Embed Size (px)
Citation preview

Más:
Las métricas pretenden asegurar la calidad de fármacos
Calibración de instrumentos
Regulación de la UE de los biosimilares
Deficiencias encontradas en las inspecciones de APIs
Volumen 12, Número 1
SÍNTESIS Y MANUFACTURA DE APIS:• Innovación en nuevas entidades
moleculares• Un vistazo a futuro para la
manufactura personalizada
PRUEBAS ANALÍTICAS PARA BIOLÓGICOS: RMN y espectroscopía de masas en el análisis de glicanos
MERCADOS EMERGENTES: Innovación biotecnológica en Israel
RESOLUCIÓN DE PROBLEMAS:Técnicas para examinar la adhesividad de las tabletas
FORMULACIÓN: Formulación de Fragmentos Fab de Anticuerpos
Implementando la QbD en la manufactura estéril
El futuro de las formas
farmacéuticas¿Cómo impactará la medicina
personalizada la entrega de fármacos?Investigación Arbitrada
• Calificación del personal para inspeccionar visualmente el equipo limpio
• Establecimiento de un período de incubación mínimo para indicadores biológicos Entrevista de empleo 2013
El optimismo de la industria supera la seguridad laboral personal
El futuro de las formas
farmacéuticas¿Cómo impactará la medicina
personalizada la entrega de fármacos?Investigación Arbitrada
• Calificación del personal para inspeccionar visualmente el equipo limpio
• Establecimiento de un período de incubación mínimo para indicadores biológicos Entrevista de empleo 2013
El optimismo de la industria supera la seguridad laboral personal
El futuro de las formas
farmacéuticas¿Cómo impactará la medicina
personalizada la entrega de fármacos?Investigación Arbitrada
• Calificación del personal para inspeccionar visualmente el equipo limpio
• Establecimiento de un período de incubación mínimo para indicadores biológicos Entrevista de empleo 2013
El optimismo de la industria supera la seguridad laboral personal
Implementando la QbD en la manufactura estéril
Implementando la QbD en la manufactura estéril
Implementando la QbD en la manufactura estéril
Implementando la QbD en la manufactura estéril
Implementando la QbD en la manufactura estéril
Implementando la QbD en la manufactura estéril

Mar
que
en la
tarje
ta d
e se
rvic
io a
l lec
tor e
l No.
4

Marque en la tarjeta de servicio al lector el No. 5

Pharmaceutical Technology en Español MARZO / ABRIL 20142
En
la P
ort
ad
aPharmaceutical Technology en Español, proporciona información importante, confiable, y oportuna sobre todos los aspectos relacionados con Desarrollo e Investigación Aplicada; y con las Tecnologías de Proceso, Fabricación, Formulación, y Empaque para la Industria Farmacéutica Convencional y la de Biotecnología.
Pharmaceutical Technology es selectivamente extraida o indexada en:Biological Sciences Database (Cambridge Scientific Abstracts)Biotechnology and Bioengineering Database (Cambridge Scientific Abstracts)Business and Management Practices (RDSI)Chemical Abstracts (CAS)Current Packaging AbstractsDECHEMADerwent Biotechnology Abstracts (Derwent Information, Ltd.)Excerpta Medica (Elsevier)International Pharmaceutical Abstracts (ASHP)Science Citation Index (Thomson)Pharmaceutical Technology está orgullosa de ser miembro asociado de DCAT, IPEC y PDA.
Aspectos
Re
INvestIGAcIÓN ARbItRAdA
PRIMERA PLANA
5 El futuro de las formas farmacéuticas¿Cómo impactará la medicina personalizada la entrega de fármacos?
INDICADORES BIOLÓGICOS
19 Establecimiento de un período mínimo de in-cubación para los indicadores biológicos La industria carece de un método aceptado para el establecimiento de un tiempo mínimo de incubación (MIt) de menos de siete días para los indicadores biológicos (Ibs). Los autores proponen un método para el MIt que proporcione un medio para la determinación reproducible del tiempo de desarrollo de los Ibs
LÍMITES DE RESIDUOS VISIBLES
28 Calificación del personal para inspeccionar visualmente el equipo limpioel autor discute varios esquemas diferentes para la calificación en la inspección visual.
ENTREVISTA DE EMPLEO 2013
54 El optimismo de la industria supera la seguridad laboral personalLa reestructuración acalla el optimismo del empleo.
RESOLUCIÓN DE PROBLEMAS
38 Uso de técnicas analíticas para examinar la adhesividad de las tabletasLas pruebas analíticas, correlacionadas con las técnicas analíticas, se utilizan para predecir el comportamiento del material.
9 Uso del LC-MS en tándem para la validación de limpiezael autor discute las ventajas y desventajas de la cromatografía de líquidos-espectrometría de masas.
SÍNTESIS Y MANUFACTURA DE APIS
62 Rastreando la innovación en nuevas entidades moleculares del 2013Una revisión de la cosecha de este año de nuevas entidades moleculares y nuevas solicitudes de autorización de biológicos aprobados por la FdA hasta ahora en el 2013.
35 Un vistazo a futuro para la manufactura personalizada y las farmacéuticasLos mercados emergentes son un motor para el crecimiento en la manufactura personalizada y en los mercados farmacéuticos.
FORMULACIÓN: FRAGMENTOS FAB DE ANTICUERPOS
26 Desafíos y estrategias para la purificación corriente abajo y la formulación de Fragmentos Fab de Anticuerpos.Los autores hacen un bosquejo de algunos de los requerimientos únicos y desafíos planteados por los fragmentos de anticuerpos en términos de la recuperación, purificación y formulación.
PRUEBAS ANALÍTICAS PARA BIOLÓGICOS
50 El papel de la RMN y la espectroscopía de masas en el análisis de glicanos.el análisis RMN provee información estructural crucial de glicanos sintetizados, mientras que la Lc-Ms/Ms es ideal para la cuantificación de azúcares libres en matrices biológicas.
NovedEDITORIAL INVITADO
52Los científicos pueden compartir su tiempo y conocimiento farmacológico con los estudiantes de secundaria.
47La conferencia regulatoria de la
DEL EDITOR
41Los genéricos, la dinámica del mercado y la demanda global están cambiando los patrones en el gasto en fármacos.
PREGÚNTELE AL EXPERTO
59 Enfoque en la manufactura de fármacos: ¿Por qué la India?siegfried L, discute la situación de la ma-nufactura de fármacos en India.
SOLUCIONES ESTADÍSTICAS
44tro de UV suficientemente exacto?este artículo le da un vistazo estadístico a los requisitos de calibración para un espectrómetro de U
32 Las bases de la medición de la incertidumbre en el análisis farmacéutico¿Qué tan bueno es un valor reportable?
PERSPECTIVAS DE SUBCONTRATACIÓN
11el modelo de I+ está en transición y crea nuevas demandas para los proveedores de servicios por contrato.
MARZO/ABRIL 2014 VOLUMEN 12, NÚMERO 1
VIGILANCIA REGULATORIA EEUU
48 La regulación de opioides desafía a la FDA y a los fabricantesLas nuevas políticas y productos buscan mantener el acceso a los medicamentos para el dolor mientras se contiene el abuso desenfrenado.
42 La métrica pretende asegurar la calidad del fármacoLos reguladores trabajan con los fabricantes para mejorar los sistemas para medir el desempeño del producto y la confiabilidad en la fabricación.
14 Implementando la QbD en la Manufactura EstérilLos expertos de la industria discuten un enfoque de calidad por diseño para la manufactura estéril.
Imágenes: 4X-image/Getty Images

Pharmaceutical Technology en Español MARZO / ABRIL 2014 3
CONTENIDO
ReGULAcIÓN Y cUMpLIMIeNto
70 Cápsulas Farmacéuticas
68 Calendario de eventos
69 ¿Qué hay de nuevo?
71 Directorio Clasificado
72 Directorio de anunciantes
NovedAdes Y ANÁLIsIsEDITORIAL INVITADO
52 Inspirando a la siguiente generaciónLos científicos pueden compartir su tiempo y conocimiento farmacológico con los estudiantes de secundaria.
47 Conectando a la gente, la ciencia y la regulaciónLa conferencia regulatoria de la pdA/FdA promueve un compromiso con la calidad.
DEL EDITOR
41 Fármacos, ¿A qué costo?Los genéricos, la dinámica del mercado y la demanda global están cambiando los patrones en el gasto en fármacos.
PREGÚNTELE AL EXPERTO
59 Enfoque en la manufactura de fármacos: ¿Por qué la India?siegfried schmitt, consultor principal en pAReXeL, discute la situación de la ma-nufactura de fármacos en India.
SOLUCIONES ESTADÍSTICAS
44 Calibración de instrumentos: ¿Es su espectróme-tro de UV suficientemente exacto?este artículo le da un vistazo estadístico a los requisitos de calibración para un espectrómetro de Uv.
32 Las bases de la medición de la incertidumbre en el análisis farmacéutico¿Qué tan bueno es un valor reportable?
PERSPECTIVAS DE SUBCONTRATACIÓN
11 I+D en transiciónel modelo de I+d está en transición y crea nuevas demandas para los proveedores de servicios por contrato.
VIGILANCIA REGULATORIA EEUU
48 La regulación de opioides desafía a la FDA y a los fabricantesLas nuevas políticas y productos buscan mantener el acceso a los medicamentos para el dolor mientras se contiene el abuso desenfrenado.
42 La métrica pretende asegurar la calidad del fármacoLos reguladores trabajan con los fabricantes para mejorar los sistemas para medir el desempeño del producto y la confiabilidad en la fabricación.
VIGILANCIA REGULATORIA EUROPEA
60 Regulación de Biosimilares: un asunto de variabili-dad, similitud y comparabilidadMientras la Unión europea le da un vistazo más cercano a sus lineamientos para biosimilares, algunos elementos claves están demostrando su dificultad para resolverlos.
24 La Comunicación temprana con los reguladores es esencial para las SMEs (empresas medianas)con las sMes ganando un reconocimiento más amplio como el centro neurálgico de la investigación e innovación en europa, las agencias regulatorias están alentando a las compañías a involucrarse con los reguladores al principio del proceso de desarrollo de fármacos.
DENTRO DEL PIC/S
12 Deficiencias encontradas en las inspecciones de APIs el pIc/s revisa las deficiencias encontradas durante las inspecciones de las instalaciones de manufactura de ApIs, armoniza los estándares de las GMps y proporciona capacitación para los inspectores a nivel mundial.
REPORTES DE LOS MERCADOS EMERGENTES
57 Innovación biotecnológica y crecimiento en Israel La diversidad de la población de Israel, el sistema de salud de alta calidad y la capacidad de recuperación del estrés financiero global, hace a este país un socio fuerte para I+d, investigación clínica y crecimiento del mercado.
66 Innovación y crecimiento del mercado en Brasilbrasil es el primer país Latinoamericano en surgir como un colaborador biofarmacéutico global.
PHARMA CÁPSULAS
46 PyR con Brian Galliher, Gerente, Manufactura Se-cundaria en Cook Pharmica
18 PyR con Roger Hayes de MPI Research
EN BALANCE
67 Balanzas
seccIoNes
depARtAMeNtos / pRodUctos

Pharmaceutical Technology en Español MARZO / ABRIL 20144
James P. AgallocoPresident, Agalloco & Associates
Larry L. Augsburger, PhDProfessor, Department of Pharmaceutics, University of Maryland
David H. Bergstrom, PhDCOO, NovaDel Pharma Inc.
Phil BormanQbD Lead & Data Management & Analysis Manager GlaxoSmithKline
Rory BudihandojoDirector, Quality Systems Audit, Boehringer-Ingelheim Shanghai Pharmaceuticals Co. (China)
Todd L. CecilVice-PresidentCompendial ScienceUnited States Pharmacopeia
Metin Çelik, PhDPresident, Pharmaceutical Technologies International (PTI)
Zak T. Chowhan, PhDConsultant, Pharmaceutical Development
Suggy S. Chrai, PhDPresident and CEO,Chrai Associates, Inc.
Roger Dabbah, PhDPrincipal Consultant, Tri-Intersect Solutions
Tim FreemanManaging Director, FreemanTechnology
Sanjay Garg, PhDProfessor, Pharmaceutical Sciences, University of South Australia
R. Gary Hollenbeck, PhDChief Scientific Officer, UPM Pharmaceuticals
Ruey-ching (Richard) Hwang, PhDSenior Director, Pharmaceutical Sciences,Pfizer Global R&D
Mansoor A. Khan, PhDDirector, FDA/CDER/DPQR
Russell E. MadsenPresident, The Williamsburg Group, LLC
Heidi M. Mansour, PhDAssistant Professor,College of Pharmacy, University of Kentucky
Jim MillerPresident, PharmSource Information Services Bio/Pharmaceutical Outsourcing Report
Colin Minchom, PhDVice President Particle DesignHovione
Christine Moore, PhDDeputy Director for Science and Policy, Office of New Drug Quality Assessment, CDER, FDA
R. Christian Moreton, PhDVice-President, Pharmaceutical Sciences, Finnbrit Consulting
Fernando J. Muzzio, PhDDirector, NSF Engineering Research Center on Structured Organic Particulate Systems, Dept. of Chemical and Biochemical Engineering, Rutgers University
Moheb M. Nasr, PhDVice-President, CMC Regulatory Strategy, Global Regulatory Affairs, GlaxoSmithKline
Garnet E. Peck, PhDProfessor Emeritus of Industrial Pharmacy, Purdue University
Wendy Saffell-ClemmerDirector, Research, BioPharma Solutions
Gurvinder Singh Rekhi, PhDSenior Director,Pharmaceutical Researchand Development, Alkermes
Susan J. SchnieppVice-Precident, Quality and Regulatory Affairs, AllergyLaboratories, Inc
David R. SchonekerDirector of Global Regulatory Affairs, Colorcon
Eric B. Sheinin, PhDPresident, Sheinin and Associates
Aloka SrinivasanPrincipal Consultant, PAREXEL International
Heinz Sucker, PhDProfessor Emeritus,Pharmaceutical Institute, University of Bern
Scott Sutton, PhDMicrobiology Network
Pharmaceutical Technology en Español, V.12 No. 1 Marzo - Abril de 2014. Publica-ción bimestral editada por Revistas para la Industria, S.A. de C.V. Editor responsable: Ma. Antonieta Guerrero Paz. No. de Certificado de Reserva de Derechos al Uso Exclusivo otorgado por el Instituto Nacional del Derecho al Autor No. 04-2011-010610533100-102. No. de Certificado de Licitud de Título y Contenido otorgado por la Secretaría de Gobernación No. 15794. Domicilio de la Publicación: Av. Insurgentes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, Deleg. Benito Juárez, México, D.F. Impreso en: Polymasters de México, S.A. de C.V. - Calle Dos No. 123-C, Col. Granjas San Antonio C.P. 09070, México, D. F. Distribuida por: Revistas para la Industria, S.A. de C.V. - Av. Insurgentes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, Deleg. Benito Juárez, México, D.F.
Toda la información y conceptos que aquí aparecen son responsabilidad exclusiva de cada uno de los autores y firmas comerciales.
Esta prohibida y será castigada la reproducción total o parcial de cualquiera de los materiales que aquí aparecen.
¿Cómo el cambio a medicamentos más especializados y personalizados impactará la entrega de fármacos y las formas farmacéuticas? Pharmaceutical Technology le da un vistazo al futuro y examina algunas tecnologías emergentes.
E
La industria farmacéutica en su conjunto está experimentan-do ciertas transformaciones, como el cambio a especiali-
dades farmacéuticas dirigidas a grupos específicos de pacientes y en el largo plazo, el cambio a medicamentos per-sonalizados. Aunque este énfasis se orienta generalmente hacia el diseño a la medida de un ingrediente activo para grupos de pacientes más específicos, los regímenes personalizados/dirigidos jugarán un papel importante en la reali-zación del paradigma de medicamentos más especializados/personalizados.
“La personalización en masa ofre-ce productos individualizados para que todos hagan frente a los problemas de salud individuales con el medicamen-to hecho a la medida”, señala Francois Scheffler, vice-presidente de marketing global de la unidad de negocios de In-gredientes Farmacéuticos y Servicios de BASF. “Los medicamentos de hoy en día están principalmente unidos a la indicación, no al paciente individual o consumidor Toda / s medicación está limitada principalmente a la indicación, no al paciente o consumidor y sus nece-

Pharmaceutical Technology en Español MARZO / ABRIL 2014 5
El Futuro de las Formas Farmacéuticas
¿Cómo el cambio a medicamentos más especializados y personalizados impactará la entrega de fármacos y las formas farmacéuticas? Pharmaceutical Technology le da un vistazo al futuro y examina algunas tecnologías emergentes.
Patricia Van Arnum
El Futuro DE las Formas Farmacéuticas
La industria farmacéutica en su conjunto está experimentan-do ciertas transformaciones, como el cambio a especiali-
dades farmacéuticas dirigidas a grupos específicos de pacientes y en el largo plazo, el cambio a medicamentos per-sonalizados. Aunque este énfasis se orienta generalmente hacia el diseño a la medida de un ingrediente activo para grupos de pacientes más específicos, los regímenes personalizados/dirigidos jugarán un papel importante en la reali-zación del paradigma de medicamentos más especializados/personalizados.
“La personalización en masa ofre-ce productos individualizados para que todos hagan frente a los problemas de salud individuales con el medicamen-to hecho a la medida”, señala Francois Scheffler, vice-presidente de marketing global de la unidad de negocios de In-gredientes Farmacéuticos y Servicios de BASF. “Los medicamentos de hoy en día están principalmente unidos a la indicación, no al paciente individual o consumidor Toda / s medicación está limitada principalmente a la indicación, no al paciente o consumidor y sus nece-
sidades específicas. Un requisito previo para un enfoque de medicamento per-sonalizado es probablemente una eva-luación genética y fenotípica a costos accesibles, buscando básicamente en el cuerpo humano y pronosticando la probabilidad de posibles problemas de salud a lo largo de la vida.”
En su reciente reporte, Prepara-ción de las Bases para la Medicina Personalizada, el Papel de la FDA en una Nueva Era de Desarrollo de Pro-ductos Médicos, la FDA señala que el término “medicina personalizada” es a menudo descrito como proveer al “pa-ciente correcto con el medicamento adecuado en la dosis correcta y en el momento correcto “(1), aunque en tér-minos más ampliamente como “puede pensarse en la medicina personalizada como la adaptación de los tratamientos médicos a las características individua-les, necesidades y preferencias de un paciente durante todas las etapas de la atención, incluyendo la prevención, el diagnóstico, el tratamiento y el segui-miento “(1). La medicina personaliza-da generalmente implica el uso de dos productos médicos -un dispositivo de
diagnóstico y un producto terapéuti-co- para mejorar los resultados de los pacientes. “Aunque actualmente en la medicina personalizada se le está po-niendo una atención considerable al uso de pruebas genéticas para orientar las decisiones terapéuticas, una amplia variedad de dispositivos médicos pue-den ser utilizados en un enfoque per-sonalizado para mejorar los resultados del paciente” (1). Muchas terapias de dispositivos médicos se pueden adap-tar a las características específicas del paciente, tales como su anatomía (p.ej., tamaño), fisiología (p.ej., sistemas ner-vioso y cardiovascular, metabolismo y reproducción) y entorno (p.ej., unidad de cuidados intensivos, uso doméstico). También, los sensores fisiológicos pue-den pronosticar las respuestas al trata-miento para los pacientes individuales, tales como la impresión tridimensional (3D), para crear dispositivos médicos personalizados basados en la imagen de la anatomía de un paciente (1).
La confluencia de la terapéutica tra-dicional con dispositivos proporciona la base para la industria farmacéutica del futuro. La personalización se está arraigando a través de las tecnologías emergentes, tales como medicinas bioelectrónica y la impresión 3D de los productos farmacéuticos, a través de se-lección de inversiones por parte de las empresas farmacéuticas, capitalistas de riesgo, e investigadores académicos.
Haciendo inversionesEn Agosto de 2013, GlaxoSmithKli-ne (GSK) lanzaron el Action Potential Venture Capital (APVC) Limited, un nuevo fondo de capital de riesgo es-tratégico de %50 mdd que invertirá en compañías que desarrollen medicamen-tos y tecnologías bioelectrónicos (2). La primera inversión del fondo está en SetPoint Medical, una empresa con sede en Valencia, California, la cual ha desarrollado dispositivos implantables de neuromodulación patentados que estimulan el nervio vago del cuerpo para tratar enfermedades inflamato-rias, como la enfermedad de Crohn y la artritis reumatoide (2, 3). En Agos-to de 2013, SetPoint aseguró $27 mdd en financiamiento de los inversores, el cual incluyó el Action Potential Venture de GSK, Covidien Ventures y Boston Scientific, para financiar los desarrollos

Pharmaceutical Technology en Español MARZO / ABRIL 20146
clínicos en curso del esquema de tera-pia bioelectrónica de SetPoint en artritis reumatoide y enfermedad de Crohn y el desarrollo avanzado de su plataforma de neuromodulación (2).
APVC complementa el trabajo de la unidad de I+D de Bioelectrónicos de GSK, la cual fue establecida en 2012. El nombre del fondo viene de las se-ñales eléctricas llamadas potenciales de acción que pasan a lo largo de los nervios del cuerpo. Pueden ocurrir pa-trones irregulares o alterados de estos impulsos en asociación con un amplio rango de enfermedades (2). GSK piensa que pueden diseñarse los dispositivos miniaturizados o los medicamentos bioelectrónicos para leer estos patrones. Los dispositivos podrían diseñarse para intercomunicarse entre el sistema ner-vioso periférico y órganos específicos para leer, cambiar o generar impulsos eléctricos que ayuden a tratar los tras-tornos, tales como la enfermedad de intestino inflamado, la artritis reuma-toide y las enfermedades respiratorias y metabólicas (2). Además de trabajar con el nuevo fondo de capital de riesgo, I+D Bioelectrónico de GSK ofrecerá hasta 20 nuevos otorgamientos para in-vestigación exploratoria y crear una red de investigadores. El trabajo está cen-trado en la relación entre los nervios en el cuerpo y un rango de enfermedades, el patrón particular de los impulsos a lo largo de estos nervios, y las tecnologías que puedan intercomunicarse con fibras nerviosas individuales (2). En Diciem-bre de 2013, GSK anunció un premio de $1 mdd en investigación bioelectró-nica para la creación de un dispositivo miniaturizado, completamente implan-table, para leer, escribir y bloquear las señales eléctricas del cuerpo para tratar la enfermedad.
Action Potential Venture Capital pretende crear un portafolio de cinco a siete compañías durante los próximos cinco años. El financiamiento se cen-trará en inversiones en tres áreas: em-presas de nueva creación que busquen
promover la visión de los medicamen-tos bioelectrónicos, empresas existentes con tecnologías que están interactuando con el sistema nervioso periférico a tra-vés de dispositivos de primera genera-
ción que puedan estimular o bloquear los impulsos eléctricos, y empresas que estén avanzando en plataformas tecno-lógicas que servirán de base a estas mo-dalidades de tratamiento (2).
En Mayo de 2013, Proteus Digital Health, una compañía con sede en Red-wood City, California que desarrolla medicamentos digitales, recaudó $62.5 mdd, los cuales incluyeron a un nuevo inversor corporativo, Oracle, así como inversiones de inversores anteriores Otsuka, Novartis, y Sino Portfolio. El objetivo de Proteus está en los medi-camentos digitales basados en la detec-ción de ingeribles (4). En Julio del 2012, Proteus recibió la aprobación de la FDA de su sensor ingerible para ser comer-cializado como un dispositivo médico (5). El sensor ingerible de Proteus (es decir, el marcador del evento ingerible) es un dispositivo de prescripción usa-do para registrar los acontecimientos en el paciente con registro de tiempo. El componente ingerible se vincula inalámbricamente a través de comuni-cación intra-corporal a un registrador externo que registra la fecha y la hora de la ingestión. El monitor personal de la compañía (es decir, un parche) es un sensor que se trae puesto en el cuerpo y que recoge las métricas fisiológicas y de comportamiento, incluyendo frecuencia cardíaca, ángulo corporal, y eventos en el usuario con hora impresa generados cuando un usuario marca un evento cuando traga un marcador del evento de ingestión u oprimiendo manualmente un botón marcador del evento en el par-che, el cual almacena y envía inalám-bricamente los datos del mercador del evento de ingestión a un dispositivo de computación general. El marcador del evento de ingestión está unido a un ex-cipiente/tableta para facilitar el manejo y la deglución. El programa empareja
GSK está invirtiendo en medicamentos bioelectrónicos a través de un nuevo fondo de capital de riesgo.
el parche con un dispositivo de compu-tación móvil que organiza y despliega los eventos de ingestión (6). Proteus recibió una aprobación de Marca CE (Conformidad Europea) por su sistema de monitoreo fisiológico personal del sensor ingerible en la Unión Europea en el 2010 (7). En su negociación, Oracle y Proteus trabajarán juntos para darles a los investigadores clínicos la capaci-dad de medir la información acerca de la ingestión del medicamento, la sincro-nización de la dosis y la respuesta fisio-lógica asociada. Las dos compañías in-tegrarán el sensor ingerible de Proteus con los productos del estudio clínico de Oracle (8).
En Octubre de 2012, Qualcomm, un proveedor de tecnología inalámbrica y servicios, le otorgó $3.75 mdd al Ins-tituto de Ciencia Traslacional Scripps para avanzar en los estudios clínicos de sistemas de biosensor inalámbrico, pruebas de diagnóstico farmacogenó-mico rápidas, aplicaciones, y sensores integrados para rastreo y predicción de enfermedades. El dinero apoyará el per-sonal adicional y otros recursos para un programa de tres años, llamado Scripps Digital Medicine, diseñado para conti-nuar el avance de aplicaciones médicas prometedoras y dispositivos para de-sarrollo clínico. Algunas empresas de nueva creación involucradas son: AirS-trip Technologies (San Antonio, Texas), un proveedor de programas de salud móviles, y DNA Electronics (Londres), una prolongación del Centro de Tecno-logía Bio-Inspirada que se encuentra en el Instituto de Ingeniería Biomédica en el Imperial College de Londres y pro-veedor de un dispositivo de genotipifi-cación manual (es decir, Genalysis) que puede ser usado para adaptar mejor la prescripción a los pacientes individua-les. Otros proyectos involucran un sis-tema biosensor de transmisiones por sangre y una aplicación móvil para sen-sores integrados (9).
“Los sistemas de retroalimentación de salud digitales son una consecuencia lógica de la tendencia hacia la medica-ción preventiva y a la vida más saluda-ble,” señala Scheffler. “La influencia en la formulación de los fármacos y, por lo tanto, el uso de excipientes e ingredien-tes y su interacción será significativa. Por ejemplo, no sólo significa medir
El Futuro DE las Formas Farmacéuticas

ciertas reacciones corporales sino abor-darlas tan rápido, tan precisamente y tan al inicio como sea posible. Un incre-mento en la calidad de vida y significa-tivos ahorros en el costo serán los prin-cipales beneficios de esta tecnología.” Como un caso, Metanomics Health, una empresa de BASF, está especializa-da en traducir indicadores fenológicos en disfunciones potenciales del cuerpo que pueden entonces ser manejadas al inicio con cambios en los hábitos o con medicamentos.
Una revisión de la literatura mues-tra algún trabajo reciente utilizando tabletas digitales. En un estudio, un sistema de retroalimentación de salud digital utilizó una tableta digital, que consistió en un sensor para ingestión que fue integrado en una tableta conte-niendo excipientes no farmacológicos, la cual co-ingirieron los sujetos (12 adultos con trastorno bipolar y 16 adul-tos con esquizofrenia) con sus medica-mentos prescritos con regularidad (9). La formulación de esta tableta digital permitió la separación del sensor para
ingestión y la activación de los fluidos estomacales después de la ingestión, se-guido por la comunicación de una señal de identificación única desde el sensor de ingestión a un sensor adhesivo usado en el torso, el cual automáticamente re-gistró la fecha y la hora de la ingestión de cada tableta digital. El sensor usable también colectó las mediciones fisioló-gicas, incluyendo la actividad y la fre-cuencia cardíaca (10).
MicroCHIPs ha desarrollado una alternativa para las inyecciones diarias: un microchip programable, controlado inalámbricamente con un dispositivo implantable que permite que los fár-macos sean liberados dentro del cuer-po sin conexiones percutáneas dentro o sobre el paciente. Un dispositivo de microchip implantable también ofrece el potencial para el rastreo del esquema de dosis en tiempo real y para que los médicos ajusten a control remoto los es-quemas de tratamiento. Las tecnologías usan arreglos de micro-reservorios para almacenar y proteger herméticamente los farmacéuticos o los sensores durante
períodos de tiempo prolongados. El mi-crochip está controlado por microproce-sadores, comunicaciones inalámbricas, o loops de retroalimentación del sensor para el control dinámico del suministro o detección del fármaco. MicroCHIPs obtuvo la licencia de la tecnología del Instituto de Tecnología de Massachu-setts, el cual reportó en 2012 un exitoso estudio clínico en humanos para un chip de suministro de fármacos, controlado inalámbricamente. En el estudio, la te-riparatida humana, un fragmento de la hormona paratiroidea [hPTH(1-34)] y tratamiento de la osteoporosis anabó-lica, fue suministrada desde el dispo-sitivo in vivo. Los dispositivos basados en microchips contenían dosis discretas del hPTH(1-34) liofilizado y fueron im-plantados en ocho mujeres post-meno-páusicas osteoporóticas durante cuatro meses y programado inalámbricamente para liberar dosis desde el dispositivo una vez al día hasta por 20 días. Un pro-gramados basado en computadora esta-bleció una comunicación bidireccional inalámbrica unida con el implante para
Mar
que
en la
tarje
ta d
e se
rvic
io a
l lec
tor e
l No.
6
Visítanos en
en el Stand
1025

Pharmaceutical Technology en Español MARZO / ABRIL 20148
programar el esquema de dosis y recibir el estatus de confirmación del implante (11-13).
Impresión 3D de farmacéuticosLa impresión 3D de farmacéuticos es otra visión. “La impresión 3D es pro-bablemente la idea más innovadora en la industria farmacéutica desde hace décadas,” dice Scheffler. “Cambiará nuestras vidas y jugará un papel princi-pal, de hecho, en literalmente todas las industrias. Las nuevas formulaciones con excipientes e ingredientes existen-tes y aprobados se beneficiarán de estas nuevas maneras creativas porque po-dremos personalizar los medicamentos para cada paciente individual. El efecto es menos desperdicio, menos costos y menos complejidad para los pacientes.”
Un reciente artículo de revisión en el Journal of the American Pharmacists Association resumió la literatura publi-cada sobre las tecnologías existentes de la impresión 3D para la manufactura farmacéutica y detalló las limitaciones y el potencial de estas tecnologías (14). Una búsqueda estructurada del PubMed y el Embase identificaron artículos pu-blicados entre Enero 1, 1990 y Agosto 31, 2012. Los términos de la búsqueda incluyeron impresión de fármacos, im-presión 3D de fármacos, e impresión tri-dimensional de fármacos. En la re-visión se incluyeron 21 de 511 referen-cias identificadas. La impresión basada en chorro de tinta y polvo fueron las tecnologías primarias de impresión usa-das para el desarrollo y fabricación de fármacos. Once artículos describieron un sistema de suministro de polvo, y 10 identificaron la impresión con chorro de tinta. Estas tecnologías de impresión permiten ciertas ventajas, tales como el control preciso del tamaño de la gota, la alta reproducibilidad, los complejos perfiles de liberación del fármaco, y la terapia con medicación personalizada (14). El artículo especificó que las for-
mas farmacéuticas imprimibles en pa-pel pueden ser más fáciles de suminis-trar que las formas impresas basadas en
polvo. Los medicamentos con índices terapéuticos estrechos o con una mayor probabilidad de ser influidos por poli-morfismos genéticos pueden ser las pri-meras en ser impresas a través de esta tecnología (14).
Un equipo de investigación enca-bezado por Simon Gaisford, lector en farmacia en la Escuela de Farmacia del University College London, ha aplica-do la tecnología de impresión térmica con chorro de tinta para hacer pelícu-las orales con dosis personalizada de sulfato de salbutamol reemplazando el papel en la impresora con una hoja de película polimérica que permitió que el fármaco fuera disparado sobre la super-ficie (15-17). Se modificó un cartucho de impresora de manera que la solución acuosa del fármaco reemplazara la tinta (15-17). Se cortaron tiras de película. Variando la concentración de la solu-ción del fármaco, el área impresa o el número de pasadas de impresión se pudo controlar la dosis (15-17). La vis-cosidad de la solución para impresión y la tensión superficial fueron usadas para determinar el desempeño de la impreso-ra. Se preparó una curva de calibración para el sulfato de salbutamol, la cual mostró que la deposición del fármaco sobre una película de acetato variaba linealmente con la concentración. La impresora fue usada para depositar sul-fato de salbutamol sobre una película oral hecha de almidón de papa. Los in-vestigadores encontraron que cuando se depositaba la dosis en una sola pasada bajo la cabeza de la impresora, la dosis medida concordaba con la dosis teórica. Con múltiples pasadas, la dosis medida fue siempre significativamente menor que la dosis teórica (15-17). Los inves-tigadores conjeturaron que las pérdidas eran resultado de la erosión por fuerzas
de cizallamiento del área impresa du-rante el manejo del papel.
Referencias1. FDA, Paving the Way for Personalised
Medicine: FDA’s Role in a New Era of Medi-cal Product Development (Rockville, MD, Oct. 2013).
2. GlaxoSmithKline, “GSK Launches $50 Million Venture Capital Fund to Invest in Pioneering Bioelectronic Medicines and Technologies,” Press Release, Aug. 8, 2013.
3. SetPoint Medical, “SetPoint Medical Secures $27 Million Financing, Adding Action Potential Venture Capital/GSK, Boston Scientific and Covidien Ventures as New Investors,” Press Release, Aug. 8, 2013.
4. Proteus Digital Health, “Proteus Digital Health Completes $62.5 Million Financing,” Press Release, May 1, 2013.
5. Proteus Digital Health, “Proteus Digital Health Announces FDA Clearance of Ingestible Sensor, Press Release, July 30, 2013.
6. FDA, “Evaluation of Automatic Class II Designation (De Novo) for Proteus Personal Monitor Including Ingestion Marker,” De-cision Summary (Rockville, MD, May 14, 2012), www.accessdata.fda.gov/cdrh_docs/reviews/K113070.pdf, accessed 17 Dec. 2012.
7. Proteus Proteus Digital Health, “Proteus Biomedical Announces European CE Mark Approval of Ingestible Sensor and Monitor System,” Press Release, Aug. 13, 2010.
8. Proteus Digital Health, “Oracle Invests in Proteus Digital Health and Its FDA-Ap-proved Ingestible Sensor Platform,” Press Release, May 1, 2013.
9. Scripps Translational Science Institute, “Scripps Creates Digital Medicine Program with Qualcomm,” Press Release, Oct. 13. 2013.
10. J.M. Kane, Clin. Pyschiatry 74 (6), 533-540 (2013).
11. R. Farra et al., Sci. Transl. Med. online, DOI: 10.1126/scitranslmed.3003276, Feb. 16, 2012.
12. A. Trafton, “Successful Human Tests for First Wirelessly Controlled Drug-Delivery Chip,” Press Release, May 16, 2013.
13. J. Santini, M. Cima, and R. Langer, Nature 397 (6717), 335-338 (1999).
14. I.D. Ursan, L. Chiu,and A. Pierce, J. Am. Pharm. Assoc. 53, 136-144 (2013).
15. P. Van Arnum, Pharm. Technol. 37 (5) 58 (2013).
16. UCL, “Profile (S. Gaisford): Research Summary,” http://iris.ucl.ac.uk/iris/browse/profile?upi=SGAIS88, accessed 17 Dec. 2013.
17. S. Gaisford et al., Pharm Res. 28 (10) 2386-92 (2011). PT
Los investigadores aplicaron la tecnología de impresión con chorro de tinta térmico para hacer películas orales con dosis personalizada de sulfato de salbutamol.
RESOLUCIÓN DE PROBLEMASEl Futuro DE las Formas Farmacéuticas

Pharmaceutical Technology en Español MARZO / ABRIL 2014 9
de cizallamiento del área impresa du-rante el manejo del papel.
FDA, Paving the Way for Personalised Medicine: FDA’s Role in a New Era of Medi-cal Product Development (Rockville, MD, Oct. 2013).GlaxoSmithKline, “GSK Launches $50 Million Venture Capital Fund to Invest in Pioneering Bioelectronic Medicines and Technologies,” Press Release, Aug. 8, 2013.SetPoint Medical, “SetPoint Medical Secures $27 Million Financing, Adding Action Potential Venture Capital/GSK, Boston Scientific and Covidien Ventures as New Investors,” Press Release, Aug. 8, 2013. Proteus Digital Health, “Proteus Digital Health Completes $62.5 Million Financing,” Press Release, May 1, 2013. Proteus Digital Health, “Proteus Digital Health Announces FDA Clearance of Ingestible Sensor, Press Release, July 30, 2013. FDA, “Evaluation of Automatic Class II Designation (De Novo) for Proteus Personal Monitor Including Ingestion Marker,” De-cision Summary (Rockville, MD, May 14, 2012), www.accessdata.fda.gov/cdrh_docs/reviews/K113070.pdf, accessed 17 Dec. 2012. Proteus Proteus Digital Health, “Proteus Biomedical Announces European CE Mark Approval of Ingestible Sensor and Monitor System,” Press Release, Aug. 13, 2010. Proteus Digital Health, “Oracle Invests in Proteus Digital Health and Its FDA-Ap-proved Ingestible Sensor Platform,” Press Release, May 1, 2013. Scripps Translational Science Institute, “Scripps Creates Digital Medicine Program with Qualcomm,” Press Release, Oct. 13. 2013.
74 (6), 533-540 (2013).
. online, DOI: 10.1126/scitranslmed.3003276, Feb. 16, 2012.A. Trafton, “Successful Human Tests for First Wirelessly Controlled Drug-Delivery Chip,” Press Release, May 16, 2013. Nature
I.D. Ursan, L. Chiu,and A. Pierce, J. Am.
37 (5) 58 (2013). UCL, “Profile (S. Gaisford): Research Summary,” http://iris.ucl.ac.uk/iris/browse/profile?upi=SGAIS88, accessed 17 Dec. 2013.
RESOLUCIÓN DE PROBLEMASEquipo Y Proceso
Uso de la CL-MS en tándem para la validación de limpiezaGeoff Carr
El autor describe cómo la cromatografía de líquidos-espectrometría de masas trabaja y explica algunas de sus ventajas y desventajas.
GLo
wIM
AGES
/GE
TTY
IMAG
ES
Dentro de cualquier instala-ción de manufactura farma-céutica es crucial asegurar que después de la manufac-
tura de un producto, el equipo ha sido meticulosamente limpiado para evitar riesgos de cualquier arrastre al siguien-te producto. El proceso mediante el cual se logra esto se llama validación de la limpieza, y un enfoque para reali-zar tales validaciones es desarrollar una secuencia de operaciones que probable-mente requieran de la aplicación de sol-ventes o detergentes y enjuagues. Debe demostrarse que esta secuencia de ope-raciones puede remover las trazas de residuos del producto a niveles que no presenten riesgos para el paciente que puede recibirlos como componentes del arrastre en el siguiente producto fabri-cado en el mismo tren del equipo. Para colectar los datos, se analizan muestras de hisopo tomadas de las superficies del equipo o de los enjuagues (es decir, la-vados que se utilizan para el enjuagado del equipo después de que ha sido trata-do con detergente). Típicamente, se to-man las muestras de los lavados finales para el análisis.
Si no se ha desarrollado un proce-dimiento de limpieza validado, es de esperar que el análisis del hispo o del enjuague sea realizado en cada ocasión para demostrar la eficiencia del proce-
dimiento de limpieza; esta práctica se conoce como verificación de la limpie-za.
Tanto para las validaciones de lim-pieza como para las verificaciones, el estudio analítico de las muestras es un paso crucial. Típicamente, se aplican los procedimientos analíticos que son específicos para los APIs que fueron usados para la manufactura de produc-tos previos. Se espera que se desarrolle y validare un procedimiento analítico separado para cada API individual que sea manejado en cualquier planta de ma-nufactura. La cromatografía de líquidos de alta resolución (HPLC) es probable-mente el procedimiento analítico más ampliamente usado para esta aplicación con la cromatografía de líquidos de ul-tra alta resolución (UHPLC) ganando cada vez más popularidad, aunque la cromatografía de líquidos-espectrome-tría de masas (LC-MS) también ofrece oportunidades potenciales.
Cómo trabaja la detección por LC-MSPara que la detección de masas fun-cione, varios componentes del analito que eluye de la columna de cromato-grafía deben volverse ionizados. La ionización puede lograrse utilizando ionización con electro-espray (ESI). En la Figura 1 se describe un ejemplo de un detector de MS (MDS SCIEX, AB Sciex), y la Figura 2 describe la dispo-sición del electro-espray de un detector de MS. Según se muestra en la Figura 2, el efluente de la columna de HPLC se dirige a través de un nebulizador que se mantiene bajo un voltaje alto. El voltaje puede seleccionarse ya sea para polari-
dad positiva o negativa dependiendo de las condiciones requeridas por los com-ponentes de interés. Las gotas cargadas son sometidas a calor para remover el solvente, el cual crea un aerosol carga-do que se dirige al cuadrupolo del MS.
Un sistema de cuadrupolo provee el mecanismo para discriminar diferentes componentes dentro de la muestra de prueba sobre la base de los valores M/z, en donde M es el peso molecular y z es la magnitud de la carga. Por ejemplo, el proceso ESI podría llevar a especies de una o de doble carga o incluso mayores dependiendo de la estructura química de los componentes involucrados. Va-riando el campo magnético dentro del sistema cuadrupolo, las especies de di-ferentes valores M/z pueden ser ya sea eliminadas o dirigidas al detector del instrumento.La configuración descrita en la Figura 2 es para un instrumento de un solo cua-drupolo (es decir, LC-MS). Un sistema incluso más discriminatorio se da con un enfoque de triple cuadrupolo (es decir., LC-MS-MS). En este caso, las especies que son seleccionadas por el primer cuadrupolo (Q1) se dirigen al segundo cuadrupolo (Q2), el cual sirve para colectar las especies particulares M/z de interés y después se dirigen a un tercer cuadrupolo (Q3). Dentro del Q3, las especies se someten a colisiones con otros átomos o moléculas que se dan con frecuencia permitiendo un pe-queño volumen de nitrógeno dentro del sistema. Las colisiones llevan a frag-mentaciones y los iones del fragmento resultante pueden entonces ser usados como una base del análisis requerido.
Ventajas y desventajas del LC-MSCosto. El precio de un sistema típico de HPLC con detección UV es aproxima-
Geoff Carr, PhD,es director de Desarrollo Analítico en Patheon, Mississauga, Ontario. [email protected]

Pharmaceutical Technology en Español MARZO / ABRIL 201410
Voltaje del aerosol atomizado Corriente del LC
Spray Gas
Gas secante
Aerosol
Gota cargada
Evaporación del solvente DesaglomeraciónCortina de placa
Ori�cio Cuadrupolo
Cortina de gas
SolventeGotaAnalito
damente de 77,000 dólares. El precio de un detector típico de MS de triple cuadrupolo es de aproximadamen-te 193,000 dólares y el usuario debe comprar todavía un extremo frontal de HPLC para el sistema. Además, se re-comiendan al menos dos sistemas para cualquier organización que considere el uso del enfoque LC-MS de manera que se disponga de un sistema de respaldo. Las instalaciones de manufactura far-macéutica están programadas para lími-tes de tiempo muy cerrados. Cualquier tiempo muerto, como el que se dedica a la limpieza después de la fabricación de un lote y al estudio analítico del hi-sopo y a las formalidades de liberación del equipo antes de la fabricación del siguiente producto, deben mantenerse en el mínimo; los retrasos debidos a las descomposturas del equipo analítico no pueden ser toleradas.
Aplicabilidad. El HPLC se aplica con más frecuencia utilizando detec-tores de absorción de luz ultravioleta (UV), los cuales requieren que las es-tructuras químicas de los analitos de in-terés incluyan un cromóforo (es decir, características químicas estructurales que absorben luz UV). Para la mayo-
ría de los compuestos de interés farma-céutico, éste es frecuentemente el caso, aunque existen excepciones que pueden requerir otros tipos de detección. La de-tección MS es apropiada para cualquier analito, siempre y cuando pueda ser ionizado. Como la mayoría pueden ser ionizados, este enfoque tiene casi apli-cabilidad universal.
Sensibilidad. El análisis para los re-siduales de la limpieza es un ejemplo de una aplicación que requiere muy alta sensibilidad. La LC-MS es capaz de proveer cuantificación confiable de los componentes traza desde 100 hasta 1000 veces más bajos que el HPLC con detección UV.
Velocidad. Los tiempos de corrida del HPLC son extremadamente varia-bles dependiendo del método particular involucrado. El tiempo de corrida es el tiempo desde la inyección de la solu-ción analítica en el cromatógrafo hasta el final del cromatograma resultante. Los tiempos de corrida de 5-10 o inclu-
so de 20 min, no son raros. Con la LC-MS, los tiempos de corrida típicos son de uno a dos minutos. El tiempo de pre-paración del sistema es también muy reducido para LC-MS, ya que la misma columna de LC y la fase móvil pueden utilizarse para muchas aplicaciones. Cada análisis involucrará una secuencia de inyecciones, incluyendo las requeri-das para la verificación del sistema, las soluciones estándar y las soluciones de la muestra. Los cromatogramas necesi-tan entonces ser procesados en reportes analíticos y los datos deben ser revisa-dos y recibir la aprobación del laborato-rio. En general, Patheon ha encontrado que el tiempo de recuperación para el análisis de hisopos es de alrededor de 24 h con HPLC y alrededor de 8 h con LC-MS.
ReconocimientosEl autor desea agradecer la valiosa ayu-da que recibió de Heng Yao en la prepa-ración de este artículo. PT
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486E-mail: [email protected]
Figura 1: Cromatógrafo de líquidos de alta resolución (Agilent 1200) con espectrómetro de masas con triple cuadrupolo (MDS SCIEX).
Figura 2: Diagrama que muestra la disposición del sistema de electroatomización de un espectrómetro de masas; LC es cromatografía de líquidos.
PERSPECTIVAS DE LA SUBCONTRATACIÓN
Jim Miller
El modelo de I+D está en transición y creando nuevas demandas con los proveedores de servicio por contrato.
RESOLUCIÓN DE PROBLEMAS

