Embed Size (px)
Citation preview

INSENSIBILIDAD ANDROGENICA
Juan sebastian lopez

M O R R I S S Y N D R O M E
El síndrome de insensibilidad Androgenica (SIA) es
una enfermedad relacionada con la inactivación de la
AR, debido a una mutación que inactiva la
diferenciación sexual masculina, y causa un espectro
de anomalías fenotípicas que tienen como elemento
común la pérdida de las características
reproductivas.1

El síndrome de Morris es una enfermedad recesiva ligada al cromosoma X
debido a una falta de sensibilidad total o parcial a los andrógenos, lo que resulta
en un fracaso de la masculinización normal de los genitales externos en los
individuos cromosómicamente macho. Esta falta de virilización puede ser
completa o parcial en función de la cantidad de función residual del receptor de
andrógenos. El fenotipo de las personas con síndrome parcial de insensibilidad a
los andrógenos puede variar de leve virilized genitales externos femeninos a
ligeramente undervirilized genitales externos masculinos. Se describe un caso de
síndrome de insensibilidad parcial a los andrógenos en un paciente de 21 años de
edad, con 46, cariotipo XY, las masas inguinales bilaterales, agrandamiento del
clítoris y parcial de fusión labial posterior. La atención quirúrgica consistió en
orquiectomía bilateral y la cirugía plástica de los genitales externos. El paciente
fue sometido a la terapia de reemplazo de estrógeno.2

Reporte de un caso.
Un hombre de 49 años de edad, con características undermasculinized y antecedentes de
criptorquidia y azoospermia.
INTERVENCIÓN (S):
La evaluación hormonal y las pruebas genéticas del gen del receptor de andrógenos (AR).
La variable principal (S):
Los niveles hormonales y los cromatogramas de secuencia de la persona afectada y su madre.
RESULTADO (S):
Hemos encontrado T total en el rango normal y altos niveles de gonadotropinas. Cariotipo fue 46,
XY. Las pruebas genéticas identificó una nueva mutación del exón 1 del AR, lo que resultó en una
alanina a la sustitución de la serina en el dominio de transactivación en el codón 240 (A240S).
Catorce otras mutaciones del exón 1 de la RA se han asociado con MAIS hasta la fecha.
CONCLUSIÓNES (S):
Los A240S nueva mutación de la AR participa en MAIS, un síndrome asociado con azoospermia.3

Se estudió una familia con dos primos que fueron diagnosticados con el síndrome
de insensibilidad a los andrógenos completo, un trastorno ligado al cromosoma X
causada por mutaciones en el gen receptor de andrógenos. Un análisis de pedigrí
y un estudio molecular por PCR y secuenciación del ADN aclaró la situación de
cada mujer de la familia del receptor de andrógenos y reveló una mutación que
consiste en la deleción del exón 2 y los intrones que rodean el gen receptor de
andrógenos. Sobre la base de las posiciones de nucleótidos relativos, llegamos a la
conclusión de que la mutación de deleción en el exón 2 y sus alrededores intrones
fue de aproximadamente 6000 a 7000 pb. Esta mutación no, previamente
caracterizado completamente mediante la secuenciación del ADN, fue responsable
del síndrome de insensibilidad a los andrógenos completo en esta familia. Análisis
de pedigrí con un estudio molecular del gen receptor de andrógenos en las
familias afectadas facilita el consejo genético proporcionado a miembros de la
familia.

MÉTODOS:
Se revisaron retrospectivamente las historias clínicas de pacientes seguidos durante los últimos tres años.
Los sujetos fueron divididos en tres grupos etiológicos de acuerdo a sus cariotipos. Los diagnósticos
definitivos en cada subgrupo se establecieron en las investigaciones clínicas y de laboratorio que incluyen
imágenes abdominopélvica, así como basal y estimulada medidas de la hormona. Prueba genética molecular, a
excepción de CYP21A2 gen, no pudo realizarse.
RESULTADOS:
De un total de 95 pacientes, 26 tenían el cromosoma sexual DSD, de 45 años tenía 46, XY DSD y 24 tenían 46,
XX DSD. Las causas más comunes de DSD fueron el síndrome de Turner (ST), hiperplasia suprarrenal
congénita (CAH) y el síndrome de insensibilidad a los andrógenos (SIA). Hubo una amplia variación en la edad
de presentación que van desde 1 día hasta 17,5 años con una media de 6,5 ± 6,5 años. Las quejas más
frecuentes a la presentación genitales ambiguos, hipospadias perineal aislada y baja estatura.
CONCLUSIÓN:
Los resultados de nuestro estudio demuestran que el nuevo sistema de clasificación de DSD conduce a un
cambio importante en la distribución de los diagnósticos etiológicos de DSD, que se ejemplifica por las
frecuencias significativas de TS y el síndrome de los testículosa desaparecidos. Esta alteración se amplía el
espectro clínico y aumenta la edad media al diagnóstico. Sin embargo, las causas más comunes de los
genitales ambiguos, tales como hiperplasia suprarrenal congénita y AIS, permanecen sin cambios. Otros
estudios utilizando los análisis genéticos moleculares son necesarios para dar una distribución más precisa de
las etiologías de DSD, especialmente en el 46, los pacientes XY.4

