Embed Size (px)
Citation preview

0
Gyűrűs kalkonszármazékok citotoxikus és
daganatellenes hatása molekuláris
mechanizmusának vizsgálata
Egyetemi doktori (Ph.D.) értekezés
Dr. Rozmer Zsuzsanna
Doktori Iskola vezetője: Prof. Dr. Pintér Erika
Programvezető: Prof. Dr. Gregus Zoltán
Témavezető: Prof. Dr. Perjési Pál
Konzulens: Prof. Dr. Gregus Zoltán
Pécsi Tudományegyetem, Általános Orvostudományi Kar
Gyógyszerészi Kémiai Intézet
Pécs
2014.

1
Tartalomjegyzék
I. RÖVIDÍTÉSEK JEGYZÉKE ................................................................................ 3
II. BEVEZETÉS, IRODALMI ÁTTEKINTÉS ......................................................... 5
II.1. TERMÉSZETES KALKONOK ÉS BIOLÓGIAI HATÁSAIK .......................................... 7
II.2. SZINTETIKUS KALKONSZÁRMAZÉKOK ............................................................. 10
II.2.1. Kalkonok szintetikus előállítása ........................................................... 10
II.2.2. Szintetikus kalkonszármazékok biológiai vizsgálatának célja
és háttere ............................................................................................... 11
II.2.3. Gyűrűs kalkonanalógok szintézise és szerkezete .................................. 15
II.2.4. Gyűrűs kalkonszármazékok citotoxikus hatása és szerkezete
közötti kapcsolat ................................................................................... 17
II.2.5. Gyűrűs kalkonszármazékok daganatellenes hatása .............................. 19
III. CÉLKITŰZÉSEK .................................................................................................. 21
IV. VIZSGÁLATI MÓDSZEREK ............................................................................. 24
IV.1. KALKONSZÁRMAZÉKOK LOGP ÉRTÉKEINEK MEGHATÁROZÁSA ...................... 24
IV.1.1. Vizsgált anyagok ................................................................................... 24
IV.1.2. Fordított-fázísú vékonyréteg-kromatográfiás módszer ......................... 25
IV.2. GYŰRŰS KALKONSZÁRMAZÉKOK HATÁSMECHANIZMUSÁNAK VIZSGÁLATA ... 25
IV.2.1. Kísérleti anyagok és sejtkultúra ............................................................ 25
IV.2.2. Sejtkezelési protokoll ............................................................................ 26
IV.2.3. „Túlélés”-vizsgálat ................................................................................ 26
IV.2.4. Áramlási citometriai módszerek ........................................................... 26
IV.2.4.1. Az áramlási citometria ......................................................................................... 26
IV.2.4.2. Sejtszaporodásra gyakorolt hatás vizsgálata ........................................................ 27
IV.2.4.3. Sejtciklus-vizsgálat .............................................................................................. 28
IV.2.5. A vegyületek in vitro antioxidáns hatásának vizsgálata
dezoxiribóz degradációs módszerrel ..................................................... 28
IV.2.6. A kalkonanalógok celluláris redox-homeosztázisra gyakorolt
hatásának vizsgálata .............................................................................. 29
IV.2.6.1. Az intracelluláris ROS-aktivitás meghatározása .................................................. 29
IV.2.6.2. Mitokondriális funkciókra gyakorolt hatás vizsgálata .......................................... 30

2
IV.2.6.3. A celluláris tiolszint meghatározása ..................................................................... 30
IV.2.6.4. A celluláris redukált és oxidált glutationszint vizsgálata ..................................... 31
IV.2.7. A kalkonszármazékok glutationnal való reaktivitásának
vizsgálata .............................................................................................. 33
IV.2.7.1. A kalkon-glutation reakció vizsgálata sejtmentes körülmények között ............... 33
IV.2.7.2. A kalkon-glutation reakció vizsgálata Jurkat T sejtekben HPLC-MS módszerrel 33
V. EREDMÉNYEK ÉS KÖVETKEZTETÉSEK .................................................... 35
V.1. KALKONANALÓGOK LIPOFILITÁSÁNAK JELLEMZÉSE ....................................... 35
V.2. GYŰRŰS KALKONSZÁRMAZÉKOK HATÁS-VIZSGÁLATÁNAK EREDMÉNYEI ....... 41
V.2.1. Az (E)-2-(4-metoxibenzilidén)-1-benzoszuberon és
4-metilanalógjának sejtproliferációra és sejtciklusra gyakorolt
hatása .................................................................................................... 41
V.2.1.1. A kalkonanalógok apoptózist indukálnak Jurkat T sejtekben ................................ 41
V.2.1.2. A vizsgált származékok eltérő módon befolyásolják a sejtciklust ......................... 45
V.2.2. A kalkonszármazékok in vitro antioxidáns hatással bírnak .................. 50
V.2.3. A kalkonszármazékok hatással vannak a Jurkat T sejtek
redox státuszára ..................................................................................... 53
V.2.3.1. A vegyületek befolyásolják a ROS-aktivitást ........................................................ 53
V.2.3.2. A kalkonszármazékok hatással vannak a mitokondriális funkciókra .................... 55
V.2.3.3. A kalkonanalógok befolyásolják a celluláris SH-szintet ....................................... 57
V.2.3.4. A kalkonszármazékok hatása a Jurkat sejtek glutation státuszára ......................... 58
V.2.4. A kalkon-glutation konjugációs reakció vizsgálata .............................. 61
V.2.4.1. A vizsgált kalkonszármazékok intrinsic aktivitást mutatnak glutationnal ............. 61
V.2.4.2. A kalkonanalógok és glutation konjugációs reakciójának vizsgálata Jurkat T
sejtekben ................................................................................................................ 63
VI. ÚJ EREDMÉNYEK ÖSSZEFOGLALÁSA, MEGBESZÉLÉS ........................ 67
VII. IRODALOMJEGYZÉK ...................................................................................... 71
VIII. SAJÁT PUBLIKÁCIÓK .................................................................................... 85
IX. KÖSZÖNETNYILVÁNÍTÁS ............................................................................... 89

3
I. Rövidítések jegyzéke
ATP adenozin-5’-trifoszfát
ADP adenozin-5’-difoszfát
BCA bicinkoninsav
BSA marha szérum albumin (bovine serum albumin)
CHI kalkon-izomeráz
CHS kalkon-szintáz
CYP citokróm-P450 enzimrendszer
C4H fahéjsav-4-hidroxiláz
DAD diódasoros detektor
DCFH-DA 2’,7’-diklorofluoreszcein-diacetát
DMBA 7,12-dimetil-benz[α]antracén
DMSO dimetil-szulfoxid
DNS dezoxiribonukleinsav
DTNB 5,5'-ditiobisz-(2-nitrobenzoesav)
EDTA etiléndiamin-tetraecetsav
EpRE electrophile response element
ESI elektrospray-ionizáció
FITC fluoreszcein-izotiocianát
FCS fötális borjú savú (fetal calf serum)
FSC besugárzó fény irányával megeegyező irányú fényszóródás
(forward scatter)
GC gázkromatográfia
GCL glutamát-cisztein-ligáz
GGT gamma-glutamil-transzpeptidáz
GSH redukált glutation
GSSG oxidált glutation
GST glutation-S-transzferáz
HPLC nagyhatékonyságú folyadékkromatográfia
IR infravörös spektroszkópia
KE karboxiláz enzim
Keap1 Kelch-like ECH-associated protein 1

4
logP megoszlási hányados logaritmusa
MALDI mátrix közvetítésével végzett lézer deszorpciós ionizáció
MS tömegspektrometria
NADPH redukált nikotinamid-adenin-dinukleotid-foszfát
NEM N-etilmaleimid
NMR mágneses magrezonancia
Nrf2 nuclear factor erythroid 2-related factor 2
OPA orto-ftálaldehid
PAH policiklusos aromás szénhidrogén
PAL fenilalanin-ammónia-liáz
PBS NaCl-tartalmú foszfát-tompítóoldat (phosphate buffered saline)
PGEE prosztaglandin E1-etilészter
PI propidium-jodid
RNS ribonukleinsav
ROS reaktív oxigénszármazék
RP-HPLC fordított fázisú nagyhatékonyságú folyadékkromatográfia
RP-TLC fordított fázisú vékonyréteg-kromatográfia
RPMI Roswell Park Memorial Institute sejttenyésztő médium
SF shake-flaske method
SH tiolcsoport
SDS nátrium-laurilszufát
SSC fény oldalirányú szóródása (side scatter)
TBA tiobarbitursav
TGSH totál glutation
TOF-MS time of flight / repülési idő tömegspektrometria
UDP uridin-5’-difoszfát
VRK vékonyrétegkromatográfia
4CL 4-kumarát-koenzimA-ligáz
γGCS gamma-glutamil-cisztein-szintáz

5
II. Bevezetés, irodalmi áttekintés
A daganatos megbetegedések korunk egyik legaggasztóbb népegészségügyi problémája.
Magyarországon, mint az iparilag fejlett országokban általában, a legtöbb ember a szív
és keringési rendszert érintő betegségekben hal meg, de a daganatos betegségek a
halálokok gyakoriságának sorrendjében a második helyen állnak, az összhalálozás közel
egynegyedét teszik ki. A főbb daganatos halálokok nemenként különbözőek: férfiak
esetén a daganatos halálokok több mint háromnegyedét az emésztőrendszeri és a
légzőszervi daganatok teszik ki, míg a nők között az emésztőszervi, a légzőszervi,
valamint az emlő- és méhdaganatok közel egyenlő százalékban okoznak halált. A
daganatos halálozás abszolút számait tekintve a hazai érték mindkét nem körében
magasabb, mint az európai átlag. (1) Mindezek a jelenségek előtérbe helyezik a
daganatos megbetegedések megelőzésére és kezelésére irányuló lehetőségek és
módszerek az eddigieknél még hatékonyabb kutatását.
A daganatos megbetegedések kialakulása, vagyis a karcinogenezis egy hosszú,
többlépcsős folyamat, melynek során a daganatsejtben mind több mutáció és hiba
halmozódik fel, és egyre jobban elvész a sejszaporodás kontrollja. A folyamat első fő
stádiuma az iniciáció, amikor spontán, vagy mutagén behatások mutációt váltanak ki a
genomban. A promóció során rögzülnek a genetikus hibák, hiszen promóterek hatására
a károsodott DNS expresszálódik, sejt-elfajulásokat okozva. Az invázió a rögzült
genetikus hibáknak az utódsejtek egyre nagyobb számában történő megjelenése,
malignus tumorok kialakulása. A progresszió egy komplex folyamat, amely ahhoz
vezet, hogy az elfajult sejtek elszaporodnak, klinikai daganat alakul ki, metasztázisok
jelennek meg. (2; 3)
(1. ábra)
A karcinogenezis folyamatába több ponton történhet beavatkozás. (1. ábra) A
daganatos megbetegedések elsődleges megelőzését a kockázati tényezők elkerülése,
vagy legalábbis csökkentése, a kemoprevenció és az egészségnevelés jelentik. A
kemoprevenció a DNS állomány károsítására alkalmas karcinogén formák, a DNS
nukleofil centrumaival szemben nagy reakciókészséget mutató elektrofil tulajdonságú
részek képződésének gátlása és/vagy eliminációjuk elősegítése, vagyis a genommal
történő interakciójuk meggátlása.
A tudatos kemoprevenció lényege, hogy természetes eredetű vagy szintetikus
anyagokat hosszú időn keresztül fogyasztunk annak érdekében, hogy a daganat

6
kialakulását megelőzzük, vagy a még reverzibilis folyamatot visszafordítsuk. Számos
epidemiológiai tanulmány döntő bizonyítékot nyújt néhány étrendi összetevő (pl.
vitaminok, fenolos és nem-fenolos antioxidánsok, omega-3-zsírsavak stb.)
kemopreventív hatásáról. Többek között a friss zöldségekben és gyümölcsökben nagy
mennyiségben előforduló polifenolok (főként flavonoidok) is csökkentik a rák
kialakulásának rizikóját. (4; 5)
1. ábra A karcinogenezis többlépcsős folyamata –
beavatkozási lehetőségek, preventív stratégiák (3)
A karcinogenezis folyamatának egy másik stádiumába történő beavatkozási
lehetőséget jelentenek a kemoterápiás gyógyszerek. A citosztatikumok különböző
hatásmechanizmus révén, de végső soron apoptózis indukciója és ezáltal a
daganatnövekedés gátlása útján hatnak. Szelektív toxicitásuk alapja, hogy a daganatos
sejtek osztódása gyorsabb, mint az egészséges sejteké, de alkalmazásuk során jelentős
mellékhatásokkal kell számolni. Bár folyamatosan számos új kemoterápiás szer kerül

7
bevezetésre, a hatékonyságot és a vegyületek szelektív toxicitását növelő kutatások
továbbra is a daganatellenes kutatások intenzív területét képezik.
II.1. Természetes kalkonok és biológiai hatásaik
A természetes növényi hatóanyagok egyik gyógyászati szempontból is széles körben
vizsgált csoportját képezik a flavonoidok. Nagyobbrészt ezek okozzák a virágok és
termések sárga, piros és kék színét. A flavonoidok formailag a difenilpropánból
levezethető (C6-C3-C6 alapszénváz) vegyületek, a két benzolgyűrű általában egy
oxigéntartalmú heterocikluson keresztül kapcsolódik. (6) A flavonoidok csoportján
belül számos alcsoportot (flavonok, flavonolok, flavanonok, flavanonolok,
antocianidinek, proantocianidinek, katechinek, kalkonok, auronok) különböztethetünk
meg, ugyanis az alapszerkezet nagy változatosságot biztosít mind a gyűrűk kapcsolódási
típusa, mind az összekapcsoló gyűrű szerkezete, mind a szubsztituensek tekintetében.
(2. ábra)
2. ábra A flavonoidok kapcsolódási típusai
A flavonoidok főként fenolos hidroxil- és metoxicsoportokat tartalmazó
természetes vegyületek, az alapvázhoz (aglikon) különböző – minőségű, számú és
helyzetű – cukormolekulák kapcsolódhatnak, így glükozidokat hozhatnak létre, amelyek
sokkal gyakrabban fordulnak elő a természetben, mint aglikonjaik.
A növényi flavonoidok szerepéről, kémiai szerkezetéről, élettani hatásairól
rendszeresen jelennek meg összefoglaló tanulmányok. (7; 8; 9; 10; 11) Igen széles körű
biológiai aktivitással rendelkeznek, például a citrusfélékben nagy mennyiségben
megtalálható heszperidin antioxidáns és gyulladáscsökkentő hatása mellett értonizáló.
(12) A máriatövis hatóanyagai (pl. szilimarin, szilibin) májvédő hatásúak (13), a kerti
ruta (Ruta graveolens L., hatóanyaga a rutin, ami a kvercetin rutinózzal alkotott
glikozidja) gyulladáscsökkentő és antioxidáns hatása révén szintén régóta használt
reumás megbetegedések, dermatitisz kezelésében, valamint görcsoldóként. (14)

8
A flavonoidok jellegzetes szerkezetű csoportját képezik a kalkonok. A kalkonok
szerkezetüket tekintve 1,3-diaril-2-propén-1-on alapvázzal rendelkező vegyületek.
(3. ábra)
3. ábra A kalkon szerkezete
A növényi kalkonok további átalakulásaik révén a flavonoidok előanyagainak is
tekinthetők. A kalkonok és flavonoidok biogenezisére a legvalószínűbbnek és
általánosan elfogadottnak a 4. ábra vázolt bioszintetikus út tekinthető. (15; 16; 17)
A kalkonok bioszintézisének első lépéseiben fenilalaninból, fenilalanin-ammónia-
liáz (PAL) enzim által katalizált dezaminálással fahéjsav képződik, melynek
hidroxilezését membránhoz kötött P450 monooxigenáz (fahéjsav-4-hidroxiláz, C4H)
végzi. Az így kialakult p-hidroxikumarilsav aktiválását 4-kumaroil-CoA-vá a 4-
kumarát-koenzimA-ligáz (4CL) végzi. A kalkonok B-gyűrűje 4-kumaroil-CoA-ból a
kalkon-szintáz segítségével (CHS) képződik, míg az A-gyűrű három – acetil-CoA-ból
és CO2-ból acetil-CoA-karboxiláz enzim (KE) hatására képződő – malonil-CoA
egységekből épül fel. (18)
A kalkonok sztereospecifikus gyűrűzáródása a kalkon-izomeráz (CHI) enzim
hatására következik be, és flavanonszármazékok keletkeznek, melyek megfelelő
enzimkatalízissel további flavonoidokká és izoflavonoidokká alakulhatnak tovább.
A kalkonszármazékok előfordulása általános a magasabbrendű növényekben. Irodalmi
adatok alapján a Leguminosae család több mint 70 fajtájából izoláltak különböző
szerkezetű kalkonokat, de az Asteraceae és Moraceae családok számos képviselőjében
is megtalálhatóak. (18) Ugyanakkor egyes növényfajták igen nagyszámú, különböző
kalkont tartalmaznak egymás mellett. A Cannabaceae családba tartozó Humulus lupus
gyantájában például 18 fajta kalkonszármazék fordul elő. A Glycyrrhiza inflata
gyökeréből 15 féle, az Angelica keiskei gyökeréből 25 kalkonanalógot különítettek el.
(18)

9
4. ábra A kalkonok bioszintézise
A növényi kalkonok főként hidroxil- és metoxiszubsztituált származékok, de
metil-, prenil-, geranilcsoportokat hordozó analógok, kromén- és benzofurán
heterociklusokat tartalmazó származékok, valamint kalkon-dimerek és dihidrokalkon-
származékok is előfordulnak a természetben.
A vegyületek sokrétű biológiai hatással rendelkeznek. Kalkonban gazdag növényi
drogokat, például az édesgyökeret (Liquorice), évszázadok óta használják Kínában
gyomor- és duodenális fekély kezelésére, különböző bőrproblémák, többek között
ekcéma és urtikária, valamint asztma terápiája során. (19) A távol-keleti országokban
tradícionálisan használják a buteint fájdalomcsillapításra, gasztritisz kezelésére,
különböző paraziták okozta fertőzésekben. (20; 21) A leggyakrabban vizsgált
természetes származékok az izoliquiritigenin, a butein, az izobavakalkon, a

10
xantoangelol, a likokalkon-A és a 4-hidroxi-lonhokarpin. (18) A természetes kalkonok
között megtalálhatóak antibakteriális (22), gombaellenes (23), vírusellenes (24),
protozonellenes (25) és tumorellenes (citotoxikus) hatású (26; 27) származékok,
úgymint kemoprotektív (28), kardioprotektív (29), antidiabetikus (30) hatással bíró
vegyületek. Az irodalmi eredmények alapján a leghatékonyabb tumorellenes hatású
származékoknak az izoliquiritigenin és butein bizonyultak.
II.2. Szintetikus kalkonszármazékok
II.2.1. Kalkonok szintetikus előállítása
A kalkonszármazékok laboratóriumi előállítására több lehetőség is rendelkezésre áll.
Kalkonok szintézise történhet Claisen-Schmidt féle kondenzációs reakció útján.
(5. ábra) (31) A reakció keresztezett aldoldimerizációs reakciónak tekinthető, amelynek
során a megfelelő keton és aldehid között nukleofil addíció játszódik le, és
vízkihasadással a megfelelő kalkonszármazék keletkezik.
5. ábra Claisen-Schmidt féle kondenzációs reakció
A kalkonok szintézisére alkalmas ún. Makaiyama-típusú aldolkondenzációs
reakció a Claisen-Schmidt féle reakcióhoz hasonló mechanizmussal játszódik le. (32)
A Suzuki-reakción alapuló eljárások szintén lehetőséget teremtenek kalkonok
előállítására. (6. ábra) (33) A Suzuki-reakció cinnamoil-klorid és fenil-bórsav, illetve
benzoil-klorid és fenilvinil-bórsav között, palládium katalizátor jelenlétében lejátszódó
keresztkapcsolási reakció.

11
6. ábra A Suzuki-féle kalkonszintézis egyenletei
Dihidrokalkonok szintézise, palládium katalizátor jelenlétében, 1-aril-2-propén-1-
ol és orto-halogénszubsztituált-fenol összekapcsolása révén történhet. (7. ábra) (34)
7. ábra Dihidrokalkonok szintézisének egyenlete
II.2.2. Szintetikus kalkonszármazékok biológiai vizsgálatának célja és
háttere
A növényekből izolált kalkonok vizsgálata mellett nagyszámú szintetikus származék
biológiai vizsgálatáról is beszámoltak, több összefoglaló közlemény is megjelent a
témában. (22; 35; 36; 37)
A szerteágazó biológiai hatások közül kiemelhető a citotoxikus és kemoprotektív
hatás. A szintetikus kalkonszármazékok szintézisének és biológiai vizsgálatainak egyik
előnye, hogy a rokon szerkezetű vegyületek összehasonlító vizsgálata során adatok
nyerhetők a szerkezet-hatás közötti kapcsolatról. Így a szintetikus származékok kiemelt
kutatási célja a természetes vegyületeknél hatékonyabb, főként a daganatterápiában
eredményesen alkalmazható analógok előállítása.
Az erre irányuló kutatások kémiai hátterében az áll, hogy a kalkonok, mint α,β-
telítetlen karbonilvegyületek (3. ábra) lágy elektrofilek, viszonylag könnyen reakcióba
léphetnek celluláris tiolokkal (lágy nukleofilek), ugyanakkor nem reagálnak amino- és
hidroxilcsoportokkal. (38) Mivel kémiailag nem reagálnak nukleinsavakkal, így a

12
kalkonszármazékok nem rendelkeznek genotoxikus hatással, ami a daganatterápiában
alkalmazott alkiláló típusú szerek egyik nemkívánt mellékhatása. (39)
A glutation szerepe
A sejtekben levő tiol (SH)-csoportok nagy többsége glutationból (GSH) származik. A
GSH intracellulárisan viszonylag nagy mennyiségben jelen levő nem-protein típusú tiol,
glicint, ciszteint és glutaminsavat tartalmazó tripeptid. (8. ábra)
8. ábra A glutation (GSH) szerkezete
A sejtek redox homeosztázisában igen fontos az intracelluláris redukált glutation-
szint fenntartása. A GSH kofaktora az oxigéntartalmú szabadgyökök, hidrogén-peroxid,
illetve egyéb organikus peroxidok eliminációjában részt vevő glutation-peroxidáz
enzimnek. A redukált glutation (GSH) oxidációja során két glutationmolekula diszulfid
híddal összekapcsolódhat és oxidált glutation (GSSG) keletkezik. A GSSG redukcióját
glutation-reduktáz enzim katalizálja NADPH felhasználásával. Normál körülmények
között az egyensúly a redukált forma irányába tolódik 500:1 arányban. A redukált
glutation koncentrációja magas a sejtekben, akár 5 mM is lehet. (40)
A glutation továbbá részt vesz a májban történő detoxikációs folyamatokban is, a
szervezetbe kerülő elektrofil vegyületek egy része a májban glutationnal konjugálódik.
A két szakaszban zajló folyamat első lépése a glutationnal való kapcsolódás, amit a
glutation-S-transzferáz enzim katalizál. A konjugáció a cisztein tiolcsoportján keresztül
valósul meg. Ezt követően előbb a γ-glutamil-transzpeptidáz hatására a glutaminsav,
majd a dipeptidáz hatására a glicin is lehasad a molekuláról, és a megmaradt konjugált
cisztein aminocsoportja acetilálódhat. A konjugáció révén az eredetileg toxikus
metabolitok vagy oldhatatlan vegyületek kiválasztásra kerülhetnek. (40)

13
A GSH funkciója tehát igen összetett: többek között detoxikáló folyamatokban
vesz részt, antioxidáns, cisztein-rezervoár és szabályozó szerepe van jelátviteli
folyamatokban. (41)
A kalkonok citotoxikus hatása
A kalkonok citotoxikus hatásának hátterében egyrészt a sejtek glutation-státuszának a
befolyásolása állhat. A kalkonszármazékok glutationnal történő reakcióját több
kutatócsoport is vizsgálta. (42; 43)
A konjugált karbonil-funkcióval rendelkező vegyületek jellegzetes Michael-
reakciós akceptorok, így könnyen reakcióba lépnek glutationnal. (44) A kalkonok -
szerkezetükből adódóan - feltételezhetően szintén reakcióba lépnek GSH-val, főként
konjugációs reakció révén. (42) (9. ábra)
9. ábra A kalkon, mint Michael-reakciós akceptor reakciója nukleofil tiollal (GSH)
Ugyanakkor, főként a hidroxilszubsztituált származékok, és a belőlük képződő
fenoxigyökök, oxidálhatják a GSH-t oxidált glutationná (GSSG), így előidézve a GSH-
szint csökkenését. (43) A GSH-szint megváltozása az intracelluláris redox-státusz
változását vonhatja maga után, ami befolyásolhatja többek között a DNS szintézis
folyamatát, a sejtciklus szabályozását, valamint enzimaktivációt, szelektív gén
expressziót válthat ki. (45)
A kalkonok citotoxikus aktivitásának hátterében még számos más tényező is
állhat. A B-gyűrűn elektronszívó szubsztituenst is hordozó hidroxikalkon-analógok
kiemelkedő tumorellenes hatást mutattak, amihez hozzájárul az angiogenezist gátló

14
hatásuk is. (46) Ugyanakkor, számos vegyület apoptózist indukál (47; 48), befolyásolja
a sejtciklust (48; 49; 50), valamint a mitokondriális membránpotenciál összeomlását
eredményezi (43).
Az antioxidáns hatású flavonoidok – ideértve a kalkonokat is – citotoxikus hatása
összefüggésbe hozható azok esetleges prooxidáns hatásával is. (35; 37; 51) A Fenton-
reakcióban keletkező hidroxilgyökök (HO˙) és aromás vegyületek reakciója során
fenolos-származékok keletkezhetnek. (52) A keletkező metabolitok további
biotranszformációja egy ún. „redox-cycling” reakcióban reaktív oxigén származékok
(ROS) képződését eredményezheti, ami oxidatív stressz kialakulásához vezethet, és így
az eredetileg antioxidáns vegyület prooxidáns hatású metabolittá válhat. (53)
Összeségében elmondható, hogy számos kalkonszármazék mikromólos
tartományban (IC50 < 50 μM) citotoxikus aktivitást mutat különböző tumor sejtvonallal
szemben. (37)
A kalkonok citoprotektív hatása
A daganatsejtekkel szemben mutatott toxicitásuk mellett, a kalkonok citoprotektív
hatása is jól dokumentált. A citoprotektív hatás hátterében egyrészt az állhat, hogy a
kalkonok és rokon szerkezetű Michael-reakciós akceptorok egyes, a fázis 2
biotranszformációkban részt vevő enzimeket indukálnak. (10; 54; 55) Ezen enzimek,
mint például a glutation-S-transzferáz (GST), az UDP-glükuronil-transzferáz, a
NAD(P)H:kinon-oxidoreduktáz (fázis 1 enzim) a celluláris védekező mechanizmus
meghatározó enzimei, és alapvető szerepet töltenek be a sejtek makromolekuláinak
kémiai módosítására irányuló elektrofil vegyületek reaktivitásának eliminálásában. (56)
Az antioxidáns és citoprotektív enzimek, fehérjék megnövekedett expressziójának
hátterében főként az Nrf2 (nuclear factor erythroid 2-related factor 2) által aktivált
folyamatok állhatnak. Normál fiziológiás körülmények között az Nrf2 a citoplazmában
a Keap1 (Kelch-like ECH-associated protein 1), ciszteinben gazdag, homodimer cink
metalloproteinhez kapcsolódik, és inaktív formában van. (57; 58) Oxidatív stressz
hatására az Nrf2-Keap1 komplex felbomlik, és az Nfr2 aktív formába kerül.
Ugyanakkor lágy elektrofil molekulák, mint az α,β-telítetlen aldehidek és ketonok is,
kovalens kötéssel a Keap1 fehérjék tiolcsoportjaihoz kötődnek, ami az Nrf2-Keap1
komplex bomlásához vezet. A szabad Nrf2 molekulák mennyisége megnő, a sejtmagba
kerülnek, ahol Maf proteinekkel heterodimert képeznek és a promóter szakaszukban