Pharmaceutical Technology en Español MARZO / ABRIL 2014 11
so de 20 min, no son raros. Con la LC-MS, los tiempos de corrida típicos son de uno a dos minutos. El tiempo de pre-paración del sistema es también muy reducido para LC-MS, ya que la misma columna de LC y la fase móvil pueden utilizarse para muchas aplicaciones. Cada análisis involucrará una secuencia de inyecciones, incluyendo las requeri-das para la verificación del sistema, las soluciones estándar y las soluciones de la muestra. Los cromatogramas necesi-tan entonces ser procesados en reportes analíticos y los datos deben ser revisa-dos y recibir la aprobación del laborato-rio. En general, Patheon ha encontrado que el tiempo de recuperación para el análisis de hisopos es de alrededor de 24 h con HPLC y alrededor de 8 h con LC-MS.
I+D en transición
JoN
ATH
AN
EVA
NS
/PH
oTo
DIS
C/
GE
TTY
IMAG
ES
PERSPECTIVAS DE LA SUBCONTRATACIÓN
Jim Miller
El modelo de I+D está en transición y creando nuevas demandas con los proveedores de servicio por contrato.
La investigación y el desarro-llo es la sangre vital de la industria bio/farmacéutica. Aunque las estrategias de ci-
clo de vida y sistemas de entrega nove-dosos pueden disminuir la caída de los ingresos a raíz de la expiración de las patentes, las compañías farmacéuticas pueden sobrevivir sólo por un tiempo con la reformulación de productos más antiguos. La industria debe poder intro-ducir nuevas terapias para mantener su crecimiento y rentabilidad.
La presión en las operaciones de I+D de las compañías bio/farmacéuti-cas nunca ha sido mayor. Las compa-ñías necesitan nuevos productos exito-sos para reavivar el crecimiento y re-frenar la pérdida de ingresos y trabajos después de la expiración de las paten-tes. Como la nueva ciencia clínica está produciendo productos que están más dirigidos que los productos estrella del mercado, a los que están reemplazando, los laboratorios de I+D deben producir un mayor número de nuevos productos que nunca antes.
Decaimiento de I+DA pesar del imperativo para acelerar el desarrollo de nuevos productos, los in-dicadores nos dicen que el número de compuestos que entran al desarrollo clí-nico ha estado declinando actualmente.
El número de sometimientos de nuevos productos en investigación (IND) en la FDA ha estado declinando desde el 2008, estando ahora alrededor de un tercio por debajo. Además, el registro de nuevos estudios clínicos Fase II y Fase III en la base de datos clinical-trials.gov del gobierno, ha caído 15%, con nuevos sometimientos por parte de compañías bio/farmacéuticas globales por debajo en más del 20%.
¿Qué hay detrás de la aparente de-clinación en la actividad de I+D? Pare-ce ser que es la confluencia de un nú-mero de eventos que han trastornado la productividad de I+D, tales como:
•Las compañías bio/farmacéuticas globales están rediseñando su mo-delo de I+D estrechando su foco terapéutico, despidiendo a los cien-tíficos, desmantelando campus de investigación aislados en favor de instalaciones de investigación en centros de investigación médica tales como Boston y San Francis-co, y en la concesión de licencias o adquiriendo más candidatos a fár-macos de compañías nuevas.
•La investigación bio/farmacéutica está estableciendo una nueva base científica basada en la genómi-ca y los biológicos. La genómica
está todavía en proceso de madu-ración mientras que la mayoría de las compañías bio/farmacéuticas todavía están construyendo su ex-periencia e infraestructura para de-sarrollar biofarmacéuticos.
•Las compañías bio/farmacéuticas están matando a los candidatos des-de el inicio, antes de que incurran en altos costos de desarrollo clí-nico. La práctica refleja, en parte, el mayor entendimiento de lo que hace tóxicos a los compuestos y mejores herramientas para evaluar la toxicidad en el desarrollo inicial. También refleja el hecho de que las aseguradoras y los gobiernos están insistiendo en que los nuevos fár-macos aporten mayor eficacia que la de los productos de que se dis-pone actualmente. Los pagadores ya no quieren pagar extensas te-rapias que proporcionen sólo unas cuantas semanas más de vida a un paciente con cáncer o a una reduc-ción marginal en los niveles de las LDL (lipoproteínas de baja densi-dad), por ejemplo, como fármaco anti-colesterol. Sólo los fármacos que entregan beneficios clínicos sustanciales son los que avanzan en los proyectos de desarrollo.
•El desarrollo clínico en la última etapa se está volviendo más cos-toso. Las compañías bio/farma-céuticas están ahorrando dinero retrasando formulaciones costosas, desarrollo de métodos y análisis toxicológicos hasta después de que se haya demostrado la prueba de
Las compañías necesitan productos nuevos exitosos para reavivar el crecimiento.
“I+D en transición” Continúa en la pág. 22
Jim Miller es presidente de PharmSource Information Services, Inc., y editor de Bio/Pharmaceutical Outsourcing Report, tel. 703.383.4903, Twitter@JimPharmSource, [email protected], www.pharmsource.com

Pharmaceutical Technology en Español MARZO / ABRIL 201412
DENTRO DEL PIC/S
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
Deficiencias encontradas en las investigaciones de los APIsEl PIC/S revisa las deficiencias encontradas durante las inspecciones de plantas de manufactura de APIs, armoniza los estándares de GMP, y provee capacitación para los inspectores en todo el mundo.
Carmelo Rosa es director del Círculo de Expertos del PIC/S y director, División de
Calidad Internacional de Fármacos, FDA EEUU. Paula R. Katz, jefe interino de la rama
para la Política Regulatoria y Colaboración, y Alicia Mozzachio, jefe interino, División
de Calidad Internacional de Fármacos, ambas con en la FDA EEUU, la CDER/Oficina
de Cumplimiento/Oficina de Manufactura y Calidad del Producto, contribuyeron a
este artículo.
La Convención de Inspección Farmacéutica y el Es-quema de Cooperación para Inspección Farmacéu-tica (OIC/S) juega un importante papel en reunir a los reguladores de todo el mundo para desarrollar
mejores maneras de intercambiar información, lograr y man-tener la confianza mutua y la uniformidad, compartir planes de inspección y hallazgos, y proporcionar capacitación espe-cializada y avanzada en GMPs/técnica. Este artículo se enfoca en los esfuerzos del PIC/S para capacitar inspectores de todo el mundo sobre los APIs y describe algunas de las deficien-cias más comunes encontradas por los miembros del PIC/S (inspectores) durante las inspecciones de las plantas de ma-nufactura de APIs.
El PIC/S a través de sus diferentes grupos de trabajo, cír-culos de expertos y foros de capacitación, ha evolucionado como organización haciendo una contribución importante en la capacitación de inspectores en GMPs manteniendo el ni-vel más alto de competencia técnica del grupo de inspectores. Aunque las 43 autoridades miembros del PIC/S son respon-sables de tener su propia capacitación local, el PIC/S facilita la capacitación técnica internacional sobre tópicos de interés relevantes. Uno de los cuerpos más activos dentro del PIC/S es el Círculo de Expertos en APIs. Sus esfuerzos y plan estra-tégico están enfocados en cuatro áreas clave: identificar regio-nes y países donde el grupo de inspectores y la industria ten-gan mayores necesidades de capacitación de GMPs en APIs; proporcionándole a los inspectores información actualizada sobre procesos de manufactura de APIs avanzados o com-plejos; a través de sociedades con organizaciones técnicas y procedimientos establecidos, asegurando que los inspectores estén expuestos a nueva tecnología mientras que la industria recibe información relevante con respecto a los hallazgos de inspección y reguladores actuales, expectativas en el área de los APIs y; ofreciendo discusión y capacitación sobre temas regulatorios emergentes para los APIs. Hasta ahora, Se han realizado cinco Círculos de Expertos en APIs, el más reciente en Septiembre de 2012, en Washington DC, donde aproxima-damente se capacitaron 130 inspectores sobre los APIs estéri-les y manufactura biotecnológica. El sexto Círculo de Exper-tos en APIs se llevará a cabo en Roma del 19-21 de Mayo del 2014 sobre el tema de validación del proceso del API y temas contemporáneos. Aunque estos Círculos de Expertos son sólo diseñados y organizados para inspectores, están en curso otros esfuerzos. PIC/S y la Asociación de Fármacos Parenterales (PDA) han desarrollado un curso de capacitación para la in-dustria y los reguladores en la Conferencia Internacional de Armonización (ICH) Q7, Buenas Prácticas de Manufactura para Ingredientes Activos Farmacéuticos (1). El siguiente
curso está programado para que tenga lugar en Johannesburgo durante Marzo 18-19, 2014 (se han realizado capacitaciones similares en Lisboa, Portugal y en China). Se están planeando otras capacitaciones y seminarios con la PDA y otras organi-zaciones para que haya diversidad.
Deficiencias comunes encontradasDesde la implementación del Q7, la mayoría de los fabrican-tes de APIs parecen tener un mejor entendimiento de los prin-cipios de las GMPs. Muchas empresas tienen buenos sistemas de calidad, comprensión del proceso, buscan oportunidades para la optimización, tienen sistemas robustos de control de cambios, y tienen buenos controles de laboratorio y procedi-mientos para facilitar la gestión del conocimiento. Por otro lado, los inspectores también revelan que algunos fabricantes de APIs continúan luchando para lograr un cumplimiento sus-tentable con los requerimientos de las GMPs.
Las inspecciones de las instalaciones de APIs, realizadas por miembros del PIC/S, han estado reportando recientemen-te deficiencias críticas relacionadas con los controles de labo-ratorio, los registros/investigaciones, los sistemas de calidad, la limpieza/ mantenimiento del equipo y la validación del proceso.
Las principales deficiencias en GMPs citadas por la FDA en 2012 fueron:
• Controles de laboratorio. Validación de métodos carente o inadecuada; falla para tener especificaciones científica-mente válidas y apropiadas y procedimientos de prueba; fallas para investigar adecuadamente los resultados fuera de especificación; fallas para documentar actividades en el momento del desempeño; y falla para tener adecuados programas de estabilidad para evaluar los atributos de ca-lidad del API a lo largo de su fecha de caducidad.
• Sistema de calidad. falla de la unidad de calidad para liberar/rechazar APIs; falla de la unidad de calidad para revisar y aprobar todos los documento relacionados con la calidad; falla para asegurar que todas las quejas rela-cionadas con la calidad sean investigadas; falla para rea-lizar revisiones de calidad regulares de los APIs; y fallas para evaluar el impacto de los cambios en la calidad de los APIs.
• Equipo, limpieza, mantenimiento y validación. El equipo no se mantiene apropiadamente; procedimientos de lim-pieza deficientes; falla para validar los procedimientos de limpieza; falla para limpiar, almacenar o sanitizar el equipo para evitar contaminación o arrastre que pudiera alterar la calidad de los APIs; y calificación inadecuada del equipo crítico.

Pharmaceutical Technology en Español MARZO / ABRIL 2014 13
es director del Círculo de Expertos del PIC/S y director, División de
Calidad Internacional de Fármacos, FDA EEUU. , jefe interino de la rama
para la Política Regulatoria y Colaboración, y , jefe interino, División
de Calidad Internacional de Fármacos, ambas con en la FDA EEUU, la CDER/Oficina
de Cumplimiento/Oficina de Manufactura y Calidad del Producto, contribuyeron a
este artículo.
• Registros y reportes. Falla para preparar registros de lote adecuados; falla para incluir datos completos de todas las pruebas realizadas; falla para establecer procedimientos escritos relacionados con la producción, calidad, contro-les de laboratorio y manejo del material.Durante 2011, 12 de 20 Cartas de Advertencia emitidas
por la FDA fueron emitidas a los fabricantes de APIs; mien-tras que en 2012, 7 de 23 Cartas de Advertencia fueron emiti-das a los sitios de APIs. Una de las siete fue a un sitio que pro-ducía tanto API como producto terminado. A partir de Agosto 2013, 6 de las 26 Cartas de Advertencia fueron emitidas a fabricantes de APIs.
También se han encontrado hallazgos similares por otros miembros del PIC/S así como por la Dirección Europea para la Calidad de las Medicinas y la Salud (EDQM), quien es un copartícipe del PIC/S. En el 2012, el EDQM realizó 32 inspec-ciones de fabricantes de APIs localizados principalmente en Asia, de los cuales 13 mostraron serios hallazgos de no cum-plimiento con las GMPs o no cumplimiento del Certificado de Aptitud para las monografías de la Farmacopea Europea.
Problema de integridad de datosLos miembros y asociados del PIC/S también han señalado un incremento en los hallazgos de las prácticas de integridad de datos durante las inspecciones de los sitios de APIs. Estas deficiencias incluyen: registro de datos en bitácoras, falsifica-ción de registros de lotes y resultados de análisis, preanálisis de muestras y se ignora o no se investigan los resultados fuera de especificaciones, mezclado de lotes de APIs que no cum-plieron las especificaciones de liberación establecidas con lotes que cumplían las especificaciones finales requeridas, carencia de los controles necesarios en el manejo y gestión de datos críticos, e ingreso de actividades de manufactura en los registros antes de que hubieran ocurrido.
Aunque los principios establecidos en el Q7 no corregirán la conducta de las empresas que intencionalmente se engan-chan en una conducta decepcionante o errónea, las compañías que buscan sólo cumplir con apenas los principios mínimos, más que ir tras un marco de trabajo robusto y de calidad sus-tentable, siempre se encontrarán a sí mismas operando en un modo reactivo y es más probable que se enreden en problemas de integridad de los datos desafiantes.
Operaciones subcontratadas y la cara cambiante de la manufactura de APIsCon el incremento de la subcontratación de APIs por parte de patrocinadores/solicitantes de fármacos/propietarios de pro-ductos farmacéuticos terminados/otorgantes de contratos (la parte que compra APIs), existe más preocupación acerca de cómo se establecen y se administran los papeles y responsa-bilidades de cada parte. Las siguientes son algunas preguntas que los reguladores hacen durante las inspecciones:
• ¿Cómo asegura el propietario de la autorización de co-mercialización (solicitud de fármaco) que la planta de ma-nufactura del API está operando en un estado de control sustentable?
• ¿Qué marco de trabajo o política de calidad tiene en el lugar el sitio de manufactura del API?
• ¿El acuerdo de calidad describe la responsabilidad de cada parte?
• ¿Quién es responsable de supervisar el cumplimiento con
el acuerdo de calidad?• ¿Cómo son comunicados y manejados los problemas de
calidad?• ¿Los problemas de calidad son identificados, aislados o
reflejan un patrón más amplio?• ¿Qué métricas se usan para monitorear el desempeño del
proceso y la calidad del producto?• ¿Cómo son medidos, con la tendencia analizada, comu-
nicados y utilizados los atributos de calidad para hacer mejoras en el producto y el proceso?
• ¿Está la alta gerencia comprometida con la calidad? ¿Cómo se compromete la alta gerencia en comprender, resolver, comunicar y proporcionar recursos para los asuntos de calidad?
• ¿Está el proceso de manufactura bien diseñado y clara-mente comprendido por los operadores y el personal de calidad/producción?
• ¿Cómo se implementan los cambios críticos: como re-sultado de la innovación, a través de procesos de mejora continua, en respuesta a un pobre desempeño del proce-so o en respuesta a acción correctiva y acción preventiva (ACAP)?
• ¿Existe una fuerte comprensión científica y regulatoria del proceso para asegurar que el ACAP apropiado se ha o será implementado cuando se necesita?
ConclusiónEl papel de un regulador es determinar si las empresas están operando con cumplimiento sustentable de las GMPs para los APIs, de conformidad con el ICH Q7. En conexión con esto, el PIC/S ha revisado también recientemente cientos de pre-guntas y respuestas relacionadas con el ICH Q7 y transferido su revisión al ICH para su consideración. Durante los Círcu-los de Expertos del PIC/S, los inspectores se han capacitado en nueva tecnología, tendencias de calidad y deficiencias crí-ticas y cómo detectar problemas que pueden afectar la calidad de los APIs producidos y que puede impactar a los productos farmacéuticos terminados, y posteriormente afectar a los pa-cientes que consumen estas medicinas. Teniendo en mente que las inspecciones están limitadas por el tiempo y otras res-tricciones de recursos, los reguladores se apoyan fuertemente en los fabricantes para implementar y mantener sistemas de calidad apropiados y procesos para asegurar que todos los APIs producidos cumplan los estándares de calidad requeri-dos. A medida que se enfrentan nuevos retos con respecto al suministro y disponibilidad de APIs y fármacos en general, los reguladores necesitan alejarse del modelo de que son los que encuentran problemas para la industria. En lugar de esto, sería de esperar que las empresas diseñaran e implementaran proactivamente sistemas de calidad que conduzcan a las insta-laciones de manufactura de APIs a operar en un estado susten-table de control. Cuando los fabricantes de APIs operan bajo sistemas de calidad fragmentados, estructuras desconectadas de gestión de calidad o carencia de compromiso gerencial con una fuerte cultura de calidad, los problemas de integridad de los datos pueden ser más prevalentes y pueden impactar la calidad de los APIs producidos.
Referencias1. ICH, Q7, Good Manufacturing Practices for Active Pharmaceutical
Ingredients (ICH, November 2000). PT

Pharmaceutical Technology en Español MARZO / ABRIL 201414
QbD En la manuFactura Estéril
MESA REDONDA DE LA INDUSTRIAModerada por Susan Haigney
Implementando laen lamanufactura estérilQbD
Los expertos de la industria discuten la aplicación, los desafíos y los beneficios de un enfoque de calidad por diseño para la manufactura estéril.
Para adquirir perspectiva sobre la implementación de la calidad por diseño (QbD) en la manu-factura estéril, Pharmaceutical
Technology habló con expertos clave de la industria: Wolfgang Weikmann, vice-presidente de aseguramiento de la cali-dad en Vetter Pharma-Fertigung GmbH & Co.KG; Hal Baseman COO y director en ValSource LLC y director electo del Consejo de Directores de la Asociación de Fármacos Parenterales (PDA) y vi-cedirector de PDA Science Advisory; y James E. Akers, presidente de Akers Kennedy y Asociados.
Atributos de calidad críticosPharmTech: En la implementación de un enfoque de QbD ¿Qué identificaría Ud. como los atributos de calidad críti-cos (CQAs) en la manufactura estéril y el proceso aséptico?
Akers (Akers Kennedy y Asociados): Los atributos de calidad críticos están relacionados con el control y la preven-
ción de la contaminación, principal-mente microbiológica, con el material particulado total y con las endotoxinas aunque claramente los problemas de pureza química y uniformidad del mate-rial, que son una constante en la manu-factura farmacéutica, también aplican a los productos estériles y asépticos.
Weikmann (Vetter): Los potenciales atributos de calidad críticos del produc-to farmacéutico de un cliente en térmi-nos del procesado aséptico son determi-nados por la naturaleza del producto y por la definición de su perfil de produc-to objetivo. La mayoría de los atributos críticos comunes identificados se refie-ren a las propiedades físicas, químicas y microbiológicas. Reconocemos la es-terilidad, las endotoxinas bacterianas y la exactitud de la dosis como tres de los CQAs esenciales.
Baseman (ValSource): La identifica-ción y el conocimiento de los CQAs son esenciales para diseñar un proceso que establezca y mantenga a estos CQAs.
Éstos pueden ser ampliamente defini-dos como pureza, seguridad, potencia e identidad del producto. Por lo tanto, los CQAs asociados con el procesado aséptico y aquéllos que necesitan ser establecidos y mantenidos por el pro-ceso son:• Pureza: Aquéllos CQAs que asegu-
ran que no hay nada presente en el producto que no debería estar ahí (es decir, material extraño o contamina-ción cruzada del producto o residuos químicos).
• Seguridad: Aquéllos CQAs cuya au-sencia probablemente causaría daño al paciente y entonces incluirían ase-guramiento de la esterilidad, ausencia de endotoxinas, ausencia de contami-nación con partículas.
• Potencia: Aquéllos CQAs que deben estar presentes para asegurar la efec-tividad del producto y entonces in-cluirían potencia, ingrediente activo y funcionalidad.
• Identidad: Aquéllos CQAs que ase-guran que el producto es lo que pre-tende ser y entonces incluirían la co-rrecta y completa indicación de iden-tidad del producto, número de lote, contenido y potencia.
Parámetros críticos del procesoPharmTech: ¿Qué herramientas de me-dición se utilizan típicamente para me-dir los parámetros críticos del proceso y las entradas y salidas del proceso resul-tantes, incluyendo las herramientas de tecnología analítica de proceso (PAT), en la manufactura estéril?
Akers (Akers Kennedy y Asociados): Muchos de los parámetros clave en la manufactura aséptica y estéril pueden ser medidos directamente y en tiempo real. La esterilización de los compo-nentes, la despirogenación de algunos materiales, los volúmenes de llenado y en algunos casos, el sellado del empa-que, pueden ser medidos en tiempo real. También, los factores ambientales clave, tales como la calidad del aire en partícu-las totales, los diferenciales de presión y las características operacionales de las manejadoras de aire pueden ser medidos en tiempo real. En la moderna manufac-tura aséptica con aisladores/cámara de aire y en la esterilización terminal, estos parámetros listados son los más críticos.
Baseman (ValSource): La definición de una correlación cuantificable entre lo que puede ser medido y el resultado

Éstos pueden ser ampliamente defini-dos como pureza, seguridad, potencia e identidad del producto. Por lo tanto, los CQAs asociados con el procesado aséptico y aquéllos que necesitan ser establecidos y mantenidos por el pro-ceso son:
Aquéllos CQAs que asegu-ran que no hay nada presente en el producto que no debería estar ahí (es decir, material extraño o contamina-ción cruzada del producto o residuos químicos). Aquéllos CQAs cuya au-sencia probablemente causaría daño al paciente y entonces incluirían ase-guramiento de la esterilidad, ausencia de endotoxinas, ausencia de contami-nación con partículas. Aquéllos CQAs que deben estar presentes para asegurar la efec-tividad del producto y entonces in-cluirían potencia, ingrediente activo y funcionalidad. Aquéllos CQAs que ase-guran que el producto es lo que pre-tende ser y entonces incluirían la co-rrecta y completa indicación de iden-tidad del producto, número de lote, contenido y potencia.
¿Qué herramientas de me-dición se utilizan típicamente para me-dir los parámetros críticos del proceso y las entradas y salidas del proceso resul-tantes, incluyendo las herramientas de tecnología analítica de proceso (PAT), en la manufactura estéril?
Muchos de los parámetros clave en la manufactura aséptica y estéril pueden ser medidos directamente y en tiempo real. La esterilización de los compo-nentes, la despirogenación de algunos materiales, los volúmenes de llenado y en algunos casos, el sellado del empa-que, pueden ser medidos en tiempo real. También, los factores ambientales clave, tales como la calidad del aire en partícu-las totales, los diferenciales de presión y las características operacionales de las manejadoras de aire pueden ser medidos en tiempo real. En la moderna manufac-tura aséptica con aisladores/cámara de aire y en la esterilización terminal, estos parámetros listados son los más críticos.
La definición de una correlación cuantificable entre lo que puede ser medido y el resultado
Marque en la tarjeta de servicio al lector el No. 3

Pharmaceutical Technology en Español MARZO / ABRIL 201416
pretendido es particularmente desafian-te para el proceso aséptico. Algunas herramientas y sistemas que pueden ayudar mejor a comprender y diseñar el proceso incluyen:• Estudios de perfil de aire/humo y
estudios del flujo de materiales/per-sonal para establecer principios del primer aire y su relación con conta-minación potencialmente microbia-na. Estas herramientas son esenciales para el diseño y aseguramiento de las estrategias de control del proceso.
• El monitoreo ambiental, que incluye el monitoreo continuo de no viables y viables, para monitorear el desempe-ño del proceso.
• Las evaluaciones de riesgo para en-tender mejor la relación y el impacto de las estrategias de control del pro-ceso sobre los CQAs y los paráme-tros críticos del proceso.
• Los factores humanos y ergonómicos para ayudar a crear procesos que mi-nimicen el efecto del desempeño hu-mano y de la fatiga.
• La presión diferencial y el flujo de aire y velocidad para mantener la lim-pieza del entorno del cuarto limpio.
• El monitoreo de temperatura y hume-dad para reducir el potencial de de-sarrollo microbiológico en el cuarto limpio.
• La indumentaria y el monitoreo del personal para asegurar el control de las condiciones ambientales.
Existe, no obstante, una correlación directa y medible entre los parámetros de la esterilización, que incluye:• El monitoreo del tiempo, la tempera-
tura y la presión de los ciclos de este-rilización con autoclave para asegurar el control de la contaminación micro-biológica de las partes y superficies en contacto con el producto.
• Algunos de los procesos asépticos, como la liofilización, pueden utilizar el PAT en su manejo de formulaciones para la consistencia del proceso y el control de los resultados de los CQAs.
• Las pruebas de integridad de la fil-tración para asegurar la integridad y eficiencia de los métodos de esterili-zación del producto.
PharmTech: ¿Cómo se diseña el equipo y se modifica la operación cuan-do se implementa la QbD en la manufac-tura estéril y aséptica?
Baseman (ValSource): Los estudios
de perfil del aire/humo pueden utilizarse para determinar la forma y configuración más óptima para el equipo del procesado aséptico. Los factores humanos pueden ser usados para determinar y mejorar las intervenciones. La evaluación basada en el riesgo de las intervenciones y el uso de automatización pueden ser usados para reducir el impacto y el riesgo de las in-tervenciones sobre la variación del pro-ceso y la calidad del producto.
Weikmann (Vetter): Viendo primero el diseño del equipo, tenemos la fuerte sensación de que el proceso de revisión del diseño tiene que ser mencionado como una herramienta esencial. La re-visión del diseño es un hito decisivo dentro del proceso de desarrollo para una sola pieza del equipo. Esto se rea-liza a través del ajuste de un diseño ini-cialmente creado con sus requerimien-tos particulares. Esto es importante para controlar los resultados de las activida-des anteriores, así como para identificar y resolver cualquier problema existen-te antes del inicio del proceso de ma-nufactura del fármaco. Nosotros ante todo ejecutamos una revisión del diseño dentro de la fase de construcción para cada pieza relevante cGMP del equipo principal del proceso. Un elemento de ejemplo de esta revisión del diseño es realizar estudios en maqueta.
Viendo el lado de las operacio-nes, al servir a múltiples clientes con diferentes productos y requisitos es-pecíficos del proceso, nuestro equipo tiene que ser altamente flexible. Dicho arreglo ofrece el beneficio de que, con base en los requerimientos específicos del producto así como con base en los estudios de bombeo realizados dentro del enfoque de QbD, los componentes específicos del equipo, tales como el producto más adecuado y las bombas de llenado ajustadas al proceso, pueden ser utilizados de inmediato para la pro-ducción posterior.
Akers (Akers Kennedy y Asociados): En los proyectos en que hemos traba-jado los últimos 20 años, no creo que el énfasis actual que se le da a la QbD haya alterado lo que hacemos ya que siempre nos hemos concentrado en lle-gar al diseño más efectivo, eficiente y seguro para un proceso. Yo pienso que debido al énfasis regulatorio actual, uti-lizamos la descripción QbD o decimos esterilidad por diseño más que en el pa-
sado. Así, hay un poco de enfoque adi-cional para pensar y hablar en términos de QbD, particularmente entre algunos clientes.
Desafíos en la implementación de la QbD
PharmTech: ¿Cuáles son los desa-fíos en la aplicación de la QbD en la manufactura estéril?
Baseman (ValSource): Los desafíos son la carencia de correlación cuanti-ficable entre lo que podemos medir y monitorear, y el resultado deseado o el resultado indeseado. También, uno debe estar consciente del riesgo residual y de las consecuencias involuntarias como resultado de los cambios hechos para dirigir el riesgo y mejorar el proceso. Cada acción tomada para resolver o ma-nejar un riesgo del proceso representa un cambio que puede tener el potencial de agregar un nuevo riesgo o una conse-cuencia involuntaria. Estos nuevos ries-gos deben ser evaluados y manejados en lo posible.
Akers (Akers Kennedy y Asociados): La QbD no es realmente un nuevo re-querimiento en mi experiencia. Yo creo en la manufactura estéril; hemos hecho la QbD antes de que fuera formalmente denominada QbD. Yo creo que mucho de lo que ahora hacemos como QbD fue una vez parte de nuestras actividades de calificación del diseño. Los parámetros de diseño para las instalaciones de pro-ducción estéril en términos de cuartos limpios y diseños sanitarios han estado bien definidos durante años.
Weikmann (Vetter): En nuestra ex-periencia, los principales desafíos en la aplicación de un enfoque de QbD se de-ben a la intensidad del tiempo, el costo y los recursos en la etapa del desarrollo inicial. Otro desafío significativo es el alto grado de pericia que debe poner-se en el sitio para realizar un enfoque apropiado de la QbD.
Con respecto a los tiempos, tiene que incluirse un programa mucho más prolongado. Para los costos, los costos más elevados se deben a la variedad de estudios adicionalmente realizados. En cuanto al manejo de recursos más eleva-dos, éstos se deben al equipo, a los dife-rentes materiales, así como al personal.
En suma, esto es siempre un acto de balanceo dependiente del producto entre los riesgos y las consecuencias
QbD En la manuFactura Estéril

Pharmaceutical Technology en Español MARZO / ABRIL 2014 17
por un lado, y el costo durante la etapa de desarrollo por el otro lado. Incluso si los desafíos previamente descritos se presentan en las primeras etapas, el conocimiento adquirido probablemente resultará en un beneficio económico en el ciclo de vida comercial a largo plazo.
Beneficios de la QbDPharmTech: ¿Qué entendimiento
adicional se adquiere a través de un en-foque de QbD en comparación con un enfoque tradicional?
Weikmann (Vetter): El entendimien-to más importante adquirido realizando una operación a través de un enfoque de QbD contra el enfoque más tradi-cional, es darse cuenta de los requisitos necesarios del producto de un cliente ya desde una etapa inicial del desarro-llo para acomodar el perfil objetivo del producto. Estos requerimientos no se limitan solamente a la calidad del pro-ducto. Éstos incluyen el desempeño del proceso así como los sistemas aplicados y el entorno, por ejemplo, en ciclos op-timizados y reducidos del proceso de liofilización.
Con base en varios estudios, los diferentes casos posibles se evalúan en detalle. Este examen finalmente lleva a una mejor comprensión de las varia-ciones del proceso, resultando así en un espacio de diseño aceptable. En conse-cuencia, el uso de un enfoque de QbD ofrece la posibilidad de adquirir un me-jor conocimiento del proceso, lo que da como resultado una mejor robustez del proceso completo.
Baseman (ValSource): Se obtiene una mejor comprensión de las variables del proceso y del impacto sobre el ries-go de la calidad del producto, lo cual ayudará a diseñar el proceso y las estra-tegias de control del proceso.
Akers (Akers Kennedy y Asociados): En realidad, Joseph M. Juran acuñó la frase ‘calidad por diseño’ a principios de los 90’s, y estas ideas fueron amplia-mente adoptadas en el proceso aséptico antes de que el QbD reingresara al lé-xico de la industria hace unos cuantos años. Estimo que el 95% de la seguri-dad del producto aséptico en 2013 ema-na directamente del diseño apropiado del equipo y del entorno.
PharmTech: ¿Cómo la QbD mejora el aseguramiento de la calidad y cómo puede mitigar los problemas en la ma-
nufactura?Weikmann (Vetter): A través de la
mayor importancia que se le da a las ac-tividades de desarrollo del proceso y al gran número resultante de los estudios de desarrollo realizados dentro de un enfoque de QbD, se adquiere una mejor comprensión del proceso. Dentro del enfoque del manejo de riesgo, los ries-gos ya son identificados desde una eta-pa temprana. En consecuencia, pueden desarrollarse actividades de mitigación del riesgo y una estrategia de control basada en el riesgo, implementada muy desde el principio de la etapa comercial, que forman las bases para la mejora del aseguramiento de la calidad y la super-visión de calidad.
Akers (Akers Kennedy y Asociados): No puedo señalar a una sola área espe-cífica donde yo creo que la QbD ha te-nido un impacto en el aseguramiento de la calidad del producto. La manufactura de productos estériles siempre ha sido y continuará siendo un esfuerzo orientado al detalle. La QbD puede haber incre-mentado la conciencia de la criticalidad para obtener la instalación y el diseño del proceso correctos. Quizás tenemos una mejor apreciación de la primacía de la buena ingeniería y del desarrollo del proceso científico en las operaciones de manufactura de productos estériles.
Baseman (ValSource): Esto puede me-jorar la comprensión del proceso, inclu-yendo la identificación y el control de las variables del proceso. Esto puede llevar a un proceso mejorado y menos riesgo.
PharmTech: ¿De qué manera las prác-ticas de QbD acumulan o complementan los enfoques adoptados en relación con la liberación paramétrica y los paráme-tros de esterilización terminal relaciona-dos, necesarios para la liberación?
Akers (Akers Kennedy y Asociados): Quizás la QbD, como se señaló pre-viamente, fortalecerá el enfoque sobre el diseño y control del proceso. Sin embargo, llegar a un proceso adecua-do para la consideración de la libera-ción paramétrica ha requerido siempre ciencia e ingeniería meticulosas. Yo personalmente creo que los productos esterilizados terminalmente, dada su robustez y confiabilidad, deberían ser paramétricamente liberados con más frecuencia. La alternativa a la liberación paramétrica es la prueba de esterilidad, la cual tiene limitaciones en términos de
detección microbiológica y sensibilidad así como en el área de la estadística del muestreo.
Baseman (ValSource): La compren-sión de la relación entre los parámetros que podemos controlar, monitorear y medir, y el resultado deseado o CQA es esencial para diseñar un proceso efectivo. Puede no ser suficiente para saber cuáles parámetros afectan la calidad del produc-to y el desempeño del proceso; también debemos saber hasta qué alcance hacen esto y qué tan bien son controlados.
PharmTech: ¿De qué manera ha me-jorado la transferencia de tecnología bajo un enfoque de QbD?
Weikmann (Vetter): La transferencia de tecnología ciertamente es un área que está especialmente mejorada a través del enfoque de QbD. Lleva a posibili-dades mejoradas para monitorear y eva-luar el proceso. Al familiarizarse con un marco del producto farmacéutico y sus límites a través de realizar numerosas corridas técnicas de versiones variadas, uno puede definir la especificación del producto en la cual el proceso funciona con robustez.
Un ejemplo cotidiano para mostrar la mejora de la transferencia de tecnolo-gía a través de la QbD para los clientes es demostrando el desarrollo del ciclo de liofilización del producto de un clien-te. Por ejemplo, utilizando un enfoque QbD con la adquisición de los datos correspondientes basados en numero-sas pruebas de liofilización realizadas, nos damos cuenta de una significativa reducción del ciclo de liofilización para un producto particular del cliente. Esta reducción dio como resultado el ahorro de una alta cantidad de capital del clien-te, principalmente dentro de la produc-ción comercial a gran escala.
Akers (Akers Kennedy y Asocia-dos): Honestamente yo no veo mucho cambio. He sido lo suficientemente afortunado para trabajar con empresas que siempre toman el desarrollo de las especificaciones del requerimiento del usuario a través del análisis detallado muy seriamente.
Baseman (ValSource): Esto resulta en una mejor comprensión del proceso y de las variables del proceso, incluyendo qué pasos del proceso requieren control-medidas de control que necesitan ser incluidas en la transferencia de tecnolo-gía. PT

Pharmaceutical Technology en Español MARZO / ABRIL 201418
Roger Hayes, Vicepresidente y Gerente General de Laboratory Sciences of MPI ResearchPharmTech: ¿Cuáles son algunos desafíos en los estudios bioa-nalíticos para fármacos altamente potentes y cómo se superan éstos?
Hayes: Más allá del rendimiento y la sensibilidad cuantitati-va está el requerimiento de datos exactos y confiables. Los pa-rámetros que deben estar en control se describen en las guías de la EMA y la FDA así como en artículos (1-4). El analista debe estar bien capacitado para asegurar la realización confiable del ensayo. La automatización en el manejo de líquidos es la manera más fácil de mitigar la variabilidad del ensayo e incrementar la eficiencia (5).
Las técnicas optimizadas para la preparación de la muestra que proveen factores de alto enriquecimiento se vuelven críticos para concentraciones diluidasa de fármacos de alta potencia. La extracción en fase sólida (SPE) es una técnica común para la ex-tracción de la muestra en el bioanálisis. Los absorbentes de alta eficiencia y los nuevos diseños de placas de 96 pozos, que inclu-yen las puntas de pipeta y discos para SPE, ofrecen soluciones para eluir fármacos en muy pequeños volúmenes. Además, el modo mezclado o los absorbentes de intercambio de carga pro-veen incluso extractos de la muestra más limpios (6, 7). También, las columnas empacadas con partículas de fase estacionaria de menos de 2µm es quizás una de las contribuciones más signifi-cativas para mejorar el bioanálisis por cromatografía de líquidos-espectrometría de masas en tándem (LC-MS/MS) (8).
Adicionalmente, la tecnología de partículas de sílica de nú-cleo fundido continúa descubriendo oportunidades para el de-sarrollo de ensayos con mayor rendimiento (9). Cuando las pla-taformas bioanalíticas convencionales no tienen suficiente sen-sibilidad, algunos investigadores voltean a la espectrometría de masas con acelerador (AMS) para medir el fármaco (10). El AMS puede entregar mejoras de 1000 veces la sensibilidad en compa-ración con la mayoría de los métodos de LC-MS/MS.
PharmTech: ¿Cuáles son algunos avances recientes de la tecno-logía usados en el análisis de fármacos altamente potentes?
Hayes: Las plataformas más nuevas de ensayos de unión a ligandos (LBA) centradas en la electroquimioluminiscencia o en la fluorescencia (11) se han vuelto plataformas de elección debi-do a su inherente capacidad para incrementar la sensibilidad del
ensayo y ensanchar el rango dinámico del ensayo. Además, algunas plataformas ultra-sensibles de ensayo de Inmunoaná-lisis ligado a enzimas (ELISAs) sin etiqueta están ahora disponibles. Por ejemplo, el enfoque de la plataforma de reacción de la cadena de inmuno-polimerasa (IPCR) vincula los inmunoensayos estándar con la amplificación exponencial de la señal del PCR, incrementando así la sensibili-dad convencional del inmunoensayo de 100 a 10,000 veces.
Los modelos predictivos para la toxicidad continúan desa-rrollándose. El enfoque típico es correlacionar in vivo (animal) la concentración plasmática contra el tiempo y las curvas de toxi-cidad para la concentración in vitro contra las curvas de respues-ta a la toxicidad para un gran número de compuestos y después definir una relación matemática entre los datos de respuesta del animal y los datos de respuesta in vitro (12). El reto que perma-nece es la exactitud de la traducción de la toxicidad in vitro en efectos significativos in vivo. Un enfoque alternativo es definir los biomarcadores de toxicidad que son traducibles entre espe-cies no clínicas y pacientes. El Consorcio de Análisis Predictivo de Seguridad, una colaboración pre-competitiva de compañías farmacéuticas y la FDA, ha formado varios grupos de trabajo para abordar los retos asociados con el desarrollo y calificación de biomarcadores traducibles de toxicidad (13).
Referencias1. B. DeSilva et al., Pharm. Res. 20 (11) 1885-1900 (2003).2. J. Lee et al., AAPS J. 9 (2) E164-170 (2007).3. M. Kelley et al., AAPS J. 9 (2) E156-163 (2007).4. R.W. Abbott et al., Bioanalysis 3(8), 833–838 (2011). 5. A.B. Ahene et al., AAPS J. 14 (1) 142-153.6. J.X. Shen et al., J. Chromatogr. B. 843 (2) 275-282 (2006).7. J.X. Shen et al. Rapid Commu. Mass Spectrom. 21 (18) 3145-3155 (2007).8. L. Tolley et al. Anal. Chem. 73 (13) 2985-2991 (2001).9. R.D. Ricker et al., Chromatogr. Sci. 46 (3). 261-268 (2008).10. A. Arjomand, Bioanalysis. 2 (3) 519-541 (2010).11. J.R. Mora et al., Bioanalysis. 2 (10) 1711-1715 (2010).12. J.M. Jr. McKim, Comb. Chem. High Throughput Screening 13 (2) 188-
206 (2010).13. E. H. Dennis et al., Drug Dev. Res. 74 (2) 112-126 (2013).
P yR con
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486E-mail: [email protected]
Estableciendo un tiempo de incubación mínimo para indicadores biológicos
La industria carece de un método aceptado para establecer un tiempo mínimo de incubación (MIT) de menos de siete días para los indicadores biológicos (IBs). Los autores proponen un método de MIT que proporcione un medio para la determinación reproducible del tiempo de desarrollo de los IBs.
John R. Gillis, Philip M. Schneider, Gregg Mosley, John B. Kowalski, Garrett Krushefski, y Kurt J. McCauley
PHARMA CÁPSULAS

Pharmaceutical Technology en Español MARZO / ABRIL 2014 19
Estableciendo un tiempo de incubación mínimo para indicadores biológicos
La industria carece de un método aceptado para establecer un tiempo mínimo de incubación (MIT) de menos de siete días para los indicadores biológicos (IBs). Los autores proponen un método de MIT que proporcione un medio para la determinación reproducible del tiempo de desarrollo de los IBs.
*John R. Gillis, PhD, es presidente retirado de SGM Biotech (Msa Laboratories), 10 Evergreen Dr., Bozeman, MT [email protected]. Philip M. Schneider es consultor senior con LexaMed, Toledo, oH, y es moderador de ISo/TC 198 wG4 Indicadores Biológicos. Gregg Mosley es presidente retirado de Biotest Laboratories, Minneapolis, MN. John B. Kowalski, PhD, es consultor senior de STERIS Isomedix Services, Mentor, oH. Garrett Krushefski es vicepresidente, Biological Indicator operations, y Kurt J. McCauley es director de I+D en Apex Labs, ambos en Mesa Laboratories, Bozeman, MT.
*A quien debe dirigirse la correspondencia.
Sometido: Mayo 17, 2013. Aceptado: Junio 12, 2013.
No existe un método universalmente reconocido para establecer un tiempo de incubación mínimo (MIT) aceptable de menos de siete días para los indicadores biológicos (IBs) usados para monito-
rear la eficiencia de los procesos de esterilización. Un méto-do para calificar un tiempo de incubación reducido (RIT) de menos de siete días fue publicado por la FDA en el Centro para Dispositivos y Salud Radiológica (CDRH) Guía para la Validación del Tiempo de Incubación de Indicadores Bioló-gicos en 1986 (1). La comunidad internacional, sin embargo, ha sido resistente a aceptar este método debido a la carencia de resultados publicados para respaldar los requerimientos de prueba seleccionados y los criterios de aceptación. En 2007, el Grupo de Trabajo 4 (WG4), Indicadores Biológicos del ISO/TC198 inició una Propuesta de Nuevo Tema de Trabajo (NWIP) para desarrollar una Especificación Técnica (TS) ISO que contuviera un método para la validación de un MIT para los IBs. La postura tomada por el WG4 ISO fue que cualquier método propuesto debería estar soportado por resultados pu-blicados en una revista técnica revisada por pares. A solicitud del WG4 ISO, el Grupo de Trabajo 4 de IB de la Asociación para el Avance de Instrumentación Médica (AAMI) formó un comité ad hoc en 2008 para desarrollar un plan para dicha investigación y la publicación de los resultados. A la fecha, se han publicado dos artículos que contienen resultados que soportan una base lógica para un método para determinar un MIT para los IBs (2, 3).
Este artículo es un resultado de los esfuerzos del comité ad hoc y propone un método para establecer un MIT para los IBs. El método está basado en un análisis de los tiempos de crecimiento para más de 10,000 IBs expuestos a una variedad de procesos de esterilización (2, 3). Los IBs usados en estos estudios fueron fabricados de conformidad con el ISO 11138-1 y sub-secciones específicas, según convenga y liberado para uso comercial (4). Las instrucciones de incubación del fabri-cante fueron seguidas, según fue lo apropiado.
Diseño del estudio y hallazgos claveLos estudios incluyeron varias configuraciones de los IBs, incluyendo tanto IBs auto-contenidos y tiras de esporas con-vencionales. Los portadores de esporas incluyeron tanto pa-pel como acero inoxidable. Las dos especies más comunes de endoesporas bacterianas usadas en la manufactura de IBs, Geobacillus stearothermophilus y Bacillus atrophaeus, fue-
John R. Gillis, Philip M. Schneider, Gregg Mosley, John B. Kowalski, Garrett Krushefski, y Kurt J. McCauley

