Embed Size (px)
Citation preview

EL PERFIL EL PERFIL FARMACOCINÉTICO FARMACOCINÉTICO DE FESOTERODINA DE FESOTERODINA
8MG CON 8MG CON ADMINISTRACION DE ADMINISTRACION DE
DÍA Y DE NOCHEDÍA Y DE NOCHEMaría Fernanda Castro
María Elena Cuellar Maria Camila Rodríguez
Rocío E. Villamil
OBJETIVOS
•Evaluar propiedades farmacocinéticas de la fesoterodina (8mg)
Diferenciar la farmacocinética en su administración diurna y nocturna.
Comparar parámetros farmacocinéticos con otros fármacos con actividad terapéutica similar.

•Urgencia urinaria con o sin incontinencia
•Condición crónica que ocurre en hombres y mujeres
•Aumenta su prevalencia con la edad.
VEJIGA HIPERACTIVA

FESOTERODINA
Antimuscarínico para el tratamiento de la vejiga hiperactiva (OAB).
Receptores muscarínicos expresados en la vejiga.
Los efectos adversos son debidos al antagonismo de receptores fuera de la vejiga
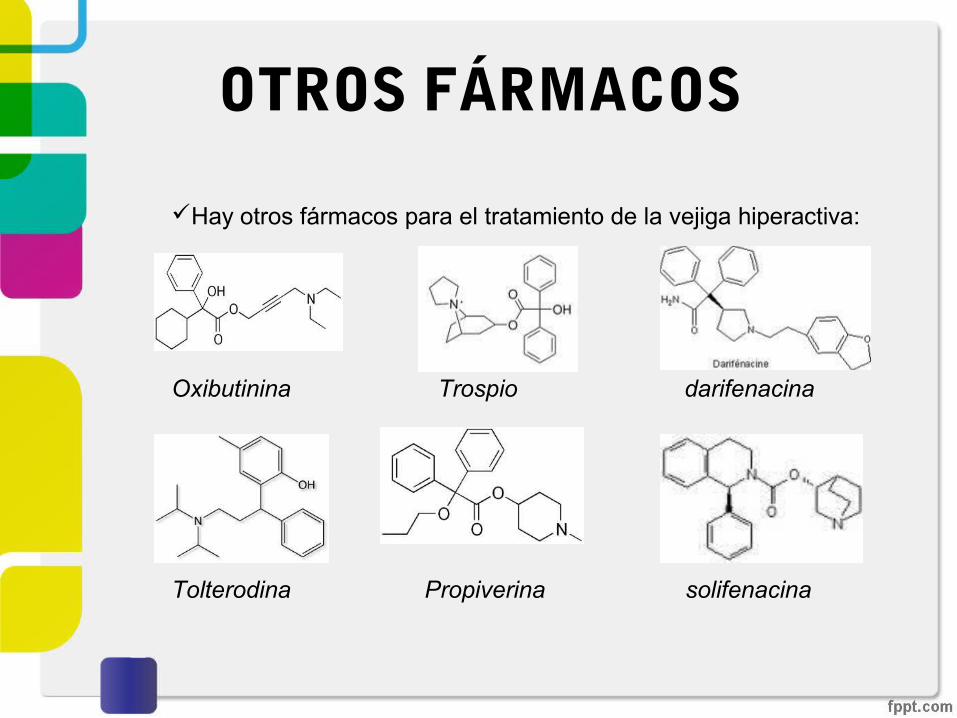
OTROS FÁRMACOS
Hay otros fármacos para el tratamiento de la vejiga hiperactiva:
Oxibutinina Trospio darifenacina
Tolterodina Propiverina solifenacina

FARMACO CARACTERISTICAS
Tolterodina • Liberación prolongada eficaz para el tratamiento de AOB (nicturia)
Oxibutinina• Liberación inmediata (IR) absorción más rápida en la mañana.• Liberación prolongada
Propiverina• IR absorción más lenta en la noche• Liberación prolongada (no comparada)
Trospio• Liberación prolongada • Menor eficacia y tolerabilidad en la administración nocturna.
