Embed Size (px)
Citation preview

Università degli Studi di Perugia
Facoltà di Medicina e Chirurgia
UniversitUniversitàà degli Studi di Perugiadegli Studi di Perugia
FacoltFacoltàà di Medicina e Chirurgiadi Medicina e Chirurgia
Le Interstiziopatie diffuse Le Interstiziopatie diffuse Le Interstiziopatie diffuse
L. CasaliL. CasaliL. Casali
Cattedra di Malattie dell’Apparato Respiratorio
Cattedra di Malattie dell’Apparato Respiratorio

Interstiziopatie DiffuseInterstiziopatie Diffuse
Gruppo di affezioni che interessano i setti interalveolari, gli alveoli, il lume e le pareti delle piccole vie aeree (dotti alveolari, bronchioli respiratori e bronchioli terminali).Già all’esordio clinico vi è il coinvolgimento di piùdi un lobo con un attacco profondo alle unità di scambio.L’elemento distintivo è rappresentato dalla proliferazione di fibroblasti e da un eccesso di deposizione di collageno sia come risposta immediata ad uno stimolo sia come risultante diun complesso processo di distruzione e riparazione.
Gruppo di affezioni che interessano i setti interalveolari, gli alveoli, il lume e le pareti delle piccole vie aeree (dotti alveolari, bronchioli respiratori e bronchioli terminali).Già all’esordio clinico vi è il coinvolgimento di piùdi un lobo con un attacco profondo alle unità di scambio.L’elemento distintivo è rappresentato dalla proliferazione di fibroblasti e da un eccesso di deposizione di collageno sia come risposta immediata ad uno stimolo sia come risultante diun complesso processo di distruzione e riparazione.
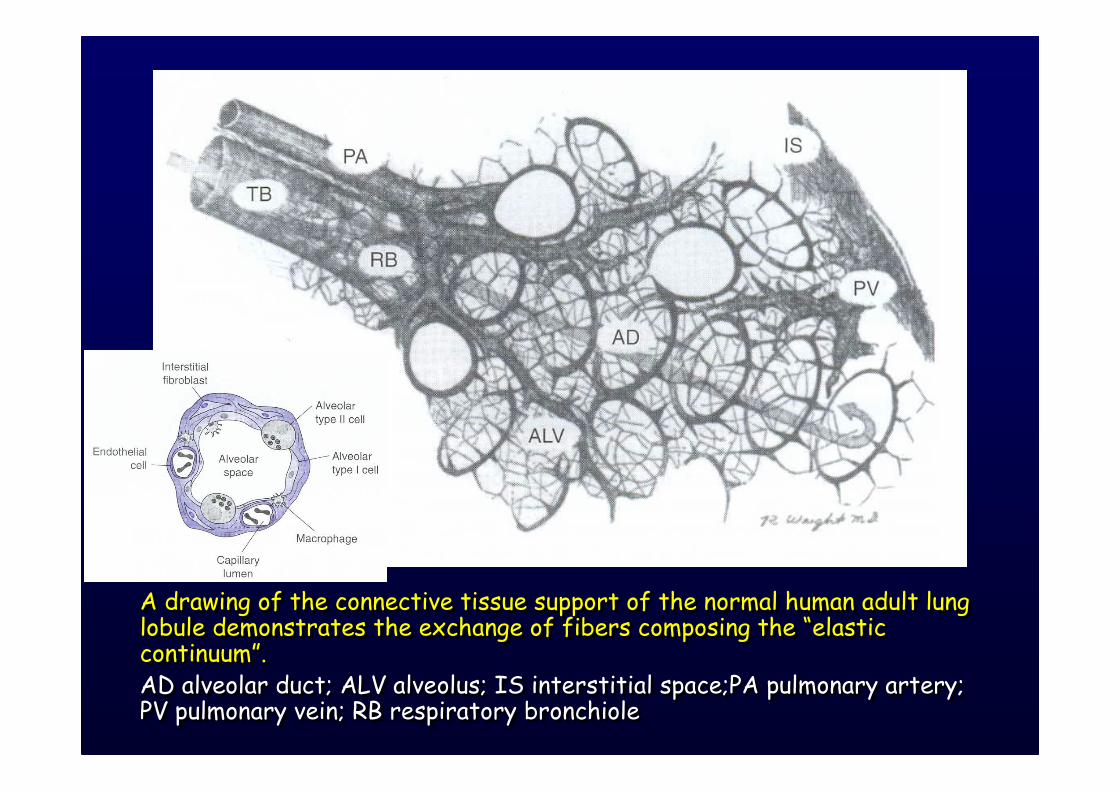
A drawing of the connective tissue support of the normal human adult lung lobule demonstrates the exchange of fibers composing the “elastic continuum”.AD alveolar duct; ALV alveolus; IS interstitial space;PA pulmonary artery; PV pulmonary vein; RB respiratory bronchiole
A drawing of the connective tissue support of the normal human adult lung lobule demonstrates the exchange of fibers composing the “elastic continuum”.AD alveolar duct; ALV alveolus; IS interstitial space;PA pulmonary artery; PV pulmonary vein; RB respiratory bronchiole
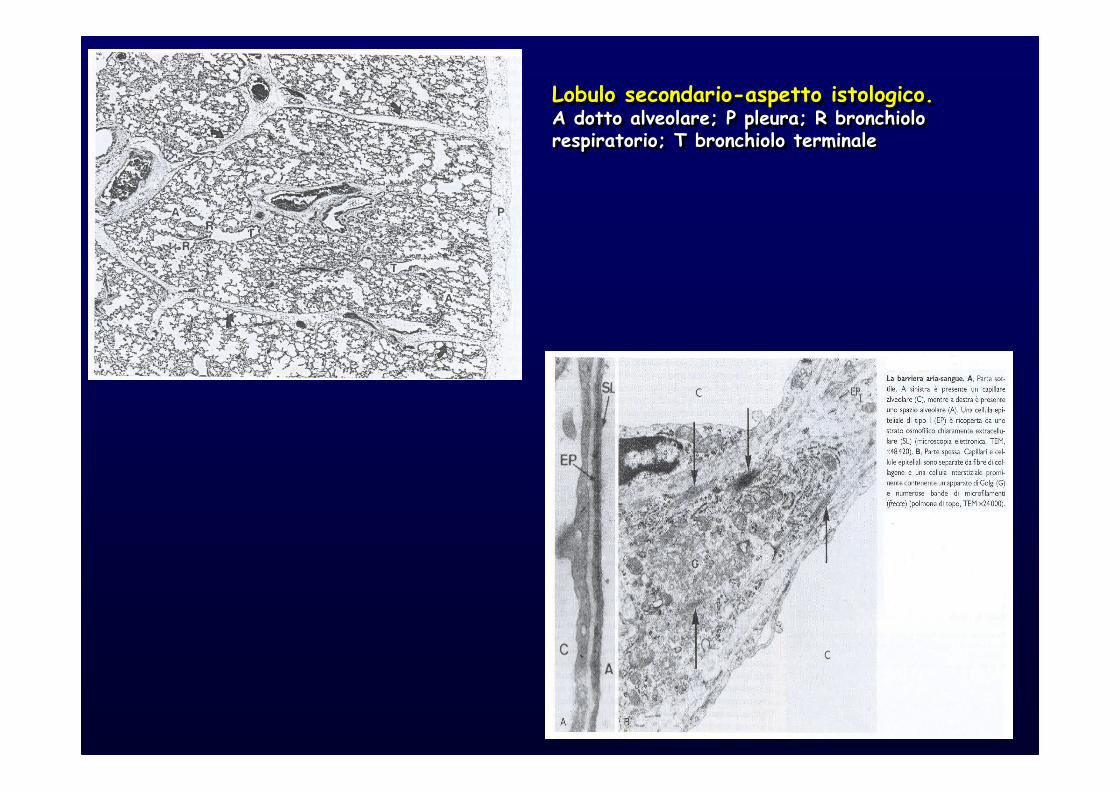
Lobulo secondario-aspetto istologico.A dotto alveolare; P pleura; R bronchiolo respiratorio; T bronchiolo terminale
Lobulo secondario-aspetto istologico.A dotto alveolare; P pleura; R bronchiolo respiratorio; T bronchiolo terminale


