Embed Size (px)
Citation preview

Interferon-a induction through Toll-like receptorsinvolves a direct interaction of IRF7 with MyD88and TRAF6
Taro Kawai1, Shintaro Sato1, Ken J Ishii1, Cevayir Coban1, Hiroaki Hemmi2, Masahiro Yamamoto2,Kenta Terai3, Michiyuki Matsuda3, Jun-ichiro Inoue4, Satoshi Uematsu2, Osamu Takeuchi1,2 & Shizuo Akira1,2
Toll-like receptors (TLRs) are involved in the recognition of microbial pathogens. A subset of TLRs, TLR7, TLR8 and TLR9,
induces antiviral responses by producing interferon-a (IFN-a). Production of IFN-a is dependent on the Toll–interleukin-1
receptor domain–containing adaptor MyD88. Here we show that MyD88 formed a complex with the transcription factor IRF7
but not with IRF3. The death domain of MyD88 interacted with an inhibitory domain of IRF7, and this interaction resulted
in activation of the IFN-a-dependent promoters. Furthermore, the adaptor molecule TRAF6 also bound and activated IRF7.
Ubiquitin ligase activity of TRAF6 was required for IRF7 activation. These results indicate that TLR-mediated IFN-a induction
requires the formation of a complex consisting of MyD88, TRAF6 and IRF7 as well as TRAF6-dependent ubiquitination.
The family of Toll-like receptors (TLRs) in vertebrates detects con-served structures found in a broad range of pathogens and triggersinnate immune responses that include production of inflammatorycytokines, chemokines and type I interferons IFN-a and IFN-b bymacrophages and dendritic cells (DCs)1,2. At present, 11 members ofthe TLR family (TLR1–TLR11) have been identified and geneticstudies have demonstrated pathogen-derived components recognizedby each TLR3–14.
Signaling pathways triggered by TLRs have been clarified2,15. AllTLRs have N-terminal leucine-rich repeats responsible for the recog-nition of pathogens and a C-terminal Toll–interleukin-1 receptor (TIR)domain that mediates intracellular signaling through homotypicinteractions between TLRs and TIR domain–containing adaptorssuch as MyD88, Trif (also known as TICAM-1), TIRAP (alsoknown as MAL) and TRAM16–26. After recognition of pathogen-derived components, individual TLRs trigger distinctive responses byrecruiting a different combination of adaptor molecules. MyD88is commonly used by all TLRs. The association of the TLRs andMyD88 in turn recruits members of interleukin 1 (IL-1) receptor–associated kinase (IRAK) family through interactions between thedeath domains. Once phosphorylated, IRAK1 and IRAK4 dissociatefrom a receptor complex, then associate with the adaptor moleculetumor necrosis factor receptor–associated factor 6 (TRAF6), a mem-ber of the TRAF family. TRAF6 forms a ubiquitin-conjugating enzymecomplex consisting of Ubc13 and Uev1A, serving as the ubiquitin E3ligase that promotes synthesis of lysine 63–linked polyubiquitin
chains27. Biochemical analysis has identified the protein kinaseTAK1 as being one of the targets of TRAF6 (ref. 27). ActivatedTAK1 then triggers activation of the transcription factor NF-kBthrough the kinase cascade involving the canonical IkB kinase com-plex (IKKa, IKKb and IKKg), resulting in the induction of inflam-matory cytokines15.
Type I interferons are induced by a subset of TLRs. Interferonregulatory factors (IRFs) such as IRF3 and IRF7 are thought to beresponsible for virus- or TLR ligand–induced induction of type Iinterferon. IRF3 and IRF7 are activated by phosphorylation of theirmultiple serine clusters in the C terminus by virus-activatedkinases28,29. Serine phosphorylation triggers dimerization and nucleartranslocation of these IRFs. IRF3 mainly regulates IFN-b induction bydouble-stranded RNA and bacterial lipopolysaccharide (LPS), whichare recognized by TLR3 and TLR4, respectively3,9,10. IRF3 activationby TLR3 and TLR4 ligands occurs independently of MyD88 butdepends on Trif22,25. Two IKK-related kinases, TANK-binding kinase1 (TBK1) and inducible IKK (IKKi), are involved in phoshorylationand activation of IRF3 (refs. 30,31). Cells from TBK1-deficient micehave a substantial decrease in the induction of the gene encoding IFN-b by LPS, demonstrating that the Trif-TBK1-IRF3–dependent pathwayregulates IFN-b induction by TLR4 (refs. 32–34). In contrast, IRF7 hasa preferential ability to activate the IFN-a promoters28,35. AlthoughIRF3 is ubiquitously expressed, most cells do not express or weaklyexpress IRF7, but inducibly express IRF7 after type I interferonstimulation35,36. However, plasmacytoid DCs constitutively express
Published online 7 September 2004; doi:10.1038/ni1118
1ERATO, Akira Innate Immunity Program, Japan Science and Technology Agency, 2Department of Host Defense, 3Department of Tumor Virology, Research Institute forMicrobial Diseases, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan. 4Division of Cellular and Molecular Biology, Department of Cancer Biology,Institute of Medical Science, University of Tokyo, Shirokanedai, Minato-ku, Tokyo 108-8639, Japan. Correspondence should be addressed to S.A.([email protected]).
NATURE IMMUNOLOGY VOLUME 5 NUMBER 10 OCTOBER 2004 1061
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y
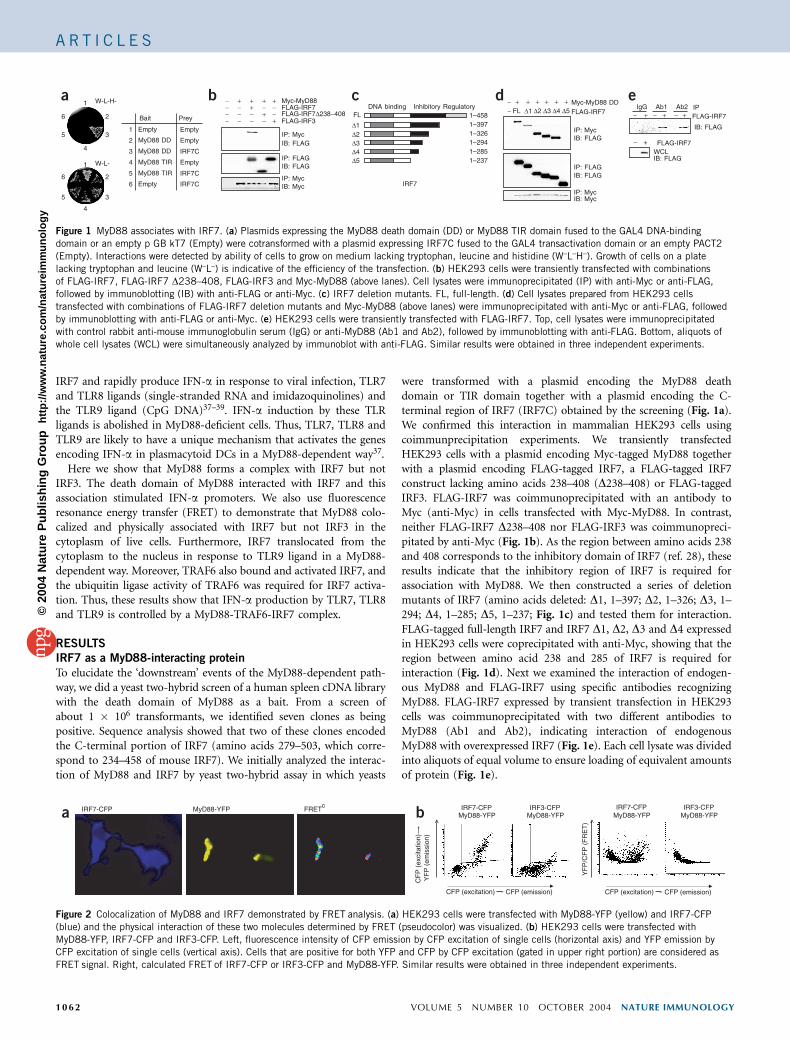
IRF7 and rapidly produce IFN-a in response to viral infection, TLR7and TLR8 ligands (single-stranded RNA and imidazoquinolines) andthe TLR9 ligand (CpG DNA)37–39. IFN-a induction by these TLRligands is abolished in MyD88-deficient cells. Thus, TLR7, TLR8 andTLR9 are likely to have a unique mechanism that activates the genesencoding IFN-a in plasmacytoid DCs in a MyD88-dependent way37.
