación de la PLC en la vía de inhibición de IRIL. Para
ello se utilizó edelfosina (ET-18-OCH3), como muestra la figura 59, para bloquear la PLC.
En este experimento se observa que IRIL control (159.81 ± 30.33 pA) no es diferente
significativamente (p = 0.71, n = 9) a IRIL (146.26 ± 21.78 pA) en presencia de edelfosina (10
µM) y de oxo-M (10 µM), como muestra las figuras 60 y 61A. El porcentaje de inhibición de
IRIL por oxo-M en presencia de edelfosina fue de 18.27 ± 9.67 %. Este porcentaje de
inhibición no fue significativo (p = 0.18, n = 9), pero es significativamente diferente (p =
0.01) al porcentaje de inhibición por oxo-M control (53.20 ± 7.10 %, ver apartado 2.1) como
se observa en la figura 61B.
La aplicación de edelfosina (Figura 60B) provocó un descenso significativo (p =
0.0011) de I(-30) de -135.79 ± 16.17 pA en 5 de las 9 neuronas registradas, mientras que en el
resto de las células no produjo cambios significativos. Con la aplicación de oxo-M también se
observó un descenso significativo (p = 0.0108) de I(-30) de -34.26 ± 3.59 pA en 3 de las 9
células registradas, mientras que en las 5 restantes no se observaron cambios significativos.
La aplicación de edelfosina sobre IRIL control en ausencia de oxo-M no provocó cambios
significativos (202.41 ± 25.20 pA y 179.39 ± 43.50 pA respetivamente, p = 0.55, n = 5).
Los resultados de este apartado indican que el enzima PLC está implicado en la
inhibición de IRIL por oxo-M.
Figura 59: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y edelfosina. Se
aplicó riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se
aplicó edelfosina (10 µM). Trascurridos 12 minutos desde la aplicación de edelfosina, se añadió oxo-M (10 µM)
durante 4 minutos previos a la aplicación de riluzol. Luego se añadió por segunda vez riluzol (300 µM), en
presencia de oxo-M y edelfosina, durante 3 minutos tras los cuales se lavó todo con solución de baño y cocktail
durante 7 minutos.
3´
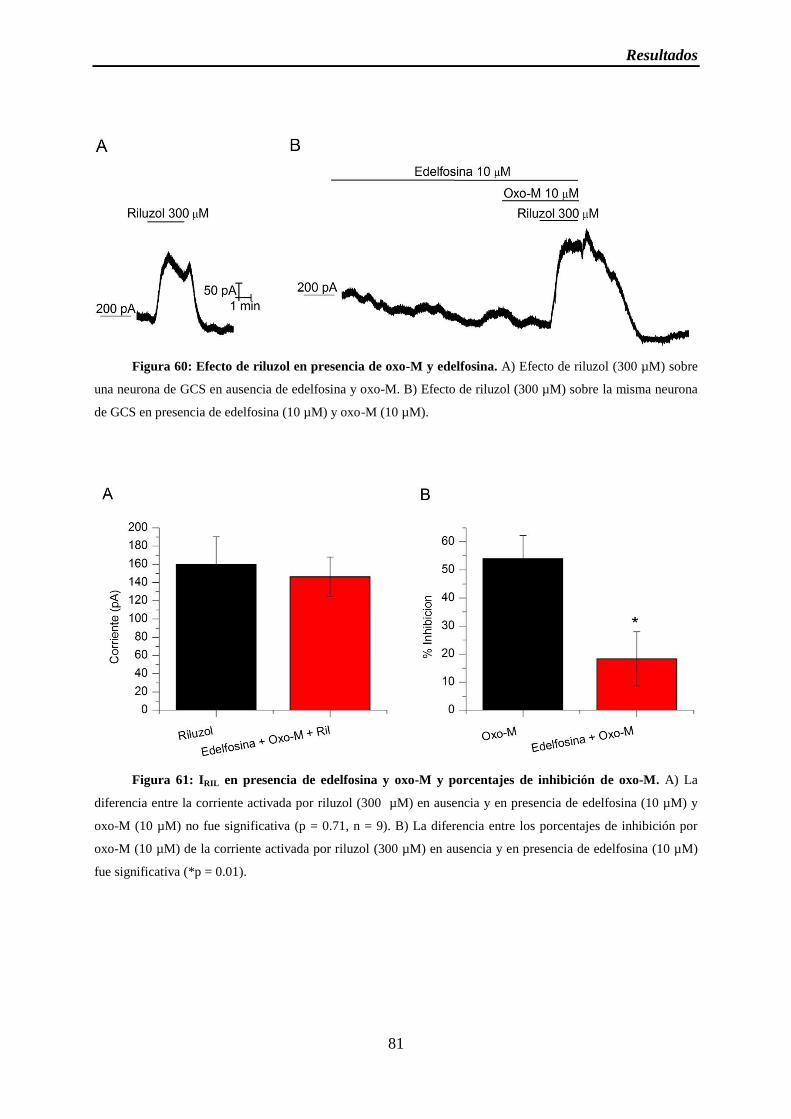
Resultados
81
Figura 60: Efecto de riluzol en presencia de oxo-M y edelfosina. A) Efecto de riluzol (300 µM) sobre
una neurona de GCS en ausencia de edelfosina y oxo-M. B) Efecto de riluzol (300 µM) sobre la misma neurona
de GCS en presencia de edelfosina (10 µM) y oxo-M (10 µM).
Figura 61: IRIL en presencia de edelfosina y oxo-M y porcentajes de inhibición de oxo-M. A) La
diferencia entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de edelfosina (10 µM) y
oxo-M (10 µM) no fue significativa (p = 0.71, n = 9). B) La diferencia entre los porcentajes de inhibición por
oxo-M (10 µM) de la corriente activada por riluzol (300 µM) en ausencia y en presencia de edelfosina (10 µM)
fue significativa (*p = 0.01).

Resultados
82
2.5 PKC no participa en la inhibición muscarínica
El enzima PLC hidroliza PIP2 para dar DAG e IP3. DAG, junto con los iones calcio,
activan la PKC, por lo que hemos investigado en este apartado la implicación de la PKC en la
inhibición muscarínica de IRIL. En este experimento se utilizó bisindolilmaleimida, como
muestra la figura 62, para bloquear PKC.
Los resultados muestran que IRIL control (172.83 ± 20.42 pA) se inhibe
significativamente (p = 0.014, n = 6) en presencia de bisindolilmaleimida (300 nM) y de oxo-
M (10 µM) (81.94 ± 15.65 pA), como se observa en las figuras 63 y 64A. El porcentaje de
inhibición de IRIL por oxo-M en presencia de bisindolilmaleimida (50.80 ± 3.81 %) es
significativo (p = 5.66*10-4
, n = 6) y no muestra diferencia significativa (p = 0.737) con el
porcentaje de inhibición por oxo-M (53.20 ± 7.10 %, ver apartado 2.1), figura 64B.
La aplicación de bisindolilmaleimida provocó un incremento no significativo (p = 0.42,
n = 6) de I(-30) (9.00 ± 1.60 pA). Posteriormente se aplicó oxo-M y se observó que I(-30) se
redujo (-46.30 ± 9.49 pA) de forma significativa (p = 0.026, n=6) (Figura 63B). Mediante otro
experimento se comprobó que la aplicación de bisindolilmaleimida (300 nM) no afectó de
forma significativa (p = 0.28, n = 4) a IRIL en ausencia de oxo-M (179.42 ± 41.20 pA y 135.65
± 24.83 pA, en ausencia y en presencia de bisindolilmaleimida respectivamente).
Se realizó un control positivo con el fin de observar la efectividad de
bisindolilmaleimida sobre la modulación muscarínica de IM. Para ello se utilizó un protocolo
de voltage-clamp de rampas de voltaje desde -30 a -100 mV y de 7 segundos de duración en
presencia de CsCl (1 mM). Los resultados muestran que la inhibición de IM por oxo-M (10
µM) en ausencia (60.05 ± 8.72 %, n = 6) y en presencia de bisindolilmaleimida (30.94 ±
5.10 %, n = 4) son diferentes significativamente (p = 0.037). Este resultado indica que
bisindolilmaleimida tiene efectos sobre las neuronas del GCS.
Los resultados de este experimento indican que la PKC no participa en la inhibición de
IRIL por oxo-M.
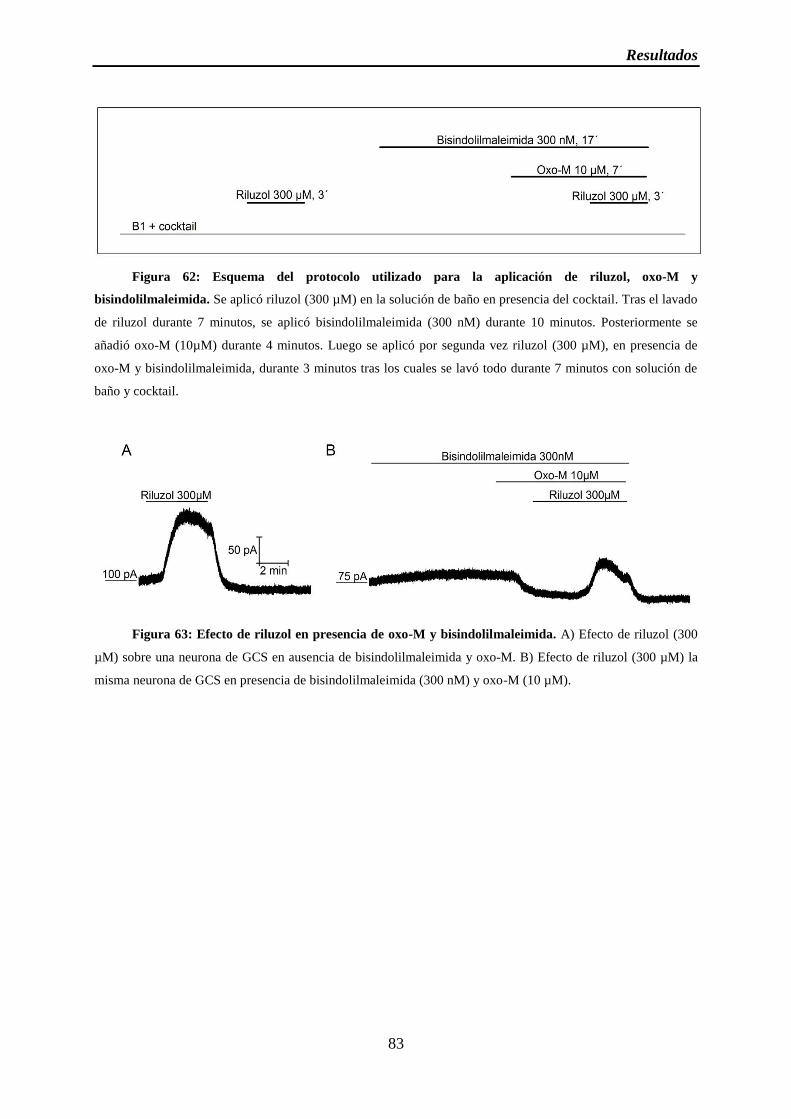
Resultados
83
Figura 62: Esquema del protocolo utilizado para la aplicación de riluzol, oxo-M y
bisindolilmaleimida. Se aplicó riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado
de riluzol durante 7 minutos, se aplicó bisindolilmaleimida (300 nM) durante 10 minutos. Posteriormente se
añadió oxo-M (10µM) durante 4 minutos. Luego se aplicó por segunda vez riluzol (300 µM), en presencia de
oxo-M y bisindolilmaleimida, durante 3 minutos tras los cuales se lavó todo durante 7 minutos con solución de
baño y cocktail.
Figura 63: Efecto de riluzol en presencia de oxo-M y bisindolilmaleimida. A) Efecto de riluzol (300
µM) sobre una neurona de GCS en ausencia de bisindolilmaleimida y oxo-M. B) Efecto de riluzol (300 µM) la
misma neurona de GCS en presencia de bisindolilmaleimida (300 nM) y oxo-M (10 µM).
3´
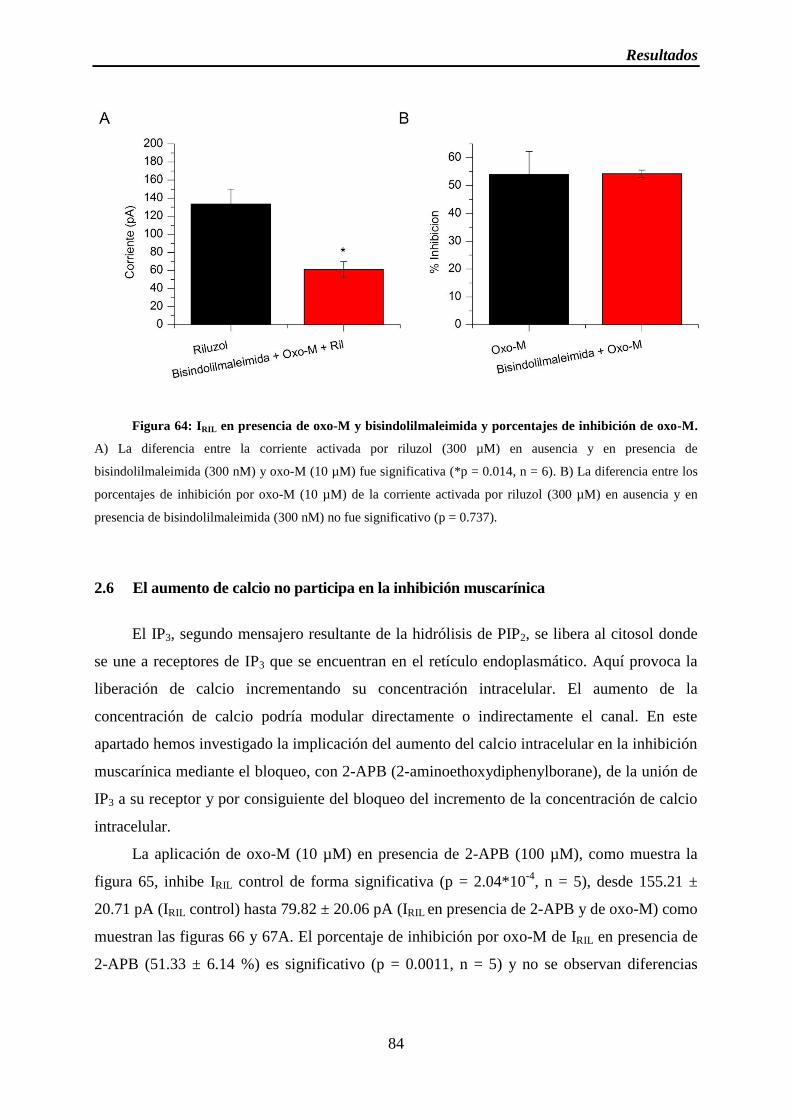
Resultados
84
Figura 64: IRIL en presencia de oxo-M y bisindolilmaleimida y porcentajes de inhibición de oxo-M.
A) La diferencia entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de
bisindolilmaleimida (300 nM) y oxo-M (10 µM) fue significativa (*p = 0.014, n = 6). B) La diferencia entre los
porcentajes de inhibición por oxo-M (10 µM) de la corriente activada por riluzol (300 µM) en ausencia y en
presencia de bisindolilmaleimida (300 nM) no fue significativo (p = 0.737).
2.6 El aumento de calcio no participa en la inhibición muscarínica
El IP3, segundo mensajero resultante de la hidrólisis de PIP2, se libera al citosol donde
se une a receptores de IP3 que se encuentran en el retículo endoplasmático. Aquí provoca la
liberación de calcio incrementando su concentración intracelular. El aumento de la
concentración de calcio podría modular directamente o indirectamente el canal. En este
apartado hemos investigado la implicación del aumento del calcio intracelular en la inhibición
muscarínica mediante el bloqueo, con 2-APB (2-aminoethoxydiphenylborane), de la unión de
IP3 a su receptor y por consiguiente del bloqueo del incremento de la concentración de calcio
intracelular.
La aplicación de oxo-M (10 µM) en presencia de 2-APB (100 µM), como muestra la
figura 65, inhibe IRIL control de forma significativa (p = 2.04*10-4
, n = 5), desde 155.21 ±
20.71 pA (IRIL control) hasta 79.82 ± 20.06 pA (IRIL en presencia de 2-APB y de oxo-M) como
muestran las figuras 66 y 67A. El porcentaje de inhibición por oxo-M de IRIL en presencia de
2-APB (51.33 ± 6.14 %) es significativo (p = 0.0011, n = 5) y no se observan diferencias
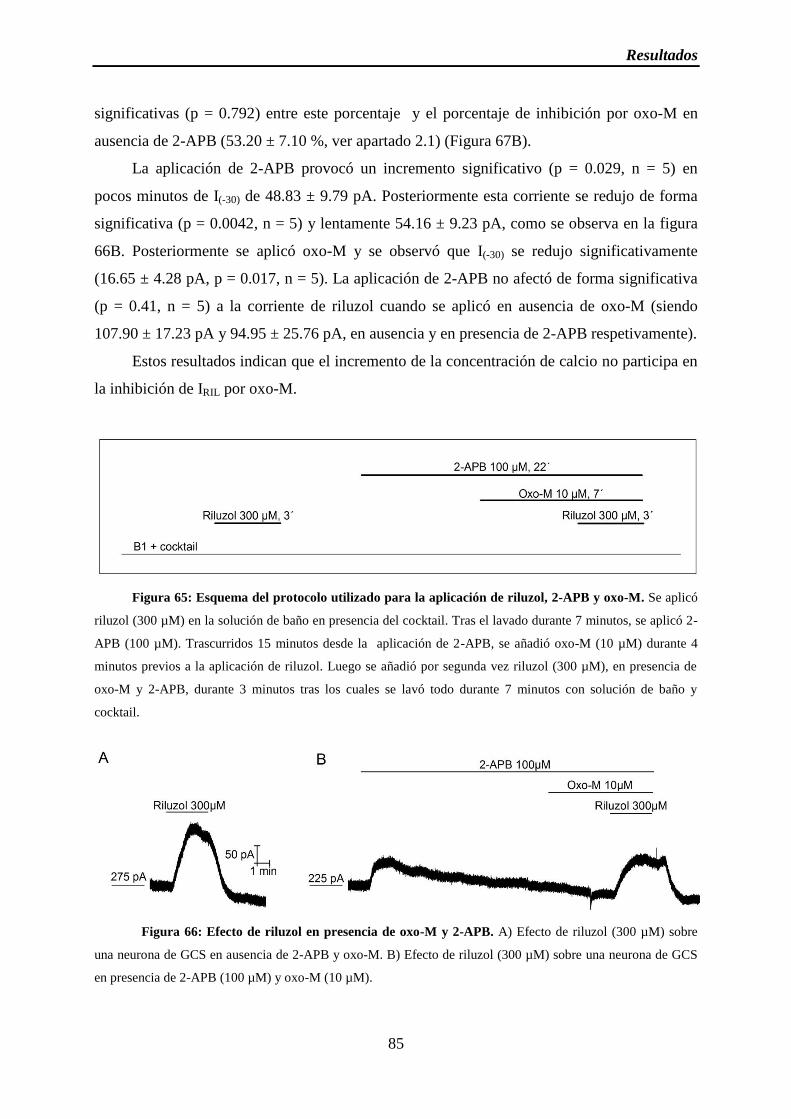
Resultados
85
significativas (p = 0.792) entre este porcentaje y el porcentaje de inhibición por oxo-M en
ausencia de 2-APB (53.20 ± 7.10 %, ver apartado 2.1) (Figura 67B).
La aplicación de 2-APB provocó un incremento significativo (p = 0.029, n = 5) en
pocos minutos de I(-30) de 48.83 ± 9.79 pA. Posteriormente esta corriente se redujo de forma
significativa (p = 0.0042, n = 5) y lentamente 54.16 ± 9.23 pA, como se observa en la figura
66B. Posteriormente se aplicó oxo-M y se observó que I(-30) se redujo significativamente
(16.65 ± 4.28 pA, p = 0.017, n = 5). La aplicación de 2-APB no afectó de forma significativa
(p = 0.41, n = 5) a la corriente de riluzol cuando se aplicó en ausencia de oxo-M (siendo
107.90 ± 17.23 pA y 94.95 ± 25.76 pA, en ausencia y en presencia de 2-APB respetivamente).
Estos resultados indican que el incremento de la concentración de calcio no participa en
la inhibición de IRIL por oxo-M.
Figura 65: Esquema del protocolo utilizado para la aplicación de riluzol, 2-APB y oxo-M. Se aplicó
riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se aplicó 2-
APB (100 µM). Trascurridos 15 minutos desde la aplicación de 2-APB, se añadió oxo-M (10 µM) durante 4
minutos previos a la aplicación de riluzol. Luego se añadió por segunda vez riluzol (300 µM), en presencia de
oxo-M y 2-APB, durante 3 minutos tras los cuales se lavó todo durante 7 minutos con solución de baño y
cocktail.
Figura 66: Efecto de riluzol en presencia de oxo-M y 2-APB. A) Efecto de riluzol (300 µM) sobre
una neurona de GCS en ausencia de 2-APB y oxo-M. B) Efecto de riluzol (300 µM) sobre una neurona de GCS
en presencia de 2-APB (100 µM) y oxo-M (10 µM).
3´