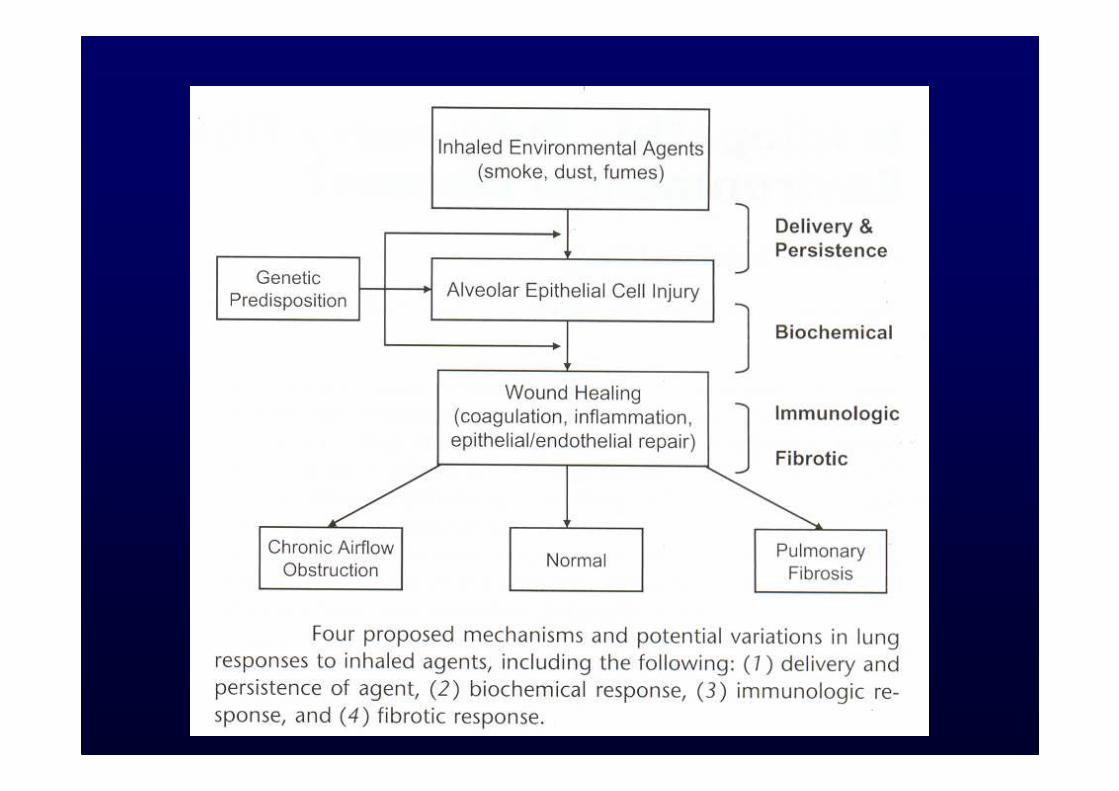
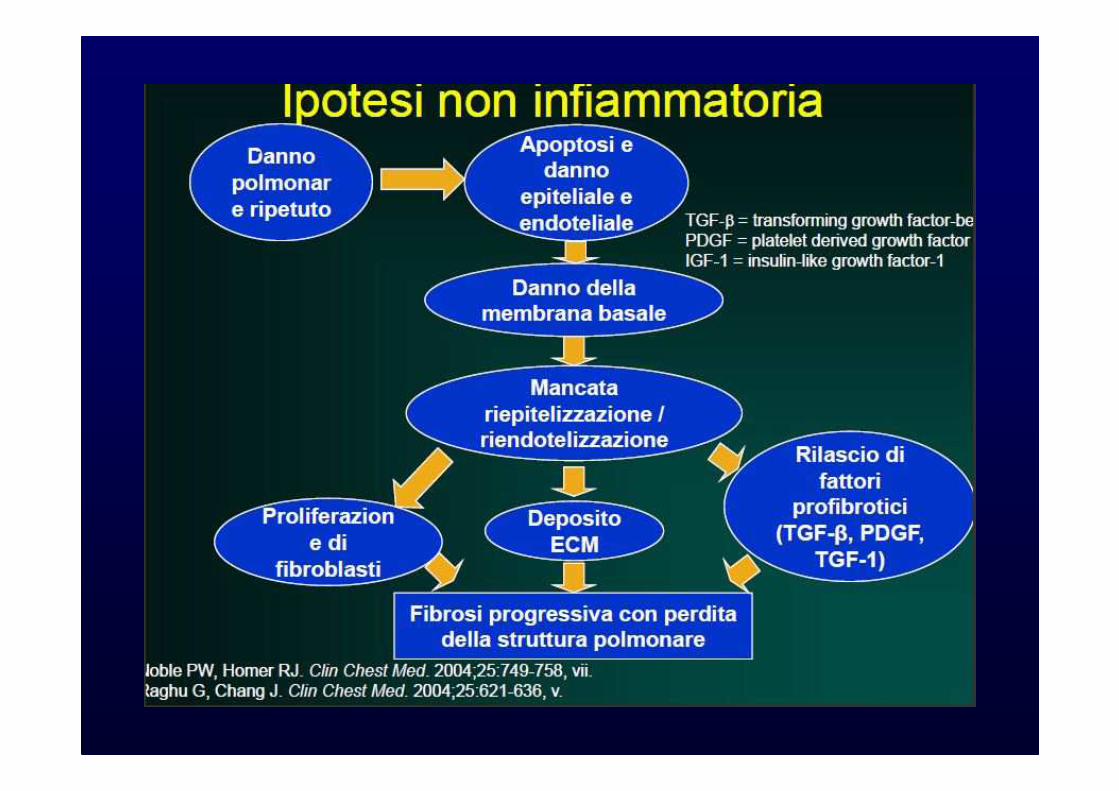
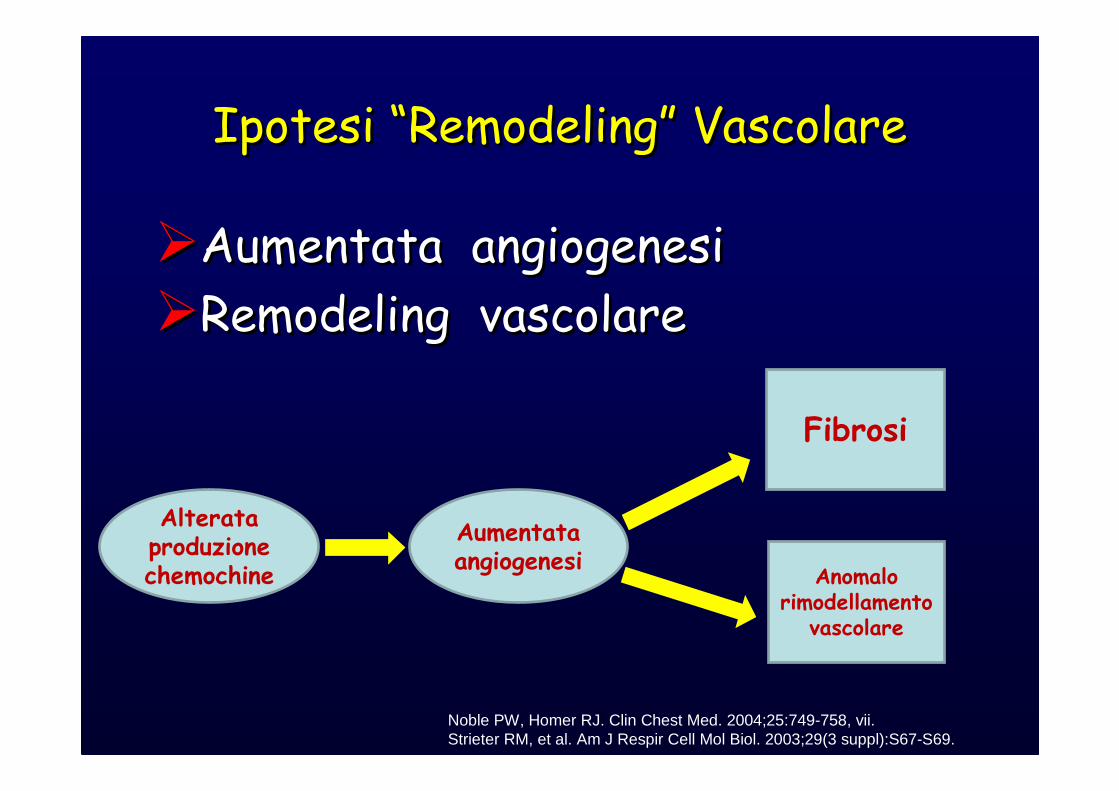
Ipotesi “Remodeling” VascolareIpotesi “Remodeling” Vascolare
�Aumentata angiogenesi
�Remodeling vascolare
�Aumentata angiogenesi
�Remodeling vascolare
Noble PW, Homer RJ. Clin Chest Med. 2004;25:749-758, vii.Strieter RM, et al. Am J Respir Cell Mol Biol. 2003;29(3 suppl):S67-S69.
Alterata produzione chemochine
Aumentata angiogenesi
Fibrosi
Anomalo rimodellamento
vascolare
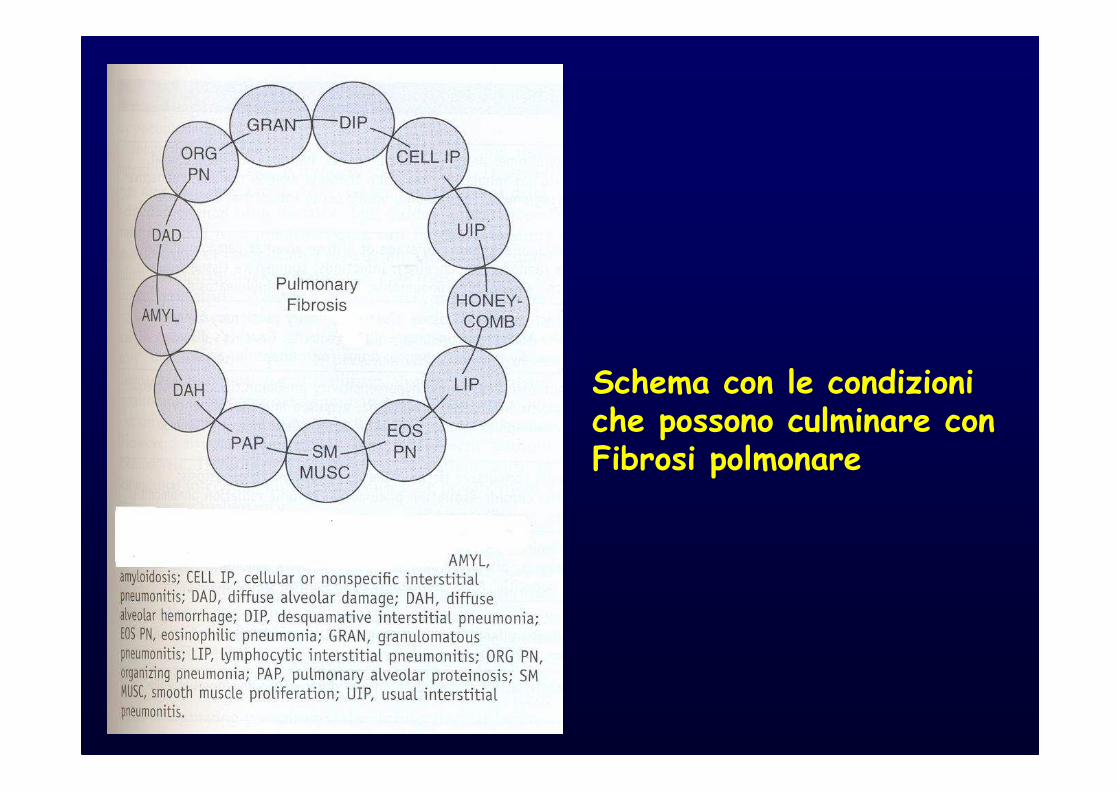
Schema con le condizioni che possono culminare con Fibrosi polmonare