Cloruro de trospio
• Liberación prolongada • Menor eficacia y tolerabilidad en la administración nocturna
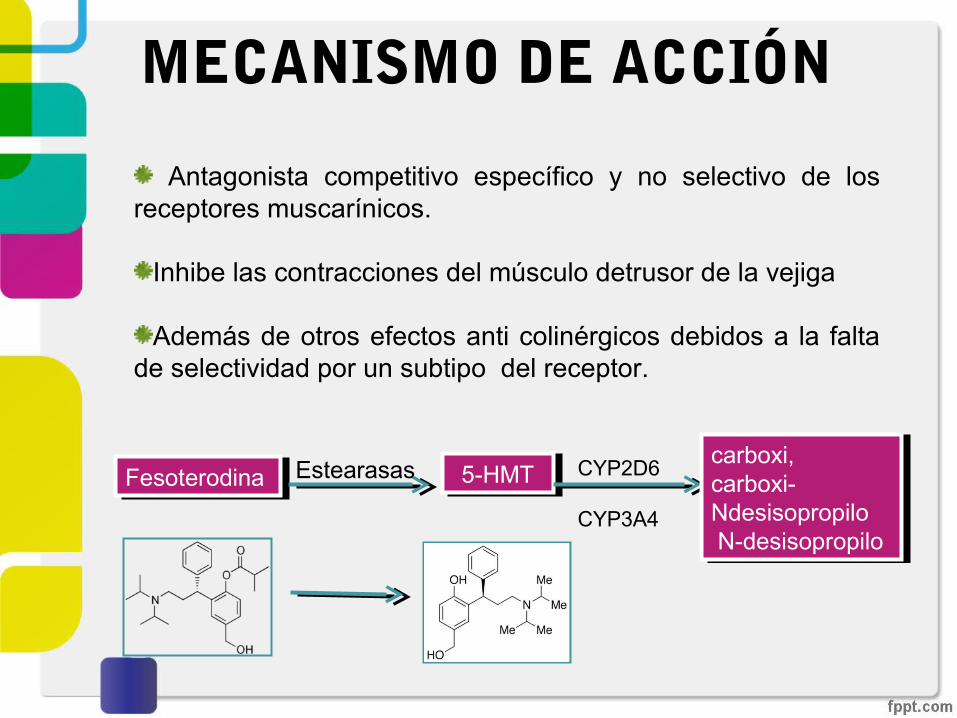
Antagonista competitivo específico y no selectivo de los receptores muscarínicos.
Inhibe las contracciones del músculo detrusor de la vejiga
Además de otros efectos anti colinérgicos debidos a la falta de selectividad por un subtipo del receptor.
MECANISMO DE ACCIÓN
FesoterodinaFesoterodina Estearasas 5-HMT5-HMT CYP2D6 CYP3A4
carboxi, carboxi-Ndesisopropilo N-desisopropilo
carboxi, carboxi-Ndesisopropilo N-desisopropilo

METODOLOGíA
Género Edad IMC Peso corporal
Hombres y mujeres Entre 18 y 55 18.30Kg/m2 >50Kg
*Criterios de exclusión
Estudio aleatorizado, de etiqueta abierta, de dos periodos, con dos tratamientos de cruce longitudinal de control experimental.
•Periodo 1 : 28 días Periodo de lavado: ≥ 60 h•Periodo 2: 28 días
Tamaño de muestra 12 Sujetos
IC (intervalo de confianza) 90%
Diferencias entre tratamientos
AUC ±0,138*
Cmax ±0,172*
Catorce sujetos fueron reclutados para permitir los posibles retiros prematuros de los estudios.