Resultados
86
Figura 67: IRIL en presencia de oxo-M y 2-APB y porcentajes de inhibición de oxo-M. A) La
diferencia entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de 2-APB (100 µM) y oxo-
M (10 µM) fue significativa (***p = 2.04*10-4
, n = 5). B) La diferencia entre porcentajes de inhibición por oxo-
M (10 µM) de la corriente activada por riluzol (300 µM) en ausencia y en presencia de 2-APB (100 µM) no fue
significativo (p = 0.792).
2.7 Implicación de PIP2 en la inhibición muscarínica
El fosfolípido de membrana PIP2 es hidrolizado por PLC en DAG y IP3, lo que provoca
una reducción de la concentración de PIP2. Algunos estudios sugieren que esta reducción en la
concentración de PIP2 provoca la inhibición de IM ya que estaría activándolo de forma
continua (Zhang et al., 2003). En este apartado pretendemos determinar la implicación de
PIP2 en la inhibición muscarínica de IRIL en neuronas del GCS de ratón en cultivo.
2.7.1 El descenso de la concentración de PIP2 inhibe IRIL
En este experimento se ha bloqueado con wortmanina la recuperación de la
concentración de PIP2 mediante el bloqueo del enzima fosfatidilinositol-3-quinasa (PI3K) y
fosfatidilinositol-4-quinasa (PI4K), manteniendo así una concentración de PIP2 muy baja en
ausencia de oxo-M, para observar si la reducción de PIP2 bloquea el canal TREK-2.
La aplicación de riluzol en presencia del bloqueante wortmanina (20 µM), como
muestra la figura 68, generó una corriente de salida (40.97 ± 12.17 pA) significativamente

Resultados
87
menor (p = 1.12*10-4
, n = 7) que IRIL control (171.47 ± 17.09 pA) como se observa en la
figura 69. El porcentaje de inhibición de IRIL debido a la aplicación de wortmanina (77.37 ±
6.99 %) es significativo (p = 3.23*10-5
, n = 7). La aplicación de wortmanina provocó una
reducción significativa (p = 9.1*10-4
, n=7) de I(-30) desde 221.50 ± 27.20 pA hasta 111.76 ±
23.63 pA (Figura 69B).
Según los resultados obtenidos podemos determinar que la reducción en la
concentración de PIP2 inhibe la corriente IRIL.
Figura 68: Esquema del protocolo utilizado para la aplicación de riluzol y wortmanina. Se aplicó
riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se aplicó
wortmanina (20 µM). Trascurridos 15 minutos desde la aplicación de wortmanina, se añadió por segunda vez
riluzol (300 µM) durante 3 minutos. A continuación se lavó todo durante 7 minutos con solución de baño y
cocktail.
3´
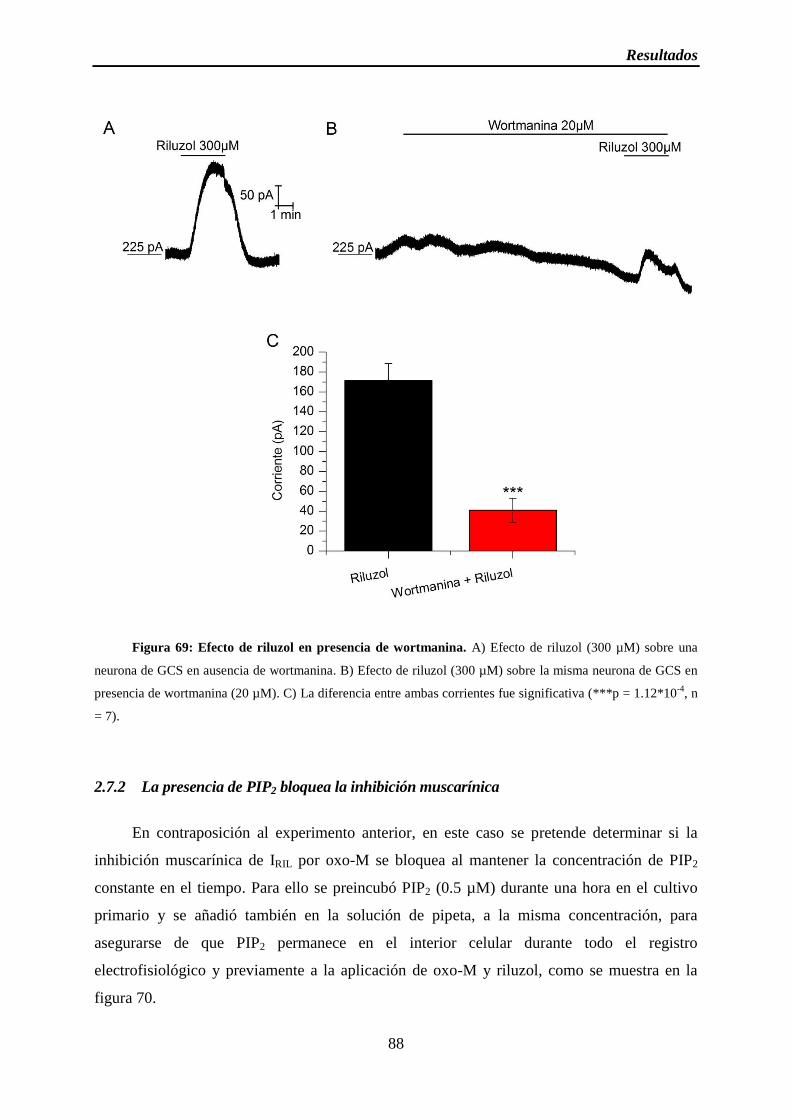
Resultados
88
Figura 69: Efecto de riluzol en presencia de wortmanina. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS en ausencia de wortmanina. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en
presencia de wortmanina (20 µM). C) La diferencia entre ambas corrientes fue significativa (***p = 1.12*10-4
, n
= 7).
2.7.2 La presencia de PIP2 bloquea la inhibición muscarínica
En contraposición al experimento anterior, en este caso se pretende determinar si la
inhibición muscarínica de IRIL por oxo-M se bloquea al mantener la concentración de PIP2
constante en el tiempo. Para ello se preincubó PIP2 (0.5 µM) durante una hora en el cultivo
primario y se añadió también en la solución de pipeta, a la misma concentración, para
asegurarse de que PIP2 permanece en el interior celular durante todo el registro
electrofisiológico y previamente a la aplicación de oxo-M y riluzol, como se muestra en la
figura 70.

Resultados
89
IRIL en presencia de oxo-M (96.72 ± 19.72 pA) no obtuvo diferencias significativas (p =
0.380, n = 7) respecto a IRIL control (111.71 ± 16.42 pA) (Figuras 71 y 72A). En este
experimento IRIL control en presencia de PIP2 es significativamente menor (p = 0.035) que IRIL
control en ausencia de PIP2 (249.43 ± 24.12 pA, ver apartado 1.1.2). El porcentaje de
inhibición por oxo-M en presencia de PIP2 (8.87 ± 15.73 %) fue significativamente diferente
(p = 0.034) al porcentaje de inhibición por oxo-M control (53.20 ± 7.10 %, ver apartado 2.1)
como se observa en la figura 72B. La aplicación de oxo-M provocó un descenso no
significativo (p = 0.20, n = 7) de I(-30) de 10.63 ± 7.47 pA, figura 71B.
Debido a que PIP2 se pre-incubó en el cultivo celular, se comparó I(-30) en ausencia y en
presencia de PIP2 de forma no pareada. No se observaron diferencias significativas (p =
0.193) entre I(-30) en ausencia (185.23 ± 27.89 pA, n = 9) y en presencia de PIP2 (134.26 ±
24.81 pA, n = 13).
Los resultados de este apartado indican que la concentración de PIP2 estable en el
tiempo mantiene la corriente de riluzol activada.
Figura 70: Esquema del protocolo utilizado para la aplicación de riluzol y oxo-M en presencia de
PIP2. Se aplicó riluzol (300 µM) en la solución de baño estándar en presencia del cocktail, pero en este caso las
células están pre-incubadas durante 1 hora con PIP2 (0.5 µM) que también se añadió a la solución de pipeta. Tras
el lavado de riluzol durante 7 minutos se aplicó oxo-M (10 µM) durante 4 minutos previos a la aplicación de
riluzol. Luego se añadió por segunda vez riluzol (300 µM) en presencia de oxo-M y de PIP2 durante 3 minutos,
tras los cuales, se lavó todo con solución de baño y cocktail durante 7 minutos.
3´
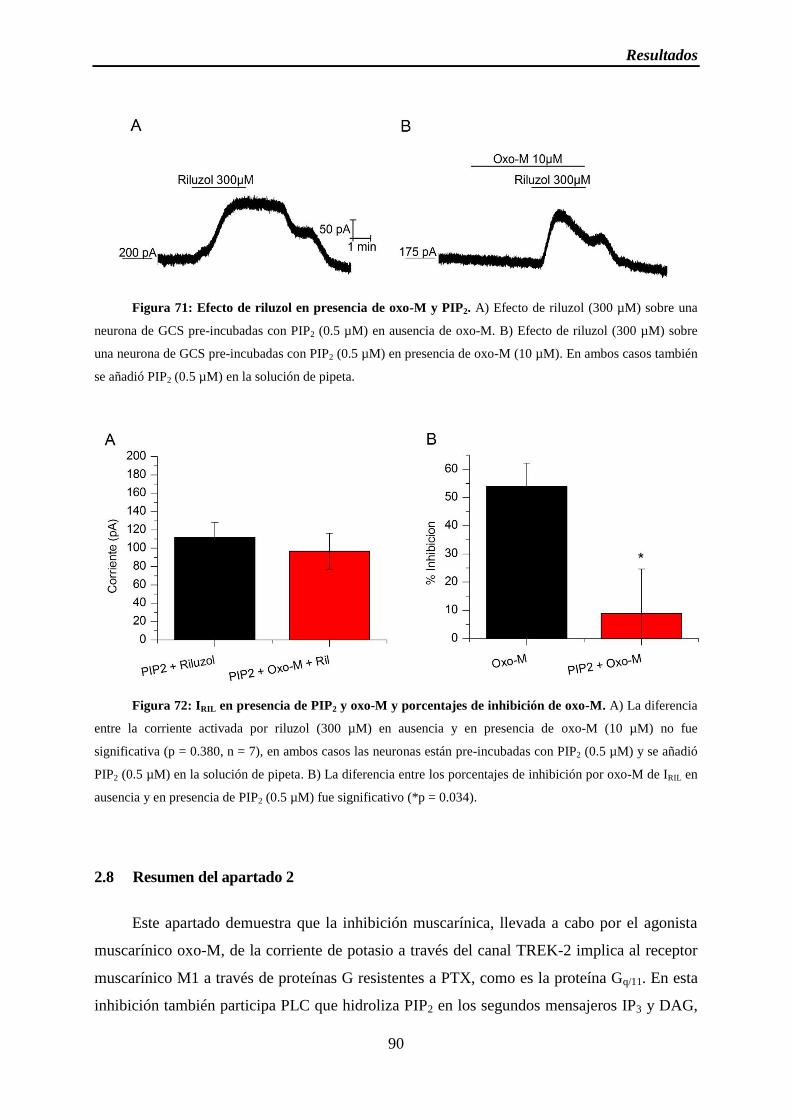
Resultados
90
Figura 71: Efecto de riluzol en presencia de oxo-M y PIP2. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS pre-incubadas con PIP2 (0.5 µM) en ausencia de oxo-M. B) Efecto de riluzol (300 µM) sobre
una neurona de GCS pre-incubadas con PIP2 (0.5 µM) en presencia de oxo-M (10 µM). En ambos casos también
se añadió PIP2 (0.5 µM) en la solución de pipeta.
Figura 72: IRIL en presencia de PIP2 y oxo-M y porcentajes de inhibición de oxo-M. A) La diferencia
entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de oxo-M (10 µM) no fue
significativa (p = 0.380, n = 7), en ambos casos las neuronas están pre-incubadas con PIP2 (0.5 µM) y se añadió
PIP2 (0.5 µM) en la solución de pipeta. B) La diferencia entre los porcentajes de inhibición por oxo-M de IRIL en
ausencia y en presencia de PIP2 (0.5 µM) fue significativo (*p = 0.034).
2.8 Resumen del apartado 2
Este apartado demuestra que la inhibición muscarínica, llevada a cabo por el agonista
muscarínico oxo-M, de la corriente de potasio a través del canal TREK-2 implica al receptor
muscarínico M1 a través de proteínas G resistentes a PTX, como es la proteína Gq/11. En esta
inhibición también participa PLC que hidroliza PIP2 en los segundos mensajeros IP3 y DAG,

Resultados
91
provocando el descenso en la concentración de PIP2. Mediante estos experimentos hemos
determinado que este descenso provoca la inhibición del canal y que el mantenimiento de la
concentración alta y estable de PIP2 permite que el canal permanezca abierto a pesar de la
presencia del agonista muscarínico. Además estos resultados nos muestran que el aumento en
la concentración de calcio, así como el enzima PKC no participan en la inhibición
muscarínica de IRIL por oxo-M (Figura 73).
Figura 73: Vía de inhibición muscarínica de TREK-2. Los resultados de este trabajos muestran que la
inhibición muscarínica por oxo-M del canal TREK-2 sucede a través de la activación del receptor muscarínico
M1, que provoca la activación de la proteína Gq/11. A su vez ésta activa el enzima PLC que provoca la reducción
en la concentración de PIP2 y por consiguiente la inhibición del canal.
3 Modulación por bradiquinina de IRIL
El objetivo de este grupo de experimentos es observar si la hormona peptídica BK
inhibe la corriente IRIL en neuronas del GCS de ratón, ya que modula el potencial de
membrana y la excitabilidad neuronal mediante la inhibición de IM en el GCS de rata (Jones et
al., 1995). Además se determinará si la inhibición muscarínica y por BK siguen la misma vía
de modulación y qué segundos mensajeros están implicados en la modulación por BK de IRIL.
Los experimentos de este apartado se realizaron mediante registros electrofisiológicos en la
modalidad de sello perforado en neuronas de GCS de ratón en cultivo mediante protocolos de
voltage-clamp con el potencial de membrana fijado a -30 mV.

Resultados
92
3.1 BK inhibe IRIL
Se ha realizado este experimento para determinar si BK inhibe IRIL, mediante la
aplicación de BK (250 nM) posteriormente a la primera aplicación de riluzol y previamente a
la segunda aplicación, como muestra la figura 74.
El resultado de este experimento muestra que IRIL control (200.19 ± 39.33 pA) e IRIL en
presencia de BK (129.83 ± 32.24 pA) son significativamente diferentes (p = 5.06*10-5
, n =
10). El porcentaje de inhibición de la corriente IRIL en presencia de BK (40.88 ± 4.33 %) fue
significativo (p = 5.75*10-6
). La aplicación de BK redujo I(-30) significativamente -83.82 ±
24.47 pA (p = 0.0075, n = 10) (Figura 75).
Este experimento muestra que la corriente de potasio de TREK-2 activada por riluzol es
sensible a BK.
Figura 74: Esquema del protocolo utilizado para la aplicación de riluzol y BK. Se aplicó riluzol (300
µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se aplicó BK (250 nM)
durante 4 minutos. A continuación se añadió por segunda vez riluzol (300 µM), en presencia de BK, durante tres
minutos tras los cuales se lavó todo durante 7 minutos con solución de baño y cocktail.
3´
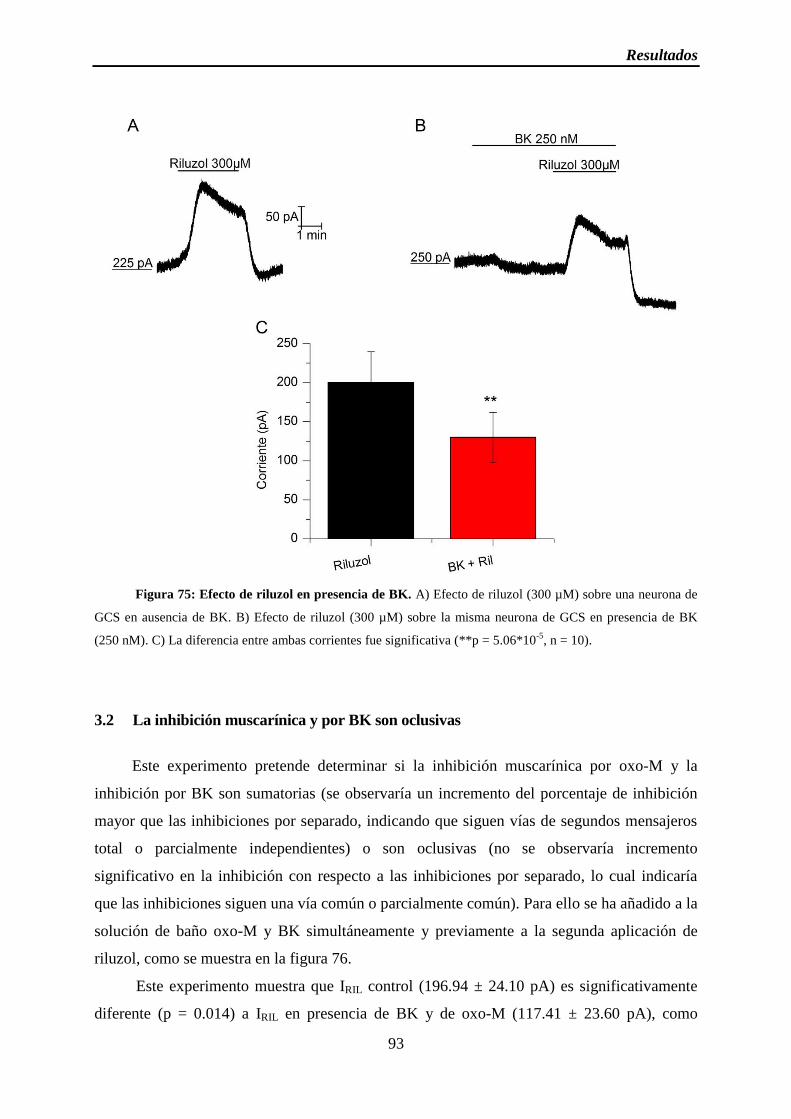
Resultados
93
Figura 75: Efecto de riluzol en presencia de BK. A) Efecto de riluzol (300 µM) sobre una neurona de
GCS en ausencia de BK. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en presencia de BK
(250 nM). C) La diferencia entre ambas corrientes fue significativa (**p = 5.06*10-5
, n = 10).
3.2 La inhibición muscarínica y por BK son oclusivas
Este experimento pretende determinar si la inhibición muscarínica por oxo-M y la
inhibición por BK son sumatorias (se observaría un incremento del porcentaje de inhibición
mayor que las inhibiciones por separado, indicando que siguen vías de segundos mensajeros
total o parcialmente independientes) o son oclusivas (no se observaría incremento
significativo en la inhibición con respecto a las inhibiciones por separado, lo cual indicaría
que las inhibiciones siguen una vía común o parcialmente común). Para ello se ha añadido a la
solución de baño oxo-M y BK simultáneamente y previamente a la segunda aplicación de
riluzol, como se muestra en la figura 76.
Este experimento muestra que IRIL control (196.94 ± 24.10 pA) es significativamente
diferente (p = 0.014) a IRIL en presencia de BK y de oxo-M (117.41 ± 23.60 pA), como