Pharmaceutical Technology en Español MARZO / ABRIL 201420
ron probadas. Los medios de crecimiento para los estudios fueron controlados e incluyeron ampolletas de medio para los IBs auto-contenidos y medios en tubos para las tiras de espora que se describieron previamente (2, 3).
Los procesos de esterilización evaluados fueron calor hú-medo a 121, 132, 134 y 135°C, gas de óxido de etileno (EO) (Oxyfume 2000), vapor de peróxido de hidrógeno (H
2O
2), y
gas de dióxido de cloro (ClO2). Estos procesos fueron usados
para el análisis de los IBs ya que representan los procesos de esterilización más comúnmente usados, monitoreados con IBs y también muestran diferentes mecanismos letales que in-cluyen transferencia de energía mediada por H
2O, alquilación
y oxidación.Se evaluaron grupos separados de IBs bajo las siguientes
condiciones:• No expuestos• Expuestos a los varios procesos de esterilización de manera
que resultaran en 30 a 80% del análisis de los IBs no esté-riles (positivos para el crecimiento). Este resultado de la zona cuantal, en la cual una fracción de los IBs son estériles y una fracción es no estéril, cumple los requerimientos del protocolo CDRH de la FDA.
• Expuestos utilizando los tiempos calculados a partir de la fórmula del Tiempo de Sobrevivencia Calculado (CST) pu-blicada en el ISO 11138, ISO 14161 y la Farmacopea de los Estados Unidos (4-6).
Todos los IBs expuestos fueron incubados hasta que fuera observado un resultado no estéril o durante un máximo de sie-te días. Un resultado no estéril estuvo basado en la evidencia de crecimiento microbiano tal como la turbidez y/o un cambio en el color del indicador de pH en el medio de crecimiento.
Los hallazgos clave de estos estudios de tiempo de creci-miento que son relevantes para la identificación de un método de MIT, basado en resultados, se listan a continuación:• Los rangos de los tiempos de crecimiento observados se
aproximan a la distribución normal sin importar el tipo de proceso de esterilización y los mecanismos letales asocia-dos o la especie de esporas en el IB.
• Existe una relación inversa entre el número de esporas so-brevivientes en un IB expuesto y el tiempo de crecimiento observado. Los IBs que tienen un mayor número de esporas sobrevivientes tienen un tiempo de crecimiento más corto que aquéllos que tienen menos esporas sobrevivientes.
• Se encontraron tiempos de crecimiento prolongados para un pequeño porcentaje (∼1.1%) de los IBs probados. Dichos tiempos de crecimiento prolongados fueron observados con todos los procesos de esterilización (calor húmedo, EO, va-por de H2
O2, y gas de ClO
2) y se presentaron cuando la ex-
posición a la esterilización dio un resultado de zona cuantal.• Los IBs que demostraron tiempos de crecimiento prolonga-
dos no fueron observados cuando se expusieron a esterili-zación en el CST. Los IBs expuestos de esta manera tuvie-ron tiempos de crecimiento más rápidos y más consistentes cuando se comparó con los IBs expuestos bajo condiciones que dieron resultados de zona cuantal.
Justificación para el método MIT propuestoUna de las cosas que afectan negativamente la amplia acepta-
ción del protocolo CDRH de la FDA es la carencia de resul-tados de la prueba para soportar el propio método y sus cri-terios de aceptación. El protocolo CDRH de la FDA requiere la realización de tres exposiciones a esterilización, cada una de las cuales usa 100 IBs y debe resultar en 30 a 80% de los análisis de los IBs no estériles. Los críticos ven como arbitra-rios los valores de 30 a 80% y el requerimiento adicional de más de 97% de correlación de los resultados del RIT con los resultados del tiempo de incubación de siete días. La comu-nidad internacional ha planteado consistentemente el asunto de porqué 30 a 80% de sobrevivencia es apropiado y por qué no se requiere una correlación del 100% para los resultados del tiempo de incubación de siete días. No se ha presentado ninguna base lógica o resultados de prueba de soporte para justificar el uso de 30 a 80% de no estéril y los valores de correlación mayores de 97%.
La exposición de microorganismos en general, y las espo-ras en particular, a un proceso de esterilización da como resul-tado un rango de efectos característicos del propio proceso de esterilización y del alcance del tratamiento. Conforme avanza el alcance del tratamiento, un IB tiene menos y menos esporas sobrevivientes hasta que, en algún nivel del tratamiento, no hay esporas sobrevivientes. En la zona cuantal, algunos de los IBs tienen sólo una espora sobreviviente y otros tienen un bajo número de sobrevivientes.
Para los IBs expuestos que tienen sólo una espora sobrevi-viente, puede existir una de dos condiciones: la espora no ha sido dañada de una manera que afecte significativamente ya sea el tiempo de germinación o la velocidad de crecimiento post-germinación, o ha ocurrido daño que afecta significati-vamente una o ambas características. Cuando una exposición a la esterilización da un resultado de zona cuantal de 50% de los IBs que probaron ser no estériles, la distribución de Poisson pronostica que aproximadamente 35 IBs tendrán sólo una espora sobreviviente y los otros 15 tendrán más de una espora sobreviviente (2). De los resultados de los estudios de tiempo de crecimiento reportados previamente, es claro que la mayoría de los IBs con sólo una espora sobreviviente y, más probablemente, todos los IBs que tengan más de una espora sobreviviente, exhiben tiempos de crecimiento dentro de un rango relativamente estrecho. También, se esperaría que un número muy bajo de IBs exhibiera un tiempo de crecimiento prolongado; los tiempos prolongados fueron observados para 2/458 (0.44%) y 3/309 (0.97%) IBs para los procesos de este-rilización con calor húmedo y con EO, respectivamente.
Los IBs que tienen tiempos de crecimiento prolongados son más probable que se presenten cuando las condiciones de exposición resultan en un menor porcentaje que son no estériles, por ejemplo, 35% contra 75%. Para un resultado no estéril de 35%, el número promedio de esporas viables por IB expuesto es 0.43 contra 1.39 cuando 75% de los IBs dieron no estériles. Conforme el porcentaje de IBs no estériles se redu-ce, proporcionalmente más de los IBs no estériles tienen dos o más esporas sobrevivientes; para un resultado no estéril de 75%, aproximadamente 40 de los IBs no estériles tienen dos o más esporas sobrevivientes (2)
Para desarrollar un método MIT viable, el uso de una exposición a la esterilización que resulte en un número con-
sistente bajo de esporas sobrevivientes es un pre-requisito importante. El tratamiento de los IBs en un resistómetro en un tiempo de exposición especificado por el cálculo de CST resultará en un método más consistente y reproducible para determinar un MIT en contraposición a la producción de 30 a 80% de IBs no estériles.
El CST, según se indica en la , es un tiempo de exposición que toma en cuenta tanto el nivel de población de esporas como el valor D y se pretende que resulte en 100% de los IBs que prueban ser no estériles (4-6). Un IB sometido a una exposición CST se esperaría que tuviera aproximada-mente 100 esporas sobrevivientes; los resultados de los estu-dios previos del tiempo de crecimiento mostraron que cuando los IBs se expusieron al CST, no se observaron tiempos de crecimiento prolongados en 1927 exposiciones CST,
CST = (valor D
donde el CST y el valor D están en minutos. El alcance de tratamiento para una exposición CST es marginalmente me-nor que la usada para obtener el 30 a 80% de resultados de no esterilidad requeridos por el protocolo CDRH de la FDA. Los IBs tratados en una exposición a la esterilización CST desafia-rán el medio de incubación y las condiciones con 100 espo-ra que han recibido una agresión letal significativa (99.99% de las esporas inactivadas debido a la acción letal del agente esterilizante). Este nivel de microorganismos es similar al usado en la prueba de promoción de crecimiento en medio microbiológico y la prueba de bacteriostasis/fungistasis don-de las inoculaciones se realizan con niveles bajos (100) de microorganismos de prueba (7).
Con base en los resultados del análisis resumido en los dos estudios previamente publicados y el análisis posterior de los datos, se propone el siguiente método para la determinación de un MIT para los IBs:•
••
•
•
•

Pharmaceutical Technology en Español MARZO / ABRIL 2014 21
ción del protocolo CDRH de la FDA es la carencia de resul-tados de la prueba para soportar el propio método y sus cri-terios de aceptación. El protocolo CDRH de la FDA requiere la realización de tres exposiciones a esterilización, cada una de las cuales usa 100 IBs y debe resultar en 30 a 80% de los análisis de los IBs no estériles. Los críticos ven como arbitra-rios los valores de 30 a 80% y el requerimiento adicional de más de 97% de correlación de los resultados del RIT con los resultados del tiempo de incubación de siete días. La comu-nidad internacional ha planteado consistentemente el asunto de porqué 30 a 80% de sobrevivencia es apropiado y por qué no se requiere una correlación del 100% para los resultados del tiempo de incubación de siete días. No se ha presentado ninguna base lógica o resultados de prueba de soporte para justificar el uso de 30 a 80% de no estéril y los valores de correlación mayores de 97%.
La exposición de microorganismos en general, y las espo-ras en particular, a un proceso de esterilización da como resul-tado un rango de efectos característicos del propio proceso de esterilización y del alcance del tratamiento. Conforme avanza el alcance del tratamiento, un IB tiene menos y menos esporas sobrevivientes hasta que, en algún nivel del tratamiento, no hay esporas sobrevivientes. En la zona cuantal, algunos de los IBs tienen sólo una espora sobreviviente y otros tienen un bajo número de sobrevivientes.
Para los IBs expuestos que tienen sólo una espora sobrevi-viente, puede existir una de dos condiciones: la espora no ha sido dañada de una manera que afecte significativamente ya sea el tiempo de germinación o la velocidad de crecimiento post-germinación, o ha ocurrido daño que afecta significati-vamente una o ambas características. Cuando una exposición a la esterilización da un resultado de zona cuantal de 50% de los IBs que probaron ser no estériles, la distribución de Poisson pronostica que aproximadamente 35 IBs tendrán sólo una espora sobreviviente y los otros 15 tendrán más de una espora sobreviviente (2). De los resultados de los estudios de tiempo de crecimiento reportados previamente, es claro que la mayoría de los IBs con sólo una espora sobreviviente y, más probablemente, todos los IBs que tengan más de una espora sobreviviente, exhiben tiempos de crecimiento dentro de un rango relativamente estrecho. También, se esperaría que un número muy bajo de IBs exhibiera un tiempo de crecimiento prolongado; los tiempos prolongados fueron observados para 2/458 (0.44%) y 3/309 (0.97%) IBs para los procesos de este-rilización con calor húmedo y con EO, respectivamente.
Los IBs que tienen tiempos de crecimiento prolongados son más probable que se presenten cuando las condiciones de exposición resultan en un menor porcentaje que son no estériles, por ejemplo, 35% contra 75%. Para un resultado no estéril de 35%, el número promedio de esporas viables por IB expuesto es 0.43 contra 1.39 cuando 75% de los IBs dieron no estériles. Conforme el porcentaje de IBs no estériles se redu-ce, proporcionalmente más de los IBs no estériles tienen dos o más esporas sobrevivientes; para un resultado no estéril de 75%, aproximadamente 40 de los IBs no estériles tienen dos o más esporas sobrevivientes (2)
Para desarrollar un método MIT viable, el uso de una exposición a la esterilización que resulte en un número con-
sistente bajo de esporas sobrevivientes es un pre-requisito importante. El tratamiento de los IBs en un resistómetro en un tiempo de exposición especificado por el cálculo de CST resultará en un método más consistente y reproducible para determinar un MIT en contraposición a la producción de 30 a 80% de IBs no estériles.
El CST, según se indica en la Ecuación 1, es un tiempo de exposición que toma en cuenta tanto el nivel de población de esporas como el valor D
10 y se pretende que resulte en 100%
de los IBs que prueban ser no estériles (4-6). Un IB sometido a una exposición CST se esperaría que tuviera aproximada-mente 100 esporas sobrevivientes; los resultados de los estu-dios previos del tiempo de crecimiento mostraron que cuando los IBs se expusieron al CST, no se observaron tiempos de crecimiento prolongados en 1927 exposiciones CST,
CST = (valor D10
) x (log10
del nivel de población - 2) Ec. 1
donde el CST y el valor D10
están en minutos. El alcance de tratamiento para una exposición CST es marginalmente me-nor que la usada para obtener el 30 a 80% de resultados de no esterilidad requeridos por el protocolo CDRH de la FDA. Los IBs tratados en una exposición a la esterilización CST desafia-rán el medio de incubación y las condiciones con ~100 espo-ra que han recibido una agresión letal significativa (~99.99% de las esporas inactivadas debido a la acción letal del agente esterilizante). Este nivel de microorganismos es similar al usado en la prueba de promoción de crecimiento en medio microbiológico y la prueba de bacteriostasis/fungistasis don-de las inoculaciones se realizan con niveles bajos (≤100) de microorganismos de prueba (7).
Método MIT propuestoCon base en los resultados del análisis resumido en los dos estudios previamente publicados y el análisis posterior de los datos, se propone el siguiente método para la determinación de un MIT para los IBs:• Obtener un mínimo de 100 IBs de cada uno de tres lotes,
los cuales son producidos de diferentes cultivos de esporas.• Determinar el valor D10 y la población para cada lote.• Calcular el tiempo de sobrevivencia para cada lote de IB
que utilice la Ecuación 1.• Exponer cada lote de IBs en el CST en corridas separadas
del resistómetro. • Utilizar métodos de recobro especificados por el fabricante
del IB (o el medio de crecimiento especificado) y las condi-ciones ambientales definidas.
• Incubar hasta que todos los IBs prueben ser no estériles o hasta que se hayan completado siete días de tiempo de incu-bación. (Nota: Si todos los IBs no prueban ser no estériles después de siete días de incubación, reconfirmar el valor D10
y la población).• Registrar el tiempo para que todos los IBs prueben ser no
estériles.• El MIT es el tiempo más prolongado encontrado para un
resultado no estéril para los tres lotes de IB.Para la determinación del CST, se recomienda que el valor
D10
sea calculado utilizando el método de Holcomb-Spear-
man-Karber (8). Este método utiliza todos los resultados de la fracción negativa de la serie de exposiciones a la esteriliza-ción para calcular el “tiempo medio para la esterilidad” y el valor D
10 asociado. Un valor D
10 calculado de esta manera es
más probable que sea el más preciso y provea el mejor valor D
10 estimado desde una perspectiva estadística.
DiscusiónEn el desarrollo de un nuevo método para la determinación del MIT, es importante considerar la evolución de las prácti-cas para el monitoreo y el control de los procesos de esteri-lización durante los pasados 25 años o más. Por ejemplo, los primeros esterilizadores con calor húmedo tenían sólo un ma-nómetro para la presión de la cámara y un termómetro en la línea de drenado para monitorear el proceso. El control elec-tromecánico activo del proceso no existía y, en consecuencia, los resultados del análisis del IB eran críticos para determinar la eficacia del proceso.
Hoy día, operamos bajo la protección de la validación del proceso. La validación requiere de extensa documentación de los aspectos físicos/químicos de cada ciclo de esteriliza-ción. La documentación debe ser suficiente para mostrar que el proceso se ejecutó como se esperaba y que se cumplió la especificación del proceso en su totalidad. Los esterilizadores modernos, en contraste con los primeros recipientes, tienen monitoreo sofisticado y sistemas de control que proporcionan registros de corrida detallados para cada proceso de esterili-zación.
En la práctica actual, los resultados del análisis del IB están subordinados a los resultados físicos/químicos registra-dos; una corrida de esterilización es no conforme si los re-querimientos físicos/químicos no se cumplen incluso aunque todos los IBs estén inactivados. El uso de IBs en el contexto actual de control del monitoreo del proceso de esterilización debe emparejarse con nuestro conocimiento actual de creci-miento del IB.
Un extenso análisis del tiempo de crecimiento para los IBs expuestos a cuatro diferentes procesos de esterilización bajo condiciones controladas ha dado un cuadro muy claro de la cinética de germinación de las esporas y el crecimiento tanto para los IBs no expuestos como para los IBs expuestos en diferentes medidas del tratamiento esterilizante. Los IBs no expuestos crecen más rápidamente y exhiben la menor varia-ción en el tiempo de crecimiento. Como sería de esperar, los IBs expuestos a un proceso de esterilización exhiben tiem-pos de crecimiento más prolongados en promedio y también muestran más variabilidad en los tiempos de crecimiento. El tiempo promedio más largo de crecimiento está claramente relacionado con la presencia de un menor número de espo-ras viables después de la exposición al esterilizante y también probablemente debido a algún daño para el aparato metabóli-co y/o genético de la espora.
Los IBs expuestos a un esterilizante que resultan en un re-sultado de zona cuantal demuestran más variabilidad que los IBs no expuestos pero también muestran una baja frecuencia de IBs con un tiempo de crecimiento prolongado. El méto-do de MIT propuesto en este artículo que usa la exposición a la esterilización CST, toma en cuenta esta variabilidad au-

Pharmaceutical Technology en Español MARZO / ABRIL 201422
mentada pero evita apropiadamente la presencia e influencia inapropiada de resultados “aberrantes”. En contraste, una consecuencia imprevista de la ventana de sobrevivencia de 30 a 80% del protocolo CDRH de la FDA es que el RIT está con-ducido por los resultados aberrantes de grandes porcentajes de IBs que tienen sólo una espora. Este efecto puede verse en el hecho de que un tiempo de incubación más corto puede lograrse si el promedio de resultados de no estériles está cer-cano al 80% contra el 30%, el cual es un resultado atribuido a la diferencia en la fracción de IBs no estériles que tienen sólo una espora sobreviviente. Esta consecuencia es claramente in-deseable para un método de prueba y demuestra una carencia de robustez y reproducibilidad.
La adopción del método MIT propuesto no estaría pre-vista para reducir la eficiencia de los IBs para el monitoreo y control de los procesos de esterilización, sino más bien para proporcionar un medio para la determinación reproducible del tiempo de crecimiento del IB.
Referencias1. FDA, Guide for the Validation of Biological Indicator Incubation
Time (Rockville, MD, June, 1986). 2. J.R. Gillis et al., Pharm. Tech. online, “Understanding Biological
Indicator Grow-Out Times,” www.pharmtech.com/BIgrowout, Jan. 2, 2010, accessed May 6, 2013.
3. J.R. Gillis, et al., Pharm. Tech. 37 (6) 52-59 (2013).4. ISO 11138-1:2006 Sterilization of Health Care Products—Biologi-
cal Indicators—Part 1: General Requirements (ISO, Geneva, 2006).5. ISO 14161:2009 Sterilization of Health Care Products—Biological
Indicators—Guidance for the Selection, Use and Interpretation of Results (ISO, Geneva, 2009).
6. USP 36-NF 31 (US Pharmacopeial Convention, Rockville, MD 2013), p. 2659-2662.
7. USP General Chapter <61>, “Microbiological Examination of Non-sterile Products: Microbial Enumeration Tests” (US Pharmaco-peial Convention, Rockville, MD 2013).
8. I.J. Pflug, R.G. Holcomb, and M.M. Gomez, “Thermal Destruction of Microorganisms,” in Disinfection, Sterilization, and Preserva-tion, S. Block, Ed. (Lippincott, Williams, & Wilkins, Philadelphia, PA, 2001), pp. 79-129. PT
concepto. Sin embargo, otros factores están dirigiendo el costo del desarrollo clínico. La necesidad de demostrar superioridad terapéutica significa que los estudios clíni-cos deben realizarse contra los productos comercializa-dos, los cuales deben ser comprados y cegados con gran-des gastos en lugar de usar placebos. La guía de la FDA con frecuencia requiere caracterizaciones del compuesto y estudios analíticos más extensos. El enrolamiento de suficientes pacientes para obtener resultados estadística-mente significativos se ha vuelto cada vez más difícil y caro.
•Las compañías pequeñas tienen miedo de gastar dinero demasiado rápido por temor a que no puedan reemplazar los fondos gastados, los cuales tienen disposición limita-da para gastarse.
Que la declinación en la actividad de I+D sea permanen-te o transitoria es una pregunta crucial para la industria bio/farmacéutica completa, y especialmente para los CROs y los CDMOs que respaldan la investigación y el desarrollo. Si la I+D no se recupera, los prospectos de crecimiento de los pro-veedores de servicios de I+D sufrirán, e incluso los CMOs comerciales verán menos oportunidades en el camino.
El futuro de I+DEs probable que la mayoría de las tendencias negativas que impactan la actividad de I+D se corrijan por sí mismas a su debido tiempo. La alteración en los programas de I+D causa-do por la eliminación de programas terapéuticos y el cierre de operaciones de I+D se calmará. La fertilización cruzada de ideas, similar a Silicon Valley, que las compañías bio/farma-céuticas están esperando instigar mediante el establecimiento
de operaciones de I+D en los centros de investigación médica, empezará a dar frutos.
Y es probable que regrese el financiamiento para I+D de alto riesgo como la re-apertura de la ventana de oferta pública inicial convencerá a los capitalistas de riesgo que todavía son posibles los éxitos rentables.
Dos factores, no obstante, continuarán creando fuertes vientos contrarios para el crecimiento de I+D: los pagadores continuarán demandando eficacia demostrable y ventajas de costo para los nuevos fármacos y el costo de I+D para demos-trar valor terapéutico y rentabilidad continuará aumentando.
Estas nuevas demandas crearán nuevas oportunidades y retos para los CDMOs y CROs. Las oportunidades vienen de las compañías bio/farmacéuticas que buscan controlar los costos de I+D volteando a ver a los proveedores de servicios por contrato, cuyos costos pueden hacerse variables, en com-paración a las capacidades internas cuyos costos son fijos. Los beneficios también aumentarán para los CDMOs y CROs que pueden salir con soluciones innovadoras, rentables para el problema de control de los costos de I+D.
Los retos surgirán de las presiones sobre los precios y los márgenes ya que las compañías bio/farmacéuticas buscan ne-gociar mejores términos para administrar sus costos. También vendrán de la rotación continua en proyectos conforme los programas de I+D sean cancelados cuando no cumplan los elevados obstáculos del desempeño.
Los CDMOs y los CROs que esperan la continuación de los “buenos viejos tiempos” de mediados de la década del 2000 no están enfrentando las realidades del ambiente de in-vestigación y desarrollo bio/farmacéutico de hoy día. El mun-do ha cambiado para siempre, y los proveedores de servicio necesitan enfrentarse a las nuevas realidades. PT
“I+D en transición” Continuación de la pág. 11

Guide for the Validation of Biological Indicator Incubation Time
, “Understanding Biological Indicator Grow-Out Times,” www.pharmtech.com/BIgrowout, Jan. 2, 2010, accessed May 6, 2013.
3.Sterilization of Health Care Products—Biologi-cal Indicators—Part 1: General RequirementsSterilization of Health Care Products—Biological Indicators—Guidance for the Selection, Use and Interpretation of Results(US Pharmacopeial Convention, Rockville, MD 2013), p. 2659-2662.
7.
I.J. Pflug, R.G. Holcomb, and M.M. Gomez, “Thermal Destruction of Microorganisms,” in
S. Block, Ed. (Lippincott, Williams, & Wilkins, Philadelphia, PA, 2001), pp. 79-129.
de operaciones de I+D en los centros de investigación médica, empezará a dar frutos.
Y es probable que regrese el financiamiento para I+D de alto riesgo como la re-apertura de la ventana de oferta pública inicial convencerá a los capitalistas de riesgo que todavía son posibles los éxitos rentables.
Dos factores, no obstante, continuarán creando fuertes vientos contrarios para el crecimiento de I+D: los pagadores continuarán demandando eficacia demostrable y ventajas de costo para los nuevos fármacos y el costo de I+D para demos-trar valor terapéutico y rentabilidad continuará aumentando.
Estas nuevas demandas crearán nuevas oportunidades y retos para los CDMOs y CROs. Las oportunidades vienen de las compañías bio/farmacéuticas que buscan controlar los costos de I+D volteando a ver a los proveedores de servicios por contrato, cuyos costos pueden hacerse variables, en com-paración a las capacidades internas cuyos costos son fijos. Los beneficios también aumentarán para los CDMOs y CROs que pueden salir con soluciones innovadoras, rentables para el problema de control de los costos de I+D.
Los retos surgirán de las presiones sobre los precios y los márgenes ya que las compañías bio/farmacéuticas buscan ne-gociar mejores términos para administrar sus costos. También vendrán de la rotación continua en proyectos conforme los programas de I+D sean cancelados cuando no cumplan los elevados obstáculos del desempeño.
Los CDMOs y los CROs que esperan la continuación de los “buenos viejos tiempos” de mediados de la década del 2000 no están enfrentando las realidades del ambiente de in-vestigación y desarrollo bio/farmacéutico de hoy día. El mun-do ha cambiado para siempre, y los proveedores de servicio necesitan enfrentarse a las nuevas realidades.
Marque en la tarjeta de servicio al lector el No. 8

Pharmaceutical Technology en Español MARZO / ABRIL 201424
VIGILANCIA REGULATORIA EUROPEA
Con las PyMEs ganando mayor reconocimiento como la fuerza motriz de la Investigación e innovación en Europa, las agencias regulatorias están empujando a las compañías a comprometerse con los reguladores desde el principio del proceso de desarrollo del fármaco.
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
La comunicación temprana con los reguladores es esencial para las PyMEs
es escritor independiente con sede en Essex, RU,
Sean Milmo
Las agencias regulatorias de Europa han estado in-crementando su apoyo a las pequeñas y medianas empresas (PyMEs), incluyendo a las micro compa-ñías, en un esfuerzo para estimular más desarrollo
de medicinas innovadoras. Se les ha ofrecido a las compa-ñías asistencia regulatoria y administrativa gratis o con un gran descuento, sesiones de capacitación, evaluación, y sobre todo, consejo científico que puede aumentar las posibilidades de un nuevo medicamento que gane la aprobación para la comercialización.
La importancia del diálogo inicial con los reguladoresLas compañías son alentadas a ponerse en contacto con las agencias tan al inicio como sea posible durante la etapa de desarrollo del fármaco para asegurar que sus procesos de manufactura, en particular, no llevarán a dificultades regu-latorias. Sin embargo, es difícil trabajar para las agencias regulatorias porque muchas compañías pequeñas (una gran proporción de las cuales son empresas fundadas por especia-listas individuales o pequeños grupos de científicos) tienden a retardar el contacto con los reguladores hasta que el desa-rrollo del producto esté cerca de la etapa del estudio preclí-nico o clínico. Para las propias agencias, la promoción de la innovación puede ser un nuevo papel con el cual necesitan familiarizarse más para cerrar cualquier brecha de comunica-ción con los empresarios.
“Las personas que dirigen las PyMEs conocen la ciencia y tecnología del área en la cual están haciendo negocios,” explica Hooshang Zavareh, fundador de ChemPharmServe, Cambridge, Inglaterra, una compañía que se especializa en servicios de desarrollo de fármacos. “Con frecuencia ellos no ven la necesidad de ayuda de los reguladores hasta que llegan al punto donde necesitan hacer una solicitud regulatoria. Si quieren ayuda exterior antes, acudirán a un consultor.”
No obstante, las agencias piensan que teniendo vínculos más estrechos entre ellos mismos y los empresarios innova-dores es esencial. Con las grandes farmacéuticas haciendo recortes en las actividades de investigación, ellos creen que gran parte del impulso de la innovación en medicamentos -al menos en las primeras etapas del desarrollo- vendrá de las compañías con bajos volúmenes de venta y pocos empleados, por ejemplo, micro compañías con menos de 10 empleados y e ingresos anuales de menos de 2 millones de euros (aproxi-madamente $2.7 mdd), los PyMEs con no más de 50 perso-
nas e ingresos anuales que no exceden 10 millones de euros, y empresas medianas de menos de 250 empleados y un vo-lumen de ventas anuales de no más de 50 millones de euros.
PyMEs a la alzaActualmente la Unión Europea está llamando a las PyMEs “la fuerza motriz” de su Unión de Innovación, un concepto detrás de su nuevo programa de investigación de siete años y 77 billones de euros llamado Horizon 2020, el cual se inicia el siguiente año. Aproximadamente el 10% del pre-supuesto del programa será asignado a la salud, el cambio demográfico y el bienestar. Un objetivo mayor es la elimina-ción de la fragmentación en los campos de la investigación e innovación científica. Entre las agencias regulatorias de la Unión Europea en farmacéuticos, la más activa en la defensa de la innovación, ha sido la Agencia de Medicinas Europea (EMA), la cual es responsable de las aprobaciones centrali-zadas de los medicamentos, incluyendo aquéllos basados en nuevas tecnologías y medicamentos huérfanos.
Las PyMEs constituyen una proporción creciente de compañías que solicitan a la EMA una evaluación inicial de un nuevo medicamento, incluyendo su proceso de manufac-tura. Casi el 30% de los solicitantes en 2012 fueron PyMEs, lo que constituyó también el 68% de las solicitudes de eva-luación para medicamentos huérfanos en comparación con el 27% el año anterior.
En 2012, el número de PyMEs registradas con la agen-cia se elevó a 62% para 1098 solicitudes, principalmente debido a las reducciones de tarifas y también debido a que las compañías podían hacer sometimientos electrónicos de la información del producto. Este año, la tasa de incremento se ha desacelerado con el total de registros a finales de Noviem-bre a la alza en 12% más para 1234 solicitudes. De este total, 43% eran compañías de tamaño micro, 12% eran académicas y 5% de las compañías PyME registradas habían sido crea-das desde Enero de 2012. Las PyMEs registradas tuvieron un total de aproximadamente 3000 productos en desarrollo.
Entendiendo la ruta para la aprobación de fármacos“De una encuesta que hicimos en el 2011, encontramos que bastantes PyMEs carecían de personal con experiencia para llevar los productos a través del sistema regulatorio. Como resultado, las empresas le dan gran importancia a tener un acceso inicial al EMA,” dice un funcionario de la agencia

Pharmaceutical Technology en Español MARZO / ABRIL 2014 25
Con las PyMEs ganando mayor reconocimiento como la fuerza motriz de la Investigación e innovación en Europa, las agencias regulatorias están empujando a las compañías a comprometerse con los reguladores desde el principio del proceso de desarrollo del fármaco.
en una entrevista con Pharmaceutical Technology. “Nuestras cifras muestran que aquéllos que se pusieron en contacto ini-cialmente tienen más probabilidad de éxito con una solicitud de autorización de comercialización.”
Uno de los servicios más valorados de la agencia entre las PyMEs ha sido la provisión de asesoramiento científico, disponible con 90% de descuento, particularmente en temas de calidad y proceso. “La documentación de calidad conti-núa siendo un área problemática para muchas PyMEs, par-ticularmente en lo que respecta a la validación del proceso de manufactura, el establecimiento de especificaciones y los datos de estabilidad.” dice la Oficina de PyMEs en su más reciente reporte anual (1). La Oficina PyME de la agencia se ha decepcionado con la baja captación de servicios por parte de las PyMEs involucradas en el desarrollo de medicamentos para terapias avanzadas (ATMPs) -tales como la terapia géni-ca, la terapia de células somáticas, y la ingeniería de tejidos. Las PyMEs han constituido el 75% del pequeño número de aprobaciones de ATMPs de la EMA, para lo cual sólo hubo tres solicitudes el año pasado.
Para darle un incentivo a más PyMEs para el desarrollo de terapias avanzadas, la EMA ha preparado un esquema para la evaluación científica y la posterior certificación de la calidad y, cuando sea necesario, datos no clínicos de los nuevos productos en el área. La certificación ayudaría a faci-litar el camino para la autorización de comercialización. Otra atracción es que ayudaría a las compañías a obtener financia-miento y a vender la licencia de sus tecnologías. Los datos de calidad a ser certificados incluirían la información sobre los materiales de inicio y las materias primas, la manufactura y caracterización de la sustancia activa y la reproducibilidad del proceso de manufactura para el producto medicinal.
Alentando vínculos más estrechos con las PyMEsSin embargo, a principios de este año, ha habido sólo dos solicitantes para la certificación, lo cual impulsó a la EMA a realizar una encuesta para encontrar las razones para la apa-rente carencia de interés. Los resultados de la encuesta mos-traron que casi dos tercios de las PyMEs que respondieron las preguntas sobre la certificación -que sumó sólo el 21% de los encuestados- consideraron que esto tenía ventajas po-sitivas. La EMA concluyó del estudio que parecía que los participantes estaban confundidos acerca del alcance de la certificación, lo cual indicaba la necesidad de que la agencia aclarara su papel en el proceso regulatorio.
Desde el punto de vista de la EMA, una historia de gran éxito con su nueva relación con las PyMEs ha sido la manera en que trabajó estrechamente, desde una etapa inicial, con la nueva empresa holandesa uniQure para permitir que su pro-ducto alipogene tiparvovec (Glybera) obtuviera la primera aprobación para comercialización de la UE para una terapia génica a finales del 2012. El producto fue desarrollado para el tratamiento de la deficiencia de lipoproteína lipasa (LPL). Para la agencia, el proceso de aprobación fue desafiado cien-
tíficamente porque el alipogene tiparvovec era un nuevo tipo de medicamento sin ninguna historia regulatoria. “Las lec-ciones aprendidas de este caso son múltiples y prepararon el camino para la aprobación de medicamentos similarmen-te complejos en el futuro,” dijo Christian Schneider, direc-tor del comité de la EMA para terapias avanzadas (CAT) y Hans-Georg Eichler, funcionario médico senior, en un artí-culo conjunto en el último reporte anual de la agencia (2).
La EMA destaca que en sus esfuerzos para forjar víncu-los más estrechos con las PyMEs, no está tratando de socavar la posición de los consultores y otros expertos en la industria farmacéutica. Su papel no es “sustituir la responsabilidad de la industria para el desarrollo de sus productos,” dijo la agencia.
Un punto similar se está dando por las agencias nacio-nales para la regulación de medicamentos, las cuales han estado estableciendo oficinas de innovación para establecer contactos con las PyMEs. Hasta ahora, de los 29 estados miembros de la UE, se han creado unidades de PyME en Alemania, Francia, RU, Suecia y Finlandia que ya se han conformado en una red para intercambiar experiencias y me-jores prácticas.
En el RU, una Oficina de innovación, abierta en Marzo por la Agencia Regulatoria de Medicinas y Productos para la Salud (MHRA) del país, está dando guía multi-disciplinaria sobre “nuevas metodologías de desarrollo, procesos de ma-nufactura o nuevos materiales que no se han usado previa-mente en alguna de las industrias farmacéuticas o de dispo-sitivos,” dice Ian Hudson, director de la división de licencias del MHRA.
Las agencias están poniendo énfasis en que sus unidades pueden ser usadas para cualquier PyME que desarrolle un producto o proceso innovador cualquiera que sea el procedi-miento o ruta regulatoria que pretendan usar. “Queremos es-timular que las PyMEs se acerquen a un regulador tan pronto como sea posible en el trabajo de desarrollo, de manera que lleguen al proceso de producción correcto desde una pers-pectiva regulatoria desde el principio,” explica Lars Dager-holt, jefe de la Oficina de Innovación en la Agencia Sueca de Medicamentos, Estocolmo. “A nosotros no nos importa que vayan a acercarse a otras agencias. Estas agencias pueden tener diferentes procedimientos y expertos con diferentes enfoques.” La idea es persuadir a las PyMEs de que el me-canismo regulatorio no es tan complejo como parece y sobre todo, que los reguladores están ahí para apoyar, más que para obstaculizar la innovación.
Referencias 1. EMA, Annual Report from the SME Office 2012, www.ema.
europa.eu/docs/en_GB/document_ l ibrary/Annual_re-port/2013/02/WC500138925.pdf, accessed Dec. 10, 2013.
2. EMA, Annual Report 2012, www.ema.europa.eu/docs/en_GB/document_library/Annual_report/2013/04/WC500142077.pdf, accessed Dec. 10, 2013. PT

Pharmaceutical Technology en Español MARZO / ABRIL 201426
Formulación: FragmEntos DE anticuErpos Fab
Desafíos y estrategias para la purificación y formulación corriente abajo de fragmentos Fab de anticuerposClaire Scanlan, Jeffrey Shumway, Juan Castano, Mark Wagner, and Ruta Waghmare
Los autores describen algunos de los requerimientos y desafíos únicos, planteados por los fragmentos de anticuerpos en términos de recobro, purificación y formulación.
Claire Scanlan es científica en desarrollo de proceso; Jeffrey Shumway es director asociado, mejora de la biodisponibilidad; Juan Castano es científico de desarrollo de proceso; Mark Wagner es especialista en tecnología de purificación y desarrollo de negocio; y Ruta Waghmare, PhD, es director asociado, biotecnología emergente, [email protected], todos en EMD Millipore, 290 Concord Road, Billerica, MA 01821.
Los anticuerpos monoclonales (mAbs) son una importan-te clase de terapéuticos. Sin embargo, en años recientes,
se ha puesto una mayor atención al de-sarrollo de la siguiente generación de terapéuticos biológicos y a las platafor-mas de manufactura. Este cambio está siendo conducido por diversos factores, incluyendo la necesidad de desarrollar procesos más eficientes y redituables, reducir efectos colaterales, e incremen-tar la especificada para incrementar la eficacia.
Los fragmentos de anticuerpos co-nocidos como fragmentos de unión al antígeno (Fab) son un ejemplo de estos biológicos de la siguiente generación que están surgiendo como alternativas creíbles al ampliamente aceptado mAbs
(ver Figura 1). El abciximab (ReoPro, Eli Lilly), ranibizumab (Lucentis, Ge-nentech) y certolizumab pegol (Cimzia, UCB) son productos Fab que están ac-tualmente en el mercado. Existen varios más que están en desarrollo por otras compañías.
Los fragmentos Fab pueden ser pro-ducidos utilizando sistemas de expre-sión más económicos (p.ej., microbio-lógicos) conservando al mismo tiempo la especificidad para el objetivo de un mAb. Otras ventajas incluyen la elimi-nación de uniones no específicas entre las porciones del fragmento cristaliza-ble (Fc) de los anticuerpos y el receptor Fc en las células, y la capacidad para construir la región Fab para una espe-cificidad mejorada y eficiencia terapéu-tica.
Los fragmentos de anticuerpos son comúnmente producidos como produc-tos intracelulares en sistemas de expre-sión microbiológica como el E. coli. Después de la fermentación, la cosecha de las células y la ruptura, el proceso de purificación corriente abajo consiste típicamente en la clarificación, captura cromatográfica, cromatografía de pu-lido y ultrafiltración/diafiltración. La formulación final y la filtración estéril
tienen lugar antes del llenado-termina-do (ver Figura 2). En este artículo, los autores describen algunos de los reque-rimientos y desafíos únicos planteados por los fragmentos de anticuerpos en términos del recobro, la purificación y la formulación.
Recobro y purificaciónLas moléculas de Fab se expresan típi-camente en E. coli o sistemas microbio-lógicos de levaduras. Para lograr títulos finales más elevados, la cantidad de sólidos en estos sistemas de expresión se incrementa, haciendo que la clari-ficación sea más desafiante. Para pro-teínas Fab solubles, el uso de métodos de clarificación por filtración con flujo normal post-lisis no es común. En lugar de esto, se usa con frecuencia la filtra-ción con flujo tangencial (TFF) en lugar de la filtración con flujo normal.
Si se desea la filtración con flujo normal, ésta se incorpora típicamente post-centrifugación, utilizando discos apilados o centrífuga tubular. Como la distribución del tamaño de partícula en el lote es pequeña (el tamaño de partí-cula es muy homogéneo y tiene impure-zas con tamaño de partícula pequeño), se emplea con frecuencia un filtro de profundidad. También puede usarse la microfiltración sin centrifugación.
Con las proteínas secretadas, las cé-lulas completas se separan del caldo de fermentación y el tamaño de partícula es mayor. Como resultado, la micro-filtración (utilizando TFF), la centrifu-gación y la filtración con flujo normal son opciones viables. También pueden añadirse agentes de endonucleasa antes de la clarificación para digerir el ADN y el ARN y para ayudar a la eficiencia del proceso de clarificación.
La proteína A, seguida por el inter-cambio catiónico y el intercambio anió-nico, puede usarse con éxito para la pu-rificación de los Fabs que contienen la región Fc. Para un Fab que no contiene una región Fc, la captura se logra típica-mente vía intercambiadores catiónicos o en el modo de resinas mezcladas en el modo de unión/elución. Las ventajas del modo mezclado son una ventana de operación más amplia para la unión en varios escenarios de conductivi-dad y pH. Con el modo mezclado, sin embargo, puede requerirse un período del proceso más prolongado para desa-
rrollar una estrategia de elución. Para superar esta desventaja, se están desa-rrollando resinas en modo simple que operan en conductividades mayores con altas capacidades de unión y procesos de elución fáciles. Un paso posterior de pulido, que usa típicamente un tamaño de esfera más pequeño, generalmente sigue al paso de captura con el objetivo de mejorar la resolución. Este paso de pulido podría ser el intercambio iónico (IEX) o la cromatografía de interacción hidrofóbica (HIC) dependiendo del paso previo y de la proteína de interés. Si requiere purificación adicional, po-dría implementarse un tercer paso con HIC o IEX dependiendo de los pasos previos. Una purificación típica para fragmentos que no contienen Fc sería por lo tanto intercambio catiónico > HIC > intercambio aniónico.
Las moléculas de Fab son típica-mente pequeñas en comparación con los mAbs (aproximadamente 10 kD - 80 kD), y como resultado, se utilizan comúnmente dispositivos de flujo tan-gencial más apretados para el paso de ultrafiltración/diafiltración (UF/DF).