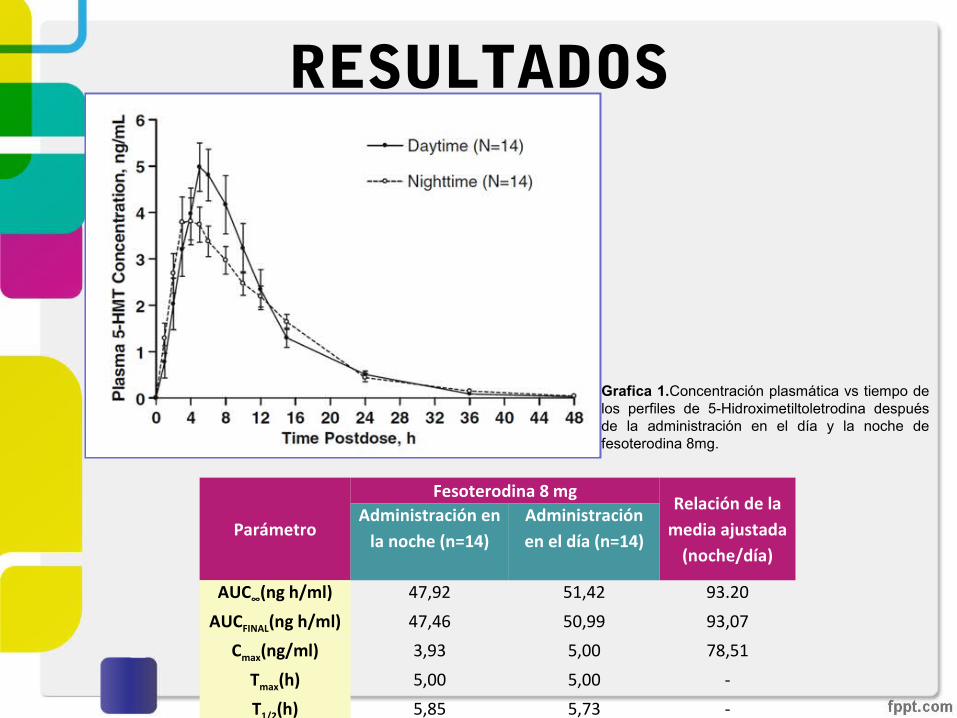
RESULTADOS
Parámetro
Fesoterodina 8 mgRelación de la
media ajustada (noche/día)
Administración en la noche (n=14)
Administración en el día (n=14)
AUC∞(ng h/ml) 47,92 51,42 93.20
AUCFINAL(ng h/ml) 47,46 50,99 93,07
Cmax(ng/ml) 3,93 5,00 78,51
Tmax(h) 5,00 5,00 -
T1/2(h) 5,85 5,73 -
Grafica 1.Concentración plasmática vs tiempo de los perfiles de 5-Hidroximetiltoletrodina después de la administración en el día y la noche de fesoterodina 8mg.
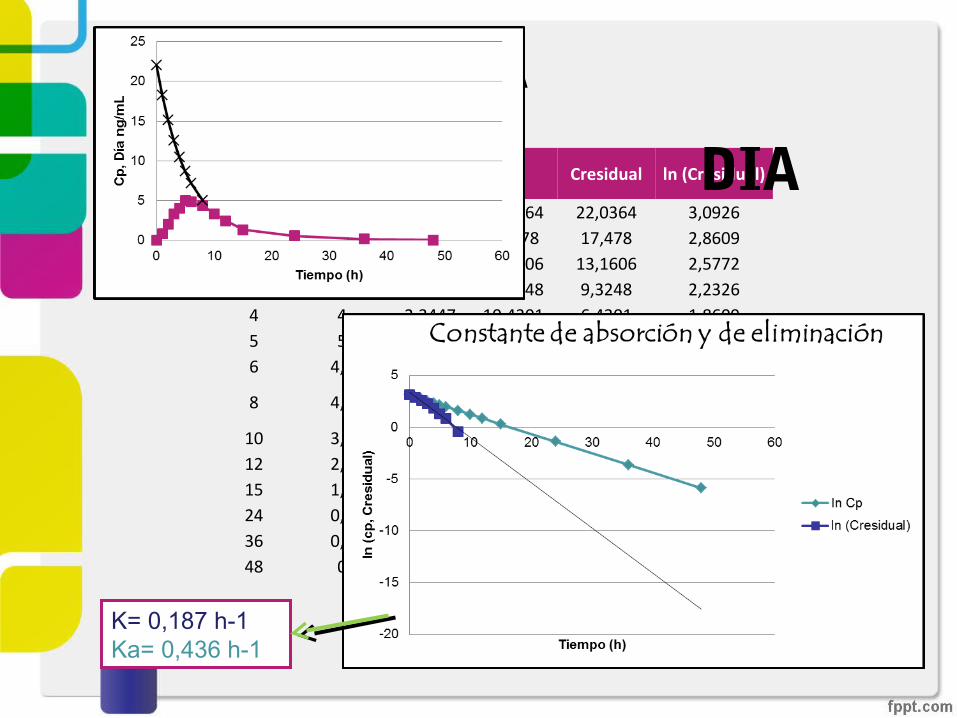
DIATiempo
(h)Diang/mL lnCp ĉp Cresidual ln (Cresidual)
0 0 3,0927 22,0364 22,0364 3,09261 0,8 2,9057 18,278 17,478 2,86092 2 2,7187 15,1606 13,1606 2,57723 3,25 2,5317 12,5748 9,3248 2,23264 4 2,3447 10,4301 6,4301 1,86095 5 2,1577 8,6512 3,6512 1,29506 4,8 1,9707 7,1756 2,3756 0,8652
8 4,3 1,5967 4,9367 0,6367 -0,4514
10 3,3 1,222712 2,4 0,848715 1,3 0,287724 0,5 -1,395336 0,1 -3,639348 0 -5,8833
K= 0,187 h-1Ka= 0,436 h-1