15
EpRE (electrophile response element) részt tartalmazó, a detoxikációs folyamatokban
fontos szerepet betöltő enzimeket kódoló géneket aktiválnak. (59; 60)
A kalkonok kemopreventív hatásában az is szerepet játszhat, ha a vegyület gátolja
azon onkogéneknek, például a c-myc onkogén expresszióját, amelyek megnövekedett
megjelenése hozzájárul a karcinogenezis folyamatához. (61)
Számos xenobiotikum toxikus hatása abból fakad, hogy citokróm P450 1A enzim
aktiválásán keresztül elősegítik promutagének metabolikus aktivációját (10), illetve
megnövelik a reaktív oxigéngyökök mennyiségét, ezzel oxidatív stresszt előidézve.
Főként a hidroxi- és metoxiszubsztituált kalkonszármazékok gátolják a CYP1A
aktivitását, antioxidáns hatással rendelkeznek, valamint pozitív hatással vannak az
intercelluláris kommunikációra (gap junctional intercellular communication, GJIC), így
ígéretes kemoprotektív hatással bíró molekulák. (62)
Az antioxidáns aktivitás hátterében elsősorban a reaktív oxigéngyök-fogó
tulajdonság állhat, ami főként a hidroxiszubsztituált származékok esetén kifejezett. (37)
Az α- és β-szénatom közötti kettőskötés geometriai izomerek jelenlétét feltételezi. Az
(E)-izomer termodinamikai szempontból stabilisabb, szinte minden kalkon ebben a
formában izolálható. A 3-hidroxi-3’-metilkalkon (E)-izomerjének metanolos oldatában
ugyanakkor fény hatására fotoizomerizáció történik, és a (Z)-forma jelenik meg. (63) A
(Z)-izomer antitumor aktivitása nagyobb, mint az eredeti (E)-izomeré, tehát
fotoizomerizáció hatására is megváltozhat a vegyületek biológiai aktivitása.
II.2.3. Gyűrűs kalkonanalógok szintézise és szerkezete
Korábbi kísérleti eredmények szerint a két aromás gyűrű egymáshoz viszonyított térbeli
helyzete alapvetően befolyásolja a kalkonok bioaktivitását. Bár a kalkonok konjugált
molekulák, eléggé flexibilisek ahhoz, hogy oldatban több konformációt is felvegyenek.
A kalkonok vizsgálata így nem teszi lehetővé az optimális térszerkezet és biológiai
hatás közötti kapcsolat vizsgálatát.
Ezen kapcsolat alaposabb megismerése céljából történt a kalkonokkal szerkezeti
rokonságban álló gyűrűs telítetlen karbonilvegyületek szintézise, kémiai
reaktivitásának, elektron- és térszerkezetének, valamint biológiai hatásainak vizsgálata.
(64)

16
A kalkon (1) kevéssé flexibilis analógjainak tekinthetők az (E)-2-benzilidén-1-
indanonok (2), -tetralonok (3) és -benzoszuberonok (4). (10. ábra)
10. ábra A kalkon és gyűrűs származékainak szerkezete
A 2-4 vegyületek és B-gyűrűn szubsztituált származékaik 1-indanon, 1-tetralon és
1-benzoszuberon, valamint a megfelelő aromás aldehid Claisen-Schmidt
kondenzációjával állíthatók elő. (11. ábra)
11. ábra Gyűrűs kalkonszármazékok szintézise Claisen-Schmidt kondenzációval
(1-indanon: n=1; 1-tetralon: n=2; 1-benzoszuberon: n=3)
A vegyületek szerkezetét, térszerkezetét (konfiguráció és konformáció)
elemanalízissel, valamint IR, 1H NMR és
13C NMR módszerekkel igazolták. (64; 65;
66) A gyűrűs kalkonanalógok nukleofil reaktánsokkal szemben mutatott eltérő
reaktivitásának értelmezése céljából Perjési és mtsai megvizsgálták a vegyületek tér- és
elektronszerkezetét. Az infravörös (IR) és 13
C-NMR spektroszkópiai módszerek
segítségével megállapították, hogy az arilidéncsoport elektronküldő, illetve
elektronszívó szubsztituensei hatással vannak a vegyületek telítetlen karbonil (enon)-

17
fragmensének elektroneloszlására, így annak nukelofil reagensekkel szemben mutatott
reaktivitására. (67; 68)
A két aromás gyűrű egymáshoz viszonyított térbeli helyzetén kívül az aromás
gyűrű szubsztituenseinek szterikus, elektronszerkezeti és hidrofób tulajdonságai szintén
fontos szerepet játszanak a biológiai hatás kialakításában.
II.2.4. Gyűrűs kalkonszármazékok citotoxikus hatása és szerkezete
közötti kapcsolat
A különböző szubsztituensekkel bíró gyűrűs kalkonszármazékok in vitro
citotoxicitásának vizsgálata P388, L1210 leukémia sejtekkel, valamint Molt 4/C8 és
CEM T-limfocitákkal szemben történt. (64; 69) A 2-4 sorozat vegyületeinek vizsgált
sejtekkel szemben meghatározott IC50 (a sejtek szaporodásának 50%-os gátlásához
szükséges koncentráció; μM) értékei alapján számított ún. „Átlagos Potenciál” (ÁP,
μM) értékeket az 1. táblázat tartalmazza.
A négy teszt alapján számított ÁP értékek összehasonlítása alapján
megállapították, hogy a 7-tagú gyűrűs (E)-2-(X-benzilidén)-1-benzoszuberon (4)
származékok a leghatékonyabb citotoxikus analógok.
Sorozat P388 sejtek L1210 sejtek Molt 4/C8
limfociták
CEM
limfociták
Indanon (2) 42,9 127 267 155
Tetralon (3) 20,9 135 158 115
Benzoszuberon (4) 11,2 85,2 66 47
1. táblázat A 2-4 sorozat „Átlagos Potenciál” (ÁP; μM) értékei
a vizsgált egér és humán daganatos sejtvonalakkal szemben (64)
Dimmock és mtsai kvantumkémiai számításokkal is meghatározták a
szubsztituálatlan ciklikus kalkonszármazékok energetikailag legkedvezőbb
konformációját az aromás gyűrűk térbeli elhelyezkedésének vizsgálata céljából. (64) Az
öttagú-gyűrűs analóg esetén az arilidéncsoport térbeli elhelyezkedése lényegesen eltér a
másik két vegyületétől, ami alapjául szolgálhat a 2, valamit a 3 és 4 sorozat tagjai eltérő
citotoxikus hatásának.

18
A vegyületek térszerkezete mellett az in vitro citotoxikus hatás nagymértékben
függ a cinnamoil molekulafragmensen található szubsztituens természetétől és
helyzetétől. (2. táblázat) Ezt támasztja alá, hogy minden vizsgált tumorsejtvonallal
szemben a 4-es, vagyis para-helyzetben metoxi-, valamint dimetilamino-szubsztituenst
hordozó 2-(X-benzilidén)-1-benzoszuberonok bizonyultak a sorozat leghatékonyabb
származékainak. (64)
Vegyület (X) P388 sejtek L1210
sejtek
Molt 4/C8
limfociták
CEM
limfociták
H 12,7 106,0 42,7 28,9
4-CH3 11,8 25,0 21,3 11,4
3-Cl 8,2 52,3 23,8 9,44
4-Cl 9,5 51,8 33,0 15,1
4-F 11,1 60,3 53,6 20,0
4-OCH3 1,6 0,34 0,47 0,35
4-N(CH3)2 1,9 1,87 5,25 3,05
melfalán 0,22 2,13 3,24 2,47
2. táblázat Néhány (E)-2-(X-benzilidén)-1-benzoszuberon (4) in vitro IC50 értéke (μM)
(64)
A 4 sorozat szubsztituensei elektronszerkezeti, hidrofób és térbeli
tulajdonságainak vizsgálata során megállapították, hogy a vegyületek P388 és L1210
sejtekkel, valamint humán daganatos sejtekkel szemben mutatott citotoxicitása negatív
korrelációt mutatott az arilidén molekularész szubsztituenseinek Hammett-szigma
értékeivel, ugyanakkor pozitív korrelációt mutatott a szubsztituensek moláris
refraktivitás (MR) értékeivel. (64) A vegyületek citotoxicitása egyetlen vizsgált
sejtvonalon sem mutatott korrelációt a szubsztituensek lipofilitását jellemző Hansch-
féle π-értékkel. Röntgenkrisztallográfiás vizsgálatok támasztották alá, hogy a két
aromás gyűrű által bezárt szög – amit szintén befolyásol a B-gyűrűn levő szubsztituens
jellege – növekedése, vagyis a molekula koplanaritásának csökkenése a citotoxikus
hatás növekedését eredményezi. A legaktívabb 7-tagú gyűrűs származékok (4-OCH3 és
4-N(CH3)2-vegyületek) esetén mért értékek (19° illetve 23°) a hatás szempontjából az
optimális értékeket jelenthetik. Összességében az mondható el, hogy a vizsgált
vegyületek közül a para-helyzetben nagyméretű, elektronban gazdag szubsztituens
hordozó 7-tagú gyűrűs analógok bizonyultak a leghatásosabbnak.

19
Néhány reprezentatív vegyület (4-CH3; 4-OCH3 és 4-N(CH3)2-szubsztituált 2-(X)-
benzilidén-1-benzoszuberon) hatásmechanizmusának előzetes vizsgálati eredményei azt
mutatták, hogy in vitro humán Jurkat-T sejttenyészetben gátolják az RNS- és a
fehérjeszintézist és a sejtek apotózisát váltották ki. A vizsgált koncentrációban (IC80) a
vegyületek RNS- és fehérjeszintézist gátló hatása, valamint azok Jurkat T-sejtekkel
szemben mutatott citotoxicitása között azonban nem állapítható meg korreláció. (64)
II.2.5. Gyűrűs kalkonszármazékok daganatellenes hatása
A kiemelkedő citotoxikus hatást mutató 7-tagú gyűrűs, 4-helyzetben metoxiszubsztituált
kalkonanalóg in vivo daganatellenes hatását vizsgálták Perjési és mtsai. Az
experimentális toxikológiai kísérletek során az egyik gyakran alkalmazott ún.
policiklusos aromás szénhidrogén (PAH) a 7,12-dimetilbenz[α]antracén (DMBA),
amely állatkísérletekben erősen rákkeltőnek bizonyult, és feltételezett humán
karcinogén. A kísérleti állatok DMBA-val történő kezelése emlőtumort, tüdőtumort,
hólyagtumort és leukémiát eredményezhet. (70) Az (E)-2-(4-metoxibenzilidén)-1-
benzoszuberon in vivo daganatellenes hatásának vizsgálata során a vegyület – a rövid
idejű „short-term” (24 és 48 órás) eredmények alapján – hatékonyan csökkentette
onko/szupresszor gének (H-ras, c-myc és p53) DMBA-indukálta expresszióját a kísérleti
állatok (CBA/Ca egér) májában, tüdejében, veséjében és nyirokcsomójában, különösen
akkor, ha azt a DMBA adását megelőzően, vagy azzal egyidőben alkalmazták. (71; 72)
A policiklusos aromás szénhidrogének, így a DMBA is, genotoxikus hatású
karcinogének. A genotoxikus hatás kialakulásának előfeltétele a vegyületek citokróm P-
450 enzimek által katalizált metabolizmusa, melynek eredményeképpen az aromás
szénhidrogének reaktív dihidrodiol-epoxid származékokká alakulnak át, és kémiai
reakcióba lépnek a sejt nukleofil molekuláival, így fehérjékkel és DNS bázisokkal. (73;
74) A DMBA genotoxikus hatásának másik lehetséges mechanizmusa a DMBA
metabolizmusa során keletkező 7-OH-DMBA származék – ami a citoszolban található
szulfotranszferáz enzim szubsztrátja - elektrofil szulfát-észterének keletkezése, ami a
DNS adenin és guanin bázisainak aminocsoportjait támadja meg. (75; 76) Monostory és
mtsai igazolták, hogy az (E)-2-(4-metoxibenzilidén)-1-benzoszuberon DMBA-indukálta
génexpresszióra gyakorolt hatásának kialakulásában szerepet játszhat a vegyületnek a
DMBA-t metabolizáló CYP1A izoenzimre gyakorolt gátló hatása. (77)

20
Ezen kísérleti tapasztalatok alapján a vizsgált metoxiszármazék, valamint
analógjai a policiklusos aromás szénhidrogének okozta karcinogenezissel szemben
alkalmazható kemopreventív szerek lehetnek.

21
III. Célkitűzések
Az irodalmi adatok, valamint a korábbi kísérleti eredmények arra engednek
következtetni, hogy a kalkonok (1) és gyűrűs kalkonszármazékok (2-4) ígéretes
kiindulási anyagok lehetnek a citotoxikus és/vagy citoprotektív hatással rendelkező
vegyületek kutatásában.
A számos, különbözőképpen szubsztituált és eltérő gyűrűtagszámú kalkonanalóg
előzetes, in vitro sejtproliferációt gátló hatásának vizsgálata során nyilvánvalóvá vált,
hogy a vegyületek biológiai hatása nagymértékben függ a gyűrűtagszámtól, valamint a
szubsztituens pozíciójától és jellegétől, ami a vegyület elektron- és térszerkezetét
alapvetően befolyásolja. Munkánk során fő célul a gyűrűs kalkonszármazékok fizikai-
kémiai tulajdonságainak és a citotoxikus/citoprotektív hatás hátterében álló molekuláris
mechanizmusoknak alaposabb megismerését tűztük ki.
Az elektron- és térszerkezeti jellemzők mellett a vegyületek biológiai hatásának, illetve
hasznosíthatóságának fontos paramétere a molekulák lipofilitása, melynek számszerű
jellemzésére leggyakrabban a vegyületek oktanol-víz megoszlási hányadosának
logaritmusát (logP) használják. (78) A logP meghatározásának több kísérleti módszere
is ismeretes az irodalomban. (79) Célunk az 1-4 sorozat különböző szubsztituenseket
hordozó tagjai logP értékének meghatározása egy korábban beállított, ún. „helyettesítő”
módszer, alapvetően fordított fázisú vékonyrétegkromatográfiás technika (RP-TLC)
alkalmazásával. (80) A logP értékek ismeretében a molekulák lipofilitása és szerkezete
közötti esetleges összefüggéseket kívánjuk vizsgálni.
A gyűrűs kalkonszármazékok citotoxikus és daganatellenes hatásának kialakulásában
szerepet játszó mechanizmusok alaposabb megismerése céljából a korábbi in vitro
toxicitásvizsgálatok során kiemelkedő hatást mutató 7-tagú gyűrűs (E)-2-(4-
metoxibenzilidén)-1-benzoszuberont (4a) és kevéssé toxikus 4-metilszubsztituált
analógját (4b) választottuk a tervezett vizsgálatok elvégzéséhez. (12. ábra)

22
12. ábra A vizsgált kalkonszármazékok szerkezete
Munkánk során az alábbi célokat tűztük ki:
1. A vizsgált vegyületek in vitro sejtszaporodást gátló hatásának vizsgálata humán
Jurkat T sejtekkel szemben a vegyületek közel ekvitoxikus dózisával történő
kezelést követően, tripánkékkel történő festési eljárással. Az élő, a korai
apoptotikus fázisban levő és az apoptotikus/nekrotikus sejtek
mennyiségének/arányának meghatározása áramlási citometriai módszerrel.
2. A vegyületek sejtosztódásra, sejtciklusra gyakorolt hatásainak áramlási citometriai
módszerrel történő vizsgálata Jurkat T sejtekben, a vegyületekkel történő 8, 24 és
48 órás kezelést követően.
3. A vizsgált kalkonanalógok in vitro antioxidáns (hidroxilgyök „scavenger”),
illetőleg esetleges prooxidáns hatásának vizsgálata, annak időbeli függése a Fenton-
reakció inicializálta dezoxiribóz degradációs teszt alkalmazásával. A vizsgálatok
során kísérletet kívánunk tenni a vegyületek hidroxilgyökökkel lejátszódó
reakciójában esetlegesen képződő termékek azonosítására is.
4. A vegyületek antioxidáns/prooxidáns jellegének tanulmányozása céljából sejtes
körülmények között, Jurkat T sejtekben is vizsgáljuk az analógok ROS (reaktív
oxigénszármazék)-aktivitásra, vagyis a celluláris redox-státuszra gyakorolt hatását.
A vizsgálatokhoz a diklorofluoreszcein-tesztet kívánjuk alkalmazni.
5. A mitokondriális funkciók alapvetően befolyásolják a sejtek működését.
Xenobiotikumok citotoxikus és antineoplasztikus aktivitásához hozzájárulhat a
mitokondriális funkciók megváltozása. Vizsgálni kívánjuk a kalakonanalógok
mitokondriális respirációra és ATPáz-aktivitásra gyakorolt hatását.

23
6. Az élő sejtek redox-homeosztázisát alapvetően meghatározza a sejt SH-státusza.
Célunk a vegyületek SH-szintre, majd glutation-szintre gyakorolt hatásának
vizsgálata Jurkat T sejtekben. A redox-státusz jellemzésére a sejtek redukált és
oxidált glutation arányának, illetve azok mennyiségének a meghatározása is
szükséges.
7. A vegyületek kémiai szerkezete alapján feltételezhető, hogy tiolcsoportokkal
konjugációs reakcióba is léphetnek, ezért vizsgálni kívánjuk a vegyületek redukált
glutationnal szemben mutatott reaktivitását, mind sejtmentes, mind sejtes
körülmények között. Az esetlegesen képződő konjugátumok jelenlétét
vékonyrétegkromatográfiás módszerrel, ún. „kettős-előhívást” alkalmazva kívánjuk
kimutatni. Az esetleges pozitív reakciót követően RP-HPLC-hez csatolt
tömegspektrometriás eljárással kivánjuk a létrejövő konjugátumok szerkezetét
igazolni.

24
IV. Vizsgálati módszerek
IV.1. Kalkonszármazékok logP értékeinek meghatározása
A 1-4 sorozat különféle szubsztituensekkel bíró tagjai logP értékének meghatározása
egy korábbi, a Semmelweis Egyetem Gyógyszerészi Kémiai Intézetével kialakult
kooperációs munka során alkalmazott fordított-fázisú vékonyréteg-kromatográfiás
technika alkalmazásával történt. (80)
IV.1.1. Vizsgált anyagok
A vizsgált vegyületek az 1-4 sorozatok tagjai, szintézisük és tisztításuk korábban leírtak
szerint történt. (64) A kalibrációs sorozat tagjainak szerkezete a 3. táblázatban látható.
(81) A módszer validálásához használt progeszteron és diazepam gyógyszerkönyvi
minőségűek, a prosztaglandin E1-etilésztert (PGEE) a Sanofi-Synthelabo-Chinoin Zrt.
(Budapest, Hungary) bocsátotta rendelkezésünkre. A vizsgálatokhoz használt metanol
és n-oktanol HPLC minőségűek, a többi vegyszer analitikai tisztaságú.
R1 R2 R3
CH3 CH2-C6H5 H
CH2- CH2- CH2- CH2- CH2 H
H H CH3
H C6H5 CH3
H C6H5 H
CH3 C2H5 H
3. táblázat A logP meghatározáshoz használt kalibrációs sorozat tagjainak szerkezete

25
IV.1.2. Fordított-fázísú vékonyréteg-kromatográfiás módszer
A korábbi meghatározások (80) folytatásaként 29 kalkonszármazék vizsgálata történt. A
meghatározáshoz szilanizált szilikagél 60F254 (20 cm x 20 cm) rétegeket (Merck,
Germany) használtunk. A vizsgálatok előtt a rétegeket metanollal mostuk, szárítottuk,
majd 1 órán keresztül 160 °C-on aktíváltuk. A vizsgálandó anyagokat, valamint a
kalibrációs és validációs sorozat tagjait metanol-kloroform 1:1 arányú elegyében
oldottuk (2 mg/ml), majd vittük fel a lemezre. Mozgófázisként metanol-víz (60+40)
elegyet használtuk. A rétegek kifejlesztése előtt a szűrőpapírral bélelt futtató kádakat fél
órán át telítettük a mozgófázissal. A kifejlesztést (150 mm) követően a foltok előhívása
UV-fényben (λ = 254 nm), valamint jód-gőzben történt. Minden mérési eredmény
három párhuzamos meghatározásból származik.
A para-szubsztituált vegyületek logPCLOGP értékének számolásához a Chem3D
Ultra 9.0.1. szoftver (CambridgeSoft, Cambridge, MA, USA) CLOGP alkalmazását
használtuk.
IV.2. Gyűrűs kalkonszármazékok hatásmechanizmusának
vizsgálata
IV.2.1. Kísérleti anyagok és sejtkultúra
A vizsgálni kívánt gyűrűs kalkonszármazékok (4a és 4b) szintézise és tisztítása a
korábban leírtak szerint történt. (64) 4a vegyület összegképlete: C19H18O2, moláris
tömege: 278,35 g/mol. 4b vegyület összegképlete: C19H18O, moláris tömege:
262,35 g/mol. Mindkét származék halványsárga, tűkristályos vegyület, amely
metanolban jól oldódik.
A használt vegyszerek mindegyike analitikai tisztaságú volt, és – amennyibben az
külön nincs jelezve – beszerzése a Sigma-Aldrich (Budapest, Hungary) cégtől történt.
A humán Jurkat T limfocita sejtek (ATCC, E6-1 klón) az LGC Promochemtől
(Teddington, UK) származnak. A Jurkat T sejtek tenyésztése 37 °C-on, megfelelő pára-
és 5% CO2-tartalmú környezetben, RPMI 1640 tápoldatban (pH = 7,4) történt, amit
kiegészítettünk 10% FCS-el (magzati borjú szérum, Gibco), penicillinnel (100 U/ml) és
sztreptomicinnel (100 μg/ml).

26
IV.2.2. Sejtkezelési protokoll
A Jurkat T sejtek kezelése (5 x 105 db sejt / 1,0 ml RPMI 1640 tápoldat) a 4a (5,6 μM),
illetve 4b (23,3 μM) vegyületekkel a vizsgálati módszereknek megfelelően 1, 4, 8, 24
illetve 48 órán keresztül történt. A koncentrációértékek az előzetesen meghatározott
IC80 dózisértékeknek felelnek meg. (64) A kalkonszármazékokat DMSO-ban oldottuk,
és az elkészült DMSO-oldatból 10 μl-t adtunk 1,0 ml sejteket tartalmazó tápoldathoz,
így mindig 1% DMSO-t tartalmazott a kezelt sejtszuszpenzió. A kontroll
sejtszuszpenzókat nem kezeltük, illetve minden esetben készítettünk egy olyan kontrollt
is, ami csak 1% DMSO-t tartalmazott, kalkon nélkül. (Jelölése: DMSO 1%)
IV.2.3. „Túlélés”-vizsgálat
A Jurkat T sejtek kezelése (5 x 105 db sejt / 1,0 ml RPMI 1640 tápoldat) a 4a (5,6 μM),
illetve 4b (23,3 μM) vegyületekkel a protokoll (IV.2.2) szerint történt. A 8, 24 és 48
órás sejtkezelést követően az összes sejt, valamint az életképes sejtek számolása a sejtek
tripánkékkel történő festését követően hemocitométer használatával történt. (82) Az
eredmények négy párhuzamos mérésből származnak.
IV.2.4. Áramlási citometriai módszerek
IV.2.4.1. Az áramlási citometria
Az áramlási citometria egy olyan eljárás, amely alkalmas vegyes sejtpopulációkban az
egyes sejtcsoportok elkülönítésére azok nagysága és a sejtalkotók összetettsége
(granularitás) alapján, valamint egy-sejt szinten, több hullámhosszon mérve, azok
fluoreszcens jelének detektálására. (83)
A vizsgált sejtszuszpenziót a készülékben a köpenyfolyadék egy kapillárisban, az
ún. áramlási cellában áramoltatja. A kapillárisban haladó sejteket a készülék nagyságtól
függetlenül számlálja. Ezzel párhuzamosan hagyományos fényforrással megvilágítva a
sejteket kétféle jelet detektálunk: a besugárzó fény irányával megeegyező irányú
fényszóródás (forward scattered light, FSC) a sejtek nagyságáról ad információt, a fény
oldalirányú szóródása (side scattered light, SSC) pedig a sejtek granularitásával
arányos. A jeleket két detektor érzékeli és alakítja digitális jellé, amit egy számítógépes
program diagrammon jelenít meg.