Pharmaceutical Technology en Español MARZO / ABRIL 2014 27
). En este artículo, los autores describen algunos de los reque-rimientos y desafíos únicos planteados por los fragmentos de anticuerpos en términos del recobro, la purificación y la formulación.
o sistemas microbio-lógicos de levaduras. Para lograr títulos finales más elevados, la cantidad de sólidos en estos sistemas de expresión se incrementa, haciendo que la clari-ficación sea más desafiante. Para pro-teínas Fab solubles, el uso de métodos de clarificación por filtración con flujo normal post-lisis no es común. En lugar de esto, se usa con frecuencia la filtra-ción con flujo tangencial (TFF) en lugar de la filtración con flujo normal.
Si se desea la filtración con flujo normal, ésta se incorpora típicamente post-centrifugación, utilizando discos apilados o centrífuga tubular. Como la distribución del tamaño de partícula en el lote es pequeña (el tamaño de partí-cula es muy homogéneo y tiene impure-zas con tamaño de partícula pequeño), se emplea con frecuencia un filtro de profundidad. También puede usarse la microfiltración sin centrifugación.
Con las proteínas secretadas, las cé-lulas completas se separan del caldo de fermentación y el tamaño de partícula es mayor. Como resultado, la micro-filtración (utilizando TFF), la centrifu-gación y la filtración con flujo normal son opciones viables. También pueden añadirse agentes de endonucleasa antes de la clarificación para digerir el ADN y el ARN y para ayudar a la eficiencia del proceso de clarificación.
La proteína A, seguida por el inter-cambio catiónico y el intercambio anió-nico, puede usarse con éxito para la pu-rificación de los Fabs que contienen la región Fc. Para un Fab que no contiene una región Fc, la captura se logra típica-mente vía intercambiadores catiónicos o en el modo de resinas mezcladas en el modo de unión/elución. Las ventajas del modo mezclado son una ventana de operación más amplia para la unión en varios escenarios de conductivi-dad y pH. Con el modo mezclado, sin embargo, puede requerirse un período del proceso más prolongado para desa-
IgG de longitud completa
F(ab’)2F(ab’)
Fermentación
Ultra�ltración/Dia�ltración
Formulación �nal Filtración estéril Llenado terminal
Cromatografía de pulido Cromatografía de captura
Cosecha de células Ruptura de células Clari�cación
rrollar una estrategia de elución. Para superar esta desventaja, se están desa-rrollando resinas en modo simple que operan en conductividades mayores con altas capacidades de unión y procesos de elución fáciles. Un paso posterior de pulido, que usa típicamente un tamaño de esfera más pequeño, generalmente sigue al paso de captura con el objetivo de mejorar la resolución. Este paso de pulido podría ser el intercambio iónico (IEX) o la cromatografía de interacción hidrofóbica (HIC) dependiendo del paso previo y de la proteína de interés. Si requiere purificación adicional, po-dría implementarse un tercer paso con HIC o IEX dependiendo de los pasos previos. Una purificación típica para fragmentos que no contienen Fc sería por lo tanto intercambio catiónico > HIC > intercambio aniónico.
Las moléculas de Fab son típica-mente pequeñas en comparación con los mAbs (aproximadamente 10 kD - 80 kD), y como resultado, se utilizan comúnmente dispositivos de flujo tan-gencial más apretados para el paso de ultrafiltración/diafiltración (UF/DF).
El corte del peso molecular de la mem-brana, recomendado para la retención adecuada es un quinto del tamaño del producto de interés. Los cortes de peso molecular de 1 kD-10 kD son usados típicamente para las moléculas del Fab. Estos cortes más pequeños pueden lle-var a flujos de permeado más bajos.
Adicionalmente, algunas moléculas de Fab están PEGiladas, lo que condu-ce a mayores viscosidades conforme se incrementa la concentración. Debido a esto, la influencia del tipo de malla (las mallas ayudan a crear turbulencia y promueven la transferencia de masas) en dispositivos planos es primordial y debe tomarse en cuenta en estudios de optimización en pequeña escala. Una consideración final para las moléculas Fab expresadas por E. coli es la remo-ción de endotoxinas corriente abajo, la cual se logra frecuentemente a través de adsorbedores de membrana aniónica o filtros de membrana cargados.
FormulaciónLa consideración primaria en el diseño de la formulación para los terapéuticos
de proteína Fab es la estabilidad. La protección de la proteína de la degrada-ción química durante el almacenamien-to así como la degradación proveniente de influencias protolíticas in vivo es crucial en la formulación para todos los terapéuticos basados en proteínas. Una segunda función igualmente importante del diseño de la formulación relaciona-do con la actividad de la proteína, es in-hibir la tendencia a la agregación.
Cuando se considera la biodispo-nibilidad, los Fabs presentan oportuni-dades y desafíos. Siendo significativa-mente menores que las moléculas del anticuerpo monoclonal nativo, los Fabs diseñados como terapéuticos para el cáncer tienen la capacidad de proveer mejor penetración en el tumor en com-paración con las especies moleculares más grandes. Por el contrario, los Fabs sufren la desventaja de circulación y vida media significativamente más cor-tas in vivo, dos características que pue-den ser abordadas en la formulación.
La PEGilación, la modificación quí-mica de una proteína mediante conju-gación con polímeros de poli(etilen gli-col) ofrece, en muchos casos, una solu-ción apropiada al desafío asociado con la circulación y la vida media acortadas del terapéutico Fab. La modificación química más común de los Fabs con PEGs involucra la reacción de un PEG-maleimido activado con residuos de cisteína en el Fab. Las proteínas PEGi-ladas ofrecen la ventaja de parecer 5-10 veces más grandes en volumen que las proteínas con peso molecular compara-tivo debido a os efectos de hidratación asociados con el PEG. La PEGilación da como resultado una estabilidad y solubilidad mejoradas manteniendo al mismo tiempo la eficacia asociada con el fragmento de anticuerpo. Los benefi-cios adicionales de la PEGilación inclu-yen toxicidad reducida, tasas de depura-ción y efecto de proteólisis reducidos, e inmunogenicidad y antigenicidad redu-cidas aunque el reciente conocimiento de los efectos de la PEGilación han in-dicado una respuesta inmune asociada con el PEG, que debe ser considerada y monitoreada (1, 2).
Otro efecto de la PEGilación es la tendencia de una proteína PEGilada
“Formulación: Fragmentos De Anti-cuerpos Fab” Continúa en la pág. 37
Figura 1: Fragmentos de unión a antígeno (Fab) de fragmentos del anticuerpo contra el anticuerpo monoclonal (mAbs) en su longitud completa
Figura 2: Proceso de producción típico del fragmento de unión al antígeno (Fab)

Pharmaceutical Technology en Español MARZO / ABRIL 201428
Calificación del personal para inspeccionar visualmente el equipo limpioRichard Forsyth
La inspección visual del equipo limpio se requiere tanto después de la limpieza como nuevamente antes del inicio de las actividades de manufactura. Históricamente, la capacitación se ha realizados para las inspecciones visuales, pero la calificación del personal para la inspección visual es un desarrollo más reciente. La calificación debe asegurar que el personal pueda ver los residuos confiablemente y repetidamente cuando están visibles en la superficie de los equipos. El nivel de residuos para la calificación debe ser indicativo de los residuos de una instalación en niveles en o cercanos al límite de residuos visibles. El autor discute varios diferentes enfoques para la calificación para inspección visual.
Richard Forsyth es consultor principal con Forsyth Pharmaceutical Consulting, 907 Shamrock Ct, Royersford, PA 19468, tel. 484.535.1688, forsyth@[email protected].
Sometido: Abril 10, 2013. Aceptado: Abril 23, 2013.
La inspección visual de la limpieza del equipo de ma-nufactura farmacéutica se requiere después de que se han completado los procedimientos de limpieza y es un pre-requisito para el inicio de las actividades
de manufactura (1). La validación de la limpieza demuestra que el procedimiento de limpieza limpia confiable y consis-tentemente el equipo de manufactura farmacéutica hasta un nivel aceptable. La inspección visual es una observación acti-va de las superficies en contacto con el producto visualmente accesibles. La inspección visual debe determinar que el equi-po esté libre de cualquier residuo visible para que la limpieza sea considerada adecuada y para permitir que comiencen las actividades de manufactura. Aunque puede haber otras medi-das de limpieza de equipo adecuadas, tales como el muestreo con hisopo, la inspección visual es la primera medida de lim-pieza del equipo.
Las inspecciones visuales son realizadas por una variedad del personal del sitio. El personal de limpieza del equipo ins-pecciona rutinariamente el mismo durante un procedimiento de limpieza manual y después del término. Ellos inspeccio-nan las partes que hayan sido limpiadas en una lavadora de partes. Inspeccionan el equipo grande después de la limpieza manual o de los procedimientos de limpieza en el sitio (CIP). Finalmente, inspeccionan el equipo de sistema cerrado que se limpia utilizando un patín de limpieza que bombea agentes limpiadores a través de tuberías al equipo. La limpieza del equipo se documenta en el registro de limpieza para el equipo.
Además del personal de limpieza del equipo, la limpieza visual del equipo limpio se confirma rutinariamente y se do-cumenta en el registro de limpieza por una segunda persona. Previo a la manufactura del lote subsiguiente, se inspecciona rutinariamente la limpieza visual del equipo por parte del per-sonal de manufactura y se documenta en el registro del lote de manufactura. El personal de calidad y validación también inspecciona el equipo limpio periódicamente como parte de su función.
La capacitación del personal para la inspección visual del equipo limpio, históricamente ha consistido de capacitación sobre la marcha en el trabajo realizada por personal con expe-riencia. La documentación de la capacitación típicamente no estaba en un registro único, por separado, de la capacitación, sino como parte de la capacitación general para la limpieza, operaciones o calidad.
Como parte de un programa de validación de limpieza, el personal que tomaba muestras con hisopo ha sido califi-cado rutinariamente para determinar que ellos pueden recu-perar adecuadamente residuos de las superficies del equipo. El personal de laboratorio está calificado para el análisis de
las muestras de la limpieza. Los procedimientos de limpieza y análisis están validados para demostrar la ejecución consis-tente. Un criterio de calificación para la inspección visual del equipo limpio por parte del personal es una extensión lógica de esta filosofía como parte de una ejecución de validación de la limpieza, y la calificación de todo el personal que ins-pecciona visualmente el equipo limpio sería un resultado es-perado.
La implementación de un programa de calificación para la inspección visual en un sitio presenta algunas complicaciones logísticas. La calificación del personal de validación para los procedimientos de validación de la limpieza generalmente se realiza por un pequeño grupo y puede hacerse en un labora-torio. Sin embargo, para la calificación de todo el personal necesario para la inspección visual, sería más práctico llevar el análisis visual al personal en sala de juntas general o en un área de capacitación. Una estación de visualización con las muestras secas montadas para visualizarlas sobre una mesa o colgadas en una pared sería ideal.
Los parámetros de análisis para la calificación de la inspec-

Pharmaceutical Technology en Español MARZO / ABRIL 2014 29
Figura 1: Uso de muestras de ensayo para representar los materiales de construcción del equipo para la calificación visual.
Tabla I. Capacitación para la calificación en inspección visual
Paso Capacitación
1 Revisión del procedimiento estándar de operación (PNO) para la inspección visual de equipo de manufactura limpio
2 Revisión del PNO para calificación del personal para la inspección de las áreas de contacto con el producto
3 Revisión de los diagramas del equipo que muestran las áreas difíciles de limpiar y las áreas de acumulación del producto
4 Revisión de ejemplos de límites de residuos visibles (VRL)
5 Discusión de los VRL contra el límite de limpieza
6 Capacitación sobre la marcha
a inspección visual de la limpieza del equipo de ma-nufactura farmacéutica se requiere después de que se han completado los procedimientos de limpieza y es un pre-requisito para el inicio de las actividades
de manufactura (1). La validación de la limpieza demuestra que el procedimiento de limpieza limpia confiable y consis-tentemente el equipo de manufactura farmacéutica hasta un nivel aceptable. La inspección visual es una observación acti-va de las superficies en contacto con el producto visualmente accesibles. La inspección visual debe determinar que el equi-po esté libre de cualquier residuo visible para que la limpieza sea considerada adecuada y para permitir que comiencen las actividades de manufactura. Aunque puede haber otras medi-das de limpieza de equipo adecuadas, tales como el muestreo con hisopo, la inspección visual es la primera medida de lim-pieza del equipo.
Las inspecciones visuales son realizadas por una variedad del personal del sitio. El personal de limpieza del equipo ins-pecciona rutinariamente el mismo durante un procedimiento de limpieza manual y después del término. Ellos inspeccio-nan las partes que hayan sido limpiadas en una lavadora de partes. Inspeccionan el equipo grande después de la limpieza manual o de los procedimientos de limpieza en el sitio (CIP). Finalmente, inspeccionan el equipo de sistema cerrado que se limpia utilizando un patín de limpieza que bombea agentes limpiadores a través de tuberías al equipo. La limpieza del equipo se documenta en el registro de limpieza para el equipo.
Además del personal de limpieza del equipo, la limpieza visual del equipo limpio se confirma rutinariamente y se do-cumenta en el registro de limpieza por una segunda persona. Previo a la manufactura del lote subsiguiente, se inspecciona rutinariamente la limpieza visual del equipo por parte del per-sonal de manufactura y se documenta en el registro del lote de manufactura. El personal de calidad y validación también inspecciona el equipo limpio periódicamente como parte de su función.
La capacitación del personal para la inspección visual del equipo limpio, históricamente ha consistido de capacitación sobre la marcha en el trabajo realizada por personal con expe-riencia. La documentación de la capacitación típicamente no estaba en un registro único, por separado, de la capacitación, sino como parte de la capacitación general para la limpieza, operaciones o calidad.
Como parte de un programa de validación de limpieza, el personal que tomaba muestras con hisopo ha sido califi-cado rutinariamente para determinar que ellos pueden recu-perar adecuadamente residuos de las superficies del equipo. El personal de laboratorio está calificado para el análisis de
las muestras de la limpieza. Los procedimientos de limpieza y análisis están validados para demostrar la ejecución consis-tente. Un criterio de calificación para la inspección visual del equipo limpio por parte del personal es una extensión lógica de esta filosofía como parte de una ejecución de validación de la limpieza, y la calificación de todo el personal que ins-pecciona visualmente el equipo limpio sería un resultado es-perado.
La implementación de un programa de calificación para la inspección visual en un sitio presenta algunas complicaciones logísticas. La calificación del personal de validación para los procedimientos de validación de la limpieza generalmente se realiza por un pequeño grupo y puede hacerse en un labora-torio. Sin embargo, para la calificación de todo el personal necesario para la inspección visual, sería más práctico llevar el análisis visual al personal en sala de juntas general o en un área de capacitación. Una estación de visualización con las muestras secas montadas para visualizarlas sobre una mesa o colgadas en una pared sería ideal.
Parámetros de calificaciónLos parámetros de análisis para la calificación de la inspec-
ción visual deben estar basados en los parámetros de visua-lización del equipo limpio en un sitio. Para el equipo que se limpia manualmente o que se limpia en una lavadora de partes, las inspecciones son generalmente en el rango de 1-2 pies (30.5 - 61.0 cm). El ángulo de visión es sin restricción y la inspección se realiza bajo luz ambiental. Para el equipo de CIP, más grande, estacionario, la distancia para la visua-lización es mayor; el ángulo de visión estará restringido y el nivel de iluminación podría ser más bajo en las superficies interiores del equipo limpio. Una evaluación del equipo y la instalación en el sitio señalará la distancia de visualización, el ángulo de visualización y las condiciones de iluminación de los parámetros de visualización para la calificación visual. Los lineamientos sugeridos para los parámetros incluyen: < 10 pies (3 m) de distancia de visualización; > 30° de ángu-lo de visualización y > 200 lux de nivel de iluminación (2).
Los materiales de construcción (MOC) para la calificación de la inspección visual deben ser los que más prevalecen en el sitio, y que en muchos casos será el acero inoxidable según se muestra en la Figura 1. Pueden incluirse otros MOC represen-tativos si se considera apropiado y deberán considerarse sus características de visualización. Los parámetros de visualiza-ción óptimos para el vidrio, por ejemplo, proporcionan con-diciones adicionales que incluyen sostener el vidrio y ver a través de él en un ángulo ligeramente inclinado con respecto a la fuente de luz. Otros MOC como el Teflón, que hace que sea mucho más difícil discernir los residuos, no tienen un valor agregado para la calificación de la inspección visual. Estos MOCs necesitan ser abordados en un formato diferente, tal como la capacidad de limpieza. Los problemas para la inspec-ción visual del MOC necesitan ser abordados ya sea experi-mentalmente (4) o durante la capacitación en el trabajo.
Los residuos para la calificación de la inspección visual deben incluir a todos los productos fabricados en el sitio. Pue-de usarse una sub-serie de residuos del portafolio del producto en el sitio con la justificación basada en la evidencia experi-mental de similitud de aparición del residuo. Si los datos dis-ponibles muestran (5) que el VRL del API es tan bajo como el de la formulación, entonces podrían usarse para la calificación las muestras del residuo del API, que son con frecuencia más fáciles de preparar. Los residuos probados para la calificación de la inspección visual deben ser indicativos de que el inspec-tor calificado será capaz de detectar residuos en el equipo de manufactura durante una inspección visual ya sea después de la limpieza y nuevamente antes de la manufactura.
Los niveles de residuo para la calificación de la inspec-ción visual deberán estar basados en los límites de residuo visible (VRL) de los APIs o productos en el portafolio de ma-nufactura del sitio. Como mínimo, debe establecerse el VRL para todos los productos fabricados en el sitio utilizado para la calificación. Una evaluación más profunda establecería VRLs separados para todos los productos, APIs, excipientes y de-tergentes usados en la manufactura o limpieza en el sitio (5).
EnfoquesExisten varios enfoques alternativos para la calificación del personal para la inspección visual. La primera opción y la más

Pharmaceutical Technology en Español MARZO / ABRIL 201430
obvia, es presentar materiales de acero inoxidable adiciona-dos con un número (p.ej., 10) de productos representativos en el VRL, mezclados con un número igual de blancos, mate-riales sin adicionar y presentados en un orden aleatorio para la calificación, como se muestra en la Figura 2. El número de productos dependería del portafolio de productos en el sitio, aunque idealmente todos los residuos de productos deberían ser presentados. El personal vería el residuo en los materiales adicionados o no vería el residuo en los materiales blancos. La ventaja de esta técnica es que es la más directa y presenta materiales adicionados y blancos en una situación de ‘pasa o no pasa’.
Este enfoque, no obstante, no capacita al personal en la aparición de los residuos conforme su concentración se apro-xima al VRL. El personal que se somete a calificación podría identificar erróneamente un material adicionado como blanco y un blanco como adicionado. Una variación en esta técnica presentaría a los APIs así como a los productos para situacio-nes en donde el API está solamente involucrado en uno o más de los pasos de manufactura o donde el portafolio de produc-tos del sitio está limitado sólo a unos cuantos productos. Otra variación podría incluir VRLs en tipos de materiales de MOC adicionales si esto se considerara necesario. Finalmente, un sitio con un portafolio de productos limitado podría presen-tar múltiples materiales adicionados del mismo producto para completar la organización de la calificación.
Una segunda alternativa sería presentar materiales de prueba adicionados en concentración descendente para imi-tar una determinación de VRL. Los materiales adicionados se mezclarían con materiales blanco y se presentarían aleato-riamente para la calificación como se muestra en la Figura 3. Este enfoque le daría al personal una idea de cómo se ven los residuos en concentraciones más altas y el criterio de ‘pasa o no pasa’ podría basarse en el VRL del producto o del material adicionado en el siguiente nivel más alto al VRL. El núme-ro de materiales requeridos sería mucho mayor o el número de productos probados estaría limitado. Una variación sobre este mismo enfoque presentaría los materiales del VRL y las concentraciones aproximadamente 25% por arriba del VRL o presentar la concentración más alta de 25% para la cali-ficación. Este es un enfoque de bajo riesgo cuando el VTL establecido es significativamente más bajo que el límite de residuo permitido (ARL) basado en los problemas de seguri-dad para el paciente.
Esta técnica ha sido usada para establecer niveles de VRL (6). Los materiales fueron presentados tanto en orden seriado desde alto hasta bajo y en orden aleatorio para la determi-nación del VRL. Los datos concluyeron que el orden de la presentación no tenía impacto en la capacidad del observador para discernir el VRL. Los observadores en ese estudio, sin embargo, tenían experiencia previa con las determinaciones del VRL.
Utilizado niveles de adición mayores que el VRL para la calificación de la inspección visual, ya sea de múltiple nivel o de un solo nivel, es difícil de justificar si los VRLs estable-cidos son robustos. Los VRLs están establecidos de manera que existe un nivel de confianza de que todo el personal podrá
ver el nivel del VRL. El uso de múltiples niveles de residuo para la calificación de la inspección visual indica claramente una carencia de confianza en los VRLs establecidos o de la capacidad del personal para detectarlos.
Criterios de aceptación de la calificaciónLos criterios de aceptación lógicos para la calificación de la inspección visual es que el capacitado identifica todos los materiales adicionados como visibles y todos los materiales blanco como blancos. Si un capacitado identifica erróneamen-te un material de prueba, ya sea adicionado o blanco, ¿nece-sariamente deberá fracasar en la calificación? Para un material adicionado, un material erróneamente identificado tendrá que ser considerado como una falla en la calificación. Un material de prueba blanco considerado visible es un error por el lado de la precaución y podría ser tolerado. Identificar múltiples mate-riales de prueba blancos como visibles podría ser problemática porque podría llevar a investigaciones innecesarias de un equi-po limpiado apropiadamente y que sea declarado no limpio visiblemente. Los criterios de aceptación para la inspección visual podrían permitir que uno o más materiales de prueba blancos fueran identificados erróneamente como visibles. El número aceptable de blancos identificados erróneamente per-mitidos (p.ej., 0-3 de 10 blancos) sería decidido en el sitio con base en una evaluación del riesgo de la robustez del VRL y su relación con el límite de seguridad permitido para la limpie-za. A mayor diferencia entre los niveles del VRL y del ARL, mayor seguridad tiene uno de que el equipo de manufactura farmacéutica visualmente limpio está suficientemente limpio y menor el riesgo de una falla en la limpieza como resultado de una inspección visual inadecuada.
Capacitación para la calificación de la inspección visualLa calificación para las inspecciones visuales del equipo lim-pio es sólo una parte de la capacitación requerida para asegu-rar una inspección visual confiable y profunda del equipo de manufactura farmacéutica limpiado. Según se muestra en la Tabla I, la capacitación adecuada debe iniciar con una revi-sión de la inspección visual y con los PNOs de calificación de la inspección visual, los cuales describen el proceso para inspección y documentación del equipo limpio y el proceso de calificación de la inspección visual, respectivamente. Si se dispone de diagramas del equipo que muestren dimensiones y ubicaciones para el muestreo con hisopo con la justificación asociada, éstos pueden ser revisados para que los capacita-dos se familiaricen con el equipo de manufactura y con las áreas problemáticas para la limpieza. Previo a la calificación, la capacitación en la determinación del VRL debe consistir en varios ejemplos de determinaciones del VTL con el fin de poner al corriente al personal con la apariencia de bajos niveles de residuo para la limpieza. La capacitación en los VRLs de diferentes MOCs debe incluirse si existen múltiples MOCs en el sitio y estos son parte de la calificación: Si el personal está involucrado en la determinación de los VRLs, entonces la revisión de un ejemplo de VRL podría no ser ne-cesaria. Una discusión de la relación del VRL con el límite
de limpieza o ARL y la relación de la capacidad de limpieza de los diferentes residuos de los diversos MOCs, impartirían una comprensión del nivel de confianza en el procedimiento de limpieza y la inspección visual. La capacitación en el tra-bajo debe consistir en la inspección supervisada del equipo de manufactura, incluyendo las ubicaciones difíciles de limpiar y aquéllas ubicaciones que más probablemente tengan acu-mulación de producto, para cada pieza de equipo limpiado, identificado por la validación de la limpieza. El paso final des-pués de la capacitación y la calificación debe consistir de una inspección visual monitoreada de una pieza de equipo limpia, asegurando que los procedimientos apropiados sean seguidos, que una inspección visual profunda sea realizada en el equipo y que la inspección visual sea apropiadamente documentada.
La calificación del personal para la inspección visual de su-perficies en contacto con el producto, del equipo de manufac-

Pharmaceutical Technology en Español MARZO / ABRIL 2014 31
de limpieza o ARL y la relación de la capacidad de limpieza de los diferentes residuos de los diversos MOCs, impartirían una comprensión del nivel de confianza en el procedimiento de limpieza y la inspección visual. La capacitación en el tra-bajo debe consistir en la inspección supervisada del equipo de manufactura, incluyendo las ubicaciones difíciles de limpiar y aquéllas ubicaciones que más probablemente tengan acu-mulación de producto, para cada pieza de equipo limpiado, identificado por la validación de la limpieza. El paso final des-pués de la capacitación y la calificación debe consistir de una inspección visual monitoreada de una pieza de equipo limpia, asegurando que los procedimientos apropiados sean seguidos, que una inspección visual profunda sea realizada en el equipo y que la inspección visual sea apropiadamente documentada.
ConclusiónLa calificación del personal para la inspección visual de su-perficies en contacto con el producto, del equipo de manufac-
A1 A2 A3 A4 A5 B1 B2 B3 B4 B5
B6 B7 B8 B9 B10
P2 B6 B7 A4 B8
P1 B1 P3 A1 B2
B9 A2 P4 B10 P5
B3 A3 B4 B5 A5
P1 P2 P3 P4 P5
Analitos, nivel VRL
Productos terminados, nivel VRL
(b) ) Presentación aleatoria de materiales de prueba adicionados
Blancos(a) (a)
A1 A2 A3
Producto terminado A, niveles 1-5
Producto terminado C, niveles 1-5
(b) Presentación aleatoria de materiales de prueba adicionados
Blancos
A4 A5
B6 A4 B7 C4 B8
C2 B9 A2 B10 C5
C1 B1 C3 A1 B2
B3 A3 B4 B5 A5
B1 B2 B3 B4 B5
B6 B7 B8 B9 B10
C1 C2 C3 C4 C5
Figura 2: Calificación de la inspección visual utilizando una presentación aleatoria de materiales de prueba adicionados con productos representativos en el límite de residuos visibles (VRL).
tura farmacéutica limpio es un complemento necesaria tanto para un programa de validación de limpieza como para las operaciones de manufactura en un sitio. La calificación de la inspección visual es sólo una faceta de un programa de capa-citación global para asegurar que el personal esté equipado para aportar una evaluación consistente, profunda y precisa de las superficies limpiadas para las operaciones farmacéuticas continuas. Referencias1. CFR Title 211.67 (Government Printing Office, Washington, DC).2. R. J. Forsyth and V. Van Nostrand, Pharm. Tech. 29 (10) 152-161
(2005).3. R. J. Forsyth, K. Bader, and K. Jordan, Pharm. Tech. 37 (10) 86-91
(2013).4. R. J. Forsyth, K. Bader, and K. Jordan, “Cleanability of Pharma-
ceutical Soils from Different Materials of Construction,” in press.5. R. J. Forsyth, Pharm. Tech. 33 (3) 102-111 (2009).6. R. J. Forsyth, Pharm. Tech. 35 (3), 122-128 (2011). PT
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486E-mail: [email protected]
Figura 3: Calificación de la inspección visual utilizando múltiples niveles de adición para imitar una determinación del límite de residuos visibles.

Pharmaceutical Technology en Español MARZO / ABRIL 201432
SOLUCIONES ESTADÍSTICAS
Las bases de la incertidumbre de la medición en el análisis farmacéuticoChris Burgess
¿Qué tan bueno es un valor reportable?
rea común en el laboratorio, pero pocos calculan qué tan buenas son sus solu-ciones estándar.
El estándar de referencia comprado tiene una pureza certificada de 99.46 ± 0.25. Se pesan aproximadamente 100 mg de este estándar de referencia, por diferencia, exactamente utilizando una balanza analítica de cinco lugares de-cimales. El estándar de referencia se disuelve en agua y se prepara una solu-ción hasta la marca con agua en un ma-traz volumétrico con capacidad de 100 mL Grado A a la temperatura ambiente del laboratorio. Se asume que la tem-peratura del laboratorio está controlada pero puede variar entre 16°C y 24°C. El primer paso es trazar un diagrama de flujo del proceso analítico utilizado para preparar la solución estándar. Este diagrama se muestra en la
.En este caso, el mesurando (C) es la concen-tración del material de referencia en la solución estándar en mg l-1 y está defi-nido por la ecuación:
donde m es la masa del material de refe-rencia en mg, P es la pureza como frac-ción de masa del estándar y V es el vo-lumen del matraz volumétrico en mL.
Identificar las fuentes de error. Con base en el flujo del proceso analítico (ver ), uno puede ahora identi-ficar tres áreas principales de error, es decir, el propio estándar de referencia, el proceso de pesado y la solución y el volumen final de la solución. Es valio-so usar un diagrama de Ishikawa para ayudar a la identificación y agrupar las fuentes de error. Para este ejemplo, el diagrama de Ishikawa se muestra en la , se muestrean las posibles fuentes de error para cada uno de los tres grupos. En este ejemplo, se asume que el estándar de referencia es suficientemente homogéneo para ig-norar cualquier contribución del error y es muy soluble en agua.
Obsérvese que el volumen de la so-lución tiene tres distintos componentes de incertidumbre que necesitan ser to-mados en cuenta:•
•
Todas las mediciones están sujetas al error. Cuando un valor reportable se deriva de una medición o serie de me-
diciones, este valor es sólo un estimado del valor “real” y tiene un rango a su al-rededor asociado con qué tanta confian-za tiene uno de que el valor real caiga dentro de este rango. Tradicionalmente en la industria farmacéutica, se selec-ciona un rango correspondiente al 95% de confianza (1).
Calidad de los datos de valor re-portableLa calidad de un valor reportable o de un resultado analítico depende del ta-maño del intervalo de confianza. A más pequeño es el intervalo de confianza, mayor es la confianza se tiene en apo-yarse en el valor reportable o el resul-tado analítico. Desafortunadamente, también por razones históricas que se relacionan principalmente con conside-raciones metrológicas físicas, la Orga-nización Internacional de Estandariza-ción (ISO) utiliza el término “medición de la incertidumbre” (MU) para el mis-mo concepto (2).
Una diferencia entre el enfoque de la MU de ISO y la Conferencia Interna-cional de Armonización (ICH) Q2(R1) y los esquemas de la Farmacopea de los Estados Unidos (USP) es que en esta última, los efectos de la imprecisión y el sesgo se consideran por separado (3).
Cabe hacer notar, sin embargo, que el Capítulo General <1225> de la USP, “Validación de Procedimientos Com-pendiales,” y los Capítulos Generales <1224> “Transferencia de Procedi-mientos Analíticos,” y <1226>, “Veri-ficación de Procedimientos Compen-diales, relacionados, están en revisión actualmente (4-6).
El Capítulo General <1010> de la USP, Datos Analíticos - Interpretación y Tratamiento, establece claramente que la exactitud tiene un diferente sig-nificado del ISO (7). La USP estable-ce, “En el ISO, la exactitud combina los conceptos de insesgamiento (deno-minado legitimidad) y precisión,” y la USP además define un intervalo de con-fianza convencional de 95% alrededor de la media de
El término es el error estándar de la media y se llama la incertidumbre estándar en el ISO.
se llama el factor de cobertura.
se llama la incertidumbre ex-pandida en el ISO.
Otra diferencia es la manera en la cual se calcula la desviación estándar (s). El enfoque de ISO es por medio de un presupuesto de error calculado (8), mientras que el de ICH Q2(R1) se apoya en información derivada de un estudio analítico diseñado experimen-talmente (3). Teóricamente, estos dos enfoques deben dar resultados simila-res. En la práctica, sin embargo, éste no es siempre el caso. El ISO también uti-liza una diferente nomenclatura que el ICH. Lo que habitualmente se llamaría la medición analítica o resultados en el
ISO se llama el mesurando. Este mesu-rando es la cantidad particular sujeta a medición y está relacionada con la fun-ción de la respuesta analítica medida por medio de una ecuación de la misma manera que un resultado analítico.
Concepto de un presupuesto de errorLa idea detrás de un presupuesto de error es que si se conocen todas las fuentes de error, es posible calcular un estimado de la incertidumbre del mesurando o el valor reportable basa-do en la conversión de todos los erro-res a desviaciones estándar y después combinando las varianzas. Si todos los procesos de error son independientes, entonces un presupuesto de error puede definirse en cinco pasos:• Definir todos los elementos del pro-
ceso involucrado y sus interrelacio-nes
• Definir el mesurando en términos de estos elementos del proceso
• Identificar todas las fuentes de error y agruparlas según se requiera
• Estimar sus contribuciones indivi-duales y convertirlas a desviaciones estándar y combinarlas para producir un estimado general de la desviación estándar
• Estimar la incertidumbre global utili-zando un factor de cobertura apropia-do según se describe anteriormente.
La Figura 1 muestra el proceso de presupuesto del error en forma de dia-grama.
Un ejemplo de un presupuesto del error simple para una solución estándar.El enfoque del presupuesto del error puede parecer bastante desalentador, pero un simple ejemplo de la prepara-ción de una solución estándar hará más claras las cosas. Este ejemplo es una ta-
X ± t(0.05, n-1)S
√n
S√n
t(0.05, n-1)
t(0.05, n-1)S√n
PHO
TOD
ISC
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 2014 33
rea común en el laboratorio, pero pocos calculan qué tan buenas son sus solu-ciones estándar.
El estándar de referencia comprado tiene una pureza certificada de 99.46 ± 0.25. Se pesan aproximadamente 100 mg de este estándar de referencia, por diferencia, exactamente utilizando una balanza analítica de cinco lugares de-cimales. El estándar de referencia se disuelve en agua y se prepara una solu-ción hasta la marca con agua en un ma-traz volumétrico con capacidad de 100 mL Grado A a la temperatura ambiente del laboratorio. Se asume que la tem-peratura del laboratorio está controlada pero puede variar entre 16°C y 24°C. El primer paso es trazar un diagrama de flujo del proceso analítico utilizado para preparar la solución estándar. Este diagrama se muestra en la Figura 2.
Identidad del mesurando.En este caso, el mesurando (C) es la concen-tración del material de referencia en la solución estándar en mg l-1 y está defi-nido por la ecuación:
donde m es la masa del material de refe-rencia en mg, P es la pureza como frac-ción de masa del estándar y V es el vo-lumen del matraz volumétrico en mL.
Identificar las fuentes de error. Con base en el flujo del proceso analítico (ver Figura 2), uno puede ahora identi-ficar tres áreas principales de error, es decir, el propio estándar de referencia, el proceso de pesado y la solución y el volumen final de la solución. Es valio-so usar un diagrama de Ishikawa para ayudar a la identificación y agrupar las fuentes de error. Para este ejemplo, el diagrama de Ishikawa se muestra en la Figura 3. En la Figura 3, se muestrean las posibles fuentes de error para cada uno de los tres grupos. En este ejemplo, se asume que el estándar de referencia es suficientemente homogéneo para ig-norar cualquier contribución del error y es muy soluble en agua.
Obsérvese que el volumen de la so-lución tiene tres distintos componentes de incertidumbre que necesitan ser to-mados en cuenta:• La incertidumbre en el volumen de
calibración marcado del matraz volu-métrico a 20°C
• La diferencia entre la temperatura de
Paso 1Descripción del procesode medición
Diagrama de �ujo con descripcióndetallada de todos lospasos del procedimiento
De�nir la relación entre el mesurandoy las variables del procedimiento
Trazar un diagrama de causa y efecto para identi�car las incertidumbresde cada variable
Combinar las incertidumbres de cada variable para dar una incertidumbre total
Paso 2Especi�cación delmesurando
Paso 4 y 5Cuanti�cación y combinación de las incertidumbres
Paso 3Identi�cación de lasfuentes de incertidumbre
Transferir aproximadamente100 mg del estándar de referencia a una nave de
pesado
Pesar en una balanzaanalítica con cinco lugares decimales
Transferir el materiala un matraz
volumétrico de 100mL, Grado A
Volver a pesar enla misma balanzaanalítica de cincolugares decimales
Disolver en agua
Llevar al volumencon agua
Calcular la concentracióndel estándarde referencia
PESADO PORDIFERENCIA
calibración del matraz y la tempera-tura a la cual se preparó la solución
• La incertidumbre asociada con el llenado del matraz hasta la marca de calibración.
No todas las contribuciones al error son de igual importancia. Para encon-
trar qué contribuciones al error son im-portantes, sin embargo, es esencial con-vertir todos los errores a desviaciones estándar (8).
Procesos para convertir especifica-ciones, rangos y datos de medición en una desviación estándar. El modo más
C = 1000 mg1-1mPV
Figura 1: Proceso del presupuesto del error
Figura 2: Flujo de proceso analítico para la preparación de la solución estándar en nuestro ejemplo
PHO
TOD
ISC
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 201434
Estándar de referencia
Pureza
Homogeneidad
Exactitud
Precisión
Diferencias de temperaturaentre la temperatura de calibración
y la temperatura de la solución
Incertidumbre en elvolumen certi�cado
del matraz Variación en el aforo
Volumen de la solución
Incertidumbre dela concentración
Pesado
Tabla I: Incertidumbre combinada para el mesurando C
Descripción x Valor x Ux
ux
X
Pureza del estándar de referencia
P 0.9946 UP0.001443 uP
P
0.001443
Masa del estándar dereferencia, mg
m 100.28 Um0.05 um
m
0.0005
Volumen en el matraz, mL
V 100.0 UV0.07 uv
V
0.0007
fácil para evaluar la desviación estándar es mediante el análisis estadístico de series de observaciones y asumir una distribución normal. En el ejemplo, esté método sería usado en la determinación de la incertidumbre del llenado del ma-traz volumétrico a la marca. Esta de-terminación directa es conocida como incertidumbre Tipo A.
Las incertidumbres Tipo B se deri-van de dos enfoques:• Convertir los rangos del certificado
cuando no hay conocimiento de la forma de la distribución de manera que se asume distribución rectangu-lar. Para un rango de ±a, el estimado correspondiente para la desviación estándar sería . En el ejemplo, la incertidumbre en la pureza de ±0.25 se convertiría utilizando la distribu-ción rectangular.
• Es más probable que el valor quede cerca del valor central, entonces se asume la distribución rectangular. Para un rango de ± a, el estimado correspondiente para desviación es-tándar sería . En el ejemplo, la in-certidumbre en el matraz volumétrico
grado A de ± 0.10 sería convertida utilizando la distribución triangular.
Contribuciones de incertidumbre en el ejemplo.Ahora podemos proceder a cuan-tificar todas las incertidumbres en nuestro proceso analítico de la siguiente manera:
Incertidumbre del estándar de referencia,uP. Utilizando la distribución rectangular tenemos:
Note que la pureza y su incertidum-bre han sido convertidas a fracciones de masa. Incertidumbre del pesado, um. Utilizando los datos del fabricante de la balanza (Tipo A) tenemos:
Note que nuestro valor real del ma-terial pesado fue 100.28 mg.
Incertidumbre volumétrica (uV. ).Aquí tenemos tres diferentes contribuciones a la uv: • El propio matraz que usa la distribu-
ción triangular:
• El efecto de temperatura asumiendo el coeficiente de expansión del agua de 0.00021°C-1 y asumiendo la dis-tribución rectangular:
y la incertidumbre Tipo A asociada con el llenado del matraz hasta el aforo. Esto fue determinado mediante la repetibili-dad del llenado para n = 6 asumiendo una distribución normal u
vr = 0.02 mL.
Se puede ahora combinar estas tres desviaciones estándar para llegar a la incertidumbre volumétrica general
Finalizando el presupuesto del error.Ahora que todas las incertidumbres han sido convertidas a desviaciones están-dar, pueden ser combinadas para pro-ducir una incertidumbre para el mesu-rando C como se muestra en la Tabla I y utilizando la ecuación de combinación de varianzas:
Es importante señalar que la contri-bución de la incertidumbre del estándar de referencia es mayor que la de los errores de pesado o volumétricos.
Expresión de confianza: cálculo del valor reportable y su incertidumbre.La concentración de la solución del están-dar de referencia está directamente dis-ponible de la ecuación del mesurando:
“Las bases de la incertidumbre de la medición en el análisis farmacéutico” Continúa en la pág. 40
Los mercados emergentes son una maquinaria para el crecimiento en los mercados de manufactura personalizada y farmacéutica.
√3a
√6a
uP = = 0.0014430.0025
√3
um = 0.05 mg
uvc = = 0.04 mL0.10√6
uvT = = 0.05 mL
Volume variation =±(100(4)(0.00021)) =±0.084 mL
0.084√3
Variación de volumen =± (100(4)(0.00021))=± 0.084 mL
uv =√u2 + u2 + u2vp vr vT
=√(0.04)2 + (0.02)2+ (0.05)2
= 0.07 mL
uc
C
=√(0.001443)2 + (0.0005)2+ (0.0007)2
+√ ( )=
= 0.00168
up
P
2
+( )um
m
2
( )uv
V
2
= 1000
= 997.4 mg1-1
mPVC = 1000
100.28(0.9946)100.0
Figura 3: Diagrama de Ishikawa para nuestro proceso analítico
SOLUCIONES ESTADÍSTICAS

Pharmaceutical Technology en Español MARZO / ABRIL 2014 35
El efecto de temperatura asumiendo el coeficiente de expansión del agua de 0.00021°C-1 y asumiendo la dis-tribución rectangular:
y la incertidumbre Tipo A asociada con el llenado del matraz hasta el aforo. Esto fue determinado mediante la repetibili-dad del llenado para n = 6 asumiendo una distribución normal u = 0.02 mL.
Se puede ahora combinar estas tres desviaciones estándar para llegar a la incertidumbre volumétrica general
Patricia Van Arnum
síntEsis Y manuFactura DE apis
Los mercados emergentes son una maquinaria para el crecimiento en los mercados de manufactura personalizada y farmacéutica.
Conforme la industria bio/far-macéutica entra a un nuevo año, ¿cuál es el panorama? Los mercados emergentes
continuarán jugando un importante pa-pel en el mercado más amplio de sínte-sis farmacéutica y personalizada.
Haciendo cuentasUn reciente análisis de la Asociación Química Farmacéutica de Genéricos (CPA), la cual representa a los fabrican-tes de APIs genéricos de Italia, apunta al fuerte crecimiento para la industria de investigación farmacéutica por con-trato y de servicios de manufactura (PCRAMS). La industria global mun-dial PCRAMS, la cual incluye ingresos provenientes de las organizaciones de investigación por contrato (CROs) y or-ganizaciones de manufactura por con-trato (CMOs) fue valuada en $ 72,000 mdd en 2012, $ 21,000 mdd más que en el 2000. La manufactura por contrato representó dos tercios del mercado en 2012, o $47,000 mdd, y la investigación
por contrato los $25,000 mdd restantes, de acuerdo al reporte de la CPA, La In-dustria Mundial PCRAMS (1).
Del negocio de manufactura global por contrato, la manufactura de APIs e intermedios por contrato representó el sector más grande para la manufactura por contrato en 64% o $30,000 mdd. La manufactura por contrato de las formas farmacéuticas terminadas representó el 9.6% o $4,500 mdd, de acuerdo al aná-lisis de la CPA. El desarrollo de formas farmacéuticas terminadas representó aproximadamente el 15% del mercado de manufactura por contrato, o %7,000 mdd, y el acondicionado/etiquetado y otros servicios representan el restante o $5,500 mdd.
Del mercado para la manufactura de APIs e intermedios por contrato, la sín-tesis personalizada (principalmente en-focada en nuevos APIs de marca) repre-senta el 40% o $12,000 mdd, y la manu-factura por terceros representa el 60% restante o $18,000 mdd. Para propósi-tos del análisis, “síntesis personalizada” significa cuando una compañía subcon-trata la manufactura de un intermedio o de un API sin indicar las modalidades de la operación. “Manufactura por ter-ceros” significa que la producción del intermedio o el API se lleva a cabo por el proveedor externo de acuerdo a las modalidades operacionales establecidas
por la compañía patrocinadora.En general, el mercado global
PCRAMS está proyectado para crecer en un promedio de 13.6% anualmente durante los próximos cinco años, para alcanzar $136,000 mdd para el 2017 desde los $72,000 mdd en 2012, de acuerdo al análisis del CPA. El nego-cio global del CRO se espera que se incremente desde $25,000 mdd hasta $43,000 mdd en 2017, representan-do un crecimiento promedio anual de 11.4%, de acuerdo al CPA. El negocio global del CMO está proyectado que se incremente desde $47,000 mdd en 2012 hasta $93,000 mdd en 2017, un prome-dio de 14.6% en el crecimiento anual, de acuerdo a los estimados del CPA. La región Asia Pacífico mostrará el creci-miento más fuerte en el mercado glo-bal del PCRAMS, con un crecimiento particularmente fuerte para la India y China. La participación de la India en el mercado mundial del PCRAMS casi se triplicará en los próximos cinco años, y para el 2017, la India será el segundo mercado más grande para el PCRAMS, representando el 21.3% del mercado global en comparación con sólo 8.3% en 2012. La India sólo será rebasada por los Estados Unidos, el cual se es-pera que mantenga el 24.9% del mer-cado global del PCRAMS en 2017 en comparación con una participación de 33.7% en 2012. Así como la participa-ción de EEUU en el mercado global del PCRAMS declinará, lo mismo sucederá con la participación de Europa Occiden-tal desde 25.0% en 2012 hasta 17.1% en 2017. La participación de China en el mercado global de PCRAMS se in-crementará desde 12.2% en 2012 hasta 19.2% para 2017, de acuerdo al CPA.
Perspectiva del mercado farma-céuticoLos patrones de crecimiento en el mer-cado de la manufactura personalizada van paralelos al mercado farmacéutico como un todo, lo cual refleja un creci-miento incrementado en los mercados emergentes. El gasto global en me-dicamentos se espera que alcance el umbral de $1,000,000 mdd en 2014 y que alcance $1,170,000 mdd para 2017, de acuerdo al reporte de Informática del IMS Institute for Healthcare, El Uso Global de Medicinas: Perspecti-
Un vistazo a futuro para la manufactura personalizada y las farmacéuticas
Patricia Van Arnum es editora ejecutiva del Pharmaceutical Technology, 485 Route One South, Edif. F, Primer Piso, Iselin, NJ 08830; tel. 732.346.3072, [email protected].