DIA
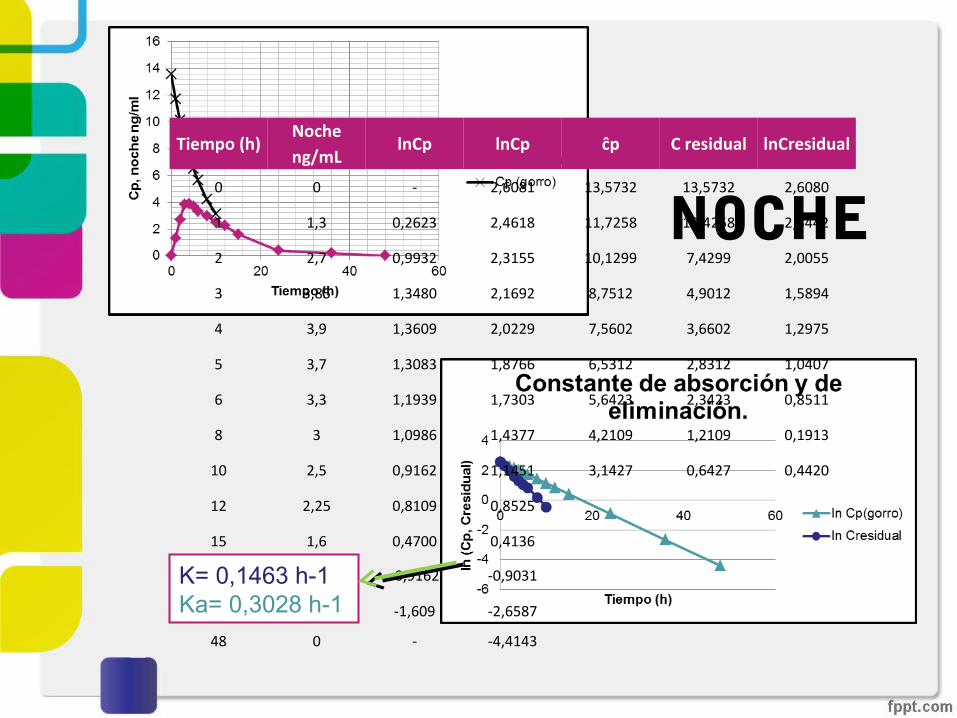
NOCHETiempo (h)
Noche ng/mL
lnCp lnCp ĉp C residual lnCresidual
0 0 - 2,6081 13,5732 13,5732 2,6080
1 1,3 0,2623 2,4618 11,7258 10,4258 2,3442
2 2,7 0,9932 2,3155 10,1299 7,4299 2,0055
3 3,85 1,3480 2,1692 8,7512 4,9012 1,5894
4 3,9 1,3609 2,0229 7,5602 3,6602 1,2975
5 3,7 1,3083 1,8766 6,5312 2,8312 1,0407
6 3,3 1,1939 1,7303 5,6423 2,3423 0,8511
8 3 1,0986 1,4377 4,2109 1,2109 0,1913
10 2,5 0,9162 1,1451 3,1427 0,6427 0,4420
12 2,25 0,8109 0,8525
15 1,6 0,4700 0,4136
24 0,4 -0,9162 -0,9031
36 0,2 -1,609 -2,6587
48 0 - -4,4143
K= 0,1463 h-1Ka= 0,3028 h-1
NOCHE

PARÁMETROS FARMACOCINÉTICOS
PARAMETROS FARMACOCINETICOS
Vd (L) K (h-1) Ka (h-1) t1/2 (h) Tmax (h)Cmax
(ng/mL)AUC
(ng.h/mL)
Día 169 0,1870 0,4360 3,7067 3,3997 5 57,10
Noche 169 0,1463 0,3028 4,7378 4,6480 3,8 52,57
La comprobación de los parámetros farmacocinéticos hallados para administración noche y día son:

ANÁLISIS