Resultados
94
muestran las figuras 77 y 78A. El porcentaje de inhibición (38.41 ± 9.17 %) es significativo (p
= 0.0018, n = 11) y no presenta diferencias significativas (p = 0.82) con el porcentaje de
inhibición en presencia solamente de BK (40.88 ± 4.33 %, ver apartado anterior), ni tampoco
presenta diferencias significativas (p = 0.28) con el porcentaje de inhibición en presencia de
oxo-M sola (53.20 ± 7.10 %, ver apartado 2.1), figura 78B. Se ha determinado que tampoco
hay diferencias significativas (p = 0.14) entre el porcentaje de inhibición de BK sola (40.88 ±
4.33 %) y el porcentaje de inhibición de oxo-M sola (53.20 ± 7.10 %).
La aplicación de BK (250 nM) y de oxo-M (10 µM) simultáneamente provocó un
descenso significativo (p = 0.0019, n = 11) de I(-30) (64.10 ± 15.34 pA, n = 11), figura 77B.
Estos resultados indican que la inhibición muscarínica por oxo-M y la inhibición por
BK son oclusivas, es decir, la vía de segundos mensajeros es común de forma parcial o
completa.
Figura 76: Esquema del protocolo utilizado para la aplicación de riluzol, BK y oxo-M. Se aplicó
riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se aplicó BK
(250 nM) y oxo-M (10 µM) simultáneamente. Trascurridos 4 minutos desde la aplicación de BK y oxo-M se
aplicó por segunda vez riluzol (300 µM) durante 3 minutos tras los cuales se lavó todo durante 7 minutos con
solución de baño y cocktail.
Figura 77: Efecto de riluzol en presencia de oxo-M y BK. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS en ausencia de BK y oxo-M. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en
presencia de BK (250 nM) y oxo-M (10 µM).
3´
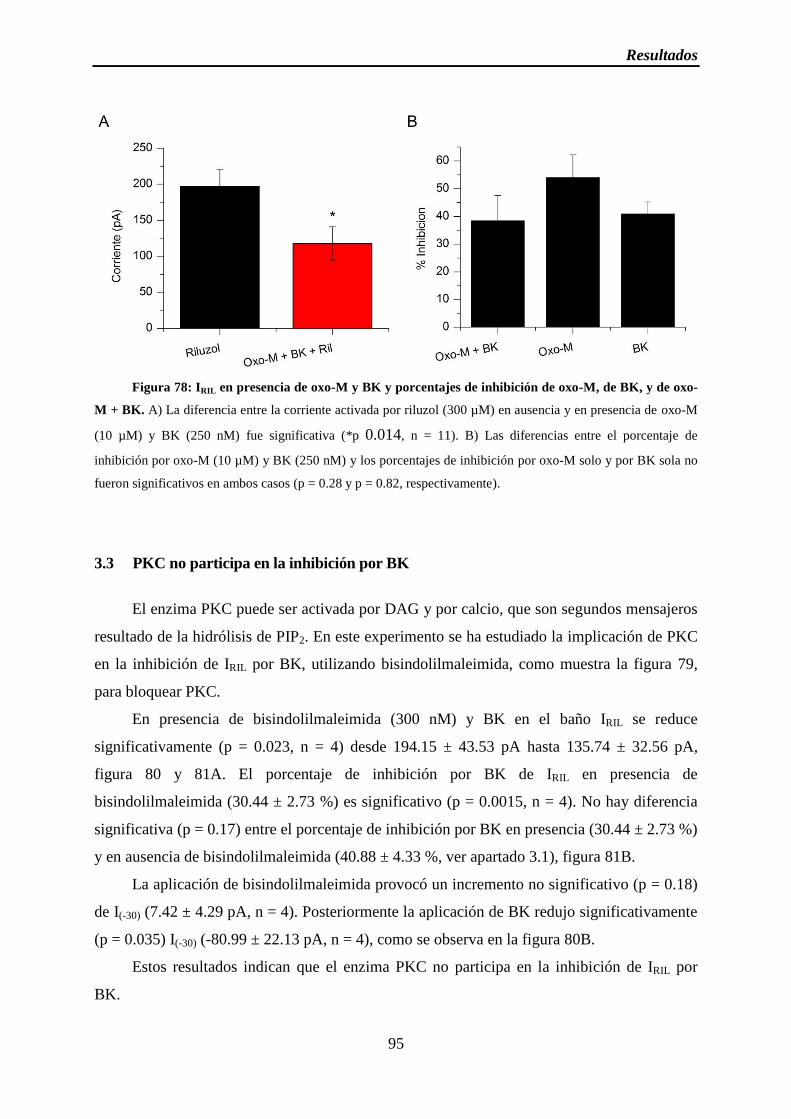
Resultados
95
Figura 78: IRIL en presencia de oxo-M y BK y porcentajes de inhibición de oxo-M, de BK, y de oxo-
M + BK. A) La diferencia entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de oxo-M
(10 µM) y BK (250 nM) fue significativa (*p 0.014, n = 11). B) Las diferencias entre el porcentaje de
inhibición por oxo-M (10 µM) y BK (250 nM) y los porcentajes de inhibición por oxo-M solo y por BK sola no
fueron significativos en ambos casos (p = 0.28 y p = 0.82, respectivamente).
3.3 PKC no participa en la inhibición por BK
El enzima PKC puede ser activada por DAG y por calcio, que son segundos mensajeros
resultado de la hidrólisis de PIP2. En este experimento se ha estudiado la implicación de PKC
en la inhibición de IRIL por BK, utilizando bisindolilmaleimida, como muestra la figura 79,
para bloquear PKC.
En presencia de bisindolilmaleimida (300 nM) y BK en el baño IRIL se reduce
significativamente (p = 0.023, n = 4) desde 194.15 ± 43.53 pA hasta 135.74 ± 32.56 pA,
figura 80 y 81A. El porcentaje de inhibición por BK de IRIL en presencia de
bisindolilmaleimida (30.44 ± 2.73 %) es significativo (p = 0.0015, n = 4). No hay diferencia
significativa (p = 0.17) entre el porcentaje de inhibición por BK en presencia (30.44 ± 2.73 %)
y en ausencia de bisindolilmaleimida (40.88 ± 4.33 %, ver apartado 3.1), figura 81B.
La aplicación de bisindolilmaleimida provocó un incremento no significativo (p = 0.18)
de I(-30) (7.42 ± 4.29 pA, n = 4). Posteriormente la aplicación de BK redujo significativamente
(p = 0.035) I(-30) (-80.99 ± 22.13 pA, n = 4), como se observa en la figura 80B.
Estos resultados indican que el enzima PKC no participa en la inhibición de IRIL por
BK.

Resultados
96
Figura 79: Esquema del protocolo utilizado para la aplicación de riluzol, bisindolilmaleimida y BK.
Se aplicó riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se
aplicó bisindolilmaleimida (300 nM). Trascurridos 10 minutos desde su aplicación, se añadió BK (250 nM)
durante 4 minutos previos a la aplicación de riluzol. Luego se añadió por segunda vez riluzol (300 µM), en
presencia de BK y bisindolilmaleimida, durante 3 minutos tras los cuales se lavó todo durante 7 minutos con
solución de baño y cocktail.
Figura 80 Efecto de riluzol en presencia de bisindolilmaleimida y BK. A) Efecto de riluzol (300 µM)
sobre una neurona de GCS en ausencia de bisindolilmaleimida y BK. B) Efecto de riluzol (300 µM) sobre la
misma neurona de GCS en presencia de bisindolilmaleimida (300 nM) y BK (250 nM).
3´
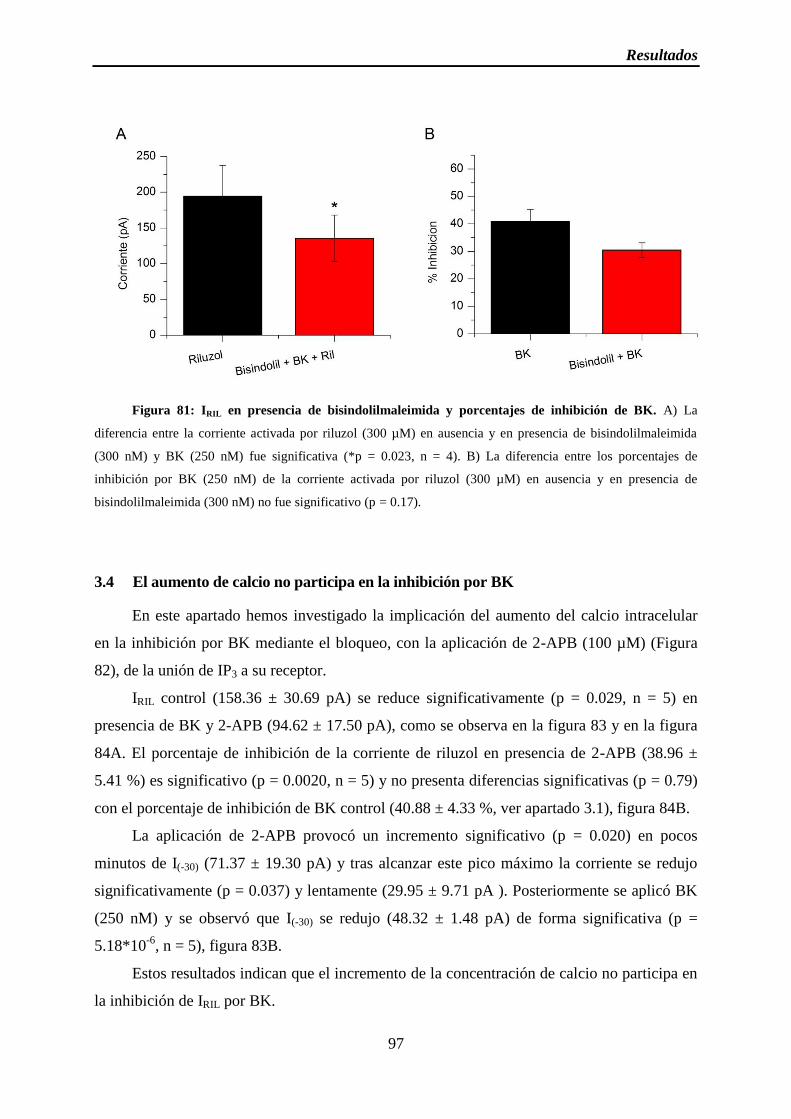
Resultados
97
Figura 81: IRIL en presencia de bisindolilmaleimida y porcentajes de inhibición de BK. A) La
diferencia entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de bisindolilmaleimida
(300 nM) y BK (250 nM) fue significativa (*p = 0.023, n = 4). B) La diferencia entre los porcentajes de
inhibición por BK (250 nM) de la corriente activada por riluzol (300 µM) en ausencia y en presencia de
bisindolilmaleimida (300 nM) no fue significativo (p = 0.17).
3.4 El aumento de calcio no participa en la inhibición por BK
En este apartado hemos investigado la implicación del aumento del calcio intracelular
en la inhibición por BK mediante el bloqueo, con la aplicación de 2-APB (100 µM) (Figura
82), de la unión de IP3 a su receptor.
IRIL control (158.36 ± 30.69 pA) se reduce significativamente (p = 0.029, n = 5) en
presencia de BK y 2-APB (94.62 ± 17.50 pA), como se observa en la figura 83 y en la figura
84A. El porcentaje de inhibición de la corriente de riluzol en presencia de 2-APB (38.96 ±
5.41 %) es significativo (p = 0.0020, n = 5) y no presenta diferencias significativas (p = 0.79)
con el porcentaje de inhibición de BK control (40.88 ± 4.33 %, ver apartado 3.1), figura 84B.
La aplicación de 2-APB provocó un incremento significativo (p = 0.020) en pocos
minutos de I(-30) (71.37 ± 19.30 pA) y tras alcanzar este pico máximo la corriente se redujo
significativamente (p = 0.037) y lentamente (29.95 ± 9.71 pA ). Posteriormente se aplicó BK
(250 nM) y se observó que I(-30) se redujo (48.32 ± 1.48 pA) de forma significativa (p =
5.18*10-6
, n = 5), figura 83B.
Estos resultados indican que el incremento de la concentración de calcio no participa en
la inhibición de IRIL por BK.
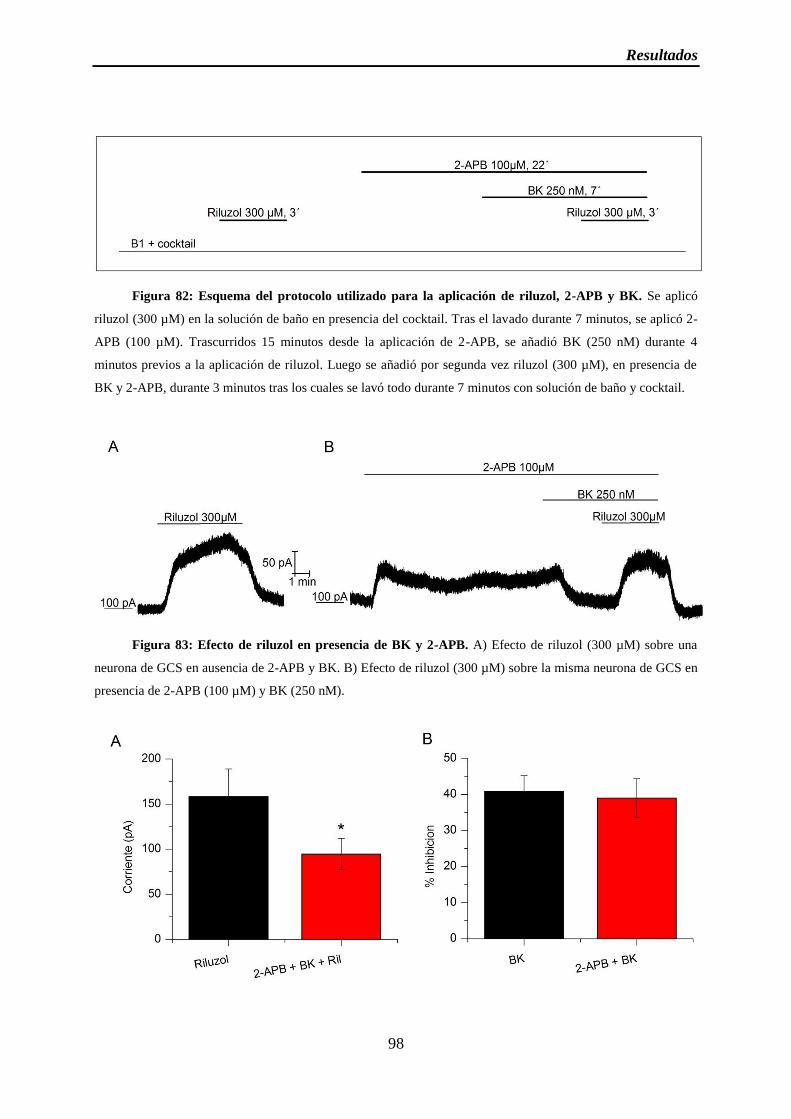
Resultados
98
Figura 82: Esquema del protocolo utilizado para la aplicación de riluzol, 2-APB y BK. Se aplicó
riluzol (300 µM) en la solución de baño en presencia del cocktail. Tras el lavado durante 7 minutos, se aplicó 2-
APB (100 µM). Trascurridos 15 minutos desde la aplicación de 2-APB, se añadió BK (250 nM) durante 4
minutos previos a la aplicación de riluzol. Luego se añadió por segunda vez riluzol (300 µM), en presencia de
BK y 2-APB, durante 3 minutos tras los cuales se lavó todo durante 7 minutos con solución de baño y cocktail.
Figura 83: Efecto de riluzol en presencia de BK y 2-APB. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS en ausencia de 2-APB y BK. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en
presencia de 2-APB (100 µM) y BK (250 nM).
3´

Resultados
99
Figura 84: IRIL en presencia de BK y 2-APB y porcentajes de inhibición de BK. A) La diferencia
entre la corriente activada por riluzol (300 µM) en ausencia y en presencia de 2-APB (100 µM) y BK (250 nM)
fue significativa (*p = 0.029, n = 5). B) La diferencia entre porcentajes de inhibición por BK (250 nM) de la
corriente activada por riluzol (300 µM) en ausencia y en presencia de 2-APB (100 µM) no fue significativo (p =
0.79).
3.5 La presencia de PIP2 bloquea la inhibición por BK
Se pretende determinar si la inhibición por BK de IRIL se bloquea al mantener la
concentración de PIP2 constante en el tiempo, interrumpiendo así la hidrólisis de éste. Para
ello se pre-incubó PIP2 (0.5 µM) durante una hora en el cultivo y se añadió también en la
solución de pipeta, con la misma concentración, para asegurarse de que PIP2 permanece en el
interior celular durante todo el registro electrofisiológico. Posteriormente se aplicó riluzol en
ausencia y en presencia de BK como muestra la figura 85.
En presencia de PIP2, IRIL control (161.26 ± 21.41 pA) e IRIL en presencia de BK (250
nM) (135.54 ± 9.26 pA) no muestran diferencias significativas (p = 0.20, n = 5), como se
muestra en las figuras 86 y 87A. El porcentaje de inhibición por BK en presencia de PIP2
(10.53 ± 10.62 %) fue significativamente diferente (p = 0.0072) al porcentaje de inhibición
por BK control (40.88 ± 4.33 %) como se observa en la figura 87B.
La aplicación de BK en presencia de PIP2 provocó un descenso significativo (p =
0.0067, n = 5) de I(-30) (113.36 ± 22.02 pA), figura 86B.
Los resultados de este apartado indican que la concentración de PIP2 estable en el
tiempo mantiene IRIL activada, a pesar de la presencia de BK.
Figura 85: Esquema del protocolo utilizado para la aplicación de riluzol y BK en presencia de PIP2.
Se aplicó riluzol (300 µM) en la solución de baño en presencia del cocktail, pero en este caso las células están
pre-incubadas con PIP2 (0.5 µM) que también se añadió a la solución de pipeta. Tras el lavado de riluzol durante
7 minutos se aplicó BK (250 nM) durante 4 minutos previos a la segunda aplicación de riluzol. Luego se añadió
por segunda vez riluzol (300 µM) en presencia de BK y de PIP2 durante 3 minutos, tras los cuales, se lavó todo
con solución de baño y cocktail durante 7 minutos.
3´
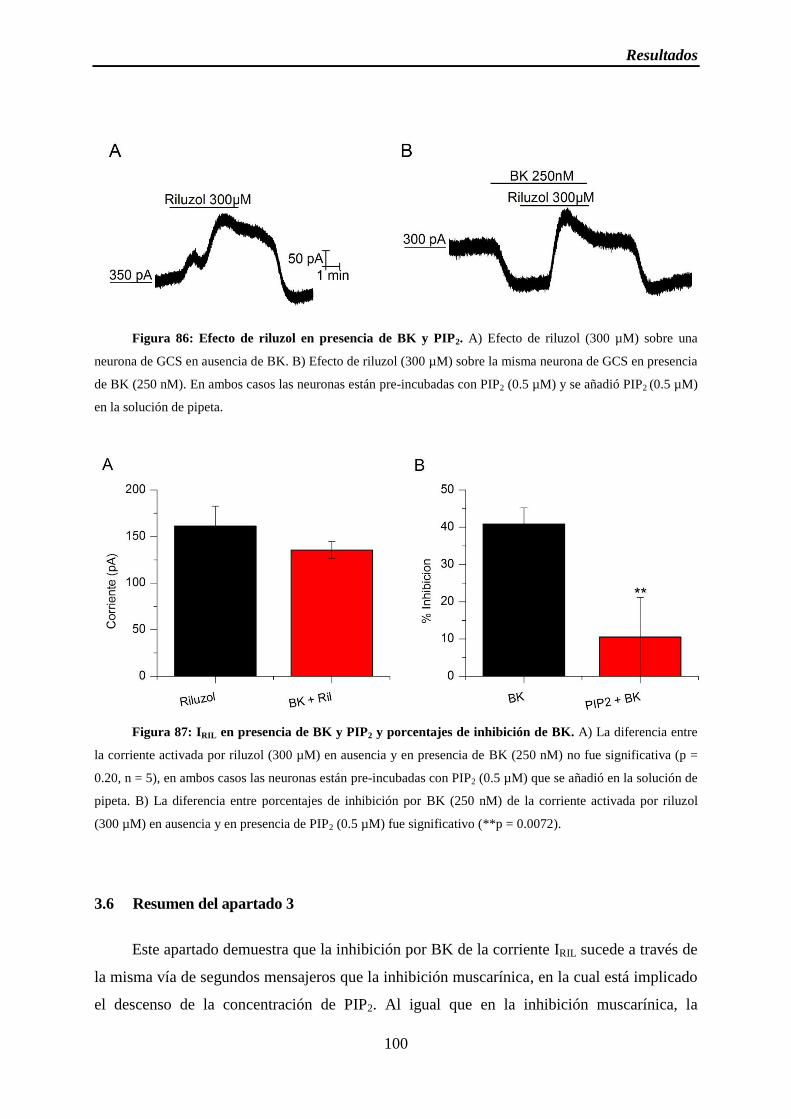
Resultados
100
Figura 86: Efecto de riluzol en presencia de BK y PIP2. A) Efecto de riluzol (300 µM) sobre una
neurona de GCS en ausencia de BK. B) Efecto de riluzol (300 µM) sobre la misma neurona de GCS en presencia
de BK (250 nM). En ambos casos las neuronas están pre-incubadas con PIP2 (0.5 µM) y se añadió PIP2 (0.5 µM)
en la solución de pipeta.
Figura 87: IRIL en presencia de BK y PIP2 y porcentajes de inhibición de BK. A) La diferencia entre
la corriente activada por riluzol (300 µM) en ausencia y en presencia de BK (250 nM) no fue significativa (p =
0.20, n = 5), en ambos casos las neuronas están pre-incubadas con PIP2 (0.5 µM) que se añadió en la solución de
pipeta. B) La diferencia entre porcentajes de inhibición por BK (250 nM) de la corriente activada por riluzol
(300 µM) en ausencia y en presencia de PIP2 (0.5 µM) fue significativo (**p = 0.0072).
3.6 Resumen del apartado 3
Este apartado demuestra que la inhibición por BK de la corriente IRIL sucede a través de
la misma vía de segundos mensajeros que la inhibición muscarínica, en la cual está implicado
el descenso de la concentración de PIP2. Al igual que en la inhibición muscarínica, la