Interstitial Lung Diseases and Underlying Clinical Entities
Interstitial Lung Diseases and Underlying Clinical Entities
Usual interstitial pneumonia (UIP)–like pattern: idiopathic (UIP), collagen vascular disease (scleroderma), asbestos interstitial lung disease.
Desquamative interstitial pneumonitis (DIP): idiopathic.
Lymphoid interstitial pneumonitis (LIP): idiopathic, collagen vascular disease (Sjögren's disease), AIDS, immunosuppression (acquired or hereditary), viral (Epstein-Barr virus [EBV] infection).
Nonspecific interstitial pneumonitis (NSIP): idiopathic; NSIP-like due to hypersensitivity pneumonitis, NSIP-like due to collagen vascular disease (i.e., scleroderma).
Giant cell interstitial pneumonitis (GIP): heavy metal exposure.
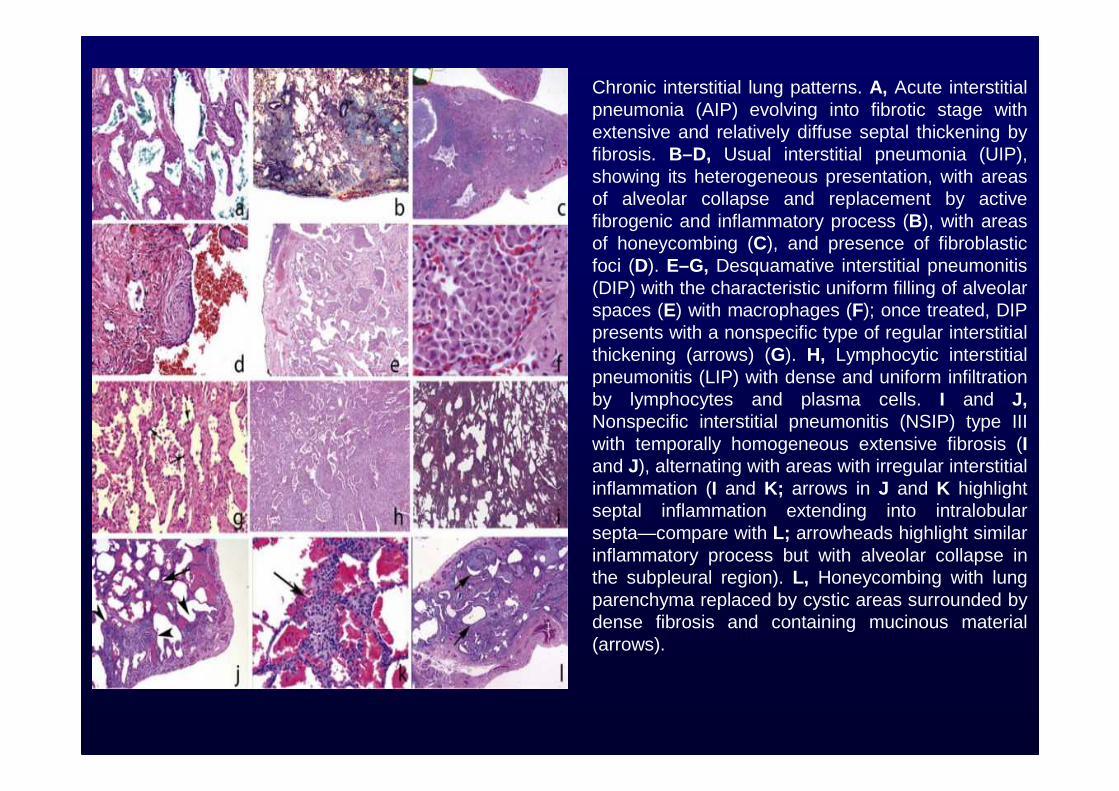
Chronic interstitial lung patterns. A, Acute interstitial pneumonia (AIP) evolving into fibrotic stage with extensive and relatively diffuse septal thickening by fibrosis. B–D, Usual interstitial pneumonia (UIP), showing its heterogeneous presentation, with areas of alveolar collapse and replacement by active fibrogenic and inflammatory process (B), with areas of honeycombing (C), and presence of fibroblastic foci (D). E–G, Desquamative interstitial pneumonitis (DIP) with the characteristic uniform filling of alveolar spaces (E) with macrophages (F); once treated, DIP presents with a nonspecific type of regular interstitial thickening (arrows) (G). H, Lymphocytic interstitial pneumonitis (LIP) with dense and uniform infiltration by lymphocytes and plasma cells. I and J,Nonspecific interstitial pneumonitis (NSIP) type III with temporally homogeneous extensive fibrosis (Iand J), alternating with areas with irregular interstitial inflammation (I and K; arrows in J and K highlight septal inflammation extending into intralobular septa—compare with L; arrowheads highlight similar inflammatory process but with alveolar collapse in the subpleural region). L, Honeycombing with lung parenchyma replaced by cystic areas surrounded by dense fibrosis and containing mucinous material (arrows).
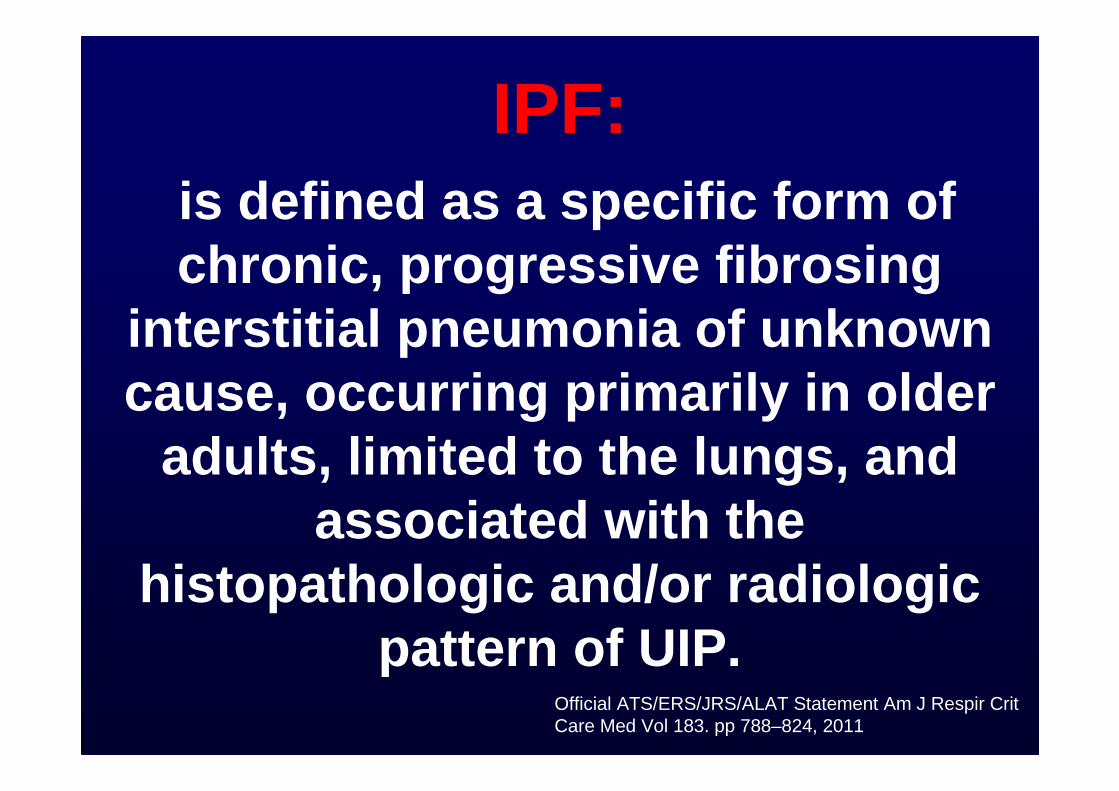
IPF:is defined as a specific form of chronic, progressive fibrosing
interstitial pneumonia of unknown cause, occurring primarily in older
adults, limited to the lungs, and associated with the
histopathologic and/or radiologic pattern of UIP.
Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011

Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
IPF is a fatal lung disease; the natural history is variable and unpredictable:
a.Most patients with IPF demonstrate a gradual worsening of lung function over years; a minority of patients remains stable or declines rapidly.
a.Some patients may experience episodes of acute respiratory worsening despite previous stability.
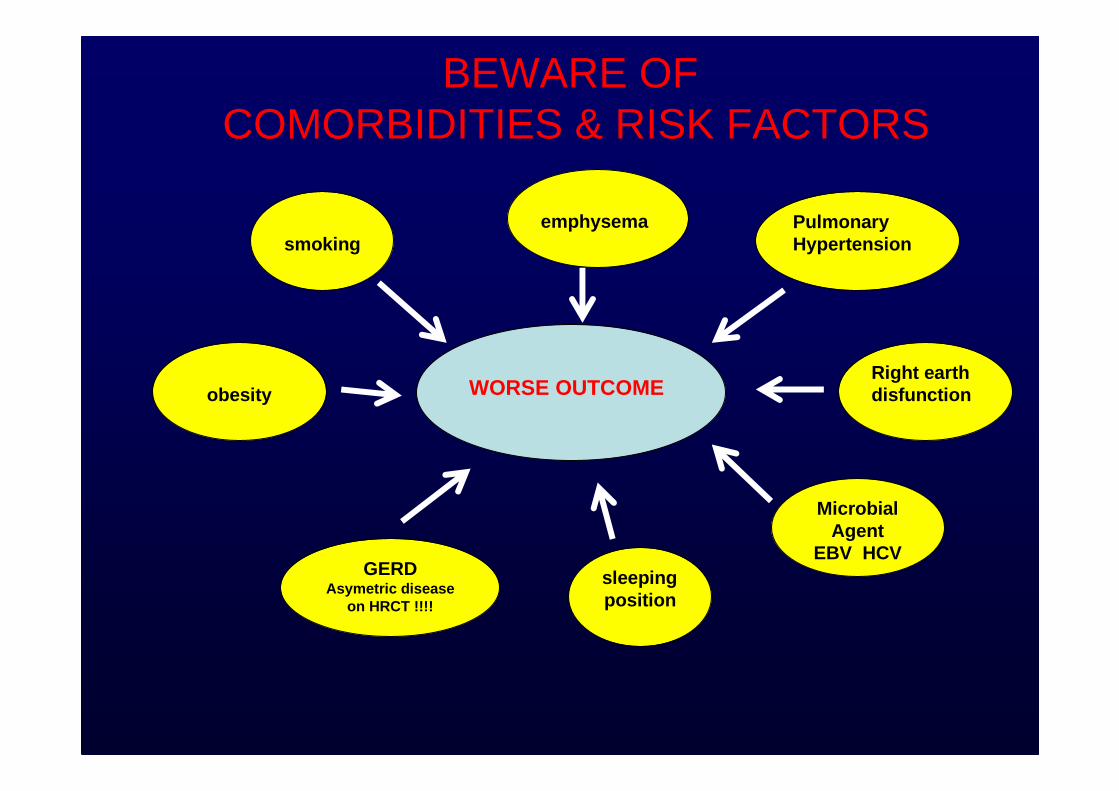
BEWARE OF COMORBIDITIES & RISK FACTORS
WORSE OUTCOME
smoking
sleepingposition
Right earth disfunction
Pulmonary Hypertension
GERDAsymetric disease
on HRCT !!!!
obesity
emphysema
MicrobialAgent
EBV HCV

Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
Disease progression is manifested by increasing
respiratory symptoms, worsening pulmonary function test results,
progressive fibrosis on HRCT, acute respiratorydecline, or death.