El volumen de distribución reportado por algunos autores fue de 169 L.
Se determinaron las constantes de eliminación y absorción tanto para día y noche, a pesar que estas no se reportaban por los autores; de esta manera se puede comparar el comportamiento del fármaco en el día y en la noche
Es vital determinar los demás factores farmacocinéticos para poder sacar conclusiones concisas.
Ka
Día 0.4360 h-1durante el día el fármaco ejerce su
acción mas rápidamente puesto que se absorbe con mayor
velocidadNoche 0.3028 h-1
K
Día 0,1870 h-1Fármaco se elimina un poco
mas lento en la nocheIntoxicación en caso de tener una constante de eliminación muy baja.Noche 0,1463 h-1
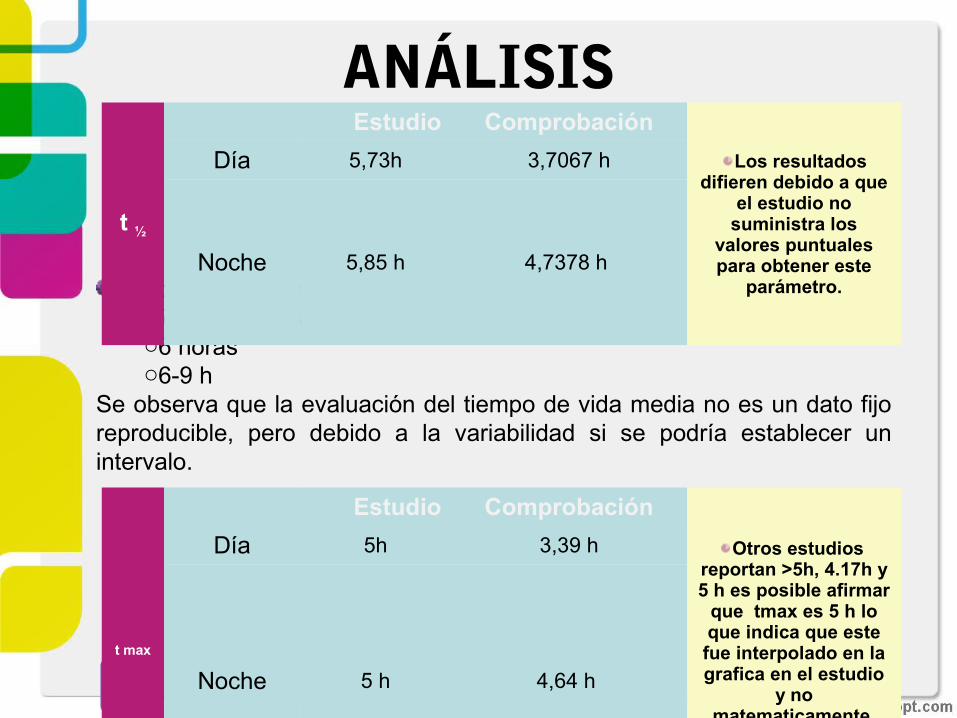
ANÁLISIS
En bibliografía se reporta gran variedado7 horas a dosis única, o6 horas o6-9 h
Se observa que la evaluación del tiempo de vida media no es un dato fijo reproducible, pero debido a la variabilidad si se podría establecer un intervalo.