Resultados
101
inhibición por BK es independiente del enzima PKC y del aumento en la concentración de
calcio intracelular (Figura 88).
Figura 88: Vía de inhibición por BK de TREK-2. Los resultados de este apartado muestran que la
inhibición muscarínica por BK de IRIL a través de la activación del receptor B2 sigue la misma vía de segundos
mensajeros que la inhibición muscarínica. La activación de este receptor B2, través de Gq y de PLC, provoca la
hidrólisis de PIP2 que estaría activando el canal permanentemente. Al igual que en la modulación muscarínica, el
enzima PKC y el aumento de la concentración de calcio no están implicados en esta inhibición.
4 Implicación del canal TREK-2 en el potencial de membrana en reposo y la
excitabilidad neuronal
El objetivo de este apartado es determinar la implicación de TREK-2 en el
mantenimiento del potencial de membrana en reposo y en la excitabilidad neuronal en
condiciones de temperatura fisiológica, pero también a temperatura ambiente a la cual se han
realizado la mayoría de los experimentos de este trabajo. Para ello se han realizado distintos
experimentos mediante protocolos de current-clamp y de voltaje-clamp en neuronas del GCS
de ratón en cultivo.

Resultados
102
4.1 Implicación del canal TREK-2 en el potencial de membrana
En este grupo de experimentos se pretende determinar cómo cambia el potencial de
membrana en reposo a temperatura ambiente y a temperatura fisiológica cuando se inhibe el
canal TREK-2. Los experimentos se realizaron utilizando protocolos de current-clamp con el
potencial de membrana de la célula fijada a -60 mV (este valor es el valor medio del potencial
de membrana en reposo de las neuronas del GCS de ratón en cultivo (Romero et al., 2004)).
4.1.1 La inhibición de TREK-2 no modifica el potencial de membrana a temperatura
ambiente
En este experimento se utilizó oxo-M (10 µM) en presencia de cocktail, del modo en
que se muestra en la figura 89, para bloquear el canal TREK-2 a temperatura ambiente. La
aplicación de oxo-M provocó una ligera despolarización (2.71 ± 1.76 mV), no significativa (p
= 0.1, n = 8) desde -61.20 ± 6.38 mV a -58.49 ± 7.05 mV a temperatura ambiente (23 ºC),
como se observa en la figura 90.
Este experimento demuestra que la inhibición de TREK-2 a temperatura ambiente no
afecta significativamente al potencial de membrana en reposo de las neuronas del GCS de
ratón.
Figura 89: Esquema del protocolo utilizado para la aplicación de oxo-M. Se aplicó cocktail en la
solución de baño durante 4 minutos previos a la aplicación de oxo-M (10 µM) que se aplicó durante 7 minutos,
utilizando un protocolo de current-clamp cuando el potencial de membrana permanece en reposo a temperatura
ambiente (23 ºC).
Figura 90: Efecto de oxo-M sobre el potencial de membrana en reposo. La aplicación de oxo-M (10
µM) en presencia de cocktail a temperatura ambiente (23 ºC) no afectó significativamente (p = 0.1, n = 8) al
potencial de membrana en reposo de las neuronas del GCS.
3´

Resultados
103
4.1.2 La inhibición de TREK-2 modifica el potencial de membrana a temperatura fisiológica
En este experimento se pretende determinar la implicación del canal TREK-2 y del
canal de potasio tipo M en el potencial de membrana en reposo a temperatura fisiológica (37
ºC), como se observa en la figura 91.
Se aplicó TEA (15 mM), en presencia de TTX (0.5 µM) y CsCl (1 mM), a temperatura
fisiológica y con el potencial de membrana fijado a -60 mV, mediante un protocolo de
current-clamp. La aplicación de TEA provocó una despolarización significativa (p = 0.012, n
= 6) de 5.55 ± 1.46 mV (Figura 92) desde -60.46 ± 1.22 mV hasta -54.90 ± 2.31 mV.
Posteriormente se fijó el potencial de membrana en -60 mV nuevamente. A continuación se
aplicó XE991 (3 µM), en presencia del cocktail, utilizando un protocolo de voltage-clamp con
el potencial de membrana fijado a -30 mV, ya que XE991es voltaje dependiente (Romero et
al., 2004). Posteriormente se observa el potencial de membrana en reposo mediante el
protocolo de current-clamp y se observa que la aplicación de XE991 provocó una
despolarización significativa (7.45 ± 1.99 mV, n = 4, p = 0.034) respecto al potencial de
membrana en reposo fijado a -60 mV (Figura 92).
Este experimento indica que la inhibición de IM con TEA y la inhibición de la
corriente de TREK-2 con XE991 a temperatura fisiológica despolarizan el potencial de
membrana en reposo de las neuronas del GCS.
Figura 91: Esquema del protocolo utilizado para la aplicación de TEA y XE991 a temperatura
fisiológica. Se aplicó TEA (15 mM) durante 5 minutos en presencia de TTX (0.5 µM) y CsCl (1 mM) utilizando
un protocolo de current-clamp con el potencial de membrana en reposo a 37 ºC. Posteriormente se aplicó XE991
(3 µM) en presencia de TEA mediante un protocolo de voltage-clamp con el potencial de membrana fijado a -30
mV. Tras 5 minutos de aplicación de XE991 se observó el valor del potencial de membrana utilizando
nuevamente el protocolo de current-clamp.
3´

Resultados
104
Figura 92: Efecto de TEA y de TEA + XE991 sobre el potencial de membrana en reposo. La
aplicación de TEA (15 mM) y la aplicación de XE991 (3 µM) en presencia de TEA (en ambos casos en
presencia de TTX (0.5 µM) y CsCl (1 mM)) provocaron la despolarización del potencial de membrana en reposo
a temperatura fisiológica. En ambos casos estas despolarizaciones fueron significativas (5.55 ± 1.46 mV, p =
0.012, n = 6 y 7.45 ± 1.99 mV, p = 0.034, n = 4, respectivamente). Las dos barras verticales indican que la
aplicación de XE991 se realizó utilizando un protocolo de voltage-clamp con el potencial de membrana fijado a -
30 mV.
4.2 Implicación del canal TREK-2 en la excitabilidad neuronal
En este apartado el objetivo es determinar la implicación del canal TREK-2 en la
excitabilidad de las neuronas del GCS. Para ello hemos realizado protocolos de current-clamp
aplicando estímulos de distintas intensidades. También se ha utilizado protocolos de saltos de
voltaje, inicialmente con la célula fijada a -30 mV, mediante la modalidad de voltage-clamp
para determinar la constante de activación de IM a distintas temperaturas.
4.2.1 TREK-2 e IM reducen la excitabilidad a temperatura fisiológica
En este apartado queremos determinar la participación del canal TREK-2 en la
adaptación cuando la temperatura es la fisiológica. Para ello se han aplicado estímulos
eléctricos desde -50 pA a 175 pA cada 25 pA durante 1 segundo, mediante un protocolo de
current-clamp intensidad-voltaje (IV) con el potencial de membrana fijado en -60 mV
(potencial de membrana en reposo de las neuronas del GCS de ratón en cultivo (Romero et
al., 2004)) con el fin de determinar el número de potenciales de acción, la latencia y la
frecuencia entre los dos primeros potenciales de acción a temperatura ambiente, a temperatura

Resultados
105
fisiológica y posteriormente con la aplicación de XE991 (3 µM) a temperatura fisiológica
como muestra la figura 93.
Los resultados muestran que el número de potenciales de acción se reduce de forma
significativa (en la mayoría de las intensidades de estímulo eléctrico aplicados) al incrementar
la temperatura desde la temperatura ambiente (23 ºC) hasta la temperatura fisiológica (37 ºC).
Posteriormente se añadió XE991 cuando la temperatura es de 37 ºC y se observó que el
número de potenciales de acción se incrementa significativamente (Figura 94 y 95). La
aplicación de XE991 se realizó con el potencial de membrana fijado a -30 mV durante cuatro
minutos mediante un protocolo de voltage-clamp y posteriormente se cambió al protocolo de
current-clamp con el potencial de membrana en reposo para continuar con el experimento. La
figura 95 nos muestra un ejemplo del patrón de disparos a temperatura ambiente, a
temperatura fisiológica y en presencia de XE991 a temperatura fisiológica cuando el estímulo
eléctrico aplicado es de 175 pA.
Se ha observado la latencia a temperatura ambiente (7.68 ± 0.72 ms), a temperatura
fisiológica (5.98 ± 0.92 ms) y a temperatura fisiológica en presencia de XE991 (4.64 ± 0.60
ms) cuando el estímulo eléctrico aplicado es de 175 pA. Se ha observado que cuando se
incrementa la temperatura se reduce la latencia de forma significativa (p = 0.0063, n = 5), sin
embargo la aplicación de XE991 no provoca una reducción significativa (p = 0.11, n = 5),
como se observa en la figura 96A.
Se ha determinado la frecuencia entre el primer y el segundo potencial de acción a
temperatura ambiente (45.28 ± 2.70 Hz), a temperatura fisiológica (73.2 ± 6.38 Hz) y a
temperatura fisiológica en presencia de XE991 (97.88 ± 6.92 Hz) cuando el estímulo eléctrico
aplicado es de 175 pA. Se ha observado que cuando se incrementa la temperatura y
posteriormente se aplica XE991 (3 µM) aumenta la frecuencia de disparo de forma
significativa en ambos casos (p = 0.021 y p = 0.046, respectivamente, n = 5), como se observa
en la figura 96B.
Este conjunto de experimentos determina que el aumento de la temperatura a valores
fisiológicos aumenta la adaptación de las neuronas del GCS en gran parte debido a la
activación de los canales de TREK-2 y de potasio tipo M, ya que la adaptación se reduce
cuando se bloquean dichos canales con el bloqueante XE991. La inhibición de la corriente de
TREK-2 e IM a temperatura fisiológica con XE991 incrementa de forma significativa la
frecuencia de disparo, sin embargo la latencia no cambia significativamente aunque tiene
tendencia a disminuir.
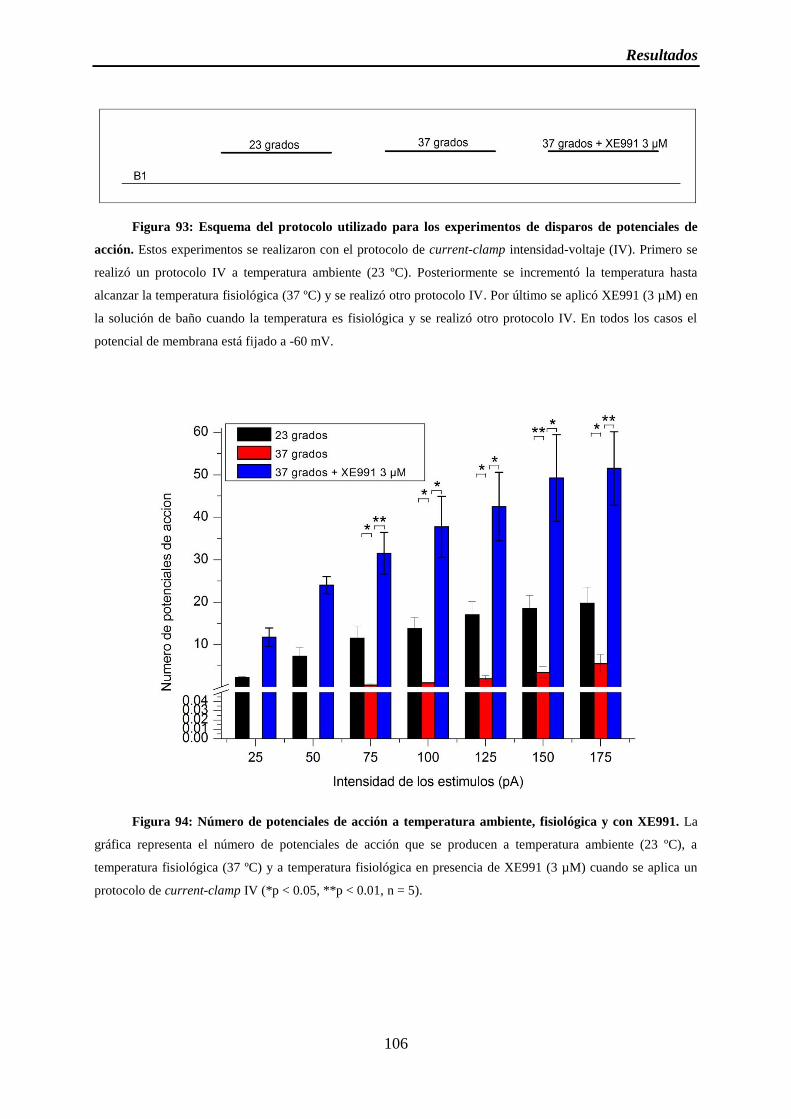
Resultados
106
Figura 93: Esquema del protocolo utilizado para los experimentos de disparos de potenciales de
acción. Estos experimentos se realizaron con el protocolo de current-clamp intensidad-voltaje (IV). Primero se
realizó un protocolo IV a temperatura ambiente (23 ºC). Posteriormente se incrementó la temperatura hasta
alcanzar la temperatura fisiológica (37 ºC) y se realizó otro protocolo IV. Por último se aplicó XE991 (3 µM) en
la solución de baño cuando la temperatura es fisiológica y se realizó otro protocolo IV. En todos los casos el
potencial de membrana está fijado a -60 mV.
Figura 94: Número de potenciales de acción a temperatura ambiente, fisiológica y con XE991. La
gráfica representa el número de potenciales de acción que se producen a temperatura ambiente (23 ºC), a
temperatura fisiológica (37 ºC) y a temperatura fisiológica en presencia de XE991 (3 µM) cuando se aplica un
protocolo de current-clamp IV (*p < 0.05, **p < 0.01, n = 5).
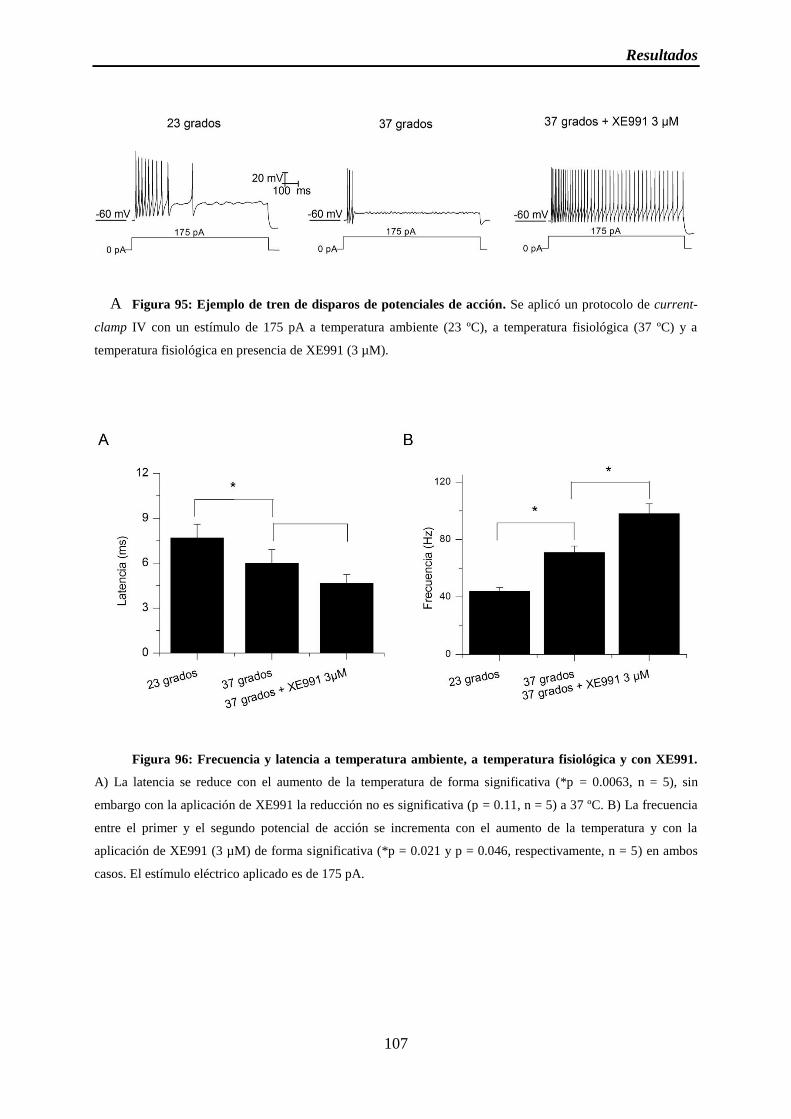
Resultados
107
Figura 95: Ejemplo de tren de disparos de potenciales de acción. Se aplicó un protocolo de current-
clamp IV con un estímulo de 175 pA a temperatura ambiente (23 ºC), a temperatura fisiológica (37 ºC) y a
temperatura fisiológica en presencia de XE991 (3 µM).
Figura 96: Frecuencia y latencia a temperatura ambiente, a temperatura fisiológica y con XE991.
A) La latencia se reduce con el aumento de la temperatura de forma significativa (*p = 0.0063, n = 5), sin
embargo con la aplicación de XE991 la reducción no es significativa (p = 0.11, n = 5) a 37 ºC. B) La frecuencia
entre el primer y el segundo potencial de acción se incrementa con el aumento de la temperatura y con la
aplicación de XE991 (3 µM) de forma significativa (*p = 0.021 y p = 0.046, respectivamente, n = 5) en ambos
casos. El estímulo eléctrico aplicado es de 175 pA.
A

