Embed Size (px)
Citation preview

Guía de farmacovigilancia para establecimientos farmacéuticos
distribuidores mayoristas
SECRETARÍA DE SALUD


Alcaldía Mayor de BogotáSecretaría Distrital de SaludHospital del Sur ESE
Alcalde Mayor de BogotáGustavo Francisco Petro Urrego
Secretario Distrital de SaludAldo Enrique Cadena Rojas
Subsecretario Distrital de SaludHelver Giovanni Rubiano García
Director de Salud PúblicaJaime Hernán Urrego
Profesional especializada del Área de Vigilancia en Salud PúblicaPatricia Arce Guzmán
AutoresLínea de Acción Medicamentos Seguros
Gerente Hospital del Sur ESE Ricardo Beira Silva
Convenio Interadministrativo N° 1035 de 2012Proyecto de Vigilancia Intensificada del Programa DistritalFarmacovigilancia
© Secretaría Distrital de Salud de Bogotá, D. C.Dirección de Salud PúblicaÁrea de Vigilancia en Salud PúblicaCarrera 32 # 12-81, cuarto pisoBogotá-Colombiawww.saludcapital.gov.co
Coordinación editorialOriana Obagi OrozcoJefe de la Oficina Asesora de Comunicaciones
Corrección de estilo, diseño y diagramaciónGustavo Patiño DíazJimmy RosasCamilo Patiño
Impresión
Primera edición
Bogotá, agosto de 2013

5 Presentación5 Objetivo5 Introducción6 Farmacovigilancia6 Objetivos de la farmacovigilancia7 Métodos utilizados en farmacovigilancia7 Marco legal de la farmacovigilancia en Colombia8 Programa Distrital de Farmacovigilancia (PDFV)8 Red Distrital de Farmacovigilancia9 Nodos de la red nacional de farmacovigilancia10 Diagrama 1. Flujo de notificación de sospechas de reac-
ciones adversas a medicamentos y problemas relacio-nados con el uso de medicamentos
11 Diagrama 2. Análisis de las notificaciones14 Prohibiciones para los dependientes y los propietarios
de los depósitos de drogas15 Glosario 18 Referencias19 Anexo 1. Formato de reporte20 Instructivo de diligenciamiento de Formato de reporte
de sospecha de reacción adversa a medicamento (Foram)
Contenido

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
5
Presentación
El Programa Distrital de Farmacovigilancia de Bogotá, D. C. (PDFV), enmarcado den-tro de la Línea de Medicamentos Seguros de Salud Pública de la Secretaría Distrital de Salud (SDS), se ha propuesto integrar a los prestadores de servicios de salud, los
establecimientos farmacéuticos y la comunidad en general en una Red Distrital de Farma-covigilancia.
En aras de este propósito, se presenta esta cartilla con información básica en farmaco-vigilancia, para proporcionar un elemento de consulta a los dependientes y los propietarios de establecimientos farmacéuticos mayoristas, e invitarlos a convertirse en nodos locales del PDFV.
ObjetivoDar a conocer lineamientos básicos en farmacovigilancia, que permitan articular a los
establecimientos farmacéuticos mayoristas como reportantes activos al PDFV.
IntroducciónLos medicamentos están diseñados, fundamentalmente, para tratar las enfermedades o
las diversas alteraciones del estado de salud; sin embargo, pese a las ventajas que ofrecen, hay evidencia de que los efectos adversos a los medicamentos pueden causar enfermedad, discapacidad, o, incluso, la muerte.
En Colombia la mayoría de los pacientes y los distribuidores de drogas, y hasta los pro-pios profesionales de la salud, desconocen que cuando un medicamento produce eventos adversos, aun los mencionados en el prospecto, deben ser notificados al PDFV. Así mismo, cuando el medicamento no produce efecto terapéutico deseado debe reportarse.
Frente a lo anterior, la administración distrital se ha propuesto como meta del plan te-rritorial “Garantizar el funcionamiento de la Red Distrital de Farmacovigilancia integrada por 100% de los prestadores de servicios de salud, establecimientos farmacéuticos y comu-nidad en general a 2016”, y así allanar el camino para que los ciudadanos se informen sobre la posibilidad de reportar estos eventos adversos y problemas relacionados con el uso de los medicamentos y sean parte activa en el seguimiento del uso de los fármacos.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
6
Es el estudio del uso y los efectos de los medicamentos en grandes poblaciones. Incluye estudios de utilización de medicamentos, ensayos clínicos y farmacovigilancia.(1)
FarmacovigilanciaLa Organización Mundial de la Salud (OMS) la define como la ciencia y las actividades
relativas a la detección, la evaluación, la comprensión y la prevención de los efectos adversos de los medicamentos (EAM) o cualquier otro problema de salud relacionado con ellos (1).
Objetivos de la farmacovigilanciaLos objetivos finales de la farmacovigilancia son: • El uso racional y seguro de los medicamentos.• La evaluación y la comunicación de los riesgos y los beneficios de los medicamentos
comercializados.• La educación y la información sobre medicamentos a los profesionales de la salud y
a la comunidad en general.