Elementi prognostici predittiviElementi prognostici predittivi
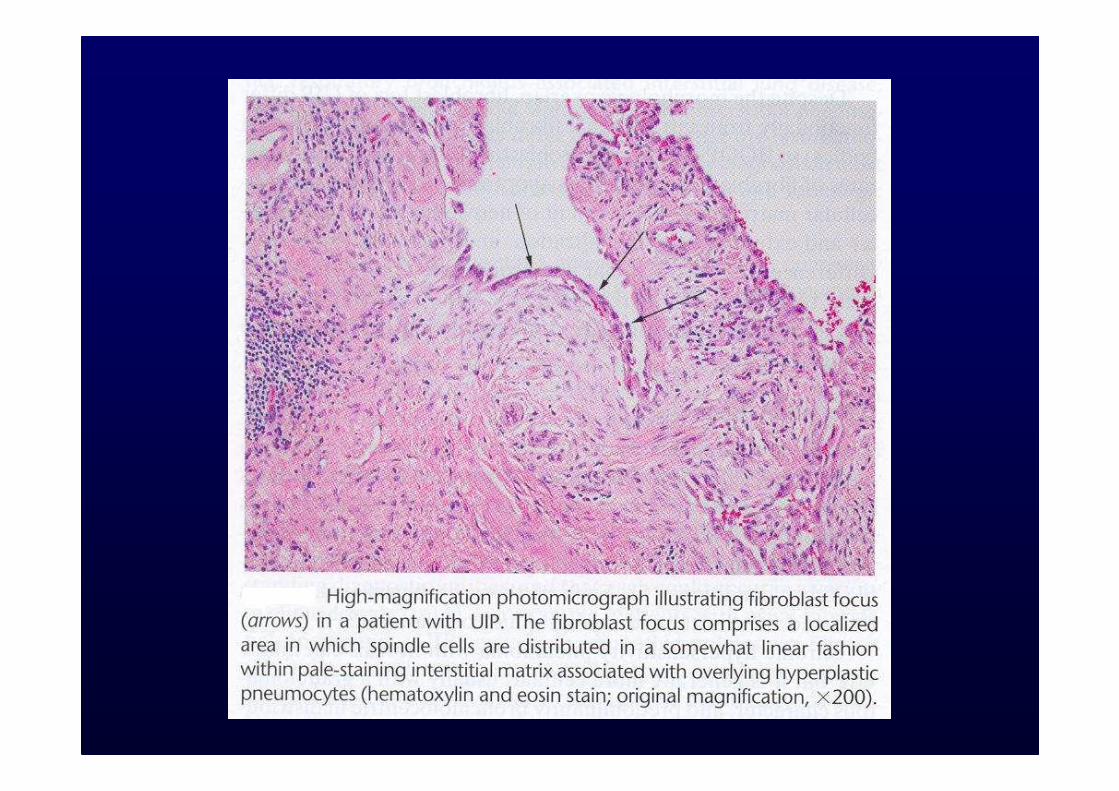
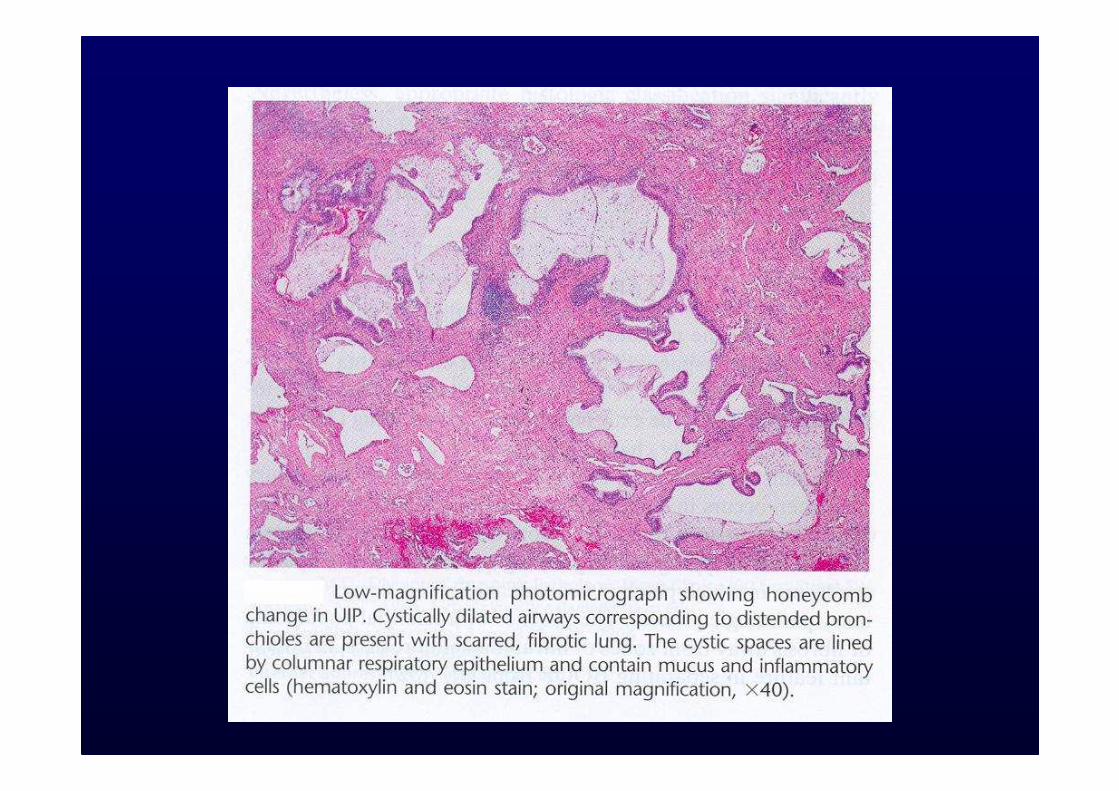
Prognosi scadente:Età avanzata, sesso maschile, grado di dispnea, fumo, alterazioni funzionali, neutrofilia e/o eosinofilia al BAL, polmone a nido d’ape, presenza di foci fibroblastici nei campioni bioptici.
Prognosi scadente:Età avanzata, sesso maschile, grado di dispnea, fumo, alterazioni funzionali, neutrofilia e/o eosinofilia al BAL, polmone a nido d’ape, presenza di foci fibroblastici nei campioni bioptici.

Caratteristiche cliniche, radiologiche e funzionali generali delle InterstiziopatieCaratteristiche cliniche, radiologiche e funzionali generali delle Interstiziopatie
a) Dispnea ingravescente e tosse secca
b) Alterazioni radiografiche
c) Prove funzionali respiratorie alterate
Non sempre queste caratteristiche sono presenti: 5-10% dei pz. possono presentare una Rx normale.
Pz. sintomatici possono avere prove di funzionalità respiratoria di routine entro i limiti della norma
a) Dispnea ingravescente e tosse secca
b) Alterazioni radiografiche
c) Prove funzionali respiratorie alterate
Non sempre queste caratteristiche sono presenti: 5-10% dei pz. possono presentare una Rx normale.
Pz. sintomatici possono avere prove di funzionalità respiratoria di routine entro i limiti della norma