t ½
Estudio Comprobación
Los resultados difieren debido a que
el estudio no suministra los
valores puntuales para obtener este
parámetro.
Día 5,73h 3,7067 h
Noche 5,85 h 4,7378 h
t max
Estudio Comprobación
Otros estudios reportan >5h, 4.17h y 5 h es posible afirmar
que tmax es 5 h lo que indica que este fue interpolado en la grafica en el estudio
y no matematicamente.
Día 5h 3,39 h
Noche 5 h 4,64 h
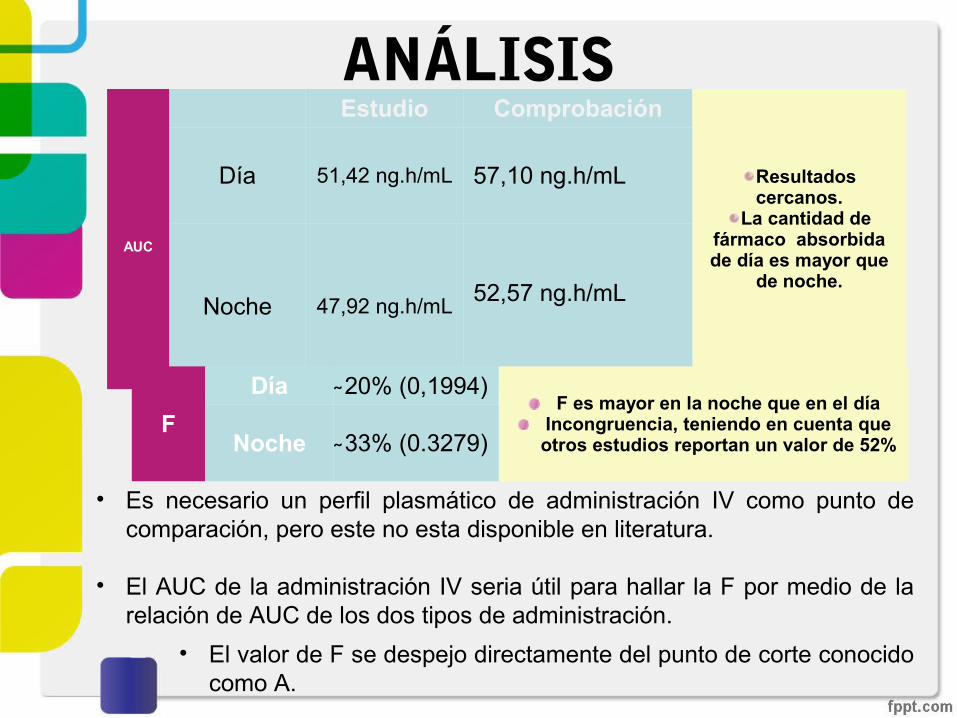
ANÁLISIS
• Es necesario un perfil plasmático de administración IV como punto de comparación, pero este no esta disponible en literatura.
• El AUC de la administración IV seria útil para hallar la F por medio de la relación de AUC de los dos tipos de administración.
AUC
Estudio Comprobación
Resultados cercanos.
La cantidad de fármaco absorbida de día es mayor que
de noche.