Here we show that MyD88 forms a complex with IRF7 but notIRF3. The death domain of MyD88 interacted with IRF7 and thisassociation stimulated IFN-a promoters. We also use fluorescenceresonance energy transfer (FRET) to demonstrate that MyD88 colo-calized and physically associated with IRF7 but not IRF3 in thecytoplasm of live cells. Furthermore, IRF7 translocated from thecytoplasm to the nucleus in response to TLR9 ligand in a MyD88-dependent way. Moreover, TRAF6 also bound and activated IRF7, andthe ubiquitin ligase activity of TRAF6 was required for IRF7 activa-tion. Thus, these results show that IFN-a production by TLR7, TLR8and TLR9 is controlled by a MyD88-TRAF6-IRF7 complex.
RESULTS
IRF7 as a MyD88-interacting protein
To elucidate the ‘downstream’ events of the MyD88-dependent path-way, we did a yeast two-hybrid screen of a human spleen cDNA librarywith the death domain of MyD88 as a bait. From a screen ofabout 1 � 106 transformants, we identified seven clones as beingpositive. Sequence analysis showed that two of these clones encodedthe C-terminal portion of IRF7 (amino acids 279–503, which corre-spond to 234–458 of mouse IRF7). We initially analyzed the interac-tion of MyD88 and IRF7 by yeast two-hybrid assay in which yeasts
were transformed with a plasmid encoding the MyD88 deathdomain or TIR domain together with a plasmid encoding the C-terminal region of IRF7 (IRF7C) obtained by the screening (Fig. 1a).We confirmed this interaction in mammalian HEK293 cells usingcoimmunprecipitation experiments. We transiently transfectedHEK293 cells with a plasmid encoding Myc-tagged MyD88 togetherwith a plasmid encoding FLAG-tagged IRF7, a FLAG-tagged IRF7construct lacking amino acids 238–408 (D238–408) or FLAG-taggedIRF3. FLAG-IRF7 was coimmunoprecipitated with an antibody toMyc (anti-Myc) in cells transfected with Myc-MyD88. In contrast,neither FLAG-IRF7 D238–408 nor FLAG-IRF3 was coimmunopreci-pitated by anti-Myc (Fig. 1b). As the region between amino acids 238and 408 corresponds to the inhibitory domain of IRF7 (ref. 28), theseresults indicate that the inhibitory region of IRF7 is required forassociation with MyD88. We then constructed a series of deletionmutants of IRF7 (amino acids deleted: D1, 1–397; D2, 1–326; D3, 1–294; D4, 1–285; D5, 1–237; Fig. 1c) and tested them for interaction.FLAG-tagged full-length IRF7 and IRF7 D1, D2, D3 and D4 expressedin HEK293 cells were coprecipitated with anti-Myc, showing that theregion between amino acid 238 and 285 of IRF7 is required forinteraction (Fig. 1d). Next we examined the interaction of endogen-ous MyD88 and FLAG-IRF7 using specific antibodies recognizingMyD88. FLAG-IRF7 expressed by transient transfection in HEK293cells was coimmunoprecipitated with two different antibodies toMyD88 (Ab1 and Ab2), indicating interaction of endogenousMyD88 with overexpressed IRF7 (Fig. 1e). Each cell lysate was dividedinto aliquots of equal volume to ensure loading of equivalent amountsof protein (Fig. 1e).
W-L-H-
W-L-
Empty
MyD88 DD
MyD88 TIR
MyD88 DD
MyD88 TIR
Empty
Empty
Empty
Empty
IRF7C
IRF7C
IRF7C
1
2
4
3
5
6
Bait Prey
1
2
4
35
6
1
2
4
35
6
FLAG-IRF3FLAG-IRF7∆238−408
IP: Myc
IB: FLAGIP: FLAG
IP: Myc
++
IB: Myc
IB: FLAG
FLAG-IRF7Myc-MyD88++
++ +
1–3971–458
DNA binding Inhibitory Regulatory
1–2941–2851–237
FL
∆1∆2∆3∆4∆5
FLAG-IRF7Myc-MyD88 DD++++++
∆1 ∆2 ∆3 ∆4 ∆5
IB: FLAGIP: Myc
IP: FLAGIB: FLAG
IP: MycIB: Myc
1–326
IB: FLAGWCL
IB: FLAG
FLAG-IRF7+ ++
+
Ab1 Ab2IgG IP
FLAG-IRF7
−− FL
IRF7
−−−−
−−−
−−
− −−
−
− −−−
a b c d e
Figure 1 MyD88 associates with IRF7. (a) Plasmids expressing the MyD88 death domain (DD) or MyD88 TIR domain fused to the GAL4 DNA-binding
domain or an empty p GB kT7 (Empty) were cotransformed with a plasmid expressing IRF7C fused to the GAL4 transactivation domain or an empty PACT2
(Empty). Interactions were detected by ability of cells to grow on medium lacking tryptophan, leucine and histidine (W–L–H–). Growth of cells on a plate
lacking tryptophan and leucine (W–L–) is indicative of the efficiency of the transfection. (b) HEK293 cells were transiently transfected with combinations
of FLAG-IRF7, FLAG-IRF7 D238–408, FLAG-IRF3 and Myc-MyD88 (above lanes). Cell lysates were immunoprecipitated (IP) with anti-Myc or anti-FLAG,
followed by immunoblotting (IB) with anti-FLAG or anti-Myc. (c) IRF7 deletion mutants. FL, full-length. (d) Cell lysates prepared from HEK293 cells
transfected with combinations of FLAG-IRF7 deletion mutants and Myc-MyD88 (above lanes) were immunoprecipitated with anti-Myc or anti-FLAG, followed
by immunoblotting with anti-FLAG or anti-Myc. (e) HEK293 cells were transiently transfected with FLAG-IRF7. Top, cell lysates were immunoprecipitated
with control rabbit anti-mouse immunoglobulin serum (IgG) or anti-MyD88 (Ab1 and Ab2), followed by immunoblotting with anti-FLAG. Bottom, aliquots of
whole cell lysates (WCL) were simultaneously analyzed by immunoblot with anti-FLAG. Similar results were obtained in three independent experiments.
IRF7-CFP IRF7-CFPMyD88-YFP
IRF3-CFPMyD88-YFP
CF
P (
exci
tatio
n)Y
FP
(em
issi
on)
MyD88-YFP
CFP (excitation) CFP (emission)
IRF7-CFPMyD88-YFP
IRF3-CFPMyD88-YFP
YF
P/C
FP
(F
RE
T)
FRETc
CFP (excitation) CFP (emission)
a b
Figure 2 Colocalization of MyD88 and IRF7 demonstrated by FRET analysis. (a) HEK293 cells were transfected with MyD88-YFP (yellow) and IRF7-CFP
(blue) and the physical interaction of these two molecules determined by FRET (pseudocolor) was visualized. (b) HEK293 cells were transfected with
MyD88-YFP, IRF7-CFP and IRF3-CFP. Left, fluorescence intensity of CFP emission by CFP excitation of single cells (horizontal axis) and YFP emission by
CFP excitation of single cells (vertical axis). Cells that are positive for both YFP and CFP by CFP excitation (gated in upper right portion) are considered as
FRET signal. Right, calculated FRET of IRF7-CFP or IRF3-CFP and MyD88-YFP. Similar results were obtained in three independent experiments.