IPF- Quadro clinicoIPF- Quadro clinico
Sintomi principali:Dispnea ingravescente- Tosse seccaSintomi meno frequenti:Astenia, calo ponderale, malessere- Dolore
toracicoSegno clinico principale:Rantoli “a velcro”Segni aggiuntivi:Dita a bacchetta di tamburo- Cianosi
Sintomi principali:Dispnea ingravescente- Tosse seccaSintomi meno frequenti:Astenia, calo ponderale, malessere- Dolore
toracicoSegno clinico principale:Rantoli “a velcro”Segni aggiuntivi:Dita a bacchetta di tamburo- Cianosi
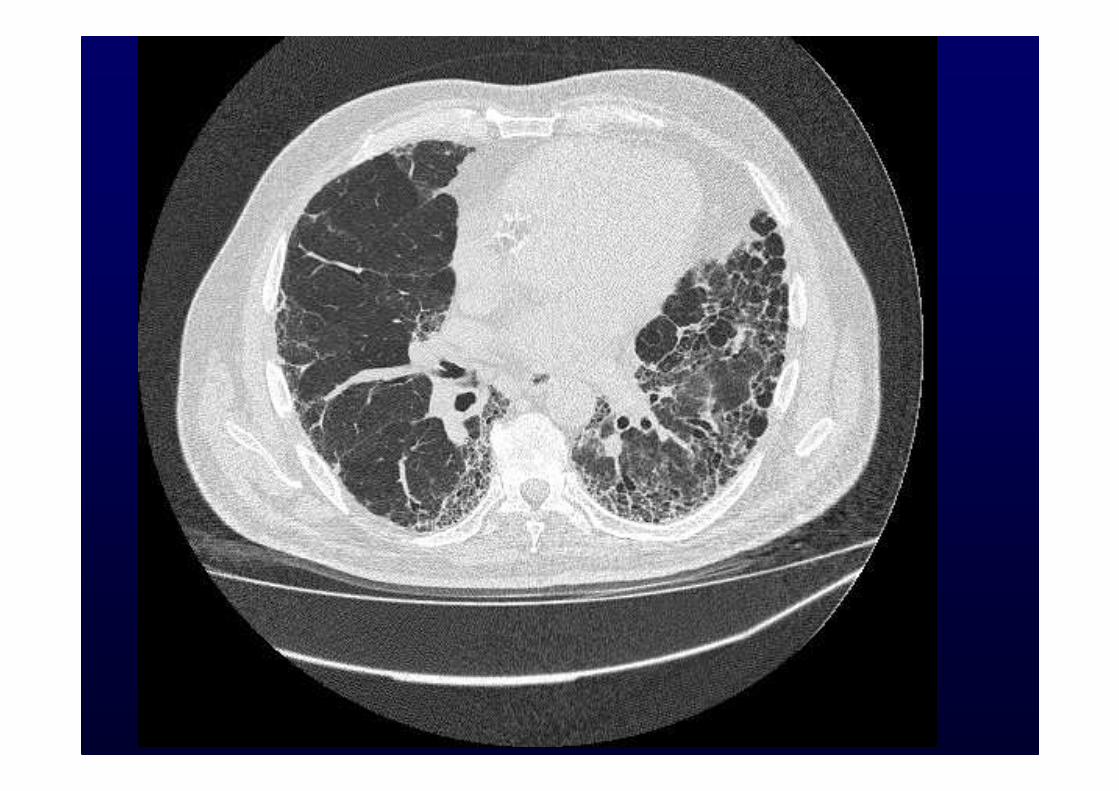
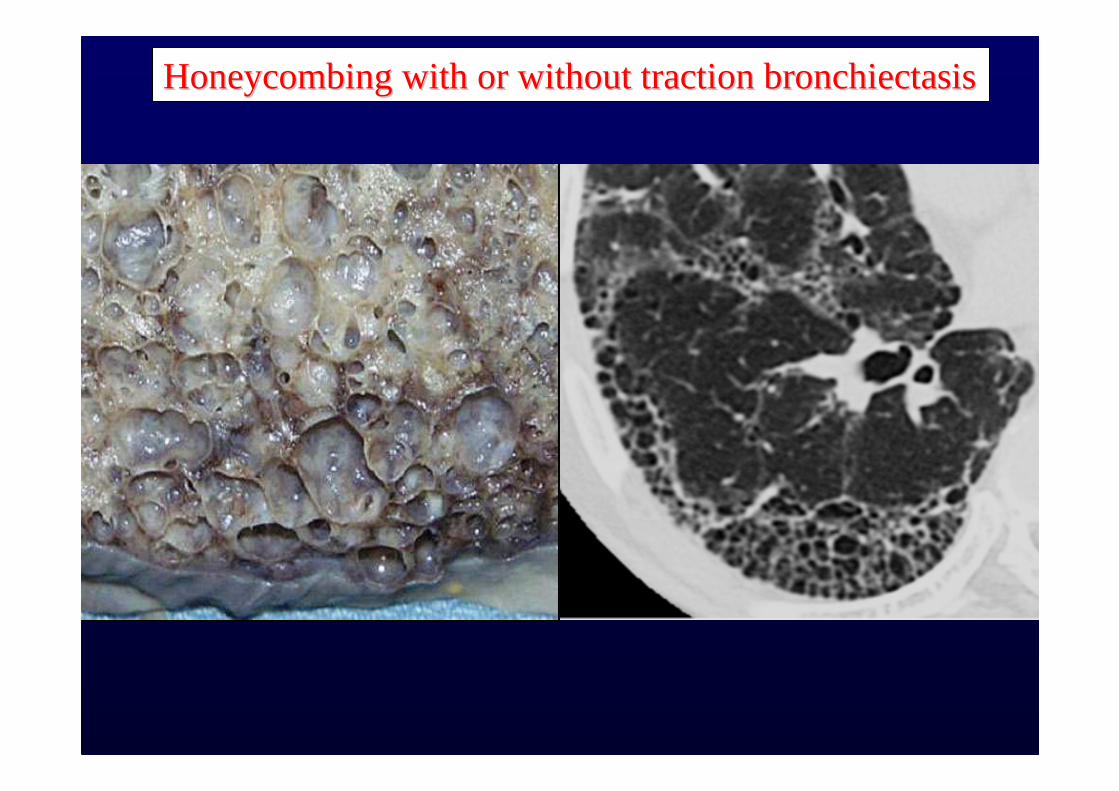