¿Por qué la farmacovigilancia?La información sobre un fármaco reunida durante la fase de precomercialización es
inevitablemente incompleta respecto a las posibles reacciones adversas. Las pruebas de fármacos en animales son insuficientes para predecir la seguridad de su
uso en seres humanos.• En los ensayos clínicos los pacientes son seleccionados y se limita su número; ade-
más, las condiciones de uso difieren de las de la práctica médica habitual y la dura-ción de los ensayos es limitada.
• La información a menudo es incompleta, o no está disponible sobre reacciones adversas graves y poco comunes, toxicidad crónica, uso en grupos especiales (niños, ancianos o mujeres embarazadas), o respecto a interacciones farmaco-lógicas.
La farmacovigilancia es necesaria en cada país, ya que hay diferencias entre países (y aun entre regiones de algunos países) en cuanto a la manifestación de reacciones adversas a medicamentos (RAM) y otros problemas relacionados con los medicamentos (PRM). Todo esto puede deberse a diferencias en cuanto a:
• La producción de medicamentos.• La distribución y el uso de los medicamentos (por ejemplo: indicaciones, dosis, dis-
ponibilidad).• La genética, la dieta, las tradiciones de la población.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
7
• La calidad y la composición (excipientes) de los productos farmacéuticos fabricados localmente.
• El uso de medicamentos no ortodoxos (por ejemplo, plantas medicinales), que pue-den presentar problemas toxicológicos cuando se los usa bien sea solos o en combi-nación con otros medicamentos.
Por otra parte, normatividades propias de la vigilancia internacional, como el Progra-ma Internacional de Farmacovigilancia de la OMS, pueden proporcionar información sobre aspectos de seguridad de medicamentos que aún no se hayan detectado en el país.
Métodos utilizados en farmacovigilanciaPara desarrollar actividades de farmacovigilancia se emplean diversos métodos:• Un sistema de notificaciones espontáneas basado en la identificación y la detección
de las reacciones adversas sospechosas, por parte de los profesionales de la salud en su práctica diaria, y el envío de esta información a un organismo que la centraliza. Es la metodología utilizada por los centros participantes del Programa Internacional de Farmacovigilancia de la OMS.
• Procedimientos de farmacovigilancia intensiva, basados en la recolección sistemáti-ca y detallada de datos sobre todos los efectos perjudiciales que pueden suponerse inducidos por medicamentos en determinados grupos de población. Estos métodos se dividen en dos grandes grupos:
- Sistemas centrados en el medicamento. - Sistemas centrados en el paciente.
Estudios epidemiológicos, cuya finalidad es comprobar una hipótesis; es decir, estable-cer una causalidad entre la presencia de RAM y su empleo. Pueden ser:
• Estudios de cohorte.• Estudios de casos y control.
La notificación sistemática de reacciones adversas y su análisis estadístico permanente permiten generar señales sobre el comportamiento de los medicamentos en la población colombiana. El éxito o fracaso de cualquier actividad de farmacovigilancia dependen de la notificación de sospechas de reacciones adversas. (2)
Marco legal de la farmacovigilancia en ColombiaLa Resolución 1403 de 2007 establece los lineamientos básicos para la farmacovigilancia
y establece sus correspondientes responsabilidades al fabricante, a los integrantes del Sistema

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
8
General de Seguridad Social en Salud (SGSSS), a los establecimientos farmacéuticos, a los profesionales de la salud, al personal técnico que maneje medicamentos, a los pacientes, a las autoridades de control y del sector y a la comunidad en general.
La misma resolución asigna competencia a la entidad territorial en el tema de farma-covigilancia para recibir los reportes de sospechas de eventos adversos a medicamentos (EAM) dentro de las 72 horas siguientes a la ocurrencia del evento adverso serio y durante los primeros 5 días hábiles del mes siguiente a la ocurrencia del evento no serio (3).
Programa Distrital de Farmacovigilancia (PDFV)En Bogotá, el Programa Distrital de Farmacovigilancia (PDFV) trabaja en pro del uso
seguro de los medicamentos, mancomunadamente con los programas institucionales. El PDFV tiene como objetivos principales recibir, analizar, retroalimentar a los reportantes, consolidar y transmitir la información relacionada con los EAM y los problemas relaciona-dos con el uso de medicamentos (PRM) al Invima, a través del PNFV, y este, a su vez, tras-mite la información al Centro Internacional de Monitoreo de Uppsala de la OMS; también, socializar información relevante para el uso adecuado de los medicamentos en Colombia.
El PNFV constituye el nodo central de la Red Nacional de Farmacovigilancia, la cual cuenta con dos grupos de actores:
• Personas (pacientes, cuidadores y profesionales de la salud [médicos tratantes, odon-tólogos, enfermeros, químicos farmacéuticos, expendedores de drogas, etc.]).
• Instituciones (hospitales, clínicas, autoridades seccionales de salud, laboratorios farma-céuticos, importadores y comercializadores de medicamentos, así como el Invima) (4).
El PDFV permite conocer los eventos adversos o PRM en Bogotá, para, de esta forma, conocer o ampliar la información sobre la seguridad de los medicamentos, y así promover el uso seguro y adecuado de estos.
Red Distrital de FarmacovigilanciaLa Red Distrital de Farmacovigilancia es el conjunto de personas e instituciones que
mantienen contacto entre sí, a través de reportes de EAM y PRM, además, de comunica-ciones e información z en relación con problemas de seguridad o uso correcto de medica-mentos.