1062 VOLUME 5 NUMBER 10 OCTOBER 2004 NATURE IMMUNOLOGY
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y
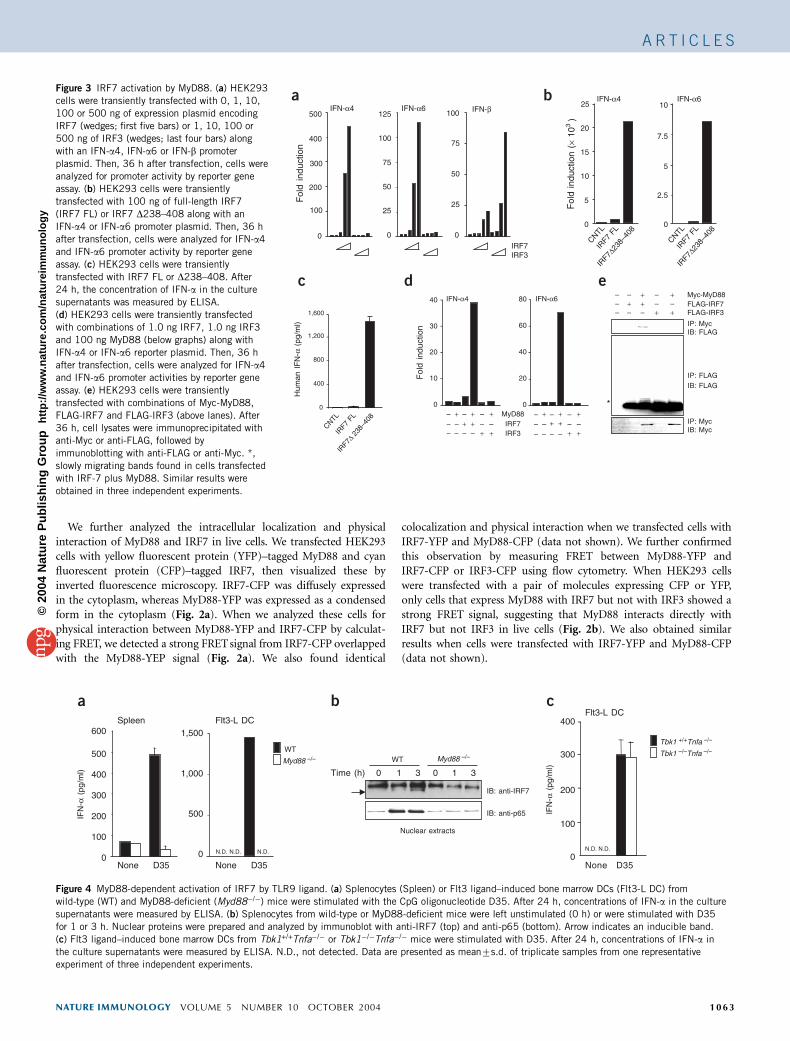
We further analyzed the intracellular localization and physicalinteraction of MyD88 and IRF7 in live cells. We transfected HEK293cells with yellow fluorescent protein (YFP)–tagged MyD88 and cyanfluorescent protein (CFP)–tagged IRF7, then visualized these byinverted fluorescence microscopy. IRF7-CFP was diffusely expressedin the cytoplasm, whereas MyD88-YFP was expressed as a condensedform in the cytoplasm (Fig. 2a). When we analyzed these cells forphysical interaction between MyD88-YFP and IRF7-CFP by calculat-ing FRET, we detected a strong FRET signal from IRF7-CFP overlappedwith the MyD88-YEP signal (Fig. 2a). We also found identical
colocalization and physical interaction when we transfected cells withIRF7-YFP and MyD88-CFP (data not shown). We further confirmedthis observation by measuring FRET between MyD88-YFP andIRF7-CFP or IRF3-CFP using flow cytometry. When HEK293 cellswere transfected with a pair of molecules expressing CFP or YFP,only cells that express MyD88 with IRF7 but not with IRF3 showed astrong FRET signal, suggesting that MyD88 interacts directly withIRF7 but not IRF3 in live cells (Fig. 2b). We also obtained similarresults when cells were transfected with IRF7-YFP and MyD88-CFP(data not shown).
IRF7IRF3
a b
c
IFN-β
400
200
0 0
Hum
an I
FN
-α (
pg/m
l)
0
400
800
1,200
1,600
5
2.5
7.5
0
5
10
15
20
0
FLAG-IRF7FLAG-IRF3
Myc-MyD88
IP: Myc
IB: FLAGIP: FLAG
IP: Myc
+–
+–+–
––+
+–+
IB: Myc
IB: FLAG
––
–
*
ed
0
40
20
60
IFN-α4 IFN-α6
IFN-α4 IFN-α6IFN-α4 IFN-α6
IRF7MyD88
IRF3
–+++–
–+–
––– –
–––+
++
–+++–
–+–
––– –
–––+
++
IRF7
FL
IRF7
∆238–
408
CNTL
IRF7
FL
IRF7
∆238–
408
CNTL
Fol
d in
duct
ion
Fol
d in
duct
ion
IRF7
FL
IRF7
∆ 238
–408
CNTL
Fol
d in
duct
ion
(× 1
03 )
500
300
100
0
100
50
75
125
50
25
75
100
25
25 10
40
30
20
10
0
80
Figure 3 IRF7 activation by MyD88. (a) HEK293
cells were transiently transfected with 0, 1, 10,
100 or 500 ng of expression plasmid encoding
IRF7 (wedges; first five bars) or 1, 10, 100 or
500 ng of IRF3 (wedges; last four bars) along
with an IFN-a4, IFN-a6 or IFN-b promoter
plasmid. Then, 36 h after transfection, cells were
analyzed for promoter activity by reporter geneassay. (b) HEK293 cells were transiently
transfected with 100 ng of full-length IRF7
(IRF7 FL) or IRF7 D238–408 along with an
IFN-a4 or IFN-a6 promoter plasmid. Then, 36 h
after transfection, cells were analyzed for IFN-a4
and IFN-a6 promoter activity by reporter gene
assay. (c) HEK293 cells were transiently
transfected with IRF7 FL or D238–408. After
24 h, the concentration of IFN-a in the culture
supernatants was measured by ELISA.
(d) HEK293 cells were transiently transfected
with combinations of 1.0 ng IRF7, 1.0 ng IRF3
and 100 ng MyD88 (below graphs) along with
IFN-a4 or IFN-a6 reporter plasmid. Then, 36 h
after transfection, cells were analyzed for IFN-a4
and IFN-a6 promoter activities by reporter gene
assay. (e) HEK293 cells were transiently
transfected with combinations of Myc-MyD88,
FLAG-IRF7 and FLAG-IRF3 (above lanes). After36 h, cell lysates were immunoprecipitated with
anti-Myc or anti-FLAG, followed by
immunoblotting with anti-FLAG or anti-Myc. *,
slowly migrating bands found in cells transfected
with IRF-7 plus MyD88. Similar results were
obtained in three independent experiments.
Nuclear extracts
IB: anti-IRF7
WT
Myd88 –/– Myd88 –/–
0
100
200
300
400
500
Spleen Flt3-L DC
None D35
0 1 30 1 3Time (h)
IB: anti-p65
Flt3-L DC
0
100
200
300
Tbk1 +/+Tnfa –/–
Tbk1 –/–Tnfa –/–
None D35
IFN
-α (
pg/m
l)
IFN
-α (
pg/m
l)
WT
600
0
500
1,000
1,500
None D35
N.D. N.D.N.D.
400
N.D. N.D.