IMAGEN 1
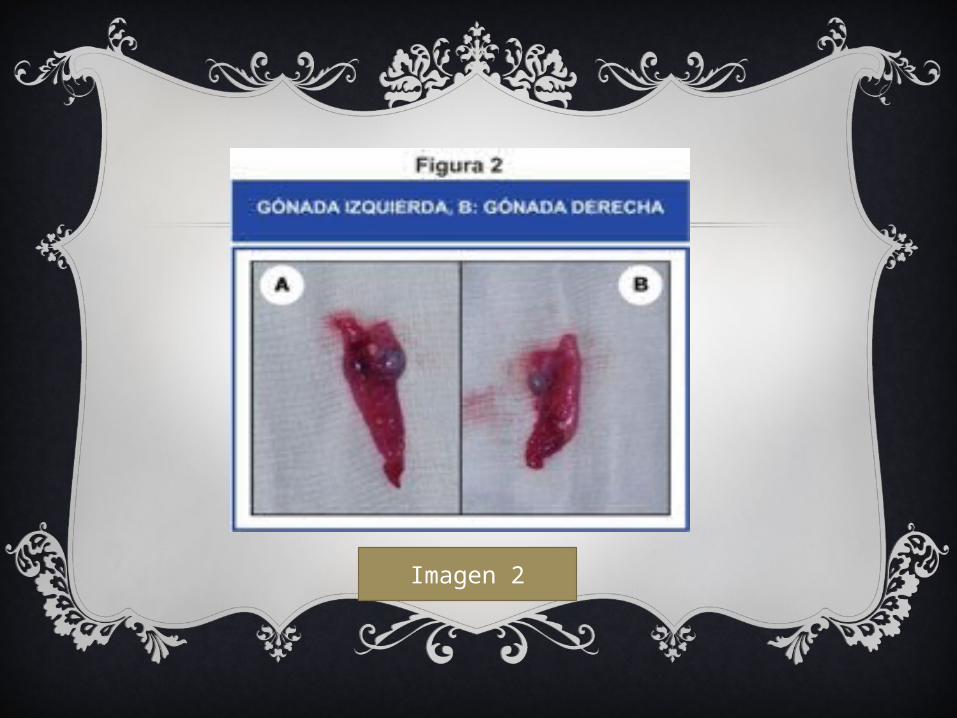
Imagen 2
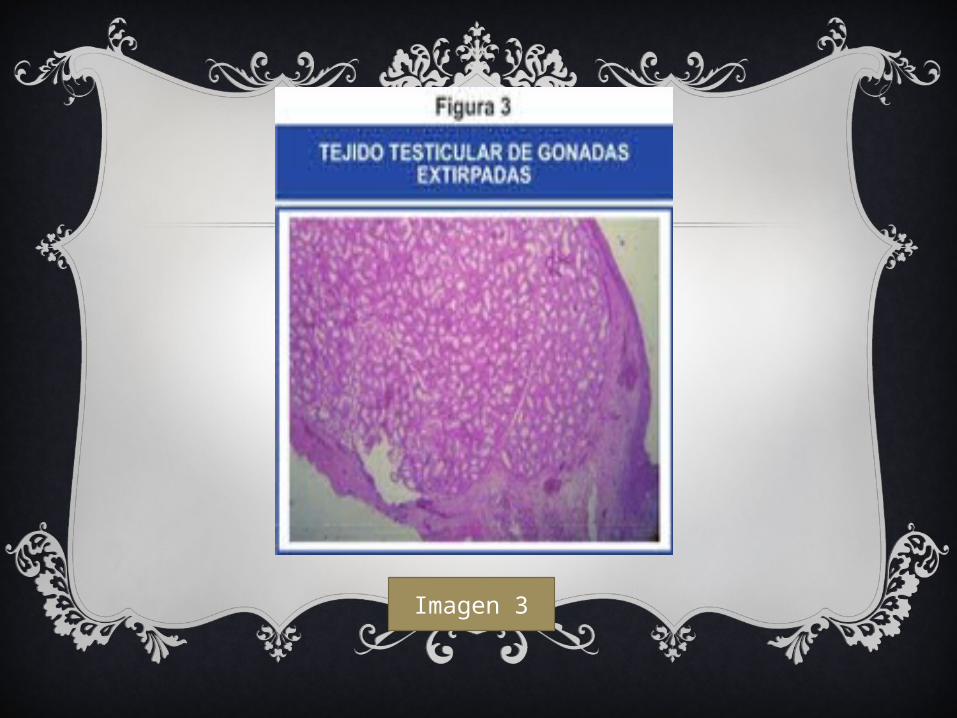
Imagen 3
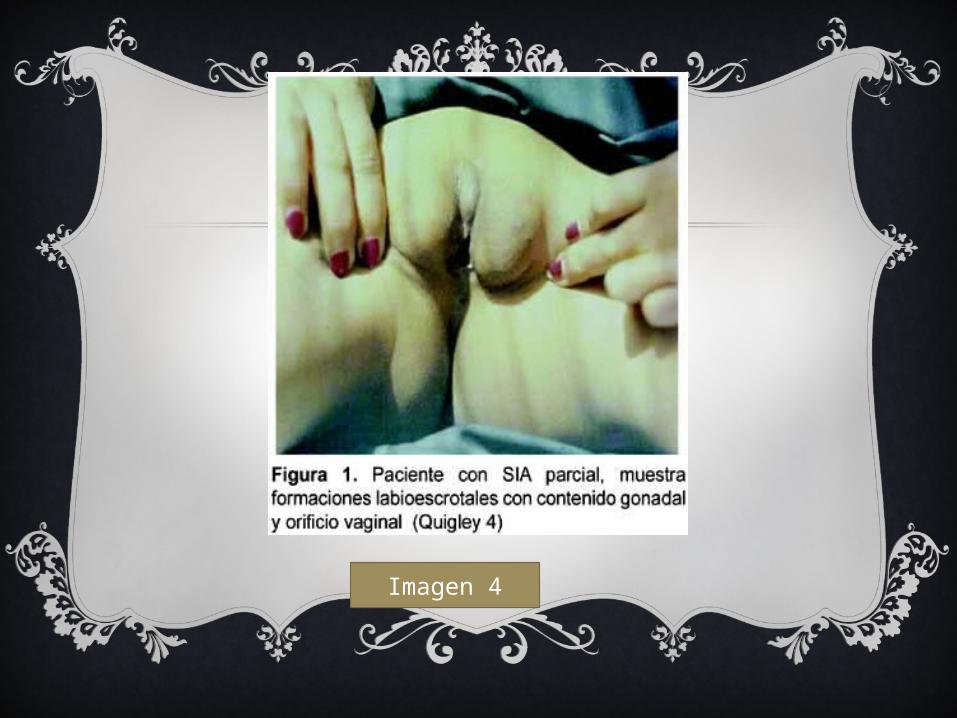
Imagen 4
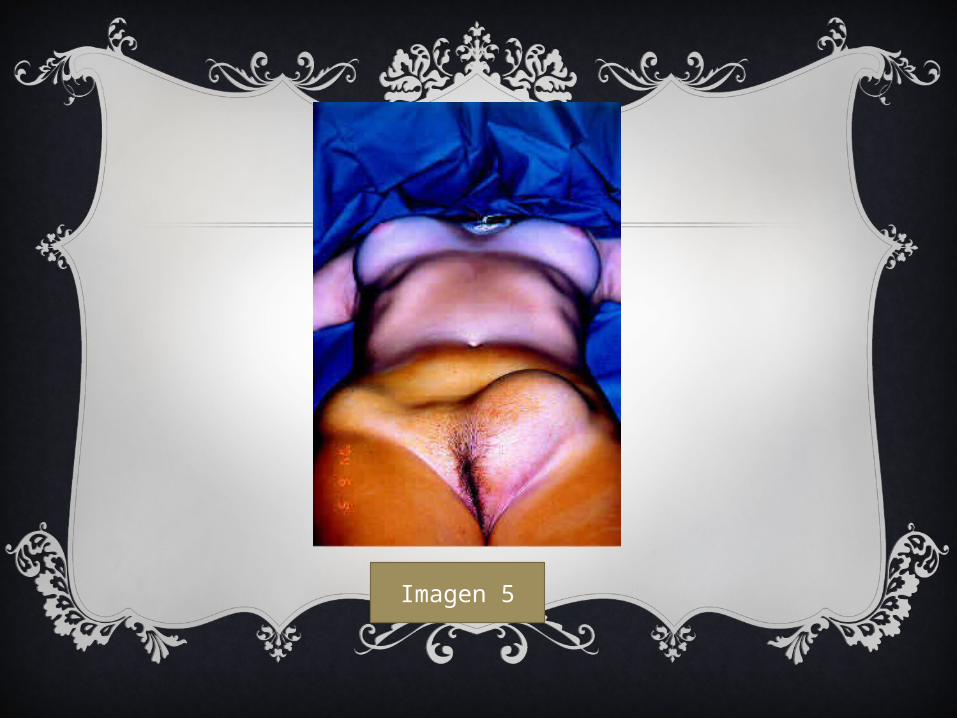
Imagen 5