La red incorpora a personas o instituciones que han establecido contactos electrónicos, telefónicos o escritos con el PDFV.
¿Por qué una Red Distrital de Farmacovigilancia? La Red Distrital de Farmacovigilancia es una respuesta a la necesidad de intercambiar y
transferir información, conocimientos y experiencias que generen capacidad, autonomía y po-der social en la gestión del riesgo de aparición de eventos adversos u otros PRM.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
9
La participación en la red de farmacovigilancia garantiza el apoyo técnico por parte de los profesionales del PDFV y el acceso al archivo plano para el manejo de reportes, así como consultas específicas a la base de datos distrital.
Además de lo anterior, participar en la red les permite a individuos e instituciones for-talecer su formación, su capacidad y su autonomía en el análisis y la resolución de casos de EAM y de PRM, con el fin de desarrollar políticas institucionales orientadas al uso adecua-do de medicamentos.
La estrategia de red no pretende únicamente el fortalecimiento de la notificación espon-tánea de eventos adversos, sino también, la implementación de iniciativas de investigación y vigilancia activa, la búsqueda y el desarrollo de conocimiento propio que permita identi-ficar, caracterizar y solucionar problemas de seguridad y de uso inadecuado de los medica-mentos, según la realidad de la ciudad.
Nodos de la red nacional de farmacovigilanciaSon nodos de la red todas las entidades o instituciones que hayan establecido más de
un contacto por vía electrónica, escrita o telefónica con el programa nacional del Invima para el envío de información científica, comunicaciones de interés o reportes de eventos adversos (4).
• El nodo central será el Invima.• Según el grado de desarrollo de los programas se establecerán nodos regionales
(el centro distrital de farmacovigilancia es un nodo regional).• Los nodos locales, individuales o institucionales serán las entidades promotoras de
salud (EPS), las instituciones prestadoras de servicios de salud (IPS), los titulares de registros sanitarios de medicamentos, las instituciones educativas, los establecimien-tos farmacéuticos y las agrupaciones de usuarios o de profesionales.
• Los individuos aislados serán parte de la red y actuarán como nodos.
La notificación o el análisis deberá darse desde los nodos notificadores a los nodos regionales y nacionales, y estos, a su vez, deben retroalimentar al reportante (diagrama 1).

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
10
Diagrama 1. Flujo de notificación de sospechas de reacciones adversas a medicamentos y problemas relacionados con el uso de medicamentos
¿Quién debe reportar EAM y PRM al PDFV?La comunidad en general, los pacientes, los propietarios y los dependientes de es-
tablecimientos farmacéuticos mayoristas y minoristas, así como el personal de la salud y las IPS.
¿Cómo se deben reportar los EAM y los PRM al PDFV?• Diligenciando el formato de reporte de EAM y PRM y enviándolo al correo elec-
trónico [email protected], o allegándolo a la SDS: Carrera 32 # 12-81, Edificio Administrativo, cuarto piso, Salud Pública (anexo 1).
• Diligenciando el formato electrónico, a través de la página web de la SDS.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
11
¿Qué se debe reportar al PDFV?Se deben notificar todas las sospechas de EAM y de PRM; es decir, síntomas y signos,
nuevos o conocidos, enfermedades nuevas o conocidas que se presentan en los pacientes durante la terapia farmacológica, y todos los PRM.
Además de lo anterior, los establecimientos farmacéuticos los distribuidores mayoristas deben actuar como nodos locales de la Red Distrital de Farmacovigilancia recolectando y analizando las notificaciones generadas por los establecimientos farmacéuticos minoristas, para luego enviarlas al PDFV y al Invima (diagrama 2).
Diagrama 2. Análisis de las notificaciones
Invima
Recibe, consolida y
transmite información sobre EAM Y
PRM
Programa distrital de farmacovigilancia
Recopilan,generan y analizan
reportes de EAN Y PRM
Generarreportes de:
* EAM
* PRM
Minoristas
Mayoristas
PRMSon aquellas situaciones que en el proceso de uso de medicamentos causan o pue-
den causar la aparición de un resultado negativo asociado a la medicación. En el Tercer Consenso de Granada se describieron los siguientes PRM, sin que la lista sea definitiva ni excluyente (5):

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
12
Problemas relacionados con el uso de los medicamentos (PRM), según el Tercer Consenso de Granada
Características personales
Conservación inadecuada
Contraindicación
Dosis, pautazo duración inadecuadas
Duplicidad
Errores en la dispensación
Errores en la prescripción
Incumplimiento
Interacciones
Otros problemas de salud que afectan el tratamiento
Probabilidad de efectos adversos
Problema de salud insuficientemente tratado
Otros
¿Cómo evitar PRM de conservación inadecuada?1. Asegurar condiciones de infraestructura física:
a. Los pisos deben ser de material impermeable y resistente, y contar con un sistema de drenaje que permita su fácil limpieza e higienización.
b. Las paredes y los muros deben ser impermeables, sólidos, de fácil limpieza y resistentes a factores ambientales como la humedad y la temperatura.
c. Los techos y los cielos rasos deben ser resistentes, uniformes y de fácil limpieza e hi-gienización.