Resultados
108
4.2.2 TREK-2 no afecta a la excitabilidad a temperatura ambiente
En este apartado el objetivo es determinar exclusivamente la implicación de la corriente
de potasio de TREK-2 en la excitabilidad neuronal. Para ello debemos eliminar lo máximo
posible IM, sin embargo, el estudio de la excitabilidad neuronal a través de protocolos de
current-clamp IV dificulta la utilización de TEA ya que éste bloquea otras corrientes de
potasio indispensables en el disparo de los potenciales de acción, tales como los canales de
potasio rectificadores tardío y los canales de potasio tipo A o transitorios (Decoursey et al.,
1984; Lipton & Tauck, 1987; Cobbett et al., 1989). Sin embargo, IM es dependiente de voltaje
y permanece cerrada en su mayoría cuando el potencial de membrana está en reposo y se abre
de forma lenta cuando el potencial de membrana se despolariza (Adams et al., 1982b; Brown
& Selyanko, 1985). Por otro lado la corriente de potasio a través de TREK-2 es poco
dependiente de voltaje, por lo que no cambiaría sustancialmente durante el tren de disparos
(Goldstein et al., 1996). Es decir, mediante los protocolos de current-clamp IV, en los
primeros milisegundos de aplicación del estímulo de corriente, los canales TREK-2 estarán
abiertos mientras que la mayoría de los canales de potasio tipo M todavía estarán cerrados.
En este grupo de experimentos el primer objetivo fue determinar la constante de tiempo
(tau) del cierre y de la apertura de IM, que nos indica el tiempo que tarda IM en alcanzar el
63.2 % de su cierre total o de su apertura total cuando se aplican pulsos eléctricos. Para ello se
han realizado experimentos de voltaje-clamp aplicando saltos de voltaje desde -30 mV a -50
mV y de -50 mV a -30 mV de la corriente sensible a TEA (sustraída de la corriente en
presencia y en ausencia de TEA, en ambos casos en presencia de TTX (0.5 µM) y CsCl (1
mM)) a temperatura ambiente (23 ºC) y a temperatura fisiológica (37 ºC) (Figura 97). La
constante de tiempo (tau) del cierre de la corriente sensible a TEA a temperatura ambiente es
de 67.61 ± 5.16 ms, sin embargo a temperatura fisiológica es 8.26 ± 1.88 ms. La constante de
tiempo de la apertura a temperatura ambiente es de 157.43 ± 12.62 ms, mientras que a 37 ºC
es de 7.18 ± 1.90 ms (n = 9) (Figura 98).
El segundo objetivo de este grupo de experimentos fue determinar cómo cambia la
latencia y la frecuencia entre los dos primeros potenciales de acción que suceden en los
primeros 60 ms, tras aplicar 175 pA como estímulo eléctrico, cuando se aplica XE991 (3 µM).
La frecuencia entre el primer y el segundo potencial de acción a temperatura ambiente
en ausencia (38.31 ± 3.71 Hz) y en presencia de XE991 (42.94 ± 3.70 Hz) no presentan
diferencias significativas (p = 0.058, n = 7). Del mismo modo, tampoco se observan
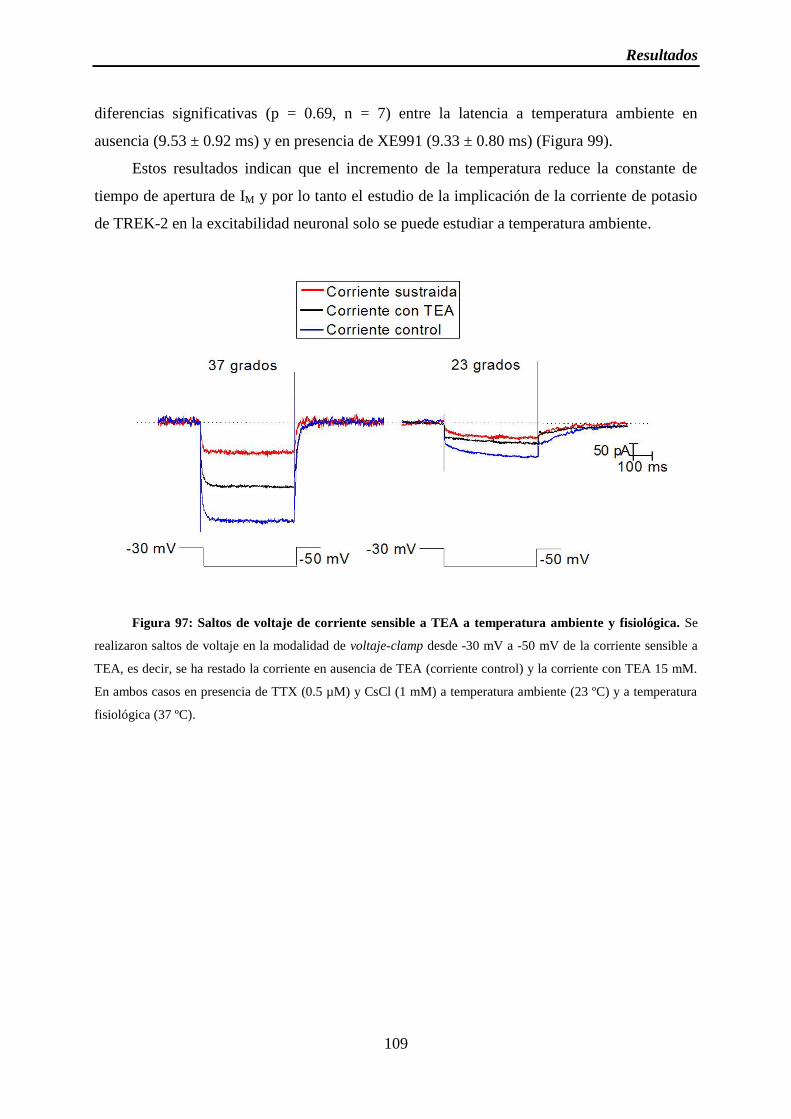
Resultados
109
diferencias significativas (p = 0.69, n = 7) entre la latencia a temperatura ambiente en
ausencia (9.53 ± 0.92 ms) y en presencia de XE991 (9.33 ± 0.80 ms) (Figura 99).
Estos resultados indican que el incremento de la temperatura reduce la constante de
tiempo de apertura de IM y por lo tanto el estudio de la implicación de la corriente de potasio
de TREK-2 en la excitabilidad neuronal solo se puede estudiar a temperatura ambiente.
Figura 97: Saltos de voltaje de corriente sensible a TEA a temperatura ambiente y fisiológica. Se
realizaron saltos de voltaje en la modalidad de voltaje-clamp desde -30 mV a -50 mV de la corriente sensible a
TEA, es decir, se ha restado la corriente en ausencia de TEA (corriente control) y la corriente con TEA 15 mM.
En ambos casos en presencia de TTX (0.5 µM) y CsCl (1 mM) a temperatura ambiente (23 ºC) y a temperatura
fisiológica (37 ºC).
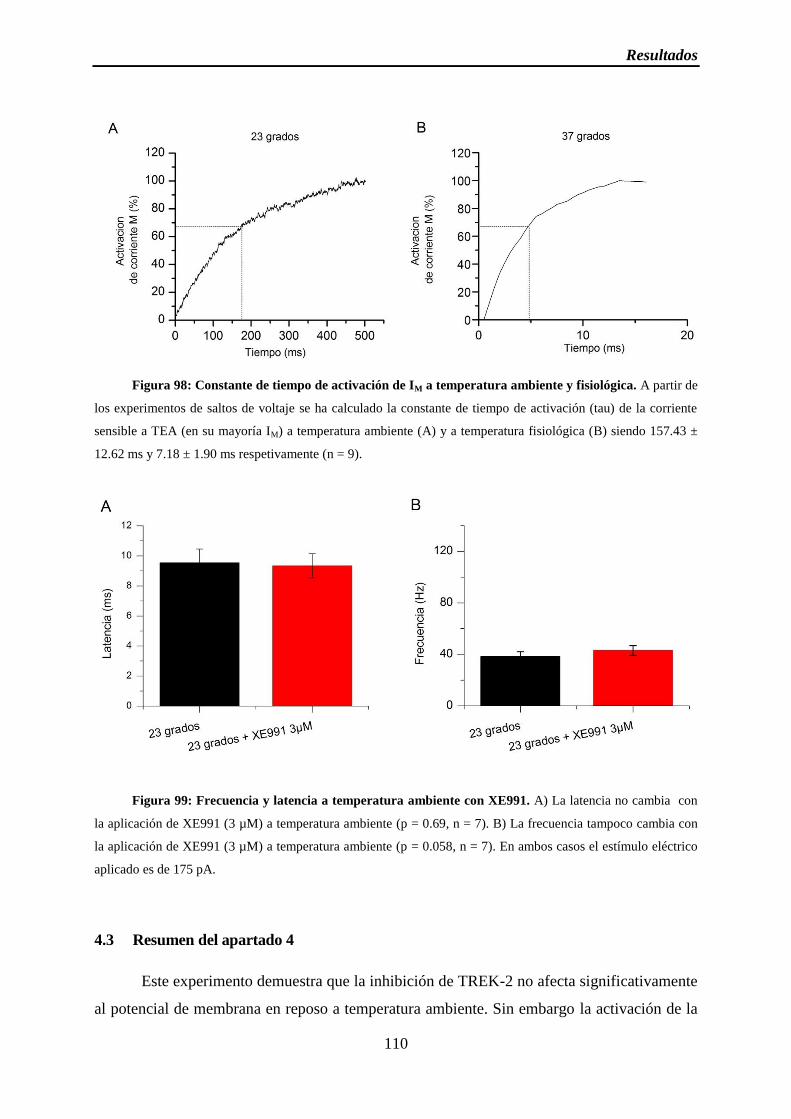
Resultados
110
Figura 98: Constante de tiempo de activación de IM a temperatura ambiente y fisiológica. A partir de
los experimentos de saltos de voltaje se ha calculado la constante de tiempo de activación (tau) de la corriente
sensible a TEA (en su mayoría IM) a temperatura ambiente (A) y a temperatura fisiológica (B) siendo 157.43 ±
12.62 ms y 7.18 ± 1.90 ms respetivamente (n = 9).
Figura 99: Frecuencia y latencia a temperatura ambiente con XE991. A) La latencia no cambia con
la aplicación de XE991 (3 µM) a temperatura ambiente (p = 0.69, n = 7). B) La frecuencia tampoco cambia con
la aplicación de XE991 (3 µM) a temperatura ambiente (p = 0.058, n = 7). En ambos casos el estímulo eléctrico
aplicado es de 175 pA.
4.3 Resumen del apartado 4
Este experimento demuestra que la inhibición de TREK-2 no afecta significativamente
al potencial de membrana en reposo a temperatura ambiente. Sin embargo la activación de la

Resultados
111
corriente de TREK-2 y de IM debido al incremento de temperatura hasta valores fisiológicos
provoca hiperpolarización en las neuronas del GCS en reposo. Además el aumento de la
temperatura aumenta la adaptación de las neuronas y dicha adaptación se reduce cuando se
bloquean ambas corrientes con XE991. Sin embargo, en la frecuencia y en la latencia, la
aplicación del bloqueante XE991 no recupera el efecto provocado por la temperatura.
También se ha visto que el incremento de la temperatura hasta valores fisiológicos
reduce la constante de tiempo de apertura de IM. Esto nos imposibilita determinar la
implicación de la corriente de potasio de TREK-2 en la excitabilidad neuronal a temperatura
fisiológica. A temperatura ambiente se observó que el canal TREK-2 no afecta a los
parámetros de latencia y frecuencia de los primeros potenciales de acción del tren de disparos.


DISCUSIÓN


Discusión
115
DISCUSIÓN
El valor del potencial de membrana en reposo y la excitabilidad neuronal dependen de
corrientes de potasio, de corrientes de sodio, de corrientes catiónicas y de la bomba
electrogénica de Na+/K
+. Sin embargo las neuronas en reposo son más permeables al ión
potasio que a los demás iones. La modulación de los canales de potasio que permanecen
abiertos durante el reposo es fundamental en el estudio del potencial de membrana en reposo.
En neuronas del GCS de rata y de ratón se ha visto que la modulación muscarínica de IM
modifica el valor del potencial de membrana en reposo y la excitabilidad neuronal (Lamas et
al., 2002; Romero et al., 2004). A partir del descubrimiento de los canales K2P como
corriente de fuga de potasio (Lesage et al., 1996) se empezó a estudiar la implicación de
dichos canales en la regulación del potencial de membrana y en la excitabilidad neuronal. En
las neuronas de GCS de ratón se ha determinado una importante presencia de los canales de
potasio TREK-2 (Cadaveira-Mosquera et al., 2012) que, en sistemas heterólogos, también son
modulados mediante agonistas muscarínicos (Kang et al., 2006).
En el presente trabajo se ha tratado de diferenciar IM de la corriente de potasio de
TREK-2 con el fin de poder estudiar la modulación de esta última y su influencia sobre el
potencial de membrana en reposo y la excitabilidad en neuronas del GCS de ratón en cultivo
primario. Los resultados indican que la corriente de potasio activada por riluzol y transportada
por los canales TREK-2 (IRIL) es independiente de IM. Sin embargo, estas dos corrientes no se
pueden distinguir según su sensibilidad a la temperatura, ya que ambas corrientes aumentan al
incrementar la temperatura. Tampoco los bloqueantes considerados hasta el momento como
específicos de IM (XE991 y linopirdina) se pueden utilizar para separar ambas corrientes ya
que también bloquean IRIL. No obstante, dada la diferencia en la cinética de activación, sí es
posible estudiar su contribución relativa a la excitabilidad.
Por tanto, activando la corriente de potasio a través de TREK-2 con riluzol e inhibiendo
casi completamente IM con TEA, hemos estudiado la modulación muscarínica y por
bradiquinina de la corriente de potasio de TREK-2. Hemos visto que oxo-M y BK inhiben IRIL
a través de una misma vía común cuyo efecto se debe al descenso de la concentración de PIP2.
Por último hemos observado la contribución de la corriente de potasio de TREK-2 sobre
la excitabilidad neuronal y sobre el valor del potencial de membrana en reposo. A temperatura
ambiente la corriente de potasio de TREK-2 no influye significativamente sobre el potencial

Discusión
116
de membrana en reposo, sin embargo, a temperatura fisiológica, la inhibición de IM y de la
corriente de TREK-2 provoca despolarización en estas neuronas. También se observa un
aumento de la adaptación de las neuronas del GCS cuando se incrementa la temperatura, que
se revierte cuando se bloquean ambas corrientes con el bloqueante XE991. Estas corrientes
además modulan la frecuencia de disparo de los potenciales de acción a temperatura
fisiológica.
Distinción entre IM y la corriente de potasio de TREK-2
Para poder estudiar la corriente de potasio de TREK-2 debemos bloquear el resto de
corrientes presentes en las neuronas del GCS. Para ello hemos utilizado un cocktail de varias
drogas compuesto por TTX que bloquea los canales de sodio voltaje dependiente (Wen et al.,
1994) y la corriente de sodio persistente (Davis & Stuart, 1988), por CsCl que bloquea la
corriente catiónica h (Fu et al., 1997) y por TEA que bloquea los canales de potasio
rectificadores tardío, de potasio tipo A o transitorio, canales de potasio dependientes de calcio
(Decoursey et al., 1984; Lipton & Tauck, 1987; Cobbett et al., 1989) y la corriente de potasio
tipo M (Storm, 1989). Como resultado de este bloqueo se observa en los experimentos una
inhibición de I-30.
Como se ha dicho anteriormente la corriente de potasio de TREK-2 e IM son corrientes
de potasio implicadas en el mantenimiento del potencial de membrana en reposo y la
excitabilidad en las neuronas del GCS. Se ha visto que la modulación muscarínica de IM
modifica el valor del potencial de membrana en reposo y la excitabilidad neuronal (Lamas et
al., 2002; Romero et al., 2004). Por ello es fundamental bloquear IM para poder estudiar la
modulación muscarínica y por bradiquinina de la corriente de TREK-2 y su implicación en el
mantenimiento del potencial de membrana en reposo y la excitabilidad neuronal. En este
trabajo se ha bloqueado IM con TEA. La aplicación de TEA, a la concentración de 15 mM,
bloquea aproximadamente el 70 % de IM. Este porcentaje de inhibición es cercano al
porcentaje de inhibición máximo que se alcanza con TEA 30 mM (Wang et al., 1998; Hadley
et al., 2003).

Discusión
117
En nuestro laboratorio se ha demostrado que en las neuronas del GCS de ratón en
cultivo el neuroprotector riluzol activa mayoritariamente el canal TREK-2 ya que es el canal
más expresado de la subfamilia TREK en estas neuronas (Cadaveira-Mosquera et al., 2011,
2012). Sin embargo riluzol inhibe otros canales iónicos como por ejemplo canales de sodio
persistente en neuronas del GCS (Lamas et al., 2009), canales de calcio en neuronas del
ganglio de la raíz dorsal (Huang et al., 1997) y canales de potasio rectificadores tardío
expresados en la línea celular CHO (Ahn et al., 2006). Además riluzol activa los canales de
potasio dependientes de calcio BK (Wang et al., 2008) y SK (Grunnet et al., 2001). La
presencia del cocktail anteriormente citado bloquea dichas corrientes y por tanto el efecto de
riluzol sobre ellas, permaneciendo solamente el efecto sobre el canal de TREK-2. Los canales
de potasio dependientes de calcio comienzan a abrirse a partir de potenciales de membrana
entre -30 y -20 mV (Marty & Nehert, 1985), por lo que estos canales están cerrados a los
potenciales de membrana utilizados en nuestros experimentos (-30 mV y -60mV) y no se
activan con la aplicación de riluzol (Cadaveira-Mosquera et al., 2011). Por ello en el presente
trabajo se ha utilizado riluzol para activar principalmente la corriente de potasio a través del
canal TREK-2. La curva de activación de IRIL es diferente a la curva de activación de la
corriente bloqueada con TEA (ITEA), lo que nos indica que son corrientes diferentes. Además
IRIL es insensible a TEA (Cadaveira-Mosquera et al., 2011) lo que confirma que son corrientes
independientes. Esto permite estudiar la corriente de potasio de TREK-2 utilizando riluzol
para activarla a temperatura ambiente mientras bloqueamos IM con TEA.
Otra hipótesis de este trabajo es diferenciar la corriente de potasio de TREK-2 e IM
dependiendo de su sensibilidad a la temperatura. Esta hipótesis se fundamenta en que la
corriente de TREK-2 aumenta de una forma muy acusada al incrementar la temperatura en
neuronas grano del cerebelo y en neuronas del ganglio de la raíz dorsal (Kang et al., 2005).
Por otro lado, la bibliografía no confirma una dependencia importante de IM con la
temperatura, sino que solamente sugiere un ligero aumento en la cinética que lleva un sutil
incremento de la conductancia de IM con la temperatura (Brown, 1988). También se ha
observado que el aumento de la temperatura, desde la temperatura fisiológica (37 ºC) hasta
temperaturas febriles (40 ºC), aumenta la expresión y la conductancia del canal homomérico
KCNQ2 expresado en la línea celular HEK293 (Yu et al., 2012). Nuestros resultados sin
embargo, muestran que tanto la corriente de TREK-2 como IM aumentan con la temperatura
con ratios de activación muy similares. Además el porcentaje de activación de ambas