a b c
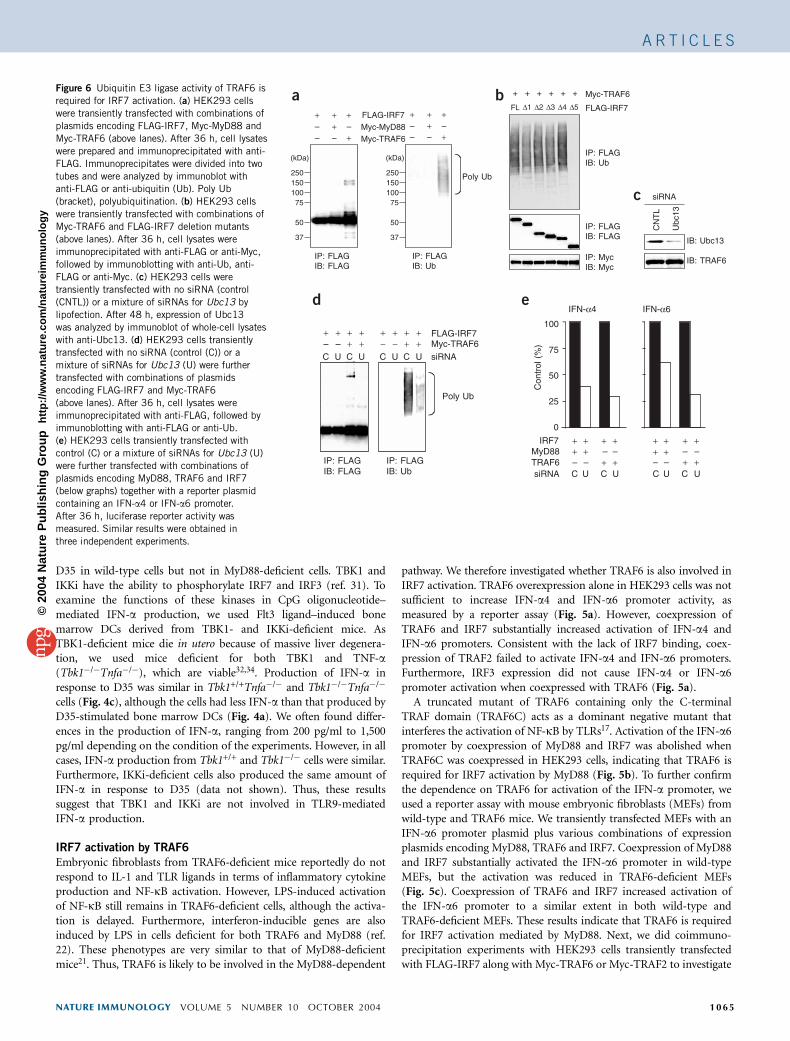
Figure 4 MyD88-dependent activation of IRF7 by TLR9 ligand. (a) Splenocytes (Spleen) or Flt3 ligand–induced bone marrow DCs (Flt3-L DC) from
wild-type (WT) and MyD88-deficient (Myd88�/�) mice were stimulated with the CpG oligonucleotide D35. After 24 h, concentrations of IFN-a in the culture
supernatants were measured by ELISA. (b) Splenocytes from wild-type or MyD88-deficient mice were left unstimulated (0 h) or were stimulated with D35
for 1 or 3 h. Nuclear proteins were prepared and analyzed by immunoblot with anti-IRF7 (top) and anti-p65 (bottom). Arrow indicates an inducible band.
(c) Flt3 ligand–induced bone marrow DCs from Tbk1+/+Tnfa�/� or Tbk1�/�Tnfa�/� mice were stimulated with D35. After 24 h, concentrations of IFN-a in
the culture supernatants were measured by ELISA. N.D., not detected. Data are presented as mean7s.d. of triplicate samples from one representativeexperiment of three independent experiments.
NATURE IMMUNOLOGY VOLUME 5 NUMBER 10 OCTOBER 2004 1063
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y

IRF7 activation by MyD88
The finding that MyD88 interacts with IRF7 prompted us to inves-tigate whether IRF7 is involved in the production of IFN-a and IFN-bby the MyD88-dependent pathway. We initially tested the ability ofIRF7 to activate IFN-a and IFN-b promoters by a reporter assay.HEK293 cells were transiently transfected with the various amounts ofan expression plasmid encoding IRF7 or IRF3 along with a reporterplasmid driven by an IFN-a4, IFN-a6 or IFN-b promoter, andmeasured luciferase activity 36 h after transfection. Expression ofIRF7 caused activation of IFN-a4 and IFN-a6 promoters in a dose-dependent way, whereas there was no induction of IFN-a4 and IFN-a6 promoters when IRF3 was overexpressed (Fig. 3a). In contrast, anIFN-b promoter was activated when IRF3 was overexpressed. IRF7overexpression also increased IFN-b promoter activity, although theactivity was lower than with IRF3 overexpression. Thus, consistentwith a previous report40, these results also indicate that IRF7 has theability to preferentially activate IFN-a promoters. Next we measuredIFN-a promoter activity in HEK293 cells using a mutant IRF7 lackingamino acid 238–285. IRF7 D238–285 enhanced IFN-a4 and IFN-a6promoter activities more strongly than did the full-length IRF7(Fig. 3b), consistent with a previous report showing that removal ofamino acids between 238 and 285 results in a constitutively activeform of IRF7 (ref. 28). To investigate whether expression of IRF7induces endogenous IFN-a production, we used enzyme-linkedimmunosorbent assay (ELISA). IFN-a was present in the culturesupernatants of cells transfected with IRF7 D238–285, indicatingthat IRF7 activation is linked to the production of IFN-a, at least inHEK293 cells (Fig. 3c).
We next tested whether MyD88 enhances the transcriptional activityof IRF7. We transiently transfected HEK293 cells with plasmidsencoding IRF7, MyD88 or in combination together with a reporterplasmid containing an IFN-a4 or IFN-a6 promoter. In these experi-ments, we used a small amount of IRF7 plasmid (1.0 ng) fortransfection, as this amount was not sufficient for IFN-a promoteractivation. Transfection with either IRF7 or MyD88 alone was notsufficient for activation of IFN-a4 and IFN-a6 promoters (Fig. 3d).However, when we transfected both plasmids together, we foundsynergistic activation of IFN-a4 and IFN-a6 promoters. In contrast,
coexpression of MyD88 and IRF3 did not cause any activation of IFN-a promoters (Fig. 3d). Next, we transiently transfected HEK293 cellswith plasmids encoding Myc-MyD88 along with FLAG-IRF7 orFLAG-IRF3. We lysed cells and immunoprecipitated proteins in thecell lysates with anti-FLAG followed by immunoblotting with anti-FLAG. We found slowly migrating forms of FLAG-IRF7 in cellscoexpressing Myc-MyD88 (Fig. 3e). Phosphatase treatment of thelysates resulted in a decrease in these slowly migrating bands, indicat-ing that IRF7 was phosphorylated by a MyD88-associated proteinkinase present in the lysates (data not shown). In contrast, we foundno migration of the IRF3 protein in cells coexpressing MyD88,consistent with the results demonstrating that IRF3 is activatedby the MyD88-independent pathway22. These results suggest thatMyD88 induces IRF7 phosphorylation and thus is likely to contributeto IRF7 activation.
IRF7 activation by TLR9 ligand
CpG oligonucleotides D19 and D35, both TLR9 ligands, can stronglyinduce IFN-a production by plasmacytoid DCs in a MyD88-dependentway37,41. We obtained splenocytes and Flt3 ligand–induced bonemarrow DCs from wild-type or MyD88-deficient mice, which containplasmacytoid DCs, stimulated these cells with D35 and measured IFN-a production by ELISA. D35 induced IFN-a production in the culturesupernatants of wild-type splenocytes and Flt3 ligand–induced bonemarrow DCs (Fig. 4a). In contrast, we found no IFN-a induction inMyD88-deficient cells. Thus, IFN-a production by TLR9 ligands isregulated by the MyD88-dependent pathway.