27
Amennyiben a vizsgálandó sejt valamely felszíni vagy citoplazmatikus
markeréhez/markereihez közvetlenül, vagy közvetett módon (monoklonális
ellenanyag(ok)hoz kötve) fluoreszcens festéket kötünk, úgy a készülékben elhelyezett
lézerek (számuktól és a gerjesztő fényük hullámhosszától függően) alkalmasak az adott
fluoreszcens festékmolekulák gerjesztésére. A fluoreszcens festékek fényemisszióját
detektorok mérik adott hullámhosszakon (FL-1, FL-2, FL-3, FL-4 csatornákon). A jelet
átalakítva és ábrázolva lehetőség nyílik egy-sejt szinten egyszerre több molekula
fluoreszcens jelének detektálására.
A módszer nagy előnye, hogy rövid idő alatt nagyszámú (105-10
6 db) sejtről
kapunk többféle információt.
IV.2.4.2. Sejtszaporodásra gyakorolt hatás vizsgálata
A korai apototikus fázisban levő, illetve apoptotikus sejtek mennyiségének
meghatározása a következők szerint történt. A Jurkat T sejteket (5 x 105 db sejt / 1,0 ml
tápoldat) a 4a (5,6 μM), illetve 4b (23,3 μM) vegyületekkel a protokoll (IV.2.2) szerinti
8, 24 és 48 órás kezelését és centrifugálását (1000g, 5 perc) követően kétszer mostuk
1,0 ml hideg PBS-pufferben, majd 1,0 ml annexin-kötő pufferben (10 mM
Hepes/NaOH; pH = 7,4; 140 mM NaCl; 2,5 mM CaCl2). A sejteket 100 μl annexin-kötő
pufferben vettük fel és 2 μl fluoreszcein-izotiocianáttal (FITC) jelölt annexin V-oldattal
(Annexin V-FITC) (BD Biosciences) és 10 μl 50 μg/ml koncentrációjú propídium-jodid
(PI)-oldattal inkubáltuk szobahőmérsékleten, fénytől védve, 15 percig. Az inkubációs
idő lejárta után a mintákat 400 μl annexin-kötő pufferrel higítottuk, majd 30 percen
belül vizsgáltuk áramlási citométerrel. A 48 órán keresztül kezelt sejtek jelölése az
előzőkhöz hasonlóan, de csak propídium-jodiddal történt.
Az áramlási citometriás mérést és analízist a FACSCalibur (Becton Dickinson
Biosciences, San Jose, CA) áramlási citométeren a CellQuest (BD Biosciences)
software segítségével végeztük. A vizsgálatkor nem használtunk kapuzást.
Az annexin V-FITC-el jelölt sejteket (zöld fluoreszcencia) az FL-1 csatornán
(525 nm), a propídium-jodiddal jelölt sejteket (piros fluoreszcencia) az FL-2 csatornán
(575 nm) detektáltuk. Mintánként 50 000 eseményt mértünk. A két emissziós spektrum
átfedésének kiküszöbölésére elektronikus kompenzációt használtunk. A kezeletlen
kontroll sejtek esetén beállított kvadráns statisztikai paramétereket használtuk a további
vizsgálatokhoz. Az FL-1/FL-2 csatornákon mérhető fluoreszcencia intenzitás alapján

28
megkülönböztethetünk élő sejteket (annexin V-negatív + PI-negatív), korai apoptotikus
fázisban levő sejteket (annexin V-pozitív + PI-negatív) és apototikus (vagy nekrotikus,
azaz elpusztult) sejteket (annexin V-pozitív + PI-pozitív). (84; 85) Az eredmények négy
párhuzamos mérésből származnak.
IV.2.4.3. Sejtciklus-vizsgálat
A kalkonanalógok sejtciklusra gyakorolt hatásának vizsgálatához a Jurkat T sejteket (5
x 105 db sejt / 1,0 ml tápoldat) a 4a (5,6 μM), illetve 4b (23,3 μM) vegyületekkel a
protokoll (IV.2.2) szerinti 8, 24 és 48 órás kezelését és centrifugálását (1000g, 5 perc)
követően kétszer mostuk 1,0 ml hideg PBS-pufferben, majd 300 μl 4% paraformalehid-
oldattal fixáltuk 20 percen keresztül. Ezt követően a sejteket kétszer mostuk 2,0 ml
PBS-pufferben, majd 1,0 ml szaponin-pufferben (PBS/0,1% BSA/0,1% szaponin),
végül 300 μl propídium-jodid-oldatban (50 μg/ml PI; 100 Kunitz units RNase A/ml
szaponin pufferben) vettük fel. 30 perc inkubálási időt követően az áramlási citometriás
mérést és analízist a FACSCalibur (BD Biosciences) áramlási citométeren a CellQuest
(BD Biosciences) software segítségével végeztük.
A jelölt sejtek DNS-mennyiségének meghatározása az FL-2 csatornán (575 nm)
történt, lineáris erősítés mellett. Mintánként 10 000 eseményt mértünk. A kezeletlen
kontroll sejtek esetén beállított kapukat használtuk a további vizsgálatokhoz. Az
eredmények négy párhuzamos mérésből származnak.
IV.2.5. A vegyületek in vitro antioxidáns hatásának vizsgálata
dezoxiribóz degradációs módszerrel
A vizsgált kalkonanalógok in vitro antioxidáns (hidroxilgyök scavenger), esetleges
prooxidáns hatásának vizsgálatához a Fenton-reakció inicializálta dezoxiribóz
degradációs tesztet alkalmaztuk. (86; 87)
30 mM foszfát-puffer (pH=7,4), 9 mM 2-dezoxi-D-ribóz, 40 mM nátrium-klorid,
30 µM vas(II)-szulfát-oldat elegyéhez adtuk a 4a, illetve 4b vegyületet az össztérfogat
1%-nak megfelelő térfogatú DMSO-ban oldva, valamint 50 µM H2O2-ot. A kalkonok
végkoncentrációja 200 µM volt. A vizsgálatokat EDTA hozzáadása nélkül, illetve
EDTA (36 μM) jelenlétében is elvégeztük. Az elegyeket jól záró kémcsövekben 37 oC-
on inkubáltuk, majd a degradációs reakció időbeli követéséhez a 20 percenként (4 órán

29
át) vett mintákban meghatároztuk a tiobarbitursav (TBA)-reaktív anyagok mennyiségét.
A kontroll-oldat a kalkonok DMSO-val készült oldata helyett csak a vizsgált anyagok
oldószereként használt DMSO-t tartalmazta.
A keletkezett TBA-reaktív anyagok meghatározása céljából az inkubátumok 1,0
ml térfogatához 0,5 ml tiobarbitursav-reagenst (1% (w/v) TBA 50 mM NaOH-ban
oldva) és 0,5 ml 5,6%-os (w/v) triklórecetsav-oldatot adtunk, majd az elegyet jól záró
kémcsőben 8 percig 100 oC-os glicerines fürdőben inkubáltuk. A szobahőmérsékletre
hűtött oldat abszorbanciáját 532 nm-en mértük, a megfelelő vak-oldat hasonló
körülmények között derivatizált oldatával szemben. A vak-oldat összetétele minden
esetben megegyezett a vizsgált minta összetételével, de nem tartalmazott vas(II)-
szulfátot és hidrogén-peroxidot. Minden oldat frissen készült, kétszeresen desztillált víz
felhasználásával. Minden mérési eredmény három párhuzamos kísérletből származik.
IV.2.6. A kalkonanalógok celluláris redox-homeosztázisra gyakorolt
hatásának vizsgálata
IV.2.6.1. Az intracelluláris ROS-aktivitás meghatározása
A vizsgált kalkonszármazékok intracelluláris reaktív oxigéngyök (ROS) termelésre
gyakorolt hatását az oxidációra érzékeny 2’,7’-diklórfluoreszcein-diacetátot (DCFH-
DA) felhasználva vizsgáltuk. (88; 89)
A Jurkat T sejteket (5 x 105 db sejt / 1,0 ml tápoldat) a 4a (5,6 μM), illetve 4b
(23,3 μM) vegyületekkel a protokoll (IV.2.2) szerinti 1, illetve és 4 órás kezelését és
centrifugálását (1000g, 5 perc) követően 1,0 ml hideg PBS-pufferben mostuk, majd 1,0
ml PBS-pufferben vettük fel. A sejteket 10 μM DCFH-DA-val, 37 °C-on 20 percig
sötétben inkubáltuk, majd kétszer 1,0 ml PBS pufferrel mostuk és 1,0 ml pufferbe
reszuszpendáltuk. A diklórfluoreszcein fluoreszcencia-intenzitásának meghatározását
FACSCalibur (BD Biosciences) áramlási citométeren a CellQuest (BD Biosciences)
software segítségével végeztük. A mérés az FL-1 csatornán történt, lineáris erősítés
mellett. Mintánként 10 000 eseményt mértünk.
Az eredmények a minták, és a kezeletlen kontroll sejtek fluoreszcencia
intenzitásának összehasonlításából származnak. Minden érték négy párhuzamos
mérésből származó átlag ± szórás. A statisztikai értékelés a Student-t teszt (p < 0,05)
használatával történt.

30
IV.2.6.2. Mitokondriális funkciókra gyakorolt hatás vizsgálata
A kalkonszármazékok mitokondriális funkciókra gyakorolt hatásainak vizsgálatát a
kassai P. J. Šafarik Egyetem Orvosi Kémiai és Biokémiai Intézetével kooperációban
végeztük.
A kísérletekhez hím Wistar patkányok (250-300 g) (Velaz, Praha, Czech
Republic) májából, Johnson és Lardy módszere alapján izolált mitokondriumot
használtunk. (90) A mitokondriális fehérjemennyiség meghatározása Lowry módosított
módszere szerint történt. (91)
Az izolált mitokondrium oxigénfogyasztásának mérése polarográfiás módszerrel,
Clark-típusú oxigénelektróddal (WTW oxi 325 (Germany)), 25°C-on, ún. respirációs
médiumban történt. A respirációs médium összetétele: 80 mM KCl, 300 mM KH2PO4,
300 mM K2HPO4, 15 mM Tris-HCl, 6 mM MgCl2, 0,78 mM EDTA desztillált vízben
oldva; pH = 7,4. A respirációs médiumhoz adtunk 20 mg/ml koncentrációban
mitokondrium-szuszpenziót, respirációs szubsztrátként 50 mM nátrium-szukcinátot, 0,5
mM ADP-t, valamint a vizsgált kalkonszármazékot, melynek mennyisége 70
nmol/mitokondriális fehérje mg. Az oxigénfogyasztás (nmol O2 x min-1
x mg-1
fehérje),
valamint az ATPáz aktivitás meghatározása 1 órás inkubáció után történt. (92) A
respirációs kontroll-hányados (RCR; respiratory control ratio) az „oxigénfogyasztás a 3-
as állapotban (state 3)” (ADP jelenlétében) és az „oxigénfogyasztás a 4-es állapotban
(state 4)” (ADP hozzáadása előtt) hányadosa. A vegyületek ATPáz-aktivitásra gyakorolt
hatásának vizsgálata Meissner szerint történt. (93) Minden érték három párhuzamos
mérésből származó átlag ± szórás. A statisztikai értékelés a Student-t teszt (p < 0,05)
használatával történt.
IV.2.6.3. A celluláris tiolszint meghatározása
A kalkonszármazékok celluláris tiol-szintet befolyásoló hatását vizsgáltuk Jurkat T
sejteken, tápanyag-mentes és tápanyagokat tartalmazó környezetben. A mért SH-szintet
a minták fehérjemennyiségére vonatkoztattuk.
A 4a (5,6 μM), illetve 4b (23,3 μM) vegyületekkel kezelt Jurkat T sejteket (2 x
106 db sejt) egyrészt 1,0 ml PBS-pufferben inkubáltuk (tápanyagmentes környezet) 1
órán át, másrészt 1,0 ml RPMI 1640-tápoldatban (10% FCS) 4 és 8 órán át. A kezelést
és centrifugálását (1000g, 5 perc) követően a sejteket kétszer mostuk 0,9%-os NaCl-

31
oldatban, majd 500 μl TRIS-pufferben (50 mM Trisz-HCl; 0,1% SDS; pH = 7,4) vettük
fel és ultrahangos szonikáció (3 perc) révén lizáltuk. A sejtlizátumokat centrifugáltuk
(13 000g, 5 perc), a tiszta felülúszót használtuk a tiol-szint, illetve a fehérjemennyiség
meghatározásához.
A szabad celluláris SH-szintet a DTNB- (5,5’-ditiobis(2-nitrobenzoesav))
módszer szerint határoztuk meg, redukált glutation használva standardként. (94) A
fehérjekoncentráció meghatározása a BCA (bicinkoninsav)-módszer alkalmazásával
történt. (95) A fehérjeméréshez BSA (bovine serum albumine) standard oldatsorozatot
használtunk.
Mindkét meghatározás esetén Dynatech MR 7000 microplate (ELISA) reader-t
használtunk abszorbancia mérésre. Minden érték négy párhuzamos mérésből származó
átlag ± szórás. A statisztikai értékelés a Student-t teszt (p < 0,05) szerint, SPSS 12.0 for
Windows szoftver használatával történt.
IV.2.6.4. A celluláris redukált és oxidált glutationszint vizsgálata
A kalkonszármazékok celluláris glutation-szintet befolyásoló hatását vizsgáltuk Jurkat
T sejteken, ami magába foglalja a redukált GSH-szint, valamint az oxidált glutation
(GSSG)-szint meghatározását. A mért glutation-szinteket a minták fehérjemennyiségére
vonatkoztattuk.
A Jurkat T sejteket (2 x 106 db sejt / 5,0 ml RPMI 1640 tápoldat) 4a (5,6 μM),
illetve 4b (23,3 μM) vegyületekkel a IV.2.2 protokoll szerint 4 órán át kezeltük. Az
inkubálást és centrifugálását (1000g, 5 perc) követően a sejteket kétszer mostuk 0,9%-
os NaCl-oldatban, majd 400 μl TRIS-pufferben (50 mM Trisz-HCl; 0,1% SDS; pH =
7,4) vettük fel és 3 egymást követő „freeze-and-thaw ciklust” alkalmazva (fagyasztás -
80 °C-on 30 percig, majd lassú olvasztás szobahőmérsékleten) lizáltuk. A
sejtlizátumokat centrifugáltuk (13 000g, 5 perc), a tiszta felülúszót használtuk a
fehérjemennyiség, illetve a glutation-szintek meghatározásához.
A fehérjemennyiség meghatározása a BCA (bicinkoninsav)-módszer szerint, BSA
standard oldatsorozat felhasználásával történt. (95) Abszorbancia mérésre Dynatech MR
7000 microplate (ELISA) reader-t használtunk.
A redukált és oxidált glutation-szint meghatározása Kand’ár és mtsai által
kidolgozott módszer kisebb változtatása szerint történt. (96) A minták
fehérjementesítéséhez a sejtfelülúszókat 6%-os perklórsavval elegyítettük (300 μl

32
felülúszó + 210 μl HClO4-oldat ), majd a kicsapódott fehérjét centrifugálással (10 000g,
4 perc) eltávolítottuk. A mintákat 1 M KOH-oldattal semlegesítettük, a kicsapódott
KClO4 csapadékot centrifugálás révén (3000g, 3 perc) eltávolítottuk. Az itt kapott
felülúszóból történt a redukált és oxidált glutation-szint meghatározása.
A redukált glutation-szint (GSH) meghatározásához 100 μl mintát 1,0 ml 0,1%
EDTA-t tartalmazó 0,1 M foszfát-pufferrel (pH = 8,0) hígítottuk. Ennek 20 μl-éhez 300
μl foszfát-puffert (0,1 M, pH = 8,0, 0,1% EDTA) és 20 μl metanollal készült 0,1%-os
orto-ftálaldehid-oldatot (OPA) adtunk. A mintákat szobahőmérsékleten, 15 percig
sötétben inkubáltuk, majd a vizsgálatig 4 °C-on tároltuk.
Az oxidált glutation-szint meghatározásához (GSSG) 200 μl mintát 200 μl 40 mM
N-etilmaleimid-oldattal (NEM), 25 °C-on, 25 percig sötétben inkubáltuk, majd 750 μl
0,1 M NaOH-oldattal hígítottuk. A hígított minta 20 μl-éhez 300 μl 0,1 M NaOH-
oldatot és 20 μl metanollal készült 0,1%-os OPA-oldatot adtunk. A mintákat
szobahőmérsékleten, 15 percig sötétben inkubáltuk, majd a vizsgálatig 4 °C-on tároltuk.
Referenciaként frissen készült, TRIS-pufferben (50 mM Trisz-HCl; 0,1% SDS;
pH 7,4) oldott, redukált és oxidált glutationt tartalmazó standard sorozatot használtunk.
Az minták GSH és GSSG-tartalmát az OPA-val történő derivatizálást követően
nagyhatékonyságú folyadékkromatográfiás módszerrel határoztuk meg. A vizsgálatokat
Agilent 1100 HPLC-készüléken, fluoreszcens detektálás mellett végeztük. Az
elválasztást Poroshell 120 EC-C18 oszlopon (150 x 4,1 mm; szemcseméret: 2,7 μm)
végeztük (előtétoszlop: TR-C-160-K1, ABLE&Jasco), 0,4 ml/perc áramlási sebesség
mellett. Az injektált térfogat 10 μl volt. Az izokratikus elválasztáshoz használt eluens
összetétele: 15% metanol és 85% 25 mM foszfát-puffer (pH 6,0). A fluoreszcens jel
intenzitásának detektálásakor az excitációs hullámhossz 350 nm, az emissziós
hullámhossz 420 nm volt. Az adatok gyűjtését és a kromatogramok értékelését Agilent
ChemStation (Rev. A. 10. 02) szoftver segítségével végeztük.
A kezeletlen és kalkonokkal kezelt minták glutation-koncentrációjának
meghatározása a standard sorozat mintáinak meghatározása során felvett kalibrációs
egyenesek segítségével történt. (GSH-standard: y(csúcs alattti terület) = 7,298x + 60,69
(μM) (r2 = 0,989); GSSG-standard: y(csúcs alattti terület) = 297,96x + 21,28 (μM) (r
2 =
0,995))
A meghatározott redukált és oxidált glutation-szinteket a minták
fehérjemennyiségére vonatkoztattuk. Minden étrék öt párhuzamos mérésből származó
átlag ± szórás. A statisztikai értékelés a Student-t teszt (p < 0,05) használatával történt.

33
Annak vizsgálatára, hogy a NEM valóban megvédi-e a GSH-t az oxidációtól, a
redukált glutation standard oldatát, a NEM-es reakciót követően, a GSSG-szint
meghatározásához használt módszer szerint derivatizáltuk és vizsgáltuk a
kromatográfiás rendszerben.
IV.2.7. A kalkonszármazékok glutationnal való reaktivitásának
vizsgálata
IV.2.7.1. A kalkon-glutation reakció vizsgálata sejtmentes körülmények
között
A vizsgált kalkonszármazékok és redukált glutation (GSH) között esetlegesen
lejátszódó spontán reakciót vizsgáltuk.
A glutation 0,01M foszfát-pufferrel (pH = 5,5; pH = 7,4 és pH = 9,0) készült
oldatát ugyanakkora mennyiségű 4a és 4b frissen készült metanolos oldatával
elegyítettük. A GSH és kalkonok végkoncentrációja 5 x 10-3
M volt. A reakcióelegyeket
37 °C, illetve 50 °C-os vízfürdőben inkubáltuk és a reakcióelegy összetételét 1 és 3 órát
követően vékonyréteg-kromatográfiás (TLC) módszerrel vizsgáltuk.
A TLC-futtatáshoz Kieselgel F254 (Merck, Germany) lapokat használtunk
állófázisként. A kifejlesztő elegy összetétele: n-butanol : ecetsav : víz (40 : 10 : 20,
v/v%). A kromatogram előhívása egyrészt UV-fényben (λ = 254 nm) történt, majd
ninhidrin-előhívó oldattal történő bepermetezés révén.
Az kalkon-GSH inkubátumok, valamint a tiszta 4a, 4b és GSH-oldatok VRK
futtatásakor kapott foltok Rf értékei a következők: GSH: Rf = 0 (UV negatív, ninhidrin
pozitív), 4a: Rf = 0,95 (UV pozitív, ninhidrin negatív), „4a-GSH konjugátum”: Rf = 0,42
(UV pozitív, ninhidrin pozitív), 4b: Rf = 0,96 (UV pozitív, ninhidrin negatív), „4b-GSH
konjugátum”: Rf = 0,46 (UV pozitív, ninhidrin pozitív).
IV.2.7.2. A kalkon-glutation reakció vizsgálata Jurkat T sejtekben HPLC-
MS módszerrel
A Jurkat T sejtekben esetlegesen lejátszódó kalkon-glutation konjugációs reakció
vizsgálata céljából a PTE ÁOK Biokémiai és Orvosi Kémiai Intézetével kooperációban
HPLC-MS módszert dolgoztunk ki.

34
A Jurkat T sejteket (2 x 106 db sejt / 5,0 ml tápoldat) a 4a (5,6 μM), illetve 4b
(23,3 μM) vegyületekkel a protokoll (IV.2.2) szerint 4 órán inkubáltuk. A kezelést és
centrifugálását (1000g, 5 perc) követően a sejteket kétszer mostuk 0,9%-os NaCl-
oldatban, majd 600 μl TRIS-pufferben (50 mM Trisz-HCl; 0,1% SDS; pH 7,4) vettük
fel. A sejtlízist ultrahangos szonikáció (3 perc) mellett végeztük. A sejtlizátumokat
centrifugáltuk (13 000g, 5 perc), a tiszta felülúszót és az üledéket -80 °C-on tároltuk a
további vizsgálatokig.
A sejtes üledék feltárását nagy energiájú ultraszonikátorban (UIS250V) (6 x 10
másodperc) végeztük, jeges hűtést alkalmazva a ciklusok között. A mintákat vortexeltük
és centrifugáltuk (10 000g, 10 perc).
A minták fehérjementesítéséhez a felülúszókat és a feltárt üledékeket centrifugális
szűrőbe (Amicon Ultra with 10K „cut off” membrane, Merck-Millipore, Germany)
tettük, és centrifugáltuk (10 000g, 30 perc). A filtrátumok fehérjetartalmát ellenőriztük,
és amennyiben fehérjét találtunk a mintában, a szűrést új szűrőn ismételtük. A
fehérjementes mintákhoz 0,1% hangyasavat adtunk a HPLC-MS vizsgálatokhoz.
A vizsgálatokat Ultimate 3000 (Dionex, Sunyvale, USA) mikro HPLC-készüléken
és Bruker micro-TOF (Bruker Daltonics, Bremen, Germany) tömegspektrométeren
végeztük. A készülékek ellenőrzése Hystar 3.2 (Bruker Daltonics, Bremen, Germany)
és Bruker micro-TOF control 1.3 szoftverekkel történt. Az adatok gyűjtését és a
spektrumok értékelését Brucker Data analysis 4.1 szoftver segítségével végeztük. Az
elválasztást Kinetex C-18 oszlopon (150 x 2,1 mm; szemcseméret: 2,6 μm)
(Phenomenex, Torrance, USA) végeztük, 0,2 ml/perc áramlási sebesség mellett. Az
injektált térfogat 1 μl volt.
A grádiens elválasztáshoz „A” eluenst (99,9% metanol + 0,1% trifluorecetsav) és
„B” eluenst (99,9% víz + 0,1% trifluorecetsav) használtunk. A grádiens-profil a
következő volt: 0-5 percig 40% „A” eluens + 60% „B” eluens; 5-15 percig lineáris
grádiens 40% → 90% „A” eluens + 60% → 10% „B” eluens; 15-20 percig 90% „A”
eluens + 10% „B” eluens.
A tömegspektrometrás mérés elektrospray-ionizáció (ESI) mellett történt. ESI
pozitív ionizációs módot, N2 ütközőgázt (áramlás: 8 dm3/perc, 201 °C) használtunk 3,8
kV véglemez potenciál mellett. Porlasztógáz nyomása 20 psi. A spektrumokat 50 –
2.000 tömeg/töltés (m/z) tartományban értékeltük.

35
V. Eredmények és következtetések
V.1. Kalkonanalógok lipofilitásának jellemzése
A lipofilitás a biológiailag aktív molekulák egyik legfontosabb, és igen régóta
használatos fizikai-kémiai paramétere, amely, mint anyagi tulajdonság, a vegyületek
apoláris (lipofil) környezethez való affinitását jellemzi. A lipofilitás az a tulajdonság,
amely nagyban befolyásolja egy vegyület felszívódását, eloszlását, fehérjekötődését,
kiürülését, másrészt a receptorral való kölcsönhatását is, tehát jelentős mértékben
meghatározza egy molekula sorsát a szervezetben. Mindezek alapján a
gyógyszertervezés kiemelt paramétere is, hiszen változtatásával befolyásolhatók az
ADME tulajdonságok. (97; 98) Egy molekula lipofilitását számszerű adatokkal
jellemezhetjük, aminek kialakításában azok a kölcsönhatások játszanak szerepet,
amelyek a szervezetben is létrejönnek a molekula és környezete között.
Egy megoszlási folyamat során a vegyület megoszlik két egymással nem
elegyedő, de bőségesen érintkező oldószer között. Az anyag megoszlási hányadosán a
két egymással nem elegyedő oldószerben mért aktivitásának (híg oldatok esetén a
koncentrációknak) az arányát értjük, mely adott hőmérsékleten és nyomáson az
egyensúlyi állapot elérése után konstans érték.
A lipofilitás általános jellemzésére választott oktanol/víz oldószer-rendszerre
vonatkozó megoszlási hányadost P betűvel jelöljük, leginkább ennek logaritmusát
alkalmazzuk (logP).
A lipofilitás meghatározására jelenleg a leggyakrabban alkalmazott eljárások az
indirekt, másnéven alternatív meghatározási módszerek. (79; 99) Az indirekt eljárások
előnye, hogy az igen nagy és nagyon kis lipofiltású vegyületek is mérhetők, a
módszerek gyorsak és egyszerűek, ami nagyon fontos szempont a gyógyszerkutatás
korai fázisában.
A kromatográfiás vizsgálatoknál alapvető olyan kromatográfiás rendszer
felállítása, ahol a vegyületek retenciója nagy pontossággal megállapítható, ugyanakkor
az adott kromatográfiás állófázissal minél hitelesebben lehessen karakterizálni a
membrán kettősréteget. (100)
A logP meghatározás elvi alapját a folyadék/folyadék megoszláson alapuló
kromatográfiás retenció és a megoszlási hányados között fennálló összefüggés adja
meg. A módszer lényege, hogy az eljárás során a lipofilitástól függő kromatográfiás

36
paramétert (a retenciós faktorral közvetlenül összefüggésben álló ún. RM értéket: RM =
log(1/Rf – 1)) határozzuk meg, majd kalibrációs egyenes felhasználásával logP értéket
számolunk (logP = aRM + b).
A kromatográfiás logP meghatározás lényege tehát, hogy egy kiválasztott
standard anyagsorozat esetén, melynek oktanol/víz logP értéke ismert, meg kell
határozni az aktuális kromatográfiás rendszerben érvényes RM értékeket, amely
értékeket felhasználva a fenti egyenlet állandói megismerhetők. Ennek alapján egyéb
vegyületek logP értéke számítható.
Vizsgálatainkhoz, standard sorozatként, az ismert logP értékekkel rendelkező
pirido[1,2-a]pirimidin-származékokat (3. táblázat) használtunk. Bár a vegyületek
kémiai szerkezete különbözik a kalkonok szerkezetétől, fizikai-kémiai tulajdonságaik
nagyon hasonlóak: lipofilek és közel semlegesek (nagyon gyenge bázisok, és
alkalmazott RP-TLC kondíciók mellett deprotonált állapotban vannak (80)). A
kalibrációs sorozat tagjai vékonyréteg-kromatográfiás futtatásának eredményeképpen
kapott kalibrációs egyenes egyenlete:
logP = 4,007 RM + 1,373
(n = 6, r = 0,9935, s = 0,09, F = 304)
Az optimalizált kromatográfiás rendszer validálása három, ismert logP értékkel
rendelkező gyógyszermolekula (diazepám, progeszteron és PGEE) felhasználásával
történt. Az RP-TLC módszer során, a fenti kalibrációs egyenes segítségével
meghatározott logPTLC értékek közel megegyeznek a vegyületek rázótölcséres-
módszerrel meghatározott logPSF értékeivel (80). (4. táblázat)
Minta logPSF logPTLC ∆ logP
Diazepám 2,77 2,80 0,03
Progeszteron 3,54 3,42 0,12
PGEE 4,08 4,02 0,06
4. táblázat A logP meghatározás során használt validációs sorozat tagjainak
rázótölcséres módszerrel (logPSF), illetve RP-TLC módszerrel (logPTLC) történt logP
értékeinek összehasonlítása