Discusión
118
corrientes con la temperatura desde la temperatura ambiente, a la cual se realizan la mayoría
de experimentos, hasta la temperatura fisiológica es similar en ambos casos, lo que nos indica
que ambas corrientes son sensibles a la temperatura de forma similar y por tanto imposibilita
separarlas y distinguirlas utilizando la temperatura.
Por último se utilizaron los bloqueantes XE991 y linopirdina, que se consideraban hasta
el momento bloqueantes específicos para el estudio de IM (Aiken et al., 1995; Lamas et al.,
1997; Costa & Brown, 1998), con el fin de eliminar dicha corriente lo máximo posible y
poder estudiar la corriente de potasio de TREK-2. Sin embargo nuestros resultados muestran
que linopirdina y XE991 también bloquean IRIL. De hecho, comparando los resultados de este
trabajo donde se observa la inhibición de TREK-2, con los obtenidos previamente en nuestro
laboratorio donde se observa la inhibición de IM en neuronas de GCS de ratón (Romero et al.,
2004), observamos que TREK-2 es más sensible a linopirdina (IC50 = 0.31 µM) que IM (IC50 =
2.56 µM). También TREK-2 es más sensible a XE991 (IC50 = 0.045 µM) que IM (IC50 = 0.26
µM). Por ello no podemos utilizar XE991 ni linopirdina para eliminar IM sin modificar la
corriente de potasio de TREK-2.
Modulación muscarínica
El potencial de membrana en reposo y la excitabilidad en las neuronas del GCS se
modulan mediante la inhibición muscarínica de IM (Romero et al., 2004). El canal TREK-2
también está implicado en el mantenimiento del potencial de membrana en reposo y la
excitabilidad neuronal (Bang et al., 2000) y por ello en este trabajo se ha estudiado su
modulación muscarínica.
Los resultados muestrna que la corriente de potasio que fluye a través del canal TREK-2
(experimentos de single-channel), al igual que la corriente activada por el neuroprotector
riluzol (IRIL), se inhiben en presencia de oxo-M. Esto sugiere que el agonista muscarínico
también inhibe a este componente de la familia K2P en cultivo primario de neuronas del GCS
de ratón.
Los receptores muscarínicos se dividen en dos grupos, al primer grupo pertenecen los
receptores M1, M3 y M5 que típicamente se unen a las proteína Gq/11 y al segundo grupo
pertenecen los receptores M2 y M4 que se acoplan a la proteína Gi/o/t (Caulfield & Birdsall,

Discusión
119
1998). Según Lopes y colaboradores (Lopes et al., 2005) el canal TREK-1 se modula a través
del receptor muscarínico M1, al igual que IM en el GCS de rata y de ratón (Hamilton et al.,
1997; Shapiro et al., 2000). Los resultados de este trabajo muestran que la inhibición por oxo-
M de IRIL se bloquea en presencia del antagonista muscarínico atropina y del antagonista
específico del receptor M1 pirenzepina. Los experimentos realizados en la modalidad de
single-channel sugieren que la inhibición muscarínica de la corriente de potasio que fluye a
través del canal TREK-2 se debe a la reducción de su probabilidad de apertura. Dicha
reducción sucede a través del receptor muscarínico M1 que es bloqueado selectivamente por
pirenzepina. El antagonista específico de los receptores M2 y M4 himbacina no afecta a la
inhibición de IRIL por oxo-M. Estos resultados sugirieren que la inhibición de IRIL por oxo-M
sucede exclusivamente a través del receptor M1. La aplicación de atropina y de pirenzepina
solas incrementó I-30. Este incremento se debe a que estos antagonistas bloquearon la
inhibición de I-30 debida a la actividad constitutiva de la proteína Gq/11 a través del receptor
M1 (Jakubík et al., 1995).
Los receptores muscarínicos del grupo M1 están acoplados a las proteínas Gq/11 y éstas a
su vez activan el enzima PLC (Hamilton et al., 1997; Caulfield & Birdsall, 1998). La PTX
bloquea las proteínas Gi, Go y Gt pero no afecta a Gq/11. En el presente trabajo se ha observado
que oxo-M en presencia de PTX no pierde su efecto, indicando que la inhibición de IRIL por
oxo-M sucede a través de proteínas resistentes a PTX como la proteína Gq/11. También hemos
visto que la inhibición de IRIL por oxo-M se redujo en presencia de edelfosina, confirmando
que el enzima PLC participa en la vía de modulación muscarínica de IRIL. Se ha descrito que
la inhibición muscarínica de la corriente a través de TREK-2 sucede a través de las proteínas
Gq/11 y del enzima PLC en células COS-7 donde expresan el canal TREK-2 y el receptor
muscarínico M3 (Kang et al., 2006). Lesage y colaboradores (Lesage et al., 2000)
demostraron que expresando el canal TREK-2 junto con la proteína Gq/11 acoplada al receptor
M1 se inhibe la corriente en presencia de glutamato a través de la PLC.
Siguiendo la cascada de segundos mensajeros, el enzima PLC hidroliza PIP2 en DAG e
IP3 y éstos activan la PKC e incrementan la concentración citosólica de Ca+2
, respectivamente
(Mathie, 2007). Una de estas vías es la fosforilación y consecuente inhibición del canal
TREK-2 por el enzima PKC (Lesage et al., 2000; Nelson & Cox, 2009). Nuestros resultados
en neuronas simpáticas de GCS de ratón en cultivo sugieren que la inhibición de IRIL es
independiente de PKC ya que, aun habiendo bloqueado PKC con bisindolilmaleimida, oxo-M

Discusión
120
consigue inhibir IRIL. Kang y colaboradores (Kang et al., 2006) determinaron que la inhibición
muscarínica de TREK-2 en las células COS-7 sucede mediante la fosforilación por PKC. Sin
embargo otros autores explican que TREK-2, en células grano de cerebelo y en sistemas
expresados, se inhibe por DAG y ácidos fosfatídicos directamente sin la implicación de PKC,
ni debido a cambios en la concentración intracelular del ion calcio (Chemin et al., 2003). El
presente trabajo también muestra que la inhibición muscarínica de TREK-2 es independiente
de los cambios producidos en la concentración intracelular del ión calcio, ya que cuando
bloqueamos el receptor de IP3 que induce la salida de calcio desde el interior del RE hasta el
citosol, la inhibición muscarínica no se ve afectada. La aplicación del bloqueante del receptor
de IP3 2-APB, en ausencia de riluzol, generó una corriente de salida transitoria. Este efecto se
puede explicar porque 2-APB activa directamente el canal TREK-2 (Beltrán et al., 2013).
Esta corriente transitoria, se reduce en pocos minutos, previamente a la aplicación de oxo-M y
de riluzol, por lo que no afecta a la modulación muscarínica de IRIL.
Zhang y colaboradores afirman que la inhibición muscarínica de IM se debe a la
reducción de la concentración de PIP2 que estaría activando permanentemente el canal (Zhang
et al., 2003). La similitud en la modulación del canal de potasio tipo M y del canal TREK-2
en el GCS indica que TREK-2 podría bloquearse también por la reducción de la
concentración de PIP2. Los resultados de este trabajo muestran que la hidrólisis de PIP2
producida por oxo-M a través de la activación de la PLC, inhiben el canal TREK-2, ya que
cuando se bloquea el enzima PI3K/PI4K cuya función es recuperar los niveles de PIP2 en
ausencia de oxo-M, la corriente de TREK-2 permanece inhibida. Además los resultados
indican que mantener los niveles altos y constantes de PIP2 es completamente necesario para
bloquear la inhibición muscarínica. Este efecto de PIP2 sobre el canal TREK-1 y sobre IM ya
fue observado previamente (Zhang et al., 2003; Chemin et al., 2005).
Conocer la vía de modulación muscarínica del canal TREK-2, así como los segundos
mensajeros que participan en ella, es fundamental para saber cómo se regula este canal, al
igual que el potencial de membrana de reposo y la excitabilidad en las neuronas del GCS.
Además observando los efectos de los agonistas y antagonistas muscarínicos como oxo-M,
atropina y pirenzepina, sobre nuestro canal TREK-2 en las neuronas de GCS en cultivo, nos
ayuda a comprender como funcionan estas sustancias in vivo.

Discusión
121
Modulación por BK
Los receptores de BK se localizan mayoritariamente en las neuronas del ganglio de la
raíz dorsal relacionadas con el dolor, pero también se localizan en las neuronas del GCS
donde la hormona BK provoca la liberación de prostaglandinas causantes del dolor (Khasar et
al., 1995). Además BK excita las neuronas del GCS de rata, sobre las cuales provoca una
ligera despolarización y modula la excitabilidad neuronal debido a la inhibición de IM (Jones
et al., 1995). Los resultados de este trabajo muestran que BK bloquea también IRIL, indicando
que la modulación del potencial de membrana en reposo y de la excitabilidad neuronal no
solo se debe a la inhibición de IM sino también a la inhibición de la corriente de potasio a
través del canal TREK-2.
La modulación por BK del potencial de membrana en neuronas del GCS de rata
sucede a través del receptor B2 (Babbedge et al., 1995), el cual activa principalmente la
proteína Gq/11 en estas neuronas (Jones et al., 1995).
En el GCS de rata la inhibición muscarínica y la inhibición por BK de IM suceden a
través de vías de segundos mensajeros diferentes (Jones et al., 1995). En el presente trabajo se
ha visto que la inhibición muscarínica y por BK de IRIL son oclusivas, es decir, cuando ambas
sustancias se aplican juntas sus efectos no se suman y por tanto siguen la misma vía de
segundos mensajeros. Para corroborarlo, se estudió la implicación de los segundos mensajeros
PKC, Ca++
y PIP2. Se observó que el enzima PKC, y el incremento en la concentración de
Ca++
intracelular no participan en la modulación por BK de la corriente de potasio de TREK-2
en las neuronas del GCS de ratón. Villarroel y colaboradores (Villarroel et al., 1989)
observaron, en neuronas simpáticas de rana, que la modulación de IM tampoco depende de
PKC. Sin embargo los cambios en la concentración de calcio intracelular y la calmodulina
están implicados en la modulación por bradiquinina de IM en neuronas simpáticas de rata
(Cruzblanca et al., 1998; Gamper & Shapiro, 2003; Gamper et al., 2005). En el GCS de ratón
la inhibición por BK del canal TREK-2 se debe al descenso de la concentración de PIP2,
según muestran los resultados del presente trabajo. Este resultado corrobora que la inhibición
por BK y la inhibición muscarínica del canal TREK-2 comparten la misma vía de segundos
mensajeros.

Discusión
122
Interacción canal-receptores
En los apartados anteriores se ha citado que la inhibición muscarínica y por BK de IM en
neuronas simpáticas utilizan distintas vías de inhibición: la inhibición muscarínica se debe a
la hidrólisis de PIP2 en sí misma (Zhang et al., 2003), mientras que la inhibición por BK se
debe al aumento de la concentración de Ca++
intracelular (Cruzblanca et al., 1998). En los
resultados de este trabajo podemos observar que la corriente I(-30) en presencia de PIP2
preincubada se reduce cuando se aplicó oxo-M, pero no cuando se aplicó BK. I(-30) está
constituida por corrientes de sodio y de potasio que no se bloquean completamente con el
cocktail, como es el caso de IM. Por tanto la diferencia que se observa en la modulación por
oxo-M y por BK de I(-30) puede deberse a la diferente vía de modulación por BK y por oxo-M
de IM y su dependencia de PIP2. Es decir, como la modulación muscarínica de IM es debida a
la hidrólisis de PIP2, en presencia de alta concentración de PIP2, oxo-M apenas inhibe IM, sin
embargo la modulación por BK de IM no se debe a la hidrólisis de PIP2, sino al aumento de
calcio y por tanto se observa una importante inhibición de I(-30).
En neuronas simpáticas la diferencia entre la modulación por BK y por oxo-M de IM se
cree que se debe a la distancia de los receptores muscarínicos y de BK respecto al receptor de
IP3 del RE. El receptor de BK está lo suficientemente cerca del receptor de IP3 como para
provocar la liberación de Ca++
y la consiguiente inhibición de IM. Además el canal de IM está
muy cerca del receptor de IP3 (Delmas et al., 2002). Pero además el incremento de Ca++
activa
el enzima PI4K cuya función es restaurar la concentración de PIP2 y por ello la inhibición de
IM por BK no se debe al descenso de la concentración de PIP2 por sí. Sin embargo el receptor
muscarínico está más lejos del receptor de IP3 presente en el RE, por lo que la liberación de
Ca++
es menor e insuficiente para restaurar la concentración de PIP2 y por lo tanto su
inhibición se debe a la hidrólisis de PIP2 (Delmas & Brown, 2005; Hernandez et al., 2008).
Los resultados de este trabajo muestran que la modulación muscarínica y por BK del
canal TREK-2 sucede a través de la misma vía en ambos casos y se debe a la hidrólisis de
PIP2 independientemente de la liberación de calcio a través de receptores de IP3 del RE. En
este caso el receptor de IP3, que se encuentra en el RE, estaría alejado del canal TREK-2, y
por ello la liberación de Ca++
debida a la activación del receptor de BK es insuficiente para
bloquear el canal.

Discusión
123
Participación del canal TREK-2 en el potencial de membrana en reposo y la
excitabilidad neuronal
El incremento en el flujo de potasio a través de canales iónicos que están abiertos a
potenciales de membrana en reposo tiende a hiperpolarizar la membrana (Lamas, 2005),
mientras que cuando se cierran estos canales las neuronas estará más despolarizadas. El
neurotransmisor Ach y los agonistas muscarínicos tienen también efecto excitatorio ya que
modulan la frecuencia de disparo y el potencial de membrana. El agonista muscarínico oxo-M
provoca despolarización en el potencial de membrana en reposo en torno a 10 mV y aumenta
el número de potenciales de acción en las neuronas del GCS de ratón, debida a la inhibición
muscarínica de IM (Romero et al., 2004). En el presente trabajo se determinó la participación
del canal TREK-2 en la modulación del potencial de membrana en reposo mediante el
agonista muscarínico oxo-M. Este experimento se realizó en presencia de TEA, con lo cual el
efecto de oxo-M es debido al bloqueo mayoritariamente del canal TREK-2. Sin embargo los
resultados muestras que la despolarización provocada por la inhibición muscarínica de
TREK-2 no es significativa (2.5 mV). Esta ligera despolarización podría deberse a que la
corriente de TREK-2 que es muy pequeña a temperatura ambiente (Maingret et al., 2000).
Previamente en nuestro laboratorio se ha visto que cuando se aplica riluzol (300 µM) las
neuronas se hiperpolarizan de forma significativa alrededor de 8 mV (Cadaveira-Mosquera et
al., 2011). Esto indica que la activación del canal TREK-2 es capaz de modular el potencial
de membrana en reposo en las neuronas del GCS de ratón. Nuestros resultados muestran que
cuando aplicamos XE991 en presencia de TEA y a temperatura fisiológica, se produce una
despolarización del potencial de membrana en reposo de casi 8 mV. Esta despolarización se
debe en su mayoría al bloqueo de TREK-2 ya que en este caso la corriente de potasio de
TREK-2 está activada con la temperatura, aunque una pequeña parte se debe al bloqueo de IM
ya que esta corriente no se inhibe completamente con TEA. Este resultado muestra que la
contribución de TREK-2 al valor del potencial de membrana en reposo a temperatura
fisiológica es similar a la aportación de IM (los resultados muestran que cuando inhibimos IM
con TEA a temperatura fisiológica, el potencial de membrana en reposo se despolariza 5.55
mV). Se ha visto que el aumento de la temperatura hiperpolariza los miocitos en el corazón
del embrión de pollo desde la temperatura ambiente (20 ºC) hasta la temperatura fisiológica
(en este caso 35 ºC) cuando el potencial de membrana está en reposo, debida en gran parte a

Discusión
124
la activación del canal TREK-1. De hecho, cuando se bloquea el canal TREK-1 el potencial
de membrana se despolariza alrededor de 30 mV, recuperándose el potencial de membrana en
reposo previo al aumento de la temperatura (Zhang et al., 2008).
En los ganglios periféricos, la diferenciación entre neuronas fásicas o tónicas (aquellas
que disparan potenciales de acción sin adaptar) lo marca la presencia de IM. De este modo la
presencia de IM es mayor en las neuronas del GCS que en las neuronas tónicas del ganglio
celiaco y del ganglio mesentérico superior (Wang & McKinnon, 1995). Nuestros resultados
muestran que el aumento de temperatura y por ello, el incremento de IM y de la corriente de
TREK-2, incrementa la adaptación de las neuronas de GCS. Además cuando se añade XE991,
y por consiguiente se bloquean ambas corrientes, se reduce la adaptación.
El aumento de temperatura desde 23 ºC (temperatura ambiente) hasta 37 ºC
(temperatura fisiológica) incrementa la corriente de potasio a través de los canales TREK-2.
Este aumento de corriente se debe al aumento en la probabilidad de apertura, sin modificación
en la conductancia del canal. De hecho a temperatura ambiente el canal TREK-2 apenas tiene
actividad, al igual que los canales TREK-1 y TRAAK (Maingret et al., 2000; Kang et al.,
2005).
El siguiente objetivo en el presente trabajo fue determinar la implicación
exclusivamente de la corriente de TREK-2 en el patrón de disparos de las neuronas del GCS.
En este caso no se pudo inhibir IM mediante la aplicación de TEA, ya que bloquea los canales
de potasio rectificadores tardío y los canales de potasio tipo A o transitorios (Decoursey et al.,
1984; Lipton & Tauck, 1987; Cobbett et al., 1989). Por ello estudiamos el patrón de disparos
en los primeros milisegundos tras la aplicación del estímulo que genera los potenciales de
acción. IM es dependiente de voltaje y permanece cerrada en su mayoría cuando el potencial
de membrana está en reposo y se abre de forma lenta cuando el potencial de membrana se
despolariza (Adams et al., 1982b). Sin embargo, la corriente de potasio a través de TREK-2
es poco dependiente de voltaje (Goldstein et al., 1996), por lo que no cambia sustancialmente
durante el tren de disparos. El incremento de temperatura modifica la cinética de los canales
iónicos y por tanto la cinética de IM también se modifica (Brown, 1988). En el presente
trabajo hemos visto que la constante de tiempo de cierre de IM a temperatura ambiente es de
aproximadamente 68 ms (70 ms en neuronas del GCS de rata según Hadley y colaboradores
(Hadley et al., 2003)), sin embargo a temperatura fisiológica es 8 ms. Los resultados de este
trabajo indican que la constante de tiempo de apertura de IM a temperatura ambiente es de 157

Discusión
125
ms y a temperatura fisiológica se reduce a 7 ms. La latencia y la frecuencia estudiadas en este
trabajo a temperatura ambiente se observaron en los primeros 60 ms desde la aplicación del
estímulo, y corresponden a los dos primeros potenciales de acción. Estudiando solamente
estos primeros 60 ms nos cercioramos de que IM todavía está cerrada y así los cambios
observados debido a la aplicación de XE991 se deben a la inhibición exclusivamente del canal
TREK-2. Sin embargo la reducción de la constante de tiempo de apertura de IM al incrementar
la temperatura nos imposibilita el estudio de la implicación de TREK-2 en la latencia y la
frecuencia a temperatura fisiológica.
Los experimentos de inhibición de TREK-2 con XE991 a temperatura ambiente nos
muestran que la latencia y la frecuencia en los primeros 60 ms no se modifican. Esto puede
deberse a que el canal TREK-2 está poco activo a temperatura ambiente (Maingret et al.,
2000).
En el presente trabajo no se pudo determinar la implicación del canal TREK-2 en la
excitabilidad neuronal debido a la contaminación de IM a temperatura fisiológica y a la falta
de actividad de TREK-2 a temperatura ambiente. En este estudio tampoco se puede utilizar
riluzol para activar la corriente a través de TREK-2 ya que este neuroprotector inhibe la
corriente de sodio persistente y la corriente de sodio transitoria (Lamas et al., 2009). Sin
embargo la acusada activación de la corriente de potasio de TREK-2 con el incremento de
temperatura y la participación de éste en la modulación del potencial de membrana en reposo,
sugieren que al igual que IM, TREK-2 podría reducir la excitabilidad neuronal e incrementar
la adaptación de las neuronas del GCS de ratón.
Implicaciones fisiológicas del canal TREK-2
La estimulación del sistema nervioso simpático, cuyo neurotransmisor en las neuronas
preganglionares es ACh (Feldman et al., 1997), tiende a aumentar la actividad del animal,
acelerar la circulación y frenar los fenómenos digestivos preparando así al animal para la
lucha o la huída (Romer & Parsons, 1981). La estimulación de las neuronas del GCS provoca
vasoconstricción de las glándulas lacrimales, dilatación pupilar, elevación del párpado
superior, participa en la acomodación en la visión lejana e inerva el lecho vascular donde
provoca vasoconstricción. Además estimula la secreción de enzimas salivares y aumenta la