d. Las áreas para el almacenamiento de medicamentos y de dispositivos médicos deben ser independientes, diferenciadas y debidamente señalizadas, además de tener condi-ciones ambientales, de temperatura y de humedad relativa controladas.
e. La infraestructura física debe poseer un sistema de iluminación natural o artificial que permita la conservación adecuada y la identificación de los medicamentos y los dispo-sitivos médicos, así como un buen manejo de la documentación.
f. Los plafones deben hallarse en buen estado; los tomacorrientes, los interruptores y el cableado deben estar debidamente protegidos.
g. Debe haber un sistema de ventilación natural o artificial que garantice la conservación adecuada de los medicamentos y de los dispositivos médicos. No deben entenderse por formas de ventilación natural las ventanas o las puertas abiertas, que podrían per-

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
13
mitir la contaminación de los medicamentos y de los dispositivos médicos con polvo y suciedad del exterior.
h. Los sitios donde se almacenen medicamentos deben contar con mecanismos que ga-ranticen las condiciones de temperatura y humedad relativa recomendadas por el fa-bricante y los registros permanentes de estas variables, y utilizar para ello termómetros, higrómetros u otros instrumentos que cumplan dichas funciones.
i. Los dispositivos médicos y los medicamentos se almacenarán según la clasificación farmacológica (medicamentos), en orden alfabético o por cualquier otro método de clasificación, siempre y cuando ello garantice el orden y minimice eventos tales como la confusión, la pérdida y el vencimiento durante el almacenamiento. El sistema de segregación de los dispositivos médicos y de los medicamentos debe garantizar que los lotes más próximos a vencerse sean los primeros en dispensarse.
2. Adecuar áreas específicas:a. Área administrativa debidamente delimitada.b. Área de recepción de medicamentos, dispositivos médicos y productos autoriza-
dos.c. Área de cuarentena de medicamentos.d. Área adecuada para el almacenamiento, teniendo en cuenta los tipos de productos que
se van a distribuir.e. Área adecuada y segura para almacenar medicamentos de control especial.f. Área de almacenamiento de materias primas y de medicamentos que requieran cadena
de frío para su conservación.g. Área debidamente identificada para almacenar medicamentos que deben ser destrui-
dos o desnaturalizados, por vencimiento o deterioro.h. Área destinada para el almacenamiento de productos rechazados, devueltos y retirados
del mercado.i. Área de alistamiento y despacho.j. Área independiente de reenvase de materia prima, en caso de que se lleve a cabo.k. Área para el manejo y la disposición de residuos, de acuerdo con la reglamentación
vigente.
3. Dirección técnicaLos depósitos de drogas estarán dirigidos por un químico farmacéutico o por un tec-
nólogo en regencia de farmacia; sin embargo, todo depósito donde se haga el reenvase de materias primas estará bajo la dirección exclusiva de un químico farmacéutico. Los depó-sitos de drogas que tengan sección de reenvase de materias primas contarán, básicamente, con los protocolos establecidos para el caso.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
14
Prohibiciones para los dependientes y los propietarios de los depósitos de drogas
• No podrán elaborar, transformar ni reenvasar ningún medicamento, de conformi-dad con lo ordenado en el artículo 440 de la Ley 09 de 1979.
• No podrán vender al detal los medicamentos, ni los dispositivos médicos ni los pro-ductos que comercializan.
• No podrán dispensar esos mismos medicamentos ni dispositivos a los pacientes afiliados a empresas administradoras de planes de beneficios, ni a quienes sean aten-didos por IPS en desarrollo de contratos de suministros (3).
Glosario • Buenas prácticas de farmacovigilancia: Conjunto de normas o recomendaciones destina-
das a garantizar: la autenticidad y la calidad de los datos recogidos para la evaluación en cada momento de los RAM; la confidencialidad de las informaciones relativas a la identidad de las personas que hayan presentado o notificado las reacciones adversas, y el uso de criterios uniformes en la evaluación de las notificaciones y en la genera-ción de señales de alerta (6).
• Causalidad (imputabilidad): El resultado del análisis de la imputabilidad y de la evalua-ción individual de la relación entre la administración de un medicamento y la apa-rición de una reacción adversa permite determinar una categoría de causalidad (2).
• Categorías de causalidad: Las categorías descritas al respecto por el Centro de Monito-reo de Uppsala son las siguientes:
- Definitiva: Un acontecimiento clínico, incluso alteraciones en las pruebas de labo-ratorio, que se manifiesta con una secuencia temporal plausible relacionada con la administración del medicamento, y que no puede ser explicado por la enfermedad concurrente ni por otros medicamentos o sustancias. La respuesta a la supresión del medicamento (retirada) debe ser plausible clínicamente. El acontecimiento debe ser definitivo desde un punto de vista farmacológico o fenomenológico, y utilizando, si es necesario, un procedimiento de reexposición concluyente.
- Probable: Acontecimiento clínico, incluso, alteraciones en las pruebas de la-boratorio, que se manifiesta con una secuencia temporal razonable respecto a la administración del medicamento; es improbable que se lo atribuya a la enfermedad concurrente o a otros medicamentos o sustancias, y al retirar el medicamento se presenta una respuesta clínicamente razonable. No se requiere tener información sobre reexposición para asignar esta definición.