37
A validálás során használt három vegyület logPTLC értékeivel kibővítettük a
kalibrációs sorozatot, így a következő kalibrációs egyenes egyenletet kaptuk, amit a
kalkonanalógok logP értékének számolásához használtunk fel.
logP = 4,115 RM + 1,362
(n = 9, r = 0,9973, s = 0,08, F = 1316)
A vizsgált (illetve korábban vizsgált) kalkonszármazékok kísérletes úton
meghatározott logPTLC értékeit az 5. táblázat tartalmazza. (101)
A vegyületek igen lipofilek, logP értékük viszonylag széles tartományba (logP =
2,53 - 5,35) esik, a gyűrű tagszámának valamint a szubsztituens minőségének,
jellegének és helyzetének függvényében. Mivel a lipofilitásbeli különbség minden
bizonnyal a biológiai hatás kialakulásában is szerepet játszik, meghatározása már a
gyógyszertervezés korai szakaszában igen fontos.
A gyűrűtagszám lipofilitást befolyásoló hatását a para-helyzetben szubsztituált
kalkonanalógok kísérletesen meghatározott logP értékeinek összehasonlítása révén
figyelhetjük meg. (6. táblázat) A gyűrű tagszámának növekedése a ciklikus
kalkonanalógok lipofilitásának növekedésével jár. Egy további metiléncsoport
beépülése a gyűrűbe átlagosan 0,4 egységgel növelte meg a logPTLC értéket. Az 1 és 2
sorozat tagjai, valamint a 3 és 4 sorozat tagjai közötti lipofilitásbeli különbség kisebb,
mint a 2 és 3 sorozat tagjai között. Mindezek arra utalnak, hogy az intra- és
intermolekuláris interakciók, valamint a térszerkezet is befolyással vannak a
lipofilitásra.
A 6. táblázat a kísérletes úton meghatározott logPTLC értékeken kívül, számolás
útján prediktált logP értékeket is tartalmaz. A CLOGP szoftver segítségével számolt
logP egy ún. kalkulált partíciós koefficiens. A számítógépes program által használt
módszer a Hansch-Leo fragmens alapú számítás, ahol az alapmolekula és a hozzá
kapcsolódó fragmensek, szubsztituensek száma, pozíciója határozza meg a kalkulált
logP értéket. A mért és számolt értékek viszonylag jól korrelálnak egymással. Az öttagú
gyűrűs és hattagú gyűrűs dimetilamino-származék esetén azonban viszonylag nagy az
eltérés a két adat között, a mért logP értékek jóval magasabbak (4,50, illetve 4,91) mint
a számolt logP értékek (3,76, illetve 4,32). A többi esetben a differencia kisebb, mint
0,45. Míg a 2 sorozat tagjainak kísérleti úton meghatározott logP értéke minden esetben
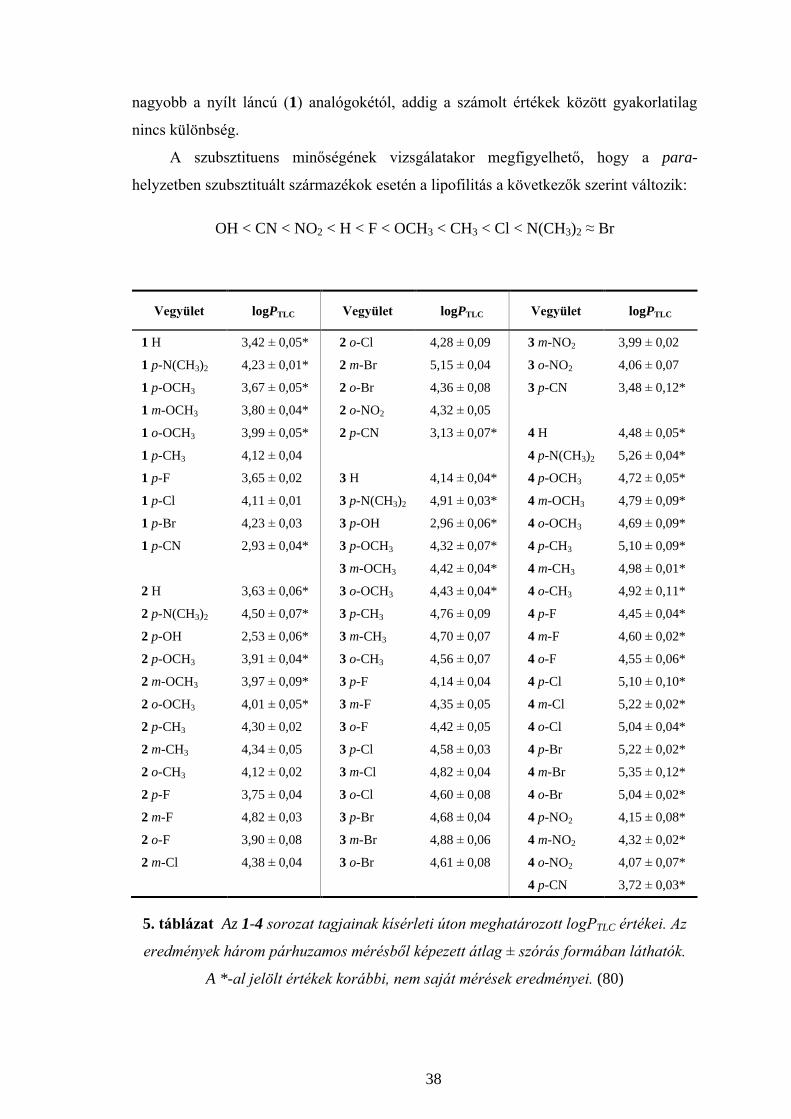
38
nagyobb a nyílt láncú (1) analógokétól, addig a számolt értékek között gyakorlatilag
nincs különbség.
A szubsztituens minőségének vizsgálatakor megfigyelhető, hogy a para-
helyzetben szubsztituált származékok esetén a lipofilitás a következők szerint változik:
OH < CN < NO2 < H < F < OCH3 < CH3 < Cl < N(CH3)2 ≈ Br
Vegyület logPTLC Vegyület logPTLC Vegyület logPTLC
1 H 3,42 ± 0,05* 2 o-Cl 4,28 ± 0,09 3 m-NO2 3,99 ± 0,02
1 p-N(CH3)2 4,23 ± 0,01* 2 m-Br 5,15 ± 0,04 3 o-NO2 4,06 ± 0,07
1 p-OCH3 3,67 ± 0,05* 2 o-Br 4,36 ± 0,08 3 p-CN 3,48 ± 0,12*
1 m-OCH3 3,80 ± 0,04* 2 o-NO2 4,32 ± 0,05
1 o-OCH3 3,99 ± 0,05* 2 p-CN 3,13 ± 0,07* 4 H 4,48 ± 0,05*
1 p-CH3 4,12 ± 0,04 4 p-N(CH3)2 5,26 ± 0,04*
1 p-F 3,65 ± 0,02 3 H 4,14 ± 0,04* 4 p-OCH3 4,72 ± 0,05*
1 p-Cl 4,11 ± 0,01 3 p-N(CH3)2 4,91 ± 0,03* 4 m-OCH3 4,79 ± 0,09*
1 p-Br 4,23 ± 0,03 3 p-OH 2,96 ± 0,06* 4 o-OCH3 4,69 ± 0,09*
1 p-CN 2,93 ± 0,04* 3 p-OCH3 4,32 ± 0,07* 4 p-CH3 5,10 ± 0,09*
3 m-OCH3 4,42 ± 0,04* 4 m-CH3 4,98 ± 0,01*
2 H 3,63 ± 0,06* 3 o-OCH3 4,43 ± 0,04* 4 o-CH3 4,92 ± 0,11*
2 p-N(CH3)2 4,50 ± 0,07* 3 p-CH3 4,76 ± 0,09 4 p-F 4,45 ± 0,04*
2 p-OH 2,53 ± 0,06* 3 m-CH3 4,70 ± 0,07 4 m-F 4,60 ± 0,02*
2 p-OCH3 3,91 ± 0,04* 3 o-CH3 4,56 ± 0,07 4 o-F 4,55 ± 0,06*
2 m-OCH3 3,97 ± 0,09* 3 p-F 4,14 ± 0,04 4 p-Cl 5,10 ± 0,10*
2 o-OCH3 4,01 ± 0,05* 3 m-F 4,35 ± 0,05 4 m-Cl 5,22 ± 0,02*
2 p-CH3 4,30 ± 0,02 3 o-F 4,42 ± 0,05 4 o-Cl 5,04 ± 0,04*
2 m-CH3 4,34 ± 0,05 3 p-Cl 4,58 ± 0,03 4 p-Br 5,22 ± 0,02*
2 o-CH3 4,12 ± 0,02 3 m-Cl 4,82 ± 0,04 4 m-Br 5,35 ± 0,12*
2 p-F 3,75 ± 0,04 3 o-Cl 4,60 ± 0,08 4 o-Br 5,04 ± 0,02*
2 m-F 4,82 ± 0,03 3 p-Br 4,68 ± 0,04 4 p-NO2 4,15 ± 0,08*
2 o-F 3,90 ± 0,08 3 m-Br 4,88 ± 0,06 4 m-NO2 4,32 ± 0,02*
2 m-Cl 4,38 ± 0,04 3 o-Br 4,61 ± 0,08 4 o-NO2 4,07 ± 0,07*
4 p-CN 3,72 ± 0,03*
5. táblázat Az 1-4 sorozat tagjainak kísérleti úton meghatározott logPTLC értékei. Az
eredmények három párhuzamos mérésből képezett átlag ± szórás formában láthatók.
A *-al jelölt értékek korábbi, nem saját mérések eredményei. (80)

39
p-X logPTLC(logPCLOGP)
1 2 3 4*
H 3,42 (3,62) 3,63 (3,60) 4,14 (4,16) 4,48 (4,72)
N(CH3)2 4,23 (3,79) 4,50 (3,76) 4,91 (4,32) 5,26 (4,88)
OCH3 3,67 (3,54) 3,91 (3,52) 4,32 (4,08) 4,72 (4,64)
CH3 4,12 (4,12) 4,30 (4,10) 4,76 (4,66) 5,10 (5,22)
F 3,65 (3,77) 3,75 (3,74) 4,14 (4,30) 4,45 (4,86)
Cl 4,11 (4,34) N.A. (4,31) 4,58 (4,87) 5,10 (5,43)
Br 4,23 (4,49) N.A. (4,46) 4,68 (5,02) 5,22 (5,58)
CN 2,93 (3,06) 3,13 (3,03) 3,48 (3,59) 3,72 (4,15)
6. táblázat Az 1-4 sorozat para-helyzetben szubsztituált származékainak kísérletes úton
meghatározott (logPTLC), valamint prediktált (logPCLOGP) logP értékeinek
összehasonlítása. A logPCLOGP értékek számolása Chem3D Ultra 9.0.1. szoftver csomag
CLOGP alkalmazásával történt. A *-al jelölt sorozat logP értékeinek meghatározása
korábban történt. (80) (N.A. = nincs adat)
A szubsztituenseknek nemcsak a minősége, hanem a helyzete is befolyásolja a
logP értékeket. (7. táblázat)
A 7-tagú gyűrűs benzoszuberon származékok (4) esetén megállapítható, hogy
adott szubsztituenst orto-, meta-, illetve para-helyzetben taralmazó vegyületek logP
értéke a következők szerint változik: logPTLC(orto) ≤ logPTLC(para) ≤ logPTLC(meta). A
2 és 3 sorozat tagjainál azonban a különböző helyzetben szubsztituált analógok logPTLC
értékeiben nem figyelhető meg hasonló szabályszerűség. Ez a megfigyelés szintén arra
utal, hogy a tér- és elektronszerkezet, amit a szubsztituensek minősége és helyzete is
nagyban befolyásol, jelentős hatással van a molekulák lipofilitására. (80; 101; 67; 68)
A kidolgozott módszer lehetőséget teremt a nagy lipofilitással bíró
kalkonanalógok logP értékeinek pontos meghatározására. A kísérleti eredmények, a
várakozásoknak megfelelően, igazolták a vegyületek lipofilitásának változását a gyűrű
tagszámának, valamint a szubsztituensek minőségének és helyzetének függvényében.
Mindezek segítséget nyújthatnak a kalkon-molekulaelemmel rendelkező, esetleges
gyógyszerjelölt molekula optimális fizikai-kémiai tulajdonságainak megtervezésében.

40
Vegyület logPTLC
orto meta para
2 OCH3 4,01 3,97 3,91
2 CH3 4,12 4,34 4,30
2 F 3,90 4,82 3,75
2 Cl 4,28 4,38 N.A.
2 Br 4,36 5,15 N.A.
3 OCH3 4,43 4,42 4,32
3 CH3 4,56 4,70 4,76
3 F 4,42 4,35 4,14
3 Cl 4,60 4,82 4,58
3 Br 4,61 4,88 4,68
3 NO2 4,06 3,99 N.A.
4 OCH3 4,69 4,79 4,72
4 CH3 4,92 4,98 5,10
4 F 4,55 4,60 4,45
4 Cl 5,04 5,22 5,10
4 Br 5,04 5,35 5,22
4 NO2 4,07 4,32 4,15
7. táblázat A szubsztituensek helyzetének hatása a 2-4 sorozat vegyületeinek kísérletes
úton meghatározott logP értékeire. A 4 sorozat tagjainak logP meghatározása korábban
történt. (80) (N.A. = nincs adat)

41
V.2. Gyűrűs kalkonszármazékok hatás-vizsgálatának eredményei
A korábbi in vitro toxicitásvizsgálatok során kiemelkedő hatást mutató 7-tagú gyűrűs
(E)-2-(4-metoxibenzilidén)-1-benzoszuberont (4a) és kevéssé toxikus 4-
metilszubsztituált analógját (4b) (12. ábra) választottuk a tervezett vizsgálatok
elvégzéséhez, a gyűrűs kalkonszármazékok citotoxikus és daganatellenes hatásának
kialakulásában szerepet játszó mechanizmusok alaposabb megismerése céljából.
V.2.1. Az (E)-2-(4-metoxibenzilidén)-1-benzoszuberon és
4-metilanalógjának sejtproliferációra és sejtciklusra gyakorolt
hatása
V.2.1.1. A kalkonanalógok apoptózist indukálnak Jurkat T sejtekben
Korábbi vizsgálatok azt mutatták, hogy a humán Jurkat T sejtek érzékenyebbek egy új
α,β-telítetlen ketonra, mint más tumorsejtvonalak, így munkánkhoz Jurkat T sejteket
használtunk. (102)
A kalkonszármazékok Jurkat T sejtek sejtproliferációjára gyakorolt hatásának
megismeréséhez elsőként a teljes sejtszám, valamint az élő sejtek arányának az
alakulását vizsgáltuk a vegyületekkel történő kezelést követően. A 4a és 4b
vegyületekkel – a Jurkat T sejtekkel szemben korábban meghatározott IC80
koncentrációértékek (4a: 5,6 μM; 4b: 23,3 μM) (64) mellett – történő 8, 24 illetve 48
órás kezelést követően a teljes sejtszámot (8. táblázat), illetve ebből az élő sejtek
arányát (13. ábra) a sejtek tripánkékkel történő festését követően, hemocitométerrel
határoztuk meg. (82)
A tripánkék festék csak az elpusztult sejtek citoplazmájába képes behatolni és
citoplazmás fehérjékhez kötődni, mivel az élő sejtek membránja nem ereszti át a
festéket, így megkülönböztethetők egymástól az élő és elpusztult sejtek.
A 8. táblázatból kiolvasható, hogy a kezeletlen sejtek, valamint az 1% DMSO-t
tartalmazó inkubátumban levő sejtek száma 24 óra alatt megkétszereződik, ami
megfelel a Jurkat T sejtek duplikációs idejének. (103) (A következő 24 óra alatt már
térbeli korlátok miatt nem tud újra osztódni.) A kalkonok oldószereként használt DMSO
tehát ekkora mennyiségben (1%), önmagában nem befolyásolja a sejtszámot. A két

42
vizsgált vegyület egymáshoz hasonló módon csökkenti a sejtek számát a 8, 24 és 48
órás időpontokban, gátló hatásuk a sejtek proliferációjára kifejezett.
Teljes sejtszám (x 105)
8 óra 24 óra 48 óra
Kontroll 5,45 ± 0,66 11,26 ± 1,67 11,07 ± 1,01
DMSO 1% 5,01 ± 0,74 10,53 ± 1,72 11,85 ± 2,44
4a (5,6 μM) 4,88 ± 0,68 8,68 ± 0,98 5,33 ± 0,83
4b (23,3 μM) 4,69 ± 0,52 7,91 ± 0,78 7,00 ± 0,76
8. táblázat 4a és 4b vegyületekkel történő sejtkezeléseket követően a teljes sejtszám
alakulása. A meghatározás tripánkékkel történő festéssel történt. Az eredmények négy
párhuzamos mérésből képezett átlag ± szórás formában láthatók.
Az életképes sejtek aránya a kontroll sejtekhez képest mindkét kalkonanalóg
esetén hasonlóan alakul. (13. ábra) 8 órás inkubációs időt követően az élő sejtek
mennyisége még közel annyi, mint a kezeletlen sejtek esetében, de 24 órás, illetve 48
órás inkubációs idő után már csaknem felére csökken a számuk. Nagy valószínűség
szerint már a 8 órás kezelési időtartam alatt is lejátszódnak olyan folyamatok, amelyek
hatással lesznek a sejtproliferáció további alakulására.
13. ábra A 4a és 4b vegyületek hatása a Jurkat T sejtek életképességére. Az élő sejtek
arányának meghatározása a kezelést követően tripánkékkel történő festési eljárással
történt. Az eredmények négy párhuzamos mérésből képezett átlag ± szórás formában
láthatók.
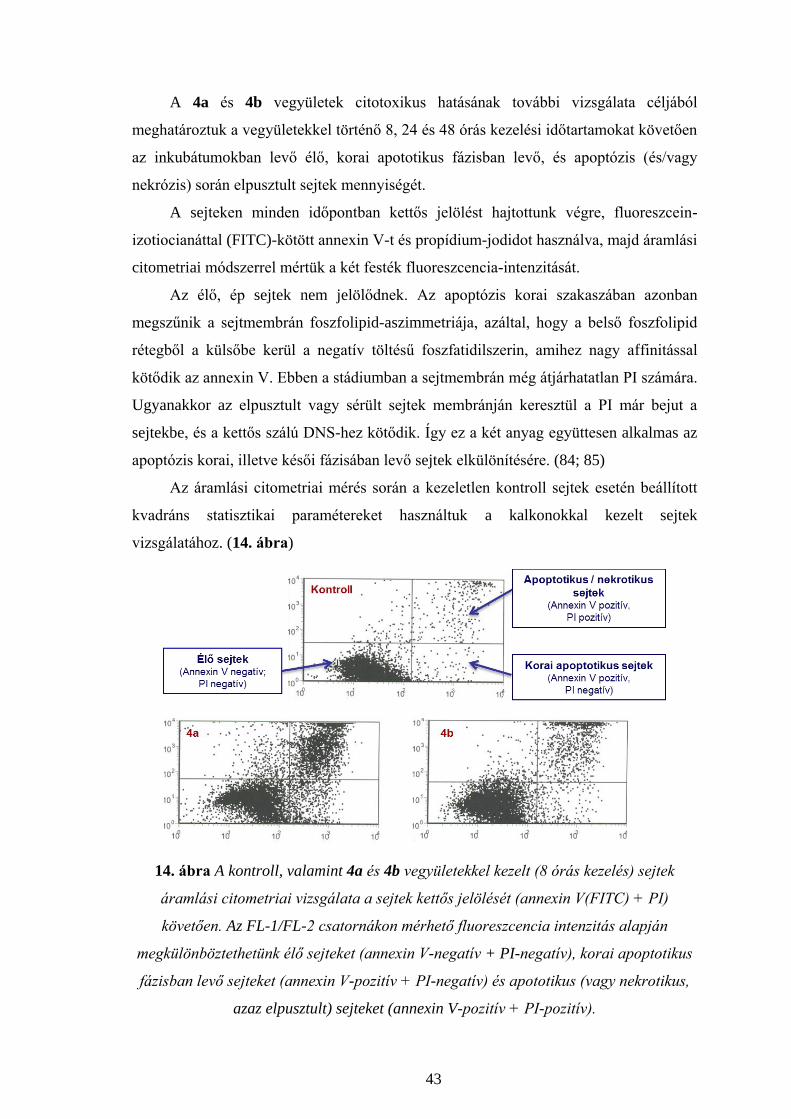
43
A 4a és 4b vegyületek citotoxikus hatásának további vizsgálata céljából
meghatároztuk a vegyületekkel történő 8, 24 és 48 órás kezelési időtartamokat követően
az inkubátumokban levő élő, korai apototikus fázisban levő, és apoptózis (és/vagy
nekrózis) során elpusztult sejtek mennyiségét.
A sejteken minden időpontban kettős jelölést hajtottunk végre, fluoreszcein-
izotiocianáttal (FITC)-kötött annexin V-t és propídium-jodidot használva, majd áramlási
citometriai módszerrel mértük a két festék fluoreszcencia-intenzitását.
Az élő, ép sejtek nem jelölődnek. Az apoptózis korai szakaszában azonban
megszűnik a sejtmembrán foszfolipid-aszimmetriája, azáltal, hogy a belső foszfolipid
rétegből a külsőbe kerül a negatív töltésű foszfatidilszerin, amihez nagy affinitással
kötődik az annexin V. Ebben a stádiumban a sejtmembrán még átjárhatatlan PI számára.
Ugyanakkor az elpusztult vagy sérült sejtek membránján keresztül a PI már bejut a
sejtekbe, és a kettős szálú DNS-hez kötődik. Így ez a két anyag együttesen alkalmas az
apoptózis korai, illetve késői fázisában levő sejtek elkülönítésére. (84; 85)
Az áramlási citometriai mérés során a kezeletlen kontroll sejtek esetén beállított
kvadráns statisztikai paramétereket használtuk a kalkonokkal kezelt sejtek
vizsgálatához. (14. ábra)
14. ábra A kontroll, valamint 4a és 4b vegyületekkel kezelt (8 órás kezelés) sejtek
áramlási citometriai vizsgálata a sejtek kettős jelölését (annexin V(FITC) + PI)
követően. Az FL-1/FL-2 csatornákon mérhető fluoreszcencia intenzitás alapján
megkülönböztethetünk élő sejteket (annexin V-negatív + PI-negatív), korai apoptotikus
fázisban levő sejteket (annexin V-pozitív + PI-negatív) és apototikus (vagy nekrotikus,
azaz elpusztult) sejteket (annexin V-pozitív + PI-pozitív).

44
A citotoxicitás vizsgálat eredményeit a 9. táblázat tartalmazza. 8 órás kezelés
alatt sem a 4a, sem a 4b vegyület nem mutat kifejezett toxikus hatást. 24 órás kezelési
időtartam után azonban az élő sejtek száma sokkal kisebb a metoxiszármazék (4a)
esetén (57,7%), mint a 4b vegyülettel történő kezelést követően (71,7%). A sejtek
jelentős része korai apoptotikus fázisban (4a: 22,8%; 4b: 7,8%), illetve késői
apoptotikus/nekrotikus fázisban van (4a: 16,8%; 4b: 17,1%).
Élő sejtek (%)
Korai
apoptotikus
sejtek (%)
Elpusztult
sejtek (%)
8 óra
Kontroll 90,33 ± 3,50 2,15 ± 1,07 3,98 ± 1,53
DMSO 1% 89,53 ± 5,60 2,67 ± 0,17 6,16 ± 2,68
4a (5,6 μM) 84,31 ± 4,24 3,15 ± 0,45 8,66 ± 2,15
4b (23,3 μM) 84,15 ± 0,52 2,81 ± 0,09 8,09 ± 0,11
24 óra
Kontroll 89,37 ± 4,08 4,40 ± 3,07 4,24 ± 0,71
DMSO 1% 88,43 ± 5,72 3,17 ± 2,32 5,71 ± 2,49
4a (5,6 μM) 57,65 ± 5,52 22,80 ± 1,55 16,82 ± 3,39
4b (23,3 μM) 72,71 ± 6,59 7,81 ± 0,98 17,11 ± 6,35
48 óra
Kontroll 90,27 ± 0,37 N.A. 9,70 ± 0,35
DMSO 1% 85,56 ± 1,26 N.A. 14,43 ± 1,24
4a (5,6 μM) 40,15 ± 1,39 N.A. 59,79 ± 1,39
4b (23,3 μM) 48,53 ± 3,18 N.A. 51,44 ± 3,18
9. táblázat 4a és 4b vegyületekkel történő 8, 24 és 48 órás sejtkezeléseket, és
annexinV(-FITC)/propídium jodiddal történő kettős jelölést követő áramlási citometriai
módszer eredményei: az élő, korai apoptotikus fázisban levő, valamint elpusztult sejtek
mennyisége %-ban kifejezve. Az eredmények négy párhuzamos mérésből képezett
átlag ± szórás formában láthatók. (N.A.=nincs adat, 48 órás kezelést követően csak
propídium-jodiddal történt jelölés)
48 órás kezelést követően csak propídium-jodidot használtunk az élő (PI-negatív)
és a késői apoptotikus/nekrotikus fázisban levő (PI-pozitív) sejtek mennyiségének
meghatározására. Ebben az időpontban az élő sejtek száma jelentősen lecsökken, a 4a
vegyület esetén 40,2%-ra, a 4b analóg esetén pedig 48,5%-ra. (9. táblázat)

45
Az eredményekből kiolvasható, hogy a vizsgált kalkonszármazékok, ezen
koncentrációértékek mellett, gátolják a sejtszaporodást, és apoptózist indukálnak. (104)
A fenti módszerekkel kapott 48 órás citotoxicitás vizsgálat eredményei
valamelyest különböznek a 4a és 4b vegyületekkel történt, nigrozin festés mellett
értékelt korábbi vizsgálatok eredményétől. (64) A két különböző festési módszerrel
kapott eltérő eredmény a tripánkék-festés relatív érzéketlenségének lehet a
következménye, ami a festék makromolekuláris szerkezetéből adódik. (105) Másrészt,
az áramlási citometriai módszerrel történt citotoxicitás vizsgálat eltérő eredménye a két
különböző eljárás és értékelési módszer következménye lehet. (106) A 48 órás
eredményeket illetően, az alkalmazott eljárásoktól függetlenül látszódik az, hogy a két
koncentrációérték (4a: 5,6 μM 4b: 23,3 μM) közel ekvitoxikus dózist képvisel.
V.2.1.2. A vizsgált származékok eltérő módon befolyásolják a sejtciklust
A kísérletekbe bevont kalkonanalógok Jurkat T sejtek sejtciklusára gyakorolt hatásának
vizsgálata a vegyületekkel történt kezeléseket (IV.2.2) követően, a DNS mennyiségi
analízise által valósult meg. A Jurkat T sejtek kezelése során különböző: 8, 24 és 48
órás időpontokban vizsgáltuk a sejteket, hogy időben is láthassuk a kalkonok
sejtciklusra gyakorolt hatását.
A DNS mennyiségi analíziséhez a sejtek jelölése minden időpontban propídium-
jodiddal (PI) történt. A PI a sejtekben a kettősszálú DNS-hez kötődik, ezért a megkötött
PI mennyisége, így az áramlási citometriai eljárás során látható fluoreszcens jel
erőssége is arányos a sejtek DNS-mennyiségével. (A kísérletben használt RNAse enzim
a kettős szálú RNS-t bontja, így a PI ahhoz nem tud kötődni, és nem torzítja el az
eredményt.)
A sejtciklus egyes fázisaiban levő sejtek egymástól való megkülönböztetetése a
kezeletlen kontroll sejtek esetén felvett ábra alapján történt, az ott meghatározott
„sávok” kerültek meghatározásra minden vizsgált vegyület és időpont esetén. (107) (15.
ábra)
A 4a és 4b vegyületek sejtciklusra gyakorolt hatásában eltérések mutatkoznak,
különösen drasztikus a változás a metoxiszubsztituált (4a) származék esetén. A
sejtciklus vizsgálat eredményeit, az egyes fázisokban levő sejtek mennyiségét, %-ban
kifejezve az 10. táblázat, valamint a 16. ábra foglalja össze.