Pharmaceutical Technology en Español MARZO / ABRIL 201436
vas hasta 2017, el cual muestra que el crecimiento en el gasto global en me-dicamentos se incrementó 2.6% hasta $965,000 mdd en 2012 y está proyec-tado para que crezca en una tasa de cre-cimiento anual compuesta (CAGR) de 3-6% para el 2017 (2). El IMS proyecta un incremento gradual en el crecimien-to del gasto global anual en medicamen-tos, el cual se espera que se eleve desde 2-3% en 2013 hasta 5-7% en 2017, el ritmo más elevado de crecimiento desde el 2009. El gasto global absoluto para los farmacéuticos se incrementará en $230-260,000 mdd sobre una base de dólar constante en comparación a los $217,000 mdd en el período anterior de cinco años. El impacto más grande en los niveles de crecimiento es el efecto continuo de muchos fármacos estrella que pierden la patente y un incremento en los genéricos alternativos de menor costo. Adicionalmente, el retorno gra-dual del crecimiento global en el pro-ducto interno bruto (PIB) a más de 4% para el 2017, contribuirá a una tenden-cia ascendente en el gasto en fármacos. “Conforme pasamos el quinto aniver-sario de la desaceleración económica global y con muchos países moviéndose hacia una cobertura de salud universal, esperamos ver una continua divergen-cia en las tasas de crecimiento entre los mercados farmacéuticos emergentes y desarrollados,” dijo Murray Aitken, director ejecutivo del IMS Institute for Healthcare Informatics, en un comuni-cado de prensa del IMS. “Las medidas de austeridad dirigidas a los presupues-tos en medicamentos, junto con la cre-ciente disponibilidad de genéricos de bajo costo, llevará a un crecimiento del gasto anual de 1-4% entre los mercados de Norteamérica, Europa y Japón. En contraste, las naciones ‘farmaemergen-tes’ experimentarán 10-13% de creci-miento en el gasto.”
En 2012, los mercados desarrolla-dos en su conjunto redujeron su gasto en medicamentos por primera vez debi-do a las caducidades de las patentes y a las medidas de austeridad, junto con cambios en las políticas para incremen-tar la penetración de fármacos genéri-cos, señala el IMS. El crecimiento en los mercados desarrollados tendrá un
rebote desde $3,000 mdd en 2012 has-ta $20,000 mdd - $25,000 mdd para el 2017. Los Estados Unidos reanudarán los niveles en gasto incrementados en 2014, después de dos años de reduc-ción debida a la expansión del acceso a la salud y a menores niveles de ca-ducidad de patentes. El crecimiento en los mercados ‘farmaemergentes’, de-finido por el IMS, se incrementará de $26,000 mdd en 2012 a entre $30,000 mdd y $50,000 mdd en 2017, principal-mente debido a un mayor acceso a los medicamentos conforme evoluciona la infraestructura y los sistemas de salud. Los países farmaemergentes incluyen a China, países del Grupo 2 (Brasil, Rusia e India) y países del Grupo 3 (México, Turquía, Venezuela, Polonia, Argentina, Arabia Saudita, Indonesia, Colombia, Tailandia, Ucrania, Sudáfrica, Egipto, Rumania, Argelia, Vietnam, Paquistán y Nigeria) (2).
Perspectiva del mercado desa-rrolladoLos mercados desarrollados de Nor-teamérica, Europa y Japón verán un modesto crecimiento de un solo dígito en el crecimiento del gasto durante los próximos cinco años debido a una com-binación de medidas de austeridad eco-nómicas y de salud y de ahorros mate-rializados a partir del crecimiento de las versiones de genéricos de menor costo.
En EEUU, los siguientes cinco años verán el mayor impacto de la implemen-tación del Acta de Cuidados Accesibles. El IMS señala que la incertidumbre ve desde el nivel de afiliación de los asegu-rados en la actualidad a la velocidad con la cual el sistema de pagos cambia de uno basado en el pago por servicio a uno basado en el desempeño y los resultados del paciente. El cambio en el modelo de entrega de servicios, que incluye la in-corporación de la atención médica y el poder de negociación relativo ente los pagadores y los proveedores, impactará el gasto en medicamentos, aunque la di-rección y magnitud de dicho impacto es incierta. Para el mercado farmacéutico de EEUU, el IMS estima un CAGR de 1 a 4% desde 2013-2017. El mercado farmacéutico de EEUU alcanzará entre $350,000 mdd y $380,000 mdd para el
2017. La caducidad de las patentes y el impacto de los genéricos de bajo costo impactarán el crecimiento del gasto a lo largo del período proyectado, lo cual tuvo su impacto más fuerte en 2012 y 2013. El menor impacto de las caduci-dades contribuye a un crecimiento de aproximadamente la mitad del creci-miento más alto del mercado en 2014 con relación al 2013.
El crecimiento del gasto farmacéu-tico en la UE 5 (Alemania, Francia, Italia, Reino Unido y España) se espera que sea entre cero y 3% para el período del 2013 al 2017 en comparación con el 2.4% para 2008-2012. El gasto far-macéutico en la UE 5 está proyectado para que alcance entre $140,000 mdd y $170,000 mdd en 2017, de acuerdo al IMS. Europa ha visto una mayor adopción de los genéricos y políticas más restrictivas que han hecho que los pacientes en casi todos los países de Europa tengan menos probabilidad de tener acceso a los medicamentos in-novadores, señala el reporte del IMS. El desempeño de la industria farma-céutica europea durante los próximos cinco años dependerá del alcance de la recuperación económica en Europa. El mayor crecimiento se anticipa para Alemania y el Reino Unido en compa-ración con España, Italia y Francia, ya que estos mercados ya tienen merca-dos eficientes de genéricos. Los cinco países europeos principales han visto una menor captación de nuevos medi-camentos en el período más reciente de cinco años. Alemania y el RU tuvieron controles de captación fuertes antes de la crisis financiera y ven el deterioro menor. En Japón, el pronóstico del cre-cimiento del gasto de 2-5% CAGR está proyectado desde 2013 a 2017, señala el IMS. En Japón, la cuestión clave es el establecimiento de un mercado de fármacos genéricos, el objetivo dirigi-do por el Ministerio de Salud, Trabajo y Bienestar (MHLW) de Japón es un incremento del 60% en el volumen de genéricos como un porcentaje de fár-macos genéricos y listados en 2018. Si el MHLW tiene éxito, significaría que los niveles de genéricos para el 2018 en Japón estarían en un nivel similar al de Francia o España.
“Formulación: Fragmentos De Anti-cuerpos Fab” Continuación de la pág. 27
síntEsis Y manuFactura DE apis

Pharmaceutical Technology en Español MARZO / ABRIL 2014 37
Crecimiento del mercado emergentePara los mercados ‘farmaemergentes’, el IMS proyecta una CAGR de 10-13% con el mercado llegando a entre $370,000 mdd y $400,000 mdd para el 2017. Para Cina, el mercado farma- emergente más grande, el IMS proyecta una CAGR de 14-17% con el merca-do alcanzando entre $160,000 mdd y $190,000 mdd para el 2017. A pesar de las saludables proyecciones, el creci-miento de la industria farmacéutica en China fue modificado a la baja debido a una desaceleración en el crecimiento del PIB, una pérdida de la confianza en los negocios, y una menor inversión. El crecimiento basado en el volumen será manejado por los esfuerzos del gobier-no para mejorar los servicios de salud y médicos, incluyendo la expansión de la
cobertura del seguro para enfermedades críticas y el uso incrementado de hos-pitales privados. Los esfuerzos para ex-pandir la lista de fármacos esenciales y la reciente investigación de la Comisión Nacional de Desarrollo y Reformas de los precios de los fármacos y el costo podrían resultar en un mercado de mar-cas originales sin patente, más económi-cas. También, una nueva modificación de la Lista Nacional de Medicamentos para Reembolso podría dar lugar a una lista más amplia de productos innova-dores que reciben reembolsos.
El crecimiento en los países del Grupo 2 (Brasil, Rusia e India) mos-trará una CAGR de 10-13% en 2013-2017 y alcanzará entre %90,000 mdd y $110,000 mdd para el 2017. El cre-cimiento en el mercado farmacéutico
de la India, el segundo mercado emer-gente más grande detrás de China y el mercado farmacéutico más grande en-tre los países del Grupo 2, mostrará una CAGR de 5-8% entre 2013 y 2017 con el mercado alcanzando entre $100,000 mdd y $130,000 mdd para el 2017. El crecimiento en los mercados farmae-mergentes del Grupo 3 estará entre 5% y 8% hasta el 2017 esperando que el mercado alcance entre $100,000 mdd y $130,000 mdd para este año (2).
Referencias1. Chemical Pharmaceutical Association,
The World Pharmaceutical Contract Re-search and Manufacturing Services Industry (Milan, September 2013).
2. IMS, The Global Use of Medicines: Outlook through 2017 (November 2013). PT
“Formulación: Fragmentos De Anti-cuerpos Fab” Continuación de la pág. 27
para demostrar conducta de sedimenta-ción más lenta, incluso con un volumen más grande comparado con el Fab nati-vo. De hecho, el PEG asociado a la pro-teína tiende a dominar las propiedades hidrodinámicas asociadas con la proteí-na unida y también puede contribuir a una reducción en los efectos relaciona-dos con la agregación.
A pesar de los beneficios anterior-mente mencionados, la PEGilación no es la única metodología que puede producir efectos estabilizantes sobre las formulaciones de proteína Fab te-rapéutica. Los detergentes no iónicos, como los polisorbatos, se emplean co-múnmente en las estrategias de formu-lación del Fab como medio adicional de promoción de la solubilidad y de reduc-ción de la agregación. También puede ser considerada la cuidadosa creación de los excipientes (sales y buffers) en términos de efectos relacionados con la viscosidad. Los efectos repulsivos de interacción de la proteína acentua-dos con las sales basados en la teoría de Debye de carga difusora indican una correlación para los efectos repulsivos mejorados asociados con sales mono-valentes (p.ej., NaCl) contrariamente a las relaciones iónicas de las divalen-
tes (p.ej., MgCl2) (3). Los cationes y/o
aniones monovalentes contra los diva-lentes también pueden ser considerados como especies estabilizadoras en la formulación. Finalmente, existen indi-caciones de que los contra-iones, tales como la meglumina, también pueden tener un efecto significativo en la miti-gación de la conducta de agregación y, por lo tanto, en la actividad mejorada en soluciones de proteína en general (4).
ConclusiónLas moléculas de Fab están generando cada vez más interés conforme continúa creciendo la demanda de terapéuticos dirigidos con eficacia mejorada. Estas moléculas presentan desafíos únicos para los procesos “tradicionales” usados para el desarrollo y la manufactura de anticuerpos monoclonales.
Cada uno de los pasos en el recobro y purificación deben ser optimizados con base en los requerimientos del proceso y las características de la molécula para asegurare un proceso de producción del Fab robusto, estable y escalable.
Las mismas consideraciones cuida-dosas deben aplicarse al desarrollo de la formulación óptima. Al igual que con todos los esfuerzos de desarrollo de for-
mulaciones de proteínas complejas, el objetivo de la formulación del Fab es la preservación de la estabilidad y eficacia del producto. Todas las tecnologías po-tenciales pueden y deben ser considera-das para asegurar el nivel apropiado de pureza y consistencia en todos los ma-teriales usados en la formulación para minimizar el impacto de las impurezas y el posible efecto sobre la estabilidad y la actividad.
ReconocimientosLos autores desean agradecer a Robert Blanck, gerente de investigación de mercados y analítica en EMD Millipo-re, por su contribución a este artículo.
Referencias1. Y. Lu, et al., J. Pharm. Sci., 97 (6) 2062–
2079 (2008). 2. R.P. Garay and J.P. Labaune, The Open
Conference Proceedings Journal, 2, 104-107 (2011).
3. A.S. Parmar and M. Muschol, Biophys J., 97 (2) 590-598 (2009).
4. Chugai Seiyaku Kabushiki Kaisha, “Sta-bilizer for protein preparation comprising meglumine and use thereof,” EP 1908482 A1, April 2008. PT

Pharmaceutical Technology en Español MARZO / ABRIL 201438
RESOLUCIÓN DE PROBLEMASEquipo Y Proceso
Uso de técnicas analíticas para examinar la adhesividad de las tabletas Abdennour Bouhroum y Rob Blanchard
Las pruebas analíticas, correlacionadas con técnicasestadísticas, se utilizan para pronosticar la conducta del material.
Abdennour Bouhroum, PhD, es colega de investigación en la Universidad de Nottingham; Rob Blanchard es gerente de investigación, desarrollo y sistemas de calidad en I Holland, tel. 44 0 115 9726153, [email protected].
Z: 686.8nm
X: 10.0µm
Y: 10.0µm
El programa Investigación An-ti-adhesividad en la Ciencia del Tableteado (TSAR) reali-zado por el fabricante de he-
rramental para tableteadoras I Holland en colaboración con la Universidad Nottingham en el RU, está enfocada a investigar el problema común en la ma-nufactura de tabletas, la “adhesividad” (es decir, acumulación de gránulos en la punta del punzón) que puede causar tiempos muertos de la tableteadora y un rendimiento reducido.
Las causas de la adhesividad son complejas. Algunos de los factores sub-yacentes incluyen lo siguiente:• Química de superficie. Los elementos
dentro de la formulación son atraídos de manera natural a los elementos dentro del material del punzón de la tableta a través de fuerzas de Van der Waals. Estas bajas fuerzas se mi-den en nanonewtons (nN), pero en el comportamiento en volumen, estas fuerzas pueden causar adhesividad.
• Contenido de humedad. La acción capilar, la cual puede estar vinculada al alto contenido de humedad, crea el potencial para la adhesividad tanto en la compresión directa como en la gra-nulación húmeda. Sin embargo, si los gránulos están muy secos, se puede formar electricidad estática y causar adhesividad.
• Morfología. La rugosidad de la super-
ficie de contacto afecta la adhesivi-dad.
• Mecánica de la deformación. Bajo compresión, el gránulo es ya sea elás-tico o plástico, y estas características influyen la adhesividad.
El proyecto TSAR está desarro-llando una herramienta de pronóstico que utilizará los parámetros de entrada (p.ej., química de superficie, temperatu-ra, humedad, tamaño del gránulo y si el gránulo es elástico o plástico) para pro-nosticar la propensión a la adhesión de una formulación dada. La herramienta puede ser usada para optimizar un pun-zón o una solución de recubrimiento del molde para las formulaciones pega-josas.
Se han utilizado varias técnicas analíticas avanzadas para comprender las causas subyacentes de la adhesión. Estas técnicas incluyen perfilometría óptica de superficie, microscopía elec-trónica de barrido (SEM), espectrosco-pía fotoelectrónica de rayos X (XPS), microscopía de fuerza atómica (AFM), y espectrometría de masas de ion secun-dario en tiempo de vuelo (TOF-SIMS).
Microscopía de fuerza atómicaLa AFM trabaja escaneando un sensor con una punta aguda sobre la superficie de la muestra para construir un mapa de alturas y topografía. La AFM utili-za una palanca óptica para monitorear el movimiento de la punta montada en el extremo de una ménsula reflexiva uti-lizando un láser que está focalizado en el reverso de la ménsula. Cuando se es-canea la muestra de superficie, la punta
experimenta movimientos ascendentes y descendentes con los cambios en la topografía de la superficie que llevan a cambios en la deflexión de un láser óptico. La punta puede mantenerse en una fuerza constante que permite la determinación en tiempo real de la in-formación de la altura o en una altura constante para determinar la fuerza de deflexión por arriba de la muestra. Las imágenes generadas desde el detector son mapas topográficos tridimensiona-les de la superficie que se construyen graficando la altura de la muestra local contra la posición horizontal de la punta del sensor (ver Figura 1).
Los investigadores del TSAR pre-pararon mapas de adhesión para cada recubrimiento a diferentes humedades para medir las diferentes fuerzas adhe-sivas sobre la cara de un punzón de la tableta. Debe entenderse que la varianza de estas fuerzas es capaz de considerar la diferencia causada por las diferencias
topográficas de la superficie.
Además de estas mediciones topográ-ficas, la AFM puede proporcionar mu-cha más información. Como la AFM se apoya en las fuerzas entre la punta y la muestra, ésta puede ser usada para me-dir las fuerzas de atracción o repulsión de rango largo entre la punta del sen-sor y la superficie de la muestra en un solo punto o a través de una superficie, elucidando de esta manera las propie-dades químicas y mecánicas locales, tales como la adhesión y la elasticidad, e incluso la fuerza de ruptura del enlace molecular. La fuerza no se mide direc-tamente, sino que se calcula midiendo la deflexión de la palanca y conociendo la rigidez (constante de muelleo) de la ménsula siguiendo la ley de Hook:
F = -kzdonde F es la fuerza, k es la cons-
tante de muelleo (la rigidez de la palan-ca) y z es la distancia de deflexión de la ménsula.
Una curva de fuerza contra distan-cia, tal como la que se muestra en la Figura 2, muestra típicamente la de-flexión del extremo libre de la ménsula del AFM conforme el extremo fijo de la ménsula se lleva verticalmente hacia la superficie de la muestra y después se aleja de la misma. Experimentalmente, esto se hace aplicando un patrón de vol-taje de onda triangular a los electrodos para el escáner del eje z. Esto causa que el escáner se expanda y después se contraiga en dirección vertical, gene-
Figura 1. Mapa topográficotridimensional obtenido utilizando microscopía de fuerza atómica.
GlO
wIM
AGES
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 2014 39
Fuerza(nN)
c
b a
e
d
0
b) c) d) e)
0
xy
z a)
Ménsula con sensor
Sustrato
Desplazamiento del piezo
Piezo
Desplazamiento (nm)
Espejo iónico
Pistola de iones
Pulsado
Focalizado
Pulverizador
ObjetivoCañón proyector de electrones
Extractor
Óptica de transporte
DetectorEspectro
experimenta movimientos ascendentes y descendentes con los cambios en la topografía de la superficie que llevan a cambios en la deflexión de un láser óptico. La punta puede mantenerse en una fuerza constante que permite la determinación en tiempo real de la in-formación de la altura o en una altura constante para determinar la fuerza de deflexión por arriba de la muestra. Las imágenes generadas desde el detector son mapas topográficos tridimensiona-les de la superficie que se construyen graficando la altura de la muestra local contra la posición horizontal de la punta del sensor (ver ).
Los investigadores del TSAR pre-pararon mapas de adhesión para cada recubrimiento a diferentes humedades para medir las diferentes fuerzas adhe-sivas sobre la cara de un punzón de la tableta. Debe entenderse que la varianza de estas fuerzas es capaz de considerar la diferencia causada por las diferencias
topográficas de la superficie.Modo de mediciones de fuerza AFM.
Además de estas mediciones topográ-ficas, la AFM puede proporcionar mu-cha más información. Como la AFM se apoya en las fuerzas entre la punta y la muestra, ésta puede ser usada para me-dir las fuerzas de atracción o repulsión de rango largo entre la punta del sen-sor y la superficie de la muestra en un solo punto o a través de una superficie, elucidando de esta manera las propie-dades químicas y mecánicas locales, tales como la adhesión y la elasticidad, e incluso la fuerza de ruptura del enlace molecular. La fuerza no se mide direc-tamente, sino que se calcula midiendo la deflexión de la palanca y conociendo la rigidez (constante de muelleo) de la ménsula siguiendo la ley de Hook:
F = -kzdonde F es la fuerza, k es la cons-
tante de muelleo (la rigidez de la palan-ca) y z es la distancia de deflexión de la ménsula.
Una curva de fuerza contra distan-cia, tal como la que se muestra en la Figura 2, muestra típicamente la de-flexión del extremo libre de la ménsula del AFM conforme el extremo fijo de la ménsula se lleva verticalmente hacia la superficie de la muestra y después se aleja de la misma. Experimentalmente, esto se hace aplicando un patrón de vol-taje de onda triangular a los electrodos para el escáner del eje z. Esto causa que el escáner se expanda y después se contraiga en dirección vertical, gene-
rando un movimiento relativo entre la ménsula y la muestra. La deflexión del extremo libre de la ménsula se mide y se grafica en muchos puntos conforme el escáner del eje z extiende la ménsula hacia la superficie y después la retracta nuevamente.
AFM con sensor coloidal. El prin-cipio de medición de fuerza del AFM con sonda coloidal es idéntico al del AFM estándar, excepto que las partí-culas están unidas a las ménsulas del AFM y son desafiadas para diferentes superficies La partícula del producto farmacéutico se monta sobre la ménsu-la utilizando una cantidad muy pequeña de pegamento. Una vez que se une a la ménsula, la partícula se pone en contac-to con el sustrato (superficie) y después se retrae para determinar la fuerza de adhesión. Esta técnica permite la cuan-tificación de la adhesión del producto farmacéutico a diferentes superficies en diferentes entornos.
Espectrometría de masas de ión secundario con tiempo de vueloEl TOF-SIMS es una técnica sensible de análisis de superficies que propor-ciona información acerca de la compo-sición, estructura molecular y enlaces químicos del material. El TOF-SIMS permite obtener un espectro de masas de una superficie del material hasta una profundidad de aproximadamente 1 a 2 nm. El TOF-SIMS también puede ser usado para convertir en ‘imágenes’ las superficies, mostrando así la distribu-
ción de los iones seleccionados sobre la superficie de una muestra. Es posi-ble hacer imágenes de áreas que van en tamaño desde unos pocos micrómetros hasta décimas de milímetro reticulando el haz iónico primario sobre la super-ficie seleccionada. Debido a la colecta simultánea de todos los fragmentos de masa, las imágenes resultantes tienen un espectro de masas completo en cada píxel, permitiendo la visualización re-trospectiva de cualquier ión en el espec-tro y su posición sobre una superficie.El TOF-SIMS tiene la capacidad de ob-tener un perfil de profundidad y carac-terizar una distribución tridimensional de los componentes del material. Un haz de iones focalizado se reticula so-bre un área rectangular definida sobre la superficie de la muestra a ser analizada (ver Figura 3). Mientras que el haz ióni-co pulverizador está creando un cráter, el haz iónico de análisis está analizan-do progresivamente el centro del fondo del cráter. Las intensidades de los iones característicos de cada capa pueden re-portarse como una función de la profun-didad, dando como resultado un cubo tridimensional que representa los mate-riales pulverizados/analizados. La masa de cada ión secundario que alcanza el detector se colecta y se almacena junto con las coordenadas espaciales x, y, y z, formando así una pila de espectros de masa, adecuada para la visualización retrospectiva de los iones selecciona-dos. El TOF-SIMS permite la visualiza-ción de la distribución química de iones
Figura 2: Típico ciclo fuerza-curva de microscopía de fuerza atómica Figura 3: Representación esquemática del perfilado de profundidad con espec-trometría de masas de ión secundario con tiempo de vuelo (TOF-SIMS) de un sistema multi-capas.
GlO
wIM
AGES
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 201440
seleccionados en volúmenes tridimen-sionales de los materiales analizados.
Otras técnicasLa espectroscopia Raman puede ser usada para la identificación química, la caracterización de estructuras molecu-lares y la identificación de los efectos de la unión, el entorno, y el estrés sobre una muestra. La XPS (espectroscopía de electrones emitidos por Rayos X) provee información de la composición cuantitativa (p.ej., cantidades de ele-mentos y entornos de enlace) desde las primeras 10 capas atómicas de la super-ficie de una muestra. La SEM (micros-copía electrónica de barrido) emplea un haz de electrones altamente energéticos para examinar objetos en una escala de
sub-micas y produce información acer-ca de la topografía de la superficie, la
morfología, la composición y la crista-lografía. La perfilometría con láser es usada para determinar un perfil de la superficie para cuantificar su rugosidad.
El proyecto TSAR utiliza estas téc-nicas para ver las partículas que se ad-hieren a la punta del punzón y analizar qué son químicamente, resolviendo así lo que son estas interacciones.
Análisis del componente princi-pal (PCA)El PCA, una técnica estadística multi-variada, se enfoca a generar correlacio-nes entre los datos del XPS, la espec-
troscopía Raman y el TOF-SIMS para mostrar la información química cuan-titativa para los sistemas individuales que podrían estar involucrados en el problema de adhesión del polvo con di-ferentes recubrimientos. El PCA puede verse como un método para reducir una gran matriz de datos (es decir, una serie de muestras con picos variables de io-nes secundarios) a unas pocas combina-ciones clave de variables que describen las tendencias más significativas en los datos e identifican una correlación entre los diferentes datos obtenidos. Esto es una manera de identificar patrones en los datos y expresar los mismos de tal manera que destaquen sus similitudes y diferencias. Como los patrones en los datos pueden ser difíciles de encontrar en datos de alta dimensión. el PCA es una herramienta poderosa para ana-lizar los datos. El PCA se utiliza para comprender las interacciones clave que están causando que una formulación se adhiera a las caras de la punta del pun-zón y para desarrollar la herramienta predictiva del TSAR. PT
El AFM funciona escaneando un sensor con una punta aguda sobre la superficie de la muestra para crear un mapa de la altura y la topografía.
La incertidumbre en la Uc del me-surando y la incertidumbre U expandi-da están ahora disponibles.
El factor de cobertura de K = 2 co-rresponde a una confianza de 95.45%
Con base en esta incertidumbre expandida, calculamos que tenemos confianza de que la incertidumbre de la solución estándar es aproximadamente 0.34.
ResumenEste artículo cubrió algunas de las ba-ses de los presupuestos de error y llevó a cabo un cálculo de una incertidumbre expandida para una solución estándar. La incertidumbre expandida es pequeña (0.34%) y está dominada por la contri-bución del propio estándar de referen-cia. Sin embargo, mientras más comple-jo es el procedimiento analítico, se cons-truirán más incertidumbres expandidas.
En los laboratorios regulados, ta-les como los Laboratorios Oficiales de Control de Medicamentos, en Europa, es un pre-requisito que las pruebas ana-líticas sean realizadas bajo un sistema de calidad que funcione apropiadamen-te, lo que significa que:• Todas las balanzas y el material volu-
métrico estén bajo control regular• Las sustancias de referencia oficiales
o las sustancias de referencia internas estén apropiadamente calificadas y almacenadas
• Los instrumentos estén regularmente calibrados
• El equipo esté regularmente recalifi-cado
• Los técnicos de laboratorio sean reca-lificados.
Las incertidumbres debidas a estas fuentes están bajo control y se asume que contribuyen poco a la incertidum-bre total del resultado de prueba (9).
Referencias1. L. Torbeck, Pharm. Tech., 34 (7) (2010).2. See, for example, NIST Reference on
Constants, Units and Uncertainty; http://
physics.nist.gov/cuu/Uncertainty/index.html, accessed Aug. 12, 2013.
3. ICH, Q2(R1) Harmonised Tripartite Guide-line, Validation of Analytical Procedures: Text And Methodology (2005).
4. USP, General Chapter <1224>, “Transfer of Analytical Procedures,” United States Pharmacopeia, 36 (US Pharmacopeial Convention, Rockville, Md, 2013).
5. USP, General Chapter <1225>, “Valida-tion of Compendial Proceduures,” United States Pharmacopeia, 36 (US Pharmaco-peial Convention, Rockville, Md, 2013).
6. USP, General Chapter <1226>, “Verifica-tion of Compendial Procedures,” United States Pharmacopeia, 36 (US Pharmaco-peial Convention, Rockville, Md, 2013).
7. USP, General Chapter <1010>, United States Pharmacopeia, 36 (US Pharmaco-peial Convention, Rockville, Md, 2013).
8. S.L.R. Ellison and A Williams (Eds), Eura-chem/CITAC guide: Quantifying Uncer-tainty in Analytical Measurement, Third edition, (2012) ISBN 978-0-948926-30-3, available for www.eurachem.org/index.php/publications/guides/quam, accessed Aug. 12, 2013.
9. PA/PH/OMCL (05) 49 DEF CORR— OMCL Guideline on Uncertainty of Mea-surement (for compliance testing) (2007), www.edqm.eu/en/EDQM-Downloads-527.html, accessed Aug. 12, 2013. PT
uc = 0.00168C = 0.00168(997.4) = 1.68mgU = ± kuc
= ± 2uc
= ± 3.36
“Las bases de la incertidumbre de la medición en el análisis farmacéutico” Continuación de la pág. 34
Fármacos, ¿A qué costo?Los genéricos, las dinámicas del mercado y la demanda global están cambiando los patrones del gasto en fármacos.
RESOLUCIÓN DE PROBLEMAS
Rita Peters

Pharmaceutical Technology en Español MARZO / ABRIL 2014 41
troscopía Raman y el TOF-SIMS para mostrar la información química cuan-titativa para los sistemas individuales que podrían estar involucrados en el problema de adhesión del polvo con di-ferentes recubrimientos. El PCA puede verse como un método para reducir una gran matriz de datos (es decir, una serie de muestras con picos variables de io-nes secundarios) a unas pocas combina-ciones clave de variables que describen las tendencias más significativas en los datos e identifican una correlación entre los diferentes datos obtenidos. Esto es una manera de identificar patrones en los datos y expresar los mismos de tal manera que destaquen sus similitudes y diferencias. Como los patrones en los datos pueden ser difíciles de encontrar en datos de alta dimensión. el PCA es una herramienta poderosa para ana-lizar los datos. El PCA se utiliza para comprender las interacciones clave que están causando que una formulación se adhiera a las caras de la punta del pun-zón y para desarrollar la herramienta predictiva del TSAR.
United States Pharmacopei
United States Pharmacopei
United States Pharmacopei
PA/PH/OMCL (05) 49 DEF CORR— OMCL Guideline on Uncertainty of Mea-surement (for compliance testing) (2007), www.edqm.eu/en/EDQM-Downloads-527.html, accessed Aug. 12, 2013.
DEL EDITOR
JOR
G G
RE
UE
L/P
HO
TOD
ISC
/GE
TT
Y IM
AG
ES
Fármacos, ¿A qué costo?Los genéricos, las dinámicas del mercado y la demanda global están cambiando los patrones del gasto en fármacos.
La cuestión subyacente para cualquier fármaco en desa-rrollo es: ¿Qué precio pueden pagar o pagarán los pacientes
por la terapia necesaria? El impacto del precio en el paciente, los programas de austeridad del gobierno, los fármacos estrella que pierden la patente, y las prácticas variadas médicas y regulato-rias en todo el mundo continúan confor-mando la perspectiva financiera de las compañías bio/farmacéuticas.
En un nuevo reporte, El uso Global de las Medicinas: Perspectivas para el 2017, el IMS Institute for Health-care Informatics proyecta que en 2014 el gasto global total en medicamentos excederá, por primera vez, $1,000,000 mdd. En 2017, el total alcanzará casi $1,200,000 mdd.
En 2012, los mercados desarrolla-dos (Estados Unidos, Japón, Alemania, Francia, Italia España, Reino Unido, Canadá y Corea del Sur) redujeron el gasto por primera vez en gran medida debido a las caducidades de las paten-tes, a las medidas de austeridad, y a los cambios hacia los fármacos genéricos. El crecimiento anual en los niveles de gasto alcanzará un punto bajo en 2013, dice el reporte, pero tendrá un rebote del negativo de $3,000 mdd en 2012 a $20,000-25,000 mdd en 2017.
En los mercados emergentes (más de 20 naciones incluyendo China, Bra-sil, India, Rusia, México y Turquía) el crecimiento se incrementará desde $26,000 mdd en 2012 hasta $30,000-50,000 mdd en 2017, debido principal-mente al mayor acceso a los medica-mentos.
En 2017, el gasto en medicamentos para enfermedades específicas diferirá entre los mercados maduros y emer-gentes, dice el reporte. En los mercados desarrollados, las poblaciones cada vez más viejas y obesas dirigirán el gasto en oncología y diabetes, una tendencia que se está incrementando en los mercados emergentes. Las especialidades farma-céuticas, que tratan enfermedades com-plejas y serias crecerán en importancia en mercados desarrollados; las terapias tradicionales dirigirán el gasto en los mercados emergentes.
El ascenso de los biológicosLos biológicos tienen un impacto cre-ciente en el mercado y precios de fár-macos. En 2002, los biológicos repre-sentaron 11% de las ventas totales de fármacos; en 2012, el número fue 18%. El IMS estima que los agentes bioló-gicos continuarán superando el creci-miento del gasto global farmacéutico y representarán 19-20% del valor total del mercado para el 2017. Los anticuerpos monoclonales y la insulina estimularán este crecimiento.
La competencia generada a través de las vías de genéricos establecidas hará descender los altos costos de los biológicos en Europa, EEUU, y algu-nos países emergentes. Sin embargo, en mercados con protección menos riguro-sa de la IP, el IMS reporta una oleada en los biológicos no originales - copias de marcas del innovador que no han sido aprobadas por una vía dedicada. Los biosimilares constituyen menos del 0.5% del gasto en biológicos en los mercados maduros; en los mercados
emergentes, los biológicos no originales representan más del 10% de todo el gas-to en biológicos.
El impacto de los medicamentos genéricosLa mezcla del gasto global total en me-dicamentos cambiará hacia los fármacos genéricos, elevándose desde 27% hasta 36% del total para 2017; sin embargo, los fármacos de marca constituirán más de dos tercios del gasto en los mercados desarrollados. En los mercados emer-gentes, los productos de fármacos gené-ricos representarán 63% del gasto.
Los farmacéuticos tradicionales usados para tratar enfermedades cró-nicas en cuidado primario serán cada vez más dispensados como genéricos; el gasto total sólo se elevará 5% para el 2017 en los países desarrollados. En los mercados emergentes, el gasto total en los farmacéuticos tradicionales se espe-ra que se eleve desde $199,000 mdd en 2012 hasta $366,000 mdd en 2017 con-forme los pacientes adquieran acceso a fármacos genéricos costeables.
El IMS reporta que el gasto en me-dicamentos de especialidad en los mer-cados desarrollados se espera que se in-cremente en 30% durante los próximos cinco años; el uso de medicamentos de especialidades en los mercados emer-gentes está en niveles muy bajos, aun-que se espera que se eleven los costos casi en un 90% entre 2012 y 2017.
El año por delanteEl reporte identifica desafíos, pero también indica oportunidades para que la industria mejore las vidas de los pa-cientes. Los editores de Pharmaceutical Technology le desean un Año Nuevo se-guro, saludable y exitoso en todos sus propósitos.
Referencias1. IMS Institute for Healthcare Informat-
ics, The Global Use of Medicines: Outlook through 2017, November 2013. PT
Rita Peters es director editorial de Pharmaceutical Technology. Envíe sus pensamientos e ideas a [email protected]
Los agentes biológi-cos continuarán superando el cre-cimiento del gasto global farmacéutico.
Rita Peters

Pharmaceutical Technology en Español MARZO / ABRIL 201442
VIGILANCIA REGULATORIA EEUU
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO La métrica pretende asegurar la calidad del fármacoLos reguladores trabajan con los fabricantes para mejorar sistemas para medir el desempeño del producto y la confiabilidad de la manufactura.
Jill Wechsler es editora en Washington de Pharmaceutical Technology,
tel. 301.656.4634, [email protected]. Lea los blogs de Jill en
PharmTech.com/wechsler.
Un objetivo principal para los funcionarios de la FDA en 2014 es establecer una base más sólida para asegurar que los fármacos y las terapias bio-tecnológicas cumplan altos estándares de calidad
a lo largo del ciclo de vida del producto. La crisis continua de desabasto de fármacos ha intensificado la conciencia públi-ca de la importancia de las operaciones de manufactura en el mantenimiento del acceso confiable a las terapias necesarias. Las muertes y las enfermedades provenientes de productos por debajo de los estándares, hechos por mezcladores de far-macias menos reguladas persuadió al Congreso de apuntalar a la autoridad de la FDA para evitar dichos fallos, y a requerir un sistema de rastreo electrónico más efectivo para mantener a los fármacos no autorizados y falsificados fuera de la cadena de suministro global.
Janet Woodcock, directora del Centro para la Evaluación e Investigación de Fármacos (CDER), ha hecho de la calidad de los fármacos la mayor prioridad para el 2014. Para cumplir los requerimientos legislativos de mejora de los sistemas para evitar el desabasto de fármacos, el CDER ha estado discutien-do opciones para la recolecta más amplia de datos de calidad en numerosas reuniones con la industria este año pasado (1). El proceso para establecer métricas informativas de la calidad del producto fue un tema principal en la Asociación de Fárma-cos Parenterales (PDA)/Conferencia Regulatoria de la FDA en Septiembre del 2013, la Conferencia Técnica en Octubre de la Asociación Farmacéutica de Genéricos y la reunión anual en Noviembre de 2013 de la Sociedad Internacional de Ingenieros Farmacéuticos (ISPE). La PDA patrocinó una con-ferencia de las Métricas de Calidad Farmacéutica en Diciem-bre de 2013 específicamente para alcanzar un acuerdo con los
funcionarios de la FDA en un documento con los “puntos a considerar” sobre el asunto.
Autorización de FDASIALos funcionarios de la FDA citan el Acta de Seguridad e Inno-vación de la FDA (FDASIA) del 2012 para proveer una firme base legal para establecer estándares más claros y para co-lectar datos para evaluar las operaciones de manufactura. La Sección 705 del FDASIA requiere que la FDA establezca un esquema basado en el riesgo para la inspección de los sitios de manufactura, la cual requiere del acceso a información más detallada sobre las instalaciones y los controles de calidad. La Sección 706 del proyecto de ley autoriza a la agencia a obte-ner información sobre los sitios de producción del fármaco y las operaciones antes de una inspección (2). Juntas, estas provisiones respaldan la estrategia de la FDA para mejorar la manera de evaluar la capacidad de un sitio de manufactura para producir consistentemente medicamentos que se ajusten al uso destinado.
Los funcionarios del CDER hacen énfasis en que no le están pidiendo a la industria información nueva y descono-cida, sino el acceso a los datos de desempeño del sistema ya colectados por los fabricantes para su uso interno. La métrica rutinaria de la empresa las pruebas para liberación del número de lote, los resultados fuera de especificaciones, y los lotes frustrados, rechazados, retrabajados, y reprocesados. Dicha información de la manufactura por lo general aparece en las revisiones anuales de producto de la compañía que están dis-ponibles para los investigadores del sitio cuando las soliciten. Pero eso es muy tarde para apoyar la planeación del CDER para las inspecciones y la supervisión post-comercialización, señaló Russell Wesdyk, coordinador científico en la Oficina de Programas Estratégicos del CDER, en la conferencia de la GPhA. Por otro lado, cada compañía tiende a colectar datos a su propio estilo, utilizando un rango de definiciones y planes de muestreo. La FDA anticipa que las métricas estándar re-ducirían las diferencias de cómo definen las empresas “lote” (granel), “lote” (producto terminado), e incluso “rechazo”.Ahora el CDER, en consecuencia, tiene que evaluar el nivel de riesgo de un sitio de manufactura basado principalmente
La FDA se ha embarcado en una iniciativa más amplia para definir la métrica ‘correcta’ para colectar y auditar.

Pharmaceutical Technology en Español MARZO / ABRIL 2014 43
La métrica pretende asegurar la calidad del fármaco
Pharmaceutical Technology ,
tel. 301.656.4634, [email protected]. Lea los blogs de Jill en
PharmTech.com/wechsler.
funcionarios de la FDA en un documento con los “puntos a considerar” sobre el asunto.
Los funcionarios de la FDA citan el Acta de Seguridad e Inno-vación de la FDA (FDASIA) del 2012 para proveer una firme base legal para establecer estándares más claros y para co-lectar datos para evaluar las operaciones de manufactura. La Sección 705 del FDASIA requiere que la FDA establezca un esquema basado en el riesgo para la inspección de los sitios de manufactura, la cual requiere del acceso a información más detallada sobre las instalaciones y los controles de calidad. La Sección 706 del proyecto de ley autoriza a la agencia a obte-ner información sobre los sitios de producción del fármaco y las operaciones antes de una inspección (2). Juntas, estas provisiones respaldan la estrategia de la FDA para mejorar la manera de evaluar la capacidad de un sitio de manufactura para producir consistentemente medicamentos que se ajusten al uso destinado.
Los funcionarios del CDER hacen énfasis en que no le están pidiendo a la industria información nueva y descono-cida, sino el acceso a los datos de desempeño del sistema ya colectados por los fabricantes para su uso interno. La métrica rutinaria de la empresa las pruebas para liberación del número de lote, los resultados fuera de especificaciones, y los lotes frustrados, rechazados, retrabajados, y reprocesados. Dicha información de la manufactura por lo general aparece en las revisiones anuales de producto de la compañía que están dis-ponibles para los investigadores del sitio cuando las soliciten. Pero eso es muy tarde para apoyar la planeación del CDER para las inspecciones y la supervisión post-comercialización, señaló Russell Wesdyk, coordinador científico en la Oficina de Programas Estratégicos del CDER, en la conferencia de la GPhA. Por otro lado, cada compañía tiende a colectar datos a su propio estilo, utilizando un rango de definiciones y planes de muestreo. La FDA anticipa que las métricas estándar re-ducirían las diferencias de cómo definen las empresas “lote” (granel), “lote” (producto terminado), e incluso “rechazo”.Ahora el CDER, en consecuencia, tiene que evaluar el nivel de riesgo de un sitio de manufactura basado principalmente
en su historia de cumplimiento, como se ve en las cartas de advertencia y los reportes de campo, más los registros sobre las recuperaciones de producto del mercado y los problemas de calidad. Pero estas no son las medidas más informativas, dijo Wesdyk. Más datos relevantes, proporcionados por an-ticipado determinarían mejor la necesidad de una visita real al sitio, así como los factores de riesgo que podrían predecir futuros desabastos de fármacos y otros problemas de calidad. La intención es no emitir “grados estilo restaurant,” recalca Wesdyk, sino aportar resultados amplios del desempeño de la industria de manera que las compañías puedan ver dónde están paradas con respecto a sus pares.Para establecer este sistema más informativo de análisis de datos de calidad del fármaco, la FDA se ha embarcado en una amplia iniciativa para definir la métrica “correcta” para colectar y auditar. Los funcionarios de la agencia y los eje-cutivos de la industria involucrados con el manejo de la plan-ta, el control de calidad, y el cumplimiento regulatorio están examinando cómo analizar las diferentes medidas de calidad y cómo debería proporcionarse esta información a los regula-dores para apoyar la evaluación de riesgo de la FDA y ayudar a las empresas a mejorar la calidad. Existe interés en exami-nar cómo la cultura de la compañía puede darle forma a estos esfuerzos, más la preocupación acerca de las consecuencias involuntarias de seleccionar las métricas erróneas. Una com-pañía podría tratar de acelerar el cierre del lote, por ejemplo, si hay más atención a la medición del tiempo del ciclo del lote. La revisión de la tendencia de las quejas podría llevar a las empresas a resolver problemas superficialmente sin ir a las causas raíz.Estas discusiones respaldan los esfuerzos de la FDA para de-sarrollar la guía sobre las métricas de calidad, la cual estaría abierta a comentarios y retroalimentación y debe estimular el desarrollo de más documentos de consenso. Con base en toda esta información de entrada, la FDA espera definir las métri-cas iniciales para finales del 2014, estableciendo la etapa para la colecta de las métricas de calidad de la industria en el 2015.Los incentivos claros para que los fabricantes logren mayor producción de calidad también son parte de la discusión. La industria ha estado decepcionada con las pasadas iniciativas de calidad de la FDA, en parte porque la agencia otorgó poco alivio regulatorio a las empresas que invirtieron en esfuerzos para la mejora de la calidad con frecuencia costosos. La FDA no ha hecho nada para refrenar los requerimientos para regu-lar los cambios post-comercialización o para reducir las ins-pecciones. Ahora los líderes del CDER indican que una infor-mación más completa sobre los establecimientos podría llevar a inspecciones en campo menos frecuentes en las empresas que puedan documentar un logro de calidad del sitio y podrían reducirse los requerimientos de sometimiento complementa-rio para los cambios en la manufactura de bajo riesgo.
Nueva organización de calidadEstas iniciativas estarán soportadas y manejadas por una nue-va Oficina de Calidad de Producto (OPQ) en el CDER, la cual espera Woodcock que entre en operaciones este año bajo su dirección personal. “No es un hecho”, advirtió Woodcock en la reunión con el ISPE, pero los planes están en marcha, y ella anticipa que la nueva organización se “hará real” en los próximos meses.Un objetivo para el OPQ es establecer “una voz para cómo regula la FDA la calidad del fármaco” -para nuevas molécu-las, fármacos genéricos, productos de libre venta, terapias biotecnológicas, y pequeñas moléculas igualmente. Un tema principal es unificar la revisión y la inspección de manera que puedan trabajar juntas para asegurar la producción de cali-dad a través del ciclo de vida de un producto. Una unidad de revisión e inspección, por ejemplo, supervisaría los APIs. Los especialistas para nuevos fármacos pueden agruparse por indicación clínica, mientras que los que monitorean genéri-cos pueden estar organizados de acuerdo a las formas farma-céuticas. Los microbiólogos para todos los productos estarán combinados para proveer un enfoque unificado para la micro-biología.Una Oficina de Supervisión en la OPQ manejará las inspec-ciones basadas en un completo inventario de las instalaciones, más las métricas para evaluar el desempeño e identificar las operaciones de calidad -todo respaldado por sistemas de in-formación extensos. Y una Oficina de Políticas central emitirá la guía y las regulaciones y pugnará por más consistencia en las acciones del CDER.“Tenemos que ser pronosticables y consistentes,” enfatizó Woodcock en la reunión con el ISPE. Ella señaló que es cru-cial para la FDA una regulación más moderna de la manufac-tura para cumplir los nuevos objetivos de la tasa para usuarios, para acelerar el desarrollo de terapias innovadoras y biosimi-lares, para evitar la escasez de fármacos, y para armonizar más los requerimientos regulatorios alrededor del mundo. Al mismo tiempo, la FDA busca darle a la industria flexibilidad en el logro de estándares para la revisión e inspección.Los reguladores no hacen fármacos, señaló Woodcock, pero pueden establecer los postes de la meta, estimulan la mejora y mantienen a la industria responsable. Además de estable-cer las métricas del desempeño que la FDA puede usar para evaluar el riesgo, la agencia quiere alentar la mejora continua por parte de la industria. Los fabricantes, señaló Woodcock, tienen que reconocer que hay un “costo por la pobre calidad.”
References 1. Wechsler J., Pharm. Techn., 37 (10) (2013). 2. FDASIA, available at www.fda.gov.
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO

Pharmaceutical Technology en Español MARZO / ABRIL 201444
SOLUCIONES ESTADÍSTICAS
Calibración de instrumentos:¿Es su espectrómetro de UV lo suficientemente exacto?Chris Burgess
Este artículo le da un vistazo estadístico a los requerimientos de calibración para un espectrómetro de UV.
Todas las calibraciones están sujetas a error. Es esencial asegurar que se empleen ins-trumentos calificados en el
ambiente regulado. Las GMPs tanto en los Estados Unidos como en Europa, requieren control demostrable de los instrumentos y sistemas. En EEUU, por ejemplo, bajo las regulaciones de las cGMP (1):
El Capítulo General <1058> sobre la Calificación de Instrumentos Analí-ticos de la Farmacopea de los Estados Unidos (USP) entró en vigor en Agos-
“Los controles de laboratorio debe-rán incluir: (b) (4) La calibración de instrumentos, aparatos, manómetros y dispositivos de registro en inter-valos adecuados de acuerdo con un programa por escrito establecido que contiene indicaciones especí-ficas, programas, límites para la exactitud y precisión, y provisiones para acciones de solución en el caso de que no se cumplan los límites de exactitud y/o precisión. Los instru-mentos, aparatos, manómetros y dis-positivos de registro que no cumplan con las especificaciones estableci-das no deberán usarse.”
Chris Burgess, PhD, es científico analítico enBurgess AnalyticalConsultancy Limited,‘Rhose Rae’, The Lendings, Startforth, Barnard Castle, Co. Durham, DL12 9AB, RU; Tel: +44 1833 637 446;[email protected];www.burgessconsultancy.com
Media observada
0.4660 95%UCL de la
media
A=0.0042
0.455 0.460 0.465 0.470 0.475 0.480
Valor del CRM 0.4702
A=0.0017
to de 2008 (2). La calificación de ins-trumentos analíticos (AIQ) describe el marco de trabajo y las actividades gene-rales necesarias para asegurar la aptitud de un instrumento analítico para el uso que se pretende.
La calibración ha sido definida como “la serie de operaciones que esta-blecen, bajo condiciones específicas, la relación entre los valores indicados por
un instrumento de medición o un siste-ma de medición, o los valores represen-tados por una medida de material, y los correspondientes valores conocidos de un estándar de referencia” (3). La cues-tión, por lo tanto, es qué tan buena debe ser la concordancia entre la medición en un espectrómetro con la certificada para el estándar de referencia. Esto, por supuesto, podría ser un simple enun-
ciado de una diferencia absoluta o una diferencia en porcentaje. Este artículo le da un vistazo desde una perspectiva estadística.
Se ha comprado un Material de Refe-rencia Certificado (CRM) para la absor-bancia, el cual tiene rastreabilidad hasta un Laboratorio Nacional de Medición (p.ej., el Instituto Nacional de Ciencia y Tecnología [NIST]) en EEUU). El error de la escala de longitud de onda del espectrómetro es lo suficientemente pequeño para no cau-sar algún error de absorbancia significativo en la longitud de onda de interés. Utilizando los procedimientos de operación recomendados, se realizan seis mediciones independientes del CRM en la longitud de onda de interés y se obtienen los siguientes datos de absorbancia: 0.464, 0.468, 0.463, 0.465, 0.467 y 0.469.
De estos seis datos de medición, se deriva un valor medio de 0.4660. En el certificado del CRM, el valor certificado está dado como 0.4702. Existe por lo tanto una diferencia entre el valor medio medido y el valor certificado de 0.0042 unidades de absorbancia según se ilustra en la ¿Es esta diferencia aceptable? ¿Es la diferencia significativa-mente diferente estadísticamente o analíticamente? Viendo la diferencia desde una perspectiva estadística, observando que el intervalo de confianza al 95%, , llamado la incerti-dumbre expandida en ISO, puede calcularse el límite supe-rior de confianza al 95% de la media, a partir de la desviación estándar estimada, s, la cual es 0.0024. El resultado se muestra gráficamente en la
Podemos ver ahora que en la confianza de 95% existe una diferencia de sólo 0.0017 unidades de absorbancia entre el UCL y el valor certificado. ¿Es esta diferencia lo suficiente-mente pequeña para que uno pueda considerar la escala de absorbancia suficientemente exacta?
El CRM ha sido certificado con los estándares ISO (4) y, por lo tanto, provee una certidumbre expandida para el valor certificado. En este ejemplo, la incertidumbre expandida, , es 0.0034, la cual corresponde con el 95.45% de confianza ya que el factor de cobertura, , es 2 (5). La desviación estándar del CRM, por lo tanto, es la mitad de este valor y es 0.0017. La puede ser ahora modificada para mostrar las dis-tribuciones calculadas para las mediciones y la CRM en la
puede verse que la media observada no está dentro de la incertidumbre expandida del CRM. La probabi-lidad acumulada de valores puede ser calculada hasta e inclu-yendo el valor medio en la distribución del CRM utilizando, por ejemplo, MS Excel, con la
Probabilidad= NORM.DIST(0.4660, 0.4702, 0.0017, TRUE)*100 = 0.67
Media observada
0.4660
A=0.0042
0.455 0.460 0.465 0.470 0.475 0.480
Valor del CRM 0.4702
Figura 1: Diferencia entre la media de las mediciones y el valor del material de referen-cia certificado (CRM).
Figura 2: Inclusión del límite de confianza superior al 9% (UCL) de la media observada.
PHO
TOD
ISC
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 2014 45
0.455 0.460 0.465
U=0.0034
0.470 0.475 0.480
A=0.0042
Media observada
0.4660
Valor del CRM 0.4702
un instrumento de medición o un siste-ma de medición, o los valores represen-tados por una medida de material, y los correspondientes valores conocidos de un estándar de referencia” (3). La cues-tión, por lo tanto, es qué tan buena debe ser la concordancia entre la medición en un espectrómetro con la certificada para el estándar de referencia. Esto, por supuesto, podría ser un simple enun-
ciado de una diferencia absoluta o una diferencia en porcentaje. Este artículo le da un vistazo desde una perspectiva estadística.
Calibración de la escala de ab-sorbancia de un espectrómetro de UVSe ha comprado un Material de Refe-rencia Certificado (CRM) para la absor-bancia, el cual tiene rastreabilidad hasta un Laboratorio Nacional de Medición (p.ej., el Instituto Nacional de Ciencia y Tecnología [NIST]) en EEUU). El error de la escala de longitud de onda del espectrómetro es lo suficientemente pequeño para no cau-sar algún error de absorbancia significativo en la longitud de onda de interés. Utilizando los procedimientos de operación recomendados, se realizan seis mediciones independientes del CRM en la longitud de onda de interés y se obtienen los siguientes datos de absorbancia: 0.464, 0.468, 0.463, 0.465, 0.467 y 0.469.
De estos seis datos de medición, se deriva un valor medio de 0.4660. En el certificado del CRM, el valor certificado está dado como 0.4702. Existe por lo tanto una diferencia entre el valor medio medido y el valor certificado de 0.0042 unidades de absorbancia según se ilustra en la Figura 1.¿Es esta diferencia aceptable? ¿Es la diferencia significativa-mente diferente estadísticamente o analíticamente? Viendo la diferencia desde una perspectiva estadística, observando que el intervalo de confianza al 95%, , llamado la incerti-dumbre expandida en ISO, puede calcularse el límite supe-rior de confianza al 95% de la media, a partir de la desviación estándar estimada, s, la cual es 0.0024. El resultado se muestra gráficamente en la Figura 2.
Podemos ver ahora que en la confianza de 95% existe una diferencia de sólo 0.0017 unidades de absorbancia entre el UCL y el valor certificado. ¿Es esta diferencia lo suficiente-mente pequeña para que uno pueda considerar la escala de absorbancia suficientemente exacta?
El CRM ha sido certificado con los estándares ISO (4) y, por lo tanto, provee una certidumbre expandida para el valor certificado. En este ejemplo, la incertidumbre expandida, U, es 0.0034, la cual corresponde con el 95.45% de confianza ya que el factor de cobertura, k, es 2 (5). La desviación estándar del CRM, por lo tanto, es la mitad de este valor y es 0.0017. La Figura 1 puede ser ahora modificada para mostrar las dis-tribuciones calculadas para las mediciones y la CRM en la Figura 3.
En la Figura 3 puede verse que la media observada no está dentro de la incertidumbre expandida del CRM. La probabi-lidad acumulada de valores puede ser calculada hasta e inclu-yendo el valor medio en la distribución del CRM utilizando, por ejemplo, MS Excel, con la Ecuación 1:
Probabilidad= NORM.DIST(0.4660, 0.4702, 0.0017, TRUE)*100 = 0.67
(Nota: Esta ecuación es del Excel en Office 2010 y 2013 donde las principales funciones estadísticas han sido actuali-zadas. NORMDIST en las versiones anteriores también tra-bajará).
Puede verse que hay solo menos de 1% de probabilidad de que los valores hasta la media observada caigan dentro de la distribución del valor certificado y también la media cae fuera de la incertidumbre expandida del CRM. ¿Esto llevaría a la conclusión de que nuestro espectrómetro es inexacto?
La respuesta es no, porque el CRM fue medido en el mis-mo espectrómetro y, por lo tanto, necesita calcularse la incerti-dumbre combinada tomando en cuenta ambas fuentes de error, el espectrómetro y el CRM, en la incertidumbre global, la cual se calcula con la Ecuación 2:
Para un factor de cobertura de k = 2, la incertidumbre expan-dida global de nuestro CRM medido es 0.0058.La Figura 3 puede ser ahora modificada para mostrar el efecto del cálculo de incertidumbre combinada en la Figura 4. Obser-ve que la distribución de la incertidumbre combinada es más amplia que la distribución medida o la del CRM.Parece ser ahora con la Figura 4 que la media observada cae no sólo dentro de la incertidumbre expandida del CRM me-dido sino también, si se repite el cálculo de probabilidad del Excel inicial utilizando el valor modificado para la desviación estándar, u
c
Probabilidad = NORM.DIST(0.4660, 0.4702, 0.00291, TRUE)*100 = 7.45% [Ec. 3]
Existe ahora un 7.5% aproximadamente de oportunidad de que la probabilidad acumulativa de valores hasta, e incluyen-do el valor medio observado, caiga dentro de la distribución modificada del valor certificado del CRM. Puede, por lo tanto, concluirse que el espectrómetro es lo suficientemente exacto.
t(0.05,n-1)s√n
X+t(0.05,n-1) = 0.4685s√n
√sMeasure + scrm
√(0.0024)2 + (0.0017)2
uc =
=
= 0.00291
2 2
[Ec. 1]
[Ec. 2]
Figura 3:Modificación de la Figura 1 para mostrar las distribuciones de las observa-ciones y del CRM.
PHO
TOD
ISC
/GE
TTY
IMAG
ES

Pharmaceutical Technology en Español MARZO / ABRIL 201446
0.455 0.460 0.465
U=0.0058
0.470 0.475 0.480
A=0.0042
Media observada
0.4660
Valor del CRM 0.4702
Referencias 1. CFR 21 Part 211.160, General require-
ments 2. USP, USP 36–NF 31, General Chapter
<1058> Analytical Instrument Quali-fication (United States Pharmacopeial Convention, Rockville, Maryland, 2012).
3. Glossary to European Union GMPs; http://ec.europa.eu/health/documents/eudralex/vol-4/, accessed Dec. 6, 2013.
4. C. Burgess, Pharm. Techn., 37 (9), 62-64, 73 (2013).
5. S.L.R Ellison and A. Williams (Eds). Eurachem/CITAC guide: Quantifying Uncertainty in Analytical Measure-ment, (3rd ed., 2012), www.eurachem.org/index.php/publications/guides/quam, accessed Dec. 6, 2013.PT
P yR con
PharmTech: ¿Cuáles son los cambios recientes o pendientes para los requerimientos compendiales y/o regulatorios con respecto a la inspección visual de parenterales o relacionados con el análisis de partículas?Galliher: Los cambios pendientes incluyen un nuevo capítulo en la Farmacopea de los EEUU, USP Capítulo General <790>, el cual provee guía con respecto a las expectativas para el entorno de inspección visual manual. La guía da las expectativas para la in-tensidad de la luz, la construcción del gabinete, el ritmo de ins-pección y la manipulación del contenedor. La guía se alineará con las expectativas de la Farmacopea Europea y de la Organiza-ción Mundial de la Salud.
PharmTech: ¿Cuáles son las ventajas/desventajas relativas de la inspección automatizada contra la manual?Galliher: Las ventajas de la inspección automatizada incluyen mayor repetibilidad y reproducibilidad. Las máquinas para la inspección automatizada inspeccionarán consistentemente el producto de acuerdo a los criterios para los que han sido pro-gramadas. También es una ventaja el mayor rendimiento para el proceso de inspección automatizada. Para productos que tienen grandes tamaños de lote, y requerimientos de volumen anual grande, la inspección automatizada puede ser necesaria para soportar la demanda. La retroalimentación en tiempo real del proceso es otra ventaja de la inspección automatizada. Los fa-bricantes del equipo original ofrecen paquetes de computadora que pueden establecer límites de rechazo y datos de tendencia en tiempo real, lo cual puede ser una ventaja, especialmente si la máquina de inspección automatizada opera en línea con la llenadora.
Las desventajas de la inspección automatizada incluyen cos-tos más elevados. La inversión inicial en las capacidades de la inspección automatizada puede ser muy costosa. Adicionalmen-te, existen costos asociados con la calificación. Una calificación
Brian Galliher, Gerente, Manufactura Secundaria en Cook Pharmicarobusta puede requerir una cantidad signi-ficativa de componentes y producto. Otra desventaja de la inspección automatizada es que las máquinas sólo inspeccionarán el producto de acuerdo a los criterios para los que están programadas. Dependiendo de las brechas en el desarrollo de la receta inicial o de las limitaciones conocidas de la máquina, existe la posibilidad de un rechazo falso elevado o la aceptación de un defecto. Finalmente, debido a la alta inversión y a los costos de la cali-ficación, la inspección automatizada puede no ser para todos los productos.
Una ventaja de la inspección manual es el hecho de que los inspectores pueden aplicar su juicio para el proceso de toma de decisiones. Los inspectores manuales pueden de-tenerse e involucrar al departamento de calidad cuando se observan defectos nuevos o cuestionables. El costo puede ser otra ventaja de la inspección manual. El costo de establecer un proceso de inspección manual y capacitar a los operadores es significativamente menor que los costos asociados con la inspección automatizada.
El rendimiento es una desventaja de la inspección manual. El rendimiento está limitado al ritmo establecido y al número de inspectores calificados. El proceso de inspección manual podría requerir una plantilla de trabajadores significativa y una gran cantidad de espacio. Otra desventaja del proceso de inspección manual es el impacto humano en el proceso de inspección. Factores tales como la fatiga del inspector y el estado de ánimo, pueden impactar el desempeño de los ins-pectores manuales. Por último, las consideraciones ergonómi-cas son una desventaja de la inspección manual. El proceso es repetitivo por diseño y plantea riesgos ergonómicos si no está apropiadamente preparado.
PHARMA CÁPSULAS
SOLUCIONES ESTADÍSTICAS EDITORIAL INVI
Richard Johnson
La conferencia regulatoria PDA/FDA promueve un compromiso con la calidad.
La Asociación de Fármacos Pa-renterales (PDA) se centra en conectar a la gente, la ciencia y la regulación. Durante más
de 60 años, los miembros de la PDA han estado activos para responder y conducir la implementación de iniciativas regula-torias globales. Los miembros de la aso-ciación no descubrieron recientemente lo que la en cGMPs significa; ellos han estado definiéndola y dándole for-ma activamente durante muchos años. En ninguna parte este compromiso y energía ha estado más evidente que en la Conferencia Regulatoria Conjunta de PDA/FDA, la cual se llevará a cabo para el vigésimo tercer aniversario del 16 al 18 de Septiembre en Washington, D.C.
Un objetivo identificado en el Plan Estratégico de la FDA -Regulación- es-tablece: “Nuestras actividades regula-torias están científica y técnicamente enfocadas, y la información vigente se comunica a nuestros miembros.”
La organización apoya esta estrate-gia con varios criterios, que incluyen:•
• El mantenimiento de relaciones va-liosas y efectivas con los reguladores globales y la educación de los miem-bros sobre las expectativas actuales.
Durante las últimas seis décadas, los miembros de la PDA han hecho comen-
Figura 4:Efecto del cálculo de incertidumbre combinado en la distribución del valor CRM.

Pharmaceutical Technology en Español MARZO / ABRIL 2014 47
EDITORIAL INVITADO
JOR
G G
RE
UE
L/P
HO
TOD
ISC
/GE
TT
Y IM
AG
ES
Conectando a la gente, la ciencia y la regulaciónRichard Johnson
La conferencia regulatoria PDA/FDA promueve un compromiso con la calidad.
Richard Johnson es presidente de la Asociación de FármacosParenterales (PDA).
La Asociación de Fármacos Pa-renterales (PDA) se centra en conectar a la gente, la ciencia y la regulación. Durante más
de 60 años, los miembros de la PDA han estado activos para responder y conducir la implementación de iniciativas regula-torias globales. Los miembros de la aso-ciación no descubrieron recientemente lo que la “c” en cGMPs significa; ellos han estado definiéndola y dándole for-ma activamente durante muchos años. En ninguna parte este compromiso y energía ha estado más evidente que en la Conferencia Regulatoria Conjunta de PDA/FDA, la cual se llevará a cabo para el vigésimo tercer aniversario del 16 al 18 de Septiembre en Washington, D.C.
Un objetivo identificado en el Plan Estratégico de la FDA -Regulación- es-tablece: “Nuestras actividades regula-torias están científica y técnicamente enfocadas, y la información vigente se comunica a nuestros miembros.”
La organización apoya esta estrate-gia con varios criterios, que incluyen:• La provisión de las entradas basadas
en ciencia y tecnología de las regu-laciones y guías relacionadas con las áreas estratégicas de la PDA, utilizan-do la base de voluntarios y miembros de la PDA.
• El mantenimiento de relaciones va-liosas y efectivas con los reguladores globales y la educación de los miem-bros sobre las expectativas actuales.
Durante las últimas seis décadas, los miembros de la PDA han hecho comen-
tarios sobre cada regulación y guía clave de las cGMPs. La PDA ha hospedado las conferencias conjuntas con regula-dores, dando capacitación especializa-da a las autoridades regulatorias, y ha incluido a los funcionarios regulatorios en sus procesos para el desarrollo de re-portes técnicos. Esta actividad llevó a la creación de la Conferencia Regulatoria Conjunta PDA/FDA desde hace casi 25 años.
No le tomó mucho tiempo a esta conferencia volverse uno de los eventos anuales más importantes albergados por la PDA para sus miembros. La asisten-cia global a la reunión y el número de asistentes y oradores que representan no sólo a la FDA, sino a los cuerpos regulatorios de todo el mundo ha ido creciendo cada año.
La Conferencia Regulatoria Con-junta PDA/FDA del 2013: Conducien-do la Calidad y el Cumplimiento a lo largo del Ciclo de Vida del Producto en un Ambiente Regulatorio Global, abre con una empresa de importancia. Janet Woodcock, MD, directora del Centro para la Evaluación e Investigación de Fármacos (CDER) de la FDA dará la perspectiva regulatoria y Daniel Kraft, MD, director ejecutivo de FutureMed, dará el punto de vista de la industria so-bre la situación de los sistemas de cali-dad, la calidad por diseño, y escasez de fármacos. La conferencia concluirá con información acerca de las iniciativas de la FDA por parte de los representantes del CDER, el Centro para la Evaluación e Investigación de Biológicos (CBER), el Centro para Dispositivos y Salud Radiológica (CDRH) y la Oficina de Asuntos Regulatorios (ORA).
La conferencia ofrece cuatro sesio-nes plenarias:
• La sesión de Cultura de Calidad y Socios explora la manera de imple-mentar la cultura de calidad, cómo impacta la cultura la eficiencia de un sistema de calidad y destaca los indi-cadores significativos de una cultura de calidad débil.
• En la sesión de Entendiendo las Bue-nas Prácticas de Manufactura, los oradores discutirán como se asegu-ra la seguridad y eficacia mediante el apego rutinario a las prácticas de aseguramiento de la calidad y control de la manufactura contenidas en las GMPs.
• Una Perspectiva del Paciente está diseñada para ofrecer conocimien-to profundo de un paciente -alguien que puede ayudar a los profesionales del desarrollo de fármacos en su mi-sión, expresando cómo los productos farmacéuticos han ayudado a salvar vidas.
• En la sesión de Actualización del Cumplimiento, los directores de cumplimiento en cada centro de la FDA informarán sobre los tópicos ac-tuales en el campo del cumplimiento.
Después de la conferencia en Sep-tiembre 18-19, hay una sesión que no querrá perderse: el Taller de Mejora de Investigaciones 2013 de la PDA/FDA. Los especialistas y expertos de la in-dustria discuten las causas raíz de las investigaciones pobres. Como Ud. debe saber, éstas están entre las principales observaciones en la inspección global-mente. Finalmente, extender su apren-dizaje en uno de seis cursos ofrecidos por el Instituto de Capacitación e Inves-tigación de la PDA.
Estas actividades ejemplifican el in-cansable enfoque sobre “Conectando a la Gente, la Ciencia y la Regulación.” PT

Pharmaceutical Technology en Español MARZO / ABRIL 201448
VIGILANCIA REGULATORIA EEUU
La regulación de opioides desafía a la FDA y a los fabricantesLas nuevas políticas y productos buscan mantener el acceso a los analgésicos mientras refrenan el abuso creciente.
Jill Wechsler es editora en Washington de Pharmaceutical Technology,
tel. 301.656.4634, [email protected]. Lea los blogs de Jill en
PharmTech.com/wechsler.
El mal uso cada vez mayor de los opioides de acción prolongada ha impulsado los esfuerzos para formu-lar terapias más seguras para pacientes que sufren de dolor mientras también se dan los pasos para
limitar la prescripción inapropiada y el uso ilegal. La FDA recientemente acordó hacer los controles más estrictos en las terapias combinadas con hidrocodona, ampliamente usadas (1). La agencia al principio pidió cambios en el etiquetado de los opioides de acción prolongada para aumentar la concien-cia de los riesgos y para limitar la prescripción (2).
La FDA también está estimulando el desarrollo de formu-laciones de opioides resistentes al abuso, como se contempla en la guía emitida al inicio de este año (2013) sobre el análisis de dichos productos. Sin embargo, las dificultades para llevar tratamientos más seguros al mercado, también impulsó a la FDA a aprobar un nuevo fármaco de hidrocodona pura para el alivio prolongado del dolor crónico, a pesar de su poten-cial para el abuso. Los funcionarios de la agencia explicaron que ellos no podían bloquear todos los nuevos analgésicos del mercado mientras esperaban por más desarrollos de tecnolo-gías limitantes para el abuso. Estos hechos ilustran el acto de malabarismos continuos que requieren las autoridades regu-ladoras y los fabricantes para abordar las serias consecuencias del uso ilegal de opioides mientras le proporcionan el trata-miento necesario para los que prescriben y para los pacientes.
Controles más estrictosLa presión sobre la FDA para que haga más para limitar el abuso con la prescripción de fármacos fue aparente en su decisión para respaldar controles más estrictos mediante la Agencia de Control de Drogas (DEA) para tratamientos con opioides que combinan la hidrocodona con analgésicos de libre venta tales como ibuprofeno o acetaminofén. Estos analgésicos ampliamente usados, que incluyen al Vicodin de AbbVie y al Lortab de UCB, se han beneficiado durante años por estar listados en los controles del Esquema III de la DEA. Los fármacos en esta categoría son más fáciles para ser pres-critos por los médicos, para que los pacientes los adquieran y para que los farmacéuticos los almacenen y dispensen. El estatus de Esquema III fue respaldado por la FDA con base en su conocimiento de que los productos combinados no son tan peligrosos como los opioides con la potencia completa.
Durante la última década, la DEA ha visto cambiar el esquema de estos fármacos combinados a la categoría más restrictiva como evidencia de que los abusadores podían ma-nipular los productos de liberación prolongada para lograr “subidas” inmediatas. En 2008, la FDA rechazó la primera solicitud de la DEA para este cambio, en parte como respues-
ta a fuertes objeciones de los farmacéuticos, los dentistas y los especialistas en cáncer y dolor así como de los fabricantes. Pero a medida que la comunidad médica y las autoridades de salud pública continuaban su campaña para restricciones más fuertes, la DEA revivió su solicitud en 2009 para que la FDA respaldara el cambio del esquema para estos fármacos, aportando evidencia adicional sobre los peligros de estos me-dicamentos para el dolor.
El Senador Joe Manchin (D-W. Va.) presionó para incluir el cambio de esquema propuesto para los opioides combina-dos en el Acta de Seguridad e Innovación de la FDA (FDA-SIA). Él se echó para atrás debido a la oposición de políti-cos y grupos de interés, pero ganó la promesa de que la FDA re-examinaría el problema más concienzudamente. La FDA realizó una reunión pública sobre el uso de opioides en Enero de 2013, donde la creciente evidencia del daño causado por el abuso y mal uso convenció a un panel de expertos para apoyar a los controles más estrictos a estos medicamentos.
Janet Woodcock, directora del Centro para Evaluación e Investigación de Fármacos (CDER), anunció en Octubre de 2013 que para finales del año la agencia le enviaría a la DEA una recomendación formal para mover los productos combi-nados con hidrocodona del Esquema III al Esquema II, un proceso que debería completarse el próximo año (1). El cam-bio restringirá las prescripciones a 90 días de los 180 días y requerirá que los pacientes lleven una prescripción a la far-macia, en lugar de llamar al médico. Al igual que con otros fármacos del Esquema II, como la oxicodona, la morfina y el fentanilo, el objetivo es hacerle más difícil a los pacientes ob-tener múltiples surtidos en un corto período de tiempo y hacer más estrictos los controles del almacenamiento y el manejo de estos fármacos por parte de los farmacéuticos.
Buscando más opcionesAl mismo tiempo, el deseo de asegurarle el acceso a los pa-cientes a terapias eficientes para el dolor estimuló una sor-presiva decisión de la FDA a aprobar un nuevo medicamen-to opioide de acción prolongada y alta potencia sin ninguna característica anti-abuso (3). Zogenix con sede en San Die-go había estado buscando la aprobación para el mercado de Zohydro ER (hidrocodona bitartrato de liberación extendida) durante varios años, pero se encontró con objeciones rela-cionadas con la seguridad del producto. Un comité consul-tor votó en Diciembre del 2012 contra la aprobación sobre la base de que el fármaco es peligroso y carece de características disuasivas del abuso. Los funcionarios de la agencia, no obs-tante, pasaron por encima de la decisión del comité.
El nuevo fármaco cae inmediatamente dentro del Esque-
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO

Pharmaceutical Technology en Español MARZO / ABRIL 2014 49
Pharmaceutical Technology,
tel. 301.656.4634, [email protected]. Lea los blogs de Jill en
PharmTech.com/wechsler.
ta a fuertes objeciones de los farmacéuticos, los dentistas y los especialistas en cáncer y dolor así como de los fabricantes. Pero a medida que la comunidad médica y las autoridades de salud pública continuaban su campaña para restricciones más fuertes, la DEA revivió su solicitud en 2009 para que la FDA respaldara el cambio del esquema para estos fármacos, aportando evidencia adicional sobre los peligros de estos me-dicamentos para el dolor.
El Senador Joe Manchin (D-W. Va.) presionó para incluir el cambio de esquema propuesto para los opioides combina-dos en el Acta de Seguridad e Innovación de la FDA (FDA-SIA). Él se echó para atrás debido a la oposición de políti-cos y grupos de interés, pero ganó la promesa de que la FDA re-examinaría el problema más concienzudamente. La FDA realizó una reunión pública sobre el uso de opioides en Enero de 2013, donde la creciente evidencia del daño causado por el abuso y mal uso convenció a un panel de expertos para apoyar a los controles más estrictos a estos medicamentos.
Janet Woodcock, directora del Centro para Evaluación e Investigación de Fármacos (CDER), anunció en Octubre de 2013 que para finales del año la agencia le enviaría a la DEA una recomendación formal para mover los productos combi-nados con hidrocodona del Esquema III al Esquema II, un proceso que debería completarse el próximo año (1). El cam-bio restringirá las prescripciones a 90 días de los 180 días y requerirá que los pacientes lleven una prescripción a la far-macia, en lugar de llamar al médico. Al igual que con otros fármacos del Esquema II, como la oxicodona, la morfina y el fentanilo, el objetivo es hacerle más difícil a los pacientes ob-tener múltiples surtidos en un corto período de tiempo y hacer más estrictos los controles del almacenamiento y el manejo de estos fármacos por parte de los farmacéuticos.
Al mismo tiempo, el deseo de asegurarle el acceso a los pa-cientes a terapias eficientes para el dolor estimuló una sor-presiva decisión de la FDA a aprobar un nuevo medicamen-to opioide de acción prolongada y alta potencia sin ninguna característica anti-abuso (3). Zogenix con sede en San Die-go había estado buscando la aprobación para el mercado de Zohydro ER (hidrocodona bitartrato de liberación extendida) durante varios años, pero se encontró con objeciones rela-cionadas con la seguridad del producto. Un comité consul-tor votó en Diciembre del 2012 contra la aprobación sobre la base de que el fármaco es peligroso y carece de características disuasivas del abuso. Los funcionarios de la agencia, no obs-tante, pasaron por encima de la decisión del comité.
El nuevo fármaco cae inmediatamente dentro del Esque-
ma II de la DEA, lo cual significa que puede ser comercializa-do inmediatamente y evitar la frecuente larga espera para que la DEA determine los controles apropiados. Pero la prescrip-ción estará restringida, y Zohydro estará sujeto a la Estrategia de Evaluación y Mitigación del Riesgo (REMS), adoptada en 2012 para todos los opioides de acción prolongada. Adicio-nalmente, Zohydro tiene que realizar el nuevo etiquetado que la FDA anunció en Septiembre de 2013 para todos los fár-macos opioides de liberación extendida y acción prolongada tales como el OxyContin (oxicodona) de Purdue Pharma y el Opana ER (oximorfona) de Endo (2). El nuevo etiquetado les recomienda a los médicos prescribir estos medicamentos para tratar sólo dolor muy grave, no moderado, como una manera de limitar el uso de estos peligrosos fármacos.
Aunque la FDA reveló la actualización del etiquetado con fanfarrias, la nueva política probablemente no tenga un gran impacto sobre el abuso de opioides. Parece improbable que los sellos de advertencias y la información de riesgos adicio-nal le ponga trabas a la prescripción inapropiada o que disua-da a los adictos de infringir la ley. Por otro lado, los estudios adicionales de post-comercialización requeridos por la FDA son complicados y costosos y pueden ser difíciles de realizar por los fabricantes.
La aprobación de la FDA del Zohydro fue sorpresiva por-que el producto carece de características diseñadas para im-pedir el abuso. La FDA publicó el proyecto de guía en Enero de 2013 recomendando a los fabricantes sobre las estrategias para examinar fármacos con características anti-abuso (4). En Abril de 2013, la FDA aprobó nuevo etiquetado que permi-te la promoción de características disuasivas del abuso del OxyContin reformulado de Purdue. La evidencia de que el producto puede impedir el abuso, además, impulsó a la FDA a declarar que no aprobaría ninguna versión genérica de la formulación original del OxyContin, que ya no es producido por Purdue Pharma (5).
Las complejidades del estatus regulatorio de estos medi-camentos para el dolor, no obstante, se pueden ver en la deci-sión contraria de la FDA un mes después, cuando decidió no otorgar esa misma clase de protección de la competencia de genéricos para el Opana ER reformulado de Endo Pharma-
ceutical. La FDA concluyó que a pesar de los cambios de formulación, el fármaco seguía siendo vulnerable al abuso y podía ser preparado para inyección e inhalación.
Los enfoques para el desarrollo y estudios de caracte-rísticas resistentes al abuso fueron además discutidos en una reunión científica en Sept. 30 - Oct. 1, 2013 organizada por el Consorcio de Responsabilidad para el Abuso entre Empresas y el Colegio sobre Problemas de Dependencia de Fármacos. Los científicos de la industria, la academia y la FDA examinaron los esquemas de estudio pre-clínico y post-comercialización para demostrar el potencial para la prevención del abuso. La FDA quiere particularmente esti-mular el desarrollo de fármacos genéricos con formulacio-nes anti-abuso para facilitar el acceso del paciente a terapias más seguras y menos costosas.
Los expertos en la reunión reconocieron que la tecnolo-gía que respalda la disuasión para el abuso todavía es nueva y no probada y que se necesita mucho trabajo sobre los mé-todos para analizar los datos clínicos sobre el abuso y el im-pacto de las nuevas formulaciones sobre las tasas de abuso. Los funcionarios de la FDA y las compañías farmacéuticas continuarán buscando más comprensión sobre los tipos de esquemas que pueden aportar una significativa disuasión para el abuso como parte de su esfuerzo para ofrecerle a los pacientes terapias para el dolor efectivas y más seguras.
Referencias1. FDA, “Statement on Proposed Hydrocodone Reclassification
from Janet Woodcock, M.D., Director, Center for Drug Evalu-ation and Research,” Oct. 24, 2013.
2. FDA, “FDA Announces Safety Labeling Changes And Post-market Study Requirements For Extended-Release And Long-Acting Opioid Analgesics,” press release (Sept. 10, 2013)
3. FDA, “FDA Approves Extended-Release, Single-Entity Hydro-codone Product,” Press Release (Oct. 25, 2013).
4. FDA, Draft Guidance, Abuse-Deterrent Opioids—Evaluation and Labeling (Silver Spring, MD, January 2013).
5. FDA, “FDA Approves Abuse-Deterrent Labeling For Refor-mulated OxyContin,” press release, (Apr. 16, 2013). PT
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486E-mail: [email protected]
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO

Pharmaceutical Technology en Español MARZO / ABRIL 201450
análisis para biológicos
El análisis por RMN proporciona infor-mación estructural crucial de glicanos sintetizados, mientras que la LC-MS/MS es ideal para la cuantificación de azú-cares libres en matrices biológicas.
El papel de la RMN y la Espectroscopía de Masas en el Análisis de GlicanosCynthia A. Challener, PhD
En los sistemas biológicos, la mayoría de los péptidos y proteínas están glicosilados. Las diferencias en la glico-
silación pueden afectar la estructura de la proteína, la estabilidad, la inmu-nogenicidad, la vida media in vivo, y la función. Los glicanos, de hecho, son importantes para la función celu-lar normal, mediando las interacciones dentro de las células y entre las células y los patógenos, haciendo posibles las respuestas inmunes e influyendo en el avance del tumor. “En los mamíferos, los azúcares se encuentran más común-mente como glicoconjugados, siendo los más abundantes las glicoproteínas, los proteoglicanos y los glicolípidos. Estas importantes sustancias biológicas están localizadas predominante-mente en las membranas celulares, aunque
también se encuentran en fluidos secre-tados en donde modulan o median una multitud de eventos en las interacciones célula-célula y célula-matriz,” señala Brady Clark, fundador y CEO de Sus-sex Research Laboratories, un CRO Canadiense enfocado en la síntesis de glicanos y glicoconjugados, incluyendo glicanos en vinculadores/espaciadores funcionalizados, glicoamino ácidos, glicopéptidos, glico-lípidos y glicanos en proteínas acarreadoras. Adicional-mente, Sussex Research está también activo en la determinación de azúcares reductores libres (o no conjugados) en muestras biológicas.
La glicosilación de fármacos bioló-gicos afecta su mecanismo de acción, la farmacocinética y la eficacia. La glicoconjugación de un bioterapéutico (péptido, proteína o anti-cuerpo) puede,
por lo tanto, conferir una mayor efica-cia terapéutica incrementando la estabi-lidad, la solubilidad acuosa y/o la bio-disponibilidad; mejorando la resolución para el blanco; y/o extendiendo la vida media in vivo. “Los glicanos están in-volucrados en casi todos los aspectos de la bioquímica de la vida, y las proteínas y péptidos en el cuerpo no pueden ser evaluadas sin ver los glicanos,” decla-ra Garnet McRae, director de servicios bioanalíticos con Sussex Research La-boratories en una entrevista con Phar-maceutical Technology. “Recientemen-te, la gente ha empezado a darse cuenta de la importancia de la glicosilación y por lo tanto, ha habido más interés en la síntesis y análisis de glicanos,” agrega.
Clark, McRae y Dennis Whitfield, quienes pertenecen al consejo consultor científico de Sussex Research, hablaron con Pharmaceutical Technology acerca de los métodos analíticos utilizados en Sussex Research para la determinación estructural de los glicanos durante los complejos procesos de síntesis y la eva-luación cuantitativa de los azúcares li-bres involucrados en los procesos bio-lógicos.
Determinación estructuralPharmTech: ¿Por qué es la determina-ción estructural tan importante durante la síntesis de glicanos?
Clark (Sussex): Es tan importante como para poder caracterizar las es-tructuras reales que creamos como lo es para conocer cómo sintetizarlas. A diferencia de los péptidos, los cuales son estructuras lineales compuestas de aminoácidos con apenas dos sitios reac-tivos, los glicanos están comprendidos de monosacáridos con hasta cinco gru-pos hidroxilo por molécula que pueden ser reactivos. Pueden, por lo tanto, ser lineales o muy ramificados, contener sitios múltiples de glicosilación y estar unidos de manera ya sea alfa o beta. Es imperativo conocer exactamente cómo se ha unido cada sacárido, lo que sig-nifica poder confirmar que la reacción ocurrió en el grupo hidroxilo correcto y con la estequiometría correcta.
Nosotros utilizamos métodos quí-micos, enzimáticos y quimioenzimáti-cos altamente efectivos y, en general, bien conocidos, para la síntesis de gli-canos complejos que con frecuencia
Para algunas de las estructuras más complejas de los glicanos que sintetizamos, no sería posible determinar dichas estructuras sin las técnicas 2D de RMN. -

Pharmaceutical Technology en Español MARZO / ABRIL 2014 51
son candidatos a fármacos. Estos mé-todos también pueden ser usados como estándares en las compañías biotecno-lógicas y biofarmacéuticas así como en laboratorios de investigación. Aunque tenemos una extensa experiencia en la síntesis de glicanos y generalmente obtenemos el producto deseado, ningún proceso es 100% a toda prueba. Por lo tanto, confirmamos ampliamente la es-tructura de cada intermedio a lo largo de una síntesis.
PharmTech: ¿Cuáles son los princi-pales instrumentos analíticos que utili-zan para las determinaciones estructu-rales de los glicanos y por qué?
Clark (Sussex): Utilizamos métodos tales como la espectroscopía de reso-nancia magnética nuclear (RMN) con protones de alta resolución y carbón 13 y espectrometría de masas, incluyendo tanto ionización por electroatomización y desorción láser con matriz asistida/io-nización con tiempo de vuelo (MALDI-TOF).
El análisis de RMN es la clave para analizar la estructura de glicanos com-plejos. Evaluando los cambios quími-cos y los detalles del acoplamiento, es posible confirmar el sitio de unión y la estereoquímica de la vinculación del sa-cárido, los cuales son los dos factores cruciales en la síntesis de glicanos. Con las avanzadas técnicas bidimensionales (2D) de RMN hoy día, es posible des-cifrar las estructuras de glicanos muy complejos.
Como la espectrometría de masas no provee información estructural real, ésta es principalmente usada para con-firmar que hemos sintetizado el com-puesto esperado ya que usamos méto-dos bien establecidos para preparar los glicanos.
Otros métodos que fueron previa-mente usados comúnmente para la de-terminación de la estructura, tales como la rotación óptica y el análisis elemen-
tal, ahora sólo se usan en muy raras oca-siones y sólo cuando es solicitado por el cliente. Puede obtenerse información mucho más útil y específica vía el aná-lisis RMN en combinación con la eva-luación de la espectrometría de masas.
PharmTech: ¿Qué análisis de RMN 2D avanzados utilizan y qué aprenden de cada uno de ellos?
Whitfield (Sussex): Para algunas de las estructuras más complejas de los gli-canos que sintetizamos, no sería posible
determinar las estructuras sin técnicas de RMN 2D. Los métodos que utiliza-mos para casi todas las muestras son es-pectroscopía 2D 1H-1H-gCOSY (espec-troscopía de correlación con gradiente), la cual toma aproximadamente 15-30 minutos, y 13C-1H-gHSQC (coherencia cuántica simple con gradiente heteronu-clear), típicamente la versión editada, la cual distingue los grupos CH y CH
3 de
los grupos CH2, y toma alrededor de 2-3
horas.Existen varios métodos adiciona-
les que son usados frecuentemente, aunque no para cada muestra. Es-tas técnicas incluyen 13C-1H-gHSQC con acoplamiento de protones para la medición de acoplamientos 13C-1H- 1-enlace (3-4 horas), espectroscopía 13C-1H-gHMBC (correlación de enlaces múltiples con gradiente heteronuclear) y 1H-1H-NOESY (espectroscopía de efecto overhauser nuclear) (o ROESY [espectroscopía de efecto de marco ro-tatorio]) para la determinación de las conectividades a través de los enlaces y posiciones de los grupos protectores (3-4 horas cada una), y 1H-1H-TOCSY (espectroscopía de correlación total) para la determinación de las conectivi-dades inter-anillo si el análi-sis COSY está incompleto (3-4 horas).
Sin embargo, hemos encontrado que la mayor digitalización de experimentos selectivos 1D (1Dsel), como el 1Dsel-1H-TOCSY y el 1Dsel-1H-NOESY (o
ROESY) permiten la determinación de constantes de acoplamiento y traba-jan mejor que el 2D para muestras de polímeros enlazados y otras muestras complicadas. Estas son por lo tanto usadas para determinar las conectivi-dades inter-anillo si el análisis COSY está incompleto (2 horas) y son particu-larmente aplicables si la anomericidad (alfa contra beta) no puede ser determi-nada de las constantes de acoplamiento y más comúnmente para derivados del ácido L-idurónico y D-manosa. Adicio-nalmente, para fosfatos u otras especies similares que contienen fósforo, la co-rrelación 31P-1H puede ser útil.
Es importante señalar que existen muchas variantes de estos espectros, y cada una es aplicable a ciertas cir-cunstancias. Adicionalmente, existen muchas técnicas de RMN rápidas que reducen grandemente el tiempo para adquirir los datos y completar estos ti-pos de experimentos, lo cual tendrá un impacto significativo en los tiempos de respuesta.
PharmTech: ¿Cuáles son algunos de los retos que enfrentan cuando llevan a cabo determinaciones estructurales de glicanos complejos?
Clark (Sussex): Los retos se pre-sentan mayormente por los propios compuestos. Los glicanos más grandes, más complejos, como aquéllos que con-tienen numerosos grupos glucosamina, pueden tener pobre solubilidad. Los glicanos con estructuras cargadas y un gran número de heteroátomos, como los ácidos idurónicos, las glucosaminas, los Osulfatos y los amino-sulfatos, también pueden ser difíciles de analizar. Con frecuencia, en estos casos, deben inves-tigarse varios solventes para encontrar el adecuado para el análisis por RMN. En algunos casos, el espectro de RMN es tan complejo que los picos individua-les no pueden distinguirse. En muchas de estas situaciones, se analizan por separado secciones más pequeñas de la molécula, y los espectros de RMN de los segmentos más pequeños se usan entonces para obtener el espectro com-pleto del glicano.
Para algunas de las estructuras más complejas de los glicanos que sintetizamos, no sería posible determinar dichas estructuras sin las técnicas 2D de RMN. - Whitfield
“El papel de la RMN y la Espectroscopía de Masas en el Análisis de Glicanos” Continúa en la pág. 65

Pharmaceutical Technology en Español MARZO / ABRIL 201452
EDITORIAL INVITADO
JOR
G G
RE
UE
L/P
HO
TOD
ISC
/GE
TT
Y IM
AG
ES
Inspirando a la siguiente generaciónTimothy J. Smith
Los científicos pueden compartir su tiempo y conocimiento farmacéutico con los estudiantes de educación media.
Aunque el foco primario de una publicación como el Pharmaceutical Technolo-gy tiene como objetivo la
discusión de tecnologías actuales para la industria farmacéutica, debe darse alguna consideración a inspirar a la si-guiente generación de científicos esti-mulando el interés en la aplicación de la ciencia farmacéutica. Este es claramen-te el caso en los Estados Unidos, donde las instituciones de educación secunda-ria quieren incrementar el interés en los campos de la ciencia, la tecnología, la ingeniería y las matemáticas (STEM).
Las tecnologías farmacéuticas son una actividad de STEM y, como tales, merecen una voz en este escenario. El voluntario que extiende las actividades de un científico farmacéutico a los es-tudiantes de educación media, ya sea a través de la filiación industrial o acadé-mica, no puede ser subestimado. Cuan-do un estudiante excepcional es invita-do a trabajar en un laboratorio acadé-mico o industrial a través de un apoyo o un proyecto de verano, la experiencia puede ser definitoria de la carrera.
Otras actividades STEM se es-tán moviendo al laboratorio escolar y, con planeación creativa y cuidadosa, se pueden incluir tecnologías farma-céuticas. A través de una de dichas
iniciativas, BioLink, las compañías de biotecnología donan suministros de la-boratorio para uso en secundaria, pro-porcionando soporte para programas educativos de secundaria críticamente sub-financiados.
Los científicos farmacéuticos pue-den proporcionar soporte a las secunda-rias a través de capacitación de maes-tros o directamente a los estudiantes conforme lo permitan el tiempo y las circunstancias. Como el típico labora-torio de secundaria puede tener un pre-supuesto limitado para instrumentación y consumibles, deben ofrecerse ejerci-cios de tecnología farmacéutica en un concepto auto-contenido o modular, del cual un científico voluntario y un maes-tro puedan seleccionar, con base en el ambiente de laboratorio/aprendizaje y los recursos disponibles. Estos módulos de enseñanza no necesitan ser costosos o sofisticados. El objetivo deben ser experiencias de aprendizaje que sean direc-tas, conceptuales y desde luego, fascinantes.
Aquí hay algunos simples módulos de enseñanza que yo he usado:
Ensayo de salicilato con cloruro fé-rrico. Este ejercicio, comúnmente usa-do en laboratorios de química orgánica para estudiantes universitarios, se deri-va del ensayo de ácido salicílico basado en el reactivo de Trinder. Como intro-ducción al análisis del fármaco, puede usarse una serie de estándares visuales para estimar la concentración del fár-maco si no se dispone de un espectro-fotómetro.
Encapsulado de alginato de calcio. En este módulo, se mezcla un químico con alginato de sodio y se agrega gota a
gota a una solución de cloruro de calcio, formando perlas de alginato de calcio, las cuales atrapan un químico dentro de la matriz del polisacárido. Cuando se mezcla colorante negro para alimentos con el alginato, se forman perlas rígidas negras con un contraste visual dramáti-co. Las perlas pueden ser rápidamente filtradas de la solución de cloruro de calcio con un colador para té y exami-narse manualmente.
Prueba de toxicidad para fármacos, por contacto con anélidos del suelo. Este módulo se derivó de procedimien-tos de análisis de pesticidas que utili-zan lombrices para evaluar el impacto ambiental. La prueba es marcadamen-te simple. Las lombrices se exponen a fármacos aplicados a papel filtro y con el tiempo, se retiran para evaluar la le-talidad de los fármacos absorbidos a través de la cutícula de las lombrices. Este es un medio relativamente directo para ilustrar la dosis-respuesta y la to-xicidad.
Estos procedimientos requieren po-cos químicos económicos y evitan el uso de agentes corrosivos, inflamables, volátiles y tóxicos y de mamíferos (en observación de las restricciones por riesgo del distrito escolar). Los recursos más valiosos son el tiempo y la expe-riencia del científico farmacéutico, in-cluyendo las discusiones después del laboratorio. Esto es especialmente crí-tico durante los años de los estudiantes jóvenes o mayores, cuando la inclina-ción a la carrera y la planeación univer-sitaria están afinándose.
La tecnología farmacéutica es su-ficientemente importante para merecer esta ruta de promoción educativa. PT
Timothy J. Smith, RPh, PhD , es profesor en el Departamento de Fisiología y Farmacología, Escuela de Farmacia y Ciencias de la Salud Thomas J. long, Universidad del Pacífico, Stockton, Calif.

Pharmaceutical Technology en Español MARZO / ABRIL 2014 53
JOR
G G
RE
UE
L/P
HO
TOD
ISC
/GE
TT
Y IM
AG
ES
Marque en la tarjeta de servicio al lector el No. 7

Pharmaceutical Technology en Español MARZO / ABRIL 201454
EntrEvista DEl EmplEo 2013
EL OPTIMISMO DE LA INDUSTRIA SOBREPASA A LAS PERSONAS EN EL TRABAJOLA REESTRUCTURACIÓN ACALLA EL OPTIMISMO. Rita Peters
En medio del ambiente de una recuperación económica gra-dual y la consolidación conti-nuada de las compañías bio/
farmacéuticas, los científicos, los técni-cos y los profesionales involucrados en el desarrollo de fármacos expresaron una mezcla de optimismo silencioso y opciones de empleo estancadas. Los lectores de Pharmaceutical Technolo-gy compartieron sus perspectivas so-bre la situación actual del empleo en la encuesta anual sobre el empleo de la revista. Las personas que respondieron representan una sección transversal de funciones de desarrollo de fármacos de moléculas grandes y pequeñas de instalaciones e Norteamérica, Europa y otros lugares. Aproximadamente el
60% de los que respondieron tienen grados de estudios avanzados, dividi-dos igualmente entre grados de Maes-tro y Doctorados.
En 2013, más de la mitad de los que respondieron dijeron que estaban in-volucrados en una fusión, adquisición, reducción o reestructuración. Esto re-presenta un salto significativo desde el 2012, cuando el 36.1% de los partici-pantes en la encuesta experimentaron cambios similares en su compañía. Esta actividad podría ser una razón para el ligero retroceso en la confianza que se tiene de la seguridad laboral.
Desde 2010, la confianza en la segu-ridad laboral ha tendido a la alza. Hace cuatro años, el 53% de los que respon-dieron dijeron que estaban menos segu-
ros de su posición contra el año anterior. En el 2013, sólo el 37.4% dijeron que estaban menos seguros en comparación con el 2012. Sin embargo, más encues-tados se sintieron menos seguros en sus posiciones en el 2013 en comparación con el 2012, lo que ilustra una ligera caí-da en la confianza de seguridad laboral.
Aproximadamente dos tercios de los encuestados (63.3%) reportaron un incremento en su salario en 2013. Mien-tras que el 43% dijo que se les pagaba bastante, casi el 35% dijo que su com-pensación estaba en el rango más bajo del valor del mercado. Casi el 20% dijo que se les pagaba por debajo del valor en el mercado. Más de la mitad reportó un incremento en el costo de los seguros de salud.
¿Qué tan seguro se siente en su trabajo en comparación con los dos años anteriores?
4.6%
Desde su punto de vista, ¿cuál es el panorama general para la industria bio/farmacéutica en el corto y largo plazo?