- Posible: Acontecimiento clínico, incluso, alteraciones en las pruebas de labo-ratorio, que se manifiesta con una secuencia temporal razonable en cuanto a

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
15
la administración del medicamento, pero puede ser explicado también por la enfermedad concurrente o por otros medicamentos o sustancias. La informa-ción respecto a la retirada del medicamento puede faltar o no ser clara.
- Improbable: Acontecimiento clínico, incluso alteraciones en las pruebas de laborato-rio, que se manifiesta con una secuencia temporal improbable relacionada con la administración del medicamento, y que puede ser explicado de forma más plausible por la enfermedad concurrente o por otros medicamentos o sustancias.
- Condicional/no clasificada: Acontecimiento clínico, incluso alteraciones en las pruebas de laboratorio, notificado como una reacción adversa, de la que es imprescindible obtener más datos para poder hacer una evaluación apropiada, o cuyos datos adicionales están bajo examen.
- No evaluable/inclasificable: Notificación que sugiere una reacción adversa, pero que no puede ser juzgada porque la información es insuficiente o contradic-toria, y la cual no puede ser verificada o completada en sus datos.
• Evento adverso a medicamento (EAM): Cualquier episodio médico desafortunado que puede presentarse durante el tratamiento con un medicamento, pero no tiene re-lación causal necesaria con ese tratamiento. Si bien se observa coincidencia en el tiempo, no se sospecha que exista relación causal. Ejemplo: dolor de cabeza, mareos, vómitos, prurito, hipertensión, etc. (2).
• Evento adverso grave: Cualquier situación médica desfavorable que a cualquier dosis causa la muerte, amenaza la vida, causa la hospitalización o la prolonga. Da como resultado incapacidad/discapacidad persistente o significativa; por ejemplo, una ano-malía congénita o defecto de nacimiento, o cualquier otra situación que se clasifique como médicamente significativa. Todos los demás eventos adversos que no cumplen con las anteriores características serán clasificados como no serios (2).
• Efecto secundario: Efecto que no surge como consecuencia de la acción farmacológica primaria de un medicamento, sino que constituye una consecuencia eventual de esta acción; por ejemplo, la diarrea asociada a la alteración del equilibrio de la flora bac-teriana normal, que es producto de un tratamiento antibiótico (2).
• Medicamento: Preparado farmacéutico obtenido a partir de principios activos, con o sin sustancias auxiliares, presentado bajo forma farmacéutica y que se utiliza para la prevención, el alivio, el diagnóstico, el tratamiento, la curación o la rehabilitación de la enfermedad. Los envases, los rótulos, las etiquetas y los empaques hacen parte integral del medicamento, por cuanto estos garantizan su calidad, su estabilidad y su uso adecuado (Decreto 677/1995) (7).
• Reacción adversa a los medicamentos (RAM): Según la OMS, “[…] reacción nociva y no deseada que se presenta tras la administración de un fármaco, a dosis utilizadas habi-

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
16
tualmente en la especie humana, para prevenir, diagnosticar o tratar una enfermedad, o para modificar cualquier función biológica”. Nótese que esta definición implica una relación de causalidad entre la administración del medicamento y la aparición de la reacción. Los cambios en los resultados de pruebas de laboratorio también se consideran RAM (2).
• RAM, según su gravedad: - Leves: Manifestaciones clínicas poco significativas o de baja intensidad, que no requie-
ren ninguna medida terapéutica importante o no justifican suspender el tratamiento. - Moderadas: Manifestaciones clínicas importantes, que no representan amenaza
inmediata para la vida del paciente, pero requieren medidas terapéuticas o la suspensión de tratamiento.
- Graves: Manifestaciones clínicas que producen la muerte, amenazan la vida del paciente, producen incapacidad permanente o sustancial, requieren hos-pitalización o prolongan el tiempo de hospitalización y producen anomalías congénitas o procesos malignos.
• Resultados negativos asociados a la medicación (RNM): Resultados en la salud del paciente no adecuados al objetivo de la farmacoterapia, y asociados al uso o al fallo en el uso de medicamentos (5).
• RNM (clasificación según el Tercer Consenso de Granada): - Según la necesidad:
- Problema de salud no tratado: El paciente sufre un problema de salud asociado al hecho de no recibir una medicación que necesita.
- Efecto de medicamento innecesario: El paciente sufre un problema de salud asocia-do al hecho de recibir un medicamento que no necesita.
- Según su efectividad: - Inefectividad no cuantitativa: El paciente sufre un problema de salud asociado a
una inefectividad no cuantitativa de la medicación. - Inefectividad cuantitativa: El paciente sufre un problema de salud asociado a una
inefectividad cuantitativa de la medicación. - Según la seguridad:
- Inseguridad no cuantitativa: El paciente sufre un problema de salud asociado a una inseguridad no cuantitativa de un medicamento.
- Inseguridad cuantitativa: El paciente sufre un problema de salud asociado a una inseguridad cuantitativa de un medicamento.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
17
• Señal: Según la OMS, en farmacovigilancia una señal es un “[…] aviso sobre la exis-tencia de información que sugiere una posible relación causal entre un evento inde-seable y la utilización de un medicamento. Usualmente se necesita más de una sola notificación para detectar una señal, dependiendo de la gravedad del evento y de la calidad de la información suministrada en la notificación”.