46
15. ábra Áramlási citometriai sejtciklus-analízis a kezeletlen kontroll sejteken (8 óra),
valamint a 4a és 4b vegyületekkel történő 8, 24 és 48 órás kezelést követően. A
sejtciklus egyes szakaszainak kijelölése a kontroll sejtek ábráján történt.
A 4a vegyület korai hatása (8 óra) abban nyilvánul meg, hogy megnöveli a G2/M
fázisban levő sejtek arányát (36,9%), ami együtt jár a G0/G1 fázisban levő sejtek
mennyiségének csökkenésével (18,6%). A metilszármazék esetén (4b) a sejtek
legnagyobb hányada, a kontroll sejtekhez hasonlóan, a G0/G1 fázisban található (36,8%).

47
Sub-G0
fázis (%)
G0 / G1 fázis
(%)
S fázis
(%)
G2 / M fázis
(%)
Poliploid
sejtek (%)
8 óra
Kontroll 3,3 ± 0,7 42,6 ± 0,9 21,2 ± 3,9 27,2 ± 0,9 5,6 ± 1,0
DMSO 1% 3,1 ± 0,3 40,8 ± 1,4 20,7 ± 3,0 26,6 ± 1,7 7,6 ± 1,3
4a (5,6 μM) 6,4 ± 0,9 18,6 ± 2,2 24,7 ± 1,0 36,9 ± 0,9 9,9 ± 2,7
4b (23,3 μM) 3,8 ± 0,4 36,8 ± 2,3 22,8 ± 3,3 25,1 ± 3,2 8,4 ± 1,1
24 óra
Kontroll 5,0 ± 1,5 44,7 ± 3,2 25,8 ± 0,5 22,7 ± 1,8 3,6 ± 0,9
DMSO 1% 5,5 ± 2,2 43,6 ± 2,6 21,1 ± 2,1 20,3 ± 1,1 4,9 ± 0,7
4a (5,6 μM) 22,4 ± 1,1 7,3 ± 0,4 9,6 ± 1,2 44,8 ± 4,9 16,2 ± 2,7
4b (23,3 μM) 14,5 ± 2,5 31,7 ± 2,5 17,3 ± 2,8 27,1 ± 2,2 8,5 ± 1,5
48 óra
Kontroll 6,4 ± 1,9 49,6 ± 2,7 21,0 ± 1,1 17,3 ± 1,5 5,2 ± 2,0
DMSO 1% 8,4 ± 1,8 51,5 ± 2,3 17,2 ± 2,0 19,0 ± 0,7 7,1 ± 2,6
4a (5,6 μM) 40,8 ± 2,2 8,5 ± 0,8 5,9 ± 2,3 8,3 ± 1,5 34,3 ± 6,5
4b (23,3 μM) 36,5 ± 2,4 24,4 ± 1,2 13,0 ± 2,4 13,9 ± 1,3 12,4 ± 2,9
10. táblázat Az áramlási citometriai módszerrel történt sejtciklus-analízis eredményei,
a 4a és 4b vegyületekkel történő 8, 24 és 48 órás kezelést és propídium-jodidos jelölést
követően. Az eredmények négy párhuzamos mérésből képezett átlag ± szórás formában
láthatók.
Hosszabb, 24 órás inkubációs időt követően a 4a vegyület hatására tovább
csökken a G0/G1 fázisban levő sejtek mennyisége (7,3%), és nő a G2/M fázisban
(44,8%), valamint a Sub-G0 (apoptotikus/nekrotikus sejtek) fázisban (22,4%) levő
sejtek aránya. Ebben az időpontban (24 óra), a 4b származékkal kezelt sejtek esetén
mérsékelt csökkenés következik be a G0/G1 fázisban levő sejtek mennyiségében
(31,7%), ami együtt jár a G2/M fázisban (27,1%), illetve a Sub-G0 fázisban (14,5%)
levő sejtek arányának kismértékű növekedésével. A 4a és 4b vegyületek eltérő hatása a
(G2/M)/(G0/G1) aránnyal is kifejezhető, ami 0,51 a kontroll, 6,1 a metoxi- (4a), és 0,85 a
metilszármazék (4b) esetén.
48 órás kezelést követően mindkét kalkonanalóg esetén további növekedés
tapasztalható a Sub-G0 fázisban, vagyis jelentősen megnő az apoptotikus/nekrotikus
sejtek aránya (4a: 40,8%; 4b: 36,5%), valamint ún. poliploid sejtek kialakulása
figyelhető meg.

48
16. ábra A kontroll, az 1%-ban DMSO-t tartalmazó, valamint a 4a és 4b vegyületekkel
történő 8, 24 és 48 órás kezelést és propídium-jodidos jelölést követő sejtciklus-analízis
eredményei - a sejtciklus egyes fázisaiban levő sejtek százalékos mennyisége szerint
ábrázolva. Az eredmények négy párhuzamos mérésből képezett átlag ± szórás formában
láthatók.

49
A sejtciklus-vizsgálat eredményeit összefoglalva elmondható, hogy a kalkonok
oldószereként használt DMSO, az alkalmazott 1%-ban nem befolyásolja a sejtciklust, a
két vizsgált vegyület hatása jelentős, de egymástól eltérő. (104) Míg a metilszármazék
(4b) esetén, a 8 órás kezelést követően a sejtek legnagyobb hányada – a kontroll
sejtekhez hasonlóan – a G0/G1 fázisban található, addig a metoxiszubsztituált analóg
(4a) esetén a G2/M fázisban. Ez együtt jár a G0/G1 fázisban levő sejtek mennyiségének
csökkenésével, ugyanakkor a Sub-G0 fázisban levő apoptotikus/nekrotikus sejtek
arányának növekedésével is. (84; 85) Mivel a 8 órás időtartam körülbelül harmada egy
sejtciklus időtartamának Jurkat T sejtekben (103), a kísérleti eredmény arra utal, hogy a
4a vegyület esetén nem működik megfelelően a G1 ellenőrző pont, aminek egyébként
meghatározó szerepe van genetikai problémák kiküszöbölésében. (108) A G1
ellenőrzőpont által „visszatartott” sejtek ugyanis részben, apoptózis vagy nekrózis során
elpusztulnak, aminek fontos szerepe van a genom integritásának biztosításában. (109)
A vegyület tulajdonképpen felfüggeszti a G1 ellenőrzőpont „munkáját”. A 4a vegyület
G2/M fázisú blokkot is okoz. A sejtciklusban bekövetkező ilyen jellegű zavar, mint
egyes flavonoidok és kalkonok lehetséges hatásmechanizmusa, az irodalomban is
dokumentált. (110; 111; 112; 113) Az észlelt változások arra utalnak, hogy a
metoxiszármazék (4a) viszonylag gyorsan DNS károsodást okozhat, aminek gyakori
velejárója a G2/M fázisú blokk. (114; 115)
A metilvegyület (4b) ekvitoxikus dózisával történő sejtkezelés estén nem
figyelhető meg hasonlóan jelentős változás a G1 ellenőrzőpontot illetően, és a G2/M
blokk sem kifejezett.
A 4a származékkal történő hosszútávú (24 és 48 órás) expozíciót követően mind a
G0/G1, az S, valamint a G2/M fázisban levő sejtek mennyisége drasztikusan csökken.
Egyidejűleg nagymértékben megnő a Sub-G0 fázisban levő (apoptotikus/nekrotikus)
sejtek, valamint a poliploid sejtek mennyisége, ami a G2 ellenőrzőpont működési
zavarára utal. Az ilyen jellegű késleltetett sejthalál, amit „mitotikus halál”-nak is
neveznek, jellegzetessége a p53 mutáns tumoroknak. (116)
A poliploid populáció proliferációs potenciálja egyelőre vitatott. Vannak, akik azt
állítják, hogy a poliploid sejtek alacsony proliferatív potenciállal rendelkeznek, mások
szerint azonban – bár késleltetett a metafázis – proliferációra képesek. (116)
A vizsgált kalkonszármazékok csökkentik az élő sejtek számát, apoptózist
indukálnak, ami minden bizonnyal összefüggésbe hozható azzal a kísérleti
eredménnyel, hogy jelentős mértékben befolyásolják a sejtciklust, különösen a 4a

50
vegyület. Az apotózis számos anti-apoptotikus, valamint pro-apototikus modulátor által
irányított folyamat. Későbbi kutatások igazolták, hogy a 4a származék csökkenti a
Bcl-2 fehérje – az anti-apoptotikus proteinek egy típusának – expresszióját, ugyanakkor
megnöveli az apotózist elősegítő Bax protein szintézisét. (117) A Bax/Bcl-2 arány
növekedése nagyban hozzájárulhat az apoptózis indukciójához.
V.2.2. A kalkonszármazékok in vitro antioxidáns hatással bírnak
A 7-tagú gyűrűs 4-metoxi- (4a) és 4-metilszubsztituált (4b) kalkonanalógok in vitro
antioxidáns (hidroxilgyök scavanger), esetleges prooxidáns hatását, illetve annak
időfüggését, a Fenton-reakció inicializálta dezoxiribóz degradációs tesztet alkalmazva
vizsgáltuk. (86; 87)
A hidroxilgyök scavanger hatás in vitro jellemzésére számos módszer ismert az
irodalomban. Ezek egyike a Halliwell és mtsai által kidolgozott módszer, melynek
lényege, hogy a vas(II)-ionok és H2O2 reakciójában keletkező hidroxilgyökök a 2-
dezoxi-D-ribózt eloxidálják. A láncszakadással járó oxidáció eredményeképpen savas
közegben tiobarbitursavval 532 nm körüli abszorpciós maximummal rendelkező
karbonilvegyületek keletkeznek. (87) (17. ábra) Ha a vizsgálatot hidroxilgyök-
scavanger tulajdonságú vegyület jelenlétében végezzük, akkor kompetitív reakció
eredményeként az 532 nm körüli abszorpciós maximummal rendelkező fragmens
vegyületek mennyisége csökken. (87)
17. ábra A Fenton-reakció, illetve a dezoxiribóz degradációs teszt lényege
Kísérleteink során Winterbourn vizsgálatai alapján optimalizált dezoxiribóz teszt
összetételt alkalmaztuk. (118) Bár a reakció pontos mechanizmusa és a reakció során
keletkező vegyületek kémiai szerkezete teljességében nem ismert, az érzékeny és
könnyen kivitelezhető módszer széles körben alkalmazott a hidroxilgyök scavanger
tulajdonságú vegyületek in vitro antioxidáns hatásának vizsgálatára. A módszer, az
alkalmazott kísérleti körülmények között alkalmas az azonnali, illetve hosszútávú

51
antioxidáns, sőt pro-oxidáns aktivitás meghatározására. (119) A hosszútávú (240 perc)
vizsgálat során ugyanis a hidroxilgyökök hatására esetlegesen keletkező redox-aktív
metabolitok további redox-reakciókat indíthatnak el (ami a vas(III)-ionok vas(II)-
ionokká történő redukcióját is magába foglalja), és „újra” hidroxilgyökök
képződhetnek. (53)
A vizsgálatokat etiléndiamin-tetraecetsav (EDTA) jelenlétében és EDTA
hozzáadása nélkül is elvégeztük. EDTA hozzáadása vas(II)-EDTA-komplex
kialakulását eredményezi (120), aminek Fenton-reakcióban való részvétele az EDTA
degradációjához vezethet. (121) EDTA hiányában a vas(II)-ionok egy része a
dezoxiribózzal képez komplexet. (122) Az ún. „hely-specifikus” Fenton-reakcióban a
hidroxilgyökök képződésük helyén azonnal elreagálnak. Tehát azok a vegyületek,
amelyek ligandként szerepelhetnek (jelen esetben az EDTA), versengenek a
dezoxiribózzal a vas(II)-ionokért, ezáltal csökken a vas(II)-ion katalizálta reakcióban
keletkező hidroxilgyök okozta degradáció.
A dezoxiribóz degradációs teszt eredményeit a 18.-19. ábra foglalja össze. A
kontroll inkubátumok esetén a keletkező TBA-reaktív anyagok mennyisége (a mért
abszorbancia) időben viszonylag állandónak mondható. Ahogy az a 18. ábraról
leolvasható, az EDTA-t nem tartalmazó rendszerben mindkét vizsgált kalkonszármazék
folyamatos antioxidáns, vagyis hidroxilgyök scavanger aktivitást mutat a 240 perces
kísérleti ídőtartam alatt. Bár a 4a vegyület antioxidáns kapacitása az inkubációs idő
120-adik perce körül minimumot mutat, az inkubációt folytatva a két vegyület hasonló
mértékben csökkentette a dezoxiribóz oxidációja során keletkező TBA-reaktív termékek
mennyiségét.
A 19. ábran láthatók az EDTA (36 μM) jelenlétében elvégzett kísérletek
eredményei. EDTA jelenlétében a reakció során keletkező TBA-reaktív termékek
mennyisége jelentősen kisebb (alacsonyabb a mért abszorbancia), mint EDTA nélkül. A
4a vegyület esetén a TBA-reaktív anyagok mennyisége megnő a 80-120 percig tartó
periódius alatt, majd jelentős csökkenésnek indul. Az észlelt prooxidáns hatás könnyebb
értelmezésének céljából a 120. percben vett inkubátum etilacetátos kivonatát GC-MS
mérésnek vetettük alá. A GC-MS analízis során azonban nem sikerült a 4a vegyület
valamely, az inkubáció során keletkező metabolitjának jelenlétét igazolni. Az észlelt
hatás hátterének megismeréséhez további vizsgálatok szükségesek. A metilszármazék
(4b) ezen kísérleti körülmények között is mindvégig antioxidáns aktivitást mutatott.

52
18. ábra A Fenton-reakció inicializálta dezoxiribóz degradációs teszt időbeli
függésének eredményei a kontroll inkubátumban, valamint 4a (200 μM) és 4b (200 μM)
jelenlétében, EDTA hozzáadása nélkül. Az abszorbancia mérése 532 nm-en történt. Az
eredmények három párhuzamos mérésből képezett átlag ± szórás formában láthatók.
19. ábra A Fenton-reakció inicializálta dezoxiribóz degradációs teszt időbeli
függésének eredményei a kontroll inkubátumban, valamint 4a (200 μM) és 4b (200 μM)
jelenlétében, 36 μM EDTA hozzáadása mellett. Az abszorbancia mérése 532 nm-en
történt. Az eredmények három párhuzamos mérésből képezett átlag ± szórás formában
láthatók.
A rákos sejtek folyamatosan, viszonylag nagy mennyiségben termelnek hidrogén-
peroxidot (H2O2). (123) A hidrogén-peroxid és vas-, illetve réz-ionok reakciója során
(általánosságban a Fenton-reakció során (124)) folyamatosan hidroxilgyökök is
termelődnek. Az aromás vegyületek reakcióba léphetnek a keletkező gyökökkel,

53
miközben hidroxilszubsztituált származékok keletkezhetnek. (52) A fenolos
metabolitok további oxidációja során reaktív oxigéngyökök keletkezhetnek, és az
eredetileg antioxidáns hatású aromás vegyületek – számos, spontán vagy
enzimkatalizált reakció eredményeként – akár oxidatív stresszt is okozhatnak. (53; 125)
Ezek ismeretében vizsgáltuk a két kalkonszármazék in vitro antioxidáns, illetve
esetleges prooxidáns hatását. A Fenton-reakció inicializálta dezoxiribóz degradációs
teszt eredményeként azt láthatjuk, hogy a vizsgált vegyületek rövidtávon antioxidáns
aktivitást mutatnak, de hosszútávú inkubációjuk során kapott eredmények függnek az
alkalmazott kísérleti körölményektől. A metilszármazék (4b) mindkét vizsgálati
protokoll (EDTA nélkül és EDTA mellett) szerint in vitro antioxidáns hatással bír. A
metoxiszubsztituált analóg (4a) az EDTA jelenlétében elvégzett kísérletben átmeneti
prooxidáns aktivitást mutatott, ami arra utal, hogy a vizsgált aktivitás függ a jelen levő
kelátkomplexképző anyagtól, ami befolyásolhatja a vasionok mikrospeciációját és
reaktivitását. (104; 126)
V.2.3. A kalkonszármazékok hatással vannak a Jurkat T sejtek redox
státuszára
V.2.3.1. A vegyületek befolyásolják a ROS-aktivitást
A vizsgált kalkonanalógok reaktív oxigéngyök (ROS) termelésre gyakorolt hatását, a
vegyületekkel történő 1 és 4 órás sejtkezelést követően, 2’,7’-diklórfluoreszcin-
diacetátot (DCFH-DA) használva vizsgáltuk. (88; 89) A DCFH-DA egy nem-
fluoreszkáló molekula, amely képes a sejtek membránján áthatolni, és az intracelluláris
térbe kerülni. A jelen levő észteráz enzimek lehasítják a molekuláról az
acetilcsoportokat és 2’,7’-diklórfluoreszcin (DCFH) keletkezik, amely még nem
fluoreszkál. Reaktív oxigénszármazékok (főként H2O2) jelenlétében azonban
fluoreszcens 2’,7’-diklórfluoreszceinné (DCF) oxidálódik. A fluoreszcens jel intenzitása
– többek között – áramlási citométerrel detektálható. Az FL-1 csatornán mért
jelintenziás tehát végső soron az intracelluláris ROS-koncentrációval arányos.
Az áramlási citometriai analízis alkalmazásával láthatóvá vált, hogy a metil-
analóg (4b) jelentősen csökkenti a kezelt sejtekben a ROS termelést, ugyanakkor a
metoxiszármazék (4a) a kontroll sejtek analízisével hasonló képet mutat, vagyis nem
befolyásolja ezen kísérleti körülmények között a ROS produkciót. (20. ábra)

54
20. ábra A kalkonszármazékok celluláris ROS termelésre gyakorolt hatása. Áramlási
citometriai (FACScan) analízis a Jurkat T sejtek 4a és 4b vegyületekkel történő 4 órás
inkubációját, és DCFH-DA-val (10 μM) történő 20 perces jelölését követően.
A 21. ábra foglalja össze a ROS-mérés eredményeit, a kezeletlen kontroll
sejtekhez viszonyított relatív fluoreszcencia intezitás (%) értékek láthatók.
21. ábra A kalkonszármazékok celluláris ROS termelésre gyakorolt hatása. A kezelt
sejtek (DMSO 1%, 4a és 4b) fluoreszcencia intenzitását a kezeletlen kontroll sejtek
esetén mért fluoreszcenzia intenzitáshoz viszonyítjuk (%). Az eredmények négy
párhuzamos mérésből képezett átlag ± szórás formában láthatók. A kezeletlen kontroll
sejtekhez képest szignifikáns különbség (Student-t teszt, p < 0,05) *-al jelölve.

55
A 4b vegyület szignifikánsan csökkentette a DCF jelének intenzitását, mind az 1
órás, mind a 4 órás kezelést követően (80,20 ± 10,35% és 71,35 ± 16,51% a kontrollhoz
viszonyítva). A 4a származék esetén nem figyelhető meg szignifikáns változás.
A korábbiakban láthattuk, hogy a vizsgált kalkonszármazékok apoptózist
indukálnak. Az apoptózis olyan szigorúan szabályozott morfológiai változások sora,
amelyek a sejtek halálához vezetnek, de az apotózist nemcsak természetes, hanem
mesterséges stimulusok is indukálhatják. Egyre nagyobb az egyetértés arra
vonatkozólag, hogy az oxidatív stressz és a redox-státuszt befolyásoló tényezők döntő
szerepet játszanak az apoptózis irányításában és lefolyásában. (127)
Néhány kalkonszármazék citotoxikus hatásának hátterében az állhat, hogy
oxidatív stresszt okoznak (128), ugyanakkor mások a kalkonok antioxidáns hatásáról
számolnak be (35; 37), de azt is feltételezik, hogy a kalkonok toxikus hatása a pro-
oxidáns tulajdonságnak tudható be (35; 51). A 4a és 4b vegyületek ROS termelésre
gyakorolt hatását azért vizsgáltuk, hogy vajon oxidatív stresszt is okozva indukálnak
apoptózist. A vizsgálati eredmények arra utalnak (21. ábra), hogy a vegyületek nem
növelik meg az intracelluláris ROS aktivitást, sőt a metilszubsztituált analóg (4b)
szignifikánsan csökkenti a ROS-szintet és antioxidáns aktivitást mutat. Ez utóbbi
eredmény összhangban áll az in vitro antioxidáns hatást vizsgáló dezoxiribóz teszt
eredményével (V.2.2), ami szerint a vegyületek in vitro hidroxilgyök-scavanger
aktivitással rendelkeznek. (104; 126)
V.2.3.2. A kalkonszármazékok hatással vannak a mitokondriális funkciókra
A sejtek mitokondriális működése alapvetően három folyamatot határoz meg: az
oxidatív foszforiláció, reaktív oxigénszármazékok képződése, valamint a fiziológiás
sejthalál (apoptózis) iniciációja a legfontosabb mitokondriális funkciók. (129)
Következésképpen, ha egy xenobiotikum befolyásolja a mitokondriális
tevékenységeket, az hozzájárulhat a vegyület citotoxikus vagy antineoplasztikus
hatásához.
Néhány kalkonszármazék és izolált patkány máj mitokondriális külső membrán
interakciójának fluoreszcencia polarizációs módszerrel történő vizsgálatakor a 4a
vegyület más származékoktól eltérő módon viselkedett. A 4a analóg esetén,
mitokondrium jelenlétében, folyamatosan növekedő fluoreszcencia polarizációs jelet
lehetett detektálni. (130) Ez következménye lehet annak, hogy a lipofil kalkonanalóg

56
fokozatosan beépül a mitokondriális membránba, ami csökkenti a molekula
konformációs mobilitását, ez pedig szintén hozzájárulhat a vegyület kiemelkedő
citotoxikus hatásához. (130)
A kalkonanalógok mitokondriális funkcióra gyakorolt hatásának vizsgálatát a
kassai P. J. Šafarik Egyetem Orvosi Kémiai és Biokémiai Intézetével kooperációban
végeztük.
A mitokondriális respirációra, ATPáz enzim aktivitásra, ezáltal a mitokondriális
redox státuszra gyakorolt hatás vizsgálatok izolált patkánymáj mitokondriumokon
történtek. (131)
Az izolált mitokondriumok oxigénfogyasztás sebességének meghatározása 25 °C-
on, polarográfiás módszerrel, oxigénelektróddal történt. Az ún. respirációs médumhoz
adtuk a kalkonanalógokat, ADP-t, respirációs szubsztrátként nátrium-szukcinátot,
valamint a mitokondriumot. Az oxigénfogyasztás nmol O2 x min-1
x fehérje mg-1
formában került kifejezésre, ADP hozzáadásakor (3-as állapot; state 3), illetve ADP
hozzáadása előtt (4-es állapot; state 4). A vizsgált kalkonszármazékok nem
befolyásolják az oxigénfogyasztást a 3-as állapotban, de szignifikánsan megnövelik azt
a 4-es állapotban (a 4a vegyület 50,7 %-al), vagyis ADP-mentes körülmények között.
(11. táblázat)
O2 fogyasztás a
4-es állapotban (nmol O2 x min-1 x
mg-1)
O2 fogyasztás a
3-as állapotban (nmol O2 x min-1 x
mg-1)
RCR ATPáz
(nmol x min-1 x
mg-1)
Kontroll 11,87 ± 0,83 34,57 ± 0,79 2,91 ± 0,14 0,403 ± 0,017
4a 17,89 ± 0,83* 38,44 ± 0,96 2,14 ± 0,10* 0,544 ± 0,021*
4b 15,94 ± 0,66* 40,92 ± 2,78 2,57 ± 0,21 0,457 ± 0,019
11. táblázat A 4a és 4b vegyületek mitokondriális respirációs funkciókra gyakorolt
hatása. RCR = respiratory control ratio, az „oxigénfogyasztás a 3-as állapotban” és az
„oxigénfogyasztás a 4-es állaptban” hányadosa. Az eredmények három párhuzamos
mérésből képezett átlag ± szórás formában láthatók. A kontrollhoz képest szignifikáns
különbség (Student-t teszt, p < 0,05) *-al jelölve látható (131)
A 4a vegyület szignifikánsan csökkenti az RCR-értéket (RCR = respiratory
control ratio, az „oxigénfogyasztás a 3-as állapotban” és az „oxigénfogyasztás a 4-es
állapotban” hányadosa). Mivel a vegyületek autooxidációja nem lehet felelős a
megnövekedett oxigénfogyasztásért, az „elfogyasztott” oxigén a mitokondriális

57
elektron-transzfer reakciók szubsztrátja lehet (132), ami a mitokondriális redox-státusz
megváltozásával járhat.
Annak tisztázása érdekében, hogy az oxigénfogyasztás összefüggésben áll-e az
ATP szintézis esetleges megváltozásával, az ATP szintáz (ATPáz) aktivitását
vizsgáltuk, a Meissner-féle módszer (93) szerint. A kísérlet eredménye szerint a
metoxiszármazék (4a) szignifikánsan megnöveli a mitokondriális ATPáz aktivitást,
anélkül, hogy hatással lenne az oxigénfogyasztásra a 3-as állapotban. (11. táblázat) Az
észlelt szétkapcsoló hatás főként egyes lipofil, gyengén savas karakterű kismolekulára
jellemző, amikor a megnövekedett O2-fogyasztás ellenére nem történik ATP szintézis.
(133) Miután a vizsgált vegyületek lipofil molekulák, de nem bírnak gyenge savi
karakterrel, további vizsgálatok szükségesek a szétkapcsoló hatás mechanizmusának
kiderítésére.
V.2.3.3. A kalkonanalógok befolyásolják a celluláris SH-szintet
Az α,β-telítetlen karbonilvegyületek citotoxikus hatását elsősorban az élő szervezetek
esszenciális, tiolcsoportot tartalmazó molekuláival szemben mutatott reaktivitásuknak
tulajdonítják. (35; 37) Az esetlegesen lejátszódó reakciók befolyásolják az
intracelluláris redox-státuszt, és ezáltal számos más folyamatot is. (45) A 4a és 4b
vegyületek celluláris tiol-csoportokkal kialakuló lehetséges kölcsönhatását első
lépésben a vegyületek celluláris SH-szintre gyakorolt hatása révén vizsgáltuk,
tápanyagmentes és tápanyagokat tartalmazó környezetben.
Az 1 (PBS-pufferben), 4 illetve 8 óráig (RPMI tápoldatban) tartó kezeléseket
követően, Ellmann-reagenssel történő meghatározások (94) eredményeit a sejtek
fehérjemennyiségére vonatkoztattuk. Az eredményeket a 12. táblázat foglalja össze.
A 4a vegyülettel történő 1 órás, tápanyagokat nem tartalmazó PBS-pufferben
történt kezelés alatt a celluláris SH-szint szignifikánsan csökkent az 1% DMSO-val
kezelt kontroll sejtekhez képest. 4b vegyület nem okozott ilyen jellegű változást.
A hosszabb ideig (4, illetve 8 óra) tartó, RPMI-tápoldatban történő inkubáció már
lehetőséget teremt a sejteknek, hogy reagáljanak a GSH-szintet érintő esetleges
változtatásokra. (134) A metoxivegyület (4a) ezen körülmények között is szignifikánsan
csökkentette az SH-szintet, ugyanakkor 4b vegyület nem változtatta meg szignifikáns
mértékben az SH-szintet.