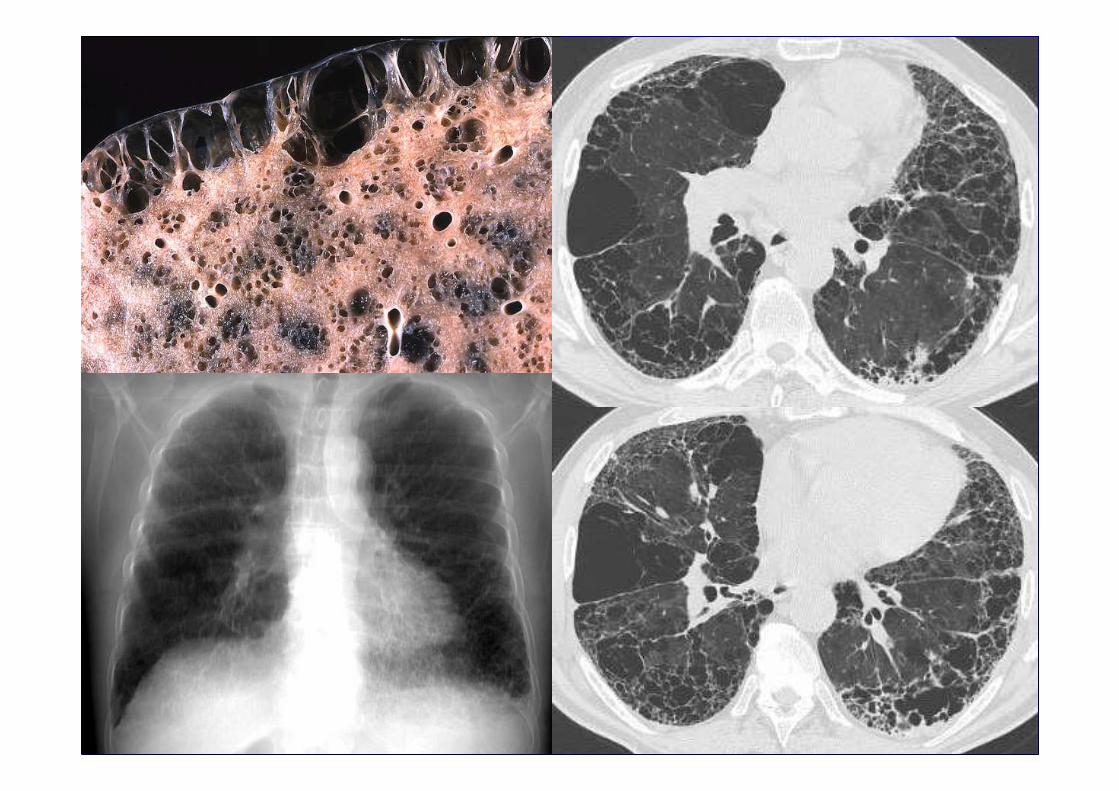
Honeycombing with or without traction bronchiectasisHoneycombing with or without traction bronchiectasis
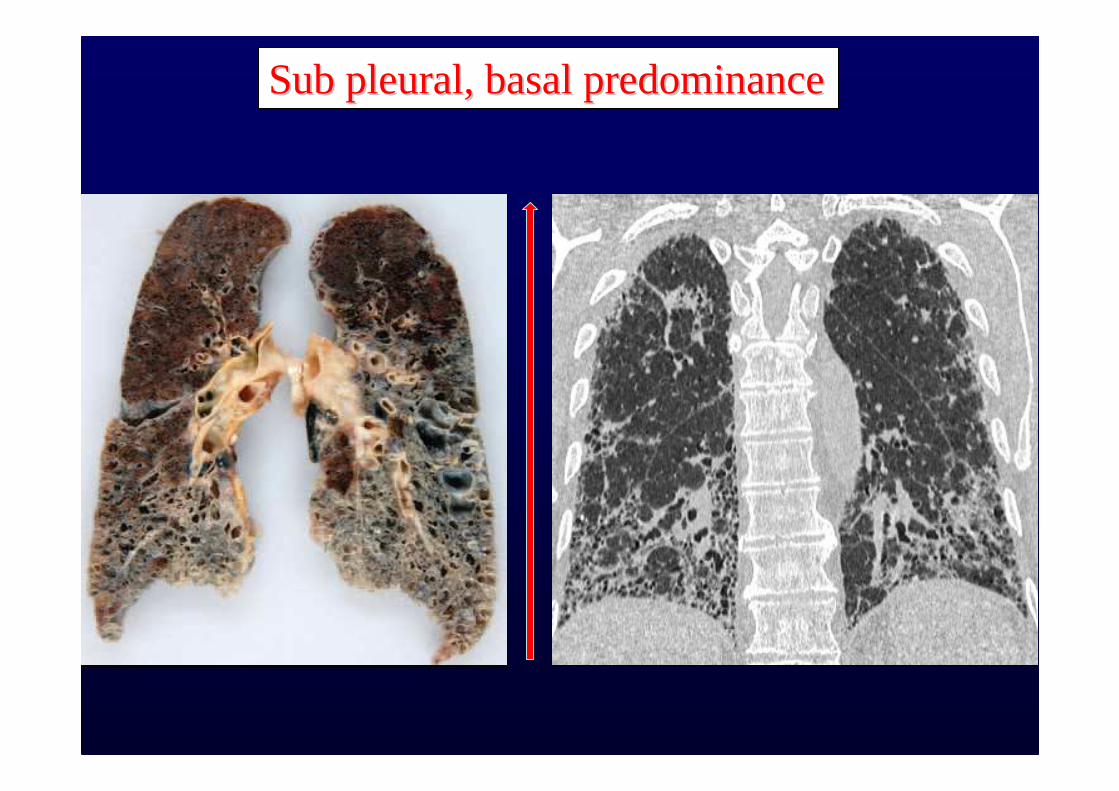
Sub pleural, basal predominanceSub pleural, basal predominance
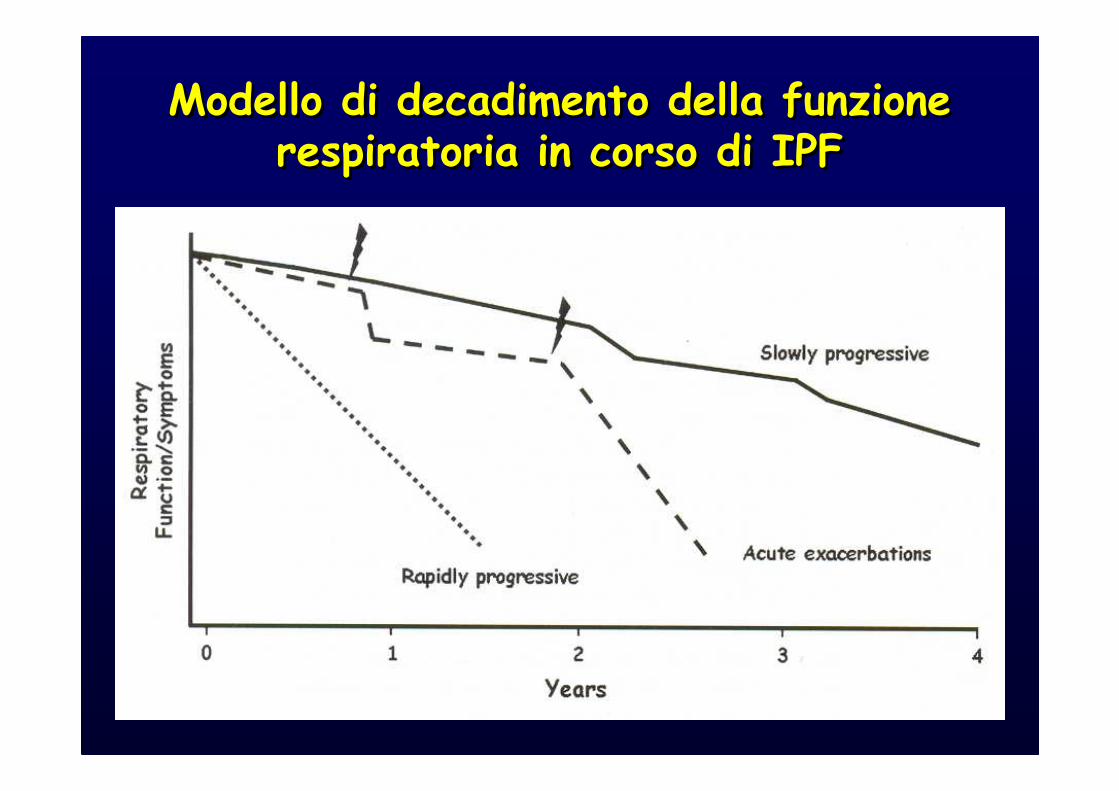
Modello di decadimento della funzione respiratoria in corso di IPF
Modello di decadimento della funzione respiratoria in corso di IPF
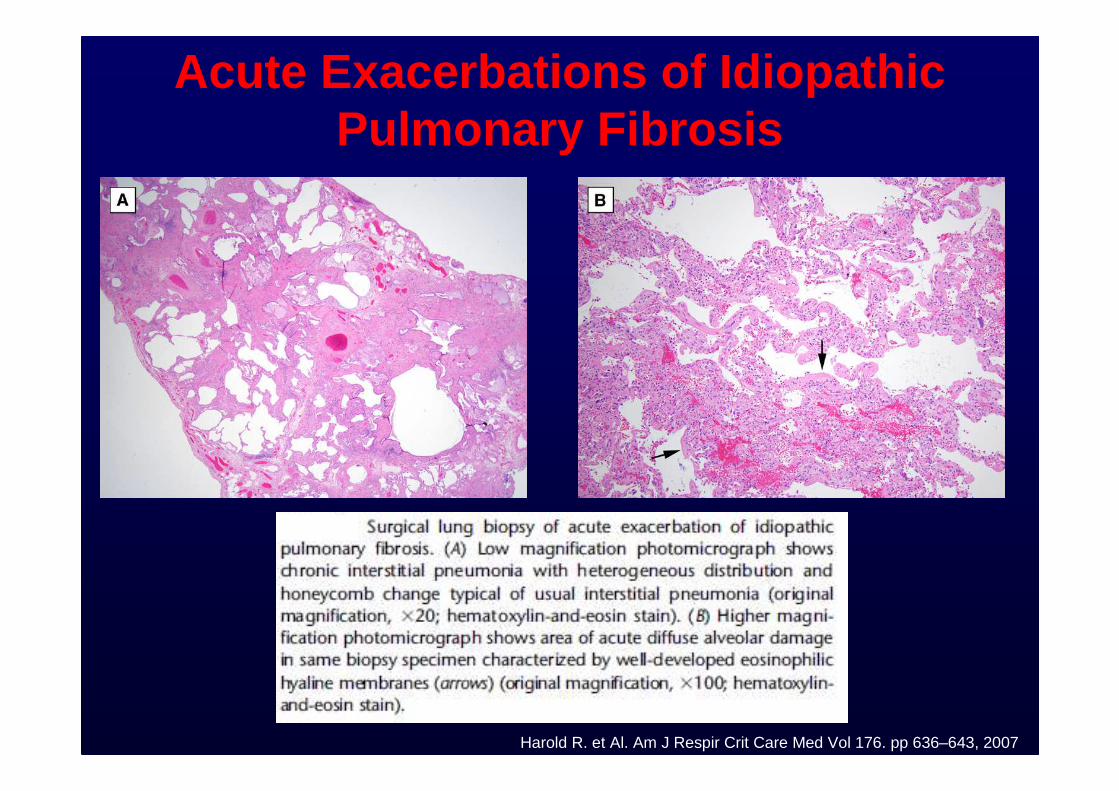
Acute Exacerbations of Idiopathic Pulmonary Fibrosis
Harold R. et Al. Am J Respir Crit Care Med Vol 176. pp 636–643, 2007

