Embed Size (px)
Citation preview

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 1/15
Fenilcetonuria
Student: Stanca Cez
Stuparu Cosm
Zăvoianu Ramo
Seria VI, Grupa 66

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 2/15
Scurt istoric
Fenilcetonuria este cunoscută ca boală metabolică ereditară din
medicul norvegian Asbjorn Folling a identificat-o în cadrul Institu
al Universităţii din Oslo.
„O mamă vine să-mi arate doi copii anormali. Ea consultase deja
fără rezultat. Nu credeam că o voi putea ajuta, dar printr-o coi
eu nu sunt numai medic, ci şi chimist.Am descoperit în urina aces
şi 7 ani), o substanţă organică pe care n-am mai întâlnit-o
fenilpiruvic”.
Ceea ce i-a atras atenţia, a fost mirosul particular al urinii cel
exclus posibilitatea unei infecții a tractului urinar și a cercetat
este explicat de prezența corpilor cetonici în urină, numai că adao
ferică în urină a dat o colorație verde nemaîntâlnită în laboratoru
Prin cercetări ulterioare, Folling a dovedit că produsul anormal
acestor bolnavi, este acidul fenilpiruvic, metabolic anorm
tulburarea metabolismului fenilalaninei.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 3/15
PKU - Biochimie
Fenilcetonuria (PKU) este cauzată de o deficiență de fenilalanin-hidroxilază. Din punct de vedere clinic, PKU
reprezintă cel mai frecvent întâlnit defect ereditar al metabolismului aminoacizilor. Prevalența bolii este în
medie de 1:15.000, cu valori foarte mari în Turcia (1:2.600) și foarte scăzute în Japonia și Finlanda
(<1:100.000).
Din punct de vedere biochimic, PKU se caracterizează prin acumularea de fenilalanină (și un deficit de
tirozină). Hiperfenilalaninemia poate fi cauzată și de deficiențe ale oricăreia dintre enzimele necesare
sintezei de BH4 (tetrabiopterină) sau de defectul enzimei dihidropteridin (BH2) reductază, care regenerează
BH4 din BH2. Astfel de deficiențe cresc indirect concentrațiile de fenilalanină, deoarece Phe-hidroxilaza are
nevoie de BH4 drept coenzimă.
BH4 este un cofactor necesar și enzimelor tirozin-hidroxilază și triptofan-hidroxilază, care catalizează
reacțiile ce conduc la sinteza unor neurotransmițători precum serotonina și catecolaminele. Simpla limitare
a aportului alimentar de Phe nu contractarează efectele produse de deficiențele neurotransmițătorilor la
nivelul SNC. Terapia de substituție cu BH4 sau cu L-DOPA și 5-hidroxitriptofan (produși ai reacțiilor catalizate
de Tyr-hidroxilază și Trp-hidroxilază care sunt afectate) ameliorează evoluția clinică a acestor forme de
hiperfenilalaninemie, deși răspunsul la tratament este imprevizibil.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 4/15
Boala este caracterizată prin mutația genei PAH, responsabilă de sintetizarea enzimei hepatice fenilalanin-
hidroxilază implicată în transformarea aminoacidului fenilalanină, în exces, în tirozină.
Fenilcetonuria sau PKU este o boală genetică transmisă autozomal recesiv, ceea ce înseamnă că cel puțin
unul dintre părinți trebuie să aibă cel puțin o alelă mutantă a genei PAH. Gena care determină sinteza
enzimei fenilalanin-hidroxilază este localizată pe cromozomul uman 12 (12q22-q24.1), are o lungime de 90kb și conține 13 exoni separați de introni. Au fost identificate peste 500 de alele mutante ale acestei gene,
determinând astfel forme mai mult sau mai puțin grave ale bolii.
Când activitatea PAH este redusă, fenilalanina se acumulează și este convertită în acidul fenilpiruvic,
acidul fenillactic și feniletilamină (fenilcetone), ce pot fi detectate în urină (test diagnostic). Phe este
metabolizată pe calea minoră, a transaminazelor până la fenilcetone.
PKU - Genetică