58
Teljes celluláris tiol (SH-csoport) mennyiség
(nmol/mg fehérje)
1 óra (PBS) 4 óra (RPMI) 8 óra (RPMI)
Kontroll (DMSO 1%) 33,08 ± 0,35 124,42 ± 3,21 122,01 ± 3,89
4a (5,6 μM) 24,01 ± 0,53* 107,92 ± 2,30* 108,03 ± 2,05*
4b (23,3 μM) 31,84 ± 0,63 129,19 ± 1,52 114,08 ± 1,07
12. táblázat Celluláris SH-szint Jurkat T sejtekben a 4a és 4b vegyületekkel történő
sejtkezeléseket követően. A PBS-pufferben történt meghatározás tápanyagmentes
környezetet, az RPMI-tápoldatban történt mérés tápanyagokat tartalmazó környezetet
jelent. Az eredmények három párhuzamos mérésből képezett átlag ± szórás formában
láthatók. Az 1% DMSO-val kezelt kontroll sejtekhez képest szignifikáns különbség
(Student-t teszt, p < 0,05) *-al jelölve látható
Sejtes körülmények között a gyűrűs kalkonszármazékok egyrészt enzim-katalizált
konjugációs reakciók révén és/vagy a redukált SH-csoportok diszulfiddá történő
oxidációja révén képesek a tiol-státuszt befolyásolni. (56; 135) A kísérlet eredményei
arra utalnak, hogy a 4a vegyület, glutation-S-transzferáz enzim katalízise mellett,
nagyobb reaktivitást mutat SH-csoportokkal, mint 4b vegyület. (135)
V.2.3.4. A kalkonszármazékok hatása a Jurkat sejtek glutation státuszára
A kalkonanalógok SH-reaktivitásának vizsgálatát tovább folytatva, a Jurkat T sejtek 4a
és 4b vegyületekkel történő kezelését követően, a redukált glutation-szintet (GSH) és az
oxidált glutation-szintet (GSSG) orto-ftálaldehiddel történő derivatizálást követően,
nagyhatékonyságú folyadékkromatográfiás módszerrel határoztuk meg. (96)
A mintaelőkészítés során a sejtlízist három, egymást követő „freeze-and-thaw
ciklus” révén valósítottuk meg (136), nem az egyébként használatos ultrahangos
szonikálás révén, ugyanis az megnövelheti a sejtek GSSG-tartalmát. (137)
Az önmagában nem fluoreszkáló orto-flálaldehid, mint bifunkcionális reagens
egyszerre reagál a GSH molekula tiol- és primer aminocsoportjával, fluoreszkáló
izoindolszármazékot képezve. A keletkező termék 348 nm gerjesztési hullámhossz,
valamint 450 nm emissziós hullámhossz mellett detektálható. (138) A reakció pH-
függő, a redukált glutation OPA-val lejátszódó reakciójának optimális pH-értéke 8,0.
Ezen a pH-értéken az OPA nem reagál oxidált glutationnal, vagyis a GSH szelektíven

59
mérhető. Magas pH-érték mellett (≈ pH 12) azonban a GSSG is reakcióba lép OPA-val.
(133) Így pH 12 mellett a GSSG-tartalom is meghatározható. (138; 139; 140)
Annak érdekében, hogy az oxidált glutationmennyiség is szelektíven mérhető
legyen, a GSSG mérésekor a mintákban levő redukált glutationt N-etilmaleimiddel
(NEM) reagáltatjuk, amely a GSH tiolcsoportjához kapcsolódik. Ezáltal a GSH nem tud
reakcióba lépni OPÁ-val, illetve a konjugáció megvédi az oxidációtól is. (141) Ezt
alátámasztja az a vizsgálat is, amikor csak GSH-t tartalmazó standard oldathoz NEM-et
adtunk, majd azt a GSSG-szint meghatározásához használt módszer (IV.2.6.4) szerint
derivatizáltuk és vizsgáltuk. A kromatogramon nem láttunk csúcsot, igazolva a GSH
teljes „eliminálását” a mintából.
A kezeletlen és kalkonokkal kezelt sejtes minták glutation-koncentrációjának
meghatározása az OPA-val történő derivatizálást követően, a kísérleti részben leírt
fordított fázisú HPLC kromatográfiás kondíciók mellett történt. (IV.2.6.4) A
koncentráció meghatározása a GSH-t és GSSG-t tartalmazó standard sorozat mintáinak
mérése során felvett kalibrációs egyenesek segítségével történt. A meghatározott
redukált és oxidált glutation-szinteket a minták fehérjemennyiségére vonatkoztattuk. Az
eredmények (nmol/mg fehérje) a 22. ábra láthatók.
22. ábra A kalkonszármazékok celluláris glutation-szintre gyakorolt hatása Jurkat T
sejtekben a 4a és 4b vegyületekkel történő 4 órás kezelést követően. A kezeletlen és
kezelt sejtek (DMSO 1%, 4a és 4b) redukált glutation, valamint oxidált glutation
mennyiségének (nmol/mg fehérje) meghatározása OPA-val történő derivatizálást
követően HPLC-módszerrel történt. Az eredmények öt párhuzamos mérésből képezett
átlag ± szórás formában láthatók. A kezeletlen kontroll sejtekhez képest szignifikáns
különbség (Student-t teszt, p < 0,05) *-al jelölve.

60
A kísérleti eredmények alapján a metoxi- (4a), és a metilszubsztituált (4b)
kalkonszármazék eltérő módon befolyásolja a Jurkat sejtekben a glutation-szintet 4 órás
kezelést követően. A 4a vegyület nem befolyásolja jelentősen a szabad glutation-szintet
(GSH), de szignifikáns mértékben megnöveli az oxidált glutation (GSSG) mennyiségét
(0,67 ± 0,26 nmol/mg fehérje a kontroll sejtek esetén, 1,15 ± 0,33 nmol/mg fehérje 4a
esetén). A 4b származék azonban a redukált glutation mennyiségét növeli meg kis
mértékben (35,3 ± 6,6 nmol/mg fehérje mennyiségről 42,4 ± 7,2 nmol/mg fehérje-re),
de nem befolyásolja jelentősen a GSSG szintet a kezeletlen kontroll, illetve 1% DMSO-
val inkubált sejtekhez viszonyítva. (22. ábra)
A glutation-rendszer az egyik legfontosabb celluláris védekező mechanizmus az
élő sejtben. A glutation önmagában ROS scavengerként működik, de az intracelluláris
redox-homeosztázis szabályozásában is kiemelt szereppel bír. (127; 139) A sejt azon
képessége, hogy milyen mértékben képes glutationt termelni (akár a GSSG redukciója
révén, vagy de novo GSH szintézis révén) alapvetően meghatározza a sejt
hatékonyságát az oxidatív stressz kezelésében.
A 4 órás kezelési időtartam elég hosszú ahhoz, hogy a sejt reagáljon a glutation-
szintet érintő változtatásokra. (134) A metilszármazék (4b) az ismert kondíciók mellett
képes a GSH szintet kis mértékben megnövelni. Korábban is beszámoltak már arról,
hogy néhány elektrofil tulajdonsággal rendelkező Mannich bázis megnövelte a celluláris
tiol-szintet Jurkat T sejtekben (134), illetve HeLa sejtvonalakban (142). A
megnövekedett GSH szint nagy valószínűséggel az azt befolyásoló tényezők hatására
fellépő feedback mechanizmus következtében, de novo szintézis eredményeként
képződött GSH-nak tudható be. (134) A GSH-szint növekedése mögött az is állhat,
hogy a kalkon, mint α,β-telítetlen keton, a Keap1 fehérjék inaktiválása révén, aktiválja
az Nrf2 protein által irányított expressziós folyamatokat, többek között a γ-glutamil-
cisztein-szintetáz (γ-GCS), glutation-szintetáz, glutation-reduktáz és glükóz-6-foszfát-
dehidrogenáz enzimek fokozott kifejeződése révén megnöveli a GSH szintézist. (59)
Ezzel ellentétben a metoxianalóg (4a) a sejt oxidált GSSG-tartalmát növelte meg
szignifikáns mértékben, ami utalhat arra, hogy a vegyület valamilyen módon oxidatív
stresszt idéz elő. A GSSG-szint fiziológiás indikátora a ROS elleni intracelluláris
védekező mechanizmus aktivitásának, így alkalmas az oxidatív stressz in vivo
monitorozására is. (143; 144) A GSSG-szint emelkedésével párhuzamosan a GSH-szint
csökkenése nem volt megfigyelhető. Ez adódhat abból, hogy a kettő mennyisége között
nagyságrendbeli különbség van, és a GSH mennyiségben esetlegesen bekövetkező

61
változás nem érzékelhető ilyen módon. Ugyanakkor szerepe lehet annak is, hogy megnő
a glutation-reduktáz enzim aktivitása és/vagy megnő a γ-GCS aktivitása, annak
érdekében, hogy a sejt próbálja fenntartani a megfelelő glutation szintet. (128)
V.2.4. A kalkon-glutation konjugációs reakció vizsgálata
V.2.4.1. A vizsgált kalkonszármazékok intrinsic aktivitást mutatnak
glutationnal
A celluláris glutation-státusz megváltoztatásának a redox reakción kívül egy másik
lehetséges, a gyűrűs kalkonanalógok kémiai szerkezete alapján feltételezhető
mechanizmusa a vegyületek redukált glutationnal lejátszódó spontán és/vagy glutation-
S-transzferáz enzim által katalizált konjugációs reakciója.
Első lépésben a spontán lejátszódó reakciót vizsgáltuk sejtmentes körülmények
között. Ehhez a kalkonanalógokat és redukált glutationt ekvimoláris mennyiségben
tartalmazó, különböző pH-jú (pH = 5,5; 7,4; 9,0) oldatokat inkubáltunk 37 °C-on,
illetve 50°C-on, 1 és 3 órán keresztül. Az inkubátumok összetételét vékonyréteg-
kromatográfiás módszerrel, a kísérleti részben (IV.2.7.1) leírtak szerint vizsgáltuk. A
pH 7,4-es, illetve a pH 9,0-es oldatok esetén az 50 °C-os inkubátum, valamint a pH 9,0-
es, 37 °C-os inkubátum analízise során, a kettős előhívást követően, a kalkon, valamint
a GSH foltján kívül egy harmadik folt is láthatóvá vált. Ez a folt egy addukt jelenlétét
feltételezi, hiszen mind a két előhívási forma szerint látható volt, vagyis az aromás
vegyületekre jellemző UV-aktivitást is mutatja és a peptid (GSH) jelenlétére utaló
színreakciót is adja.
A GSH UV-fénnyel történő előhívás során nem ad jelet, azonban ninhidrinnel
ibolyáskék színreakciót ad, ugyanis a GSH három aminosavból álló tripeptid és az α-
aminosavak zöme semleges közegben ninhidrinnel igen érzékeny, kvantitatív célra is
alkalmas színreakciót nyújt. A kalkonszármazékok csak UV fényben történő előhívás
után adnak jelet. Mivel a konjugátumok GSH-t és a kiindulási kalkon vegyületet is
tartalmazzák, ezért ezek UV fényben, valamint ninhidrinnel történő előhívás során is
pozitív reakciót adnak. (104) Mindkét vizsgált származék (4a és 4b) esetén hasonlóak
voltak a tapasztalatok.

62
Gyengén savas körülmények között (pH = 5,5), ami egyébként jellemző kémhatás
tumoros sejtek környezetében, a vizsgált származékok nem mutatnak intrinsic aktivitást
glutationnal.
A kalkonszármazékok és GSH között lejátszódó nem enzim-katalizált reakció
további vizsgálata céljából egy fordított-fázisú nagyhatékonyságú-
folyadékromatográfiás módszer (RP-HPLC) kifejlesztése is megtörtént. (145) A
reakcióelegyben a GSH végső koncentrációja 5 x 10-2
M, a kalkon koncentrációja 5 x
10-3
M volt. A reakcióelegy pH értékének beállítása pH 8-ra történt, amely pH értéken a
GSH kb. 6%-a a sokkal erősebb nukleofil tiolát-anion formában található. (A GSH tiol-
csoportjának pKa értéke 9,2.) (146) Az inkubációs idő lejárta után (150, illetve 330
perc) a komponensek elválasztása fordított fázisú C-18 oszlopon történt UV-detektálás
mellett (λ = 260 nm). (145) Ezen inkubálási körülmények és kromatográfiás
paraméterek mellett a kromatogramon a kalkonanalógok (4a, illetve 4b) jele mellett,
további két csúcs jelent meg, feltételezhetően a kalkon-glutation konjugátum
diasztereomer párjának jele. (145)
A vékonyréteg-kromatográfiás eljárás, illetve az RP-HPLC vizsgálat (145)
eredményei arra utalnak, hogy a vizsgált vegyületek (4a és 4b) és redukált glutation
között spontán konjugációs reakció játszódik le, aminek során két egymással
diasztereomer viszonyban álló termékek keletkeznek. (23. ábra)
23. ábra A kalkonszármazékok redukált glutationnal (GSH) lejátszódó Michael-típusú
konjugációs reakciója
Az irodalomban is találhatók arra vonatkozó adatok, hogy α,β-telítetlen
karbonilvegyületek és glutation konjugációs reakciójában diasztereomer adduktumok

63
keletkeznek, például a nyílt láncú analóg, (E)-4-fenil-3-butén-2-on és GSH
reakciójában. (147) Hasonlóképpen, néhány ciklikus kalkonszármazék ((E)-2-
benzilidén-1-tetralon és -1-benzoszuberon) és ditiokarbaminsav (szintén kén-nukleofil
reagens) savkatalizálta reakciója során szintén két diasztereomer konjugátum
keletkezett, amit 1H-NMR vizsgálatokkal is alátámasztottak. (148)
A folyadékkromatográfiás vizsgálatok megerősítése céljából, illetve a feltételezett
GSH-adduktok szerkezetének igazolása céljából MALDI-TOF-MS, illetve
elektronspray ionizáció segítségével HPLC-ESI-MS vizsgálatok is történtek. A mérések
megerősítették a DAD-HPLC vizsgálatok eredményeit, miszerint az inkubátumokban
4a-GSH, illetve 4b-GSH adduktok (a protonált molekulatömegek 586,18 és 570,23)
keletkeznek spontán reakció révén. (145)
V.2.4.2. A kalkonanalógok és glutation konjugációs reakciójának vizsgálata
Jurkat T sejtekben
A 4a és 4b vegyületek sejtmentes körülmények között aktivitást mutatnak redukált
glutationnal szemben. A kifejlesztett RP-HPLC és csatolt ESI-MS eljárással (IV.2.7.2)
azt kívántuk vizsgálni, hogy a konjugációs reakció a Jurkat T sejtekben is lejátszódik-e,
hozzájárulva a vegyületek toxikus hatásának kialakításához.
A protokoll szerint (IV.2.2) kezelt sejteket lizáltuk, majd külön vizsgáltuk a
centrifugálás során kapott felülúszót és üledéket is. A megfelelő fehérjementesítés és
előkészítési eljárást követően felvett HPLC kromatogramokat (24. ábra), valamint ESI
tömegspektrumokat (25. ábra) összehasonlítottuk a kalkonanalógokat és glutationt
tartalmazó inkubátumok vizsgálatakor (145) kapott kromatogramokkal, illetve
spektrumokkal.
A vizsgálatok arra utalnak, hogy a 4a származék (279,2 Da protonált
molekulatömeggel) a sejtkezeléseket követően megtalálható mind a sejt felülúszóban,
mind a sejtes üledékben. A vegyület tehát a sejtkezelés során a Jurkat sejtek
membránján átjut és hatását intracellulárisan fejti ki. A 4a származékot és GSH-t
tartalmazó inkubátumban igazoltan keletkező 4a-GSH diasztereomerpár adduktok (145)
jelenléte azonban sem a sejt felülúszóból sem az üledékből nem bizonyítható.
A 4b vegyülettel kapott eredmények teljesen azonosak a 4a vegyület analízisekor
kapott eredményekkel. A 4b analóg jelenléte (m/z = 263,2) igazolható a felülúszóból, és
az üledékből is, de 4b-GSH konjugátumok ebben az esetben sem mutathatók ki.

64
Az előzetes, sejtmentes körülmények között elvégzett mérések eredményi arra
engedtek következtetni, hogy a vizsgált ciklikus kalkonanalógok citotoxikus hatásában
a redukált glutationnal lejátszódó konjugációs reakció is szerepet kap. A HPLC-ESI-MS
mérések azonban arra utalnak, hogy a feltételezett in vivo GST-katalizált reaktivitás 4a
illetve 4b vegyületek és GSH között nem járul hozzá a vegyületek apoptózist indukáló
hatásához.
24. ábra A Jurkat T sejtek 4a (5,6 μM) vegyülettel történő kezelése (4 óra), és
centrifugálása után kapott sejtfelülúszó és sejtes üledék vizsgálata során kapott
szelektált ion-kromatogramok (m/z = 279,2), valamint a 4a vegyületet (5 x 10-3
M) és
GSH-t (5 x 10-2
M) tartalmazó inkubátum (pH = 8,0; 50 °C) analízisekor kapott (145)
egymásra helyezett ion-kromatogramok (m/z = 279,2 és m/z = 586,2)

65
25. ábra A Jurkat T sejtek 4a (5,6 μM)(279,2 Da) vegyülettel történő kezelése (4 óra),
és centrifugálása után kapott sejtfelülúszó és sejtes üledék vizsgálata során kapott ESI
tömegspektrumok, valamint a 4a vegyületet (5 x 10-3
M) és GSH-t (5 x 10-2
M) tartalmazó
inkubátum (pH = 8,0; 50 °C) analízisekor kapott ESI tömegspektrumok (4a vegyület
(279,2 Da), valamint 4a-GSH addukt (586,2 Da)). (m/z = 50 – 2.000)
Ugyanakkor az MS mérések eredményei szerint nem keletkeznek olyan
molekulatömegű adduktumok sem, amelyek a vegyületek ciszteinil-glicin-, cisztein-
vagy esetleg N-acetil-cisztein-konjugátumai lennének, illetve a már előzetesen

66
esetlegesen metabolizálódott vegyület (pl. demetilált-, vagy hidroxilált-származék)
glutationnal képezett adduktumai.
Az eredmények mögött állhat a glutation-S-transzferáz enzim esetleges gátlása is.
Az irodalomban több esetben is beszámoltak róla, hogy α,β-telítetlen
karbonilvegyületek, közöttük kalkonok is, gátolják a GST enzim aktivitását. (149; 150;
151; 152) A GST enzim gátlása Jurkat T sejtekben sejtciklus gátlást és apoptózist
indukál. (153) A glutation-S-transzferáz enzim az egyik legfontosabb képviselője a
detoxikációs folyamatokban kulcsfontosságú szerepet játszó fázis 2 enzimeknek,
ugyanakkor számos humán daganatos sejtben túlexpresszálódik, és hozzájárul a
tumorsejtek kemoterápiás szerekkel szembeni rezisztenciájának kialakulásához. (154)
Az enzim gátlása révén gyógyszer-rezisztens tumorok újra érzékennyé tehetők a terápia
iránt. (149) A vizsgált kalkonszármazékok GST enzimaktivitásra gyakorolt hatása még
további vizsgálatokat igényel.

67
VI. Új eredmények összefoglalása, megbeszélés
Az előtanulmányok során összegyűjtött kísérleti tapasztalatok, valamint irodalmi adatok
alapján a kalkonok (1), valamint gyűrűs analógjaik (2-4) ígéretes kísérleti anyagok
lehetnek egy új szerkezeti típushoz tartozó, antineoplasztikus (citotoxikus és/vagy
citoprotektív) hatással rendelkező anyagok kutatására.
A természetben előforduló kalkonok és szintetikus analógjaik jól dokumentált
antibakteriális, antifungális, antiprotozoális, kemopreventív és citotoxikus aktivitással
rendelkeznek. A vizsgált anyagok és a kapcsolódó publikációk nagy száma ellenére az
irodalomban kevés olyan adat található, amelyek a megfigyelt biológiai hatások
molekuláris hátterének, a hatás szempontjából releváns molekuláris kölcsönhatások
szerkezet-hatás összefüggéseinek vizsgálatával foglalkoznak.
A téma kidolgozásának előzményeként mintegy száz, különbözőképpen
szubsztituált kalkon és ciklikus kalkonszármazék in vitro sejtproliferációt gátló
hatásának (IC50) meghatározása történt egér és humán daganatos sejtvonalakkal
szemben. A vegyületek biológiai hatását nagymértékben befolyásolja azok tér- és
elektronszerkezét meghatározó gyűrűtagszám és a kalkon molekulrész B-gyűrűjén lévő
szubsztituens elektronküldő, illetve elektronvonzó tulajdonsága. A vizsgált vegyületek
szerkezet-hatás összefüggése alapján a leghatékonyabbnak a héttagú gyűrűs (E)-2-(4’-
metoxibenzilidén)-1-benzoszuberon és az (E)-2-(4’-(dimetilaminobenzilidén)-1-
benzoszuberon származékok bizonyultak. (64)
Az e téren folytatandó további kutatómunka fő célkitűzése a gyűrűs
kalkonszármazékok fizikai-kémiai tulajdonságainak, valamint citotoxikus és
citoprotektív hatása molekuláris mechanizmusának alaposabb megismerése volt. A
gyűrűs származékok lipofilitásának jellemzésére számos, a 2-4 sorozatba tartozó
vegyületet vizsgáltunk, a hatásmechanizmus-vizsgálatokhoz a 7-tagú gyűrűs 4-metoxi-
(4a), valamint a 4-metilszubsztituált (4b) analógokat választottuk. (12. ábra)
Munkánk során kapott új eredmények:
1. Fordított fázisú vékonyréteg-kromatográfiás módszerrel számos, különböző módon
szubsztituált, öt-, illetve hattagú gyűrűs kalkonanalóg logP értékét határoztuk meg,
kiegészítve a korábban vizsgált 7-tagú gyűrűs vegyületek vizsgálata során kapott
eredményeket. Összefüggéseket kerestünk a vegyületek gyűrűtagszáma, a
szubsztituensek minősége és helyzete, valamint a molekulák lipofilitása között.