Acute Exacerbations of Idiopathic Pulmonary Fibrosis
Harold R. et Al. Am J Respir Crit Care Med Vol 176. pp 636–643, 2007

SELECTED FEATURES ASSOCIATED WITH INCREASED RISK OF MORTALITY IN IDIOPATHIC PULMONARY
FIBROSIS
Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
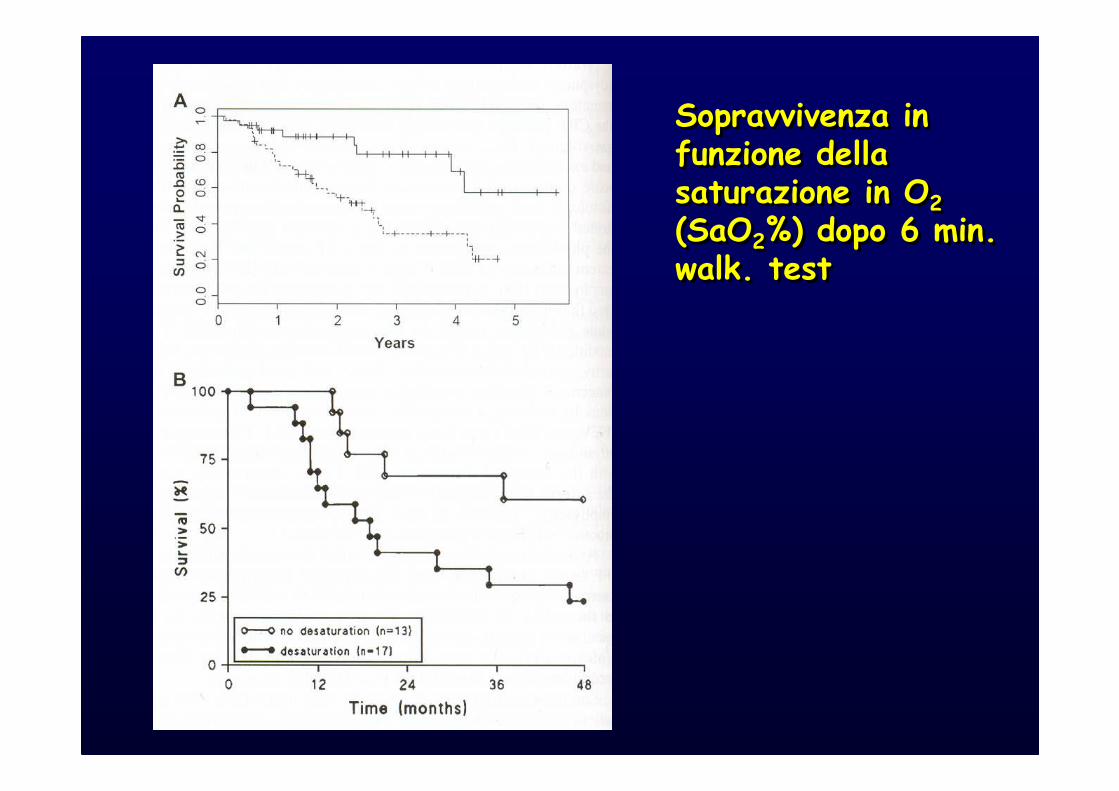
Sopravvivenza in funzione della saturazione in O2
(SaO2%) dopo 6 min. walk. test
Sopravvivenza in funzione della saturazione in O2
(SaO2%) dopo 6 min. walk. test
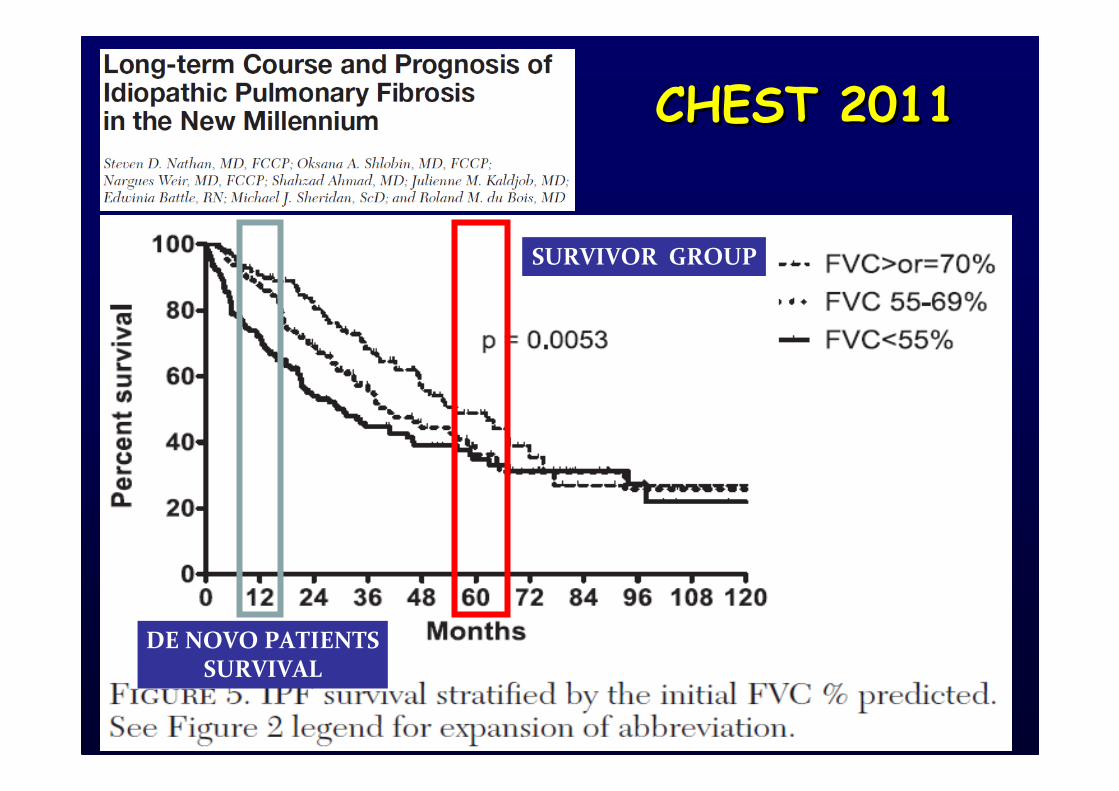
CHEST 2011CHEST 2011
SURVIVOR GROUP
DE NOVO PATIENTS
SURVIVAL
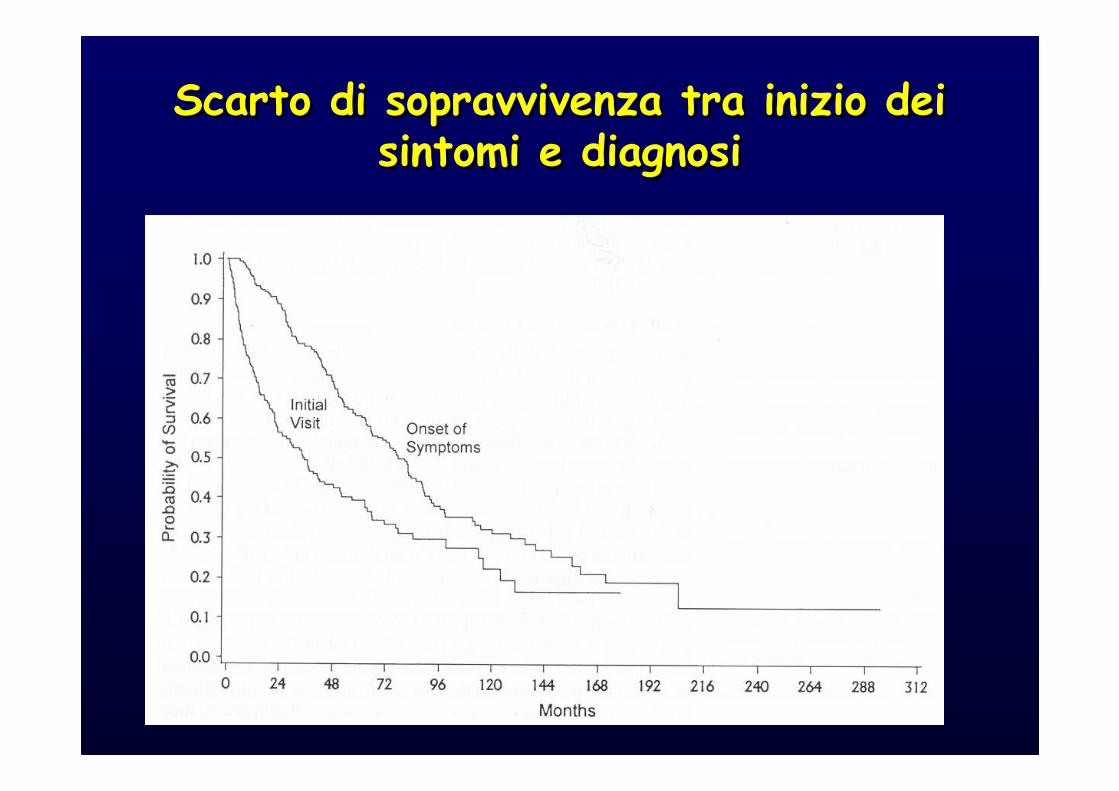
Scarto di sopravvivenza tra inizio dei sintomi e diagnosi
Scarto di sopravvivenza tra inizio dei sintomi e diagnosi
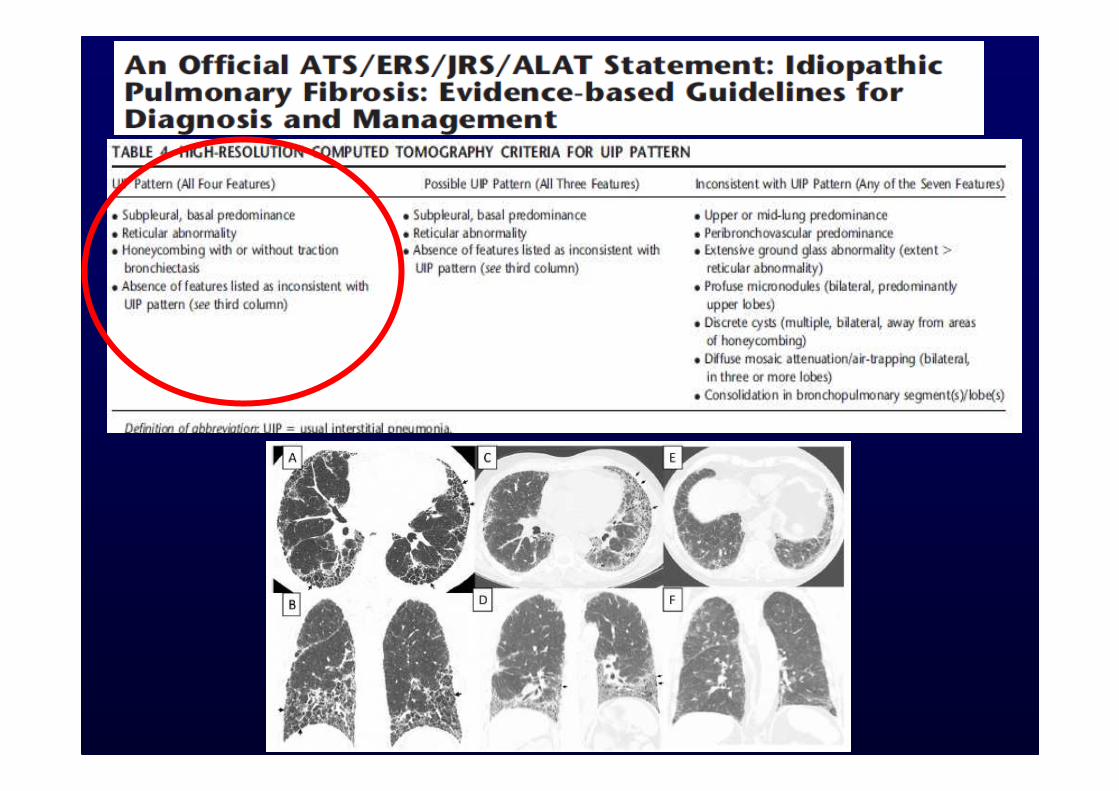
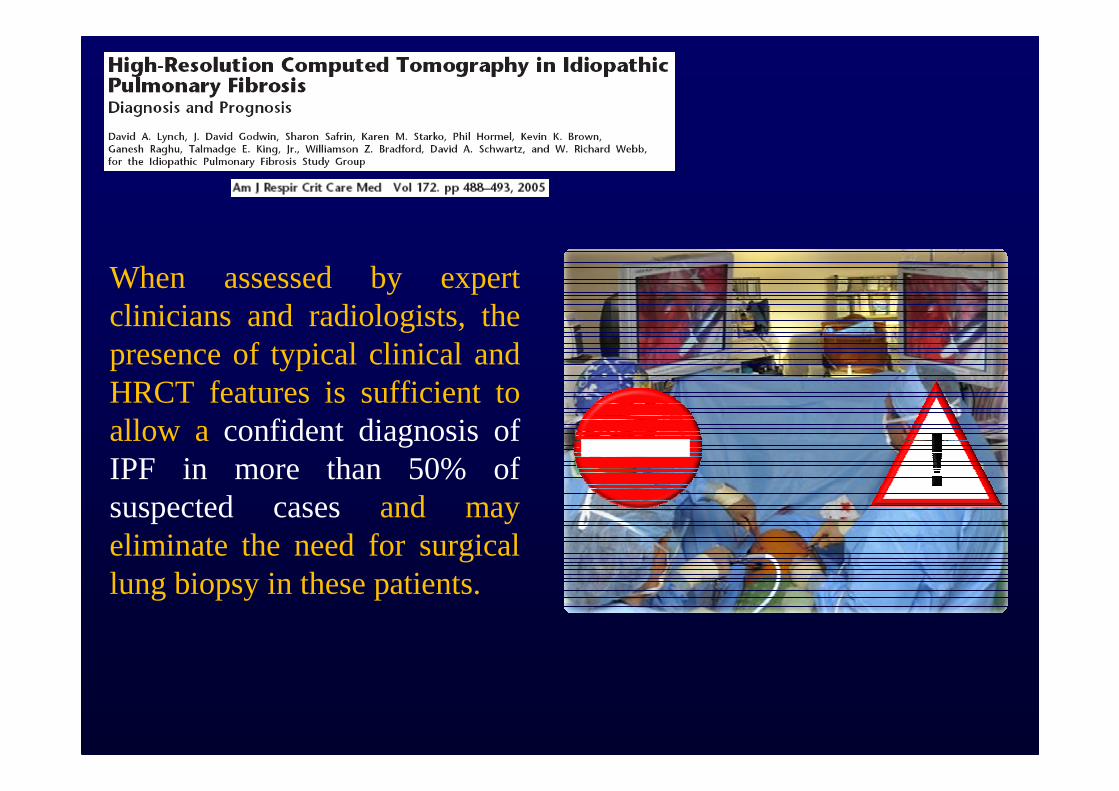
When assessed by expert clinicians and radiologists, the presence of typical clinical and HRCT features is sufficient to allow a confident diagnosis of IPF in more than 50% of suspected cases and may eliminate the need for surgical lung biopsy in these patients.