• Suplemento dietario: Producto cuyo propósito es adicionar la dieta normal, y que es fuente concentrada de nutrientes y otras sustancias con efecto fisiológico o nutricio-nal y puede contener vitaminas, minerales, proteínas, aminoácidos, otros nutrientes y derivados de nutrientes, plantas, concentrados y extractos de plantas solas o en com-binación (7).
• Producto fitoterapéutico: Producto medicinal empacado y etiquetado, cuyas sustancias ac-tivas provienen de material de plantas medicinales o asociaciones de estas, presentado en estado bruto o en forma farmacéutica, y que se utiliza con fines terapéuticos. Tam-bién puede provenir de extractos, tinturas o aceites. No podrá contener en su formula-ción principios activos aislados y químicamente definidos. Los productos obtenidos de material de la planta medicinal que hayan sido procesados y obtenidos en forma pura no serán clasificados como productos fitoterapéuticos (8).
• Establecimiento farmacéutico: Todo establecimiento dedicado a la producción, el almace-namiento, la distribución, la comercialización, la dispensación, el control o el asegu-ramiento de la calidad de los medicamentos, los dispositivos médicos o las materias primas necesarias para su elaboración, y los demás productos autorizados por ley para la comercialización en dicho establecimiento (3).
• Establecimientos farmacéuticos mayoristas: Son los laboratorios farmacéuticos, las agen-cias de especialidades farmacéuticas y los depósitos de drogas (3).
• Establecimientos farmacéuticos minoristas: Son las farmacias-droguerías y las droguerías (3). • Depósitos de drogas: Establecimientos comerciales dedicados exclusivamente a la venta
al por mayor de medicamentos, dispositivos médicos y productos autorizados (3). • Buenas prácticas de almacenamiento (BPA): Conjunto de normas mínimas en toda la ca-
dena de abastecimiento: almacenamiento, transporte, distribución y dispensación, destinadas a garantizar el mantenimiento de las características y las propiedades de los medicamentos (9).

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
18
Referencias
1. Organización Mundial de la Salud (OMS). Vigilancia de la seguridad de los medicamentos. Guía para la instalación y puesta en funcionamiento de un Centro de Farmacovigilancia. Uppsala: OMS; 2001.
2. Red Panamericana de Armonización de la Reglamentación Farmacéutica (PARF). Buenas Prácticas de Farmacovigilancia para las Américas. Documento Técnico No. 5. Washington: Red PARF; 2010.
3. Colombia, Ministerio de Protección Social (MPS). Resolución 1403/2007 Por la cual se determina el Modelo de Gestión del Servicio Farmacéutico, se adopta el Manual de Con-diciones Esenciales y Procedimientos y se dictan otras disposiciones. Bogotá: MPS; 2007.
4. Colombia, Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima). Bole-tín No. 13. Bogotá: Invima; 2006.
5. Tercer Consenso de Granada sobre Problemas Relacionados con Medicamentos. Comité de consenso GIAF-UGR, GIFAF-USE, GIF-UGR, (PRM) y Resultados Negativos aso-ciados a la Medicación (RNM). Ars Pharm. 2007;48:5-17.
6. Colombia, Presidencia de la República. Decreto 677 de 1995, por el cual se reglamenta parcia-mente el régimen de vigilancia sanitaria y farmacéutica. Bogotá: Diario Oficial 41827; 1995.
7. Colombia, Cámara de Representantes. Proyecto de ley 065 de 2008, por la cual se prohíbe la publicidad de medicamentos, productos naturales, fitosanitarios y se dictan otras dispo-siciones. Bogotá; Secretaría General; 2008.
8. Colombia, Presidencia de la República. Decreto 2266 de 2004, por el cual se reglamentan los regímenes de registros sanitarios. Bogotá: Diario Oficial 45610; 2004.
9. Colombia, Consejo Nacional de Política Económica y Social. Documento Conpes 155: Política Farmacéutica Nacional. Bogotá: Conpes; 2012.
Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
19
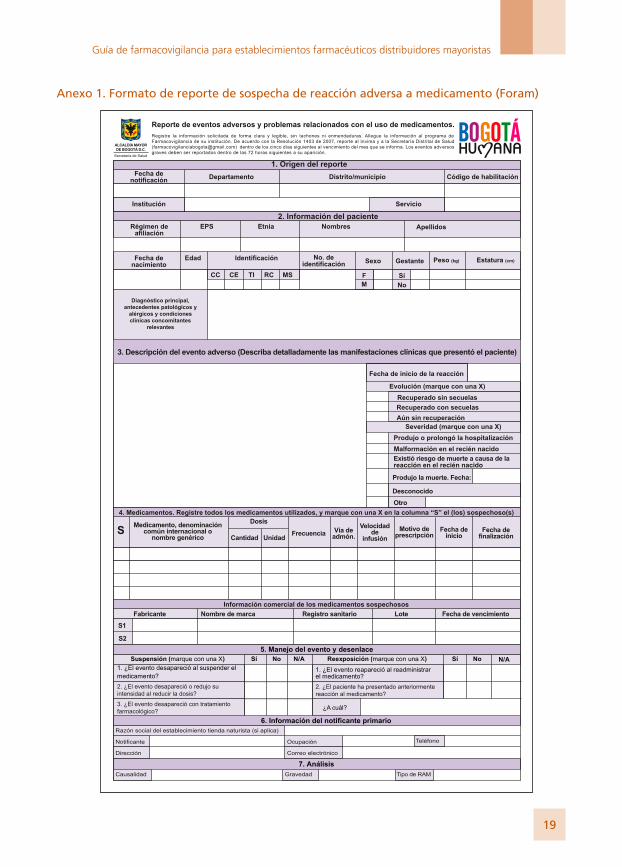
Anexo 1. Formato de reporte de sospecha de reacción adversa a medicamento (Foram)

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
20
Instructivo de diligenciamiento de Formato de reporte de sospecha de reacción adversa a medicamento (Foram)
1. Origen del reporte• Fecha de notificación: Indicar la fecha en la que se diligencia el formato.• Origen: Indicar el departamento y el distrito o municipio donde se encuentra la IPS.• Institución: Indicar el nombre de la institución que genera el reporte (IPS, EPS, profesio-
nal independiente, etc.). • Servicio: Registrar el servicio asistencial de la IPS donde se presenta el evento.• Código de habilitación: Indicar el código de habilitación de la IPS.