68
2. In vitro kísérletek során áramlásos citometriai módszerrel vizsgáltuk a vegyületek
sejtszaporodást gátló, valamint sejtciklusra gyakorolt hatását Jurkat T sejtekkel
szemben. A Jurkat T sejteknek a vegyületek közel ekvitoxikus dózisával történő 8
órás kezelést követően a metilszármazék (4b) esetén a sejtek legnagyobb hányada –
a kontroll sejtekhez hasonlóan – a sejtciklus G0/G1 fázisában, míg a
metoxivegyülettel (4a) történt kezelés esetén a G2/M fázisban volt található. A
metoxivegyülettel (4a) történt 24 órás kezelést követően a sejtek közel fele a G2/M
fázisban volt található, míg a metilszármazékkal (4b) kezelt sejtek csak kisebb
hányada volt ebben a fázisban. Hosszabb inkubálás (48 óra) során, mindkét kezelés
estén, jelentősen megnő az apoptotikus sejtek aránya, ugyanakkor poliploid sejtek
képződése is megfigyelhető. A poliploid sejtek aránya a metoxivegyület (4a)
esetében közel háromszor olyan magas volt, mint a metilszármazék esetén.
Ezek alapján megállapíthattuk, hogy az in vitro citotoxicitás azon alapszik, hogy a
vegyületek erős apoptózist indukáló szerek, a hatékonyabb vegyület (4a) esetén
megfigyelhető a G2/M fázisú sejtciklus gátlás, és jellemző a poliploid sejtek
képződése.
A vegyületek sejtciklusra gyakorolt hatása alapján feltételezhető, hogy azok
kölcsönhatást alakítanak ki celluláris makromolekulákkal (tubulinnal, DNS-sel),
valamint redukált glutationnal, és befolyást gyakorolnak a sejtek redox-státuszára.
3. A vizsgálandó vegyületek in vitro antioxidáns hatását úgy jellemeztük, hogy
vizsgáltuk, a vegyületek mennyire képesek gátolni a hidroxilgyök – a biológiai
rendszerekben képződő legreaktívabb oxigénszármazék – toxicitását. Vizsgáltuk a
vegyületek lehetséges pooxidáns aktivitását is, a dezoxiribóz degradációra
gyakorolt hatásuk révén. A 4b vegyület az alkalmazott tesztrendszerben mindvégig
antioxidáns aktivitást mutatott, a 4a vegyület a rövid távú antioxidáns hatás után
(főként az EDTA-t is tartalmazó környezetben) növelte a dezoxiribóz degradációját,
és átmenetileg prooxidáns aktivitást mutatott.
4. A vegyületek celluláris redox-státuszra gyakorolt hatásának vizsgálatakor
megállapítottuk, hogy a vegyületek nem növelik meg az intracelluláris ROS
aktivitást, sőt a metilszubsztituált analóg (4b) szignifikánsan csökkenti a ROS-
szintet és antioxidáns aktivitást mutat. Ez utóbbi eredmény összhangban áll az in

69
vitro antioxidáns hatást vizsgáló dezoxiribóz teszt eredményével, ami szerint a
vegyület in vitro hidroxilgyök-scavanger aktivitással rendelkezik.
A vegyületek apoptózist okozó hatásában az oxidatív stressz indukciója nagy
valószínűséggel nem játszik szerepet.
5. A vegyületek izolált patkánymáj mitokondriumok funkcióira gyakorolt hatásának
vizsgálatakor azt tapasztaltuk, hogy a 4a vegyület megnöveli a mitokondriális
oxigénfogyasztást és emeli az ATPáz aktivitást. A mitokondriális funkciók
megváltozása szerepet játszhat a vegyület citotoxikus hatásának kialakításában.
6. A celluláris redox-homeosztázist alapvetően befolyásolja a celluláris tiol-szint,
illetve a glutation-státusz. A gyűrűs kalkonanalógok celluláris tiolokkal, ezen belül
a sejtekben nagy mennyiségben jelen levő redukált glutationnal szemben mutatott
esetleges kölcsönhatását vizsgáltuk.
Megállapítottuk, hogy míg a 4a vegyület az oxidált glutation mennyiségét növelte
meg, és a redukált glutation-szintet nem befolyásolta, addig a 4b származék a
redukált glutation szintet (és a totál SH-szintet is) növelte. A redukált glutation-
szint növelése magyarázatot adhat a korábbi vizsgálatok során tapasztalt
antioxidáns aktivitás (ROS csökkentés) hátterére.
A 4a vegyület GSSG-szintet növelő hatása szerepet játszhat az apoptózis-
indukcióban.
7. A vegyületek glutationnal történő konjugációs reakciójának vizsgálatakor
megállapítottuk, hogy a vegyületek közel semleges és gyengén lúgos kémhatás
mellett, magasabb hőmérsékleten intrinsic aktivitást mutatnak redukált glutationnal
szemben. A reakció során keletkező kalkon-GSH adduktok jelenlétének igazolása
vékonyréteg-kromatográfiás, illetve RP-HPLC eljárásokkal, szerkezetük igazolása
HPLC-MS technika segítségével történt.
A kifejleszett módszerek lehetőséget nyújtottak a konjugációs reakció sejtes
körülmények közötti vizsgálatára is. A Jurkat T sejtekben a kalkonokkal való
kezeléseket követően, azonban csak a vizsgált származékot sikerült kimutatni,
konjugátumok jelenlétét nem. Az eredmény arra enged következtetni, hogy a
citotoxicitásban minden bizonnyal szerepet játszó, GSH-szintet befolyásoló hatás
nem a konjugációs-reakció következménye.

70
Összességében azt láthattuk, hogy bár a két vizsgált kalkonszármazék szerkezetét
tekintve csak egy oxigénatomban tér el egymástól, hatásmechanizmusuk igen
különböző.
A kevésbé toxikus 4-metilanalóg (4b) apoptózist indukál Jurkat T sejtekben, de
hatása elmarad a 4a vegyület sejtciklust befolyásoló hatásától. Mind az in vitro tesztben,
mind sejtes körülmények között antioxidáns aktivitást mutat, ami összefüggésben állhat
a redukált glutation-szintet emelő hatásával.
A kiemelkedő citotoxicitást mutató 4-metoxiszármazék (4a) drasztikusan
befolyásolja a sejtciklust, apoptózist indukál, poliploid sejtek képződését váltja ki. Nem
rendelkezik kifejezett antioxidáns hatással, sőt citotoxikus hatásának hátterében a
vegyület prooxidáns hatása is valószínűsíthető. Befolyásolja a mitokondriális
funkciókat, a celluláris SH-szintet, megnöveli a sejtek oxidált glutation tartalmát, ezáltal
befolyásolja a redox homeosztázist.
A sejtciklus G2/M fázisú, a vegyületek általi gátlásának egyik biokémiai háttere a
tubulin polimerizációs-depolimerizációs folyamatára gyakorolt hatás lehet. A
citotoxikus hatás, a fent említett hatások mellett, részben a vegyületek celluláris
makromolekulákkal (tubulin, DNS) történő nem-kovalens kölcsönhatásának
eredményeképpen alakulhat ki.
A kutatási programunk eredményeképpen tovább bővültek ismereteink a gyűrűs
kalkonszármazékok kettős – citotoxikus és citoprotektív (kemopreventív) – hatásának
mechanizmusát illetően. A kísérletek eredményei alapjául szolgálnak a további
munkának: a természetes és/vagy szintetikus kalkonszármazékok daganatellenes hatás
szempontjából történő komplex vizsgálatának.

71
VII. Irodalomjegyzék
1. Döbrössy, L.: A daganatos betegségek helyzete és várható alakulása. Kopper L.,
Jeney A. (Eds.): Onkológia - a géntől a betegágyig. Medicina Kiadó, Budapest.
2003.
2. Szeberényi, J.: Molekuláris sejtbiológia. Dialóg-Campus Kiadó, Budapest-Pécs.
1999
3. Ember, I.: Népegészségügyi orvostan. Dialog-Campus Kiadó, Budapest-Pécs. 2007.
4. Hoensch, H.P., Kirch, W., 2005. Potential role of flavonoids in the prevention of
intestinal neoplasia. International Journal of Gastrointestinal Cancer 35, 187-196.
5. Cutler, G.J., Nettleton, J.A., Ross, J.A., Harnack, L.J., Jacobs, D.R., Scrafford,
C.G., Barraj, L.M., Mink, P.J., Robien, K., 2008. Dietary flavonoid intake and risk
of cancer in postmenopausal women: The Iowa Women's Health Study.
International Journal of Cancer 123, 664-671.
6. Petri, G.: Gyógynövény és drogismeret. Medicina, Budapest. 1991.
7. Carlo, G.D., Mascolo, N., Izzo, A.A., Capasso, F., 1999. Flavonoids: Old and new
aspects of a class of natural therapeutic drugs. Life Sciences 65, 337-353.
8. Crozier, A., Burns, J., Aziz, A.A., Stewart, A.J., Rabiasz, H.S., Jenkins, G.I.,
Edwards, C.A., Lean, M.E., 2000. Antioxidant flavonols from fruits, vegetables and
beverages: measurments and bioavailability. Biological Research 33, 79-88.
9. Havsteen, B.H., 2002. The biochemistry and medical significance of the flavonoids.
Pharmacology and Therapeutics 96, 67-202.
10. Moon, Y.J., Wang, X., és Morris, M.E., 2006. Dietary flavonoids: Effects on
xenobiotic and carcinogen metabolism. Toxicology in Vitro 20, 187-210.
11. Cushnie, T.P.T. és Lamb, A.J., 2005. Antimicrobal activity of flavonoids.
International Journal of Antimicrobial Agents 26, 343-356.
12. Garg, A., Garg, S., Zaneveld, L.J.D., Singla, A.K., 2001. Chemistry and
pharmacology of the citrus bioflavonoid hesperidin. Phytotherapy Research 15,
655-669.
13. Jacobs, B.P., Dennehy, C., Ramirez, G., Sapp, J., Lawrence, V.A., 2002. Milk
thistle for the treatment of liver disease: a systematic review and meta-analysis. The
American Journal of Medicine 113, 506-515.

72
14. Ratheesh, M., Shyni, G.L., Sindhu, G., Helen, A., 2011. Inhibitory effect of Ruta
graveolens L. on oxidative damage, inflammation and aortic pathology in
hypercholesteremic rats. Experimental and Toxicologic Pathology 63, 285-290.
15. Seigler, D.S.: Plant secondary metabolism. Kluwer Academic Publishers, 1998.
16. Dixon, R.A., Steele, C.L., 1999. Flavonoids and isoflavonoids - a gold mine for
metabolic engineering. Trend in plant science (reviews) 4, 394-400.
17. Winkel-Shirley, B., 2001. Flavnoid biosynthesis. A colorful model for genetics,
biochemistry, cell biology and biotechnology. Plant Physiology 126, 485-493.
18. Rozmer, Zs. és Perjési, P. Naturally occuring chalcones and their biological
activity. Phytochemistry Reviews Közlésre beküldve.
19. Fenwick, G.R., Lutomski, J., Nieman, C., 1990. Liquorice, Glycyrrhiza glabra -
Composition, usues and analysis. Food Chemistry 38, 119-143.
20. Kang, D.G., Lee, A.S., Mun, Y.J., Woo, W.H., Kim, J.C., Sohn, E.J., Moon, M.K.,
Lee, H.S., 2004. Butein ameliorates renal concentrating ability in cisplatin-induced
acutes renal failure in rats. Biological and Pharmaceutical Bulletin 27, 366-370.
21. Lee, J.C., Lee, K.Y., Kim, J., Na, C.S., Jung, N.C., Chung, G.H., Jang, Y.S., 2004.
Extract from Rhus verniciflua Stokes is capable of inhibiting the growth of human
lymphoma cells. Food Chemistry and Toxicology 42, 1383-1388.
22. Nowakowska, Z., 2007. A review of anti-infective and anti-inflammatory
chalcones. European Journal of Medicinal Chemistry 42, 125-137.
23. ElSohly, H.N., Joshi, A.S., Nimrod, A.C., Walker, L.A., Clark, A.M., 2001.
Antifungal chalcones from Maclura tinctoria. Planta Medica 67, 87-89.
24. Buckwold, V.E., Wilson, R.J.H., Nalca, A., Beer, B.B., Voss, T.G., Turpin, J.A.,
Buckheit, R.W., Wei, J., Wenzel-Mathers, M., Walton, E.M., Smith, R.J., Pallansh,
M., Ward, P., Wells, J., Chuvala, L., Sloane, S., Paulman, R., Russell, J., Hartman,
T., Ptak, R., 2004. Antiviral activity of hop constituents against a series of DNA
and RNA viruses. Antiviral Research 61, 57-62.
25. Sen, R., Chatterjee, M., 2011. Plant derived therapeutics for the treatment of
Leishmaniasis. Phytomedicine 18, 1056-1069.
26. Zhang, X., Yeung, E.D., Wang, J., Panzhinskiy, E.E., Tong, C., Li, W., Li, J., 2010.
Isoliquiritigenin, a natural anti-oxidant, selectively inhibits the proliferation of
prostate cancer cells. Clinical and Experimental Pharmacology and Physiology 37,
841-847.

73
27. Szliszka, E., Czuba, Z.P., Mazur, B., Sedek, L., Paradysz, A., Krol, W., 2010.
Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells. International
Journal of Molecular Sciences 11, 1-13.
28. Haraguchi, H., Ishikawa, H., Mizutani, K., Tamura, Y., Kinoshita, T., 1998.
Antioxidative and superoxide scavenging activities of retrochalcones in
Glycyrrhiza inflata. Bioorganic and Medicinal Chemistry 6, 339-347.
29. An, W., Yang, Y., Ao, Y., 2006. Metallothionein mediates cardioprotection of
isoliquiritigenin against ischemia-reperfusion through JAK2/STAT3 activation.
Acta Pharmacologica Sinica 27, 1431-1437.
30. Enoki, T., Ohnogi, H., Nagamine, K., Kudo, Y., Sugiyama, K., Tanabe, M.,
Kobayashi, E., Sagawa, H., Kato, I., 2007. Antidiabetic activities of chalcones
isolated from a Japanese herb, Angelica keiskei. Journal of Agricultural and Food
Chemistry 55, 6013-6017.
31. Dong, F., Jian, C., Zhenghao, F., Kai, G., Zuliang, L., 2008. Synthesis of chalcones
via Claisen-Schmidt condensation reaction catalyzed by acyclic acidic ionic liquids.
Catalysis Communications 9, 1924-1927.
32. Fan, X.S., Zhang, Y.M., 2002. A simple access to chalcones via modified
Mukaiyama aldol condensation promoted by SmI3/TMSCl. Chinese Journal of
Chemistry 20, 198-201.
33. Eddarir, S., Cotelle, N., Bakkour, Y., Rolando, C., 2003. An efficient synthesis of
chalcones based on the Suzuki reaction. Tetrahedron Letters 44, 5359-5363.
34. Briot, A., Baehr, C., Brouillard, R., Wagner, A., Mioskowski, C., 2004. Concise
synthesis of dihydrochalcones via palladium-catalyzed coupling of aryl halides and
1-aryl-2-propen-1-ols. Journal of Organic Chemistry 69, 1374-1377.
35. Dimmock, J.R., Elias, D.W., Beazely, M.A., Kandepu, N.M., 1999. Bioactivities of
chalcones. Current Medicinal Chemistry 6, 1125-1149.
36. Ni, L., Meng, C.Q., Sikorski, J.A., 2004. Recent advances in therapeutic chalcones.
Expert Oponion on Therapeutic Patents 14, 1669-1691.
37. Go, M.L., Wu, X., Liu, X.L., 2005. Chalcones: an update on cytotoxic and
chemoprotective properties. Current medicinal chemistry 12, 483-499.
38. Dimmock, J.R., Raghavan, S.K., Logan, B.M., Bigam, G.E., 1983. Antileukemic
evaluation of some Mannich bases derived from 2-arylidene-1,3-deiketones.
European Journal of Medicinal Chemistry 18, 248-254.

74
39. Benvenuto, J.A., Connor, T.H., Monteith, D.K., Laidlaw, J.L., Adams, S.C.,
Matney, T.S., Theiss, J.C., 1993. Degradation and inactivation of antitumor drugs.
Journal of Pharmaceutical Science 82, 988-991.
40. Ádám, V.: Orvosi biokémia. Medicina Könykiadó, Budapest, 2001.
41. Sen, C.K., 1997. Nutritional biochemistry of cellular glutathione. (Review).
Nutritional Biochemistry 8, 660-672.
42. Dimmock, J.R., Kandepu, N.M., Hetherington, M., Quail, J.W., Pugazhenthi, U.,
Sudom, A.M., Chamankhah, M., Rose, P., Pass, E., Allen, T.M., Halleran, S.,
Szydlowski, J., Mutus, B., Tannous, M., Manavathu, E.K., Myers, T.G., De Clercq,
E., Balzarini, J., 1998. Cytotoxic activities of Mannich bases of chalcones and
related compounds. Journal of Medicinal Chemistry 41, 1014-1026.
43. Sabzevari, O., Galati, G., Moridani, M.Y., Siraki, A., O'Brien, P.J., 2004.
Molecular cytotoxic mechanisms of anticancer hydroxychalcones. Chemico-
Biological Interactions 148, 57-67.
44. Esterbauer, H., Zollner, H., Scholz, N., 1975. Reaction of glutathione with
conjugated carbonyls. Zeitschrift für Naturforschung C 30, 466-473.
45. Powis, G., Gasdaska, J.R., Baker, J.R.: Redox signaling and the controll of cell
growth and death. In: Sies, H. (Ed.), Antioxidants in Disease Mechanism and
Therapy. In:Advances in Pharmacology, vol. 38. Academic Press, San Diego, pp
329-359. 1997.
46. Nam, N.H., Kim, Y., You, Y.J., Hong, D.H., Kim, H.M., Ahn, B.Z., 2003.
Cytotoxic 2',5'-dihydroxychalcones with unexpected antiangiogenic activity.
European Journal of Medicinal Chemistry 38, 179-187.
47. Navarini, A.L.F., Chiaradia, L.D., Mascarello, A., Fritzen, M., Nunes, R.J., Yunes,
R.A., Creczynski-Pasa, T.B., 2009. Hydroxychalcones induce apopotosis in B16-
F10 melanoma cells via GSH and ATP depletion. European Journal of Medicinal
Chemsitry 44, 1630-1637.
48. Hsu, Y.L., Kuo, P.L., Tzeng, W.S., Lin, C.C., 2006. Chalcone inhibits the
proliferation of human breast cancer cell by blocking cell cycle progression and
inducing apoptosis. Food and Chemical Toxicology 44, 704-713.
49. Rao, Y.K., Fang, S.H., Tzeng, Y.M., 2004. Differential effects of synthesized 2'-
oxygenated chalcone derivatves: modulation of human cell cycle phase distribution.
Bioorganic and Medicinal Chemistry 12, 2679-2686.

75
50. Satomi, Y., 1993. Inhibitory effects of 3'-methyl-3-hydroxy-chalcone on
proliferation of human malignant tumor cells and on skin carcinogenesis.
International Journal of Cancer 55, 506-517.
51. Middleton, E., Kandaswami , C., Theoharides, T.C., 2000. The effect of plant
flavonoids on mammalian cells: Implications for inflammation, heart disease and
cancer. Pharmacological Reviews 52, 673-751.
52. Gutteridge, J.M.C., 1987. Ferrous-salt-promoted damage to deoxyribose and
benzoate. The increased effectiveness of hydroxyl-radical scavengers in the
presence of EDTA. Biochemical Journal 243, 709-714.
53. Gregus, Z., Klaasen, C.D.: Mechanism of toxicity. In: Klaasen, C.D. (Ed.), Casarett
and Dull's Toxicology. The Basic Science of Poisons. Sixth ed. McGraw-Hill, New
York, (Chapter 3.) pp. 35-81. 2001.
54. Prochaska, H.J., Fernandes, C.L., 1993. Elevation of serum Phase II enzymes ba
anticarcinogenic enzym inducers: markers for a chemoprotrcted state?
Carcinogenesis 14, 2441-2449.
55. Dinkova-Kostova, A.T., Abeygunawardana, C., Talalay, P., 1998. Chemoprotective
properties of phenylpropenoids, bis(benzylidene)cycloalkanones, and related
Michael reaction acceptors: correlation of potencies as Phase 2 enzyme inducers
and radical scavengers. Journal of Medicinal Chemistry 41, 5287-5296.
56. Dinkova-Kostova, A.T., Massiah, M.A., Bozak, R.E., Hicks, R.J., Talalay, P.,
2001. Potency of Michael reaction acceptors as inducers of enzymes that protect
against carcinogenesis depends on their reactivity with sulfhydryl groups.
Proceedings of the National Academy of Sciences 98, 3404-3409.
57. Dinkova-Kostova, A.T., Holtzclaw, W.D., Kensler, T.W., 2005. The role of Keap1
in cellular protective responses. Chemical Research in Toxicology 18, 1779-1791.
58. Dinkova-Kostova, A.T., Holtzclaw, W.D., Wakabayashi, N., 2005. Keap1, the
sensor for electrophiles and oxidants that regulates the Phase 2 response, is a zinc
metalloprotein. Biochemistry 44, 6889-6899.
59. Motohashi, H., Yamamoto, M., 2004. Nrf2-Keap1 defines a physiologically
important stress response mechanism. Trends in Molecular Medicine 10, 549-557.
60. Izumi, Y., Matsumura, A., Wakita, S., Akagi, K., Fukuda, H., Kume, T., Irie, K.,
Takada-Takatori, Y., Sugimoto, H., Hashimoto, T., Akaike, A. 2012. Isolation,
identification and biological evaluation of Nfr2-ARE activator from the leaves of

76
green perilla (Perilla frutescens var. crispa f. viridis). Free Radical Biology and
Medicine 53, 669-679.
61. Hashimoto, Y., Kagechika, H., Kawachi, E., Shudo, K., 1987. New-type inducers
of differentation of HL-60 leukemia cells suppress c-myc expression. Chemical and
Pharmaceutical Bulletin 35, 3190-3194.
62. Forejtníková, H., Lunerová, K., Kubinová, R., Jankovská, D., Marek, R., Kares, R.,
Suchy, V., Vondrácek, J., Machala, M., 2005. Chemoprotective and toxic potentials
of synthetic and naural chalcones and dihydrochalcones in vitro. Toxicology 208,
81-93.
63. Iwata, S., Nishino, T., Inoue, H., Nagata, N., Satomi, Y., Nishino, H., Shibata, S.,
1997. Antitumorigenic activities of chalcones. (II) Photo-isomerization of
chalcones and the correlation with their biological activities. Biological and
Pharmaceutical Bulletin 20, 1266-1270.
64. Dimmock, J.R., Kandepu, N.M., Nazarali, A.J., Kowalchuk, T.P., Motaganahalli,
N., Quail, J.W., Mykiytiuk, P.A., Audette, G.F., Prasad, L., Perjési, P., Allen, T.M.,
Santos, C.L., Szydlowski, J., De Clercq, E., Balzarini, J., 1999. Conformational and
quantitative structure-activity relationship study of cytotoxic 2-
arylidenebenzocycloalkanones. Journal of Medicinal Chemistry 42, 1358-1366.
65. Perjési, P., Nusser, T., Tarczay, Gy., Sohár, P., 1999. E-2-Bezylidene-
benzocycloalkanones. Stereostructure and NMR spectroscopic investigations.
Journal of Molecular Structure 479, 13-19.
66. Tarczay, Gy., Vékey, K., Ludányi, K., Perjési, P., Sohár, P., 2000. E-2-
benzylidenebenzocyclanones. II. IR and mass spectrometric investigations. Journal
of Molecular Structure 520, 97-102.
67. Perjési, P., Perjéssy, A., Kolehmainen, E., Ősz, E., Samalikova, M., Linnato, J.,
Virtanen, E., 2004. E-2-Benzylidenebenzocycloalkanones III. Studies on
transmission of substituent effects on IR carbonyl stretching frequencies and 13C
NMR chemical shifts of E-2-(X-benzylidene)-1-indanones. Comparison with the IR
data of E-2-(X-benzylidene)-1-indanones. Journal of Molecular Structure 697, 41-
47.
68. Perjési, P., Linnanto, J., Kolehmainen, E., Ősz, E., Virtanen, E., 2005. E-2-
Benzylidenebenzocycloalkanones IV. Studies on transmission of substituent effects
on 13C NMR chemical shifts of E-2-(X-benzylidene)-1-tetralones and

77
benzosuberones. Comparison with the 13C NMR data of chalcones and E-2-(X-
benzylidene)-1-indanones. Journal of Molecular Structure 740, 81-89.
69. Dimmock, J.R., Zello, G.A., Oloo, E.O., Quail, J.W., Kraatz, H.B., Perjési, P.,
Aradi, F., Takács-Novák, K., Allen, T.M., Santos, C.L., Balzarini, J., De Clercq, E.,
Stables, J.P., 2002. Correlations between citotoxicity and topography of some 2-
arylidenebenzocycloalkanones determined by X-ray crystallography. Journal of
Medicinal Chemistry 45, 3103-3111.
70. Miyata, M., Furukawa, M., Takahashi, K., Gonzalez, F.J., Yamazoe, Y., 2001.
Mechanism of 7,12-Dimethylbenz[a]anthracene-induced immunotoxicity: Role of
metabolic activation at the target organ. Japanese Journal of Pharmacology 86,
302-309.
71. Perjési, P., Bayer, Z., Ember, I., 2000. Effect of E-2-(4'-methoxybenzylidene)-1-
benzosuberone on the 7,12-dimethylbenz[alpha]anthracene-induced onco/supressor
gene action in vivo.I: A 24-hour experiment. Anticancer Research 20, 475-481.
72. Perjési, P., Gyöngyi, Z., Bayer, Z., 2000. Effect of E-2-(4'-methoxybenzylidene)-1-
benzosuberone on the 7,12-dimethylbenz[alpha]anthracene-induced onco/supressor
gene action in vivo II: A 48-hour experiment. Anticancer Research 20, 1839-1848.
73. Pitot, H.C., Dragon, Y.P.: Chemical carcinogenesis. In: Klaassen, C.D. (Ed.),
Casarett and Dull's Toxicology. McGraw-Hill, New York, pp. 241-320. 2001.
74. Larsen, M.C., Angus, W.G.R., Brake, P.B., Eltom, S.E., Sukow, K.A., Jefcoate,
C.R., 1998. Characterization of CYP1B1 and CYP1A1 expression in human
mammary epithelial cells: Role of the aryl hydrocarbon receptor in polycyclic
aromatic hydrocarbon metabolism. Cancer Research 58, 2366-2374.
75. Michejda, C.J., Kroeger Koepke, M.B., 1994. Carcinogen activation by sulfate
conjugate formation. Advances in Pharmacology 27, 331-363.
76. Surh, Y.J., Miller, J.A., 1994. Roles of electrophilic sulfuric acid ester metabolites
in mutagenesis and carcinogenesis by some polynuclear aromatic hydrocarbons.
Chemico-Biological Interactions 92, 351-362.
77. Monostory, K., Tamási, V., Vereczkey, L., Perjési, P., 2003. A study on CYP1A
inhibitory action of E-2-(4'-methoxybenzylidene)-1-benzosuberone and some
related chalcones and cyclic chalcone analogues. Toxicology 184, 203-210.
78. Tute, M.S.: Lipophilicity: A History. In: Mannhold, R., Kubinyi, H. and
Timmerman, H. (Eds), Methods and principles in medicinal chemistry, Vol. 4,

78
Lipophilicity in Drug Action and Toxicology (Pliska, V., Testa, B. and van de
Waterbeemd, H., Eds.) VHC, Weinheim, FRG. Chap. 2., pp. 7-26. 1996.
79. Takács-Novák, K., 1997. A megoszlási hányados GLP szerinti meghatározásának
praktikus szempontjai. Acta Pharmaceutica Hungarica 67, 179-191.
80. Takács-Novák, K., Perjési, P., Vámos, J., 2001. Determination of logP for
biologically active chalcones and cyclic chalcone analogs by RPTLC. Journal of
Planar Chromatogrphy 14, 42-46.
81. Almási, J., Takács-Novák, K., Kökösi, J., Vámos, J., 1999. Characterization of
potential NMDA and cholecystokinin antagonists: II. Lipophilicity studies on 2-
methyl-4-oxo-3H-quinazoline-3-alkyl-carboxylic acid derivatives. International
Journal of Pharmaceutics 180, 13-22.
82. Tennant, J.R., 1964. Evaluation of the trypan blue technique for determination of
cell viability. Transplantation 12, 685-694.
83. Givan, A.L.: Flow Cytometry: First Principles. New York, NY-Wiley-Liss., 1992.
84. Vermes, I., Haanen, C., Steffens-Nakken, H., Reutelinsperger, C., 1995. A novel
assay for apoptosis. Flow-cytometric detection of phosphatidylserin expression on
early apoptotic cells using fluorescein labeled Annexin V. Journal of
Immunological Methods 184, 39-51.
85. Del Bino, G., Darzynkiewicz, Z., Degraef, C., Mosselmans, R., Fokan, D., Galand,
P., 1999. Comparison of methods based on annexin V binding, DNA content or
TUNEL for evaluating cell death in HL-60 and adherent MCF-7 cells. Cell
Proliferation 32, 27-37.
86. Cheng, Z., Li, Y., Chang, W., 2003. Kinetic deoxyribose degradation assay and its
application in asessing the antioxidant activities of phenolic compounds in a
Fenton-type system. Analytica Chimica Acta 478, 129-137.
87. Halliwell, B., Gutteridge, J.M.C., Auroma, O.I., 1987. The deoxyribose method: A
simple "test-tube" assay for determination of rate constants for reactions of
hydroxyl radicals. Analytical Biochemistry 165, 215-219.
88. Aronis, A., Melendez, J.A., Golan, O., Shilo, S., Dicter, N., Tirosh, O., 2003.
Potentiation of FAS-mediated apoptosis by attenuated production of mitochondria-
derived reactive oxygen species. Cell Death and Differentiation 10, 335-344.
89. Frei, G.M., Lebenthal, I., Albeck, M., Albeck, A., Sredni, B., 2007. Neutral and
positively charged thiols synergize the effect on the immunomodulator AS101 as a

79
growth inhibitor of Jurkat cells, by increasing its uptake. Biochemical
Pharmacology 74, 712-722.
90. Johnson, D., Lardy, H., 1967. Isolation of liver or kidney mitochondria. Methods in
Enzymology 10, 94-96.
91. Hartree, R.F., 1972. Determination of protein: A modification of Lowry method
that gives a linear photometric response. Analytical Biochemistry 48, 422-427.
92. Estabrook, R.E., 1967. Mitochondrial respiratory control and the polarographic
measurment of ADP/O ratios. Methods in Enzymology 10, 41-47.
93. Meissner, G., 1974. Isolation of sarcoplasmic reticulum from skeletal muscle.
Methods in Enzymology 31, 238-246.
94. Ellmann, G.L., 1958. A colorimetring method for determining low concentrations
of mercaptans. Archives of Biochemistry and Biophysics 74, 443-450.
95. Smith, P.K., Krohn, R.I., Hermanson, G.T., Mallia, A.K., Gartner, F.H.,
Provenzano, M.D., Fujimoto, E.K., Goeke, N.M., Olson, B.J., Klenk, D.C., 1985.
Measurment of protein using bicinchoninic acid. Analytical Biochemistry 150, 76-
85.
96. Kand'ár, R., Záková, P., Lotková, H., Kucera, O., Cervinková, Z., 2007.
Determination of reduced and oxidized glutathione in biological samples using
liquid chromatography with fluorimetric detection. Journal of Pharmaceutical and
Biomedical Analysis. 43, 1382-1387.
97. Testa, B., Crivori, P., Reist, M., Carrupt, P.A., 2000. The influence of lipophilicity
on the pharmacokinetic behavior of drugs: concepts and examples. Perspectives in
Drug Discovery and Design 19, 179-211.
98. Zhou, L., Wang, J., 2009. Physico-chemical characterization in drug discovery.
Trends in bio/pharmaceutical industry. Preclinical formulation 2009, 12-18.
99. Takács-Novák, K., Völgyi, G., 2005. A fizikai-kémiai jellemzés helye és módszerei
a gyógyszerkutatásban. Magyar Kémiai Folyóirat 111, 169-176.
100.Lambert, W.J., 1993. Modeling oil-water partitioning and membrane permeation
using reversed-phase chromatography. Journal of Chromatography A 656, 469-
484.
101.Rozmer, Zs., Perjési, P., Takács-Novák, K., 2006. Use of RP-TLC for
determination of logP of isomeric chalcones and cyclic chalcone analogues.
Journal of Planar Chromatography 19, 124-128.