Biopsy is required only when the HRCT scan and/or the clinical features are not typical of UIP, a situation occurring in <50% of patients with IPF.
However, HRCT maintains a role in determining the most appropriate site of biopsy, and the prognosis is further refined when histological data are integrated with HRCT and clinico-functional parameters.
2010
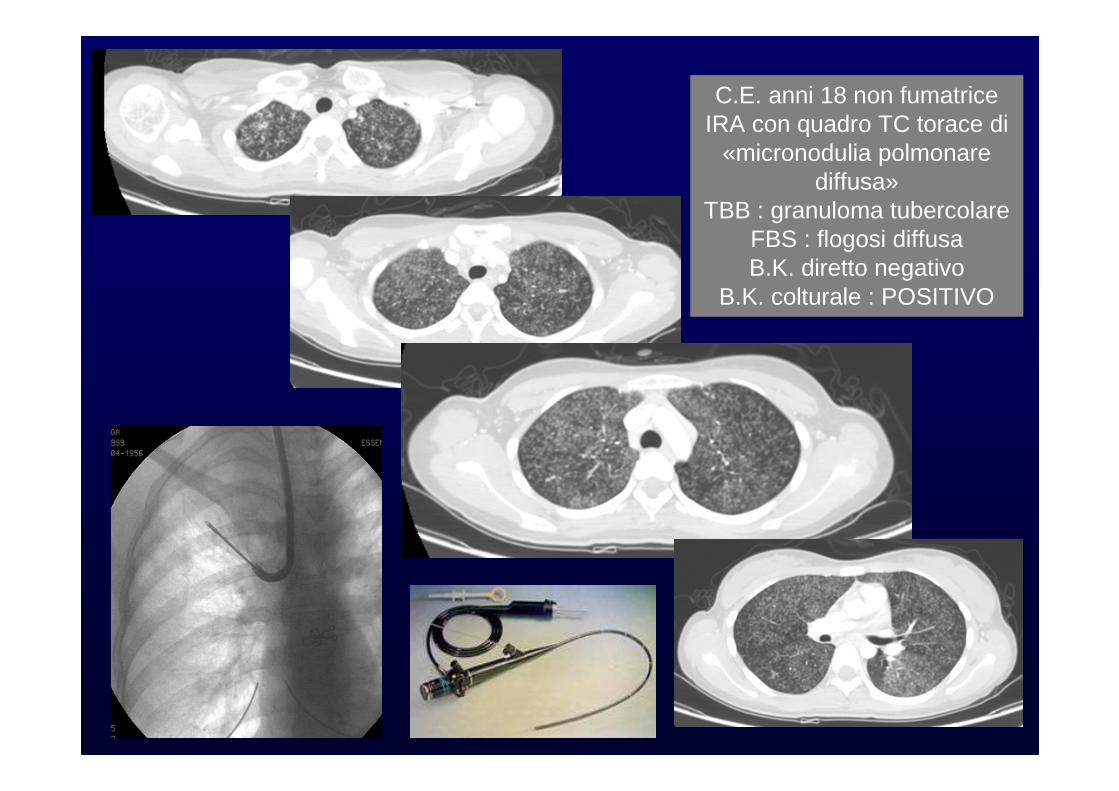
C.E. anni 18 non fumatriceIRA con quadro TC torace di
«micronodulia polmonare diffusa»
TBB : granuloma tubercolareFBS : flogosi diffusa B.K. diretto negativo
B.K. colturale : POSITIVO

Criteri diagnosticiCriteri diagnosticiDiagnosi altamente probabile (spec.>95%) quando siano soddisfatti 4 criteri maggiori ed almeno 3 dei criteri minori.Criteri maggiori:-Esclusione di altre cause di interstiziopatia-Anormalità delle prove di funzionalità respiratoria (S. restrittiva e < DLCO)-Quadro HRCT caratteristico-Assenza di ipotesi alternative su campioni bioptici o su BALCriteri minori:-Età > 50 anni-Inizio subdolo e dispnea da sforzo non altrimenti spiegabili-Durata > 3 mesi-Rantoli “a velcro”
HRCT da solo è caratteristico nel 60-75% dei casi
Diagnosi altamente probabile (spec.>95%) quando siano soddisfatti 4 criteri maggiori ed almeno 3 dei criteri minori.Criteri maggiori:-Esclusione di altre cause di interstiziopatia-Anormalità delle prove di funzionalità respiratoria (S. restrittiva e < DLCO)-Quadro HRCT caratteristico-Assenza di ipotesi alternative su campioni bioptici o su BALCriteri minori:-Età > 50 anni-Inizio subdolo e dispnea da sforzo non altrimenti spiegabili-Durata > 3 mesi-Rantoli “a velcro”
HRCT da solo è caratteristico nel 60-75% dei casi

Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
The diagnosis of IPF requires:
A.Exclusion of other known causes of interstitial lung disease (ILD) (e.g., domestic and occupational environmental exposures, connective tissue disease, and drug toxicity).
B.The presence of a UIP pattern on high-resolution computed tomography (HRCT) in patients not subjected to surgical lung biopsy.
C.Specific combinations of HRCT and surgical lung biopsy pattern in patients subjected to surgical lung biopsy.

Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
The accuracy of the diagnosis of IPF
Increases with multidisciplinary discussion between pulmonologists,
radiologists,and pathologists experienced in the
diagnosis of ILD.
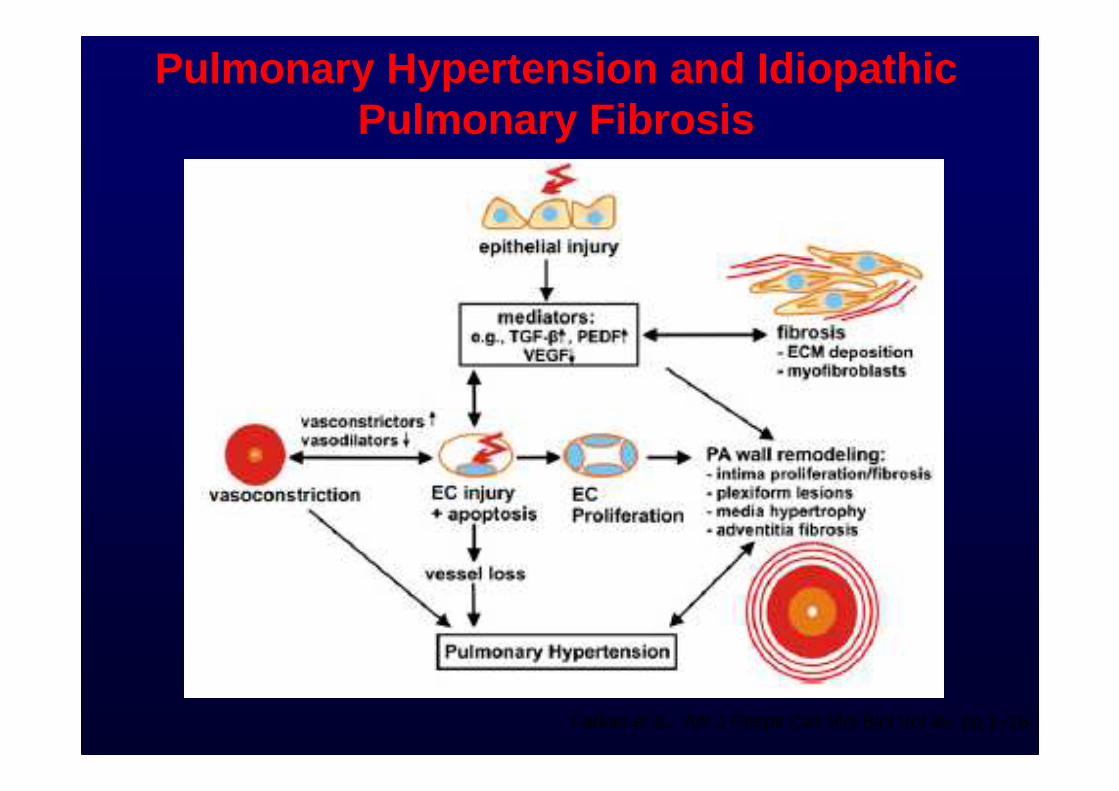
Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis
Farkas et al. Am J Respir Cell Mol Biol Vol 45. pp 1–15, 2011

Idiopathic Pulmonary Fibrosis And
Non-Specific Interstitial Pneumonia
Idiopathic Pulmonary Fibrosis And
Non-Specific Interstitial Pneumonia

Official ATS/ERS/JRS/ALAT Statement Am J Respir Crit Care Med Vol 183. pp 788–824, 2011
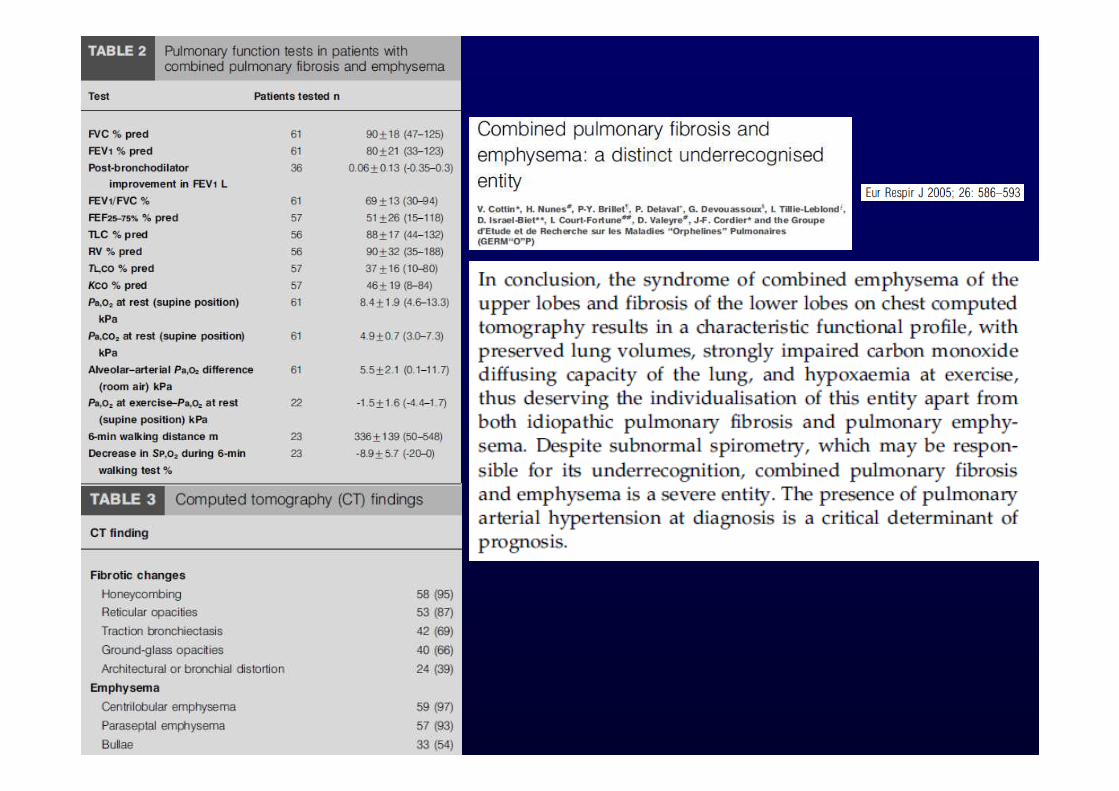
Patients with IPF may have sub -clinical or overt comorbid conditions including pulmonary hypertension, gastroesophageal reflux, obstructive sleep apnea, obesity, and emphysema. The impact of these conditions on the outcome of patients with IPF is unclear.





