IRF7 is present in the cytoplasm of unstimulated cells. After viralinfection, IRF7 is phosphorylated and translocates into the nuclei,where it regulates the expression of target genes31,35. We thereforeinvestigated whether IRF7 is activated by TLR9 ligands. We stimulatedsplenocytes from wild-type and MyD88-deficient mice with D35 andanalyzed nuclear proteins by immunoblotting with anti-IRF7 or anti–NF-kB p65. We noted translocation of IRF7 1 h after D35 stimulationof wild-type cells (Fig. 4b). In contrast, IRF7 failed to move into thenuclei in response to D35 in MyD88-deficient cells. These resultsindicate that IRF7 is activated by the TLR9-MyD88–dependent path-way. NF-kB p65 also translocated into the nuclei in response to
a
0
50
100
IFN-α4 IFN-α6 IFN-α6 IFN-α6
b
IRF7MyD88 +
+++–
++–
TRAF6C––
– +– ––
0
5
10
IRF7
TRAF6TRAF2
IRF3+
––
––
–
–
+
+
++
–– +– – – +
––– – –––
––
+–
+–
++
––
––
–
–
+
+
++
–– +– – – +
––– – –––
––
+–
+–
+
FLAG-IRF7
Myc-TRAF6++++++
FL ∆1 ∆2 ∆3 ∆4 ∆5
IB: FLAGIP: Myc
IP: FLAGIB: FLAG
IP: MycIB: Myc
dMyc-TRAF2
IB: FLAGIP: Myc
IP: FLAG
+–
IB: FLAG
–Myc-TRAF6+ ––
e
IP: Myc
IB: Myc
FLAG-IRF7+++
–––
*
**
WT Traf6 –/–
0
200
100
IRF7MyD88TRAF6
–+–
++ –––– –
– – +
++
c
250
150
100
75
50
37
(kDa)
Fol
d in
duct
ion
Fol
d in
duct
ion
Fol
d in
duct
ion
125
75
25
0
500
1,000
1,500 15 250
150
50
Figure 5 IRF7 activation by TRAF6. (a) HEK293 cells were transiently transfected with a combination of plasmids for 1.0 ng IRF7, 1.0 ng IRF3, 50 ng
TRAF2 or 50 ng TRAF6 (below graphs) along with 100 ng of IFN-a4 or IFN-a6 reporter plasmid. At 36 h after transfection, cells were analyzed for IFN-a4
and IFN-a6 promoter activity by reporter gene assay. (b) HEK293 cells were transiently cotransfected with a combination of plasmids for 1.0 ng IRF7, 50 ng
MyD88 and 50 ng TRAF6C (below graph) along with a reporter plasmid carrying the IFN-a6 promoter. At 36 h after transfection, cells were analyzed for
IFN-a6 promoter activity by reporter gene assay. (c) MEFs from wild-type and TRAF6-deficient mice were transiently transfected with a combination of
1.0 ng IRF7, 100 ng MyD88 and 100 ng TRAF6 (below graph) along with a reporter plasmid carrying the IFN-a6 promoter. At 36 h after transfection, cells
were analyzed for IFN-a6 promoter activity by reporter gene assay. (d) HEK293 cells were transiently transfected with combinations of FLAG-IRF7, Myc-
TRAF6 and Myc-TRAF2 (above lanes). After 36 h, cell lysates were immunoprecipitated with anti-Myc or anti-FLAG, followed by immunoblotting with anti-
FLAG or anti-Myc. * and **, slowly migrated bands found in cells cotransfected with IRF-7 and TRAF6. (e) HEK293 cells were transiently transfected with
combinations of Myc-TRAF6 and FLAG-IRF7 deletion mutants (above lanes). After 36 h, cell lysates were immunoprecipitated with anti-FLAG or anti-Myc,
followed by immunoblot with anti-FLAG or anti-Myc. Similar results were obtained in three independent experiments.
1064 VOLUME 5 NUMBER 10 OCTOBER 2004 NATURE IMMUNOLOGY
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y
D35 in wild-type cells but not in MyD88-deficient cells. TBK1 andIKKi have the ability to phosphorylate IRF7 and IRF3 (ref. 31). Toexamine the functions of these kinases in CpG oligonucleotide–mediated IFN-a production, we used Flt3 ligand–induced bonemarrow DCs derived from TBK1- and IKKi-deficient mice. AsTBK1-deficient mice die in utero because of massive liver degenera-tion, we used mice deficient for both TBK1 and TNF-a(Tbk1�/�Tnfa�/�), which are viable32,34. Production of IFN-a inresponse to D35 was similar in Tbk1+/+Tnfa�/� and Tbk1�/�Tnfa�/�
cells (Fig. 4c), although the cells had less IFN-a than that produced byD35-stimulated bone marrow DCs (Fig. 4a). We often found differ-ences in the production of IFN-a, ranging from 200 pg/ml to 1,500pg/ml depending on the condition of the experiments. However, in allcases, IFN-a production from Tbk1+/+ and Tbk1�/� cells were similar.Furthermore, IKKi-deficient cells also produced the same amount ofIFN-a in response to D35 (data not shown). Thus, these resultssuggest that TBK1 and IKKi are not involved in TLR9-mediatedIFN-a production.
IRF7 activation by TRAF6
Embryonic fibroblasts from TRAF6-deficient mice reportedly do notrespond to IL-1 and TLR ligands in terms of inflammatory cytokineproduction and NF-kB activation. However, LPS-induced activationof NF-kB still remains in TRAF6-deficient cells, although the activa-tion is delayed. Furthermore, interferon-inducible genes are alsoinduced by LPS in cells deficient for both TRAF6 and MyD88 (ref.22). These phenotypes are very similar to that of MyD88-deficientmice21. Thus, TRAF6 is likely to be involved in the MyD88-dependent
pathway. We therefore investigated whether TRAF6 is also involved inIRF7 activation. TRAF6 overexpression alone in HEK293 cells was notsufficient to increase IFN-a4 and IFN-a6 promoter activity, asmeasured by a reporter assay (Fig. 5a). However, coexpression ofTRAF6 and IRF7 substantially increased activation of IFN-a4 andIFN-a6 promoters. Consistent with the lack of IRF7 binding, coex-pression of TRAF2 failed to activate IFN-a4 and IFN-a6 promoters.Furthermore, IRF3 expression did not cause IFN-a4 or IFN-a6promoter activation when coexpressed with TRAF6 (Fig. 5a).
A truncated mutant of TRAF6 containing only the C-terminalTRAF domain (TRAF6C) acts as a dominant negative mutant thatinterferes the activation of NF-kB by TLRs17. Activation of the IFN-a6promoter by coexpression of MyD88 and IRF7 was abolished whenTRAF6C was coexpressed in HEK293 cells, indicating that TRAF6 isrequired for IRF7 activation by MyD88 (Fig. 5b). To further confirmthe dependence on TRAF6 for activation of the IFN-a promoter, weused a reporter assay with mouse embryonic fibroblasts (MEFs) fromwild-type and TRAF6 mice. We transiently transfected MEFs with anIFN-a6 promoter plasmid plus various combinations of expressionplasmids encoding MyD88, TRAF6 and IRF7. Coexpression of MyD88and IRF7 substantially activated the IFN-a6 promoter in wild-typeMEFs, but the activation was reduced in TRAF6-deficient MEFs(Fig. 5c). Coexpression of TRAF6 and IRF7 increased activation ofthe IFN-a6 promoter to a similar extent in both wild-type andTRAF6-deficient MEFs. These results indicate that TRAF6 is requiredfor IRF7 activation mediated by MyD88. Next, we did coimmuno-precipitation experiments with HEK293 cells transiently transfectedwith FLAG-IRF7 along with Myc-TRAF6 or Myc-TRAF2 to investigate
25015010075
50
37
(kDa)
25015010075
50
37
(kDa)
IP: FLAGIB: FLAG
––+–++
+–+
Poly Ub
Myc-MyD88FLAG-IRF7
Myc-TRAF6
IP: FLAGIB: Ub
CN
TL
Ubc
13
siRNA
IB: Ubc13
IB: TRAF6
CU
+–
Poly Ub
++
U
++
Myc-TRAF6FLAG-IRF7
siRNA
IP: FLAGIB: FLAG
IFN-α4
+
++
+–
+
–
++ +
C U C U C U C U
IRF7
TRAF6siRNA
––MyD88
100
75
50
25
IFN-α6
C
–+
IP: FLAGIB: Ub
Con
trol
(%
)
CU
+−++
U
++
C
−+
0
+
++
+–
+
–
++ + ––
FLAG-IRF7
Myc-TRAF6++++++
FL ∆1 ∆2 ∆3 ∆4 ∆5
IP: FLAGIB: Ub
IP: FLAGIB: FLAG
IP: MycIB: Myc
––+–++
+–+
a b
d e
c
Figure 6 Ubiquitin E3 ligase activity of TRAF6 is
required for IRF7 activation. (a) HEK293 cells
were transiently transfected with combinations of
plasmids encoding FLAG-IRF7, Myc-MyD88 and
Myc-TRAF6 (above lanes). After 36 h, cell lysates
were prepared and immunoprecipitated with anti-
FLAG. Immunoprecipitates were divided into two
tubes and were analyzed by immunoblot withanti-FLAG or anti-ubiquitin (Ub). Poly Ub
(bracket), polyubiquitination. (b) HEK293 cells
were transiently transfected with combinations of
Myc-TRAF6 and FLAG-IRF7 deletion mutants
(above lanes). After 36 h, cell lysates were
immunoprecipitated with anti-FLAG or anti-Myc,
followed by immunoblotting with anti-Ub, anti-
FLAG or anti-Myc. (c) HEK293 cells were
transiently transfected with no siRNA (control
(CNTL)) or a mixture of siRNAs for Ubc13 by
lipofection. After 48 h, expression of Ubc13
was analyzed by immunoblot of whole-cell lysates
with anti-Ubc13. (d) HEK293 cells transiently
transfected with no siRNA (control (C)) or a
mixture of siRNAs for Ubc13 (U) were further
transfected with combinations of plasmids
encoding FLAG-IRF7 and Myc-TRAF6
(above lanes). After 36 h, cell lysates were
immunoprecipitated with anti-FLAG, followed byimmunoblotting with anti-FLAG or anti-Ub.