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 5/15
PKU – Tablou Clinic
Nivel crescut de fenilalanină: Phe este prezentă în concentrații crescute în țesuturi,
plasmă: 15-63 μg/100ml (N:1-10 μg/100ml) și urină: 300-1000 μg/100ml (N: 30 μg/100ml).
Fenil-lactatul, fenil-acetatul și fenil-piruvatul, care în mod normal nu sunt produși în
cantități semnificative dacă Phe-hidroxilaza este prezenta și funcțională, sunt de asemenea
crescuți în PKU. Acești metaboliți conferă urinei un miros caracteristic “stătut” (“de șoarece”). [Notă: Denumirea bolii s-a datorat descoperirii unei fenilcetone (care a fost
identificată ca fenilpiruvat) în urină.];
Simptome SNC: retardul mintal, incapacitatea de a merge sau de a vorbi, convulsiile,
hiperactivitatea, tremorul, microcefalia și hipodezvoltarea staturo-ponderală sunt
manifestări clinice caracteristice pentru PKU. Pacientul cu PKU netratată manifestă în mod
tipic simptome de retard mintal înainte de împlinirea vârstei de un an, iar IQ-ul depășește rareori valoarea 50. [Notă: La ora actuală, ca urmare a programelor de screening neonatal,
aceste manifestări clinice sunt tot mai rar întâlnite.];
Hipopigmentare: Pacienții cu fenilcetonurie prezintă adesea un deficit de pigmentare (păr
depigmentat, tegumente de culoare deschisă și ochi albaștri). Hidroxilarea tirozinei de către
tirozinază, care este primul pas în formarea pigmentului melanină, este inhibată competitiv
de nivelurile crescute de Phe prezente la pacienții cu PKU.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 6/15
PKU – concentrația crescută de Phe (scăzută de Tyr) afecteazădezvoltarea SNC. Mecanism
Enzima fenilalanin-hidroxilaza transformă Phe în Tyr. Dacă reacția nu are loc, Phe se
acumulează și Tyr devine deficitară (=>conc. scăzută de Dopa => conc. scăzută de
dopamină, catecolamine, melanină)
Manifestarea clinică majoră o reprezintă retardul mental provocat de acumularea dePhe, care devine donorul major de grupări amino în activitatea aminotransferazei
ducând la depleția țesutului nervos de α-cetoglutarat. Absența α-cetoglutaratului la
nivel cerebral blochează ciclul acizilor tricarboxilici (ciclul Krebs) și deci producția
aerobă de energie, care este esențială pentru dezvoltarea cerebrală normală. Astfel,
are loc o subdezvoltare neuronală, dezvoltarea prelungirilor și sinteza de mielină fiind
afectate, având ca rezultat perturbarea transmiterii sinaptice.
În cazul pacienților cu concentrația de Phe crescută ca rezultat al deficienței de BH4
(2% din cazurile de PKU), se înregistrează un nivel foarte scăzut de dopamină cu
afectarea transmiterii sinaptice.
Alt mecanism explică competiția pentru transportor în vederea traversării barierei
hematoencefalice. Fenilalanina (aa. neutru) în exces în sânge, va satura transportorul
(LNAAT=large neutral aa. transporter), împiedicând astfel trecerea altor aa. necesari
pentru sinteza proteică și cea a neurotransmițătorilor.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 7/15
Tablou Clinic Pigmentație cutanată redusă, blonzi cu ochii alba
pigment responsabil pentru culoarea pielii și
deficitară)
Abilități sociale deficitare
Mișcări necoordonate ale membrelor
Brahicefalie și microcefalie
Convulsii, tremurături
Hiperactivitate și deficit de atenție