Pharmaceutical Technology en Español MARZO / ABRIL 2014 55
¿Qué tan seguro se siente en su trabajo en comparación con los dos años anteriores?
2011 2012 2013
38.5%
18.7%20.6%
37.4%
16.9%34%40.9%
47.3%45.7%
Aumentada
Reducida
Está igual
2011
72.7%
4.6%
22.7%
2012 2013
60.9%
29.9%
63.4%
32%
6.7%7%
Desde su punto de vista, ¿cuál es el panorama general para la industria bio/farmacéutica en el corto y largo plazo?
Me siento más seguro ahora
Me siento menos seguro ahora
Sin cambios
Dentro de los últimos dos años, ¿se ha incrementado, reducido o sigue igual su carga de trabajo?
0 10 20 30 40 50 60
Sin espera de un cambio signi�cativo
El negocio declinará internamente, pero no en el extranjero
El negocio declinará en el extranjero, pero no internamente.
El negocio mejorará internamente, pero no en el extranjero
El negocio mejorará en el extranjero, pero no internamente.
El negocio declinará.
El negocio mejorará.
2.9%
0%
5.3%
14.8%
54.3%
5.8%
16.5%
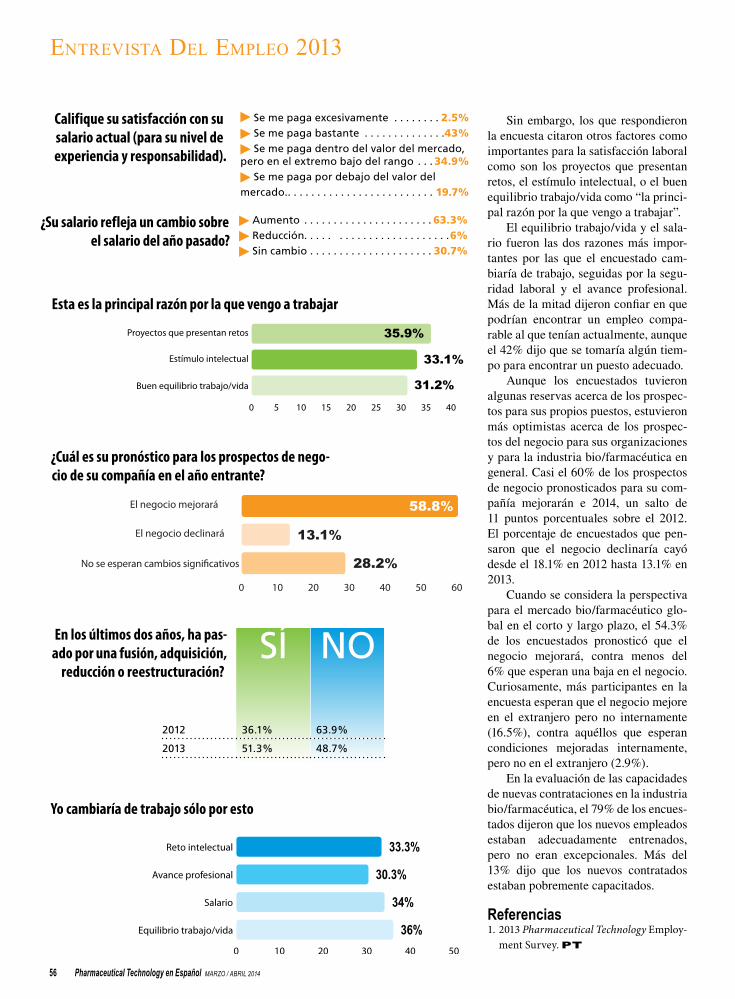
Pharmaceutical Technology en Español MARZO / ABRIL 201456
Califique su satisfacción con su salario actual (para su nivel de experiencia y responsabilidad).
Se me paga excesivamente � � � � � � � � 2.5% Se me paga bastante � � � � � � � � � � � � � �43% Se me paga dentro del valor del mercado,
pero en el extremo bajo del rango � � � 34.9% Se me paga por debajo del valor del
mercado� � � � � � � � � � � � � � � � � � � � � � � � � � 19.7%
¿Su salario refleja un cambio sobre el salario del año pasado?
Aumento � � � � � � � � � � � � � � � � � � � � � � 63.3% Reducción� � � � � � � � � � � � � � � � � � � � � � � �6% Sin cambio � � � � � � � � � � � � � � � � � � � � � 30.7%
Esta es la principal razón por la que vengo a trabajar
0 5 10 15 20 25 30 35 40
Buen equilibrio trabajo/vida
Estímulo intelectual
Proyectos que presentan retos 35.9%
31.2%
33.1%
¿Cuál es su pronóstico para los prospectos de nego-cio de su compañía en el año entrante?
En los últimos dos años, ha pas-ado por una fusión, adquisición,
reducción o reestructuración?SÍ NO
2012 36�1% 63�9%
2013 51�3% 48�7%
Yo cambiaría de trabajo sólo por esto
0 10 20 30 40 50
Equilibrio trabajo/vida
Salario
Avance profesional
Reto intelectual 33.3%
30.3%
34%
36%
0 10 20 30 40 50 60
No se esperan cambios signi�cativos
El negocio declinará
El negocio mejorará 58.8%
28.2%
13.1%
Sin embargo, los que respondieron la encuesta citaron otros factores como importantes para la satisfacción laboral como son los proyectos que presentan retos, el estímulo intelectual, o el buen equilibrio trabajo/vida como “la princi-pal razón por la que vengo a trabajar”.
El equilibrio trabajo/vida y el sala-rio fueron las dos razones más impor-tantes por las que el encuestado cam-biaría de trabajo, seguidas por la segu-ridad laboral y el avance profesional. Más de la mitad dijeron confiar en que podrían encontrar un empleo compa-rable al que tenían actualmente, aunque el 42% dijo que se tomaría algún tiem-po para encontrar un puesto adecuado.
Aunque los encuestados tuvieron algunas reservas acerca de los prospec-tos para sus propios puestos, estuvieron más optimistas acerca de los prospec-tos del negocio para sus organizaciones y para la industria bio/farmacéutica en general. Casi el 60% de los prospectos de negocio pronosticados para su com-pañía mejorarán e 2014, un salto de 11 puntos porcentuales sobre el 2012. El porcentaje de encuestados que pen-saron que el negocio declinaría cayó desde el 18.1% en 2012 hasta 13.1% en 2013.
Cuando se considera la perspectiva para el mercado bio/farmacéutico glo-bal en el corto y largo plazo, el 54.3% de los encuestados pronosticó que el negocio mejorará, contra menos del 6% que esperan una baja en el negocio. Curiosamente, más participantes en la encuesta esperan que el negocio mejore en el extranjero pero no internamente (16.5%), contra aquéllos que esperan condiciones mejoradas internamente, pero no en el extranjero (2.9%).
En la evaluación de las capacidades de nuevas contrataciones en la industria bio/farmacéutica, el 79% de los encues-tados dijeron que los nuevos empleados estaban adecuadamente entrenados, pero no eran excepcionales. Más del 13% dijo que los nuevos contratados estaban pobremente capacitados.
Referencias1. 2013 Pharmaceutical Technology Employ-
ment Survey. PT
EntrEvista DEl EmplEo 2013
Israel
Israel es una nación desarrollada e industrializada que, a pesar del conflicto continuo con los vecinos países Ára-bes, sigue comprometida para avanzar tecnológicamente y médicamente a través de la educación y altos gastos
en I+D. Durante las recientes décadas, mucho del crecimiento económico de Israel y el comportamiento en exportaciones se ha apoyado en industrias con investigación intensa. Su densa población y enfoque en la innovación posiciona bien a Israel para dirigir una cantidad significativa de crecimiento para fa-bricantes farmacéuticos y de dispositivos médicos.
La infraestructura del sistema de salud de Israel ha estado bajo escrutinio en los años recientes a medida que el gobierno de Israel continúa poniendo frenos en sus gastos de salud. Aun cuando la economía de Israel se ha recuperado de la crisis financiera global de 2008-09 mejor que las economías más avanzadas, de tamaño comparable, todavía se están ponien-do restricciones al presupuesto anual del Ministerio de Salud. En los años recientes, el Ministerio de Salud ha desarrollado fuertes capacidades en las áreas de evaluación de tecnología de salud (HTA), priorización de nuevas tecnologías y monito-reo de calidad para el cuidado comunitario para concentrarse más en el valor y minimizar el gasto ineficiente. A pesar de estos cambios, sin embargo, el mercado farmacéutico de Is-rael todavía espera crecer.
En 1995, Israel promulgó un Acta de Aseguramiento de Salud Nacional, otorgando un seguro de salud básico para to-dos los residentes de Israel, que cubre una “Canasta de Salud” que incluye tanto servicios médicos como productos médicos específicos. Los tratamientos y productos incluidos en esta lista son establecidos por el estado sobre una base anual. Los

Pharmaceutical Technology en Español MARZO / ABRIL 2014 57
Sin embargo, los que respondieron la encuesta citaron otros factores como importantes para la satisfacción laboral como son los proyectos que presentan retos, el estímulo intelectual, o el buen equilibrio trabajo/vida como “la princi-pal razón por la que vengo a trabajar”.
El equilibrio trabajo/vida y el sala-rio fueron las dos razones más impor-tantes por las que el encuestado cam-biaría de trabajo, seguidas por la segu-ridad laboral y el avance profesional. Más de la mitad dijeron confiar en que podrían encontrar un empleo compa-rable al que tenían actualmente, aunque el 42% dijo que se tomaría algún tiem-po para encontrar un puesto adecuado.
Aunque los encuestados tuvieron algunas reservas acerca de los prospec-tos para sus propios puestos, estuvieron más optimistas acerca de los prospec-tos del negocio para sus organizaciones y para la industria bio/farmacéutica en general. Casi el 60% de los prospectos de negocio pronosticados para su com-pañía mejorarán e 2014, un salto de 11 puntos porcentuales sobre el 2012. El porcentaje de encuestados que pen-saron que el negocio declinaría cayó desde el 18.1% en 2012 hasta 13.1% en 2013.
Cuando se considera la perspectiva para el mercado bio/farmacéutico glo-bal en el corto y largo plazo, el 54.3% de los encuestados pronosticó que el negocio mejorará, contra menos del 6% que esperan una baja en el negocio. Curiosamente, más participantes en la encuesta esperan que el negocio mejore en el extranjero pero no internamente (16.5%), contra aquéllos que esperan condiciones mejoradas internamente, pero no en el extranjero (2.9%).
En la evaluación de las capacidades de nuevas contrataciones en la industria bio/farmacéutica, el 79% de los encues-tados dijeron que los nuevos empleados estaban adecuadamente entrenados, pero no eran excepcionales. Más del 13% dijo que los nuevos contratados estaban pobremente capacitados.
Innovación y crecimiento en biotecnología en
IsraelJill E. Sackman y Michael KuchenreutherLa diversa población de Israel, el sistema de salud de alta calidad y la resistencia al estrés financiero global lo hacen un fuerte socio para I+D, investigación clínica y crecimiento del mercado.
Israel es una nación desarrollada e industrializada que, a pesar del conflicto continuo con los vecinos países Ára-bes, sigue comprometida para avanzar tecnológicamente y médicamente a través de la educación y altos gastos
en I+D. Durante las recientes décadas, mucho del crecimiento económico de Israel y el comportamiento en exportaciones se ha apoyado en industrias con investigación intensa. Su densa población y enfoque en la innovación posiciona bien a Israel para dirigir una cantidad significativa de crecimiento para fa-bricantes farmacéuticos y de dispositivos médicos.
La infraestructura del sistema de salud de Israel ha estado bajo escrutinio en los años recientes a medida que el gobierno de Israel continúa poniendo frenos en sus gastos de salud. Aun cuando la economía de Israel se ha recuperado de la crisis financiera global de 2008-09 mejor que las economías más avanzadas, de tamaño comparable, todavía se están ponien-do restricciones al presupuesto anual del Ministerio de Salud. En los años recientes, el Ministerio de Salud ha desarrollado fuertes capacidades en las áreas de evaluación de tecnología de salud (HTA), priorización de nuevas tecnologías y monito-reo de calidad para el cuidado comunitario para concentrarse más en el valor y minimizar el gasto ineficiente. A pesar de estos cambios, sin embargo, el mercado farmacéutico de Is-rael todavía espera crecer.
En 1995, Israel promulgó un Acta de Aseguramiento de Salud Nacional, otorgando un seguro de salud básico para to-dos los residentes de Israel, que cubre una “Canasta de Salud” que incluye tanto servicios médicos como productos médicos específicos. Los tratamientos y productos incluidos en esta lista son establecidos por el estado sobre una base anual. Los
residentes deben pagar un cierto porcentaje del ingreso (apro-ximadamente 5%) para pertenecer a uno de los cuatro provee-dores no gubernamentales llamados “fondos de enfermedad” (“kupat cholim”); en ciertos casos se requiere que hagan un co-pago para el tratamiento.
Aunque las tensas relaciones políticas y el terrorismo si-guen siento amenazas reales para la economía de Israel, in-cluyendo su mercado de salud, el sistema de salud de Israel continúa proporcionando un alto estándar de cuidado a la po-blación completa. De acuerdo a los datos globales compilados por Bloomberg, Israel está en el cuarto lugar en eficiencia de salud (1). Esta clasificación es particularmente impresionan-te considerando el nivel relativamente moderado de recursos asignados a la salud. Aun cuando Israel, que se unió a la Or-ganización para la Cooperación y el Desarrollo Económico (OCDE) en 2010, ha visto un gasto acelerado en salud desde el 2009, los gastos nacionales en salud permanecen por deba-jo de los de otras naciones miembro de la OCDE. El gasto en salud representó el 7.7% del producto interno bruto (PIB) en Israel en 2011, por debajo del promedio de 9.3% en los países de la OCDE (2).
Con aproximadamente 7.4 millones de personas en el úl-timo censo en 2008 y 311 personas por km2, la densidad de población de Israel está entre las más elevadas en el mundo Occidental. Más del 60% de la población está concentrada en la angosta franja a lo largo del Mar Mediterráneo. La alta tasa de fertilidad total de Israel ha dado lugar a una sociedad relativamente joven con 28% de la población menores de 15 años y sólo el 10% mayores de 64 años de edad (3). Para las compañías farmacéuticas que buscan expandirse en la región,
REPORTE DE MERCADOS EMERGENTES
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO

Pharmaceutical Technology en Español MARZO / ABRIL 201458
esta joven sociedad proporciona una fuerza de trabajo habi-litada, clasificándola entre los países líderes en el mundo en términos de creación de empresas de alta tecnología per cápita así como el gasto en I+D per cápita (4.40% del PIB) (4).
El ambiente de innovación y el espíritu empresarial en Is-rael ha resultado en un crecimiento sustancial en el número de compañías de ciencias de la vida desde 186 en 1996 a más de 1100 en 2012 (5). Las compañías de dispositivos médicos complementan aproximadamente el 50% de ese consorcio. Las ganancias de la producción (exportación y ventas loca-les) de la industria de dispositivos médicos de Israel en 2011 fue de $1,800 mdd (6). Israel también ocupa el segundo lugar después de Estados Unidos en las patentes biofarmacéuticas per cápita, y su mercado farmacéutico fue estimado en $1,780 mdd en 2010 y se espera que alcance $2,300 mdd para el 2020 (7).
Consideraciones regulatorias claveLos nuevos fármacos registrados en Israel, incluyendo los biológicos, deben cumplir los estándares GMP de la Unión Europea (UE) en términos de calidad y eficacia para asegurar la salud pública. Adicionalmente, los productos médicos que han sido fabricados, registrados o comercializados en otros mercados farmacéuticos mayores son elegibles para ser regis-trados en Israel. Esta elegibilidad estimula el crecimiento en el mercado permitiendo que los fármacos sean compartidos relativamente con facilidad entre Israel y otros países.
Israel se ha convertido en una ubicación cada vez más significativa para las compañías internacionales que realizan estudios clínicos farmacéuticos multinacionales. Un factor importante ha sido el hecho de que la población completa de Israel pertenece a uno de cuatro fondos de salud, lo cual mantiene una historia completa de salud de cada miembro. El sistema facilita la ubicación de pacientes y permite un buen seguimiento a lo largo del tiempo. Israel también tiene el tipo de población que se requiere con frecuencia para los estudios multinacionales, ofreciendo más diversidad étnica que muchos otros países. Adicionalmente, Israel fue uno de los primeros países en aceptar las guías de la Conferencia In-ternacional de Armonización (ICH) para las buenas prácticas clínicas (GCP), permitiendo estándares unificados entre la Unión Europea, Japón y Estados Unidos, facilitando la acep-tación mutua de los datos del estudio clínico por parte de las autoridades regulatorias.
Otras consideraciones del acceso a la salud incluyen far-macovigilancia (FV) e inclusión en la “canasta de salud.” La necesidad para una FV mejorada en Israel ha sido subcalifica-da en los años recientes por varios casos de alto perfil en los cuales los productos farmacéuticos han llegado a los consu-midores israelís con efectos colaterales adversos causados por cambios en la formulación. En respuesta a estos desarrollos, una rama de la Sociedad Internacional de Farmacovigilancia (ISoP) se está estableciendo en Israel con el objetivo de incre-mentar la conciencia acerca de la FV a lo largo de la región. Además, el Ministerio de Salud de Israel recientemente exigió por mandato que todos los productos de prescripción deben
tener etiquetas multilingües en hebreo, inglés, árabe y ruso.Históricamente, el alto precio de los fármacos no ha sido
parte del debate público en Israel, y el gobierno rara vez a usado controles de precios como es el caso en otros países. Sin embargo, la reciente tensión económica en la forma de elevación de costos militares y de defensa junto con la desace-leración de la economía global ha llevado al gobierno a buscar qué presupuestos recortar y qué impuestos elevar. Como tal, el Ministerio de Salud de Israel está ahora colocando productos nuevos y existentes bajo mayor escrutinio para limitar el gas-to. Por ejemplo, sólo 77 nuevos medicamentos y dispositivos médicos fueron agregados a la canasta de productos reembol-sados en 2012. Este número fue significativamente menor que los aproximadamente 200 nuevos fármacos y tecnologías que fueron incluidos en cada uno de los anteriores dos años (8). La inclusión de fármacos y tecnologías en la Canasta de Salud Nacional es crucial para las organizaciones que buscan éxito comercial en Israel. Aquí, los fabricantes requerirán datos de características clínicas, epidemiológicas y económicas, inclu-yendo su pronosticado impacto en el presupuesto disponible.
Ingreso del mercado exitoso y asociación en la regiónIsrael es hogar de uno de los fabricantes de fármacos genéri-cos más grande del mundo, Teva Pharmaceuticals, que a pesar de su presencia, el mercado farmacéutico israelí permanece a favor de los medicamentos patentados. En 2010, se reportó que los fármacos patentados constituían más del 50% de to-das las ventas farmacéuticas en el país y aproximadamente el 70% del mercado de fármacos de prescripción (9). Además, de acuerdo a Benny Zeevi, codirector de las Industrias de Tecnología Avanzada de Israel (IATI), la floreciente industria de biotecnología de Israel está transitando de un período de incubación a uno de madurez. El número de compañías bio-tecnológicas se ha casi triplicado desde 2003, y el número de estudios clínicos registrados continúa elevándose (10).
El gobierno estableció recientemente una nueva incubado-ra de biotecnología que reúne a más de otras 20 incubadoras o aceleradores para ayudar a atraer a grandes compañías far-macéuticas internacionales a Israel. Aunque algunas grandes compañías farmacéuticas pueden desanimarse por el tamaño de población moderado de Israel o por la percepción de un medio político volátil, otras grandes organizaciones globales reconocen las oportunidades de mercado que existen en este país. En 2009, Pfizer se asoció con Protalix Biotherapeutics y juntos desarrollaron la primera planta biológica aprobada para tratar la enfermedad de Gaucher tipo 1 (11). En 2011, la subsidiaria de Merck KGaA Merck Serono invirtió $13 mdd en una nueva incubadora para desarrollo de nuevos fármacos con el objetivo de tener al menos seis puestas en marcha de nuevos medicamentos o tecnologías para el 2018.
El mercado de capital de riesgo dinámico de Israel ha aportado una corriente de inversiones sustanciales en I+D. Con el aumento en la conciencia de enfermedades crónicas y una fuerza de trabajo altamente capacitada, se espera que los
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
“Innovación y crecimiento en biotecnología en Israel” Continúa en la pág. 61

Pharmaceutical Technology en Español MARZO / ABRIL 2014 59
tener etiquetas multilingües en hebreo, inglés, árabe y ruso.Históricamente, el alto precio de los fármacos no ha sido
parte del debate público en Israel, y el gobierno rara vez a usado controles de precios como es el caso en otros países. Sin embargo, la reciente tensión económica en la forma de elevación de costos militares y de defensa junto con la desace-leración de la economía global ha llevado al gobierno a buscar qué presupuestos recortar y qué impuestos elevar. Como tal, el Ministerio de Salud de Israel está ahora colocando productos nuevos y existentes bajo mayor escrutinio para limitar el gas-to. Por ejemplo, sólo 77 nuevos medicamentos y dispositivos médicos fueron agregados a la canasta de productos reembol-sados en 2012. Este número fue significativamente menor que los aproximadamente 200 nuevos fármacos y tecnologías que fueron incluidos en cada uno de los anteriores dos años (8). La inclusión de fármacos y tecnologías en la Canasta de Salud Nacional es crucial para las organizaciones que buscan éxito comercial en Israel. Aquí, los fabricantes requerirán datos de características clínicas, epidemiológicas y económicas, inclu-yendo su pronosticado impacto en el presupuesto disponible.
Israel es hogar de uno de los fabricantes de fármacos genéri-cos más grande del mundo, Teva Pharmaceuticals, que a pesar de su presencia, el mercado farmacéutico israelí permanece a favor de los medicamentos patentados. En 2010, se reportó que los fármacos patentados constituían más del 50% de to-das las ventas farmacéuticas en el país y aproximadamente el 70% del mercado de fármacos de prescripción (9). Además, de acuerdo a Benny Zeevi, codirector de las Industrias de Tecnología Avanzada de Israel (IATI), la floreciente industria de biotecnología de Israel está transitando de un período de incubación a uno de madurez. El número de compañías bio-tecnológicas se ha casi triplicado desde 2003, y el número de estudios clínicos registrados continúa elevándose (10).
El gobierno estableció recientemente una nueva incubado-ra de biotecnología que reúne a más de otras 20 incubadoras o aceleradores para ayudar a atraer a grandes compañías far-macéuticas internacionales a Israel. Aunque algunas grandes compañías farmacéuticas pueden desanimarse por el tamaño de población moderado de Israel o por la percepción de un medio político volátil, otras grandes organizaciones globales reconocen las oportunidades de mercado que existen en este país. En 2009, Pfizer se asoció con Protalix Biotherapeutics y juntos desarrollaron la primera planta biológica aprobada para tratar la enfermedad de Gaucher tipo 1 (11). En 2011, la subsidiaria de Merck KGaA Merck Serono invirtió $13 mdd en una nueva incubadora para desarrollo de nuevos fármacos con el objetivo de tener al menos seis puestas en marcha de nuevos medicamentos o tecnologías para el 2018.
El mercado de capital de riesgo dinámico de Israel ha aportado una corriente de inversiones sustanciales en I+D. Con el aumento en la conciencia de enfermedades crónicas y una fuerza de trabajo altamente capacitada, se espera que los
PREGUNTE AL EXPERTO
Centro de la manufactura farmacéutica: ¿Por qué la India?
Siegfried Schmitt, consultor principal en PAREXEL, discute el estado de la manufactura de fármacos en la India
P. Algunos casos recientes importantes de cuestiones de calidad en los fabricantes de la India han dado razón para examinar
más de cerca a los fabricantes. ¿Cuáles son algunas de las cues-tiones que deben ser consideradas?
R. Los fabricantes Indios de APIs y productos farma-céuticos han estado en las noticias desde hace algún tiempo, principalmente bajo encabezados negativos.
Este hecho puede llevar a alguien a pensar que hay algo fundamentalmente erróneo con ellos. Antes de saltar a las conclusiones, sin embargo, debemos ver más de cerca las cuestiones que están a la mano.
El cambio al OrienteLas continuas presiones de los costos y la mayor subcon-tratación en las décadas recientes ha resultado que muchas compañías Europeas y de EEUU contraten sus APIs de países tales como India y China, que tienen menores costos labora-les. La India fue una elección obvia por su gran combinación de mano de obra calificada y no calificada, más una industria existente química y farmacéutica. Actualmente, entre 50-70% de los APIs importados a EEUU y Europa vienen de la India (1). Esta es la razón por la que los reguladores de Europa y de EEUU han realizado muchas inspecciones en India, y la razón por la que la FDA incluso estableció oficinas permanentes en Mumbai y Nueva Deli (2). Este alto volumen de inspecciones ha resultado en un gran número de hallazgos.
De los APIs a los productos farmacéuticosLos márgenes de ganancia para los productos farmacéuticos son significativamente mayores que para los APIs, y varias compañías Indias han decidido capturar su participación de este lucrativo mercado, particularmente de los mercados de EEUU y Europa. Los requerimientos regulatorios para los productos farmacéuticos, especialmente los estériles, son mucho más demandantes que para los APIs, y aquí es donde muchos fabricantes Indios de fármacos enfrentan sus más se-rios problemas. Las cartas de advertencia y las alertas de im-portación han sido emitidas a varias compañías farmacéuticas líderes en India, disparando mucha de la publicidad negativa señalada previamente (3). Es importante señalar que los re-portes de inspección en la UE sólo se hacen públicos si son
solicitados bajo las leyes de Libertad de Información de la Au-toridad Competente Nacional, la cual conduce la inspección.
Problemas de integridad de datosAunque los hallazgos de las inspecciones en India reflejan a los citados en cualquier otro lado, lo que los hace ser algo aparte es que con frecuencia se han señalado problemas de integridad de los datos en una inspección (4). Por lo tanto, uno podría concluir que la falsificación de datos es genera-lizada. Aunque no se niega que han sido detectadas irregu-laridades en el manejo de datos, no ha habido siempre una intención maliciosa detrás de esto. En varios casos ha habido una creencia mal orientada de que los datos debían verse bien. Sin embargo, dicha manipulación de datos o invención de da-tos todavía constituye un fraude, sin importar la intención de la persona.Por último, los fabricantes de fármacos de la India están bajo la mira debido a cuestiones de incumplimiento, tales como monitoreo ambiental insuficiente, o una carencia de eviden-cia documentada. Muchas compañías en EEUU y en la UE, sin embargo, muchas veces atraen la atención por las mismas razones. Este hecho es de verdadera preocupación: ¿Por qué la madura industria farmacéutica en Europa y en EEUU aún luchan para cumplir con las regulaciones aplicables? En todo caso, la expectativa es que debería haber un nivel mucho más elevado de cumplimiento entre las compañías de EEUU y la UE en comparación con sus contrapartes Indias.Claramente, estas cuestiones deben ser resueltas por todas las compañías Indias involucradas, igual que cualquier otra. Como están en curso los esfuerzos para remediar esto, espe-ramos ver pronto más encabezados positivos que lleguen de esa parte del mundo.
Referencias1. S. Bennett, Specialty Chemicals Magazine (June 2013), www.spec-
chemonline.com/articles/view/the-falsified-medicines-directive, accessed Nov. 6, 2013.
2. A. Lal, “FDA In India: Going Global, Coming Home,” http://blogs.fda.gov/fdavoice/index.php/2013/09/fda-in-india-going-global-coming-home/, accessed Nov. 6, 2013.
3. FDA, Import Alerts, www.fda.gov/forindustry/importprogram/importalerts/default.htm, accessed Nov. 6, 2013.
4. “MHRA Cites Wockhardt Plant for GMP Noncompliance, PharmTech.com, www.pharmtech.com/pharmtech/article/ articleDetail.jsp?id=825817&topic=368, accessed Nov. 6, 2013. PT

Pharmaceutical Technology en Español MARZO / ABRIL 201460
VIGILANCIA REGULATORIA EUROPEA
Conforme la Unión Europea le da un vistazo más de cerca a sus guías de biosimilares, algunos problemas clave están demostrando ser difíciles de resolver.
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
Regulación de biosimilares: un asunto de variabilidad, similitud y comparabilidad
La Unión Europea está fortaleciendo su papel de pio-nero en la regulación de biosimilares desarrollando más las reglamentaciones básicas para determinar los niveles de compatibilidad para este grupo de
fármacos. Existen, sin embargo, algunos problemas clave que no son fáciles de resolver, como se evidenció en un reciente taller sobre biosimilares organizado por la Agencia Europea de Medicinas (EMA). La reunión, realizada en Octubre de 2013, fue convocada para considerar los problemas surgidos por en tres de las guías modificadas de la EMA sobre biosi-milares, una que abarca los principios generales, la segunda sobre temas clínicos y no clínicos, y la tercera sobre cuestio-nes de calidad. La fecha límite para las observaciones sobre las primeras dos guías fue a finales de Octubre, mientras que para el tercer asunto con respecto a la calidad, hubo todavía la necesidad de más discusiones para negociar con las dificulta-des de publicar un borrador final.
La UE quiere seguir a la cabeza del juego en la regulación de los biosimilares para darle a la región una ventaja en el establecimiento de un gran mercado interno para los medi-camentos. Con el objetivo de ayudar a estimular el desarrollo global de estas medicinas, la UE ha optado por mayor flexi-bilidad en la elección del biofarmacéutico original o producto de referencia para el cual los biofarmacéuticos tendrán que demostrar la comparabilidad en calidad, seguridad y eficacia para ser aprobados como biosimilares.
Guías modificadas de los principios generalesEn sus guías modificadas sobre los principios generales para los biosimilares, la EME establece que el producto de refe-rencia debe ser debe ser una medicina autorizada en el Área Económica Europea (EEA) (1). Sin embargo, la UE también confirma que “puede ser posible” que el biosimilar sea com-parado, “cuando sea necesario,” con un producto no autori-zado por la EEA en estudios clínicos y no clínicos. Como el producto no autorizado por la EEA tendría que seguir están-dares científicos y regulatorios similares a los de la EMA, tendría que ser en un país que fuera miembro de la Confe-rencia Internacional de Armonización (ICH), de acuerdo a la guía. También, el solicitante del biosimilar que use la ruta que no es de la EEA tendría que aportar datos puenteados del análisis estructural y funcional, y posiblemente de estudios farmacocinéticos y farmacodinámicos, para mostrar que el fármaco que no es EEA fuera “representativo” del producto de referencia de la EEA. Eugene Corretge, vocero de parte de Empresas Biofarmacéuticas Europeas (EBE), que representa tanto a los fabricantes de biológicos originales como a los de
biosimilares, dijo que la guía modificada no era clara acerca de si los datos puenteados serían necesarios cuando se rea-lizara un estudio de comparabilidad sólo parcialmente o en su totalidad con un medicamento no autorizado por la EEA.
Karl Heinz Emmert, vicepresidente del Grupo de Biosi-milares Europeo (EBG), parte de la Asociación de Medicinas Genéricas de Europa (EGA), declaró que las guías modifica-das presentaron “fuertes principios científicos” para respaldar el uso de fármacos no-EEA obtenidos de regiones ICH como productos de referencia. Podría haber mayor consistencia entre productos de diferentes jurisdicciones de concesión de licencias que aquéllos de una sola jurisdicción de concesión de licencias, explicó. “Los productos de referencia de un fa-bricante original y procedente de una diferente jurisdicción son con frecuencia no distinguibles,” dijo Emmert. “Un pro-ducto de referencia suministrado en una jurisdicción, con fre-cuencia se fabrica en diferentes instalaciones.” Con todo, no requeriría estudios de puenteo no clínicos o clínicos, añade.
La comparabilidad de los productos de referencia de un fabricante original pero de diferentes jurisdicciones podría establecerse mediante similitud o calidad basada en estrictos ensayos analíticos y biológicos, explicó Emmert. “Los estu-dios no clínicos y clínicos no son adecuados para establecer diferencias entre los ‘mismos’ productos de referencia de un fabricante original,” dijo. “Los ensayos analíticos y biológi-cos son mucho más sensibles y son, por lo tanto, mucho más adecuados.”
Determinación de los atributos de calidad críticosHubo un acuerdo general en un enfoque gradual para estable-cer las similitudes necesarias entre el fármaco del solicitante de biosimilar y el producto de referencia, el cual, efectiva-mente, en la mayoría de los casos haría superfluos los estu-dios no clínicos o clínicos. En lugar de esto, el ejercicio de comparabilidad estaría enfocado principalmente en el proce-so de manufactura, especialmente para los biosimilares más simples.
“Para productos medicinales biológicos estructuralmente más simples, puede no ser necesario un estudio comparativo de eficacia clínica si la similitud de las características fisico-químicas y de la actividad o potencia de los biosimilares y el producto de referencia pueden ser demostradas convincente-mente,” dijo Martina Weise del Instituto Federal para Fárma-cos y Dispositivos Médicos (BfArM), la agencia regulatoria alemana. A este respecto, la elaboración de un perfil de pro-ducto objetivo de calidad (QTPP), el cual resume las caracte-rísticas de calidad requeridas del biosimilar, fue considerado
es escritor independiente con sede en Essex, RU,
Sean Milmo

Pharmaceutical Technology en Español MARZO / ABRIL 2014 61
crucial. “Estaría principalmente basado en extensos datos de caracterización de los productos de referencia,” explicó Nic-klas Ekman de la Agencia de Medicinas de Finlandia. “Esto estaría detallado en una etapa temprana del desarrollo, for-mando las bases del desarrollo del producto biosimilar y su proceso de manufactura.”
Beverley Ingram, una representante del EBE, dijo que su asociación recomendaba el reforzamiento del papel del QTPP en un esquema gradual siendo esto cuantitativo por naturale-za. Sin embargo, el establecimiento de criterios cuantitativos, significativos, es un tópico técnicamente complejo, el cual su-girió que debía investigarse más en un artículo de reflexión propuesto por la EMA sobre el uso de la estadística en la com-paración de fármacos biológicos, incluyendo los biosimilares.
Joerg Windisch, director del EBG y jefe de ciencias de Sandoz Pharmaceuticals, subrayó que la comparabilidad entre biosimilares y sus productos de referencia incluía tener varia-bilidades similares, particularmente entre lotes. “Ningún lote de cualquier biológico es ‘idéntico’ a otros lotes,” explicó. “La variabilidad es natural, incluso en el cuerpo humano y habi-tualmente no es problemática.”
Las diferencias significativas serían aquéllas que tienen un impacto clínico, dijo Windisch. Por lo tanto, es importante tener una evaluación sistemática y cuantitativa de los atributos de calidad para escoger aquéllos que sean cruciales con base en su relevancia clínica. Estos atributos clínicos son uno de los elementos clave que determinan si un fármaco es acepta-ble como biosimilar, dijo él.
Otra fuente potencial de incompatibilidades inaceptables es el uso de sistemas de expresión contrastantes en proteí-nas biosimilares. Windisch señaló que tenía que hacerse una distinción entre los diferentes tipos de sistemas de expresión existentes y aquellos que fueran nuevos. Las diferentes clases de sistemas existentes podrían proveer atributos de calidad similares. También, como éstos han sido aplicados durante al-gún tiempo, estos sistemas tendrían registros de rastreo de se-guridad “iguales o mejores”. Por el otro lado, los sistemas de
expresión nuevos son, por definición, entidades desconocidas con ausencia de datos de seguridad. “El uso de sistemas de ex-presión sin registros de seguridad es bastante más desafiante y es algo que no quieres arriesgar cuando haces el desarrollo de un biosimilar, dijo Windisch. Las diferencias en un fármaco que van más allá del rango de variabilidad en el producto de referencia, particularmente cuando tiene significancia clínica, hacen probable que éste sea rechazado como biosimilar por parte de los reguladores.
Se les dijo a los asistentes al taller acerca de las complica-ciones de establecer límites generales en diferencias acepta-bles entre los biosimilares y los productos de referencia. “No puedes dar esa clase de claridad,” dijo Weise. “Sería muy di-fícil establecer un margen que se ajuste a todo. Estos límites tienen que establecerse sobre la base de caso por caso.
Con respecto a un esquema individual para los productos, algunos ponentes señalaron que con algunos biosimilares, la comparabilidad no debería estar confinada a las sustancias ac-tivas. Se debería cubrir la formulación completa, incluyendo excipientes y diluyentes debido a su efecto sobre la seguridad y eficacia del medicamento. Se sugirió un procedimiento si-milar para tratar con el tema de inmunogenicidad. “Existen moléculas en el mercado cuya inmunogenicidad no ha sido establecida,” dijo Windisch. “Depende del solicitante de la autorización de un biosimilar justificar que no son necesarios los estudios de inmunogenicidad. En la mayoría de los ca-sos, esto parece ser innecesario pero tendremos que ver cómo se desarrolla la ciencia sobre este tema.” Debido a la historia relativamente corta de los biosimilares, existe carencia de co-nocimiento acerca de algunos de los aspectos importantes de su seguridad. De esta forma, todavía hay mucho por aprender acerca de cómo podría regularse apropiadamente.
Referencias 1. EMA, Draft Guideline on Similar Biological Medicinal Products
(London, May 2013). PT
mercados farmacéuticos y de diagnóstico de Israel crezcan incluso más fuertes. Tomado en conjunto, Israel parece como un gran lugar para ver las futuras oportunidades de inversión.
Avanzando en IsraelIsrael tiene una larga historia de innovación en biotecnología y crecimiento y con una población educada y diversa, siste-ma de salud de alta calidad, y resistencia al estrés financiero global sigue siendo un fuerte socio para I+D, investigación clínica y crecimiento del mercado. Adicionalmente, Israel es un destino popular para el turismo médico. A partir del 2010, hasta 30,000 extranjeros llegan a Israel cada año para trata-miento, principalmente de Rusia (12). Israel, como EEUU, es también un frecuente “primer adoptador” de nueva tecnología e innovación médica. Los fabricantes, sin embargo, necesitan estar conscientes de que la austeridad fiscal del gobierno is-raelí y los fondos de enfermos tradicionalmente han mante-nido los fármacos de marca más costosos y los tratamientos no disponibles. Los genéricos más económicos y los salarios
bajos de los médicos y los proveedores de salud han sido usa-dos para controlar los costos globales.
Referencias1. “Israel ranks 4th globally in health care efficiency,” Times of Israel (Au-
gust 2013).2. OECD Health Data (2013).3. OECD Reviews of Health Care Quality: Israel (2012).4. The World Bank, Research and development expenditure (% of GDP)
(2013).5. M. Scudellari, The Scientist (July 2013).6. Meidata, Israel Medical Devices Industry-Market Overview 2012.7. Global Data, Research and Markets: Healthcare, Regulatory and Reim-
bursement Landscape—Israel (2012).8. Scripp Business Insights, The Pharmaceutical Market in Israel (2012).9. 9. Industry Trend Analysis, Investment In Innovation Necessary (2010).10. N. Elis, The Jerusalem Post (April 25, 2013).11. J. Fox, Nature Biotechnology (June 2012).12. “Health Ministry to probe Israel medical tourism industry following
Haaretz exposé,” haaretz.com (November 2010). PT
“Innovación y crecimiento en biotecnología en Israel” Continuación de la pág. 58

Pharmaceutical Technology en Español MARZO / ABRIL 201462
síntEsis Y manuFactura DE apis
Rastreo de la innovación en nuevas entidades moleculares del 2013Patricia Van Arnum
Revisión de la cosecha de este año de las nuevas entidades moleculares y nuevas solicitudes de licencia para biológicos aprobadas por la FDA hasta el momento en el 2013.
Patricia Van Arnum es editora ejecutiva del Pharmaceutical Technology, 485 Route one South, Edif. F, Primer Piso, Iselin, NJ 08830; tel. 732.346.3072, [email protected].
Ahora que el 2013 se apro-xima al cierre, ¿cómo le ha ido a la industria farma-céutica en la aprobación de
nuevos fármacos? Hasta Nov. 14, 2013, el Centro para la Evaluación e Investi-gación de Fármacos (CDER) de la Ad-ministración de Alimentos y Fármacos, había aprobado 24 nuevas entidades moleculares (NMEs) y solicitudes de licencia para biológicos (BLAs) (1). Aunque la actividad de fin de año de la FDA típicamente resulta en aprobacio-nes adicionales, es improbable que la industria alcance en 2013 los 39 NMEs y BLAs aprobados por la FDA en 2012, lo que fue una cifra máxima reciente. Sin embargo, la cosecha de NMEs y BLAs de este año, muestra varios de-sarrollos interesantes: la aprobación de dos nuevas terapias innovadoras, una
nueva designación por la FDA para acelerar la aprobación de fármacos que cubran las altas demandas médi-cas; tres aprobaciones de fármacos con diagnóstico acompañante y una aprobación de un conjugado fármaco-anticuerpo (ADC).
Llevando la cuentaSobre la base de una compañía, has-ta ahora en 2013, los ganadores en la carrera por aprobaciones de NMEs y BLAs por parte del CDER de la FDA son Bayer Healthcare, GlaxoSmithKli-ne (GSK), Johnson & Johnson, Roche y Takeda, cada uno con dos o más aproba-ciones de nuevos fármacos (ver Tabla I). GSK encabezó el grupo con tres apro-baciones de NME: Breo Ellipta (fluti-casona furoato y vilanterol trifenatato), Mekinist (Trametinib dimetil sulfóxi-do) y Tafinlar (dabrafenib mesilato). El Breo Ellipta es una combinación de fluticasona furoato, un corticosteroide inhalado, y vilanterol, un agonista adre-nérgico 2 de acción prolongada, utiliza-do para tratar la obstrucción de las vías aéreas y reducir las exacerbaciones en pacientes con enfermedad pulmonar
obstructiva crónica. GSK también recibió la aprobación de la FDA de dos fármacos para tratar el melanoma metastásico: Me-kinist, un inhibidor de la cinasa para tratar el melanoma no resecable o metastásico con mutaciones BRAF V600E o V600K detectadas mediante una prueba aprobada por la FDA, y Tafinlar, también un inhi-bidor de la cinasa indicado para el trata-miento del mela-noma no resecable o me-tastásico con la mutación BRAF V600E, detectada mediante una prueba aprobada por la FDA. En el 2010, GSK formó una colaboración con la compañía de diagnós-tico molecular bioMérieux para desarro-llar una prueba diagnóstica acompañan-te para detectar las mutaciones del gen BRAF V600 (V600E y V600K) encontra-das en varios cánceres, incluyendo el me-lanoma. bioMérieux recibió la aprobación previa a la comercialización de la FDA de su diagnóstico acompañante, THxID-BRAF, en mayo del 2013.
A través de sus unidades bio/far-macéuticas, Janssen Biotech y Janssen Pharmaceuticals, Johnson & Johnson tuvieron dos NMEs aprobados en 2013. Janssen Biotech se asoció con la compa-ñía biofarmacéutica Pharmacyclics para el Imbruvica (ibrutinib), un inhibidor de cinasa para tratar el cáncer de células del manto (MCL por sus siglas en inglés, Mantle-Cell Lymphoma), un raro tipo de cáncer en sangre. Imbruvica es el segun-do fármaco con la designación de terapia innovadora para recibir la aprobación de la FDA; el primero fue el Gazyva (obi-nutuzumab), de Roche, un fármaco para tratar la leucemia linfocítica crónica (CLL), también aprobado en 2013. El Acta de Seguridad e Innovación de la Ad-ministración de Alimentos y Fármacos, aprobada en julio del 2012, le dio a la FDA la capacidad de designar a un fár-maco como terapia innovadora a solicitud del patrocinador, si la evidencia clínica preliminar indica que el fármaco puede ofrecer una mejora sustancial sobre las terapias disponibles para pacientes con enfermedades graves o mortales. Además del Imbruvica, existen otros dos fármacos aprobados por la FDA para tratar el MCL: Velcade (bortezomib) por Millenium Pharmaceuticals, la compañía de onco-logía de Takeda Pharmaceuticals, y el Revlimid (lenalidomida) de Celgene (2).
La segunda aprobación de una NME de J&J fue para Invokana (canagliflozin), un inhibidor del cotransportador 2 de so-
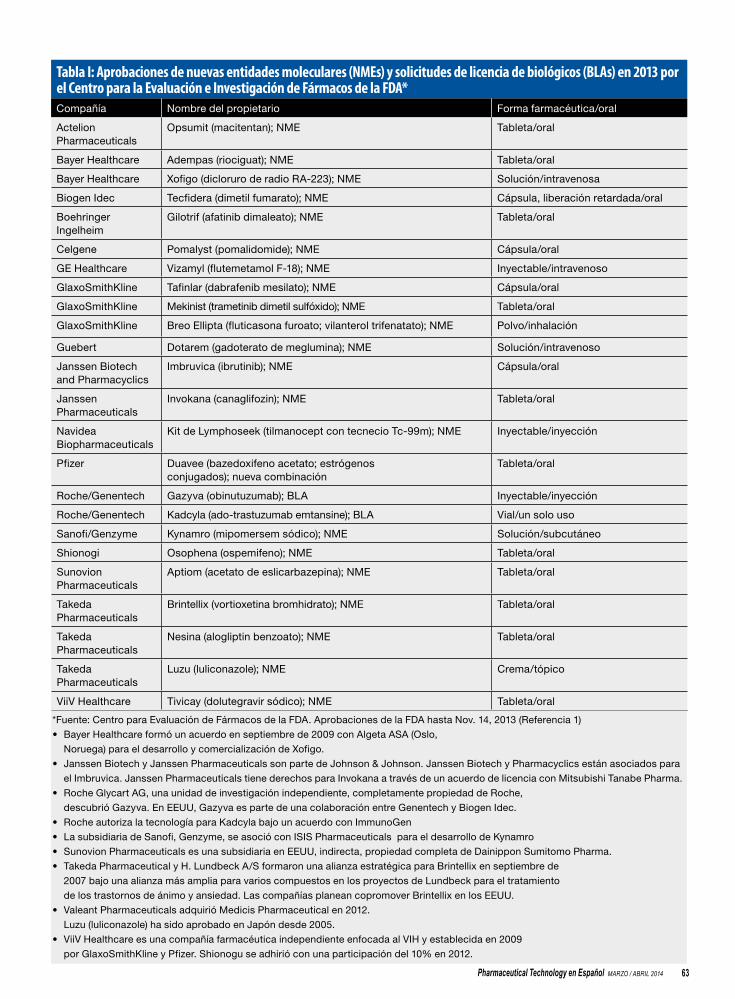
Pharmaceutical Technology en Español MARZO / ABRIL 2014 63
Tabla I: Aprobaciones de nuevas entidades moleculares (NMEs) y solicitudes de licencia de biológicos (BLAs) en 2013 por el Centro para la Evaluación e Investigación de Fármacos de la FDA*Compañía Nombre del propietario Forma farmacéutica/oral
Actelion Pharmaceuticals
Opsumit (macitentan); NME Tableta/oral
Bayer Healthcare Adempas (riociguat); NME Tableta/oral
Bayer Healthcare Xofigo (dicloruro de radio RA-223); NME Solución/intravenosa
Biogen Idec Tecfidera (dimetil fumarato); NME Cápsula, liberación retardada/oral
Boehringer Ingelheim
Gilotrif (afatinib dimaleato); NME Tableta/oral
Celgene Pomalyst (pomalidomide); NME Cápsula/oral
GE Healthcare Vizamyl (flutemetamol F-18); NME Inyectable/intravenoso
GlaxoSmithKline Tafinlar (dabrafenib mesilato); NME Cápsula/oral
GlaxoSmithKline Mekinist (trametinib dimetil sulfóxido); NME Tableta/oral
GlaxoSmithKline Breo Ellipta (fluticasona furoato; vilanterol trifenatato); NME Polvo/inhalación
Guebert Dotarem (gadoterato de meglumina); NME Solución/intravenoso
Janssen Biotech and Pharmacyclics
Imbruvica (ibrutinib); NME Cápsula/oral
Janssen Pharmaceuticals
Invokana (canaglifozin); NME Tableta/oral
Navidea Biopharmaceuticals
Kit de Lymphoseek (tilmanocept con tecnecio Tc-99m); NME Inyectable/inyección
Pfizer Duavee (bazedoxifeno acetato; estrógenos conjugados); nueva combinación
Tableta/oral
Roche/Genentech Gazyva (obinutuzumab); BLA Inyectable/inyección
Roche/Genentech Kadcyla (ado-trastuzumab emtansine); BLA Vial/un solo uso
Sanofi/Genzyme Kynamro (mipomersem sódico); NME Solución/subcutáneo
Shionogi Osophena (ospemifeno); NME Tableta/oral
Sunovion Pharmaceuticals
Aptiom (acetato de eslicarbazepina); NME Tableta/oral
Takeda Pharmaceuticals
Brintellix (vortioxetina bromhidrato); NME Tableta/oral
Takeda Pharmaceuticals
Nesina (alogliptin benzoato); NME Tableta/oral
Takeda Pharmaceuticals
Luzu (luliconazole); NME Crema/tópico
ViiV Healthcare Tivicay (dolutegravir sódico); NME Tableta/oral
*Fuente: Centro para Evaluación de Fármacos de la FDA. Aprobaciones de la FDA hasta Nov. 14, 2013 (Referencia 1)
• Bayer Healthcare formó un acuerdo en septiembre de 2009 con Algeta ASA (Oslo,
Noruega) para el desarrollo y comercialización de Xofigo.
• Janssen Biotech y Janssen Pharmaceuticals son parte de Johnson & Johnson. Janssen Biotech y Pharmacyclics están asociados para
el Imbruvica. Janssen Pharmaceuticals tiene derechos para Invokana a través de un acuerdo de licencia con Mitsubishi Tanabe Pharma.
• Roche Glycart AG, una unidad de investigación independiente, completamente propiedad de Roche,
descubrió Gazyva. En EEUU, Gazyva es parte de una colaboración entre Genentech y Biogen Idec.
• Roche autoriza la tecnología para Kadcyla bajo un acuerdo con ImmunoGen
• La subsidiaria de Sanofi, Genzyme, se asoció con ISIS Pharmaceuticals para el desarrollo de Kynamro
• Sunovion Pharmaceuticals es una subsidiaria en EEUU, indirecta, propiedad completa de Dainippon Sumitomo Pharma.
• Takeda Pharmaceutical y H. Lundbeck A/S formaron una alianza estratégica para Brintellix en septiembre de
2007 bajo una alianza más amplia para varios compuestos en los proyectos de Lundbeck para el tratamiento
de los trastornos de ánimo y ansiedad. Las compañías planean copromover Brintellix en los EEUU.
• Valeant Pharmaceuticals adquirió Medicis Pharmaceutical en 2012.
Luzu (luliconazole) ha sido aprobado en Japón desde 2005.
• ViiV Healthcare es una compañía farmacéutica independiente enfocada al VIH y establecida en 2009
por GlaxoSmithKline y Pfizer. Shionogu se adhirió con una participación del 10% en 2012.