(e) HEK293 cells transiently transfected with
control (C) or a mixture of siRNAs for Ubc13 (U)
were further transfected with combinations of
plasmids encoding MyD88, TRAF6 and IRF7
(below graphs) together with a reporter plasmid
containing an IFN-a4 or IFN-a6 promoter.
After 36 h, luciferase reporter activity was
measured. Similar results were obtained in
three independent experiments.
NATURE IMMUNOLOGY VOLUME 5 NUMBER 10 OCTOBER 2004 1065
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y

whether TRAF6 also binds IRF7. FLAG-IRF7 was coprecipitated withanti-Myc in cells expressing Myc-TRAF6 but not Myc-TRAF2(Fig. 5d). In addition, we also found slowly migrating forms ofIRF7 in cells coexpressing TRAF6 but not TRAF2 (Fig. 5d). Usingdeletion mutants of IRF7, we searched for the region responsible forinteraction with TRAF6. Myc-TRAF6 was coimmunoprecipitated withfull-length IRF7 and IRF7 D1, D2, D3 and D4 but not D5, indicatingthat the region between amino acids 238 and 285, with which MyD88also interacts, was required for interaction with TRAF6 (Fig. 5e). Wealso found interaction when a plasmid encoding the C-terminal TRAFdomain of TRAF6 was coexpressed with IRF7 D4, thus indicating thatthe TRAF domain of TRAF6 binds IRF7 (data not shown).
Ubiquitination-dependent IRF7 activation
The finding that IRF7 protein migrated slowly by SDS-PAGE whenTRAF6 was coexpressed suggested that some protein modifications arenecessary for IRF7 activation. We therefore examined if the slowmigration was due to ubiquitination, as TRAF6 reportedly has anubiquitin E3 ligase activity that catalyzes synthesis of lysine 63–linkedubiquitination42. We transiently transfected HEK293 cells with FLAG-IRF7 together with Myc-TRAF6 or Myc-MyD88, and then immuno-precipitated proteins with anti-FLAG, followed by immunoblottingwith anti-FLAG or anti-ubiquitin. Slowly migrating forms of FLAG-IRF7 approximately 150 kDa in size were strongly induced in TRAF6-overexpressing cells but not MyD88-overexpressing cells (Fig. 6a).Furthermore, we noted ubiquitination in lysates prepared from cellscoexpressing FLAG-IRF7 and Myc-TRAF6 and immunoprecipitatedwith anti-FLAG. Thus, these results strongly suggest that TRAF6induces ubiquitination of IRF7 or ubiquitinated TRAF6 associateswith IRF7. Next, we used deletion mutants of IRF7 to investigate theubiquitination. In HEK293 cells coexpressing TRAF6 along with full-length IRF7 or deletion mutants of IRF7, we found ubiquitination offull-length IRF7 and IRF7 D1, D2, D3 and D4 (Fig. 6b). Thus, theseresults indicate that the association between TRAF6 and IRF7 isrequired for TRAF6-mediated ubiquitination.
To investigate whether ubiquitination is linked to TRAF6-mediatedIRF7 activation, we used siRNA to silence UBC13, which encodes aubiquitin-conjugating enzyme required for TRAF6-mediated ubiquiti-nation27. Expression of UBC13 was reduced in HEK293 cells transientlytransfected with a mixture of siRNA oligos corresponding to humanUBC13 (Fig. 6c). We tested TRAF6-mediated ubiquitination in thiscondition. Slowly migrating forms of IRF7 were considerably reducedin HEK293 cells treated with siRNA for UBC13 compared with that ofcontrol cells (Fig. 6d). Furthermore, TRAF6-mediated ubiquitinationwas also reduced in siRNA-transfected cells (Fig. 6d, right). Next weexamined the effect of UBC13 ‘knockdown’ on the activation of IFN-apromoters. Activation of IFN-a4 and IFN-a6 promoters induced bycoexpression of IRF7 and MyD88 was reduced after UBC13 siRNAtreatment (Fig. 6e). Activation of IFN-a4 and IFN-a6 promoters wasalso reduced in cells coexpressing TRAF6 and IRF7 that had beentreated with siRNA for UBC13. Thus, these results indicate that theubiquitin ligase activity of TRAF6 is required for IRF7 activation.
DISCUSSION
Using yeast two-hybrid screening we have identified IRF7 as a proteinthat interacts with MyD88. MyD88 formed a complex with IRF7 butnot IRF3 in live cells, and MyD88 overexpression in combination withIRF7 activated IFN-a but not IFN-b promoters. As type I interferoninduction by TLR7, TLR8 and TLR9 ligands depends entirely onMyD88, the MyD88-IRF7 complex most likely mediates IFN-aproduction via TLR7, TLR8 and TLR9. Furthermore, this may account
for the mechanism by which a large amount of IFN-a is rapidlyinduced in response to TLR7, TLR8 and TLR9 ligands in plasmacytoidDCs that express IRF7 constitutively.
Type I interferon is also induced by LPS (TLR4 ligand), double-stranded RNA (TLR3 ligand) and viral infection22,32,43. However,induction of genes encoding type I interferon genes via TLR3 orTLR4 is differently regulated in that IRF3 is crucial in the primaryactivation of IFN-b. After treatment with LPS or double-strandedRNA, IRF3 is phosphorylated by the Trif-associated kinase TBK-1 andthen translocates to the nucleus, where it activates the gene encodingIFN-b30,31. These inductions are MyD88 independent22,25. Thus, basedon difference in the mechanism of initial induction of type I inter-feron, TLRs are classified into two categories: TLR7, TLR8 and TLR9activate the genes encoding IFN-a via the MyD88-IRF7 complex,whereas TLR3 and TLR4 activate the gene encoding IFN-b via theTrif-IRF3 complex. Secreted IFN-b induced by TLR3 or TLR4 ligandtriggers expression of IRF7 via the Jak-STAT signaling pathway43.Thus, newly synthesized IRF-7 is thought to participate in theinduction of IFN-a. Given that TRAF6 also binds Trif, synthesizedIRF7 might be recruited to the TRAF6-Trif complex where it isphosphorylated by Trif-associated TBK1 and IKKi44. Further studieswill be needed to clarify this point.
TLR7, TLR8 and TLR9 use only MyD88 as an adaptor to transmit asignal. In contrast, TLR2 uses MyD88 and TIRAP, and TLR4 uses fouradaptors: MyD88, TIRAP, Trif and TRAM15. Although TLR2 andTLR4 activate the MyD88-dependent pathway, they induce very littleIFN-a from plasmacytoid DCs (data not shown). Thus, it is possiblethat IRF7 or its associated protein kinase cannot be recruited to theTLR-MyD88 complex when TIRAP is associated with the receptorcomplex or that TIRAP mediates an inhibitory signal that interferesIRF7 phosphorylation.
The interaction between MyD88 and IRF7 seems to induce phos-phorylation and IRF7 activation, suggesting that MyD88 physicallyassociates with protein kinases that have the ability to phosphorylateand activate IRF7. We examined if protein kinases acting downstreamof MyD88 are involved in IRF7 activation. In our preliminaryexperiments, however, overexpression of IRAK1, IRAK4 or TAK1 incombination with IRF7 did not cause any synergistic activation ofIFN-a promoter in HEK293 cells (data not shown). This findingsuggests that overexpression of these kinases alone is not sufficient foractivation of IFN-a promoters or that some other protein kinasesmight serve as an IRF7 kinase in the MyD88-dependent pathway.TBK1 and IKKi are able to phosphorylate IRF7 as well as IRF3.However, we found IFN-a production in response to CpG oligonu-cleotide in Flt3 ligand–induced bone marrow DCs derived from micedeficient in TBK1 or IKKi. Although experiments with mice deficientin both TBK1 and IKKi will be required for precise elucidation of thefunctions of these kinases, this finding suggests that TBK1 and IKKiare not involved in the MyD88-dependent pathway.