Discusión
126
frecuencia cardíaca, la contractilidad y la vasoconstricción coronaria (Aguilar et al., 1998).
Las neuronas del GCS en cultivo son moduladas mediante agonistas muscarínicos que
simulan los efectos producidos por Ach in vivo. Hasta el momento, la modulación
muscarínica del potencial de membrana en reposo y de la adaptación en las neuronas del GCS
de ratón se asociaba fundamentalmente a la modulación muscarínica de IM (Romero et al.,
2004). Sin embargo, el presente trabajo muestra que el canal TREK-2 se inhibe mediante el
agonista muscarínico oxo-M en dichas neuronas en cultivo primario, lo que indica que este
canal se inhibe cuando a la neurona postganglionares del GCS le llega ACh in vivo. La
inhibición muscarínica de IM en el GCS de ratón provoca que las neuronas se hiperpolaricen y
reduzcan su adaptación al estímulo provocando el aumento de la actividad neuronal (Romero
et al., 2004). Este trabajo muestra que la inhibición muscarínica del canal TREK-2 también
participa en la modulación de la actividad neuronal en el sistema simpático.
La linopirdina aumenta la liberación de ACh en el terminal presináptico
independientemente de IM (Provan & Miyamoto, 1994), sin embargo provoca la liberación de
NA en la neurona postsináptica del sistema nervioso simpático mediante la inhibición de IM
(Kristufek et al., 1999). Mediante el presente trabajo se ha visto que linopirdina y XE991
también bloquean el canal TREK-2, por lo que resulta plausible que este canal también esté
relacionado con la liberación de NA en la neurona postsináptica.
Los canales TREK-2 están relacionados con la neuroprotección ya que se activan
durante procesos isquémicos tales como el estiramiento de la membrana cuando las neuronas
hinchan, cuando se acidifica el medio intracelular y cuando se incrementa la concentración de
ácidos grasos insaturados. Además son activados por el neuroprotector riluzol (Duprat et al.,
2000; Lesage et al., 2000; Cadaveira-Mosquera et al., 2011). Pero además durante procesos
isquémicos aumenta la expresión de estos canales en los astrocitos. La función de los
astrocitos es sustentar a las neuronas y mantener las concentraciones extracelulares normales
de K+, H
+, GABA y glutamato en el espacio extracelular que rodea a las neuronas (Li et al.,
2005).
El canal TREK-2 está relacionado con la sensibilidad a la temperatura ambiental y con
el dolor (Pereira et al., 2014). Además la corriente de potasio a través del canal TREK-2 se
incrementa con la temperatura, incluso por encima de la temperatura fisiológica, lo que
también los relaciona con la fiebre (Kang et al., 2005). Por este motivo, los canales TREK-2
se consideran una buena diana para el estudio de fármacos utilizados para el tratamiento

Discusión
127
contra el dolor. Se ha visto además que los analgésicos incrementan la corriente de TREK-1
(Veale et al., 2014).
En este trabajo hemos visto que a temperatura fisiológica la corriente a través del canal
TREK-2 se activa acusadamente en las neuronas del GCS de ratón. La bibliografía muestra
que la mayoría de los estudios de modulación de esta corriente se ha realizado a temperatura
ambiente. Este trabajo indica que a temperatura fisiológica la modulación del potencial de
membrana en reposo y la excitabilidad neuronal debido a la inhibición de la corriente de
TREK-2 es más acusada que a temperatura ambiente. Por consiguiente, este trabajo sugiere
que la corriente de potasio de TREK-2 es más relevante a nivel fisiológico de lo que los
experimentos in vitro proponen debido a su gran incremento a temperatura fisiológica.


CONCLUSIONES


Conclusiones
131
CONCLUSIONES
De acuerdo a nuestros resultados concluimos que:
1. La corriente de potasio a través del canal TREK-2 de neuronas del GCS de ratón
en cultivo es sensible al incremento de temperatura y a los bloqueantes
linopirdina y XE991.
2. La corriente de potasio a través del canal TREK-2 e IM de neuronas del GCS de
ratón en cultivo se distinguen activando la corriente de TREK-2 con riluzol
(IRIL) y bloqueando IM con TEA. Sin embargo no se pueden diferenciar
utilizando XE991, ni linopirdina, ni incrementando la temperatura ya que ambas
son sensibles.
3. El agonista muscarínico oxo-M inhibe IRIL a través del receptor muscarínico M1, de
la proteína Gq/11 y del enzima PLC en las neuronas del GCS de ratón. Sin embargo
no participan en dicha inhibición el enzima PKC, ni el incremento en la
concentración de Ca++
intracelular.
4. La inhibición muscarínica de IRIL en las neuronas del GCS de ratón se debe al
descenso de la concentración de PIP2, necesaria para mantener el canal activo.
5. La inhibición de IRIL por BK en las neuronas del GCS de ratón se debe al descenso
de la concentración de PIP2 y es independiente del enzima PKC y del incremento
intracelular de Ca++
.
6. La inhibición muscarínica de TREK-2 no afecta al potencial de membrana en
reposo a temperatura ambiente. Sin embargo a temperatura fisiológica, el
bloqueo de la corriente de TREK-2 provoca una despolarización del potencial de
membrana en reposo.

Conclusiones
132
7. La activación de IM y de la corriente de TREK-2 debida al aumento de
temperatura incrementa la adaptación de las neuronas del GCS de ratón.
8. El incremento de la temperatura hasta valores fisiológicos reduce la constante de
tiempo de apertura de IM. Esto nos dificulta determinar la implicación de la
corriente de potasio de TREK-2 en la excitabilidad neuronal a temperatura
fisiológica.
9. A temperatura ambiente se observó que el canal TREK-2 no afecta a los parámetros
de latencia y frecuencia de los primeros potenciales de acción del tren de disparos
de las neuronas del GCS de ratón.
10. La contribución de la corriente de TREK-2 e IM al valor del potencial de membrana
en reposo son similares a temperatura fisiológica.

BIBLIOGRAFÍA


Bibliografía
135
BIBLIOGRAFÍA
Adams, P.R., Brown, D. A, & Constanti, A (1982a) Pharmacological Inhibition of the
M-Current. J physiol, 332, 223-262.
Adams, P.R., Brown, D.A., & Constanti, A. (1982b) M-currents and other potassium
currents in bullfrog sympathetic neurones. J. Physiol., 330, 537–572.
Aguilar, E., Delgado, J., Ferrús, A., Mora, F., & Rubia, J. (1998) Sistema nervioso
autónomo. In Manual de Neurociencia, Síntesis. edn. Madrid, pp. 733–754.
Ahn, H.S., Kim, S.E., Jang, H.J., Kim, M.J., Rhie, D.J., Yoon, S.H., Jo, Y.H., Kim,
M.S., Sung, K.W., & Hahn, S.J. (2006) Interaction of riluzole with the closed
inactivated state of Kv4.3 channels. J. Pharmacol. Exp. Ther., 319, 323–331.
Aiken, S.P., Lampe, B.J., Murphy, P.A., & Brown, B.S. (1995) Reduction of spike
frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by
linopirdine (DuP 996), a neurotransmitter release enhancer. Br. J. Pharmacol., 115,
1163–1168.
Babbedge, R., Dray, A., & Urban, L. (1995) Bradykinin depolarises the rat isolated
superior cervical ganglion via B2 receptor activation. Neurosci. Lett., 193, 161–
164.
Bal, M., Zhang, J., Hernandez, C.C., Zaika, O., & Shapiro, M.S. (2010)
Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M-Type) K+
channels. J. Neurosci., 30, 2311–2323.
Bang, H., Kim, Y., & Kim, D. (2000) TREK-2, a new member of the mechanosensitive
tandem-pore K+ channel family. J. Biol. Chem., 275, 17412–17419.
Bastiani, C. & Mendel, J. (2006) Heterotrimeric G proteins in C. elegans. WormBook,
1–25.
Beltrán, L., Beltrán, M., Aguado, A., Gisselmann, G., & Hatt, H. (2013) 2-
aminoethoxydiphenyl borate activates the mechanically gated human KCNK
channels KCNK 2 (TREK-1), KCNK 4 (TRAAK), and KCNK 10 (TREK-2).
Front. Pharmacol., 4, 1–6.
Bensimon, G., Lacomblez, L., & Meininger, V. (1994) A controlled trial of riluzole in
amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med, 330,
585–591.
Brown, D. (1988) M currents. In Ion Channels, Plenum Pub. edn. pp. 55–93.
Brown, D., Buckley, N., Caulfield, M., Duffy, S., Jones, S., Lamas, J., Marsh, S.,
Robbins, J., & Selyanko, A. (1995) Coupling of muscarinic acetylcholine receptors

Bibliografía
136
to neural ion channels: closure of K+ channels. In Molecular Mechanisms of
Muscarinic Acetylcholine Receptor Function, Landes Com. edn. Austin, Texas,
U.S.A, pp. 165–182.
Brown, D.A. & Adams, P.R. (1980) Muscarinic suppression of a novel voltage-sensitive
K+ current in a vertebrate neurone. Nature, 283, 673–675.
Brown, D.A. & Constanti, A. (1980) Intracellular observations on the effects of
muscarinic agonists on rat sympathetic neurones. Br. J. Pharmacol., 70, 593–608.
Brown, D.A., Marriont, N. V, & Smart, T.G. (1989) On the transduction mechanism for
muscarine-induced inhibition of M-current in cultured rat sympathetic neurones. J.
Physiol., 413, 469–488.
Brown, D.A. & Selyanko, A. (1985) Membrane currents underlying the cholinergic
slow excitatory post-synaptic potential in the rat sympathetic ganglion. J. Physiol.,
365, 365–387.
Brown, H. & Difrancesco, D. (1980) Voltage-clamp investigations of membrane
currents underlying pace-maker activity in rabbit sino-atrial node. J. Physiol., 308,
331–351.
Buño, W., Delgado, J., Ferrús, A., Mora, F., & Rubia, J. (1998) Propiedades eléctricas
de las membranas de las células excitables. In Manual de Neurociencia, Síntesis.
edn. Madrid, pp. 121–137.
Cadaveira-Mosquera, A., Pérez, M., Reboreda, A., Rivas-Ramírez, P., Fernández-
Fernández, D., & Lamas, J.A. (2012) Expression of K2P channels in sensory and
motor neurons of the autonomic nervous system. J. Mol. Neurosci., 48, 86–96.
Cadaveira-Mosquera, A., Ribeiro, S.J., Reboreda, A., Pérez, M., & Lamas, J.A. (2011)
Activation of TREK currents by the neuroprotective agent riluzole in mouse
sympathetic neurons. J. Neurosci., 31, 1375–1385.
Caulfield, M.P. & Birdsall, N.J. (1998) International Union of Pharmacology. XVII.
Classification of muscarinic acetylcholine receptors. Pharmacol. Rev., 50, 279–
290.
Caulfield, M.P., Jones, S., Vallis, Y., Buckley, N.J., Kim, G., Milligan, G., & Brown,
D.A. (1994) Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor
inhibition of Ca++ current via Gαo in rat sympathetic neurones. J. Physiol., 477.3,
415–422.
Chemin, J., Girard, C., Duprat, F., Lesage, F., Romey, G., & Lazdunski, M. (2003)
Mechanisms underlying excitatory effects of group I metabotropic glutamate
receptors via inhibition of 2P domain K+ channels. EMBO J., 22, 5403–5411.

Bibliografía
137
Chemin, J., Patel, A.J., Duprat, F., Lauritzen, I., Lazdunski, M., & Honoré, E. (2005) A
phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO
J., 24, 44–53.
Cobbett, P., Legendre, P., & Mason, W.T. (1989) Characterization of three types of
potassium current in cultured neurones of rat supraoptic nucleus area. J. Physiol.,
410, 443–462.
Costa, A.M.N. & Brown, D.A. (1998) Inhibition of M-current in cultured rat superior
cervical ganglia by linopirdine : mechanism of action studies. Neuropharmacology,
36, 1747–1753.
Cruzblanca, H., Koh, D.S., & Hille, B. (1998) Bradykinin inhibits M current via
phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat
sympathetic neurons. Proc. Natl. Acad. Sci. U. S. A., 95, 7151–7156.
Czirják, G. & Enyedi, P. (2002) Formation of functional heterodimers between the
TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol.
Chem., 277, 5426–5432.
Czirják, G. & Enyedi, P. (2006) Zinc and mercuric ions distinguish TRESK from the
other two-pore-domain K+ channels. Mol. Pharmacol., 69, 1024–1032.
Davis, R.E. & Stuart, A.E. (1988) A persistent, TTX-sensitive sodium current in an
invertebrate neuron with neurosecretory ultrastructure. J. Neurosci., 8, 3978–3991.
Decoursey, T.E., Dempster, J., & Hutter, O. (1984) Inward rectifier current noise in frog
skeletal muscle. J Physiol, 349, 299–327.
De la Peña, E., Mälkiä, A., Vara, H., Caires, R., Ballesta, J.J., Belmonte, C. & Viana, F.
(2012) The influence of cold temperature on cellular excitability of hippocampal
networks. PloS One, 7, 1-18.
Dell’Acqua, M.L., Faux, M.C., Thorburn, J., Thorburn, A., & Scott, J.D. (1998)
Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-
bisphosphate. EMBO J., 17, 2246–2260.
Delmas, P. & Brown, D.A. (2005) Pathways modulating neural KCNQ/M (Kv7)
potassium channels. Nat. Rev. Neurosci., 6, 850–862.
Delmas, P., Wanaverbecq, N., Abogadie, F.C., Mistry, M., & Brown, D.A. (2002)
Signaling microdomains define the specificity of receptor-mediated InsP3
pathways in neurons. Neuron, 34, 209–220.
Dong, Y.Y., Pike, A.C.W., Mackenzie, A., Mcclenaghan, C., Aryal, P., Dong, L.,
Grieben, M., Goubin, S., Ruda, G.F., Clausen, M. V, Cao, L., Brennan, P.E.,
Burgess-brown, N.A., Sansom, M.S.P., & Tucker, S.J. (2015) K2P channel gating
mechanisms revealed by structures of TREK-2 and a complex with Prozac.
Science, 347, 1256–1259.

Bibliografía
138
Dray, A., Bettaney, J., Forster, P., & Perkins, M.N. (1988) Activation of a bradykinin
receptor in peripheral nerve and spinal cord in the neonatal rat in vitro. Br. J.
Pharmacol., 95, 1008–1010.
Duprat, F., Lesage, F., Patel, A., Fink, M., Romey, G., & Lazdunski, M. (2000) The
neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1
and TRAAK. Mol. Pharmacol., 57, 906–912.
Enyeart, J.J., Danthi, S.J., Liu, H., & Enyeart, J.A. (2005) Angiotensin II inhibits
bTKEK-1 K+ channels in adrenocortical cells by separate Ca2+- and ATP
hydrolysis-dependent mechanisms. J. Biol. Chem., 280, 30814–30828.
Fakler, B., Schultz, J.H., Yang, J., Schulte, U., Brandle, U., Zenner, H.P., Jan, L.Y., &
Ruppersberg, J.P. (1996) Identification of a titratable lysine residue that determines
sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO J.,
15, 4093–4099.
Faux, M.C. & Scott, J.D. (1997) Regulation of the AKAP79-protein kinase C
interaction by Ca2+/calmodulin. J. Biol. Chem., 272, 17038–17044.
Fedoroff, S. & Richardson, A. (2001) Primary cultures of sympathetic ganglia. In
Protocols for Neuronal Cell Culture, Humana Pre. edn. Totowa, New Jersey, pp.
71–94.
Felder, C.C. (1995) Muscarinic acetylcholine receptors: signal transduction through
multiple effectors. FASEB J., 9, 619–625.
Feldman, R., Meyer, L., & Quenzer, J. (1997) Acetylcholine. In Principles of
Neuropsychopharmacology, Sinauer As. edn. MassAChusetts, USA, pp. 249–276.
Fink, M., Duprat, F., Lesage, F., Reyes, R., Romey, G., Heurteaux, C., & Lazdunski, M.
(1996) Cloning, functional expression and brain localization of a novel
unconventional outward rectifier K+ channel. EMBO J., 15, 6854–6862.
Fink, M., Lesage, F., Duprat, F., Heurteaux, C., Reyes, R., Fosset, M., & Lazdunski, M.
(1998) A neuronal two P domain K+ channel activated by arachidonic acid and
polyunsaturated fatty acid. Embo J., 17, 3297–3308.
Fonnum, F. (1969) Radiochemical micro assays for the determination of choline
acetyltransferase and acetylcholinesterase activities. Biochem. J., 115, 465–472.
Franks, N.P. & Honoré, E. (2004) The TREK K2P channels and their role in general
anaesthesia and neuroprotection. Trends Pharmacol. Sci., 25, 601–608.
Fu, X.W., Brezden, B.L., & Wu, S.H. (1997) Hyperpolarization-activated inward
current in neurons of the rat’s dorsal nucleus of the lateral lemniscus in vitro. J.
Neurophysiol., 78, 2235–2245.

Bibliografía
139
Gamper, N., Li, Y., & Shapiro, M. (2005) Structural requirements for differential
sensitivity of KCNQ K+ channels to modulation by Ca2+/Calmodulin. Mol. Biol.
Cell, 16, 3538–3551.
Gamper, N. & Shapiro, M.S. (2003) Calmodulin mediates Ca2+-dependent modulation
of M-type K+ channels. J. Gen. Physiol., 122, 17–31.
Goldman, D.E. (1943) Potential, impedance, and rectification in membranes. J. Gen.
Physiol., 27, 37–60.
Goldstein, S.A., Price, L., Rosenthal, D.N., & Pausch, M.H. (1996) ORK1, a potassium
selective leak channel with wo pore domains cloned from Drosophila melanogaster
by expression in Saccharomyces cerevisiae. Pnas, 93, 13256–13261.
Goldstein, S.A., Bockenhauer, D., O’Kelly, I., & Zilberberg, N. (2001) Potassium leak
channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci., 2,
175–184.
Gray, H. (1918) Anatomy of the Human Body, Philadelph. edn. New York.
Greaves, M. & Shuster, S. (1967) Responses of skin blood vessels to bradykinin,
histamine and 5-hydroxytryptamine. J Physiol, 193, 255–267.
Grunnet, M., Jespersen, T., Angelo, K., Frøkjær-Jensen, C., Klaerke, D.A., Olesen, S.P.,
& Jensen, B.S. (2001) Pharmacological modulation of SK3 channels.
Neuropharmacology, 40, 879–887.
Gu, W., Schlichthörl, G., & Hirsch, J. (2002) Expression pattern and functional
characteristics of two novel splice variants of the two-pore-domain potassium
channel TREK-2. J Physiol, 539.3, 657–668.
Gutowski, S., Smrcka, A., Nowak, L., Wu, D., Simon, M., & Sternweis, P.C. (1991)
Antibodies to the q subfamily of guanine nucleotide-binding regulatory protein α
subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis
by hormones. J. Biol. Chem., 266, 20519–20524.
Hadley, J.K., Passmore, G.M., Tatulian, L., Al-Qatari, M., Ye, F., Wickenden, A.D., &
Brown, D.A. (2003) Stoichiometry of expressed KCNQ2/KCNQ3 potassium
channels and subunit composition of native ganglionic M channels deduced from
block by tetraethylammonium. J. Neurosci., 23, 5012–5019.
Hamill, O.P., Marty, A., Neher, E., Sakmann, B., & Sigworth, F.J. (1981) Improved
patch clamp techniques for high resolution current recording from cells and cell-
free membranes. Pflugers Arch., 391, 85–100.
Hamilton, S.E., Loose, M.D., Qi, M., Levey, A., Hille, B., McKnight, G.S., Idzerda,
R.L., & Nathanson, N.M. (1997) Disruption of the m1 receptor gene ablates
muscarinic receptor-dependent M current regulation and seizure activity in mice.
Proc. Natl. Acad. Sci. U. S. A., 94, 13311–13316.