Pharmaceutical Technology en Español MARZO / ABRIL 201464
La innovación es crucial para cualquier industria, incluyendo la industria farmacéutica, donde los avances en el desarrollo de formulaciones, la manufactura y el acondicionado son importantes para el desarrollo y éxito comercial de un fármaco. En el CPhl Mundial, organizado por UBM Live y realizado en Frankfurt en Octubre de 2013, tres compañías fueron reconocidas por la innovación, respectivamente, en desarrollo del proceso, formulación y acondicionado a través de los Premios Farmacéuticos CPhl. El ganador por desarrollo de proceso fue API Corporation (Tokio) por una síntesis práctica de farmacéuticos funcionalizados a través de la activación carbono-hidrógeno. Camurus (Lund, Suiza) ganó por la mejor innovación en formulación para FluidCrystal, una inyección de depósito patentada y la tecnología relacionada, y E-Pharma Trento (Trento, Italia) fue reconocida por la innovación en el empaque para un empaque blister de tres capas diseñado para proteger formas farmacéuticas frágiles tales como las tabletas de desintegración oral.
API Corporation estuvo entre los tres finalistas para el premio por la mejor innovación en el desarrollo de procesos, el cual también incluyó a Bachem (Bubendorf, Suiza) por una síntesis química del interferón beta-1a y Codexis (Redwood City, California) por oxidaciones biocatalíticas. En la obtención del primer premio por innovación en el desarrollo de procesos, API Corporation fue reconocida por el desarrollo de un sistema catalizador para la activación del carbón-hidrógeno que fue aplicada a la síntesis comercial de bloqueadores del receptor de angiotensina II.
Además de Camurus, los finalistas por la mejor innovación en la formulación fueron Bitop AG (Witten, Alemania) por Ectoína, un
ingrediente patentado derivado de organismos extremofílicos que tienen propiedades protectoras de las células que pueden ser usados en dispositivos médicos, productos de ciencia de la vida, y cosméticos, y EmulTech BV (Eindhoven, Holanda) por su plataforma de microfluidos y tecnología. Camurus ganó el premio principal por innovación en la formulación por su inyección de depósito FluidCrystal. El sistema, que
consiste en una solución líquida basada en lípidos que se transforma con el contacto en una matriz de gel de cristal líquido in situ con pequeñas cantidades de fluido acuoso en el sitio de la inyección, encapsula la sustancia farmacéutica para dar una liberación controlada y una mejora en la estabilidad del fármaco. El sistema puede ser usado para entrega parenteral sostenida de pequeñas moléculas, péptidos y proteínas.
E-Pharma Trento fue uno de los tres finalistas para la innovación en el empaque, lo cual también incluyó a Doctor Pack India (Bangalore, India) por el Magic Cap, una alternativa a la tapa convencional para perforación que es utilizada para perforado estéril y dosificación precisa para las gotas oculares, óticas, nasales y orales, y Neutroplast (Sobral Monte Agraco, Portugal) para un aplicador vaginal. E-Pharma Trento fue reconocida por la mejor innovación en el empaque por un empaque blister de tres capas diseñado para proteger las formas farmacéuticas frágiles tales como las tabletas de desintegración oral. El empaque permite sacar las tabletas del empaque blister empujando y protegiéndolas al mismo tiempo para que no se rompan. El avance se basa en una cápsula rígida que protege la tableta ayudando a liberarla mientras tanto del empaque blister.
JON
AT
HA
N K
ITC
HE
N/G
ET
TY
IM
AG
ES
dio-glucosa (SGLT2) para tratar la dia-betes Tipo 2. El Invokana es el primer fármaco en una nueva clase de inhibido-res SGLT2 en ser aproba-dos en Esta-dos Unidos. El Forxiga (dapaglifozin), desarrollado por Bristol-Myers Squibb y AstraZeneca y también un inhibidor del SGLT2, fue aprobado por la Agen-cia Europea de Medicamentos en no-viembre de 2012. Las compañías some-tieron una solicitud de nuevo fármaco para Forxiga a la FDA en julio de 2013.
Roche tuvo dos nuevos BLAs apro-bados por la FDA en 2013: Gazyva (obinutuzumab) y Kadcyla (ado-trastu-zumab emtansina). Gazyva es un anti-cuerpo monoclonal (mAb) anti-CD20 humanizado de la clase IgG1 para el tratamiento de CLL. Es producido por un cultivo en suspensión de células de mamífero (ovario de hámster Chino [CHO]) (3). Kadcyla es un fármaco dirigido al HER-2 del ADC (adenocar-cinoma), que contiene el anti-HER2
humanizado IgG1, trastuzumab, unido covalentemente al fármaco inhibitorio del microtúbulo DM1 (un derivado del maytansine) por medio del enlazador tioéter estable MCC (4-[N-maleimi-dometil] ciclohexano-1-carboxilato). Emtansine se refiere al complejo MCC-DM1. El trastuzumab es un mAb re-combinante bien caracterizado, produ-cido por células de mamífero CHO, y los componentes de molécula pequeña (DM1 y MCC) son producidos por sín-tesis química (4). Kadcyla es el primer fármaco para ADC de Roche aprobado por la FDA.
Takeda se anotó dos aprobaciones de NME para el 2013: Brintellix (vor-tioxetina brom-hidrato) y Nesina (alo-gliptin benzoato). Brintellix, utilizado para tratar el trastorno depresi-vo ma-yor, fue desarrollado con H. Lundbeck. Nesina es un inhibidor de la dipeptidil peptidasa-4, diseñado para desacelerar la inactivación de hormonas incretíni-
cas GLP-1 (péptido-1 similar al glu-cagon) y GIP (péptido insulinotrópico dependiente de glucosa) para el trata-miento de la diabetes Tipo 2.
Bayer tuvo dos NMEs aprobados por la FDA en 2013: Adempas (rioci-guat) y Xofigo (radium RA-223 diclo-ruro). Adempas es un estimulador so-luble de la guanilato ciclasa para tratar dos formas de hipertensión pulmonar. Xofigo es un agente terapéutico radio-activo que emite partículas alfa para tratar el cáncer de próstata resistente a la castración, la metástasis ósea sin-tomática, y la enfermedad metastásica visceral no conocida. En 2009, Bayer se asoció con Algeta para el desarrollo y comercialización de Xofigo.
Otras aprobacionesOtras grandes compañías ganaron apro-baciones de la FDA en 2013. Idec re-cibió la aprobación de la FDA para su fármaco oral para la esclerosis múltiple
Tecfidera (dimetil fumarato). Boehrin-ger Ingelheim recibió la aprobación de la FDA para Gilotrif (afatinib dimalea-to), un inhibidor de cinasa para el trata-miento del cáncer pulmonar de células no pequeñas con mutaciones del recep-tor común del factor de crecimiento epidérmico, detectados por una prueba aprobada por la FDA. Sanofi recibió la aprobación para Kynamro (mipo-mersen sódico), una sal sódica sintética de fosfo-rotioato oligonucleótido (20 nucleótidos de longitud) para tratar la hipercoleste-rolemia familiar homocigótica, un tipo raro de colesterol elevado (5).
ViiV Healthcare, una compañía farmacéutica independiente conjunta, enfocada en tratamientos para el VIH y formada por Pfizer, GSK y Shiono-gi, recibió la aprobación para Tivicay
Reconocimiento de la innovación en el desarrollo, manufactura y acondicionado de formulaciones
síntEsis Y manuFactura DE apis

Pharmaceutical Technology en Español MARZO / ABRIL 2014 65
JON
AT
HA
N K
ITC
HE
N/G
ET
TY
IM
AG
ES
cas GLP-1 (péptido-1 similar al glu-cagon) y GIP (péptido insulinotrópico dependiente de glucosa) para el trata-miento de la diabetes Tipo 2.
Bayer tuvo dos NMEs aprobados por la FDA en 2013: Adempas (rioci-guat) y Xofigo (radium RA-223 diclo-ruro). Adempas es un estimulador so-luble de la guanilato ciclasa para tratar dos formas de hipertensión pulmonar. Xofigo es un agente terapéutico radio-activo que emite partículas alfa para tratar el cáncer de próstata resistente a la castración, la metástasis ósea sin-tomática, y la enfermedad metastásica visceral no conocida. En 2009, Bayer se asoció con Algeta para el desarrollo y comercialización de Xofigo.
Otras grandes compañías ganaron apro-baciones de la FDA en 2013. Idec re-cibió la aprobación de la FDA para su fármaco oral para la esclerosis múltiple
Tecfidera (dimetil fumarato). Boehrin-ger Ingelheim recibió la aprobación de la FDA para Gilotrif (afatinib dimalea-to), un inhibidor de cinasa para el trata-miento del cáncer pulmonar de células no pequeñas con mutaciones del recep-tor común del factor de crecimiento epidérmico, detectados por una prueba aprobada por la FDA. Sanofi recibió la aprobación para Kynamro (mipo-mersen sódico), una sal sódica sintética de fosfo-rotioato oligonucleótido (20 nucleótidos de longitud) para tratar la hipercoleste-rolemia familiar homocigótica, un tipo raro de colesterol elevado (5).
ViiV Healthcare, una compañía farmacéutica independiente conjunta, enfocada en tratamientos para el VIH y formada por Pfizer, GSK y Shiono-gi, recibió la aprobación para Tivicay
(dolutregavir sódico), un inhibidor de la integrasa, indicado para usarse en com-binación con otros agentes antiretrovi-rales para tratar el VIH-1 en adultos y niños de 12 años y mayores que pesen al menos 40 kg. El Tivicay es el primer nuevo tratamiento obtenido por ViiV Healthcare, el cual se estableció en noviembre de 2009 por GSK y Pfizer. Shionogu se unió con una participación del 10% en octubre de 2012. Shionogi también recibió aprobación para Os-phena (ospemifene) para el tratamiento de dispareunia moderada a severa. Pfi-zer recibió la aprobación de su terapia combinada, Duavee (estrógenos con-jugados/bazedoxifeno), para tratar los síntomas vasomotores asociados con la menopausia y para prevenir la osteopo-rosis post-menopáusica.
Referencias1. FDA, “New Drugs at FDA: CDER’s New
Molecular Entities and New Therapeutic Biological Products of 2013” www.fda.gov/drugs/developmentapprovalprocess/druginnovation/default.htm, accessed 19 Nov. 2013.
2. FDA, “FDA Approves Imbruvica for Rare Blood Cancer,” Press Release, 13 Nov. 2013.
3. FDA, “Label for Gazy va,” w w w.ac-cessdata .fda .gov/drugsat fda _docs/label/2013/125486s000lbl.pdf, accessed 15 Nov. 2013.
4. FDA, “Label for Kadcyla,” www.ac-cessdata .fda .gov/drugsat fda _docs/label/2013/125427lbl.pdf, accessed 15 Nov. 2013.
5. FDA, “Label for Kynamro,” www.ac-cessdata .fda .gov/drugsat fda _docs/label/2013/203568s000lbl.pdf, accessed 15 Nov. 2013. PT
Análisis cuantitativo de azúcaresPharmTech: ¿Qué información puede ob-tenerse del análisis cuantitativo de azúca-res en sistemas biológicos?
McRae(Sussex): Aunque la determi-nación de la estructura de los glicanos es crucial para confirmar la síntesis de com-puestos apropiadamente glicosilados, la determinación cuantitativa de azúcares li-bres en sistemas biológicos, así como los azúcares totales (enlazados más libres, lo
cual se obtiene después de romper enla-ces de los azúcares enlazados), también puede proporcionar información valiosa acerca de los procesos biológicos. En sistemas celulares y de fermentación, se utilizan como alimento varios azúcares y otros azúcares se producen como subpro-ductos. Monitoreando el consumo y la producción de estos varios azúcares con el tiempo durante las reacciones corridas bajo varias condiciones, es posible opti-mizar un proceso biológico con el fin de obtener la cantidad máxima de producto deseado.
PharmTech: ¿Qué instrumentación utilizan para la determinación cuantitati-va de azúcares y por qué?
McRae (Sussex): Cromatografía de líquidos-espectrometría de masas (LC-
MS); en particular la LC-MS/MS, está idealmente adaptada para este tipo de análisis. La espectrometría de masas es crucial en esta aplicación porque es alta-mente específica y sensible en la detec-ción de azúcares en muy bajos niveles. Debido a que los compuestos ya son conocidos y se usan estándares para la cuantificación, la LC-MS/MS es una téc-nica dirigida muy poderosa para la cuan-tificación.
PharmTech: ¿Qué avances recientes en tecnología han sido de importancia para el análisis de carbohidratos?
McRae (Sussex): La cada vez mayor resolución y sensibilidad de los espectró-metros de masas y la creciente elección de tipos de instrumentos han tenido un impacto real en el análisis de glicanos y azúcares. También ha sido importante el desarrollo de algunos nuevos agentes derivatizantes.
PharmTech: ¿Por qué se requiere la derivatización para la determinación cuantitativa de azúcares?
McRae (Sussex): La derivatización es un paso clave en la cuantificación de azú-cares reductores libres en solución cuan-do se utiliza LC en fase reversa-MS/MS. Existen muchos azúcares isoméricos di-
La derivatización es un paso clave en la cuantificación de azúcares reductores libres en solución cuando se utiliza LC en fase reversa-MS/MS. -McRae
ferentes con el mismo peso molecular y sólo ligeras diferencias en la estructura. Existen por ejemplo, tres azúcares co-munes de seis carbones que se presentan naturalmente en los sistemas biológicos -galactosa, manosa y glucosa. Todos tie-nen cinco grupos hidroxilo, y es sólo la estereoquímica en los grupos hidroxilo (axial o ecuatorial) la diferencia a estos compuestos. Aunque esta ligera diferen-cia puede llevar a muy diferente reactivi-dad para cada molécula, no es fácil sepa-rarlas una de otra en una columna en fase reversa a menos que estén derivatizadas.
Un reactivo de derivatización común para los carbohidratos es el 1-fenil-3-me-til-5-pirazolona (PMP). No sólo están separables los derivados de los isómeros preparados utilizando este reactivo por HPLC en fase reversa, sino también pue-de lograrse hasta 100 veces más sensibili-dad durante el análisis de espectrometría de masas. La presencia de los grupos ni-trógeno en el reactivo de derivatización es importante para una mayor ionización y eficiencia de fragmentación, mientras que el carácter fenilo y alifático agregado proporciona una mayor retención en el HPLC en fase reversa.
Dos compuestos adicionales, el ácido 2-aminobenzoico (2-AA) y la 2-amino-benzamida (2-AB) también son clásica-mente usados como agentes de derivati-zación para azúcares reductores libres, particularmente para el glico-perfilado cuantitativo de biofarmacéuticos tales como las proteínas terapéuticas y los an-ticuerpos monoclonales. PT
“El papel de la RMN y la Espectroscopía de Masas en el Análisis de Glicanos” Continuación de la pág. 51

Pharmaceutical Technology en Español MARZO / ABRIL 201466
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
Innovación y crecimiento del mercado en
Brasil
REPORTE DE MERCADOS EMERGENTES
Hellen Berger
Brasil es el primer país de Latinoamérica que surge como colaborador biofarmacéutico global.
El mercado farmacéutico en Brasil ha estado crecien-do constante y fuertemente en los últimos cinco años. El país está viendo producción e innovación, apoyándose sobre el soporte clave del gobierno. De
acuerdo a las asociaciones industriales, la innovación biofar-macéutica en Brasil se incrementará en el futuro conforme el país empiece a hacer frente a sus retos y a participar global-mente con compañías locales o multinacionales.
De acuerdo a Abifina, la asociación de Industrias de Quí-micos Finos, Biotecnología y Especialidades de Brasil, los países de Latinoamérica todavía están plantando las semillas para desarrollarse y competir en el mercado biofarmacéutico. Brasil, sin embargo, juega un papel clave en el sector con una serie de universidades e institutos que promueven las accio-nes de investigación y desarrollo, un sistema de salud pública capaz de sustentar parte de la industria farmacéutica global, y una gran población de consumidores en más de 200 millones de brasileños.
Crecimiento del mercado en Brasil“Brasil ha estado definitivamente creciendo constantemente por arriba de la marca del 10% en los últimos cinco años y continuaremos viendo ventas que se incrementan anualmente en 12-15%,” le dijo Nelson Mussolini, presidente ejecutivo de Sindusfarma, a Pharmaceutical Technology. Sindusfarma es la asociación industrial de productos farmacéuticos del estado de Sao Paulo.
De acuerdo a Mussolini, citando las cifras del IMS Health durante los últimos cinco años, el crecimiento de Brasil se considera relevante si se compara con los datos de Europa y EEUU, cuyos niveles de crecimiento fueron de alrededor del 1%. Mussolini dice que el incremento se debe especialmente al hecho de que el mercado laboral local se ha calentados en
los últimos cinco años y ha estado transformando a las clases más bajas en consumidores activos de bienes y servicios. El impacto del país, no obstante, dentro del mercado de productos biológicos, está limitado a la producción y ventas y no nece-sariamente incluye innovaciones relevantes, de acuerdo a Rei-naldo Guimaraes, director de propiedad intelectual de Abifina.
“Nuestro incremento en la participación del mercado bio-farmacéutico es principalmente resultado de las inversiones públicas para el sistema de salud del gobierno, le dijo Guima-raes a Pharmaceutical Technology. El gobierno está actuando como comprador principal, actualmente responsable de tanto como 25% a 30%, con una participación estimada que alcan-zará tanto como 50% en los próximos cinco años, de acuerdo a Guimaraes. El gobierno está estableciendo sociedades clave con compañías locales y multinacionales, privadas y públicas y está financiando expansiones de capacidad de producción local para compañías públicas y privadas. Brasil también está controlando la eficacia y seguridad de los productos farmacéu-ticos a través de la regulación del mercado.
Según Guimaraes, considerando los gastos del gobierno con los productos para la salud, los biofarmacéuticos corres-ponden a sólo 5% en términos de unidades vendidas y tanto como 40% en términos de ventas.
“El gobierno está definitivamente invirtiendo en el desa-rrollo del sector biofarmacéutico local a través de sociedades y con la obtención de tecnología,” dice Mussolini.
De acuerdo al presidente ejecutivo de Sindusfarma, sin embargo, las compañías enfrentan retos para hacerse reditua-bles y todavía carecen de escalas de producción grandes. Pa-rece que las compañías con sede en Brasil buscan incrementar las escalas de producción y la rentabilidad, mientras que el
“Innovación y crecimiento del mercado en Brasil” Continúa en la pág. 69
La importancia de la correcta nivelación

Pharmaceutical Technology en Español MARZO / ABRIL 2014 67
los últimos cinco años y ha estado transformando a las clases más bajas en consumidores activos de bienes y servicios. El impacto del país, no obstante, dentro del mercado de productos biológicos, está limitado a la producción y ventas y no nece-sariamente incluye innovaciones relevantes, de acuerdo a Rei-naldo Guimaraes, director de propiedad intelectual de Abifina.
“Nuestro incremento en la participación del mercado bio-farmacéutico es principalmente resultado de las inversiones públicas para el sistema de salud del gobierno, le dijo Guima-raes a El gobierno está actuando como comprador principal, actualmente responsable de tanto como 25% a 30%, con una participación estimada que alcan-zará tanto como 50% en los próximos cinco años, de acuerdo a Guimaraes. El gobierno está estableciendo sociedades clave con compañías locales y multinacionales, privadas y públicas y está financiando expansiones de capacidad de producción local para compañías públicas y privadas. Brasil también está controlando la eficacia y seguridad de los productos farmacéu-ticos a través de la regulación del mercado.
Según Guimaraes, considerando los gastos del gobierno con los productos para la salud, los biofarmacéuticos corres-ponden a sólo 5% en términos de unidades vendidas y tanto como 40% en términos de ventas.
“El gobierno está definitivamente invirtiendo en el desa-rrollo del sector biofarmacéutico local a través de sociedades y con la obtención de tecnología,” dice Mussolini.
De acuerdo al presidente ejecutivo de Sindusfarma, sin embargo, las compañías enfrentan retos para hacerse reditua-bles y todavía carecen de escalas de producción grandes. Pa-rece que las compañías con sede en Brasil buscan incrementar las escalas de producción y la rentabilidad, mientras que el
En BalancePor: Celedonio Sáinz
Si Usted es dueño de una Balanza, comparador de ma-sas, báscula industrial de alta resolución, etc. es im-portante notar que el dispositivo no sólo sea calibra-do en su ubicación final (in situ), sino también que
sea correctamente nivelado. Hay muchas nuevas herramientas disponibles para equipos de pesaje que permiten a los usua-rios tomar conciencia de la nivelación a través de indicaciones en pantalla, gráficos y alarmas sonoras. Un equipo desnive-lado presenta condiciones perjudiciales para los resultados de un experimento, ahora las Balanzas pueden ser equipadas para documentar la nivelación a través de una “acción” me-morizada, lo que permitirá a los administradores y auditores externos ver la hora del pesaje y confirmar sí los resultados podrían estar alterados o no.
Cualquiera de nosotros que habitualmente nos vemos in-volucrados en el uso de una Balanza, sabemos que la burbuja del nivel se encuentra frecuentemente en la parte posterior del equipo en un lugar difícil de encontrar y se coloca discreta-mente como sí no fuera realmente tan importante. De hecho, es fundamental para la precisión general del resultado. Mien-tras más costoso sea su instrumento, mayor será la resolución (analíticas, semi-micro, micro, etc.) y más significativo es el resultado para Usted.
Ejemplo de nivelación de una Balanza utilizando una pantalla de nivelación asistidaLa nueva tecnología está ya disponible para la correcta ni-velación, utilizando una pantalla de nivelación asistida o el sistema motorizado de nivelación automática. Éstas permiten al usuario nivelar de forma rápida, precisa y segura con el mínimo esfuerzo.
Una visión integral de una Balanza desniveladaHay pocos fabricantes que producen Balanzas sobre el rango de resolución al microgramo = 0.001mg debido a la tecnolo-gía necesaria para mostrar con precisión un resultado repeti-
ble. Cuando estos fabricantes calibran una unidad en parti-cular, aseguran primero que el vial y burbuja de nivel estén perfectamente centrados por un proceso de posicionamiento del instrumento sobre una plantilla que se monta en una mesa que está calibrada para permanecer perfectamente nivelada (dentro de una tolerancia muy pequeña y comprobada por sis-temas de posicionamiento láser) de modo que cada Balanza se fabrica exactamente bajo el mismo punto de referencia.
Por lo tanto, al instalar una Balanza en su posición final, es fundamental que el equipo quede:
• En paralelo a la dirección de la aceleración de la gravedad• Perpendicular al sistema de pesaje
Ejemplo:
Seguir ind. hasta que la básculaesté nivelada
Menú
Volver Continuar
Nivelar la balanza
Girar cada una de las patas en
la dirección de la flecha como se
indica hasta que el icono de
nivel rojo esté centrado y se
ponga verde
a
aA = W
W
cos
En un ángulo a = 0.3 °, implica lo siguiente:A=W•cos(a)=W•0.9999875200g x 0.9999875 = 199.9975g
Esto significa que una muestra de 200 g pesa 0.0025 g me-nos en una Balanza inclinada ¡Siempre nivele su instrumento usando el indicador de nivel como guía, comprendiendo los efectos de la gravedad en la precisión del pesaje!
Figura 1: El diagrama muestra la nueva generación de Balanzas Cubis de Sartorius adicionada con nivelación asistida en la pantalla.
La importancia de la correcta nivelación

Pharmaceutical Technology en Español MARZO / ABRIL 201468
En balancE
Antes de continuar, abordando una discusión básica de los fundamentos de pesaje, es necesario entender cómo los efec-tos de la gravedad en la Balanza pueden causar errores po-tencialmente grandes en la precisión general. El Peso, como todos sabemos, se define así:
La fuerza del Peso que en realidad sentimos no es la fuer-za de la gravedad, es la fuerza normal (una fuerza de contacto hacia arriba) ejercida por la superficie que pisamos, que se opone a la gravedad y no nos deja caer al centro de la Tierra. Ésta fuerza normal, llamada el Peso aparente, es la que se mide con una Balanza.
Representada en una ecuación simple:Peso (W) = masa (m) x gravedad 9.80665 m/s 2 ó 32.174 pies/s 2 (a)
La gravedad en la ecuación anterior es un promedio para el planeta Tierra y no se debe asumir para un lugar específi-co donde se utiliza una Balanza. La gravedad puede ser, en efecto, hasta en un 0.5% diferente de un lugar a otro, incluso mover el equipo de un sótano a un tercer piso puede causar la moderación suficiente en el efecto gravitatorio que resulte
en un error sobre las Balanzas muy precisas. Por lo tanto, una Balanza debe encontrarse en su posición final antes de una calibración inicial, ya que el simple hecho de mover el instru-mento después de la calibración será, con toda probabilidad, la causa de encontrarse fuera de tolerancia.
Sin afán de ser una molestia, es fundamental darse cuenta que no sólo la nivelación sino también la ubicación final del instrumento tendrá un impacto directo sobre su rendimien-to. Una Balanza siempre es ajustada en la fábrica e incluso puede venir con un “certificado de calibración” sin embargo, éste certificado simplemente refleja un sistema de calidad del fabricante en su planta. Por lo que, una Balanza tiene que ser ajustada y calibrada con pesas certificadas en su posición úl-tima de instalación.
Otros factoresMuchos otros factores pueden comprometer la precisión ge-neral de su instrumento, incluyendo las fluctuaciones de tem-peratura y humedad, vibraciones, cargas estáticas, muestras cargadas electromagnéticamente y el empuje del aire. Los Fabricantes de Balanzas han adoptado medidas para ayudar a reducir y a veces aliviar los factores ambientales, sin embar-go, es importante tener especial cuidado en la ubicación de su instrumento de precisión. Recuerde que a las Balanzas no les gusta el cambio, así que, siempre úsela en el mismo lugar y bajo condiciones ambientales estables pero sobre todo man-téngala siempre nivelada.
Ejemplo:
g
g
g 1 2
1 2
(
( (
(≈
≈
=
(RE+4m)
g g(RE+ h)
Mover una balanza a una posición 4m másalta, implica tener una diferencia de lecturade -0,3mg (para una carga de 200g).
9.77 m/ s2
h
h
h
REart
< g < 9.83 m/ s2
4 m6 370 000 m
0.9999987
6 370 000 m
Figura 3: Cada vez que se mueva una Balanza a otro lugar, debe ser ajustada y calibrada.
CALENDARIO DE EVENTOS
Cuando usted contacte a alguno de los organizadores de éstos eventos, por favor mencione que vio su evento en
MARZO2-6 PITTCON 2014. Lugar: McCormick Place, Chicago, Il USA. Contacto / Info: [email protected] E-mail: [email protected], Teléfonos: 412-825-3220, Fax: 412-825-3224, Página Web: http://pittcon.org
4-6 EXPO MANUFACTURA 2013. Integración de nuevas tecnologías. Lugar: Cintermex, Monterrey, N.l. México. Contacto / Info: [email protected], Teléfonos: (52 55) 1087-1650 ext. 1136, Página Web: www.expomanufactura.com.mx
10-12 Duphat Dubái. Lugar: Dubai International Convention & Exhibition Center DIDEC, Sheikh Zayed Road, Dubái, EÁU. Contacto / Info: Attracta D’Silva, E-mail: [email protected], Teléfonos: +971 (0)4 3624717, Página Web: www.duphat.ae
10-13 DCAT Week 2014. Lugar: New York, NY. Contacto / Info: Diane Packard / Project Coordinator, E-mail: [email protected], Teléfonos: 1-800-640-322, Página Web: www.dcat.org/Pages/week_DCATweek.aspx
promueva su evento aquí
contrataciones (55) 5659-8880
gobierno busca menos dependencia de las importaciones y un mercado local más grande, estableciendo una fuerte sociedad farmacéutica destinada a crecer.
Con la economía todavía bajo control y regulación equi-valente a la de las naciones desarrolladas, los desafíos ahora están dentro de los altos impuestos y costos así como estrictos controles de precio, de acuerdo a Mussolini. Dichos proble-mas impactan la rentabilidad y afectan las acciones futuras de I+D así como la supervivencia a largo plazo de la mayoría de las compañías.
“Brasil compite globalmente en términos de participación del mercado… sin embargo, para volverse un competidor global en términos de nuevos productos o mejores productos, todavía tenemos problemas que enfrentar,” agrega el funcio-nario. En términos de I+D, por ejemplo, los retos se atribuyen a la carencia de capacidad tecnológica del país para producir innovación relevante, dice Guimaraes.
El gobierno de Brasil, a través de su Ministerio de Salud, tiene el objetivo de sustituir importaciones para productos producidos localmente mediante el establecimiento de nego-
El gobierno está estableciendo sociedades clave con compañías
locales y multinacionales, privadas y públicas.
“Innovación y crecimiento del mercado en Brasil” Continuación de la pág. 66
V
V
Celedonio Sáinz De Urquijolab Instruments - line ManagerNorthern, Central & Caribbean Regions of latin AmericaSartorius de Mé[email protected]
ABRIL9-11 BioPh Japan Tokio. Lugar: Tokyo Big Sight -Tokyo International Exhibition Center, 3-21-1 Ariake Kotu-ku 135-0063 Tokio, Tokio, Japón. Contacto / Info: www.events.ubm.com, E-mail: [email protected], Teléfonos: +44 (0)20 79215000, Página Web: www.cphijapan.com
16-17 CPhI Russia Lugar: lenexpo Exhibition Complex, St. Petersburg, Russia. E-mail: [email protected], Teléfonos: 30076951, Página Web: www.cphi.com/russia/
16-18 Apteka Expo Central Asia Tashkent. Lugar: UEC Uzexpocentre 107, Amir Temur street 100084 Tashkent, Taskent, Uzbekistán. E-mail: post@,ite-uzbekistan.uz, Teléfonos: +998 (0)71 1130180, Página Web: www.ite-uzbekistan.uz
24-26 Austropharm Viena. Lugar: Messe wien Messeplatz 1 A-1021 Viena, Viena, Austria. Contacto / Info: www.austropharm.at/, E-mail: [email protected], Teléfonos: +43 (0)662 44770, Página Web: www.austropharm.at/
MAYO 8-14 INTERPACK. Lugar: Messe Düsseldorf - Düsseldorf, Alemania. Contacto / Info: Tel: +52 (55) 5524549890 / +52 (55)24547235, E-mail: [email protected], Página Web: www.interpack.com/
20-22 CPhI South East Asia Yakarta. Lugar: Jakarta International Expo (JIE) Gedung Pusat Niaga lt. 1 Arena PRJ Kemayoran 10620 Yakarta, Yakarta, Indonesia. E-mail: [email protected], Teléfonos: +44 (0)20 79215000, Página Web: www.cphi-sea.com/
18-20 Interphex NY Nueva York. Lugar: Jacob K. Javits Convention Center, 655 west 34th Street, 10001 Nueva York, Nueva York, EE.UU. E-mail: [email protected], Teléfonos: +1 (2)03 8404800, Página Web: www.interphex.com
25-27 Infarma Madrid 2014. Lugar: Feria de Madrid en el recinto ferial IFEMA, en los pabellones 1 y 3. Madrid España. Contacto / Info: Clara Álvarez Fernández, E-mail: [email protected], Teléfonos: 91 406 83 81, Fax: 91 406 84 63, Página Web: www.infarma.es
26-28 Expofarma 2014 Lugar: world Trade Center Ciudad de México. Contacto / Info: Francisco Javier Padilla Arellanes. E-mail: [email protected], Teléfonos: +52 (55) 918- 32063, Página Web: www.expofarma.org.mx, www.expofarma2014.com.mx

Pharmaceutical Technology en Español MARZO / ABRIL 2014 69
gobierno busca menos dependencia de las importaciones y un mercado local más grande, estableciendo una fuerte sociedad farmacéutica destinada a crecer.
Con la economía todavía bajo control y regulación equi-valente a la de las naciones desarrolladas, los desafíos ahora están dentro de los altos impuestos y costos así como estrictos controles de precio, de acuerdo a Mussolini. Dichos proble-mas impactan la rentabilidad y afectan las acciones futuras de I+D así como la supervivencia a largo plazo de la mayoría de las compañías.
“Brasil compite globalmente en términos de participación del mercado… sin embargo, para volverse un competidor global en términos de nuevos productos o mejores productos, todavía tenemos problemas que enfrentar,” agrega el funcio-nario. En términos de I+D, por ejemplo, los retos se atribuyen a la carencia de capacidad tecnológica del país para producir innovación relevante, dice Guimaraes.
El gobierno de Brasil, a través de su Ministerio de Salud, tiene el objetivo de sustituir importaciones para productos producidos localmente mediante el establecimiento de nego-
ciaciones de transferencia de tecnología. Dichas medidas han sido tradicionalmente usadas para vacunas y más recientemen-te para fármacos sintéticos así como para productos biofarma-céuticos.
Crecimiento futuroLa crisis financiera global, que empezó en 2008, no afectó a Brasil tan severamente como afectó a EEUU y Europa; por lo tanto, el mercado farmacéutico local enfrentó un menor impac-to también. Durante los pasados ocho años (2003-2011), Brasil fue desde ocupar la décima posición entre las compañías far-macéuticas globales a ser el sexto mercado farmacéutico más grande, de acuerdo al IMS Health. Para 2016, un año en el cual el país será la sede de los Juegos Olímpicos, el IMS esti-ma que Brasil ocupará la cuarta posición, con un crecimiento estimado entre 14-15%, sólo detrás de EEUU, China y Japón y superando a los mercados altamente establecidos de Francia y Alemania.
“Yo personalmente creo que estos estimados son muy op-timistas, sin embargo, estamos ciertamente en la dirección de un 12% de crecimiento, lo que incluiría a Brasil en el quinto lugar...,” dice Mussolini.
No obstante, “estoy seguro que el IMS Health sabe exac-tamente lo que está diciendo,” agrega Mussolini. “Yo creo que no hay razones para problemas serios,” concluye Guimaraes.
-Hellen Berger es corresponsal de noticias de negocioscon sede en Sao Paulo, Brasil
El gobierno está estableciendo sociedades clave con compañías
locales y multinacionales, privadas y públicas.
“Innovación y crecimiento del mercado en Brasil” Continuación de la pág. 66

Pharmaceutical Technology en Español MARZO / ABRIL 201470
EOlIS, líder mundial en diseño, construcción y validación de sistemas críticos
(HVAC), con más de 45 años de experiencia, se dio a la tarea de demostrar el costo
real del sistema de filtración comparando filtros de diferente rango de precios.
los resultados revelaron que los filtros EOlIS pueden disminuir los gastos hasta en
un 50% en relación a otros filtros.
Es necesario tomar en cuenta que un sistema de aire puede representar un 60% del
consumo de energía de una planta, y los filtros representan el 70% de los parámetros
de diseño para el sistema HVAC.
¿CUAL ES EL VERDADERO COSTODE UN SISTEMA DE FILTRACION?
Son similares. Sus costos de operación no lo son.
Eolis llevó a cabo una prueba por 271 días en una UMA con 16 filtros clase E10 (flujo de 48.000 m3/h, motor de 40 hp), obteniendo los siguientes resultados (el estudio sólo abarcó la etapa 3 E10 @ EN1822):
1.- REDUCCION EN CONSUMO DE ENERGIA. los filtros EOlIS consumieron 10,800 kw/h menos, que en comparación con otros filtros similares representó un 35% menos energía. Este porcentaje generó disminución de los gastos de energía por $16,700 pesos en un lapso de los 271 días.
2.- TIEMPO DE VIDA. los filtros EOlIS han superado el tiempo de vida de otros filtros esperando casi duplicarlo, esto representa una disminución de aproximadamente 50% en los gastos de compra de filtros.
3.- REDUCCION EMISIONES DE CO2. la reducción de energía generó ahorros de 5.32 toneladas de CO2.
Presión Filtros Eolis vs Competencia
FILTRO COMPETENCIA CLASE 10
FILTRO EOLIS CLASE 10
A los 271 días el filtro Eolis apenas alcanza la presión inicial de otros filtros.
CONTRATOS EOLISEolis a su vez ha desarrollado una metodología para el manejo de contratos anuales con sus clientes:1.-Respaldo técnico especializado2.-Homologación filtros (Sistema Pareto) 3.-Reducción de los costos anuales de filtración y energía 4.-Programa de entregas 5.-Servicios adicionales: validaciones, mantenimiento, refacciones, contratos, pólizas.
MITOS Y VERDADES DE LOS FILTROS los contundentes resultados demostraron que es necesario comparar a detalle todas las características de los filtros antes de realizar la compra. También se demostró que filtros más baratos no necesariamente significan disminución de gastos y en ocasiones se sacrifica la calidad de los filtros y del proceso.
Eolis te invita a que solicites una propuesta de optimización.
Todos nuestros filtros cumplen con las normatividades aplicables: Ashrae 52.1, 52.2, EN1822, EN779, EUROVENT.
Tel. +52 (55) 3640 1400 www.eolis.com.mx [email protected] [email protected]

Directorio Clasificado
MAQUINARIA DE ACONDICIONAMIENTO / LLENADORAS Y TAPONADORAS
EQUIPO DE PROCESO / ENCAPSULADORAS DE GELATINA BLANDA
VARIOS / CHAT MASCOTAS
El lugar para los amantes de sus mascotas
Síguenos en...
desdeRecibe consejos e información para el cuidado de tu mascota y Consulta a Expertos que responderán a tus principales inquietudes sobre: • Comportamiento / Entrenamiento• Alimentación• Higiene• Control de plagas• Enfermedades / padecimientos comunes• y más Comparte experiencias con otros dueños de mascotas Conoce a otras mascotas como la tuya Encuentra pareja para tu mascota Compra regalos para tu mascota
Encuentra, Participa y Aprende
GRANULADORES / MOLINOS
INST. Y EQUIPO PARA INSPECCION CONTROL Y MONITOREO DE PROCESOS
MAQUINARIA DE ACONDICIONAMIENTO / BLISTERAS Y ENCARTONADORAS
Son similares. Sus costos de operación no lo son.
/h menos, que en comparación con otros filtros similares representó un 35% menos energía. Este porcentaje generó disminución de los gastos de energía por $16,700 pesos en un lapso de los 271 días.

Cuando usted contacte a alguno de estos anunciantes,
por favor mencione que vió su anuncio en
ÍND
ICE
DE
AN
UN
CIA
NT
ES
Cápsulas de Gelatina Blanda
Exposición de Distribuidores de Instrumentos para Uso Científico y M
ateriales para Laboratorio
Sistemas de Aire Acondicionado
Exposiciones
Maquinaria de Acondicionam
iento / Blisteras y Encartonadoras
Granuladores / Molinos
Varios / Estetica para Mascotas
Inst. y Equipo para Inspección. Control y Monitoreo de Procesos
Equipo de Proceso
Maquinaria de Acondicionam
iento y Embalaje
Varios / Chat mascotas
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Instrumentos y Equipos Analíticos - Biotecnología
Equipo de Proceso / Encapsuladoras de Gelatina Blanda
Sistemas de Agua: D
eionizadores, Osm
osis inversa, Destiladores, Loops, Tanques, Etc.
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
CATALEN
T
DICLA
B, A.C.
EOLIS A
MERICA
LATINA
S.A. D
E C.V.
EXPO FA
RMA
FABRIM
A - PACK &
PROCESS
FITZ PATRICK - PACK & PRO
CESS
MÁ
S QU
E PERRUQ
UERIA
OPTREL - PACK &
PROCESS
PACK & PRO
CESS - ACG
PACK FEEDER - PACK &
PROCESS
PETFACEBOO
K
ROTA
- PACK & PRO
CESS
SARTO
RIUS
SKY SOFTG
EL CO., LTD
- PACK & PRO
CESS
VEOLIA
WATER SYSTEM
S MÉXICO
S.A. D
E C.V.
WATSO
N-M
ARLO
W, S. D
E R.L. DE C.V.
4a. DE FO
RROS
531523717169711697171
2a DE FO
RROS
71
3a DE FO
RROS
7
17381114169513151241026
PR
OD
UC
TO
/SE
RV
ICIO
EM
PR
ES
AN
o. P
ÁG
INA
NO
. PA
RA
TAR
JETA
DE
S
ER
VIC
IO A
L L
EC
TO
R
ww
w.catalent.com
/ Email: consultas@
catalent.com
ww
w.expodiclab.com
/ e-mail: diclab@
diclab.com.m
x
ww
w.eolis.com
.mx y w
ww
.comsaem
te.com
ww
w.expofarm
a2014.com.m
x
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ ww
w.fitzm
ill.com
facebook Mas Q
Perruqueria, twitter @
MA
SQU
EPERRUQ
UERIA
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.petfacebook.com
.mx
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.sartorius.com
/ Email: sartorius-stedim
@sartom
ex.com.m
x
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.veoliaw
aterst.com.m
x / Email: farm
a.mexico@
veoliawater.com
ww
w.w
mflexicon.com
/ ventas@w
mpg.m
x
DIR
EC
CIÓ
N W
EB
/ CO
RR
EO
EL
EC
TR
ÓN
ICO

Marque en la tarjeta de servicio al lector el No. 2

Mar
que
en la
tarje
ta d
e se
rvic
io a
l lec
tor e
l No.
5





