Día 51,42 ng.h/mL 57,10 ng.h/mL
Noche 47,92 ng.h/mL 52,57 ng.h/mL
F
Día ̴20% (0,1994)F es mayor en la noche que en el día
Incongruencia, teniendo en cuenta que otros estudios reportan un valor de 52% Noche ̴33% (0.3279)
• El valor de F se despejo directamente del punto de corte conocido como A.

La fesoterodina, oxibutinina, propiverina y cloruro de trospio, tienen disminución de la absorción al administrarse en la noche.
• La efectividad en fesoterodina no se altera a pesar de que la Cmax se disminuye en un 21%.
• Se mejora la tolerabilidad del fármaco al igual que la tolerodina.
• Se evitan acumulaciones y demás problemas adversos.
Efectos adversos la fesoterodina Efectos adversos la fesoterodina • Menor porcentaje de sequedad en la boca y constipación.
• Son efectos dosisdependientes.
• El efecto adverso mas significativo es la cefalea, sin embargo en bibliografía la sequedad en la boca es el EA más común.

CONCLUSIONES
Fesoterodina presenta menor incidencia de efectos adversos en comparación con los otros fármacos usados para el tratamiento de AOB.
La administración nocturna presenta una disminución de la Cmax del 21%,
sin embargo no implica una disminución en su efectividad, pues la cantidad de fármaco absorbida no varía notoriamente.
La biodisponibilidad del fármaco es mayor en la noche que en el día. Es necesaria la realización y revisión de diferentes tipos de estudios con
tamaños de muestra mayores, con mayor variabilidad interindividual (edad, sexo, raza), realizados en múltiples centros pues de esta manera los resultados son más confiables, significativos y reproducibles.

BIBLIOGRAFÍA1. Chapple C, Van Kerrebroeck P, Tubaro A, Haag-Molkenteller C, Hans,
Forst H-T, Massow U, Wange J, Brodsky M, “Clinical Efficacy, Safety, and Tolerability of Once-Daily Fesoterodine in Subjects with Overactive Bladder”, European urology 52 (2007) 1204–1212
2. Simona H.U, Malhotrab B, “The pharmacokinetic profile of fesoterodine: Similarities and differences to tolterodine”, Swiss Med Wkly 2009; 139 (9–10): 146–151
3. Fesoterodina; Ficha de seguridad Pfizer Toviaz (marca comercial),Evaluacion de Novedades Terapeuticas de la Comunitat Valenciana, Junio 2009
4. Gupta K, Kaur K, Aulakh B.S, Kaushal S, “Fesoterodine for Overactive Bladder: A Review of the Literature”, Curr Ther Res Clin Exp. 2010;71:273-288
5. Pfizer laboratories “TOVIAZ®(fesoterodine fumarate) safely and effectively For oral administration” Initial U.S. Approval: 2008 (FDA) Lab-0382-9.0 Revised April 2012

6. Pfizer manufacturing deutschland gmbh. Anexo I: ficha técnica o resumen de las características del producto Fesoterodina liberación modificada 4mg y 8mg. Heinrich Mack Str. 35. Illertissen Alemania.
7. Malhotra B.K, Crownover P.H, LaBadie R, Glue P, MacDiarmid S.A, “The pharmacokinetic profile of fesoterodine 8 mg with daytime or nighttime dosing”, Eur J Clin Pharmacol (2010) 66:171–17
8. F. Morales; L. estañ. Farmacologia de la solifenacina. Arch. Esp. Urol. 2010; 63 (1): 43-52
9. Fesoterodina (DCI), Ficha de Novedad Terapeútica, TOVIAZ®, Numero 1 del año 2009
10.Wyndaele J.J, “Fesoterodine: Individualised Treatment of Urgency Urinary Incontinence Across Patient Groups”, Eurology Supplements 10 (2011) 14–22
11.Malhotra B, El-Tahtawy A, Wang E.Q, Darekar A, Cossons N, Crook T.J, Scholfield D, Reddy P,“Dose-escalating study of the pharmacokinetics and tolerability of fesoterodine in children with overactive bladder” Journal of Pediatric Urology (2012), 1e7





