Bibliografía
140
Hernandez, C.C., Zaika, O., Tolstykh, G.P., & Shapiro, M.S. (2008) Regulation of
neural KCNQ channels: signalling pathways, structural motifs and functional
implications. J. Physiol., 586, 1811–1821.
Hille, B. (1940) Ion Channels of Excitable Membranes, Sinauer As. edn. Sunderland
(USA).
Hodgkin, A.L. & Katz, B. (1949) The effect of sodium ions on the electrical activity of
the giant axon of the squid. J. Physiol., 108, 37–77.
Honoré, E., Maingret, F., Lazdunski, M., & Patel, A.J. (2002) An intracellular proton
sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO
J., 21, 2968–2976.
Hoshi, N., Zhang, J.S., Omaki, M., Takeuchi, T., Yokoyama, S., Wanaverbecq, N.,
Langeberg, L.K., Yoneda, Y., Scott, J.D., Brown, D.A., & Higashida, H. (2003)
AKAP150 signaling complex promotes suppression of the M-current by
muscarinic agonists. Nat. Neurosci., 6, 564–571.
Hsu, K., Seharaseyon, J., Dong, P., Bour, S., & Marbán, E. (2004) Mutual functional
destruction of HIV-1 Vpu and host TASK-1 channel. Mol. Cell, 14, 259–267.
Huang, C.S., Song, J.H., Nagata, K., Yeh, J.Z., & Narahashi, T. (1997) Effects of the
neuroprotective agent riluzole on the high voltage-activated calcium channels of rat
dorsal root ganglion neurons. J. Pharmacol. Exp. Ther., 282, 1280–1290.
Jakubík, J., Bacáková, L., el-Fakahany, E.E., & Tucek, S. (1995) Constitutive activity
of the M1-M4 subtypes of muscarinic receptors in transfected CHO cells and of
muscarinic receptors in the heart cells revealed by negative antagonists. FEBS
Lett., 377, 275–279.
Jobling, P. & Gibbins, I.L. (1999) Electrophysiological and morphological diversity of
mouse sympathetic neurons. J. Neurophysiol., 82, 2747–2764.
Jones, S., Brown, D.A., Milligan, G., Willer, E., Buckley, N.J., & Caulfield, M.P.
(1995) Bradykinin excites rat sympathetic neurons by inhibition of M current
through a mechanism involving B2 receptors and G alpha q/11. Neuron, 14, 399–
405.
Kang, D., Choe, C., & Kim, D. (2005) Thermosensitivity of the two-pore domain K+
channels TREK-2 and TRAAK. J. Physiol., 564, 103–116.
Kang, D., Han, J., & Kim, D. (2006) Mechanism of inhibition of TREK-2 (K2P10.1) by
the Gq-coupled M3 muscarinic receptor. Am. J. Physiol. Cell Physiol., 291, 649–
656.
Khasar, S.G., Miao, F.J.P., & Levine, J.D. (1995) Inflammation modulates the
contribution of receptor-subtypes to bradykinin-induced hyperalgesia in the rat.
Neuroscience, 69, 685–690.

Bibliografía
141
Kobilka, B.K. (2007) G protein coupled receptor structure and activation. Biochim.
Biophys. Acta, 1768, 794–807.
Koester, J., Siegelbaum, S., Kandel, E., Schwartz, J., & Jessell, T. (2001) Potencial de
membrana. In Principios de Neurociencia, McGraw-Hil. edn. Madrid, pp. 125–
139.
Kristufek, D., Koth, G., Motejlek, A., Schwarz, K., Huck, S., & Boehm, S. (1999)
Modulation of spontaneous and stimulation-evoked transmitter release from rat
sympathetic neurons by the cognition enhancer linopirdine: Insights into its
mechanisms of action. J. Neurochem., 72, 2083–2091.
Kucheryavykh, L., Kucheryavyk, Y., Inyushin, M., Shuba, Y., Sanabria, P., Cubano, L.,
Skatchkov, S., & Eaton, M. (2009) Ischemia increases TREK-2 channel expression
in astrocytes: relevance to glutamate clearence. Open Neuronci J, 3, 40–47.
Lamas, J.A. (2012) Mechanosensitive K2P Channels, TREking through the Autonomic
Nervous System. In, Mechanically Gated Channels and their Regulation, Kamkin A
& Lozinsky I. eds Springer. pp. 35-68.
Lamas, J.A., Reboreda, A., & Codesido, V. (2002) Ionic basis of the resting membrane
potential in cultured rat sympathetic neurons. Neuroreport, 13, 585–591.
Lamas, J.A., Selyanko, A., & Brown, D. (1997) Effects of a cognition-enhancer,
linopirdine (DuP 996), on M-type potassium currents (I(K(M))) and some other
voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur. J.
Neurosci., 9, 605–616.
Lamas, J.A. (1998) A hyperpolarization-activated cation current (Ih) contributes to
resting membrane potential in rat superior cervical sympathetic neurones. Pflugers
Arch. Eur. J. Physiol., 436, 429–435.
Lamas, J.A. (2005) Evolución del concepto de potencial de reposo neuronal aspectos
basicos y clinicos. Rev Neurol, 41, 538–549.
Lamas, J.A., Romero, M., Reboreda, A., Sánchez, E., & Ribeiro, S.J. (2009) A riluzole-
and valproate-sensitive persistent sodium current contributes to the resting
membrane potential and increases the excitability of sympathetic neurones.
Pflugers Arch. Eur. J. Physiol., 458, 589–599.
Lamb, J.F. & MacKinnon, M. (1971) Effect of ouabain and metabolic inhibitors on the
Na and K movements and nucleotide contents of L cells. J Physiol, 213, 665–682.
Latek, D., Modzelewska, A., Trzaskowski, B., Palczewski, K., & Filipek, S. (2012) G
protein-coupled receptors — recent advances. Acta Biochim. Pol., 59, 515–529.
Lauritzen, I., Blondeau, N., Heurteaux, C., Widmann, C., Romey, G., & Lazdunski, M.
(2000) Polyunsaturated fatty acids are potent neuroprotectors. EMBO J., 19, 1784–
1793.

Bibliografía
142
Leeb-Lundberg, L.M.F., Marceau, F., Müller-Esterl, W., Pettibone, D.J., & Zuraw, B.L.
(2005) International union of pharmacology. XLV. Classification of the kinin
receptor family: from molecular mechanisms to pathophysiological consequences.
Pharmacol Rev, 57, 27–77.
Lesage, F. (2003) Pharmacology of neuronal background potassium channels.
Neuropharmacology, 44, 1–7.
Lesage, F., Guillemare, E., Fink, M., Duprat, F., Lazdunski, M., Romey, G., &
Barhanin, J. (1996) TWIK-1, a ubiquitous human weakly inward rectifying K+
channel with a novel structure. EMBO J., 15, 1004–1011.
Lesage, F. & Lazdunski, M. (2000) Molecular and functional properties of two-pore-
domain potassium channels. J Physiol Ren. Physiol, 279, 793–801.
Lesage, F., Terrenoire, C., Romey, G., & Lazdunski, M. (2000) Human TREK2, a 2P
Domain Mechano-sensitive K+ Channel with Multiple Regulations by
Polyunsaturated Fatty Acids, Lysophospholipids, and Gs, Gi, and Gq Protein-
coupled Receptors. J. Biol. Chem., 275, 28398–28405.
Li, Z. Bin, Zhang, H.X., Li, L.L., & Wang, X.L. (2005) Enhanced expressions of
arachidonic acid-sensitive tandem-pore domain potassium channels in rat
experimental acute cerebral ischemia. Biochem. Biophys. Res. Commun., 327,
1163–1169.
Lipton, S.A. & Tauck, D.L. (1987) Voltage-dependent conductances of solitary
ganglion cells dissociated from the rat retina. J. Physiol., 385, 361–391.
Liu, C., Au, J.D., Zou, H.L., Cotten, J.F., & Yost, C.S. (2004) Potent activation of the
human tandem pore domain K channel TRESK with clinical concentrations of
volatile anesthetics. Anesth. Analg., 99, 1715–1722.
Lolicato, M., Riegelhaupt, P.M., Arrigoni, C., Clark, K.A., & Minor, D.L. (2014)
Article transmembrane helix straightening and buckling underlies activation of
mechanosensitive and thermosensitive K2P channels. Neuron, 84, 1198–1212.
Lopes, C.M.B., Rohács, T., Czirják, G., Balla, T., Enyedi, P., & Logothetis, D.E. (2005)
PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating
of two-pore domain K+ channels. J. Physiol., 564, 117–129.
Lotshaw, D.P. (2007) Biophysical, pharmacological, and functional characteristics of
cloned and native mammalian two-pore domain K+ channels. Cell Biochem
Biophys, 47, 209–256.
Macintosh, F.C. (1941) The distribution of acetylcholine in the preripheral and the
cental nervous system. J Physiol, 99, 436–442.
Maingret, F., Fosset, M., Lesage, F., Lazdunski, M., & Honoré, E. (1999) TRAAK is a
mammalian neuronal mechano-gated K+ channel. J. Biol. Chem., 274, 1381–1387.

Bibliografía
143
Maingret, F., Lauritzen, I., Patel, A.J., Heurteaux, C., Reyes, R., Lesage, F., Lazdunski,
M., & Honoré, E. (2000) TREK-1 is a heat-activated background K(+) channel.
EMBO J., 19, 2483–2491.
Malgouris, C., Bardot, F., Daniel, M., Pellis, F., Rataud, J., Uzan, a, Blanchard, J.C., &
Laduron, P.M. (1989) Riluzole, a novel antiglutamate, prevents memory loss and
hippocampal neuronal damage in ischemic gerbils. J. Neurosci., 9, 3720–3727.
Marrion, N. V, Smart, T.G., Marsh, S.J., & Brown, D.A. (1989) Muscarinic suppression
of the M-current in the rat sympathetic ganglion is mediated by receptors of the
M1-subtype. Br. J. Pharmacol., 98, 557–573.
Marty, A. & Nehert, E. (1985) Potassium channels in cultured bovine adrenal
chromaffin cells. J Physiol, 367, 117–141.
Mathews, C.K., van Holde, K.E., & Ahern, K.G. (1999) Metabolic coordination,
metabolic control, and signal transduction. In Biochemistry, Addison We. edn. San
Francisco, CA, pp. 836–842.
Mathie, A. (2007) Neuronal two-pore-domain potassium channels and their regulation
by G protein-coupled receptors. J. Physiol., 578, 377–385.
Miras, M.T., Delgado, J.M., Ferrús, A., Mora, F., & Rubia, J.R. (1998) Otros tipos de
sinapsis. Mensajeros intracelulares. In Manual de Neurociencia, Síntesis. edn.
Madrid, pp. 249–269.
Murbartián, J., Lei, Q., Sando, J.J., & Bayliss, D.A. (2005) Sequential phosphorylation
mediates receptor- and kinase-induced inhibition of TREK-1 background
potassium channels. J. Biol. Chem., 280, 30175–30184.
Neher, E. & Sakmann, B. (1976) Single-channel currents recorded from membrane of
denervated frog muscle fibres. Nature, 260, 799–802.
Nelson, C.P., Nahorski, S.R., & Challiss, R.A.J. (2006) Constitutive activity and inverse
agonism at the M2 muscarinic acetylcholine receptor. J. Pharmacol. Exp. Ther.,
316, 279–288.
Nelson, D.L. & Cox, M.M. (2009) Bioseñalización. In Lehninger. Principios de
Bioquímica, Omega. edn. Barcelona, pp. 423–438.
Noël, J., Sandoz, G., & Lesage, F. (2011) Molecular regulations governing TREK and
TRAAK channel functions. Channels (Austin), 5, 402–409.
Patel, A., Honoré, E., Lesage, F., Fink, M., Romey, G., & Lazdunski, M. (1999)
Inhalational anesthetics activate two-pore-domain background K+ channels. Nat.
Neurosci., 2, 422–426.
Patel, A.J. & Honoré, E. (2001) Properties and modulation of mammalian 2P domain
K+ channels. TRENS Neurosci., 24, 339–346.

Bibliografía
144
Patel, A.J., Honoré, E., Maingret, F., Lesage, F., Fink, M., Duprat, F., & Lazdunski, M.
(1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO
J., 17, 4283–4290.
Patel, A.J., Maingret, F., Magnone, V., Fosset, M., Lazdunski, M., & Honore, E. (2000)
TWIK-2, an inactivating 2P domain K+ channel. J. Biol. Chem., 275, 28722–
28730.
Pereira, V., Busserolles, J., Christin, M., Devilliers, M., Poupon, L., Legha, W., Alloui,
A., Aissouni, Y., Bourinet, E., Lesage, F., Eschalier, A., Lazdunski, M., & Noël, J.
(2014) Role of the TREK2 potassium channel in cold and warm thermosensation
and in pain perception. Pain, 155, 2534–2544.
Perkins, M.N. & Kelly, D. (1993) Induction of bradykinin B1 receptors in vivo in a
model of ultra-violet irradiation-induced thermal hyperalgesia in the rat. Br. J.
Pharmacol., 110, 1441–1444.
Provan, S.D. & Miyamoto, M.D. (1994) Effect of the putative cognitive enhancer,
linopirdine (DuP 996), on quantal parameters of acetylcholine release at the frog
neuromuscular junction. Br. J. Pharmacol., 111, 1103–1110.
Robbins, J. (2001) KCNQ potassium channels : physiology, pathophysiology, and
pharmacology. Pharmacol. Ther., 90, 1–19.
Romer, A. & Parsons, T. (1981) Sistema nervioso. In Anatomía Comparada, Nueva
Edit. edn. México, pp. 335–340.
Romero, M., Reboreda, A., Sànchez, E., & Lamas, J.A. (2004) Newly developed
blockers of the M-current do not reduced spike frequency adaptation in cultured
mouse sympathetic neurons. Eur. J. Neurosci., 19, 2693–2702.
Rugiero, F., Mistry, M., Sage, D., Black, J.A., Waxman, S.G., Crest, M., Clerc, N.,
Delmas, P., & Gola, M. (2003) Selective expression of a persistent tetrodotoxin-
resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J.
Neurosci., 23, 2715–2725.
Sakmann, B. & Neher, E. (1984) Patch clamp techniques for studying ionic channels in
excitable membranes. Annu. Rev. Physiol., 46, 455–472.
Sandoz, G., Tardy, M.P., Thümmler, S., Feliciangeli, S., Lazdunski, M., & Lesage, F.
(2008) Mtap2 is a constituent of the protein network that regulates twik-related K+
channel expression and trafficking. J. Neurosci., 28, 8545–8552.
Sandoz, G., Thümmler, S., Duprat, F., Feliciangeli, S., Vinh, J., Escoubas, P., Guy, N.,
Lazdunski, M., & Lesage, F. (2006) AKAP150, a switch to convert mechano-, pH-
and arachidonic acid-sensitive TREK K(+) channels into open leak channels.
EMBO J., 25, 5864–5872.

Bibliografía
145
Shapiro, M.S., Roche, J.P., Kaftan, E.J., Cruzblanca, H., Mackie, K., & Hille, B. (2000)
Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels
that underlie the neuronal M current. J. Neurosci., 20, 1710–1721.
Sharma, J.N. & Al-Sherif, G.J. (2006) Pharmacologic targets and prototype therapeutics
in the kallikrein-kinin system: bradykinin receptor agonists or antagonists.
ScientificWorldJournal., 6, 1247–1261.
Sohn, J.W., Lim, A., Lee, S.H., & Ho, W.K. (2007) Decrease in PIP(2) channel
interactions is the final common mechanism involved in PKC- and arachidonic
acid-mediated inhibitions of GABA(B)-activated K+ current. J. Physiol., 582,
1037–1046.
Springer, M.G., Kullmann, P.H.M., & Horn, J.P. (2014) Virtual leak channels modulate
firing dynamics and synaptic integration in rat sympathetic neurons: implications
for ganglionic transmission in vivo. J. Physiol., 4, 803–8232.
Storm, J.F. (1989) An after-hyperpolarization of medium duration in rat hippocampal
pyramidal cells. J. Physiol., 409, 171–190.
Taylor, P.B., Rieger, F., Shelanski, M.L., & Greene, L.A. (1981) Cellular localization of
the multiple molecular forms of acetylcholinesterase in cultured neuronal cells. J.
Biol. Chem., 256, 3827–3830.
Tippmer, S., Quitterer, U., Kolm, V., Faussner, A., Roscher, A., Mosthaf, L., Müller-
Esterl, W., & Häring, H. (1994) Bradykinin induces translocation of the protein
kinase C isoforms alpha, epsilon, and zeta. Eur. J. Biochem., 225, 297–304.
Veale, E.L., Al-Moubarak, E., Bajaria, N., Omoto, K., Cao, L., Tucker, S.J., Stevens,
E.B., & Mathie, A. (2014) Influence of the N terminus on the biophysical
properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol., 85,
671–681.
Vilardaga, J.P. (2010) Theme and variations on kinetics of GPCR
activation/deactivation. J Recept Signal Transduct Res, 30, 304–312.
Villarroel, A., Marrion, N. V, Lopez, H., & Adams, P.R. (1989) Bradykinin inhibits a
potassium M-like current in rat pheochromocytoma PC12 cells. FEBS Lett, 255,
42–46.
Wang, H.S. & McKinnon, D. (1995) Potassium currents in rat prevertebral and
paravertebral sympathetic neurones: control of firing properties. J. Physiol., 485,
319–335.
Wang, H.S., Pan, Z., Shi, W., Brown, B.S., Wymore, R.S., Cohen, I.S., Dixon, J.E., &
McKinnon, D. (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular
correlates of the M-channel. Science, 282, 1890–1893.

Bibliografía
146
Wang, Y.J., Lin, M.W., Lin, A.A., & Wu, S.N. (2008) Riluzole-induced block of
voltage-gated Na+ current and activation of BKCa channels in cultured
differentiated human skeletal muscle cells. Life Sci., 82, 11–20.
Wen, R., Lui, G.M., & Steinberg, R.H. (1994) Expression of a tetrodotoxin-sensitive
Na+ current in cultured human retinal pigment epithelial cells. J. Physiol., 476,
187–196.
Wu, N., Hsiao, C.F., & Chandler, S.H. (2001) Membrane resonance and subthreshold
membrane oscillations in mesencephalic V neurons: participants in burst
generation. J. Neurosci., 21, 3729–3739.
Yano, K., Higashida, H., Hattori, H., & Nozawa, Y. (1985) Bradykinin-induced
transient accumulation of inositol trisphosphate in neuron-like cell line NG108-15
cells. FEBS Lett., 181, 403–406.
Yu, F., Han, S., Liu, W., Wang, Y., Liu, Y., Yin, J., Wang, H., Peng, B., & He, X.
(2012) Thermal Regulation of KCNQ2 Potassium Channels. Am. J. Biomed. Sci.,
4, 333–339.
Yus-Najera, E., Santana-Castro, I., & Villarroel, A. (2002) The Identification and
Characterization of a Noncontinuous Calmodulin-binding Site in Noninactivating
Voltage-dependent KCNQ Potassium Channels. J. Biol. Chem., 277, 28545–
28553.
Zaika, O., Mamenko, M., O’Neil, R.G., & Pochynyuk, O. (2011) Bradykinin acutely
inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive
distal nephron. Am. J. Physiol. Renal Physiol., 300, 1105–1115.
Zhang, H., Craciun, L.C., Mirshahi, T., Rohács, T., Lopes, C.M.B., Jin, T., &
Logothetis, D.E. (2003) PIP2 activates KCNQ channels, and its hydrolysis
underlies receptor-mediated inhibition of M currents. Neuron, 37, 963–975.
Zhang, H., Shepherd, N., & Creazzo, T.L. (2008) Temperature-sensitive TREK currents
contribute to setting the resting membrane potential in embryonic atrial myocytes.
J. Physiol., 586, 3645–3656.
Páginas web:
www.sigmaaldrich.com
www.guidetopharmacology.org/GRAC/ReceptorFamiliesForward?type=GPCR
(IUPHAR Database)
www.medical-institution.com/g-protein-signaling/
http://www.bem.fi/book/04/04x/0427x.htm
Foto portada: (Dong et al., 2015)






