TRAF6 is a RING domain–containing ubiquitin ligase that isessential in NF-kB activation downstream of TLRs15,27. TRAF6 func-tions together with a ubiquitin-conjugating enzyme complex consist-ing of Ubc13 and Uev1A that catalyzes the synthesis of polyubiquitinlinked to lysine 63 of ubiquitin. Lysine 63–mediated ubiquitination isthought to regulate protein-protein interaction or protein functions27.TRAF6 itself is also polyubiquitinated after oligomerization or dimer-ization. TRAF6-mediated polyubiquitination triggers signal transduc-tion through association with downstream proteins. Our data haveshown that TRAF6 also associates with and activates IRF7 in anubiquitin-dependent way. Thus, in parallel with the IKK activation bythe complex consisting of ubiquitinated TRAF6 and TAK1, IRF7
1066 VOLUME 5 NUMBER 10 OCTOBER 2004 NATURE IMMUNOLOGY
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y

would be activated by the complex consisting of ubiquitinated TRAF6and a MyD88-associated kinase. Although the IRF7 kinase actingdownstream of TLR7, TLR8 and TLR9 remains to be identified, ourstudy has demonstrated that IRF7 is a key regulator in TLR7-, TLR8-and TLR9-mediated IFN-a induction through formation of a complexwith MyD88 and TRAF6.
METHODSExpression plasmids. Reporter constructs of IFN-a4 and IFN-a6 were gener-
ated by insertion of the promoter region of Ifna4 or Ifna6 into the pGL3 vector
(Promega). PCR-generated fragments of mouse genomic DNA encoding
positions �476 to +19 and �432 to +22 relative to the initiation sites of
IFN-a4 and IFN-a6 were cloned into the HindIII and NheI-HindIII sites of the
vector, respectively. The IFN-b promoter construct has been described45.
Fragments of human MyD88, MyD88 death domain (amino acids 1–151),
TRAF2, TRAF6 and TRAF6C (amino acids 290–522) were obtained by RT-PCR
and were ligated into pEF-BOS for expression of Myc-tagged proteins. Full-
length mouse IRF7, the series of deletion mutants of IRF7 and full-length
human IRF3 were obtained by RT-PCR and were ligated into the EcoRI and
SalI sites of pFLAG-CMV2 vector (Sigma). IRF7 lacking amino acids 238–408
was synthesized by PCR and was ligated into pFLAG-CMV2. Plasmids encod-
ing fusion proteins IRF7-CFP, IRF3-CFP and MyD88-YFP were constructed
essentially as described46. Human cDNA encoding MyD88 was amplified by
PCR. The cDNA fragments were introduced into the XhoI and NotI sites of
pCAGGS-Flag-m1SECFP or pCAG-His-Venus. Human MyD88, IRF3 or IRF7
obtained by PCR was inserted into the XhoI-NotI sites of pCAG and pCAGGS
vectors encoding YFP and CFP at the N and C termini of the inserted protein47.
The sequences of all constructs were confirmed with an ABI PRISM Genetic
Analyzer (PE Applied Biosystems).
Cells, antibodies and reagents. Spleen single-cell suspensions from wild-type
(129/Sv, C57BL/6 background) and Myd88�/� mice16 were cultured for 48 h in
RPMI 1640 medium (Gibco BRL) supplemented with 10% FCS (Gibco BRL).
Mouse bone marrow cells (1 � 105 cells/well) were cultured for 9 d with
Flt3 ligand (10 ng/ml; R&D Systems) in DMEM containing 10% FCS.
MEFs from wild-type and Traf6�/� mice were prepared as described22.
HEK293 cells were cultured in DMEM containing 10% FCS. Anti-MyD88
and anti-TRAF6 were purchased from ProSci and Santa Cruz, respectively.
Anti-IRF7 and anti-Ubc13 were obtained from Zymed. CpG oligonucleotide
D35 was synthesized as described41. TBK1- and TNF-a-deficient mice have
been described32,48.
Yeast two-hybrid screening. Yeast two-hybrid screening was done as
described with the Matchmaker two-hybrid system 3 (Clontech Labora-
tories)44. For construction of the bait plasmid, the N-terminal death domain
of human MyD88 (amino acids 1–151) was cloned in-frame into the GAL4
DNA-binding domain of pGBKT7. Yeast strain AH109 was transformed
with the bait plasmid plus the human spleen Matchmaker cDNA library.
After screening of 1 � 106 clones, positive clones were picked and the pACT2
library plasmids were recovered from individual clones and expanded in
Escherichia coli. The insert cDNA was sequenced and then characterized with
the BLAST program.
FRET. HEK293 cells plated on a collagen-coated glass dish were imaged as
described46,47. Cells were imaged with an inverted microscope equipped with a
cooled charge-coupled device camera and controlled by MetaMorph software
(Universal Imaging). A pair of proteins fused to YFP or CFP was expressed
in HEK293 cells. Cells were imaged with of the following filter sets: an MX0420
excitation filter and a BP470-490 emission filter (Olympus) for the CFP
images; an MX0420 excitation filter and a 535DF35 emission filter (Omega
Optical) for the FRET images; and a 510DF23 excitation filter (Omega Optical)
and a 560DF15 emission filter (Omega Optical) for the YFP images. An
XF2052 dichroic mirror (Omega Optical) was used throughout the experi-
ments. Exposure times were 200 ms for CFP and FRET images and 100 ms
for YFP images. After data acquisition, the average intensities of CFP, FRET
and YFP were measured and fluorescence was calculated through the FRET
filter set consisting of a FRET component (‘corrected’ FRET (FRETC)). The
non-FRET components were subtracted as described49. For our experimental
conditions, we used the following equation: FRETC ¼ FRET – (0.34 � CFP) –
(0.02 � YFP).
For flow cytometry for FRET, HEK293 cells transfected with CFP and/or
YFP fusion proteins as described above were resuspended in 293 expression
media (Invitrogen) and YFP (excitation, 488 nm; emission, 530 nm), CFP
(excitation, 407 nm; emission, 510 nm) and FRET (excitation, 407 nm;
emission, 535 nm) were measured with FACSAria and FACSDiVa software
(BD). FRET is presented as YFP emission obtained by CFP excitation divided
by CFP emission by CFP excitation.
Transfection, immunoprecipitation and immunoblot analysis. HEK293 cells
(1 � 106) were seeded on a 100-mm dish. Then, 12 h later, cells were transiently
transfected with a total of 6.0 mg of various plasmids with Lipofectamine 2000
(Invitrogen). Immunoprecipitation and immunoblot analysis were done
as described44.
Luciferase reporter assay. HEK293 cells seeded on 24-well plates (1 � 105 cells/
well) or MEFs seeded on six-well plates (2 � 105 cells/well) were transiently
transfected with 100 ng of the luciferase reporter plasmid together with a total
of 900 ng of various expression vectors. Then, 36 h later, the luciferase activity
in the total cell lysate was measured with the Dual-luciferase reporter assay
system (Promega). Renilla luciferase reporter gene (50 ng) was simultaneously
transfected as an internal control.
ELISA. Spleen single-cell suspensions (5 � 105 cells/well) or Flt3 ligand–
induced bone marrow cells (1 � 105 cells/well) were stimulated for 24 h with
3 mM CpG oligonucleotide D35. HEK293 cells transiently transfected with
FLAG-IRF7 or IRF7 D237–408 were cultured for 24 h. Cytokine IFN-awas measured in the supernatants by ELISA according to manufacturer’s
instructions (PBL Biomedical).
RNA interference. HEK293 cells seeded on 60-mm dishes were transfected
with 21-nucleotide complementary RNA with symmetrical 2-nucleotide over-
hangs using Lipofectamine 2000 according to the manufacturer’s instructions.
Four siRNAs targeted to Ubc13 (5¢-UCAAAGCCGAACCAGAUGA-3¢,5¢-AAGUACGUUUCAUGACCAA-3¢, 5¢-UCCUAAUGUAGACAAGUUG-3¢and 5¢-GAUCCGCACAGUUCUGCUA-3¢) were mixed for use50. After 48 h,
cells were transfected with plasmids and cultured for additional 36 h.