BIBLIOGRAFIA
1. Dell'Edera D, Malvasi A, Vitullo E, Epifania AA, Tinelli A, Laterza F, Novelli A, Pacella E, Mazzone E,
Novelli G. Androgen insensitivity syndrome (or Morris syndrome) and other associated pathologies.Eur
Rev Med Pharmacol Sci. 2010 Nov;14(11):947-57.http://www.ncbi.nlm.nih.gov/pubmed/21284344
2. Creta M, Smelzo S, Di Vito C, De Stefano G, Forchia F, Chiancone F, Imbimbo C. [Morris syndrome:
description of a case characterized by partial androgen insensitivity].Urologia. 2010 Oct-Dec;77(4):271-
3. http://www.ncbi.nlm.nih.gov/pubmed/21234872
3. Goglia U, Vinanzi C, Zuccarello D, Malpassi D, Ameri P, Casu M, Minuto F, Foresta C, Ferone
D.Identification of a novel mutation in exon 1 of androgen receptor gene in an azoospermic patient with
mild androgen insensitivity syndrome: case report and literature review.Fertil Steril. 2011
Nov;96(5):1165-9. Epub 2011 Sep 29. http://www.ncbi.nlm.nih.gov/pubmed/21962961
3. Li BK, Ding Q, Wan XD, Wang X. Clinical and genetic characterization of complete androgen
insensitivity syndrome in a Chinese family. Genet Mol Res. 2011 May 31;10(2):1022-31.
http://www.ncbi.nlm.nih.gov/pubmed/21710452