2. Información del paciente• Régimen de afiliación: Indique el régimen de afiliación en el que se encuentra el pacien-
te, como: régimen subsidiado, régimen contributivo, medicina prepagada, excepcional (fuerzas militares-policía, magisterio, etc.).
• EPS: Indique del nombre de la EPS. • Etnia: Mencione la etnia del paciente: blanco, mestizo, afrocolombiano, indígena, rom
(gitano), árabe, otro grupo.• Iniciales: Indique las iniciales correspondientes al nombre y los apellidos del paciente.• Fecha de nacimiento: Indique de la siguiente manera la fecha de nacimiento del paciente:
aaaa-mm-dd (año-mes-día).• Identificación: Indique con una X en la casilla correspondiente: cédula de ciudadanía, cedu-
la de extranjería, tarjeta de identidad, registro civil o menor sin identificación.• Número de identificación: Indique el número correspondiente al documento de identidad.• Sexo: Marque con una X en la casilla correspondiente: M (masculino) o F (femenino).• Gestante: Marque con una X en la casilla correspondiente, si es el caso.• Peso: Indique el peso del paciente en kilogramos (kg). • Estatura: Registrar la estatura del paciente.• Diagnóstico, condiciones médicas relevantes, resultados de exámenes y antecedentes: Describir el diag-
nóstico principal y los datos de importancia, como: falla hepática, falla renal, alergias, antecedentes, embarazo, resultados de exámenes clínicos y paraclínicos, entre otros.
3. Descripción del evento• Fecha de inicio del evento adverso: Indique de la siguiente manera la fecha exacta en la cual se
inició la reacción: aaaa-mm-dd.• Descripción del evento adverso: Describa detalladamente cuáles fueron los signos y los síntomas
de la reacción adversa. Si se cuenta con resultados de pruebas o exámenes diagnósticos o de procedimientos médicos, es preciso anexarlos al reporte. En caso de existir más de un evento adverso escriba la fecha de inicio de cada uno.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
21
• Evolución: Marque con una X, según la casilla correspondiente, la evolución del evento.• Severidad: Marque con una o varias X la(s) opción(es) correspondiente(s). Si el evento
produjo la muerte, indique la fecha de defunción; si produjo otro tipo de condición, descríbala.
4. Información de los medicamentos Registre todos los medicamentos utilizados, y marque con una X los que considera
sospechosos en Denominación Común Internacional (DCI) o nombre “Genérico”. • Dosis: Indicar la dosis suministrada en cantidad y unidades de medida, según la casilla
correspondiente (por ejemplo: 500 mg). • Frecuencia: Indique la frecuencia o los intervalos de administración del medicamento (por
ejemplo: cada 4-6-8-12 horas). • Vía de administración: Describa la vía de administración del medicamento (por ejemplo:
vía oral, intramuscular, intravenosa [VO, IM, IV]).• Velocidad de infusión: En caso de que el medicamento haya sido administrado por infusión,
indique la velocidad de esta.• Motivo de prescripción: Describa la indicación del medicamento.• Fecha de inicio: Indique la fecha en la que se inició el tratamiento con el medicamento.• Fecha de finalización: Indique la fecha en la que terminó el tratamiento con el medicamento.• Información comercial de los medicamentos sospechosos
- Nombre del fabricante: Registre el nombre del laboratorio farmacéutico o del titular del registro sanitario.
- Nombre de marca: Indique el nombre comercial del medicamento. - Registro sanitario, lote y fecha de vencimiento: Registre dicha información.
5. Manejo del evento y desenlace, suspensión y reexposición Indique con una X la información solicitada, según la casilla correspondiente: Sí, No, o No
aplica (N/A), cuando se desconozca tal información o cuando no se realizó la suspensión o la reexposición.
• Tratamiento farmacológico: Indicar si el evento requirió manejo farmacológico, y en caso de ser positivo, indicar cuál.