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 8/15
PKU - Diagnostic
Screening-ul neonatal si diagnosticul de PKU: Depistarea precoce a fenilcetonuriei este important
deoarece boala este tratabilă prin instituirea unor restricții alimentare. Deoarece la nou-născ
simptomele sunt absente, pentru a depista boala sunt obligatorii testele de laborator care
detecteze nivelurile serice crescute de Phe. Totuși, la naștere, copilul cu PKU are niveluri sanguinormale de Phe, deoarece organismul matern filtrează prin placentă nivelurile crescute de Phe d
sângele fătului. Nivelurile normale de Phe pot persista până în momentul în care nou-născut
primește timp de 24-48 de ore o alimentație pe bază de proteine. De aceea, se recoman
efectuarea testelor screening după acest interval pentru a evita rezultatele fals-negative. Pent
nou-născuții cu rezultat pozitiv la testul screening, diagnosticul este confirmat prin determinare
cantitativă a nivelurilor plasmatice de Phe. Testul utilizat se numește testul Guthrie și constă recoltarea de sânge din călcâi și utilizarea acestuia ca mediu de cultură pentru Bacillus subtilis.
mod normal, nu apar colonii de B. subtilis în prezența inhibitorului ß2-tienialanină, iar în caz de PK
datorită prezenței Phe în exces în sângele recoltat, coloniile cresc.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 9/15
Testul Guthrie
7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 10/15
PKU - Diagnostic
Diagnosticul antenatal al PKU: PKU clasică se referă la o clasă de afecțiuni
cauzate de una din cele peste 400 de mutații diferite ale genei care codifică Phe-
hidroxilaza (PAH). Frecvența de apariție a oricărei mutații variază în rândul
populației, iar boala este adesea dublu heterozigotă, adica gena PAH prezintă o
mutație diferită a fiecăreia din cele două alele ale aceluiași locus. În pofida
acestei complexități, diagnosticul prenatal indirect este posibil prin RFLP
(polimorfismul lungimii fragmentelor de restricție). Pentru a stabili un protocol de
diagnostic al acestei afecțiuni genetice, trebuie efectuată analiza ADN la membrii
familiei persoanei afectate. Cheia constă în idenetificarea markerilor (RFLP) caresunt în strânsă asociere cu boala. După identificarea acestor markeri, metoda
RFLP poate fi folosită pentru a stabili diagnosticul prenatal.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 11/15
PKU - Tratament
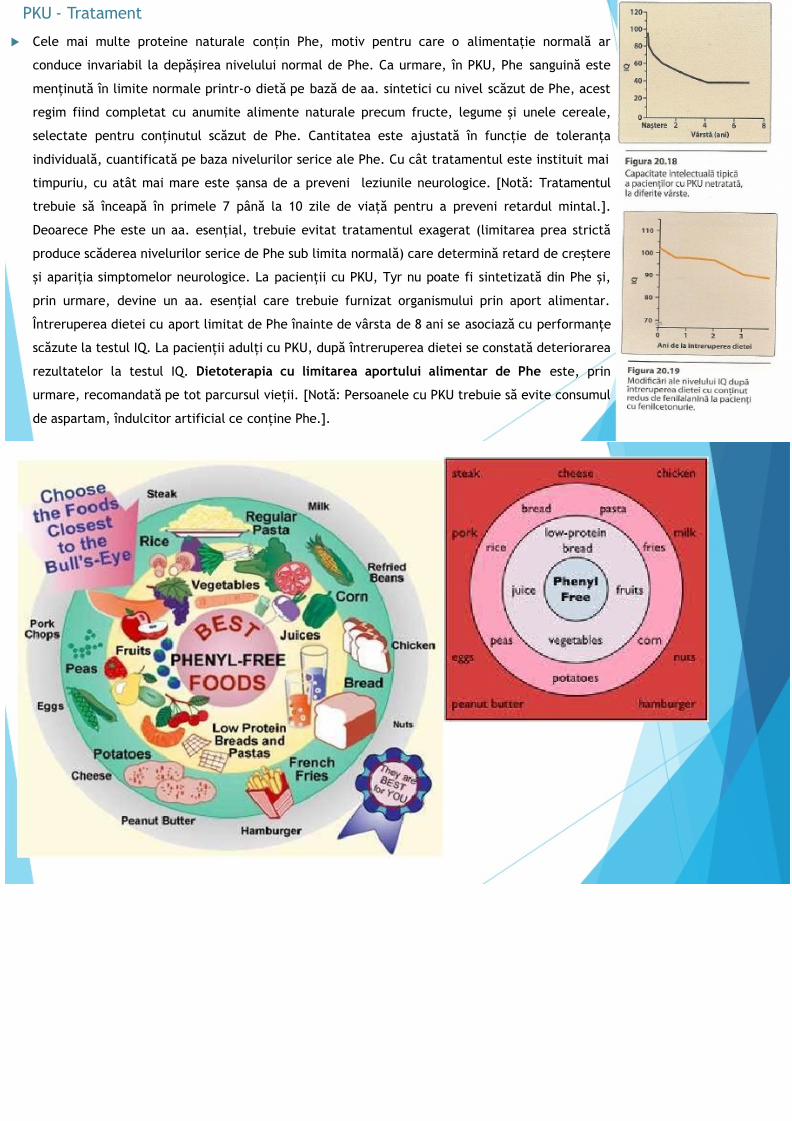
Cele mai multe proteine naturale conțin Phe, motiv pentru care o alimentație normală ar
conduce invariabil la depășirea nivelului normal de Phe. Ca urmare, în PKU, Phe sanguină este
menținută în limite normale printr-o dietă pe bază de aa. sintetici cu nivel scăzut de Phe, acest
regim fiind completat cu anumite alimente naturale precum fructe, legume și unele cereale,
selectate pentru conținutul scăzut de Phe. Cantitatea este ajustată în funcție de toleranța individuală, cuantificată pe baza nivelurilor serice ale Phe. Cu cât tratamentul este instituit mai
timpuriu, cu atât mai mare este șansa de a preveni leziunile neurologice. [Notă: Tratamentul
trebuie să înceapă în primele 7 până la 10 zile de viață pentru a preveni retardul mintal.].
Deoarece Phe este un aa. esențial, trebuie evitat tratamentul exagerat (limitarea prea strictă
produce scăderea nivelurilor serice de Phe sub limita normală) care determină retard de creștere
și apariția simptomelor neurologice. La pacienții cu PKU, Tyr nu poate fi sintetizată din Phe și, prin urmare, devine un aa. esențial care trebuie furnizat organismului prin aport alimentar.
Întreruperea dietei cu aport limitat de Phe înainte de vârsta de 8 ani se asociază cu performanțe
scăzute la testul IQ. La pacienții adulți cu PKU, după întreruperea dietei se constată deteriorarea
rezultatelor la testul IQ. Dietoterapia cu limitarea aportului alimentar de Phe este, prin
urmare, recomandată pe tot parcursul vieții. [Notă: Persoanele cu PKU trebuie să evite consumul
de aspartam, îndulcitor artificial ce conține Phe.].