4. Erdogan S, C Kara, Uçaktürk A, Aydın clasificación de M. etiológica y la evaluación
clínica de los niños y adolescentes con trastornos de sexo development.J Clin Pediatr
Res Endocrinol. 2011; 3 (2) :77-83. Epub 2011 Ago 6.http :/ /
www.ncbi.nlm.nih.gov/pubmed/21750636
Imagen1: http://web.ebscohost.com/ehost/imageQuickView?sid=183d7bab-097a-4a49-
a0c4-
e1d2c5bb3fb5@sessionmgr111&vid=4&ui=16124593&id=59289331&parentui=59289
331&tag=AN&db=lth
Imagen 2: http://web.ebscohost.com/ehost/imageQuickView?sid=183d7bab-097a-
4a49-a0c4-
e1d2c5bb3fb5@sessionmgr111&vid=4&ui=16124594&id=59289331&parentui=59289
331&tag=AN&db=lth
Imagen3http://imageserver.ebscohost.com/img/imageqv/actual/
e0c/20100901/16124595.jpg?
ephost1=dGJyMNXb4kSepq84xNvgOLCmr0qepq5Srqa4SK6WxWXS
Imagen 4:
http://revistas.concytec.gob.pe/img/revistas/rpp/v61n1/a04fig01.jpg
Imagen 5:
http://www.actasurologicas.info/v25/n04/images/2504NC04F1.jpg





