ACKNOWLEDGMENTSWe thank K. Irie, A. Yamaguchi and A. Shibano for technical support;M. Hashimoto for secretarial assistance; S. Tanaka (Nippon Becton DickinsonCompany) for FRET analysis; and F. Takeshita (Department of MolecularBiodefense Research, School of Medicine, Yokohama City University) fordiscussions and support.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 12 July; accepted 16 August 2004
Published online at http://www.nature.com/natureimmunology/
1. Medzhitov, R. & Janeway, C.J. Innate immunity: the virtues of a nonclonal system ofrecognition. Cell 91, 295–298 (1997).
2. Takeda, K., Kaisho, T. & Akira, S. Toll-like receptors. Annu. Rev. Immunol. 21, 335–376 (2003).
3. Alexopoulou, L., Holt, A.C., Medzhitov, R. & Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kB by Toll-like receptor 3. Nature 413, 732–738(2001).
4. Diebold, S.S. et al. Innate antiviral responses by means of TLR7-mediated recognitionof single-stranded RNA. Science 303, 1529–1531 (2004).
5. Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 (2001).
6. Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7and 8. Science 303, 1526–1529 (2004).
7. Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 (2002).
8. Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745(2000).
9. Hoshino, K. et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice arehyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product.J. Immunol. 162, 3749–3752 (1999).
NATURE IMMUNOLOGY VOLUME 5 NUMBER 10 OCTOBER 2004 1067
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y

10. Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice:mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
11. Takeuchi, O. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negativeand gram-positive bacterial cell wall components. Immunity 11, 443–451 (1999).
12. Takeuchi, O. et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int.Immunol. 13, 933–940 (2001).
13. Takeuchi, O. et al. Cutting edge: role of Toll-like receptor 1 in mediating immuneresponse to microbial lipoproteins. J. Immunol. 169, 10–14 (2002).
14. Zhang, D. et al. A toll-like receptor that prevents infection by uropathogenic bacteria.Science 303, 1522–1526 (2004).
15. Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511(2004).
16. Adachi, O. et al. Targeted disruption of the MyD88 gene results in loss of IL-1- andIL-18-mediated function. Immunity 9, 143–150 (1998).
17. Fitzgerald, K.A. et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4signal transduction. Nature 413, 78–83 (2001).
18. Hoebe, K. et al. Identification of Lps2 as a key transducer of MyD88-independent TIRsignalling. Nature 424, 743–748 (2003).
19. Horng, T., Barton, G.M. & Medzhitov, R. TIRAP: an adapter molecule in the Tollsignaling pathway. Nat. Immunol. 2, 835–841 (2001).
20. Horng, T., Barton, G.M., Flavell, R.A. & Medzhitov, R. The adaptor molecule TIRAPprovides signalling specificity for Toll-like receptors. Nature 420, 329–333 (2002).
21. Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 (1999).
22. Kawai, T. et al. Lipopolysaccharide stimulates the MyD88-independent pathway andresults in activation of IFN-regulatory factor 3 and the expression of a subset oflipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894 (2001).
23. Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T. & Seya, T. TICAM-1, an adaptormolecule that participates in Toll-like receptor 3–mediated interferon-b induction. Nat.Immunol. 4, 161–167 (2003).
24. Yamamoto, M. et al. Essential role for TIRAP in activation of the signalling cascadeshared by TLR2 and TLR4. Nature 420, 324–329 (2002).
25. Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptorsignaling pathway. Science 301, 640–643 (2003).
26. Yamamoto, M. et al. TRAM is specifically involved in the Toll-like receptor 4–mediatedMyD88-independent signaling pathway. Nat. Immunol. 4, 1144–1150 (2003).
27. Sun, L. & Chen, Z.J. The novel functions of ubiquitination in signaling. Curr. Opin. CellBiol. 16, 119–126 (2004).
28. Marie, I., Smith, E., Prakash, A. & Levy, D.E. Phosphorylation-induced dimerization ofinterferon regulatory factor 7 unmasks DNA binding and a bipartite transactivationdomain. Mol. Cell. Biol. 20, 8803–8814 (2000).
29. Yoneyama, M. et al. Direct triggering of the type I interferon system by virus infection:activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J.17, 1087–1095 (1998).
30. Fitzgerald, K.A. et al. IKKe and TBK1 are essential components of the IRF3 signalingpathway. Nat. Immunol. 4, 491–496 (2003).
31. Sharma, S. et al. Triggering the interferon antiviral response through an IKK-relatedpathway. Science 300, 1148–1151 (2003).
32. Hemmi, H. et al. The roles of two IkB Kinase-related kinases in lipopolysaccharide anddouble stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650(2004).
33. McWhirter, S.M. et al. IFN-regulatory factor 3-dependent gene expression is defectivein Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 101, 233–238 (2004).
34. Perry, A.K., Chow, E.K., Goodnough, J.B., Yeh, W.C. & Cheng, G. Differential require-ment for TANK-binding kinase-1 in type I interferon responses to Toll-like receptoractivation and viral infection. J. Exp. Med. 199, 1651–1658 (2004).
35. Lin, R., Mamane, Y. & Hiscott, J. Multiple regulatory domains control IRF-7 activity inresponse to virus infection. J. Biol. Chem. 275, 34320–34327 (2000).
36. Sato, M. et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 inresponse to viruses for IFN-a/b gene induction. Immunity 13, 539–548 (2000).
37. Hemmi, H., Kaisho, T., Takeda, K. & Akira, S. The roles of Toll-like receptor 9, MyD88,and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpGDNAs on dendritic cell subsets. J. Immunol. 170, 3059–3064 (2003).
38. Izaguirre, A. et al. Comparative analysis of IRF and IFN-a expression in humanplasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 74, 1125–1138(2003).
39. Kerkmann, M. et al. Activation with CpG-A and CpG-B oligonucleotides reveals twodistinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendriticcells. J. Immunol. 170, 4465–4474 (2003).
40. Yeow, W.S. et al. Reconstitution of virus-mediated expression of interferon alpha genesin human fibroblast cells by ectopic interferon regulatory factor-7. J. Biol. Chem. 275,6313–6320 (2000).
41. Verthelyi, D., Ishii, K.J., Gursel, M., Takeshita, F. & Klinman, D.M. Human peripheralblood cells differentially recognize and respond to two distinct CPG motifs. J. Immunol.166, 2372–2377 (2001).
42. Deng, L. et al. Activation of the IkB kinase complex by TRAF6 requires a dimericubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103,351–361 (2000).
43. Hoshino, K., Kaisho, T., Iwabe, T., Takeuchi, O. & Akira, S. Differential involvement ofIFN-b in Toll-like receptor-stimulated dendritic cell activation. Int. Immunol. 14,1225–1231 (2002).
44. Sato, S. et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-b (TRIF)associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, andactivates two distinct transcription factors, NF-kB and IFN-regulatory factor-3, in theToll-like receptor signaling. J. Immunol. 171, 4304–4310 (2003).
45. Yamamoto, M. et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapterthat preferentially activates the IFN-b promoter in the Toll-like receptor signaling.J. Immunol. 169, 6668–6672 (2002).
46. Mochizuki, N. et al. Spatio-temporal images of growth-factor-induced activation of Rasand Rap1. Nature 411, 1065–1068 (2001).
47. Itoh, R.E. et al. Activation of rac and cdc42 video imaged by fluorescent resonanceenergy transfer-based single-molecule probes in the membrane of living cells. Mol.Cell. Biol. 22, 6582–6591 (2002).
48. Tagawa, Y., Sekikawa, K. & Iwakura, Y. Suppression of concanavalin A-inducedhepatitis in IFN-g�/� mice, but not in TNF-a�/� mice: role for IFN-g in activatingapoptosis of hepatocytes. J. Immunol. 159, 1418–1428 (1997).
49. Sorkin, A., McClure, M., Huang, F. & Carter, R. Interaction of EGF receptor and grb2 inliving cells visualized by fluorescence resonance energy transfer (FRET) microscopy.Curr. Biol. 10, 1395–1398 (2000).
50. Zhou, H. et al. Bcl10 activates the NF-kB pathway through ubiquitination of NEMO.Nature 427, 167–171 (2004).
1068 VOLUME 5 NUMBER 10 OCTOBER 2004 NATURE IMMUNOLOGY
A R T I C L E S©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eim
mun
olog
y





