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 12/15

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 13/15
PKU - Tratament
Lofenalac este un produs făcut special pentru copiii cu PKU. Poate fi folosit toată viața ca su
de proteine fiind sărac în fenilalanină și echilibrat în ceea ce privește restul aminoacizi
esențiali.
Suplimente precum uleiul de pește (pentru că într-o alimentație fără Phe trebuie un ap
crescut de acizi grași cu lanț lung, căci ei lipsesc) vor ajuta la îmbunătățirea dezvoltă
neurologice, inclusiv coordonarea motorie fină. Alte suplimente specifice, care pot fi necesa
sunt fierul sau carnitina.
Alte modalități de tratament sunt în studiu: încărcarea cu aminoacizi neutrii care ar permi
cel puțin teoretic, diminuarea trecerii fenilmalaninei în sistemul nervos central sau creșterdegradării fenilalaninei prin administrarea unei enzime, fenilalaninamoniuligază. Terapia gen
este o altă cale importantă, testele la modele animale fiind promițătoare.

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 14/15
PKU maternă
În cazul femeilor însărcinate care au fenilcetonurie dar nu respectă o dietă cu conținut scă
fătul va prezenta “sindrom PKU matern”. Nivelurile crescute ale Phe din sângele matern dete
microcefalie, retard mintal și malformații cardiace congenitale. Unele dintre aceste
dezvoltare se produc în primele luni de sarcină. Prin urmare, controlul prin dietă al nivelulu
Phe trebuie să înceapă înainte de concepție și de asemenea trebuie continuat pe tot parcursul
Atunci când concentrația fenilalaninei este menținută scăzută pe timpul sarcinii (2-6 μg/10
nou-născutului de a avea anomalii congenitale nu este mai crescut decât cel al unui nou-
mamă fără fenilcetonurie. Nou-născuții cu fenilcetonurie pot fi alăptați la sân. Unele cercet
că alimentația nou-născuților cu PKU exclusiv la sân este posibilă doar dacă în timpul p
alăptare mama respectă un regim strict pentru a conserva o concentrație scăzută a fenilalanin

7/22/2019 Fenilcetonuria Final
http://slidepdf.com/reader/full/fenilcetonuria-final 15/15
Bibliografie
Pamela C. Champe, Richard A. Harvey, Denise R. Ferrier, Lippincott BiochimieIlustrată, Ediția a 4-a, pag. 207-272, 477;
T.W. Sadler, Langman’s Embriologie Medicală, Ediția a 10-a, pag. 117;
http://ghr.nlm.nih.gov/condition/phenylketonuria
http://www.ygyh.org/pku/
http://en.wikipedia.org/wiki/Phenylketonuria
![AMINOACIDOPATÍAS CONGÉNITAS: FENILCETONURIA · Aminoacidopatías congénitas: fenilcetonuria Corrsal Riocerezo, Lucía 7 de metabolitos tóxicos [4]. La mayoría de estas enfermedades](https://img.pdfslide.tips/doc/110x75/5e1c6a40747b1d3bc45339be/aminoacidopatas-congnitas-fenilcetonuria-aminoacidopatas-congnitas-fenilcetonuria.jpg)




