6. Información del notificante primarioNombre de la persona que diligencia el formato; profesión, dirección (de la institución),
teléfono y correo electrónico (de contacto de la persona). El objetivo de esta información es contar con los datos del notificante para solicitar mayor información, cuando se la requiera, o para el envío de la respuesta o de la retroalimentación sobre el resultado del análisis. Nota: Si el notificante primario es el paciente o un familiar de este, indicar, además, la información del médico tratante.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
22
7. Análisis • Causalidad: Se puede evaluar por algoritmo de Naranjo o el de la FDA asignándole una de las
siguientes categorías: - Probada (cierta o definitiva): Evento clínico que incluye pruebas de laboratorio anorma-
les, tiene una secuencia temporal razonable después de la administración del medica-mento, muestra un patrón de respuesta conocido que se asocia al medicamento sos-pechoso, se confirma mediante la mejoría al suspender el medicamento y reaparece después de administrarlo de nuevo, y no se puede explicar por las características de la enfermedad de base que tiene el paciente, por la administración de otros medicamen-tos o por la exposición a otros químicos.
- Probable: Evento clínico que incluye pruebas de laboratorio anormales, tiene una se-cuencia temporal razonable después de la administración del medicamento, muestra un patrón de respuesta conocido que se asocia al medicamento sospechoso, presenta una mejoría clínica razonable al suspender el medicamento, pero no reaparece des-pués de su readministración, y, probablemente, no se atribuye a las características de la enfermedad de base que tiene el paciente ni a la administración de otros medicamen-tos o la exposición a otros químicos.
- Posible: Evento clínico que incluye pruebas de laboratorio anormales, muestra una rela-ción temporal razonable después de la administración del medicamento, y puede o no seguir un patrón de respuesta conocido que se asocia al medicamento sospechoso; la información relacionada con la suspensión del medicamento puede ser incompleta o no lo suficientemente clara, y se la podría explicar por las características de la enfermedad de base que tiene el paciente o por la exposición a otros medicamentos o químicos.
- Improbable (dudosa o remota): Evento clínico que incluye pruebas de laboratorio anorma-les, sin una secuencia de temporalidad clara con el medicamento sospechoso y la en-fermedad de base; otros medicamentos y químicos pueden suministrar explicaciones razonables. Entonces, la expresión clínica estará más probablemente relacionada con otros factores que con el medicamento presuntamente implicado.
• Gravedad: Según el nivel de intensidad, las RAM se pueden clasificar como: - Leves: No interfieren con la actividad habitual del paciente, son de corta duración, son
autolimitadas y no requieren intervención del personal de la salud ni prolongación del tiempo de internamiento, y, en general, no se necesita la suspensión del medicamento.
- Moderadas: Interfieren con la actividad habitual del paciente, requiere intervención del personal de salud para una mejor solución, aumento del tiempo de estancia hospitala-ria, implica la modificación del tratamiento, aunque no necesariamente la suspensión del medicamento causante de la reacción.
- Severas o graves: Constituyen una amenaza para la vida del paciente, requieren hospitaliza-ción o la prolongación de esta, la suspensión del medicamento causante de la reacción y la administración de un tratamiento específico para el manejo clínico de la reacción.

Guía de farmacovigilancia para establecimientos farmacéuticos distribuidores mayoristas
23
• Tipo de RAM: Estas pueden ser: - Tipo A: Implican un cambio cuantitativo en la respuesta del medicamento (exage-
ración del efecto farmacológico); son comunes cuando este se administra en dosis mayores, se puede predecir su aparición a partir del conocimiento que se tiene de las acciones farmacológicas del medicamento implicado, y, por lo tanto, son previsibles o esperadas. Tienden a mejorar con una reducción de la dosis o con la suspensión del tratamiento.
- Tipo B: Implican un cambio cualitativo en la respuesta del paciente al medicamento, por lo cual originan situaciones clínicas adicionales y diferentes de sus efectos farma-cológicos esperados; no se puede predecir su aparición a partir del conocimiento de su acción farmacológica ni tienen relación con la dosis administrada. Su aparición en relación con el momento de la exposición al medicamento es variable, y para ello puede transcurrir un periodo corto o largo. No mejoran al reducir la dosis, y la admi-nistración del medicamento se debe suspender. En estas reacciones intervienen me-canismos de tipo inmunológico (hipersensibilidad o alergia), pero también se pueden presentar debido a una constitución genética atípica del paciente (idiosincrasia).
- Tipo C: Son aquellas que aparecen relacionadas con tratamientos farmacológicos pro-longados; son conocidas y previsibles, y resultan de procesos adaptativos en las células de un tejido por la continua exposición al medicamento.
- Tipo D: Están asociadas a la mutagénesis o a las anomalías del desarrollo embrionario o fetal (incluyen la dismorfogénesis o la teratogénesis) y aparecen tiempo después de la administración del medicamento.
- Tipo E: Aparecen como resultado de la suspensión súbita o repentina de la admi-nistración prolongada de un medicamento. También se conocen con el nombre de “efecto de rebote” y son poco comunes.
- Tipo F: Su aparición se relaciona con sustancias químicas diferentes del principio acti-vo del medicamento (excipientes, impurezas, contaminantes). Esta categoría también se refiere a la ausencia de respuesta clínica al medicamento o a la falla terapéutica, y son relativamente comunes.
Información para el envío de los reportes al PDFVDirección: carrera 32 D # 12-81, Bogotá, D. C. Teléfono: 364 9090Correo electrónico: [email protected]






