80
102.Vashishtha, S.C., Nazarali, A.J., Dimmock, J.R., 1998. Application of fluorescence
microscopy to measure apoptosis in Jurkat T cells after treatment with new
investigational anticancer agent. Cellular and Molecular Neurobiology 18, 437-
445.
103.Schoene, N.W., Kamara, K.S., 1999. Population doubling time, phosphatase
activity, and hydrogen peroxide generation in Jurkat cells. Free Radical Biology
and Medicine 27, 364-369.
104.Rozmer, Zs., Berki, T., Perjési, P., 2006. Different effects of two cyclic chalcone
analogues on cell cycle of Jurkat T cells. Toxicology in Vitro 20, 1354-1362.
105.Cook, J.A., Mitchell, J.B., 1989. Viability measurments of mammalian cell
systems. Analytical Biochemistry 179, 1-7.
106.Mesner, P.W., Kaufman, S.H.: Methods utilized in the study of apoptosis. In:
Kaufman S.H. (ed.), Apoptosis. Pharmacological Implications and Therapeutic
Oppurtunities. In: Advances in Pharmacology, Vol. 41. Academic Press, San
Diego, pp. 57-87.
107.Dean, P.N.: Data analysis in cell kinetics research. In: Gray, J.W., Darzynkiewicz,
Z.( Eds.) Techniques In Cell Cycle Research. Humana Press, Clifton, New Jersey,
pp. 207-253. 1987.
108.Murray, A., 1994. Cell cycle checkpoints. Current Opinion in Cell Biology 6, 872-
876.
109.Diffey, J.F.X., Evan, G., 2000. Oncogenes and cell proliferation. Cell cycle,
genome integrity and cancer - a millennial view. Current Opinion in Genetics and
Development 10, 13-16.
110.Lepley, D.M., Li, B., Birt, D.F., Pelling, J.C., 1996. The chemopreventive
flavonoid apigenin induces G2/M arrest in keratinocytes. Carcinogenesis 17, 2367-
2375.
111.Rao, Y.K., Fang, S.H., Tzeng, Y.M., 2004. Differential effects of synthesized 2'-
oxygenated chalcone derivatives: modulation of human cell cycle phase
distribution. Bioorganic and Medicinal Chemistry 12, 2679-2686.
112.Rao, Y.K., Fang, S.H., Tzeng, Y.M., 2005. Synthesis, growth inhibition, and cell
cycle evaluations of novel flavonoid derivatives. Bioorganic and Medicinal
Chemistry 13, 6850-6855.

81
113.Hsu, Y.L., Kuo, P.L., Tzeng, W.S., Lin, C.C., 2006. Chalcone inhibits the
proliferation of human breast cancer cell by blocking cell cycle progression and
inducing apoptosis. Food and Chemical Toxicology 44, 704-713.
114.Kimler, B.F., Schneiderman, M.H., Leeper, D.B., 1978. Induction of concentration-
dependent blockade in the G2 phase of the cell cycle by cancer chemotherapeutic
agents. Cancer Research 38, 809-814.
115.Konopa, J., 1988. G2 block induced by DNA crosslinking agents and its possible
consequences. Biochemical Pharamcology 37, 2303-2309.
116.Erenpreisa, J., Cragg, M.S., 2001. Mitotic death: a mechanism of survival? A
review. Cancer Cell International 1, 1-7.
117.Pilatova, M., Varinska, L., Perjési, P., Sarissky, M., Mirossay, L., Solar, P., Ostro,
A., Mojzis, J., 2010. In vitro antiproliferative and antiangiogenic effects of
synthetic chalcone analogues. Toxicology in Vitro 24, 1347-1355.
118.Winterbourn, C.C., 1991. Factors that influence the deoxyribose oxidation assay for
Fenton reaction products. Free Radical Biology and Medicine 11, 353-360.
119.Rozmer, Zs., Perjési, P., 2005. Néhány nem-szteroid gyulladáscsökkentő hatásának
vizsgálata a 2-deoxi-D-ribóz Fenton-reakció inicializálta degradációjára. Acta
Pharmaceutica Hungarica 75, 69-75.
120.Galey, J.B.: Antioxidants in Disease Mechanism and Therapy. Sies H. (Ed.)
Academic Press, San Diego, pp. 167-203. 1997.
121.Halliwell, B.: Antioxidants in Disease Mechanism and Therapy. Sies H. (Ed.)
Academic Press, San Diego, pp. 3-20. 1997.
122.Chobot, V., 2010. Simultaneous detection of pro- and antioxidative effects in the
variants of the deoxyribose degradation assay. Journal of Agricultural and Food
Chemistry 58, 2088-94.
123.Loo, G., 2003. Redox-sensitive mechanism of phytochemical-mediated inhibition
of cancer cell proliferation. Journal of Nutritional Biochemistry 14, 64-73.
124.Croft, S., Gilbert, B.C., Lidsay Smith, J.R., Whithwood, A.C., 1992. An ESR
investigation of the reactive intermediate generated in the reaction between iron and
hydrogen peroxide in aqueous solution. Direct evidence for formation of the
hydroxyl radicals. Free Radical Research Communication 17, 21-39.
125.Prokai, L., Prokai-Tátrai, K., Perjési, P., Zharikova, A.D., Perez, E.J., Liu, R.;
Simpkins, J.W., 2003. Quinol-based cyclic antioxidant mechanism in estrogen

82
neuroprotection. Proceedings of the National Academy of Sciences 100, 11741-
11746.
126.Perjési, P., Rozmer, Zs., 2011. Kinetic analysis of some chalcones and synthetic
chalcone analogues on the Fenton-reaction initiated deoxyribose degradation assay.
The Open Medicinal Chemistry Journa. 5, 61-67.
127.Curtin, J.F., Donovan, M., Cotter, T.G., 2002. Regulation and measurment of
oxidative stress in apoptosis. Journal of Immunological Methods 265, 49-72.
128.Winter, E., Chiaradia, L.D., Cordova, C.A.S., Nunes, R.J., Yunes, R.A.;
Creczynski-Pasa, T.B., 2010. Naphthylchalcones induce apoptosis and caspase
activation in a leukemia cell line: The relationship between mitochondrial damage,
oxidative stress, and cell death. Bioorganic and Medicinal Chemistry 18, 8026-
8034.
129.Wallace, D.C., 1999. Mitochondrial diseases in man and mouse. Science 283, 1148-
1482.
130.Tomeckova, V., Perjési, P., Guzy, J., Kusnir, J., Chovakova, Z., Chavkova, Z.,
Marekova, M., 2004. Comparison of effect of selected synthetic chalcone
analogues on mitochondrial outer membrane determined by fluorescence
spectroscopy. Journal of Biochemical and Biophysical Methods 61, 135-141.
131.Perjési , P., Kubalkova, J., Chovanova, Z., Marekova, M., Rozmer, Zs., Fodor, K.,
Chavkova, Z., Tomeckova, V., Guzy, J., 2008. Comparison of effects of some
cyclic chalcone analogues on selected mitochondrial functions. Pharmazie 63, 899-
903.
132.Hodnick, W.F., Milosavljevic, E.B., Nelson, J.H., Pardini, R.S., 1988.
Electrochemistry of flavonoids. Relationships between redox potentials, inhibition
of mitochondrial respiration and production of oxygen radicals of flavonoids.
Biochemical Pharmacology 37, 2601-2611.
133.Trumbeckaite, S., Bernatoniene, J., Majiene, D., Jakstas, V., Savickas, A., Toleikis,
A., 2006. The effects of flavonoids on rat heart mitochondrial function.
Biomedicine and Pharmacotherapy 60, 245-248.
134.Gul, M., Gul, H.I., Hanninen, O., 2002. Effects of Mannich bases on cellular
glutathione and related enzymes on Jurkat cells in culture conditions. Toxicology in
Vitro 16, 107-112.

83
135.Galati, G., Sabzevari, O., Wilson, J.X., O'Brien, P.J., 2002. Prooxidant activity and
cellular effects of the phenoxyl radicals of dietary flavonoids and other
polyphenolics. Toxicology 177, 91-104.
136. Michaelsen, J.T., Dehnert, S., Giustarini, D., Beckmann, B., Tsikas, D., 2009.
HPLC analysis of human erythrocytic glutathione forms using OPA and N-acetyl-
cysteine ethyl ester: Evidence for nitrite-induced GSH oxidation to GSSG. Journal
of Chromatography B. 877, 3405-3417.
137. Ridnour, L.A., Winters, R.A., Ercal, N., Spitz, D.R., 1999. Measurement of
glutathione, glutathione disulfide, and other thiols in mammalian cell and tissue
homogenates using high-performance liquid chromatography separation of N-(1-
pyrenyl)maleimide derivatives. Methods in Enzymology. 299, 258-264.
138. Hissin, P.J., Hilf, R., 1976. A fluorometric method for determination of oxidized
and reduced glutathione in tissues. Analytical Biochemistry. 74, 214-226.
139.Camera, E., Picardo, M., 2002. Analytical methods to investigate glutathione and
related compounds in biological and pathological processes. Journal of
Chromatography B 781, 181-206.
140. Lenton, K.J., Therriault, H., Wagner, J.R., 1999. Analysis of glutathione and
glutathione disulfide in whole cells and mitochondria by postcolumn derivatization
high-performance liquid chromatography with ortho-phthalaldehyde. Analytical
Biochemistry. 274, 125-130.
141. Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F., 2003. Analysis of glutathione:
implication in redox and detoxification. Clinica Chimica Acta. 333, 19-33.
142. Lóránd, T., Kocsis, B., Sohár, P., Nagy, G., Kispál, G., Krane, H.G., Schmitt, H.,
Weckert, E., 2001. Synthesis and antibacterial study of unsaturated Mannich
ketones. European Journal of Medicinal Chemistry 36, 705-717.
143.Gitler, C., Mogyoros, M., Kalef, E., 1994. Labeling of protein vicinal dithiols: Role
of protein-S2 to protein-(SH)2 conversion in metabolic regulation and oxidative
stress. Methods in Enzymology 233, 403-415.
144.Hughes, H., Taeschke, H., Mitchell, J.R., 1990. Measurment of oxidant stress in
vivo. Methods in Enzymology 186, 681-685.
145.Perjési, P., Maász, G., Reisch, R., Benkő, A., 2012. (E)-2-Benzylidene-
benzocyclanones: Part VII. Investigation of the conjugation reaction of two
cytotoxic cyclic chalcone analogues with glutathione: an HPLC-MS study.
Monatshefte für Chemie 143, 1107-1114.

84
146. Tang, S.S., Chang, G.G., 1996. Kinetic characterization of the endogenous
glutathione transferase activity of Octopus lens S-Crystallin. The Journal of
Biochemistry 119, 1182-1188.
147.Kubo, Y., Armstrong, R.N., 1989. Observation of a substituent effect on the
stereoselectivity of glutathione S-transferase toward para-substituted 4-phenyl-3-
buten-2-ones. Chemical Research in Toxicology 2, 144-145.
148. Perjési, P., Batta, G., Földesi, A., 1994. Reaction of benzylidenebenzocyclanones
with dithiocarbamic acid and thiourea. Monatshefte für Chemie 125, 433-439.
149.Wang, J., Wang, S., Song, D., Zhao, D., Sha, Y., Jiang, Y., Jing, Y., Cheng, M.,
2009. Chalcone derivatives inhibit glutathione S-transferase P1-1 activity: Insights
into the interaction mode of α,β-unsaturated carbonyal compounds. Chemical
Biology and Drug Design 73, 511-514.
150.Miyamoto, T, Silva, M., Hammock, B.D., 1987. Inhibition of epoxide hydrolases
and glutathione S-transferases by 2-, 3-, and 4-substituted derivatives of 4'-
phenylchalcone and its oxide. Archives of Biochemistry and Biophysics 254, 203-
213.
151.Huang, H., Niu, J., Zhao, L., Lu, Y., 2011. Reversal effect of 2',4'-dihydroxy-6'-
methoxy-3',5'-dimethylchalcone on multi-drug resistance in resistant human
hepatocellular carcinoma cell line BEL-7402/5-FU. Phytomedicine 18, 1086-1092.
152.Zhang, K., Das, N.P., 1994. Inhibitory effects of plant polyphenols on rat liver
glutathione S-transferase. Biochemical Pharmacology 47, 2063-2068.
153.Mccaughan, F.M., Brown, A.L., Harrison, D.J., 1994. The effect of inhibition of
glutathione S-transferase P on the growth of the Jurkat human T cell line. Journal
of Pathology 172, 357-362.
154.Townsend, D.M., Tew, K.D., 2003. The role of glutathione S-transferase in anti-
cancer drug resistance. Oncogene 22, 7369-7375.

85
VIII. Saját publikációk
A tézis alapját képező közlemények
1. Zs. Rozmer, T. Berki, G. Maász, P. Perjési: Different effects of two cyclic
chalcone analogues on redox status of Jurkat cells. Toxicology in Vitro. Közlésre
elfogadva.
2. Zs. Rozmer, P. Perjési: Naturally occurring chalcones and their biological activity.
Review. Phytochemistry Reviews. Közlésre beküldve.
3. P. Perjési, Zs. Rozmer: Kinetic analysis of some chalcones and synthetic chalcone
analogues on the Fenton-reaction initiated deoxyribose degradation assay.
Open Med. Chem. J. (2011) 142, 463-468
IF: - (2010)
4. P. Perjesi, J. Kubalkova, Z. Chovanova, M. Marekova, Zs. Rozmer, K. Fodor, Z.
Chavkova, V. Tomecková, J. Guzy: Comparison of effects of some cyclic chalcone
analogues on selected mitochondrial functions.
Pharmazie (2008) 63, 899-903
IF: 0.858 (2008)
5. Zs. Rozmer, P. Perjési, K. Takács-Novák: Use of RP-TLC for Determination of
logP of Isomeric Chalcones and Cyclic Chalcone Analogues.
J. Planar Chrom. – Modern TLC (2006) 19, 124-128.
IF: 1,153 (2006)
6. Zs. Rozmer, T. Berki, P. Perjési: Different effects of two cyclic chalcone
analogues on cell cycle of Jurkat T cells.
Toxicol. in Vitro (2006) 20, 1354-1362.
IF: 2,045 (2006)
Egyéb közlemények
1. Rozmer Zs., Perjési P.: (E)-2-benzylidenebenzocyclanones: Part X. Determination
of logP of (E)-3-benzylidene-2,3-dihydro-1-benzopyran-4-ones by RP-TLC. Effect
on logP of incorporation of oxygen atom into carbocyclic chalcone analogues.
J. Planar Chrom. - Modern TLC (2013) 26, 284-288.
IF: 0,955

86
2. P. Perjési, K. Takács-Novák, Zs. Rozmer, P. Sohár, R. E. Bozak, T. M. Allen:
Comparison of structure, logP and P388 cytotoxicity of some phenyl and ferrocenyl
cyclic chalcone analogues. Application of RP-TLC for logP determination of the
ferrocenyl analogues.
Cent. Eur. J. Chem. (2012) 10, 1500-1505.
IF: 1,073 (2011)
3. J. Guzy, J. Vasková-Kubálková, Zs. Rozmer, K. Fodor, M. Marekova, M.
Poskrobová, P. Perjési: Activation of oxidative stress response by hydroxyl
substituted chalcones and cyclic chalcone analogues in mitochondria.
FEBS Lett. (2010) 584, 567-70.
IF: 3.601 (2010)
4. P. Perjési, U. Das, E. De Clerq, J. Balzarini, M. Kawase, H. Sakagami, J. P.
Stables, T. Loránd, Zs. Rozmer, J. R. Dimmock: Design, synthesis and
antiproliferative activity of some 3-benzylidene-2,3-dihydro-1-benzopyran-4-ones
which display selective toxicity for malignant cells
Eur. J. Med. Chem. (2008) 43, 839-845-
IF: 2,882 (2008)
5. P. Perjési, I. Ember, R. E. Bozak, E. Nádasi, Zs. Rozmer, T. Varjas, R. J. Hicks:
Effect of the Chalcone Analogue E,E-bis(2-Hydroxybenzylidene)acetone on the
7,12-Dimethylbenz[a]anthracene-Induced Ha-ras Gene Action in Vivo.
In Vivo (2006) 20, 141-146.
IF: 1,273 (2006)
6. Rozmer Zs., Perjési P.: Néhány nem-szteroid gyulladáscsökkentő hatásának
vizsgálata a 2-deoxi-D-ribóz Fenton-reakció iniciálta degradációjára
Acta Pharm. Hung. (2005) 75, 87-93.
IF: - (2005)
Idézhető absztraktok
1. Rozmer Zs., Marton E., Perjési P.: Gyűrűs kalkonanalógok celluláris
makromolekulákkal kialakuló kölcsönhatásának vizsgálata spektroszkópiai és
kromatográfiás módszerekkel.
Congressus Pharmaceuticus Hungaricus XV., Budapest, 2014.
Gyógyszerészet Suppl. 2014/14. P-18

87
2. Rozmer Zs., Berki T., Maász G., Perjési P.: Gyűrűs kalkonszármazékok celluláris
redox státuszra gyakorolt hatásának vizsgálata Jurkat T sejtekben.
Congressus Pharmaceuticus Hungaricus XV., Budapest, 2014.
Gyógyszerészet Suppl. 2014/14. P-94
3. P. Perjési, M. Kuzma, K. Fodor, Zs. Rozmer: Application of crocin bleaching and
deoxyribose degradation tests to assess antioxidant capacity of capsaicinoids and
some selected flavonoids.
2nd BBBB Conference on Pharmaceutical Sciences, Tartu, 2007.
Eur. J. Pharm. Sci. 32 (S1), 39, 2007.
4. Csékei J., Szitter I., Rozmer Zs., Perjési P.: Flavonoid tartalmú teakeverékek
flavonoidtartalmának meghatározása és hidroxilgyök scavenger hatásának in vitro
jellemzése a dezoxiribóz degradációs teszt alkalmazásával.
Magyar Molekuláris és Prediktív Epidemiológiai Társaság III. Nemzetközi
Kongresszusa, Pécs, 2006.
Magyar Epidemiológia 3 (S), S33, 2006.
5. Rozmer Zs., Perjési P.: Kalkonszármazékok és fenolos nem-szteroid
gyulladáscsökkentők hidroxilgyök scavenger hatásának in vitro jellemzése a
dezoxi-ribóz degradációs teszt alkalmazásával.
Magyar Molekuláris és Prediktív Epidemiológiai Társaság II. Nemzetközi
Kongresszusa, Pécs, 2005.
Magyar Epidemiológia 2 (S), S76. 2005.
6. Nádasi E., Rozmer Zs., Bozak R. E., Hicks R. J., Perjési P.: A synthetic curcumin
analogue: chemopreventive or genotoxic effect?
Magyar Molekuláris és Prediktív Epidemiológiai Társaság II. Nemzetközi
Kongresszusa, Pécs, 2005.
Magyar Epidemiológia 2 (S), S63. 2005.
Poszterek
1. Rozmer Zs., Berki T., Maász G., Perjési P.: Different effects of two cyclic
chalcone analogues on redox status of Jurkat T cells.
2nd International Doctoral Workshop on Natural Sciences, Pécs, 2013.

88
2. Perjési P., Kuzma M., Fodor K., Rozmer Zs.: Studies on in vitro antioxidant effect
of salicylic acid and its hydroxylated metabolites.
9th International Symposium on Instrumental Analysis, Pécs, 2008.
3. Kuzma M., Fodor K., Rozmer Zs., Perjési P.: Néhány kapszaicinoid és flavonoid
antioxidáns hatásának vizsgálata krocin oxidációs teszt és deoxiribóz degradációs
teszt alkalmazásával.
Magyar Szabadgyök Kutató Társaság IV. Kongresszusa, Pécs, 2007.
4. Rozmer Zs., Berki T., Perjési P.: Gyűrűs kalkonanalógok citotoxicitásának
vizsgálata.
Congressus Pharmaceuticus Hungaricus XIII, Budapest, 2006.
5. J. Guzy, M. Mareková, Z. Chavková, J. Kubálková, V. Tomečková, Zs. Rozmer, P.
Perjési: Comparison of effect of some cyclic chalcone analogues on selected
mitochondrial functions.
8th Symposium on Instrumental Analysis, Graz, 2005.
6. Rozmer Zs., T. Novák K., Perjési P.: Kalkonok és gyűrűs kalkonszármazékok logP
értékeinek meghatározása fordított fázisú vékonyréteg kromatográfiás módszerrel.
Az MGYT Gyógyszerkutatási Szakosztály tudományos ülése, Pécs, 2005.
7. Zs. Rozmer, P. Perjési: Application of deoxyribose degradation to assess hydroxyl
radical scavanger activity of chalcones and cyclic chalcone analogues.
8th
Symposium on Instrumental Analysis, Graz, 2005.
8. Zs. Rozmer, T. Berki, P. Perjési: Cytotoxic effect of chalcone analogues in human
T-cell leukemia.
1st BBBB Conference on Pharmaceutical Sciences, Siófok, 2005.
Előadások
1. Rozmer Zs., Perjési P.: Flavonoidok sejtciklusra gyakorolt hatása,
Magyar Biológiai Társaság Pécsi Csoportja szakülése, Pécs, 2006
2. Rozmer Zs., Berki T., Perjési P.: Gyűrűs kalkonanalógok daganatsejt-toxikus
hatásának vizsgálata, MTA Gyógyszerésztudományi Osztályközi Komplex
Bizottságának ülése, Pécs, 2005
3. Rozmer Zs.: Gyűrűs kalkonanalógok citotoxicitásának és citoprotektív hatásának
vizsgálata. VII. Clauder Otto Emlékverseny, Visegrád, 2004

89
IX. Köszönetnyilvánítás
Ezúton szeretném köszönetemet kifejezni mindazoknak, akik az elmúlt évek során
segítették a munkámat.
Köszönettel tartozom témavezetőmnek, Dr. Perjési Pál Professzor Úrnak, hogy
szakmai tevékenységemet mindvégig figyelemmel kísérte, kutató munkámat irányította
és segítette. Köszönöm Dr. Gregus Zoltán Professzor Úrnak szakmai tanácsait,
disszertációm gondos átolvasását, és hogy kutatómunkámat az általa vezetett Ph.D.
program keretein belül végezhettem.
Szeretnék köszönetet mondani Dr. Berki Tímea Professzor Asszonynak, hogy
lehetőséget biztosított a sejtkezelések és áramlásos citometriai kísérletek elvégzésére az
Immunológiai és Biotechnológiai Intézetben, valamint köszönöm segítségét a kísérletek
megtervezésében és elvégzésében, szakmai és személyes tanácsait. Az intézetben
dolgozó asszisztenseknek külön köszönöm a sejtek előkészítésében nyújtott
segítségüket.
Köszönöm Dr. Maász Gábornak a tömegspektrometriás mérésekben nyújtott
segítségét.
Köszönöm Takácsné Dr. Novák Krisztina Professzor Asszonynak a logP
meghatározás módszerének elsajátításában nyújtott segítségét, valamint személyes
példamutatását. A kooperációban végzett munkáért köszönetemet szeretném kifejezni a
a kassai P. J. Šafarik Egyetem Orvosi Kémiai és Biokémiai Intézet munkatársainak.
Köszönettel tartozom kollégáimnak szakmai tanácsaikért, valamint baráti
támogatásukért. Köszönöm a Gyógyszerészi Kémiai Intézet minden munkatársának az
évek során nyújtott segítségét.
Végül köszönöm barátaim biztatását, családom támogatását, megértő türelmét és
mindazt a szeretetet, amit tőlük kaptam.





























